Изобретение относится к способу получения N-фосфонометилглицина путем взаимодействия гексагидротриазинового соединения с триацилфосфитом в органическом растворителе и омыления образующегося при этом фосфонового соединения после предыдущей экстракции в водную среду и отделения от органической фазы.
N-фосфонометилглицин (Глифосат) представляет собой широко применяемый гербицид тотального действия. Известны многочисленные способы получения фосфонометилглицина. Одна возможность получения заключается в том, что производные гексагидротриазина подвергают реакции со сложными эфирами фосфористой кислоты. Патент US 4181800 описывает, например, получение гексагидротриазинов формулы:

и патент US 4053505 описывает взаимодействие этих гексагидротриазинов со сложными диэфирами фосфористой кислоты и последующий гидролиз полученного продукта с получением фосфонометилглицина. Было установлено, что выход и селективность монофосфонированных продуктов оставляет желать лучшего. Кроме того, сложные диэфиры фосфористой кислоты очень дороги.
Европейская патентная заявка ЕР-А-104775 (соответственно, US 4425284, US 4482504 и US 4535181) описывает взаимодействие вышеприведенных гексагидротриазинов с ацилгалогенидом и фосфонирование сложным триэфиром фосфористой кислоты и омыление с получением фосфонометилглицина согласно следующему уравнению:

Таким образом получают правда фосфонометилглицин с относительно хорошим выходом, однако способ требует наряду с применением дорогих сложных эфиров фосфористой кислоты еще и дополнительного применения хлорангидрида карбоновой кислоты. К тому же хлорангидрид карбоновой кислоты во всех случаях мог бы регенерироваться в форме свободной кислоты и потом на отдельной стадии снова переводится в хлорангидрид, что значительно повышает затраты на способ. Далее спирт, с помощью которого этерифицирована фосфористая кислота, не может полностью повторно использоваться, так как при реакции образуется эквивалент соответствующего алкилхлорида, который к тому же токсически не безопасен.
Патент US 4428888 (соответственно, европейская патентная заявка ЕР-А-149294) описывает взаимодействие вышеприведенного гексагидротриазина с хлорангидридом фосфористой кислоты в присутствии сильной безводной кислоты, например хлорводородной кислоты и карбоновой кислоты с от 1 до 6 атомов углерода, например уксусной кислоты. Таким образом получают многочисленные неопределенные побочные продукты, которые снижают выход фосфонометилглицина и требуют сложной очистки продукта.
Патент US 4442044 описывает взаимодействие гексагидротриазина формулы 5 со сложным триэфиром фосфористой кислоты с получением соответствующего фосфонатного соединения, которое применяется в качестве гербицида.
В DD-A-141929 и DD-A-118435 описывается взаимодействие соли щелочного металла вышеприведенного гексагидротриазина (при котором R означает, например, Na) со сложным диэфиром фисфористой кислоты. Вследствие плохой растворимости соли щелочного металла получают, однако, очень низкий выход.
Патент US 5053529 описывает получение фосфонометилглицина взаимодействием гексагидротриазинов со сложными триэфирами фосфористой кислоты в присутствии титантетрахлорида и последующим омылением полученного продукта. Применение титантетрахлорида значительно повышает стоимость способа. Кроме того, выход фосфонометилглицина не удовлетворителен.
Патенты US 4454063, US 4487724 и US 4429124 описывают получение фосфонометилглицина посредством взаимодействия соединения формулы

где R1 и R2 означают ароматические или алифатические группы, с RCOX (X=Cl, Br, I) с получением соединения формулы

реакции этого соединения с цианидом металла и гидролиза полученного продукта. Недостатки этого способа приведены выше в связи с применением хлорангидрида кислоты.
Известны также другие возможности синтеза, которые исходят из замещенного цианометилом гексагидротриазина формулы

Патенты US 3923877 и US 4008296 описывают взаимодействие этого производного гексагидротриазина с диалкилфосфонатом в присутствии кислотного катализатора, такого как хлористый водород, кислоты Льюиса, хлорангидрида или ангидрида карбоновой кислоты с получением соединения следующей формулы:

Последующий гидролиз дает фосфонометилглицин, причем образуется от 8 до 10% два раза фосфонометилированного продукта.
