Настоящее изобретение относится к способу получения N-фосфонометилглицина, осуществляемому взаимодействием гексагидротриазинового соединения с триацилфосфитом, а также к промежуточным продуктам для использования в этом способе.
N-фосфонометилглицин (глифосат) представляет собой широко распространенный гербицид сплошного (общеистребительного) действия. Известны многочисленные способы получения фосфонометилглицина. Одной из таких возможностей его получения является реакция, осуществляемая взаимодействием производных гексагидротриазина с эфирами фосфористой кислоты. Так, в частности, в патенте US 4181800 описано получение гексагидротриазинов формулы
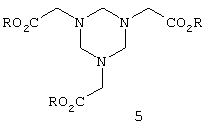
а в патенте US 4053505 описывается взаимодействие этих гексагидротриазинов с диэфирами фосфористой кислоты и последующий гидролиз полученного продукта с получением в результате фосфонометилглицина. Однако было установлено, что как выход, так и избирательность действия требуют дальнейшего улучшения касательно монофосфорирования продукта. Кроме того, диэфиры фосфористой кислоты являются крайне дорогостоящими продуктами.
В заявке ЕР-А 104775 (соответственно в патентах US 4425284, US 4482504 и US 4535181) описывается реакция, осуществляемая взаимодействием вышеназванных гексагидротриазинов с соответствующим ацилгалогенидом, и последующее фосфонирование с помощью триэфира фосфористой кислоты и омыление с получением в результате фосфонометилглицина согласно следующей схеме:
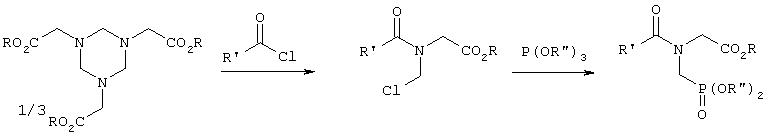
Хотя таким путем фосфонометилглицин и удается получать с относительно высоким выходом, способ тем не менее наряду с использованием дорогостоящих эфиров фосфористой кислоты предусматривает также дополнительное применение хлорангидрида карбоновой кислоты. Помимо этого, возможность рекуперации хлорангидрида карбоновой кислоты при необходимости в виде свободной кислоты и последующий перевод на отдельной стадии снова в хлорангидрид кислоты также значительно удорожает способ. Кроме того, не представляется возможным полностью рекуперировать спирт, которым этерифицируют фосфористую кислоту, поскольку во время реакции образуется эквивалент соответствующего алкилхлорида, который помимо прочего токсикологически небезопасен.
В патенте US 4428888 (соответственно ЕР-А 149294) описана реакция, осуществляемая взаимодействием вышеуказанного гексагидротриазина с хлорангидридом фосфористой кислоты в присутствии сильной безводной кислоты, например хлористого водорода, и C1-С6карбоновой кислоты, такой как уксусная кислота. При проведении этой реакции образуются многочисленные не поддающиеся точному определению побочные продукты, снижающие выход фосфонометилглицина и обусловливающие существенные затраты на очистку получаемого продукта.
В патенте US 4442044 описывается реакция, осуществляемая взаимодействием гексагидротриазина формулы 5 с триэфиром фосфористой кислоты с получением в результате соответствующего фосфонатного соединения, применяемого в качестве гербицида.
В DD-A 141929 и DD-A 118435 описана реакция, осуществляемая взаимодействием соли щелочного металла вышеназванного гексагидротриазина (R обозначает, например, Na) с диэфиром фосфористой кислоты. Однако из-за в принципе плохой растворимости солей щелочных металлов не удается достичь удовлетворительной степени химического превращения.
В патенте US 5053529 описывается получение фосфонометилглицина взаимодействием вышеназванных гексагидротриазинов с триэфирами фосфористой кислоты в присутствии тетрахлорида титана и последующим омылением полученного продукта. Однако применение тетрахлорида титана существенно удорожает технологический процесс. Кроме того, выход фосфонометилглицина остается неудовлетворительным.
В патентах US 4454063, US 4487724 и US 4429124 описывается получение фосфонометилглицина взаимодействием соединения формулы
где R1 и R2 представляют собой ароматические или алифатические группы с RCOX (X обозначает Cl, Br, I) с получением соединения формулы
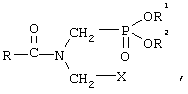
последующей реакцией этого соединения с цианидом металла и гидролизом полученного продукта. Недостатки этого способа связаны, как и в рассмотренном выше случае, с применением хлорангидрида кислоты.
В литературе описаны также другие варианты синтеза, осуществляемого исходя из замещенного цианометилом гексагидротриазина формулы
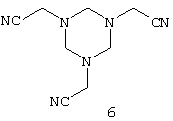
Так, в частности, в патентах US 3923877 и US 4008296 описывается реакция, осуществляемая взаимодействием этого производного гексагидротриазина с диалкилфосфонатом в присутствии кислотного катализатора, такого как хлористый водород, кислоты Льюиса, хлорангидрида либо ангидрида карбоновой кислоты с получением соединения формулы

В результате последующего гидролиза получают фосфонометилглицин, при этом образуется от 8 до 10% дважды фосфонометилированного продукта.
