Область, к которой относится изобретение
Данное изобретение относится к циклическим соединениям и их солям, к способам применения таких соединений для лечения расстройств, обусловленных протеинтирозинкиназами, таких как иммунологические или онкологические расстройства, и к фармацевтическим композициям, содержащим такие соединения.
Предпосылки создания изобретения
Протеинтирозинкиназы (РТК, ПТК) представляют собой ферменты, которые в соединении с АТР (АТФ) в качестве субстрата фосфорилируют остатки тирозина в пептидах и белках. Эти ферменты являются ключевыми элементами в регуляции передачи сигнала в клетках, включая пролиферацию и дифференцировку клеток. РТК включают, среди прочего, рецепторные тирозинкиназы (RPTK), включая члены семейства киназ эпидермального фактора роста (например, HER1 и НЕR2), тромбоцитарного фактора роста (PDGF) и киназы, которые играют роль при ангиогенезе (Tic-2 и KDR); кроме того, нерецепторные тирозинкиназы, включая члены семейств Syk, JAK и Src (например, Src, Fyn, Lyn, Lck и Blk) (см. Bolen, J.B., Rowley, R.B., Spana, C., and Tsygankov, A.Y., “The src family of tyrosine protein kinases in hemopoietic signal transduction” (“Src-семейство протеинтирозинкиназ в кроветворной сигнальной трансдукции”), FASEB J., 6, 3403-3409 (1992); Ullrich, A. and Schlessinger, J., “Signal transduction by receptors with tyrosine kinase activity” (“Сигнальная трансдукция под действием рецепторов с тирозинкиназной активностью”), Cell, 61, 203-212 (1990); и Ihle, J.N., “The Janus protein tyrosine kinases in hematopoietic cytokine signaling” (“Янус-протеинтирозинкиназы в передаче сигнала в кроветворных цитокинах”), Sem. Immunol., 7, 247-254 (1995).
Повышенная активность РТК влечет за собой ряд злокачественных и незлокачественных пролиферативных заболеваний. Кроме того, РТК играют главную роль в регуляции клеток иммунной системы. Ингибиторы РТК могут, следовательно, влиять на большой ряд онкологических и иммунологических расстройств. Такие расстройства можно уменьшать (ослаблять) путем селективного ингибирования определенной рецепторной или нерецепторной РТК, такой как Lck, или, вследствие гомологии между классами РТК, путем ингибирования более одной РТК с помощью ингибитора. РТК, представляющей особый интерес, является Lck, обнаруженная в Т-клетках, где она входит в состав фосфорилирующих ключевых белковых субстратов. Она необходима для продуктивной антиген-рецепторной передачи сигнала и активации клеток. В отсутствие Lck-активности дзета(ξ)-цепь Т-клеточного рецептора (ТСК) не фосфорилируется, киназа ZAP-70 не активизируется и не происходит иммобилизации Са2+, существенно важной для активации Т-клеток (см. Weiss, A. and Zittman, D. R., “Signal transduction by lymphocyte antigen receptors” (“Сигнальная трансдукция с помощью рецепторов антигенов лимфоцитов”). Cell, 76, 263-274 (1994); Iwashima, M., Irving, В. A., van Oers, N.S.C., Chan, A. C., and Weiss, A., “Sequential interactions of the TCR with two distinct cytoplasmic tyrosine kinases” (“Последовательное взаимодействие TCR с двумя различными цитоплазматическими тирозинкиназами”), Science, 263, 1136-1139 (1994), и Chan, А. С., Dalton, M., Johnson, R., Kong, G., Wang, Т., Thoma, R., and Kurosaki, Т., “Activation of ZAP-70 kinase activity by phosphorilation of tyrosine 493 is required for lymphocyte antigen receptor function” (“Для рецепторной функции антигена лимфоцита требуется активация ZAP-70-киназной активности с помощью фосфорилирования тирозина 493”), EMBO J., 14, 2499-2508 (1995). Таким образом, ингибиторы Lck применимы для лечения нарушений, опосредуемых Т-клетками, таких как хронические заболевания с важным Т-клеточным компонентом, например ревматоидный артрит, рассеянный склероз и волчанка, а также острые заболевания, в которых, как известно, Т-клетки играют существенную роль, например острое отторжение трансплантата и реакции замедленной гиперчувствительности (ДТН).
Сущность изобретения
Данное изобретение охватывает циклические соединения нижеприведенной формулы I и их соли, применяемые в качестве ингибиторов протеинтирозинкиназ
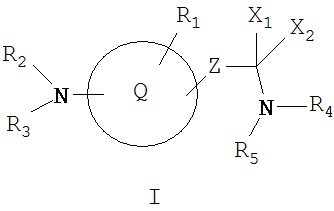
где Q обозначает:
(1) 5-членный гетероарильный цикл;
(2) 6-членный гетероарильный цикл или
(3) арильный цикл;
при необходимости, замещенный одной или более групп R1;
Z обозначает:
(1) простую связь;
(2) -R15C=CH- или
(3) -(CH2)m-, где m обозначает 1-2;
X1 и Х2, каждый, обозначает водород или вместе образуют =O или =S;
R1 обозначает:
(1) водород или R6,
где R6 обозначает алкил, алкенил, алкинил, циклоалкил, циклоалкилалкил, циклоалкенил, циклоалкенилалкил, арил, арилалкил, гетероцикло или гетероциклоалкил, каждый из которых является незамещенным или замещенным Z1, Z2 и одной или более (предпочтительно, одной или двумя) группами Z3;
(2) -ОН или -OR6;
(3) -SH или -SR6;
(4) -С(O)2Н, -C(O)qR6 или -O-C(O)qR6, где q обозначает 1 или 2;
(5) -SO3Н или -S(O)qR6;
(6) галоид (галоген);
(7) циано;
(8) нитро;
(9) -Z4-NR7R8;
(10) -Z4-N(R9)-Z5-NR10R11;
(11) -Z4-N(R12)-Z5-R6;
(12) -Р(O)(OR6)2;
R2 и R3, каждый независимо, обозначает
(1) водород или R6,
(2) -Z4-R6 или
(3) -Z13-NR7R8;
R4 и R5:
(1) каждый независимо, обозначает водород или R6;
(2) -Z4-N(R9)-Z5-NR10R11;
(3) -N(R9)Z4R6 или
(4) вместе с атомом азота, с которым они связаны, образуют 3-8-членный насыщенный или ненасыщенный гетероцикл, незамещенный или замещенный с заместителями Z1, Z2 и Z3, причем этот гетероцикл может, при необходимости, быть конденсирован с бензольным кольцом, которое, в свою очередь, является незамещенным или имеет заместители Z1, Z2 и Z3;
R7, R8, R9, R10, R11 и R12:
(1) каждый независимо, обозначает водород или R6;
(2) R7 и R8 могут вместе обозначать алкилен, алкенилен или гетероалкил, образующий 3-8-членный насыщенный или ненасыщенный цикл с атомом азота, с которым они связаны, причем цикл является незамещенным или замещенным с заместителями Z1, Z2 и Z3, или
(3) любые два из R9, R10 и R11 могут вместе обозначать алкилен или алкенилен, образующий 3-8-членное насыщенное или ненасыщенное кольцо вместе с атомами азота, с которыми они связаны, причем это кольцо является незамещенным или замещенным с заместителями Z1, Z2 и Z3;
R13 обозначает:
(1) циано;
(2) нитро;
(3) -NH2;
(4) -NHалкил;
(5) -ОН;
(6) -NHOарил;
(7) -NHCOOалкил;
(8) -NHCOOарил;
(9) -NHSO2алкил;
(10) -NHSO2арил;
(11) арил;
(12) гетероарил;
(13) -Оалкил или
(14) -Оарил;
R14 обозначает:
(1) -NO2;
(2) -СООалкил или
(3) -СООарил;
R15 обозначает:
(1) водород;
(2) алкил;
(3) арил;
(4) арилалкил или
(5) циклоалкил;
Z1, Z2 и Z3, каждый независимо, обозначает:
(1) водород или Z6, где Z6 обозначает (i) алкил, алкенил, алкинил, циклоалкил, циклоалкилалкил, циклоалкенил, циклоалкенилалкил, арил, аралкил, алкиларил, циклоалкиларил, гетероцикло или гетероциклоарил; (ii) группу (i), которая сама имеет в качестве заместителей или более одинаковых или различных групп (i); или (iii) группу (i) или (ii), которая имеет в качестве заместителей одну или более следующих групп, (2)-(16), обозначающих Z1, Z2 и Z3;
(2) -ОНили-OZ6;
(3) -SH или -SZ6;
(4) -C(O)qH, -C(O)qZ6 или -O-C(O)qZ6;
(5) -SO3Н, -S(O)qZ6; или -S(O)qN(Z9)Z6;
(6) галоид (галоген);
(7) циано;
(8) нитро;
(9) -Z4-NZ7Z8;
(10) -Z4-N(Z9)-Z5-NZ7Z8;
(11) -Z4-N(Z10)-Z5-Z6;
(12) -Z4-N(Z10)-Z5-H;
(13) оксо;
(14) -O-C(O)-Z6;
(15) любые два из Z1, Z2 и Z3 могут вместе обозначать алкилен или алкенилен, образующий вместе с атомами азота, с которым они связаны, насыщенный или ненасыщенный цикл, или
(16) любые два из Z1, Z2 и Z3 могут вместе обозначать -O-(СН2)r-O, где r обозначает 1-5, образующие насыщенный или ненасыщенный 4-8-членный цикл вместе с атомами, с которыми они связаны;
Z4 и Z5, каждый независимо, обозначает:
(1) простую связь;
(2) -Z11-S(O)q-Z12-;
(3) -Z11-C(O)-Z12-;
(4) -Z11-C(S)-Z12-;
(5) -Z11-O-Z12-;
(6) -Z11-S-Z12-;
(7) -Z11-O-C(O)-Z12- или
(8) -Z11-C(O)-O-Z12-;
Z7, Z8, Z9 и Z10:
(1) каждый независимо, обозначает водород или z6;
(2) Z7 и Z8 или Z6 и Z10 могут вместе обозначать алкилен или алкенилен, образующий 3-8-членный насыщенный или ненасыщенный цикл вместе с атомами, с которым они связаны, причем этот цикл является незамещенным или замещенным с заместителями Z1, Z2 и Z3, или
(3) Z7 или Z8 вместе с Z9 могут обозначать алкилен или алкенилен, образующий 3-8-членный насыщенный или ненасыщенный цикл вместе с атомами азота, с которым они связаны, причем этот цикл является незамещенным или замещенным с заместителями Z1, Z2 и Z3;
Z11 и Z12, каждый независимо, обозначает:
(1) простую связь;
(2) алкилен;
(3) алкенилен или
(4) алкинилен;и
Z13 обозначает:
(1) простую связь;
(2) -Z11-S(O)q-Z12-;
(3) -Z11-C(O)-Z12-;
(4) -Z11-C(S)-Z12-;
(5) -Z11-O-Z12-;
(6) -Z11-S-Z12-;
(7) -Z11-O-C(O)-Z12-;
(8) -Z11-C(O)-O-Z12-;
(9) -C(NR13)-;
(10) -C(CHR14)- или
(11) -C(C(Rl4)2)-.
Соединения, отвечающие формуле I, включают соединения нижеследующей формулы II и их соли
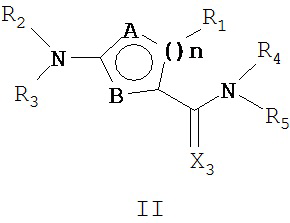
где n обозначает 1 или 2;
А выбирают из углерода и азота;
В выбирают из азота, кислорода и серы;
Х2 обозначает кислород или серу и
R1, R2, R3, R4 и R5 имеют указанные выше значения.
Подробное описание изобретения.
В настоящем описании используются следующие определения.
Начальное определение группы или термина в данном описании применяется к этой группе или термину по всему данному описанию, отдельно или как части другой группы, если не указано иначе.
Термин “алк” или “алкил” относится к углеводородным группам с линейной или разветвленной цепью, содержащим 1-12 атомов углерода, предпочтительно 1-8 атомов углерода. Выражение “низший алкил” относится к алкильным группам с 1-4 атомами углерода.
Термин “алкенил” относится к с линейным или разветвленным углеводородным группам из 2-10, предпочтительно 2-4 углеродных атомов, содержащих, по меньшей мере, одну двойную связь. Если алкенильная группа связана с атомом азота, предпочтительно, чтобы такая группа не была связана непосредственно через атом углерода при двойной связи.
Термин “алкинил” относится к с линейным или разветвленным углеводородным группам из 2-10, предпочтительно 2-4 атомов углерода, содержащих, по меньшей мере, одну тройную связь. Если алкинильная группа связана с азотом, предпочтительно, чтобы такая группа не была связана непосредственно через атом углерода при тройной связи.
Термин “алкилен” относится к линейному цепному мостику из 1-5 атомов углерода, соединенному ординарными связями (например, -(СН2)х-, где х обозначает 1-5), которые могут иметь в качестве заместителей 1-3 низших алкильных группы.
Термин “алкенилен” относится к линейному мостику из 2-5 атомов углерода, содержащему одну или две двойные связи, который соединен ординарными связями и может иметь в качестве заместителей 1-3 низших алкильных группы. Примерами алкениленовых групп являются -СН=СН-СН=СН-, -СН2-СН=СН-, -CH2-CH=CH-CH2-, -С(СН3)2СН=СН-и -СН(С2Н5)-СН=СН-.
Термин “алкинилен” относится к линейному мостику из 2-5 атомов углерода, содержащему тройную связь, связанному ординарными связями и могущему иметь в качестве заместителей 1-3 низших алкильных группы. Примерами алкиниленовых групп являются -СН≡С-, -CH2-C≡С-, -СН(СН3)-C≡С-и -C≡С-СН(С2Н5)СН2-. Термин “ar” (“ар”) или “арил” относится к ароматическим циклическим группам (например, 6-членным моноциклическим, 10-членным бициклическим или 14-членным трициклическим системам), которые содержат 6-14 атомов углерода. Примеры арильных групп охватывают фенил, нафтил, бифенил и антрацен.
Термин “циклоалкил” или “циклоалкенил” относится к циклическим углеводородным группам из 3-12 атомов углерода.
Термины “галоген” и “галоид” (“гало”) относятся к фтору, хлору, брому или иоду. Термин “ненасыщенный цикл” охватывает частично ненасыщенные и ароматические циклы.
Термины “гетероцикл”, “гетероциклический” и “гетероцикло” относятся к полностью насыщенным или ненасыщенным, включая ароматические (то есть “гетероарил”) циклические группы, например 4-7-членные моноциклические, 7-10-членные бициклические или 10-15-членные трициклические системы, которые содержат, по меньшей мере, один гетероатом в, имеющем, по меньшей мере, один атом углерода кольце. Каждый цикл гетероциклической группы, содержащей гетероатом, может иметь 1, 2, 3 или 4 гетероатома, выбранных из атомов азота, атомов кислорода и/или серы, где азот и сера могут, при необходимости, быть окисленными, а азот может, при необходимости, быть кватернизован. Гетероциклическая группа может быть связана с любым гетероатомом или атомом углерода цикла или циклической системы. Примеры моноциклических гетероциклических групп включают пирролидинил, пирролил, пиразолил, оксетанил, пиразолинил, имидазолил, имидазолинил, имидазолидинил, оксазолил, оксазолидинил, изоксазолинил, изоксазолил, тиазолил, тиадиазолил, тиазолидинил, изотиазолил, изотиазолидинил, фурил, тетрагидрофурил, тиенил, оксадиазолил, пиперидинил, пиперазинил, 2-оксопиперазинил, 2-оксопиперадинил, 2-оксопирролидинил, 2-оксоазепинил, азепинил, 4-пиперидонил, пиридинил, пиразинил, пиримидинил, пиридазинил, тетрагидропиранил, морфолинил, тиаморфолинил, тиаморфолинилсульфоксид, тиаморфолинилсульфон, 1,3-диоксолан и тетрагидро-1,1-диоксотиенил, триазолил, триазинил и т.п.
Примеры бициклических гетероциклических групп включают индолил, бензотиазолил, бензоксазолил, бензодиоксолил, бензотиенил, хинуклидинил, хинолинил, тетрагидроизохинолинил, изохинолинил, бензимидазолил, бензопиранил, индолизинил, бензофурил, хромонил, кумаринил, бензопиранил, циннолинил, хиноксалинил, индазолил, пирролопиридил, фуропиридинил (такой как фуро [2,3-с] пиридинил, фуро[3,2-b]пиридинил или фуро[2,3-b]пиридинил), дигидроизоиндолил, дигидрохиназолинил (такой как 3,4-дигидро-4-оксохиназолинил), тетрагидрохинолинил и т.п.
Примеры трициклических гетероциклических групп включают карбазолил, бензиндолил, фенантролинил, акридинил, фенантридинил, ксантенил и т.п. Термин “гетероарил” относится к ароматическим гетероциклическим группам. Примеры гетероарильных групп включают пирролил, пиразолил, имидазолил, оксазолил, изоксазолил, тиазолил, тиадиазолил, изотиазолил, фурил, тиенил, оксадиазолил, пиридинил, пиразинил, пиримидинил, пиридазинил, триазолил, триазинил и т.п.
Если q обозначает 1 или 2, “-C(O)qH” обозначает -С(O)-Н или -С(O)-ОН; “-C(O)qR6” или “-C(O)qZ6” обозначают, соответственно, -С(O)-R6 или -С(O)-OR6, или -С(O)-Z6 или -С(O)-OZ6; “-O-C(O)qR6” или “-O-C(O)qZ6” обозначают, соответственно, -O-С(O)-R6 или -O-С(O)-OR6, или -O-С(O)-Z6 или -O-С(O)-OZ6; и “-S(O)qR6” или “-S(O)qZ6” обозначают, соответственно, -SO-R6 или -SO2-R6, или -SO-Z6 или -SO2-Z6.
Соединения формулы I могут в некоторых случаях образовывать соли, которые также входят в объем данного изобретения. Понятно, что ссылка на соединение формулы I в данном описании включает ссылку на его соли, если не указано иначе. Термин “соль(и)”, как он применяется в данном описании, обозначает кислые и/или основные соли, образованные неорганическими и/или органическими кислотами и основаниями. Цвиттерионы (внутренние соли) включены в термин “соль(и)” по данному описанию (и могут образовываться, например, когда заместитель R содержит остаток кислоты, такой как карбоксильная группа). Также в данное описание включены четвертичные аммониевые соли, такие как соли алкиламмония. Фармацевтически приемлемые (т.е. нетоксичные, физиологически приемлемые) соли предпочтительны, хотя применяют и другие соли, например, на стадиях выделения и очистки, которые могут применяться при получении. Соли соединений формулы I могут образовываться, например, по реакции соединения I с некоторым количеством кислоты или основания, таким как эквивалентное количество, в среде, такой как среда, в которой соль осаждается, или в водной среде с последующей лиофилизацией.
Примеры солей присоединения кислот включают ацетаты (такие как соли, образующиеся с уксусной кислотой или тригалоидуксусной кислотой, например с трифторуксусной кислотой), адипинаты, альгинаты, аскорбаты, аспартаты, бензоаты, бензолсульфонаты, бисульфаты, бораты, бутираты, цитраты, камфораты, камфорсульфонаты, циклопентанпропионаты, диглюконаты, додецилсульфаты, этансульфонаты, фумараты, глюкогептаноаты, глицерофостфаты, гемисульфаты, гептаноаты, гексаноаты, гидрохлориды, гидробромиды, гидроиодиды, 2-гидроксиэтансульфонаты, лактаты, малеаты, метансульфонаты, 2-нафталинсульфонаты, никотинаты, нитраты, оксалаты, пектинаты, персульфаты, 3-фенилпропионаты, фосфаты, пикраты, пивалаты, пропионаты, салицилаты, сукцинаты, сульфаты (такие как сульфаты, образующиеся с серной кислотой), сульфонаты (такие как упоминавшиеся в данном описании), тартраты, тиоцианаты, толуолсульфонаты, ундеканоаты и т.п.
Примеры основных солей (образующихся, например, если заместители содержат остаток кислоты, такой как карбоксильная группа) включают аммониевые соли, соли щелочных металлов, такие как соли натрия, лития и калия, соли щелочноземельных металлов, такие как соли кальция и магния, соли органических оснований (например, органических аминов), таких как бензатины, дициклогексиламины, гидрабамины, N-метил-D-глюкамины, N-метил-D-глюкамиды, трет-бутиламины и соли с аминокислотами, такими как аргинин, лизин и т.п.Основные азотсодержащие группы могут кватернизоваться под действием таких агентов, как низшие алкилгалогениды (например, метил-, этил-, пропил- и бутилхлориды, бромиды и иодиды), диалкилсульфаты (например, диметил-, диэтил-, дибутил-и диамилсульфаты), длинноцепочечные галогениды (например, децил-, лаурил-, миристил- и стеарилхлориды, бромиды и иодиды), аралкилгалогениды (например, бензил-и фенилбромиды) и другие.
В данном описании также рассматриваются пролекарства и сольваты соединений по изобретению. Термин “пролекарство”, как он употребляется в данном описании, обозначает соединение, которое при введении его субъекту, претерпевает химическое превращение за счет метаболических или химических процессов, давая соединение формулы I или его соль и/или сольват. Сольваты соединений формулы I предпочтительно представляют собой гидраты.
Все стереоизомеры данных соединений, такие как стереоизомеры, которые могут существовать благодаря наличию асимметрических атомов углерода в заместителе R соединения формулы I, включая энантиомерные и диастереомерные формы, рассматриваются в объеме данного изобретения. Индивидуальные стереоизомеры соединений по изобретению могут, например, быть практически свободными от других изомеров или могут быть смешаны, например, в виде рацематов или смешиваться со всеми другими, или другими выбранными стереоизомерами. Хиральные центры по данному изобретению могут иметь S- или R-конфигурацию, определяемую согласно Рекомендациям IUPAC 1974.
По всему описанию группы и заместители в них выбирают так, чтобы образовывались стабильные фрагменты и соединения.
Предпочтительные соединения.
Предпочтительными соединениями по данному изобретению являются соединения формулы I и их соли, в которых Q обозначает тиазол и в которых один или более и особенно все из Z, X1, Х2, R1, R2, r3, R4 и R5 выбирают из следующих значений:
Z обозначает ординарную (простую) связь;
R1 выбирают из водорода, галоида, алкила, арила, алкокси, алкоксикарбонила или арилоксикарбонила, и более предпочтительным является водород;
Х1 и Х2 вместе образуют =O или =S и более предпочтительно образуют =O;
R2 обозначает водород;
R3 выбирают из -Z4-R6 или -Z13-NR7R8 и более предпочтительным является -Z4-R6, где Z4 обозначает простую связь, а R6 обозначает арил или гетероарил, незамещенный или замещенный с заместителями Z1, Z2 и с одной или более (предпочтительно одной или двумя) группами Z3;
R4 обозначает водород и
R5 выбирают из арильных групп или гетероарильных групп, которые имеют заместители Z1, Z2 и одну или более (например, одну или две) группы Z3.
Способы получения.
Соединения формулы I можно получать такими методами, которые проиллюстрированы на нижеприведенных схемах А-Е и I-XI. Растворители, температуры, давления и другие условия реакций может легко выбрать рядовой специалист в данной области техники. Все цитированные материалы вводятся в данное описание в качестве ссылок во всей полноте. Исходные вещества выпускаются промышленностью или их легко может получить рядовой специалист в данной области техники. Состав соединений дан либо в описании, либо конкретно приведен в схеме.
Методы, приведенные в данном описании, можно осуществлять, проводя реакции исходных веществ и/или реагентов в растворе или же, где это целесообразно, одно или более исходных веществ или реагентов может быть на твердой подложке (см. (1) Thompson, L. A., Ellman, J. A., Chemical Reviews, 96, 555-600 (1996); (2) Terrett, N. К., Gardner, M., Gordon, D. W., Kobylecki, R. J., Steele, J., Tetrahedron, 51, 8135-8173 (1995); (3) Gallop, M. A., Barrett, R. W., Dower, W. J., Fodor, S. P. A., Gordon, M. E., Journal of Medicinal Chemistry, 37, 1233-1251 (1994); (4) Gordon, M. E., Barrett, R. W., Dower, W. J., Fodor, S. P. A., Gallop, M. A., Journal of Medicinal Chemistry, 37, 1385-1401 (1994); (5) Balkenhohl, F., von dem 

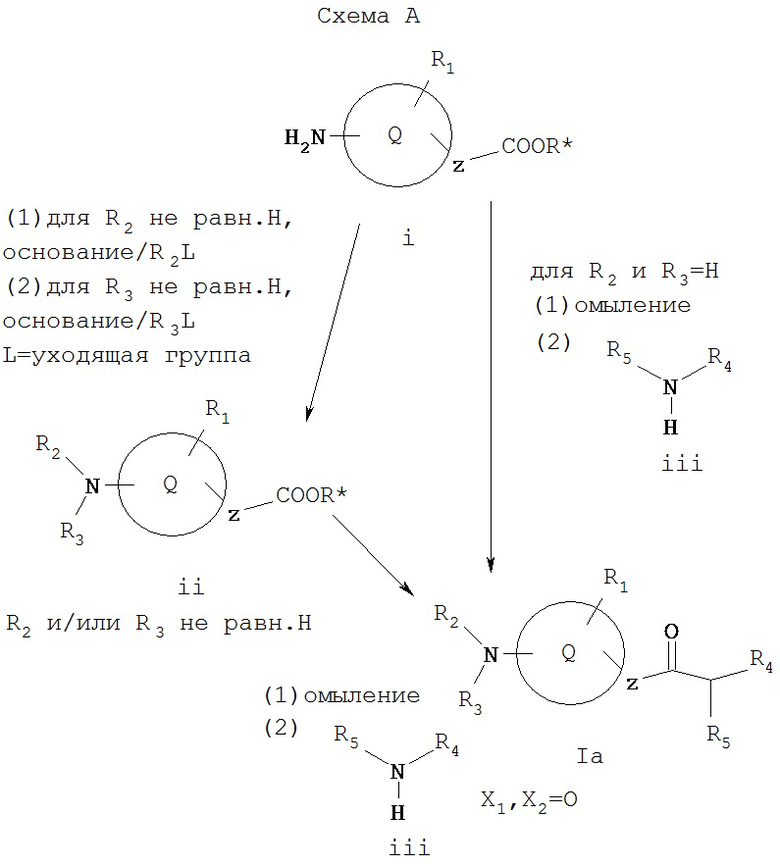
Схема А иллюстрирует общий метод получения соединения Ia, которое представляет собой соединение формулы I, где X1 и Х2 вместе образуют =O. Как показано на Схеме А, соединение Ia, где R2 и R3 обозначают водород, может образовываться при омылении i (R* обозначает карбоксил-защитную группу, такую как алкил или арилалкил) с последующей реакцией с амином iii известными из уровня техники способами. Или же i может реагировать с R2L, где L обозначает уходящую группу, такую как галоген (например, в эквимолярных соотношениях), при необходимости, с последующей реакцией с R3L (например, в эквимолярных соотношениях), образуя ii. Или же i может подвергаться восстановительному аминированию с применением соответствующего альдегида или кетона с образованием ii. Соединение ii затем может омыляться и реагировать с амином iii в условиях, известных специалистам в данной области техники, образуя Ia, где R2 и/или R3 обозначают иное, нежели водород.
Способы получения предпочтительных заместителей в соединениях I проиллюстрированы на данных ниже Схемах I-XI
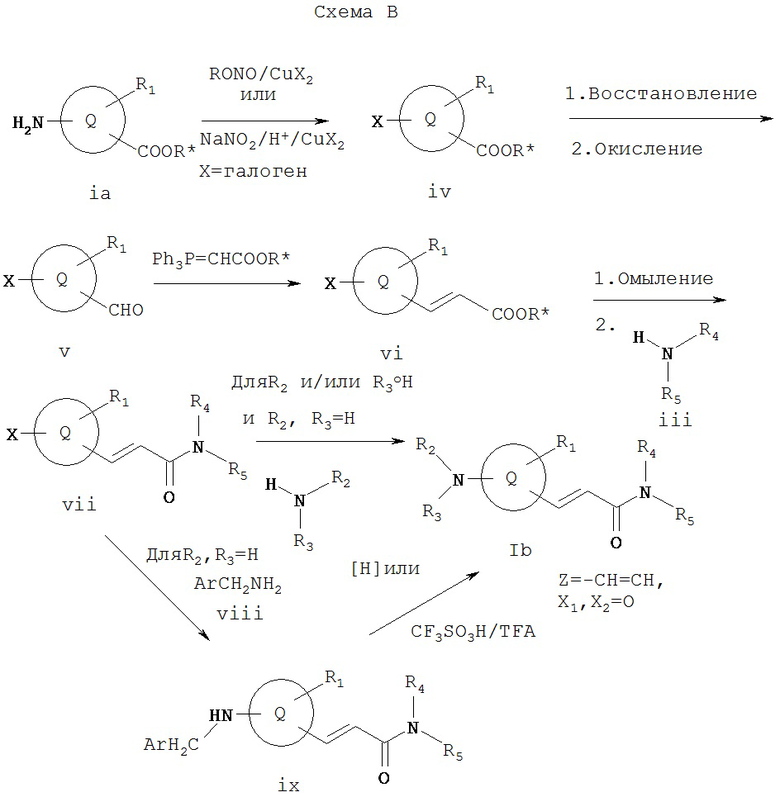 Схема В иллюстрирует общий метод получения соединения Ib, которое представляет собой соединение формулы I, где Z обозначает -СН=СН-, а X1 и Х2 вместе образуют =O. Как показано на Схеме В, 2-галоидсоединение vi можно получить реакцией соответствующего замещенного 2-аминосоединения ia с галогенидом меди (ii) и алкилнитратом, таким как трет-бутилнитрит, в апротонном растворителе, таком как ацетонитрил, образуя 2-галоидсоединение iv (см. J. Het. Chem. 22, 1621 (1985)). Соединение iv можно восстановить с помощью такого восстанавливающего агента, как натрийборгидрид в этаноле или в водном тетрагидрофуране, с образованием спирта, который можно окислить таким окислителем, как хлорхромат пиридиния или дихромат пиридиния, с образованием альдегида v. Соединение v может реагировать с алкил(трифенилфосфорилиден)ацетатом с образованием карбоксилата vi. Соединение vi может омыляться, а затем реагировать с амином iii известными специалистам в данной области техники способами с образованием vii. Соединение vii может реагировать с амином R2R3NH с образованием Ib, где Z обозначает -СН=СН-, a X1 и X2 вместе образуют =O. Или же соединения формулы Ib, где R1 и R2, обозначают Н, можно получать по реакции соединения vii с соответствующим замещенным бензиламином, таким как 4-метоксибензиламин, с образованием соединения ix, которое можно подвергать гидрогенолизу или обрабатывать кислотой, такой как трифторметансульфокислота и трифторуксусная кислота, в присутствии анизола с образованием Ib, где R1 и R2 обозначают водород.
Схема В иллюстрирует общий метод получения соединения Ib, которое представляет собой соединение формулы I, где Z обозначает -СН=СН-, а X1 и Х2 вместе образуют =O. Как показано на Схеме В, 2-галоидсоединение vi можно получить реакцией соответствующего замещенного 2-аминосоединения ia с галогенидом меди (ii) и алкилнитратом, таким как трет-бутилнитрит, в апротонном растворителе, таком как ацетонитрил, образуя 2-галоидсоединение iv (см. J. Het. Chem. 22, 1621 (1985)). Соединение iv можно восстановить с помощью такого восстанавливающего агента, как натрийборгидрид в этаноле или в водном тетрагидрофуране, с образованием спирта, который можно окислить таким окислителем, как хлорхромат пиридиния или дихромат пиридиния, с образованием альдегида v. Соединение v может реагировать с алкил(трифенилфосфорилиден)ацетатом с образованием карбоксилата vi. Соединение vi может омыляться, а затем реагировать с амином iii известными специалистам в данной области техники способами с образованием vii. Соединение vii может реагировать с амином R2R3NH с образованием Ib, где Z обозначает -СН=СН-, a X1 и X2 вместе образуют =O. Или же соединения формулы Ib, где R1 и R2, обозначают Н, можно получать по реакции соединения vii с соответствующим замещенным бензиламином, таким как 4-метоксибензиламин, с образованием соединения ix, которое можно подвергать гидрогенолизу или обрабатывать кислотой, такой как трифторметансульфокислота и трифторуксусная кислота, в присутствии анизола с образованием Ib, где R1 и R2 обозначают водород.