Публикации US 4067719, US 4083898, US 4089671 и DE-A-2751631 описывают взаимодействие замещенного цианометилом гексатриазина с диарил-фосфонатом без катализатора с получением соединения формулы 9, при котором R" означает арил. Этот способ имеет те же недостатки, что и возникающие при применении карбоксизамещенного гексагидротриазина формулы 5.
Заявка ЕР-А-097522 (соответствует US 4476063 и US 4534902) описывает взаимодействие гексагидротриазина формулы 6 с ацилгалогенидом с получением соединения формулы 10, последующее фосфонирование сложным три- или диэфиром фосфористой кислоты с получением соединения формулы 11 и последующее омыление с получением фосфонометилглицина согласно следующему уравнению реакции:

Также и здесь можно наблюдать те же недостатки, что и при способе с применением карбоксизамещенных производных гексагидротриазинов.
Патент US 4415503 описывает взаимодействие замещенного цианометилом гексагидротриазина аналогично описанному в патенте US 4428888 способу. Также и в этом случае можно наблюдать повышенное образование побочных продуктов.
Европейская патентная заявка ЕР 164923 А описывает усовершенствованный гидролиз соединения формулы 11.
Заявка DE 19962601 описывает способ получения N-фосфонометилглицина, при котором
а) производное гексагидротриазина формулы II
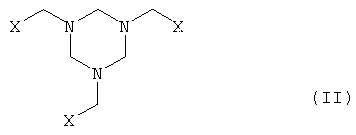
где Х означает CN, COOZ, CONR1R2 или CH2OY,
Y означает Н или остаток, который легко заменим на Н,
Z означает Н, щелочной металл, щелочноземельный металл, С1-С18-алкил или арил, который необязательно может быть замещен С1-С4-алкилом, NO2 или ОС1-С4-алкилом;
R1 и R2, могут быть одинаковыми или различными и означают Н или С1-С4-алкил,
подвергают взаимодействию с триацилфосфитом формулы III

где остатки R3, которые могут быть одинаковыми или различными, означают C1-C18-алкил или арил, который необязательно замещен С1-С4-алкилом, NO2 или ОС1-С4-алкилом,
с получением соединения формулы I

где R3 и Х имеют вышеприведенные значения, и
б) соединение формулы I гидролизуют и, если Х означает CH2OY, окисляют.
Стадия (а) способа проводится предпочтительно в инертном органическом растворителе, особенно предпочтительно в галогенированном растворителе, таком как например, 1,2-дихлорэтан. Гидролиз согласно стадии (б) (омыление) может осуществляться как в щелочной, так и в нейтральной или кислотной среде.
После омыления N-фософонометилглицин должен предпочтительно находиться в водном растворе. Применяемый на стадии (а) растворитель должен отделяться до или после омыления. Если он присутствует при омылении, в частности, для снижения затрат, он не должен расходоваться или разлагаться. Поэтому согласно одному варианту способа применяемый на стадии (а) растворитель полностью или частично отгоняется перед омылением, т.е. перед стадией (б), и остаток гидролизуется. Согласно еще одному предпочтительному варианту выполнения способа гидролиз осуществляется в водной/органической двухфазной системе. После окончания гидролиза обе фазы разделяются и перерабатываются. N-фосфонометилглицин находится в водной фазе.
Если растворитель отделяется дистилляцией, то расход энергии при проведении способа очень высок, поэтому отделение после омыления разделением фаз является более экономичным. Если в качестве растворителя применяют хлорированные углеводороды, например, 1,2-дихлорэтан, то растворитель при условиях омыления имеет только органиченную стабильность.
Недостаточная стабильность хлорированных углеводородов при условиях омыления известна уже продолжительное время. Так, например, в публикации H.Bahr и Н.Zieler "Angew. Chem." 1930, 43, стр.286, указывается, что дихлорэтан при температуре выше 150°С заметно превращается в этиленгликоль. Из патентной заявки DE 537448 известно омыление дихлорэтана в гликоль водой при температуре выше 120°С, причем концентрация образующейся соляной кислоты поддерживается ниже 4%. В заявке DE 540513 описывается получение гликоля из циангидрина посредством гидролиза в присутствии щелочи при температуре от 80 до 100°С.