В патентах US 4067719, US 4083898, US 4089671 и в заявке DE-A 2751631 описана реакция, осуществляемая взаимодействием замещенного цианометилом гексагидротриазина с диарилфосфонатом без использования катализатора с получением соединения 9, где R" обозначает арил. Этому способу присущи те же недостатки, что и в случае применения замещенного карбоксигруппой гексагидротриазина в приведенной выше формуле 5.
В заявке ЕР-А 097522 (соответственно в патентах US 4476063 и US 4534902) описывается реакция, осуществляемая согласно приведенной ниже схеме взаимодействием гексагидротриазина 6 с ацилгалогенидом с получением соединения 10, последующим фосфонированием с помощью триэфира либо диэфира фосфористой кислоты с получением соединения 11 и завершающим омылением с получением фосфонометилглицина
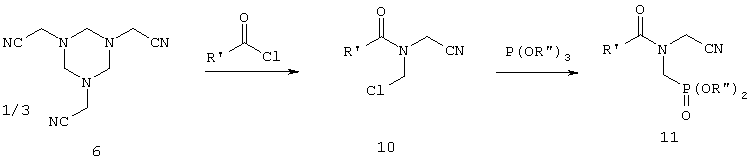
Для этого варианта характерны такие же недостатки, которые присущи способу, где применяют замещенные карбоксигруппой производные гексагидротриазина.
В заключение следует назвать патент US 4415503, где описывается превращение замещенного цианометилом гексагидротриазина аналогично способу, представленному в патенте US 4428888. В этом случае также наблюдается значительное образование побочных продуктов.
В заявке ЕР 164923 А описывается более совершенный гидролиз соединения формулы 11.
С учетом вышеизложенного в основу настоящего изобретения была положена задача разработать простой в осуществлении и экономичный способ получения фосфонометилглицина, который помимо прочего обеспечивал бы высокую степень чистоты этого целевого продукта.
Неожиданным образом было установлено, что указанную задачу удается решить взаимодействием производного гексагидротриазина с триацилфосфитом и последующим омылением полученного продукта с получением в результате требуемого фосфонометилглицина.
В соответствии с этим настоящее изобретение относится к способу получения N-фосфонометилглицина, заключающемуся в том, что
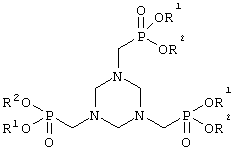
а) производное гексагидротриазина формулы II
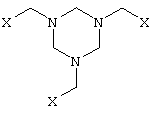
в которой
Х представляет собой CN, COOZ, CONR1R2 или CH2OY, где
Y представляет собой Н или остаток, легко заменяемый на Н,
Z представляет собой Н, щелочной металл, щелочноземельный металл,
С1-С18алкил или арил, необязательно замещенный С1-С4алкилом, NO2 либо
OC1-С4алкилом,
R1 и R2, которые могут иметь идентичные либо разные значения, обозначают Н
или С1-С4алкил,
подвергают взаимодействию с триацилфосфитом формулы III

в которой радикалы R3 могут иметь идентичные либо разные значения и обозначают С1-С18алкил или арил, необязательно замещенный С1-С4алкилом, NO2 либо ОС1-С4алкилом, с получением соединения формулы I
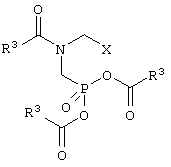
в которой R3 и Х имеют указанные выше значения, и
б) соединение формулы I гидролизуют и, если Х представляет собой CH2OY, окисляют.
Изобретение относится далее к соединениям формулы I, а также к их получению согласно стадии а) способа получения фосфонометилглицина.
Под алкилом подразумевается неразветвленная либо разветвленная алкильная цепь предпочтительно с 1-8 атомами углерода и прежде всего с 1-4 атомами углерода. В качестве примеров алкила можно назвать метил, этил, н-пропил, изопропил, н-бутил, изобутил, втор-бутил, трет-бутил, н-гексил, 2-этилгексил и т.д.
Под арилом имеются в виду предпочтительно фенил и нафтил.
Х представляет собой предпочтительно CN или COOZ.
Z представляет собой предпочтительно Н, щелочной металл или C1-С18алкил.
Если Y представляет собой остаток, легко заменяемый на Н, то имеется в виду предпочтительно алифатический или ароматический ацильный остаток либо C1-С6алкильная группа. Алифатический остаток представляет собой предпочтительно C1-С6-СО-остаток, а ароматический ацильный остаток представляет собой предпочтительно бензоил.
R1 и R2 обозначают предпочтительно Н.
R3 особенно предпочтительно представляет собой арильный остаток, который необязательно, как указывалось выше, может быть замещенным. Особенно предпочтительными значениями R3 являются фенил, n-толил и n-нитрофенил.