Способы получения предпочтительных заместителей в соединениях I проиллюстрированы на приведенных ниже Схемах I-XI.
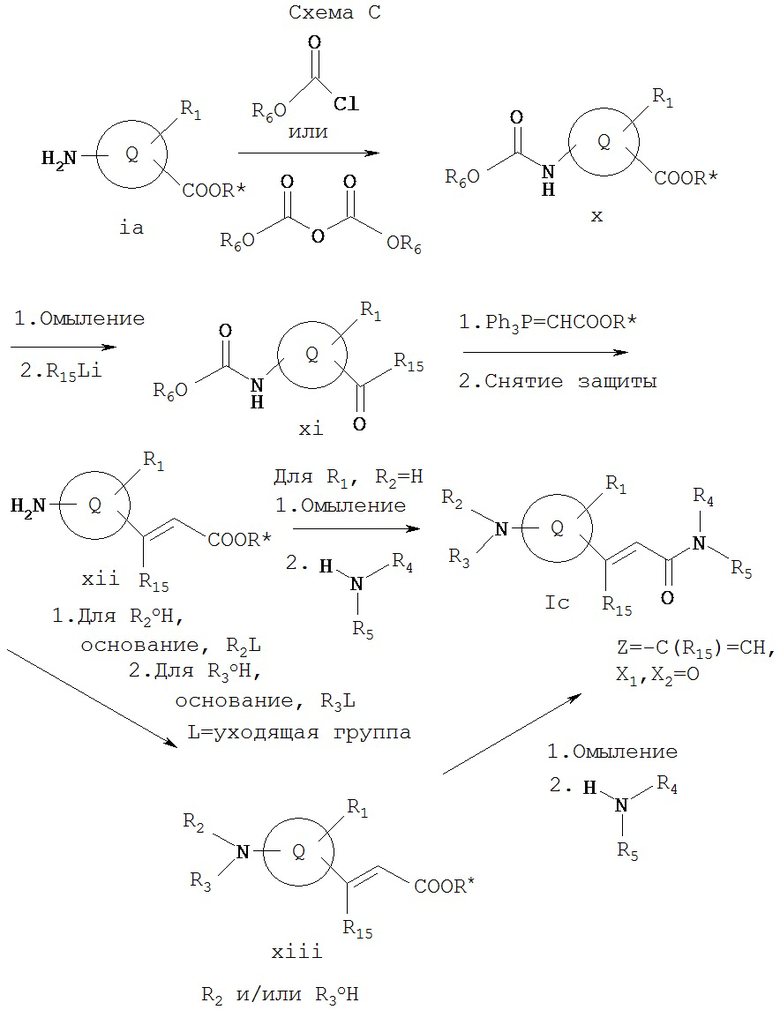
Схема С иллюстрирует общий метод получения соединения Iс, которое представляет собой соединение формулы I, где Z обозначает -R15C=CH-, а X1 и Х2 вместе образуют =O. Как показано на Схеме С, 2-аминосоединение ia может реагировать с хлорформиатом или дикарбонатом, образуя x, который можно омылить и обработать литийорганическим реагентом с образованием соединения xi. Соединение xi может реагировать с алкил(трифенилфосфорилиден)ацетатом с последующим снятием карбаматной защитной группы с образованием xii. Или же соединение Ic, где R2 и R3 обозначают водород, можно получать омылением xii с последующей реакцией с амином R4R5NH, осуществляемыми известными специалистам в данной области техники методами. Или же соединение xii может реагировать с R2L, где L обозначает уходящую группу, такую как галоген (например, в эквимолярных соотношениях), при необходимости, с последующей реакцией с R3L (например, в эквимолярных соотношениях) с образованием xiii, который может омыляться и реагировать с амином R4R5NH известными специалистам в данной области техники методами с образованием Ia, где значение R2 и/или R3 иное, нежели водород.
Способы получения предпочтительных заместителей в соединениях I проиллюстрированы на приведенных ниже Схемах I-XI.
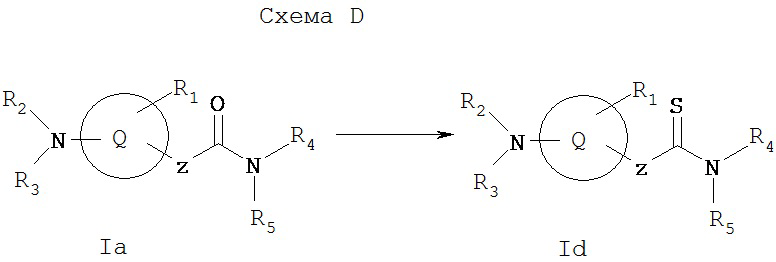
Схема D иллюстрирует общий метод получения соединения Id, которое представляет собой соединение формулы I, где X1 и Х2 вместе образуют =S. Соединения формулы Iа, получаемые по Схеме А, можно превратить в соответствующий тиоамид Id с применением такого реагента, как реагент Лавессона (2,4-бис(4-метоксифенил)-1,3-дитиа-2,4-дифосфэтан-2,4-дисульфид (см. Bull. Soc. Chim. Belg.,87, 223 (1978)).
Способы получения предпочтительных заместителей соединений I проиллюстрированы на представленных ниже Схемах I-XI.
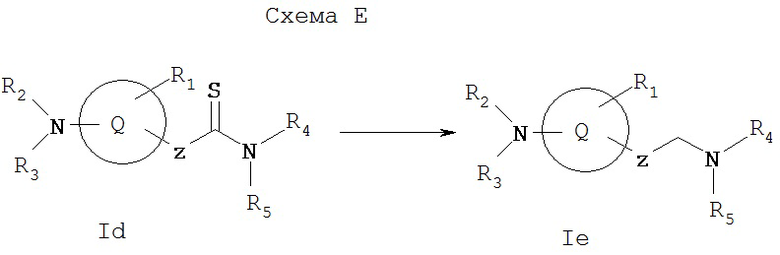
Схема Е иллюстрирует общий метод получения соединения Iе, которое представляет собой соединение формулы I, где X1 и Х2, каждый, обозначает водород. Как показано на Схеме Е, соединение формулы Id, получаемое по Схеме D, можно превратить в соответствующий амин Ie восстановлением, например, на никеле Ренея. Способы получения предпочтительных заместителей в соединениях I проиллюстрированы на данных ниже Схемах I-XI.
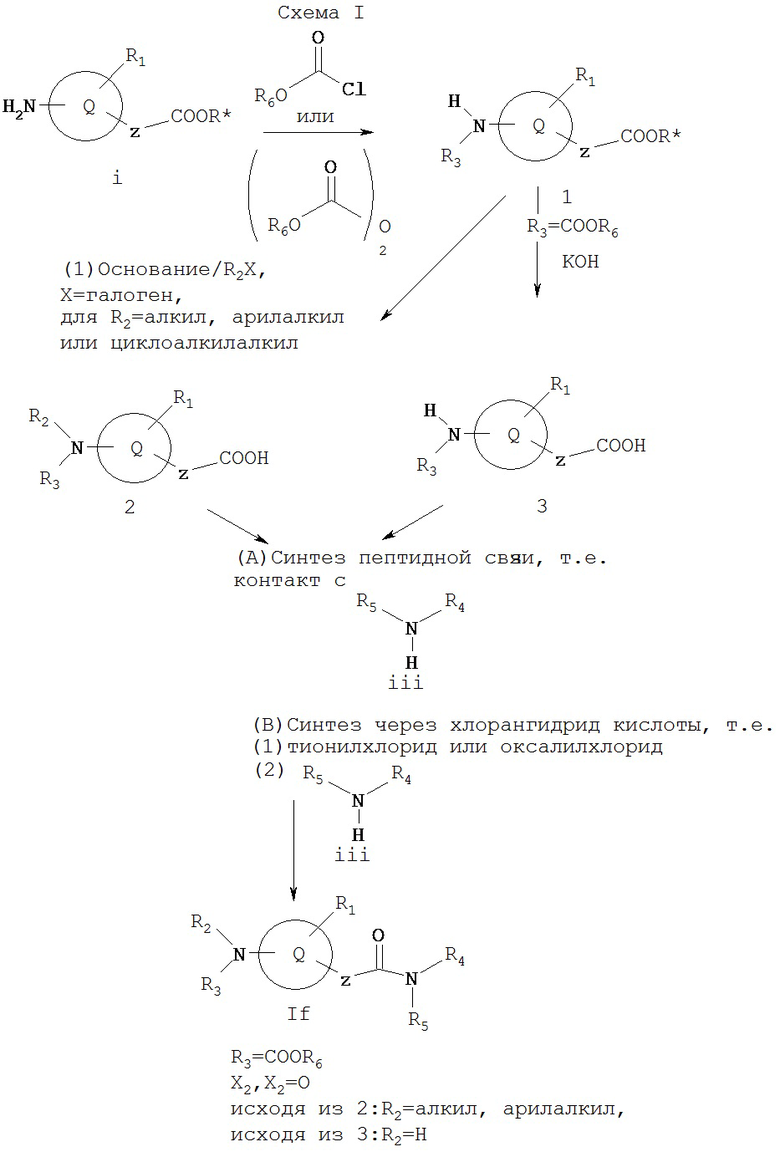
Как показано на Схеме I, карбоксилат i может реагировать с хлорформиатом или дикарбонатом с образованием 1. Соединение 1 можно обрабатывать основанием, таким как гидрид натрия, натрий/калий гексаметилдисилазид или диизопропиламид лития (LDA), и алкилирующим агентом R2Х, где Х обозначает галоген, a R2 предпочтительно представляет собой алкил, арилалкил или циклоалкилалкил, а затем омылить водным основанием, таким как гидроокись калия, с образованием 2. Или же 1 может подвергаться восстановительному аминированию, с применением соответствующего альдегида или кетона, и омылению водным основанием, таким как гидроксид калия, с образованием 2. Или же соединение 1 можно легко омылять таким водным основанием, как гидроксид калия, с образованием 3, где R2 обозначает водород.
Кислота 2 может реагировать с амином iii в условиях, хорошо известных из уровня техники для синтеза пептидной связи (см., например, Bodanszky and Bodanszky, The Practice of Peptide Chemistry, Springer-Verlag, 1984; Bodanszky, Principles of Peptide Synthesis, Springer-Verlag, 1984) с образованием соединения Id, которое представляет собой соединение формулы I, где X1 и X2 вместе образуют =O, R3 обозначает COOR6, и так как 2 представляет собой исходное соединение, R2 обозначает алкил, арилалкил или циклоалкилалкил. Например, реагенты, которые активируют карбоксильную группу 2 для реакции с амином iv, включают бис(2-оксо-3-оксазолидинил)фосфинхлорид) (ВОР хлорид), бензотриазол-1-илокси-трис(диметиламино)фосфония гексафторфосфат (ВОР реагент), [O-(7-азабензотриазол-1 -ил)-1,1,3,3 -тетраметилуроний] гексафторфосфат (HTAU) и карбодиимиды, такие как дициклогексилкарбодиимид (DCC) или 3-этил-3'-(диметиламино)пропилкарбодиимид (EDCI), либо одни, либо в сочетании с гидроксибензотриазолом. Или же активированный промежуточный сложный эфир можно выделить, а затем обрабатывать соответствующим амином iv в непротонном растворителе, таком как тетрагидрофуран (ТГФ) или диметилформамид (ДМФА) в присутствии основания, например органического основания, такого как гексаметилдисилазид натрия/калия, триэтиламин, диизопропилэтиламин или 1,8-диазабицикло[5.4.0]ундек-7-ен (DBU), или неорганического основания, такого как карбонат натрия, калия или цезия или гидрид натрия или калия. Или же хлорангидрид кислоты 2 можно получать, например, реакцией с тионилхлоридом или оксалилхлоридом с последующей реакцией с амином iii, что приводит к соединению If, которое представляет собой соединение формулы I, где R3 обозначает COOR6, X1 и Х2 вместе образуют =O и R2 обозначает алкил, арилалкил или циклоалкилалкил.
Реакции, подобные реакциям, применяемым выше для превращения 2 в If, можно использовать для превращения 3 в If, где R3 обозначает COOR6, X1 и X2 вместе образуют =O и R2 обозначает водород.
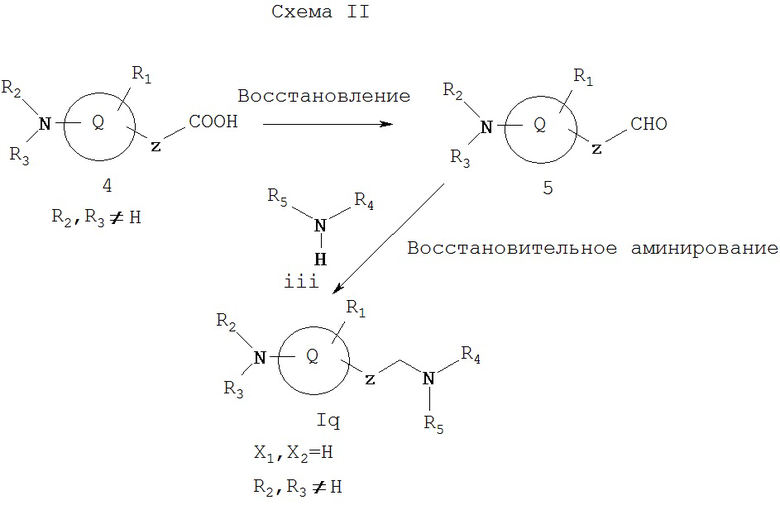
Как показано на Схеме II, кислота 4, где R2 и R3 не представляют собой водород, а выбираются так, что азот, с которым они связаны, не является основным, восстанавливается до альдегида 5 методами, хорошо известными из уровня техники (см. March, Advanced Organic Chemistry, Wiley, 1985). Например, кислоту 4 можно превратить в соответствующий сложный эфир с последующим восстановлением диизобутилалюминийгидридом. Или же кислоту 4 можно восстановить до соответствующего спирта, например, обработкой бораном/ТГФ, LiAlH4 или восстановлением смешанного ангидрида с последующим окислением до альдегида 5 с применением Cr(VI) (например, пиридинийхлорхромат, “РСС”) или в условиях реакций Сверна или Моффата (например, с (COCl)2/диметилсульфоксидом). Исходную кислоту 4 можно получать, например, омылением ii.
Восстановительное аминирование (см. Hudlicky, Reduction in Organic Chemistry, Wiley, 1984) альдегида 5 амином iii в присутствии восстановителя, такого как NaBH3CN, NaBH3(ОАс)3 (Ас=ацетил), или водородом на палладиевом катализаторе дает амин Ig, который представляет собой соединение формулы I, где X1 и Х2 каждый обозначает водород и R2 и R3, каждый, не является атомом водорода.
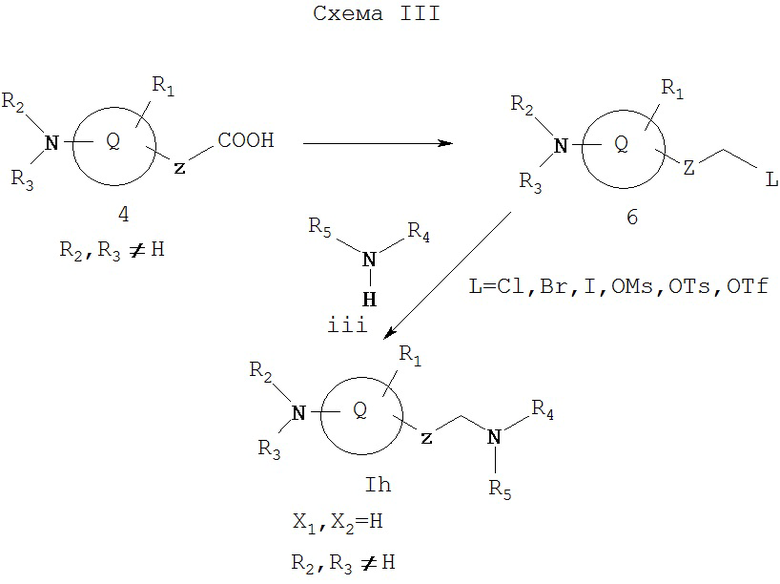
Как показано на Схеме III, восстановление кислоты 4 в первичный спирт (например, обработкой реагентом боран/тетрагидрофуран, LiAlH4 или восстановлением смешанного ангидрида) с последующим превращением методами, хорошо известными из уровня техники (см. March, Advanced Organic Chemistry, Wiley, 1985), дает 6, который содержит уходящую группу, такую как галоид, тозилат (OTs), мезилат (OMs) или трифталат (OTf). Группы R2 и R3 выбирают так, что результирующий азот, с которым они связаны, не является основным. Соединение 6 можно затем превратить в соединение Ih, представляет собой соединение формулы I, где X1 и Х2, каждый, обозначает водород, а R2 и R3, каждый, не является атомом водорода, реакцией замещения амином iii, предпочтительно, когда берется избыток амина iii.
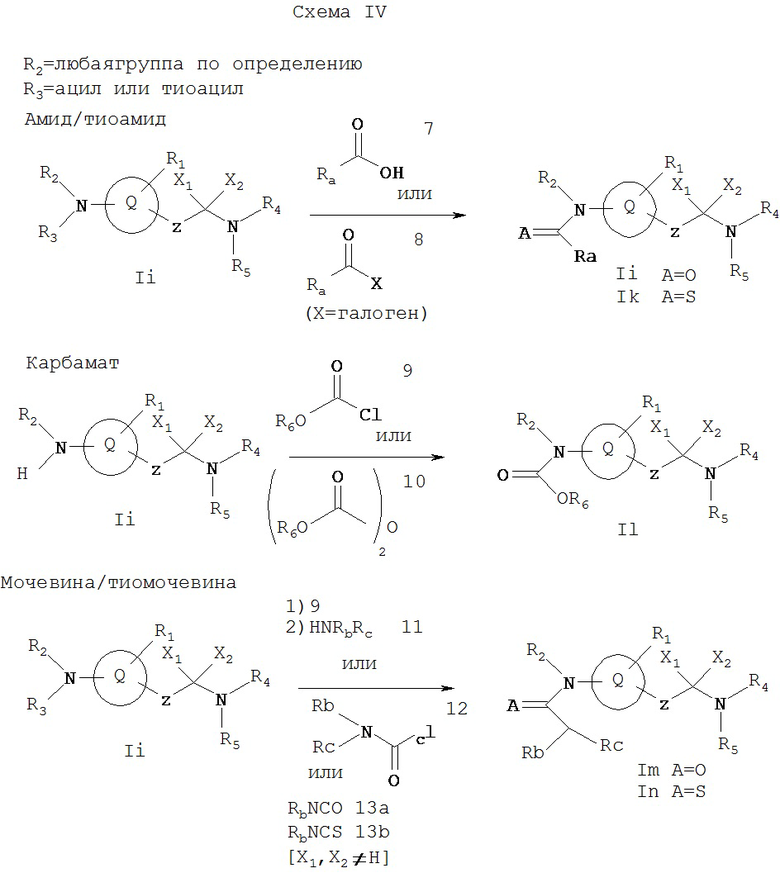
Схема IV иллюстрирует методы, которые можно применять для получения соединений Ij, Ik, Il, Im и In. Ij, ik, Il, Im и In представляют собой соединения формулы I, где R2 обозначает любую группу по определению, R3 обозначает ацильную или тиоацильную группу, X1 и Х2 не являются атомами водорода и R1 не является первичным или вторичным амином. Ij, Ik, Il, Im и In имеют другие специфические заместители, определение которых дано на этой Схеме и ниже. Исходное соединение Ii можно получить соответствующими методами, описанными на Схемах А и D.
Амид Ij можно получать обработкой амина Ii карбоновой кислотой 7 в присутствии реагентов, которые активизируют карбоксильную группу в вышеописанных реакциях, например ВОР реагент, HATU и карбодиимиды, такие как DCC или EDCI, либо одни, либо в сочетании с гидроксибензтриазолом. Или же хлорангидрид кислоты 8 может реагировать с амином Ii в присутствии “улавливателя” кислоты, такого как диизопропилэтиламин. Соответствующий тиоамид Ik можно получать обработкой амида Ii (где X1, X2 не равны 0) реагентом Лавессона, как описано выше.
Карбамат II можно получать обработкой амина Ii хлорформиатом 9 или дикарбонатом 10 в присутствии “кислотоуловителя” (нейтрализатора), такого как диизопропилэтиламин. Мочевину Im можно получать обработкой амина Ii либо 1) хлорформиатом 9, таким как фенилхлорформиат, с последующей реакцией с амином 11;
2) карбамоилхлоридом 12 в присутствии “кислотоуловителя”, такого как диизопропилэтиламин; или 3) реакцией с изоцианатом 13а (где Rc в Im=Н). Соответствующую тиомочевину In можно получать обработкой амина Ii тиоизоцианатом 13b.
Ra выбирают из групп, входящих в определение R6, так что группа -C(=A)-Ra представляет собой ацильную или тиоацильную группу, подпадающую под определение (обозначение) R3. Rb или Rc выбирают из групп, подпадающих под определение R7 и R8, так что группа -C(=A)-N(Rb)(Rc) представляет собой ацильную или тиоацильную группу, подпадающую под определение R3.
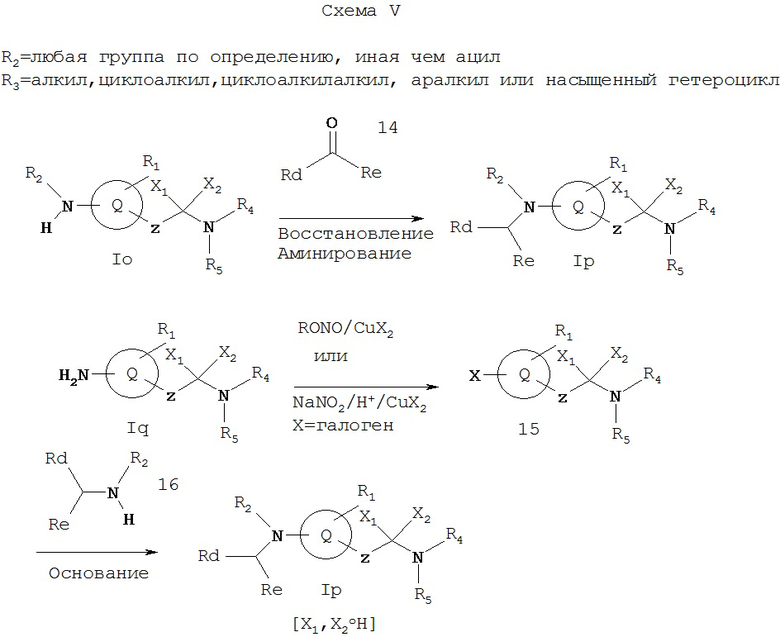
Схема V иллюстрирует метод, который можно использовать для получения Iр, которое представляет собой соединение формулы I, где R2 обозначает любую группу по определению, иную, нежели ацил, и которую выбирают так, что азот, с которым она связана, является основным, R3 обозначает алкил, циклоалкил, циклоалкилалкил, циклоалкенилалкил, аралкил или насыщенный гетероцикл, a X1 и X2 обозначают не водород. Исходные соединение Io и Iq можно получать соответствующими методами, описанными на Схемах А и D.
Как показано на Схеме V, амин Io реагирует с альдегидом или кетоном 14 в условиях восстановительного аминирования, описанных выше, дает амин Ip. Соединение Ip может быть также получено обработкой амина Iq, где R2 и R3 обозначают водород, трет-бутилнитритом или нитритом натрия в присутствии галогенида меди(II) с образованием галоидзамещенного соединения 15 с последующим замещением на амин 16 в присутствии основания, например гидрида натрия или калия и т.п. (см. Lee et al., J. Heterocyclic Chemistry, 22,1621 (1985)).
Rd и Re независимо выбирают из водорода, алкила, арила, циклоалкила или циклоалкенила, или вместе они обозначают алкилен или алкенилен, образующий 3-8-членный насыщенный или ненасыщенный цикл, так что -(CH)(Rd)(Re) обозначает группу, подпадающую под определение R3.
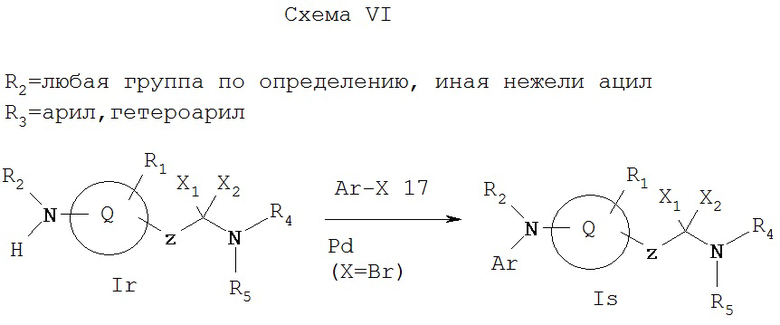
Как показано на Схеме VI, когда R2 обозначает любую группу по определению, иную, нежели ацил, и выбирается так, что азот, с которым она связана, является основным, R3 обозначает арил или гетероарил, a X1 и Х2 не обозначают водород, амин Ir, может реагировать с галоидфенильной или галоидгетероароматической группой 17 в присутствии палладиевого (0) катализатора (см. J. Am. Chem. Soc., 118, 7215 (1996)), давая амин Is, который представляет собой соединение формулы I, имеющее специфические заместители, описанные в этой схеме. Исходное соединение Ir можно получать соответствующими методами, описанными на схемах А и D.
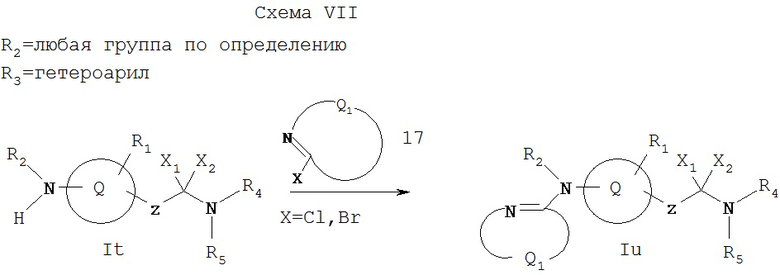
Как показано на Схеме VII, когда R2 обозначает любую группу по определению, а R3 обозначает гетероароматическую группу, амин It может реагировать, если необходимо, в присутствии основания с 2-галоидзамещенным гетероароматическим соединением 17, где Q вместе с атомами, с которыми он связан, образует 5- или 6-членную моноциклическую или 10-12-членную бициклическую гетероароматическую группу (например, образует 2-хлорпиридин или 2-хлорпиримидин), давая амин Iu, где Iu является соединением формулы I, имеющий специфические заместители, описанные в этой схеме. Исходное соединение It можно получать соответствующими методами, описанными на схемах А и D.
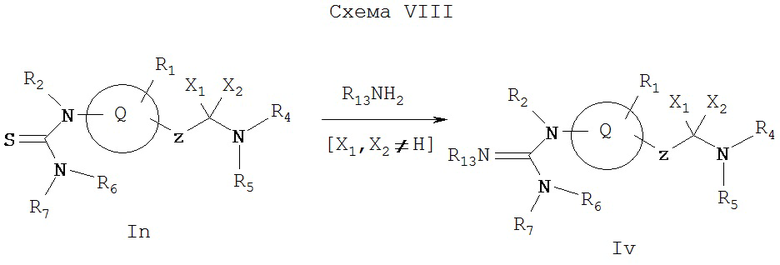
Как показано на Схеме VIII, тиомочевина In (где X1 и Х2 не обозначают водород) может реагировать с соответствующим амином в присутствии бис(2-оксо-3-оксазолидинил)фосфинхлорида (ВОР-хлорид), бензотриазол-1-илокси-трис(диметиламино)фосфонийгексафторфосфата (ВОР-реагент), [O-(7-азабензотриазол-1-ил)-1,1,3,3-тетраметилуроний]гексафторфосфата (HATU) и карбодиамида, такого как дициклогексилкарбодиимид (DCC), или 3-этил-3'-(диметиламино)пропилкарбодиимид (EDCI), или диизопропилкарбодиимид (DIC), в присутствии органического основания, такого как триэтиламин, диизопропилэтиламин или диметиламинопиридин в растворителях, таких как диметилформамид, дихлорметан или тетрагидрофуран, с образованием соединения Iv, которое представляет собой соединение формулы I, имеющее специфические заместители, описанные в этой Схеме. Или же соединение In может реагировать с соответствующим амином в присутствии соли ртути (II), такой как хлорид ртути (II), или другими известными из литературы методами, образуя Iv.
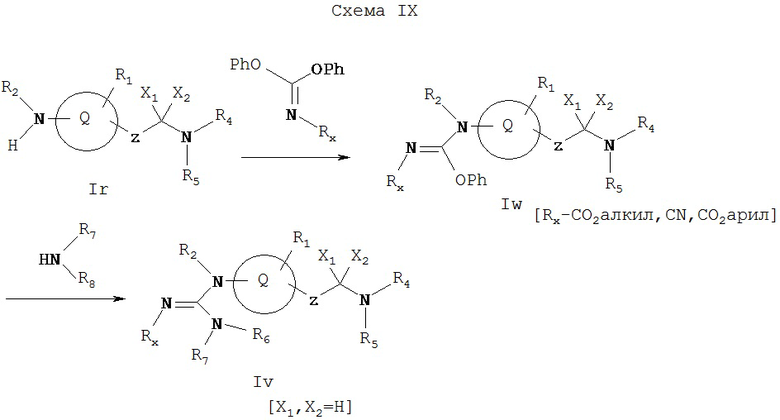
Как показано на Схеме IX, амин Ir (где X1 и X2 не обозначают водород) может реагировать с дифенилцианокарбонимидатом либо один, либо в присутствии основания, такого как гидрид натрия, гексаметилдисилазид натрия или диметиламинопиридин, в ацетонитриле, тетрагидрофуране или диметилформамиде при комнатной или повышенной температуре с образованием соединения Iw. Соединение Iw может реагировать с амином R7R8NH с образованием с Iv, которое представляет собой соединение формулы I, имеющее специфические заместители, описанные в этой Схеме.
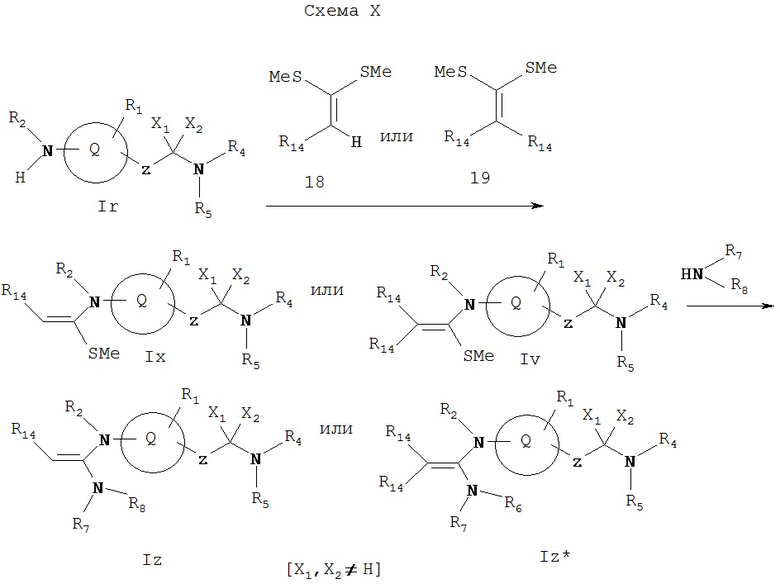
Как показано на Схеме X, соединение Ir (где X1 и Х2 не обозначают водород) может реагировать с 18 или 19 либо само, либо в присутствии основания, такого как гидрид натрия, гексаметилдисилазид натрия или диметиламинопиридин в диметилформамиде или тетрагидрофуране при комнатной или повышенной температуре с образованием соединений Ix или Iy, соответственно, которые реагируют с амином R7R8NH при комнатной или повышенной температуре с образованием соединений Iz или Iz*, соответственно. Соединение Iz представляет собой соединение формулы I, имеющее специфические заместители, описанные на этой Схеме. Соединение Iz* представляет собой соединение формулы I, имеющее конкретные заместители, описанные на этой Схеме.