Из приведенных примеров можно заключить, что особенно при применении нейтральных или щелочных условий омыления следует учитывать разложение растворителя. Из соображений экономии расходов следует отдавать предпочтение нейтральным условиям омыления, при которых образуется моноамониевая соль, и щелочным условиям омыления, при которых образуется, например, при применении натрового щелока, мононатриевая соль N-фосфонометилглицина. Обе соли хорошо растворимы в воде, и из их растворов можно осаждать N-фосфонометилглицин добавкой только эквивалента кислоты. Таким образом образуется только 1 моль соли на 1 моль N-фосфонометилглицина. При кислотном омылении требуется более высокий избыток кислоты, что приводит к большей нагрузке солью отходящих вод и к более высоким расходам. С другой стороны, вследствие склонности растворителя к разложению, нейтральное и щелочное омыление должно проводиться при очень мягких условиях, что значительно повышает время реакции.
Задачей изобретения является разработка усовершенствованного с экономической и экологической точки зрения способа получения N-фосфонометилглицина.
Задачей изобретения является также разработка способа получения N-фосфонометилглицина, при котором предотвращается разложение органического растворителя и обеспечивается получение продукта с высокой чистотой.
Таким образом изобретение относится к способу получения N-фосфонометилглицина, при котором
а) производное гексагидротриазина формулы II
где Х означает CN, COOZ, CONR1R2 или CH2OY,
Y означает Н или остаток, который легко заменим водородом Н;
Z означает H, щелочной металл, щелочноземельный металл, С1-С18-алкил или арил, который необязательно замещен С1-С4-алкилом, NO2 или ОС1-С4-алкилом;
R1 и R2, которые могут быть одинаковыми или различными, означают Н или С1-С4-алкил,
подвергают взаимодействию в органическом растворителе с триацил-фосфитом формулы III
где остатки R3, которые могут быть одинаковыми или различными, означают C1-C18-алкил или арил, который необязательно может быть замещен С1-С4-алкилом, NO2 или ОС1-С4-алкилом,
с получением соединения формулы I

где R3 и X имеют вышеприведенные значения,
б) полученную реакционную смесь экстрагируют водой или водным раствором кислоты или водным раствором основания,
в) фазы разделяют и
г) содержащийся в водной фазе продукт гидролизуют или далее гидролизуют.
После стадии (г) N-фосфонометилглицин получают из водной фазы.
Прочие содержащиеся в водной фазе, способные к дальнейшему применению или регенерируемые компоненты отделяют из водной фазы и, в случае необходимости, возвращают в процесс.
В том случае, если Х в формуле II означает CH2OY, полученный после гидролиза продукт следует еще окислять.
Алкил означает разветвленные или неразветвленные алкильные цепи, преимущественно с 1 до 8 атомами углерода, и в частности, с 1 до 4 атомов углерода. Примерами для алкила являются метил, этил, н-пропил, и-пропил, н-бутил, и-бутил, втор-бутил, трет-бутил, н-гексил, 2-этилгексил и т.д.
Арил означает, предпочтительно, фенил и нафтил.
Х означает, предпочтительно, CN или COOZ.
Z означает, предпочтительно, Н, щелочной металл или С1-С18алкил.
Если Y означает остаток, который легко заменим водородом, то речь идет об ароматическом или алифатическом ацильном остатке или об C1-С6-алкильной группе. Алифатический ацильный остаток представляет собой предпочтительно остаток C1-C6-CO, при ароматическом ацильном остатке речь идет предпочтительно о бензоильном остатке.
R1 и R2 означают предпочтительно водород Н.
При остатке R3 речь идет, в частности, об арильном остатке, который необязательно может быть замещен указано выше. Особенно пригодными остатками R3 являются фенил, п-толуол и п-нитрофенил.
Соединения формулы II известны и могут быть получены известным образом или аналогично известным способом, приведенным в вышеописанных аналогах. Например, можно подвергнуть взаимодействию амин X-CH2-NH2 с источником формальдегида, водным формалиновым раствором или параформальдегидом, например, растворением первичного амина в водном формалиновом растворе. Гексагидротриазин может быть получен кристаллизацией или отпариванием воды. Этот способ описан в заявке на патент DE-A-2645085, соответствующей патенту US 4181800, на который здесь дается ссылка в полном объеме. Соединение формулы II, где Х означает CN, может быть получено синтезом Штреккера, т.е. взаимодействием аммиака, синильной кислоты и источника формальдегида. Подобный способ описан, например, в патенте US 2823222, на который дается ссылка в полном объеме.