Соединения формулы II известны и их можно получать известным образом или же аналогично известным способам (см., например, вышеприведенный уровень техники). Так, в частности, амин Х-СН2-NH2 можно подвергать взаимодействию с источником формальдегида, таким как водный раствор формалина или параформальдегид, например, растворяя первичный амин в водном растворе формалина. Требуемый гексагидротриазин можно затем получать путем кристаллизации или выпаривания воды. Этот способ описан в заявке DE-A 2645085, соответственно в патенте US 4181800, при этом указанные публикации в полном объеме включены в настоящее описание в качестве ссылки.
Соединения формулы II, в которой Х представляет собой CN, можно получать по реакции Штреккера, т.е. взаимодействием аммиака, синильной кислоты и источника формальдегида. Способ такого типа описан, например, в патенте US 2823222, который в полном объеме включен в настоящее описание в качестве ссылки.
Соединения формулы III можно получать с помощью нескольких способов. Одной из таких возможностей является реакция, осуществляемая взаимодействием соответствующей соли карбоновой кислоты R3СООН с тригалогенидом фосфора, прежде всего с трихлоридом фосфора. В качестве соли карбоновой кислоты используют предпочтительно соль щелочного или щелочноземельного металла, прежде всего соль натрия, калия либо кальция, или соль аммония. Указанную реакцию можно проводить без использования растворителя и полученный продукт непосредственно применять на стадии а). Предпочтительно, однако, работать в инертном органическом растворителе, прежде всего в простом эфире, таком как диоксан, тетрагидрофуран и т.п., в галогенированном, прежде всего хлорированном либо фторированном органическом растворителе, таком как дихлорметан, 1,2-дихлорэтан, 1,2-дихлорпропан, 1,1,1-трихлорэтан, 1,1,2-трихлорэтан, 1,1,2,2-тетрахлорэтан, хлорбензол или 1,2-дихлорбензол, в алифатическом или ароматическом углеводороде, таком как н-октан, толуол, ксилол или нитробензол. Предпочтительно использовать тот же растворитель, что и предусмотренный для стадии а). Особенно предпочтительно использовать хлорированный углеводород.
Образующуюся при проведении реакции соль, например хлорид натрия в случае использования трихлорида фосфора и натриевой соли применяемой карбоновой кислоты, по завершении реакции можно удалять. Если в качестве соли получают хлорид аммония или какой-либо другой галогенид аммония, то используемый аммиак можно рекуперировать за счет установления рН водного раствора соли сильным основанием, например едким натром, на щелочное значение (рН 11-14) и последующей отгонки аммиака обычным путем. Полученный таким образом аммиак после сушки, например перегонкой в жидком или газообразном состоянии, либо в виде водного раствора можно повторно возвращать в технологический цикл и использовать для получения аммониевой соли карбоновой кислоты.
Еще одна возможность получения соединений формулы III состоит в том, что карбоновую кислоту R3СООН подвергают взаимодействию с тригалогенидом фосфора в присутствии соответствующего амина. В качестве амина используют прежде всего алифатические либо циклоалифатические ди- или триамины, такие как триэтиламин, трибутиламин, диметилэтиламин либо диметилциклогексиламин, а также пиридин. Как правило, в способе такого типа работают в органическом растворителе. Пригодны для использования в этих целях растворители, указанные выше для первого варианта получения соединений формулы III. Предпочтительно использовать диоксан, 1,2-дихлорпропан, 1,2-дихлорэтан, нитробензол или толуол. При использовании растворителя образующийся гидрохлорид амина выпадает в осадок и его можно отфильтровывать. При обработке гидрохлоридов амина сильным основанием, например водным раствором едкого натра, амины выделяются в свободном виде из гидрохлорида. Летучие амины можно затем рекуперировать путем перегонки или экстракции. Нелетучие амины можно рекуперировать путем экстракции либо в случае образования при выделении аминов двухфазной смеси за счет разделения фаз, твердые амины можно рекуперировать путем отфильтровывания. Рекуперированные амины, необязательно после сушки, можно возвращать в процесс и повторно использовать в способе.
В другом варианте получения соединения формулы III осуществляют взаимодействие карбоновой кислоты R3СООН с тригалогенидом фосфора, прежде всего с трихлоридом фосфора, без добавления основания. При проведении этой реакции образующийся галогеноводород необходимо удалять из реакционной смеси. Эту операцию можно осуществлять обычным образом, например пропусканием потока инертного газа, такого как азот. Выделенный в свободном виде галогеноводород можно затем использовать в виде водного раствора для гидролиза на стадии б).