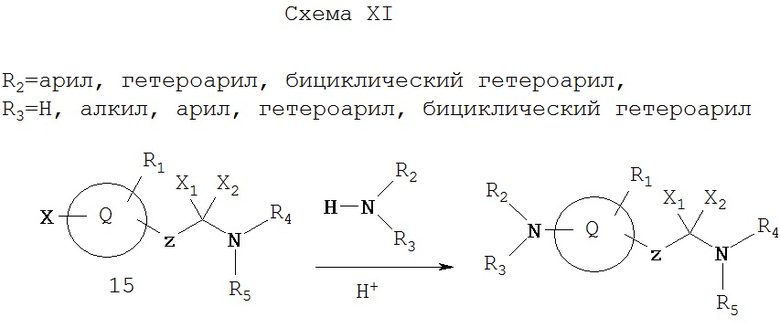
Как показано на Схеме XI, соединения формулы I также можно получать из 15 обработкой определенным амином в присутствии кислотного катализатора (см., например, Gunzenhauser et al., Helv. Chim. Acta, 71, 33 (1988)).
Применение.
Соединения по данному изобретению ингибируют протеинтирозинкиназы, в частности киназы семейства Src, такие как Lck, Fyn, Lyn, Src, Yes, Hck, Fgr и Bik, и, следовательно, применимы для лечения, включая предупреждение и терапию, обусловленных протеинтирозинкиназами расстройств, таких как иммунологические и онкологические нарушения. Соединения также ингибируют рецепторные тирозинкиназы, включая HER1 и HER2 и, следовательно, применимы для лечения пролиферативных нарушений, таких как псориаз и рак. Способность этих соединений ингибировать HER1 и другие рецепторные киназы также позволяют применять их в качестве антиангиогенных агентов для лечения расстройств, таких как рак и диабетическая ретинопатия. “Нарушения (расстройства), обусловленные протеинтирозинкиназами”, представляют собой такие расстройства, которые возникают вследствие аберрации тирозинкиназной активности и/или которые ослабляются при ингибировании одного или более этих ферментов. Например, Lck-ингибиторы важны для лечения ряда таких нарушений (например, при лечении аутоиммунных заболеваний), так как ингибирование Lck блокирует активацию Т клеток. Лечение опосредуемых Т-клетками заболеваний, включающее ингибирование активации и пролиферации Т клеток, является особенно предпочтительным вариантом данного изобретения. Соединения, которые селективно блокируют Т-клеточную активацию и пролиферацию, являются предпочтительными. Соединения по данному изобретению, которые блокируют активацию РТК эндотелиальных клеток за счет окислительного стресса, тем самым ограничивая поверхность экспрессии или адгезии молекул, которые индуцируют связывание нейтрофилов и которые ингибируют РТК, необходимую для активации нейтрофилов, применимы, например, для лечении ишемической и реперфузионной травмы.
Данное изобретение, следовательно, охватывает способы лечения расстройств, обусловленных протеинтирозинкиназами, включающие стадию введения субъекту, нуждающемуся в этом, по меньшей мере, одного соединения формулы I в количестве, эффективном для этого. Другие терапевтические агенты, такие как описанные ниже, могут использоваться с соединениями по изобретению по настоящим методам. В методах по данному изобретению такой(ие) другой(ие) терапевтический(ие) агент(ы) можно вводить перед введением соединения(ий) по данному изобретению, одновременно или после него.
Применение соединений по данному изобретению при лечении расстройств, обусловленных протеинтирозинкиназой, поясняется в качестве примера, без ограничения, лечением ряда таких расстройств, как отторжение трансплантата (например, трансплантата органа, срочного трансплантата, гетеро- или гомотрансплантата (такого, который применяют при ожогах)); защита от ишемической или реперфузионной травмы, такой как ишемическая или реперфузионная травма в ходе трансплантации органа, инфаркт миокарда, удар или другие факторы; индуцирование толерантности к трансплантату; артрит (например, ревматоидный артрит, псориатический артрит или остеоартрит); рассеянный склероз; хроническая закупорка легких (хроническое легочное обструктивное заболевание (COPD)), такая как эмфизема легких; воспалительное заболевание кишечника, включая язвенный колит и болезнь Крона (Crohn); волчанка (системная красная волчанка); болезнь “трансплантат против хозяина”; аллергические реакции, опосредуемые Т-клетками, включая контактную аллергическую реакцию, аллергическую реакцию замедленного типа и глютеновую энтеропатию (целиакия глютенчувствительная); псориаз; контактный дерматит (включая дерматит при контакте с сумахом укореняющимся, ядовитым плющом); тиреоидит Хашимото; синдром Шегрена; аутоиммунный гипертиреоз, такой как болезнь Грейвса (Graves); болезнь Аддисона (аутоиммунное заболевание надпочечников); аутоиммунная плюригландулярная болезнь (также известная как аутоиммунный плюригландулярный синдром); аутоиммунное облысение; пернициозная анемия; витилиго; гипопитуитаризм; синдром Гийена-Барре; другие аутоиммунные заболевания; злокачественные опухоли, включая злокачественные опухоли, при которых Lck или другие киназы Src-семейства, такие как Src, активируются и сверхэкспрессируют, например рак толстой кишки и тимома, и злокачественные опухоли, в которых активность киназ семейства Src облегчает рост или жизнеспособность опухоли; гломерулонефрит; сывороточная болезнь; крапивница; аллергические заболевания, такие как респираторные аллергические заболевания (астма, сенная лихорадка, аллергический ринит) или кожные аллергические заболевания; склерасиерма (scleracierma); грибовидный микоз; острые воспалительные реакции (такие как острый респираторный дистресс-синдром и ишемическая/реперфузионная травма); дерматомиозит; гнездная алопеция; хронический лучевой дерматит; экзема; болезнь Бехчета; пустулезный акродерматит; гангренозная пиодермия; синдром Сезари; диффузный нейродермит; системный склероз и кольцевидная склеродермия. Данное изобретение также включает способ лечения вышеуказанных расстройств, таких как диффузный нейродермит (атопический дерматит), введением любого соединения, способного ингибировать протеинтирозинкиназу.
Киназы Src-семейства, иные нежели Lck, такие как Hck и Fgr, важные для Fс-гамма рецепторных реакций моноцитов и макрофагов. Соединения по данному изобретению ингибируют Fc гамма-зависимое продуцирование TNF-альфа в клеточной линии моноцитов ТНР-1, которая не экспрессирует Lck. Способность ингибировать Fc гамма-рецептор-зависимый моноцитарный и макрофагальный ответы сообщает в результате дополнительную противовоспалительную активность данным соединениям помимо их действия на Т-клетки. Эта активность особенно важна, например, для лечения воспалительных заболеваний, таких как артрит или воспалительное заболевание кишечника. В частности, данные соединения важны для лечения аутоиммунного гломерулонефрита и других случаев гломерулонефрита, вызванных отложением иммунных комплексов в почке, которые “запускают” Fc гамма рецепторный ответ, приводящий к поражению почки.
Кроме того, киназы Src-семейства, иные нежели Lck, такие как Lyn и Src, важны для вызываемой Fc эпсилон рецептором дегрануляции тучных клеток и базофилов, которые играют роль при астме, аллергическом рините и других аллергических заболеваниях. Fc эпсилон рецепторы стимулируются IgE-антигенными комплексами. Соединения по данному изобретению ингибируют индуцируемую Fc эпсилон дегрануляцию, включая клеточную линию базофилов RBL, которая не экспрессирует Lck. Способность ингибировать Fc эпсилон-рецептор-зависимый ответ тучных клеток и базофилов приводит к дополнительной противовоспалительной активности данных соединений, помимо их действия на Т-клетки. В частности, данные соединения важны для лечения астмы, аллергического ринита и других аллергических заболеваний.
Комбинированная активность данных соединений в отношении моноцитов, макрофагов, Т-клеток и т.д. может быть важной для лечения любого из вышеуказанных расстройств. В конкретном варианте изобретения соединения по данному изобретению применяются для лечения приведенных выше в качестве примеров расстройств, вне зависимости от их этиологии, например для лечения отторжения трансплантата, ревматоидного артрита, рассеянного склероза, хронической закупорки легких, воспаления кишечника, волчанки, болезни “трансплантат против хозяина”, опосредуемой Т-клетками повышенной чувствительности, псориаза, тиреоидита Хашимото, синдрома Гийена-Барре, злокачественных опухолей, контактного дерматита, аллергических заболеваний, таких как аллергический ринит, астма, ишемическая и реперфузионная травма, атонический дерматит, обусловленных или не обусловленных РТК.
В силу их способности ингибировать киназы HER1 и HER2 соединения по данному изобретению можно также использовать для лечения пролиферативных заболеваний, включая псориаз и рак. Было показано, что рецепторная киназа HER1 экспрессирует и активируется во многих твердых опухолях, включая немелкоклеточный рак легких, прямой кишки и молочной железы. Аналогично HER2 рецепторная киназа, как было показано, сверхэкспрессирует при раке молочной железы, яичников, легких и желудка. Моноклональные антитела, которые негативно модулируют распространение HER2-рецептора или ингибируют передачу сигнала рецептором HER2, как было показано преклиническими и клиническими исследованиями, обладают противоопухолевой активностью. Следовательно, ожидается, что ингибиторы киназ HER1 и HER2 будут эффективны при лечении опухолей, которые зависят от передачи сигнала от любого из двух рецепторов. Ожидается, что эти соединения эффективны либо в качестве отдельного реагента, либо в сочетании с другими химиотерапевтическими агентами, такими как плаклитаксел (Taxol, таксол), доксорубицин гидрохлорид (адриамицин) и цисплатин (Platinol, платинол). См. следующие материалы и ссылки, цитируемые в данном описании: Cobleigh, M. A., Vogel, С. L., Tripathy, D., Robert, N. J., Scholl, S., Fehrenbacher, L., Wolter, J. M., Paton, V., Shak, S., Lieberman, G., and Slamon, D. J., “Multinational study of the efficacy and safety of humanized anti-HER2 monoclonal antibody in women who have HER2-overexpressing metastatic breast cancer that has progressed after chemotherapy for metastatic disease” (“Международное исследование эффективности и безопасности “гуманизированного” анти-НЕR2 моноклонального антитела у женщин, больных НЕR2-сверхэкспрессирующим метастатическим раком молочной железы, который прогрессирует после химиотерапии в заболевание с метастазами”), J. of Clin. OncoL, 17(9), р. 2639-2648 (1999); Baselga, J., Pfister, D., Cooper, M. R., Cohen, R., Burtness, В., Bos, M., D'Andrea, G., Seidman, A., Norton, L., Gunnett, K., Falcey, J., Anderson, V., Waksal, H., and Mendelsohn, J., “Phase I studies of anti-epidermal growth factor receptor chimeric antibody C225 alone and combination with cisplatin” (“Исследование фазы I рецептора антиэпидермального фактора роста химерного антитела C225, одного и в сочетании с цисплатином”), J. Clin. OncoL, 18(4), р. 904-914 (2000).
Данное изобретение также охватывает фармацевтические композиции, содержащие, по меньшей мере, одно из соединения формулы I, способное лечить обусловленное протеинтирозинкиназой расстройство в количестве, эффективном для этой цели, и фармацевтически приемлемый носитель или разбавитель. Композиции по данному изобретению могут содержать другие терапевтические агенты, как описано ниже, и могут готовиться, например, с применением обычных твердых или жидких носителей или разбавителей, а также фармацевтических добавок, соответствующих данному способу введения (например, эксципиенты, связующие, консерванты, стабилизаторы, ароматизаторы и т.д.) по методам, хорошо известным из уровня техники по фармацевтическим препаратам.
Соединения формулы I можно вводить любым приемлемым способом, например перорально, в виде таблеток, капсул, гранул или порошков; подъязычно; трансбукально; парентерально, например, в виде подкожной, внутривенной, внутримышечной или ягодичной инъекции или инфузии (например, в виде стерильных водных или неводных растворов или суспензий для инъекции); интраназально, например, в виде спрея для ингаляции; местно, например, в виде крема или мази; ректально, например, в виде суппозиториев; в виде стандартных доз, содержащих нетоксические фармацевтически приемлемые носители или разбавители. Данные соединения можно, например, вводить в форме, пригодной для мгновенного или пролонгированного действия. Мгновенное или пролонгированное действие достигается применением подходящих фармацевтических композиций, содержащих данное соединение, или, в частности, в случае пролонгированного действия, применением устройств, таких как подкожные имплантаты или осмотические насосы. Данные соединения можно также вводить с помощью липосом.
Примеры композиций для перорального введения включают суспензии, которые могут содержать, например, микрокристаллическую целлюлозу для придания объема, альгиновую кислоту или альгинат натрия в качестве суспендирующего агента, метилцеллюлозу для увеличения вязкости и подсластители или вкусовые добавки, известные из уровня техники; таблетки мгновенного действия, которые могут содержать, например, микрокристаллическую целлюлозу, дикальцийфосфат, крахмал, стеарат магния и/или лактозу и/или другие эксципиенты, связующие, наполнители, вещества, способствующие измельчению, разбавители и смазки, например, известные из уровня техники. Данные соединения можно доставлять через полость рта с помощью подъязычного и/или трансбукального (защечного) введения. Расплавленные таблетки, прессованные таблетки или лиофилизированные таблетки представляют собой примеры форм, которые могут использоваться. Примеры композиций включают композиции, включающие данное(ые) соединение(я) с быстро растворяющимися разбавителями, такими как маннит, лактоза, сахароза и/или циклодекстрины. Также в такие рецептуры могут входить высокомолекулярные эксципиенты, такие как целлюлозы (avicel) или полиэтиленгликоли (PEG, ПЭГ). Такие рецептуры могут также содержать эксципиент, способствующий прилипанию к слизистой оболочке, такой как гидроксипропилцеллюлоза (ГПЦ, НРС), гидроксипропилметилцеллюлоза (НРМС), натрийкарбоксиметилцеллюлоза (SCMC), сополимер малеинового ангидрида (например, Gantrez), и агенты для регуляции выделения (действия), такие как полиакриловый сополимер (например, Carbopol 934). Смазки, вещества, способствующие проглатыванию, вкусовые добавки, красители и стабилизаторы также можно добавлять для простоты изготовления и применения.
Примеры композиций для введения с помощью назальных аэрозолей или ингаляций включают растворы в физиологическом растворе, которые могут содержать, например, бензиловый спирт или другие соответствующие консерванты, промоторы всасывания для повышения биодоступности и/или другие солюбилизирующие или диспергирующие агенты, такие, например, как известные из уровня техники.
Примеры композиций для парентерального введения включают растворы или суспензии для инъекций, которые могут содержать, например, подходящие нетоксические парентерально приемлемые разбавители или растворители, такие как маннит, 1,3-бутандиол, вода, раствор Рингера, изотонический раствор хлорида натрия или другие подходящие диспергирующие или увлажняющие и суспендирующие агенты, включая синтетические моно- или диглицериды, и жирные кислоты, включая олеиновую кислоту. Примеры композиций для ректального введения включают суппозитории, которые могут содержать, например, подходящий не раздражающий эксципиент, такой как масло какао, синтетические глицериды или полиэтиленгликоли, твердые при обычных температурах, но ожижающиеся и/или растворяющиеся в ректальной полости с выделением лекарственного вещества.
Примеры композиций для местного введения включают местный носитель, такой как Plastibase (минеральное масло, образующее гель с полиэтиленом).
Эффективное количество соединения по данному изобретению может определить рядовой специалист в данной области техники, оно включает примерные дозировки для взрослого человека около 0,1-100 мг/кг веса тела активного соединения в день, которое можно вводить в виде однократной дозы или в виде индивидуальных небольших (раздельных) доз, например, 1-4 раза в день. Понятно, что конкретный уровень доз и частота приема доз для каждого конкретного субъекта может варьироваться и зависит от ряда факторов, включая активность конкретного применяемого соединения, метаболической стабильности и продолжительности действия этого соединения, вида, возраста, веса тела, общего состояния здоровья, пола, диеты больного, способа и времени введения, скорости выведения, комбинации лекарственных веществ и тяжести конкретного состояния. Предпочтительные субъекты для лечения включают животных, наиболее предпочтительны млекопитающие, такие как человек и домашние животные, такие как собаки, кошки и т.п., подверженные расстройствам, обусловленным протеинтирозинкиназами.
Соединения по данному изобретению можно применять отдельно или в сочетании друг с другом и/ или с другими подходящими терапевтическими агентами, пригодными для лечения расстройств, вызванных протеинтирозинкиназами, такими агентами, как ингибиторы РТК, иные, нежели ингибиторы РТК по данному изобретению, противовоспалительные, антипролиферативные и химиотерапевтические агенты, иммунодепрессанты, противораковые агенты и цитотоксические агенты.
Примеры таких иных терапевтических агентов включают следующие: циклоспорины (например, циклоспорин A), CTLA4-Ig, антитела, такие как анти-ICAM-3, рецептор анти-IL-2 (Анти-Тас), анти-СD45RВ, анти-СD2, анти-СD3 (ОКТ-3), анти-СD4, анти-CD80, анти-СD86, моноклональное антитело ОКТ3, агенты, блокирующие взаимодействие между CD40 и gp39, такие как антитела, специфические к CD40 и/или gp39 (т.е. CD154), слитые белки, конструированные из CD40 и gp39 (CD40Ig и CD8gp39), ингибиторы, такие как ингибиторы ядерной (нуклеарной) транслокации, NF-каппа В функции, такие как дезоксиспергуалин (DSG), нестероидные противовоспалительные лекарственные вещества (NSAID), такие как ибупрофен, стероиды, такие как преднизон или дексаметизон, соединения золота, антипролиферативные агенты, такие как метотрексат, FK506 (такролимус, Prograf), микофенолят мофетил, цитотоксические лекарственные вещества, такие как азатиприн и циклофосфамид, ингибиторы TNF-α, такие как тенидап, антитела против TNF или растворимый рецептор TNF, такой как этанерцепт (Enbrel), рапамицин (сиролимус или Rapamune), лефлунимид (Arava), и ингибиторы циклооксигеназы-2 (СОХ-2), такие как целекоксиб (Celebrex) и рофексиб (Vioxx), или их производные, и ингибиторы РТК, описанные в следующих патентных заявках США, вводимых ссылками в данное описание во всей полноте: Серийный №60/056770, поданная 25.08.97; Серийный №60/069159, поданная 09.12.97; Серийный №09/097338, поданная 15.06.98; Серийный №60/056797, поданная 25.08.97; Серийный №09/094797, поданная 15.06.98; Серийный №60/065042, поданная 10.11.97; Серийный №09/173413, поданная 15.10.98; Серийный №60 076 789, поданная 04.03.98, и Серийный №09 262 525, поданная 04.03.99 (Заявки у одного и того же патентного поверенного за номерами QA202*, QA202a*, QA202b, QA205*, QA205a, QA207*, QA202a, QA208* и QA208a. См. следующие материалы и ссылки, цитируемые далее: Hollenbauch, D., Douthwright, J., McDonald, V., and Aruffo, A., “Cleavable CD40Ig fusion proteins and the binding to sgp39” (“Расщепляемые CD40Ig белки и связывание с sgp39”), J. Immunol. Methods (Netherlands), 188(1), p. 1-7 (Dec. 15 1995); Hollenbauch, D., Grosmaire, L. S., Kullas, C. D., Chalupny, N. J., Braesch-Andersen, S., Noelle, R. J., Stamenkovic, I., Ledbetter, J. A., and Aruffo, A., “The human Т cell antigen gp39, a member of the TNF gene family, is a ligand for the CD40 receptor: expression of soluble form of gp39 with В cell costimulatory actinity” (“Человеческий Т-клеточный антиген gp39, член семейства гена TNF, представляет собой лиганд рецептора CD40: экспрессия растворимой формы gp39 с В-клеточной костимуляторной активностью”); ЕМВО J (England), 11(12), р. 4313-4321 (Dec 1992); and Moreland, L. W., et al., “Treatment of rheumatoid arthritis with a recombinant human tumor necrosis factor receptor (p75)-Fc fusion protein” (“Лечение ревматоидного артрита слитым белком: рецептор рекомбинантного человеческого фактора некроза опухолевых клеток (p75)-Fc”.
Примеры классов противораковых и цитотоксических агентов включают, без ограничения: алкилирующие агенты, такие как азотсодержащие горчичные масла, алкилсульфонаты, нитрозомочевины, этиленимины и триазены; антиметаболиты, такие как антагонисты фолата, аналоги пурина и пиримидина; антибиотики, такие как антрациклины, блеомицины, митомицин, дактиномицин и пликамицин; ферменты, такие как L-аспарагиназа; ингибиторы фарнезилпротеин-трансферазы; гормональные агенты, такие как глюкокортикоиды, эстрогены (антиэстрогены, андрогены/антиандрогены, прогестины и антагонисты лютеинизирующего гормона - рилизинг-гормона, октреотидацетат; агенты, разрушающие микротрубочки, такие как эктеинасциниды или их аналоги и производные; агенты, стабилизирующие микротрубочки, такие как паклитаксель (Taxol®), доцетаксель (Taxotere®), и эпотилоны A-F или их аналоги или производные; продукты растительного происхождения, такие как винка-алкалоиды, эпиподофиллотоксины, таксаны; и ингибиторы топоизомеразы; ингибиторы пренилпротеинтрансферазы; и смешанные агенты, такие как гидроксимочевина, прокарбазин, митотан, гексаметилмеламин, координационные комплексы платины, такие как цисплатин и карбоплатин; и другие агенты, применяемые в качестве противоопухолевых и цитотоксических агентов, такие как биомодуляторы, фактора роста; иммуномодуляторы и моноклональные антитела. Соединения по изобретению могут применяться также в сочетании с лучевой терапией. Репрезентативно примеры этих классов противоопухолевых (противораковых) и цитотоксических агентов включают, без ограничения, меклорэтамин гидрохлорид, циклофосфамид, хлорамбуцил, мелфалан, ифосфамид, бисульфан, кармустин, ломустин, семустин, стрептозоцин, тиотепа, дакарбазин, метотрексат, тиогуанин, меркаптопурин, флударабин, пентастатин, кладрибин, цитарабин, фторурацил, доксорубицин гидрохлорид, даунорубицин, идарубицин, блеомицин сульфат, митомицин С, актиномицин D, сафрацины, сафрамицины, хинокарцины, дискодермолиды, винкристин, винбластин, винорелбин, тартрат, этопозид, тенипозид, паклитаксел, тамоксифен, эстрамустин, эстрамустиннатрийфостфат, флутамид, бузерелин, леупролид, птеридины, диинезы, левамизол, афлакон, интерферон, интерлейкины, альдеслейкин, фильграстим, сарграмостим, ритуксимаб (rituximab), BCG, третиноин, иринотекана гидрохлорид, бетаметазон, гемцитабина гидрохлорид, альтретамин и топотека и их любые аналоги и производные.
Предпочтительные представители этих классов включают, без ограничения, паклитаксель, цисплатин, карбоплатин, доксорубицин, карминомицин, даунорубицин, аминоптерин, метотрексат, метоптерин, митомицин С, эктеинасцидин 743, порфиромицин, 5-фторурацил, 6-меркаптопурин, гемцитабин, цитозин арабинозид, подофиллотоксин или производные подофиллотоксина, такие как этопозид, этопозид фосфат или тенипозид, мельфалан, винбластин, винкристин, леурозидин, виндезин и леурозин (leurosine).
Примеры противоопухолевых (противораковых) и других цитотоксических агентов включают следующие: производные эпотилона, как показано в патентной заявке США Серийный №09/506481, поданной 17 февраля 2000 г. (No LD186 у патентного поверенного); патенте ФРГ 4138042.8; Международных заявках WO 97/19086, WO 98/22461, WO 98/25929, WO 98/38192, WO 99/01124, WO 99/02224, WO 99/02514, WO 99/03848, WO 99/07692, WO 99/27890, WO 99/28324, WO 99/43653, WO 99/54330, WO 99/54318, WO 99/54319, WO 99/65913, WO 99/67252, WO 99/67253 и WO 00/00485; ингибиторы циклин-зависимых киназ, как найдено в Международной заявке WO 99/24416, и ингибиторы пренил-протеинтрансферазы, как найдено в Международных заявках WO 97/30992 и WO 98/54966.
Другие терапевтические агенты, при употреблении в сочетании с соединениями по данному изобретению, могут применяться, например, в количествах, указанных в Справочнике врача (Physicians' Desk Reference (PDR)) или каким-либо другим способом, определяемым рядовым специалистом в данной области техники.
Следующие анализы можно использовать для выяснения степени активности соединения (“испытуемое соединение”) в качестве ингибитора РТК.
Соединения, описанные в приведенных ниже примерах, испытывались одним или более из этих аналитических методов и оказались активными.
Ферментный анализ с применением Lck, Fyn, Lyn, Hck, Fgr, Src. Bek или Yes
Следующий анализ проводят с применением протеинтирозинкиназ Lck, Fyn, Lyn, Hck, Fgr, Src, Bek и Yes.
Нужную протеинтирозинкиназу инкубируют в буфере для киназ (20 мМ MOPS, pH 7, 10 мМ MgCl2) в присутствий испытуемого соединения. Реакцию инициируют, добавляя субстраты до конечной концентрации 1 мкМ АТР, 3,3 мкКи/мл [33Р] гамма-АТР, и 0,1 мг/мл денатурированной кислотой енолазы (приготовлен, как описано в Cooper, J. А., Esch, F. S., Taylor, S. S., and Hunter, Т., “Phosphorylation sites in enolase and lactate dehydrogenase utilized by tyrosine protein kinases in vivo and in vitro” (“Сайты фосфорилирования в енолазе и лактат-дегидрогеназе, используемые протеинтирозинкиназами in vivo и in vitro”), J. Biol. Chem., 259, 7835-7841 (1984)). Реакцию прекращают через 10 минут, добавляя 10% трихлоруксусную кислоту, 100 мМ пирофосфата натрия, а затем 2 мг/мл альбумина бычьей сыворотки. Белковый субстрат меченой енолазы осаждается при 4 градусах, собирается на планшеты Packard Unifilter и считается на сцинтилляционном счетчике Topcount для того, чтобы удостовериться в ингибирующей протеинтирозинкиназу активности испытуемого соединения (активность обратно пропорциональна количеству полученного белка с меченой енолазой). Точная концентрация реагентов и количество радиоактивной метки при необходимости может меняться.
Этот анализ является предпочтительным, так как в нем используется экзогенный субстрат (енолаза) для более точного изучения кинетики фермента, и он может осуществляться в 96-луночном формате, который легко автоматизируется. Кроме того, меченые His протеинтирозинкиназы (описанные ниже) обеспечивают намного более высокие выходы продукции и чистоту по сравнению со слитым белком GST-протеинтирозинкиназа.
Протеинтирозинкиназу можно получать из промышленных источников или описанными ниже рекомбинантными методами. Для получения рекомбинантной Lck человеческую Lck получают в виде меченого His слитого белка, используя вектор бакуловируса (Life Technologies, Gibco) pFastBac Hta (выпускаемый промышленностью) в клетках насекомых. кДНК, кодирующую человеческую Lck, выделенную методом PCR (полимеразной цепной реакцией), встраивают в вектор и белок экспрессируют, применяя методы, описанные производителем. Lck очищают аффинной хроматографией. О продуцировании Lck в клетках насекомых с использованием бакуловируса, см. Spana, С., O'Rourke, Е. С., Bolen, J. В., and Fargnoli, J., “Анализ тирозинкиназы p561ck, экспрессирующей как белок глутатион S-трансфераза в клетках Spodoptera frugiperda”, “Protein expression and purification, Vol.4, p. 390-397 (1993). Сходные методы можно применять для рекомбинантного получения других киназ семейства Src.
Ферментный анализ с применением HER1 или HER2
Нужные соединения анализируют в буфере для киназ, который содержит 20 мМ Tris-HCl, pH 7,5, 10 мМ MnCl2, 0,5 мМ дитиотреитола, альбумин бычьей сыворотки 0,1 мг/мл, поли(glu/tyr, 4:1) с 0,1 мг/мл, 1 мкМ АТР и 4 мкКи/мл [гамма-33P]ATP. Поли(glu/tyr, 4:1) представляет собой синтетический полимер, который служит в качестве фосфорильного акцептора и поставляется Sigma Chemicals. Реакцию киназы инициируют, добавляя фермент, и реакционные смеси инкубируют при 26°С в течение 1 часа. Реакцию прекращают, добавляя EDTA до 50 мМ, и белок осаждают, добавляя трихлоруксусную кислоту до 5%. Осажденные белки извлекают, фильтруя через планшет Packard Unifilter и с помощью сцинтилляционного счетчика Topcount определяют радиоактивность количественно.
Для получения рекомбинантного HER1 цитоплазматическую последовательность рецептора экспрессируют в клетках насекомых в виде слитого белка с GST, который очищают аффинной хроматографией, как описано выше для Lck. Цитоплазматическую последовательность HER2 субклонируют в экспрессирующий вектор бакуловируса pBlueBac4 (Invitrogen) и экспрессируют в виде немеченого белка в клетках насекомых. Рекомбинантный белок частично очищают ионообменной хроматографией.
Клеточные анализы
(1) Клеточное фосфорилирование тирозина
Т-клетки Jurkat инкубируют с испытуемым соединением и затем стимулируют, добавляя антитело к CD3 (моноклональное антитело G19-4). Клетки лизируют через 4 минуты или через другой промежуток времени, добавляя буфер для лизина, содержащего детергент NP-40. Фосфорилирование белка обнаруживают иммуноблоттингом анти-фосфотирозина. Обнаружение фосфорилирования специфических белков, представляющих интерес, таких как ZAP-70, проводят иммунопреципитацией с антителом против ZAP-70 с последующем иммуноблотингом анти-фосфотирозина. Такие методики описаны в Schieven, G. L., Mittler, R. S., Nadler, S. G., Kirihara, J. M., Bolen, J. В., Kanner, S. В., and Ledbetter, J. A., “ZAP-70 tyrosine kinase, CD45 and Т cell receptor involvement in UV and H2O2 induced Т cell signal transolution” (“Участие ZAP-70 тирозинкиназы, CD45 и Т-клеточного рецептора в индуцированной УФ и Н2О2 сигнальной трансдукции Т клеток”), J. BioL Chem., 269, 20718-20726 (1994) и в приведенных в этой статье ссылках. Lck-ингибиторы ингибируют фосфорилирование тирозина клеточных белков, индуцируемое антителами против CD3.
О получении G19-4 см. Hansen, J. A., Martin, P. J., Beatty, P. G., dark, E. A., and Ledbetter, J. A., “Human T lymphocyte cell surface molecules defined by the workshop monoclonal antibodies” (“Молекулы поверхности человеческих Т лимфоцитов, определяемые “цехом” моноклональных антител”) в Leukocyte Typing I, A. Bernard, J. Boumsell, J. Dausett, C. Milstein, and S. Schlossman, eds. (New York: Springer Verlag), p. 195-212 (1984); и Ledbetter, J. A., June, C. H., Rabinovitch, P. S., Grossman A., Tsu, Т. Т., and Imboden, J. В., “Signal transduction through CD4 receptors: stimulatory vs. inhibitory activity is regulated by CD4 proximity to the CD3/T cell receptor” (“Сигнальная трансдукция с помощью рецепторов CD4: соотношение стимулирующая/ингибирующая активность регулируется близостью CD4 к CD3/T-клеточный рецептор”), Eur. J. ImmunoL, 18, 525 (1988).
(2) Анализ кальция
Ингибиторы Lck блокируют мобилизацию кальция в Т-клетках, стимулируемую антителами против CD3. Клетки нагружают кальциевым индикаторным красителем индо-1, обработанным антителом против CD3, таким как моноклональное антитело G19-4. и мобилизацию измеряют проточной цитометрией, регистрируя изменения в соотношении голубой/фиолетовый для индо-1, как описано в Schieven, G. L., Mittler, R.S., Nadler, G. S., Kirihara, J. M., Bolen, J. В., Kanner, S. В., and Ledbetter, J. A., “ZAP-70 tyrosine kinase, CD45 and t cell receptor involvement in UV and H2O2 induced Т cell signal transolution” (“Участие ZAP-70 тирозинкиназы, CD45 и Т-клеточного рецептора в индуцировании УФ и H2O2 сигнальной трансдукции Т клеток”), J. Biol. Chem., 269, 20718-20726 (1994) и в приведенных в этой статье ссылках.