Соединения формулы III могут быть получены различными способами. Первая возможность заключается в реакции взаимодействия соли карбоновой кислоты R3COOH с тригалогенидом фосфора, в частности с трихлоридом фосфора. В качестве соли карбоновой кислоты применяют предпочтительно соль щелочного или щелочноземельного металла, в частности натриевую, калиевую или кальциевую соль, или аммониевую соль. Это взаимодействие можно проводить без применения растворителя и полученный продукт применять непосредственно на стадии (а). Предпочтительно работают в инертном органическом растворителе, в частности в простом эфире, таком как диоксан, тетрагидрофуран и т.п., в галогенированном, в частности, хлорированном или фторированном органическом растворителе, таком как дихлорметан, 1,2-дихлорэтан, 1,2-дихлорпропан, 1,1,1-трихлорэтан, 1,1,2-трихлорэтан, 1,1,2,2-тетрахлорэтан, хлорбензол или 1,2-дихлорбензол, в алифатическом или ароматическом углеводороде, таком как н-октан, толуол, ксилол или нитробензол. Предпочтительно применяют тот же растворитель, что и на стадии (а). Особенно предпочтительно применение хлорированного углеводорода.
Образующаяся при реакции взаимодействия соль, например хлорид натрия, при применении трихлорида фосфора и соли натрия используемой карбоновой кислоты может после взаимодействия выводиться. Если в качестве соли получают хлорид аммония или другой галогенид аммония, то использованный аммиак может регенерироваться за счет того, что водный раствор соли подщелачивают сильным основанием, например натровым щелоком (рН от 11 до 14), и затем аммиак отгоняют обычным образом. Полученный таким образом аммиак после сушки, например, дистилляцией в жидком или газообразном состоянии или в качестве водного раствора может снова возвращаться и применяться для получения аммониевой соли карбоновой кислоты.
Другой возможностью получения соединений формулы III является реакция взаимодействия карбоновой кислоты R3COOH с тригалогенидом фосфора в присутствии амина. В качестве амина применяют, в частности, алифатические или циклоалифатические ди- или триамины, такие как триэтиламин, трибутил-амин, диметилэтиламин или диметилциклогексиламин, или пиридин. В общем при таком способе реакцию осуществляют в органическом растворителе. Пригодные растворители приведены выше в связи с первой возможностью получения соединений формулы III. Предпочтительно применяют диоксан, 1,2-дихлорпропан, 1,2-дихлорэтан, нитробензол или толуол. При применении растворителя образовавшийся амингидрохлорид осаждается и может отфильтровываться. Если амингидрохлорид обрабатывают сильным основанием, например водным натровым щелоком, тогда амины высвобождаются из гидрохлорида. Летучие амины можно регенерировать посредством дистилляции или экстракции. Нелетучие амины можно регенерировать экстракцией или, если при высвобождении амина получают двухфазную смесь, посредством разделения фаз. Твердые амины могут быть регенерированы фильтрацией. Регенерированные амины можно, в случае необходимости, после сушки, снова возвращать в процесс.
Другой возможностью получения соединений формулы III является взаимодействие карбоновой кислоты R3COOH с тригалогенидом фосфора, в частности, с трихлоридом фосфора без добавки основания. При таком взаимодействии требуется удаление образующегося галогеноводорода из реакционной смеси. Это может осуществляться обычным образом, например пропусканием инертного газа, например азота. Высвободившийся галогеноводород можно, если предстоит кислотное омыление, применять в форме водного раствора для гидролиза на стадии (г).
Стадия (а) способа по изобретению осуществляется в инертном растворителе, при котором речь может идти о смешиваемом с водой и предпочтительно не смешиваемом с водой растворителе. Примерами для подходящих растворителей являются углеводороды, такие как толуол или ксилол, простые эфиры, такие как тетрагидрофуран, диоксан или дибутилэфир, нитробензол и т.п. особенно предпочтительно работают с галогенированным растворителем, предпочтительно с хлорированным и/или фторированным алифатическим углеводородом, таким как дихлорметан, 1,2-дихлорэтан, 1,2-дихлорпропан, 1,1,1-трихлорэтан, 1,1,2,2-тетрахлорэтан, хлорбензол или 1,2-дихлорбензол. Особенно предпочтителен 1,2-дихлорэтан.