Стадию а) способа по изобретению можно проводить с использованием растворителя или без такового, например в расплаве. Предпочтительно, однако, использовать инертный органический растворитель, например углеводород, такой как толуол или ксилол, простой эфир, такой как тетрагидрофуран, диоксан или дибутиловый эфир. Особенно предпочтительно работать в галогенированном растворителе, прежде всего в хлорированном, предпочтительно в хлорированном и/или фторированном алифатическом углеводороде, таком как дихлорметан, 1,2-дихлорэтан, 1,2-дихлорпропан, 1,1,1-трихлорэтан, 1,1,2-трихлорэтан, 1,1,2,2-тетрахлорэтан, хлорбензол или 1,2-дихлорбензол. Компоненты реакции целесообразно применять в преимущественно стехиометрических количествах. Вместе с тем тот или иной компонент можно использовать с некоторым избытком, например вплоть до 10%. Температура реакции находится, как правило, в интервале от -10 до +140°С, предпочтительно в интервале от комнатной температуры до 100°С. При соблюдении этих условий для реакции требуется лишь короткий промежуток времени, как правило, реакция в основном полностью завершается по истечении 10-30 мин.
Получаемые на стадии а) соединения формулы I представляют собой важные промежуточные продукты для получения фосфонометилглицина. С этой целью соединения формулы I подвергают гидролизу, который можно проводить в кислых или щелочных условиях, предпочтителен кислотный гидролиз. В качестве кислот применяют при этом прежде всего неорганические кислоты, такие как соляная кислота, серная кислота или фосфорная кислота. Щелочной гидролиз осуществляют, как правило, с использованием гидроксидов щелочных или щелочноземельных металлов, прежде всего гидроксида натрия или калия.
Гидролиз целесообразно осуществлять с помощью водных кислоты или основания. При этом водные кислоту или основание, как правило, добавляют к реакционной смеси, полученной на стадии а). Гидролиз можно осуществлять без растворителя или в присутствии смешивающегося с водой, частично смешивающегося либо не смешивающегося с водой инертного органического растворителя. Предпочтительно использовать растворитель, применяемый на стадии а). В случае применения растворителя на стадии а) получаемую на этой стадии а) реакционную смесь при необходимости после удаления, например, путем отгонки части растворителя целесообразно использовать непосредственно. Вместе с тем возможен и иной подход, а именно: применяемый на стадии а) растворитель удалять полностью и остаток подвергать гидролизу. Рекуперируемый из реакционной смеси растворитель можно повторно использовать при получении соединений формулы III или на стадии а).
Особенно предпочтительно осуществлять гидролиз в двухфазной системе (водная фаза/органическая фаза). При этом используют частично смешивающийся с водой либо не смешивающийся с водой органический растворитель, предпочтительно углеводород, такой как толуол или ксилол, простой эфир, такой как дибутиловый эфир, но прежде всего галогенированный углеводород из числа указанных выше в качестве растворителей для стадии а). Гидролиз осуществляют при интенсивном перемешивании обеих фаз с помощью обычных для этих целей устройств, например реакторов с мешалкой, циркуляционных реакторов или предпочтительно смесителей без дополнительных перемешивающих приспособлений. По завершении процесса гидролиза фазы разделяют и подвергают описанной ниже переработке.
Особенно предпочтительным вариантом выполнения изобретения является способ, в котором стадию а) проводят в галогенированном растворителе, растворитель при необходимости частично удаляют и полученное соединение формулы I подвергают гидролизу путем обработки полученной на стадии а) реакционной смеси водными кислотой или основанием.
Согласно другому варианту гидролиз соединения формулы I с получением фосфонометилглицина можно осуществлять также ферментативным путем, например с помощью эстеразы и нитрилазы.
Кислоту или основание для гидролиза применяют в по меньшей мере эквивалентных количествах, предпочтительно, однако, в избытке, прежде всего в количестве не менее 2 эквивалентов.
Температура, при которой проводят гидролиз, находится, как правило, в интервале от приблизительно 10 до 180°С, предпочтительно от 20 до 150°С.
Если Х представляет собой СН2OY, то полученный в результате гидролиза продукт необходимо еще и окислять. При этом исходят прежде всего из соединения, в котором Х представляет собой СН2ОН. Окисление до фосфонометилглицина осуществляют обычным, известным специалистам в данной области образом, например путем каталитического дегидрирования в присутствии медных катализаторов.
Если Х представляет собой CH2OY, где Y представляет собой ацильный остаток, то при гидролизе продукта из стадии а) этот ацильный остаток отщепляют с образованием в результате соответствующего соединения, в котором Х обозначает СН2ОН, после чего это соединение окисляют, как указано выше, до фосфонометилглицина.
Если Х представляет собой СН2OY, где Y представляет собой алкильный остаток, то отщепление эфира происходит обычно одновременно в условиях кислотного гидролиза продукта из стадии а). Полученное соединение, где Х обозначает СН2ОН, окисляют, как указано выше, до фосфонометилглицина.