(3) Определение пролиферации
Ингибиторы Lck ингибируют пролиферацию нормальных человеческих Т клеток периферической крови, рост которых стимулируется антителами против CD3 плюс антителами против CD28. 96-луночный планшет покрывают моноклональным антителом к CD3 (таким как G19-4), дают антителу возможность связываться, а затем планшет отмывают. Антитело, связанное с планшетом, служит для стимуляции клеток. В лунки добавляют нормальные человеческие Т-клетки периферической крови вместе с испытуемым соединением плюс антитела против CD28 для обеспечения костимуляции. Через заданный период времени (например, 3 дня) к клеткам добавляют [3H]-тимидин и после дополнительной инкубации для встраивания метки во вновь синтезированную ДНК клетки собирают и считают с помощью сцинтилляционного счетчика для определения пролиферации клеток.
Следующие примеры иллюстрируют варианты настоящего изобретения и не претендуют на ограничение объема изобретения.
Аббревиатуры, применяемые в примерах, поясняются ниже. Соединения в примерах обозначаются номерами примера и стадии, на которой они получены (например, 1А означает: заглавное соединение стадии А примера 1), или только номером примера, если соединение является заглавным соединением примера (например, 2 обозначает заглавное соединение примера 2).
Аббревиатуры
аq.=вод.=водный
конц.=концентрированный
ДМСО=диметилсульфоксид
EtOAc=этилацетат
Et2O=диэтиловый эфир
час, ч=часы
HATU=N-[диметиламино-1Н-1,2,3-триазоло[4,5-b]пиридин-1-илметилен]-N-
метилметанаминий гексафторфосфат N-оксид
МеОН=метанол
MOPS=4-морфолинпропансульфокислота
MS=масс-спектрометрия
Ret Time=время удерживания
rt=комнатная температура (~ 20°С)
нас.=насыщенный
TFA=трифторуксусная кислота
ТГФ=тетрагидрофуран
ДМФА=N,N-диметилформамид
Пример 1.
Получение 1,1-диметилэтилового эфира [5-[[(2,4,6-триметилфенил)амино]карбонил]-4-метил-2-тиазолил] карбаминовой кислоты.
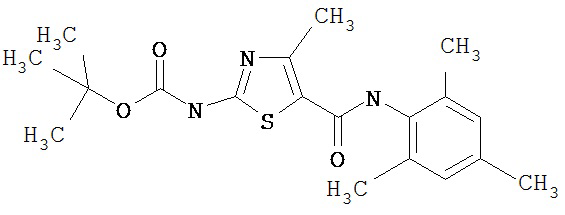
А. Этил-2-трет-бутоксикарбонилоксиамино-4-метил-тиазол-5-карбоксилат.
Суспензию этил-2-амино-4-метилтиазол-5-карбоксилата (18,6 г, 100 ммол), ди-трет-бутилдикарбоната (26,2 г, 120 ммол) и 4-диметиламинопиридина (800 мг, 6,55 ммол) в сухом тетрагидрофуране (300 мл) перемешивают под азотом 18 час. Растворитель упаривают в вакууме. Остаток суспендируют в хлористом метилене (1 л) и фильтруют через слой целита. Фильтрат промывают 1N водной HCl (300 мл, 2х), водой и рассолом, сушат (MgSO4) и упаривают в вакууме. Остаток растирают с гексанами. Твердое вещество фильтруют и сушат в вакууме, получают заглавное соединение (20 г, 72%) в виде рыжевато-коричневого твердого вещества.
В. 2-трет-Бутоксикарбонилоксиамино-4-метилтиазол-5-карбоновая кислота.
Перемешиваемый раствор этил-2-трет-бутоксикарбонилоксиамино-4-метилтиазол-5-карбоксилата (10 г, 34,95 ммол) в смеси тетрагидрофуран/этанол (250 мл, 2:3) обрабатывают 6N КОН (250 мл). Смесь греют при 55°С в течение ночи. Раствор охлаждают до 0°С и подкисляют конц. HCl до рН 1. Растворитель упаривают в вакууме. Остаток промывают водой, диэтиловьм эфиром, сушат в вакууме над безводным фосфорньм ангидридом, получают заглавное соединение (6 г, 89%) в виде белого твердого вещества.
С. 2-трет-Бутоксикарбонилоксиамино-4-метилтиазол-5-карбоновой кислоты хлор-ангидрид.
2 М раствор оксалилхлорида в хлористом метилене (22,5 мл, 45 ммол) по каплям добавляют к перемешиваемой суспензии 2-трет-бутоксикарбонилоксиамино-4-метилтиазол-5-карбоновой кислоты (10 г, 38,72 ммол) в хлористом метилене (150 мл) и N,N-диметилформамиде (150 мл) при 0°С. Суспензия постепенно становится гомогенной после окончания прибавления. Раствор нагревается до комнатной температуры и перемешивается при rt 1,5 часа. Растворитель упаривают в вакууме и остаток упаривают вместе с толуолом (300 мл, 2х), а затем сушат в вакууме, получая заглавный хлорангидрид кислоты (10,7 г, 99%) в виде рыжевато-коричневого твердого вещества.
D. 1,1-Диметилэтиловый эфир [5-[[(2,4,6-триметилфенил)амино]карбонил]-4-метил-2-тиазолил]карбаминовой кислоты.
2,4,6-Триметиланилин (6,3 мл, 38,66 ммол) добавляют по каплям к перемешиваемому раствору хлорангидрида 2-трет-бутоксикарбонилоксиамино-4-метилтиазол-5-карбоновой кислоты (10,7 г, 38,66 ммол) в хлористом метилене (150 мл) при 0°С. Через 20 минут по каплям добавляют диизопропилэтиламин (8,8 мл, 44,88 ммол). Раствор доводят до ~20°С и перемешивают еще 2 часа. Растворитель упаривают в вакууме. Остаток суспендируют в EtOAc (700 мл), промывают 1 N водн. HCl (300 мл, 2х), водой и рассолом; сушат (MgSO4), фильтруют и упаривают. Остаток затирают эфиром, получают заглавное соединение (12,5 г, 86%) в виде рыжевато-коричневого твердого вещества.
Пример 2.
Получение 2-амино-N-(2,4,6-триметилфенил)-4-метил-5-тиазолкарбоксамида.
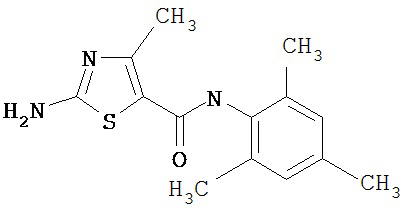
Раствор 1,1-диметилового эфира [5-[[(2,4,6-триметилфенил)амино]карбонил]-4-метил-2-тиазолил]карбаминовой кислоты (10 г, 26,63 ммол) в трифторуксусной кислоте (100 мл) перемешивают при rt 3 часа. Раствор упаривают в вакууме и остаток разбавляют EtOAc (700 мл), промывают 5% водн. КНСО3 (400 мл, 2х), водой и рассолом; сушат (MgSO4), фильтруют и упаривают. Остаток промывают эфиром (200 мл) и ацетонитрилом (100 мл), получают заглавное соединение (6,7 г, 91%) в виде белого твердого вещества.
Пример 3.
Получение 1,1-диметилэтилового эфира [5-[[(2,4,6-триметилфенил)амино]карбонил]-4-трифторметил-2-тиазолил] карбаминовой кислоты.
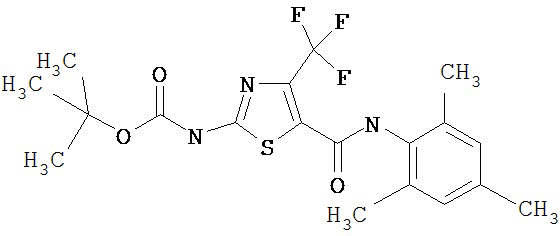
А. Этил-2-трет-бутоксикарбонилоксиамино-4-трифторметилтиазол-5-карбоксилат.
Суспензию этил-2-амино-4-трифторметилтиазол-5-карбоксилата (5,05 г, 21,02 ммол), ди-трет-бутилдикарбоната (4,82 г, 22,07 ммол) и 4-диметиламинопиридина (260 мг, 2,1 ммол) в хлористом метилене (209 мл) перемешивают под азотом 1,5 часа. Растворитель упаривают в вакууме. Остаток хроматографируют на колонке с силикагелем. Элюируют 5% EtOAc в смеси гексанов, затем 15% EtOAc в смеси гексанов, получают заглавное соединение (6,57 г, 92%) в виде белого твердого вещества.
В. 2-трет-Бутоксикарбонилоксиамино-4-трифторметилтиазол-5-карбоновая кислота.
Перемешиваемый раствор этил-2-трет-бутоксикарбонилоксиамино-4-трифтор-метилтиазол-5-карбоксилата (6,5 г, 19,1 ммол) в метаноле (100 мл) обрабатывают 1 N водн. NaOH (573 мл). Смесь перемешивают при ~20°С в течение ночи. Раствор охлаждают до 0°С и подкисляют 1 М HCl (водн.) до рН 1 и экстрагируют хлороформом (150 мл, 6х). Хлороформные вытяжки объединяют, сушат (Na2SO4), фильтруют и упаривают в вакууме, получают заданную кислоту (5,75 г, 96%) в виде белого твердого вещества.
С. 1,1-Диметилэтиловый эфир [5-[[(2,4,6-триметилфенил)амино]карбонил]-4-трифторметил-2-тиазолил]карбаминовой кислоты.
4-Метилморфолин (40 мкл, 0,39 ммол) добавляют к смеси 2-трет-бутоксикарбонилоксиамино-4-трифторметилтиазол-5-карбоновой кислоты (100 мг, 0,32 ммол), 2,4,6-триметиланилина (45 мкл, 0,32 ммол) и бензотриазол-1-илокси-трис-(диметиламино)фосфонийгексафторфосфата (ВОР-реагент, 380 мг, 0,4 ммол) в ДМФА (2 мл). Раствор перемешивают при ~20°С в течение 72 час, разбавляют хлористым метиленом и промывают 0,25 М водн. KHSO4, а затем нас.водн. КНСО3. Отделяют органический (СН2Cl2) слой, сушат (Na2SO4), фильтруют и упаривают. Остаток хроматографируют на колонке с силикагелем и элюируют 5% EtOAc в смеси гексанов, а затем 10% EtOAc в смеси гексанов, получая заглавное соединение (90 мг, 65%) в виде белого твердого вещества.
Пример 4.
Получение 2-амино-N-(2,4,6-триметилфенил)-4-трифторметил-5-тиазолкарбоксамида трифторацетата (1:1).
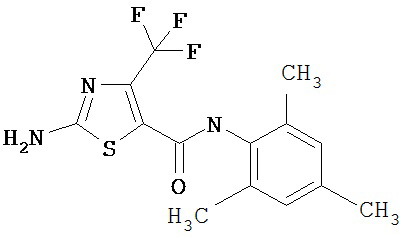
Рвствор 1,1-диметилэтилового эфира [5-[[(2,4,6-триметилфенил)амино]карбонил]-4-трифторметил-2-тиазолил]карбаминовой кислоты (120 мг, 0,28 ммол) в трифторуксусной кислоте (5 мл) перемешивают при 0°С 1 час. Раствор упаривают в вакууме и остаток упаривают вместе с эфиром, получая желтое твердое вещество, которое растирают со смесью гексанов, получают заглавное соединение (96 мг, 76%) в виде светло-желтого твердого вещества.
Пример 5.
Получение 1,1-диметилэтилового эфира [5-[[(2,4,6-триметилфенил)амино]карбонил]-4-фенил-2-тиазолил] карбаминовой кислоты.
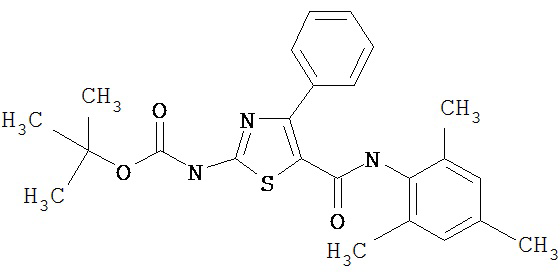
А. Этил-2-трет-бутоксикарбонилоксиамино-4-фенилтиазол-5-карбоксилат.
Соединение 5А получают аналогично 3А, но используют этил-2-амино-4-фенилтиазол-5-карбоксилат и получают заглавное соединение 5А в виде белого твердого вещества (90,5%).
В. 2-трет-Бутоксикарбонилоксиамино-4-фенилтиазол-5-карбоновая кислота.
Соединение 5В получают аналогично 3В, но используя 5А, и получают заглавное соединение 5В в виде белого твердого вещества (99%).
С. Хлорангидрид 2-трет-бутоксикарбонилоксиамино-4-фенилтиазол-5-карбоновой кислоты.
Соединение 5С получают аналогично 1С, но применяя 5В, и получают заглавное соединение 5С в виде белого твердого вещества (90%).
D. 1,1-Диметилэтиловый эфир [5-[[(2,4,6-триметилфенил)амино]карбонил]-4-фенил-2-тиазолил]карбаминовой кислоты.
Соединение 5D получают аналогично 1D, но применяя 5С, и получают заглавное соединение 5D в виде светло-желтого твердого вещества (93%).
Пример 6.
Получение 2-амино-N-(2,4,6-триметилфенил)-4-фенил-5-тиазолкарбоксамида, трифторацетата (1:1).
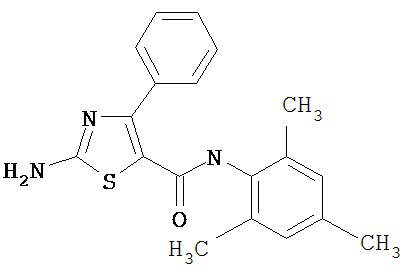
Соединение 6 получают аналогично 4, но берут 5D, получая заглавное соединение 6 в виде белого твердого вещества (68%).
Пример 7.
Получение 1,1-диметилэтилового эфира [5-[[фениламино]карбонил]-4-метил-2-тиазолил]карбаминовой кислоты.
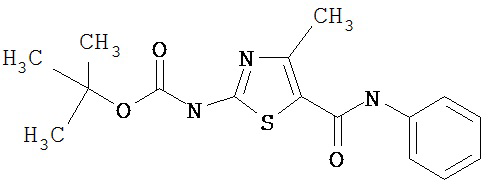
Соединение получают аналогично 1D, но берут анилин вместо 2,4,6-триметиланилина и триэтиламин вместо диизопропилэтиламина, получая заглавное соединение 7 в виде грязно-белого твердого вещества (76%).
Пример 8.
Получение 2-амино-N-(фенил)-4-метил-5-тиазолкарбоксамида, трифторацетата (1:1).
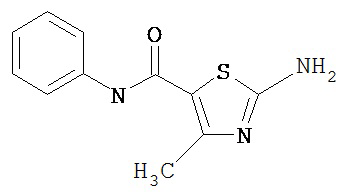
Соединение 8 получают аналогично 4, но берут 7, получая заглавное соединение 8 в виде белого твердого вещества (68%).
Пример 9.
Получение 1,1-диметилэтилового эфира [5-[[(2,4-дихлорфенил]амино]карбонил]-4-метил-2-тиазолил]карбаминовой кислоты.
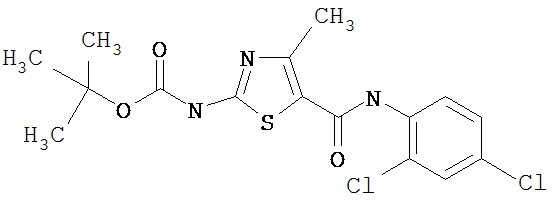
Соединение 9 получают аналогично 1D, но берут 2,4-дихлоранилин, получая заглавное соединение 9 в виде белого твердого вещества (28%).
Пример 10.
Получение 2-амино-N-(2,4-дихлорфенил)-4-метил-5-тиазолкарбоксамида, трифторацетата (1:1).
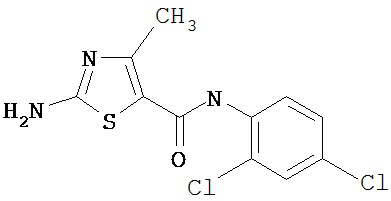
Соединение 10 получают аналогично 4, за исключением того, что берут 9, получая 10 в виде белого твердого соединения (100%).
Пример 11.
Получение 1,1-диметилэтилового эфира [5-[[(2,4,6-триметилфенил]амино]-карбонил]-2-тиазолил]карбаминовой кислоты.
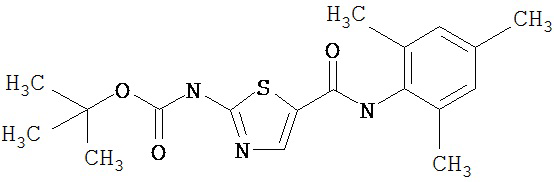
А. Этил-2-трет-бутоксикарбонилоксиаминотиазол-5-карбоксилат.
Соединение 11А получают аналогично 3А, но берут этил-2-аминотиазол-5-карбоксилат, получая заглавное соединение 11А в виде белого твердого вещества (79,5%).
В. 2-трет-Бутоксикарбонилоксиаминотиазол-5-карбоновая кислота.
Соединение 11В получают аналогично 3В, но берут 11А, получая заглавное соединение 11В в виде белого твердого вещества (95,5%).
С. Хлорангидрид 2-трет-бутоксикарбонилоксиаминотиазол-5-карбоновой кислоты.
Соединение 11С получают аналогично 1С, но берут 11В, получая заглавное соединение 11С.
D. 1,1-Диметилэтиловый эфир [5-[[(2,4,6-триметилфенил)амино]карбонил]-2-тиазолил]карбаминовой кислоты.
Соединение 11D получают аналогично 1D, но берут 11С, получая заглавное соединение 11D в виде грязно-белого твердого вещества (70%).
Пример 12.
Получение 2-амино-N-(2,4,6-триметилфенил)-4-фенил-5-тиазолкарбоксамида, трифторацетата (1:1).
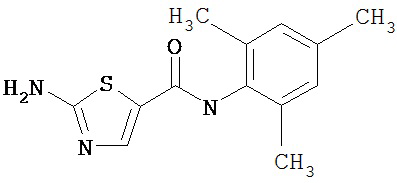
Соединение 12 получают аналогично 4, но берут 11D, получая заглавное соединение 12 в виде светло-желтого твердого вещества (88%).
Примеры 13-53.
Общая методика.
Соединения 13-53 получают по описанной ниже методике. Соответствующие амины (0,40 ммол) и диизопропилэтиламин (79 мкл, 0,40 ммол) добавляют к суспензии 1С (100 мг, 0,36 ммол) в хлористом метилене (3 мл). Раствор перемешивают механически в запаянной трубке при ~20°С в течение 16 час. Реакционные смеси разбавляют метанолом (200 мл) и загружают на Varian SCX-ионнообменные колонки (2 г/6 см), предварительно обработанные метанолом - хлористым метиленом (8 мл, 1:1), а затем хлористым метиленом (8 мл). Фильтрование на SCX-колонке осуществляют с помощью автоматического блока (робота) Гильсона (Gilson). Колонку промывают последовательно хлористым метиленом (9 мл), хлористым метиленом - метанолом (9 мл, 4:1), хлористым метиленом - метанолом (9 мл, 1:1), метанолом (9 мл), 0,01 М гидроксида аммония в метаноле (9 мл), 0,05 М гидроксида аммония в метаноле (9 мл). Элюаты собирают отдельно с помощью автомата (робота) и затем упаривают на роторном испарителе (speed vac.). Фракции, содержащие продукты, объединяют.
“ВЭЖХ время удерж.” обозначает время удерживания ВЭЖХ в следующих условиях: балластная колонка YMC S5 ODS 4,6⋅х50 мм; градиент 4 мин, начиная со 100% растворителя А (10% МеОН, 90% H2O, 0,2% Н3PO4) до 100% растворителя В (90% МеОН, 10% H2O, 0,2% Н3PO4), скорость потока 4 мл/мин, λ=220 нм.
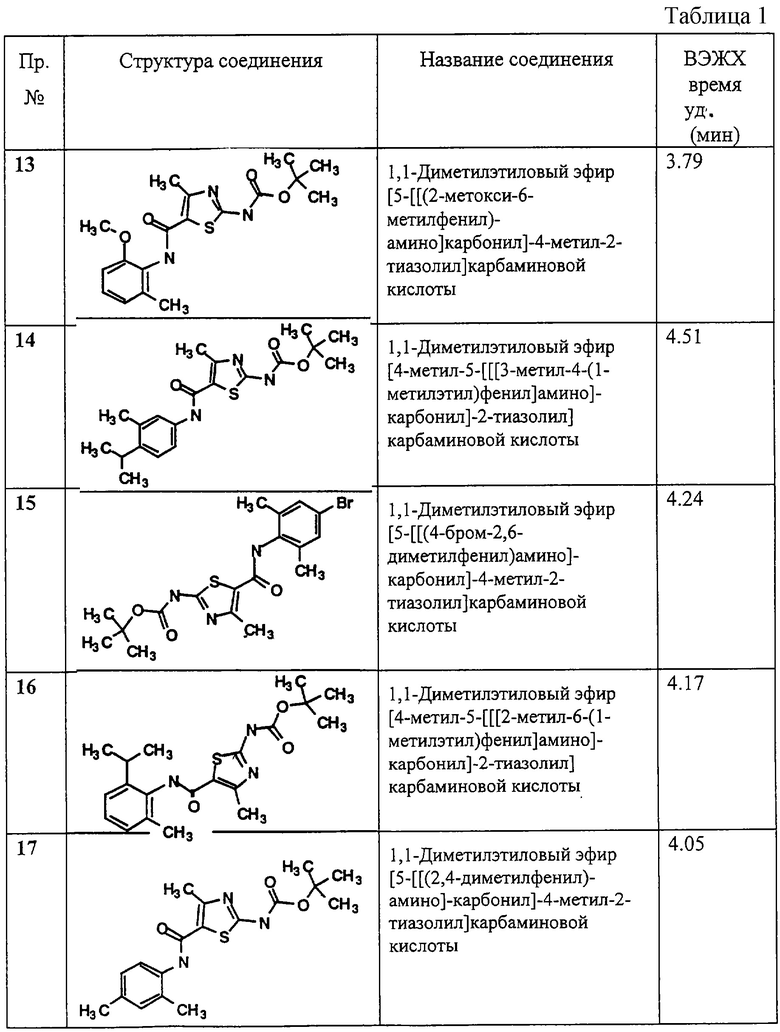
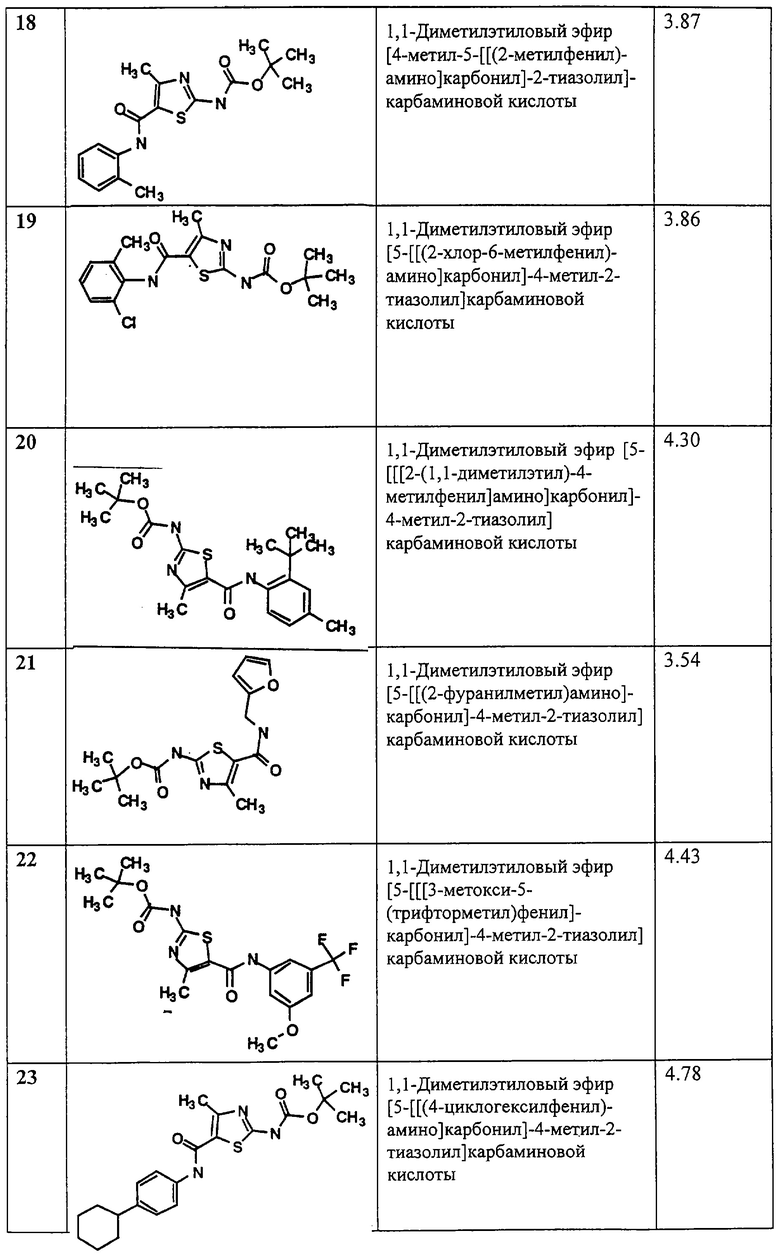
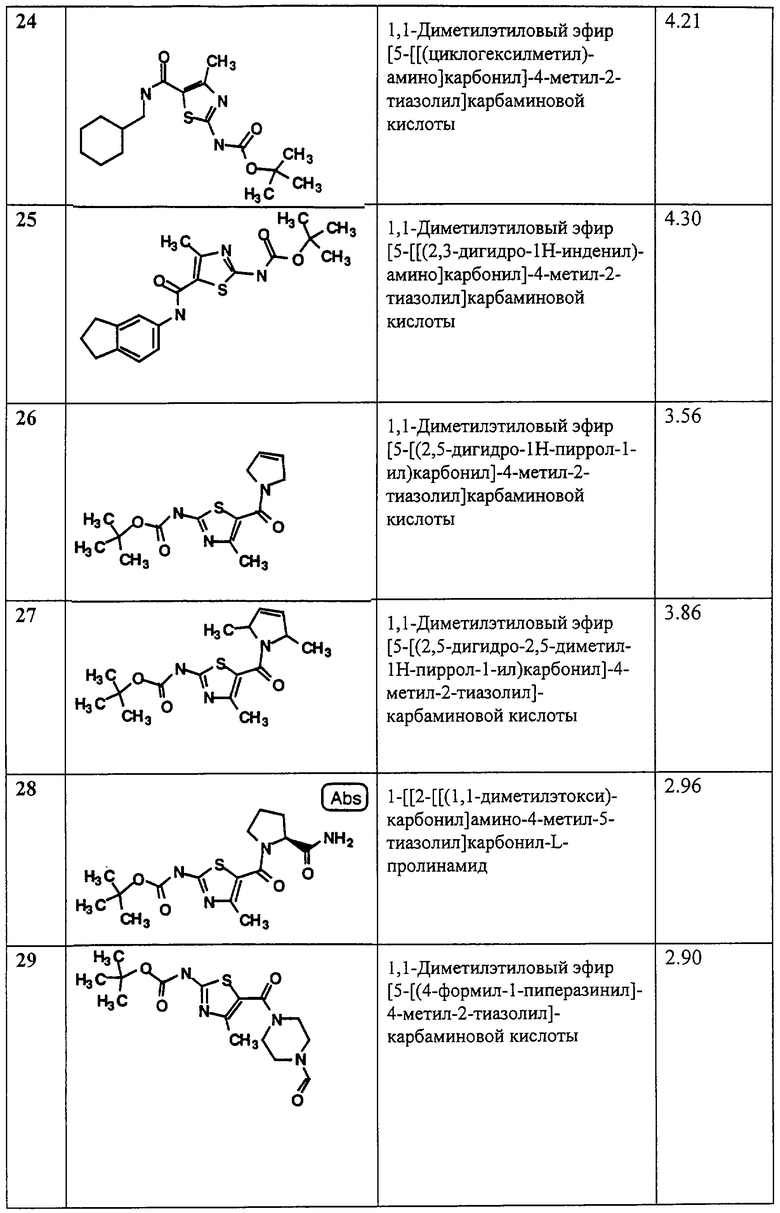
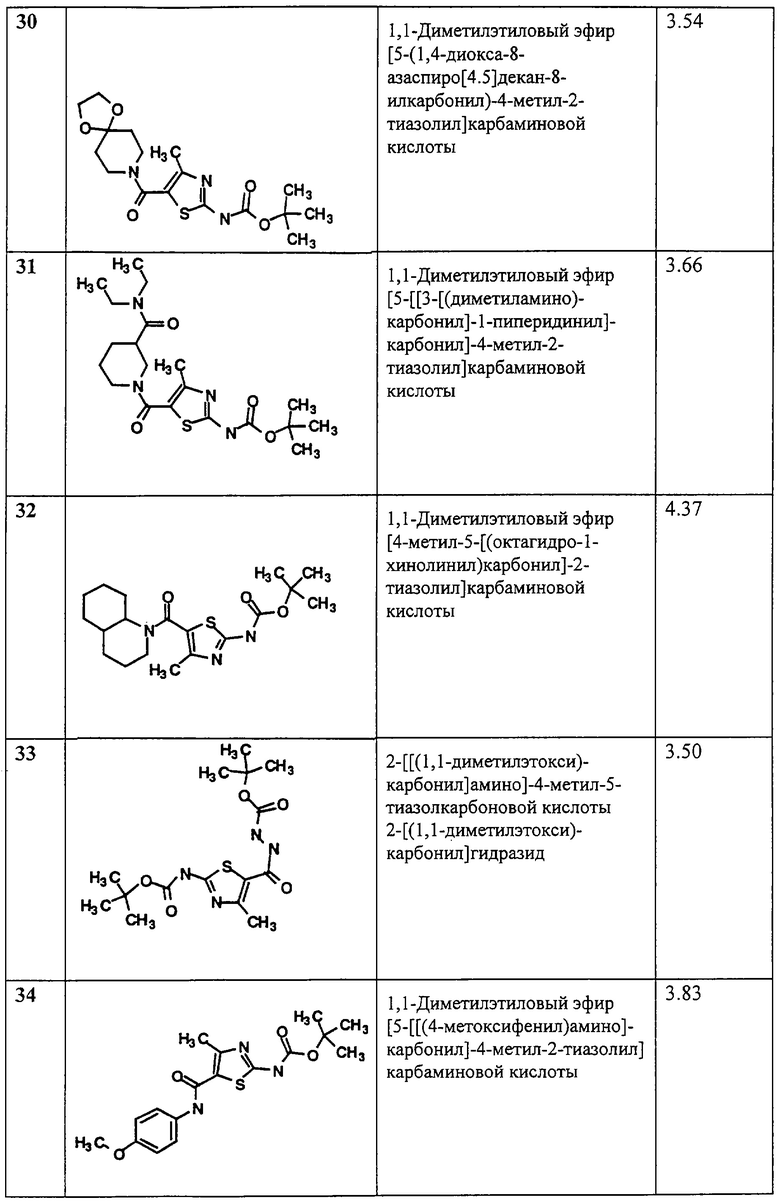
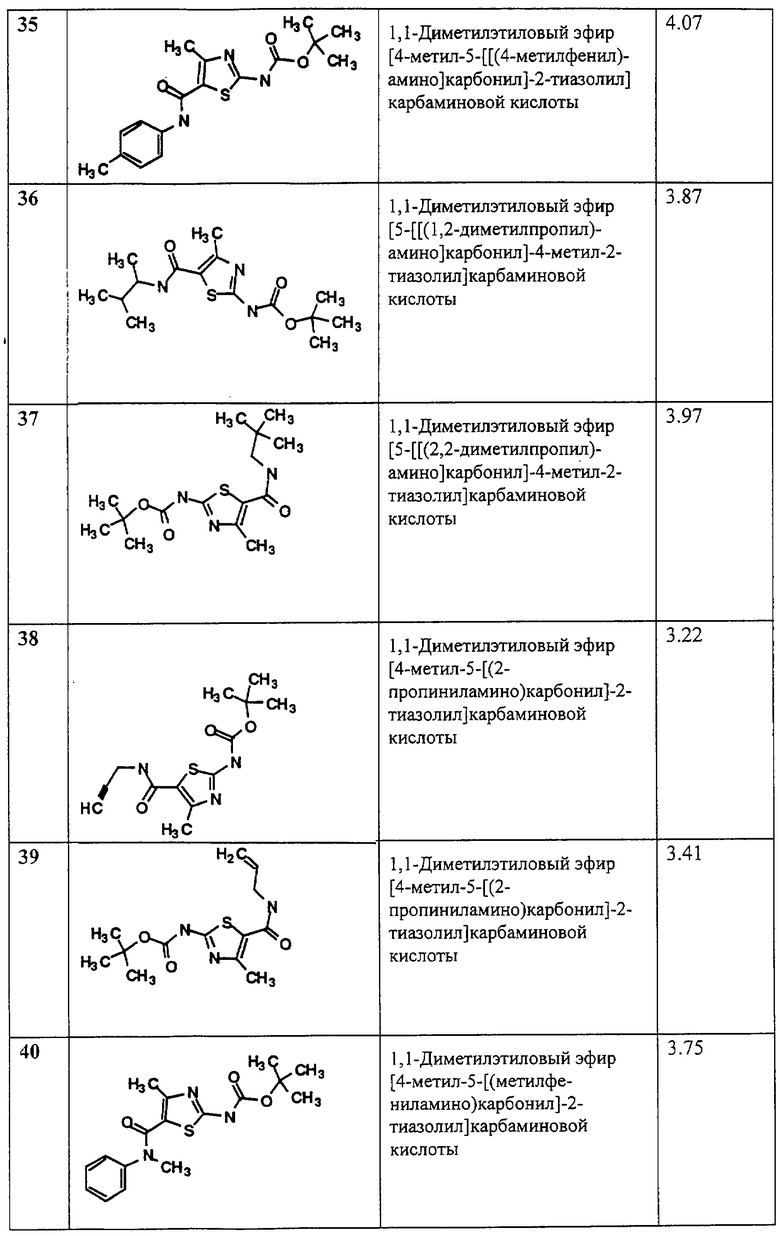

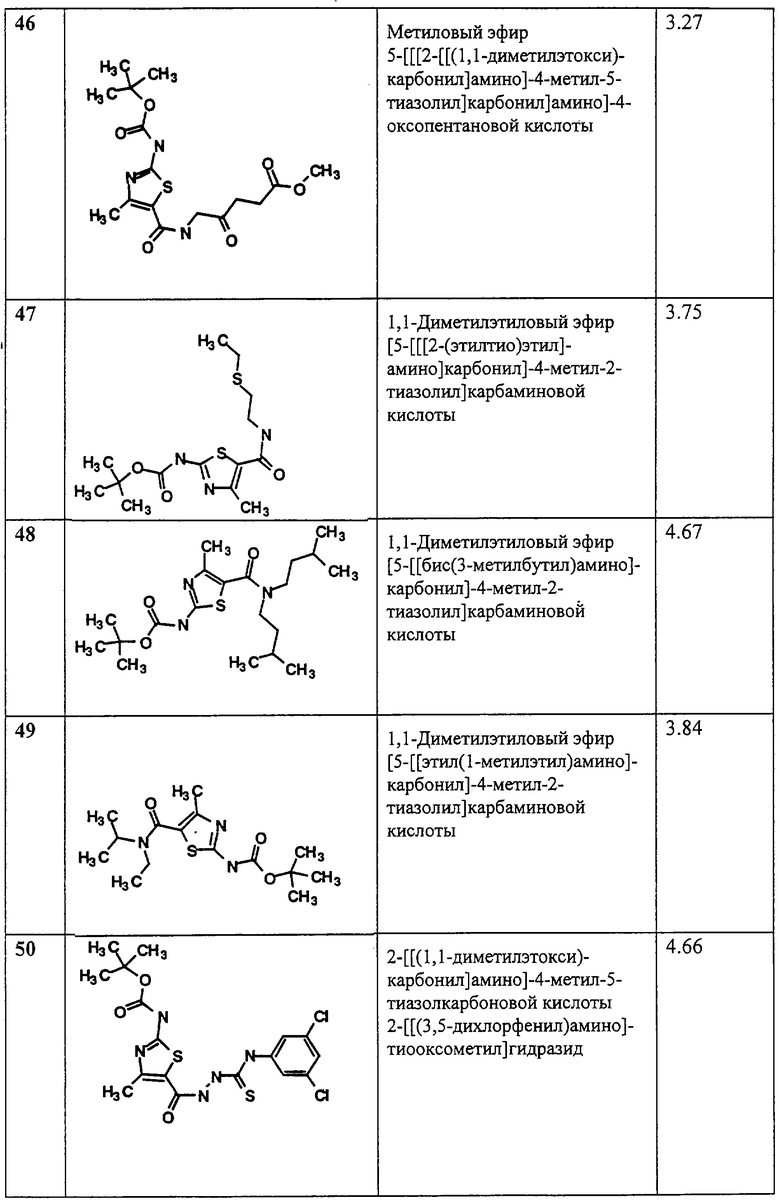
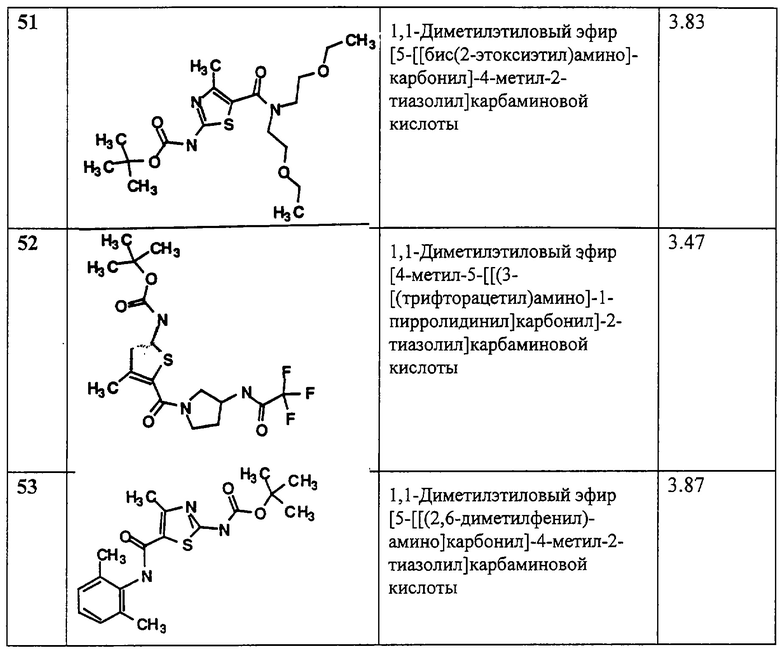
Примеры 54-129.