Сокомпоненты реакции применяются целесообразным образом в основном в стехиометрическом количестве. Можно применять также и избыток, например, до 10% одного или другого сокомпонента. Температура реакции составляет в общем от -10 до 140°С, предпочтительно она лежит в интервале от комнатной температуры и до 100°С. При этих условиях требуется только короткое время реакции, в общем реакция уже заканчивается через промежуток времени от 10 до 30 минут.
Полученные на стадии (а) соединения формулы I являются промежуточными продуктами для получения N-фосфонометилглицина. Для перевода в фосфонометилглицин полученную на стадии (а) реакционную смесь подвергают сначала экстракции (стадия (б)). Такая экстракция может осуществляться в кислотной, нейтральной или щелочной среде. Условия рН могут при этом соответствовать желаемым условиям при последующем омылении, однако можно экстрагировать и в другом диапазоне рН, чем на стадии омыления. Так, например, можно экстрагировать в кислотной или нейтральной среде, потом добавлять основание и производить щелочное омыление.
Экстракцию осуществляют предпочтительно при температуре в пределах от комнатной температуры и до температуры кипения с обратным холодильником реакционной смеси, в частности, предпочтительно, по меньшей мере, при 50°С. Переход фосфонового соединения в водную фазу протекает очень быстро. При соединении формулы I, при котором Х означает CN и R3 означает фенил, во время экстракции в воде при 50°С уже через 10 минут более 95% общего фосфора находится в водной фазе, при температуре кипения с обратным холодильником через 10 минут уже почти 99%.
В общем в зависимости от температуры достаточно время экстракции в несколько минут, например начиная от 5 минут. Предпочтительно время экстракции составляет по меньшей мере 10 минут, особенно предпочтительно по меньшей мере 1 час. В частности, при экстракции при низких температурах может требоваться более продолжительное время экстракции, например по меньшей мере 2 часа.
Во время экстракции, по меньшей мере, часть фосфонового соединения формулы I, как правило, уже частично омыляется. Под частичным омылением следует понимать то, что только часть содержащихся в продукте стадии (а) R3CO-остатков отщепляется. Степень омыления зависит от самого фосфонового соединения и от выбранных условий экстракции. При соединении формулы I, при котором Х означает CN и R3 означает фенил, после 10-минутной экстракции в зависимости от условий экстракции уже от 45 до 50% общей бензойной кислоты имеется в свободной форме.
В качестве кислоты применяют при экстракции в основном неорганические кислоты, такие как соляная кислота, серная кислота или фосфорная кислота. Щелочная экстракция осуществляется в общем с применением щелочного или щелочноземельного гидроксида, в частности, при применении гидроксида натрия или калия.
Разложение применяемого на стади (а) растворителя при экстракции в основном не имеет места также и тогда, когда при этом речь идет о особенно чувствительном к разложению хлорированном углеводороде.
В заключение водная и органическая фазы отделяются друг от друга (стадия (в)). Получают органическую фазу, которая в случае необходимости содержит растворимые в ней примеси, которые могут быть простым образом отделены от продукта. Водная фаза содержит продукт стадии (а) и, в случае необходимости, частично его омыленный продукт. Разделение фаз осуществляется обычным, известным специалисту в данной области образом. Находящееся в водной фазе соединение формулы I, соответственно частично омыленный продукт потом гидролизуют (стадия (г)). К водной фазе можно добавлять в зависимости от желаемых условий омыления кислоты или основания. Вследствие высокого требуемого избытка кислоты при кислотном омылении, омыление при нейтральных или щелочных условиях является предпочтительным.
Омыление осуществляется при повышенном давлении, чтобы достичь желаемой температуры реакции. Предпочтительно температура при омылении лежит выше, чем при экстракции. В общем температура реакции омыления составляет, по меньшей мере, на 20°С, в частности, по меньшей мере, на 30°С выше, чем при экстракции. Предпочтительная температура реакции составляет от 100 и до 180°С, особенно предпочтительно, от 130 до 150°С. Время реакции составляет предпочтительно от прибл. 5 минут до 4 часов, в частности, преимущественно от 10 минут до 2 часов, особенно предпочтительно прибл. 20 минут.