Полученный при гидролизе с использованием избытка кислоты или основания фосфонометилглицин представлен в растворенном виде в водной фазе. Карбоновая кислота R3СООН образуется при гидролизе с использованием избытка кислоты непосредственно, а при основном гидролизе - после подкисления сильной кислотой, предпочтительно до достижения значения рН<0,5. Затем карбоновую кислоту отделяют обычным образом, например путем отфильтровывания этой кислоты, выпавшей в твердой форме в осадок, путем перегонки или экстракции не смешивающимся с водной фазой органическим растворителем. При двухфазном гидролизе карбоновая кислота при определенных условиях представлена в растворенном виде в органической фазе. В этих случаях карбоновую кислоту удаляют, отделяя органическую фазу, после чего ее можно при необходимости рекуперировать из последней обычным путем. Кислоту получают с высокой степенью чистоты и ее без проблем можно повторно использовать для получения соединения формулы III. Образующий органическую фазу растворитель можно возвращать в технологический цикл и повторно использовать при получении соединений формулы III или на стадии а). Предварительно, однако, растворитель с целью удаления из него примесей, таких как водорастворимые или водонерастворимые спирты, фенолы, аммониевые соли и/или карбоновые кислоты, подвергают, как правило, перегонке, экстракции, фильтрации и/или упариванию.
Фосфонометилглицин можно осаждать, устанавливая рН водной фазы на значение от 0,5 до 2,0, прежде всего от 0,8 до 1,5, например, добавлением соответствующих кислоты либо основания, например HCl, Н2SO4 или NaOH, КОН, Са(ОН)2, и необязательно путем концентрирования водной фазы и/или добавлением вспомогательного осадительного реагента, и получать его (фосфонометилглицин) обычным образом, например путем фильтрации. В качестве вспомогательного осадительного реагента предпочтительно использовать не смешивающийся с водой растворитель, такой как метанол, этанол, изопропанол, ацетон и т.п. Растворители можно рекуперировать из маточного раствора путем перегонки и затем повторно использовать их.
Образующиеся при омылении аммиак или хлорид аммония можно возвращать в способ за счет установления при определенных условиях щелочного рН и рекуперации аммиака путем отгонки.
При необходимости полученный фосфонометилглицин можно обычным образом обесцвечивать. Эту операцию можно осуществлять путем обработки небольшими количествами обесцвечивающего средства, например окислителя, такого как пербораты или Н2О2, или адсорбентами, такими как активированный уголь. Количество обесцвечивающего средства зависит от степени обесцвечивания и может быть определено специалистами в данной области простым путем. Обработку обесцвечивающим средством можно проводить в любом месте по завершении гидролиза и простыми методами. Целесообразно добавлять обесцвечивающее средство до осаждения фосфонометилглицина.
Предлагаемый в изобретении способ, соответственно каждую из его стадий как таковую можно осуществлять в непрерывном, периодическом либо полупериодическом режиме. В этих целях используют обычные реакционные емкости, такие как реакторы с мешалкой, трубчатые реакторы, при необходимости с размещенными перед ними смесительными устройствами или встроенными в трубчатый реактор смесительными элементами.
Таким образом, предлагаемый в изобретении способ отличается простотой в осуществлении технологического процесса и использованием недорогих исходных веществ. Недостаток способа заключается лишь в образовании идущего в отходы неорганического хлорида. Что же касается защитных групп, а именно ацильных остатков триацилфосфита формулы III, то их без проблем можно возвращать в процесс. Способ позволяет получать фосфонометилглицин при предельно малой продолжительности реакции и с высоким выходом более 90% исходя из гексагидротриазина формулы II.
Ниже изобретение более подробно поясняется на примерах, которые никоим образом не ограничивают его объем.
Пример 1
0,2 моля Na-бензоата в условиях, исключающих доступ влаги, предварительно помещают при комнатной температуре в 50 мл 1,4-диоксана. Затем по каплям добавляют 0,0667 моля трихлорида фосфора и смесь в течение 20 мин перемешивают при 85°С. При этом образуется бесцветная суспензия. Далее добавляют 0,0222 моля гексагидротриазина 6 и смесь продолжают перемешивать в течение последующих 20 мин при 85-90°С (жидкая суспензия с хорошей перемешиваемостью). Затем диоксан отгоняют при 40°С в вакууме. К остатку добавляют 100 мл концентрированной соляной кислоты и в течение 4 ч нагревают с обратным холодильником. После охлаждения бензойную кислоту отфильтровывают, после чего проводят промывку (небольшим количеством холодной воды) и сушку. Объединенные фильтраты упаривают досуха. Для выделения фосфонометилглицина остаток от упаривания растворяют в небольшом количестве воды и осаждают на холоде добавлением NaOH до достижения значения рН 1,5. Полное осаждение достигают добавлением небольшого количества метанола. В завершение фосфонометилглицин отфильтровывают и сушат.
Выход: 10,3 г фосфонометилглицина (чистота 95,3% согласно ЖХВР), что соответствует выходу 91% в пересчете на PCl3. В маточном растворе в результате кристаллизации содержится еще 1,8 мас.% фосфонометилглицина.