Общая методика.
Соединения 54-129 получают по следующей методике. Диизопропилэтиламин (60 мкл, 0,34 ммол) добавляют к смеси амина 2 (30 мг, 0,11 ммол), соответствующей карбоновой кислоты (0,13 ммол), 1-гидрокси-7-азабензотриазола (19,5 мг, 0,14 ммол) и этил-3-(3-диметиламино)-пропилкарбодиимида гидрохлорида (26,8 мг, 0,14 ммол) в ТГФ (0,4 мл). Смесь нагревают в запаянной трубке под аргоном при 45°С в течение 14 часов. Реакционную смесь разбавляют хлористым метиленом (4 мл) и промывают 2 N водн. раствором HCl (2 мл, 3х). Раствор в хлористом метилене пропускают через Varian SCX-катионообменную колонку (2 г, 6 см3) на автомате (роботе) Гильсона. Колонку элюируют последовательно ацетонитрил-метанолом (10 мл, 4:1), метанолом - 2М аммиаком в метаноле (3 мл, 4:1) и 2 М раствором аммиака в метаноле (3 мл, 4х). Фракции собирают отдельно, используя робот Гильсона. Фракции, содержащие продукт, упаривают и сушат в вакууме. “ВЭЖХ время уд.” обозначает ВЭЖХ - время удерживания в следующих условиях: YMS S5 OD 4,6×50 мм балластная колонка, 4-минутный градиент, начиная со 100% растворителя А (10% МеОН, 90% H2O, 0,2% Н3PO4) до 100% растворителя В (90% МеОН, 10% Н2O, 0,2% Н3PO4), скорость потока 4 мл/мин, λ=220 нм для соединений 54-127. Для соединений 128-129 условия ВЭЖХ следующие: короткая колонка Zorbax S8-C18 4,5 мм × 7,5 см, градиент 8 минут, начиная со 100% растворителя А (10% МеОН, 90% Н2О, 0,2% Н3PO4) до 100% растворителя В (90% МеОН, 10% Н2O, 0,2% Н3PO4), скорость потока 2,5 мл/мин, λ=217 нм.
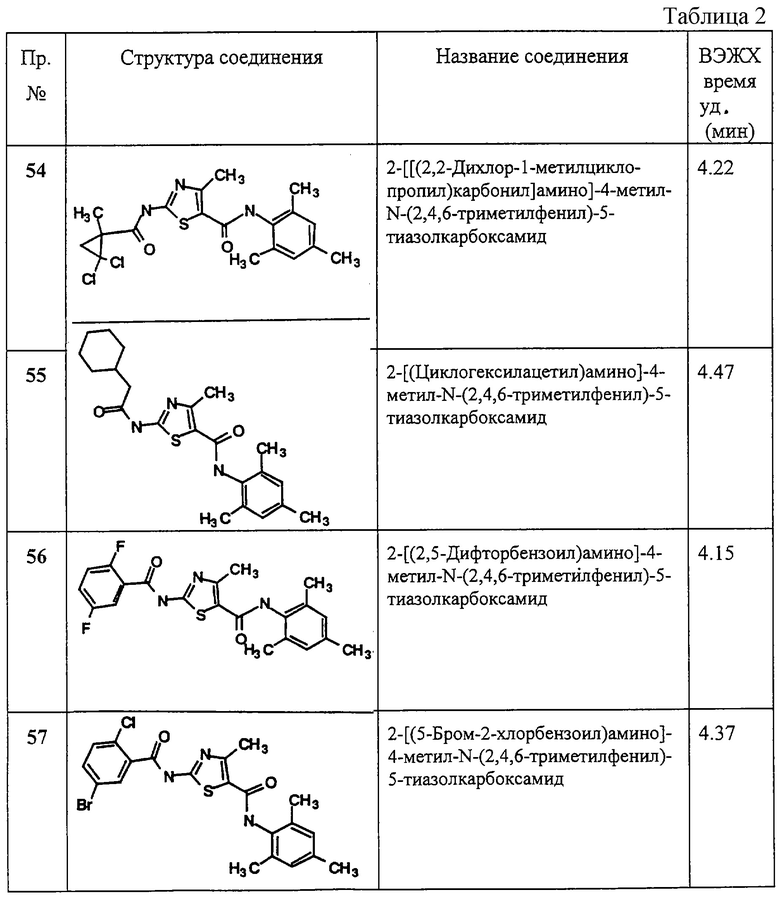
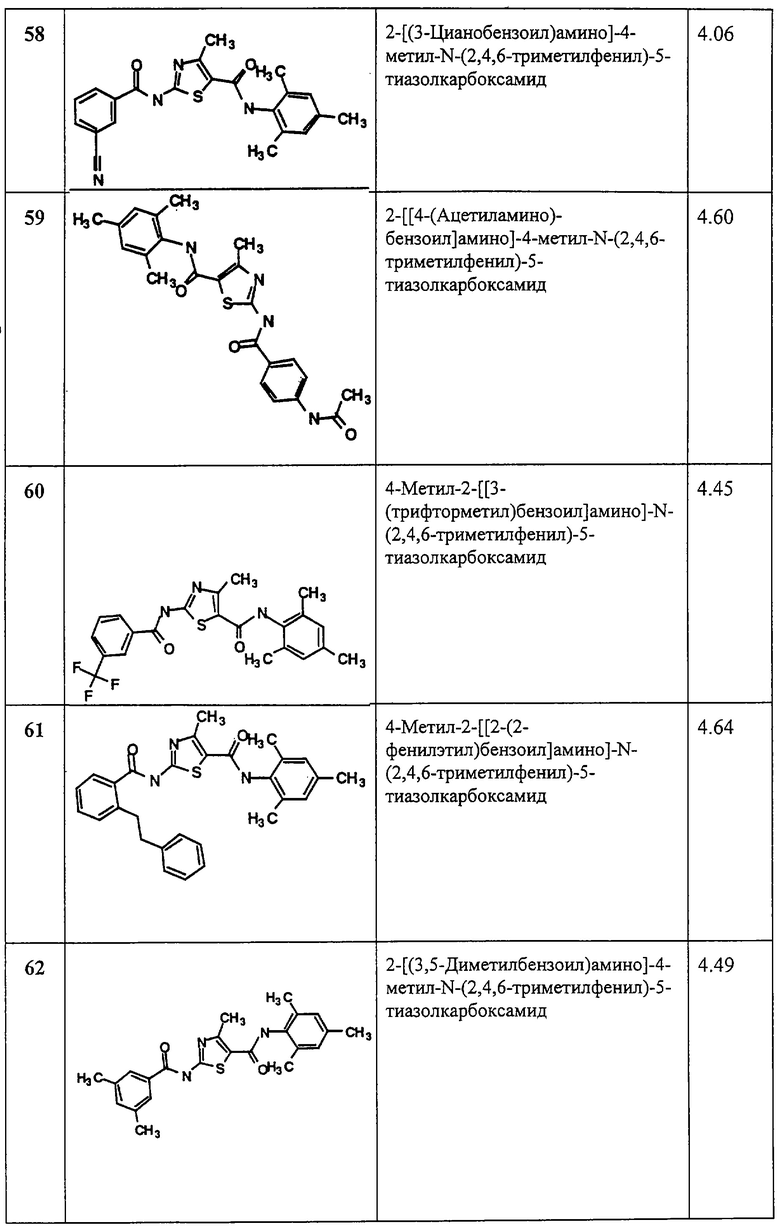
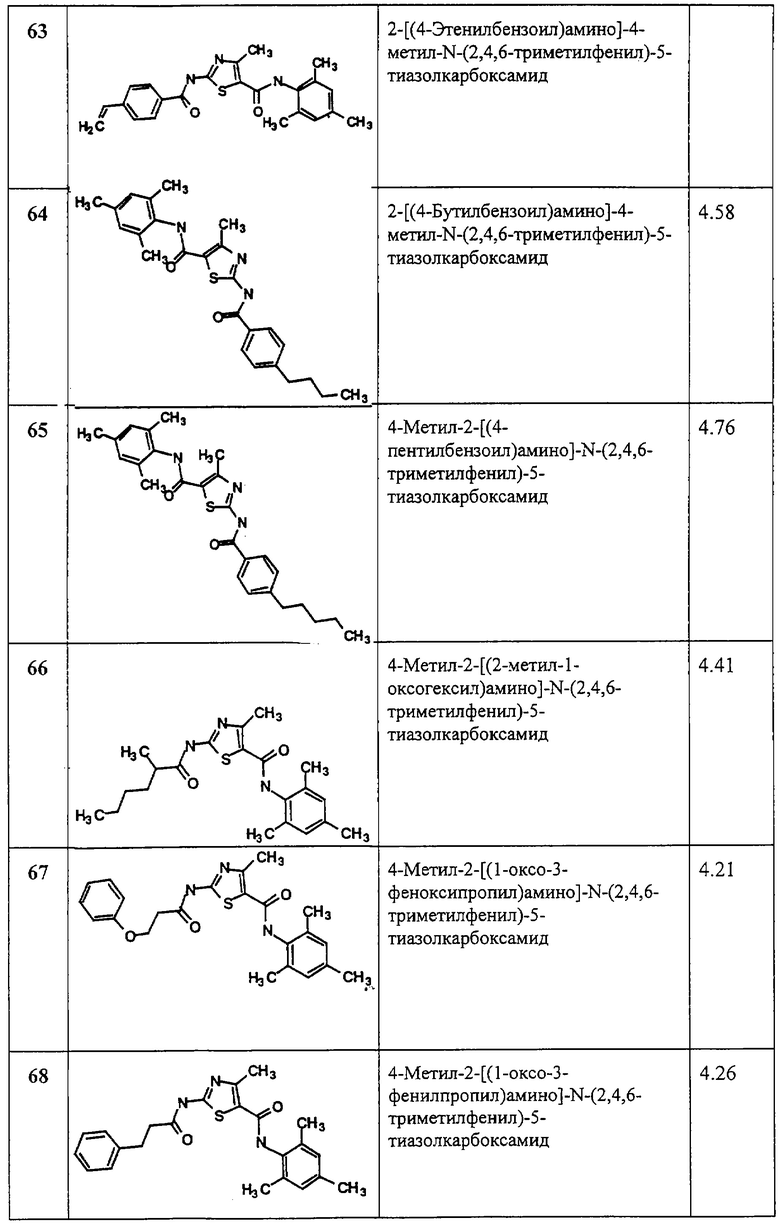
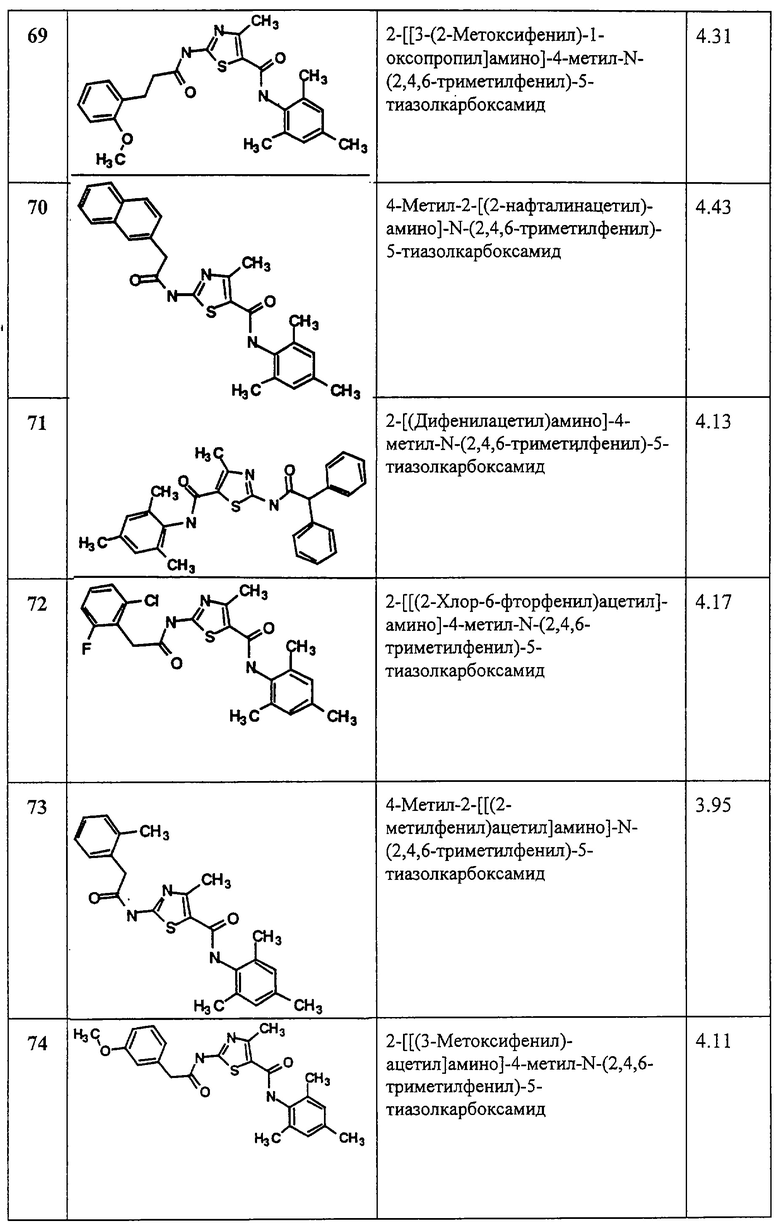
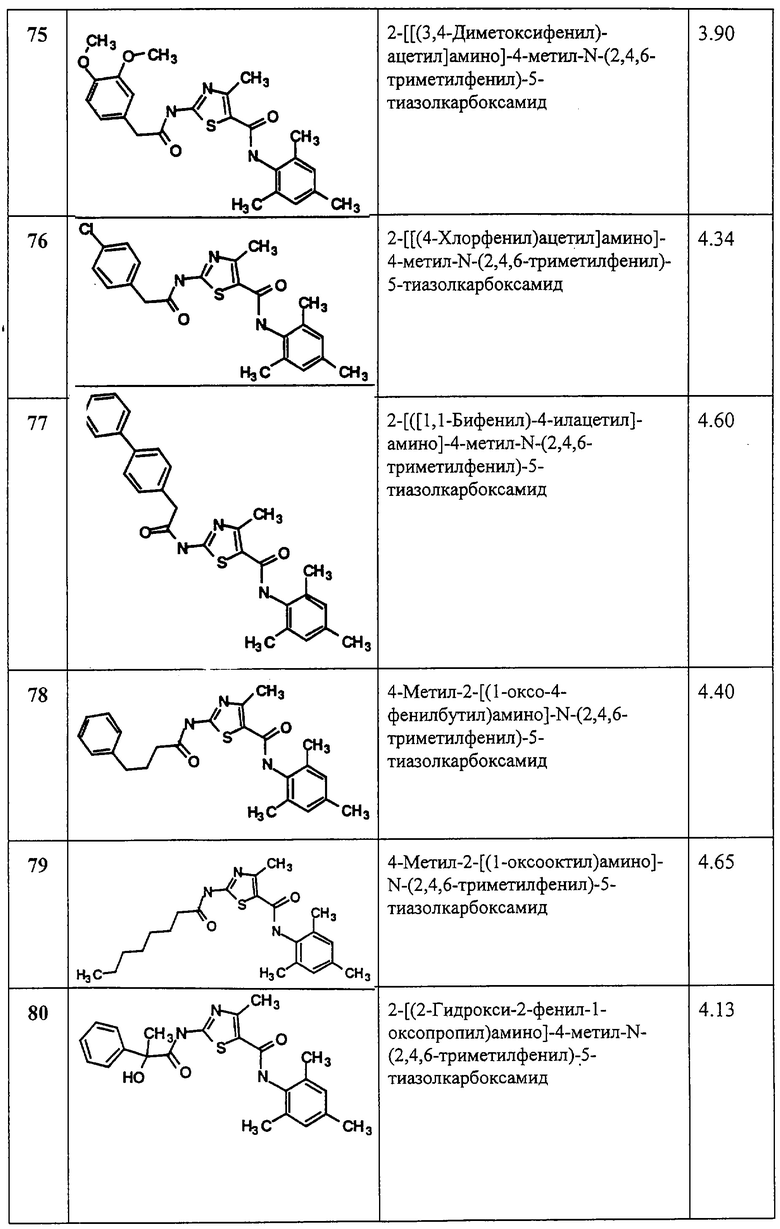
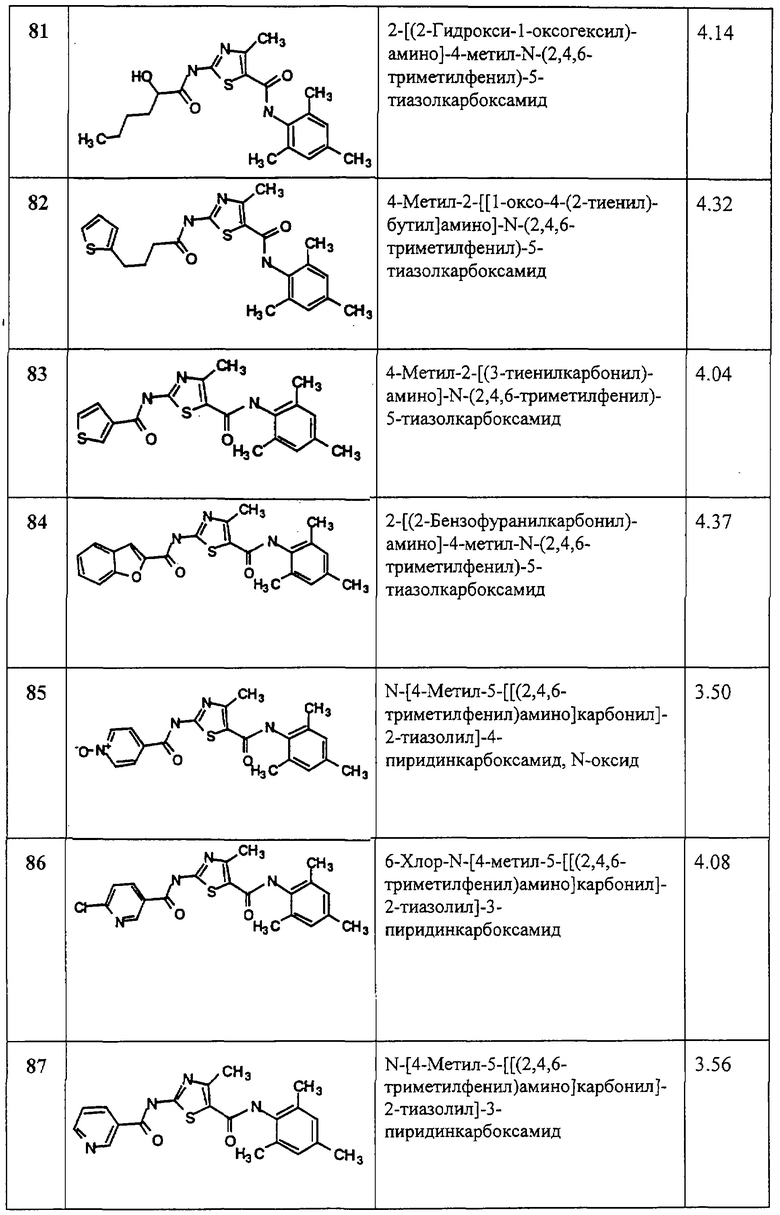
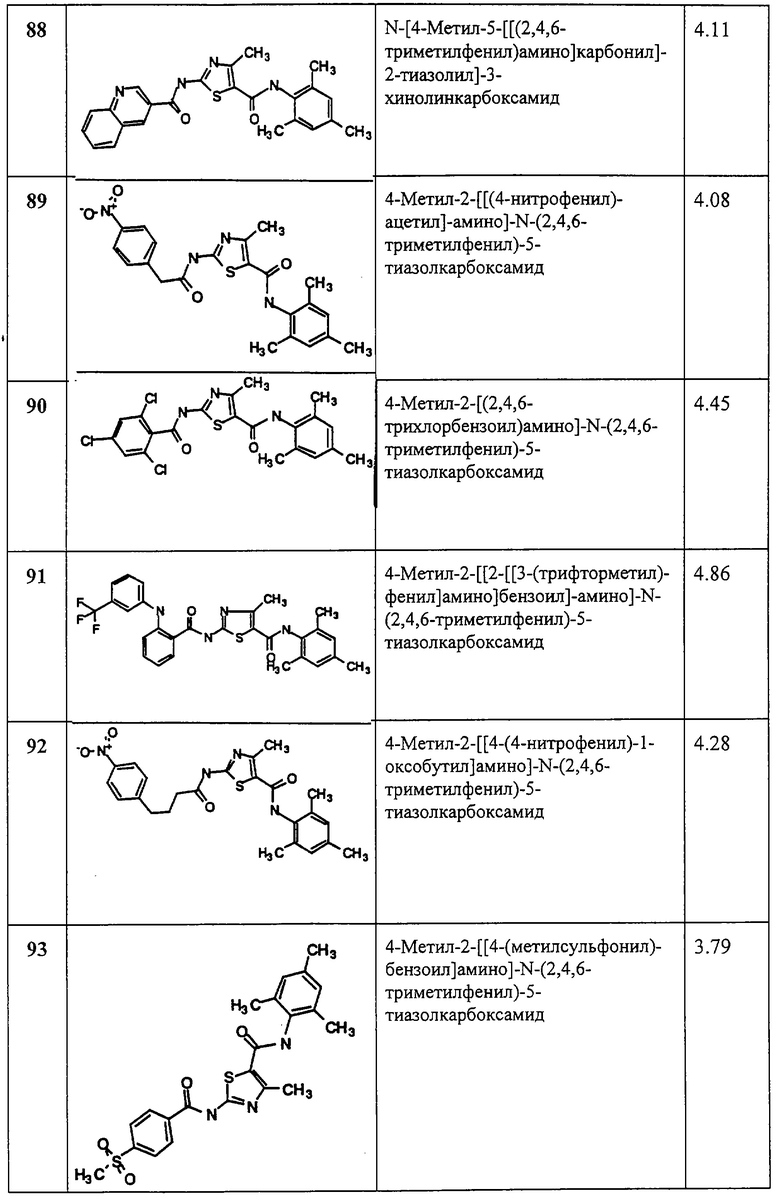
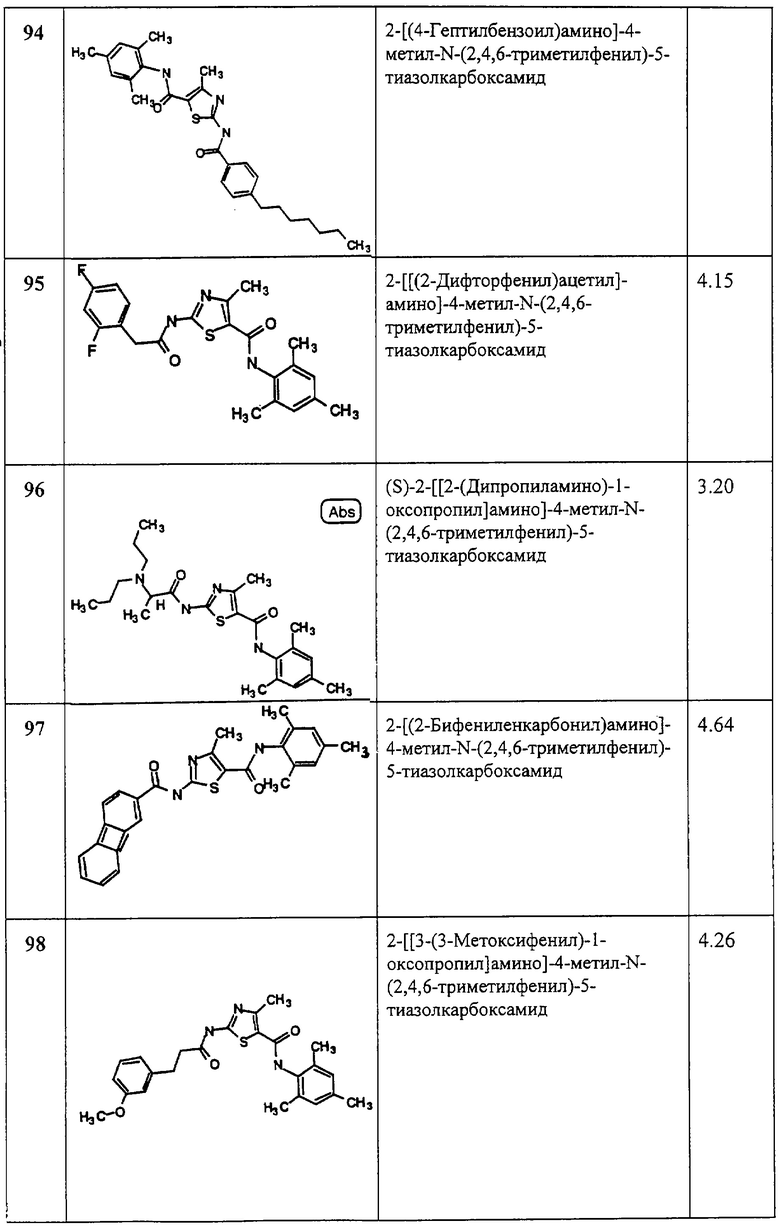
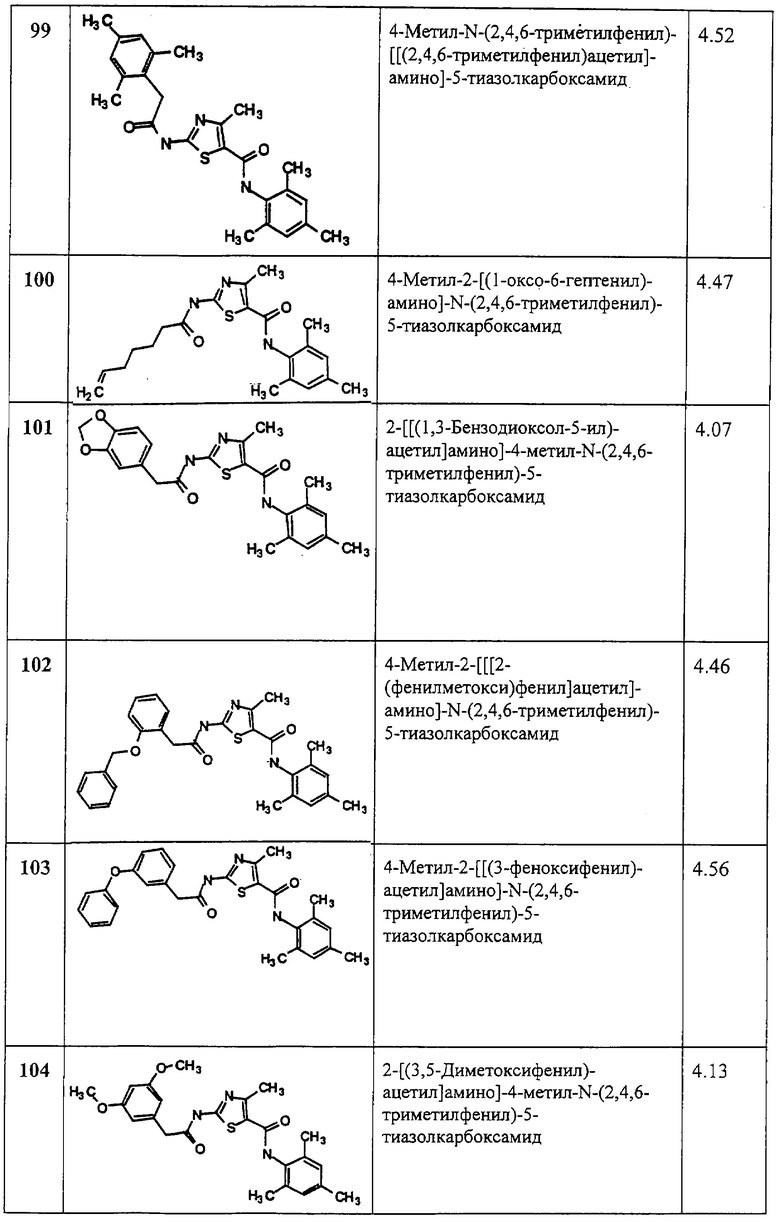
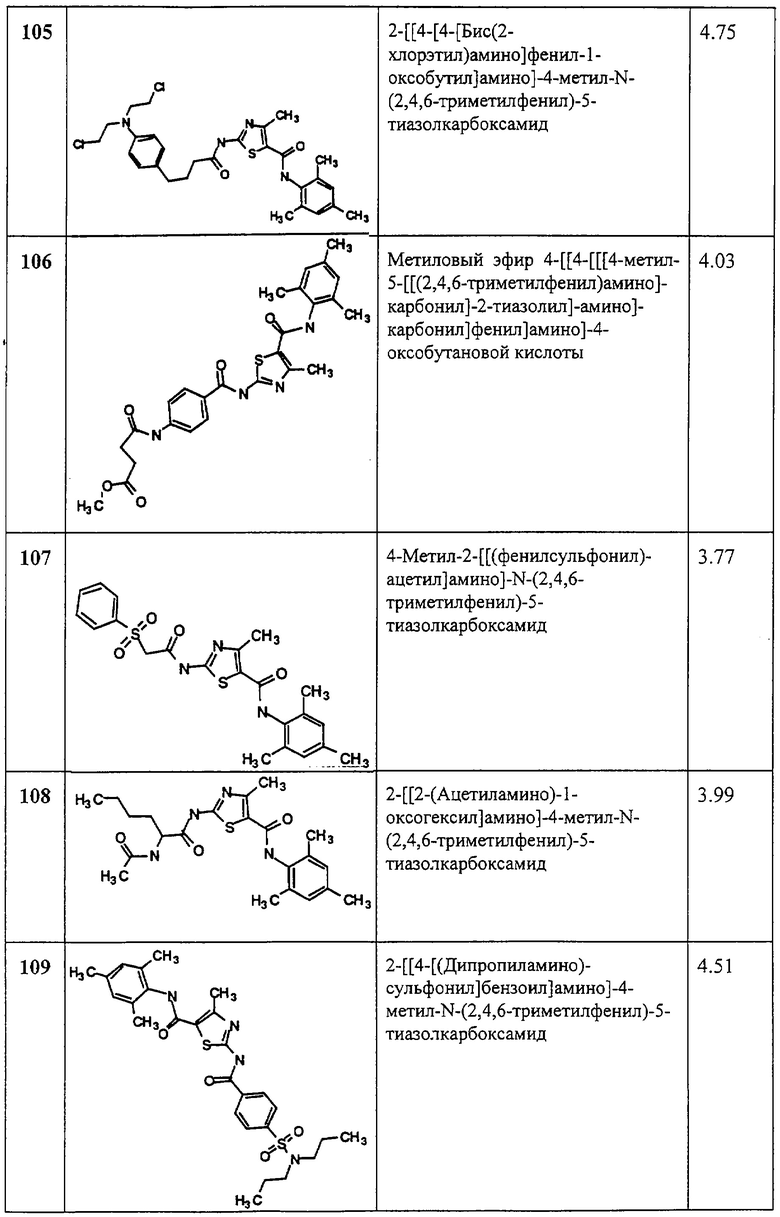
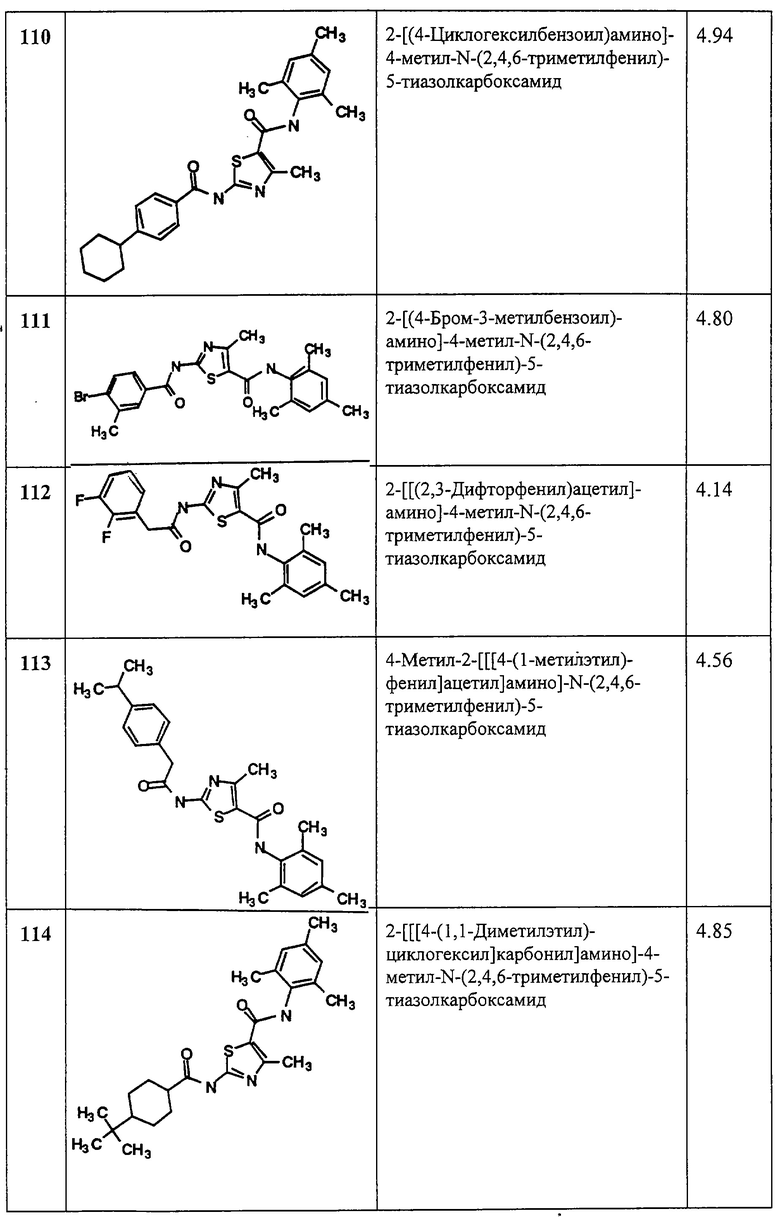
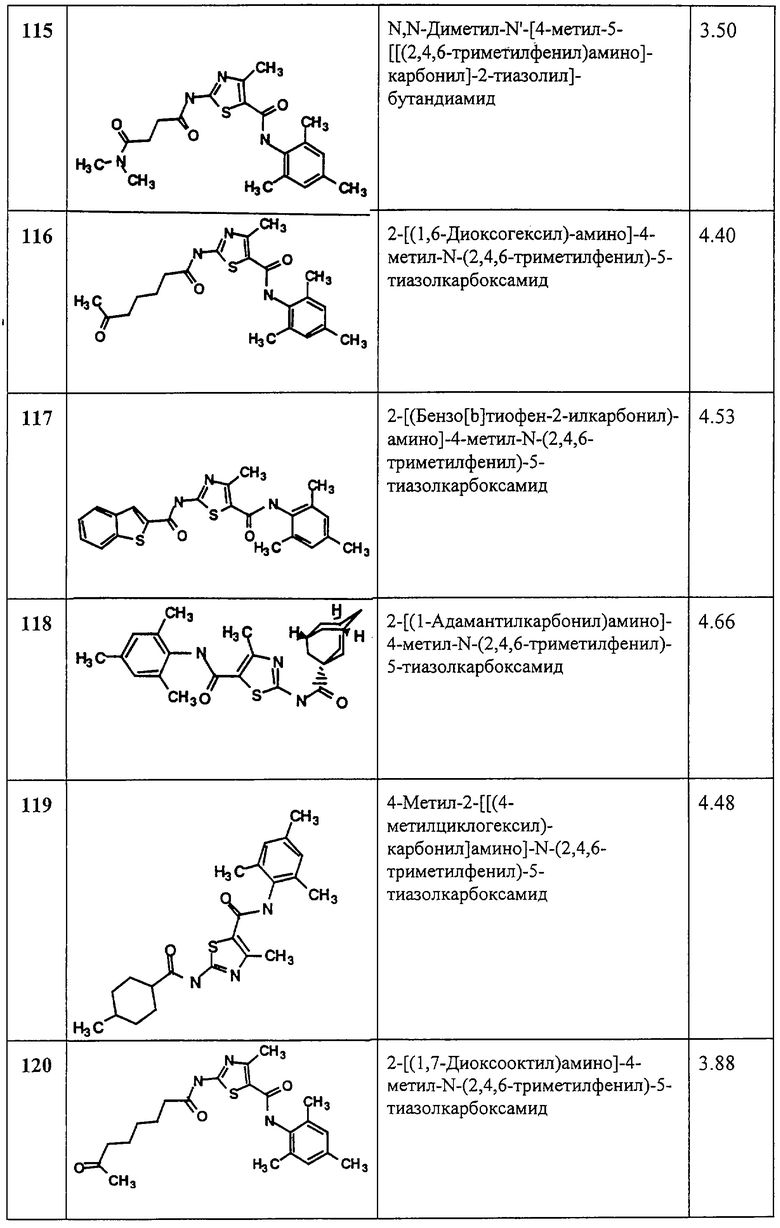
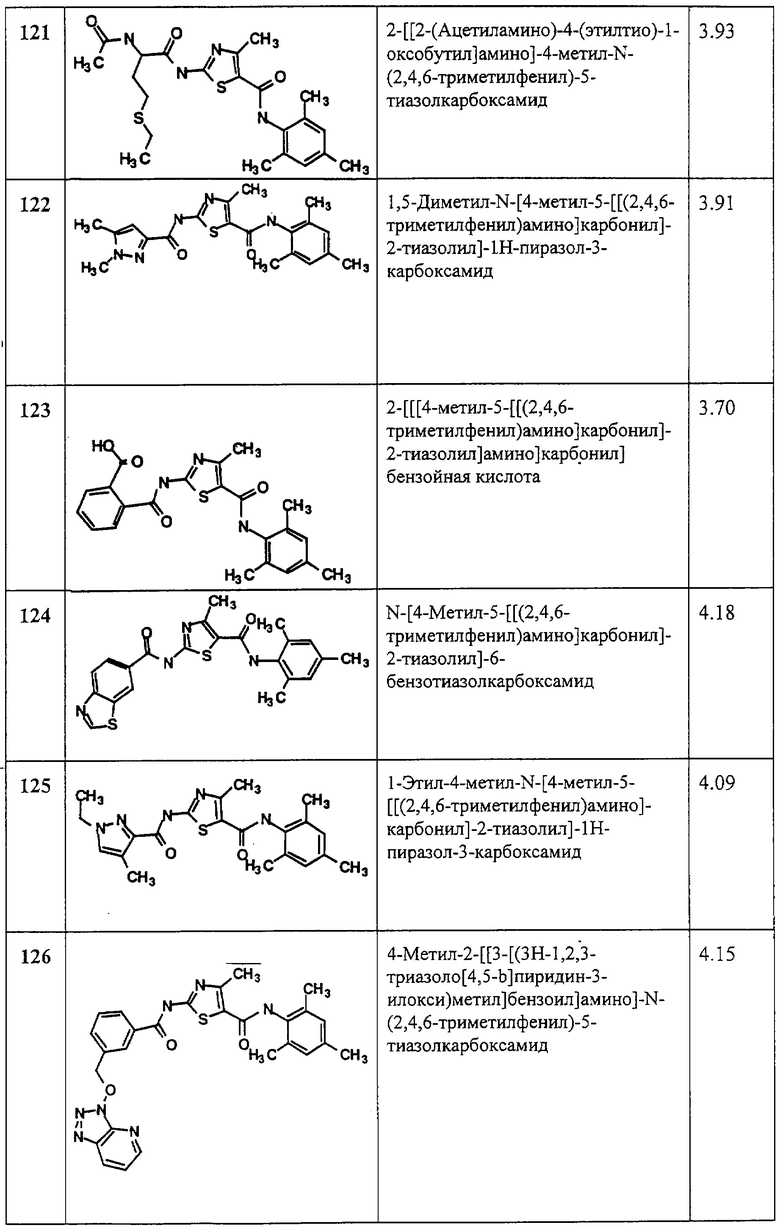
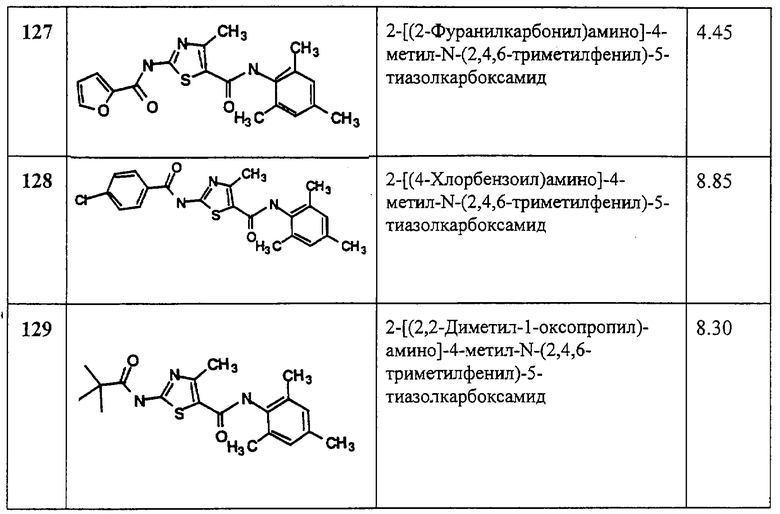
Пример 130.
Получение 1,1-диметилэтилового эфира [4-метил-5-[[(2-нитрофенил)амино]карбонил]-2-тиазолил]карбаминовой кислоты
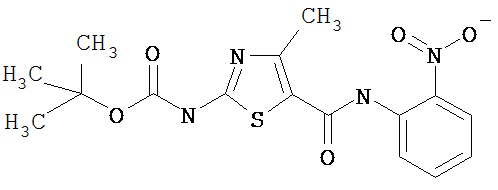
К раствору хлорангидрида 2-трет-бутоксикарбонилоксиамино-4-метилтиазол-5-карбоновой кислоты 1С (100 мг, 0,36 ммол) в хлористом метилене (3 мл) при перемешивании по каплям прибавляют 2-нитроанилина (55 мг, 0,4 ммол) и диизопропилэтиламина (70 мкл, 0,4 ммол). Через 16 часов при ~20°С добавляют 4,N,N-диметиламинопиридин (22 мг, 0,18 ммол) и смесь перемешивают еще 3,5 часа. Растворитель упаривают в вакууме. Остаток хроматографируют на колонке с силикагелем. Элюция 5% EtOAc в смеси гексанов с последующей элюцией 20% EtOAc в смеси гексанов дает заданное соединение (15 мг, 11%) в виде желтого твердого вещества
Пример 131.
Получение фенилметилового эфира [4-метил-5-[[(2,4,6-триметилфенил)амино]-карбонил]-2-тиазолил]карбаминовой кислоты.
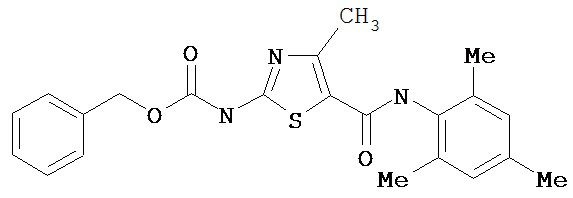
А. Этил-2-бензоилоксикарбониламино-4-метилтиазол-5-карбоксилат.
К раствору этил-2-амино-4-метилтиазол-5-карбоксилата (372 мг, 2 ммол) в ТГФ (20 мл) при 0-5°С при перемешивании добавляют 3 М aq. NaHCO3 (10 мл, 30 ммол). Добавляют бензоилхлорформиат (500 мкл). Через 2 часа добавляют еще 500 мкл бензоилхлорформиата и двухфазную систему перемешивают еще 2 часа при 0-5°С. Смесь разбавляют хлористым метиленом (50 мл) и водой (30 мл). Органический слой отделяют, сушат (MgSO4), фильтруют и упаривают. Остаток хроматографируют на колонке с силикагелем. Элюируют 10% EtOAc в смеси гексанов, а затем 20% и 30% EtOAc в смеси гексанов, получают заглавное соединение (310 мг, 48%) в виде белого твердого вещества.
В. 2-Бензилоксикарбонилоксиамино-4-метилтиазол-5-карбоновая кислота.
Соединение 131В получают аналогично 3В, но берут 131А и получают 131В в виде белого порошка (77%).
С. Фенилэтиловый эфир [4-метил-5-[[(2,4,6-триметилфенил)амино]карбонил]-2-тиазолил]карбаминовой кислоты.
К раствору 131В (100 мг, 0,34 ммол), 2,4,6-триметиланилина (60 мкл, 0,41 ммол) и [O-(7-азабензотриазол-1-ил)-1,1,3,3-тетраметилуроний]гексафторфостфата (HATU, 160 мг, 0,41 ммол) добавляют диизопропилэтиламин (70 мкл, 041 ммол). Смесь перемешивают при rt 24 часа, разбавляют EtOAc (20 мл) и промывают 2N aq. HCl (3х), рассолом, сушат (Na2SO4), фильтруют и упаривают. Остаток растирают с эфиром (40 мл), получают заглавное соединение (100 мг, 77%) в виде грязно-белого твердого вещества.
Пример 132.
Получение 1,1-диметилэтилового эфира метил-[4-метил-5-[[(2,4,6-триметилфенил)амино]карбонил] -2-тиазолил] карбаминовой кислоты.
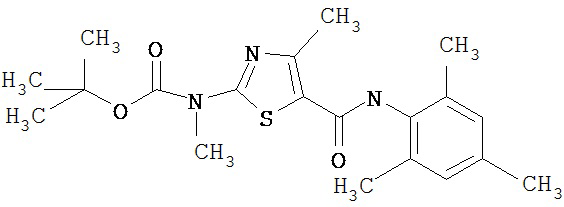
Соединение 132 получают аналогично 1, но берут этил-2-трет-бутоксикарбонилоксиаминометил-4-метилтиазол-5-карбоксилат и получают заглавное соединение 132 в виде рыжевато-коричневого твердого вещества.
Пример 133.
Получение 4-метил-2-(метиламино)-N-(2,4,6-триметилфенил)-5-тиазолкарбоксамида, трифторацетата (1:1).
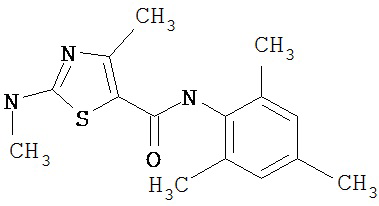
Соединение 133 получают аналогично 4, но берут 132, получая заглавное соединение 133 в виде белого твердого вещества (91%).
Пример 134.
Получение 1,1-диметилэтилового эфира [4-метил-(2,4,6-триметилфенил)амино]карбонил]-2-тиазолил]карбаминовой кислоты.
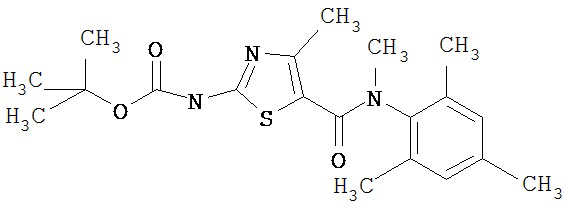
Соединение 134 получают аналогично 1, но применяют N-метил-2,4,6-триметиланилин, получая заглавное соединение 134 в виде белого твердого вещества (60%).
Пример 135.
Получение 2-амино-N,4-диметил-N-(2,4,6-триметилфенил)-5-тиазолкарбоксамид, трифторацетат (1:1).