При омылении предпочтительны нейтральные или щелочные условия. При основном омылении применяют основание преимущественно в эквивалентных количествах.
В качестве кислот и оснований для омыления применяют в общем приведенные выше в связи с экстракцией кислоты или основания.
Нет необходимости соблюдать умеренные условия омыления, так как при этом не присутствует органический растворитель, который может разлагаться.
В том случае, если Х означает CH2OY, полученный после гидролиза продукт следует еще окислять. В частности, при этом исходят из соединения, при котором Х означает СН2OH. Окисление с получением фосфонометилглицина осуществляется обычным, известным специалисту в данной области образом, например каталитическим дегидрированием с катализом на меди.
В том случае, если Х означает CH2OY и Y означает ацильный остаток при гидролизе продукта со стадии (а), происходит отщепление ацильного остатка при образовании соответствующего соединения с Х=CH2OH. Это соединение окисляют как указано выше с получением фосфонометилглицина.
Если Х означает CH2OY и Y означает алкильный остаток, осуществляется отщепление эфира обычно одновременно при условиях кислотного гидролиза продукта со стадии (а). Полученное соединение, при котором Х означает СН2ОН, окисляют как указано выше с получением фосфонометилглицина.
После завершенного гидролиза N-фосфонометилглицин, который после гидролиза имеется в форме соли, отделяется от водной фазы. Это происходит посредством соответствующей установки значения рН.
Фосфонометилглицин может осаждаться установкой водной фазы на значение рН в области от 0,5 до 2,0, в частности от 0,8 до 1,5, например добавкой кислоты или основания, например, HCl, H2SO4 или NaOH, КОН, Са(ОН)2, и, в случае необходимости, концентрацией водной фазы и/или добавкой вспомогательного средства осаждения и может получаться обычным образом, например фильтрацией. В качестве вспомогательного средства осаждения применяют предпочтительно смешиваемый с водой растворитель, такой как метанол, этанол, изопропанол, ацетон и т.п. Растворители могут регенерироваться дистилляцией из маточного щелока и снова применяться.
Образовавшийся при омылении аммиак или хлорид аммония может снова возвращаться в процесс, в случае необходимости, посредством подщелачивания и выпаривания.
В случае необходимости, полученный фосфонометилглицин может быть обесвечен обычным образом. Это может осуществляться, например, обработкой небольшим количеством обесцвечивающего средства, например, окисляющего средства, такого как перборат или Н2O2, или адсорбентом, таким как активный уголь. Количество обесцвечивающего средства зависит от степени окраски и может простым образом определяться специалистом в данной области. Обработка обесцвечивающим средством может осуществляться в любом месте после гидролиза и обычным образом. Целесообразно добавлять обесцвечивающее средство перед осаждением фосфонометилглицина.
Образующаяся при омылении реакционная смесь содержит наряду с N-фосфонометилглицином другие продукты, соответственно, побочные продукты. Карбоновая кислота R3COOH образуется при гидролизе с избытком кислоты или непосредственно при нейтральном гидролизе, или при основном гидролизе после подкисления сильной кислотой, предпочтительно на значение рН<0,5. Отделение карбоновой кислоты происходит тогда обычным образом, например фильтрацией выпавшей в твердой форме карбоновой кислоты, дистилляцией или экстракцией несмешиваемым с водной фазой органическим растворителем. Карбоновая кислота образуется с высокой чистотой и может без проблем снова применяться для получения соединения формулы III. Также и органический растворитель со стадии (а) может без проблем регенерироваться после экстракции соединения формулы I. Перед этим растворитель однако подвергают дистилляции, экстракции и/или отпариванию, чтобы удалить примеси. Способ по изобретению, соответственно каждая стадия отдельно может осуществляться непрерывно, прерывно или как полупериодический способ. Применяются обычные для такой цели емкости, такие как котел с мешалкой или трубчатый реактор, экстракционные колонны, отстойник-смеситель или отделитель фаз, в случае необходимости, с предвключенными смесительными устройствами или встроенными в трубчатый реактор смесительными элементами.
Способ по изобретению характеризуется простым ведением процесса и применением недорогих исходных веществ. В качестве отходов образуется только неорганический хлорид и защитные группы, ацильные остатки триацилфосфита формулы III могут регенерироваться простым образом. Способ обеспечивает получение N-фосфонометилглицина в очень короткое время реакции и с высоким выходом более чем 90%, в пересчете на гексагидротриазин формулы II.