Пример 2
0,2 моля Na-бензоата в условиях, исключающих доступ влаги, предварительно помещают при комнатной температуре в 50 мл 1,4-диоксана. Затем по каплям добавляют 0,0667 моля трихлорида фосфора и смесь в течение 20 мин перемешивают при 85°С. При этом образуется бесцветная суспензия. Далее фильтруют, исключая доступ влаги, и остаток промывают небольшим количеством диоксана. К фильтрату, продолжая исключать доступ влаги, добавляют 0,0222 моля гексагидротриазина 6 и смесь перемешивают еще в течение 20 мин при 85-90°С. Затем диоксан отгоняют при 40°С в вакууме. К остатку добавляют 100 мл концентрированной соляной кислоты и в течение 4 ч нагревают с обратным холодильником. После охлаждения выпавшую в осадок бензойную кислоту отфильтровывают, а затем проводят промывку (небольшим количеством холодной воды) и сушку. Объединенные фильтраты упаривают досуха. Для выделения фосфонометилглицина остаток от упаривания растворяют в небольшом количестве воды и осаждают на холоде добавлением NaOH до достижения значения рН 1,5. Полное осаждение достигают добавлением небольшого количества метанола. В завершение фосфонометилглицин отфильтровывают и сушат.
Выход: 10,5 г фосфонометилглицина (чистота 94,1% согласно ЖХВР), что соответствует выходу 93% в пересчете на PCl3. В маточном растворе в результате кристаллизации содержится еще 1,9 мас.% фосфонометилглицина.
Пример 3
К раствору 0,04 моля гексагидротриазина 6 в 80 мл диоксана добавляют при комнатной температуре раствор 0,12 моля триацетилфосфита в 50 мл диоксана. Раствор в течение 2 ч перемешивают при 100°С, после чего растворитель отгоняют сначала при 40°С при нормальном давлении, а затем в вакууме. К остатку добавляют 100 мл концентрированной соляной кислоты и в течение 4 ч нагревают с обратным холодильником. Далее реакционную смесь упаривают досуха. Для выделения фосфонометилглицина остаток от упаривания растворяют в небольшом количестве воды и осаждают на холоде добавлением NaOH до достижения значения рН 1,5. Полное осаждение достигают добавлением небольшого количества метанола. В завершение фосфонометилглицин отфильтровывают и сушат.
Выход: 15,4 г фосфонометилглицина (чистота 98,7% согласно ЖХВР), что соответствует выходу 76% в пересчете на PCl3. В маточном растворе в результате кристаллизации содержится еще 1,6 мас.% фосфонометилглицина.
Пример 4
В смесительную колбу объемом 2 л, снабженную лопастной мешалкой (с лопастями из тефлона) и обратным холодильником, предварительно помещают 284 г бензоата аммония в 1000 мл 1,2-дихлорэтана и в атмосфере азота в течение 30 мин по каплям добавляют 91,5 г трихлорида фосфора. Температура при этом повышается до максимум 36°С. Затем перемешивание продолжают еще в течение 30 мин при 25-36°С. Далее смесь фильтруют через напорный нутч-фильтр и осадок на фильтре еще дважды промывают в атмосфере азота дихлорэтаном порциями по 500 г (2054 г фильтрата).
Фильтрат предварительно загружают при комнатной температуре в смесительную колбу объемом 2 л, снабженную лопастной мешалкой (с лопастями из тефлона) и обратным холодильником, и добавляют 45,54 г гексагидротриазина 6. При перемешивании нагревают в течение 30 мин до 80°С и продолжают перемешивать в течение 30 мин при 80°С. Затем раствору дают остыть, после чего непосредственно гидролизуют.
С этой целью используемые вещества загружают дозированными порциями при 130°С и давлении 8 бар в трубчатый реактор (объемом примерно 600 мл) с размещенным перед ним смесителем без дополнительных перемешивающих устройств (1265 г/ч раствора дихлорэтана из предыдущей стадии, 207 г/ч 20%-ной HCl). Продолжительность пребывания материала в аппарате составляет 30 мин. Первый (головной) погон отбрасывают. Для дальнейшей переработки полученную двухфазную смесь улавливают в течение 60 мин. Затем фазы разделяют при 60°С и водную фазу дважды экстрагируют дихлорэтаном порциями по 100 г.
В круглодонной колбе с лопастной мешалкой (с лопастями из тефлона) сначала пропусканием в течение 1 ч потока азота при 60°С отгоняют остатки содержащегося еще в водной фазе дихлорэтана, после чего в течение 15 мин с помощью 50%-ного едкого натра при 40-60°С устанавливают рН на значение 1,0. Затем образовавшуюся суспензию перемешивают в течение последующих 3 ч при 40°С, охлаждают до комнатной температуры, выпавший в осадок продукт отфильтровывают вакуум-фильтрацией и в завершение промывают 150 г ледяной воды. Полученное твердое вещество в течение 16 ч сушат при температуре 70°С и давлении 50 мбар.
Выход: 54,6 г фосфонометилглицина (чистота 96,2% согласно ЖХВР), что соответствует выходу 80% в пересчете на PCl3. В маточном растворе в результате кристаллизации содержится еще 2,1 мас.% фосфонометилглицина.