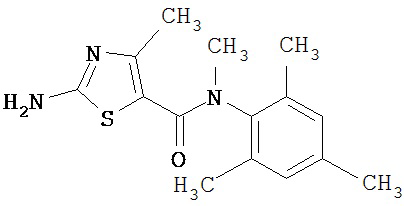
Соединение 135 получают аналогично 4, но берут 134, получая заглавное соединение 135 в виде белого твердого вещества (97%).
Пример 136.
Получение метилового эфира [4-метил-5-[[(2,4,6-триметилфенил)амино]карбонил]-2-тиазолил]карбаминовой кислоты.
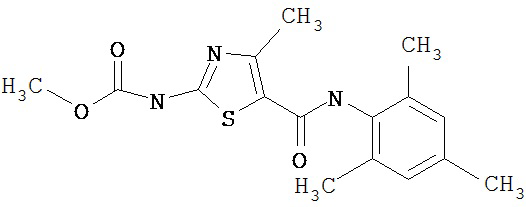
Смесь 2 (100 мг, 0,36 ммол), пиридина (87 мкл, 1,08 ммол), метилхлорформиата (111 мкл, 1,44 ммол) в хлористом метилене (3 мл) перемешивают при rt 1,5 часа. Раствор разбавляют хлористым метиленом и промывают aq. NaHCO3 (20 мл, 2х), рассолом; сушат (MgSO4), фильтруют и упаривают. Остаток растирают эфиром и получают заглавное соединение (88 мг, 82%) в виде белого твердого вещества.
Пример 137.
Получение 1,1-диметилэтилового эфира [4-этил-5-[[(2,4,6-триметилфенил)амино]карбонил]-2-тиазолил]карбаминовой кислоты.
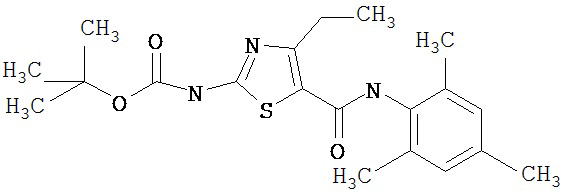
Соединение 137 получают аналогично 1, но берут метил-2-амино-4-этилтиазол-5-карбоксилат, получая заглавное соединение 137 в виде белого твердого вещества (70%).
Пример 138.
Получение 2-амино-4-этил-N-(2,4,6-триметилфенил)-5-тиазолкарбоксамида, трифторацетата.
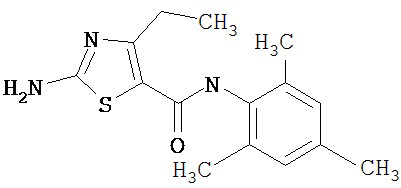
Соединение 138 получают аналогично 4, но берут 137, получая заглавное соединение 138 в виде белого твердого вещества (89%).
Пример 139.
Получение 1,1-диметилэтилового эфира [5-[[(2,6-дихлорфенил)амино]карбонил]-4-метил-2-тиазолил]карбаминовой кислоты.
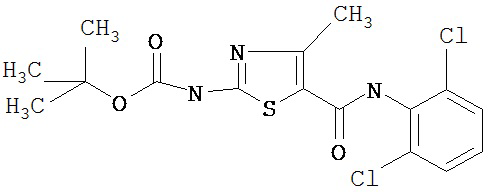
1 М раствор натрий-бис-триметилсилиламида (290 мкл, 0,29 ммол) добавляют к перемешиваемому раствору 2,6-дихлоранилина (13,4 мг, 0,08 ммол) в ТГФ (1 мл). Через 30 минут смесь охлаждают до 0°С, добавляют сразу все количество (30 мг, 0,11 ммол) соединения 1С. Смесь нагревают до комнатной температуры и перемешивают 16 часов. Раствор разбавляют хлористым метиленом и промывают 2N HCl aq. (2 мл × 3), сушат (MgSO4), фильтруют и упаривают. Остаток хроматографируют на колонке с силикагелем и элюируют 30% EtOAc в смеси гексанов, получая заглавное соединение (20 мг, 45%) в виде светло-желтого твердого вещества.
Пример 140.
Получение 2-амино-N-(2,6-диметилфенил)-4-метил-5-тиазолкарбоксамида, трифторацетат (1:1).
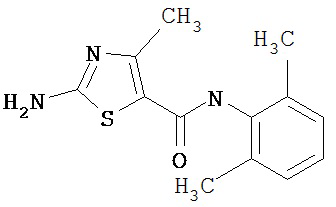
Соединение 140 получают аналогично 4, но берут 53, получая заглавное соединение 140 в виде светлого рыжевато-коричневого твердого вещества (100%).
Пример 141.
Получение 2-амино-N-(2-метокси-6-метилфенил)-4-метил-5-тиазолкарбоксамида, трифторацетата (1:1).
Соединение 141 получают по аналогии с 4, но берут 13, получая соединение 141 в виде грязно-белого твердого вещества (100%).
Пример 142.
Получение 2-амино-N-(2-метилфенил)-4-метил-5-тиазолкарбоксамида, трифторацетата (1:1).
Соединение 142 получают по аналогии с 4, за исключением того, что но берут 18, получая заглавное соединение 142 в виде легкого рыжевато-коричневого твердого вещества (90%).
Пример 143.
Получение 2-амино-N-(2,6-диметил-4-бромфенил)-4-метил-5-тиазолкарбоксамида, трифторацетата (1:1).
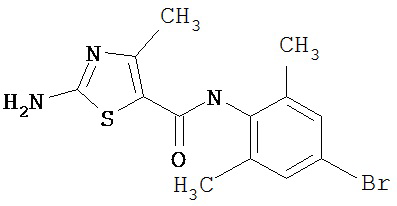
Соединение 143 получают аналогично 4, но используют 15, получая заглавное соединение 143 в виде легкого рыжевато-коричневого твердого вещества (70%).
Пример 144.
Получение 2-амино-N-(2-хлор-6-метилфенил)-4-метил-5-тиазолкарбоксамида, трифторацетата (1:1).
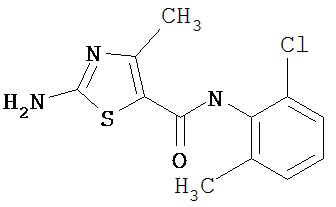
Соединение 144 получают аналогично 4, но берут 19, получая заглавное соединение 144 в виде легкого рыжевато-коричневого твердого вещества (81%).
Пример 145.
Получение 2-амино-N-(2,4-диметилфенил)-4-метил-5-тиазолкарбоксамида, трифторацетата (1:1).
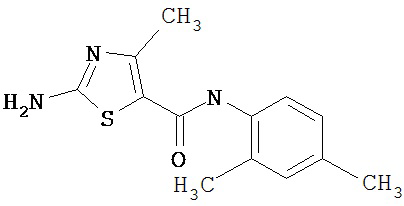
Соединение 145 получают аналогично 4, но берут 17, получая заглавное соединение 145 в виде легкого рыжевато-коричневого твердого вещества (68%).
Пример 146.
Получение 2-амино-N-(2-метил-6-изопропилфенил)-4-метил-5-тиазолкарбоксамида, трифторацетата (1:1).
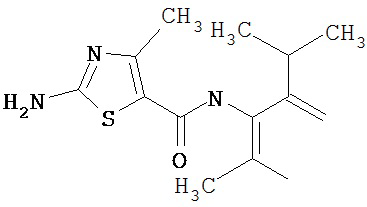
Соединение 146 получают аналогично 4, но берут 16 и получают заглавное соединение 146 в виде легкого рыжевато-коричневого твердого вещества (100%).
Пример 147.
Получение 2-(ацетиламино)-4-метил-N-(2,4,6-триметилфенил)-5-тиазолкарбоксамида.
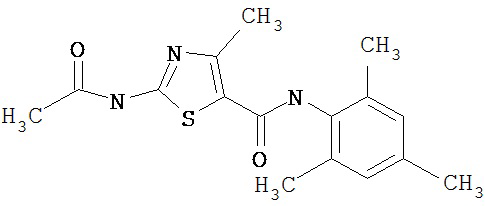
Смесь 2 (54 мг, 0,2 ммол), уксусного ангидрида (22 мкл, 0,23 ммол), диметиламинопиридина (3 мг) в хлористом метилене (4,5 мл) перемешивают при rt 4,5 часа. Смесь разбавляют хлористым метиленом (65 мл) и промывают 1 N HCl aq. (20 мл), водой; сушат (MgSO4), фильтруют и упаривают Остаток хроматографируют на колонке с силикагелем и элюируют 35% EtOAc, получая заглавное соединение (43 мг, 69%) в виде белого твердого вещества.
Пример 148. Получение 2-(бензоиламино)-4-метил-N-(2,4,6-триметилфенил)-5-тиазолкарбоксамида.
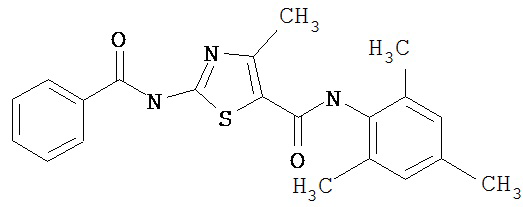
Раствор 2 (100 мг, 0,36 ммол) и бензойного ангидрида (226 мг, 1 ммол) в хлористом метилене (10 мл) и пиридине (2 мл) перемешивают при rt в течение ночи. Смесь разбавляют хлористым метиленом (50 мл) и промывают 2 N HCl aq. (15 мл, 2х), 10% NaHCO3 aq. (20 мл, 2х); сушат (MgSO4), фильтруют и упаривают Остаток хроматографируют на колонке с силикагелем и элюируют 30% EtOAc в смеси гексанов, а затем 50% EtOAc в смеси гексанов, получают заданное соединение с примесью бензойной кислоты. Твердое соединение растворяют в EtOAc (40 мл) и промывают нас. раствором КНСО3 (15 мл, 4х), сушат (MgSO4), фильтруют и упаривают, получают заглавное соединение (110 мг, 80%) в виде белого твердого вещества.
Пример 149.
Получение 4-метил-2-[(1-оксопропил)амино] -N-(2,4,6-триметилфенил)-5-тиазолкарбоксамида.