Экстракцией целевого продукта в водную фазу перед омылением предотвращается разложение применяемого на стадии (а) органического растворителя. Не требуется умеренных условий омыления и может применяться недорогое и с экологической точки зрения предпочтительное нейтральное или щелочное омыление, при котором образуется моноаммониевая соль, соответственно мононатриевая соль N-фосфонометилглицина. Обе соли могут осаждаться добавкой только эквивалента кислоты, что снижает нагрузку солью сточных вод и затраты на способ.
Нижеприведенные примеры поясняют изобретение, не ограничивая его.
Пример 1: Взаимодействие триазина с фосфитом
В 2-литровую колбу с тефлоновой лопастной мешалкой и обратным холодильником подают 284 г бензоата аммония в 1000 мл 1,2-дихлорэтана и под азотной атмосферой в течение 30 минут прикапывают 91,5 г трихлорида фосфора. Температура реакционной смеси повышается при этом максимально до 36°С. После этого реакционную смесь перемешивают в течение еще 30 минут при температуре от 25 до 36°С. Реакционную смесь фильтруют на нутч-фильтре и фильтровальный осадок промывают под азотом еще 2 раза каждый раз посредством 500 г дихлорэтана (2054 г фильтрата).
В 2-литровую колбу с тефлоновой лопастной мешалкой и обратным холодильником подают фильтрат при комнатной температуре и добавляют гексагидротриазин 6 (при Х=CN, 45,54 г). При перемешивании реакционную смесь нагревают в течение 30 минут до 80°С и продолжают перемешивать еще 30 минут при 80°С. Дают реакционной смеси охладиться и после этого подают непосредственно на дальнейшее превращение.
Пример 2: Стадия экстракции
150 г полученного в примере 1 раствора (содержащего 0,051 моль Р) перемешивают с 28 г воды или водного раствора как указано в таблице 1. После этого разделяют фазы и анализируют по отдельности элементным анализом Р и количественной высокопроизводительной хромотографией для определения безойной кислоты. Получают приведенные в таблице 1 результаты.
Столбец "свободная бензойная кислота" показывает, сколько процентов поданного теоретически в форме фосфита количества бензойной кислоты уже после экстракции имеется в несвязанной форме. Столбец "водная фаза" показывает, какая ее доля еще не растворена в водной фазе. Последний столбец показывает, какая доля общего количества фосфора после экстракции остается еще в органической фазе.
Пример 3: Стадия омыления в воде
Полученную согласно примеру 2, тест №2 водную фазу нагревают в 10 мл-вом реакторе, работающем под давлением. Условия реакции можно видеть в таблице 2. Реакционную смесь экстрагируют два раза при 50°С толуолом и затем анализируют на наличие фосфонометилглицина посредством количественной высокопроизводительной жидкостной хроматографии и количественного анализа 1H-ЯМР. Результаты представлены в таблице 2. Для выделения N-фосфонометилглицина реакционную смесь устанавливают на значение рН 1,0, N-фосфонометилглицин отфильтровывают и промывают.
Пример 4: Стадия омыления в щелочных условиях
К полученной согласно примеру 2, тест №2 водной фазе добавляют 0,051 моль 50%-ного натрового щелока и реакционную смесь нагревают в реакторе под давлением. Условия реакции приведены в таблице 3. Выгружаемую реакционную смесь анализируют количественной высокопроизводительной жидкостной хроматографией и количественным анализом 1H-ЯМР на наличие фосфонометилглицина (выход синтеза). Результаты приведены в таблице 3. Для выделения фосфонометилглицина реакционную смесь экстрагируют три раза при 60°С и значении рН 2,5 посредством 1,2-дихлорэтана, устанавливают значение рН на 1,0, затем добавляют немного метанола и перемешивают в течение 3 часов при 40°С, отфильтровывают фосфонометилглицин и промывают остаток ледяной водой.
| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБ ПОЛУЧЕНИЯ N-ФОСФОНОМЕТИЛГЛИЦИНА И ПРОМЕЖУТОЧНЫЙ ПРОДУКТ ДЛЯ ЕГО ПОЛУЧЕНИЯ | 2000 |
|
RU2260010C2 |
| СПОСОБ ПОЛУЧЕНИЯ N-ФОСФОНОМЕТИЛГЛИЦИНА | 2002 |
|
RU2274641C2 |
| СПОСОБ ПОЛУЧЕНИЯ N-ЗАМЕЩЕННЫХ САЛИЦИЛАМИДОВ | 2005 |
|
RU2404158C2 |
| СПОСОБ ПРОИЗВОДСТВА ПРОИЗВОДНЫХ ЗАМЕЩЁННОЙ 5-МЕТОКСИМЕТИЛПИРИДИН-2, 3-ДИКАРБОНОВОЙ КИСЛОТЫ | 2009 |
|
RU2549894C2 |
| СПОСОБ ПОЛУЧЕНИЯ МАКРОЦИКЛИЧЕСКИХ СОЕДИНЕНИЙ | 2006 |
|
RU2456296C2 |
| СПОСОБ ПОЛУЧЕНИЯ ЗАМЕЩЕННЫХ 3-ПИРИДИЛМЕТИЛ АММОНИЙ БРОМИДОВ | 2009 |
|
RU2549896C2 |
| СПОСОБ ПОЛУЧЕНИЯ СЛОЖНЫХ ЭФИРОВ 5- И/ИЛИ 6-ЗАМЕЩЕННОЙ 2-ОКСИБЕНЗОЙНОЙ КИСЛОТЫ | 2000 |
|
RU2245325C2 |
| ПРОИЗВОДНЫЕ ЗАМЕЩЕННОЙ ГЕТЕРОЦИКЛОМ ФЕНИЛ-ЦИКЛОГЕКСАН-КАРБОНОВОЙ КИСЛОТЫ, СМЕСЬ ИХ ИЗОМЕРОВ ИЛИ ОТДЕЛЬНЫЕ ИЗОМЕРЫ И ИХ СОЛИ | 1994 |
|
RU2125990C1 |
| Способ получения аминометилированных бисерных полимеров | 2015 |
|
RU2697528C2 |
| ПОЛИФУНКЦИОНАЛЬНЫЙ ОРГАНОСИЛАН, ПРИГОДНЫЙ ДЛЯ ИСПОЛЬЗОВАНИЯ В КАЧЕСТВЕ СВЯЗУЮЩЕГО АГЕНТА, И СПОСОБ ЕГО ПОЛУЧЕНИЯ | 2001 |
|
RU2272042C2 |
Данное изобретение касается нового способа получения N-фосфонометилглицина взаимодействием гексагидротриазинового соединения с триацилфосфитом в органическом растворителе, омылением образовавшегося при этом фосфонового соединения после предыдущей экстракции в водную фазу с отделением органической фазы и осаждением N-фосфонометилглицина путем установки значения рН на величину в интервале от 0,5 до 2,0. Технический результат - предотвращение разложения органического растворителя при омылении. 7 з.п. ф-лы, 3 табл.
а) производное гексагидротриазина формулы II

где Х означает CN;
подвергают взаимодействию с триацилфосфитом формулы III

где остатки R3, которые могут быть одинаковыми или различными, означают C1-C18-алкил или фенил, который необязательно замещен С1-С4-алкилом, NO2 или ОС1-С4-алкилом, в органическом растворителе, являющемся несмешиваемым с водой;
б) полученную реакционную смесь экстрагируют водой или водным раствором кислоты или водным раствором основания при температуре в интервале от комнатной температуры до температуры флегмы реакционной смеси;
в) разделяют фазы;
г) содержащийся в водной фазе продукт гидролизуют при более высокой температуре, чем экстракцию согласно стадии (б); и
д) N-фосфонометилглицин получают из водной фазы осаждением путем установки значения рН на величину в интервале от 0,5 до 2,0.
| Свайный ползучий гидравлический домкрат | 1955 |
|
SU104775A1 |
| Полуавтомат конвейерного типа для пайки блоков на печатном монтаже | 1961 |
|
SU149294A1 |
| АВТОМАТИЧЕСКИЙ ЗАТВОР ДЛЯ ГИДРОТЕХНИЧЕСКИХ СООРУЖЕНИЙ | 1953 |
|
SU97522A1 |
| US 5053529 А, 01.10.1991 | |||
| Способ получени -фосфонометилглицина | 1977 |
|
SU662015A3 |