Пример 5
Из остатка хлорида аммония, образовавшегося при синтезе трибензоилфосфита в примере 4, приготавливают насыщенный раствор в воде. Этот раствор объединяют с маточным раствором в результате кристаллизации фосфонометилглицина согласно примеру 4 и избытком едкого натра рН устанавливают на значение 14. Затем аммиак потоком азота отгоняют из реакционной смеси и для анализа газа улавливают с помощью ГХ (чистота 99%). Объединенные дихлорэтановые фазы из стадии омыления сушат путем отгонки азеотропа дихлорэтан/вода. В дихлорэтан подают сухой аммиак до полного превращения бензойной кислоты в бензоат аммония и образовавшуюся суспензию бензоата аммония в 1,2-дихлорэтане повторно используют в процессе синтеза.
Выход (первый рецикл): 54,0 г фосфонометилглицина (чистота 97,0% согласно ЖХВР), что соответствует выходу 79% в пересчете на PCl3.
Выход (второй рецикл): 55,1 г фосфонометилглицина (чистота 95,5% согласно ЖХВР), что соответствует выходу 81% в пересчете на PCl3.
Пример 6
Реакцию проводят аналогично примеру 1 с тем, однако, отличием, что в качестве растворителя вместо 1,2-дихлорэтана используют нитробензол.
Выход: 56,2 г фосфонометилглицина (чистота 97,4% согласно ЖХВР), что соответствует выходу 82% в пересчете на PCl3. В маточном растворе в результате кристаллизации содержится еще 2,0 мас.% фосфонометилглицина.
Пример 7
Реакцию проводят аналогично примеру 4 с тем, однако, отличием, что в качестве растворителя вместо 1,2-дихлорэтана используют 1,2-дихлорпропан.
Выход: 54,0 г фосфонометилглицина (чистота 96,92% согласно ЖХВР), что соответствует выходу 79% в пересчете на PCl3. В маточном растворе в результате кристаллизации содержится еще 2,1 мас.% фосфонометилглицина.
Пример 8
Реакцию проводят аналогично примеру 1 с тем, однако, отличием, что в качестве растворителя вместо диоксана используют 1,2-дихлорэтан. Фосфонометилглицин в этом варианте получают с выходом 75%.
Пример 9
Реакцию проводят аналогично примеру 1 с тем, однако, отличием, что в качестве растворителя вместо диоксана используют толуол. Фосфонометилглицин в этом варианте получают с выходом 68%.
Пример 10: Получение фосфита из карбоновой кислоты, амина и PCl3
0,05 моля трихлорида фосфора в 15 мл толуола по каплям добавляют при 0°С к раствору из 0,15 моля бензойной кислоты и 0,15 моля диметилциклогексиламина в 90 мл толуола. Далее перемешивают в течение 15 мин при 0°С и затем оставляют для нагревания до комнатной температуры. Выпавший в осадок гидрохлорид отфильтровывают, исключая доступ влаги, через напорный нутч-фильтр. Характеристики трибензоилфосфита (выход 99%) определяют анализом фильтрата методом 1Н-ЯМР и 31Р-ЯМР. Если остаток, полученный после отгонки толуола из фильтрата, растворить в 0,15 моля 10%-ного NaOH, то путем разделения фаз и последующей экстракцией толуолом можно обеспечить рекуперацию диметилциклогексиламина с количественным выходом.
Пример 11
0,2 моля Na-бензоата при комнатной температуре, исключая доступ влаги, предварительно помещают в 50 мл 1,4-диоксана, после чего по каплям добавляют 0,0667 моля трихлорида фосфора и смесь в течение 20 мин перемешивают при 85°С (бесцветная суспензия). Затем добавляют 0,0222 моля гексагидротриазина 1 (X обозначает CN) и смесь продолжают перемешивать еще в течение 20 мин при 85-90°С (жидкая суспензия, хорошо перемешиваемая). Далее диоксан отгоняют при 40°С в вакууме, к остатку добавляют 100 мл концентрированной соляной кислоты и нагревают в течение 4 ч с обратным холодильником. После охлаждения бензойную кислоту отфильтровывают и промывают (небольшим количеством воды). Объединенные фильтраты дважды экстрагируют толуолом порциями по 30 мл, центрифугируют до образования сухого остатка и для удаления избыточной соляной кислоты еще трижды центрифугируют с использованием этанола. Толуоловую фазу концентрируют и остаток объединяют с рекуперированной бензойной кислотой.
Для выделения фосфонометилглицина из остатка водной фазы последний можно растворять в небольшом количестве воды и осаждать на холоде при рН 1,0 (добавление NaOH). Полное осаждение достигается добавлением небольшого количества метанола, который рекуперируют из маточного раствора за счет перегонки. Выход составляет 91%.
Рекуперированную бензойную кислоту (0,2 моля, чистота выше 99% согласно ЖХВР) растворяют в 0,2 моля 5%-ного NaOH, после чего воду отгоняют и остаток сушат. Полученный таким путем бензоат натрия вместе с рекуперированным диоксаном повторно используют в процессе синтеза.