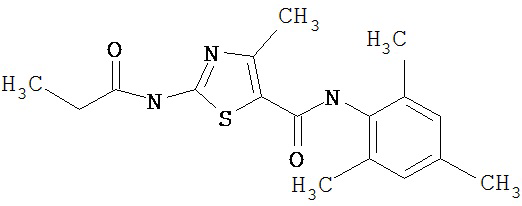
Смесь 2 (100 мг, 0,36 ммол), пропионового ангидрида (332 мкл, 2,58 ммол) в хлористом метилене (10 мл) и пиридине (4 мл) перемешивают при rt 3 часа. Добавляют диметиламинопиридин (122 мг, 1 ммол) и смесь перемешивают еще 1,5 часа. Смесь разбавляют хлористым метиленом и промывают 1 N aq. HCl (25 мл, 3х), NaHCO3 aq. (20 мл, 2х), водой (20 мл), рассолом; сушат (MgSO4), фильтруют и упаривают. Остаток хроматографируют на колонке с силикагелем и элюируют 20% EtOAc в смеси гексанов, получают заглавное соединение (81 мг, 68%) в виде белого твердого вещества.
Пример 150.
Получение 4-метил-2-[(1-оксобутил)амино]-N-(2,4,6-триметилфенил)-5-тиазолкарбоксамида.
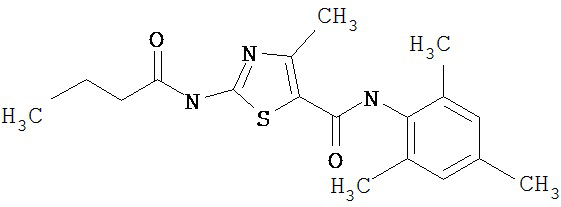
Соединение 150 получают аналогично соединению 149, но берут масляный ангидрид и получают заглавное соединение 150 в виде белого твердого вещества (76%).
Пример 151.
Получение 4-метил-2-[(оксопентил)амино]-N-(2,4,6-триметилфенил)-5-тиазолкарбоксамида.
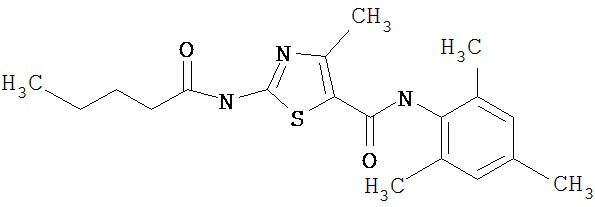
Соединение 151 получают аналогично соединению 149, но берут валериановый ангидрид, получая заглавное соединение 151 в виде белого твердого вещества (77%).
Пример 152.
Получение 4-метил-2-[(1-оксогексил)амино]-N-(2,4,6-триметилфенил)-5-тиазолкарбоксамида.
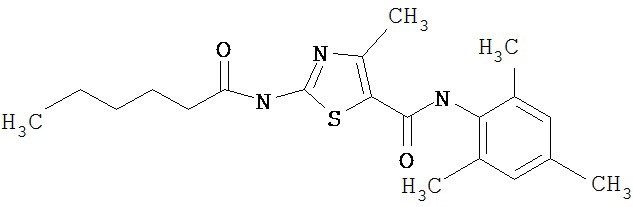
Соединение 152 получают аналогично соединению 149, но берут гексановую кислоту и получают заглавное соединение 152 в виде белого твердого вещества.
Пример 153.
Получение 4-метил-2-[(фенилацетил)амино]-N-(2,4,6-триметилфенил)-5-тиазолкарбоксамида.
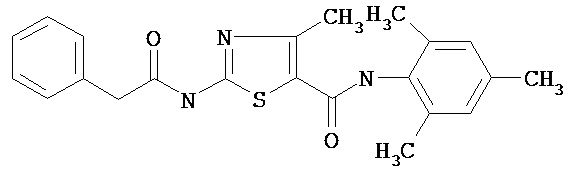
Раствор амина 2 (50 мг, 0,18 ммол), диизопропилэтиламина (101 мкл, 0,58 ммол), фенилуксусной кислоты (27,2 мг, 0,20 ммол), 1-гидрокси-7-азабензотриазола (29,4 мг, 0,22 ммол) и этил-3-(3-диметиламино)пропилкарбодиимида гидрохлорида (42,2 мг, 0,22 ммол) в хлористом метилене (0,62 мл) механически перемешивают в запаянной ампуле 16 часов. Реакционную смесь пропускают через ионообменную колонку Varian SCX (2 г/6 см3) и элюируют смесью ацетонитрил - метанол (10 мл, 4:1), затем 2 М раствором аммиака в метаноле (9 мл). Фракции, содержащие продукт, объединяют и затем упаривают. Остаток растворяют в хлористом метилене и промывают 2 N aq. HCl (3х), сушат (Na2SO4), фильтруют и упаривают, получая заглавное соединение (39 мг, 55%) в виде рыжевато-коричневого твердого вещества.
Пример 154.
Получение 2-[[(ацетиламино)ацетил]амино]-4-метил-N-(2,4,6-триметилфенил)-6-тиазолкарбоксамида.
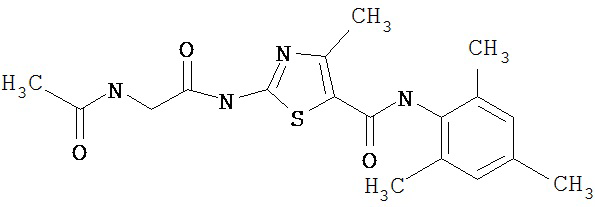
Раствор амина 2 (50 мг, 0,18 ммол), диизопропилэтиламина (400 мкл, 2,3 ммол), N-ацетилглицина (42 мг, 0,36 ммол), 1-гидрокси-7-азабензотриазола (49 мг, 0,36 ммол) и этил-3-(3-диметиламино)пропилкарбодиимида гидрохлорида (72 мг, 0,36 ммол) в ТГФ (5 мл) нагревают при 50°С в течение ночи. Смесь охлаждают, разбавляют хлористым метиленом (60 мл) и промывают 2N HCl aq. (20 мл), насыщенным водн. раствором КНСО3 (20 мл), сушат (MgSO4), фильтруют и упаривают. Сырой твердый продукт растирают с эфиром (10 мл), фильтруют и промывают эфиром (5 мл, 3х), получая заглавное соединение в виде грязно-белого твердого вещества.
Пример 155.
Получение 2-амино-4-метил-N-(2,4,6-триметилфенил)-5-тиазолкарботиоамида.
Суспензию 2 (50 мг, 0,18 ммол) и реагента Лавессона (44 мг, 0,11 ммол) в толуоле (0,23 мл) нагревают 4 часа при 100°С. Добавляют еще реагента Лавессона (44 мг, 0,11 ммол) и смесь нагревают еще 3,5 часа. Сырую смесь хроматографируют на колонке с силикагелем и элюируют 50% EtOAc в смеси гексанов, а затем 70% EtOAc в смеси гексанов, получая желтое твердое вещество, которое растирают в смеси гексанов (6 мл), получая заглавное соединение (11 мг, 21%), желтое твердое вещество.
Примеры 156-170.
Общая методика.
Соединения 156-170 получают по описанной ниже методике. Диизопропилэтиламин (60 мкл, 0,34 ммол) добавляют к смеси амина 2 (30 мг, 0,11 ммол), соответствующей карбоновой кислоты (0,13 ммол), 1-гидрокси-7-азабензотриазола (19,5 мг, 0,14 ммол) и этил-3 -(3-диметиламино)пропилкарбодиимида гидрохлорида (26,8 мг, 0,14 ммол) в ТГФ (1 мл). Смесь нагревают в запаянной трубке под аргоном при 45°С в течение 24 час. Реакционную смесь разбавляют хлористым метиленом (4 мл) и промывают 2N aq. HCl (2 мл, 3х), сушат (Na2SO4) и упаривают на роторе (speedvac.). Сырые продукты либо растирают со смесью хлористый метилен - эфир (5 мл, 1:1), либо чистят хроматографией на силикагеле (элюент: 50% EtOAc в смеси гексанов и EtOAc). “ВЭЖХ время удерж.” представляет собой время удерживания при следующих условиях: балластная колонка YMC S5 ODS 4,6×50 мм; 4-минутный градиент, начиная со 100% растворителя А (10% МеОН, 90% Н2O, 0,2% Н3PO4) до 100% растворителя В (90% МеОН, 10% H2O, 0,2% Н3PO4), скорость потока 4 мл/мин, λ=220 нм.
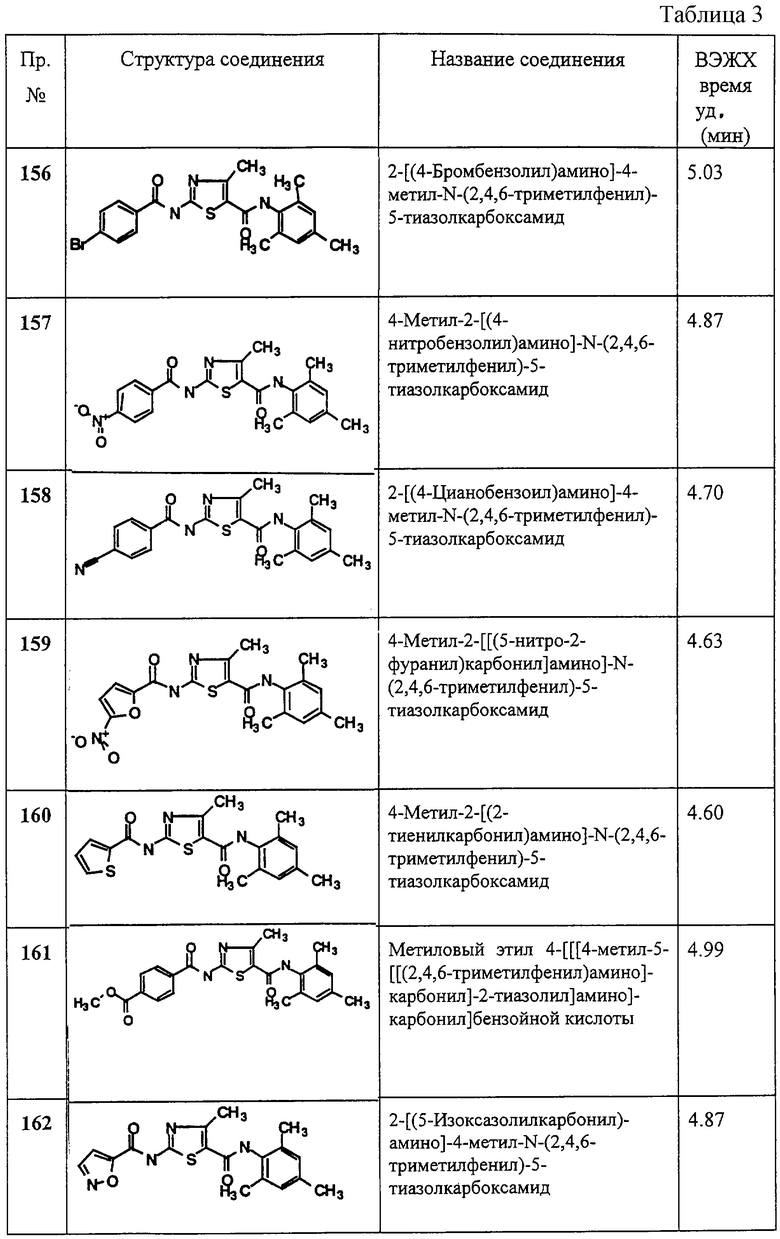
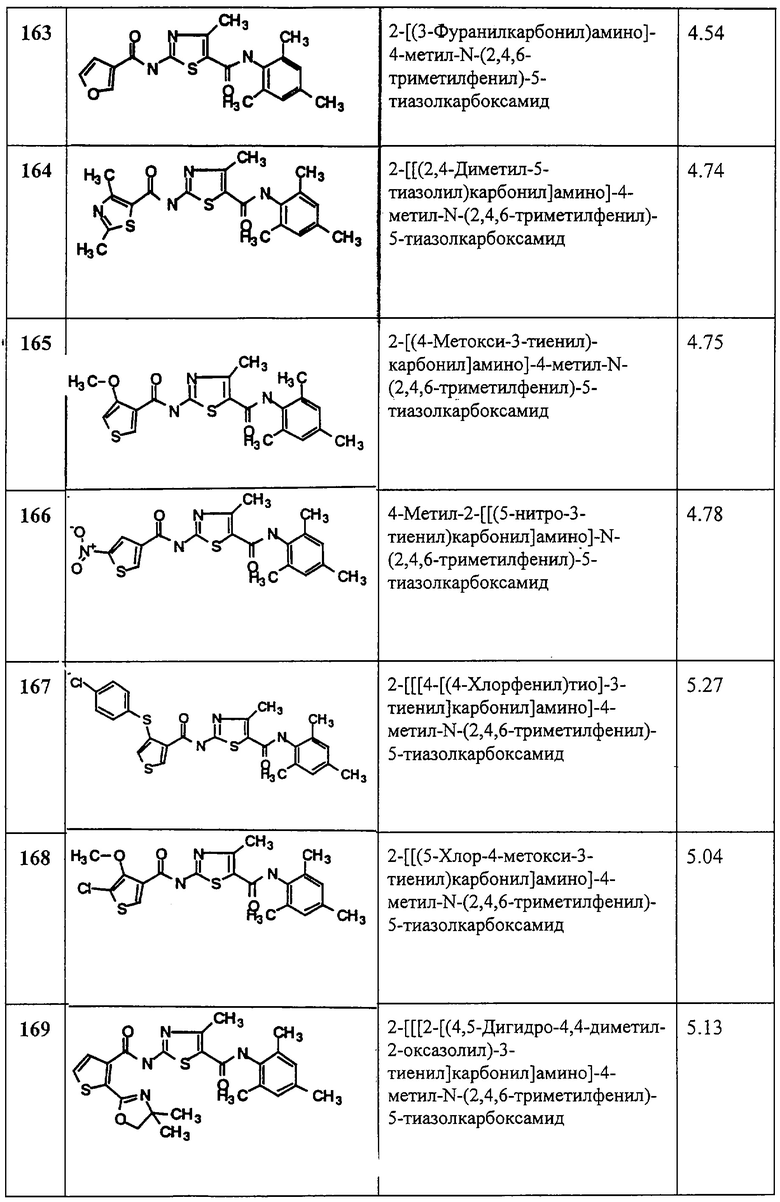
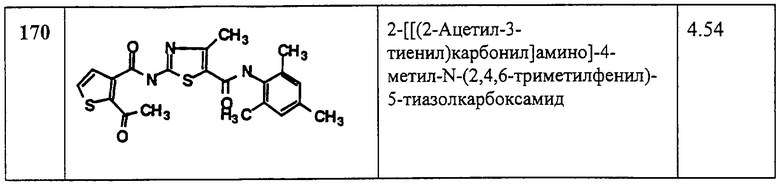
Примеры 171-180.
Общая методика.
Соединения 171-180 получают по описанной ниже методике. Смесь 2 (80 мг, 0,29 ммол), соответствующего изоцианата (0,87 ммол) и пиридина (2 мл) в ТГФ (3,5 мл) перемешивают при комнатной температуре в течение ночи. В некоторых случаях реакционную смесь нагревают при 60-70°С в течение 5 час. Некоторые из этих реакций проводят при комнатной температуре в течение ночи в присутствии катализатора N,N-диметиламинопиридина (каталитических количеств). Реакционную смесь разбавляют хлористым метиленом и промывают 1N HCl aq. (3x), водой, рассолом; сушат (MgSO4), фильтруют и упаривают. Сырой продукт очищают, либо растирая с эфиром или со смесью эфир - гексаны, либо хроматографией на колонке с силикагелем (элюент: 20-40% EtOAc в смеси гексанов) с последующим растиранием или пропусканием через катионообменный Varian SCX-картридж и последовательным элюированием метанолом (5 мл), хлористым метиленом (5 мл), смесью ацетонитрил - метанол (10 мл, 4:1) и смесью метанол -2 М раствор аммиака в метаноле (10 мл, 4:1) с целью получения заглавного соединения. “ВЭЖХ время удерж.” обозначает время удерживания ВЭЖХ при следующих условиях: для соединений 171-172, 175 и 177 условия ВЭЖХ суть: Zorbax S8-C18 4,5 мм × 7,5 см короткая колонка, 30-минутный градиент, начиная от 100% растворителя А (10% МеОН, 90% Н2О, 0,2% Н3PO4) до 100% растворителя В (90% МеОН, 10% Н2O, 0,2% Н3PO4), скорость потока 2,5 мл/мин, λ=217 нм. Для других соединений условия ВЭЖХ суть: короткая колонка Zorbax S8-C18 4,5 мм × 7,5 см, 8-минутный градиент, начиная со 100% растворителя А (10% МеОН, 90% Н2O, 0,2% Н3PO4) до 100% растворителя В (90% МеОН, 10% Н2O, 0,2% Н3PO4), скорость потока 2,5 мл/мин, λ=217 нм.
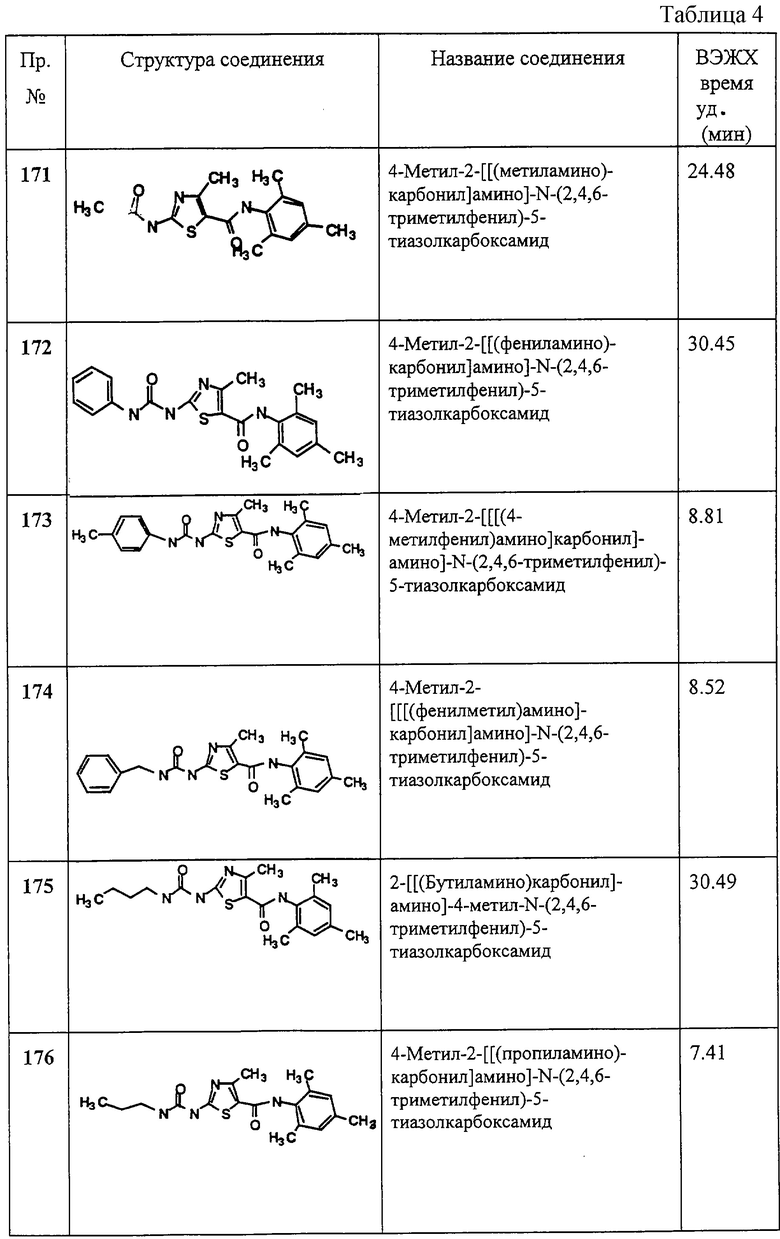
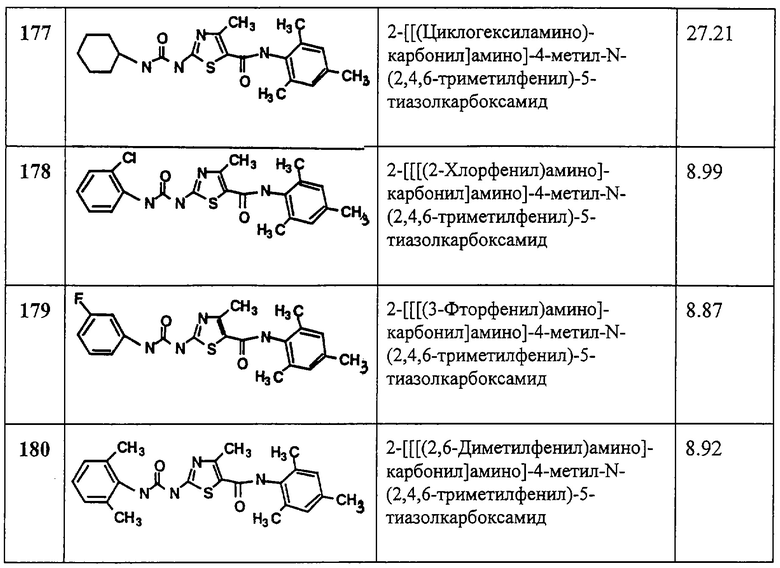
Пример 181.
Получение фенилового эфира [5-[[(2,4,6-триметилфенил)амино]карбонил]-4-метил-2-тиазолил]карбаминовой кислоты.
10% водный раствор КНСО3 добавляют при перемешивании к раствору 2 (1,02 г, 3,7 ммол) в ТГФ (130 мл). Фенилхлорформиат (1,39 мл, 11,1 ммол) добавляют по каплям. Двухфазную смесь перемешивают при комнатной температуре в течение ночи, разбавляют хлористым метиленом (200 мл) и промывают водой и рассолом. Органический слой отделяют, сушат (MgSO4), фильтруют и упаривают. Остаток хроматографируют на колонке с силикагелем 10% EtOAc в смеси гексанов с целью получения заглавного соединения (980 мг, 69%) в виде твердого вещества.
Примеры 182-236.
Общая методика.
Соединения 182-236 получают по описанной ниже методике.
Раствор фенилкарбамата 181 (20 мг, 0,054 ммол) и соответствующего амина (0,08 ммол) в смеси ТГФ - ацетонитрил (3 мл, 1:1) перемешивают при ~20°С в течение ночи. В некоторых из этих реакций требуется нагревание при 60°С от 4 часов до периода в течение ночи. Смесь разбавляют хлористым метиленом (4 мл) и промывают 1 N HCl aq. (1,5 мл, 2х), 1 N NaOH aq. (1,5 мл, 2х). Слой в дихлорметане отделяют, сушат (MgSO4), фильтруют и упаривают, чтобы получить заглавный продукт. “ВЭЖХ время уд.” обозначает время удерживания ВЭЖХ в следующих условиях: для соединений 182-192 условия ВЭЖХ таковы: короткая колонка Zorbax SB-C18 4,5 мм × 7,5 см, градиент 8 минут, начиная от 100% растворителя А (10% МеОН, 90% Н2O, 0,2% Н3PO4) до 100% растворителя В (90% МеОН, 10% Н2О, 0,2% Н3PO4), скорость потока 2,5 мл/мин, λ=217 нм. Для соединений 193-236 ВЭЖХ условия таковы: балластная колонка YMC S5 ODS 4,6 мм × 50 мм, градиент 4 минуты, начиная со 100% растворителя А (10% МеОН, 90% Н2O, 0,2% Н3PO4) до 100% растворителя В (90% МеОН, 10% H2O, 0,2% Н3PO4), скорость потока 4 мл/мин, λ=220 нм.