Выход (первый рецикл): 90%,
выход (второй рецикл): 84%,
выход (третий рецикл): 88%.
| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБ ПОЛУЧЕНИЯ N-ФОСФОНОМЕТИЛГЛИЦИНА | 2002 |
|
RU2311420C2 |
| СПОСОБ ПОЛУЧЕНИЯ N-ФОСФОНОМЕТИЛГЛИЦИНА | 2002 |
|
RU2274641C2 |
| СПОСОБ СИНТЕЗА АМИНОАЛКИЛЕНФОСФОНОВОЙ КИСЛОТЫ | 2013 |
|
RU2694047C2 |
| СПОСОБ ПОЛУЧЕНИЯ БИФОСФОНОВЫХ КИСЛОТ И ИХ СОЛЕЙ | 2007 |
|
RU2425049C2 |
| Способ синтеза альфа-аминоалкиленфосфоновой кислоты | 2013 |
|
RU2674022C9 |
| СПОСОБ ПОЛУЧЕНИЯ ФОСФОНОАЛКИЛИМИНОДИУКСУСНОЙ КИСЛОТЫ | 2010 |
|
RU2553683C2 |
| СПОСОБ СИНТЕЗА N-(ФОСФОНОМЕТИЛ)ГЛИЦИНА | 2013 |
|
RU2674021C9 |
| СПОСОБ СИНТЕЗА N-(ФОСФОНОМЕТИЛ)ГЛИЦИНА | 2013 |
|
RU2674023C9 |
| СПОСОБ ПОЛУЧЕНИЯ N-ФОСФОНОМЕТИЛГЛИЦИНА | 1989 |
|
RU2032690C1 |
| СПОСОБ ПОЛУЧЕНИЯ ДИАЛКИЛФОСФИТОВ | 2010 |
|
RU2528053C2 |
Изобретение относится к усовершенствованному способу получения N-фосфонометилглицина, который заключается в том, что производное гексагидротриазина ф-лы (II)
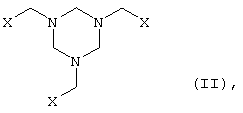
где Х представляет CN, COOZ, СН2OY и др.,
Z и Y представляют водород и др., подвергают взаимодействию с триацилфосфитом ф-лы Р(OCOR3)3 (III), где R3 обозначает C1-C18 алкил или арил, который может иметь заместители, полученный продукт гидролизуют и, если Х представляет собой СН2OY, окисляют. Описываемый способ является простым в осуществлении, экономичен и обеспечивает высокую степень чистоты целевого продукта. 2 н. и 17 з.п. ф-лы.
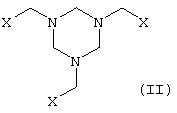
в которой Х представляет собой CN, COOZ, CONR1R2 или CH2OY, где
Y представляет собой Н, алифатический или ароматический ацильный остаток или C1-С6алкильную группу,
Z представляет собой Н, щелочной металл, щелочноземельный металл, С1-С18алкил или арил, необязательно замещенный С1-С4алкилом, NO2 или ОС1-С4алкилом,
R1 и R2, которые могут иметь идентичные либо разные значения, обозначают Н или С1-С4алкил,
подвергают взаимодействию с триацилфосфитом формулы III

в которой радикалы R3 могут иметь идентичные либо разные значения и обозначают С1-С18алкил или арил, необязательно замещенный С1-С4алкилом, NO2 или ОС1-С4алкилом, и
б) полученный продукт гидролизуют и, если Х представляет собой СН2OY, окисляют.

в которой R3 имеет значения, указанные в п.1, или ее соли с тригалогенидом фосфора.
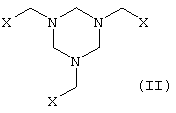
в которой Х представляет собой CN, COOZ, CONR1R2 или CH2OY, где
Y представляет собой Н, алифатический или ароматический ацильный остаток или С1-С6алкильную группу.
Z представляет собой Н, щелочной металл, щелочноземельный металл, С1-С18алкил или арил, необязательно замещенный С1-С4алкилом, NO2 или ОС1-С4алкилом,
R1 и R2, которые могут иметь идентичные либо разные значения, обозначают Н или С1-С4алкил,
подвергают взаимодействию с триацилфосфитом формулы III
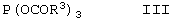
в которой радикалы R3, которые могут иметь идентичные либо разные значения, обозначают С1-С18алкил или арил, необязательно замещенный С1-С4алкилом, NO2 или ОС1-С4алкилом.
| Способ получени -фосфонометилглицина | 1977 |
|
SU662015A3 |
| Свайный ползучий гидравлический домкрат | 1955 |
|
SU104775A1 |
| Полуавтомат конвейерного типа для пайки блоков на печатном монтаже | 1961 |
|
SU149294A1 |
| АВТОМАТИЧЕСКИЙ ЗАТВОР ДЛЯ ГИДРОТЕХНИЧЕСКИХ СООРУЖЕНИЙ | 1953 |
|
SU97522A1 |