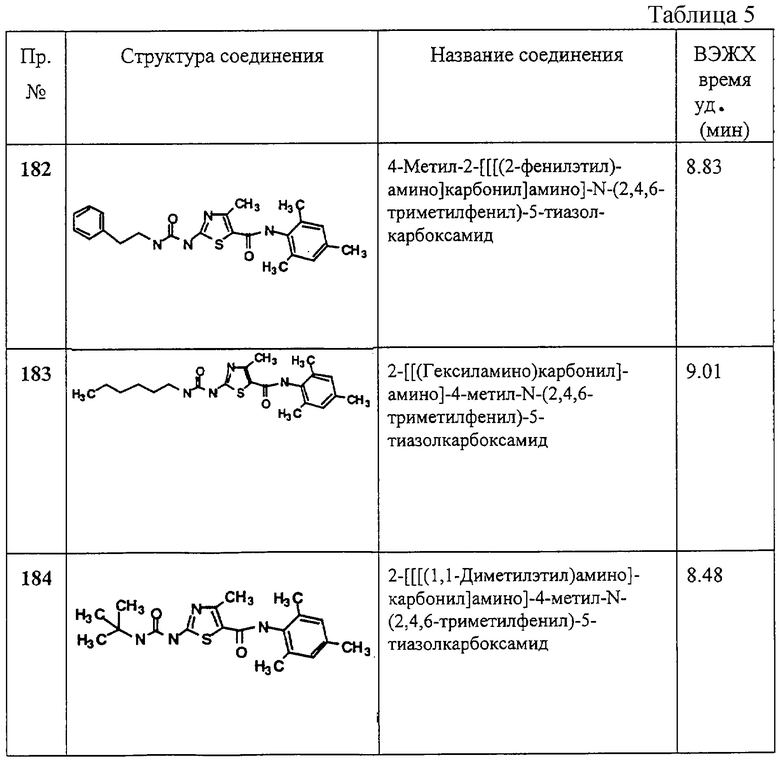
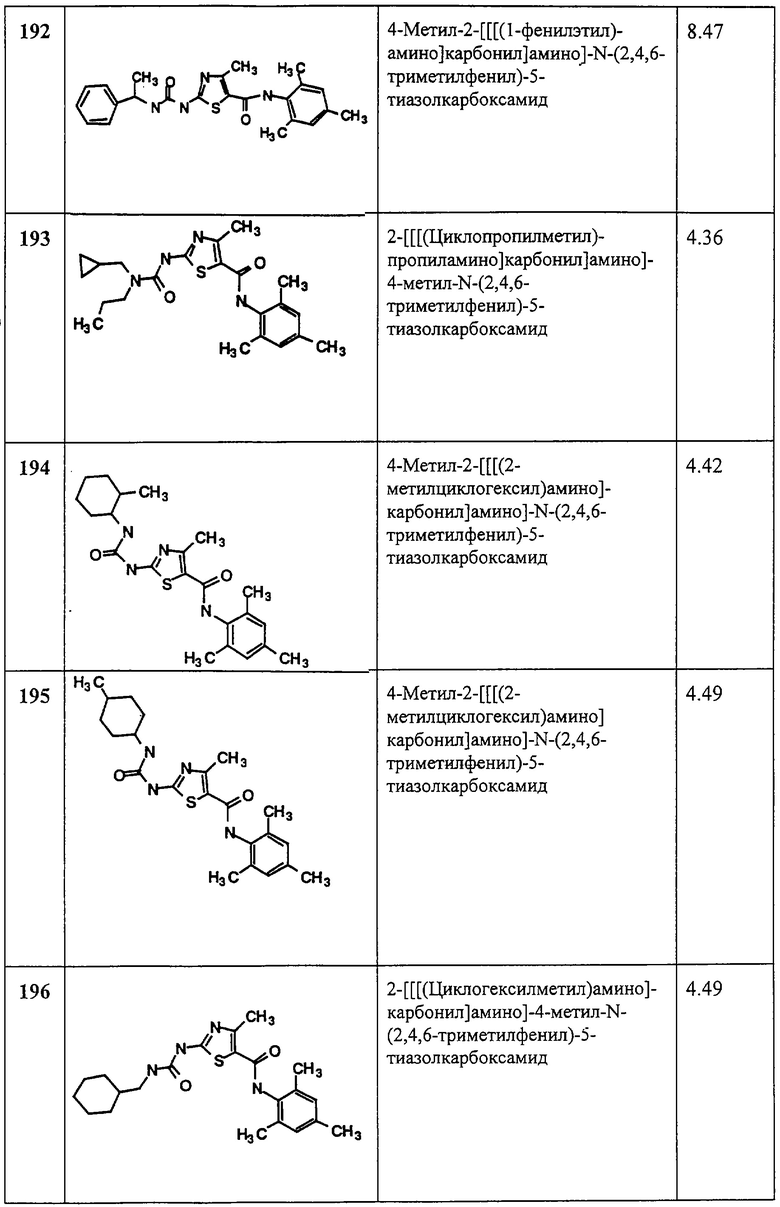
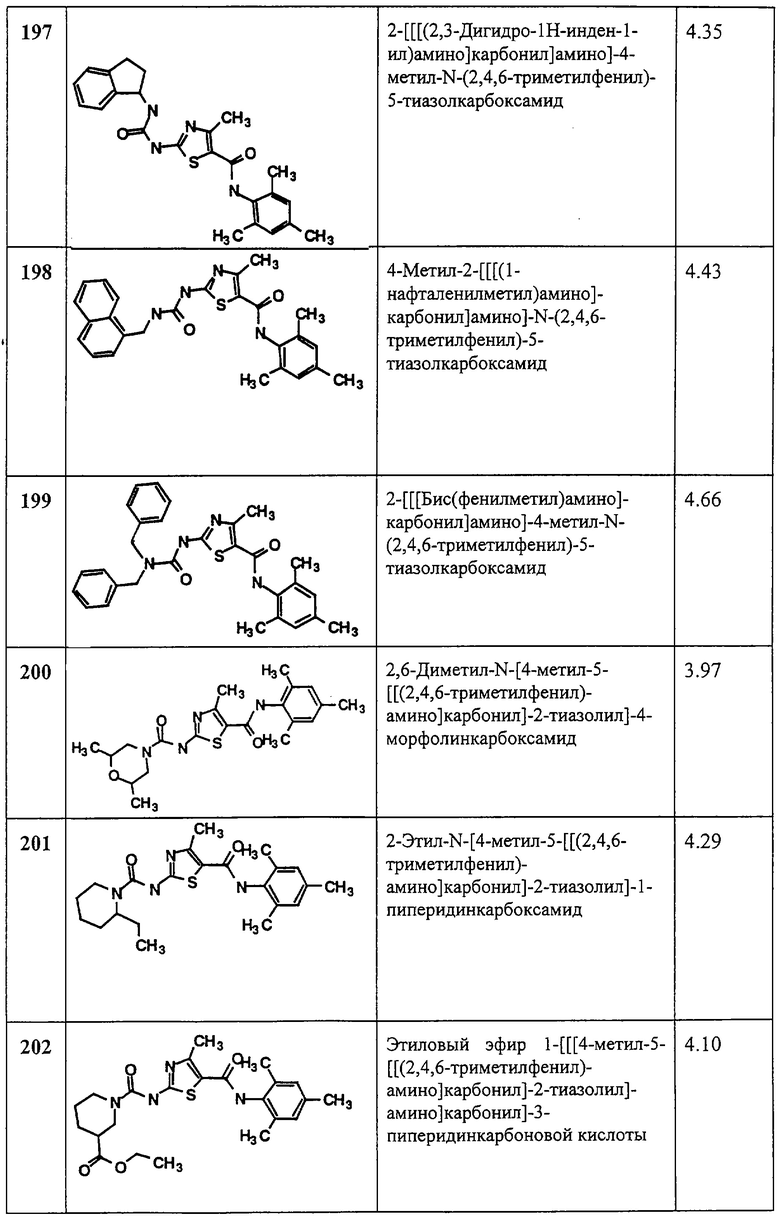
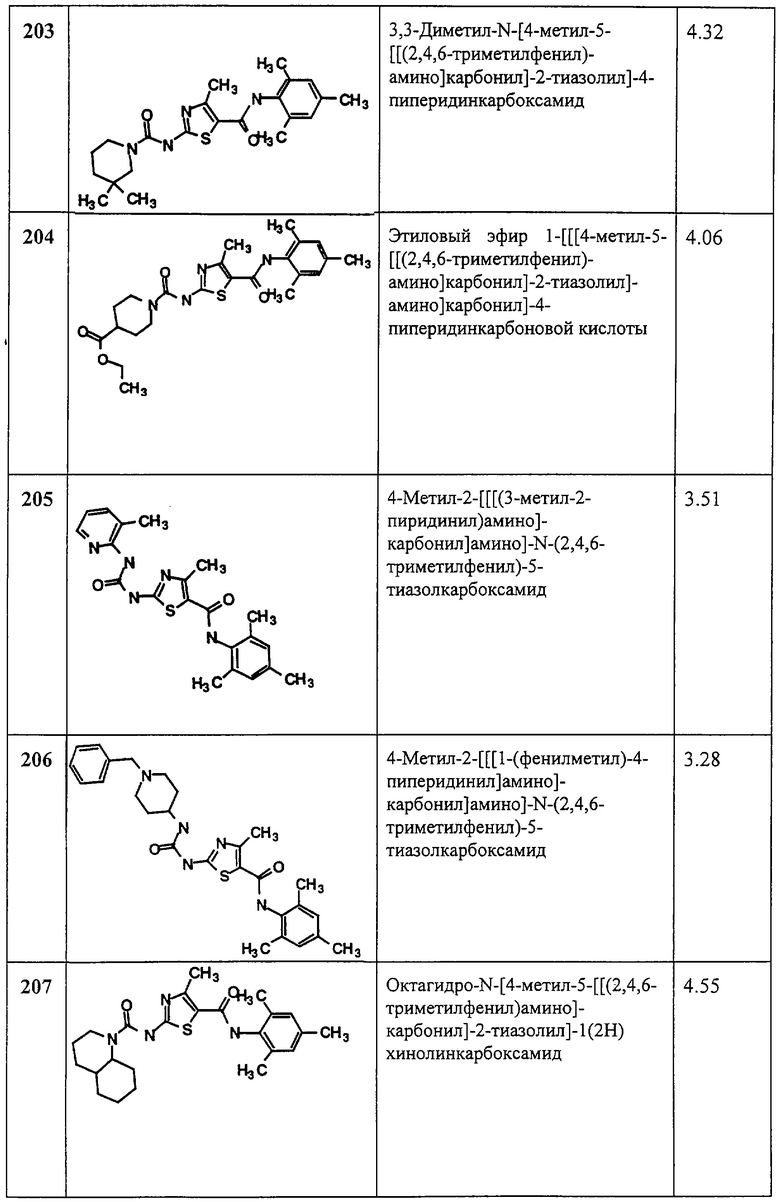
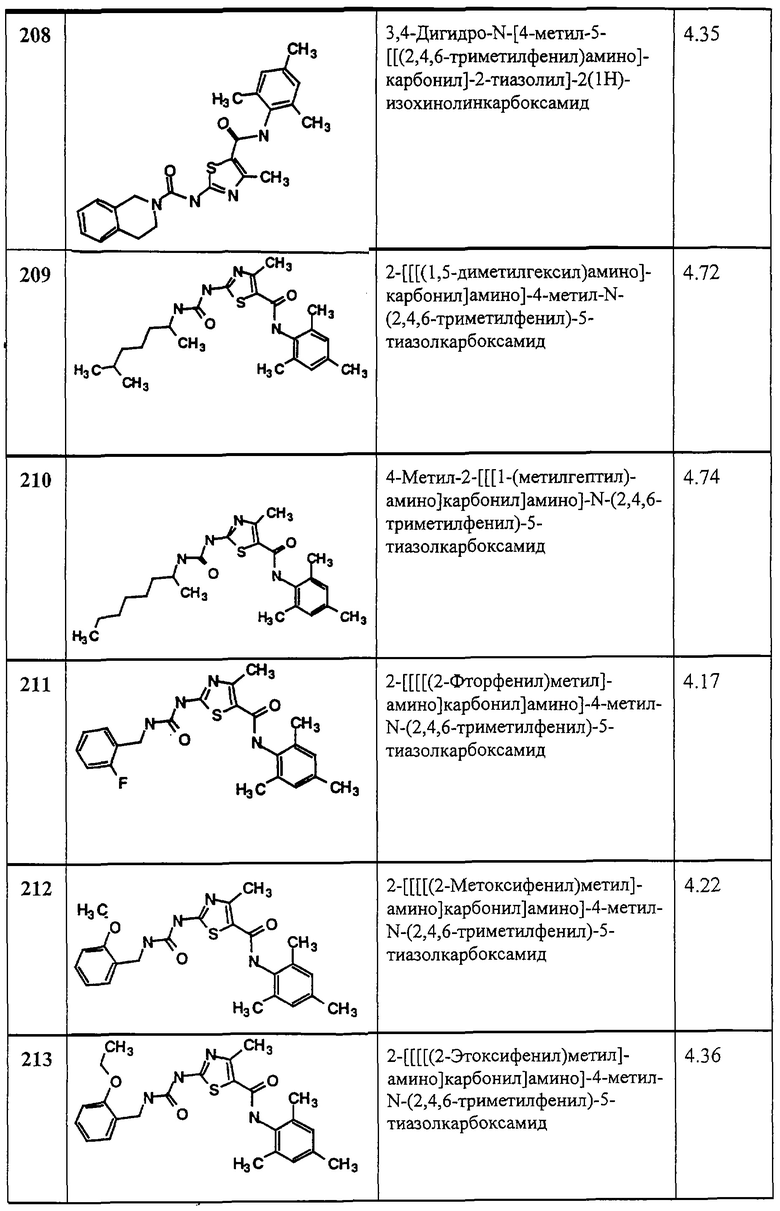
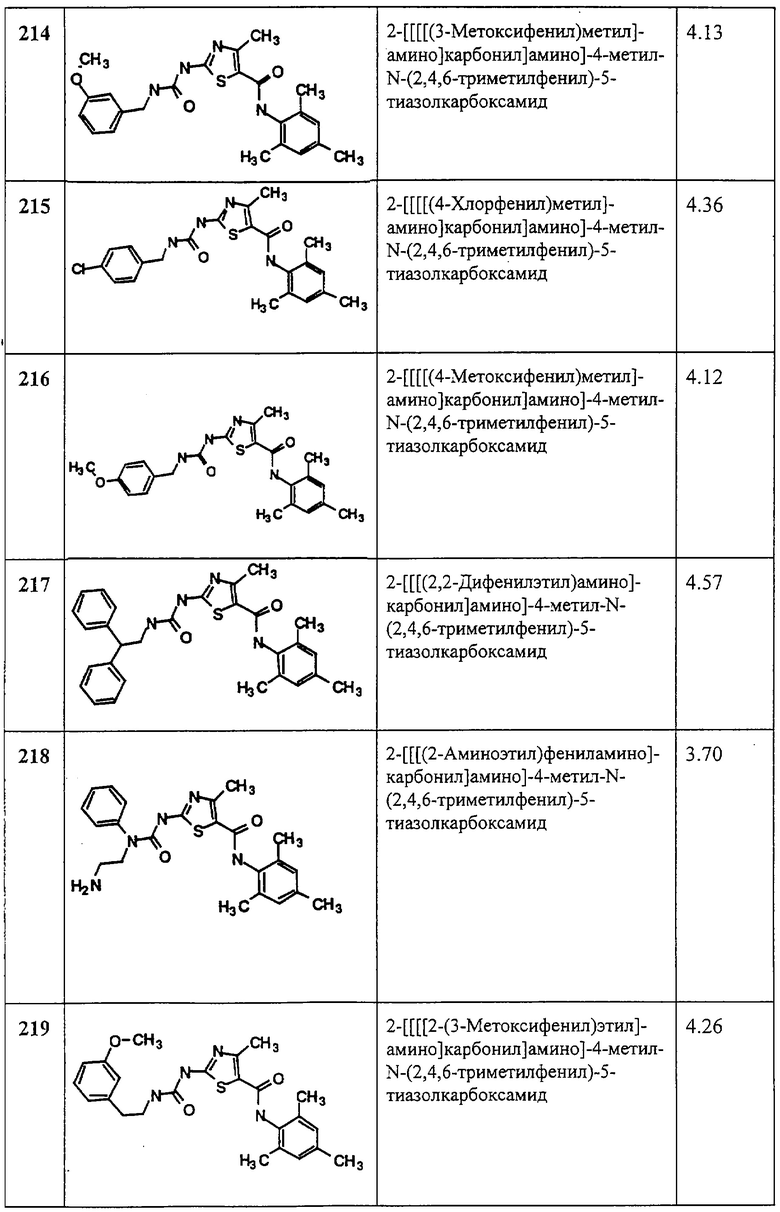
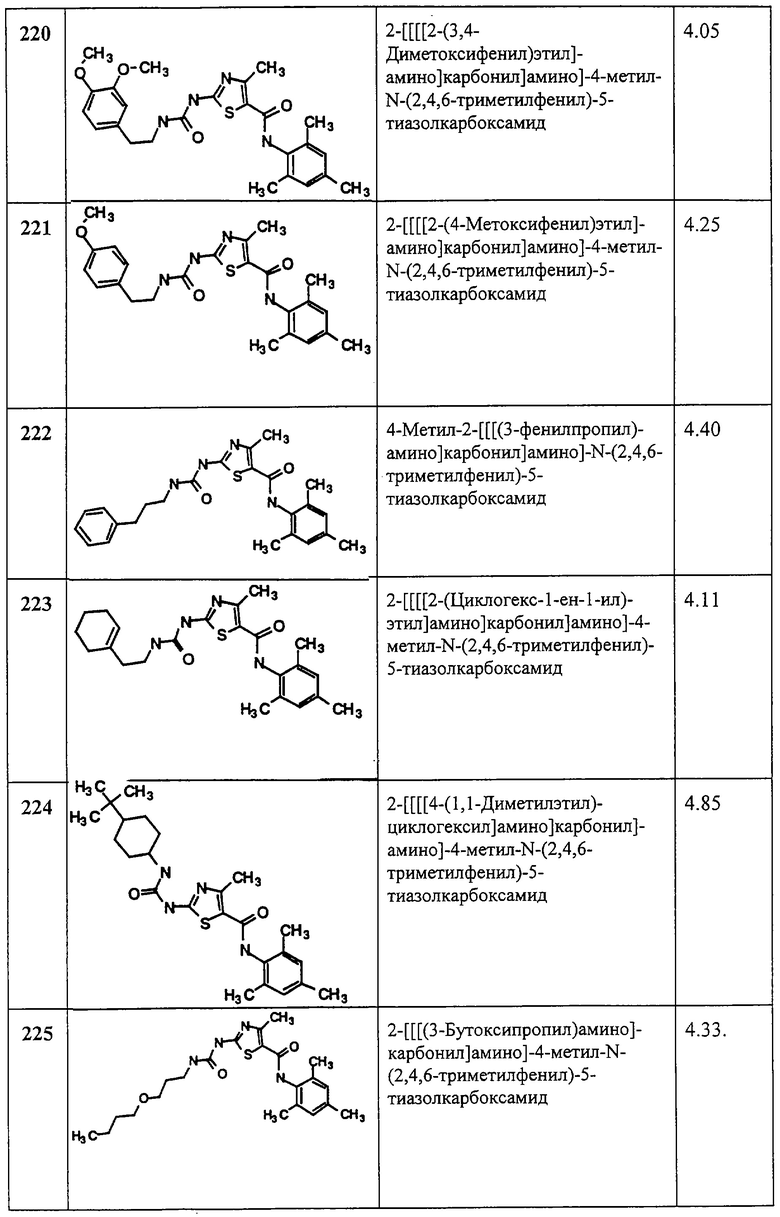
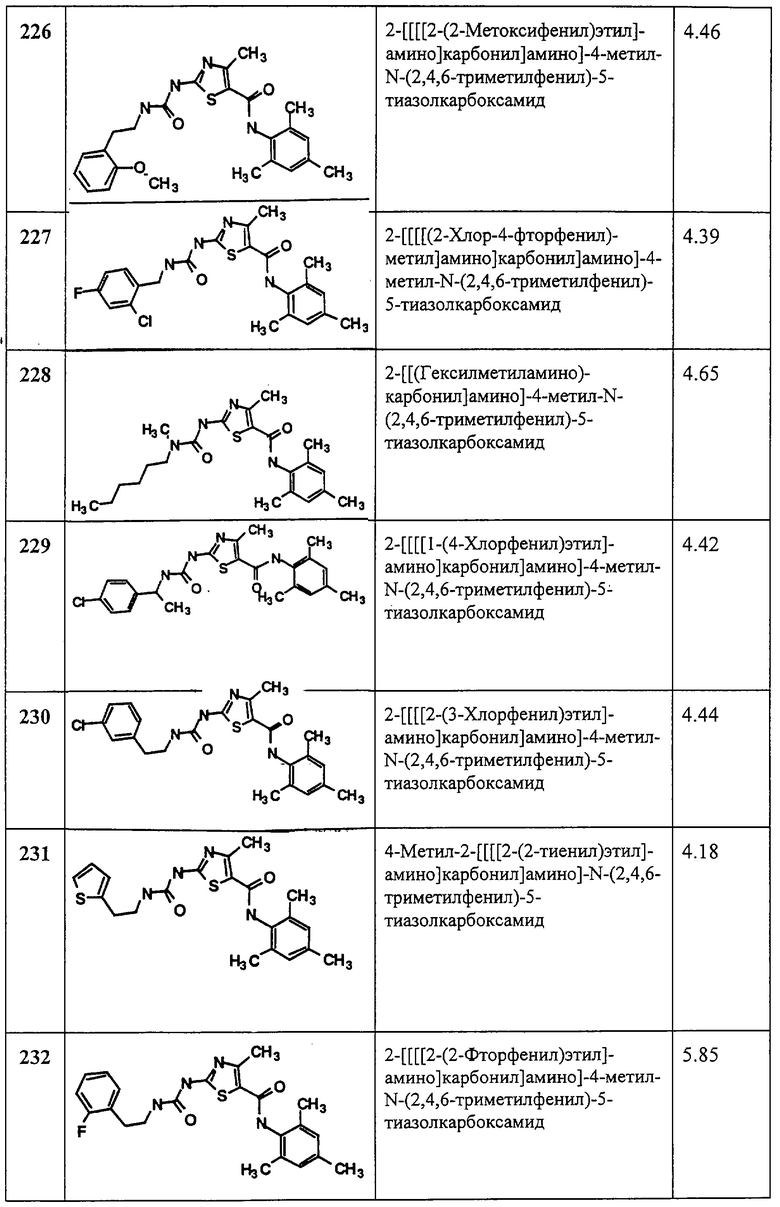
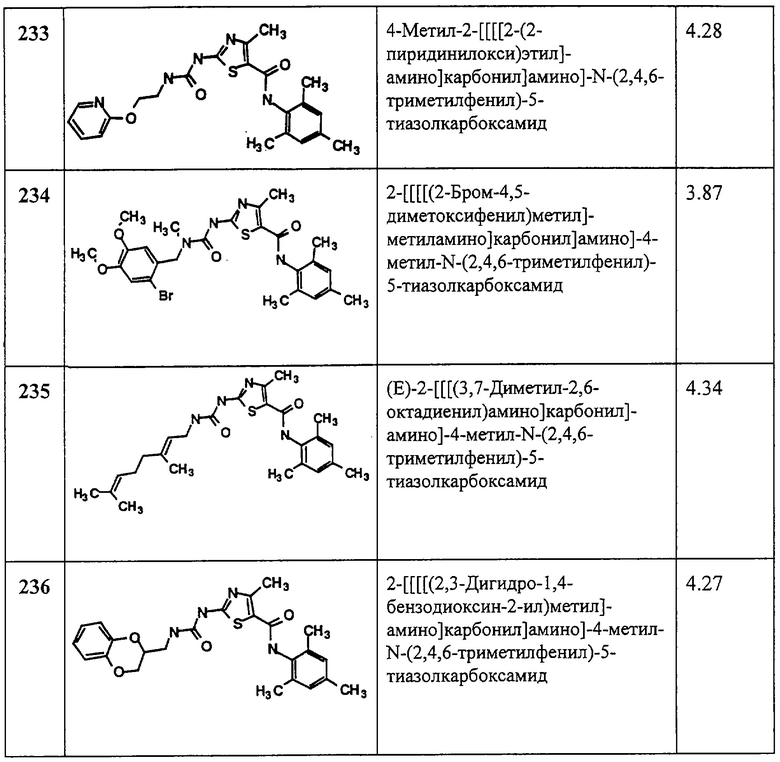
Примеры 237-285.
Общая методика.
Соединения 237-285 получают по описанной ниже методике. Раствор фенилкарбамата 181 (20 мг, 0,054 ммол) и соответствующего амина (0,08 ммол) в ТГФ-ацетонитриле (3 мл, 1:1) перемешивают при комнатной температуре. Смесь разбавляют хлористым метиленом (4 мл) и промывают 1 N HCl aq. (1,5 мл, 2х), 1 N NaOH aq. (1,5 мл, 2х). Отделяют органический (хлористый метилен) слой, сушат (MgSO4), фильтруют и упаривают и получают заглавный продукт. “ВЭЖХ время уд.” обозначает ВЭЖХ время удерживания в следующих условиях: для соединений 237-278 условия ВЭЖХ суть: балластная колонка YMC S5 ODS 4,6×50 мм, градиент 4 минуты, начиная со 100% растворителя А (10% МеОН, 90% H2О, 0,2% Н3PO4) до 100% растворителя В (90% МеОН, 10% H2O, 0,2% Н3PO4), скорость потока 4 мл/мин, λ=220 нм. Для соединений 279-285 условия ВЭЖХ суть: короткая колонка Zorbax S8-C18 4,5 мм × 7,5 см, градиент 8 минут, начиная со 100% растворителя А (10% МеОН, 90% H2О, 0,2% Н3PO4) до 100% растворителя В (90% МеОН, 10% Н2O, 0,2% Н3PO4), скорость потока 2-5 мл/мин, λ=217 нм.
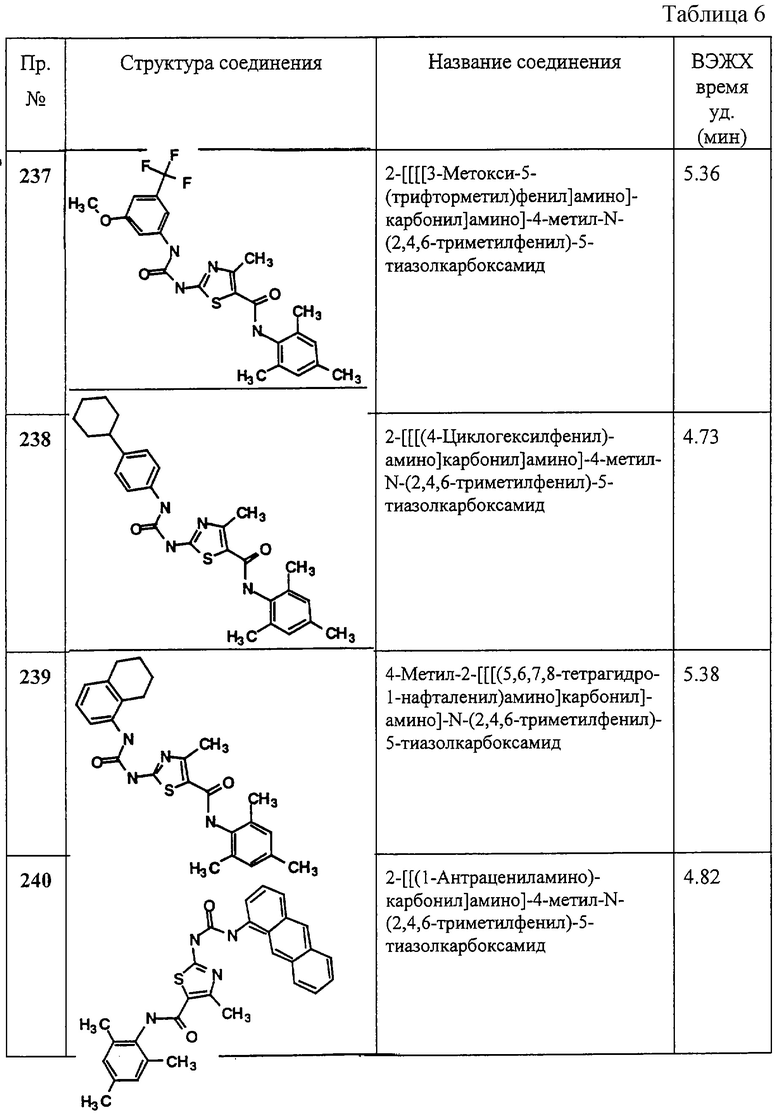
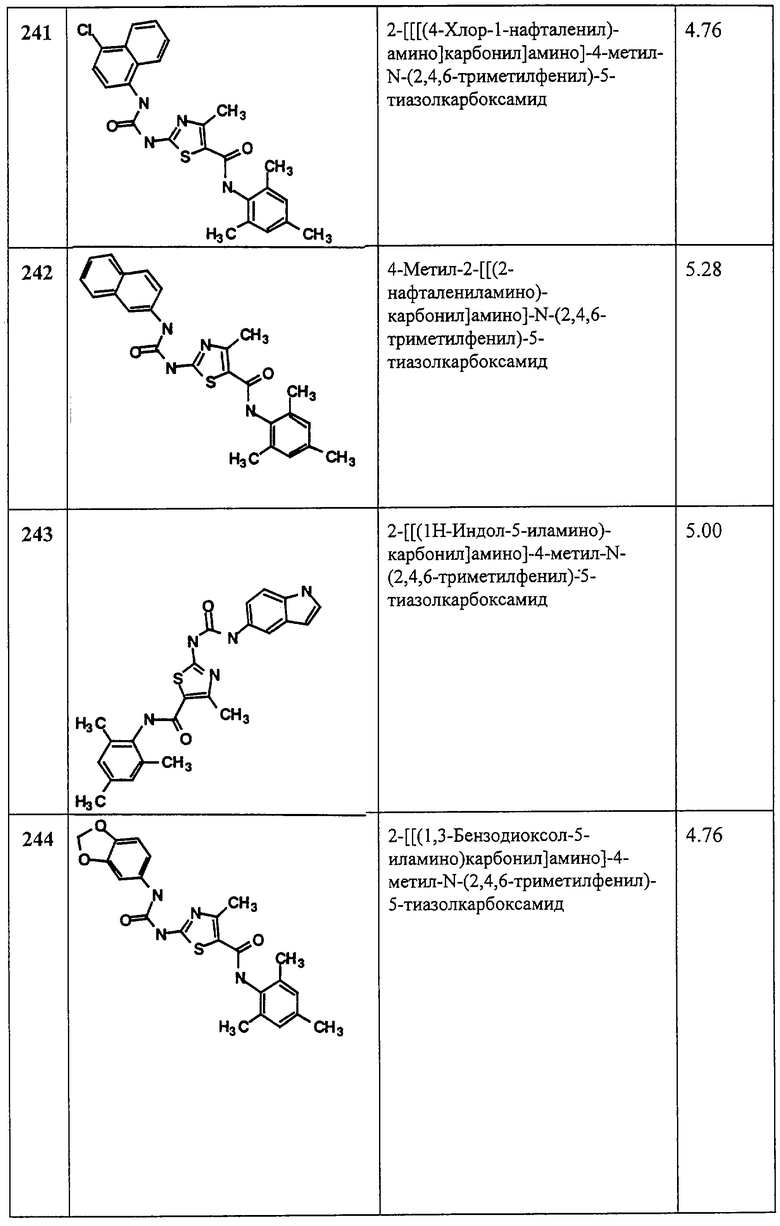
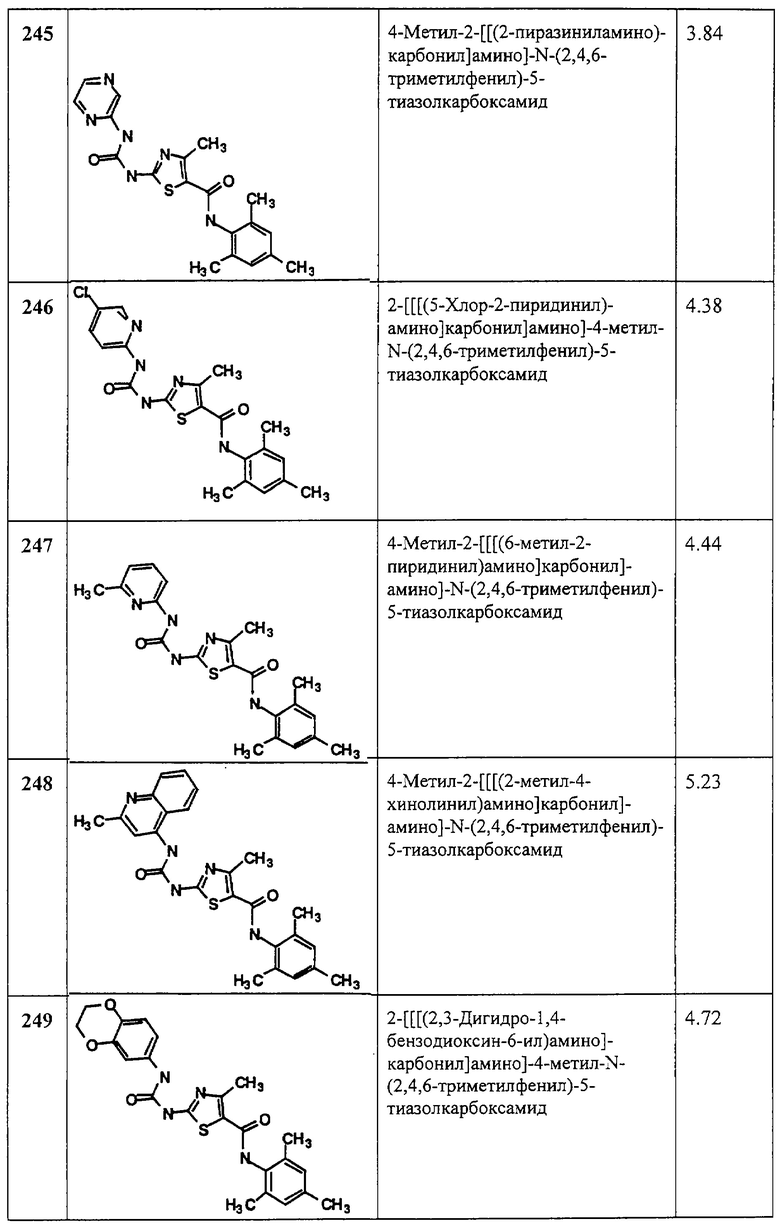
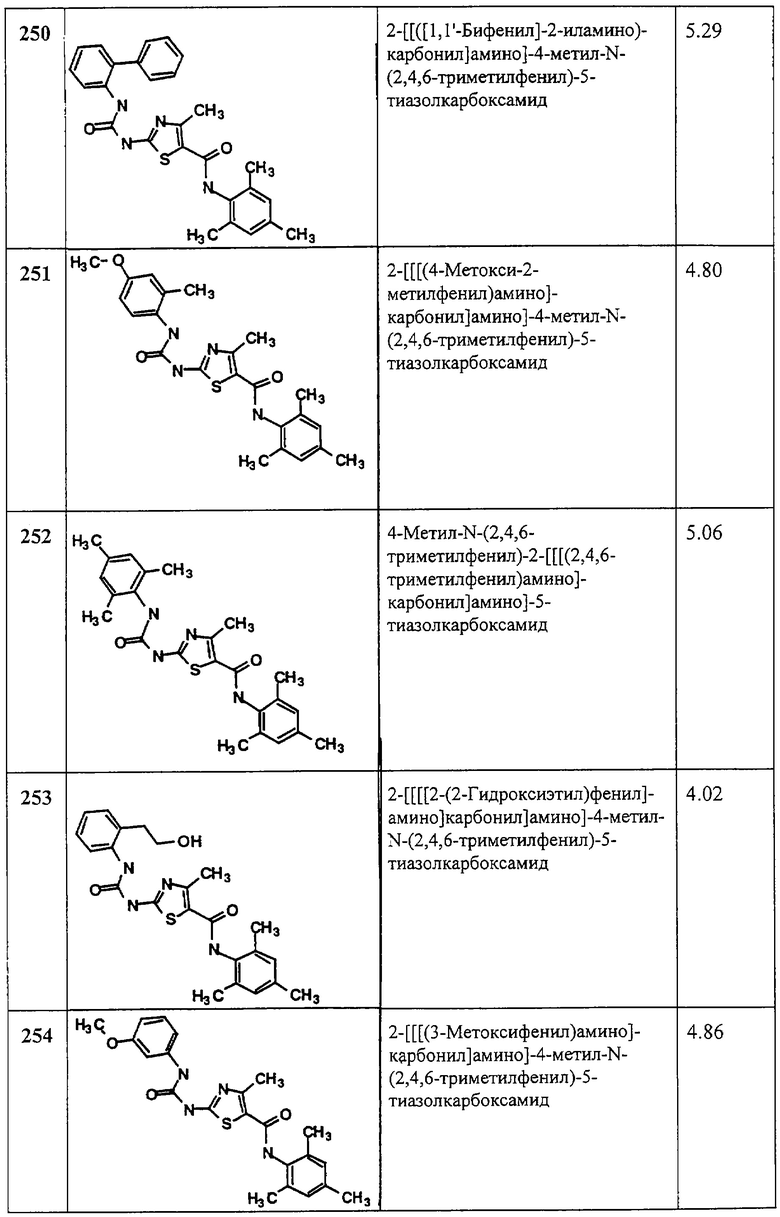
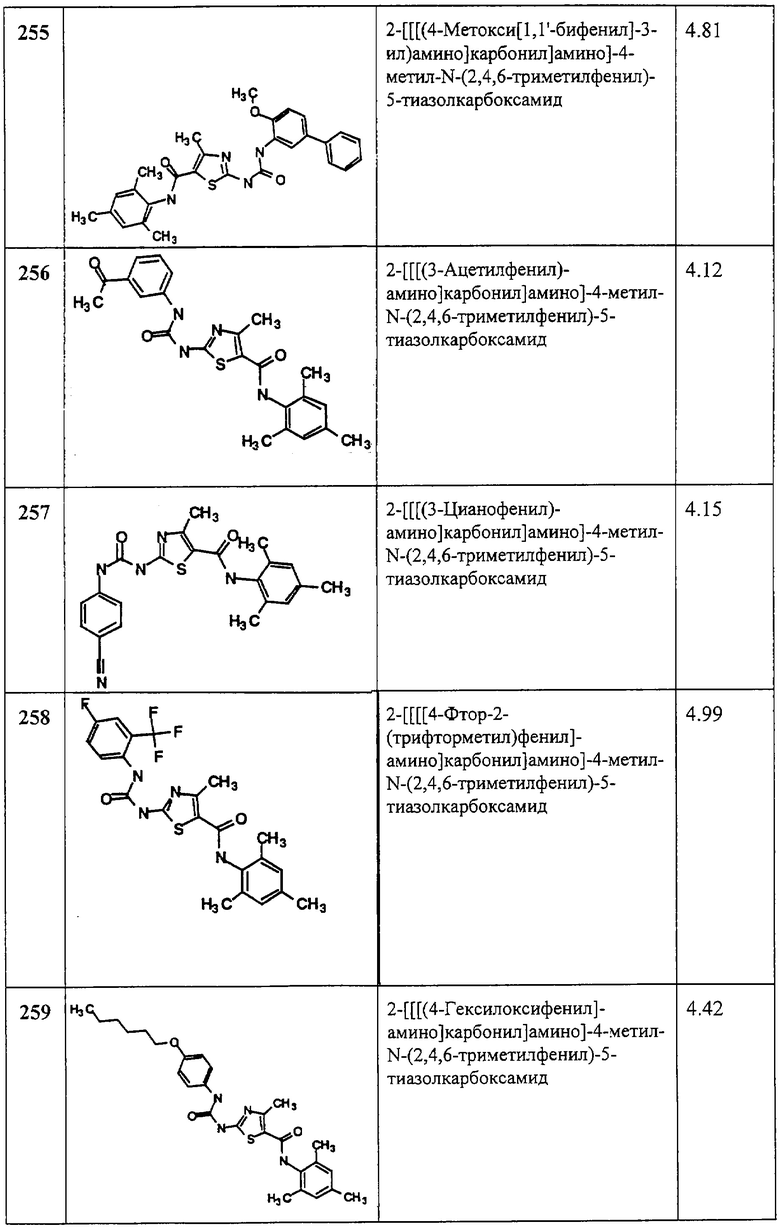
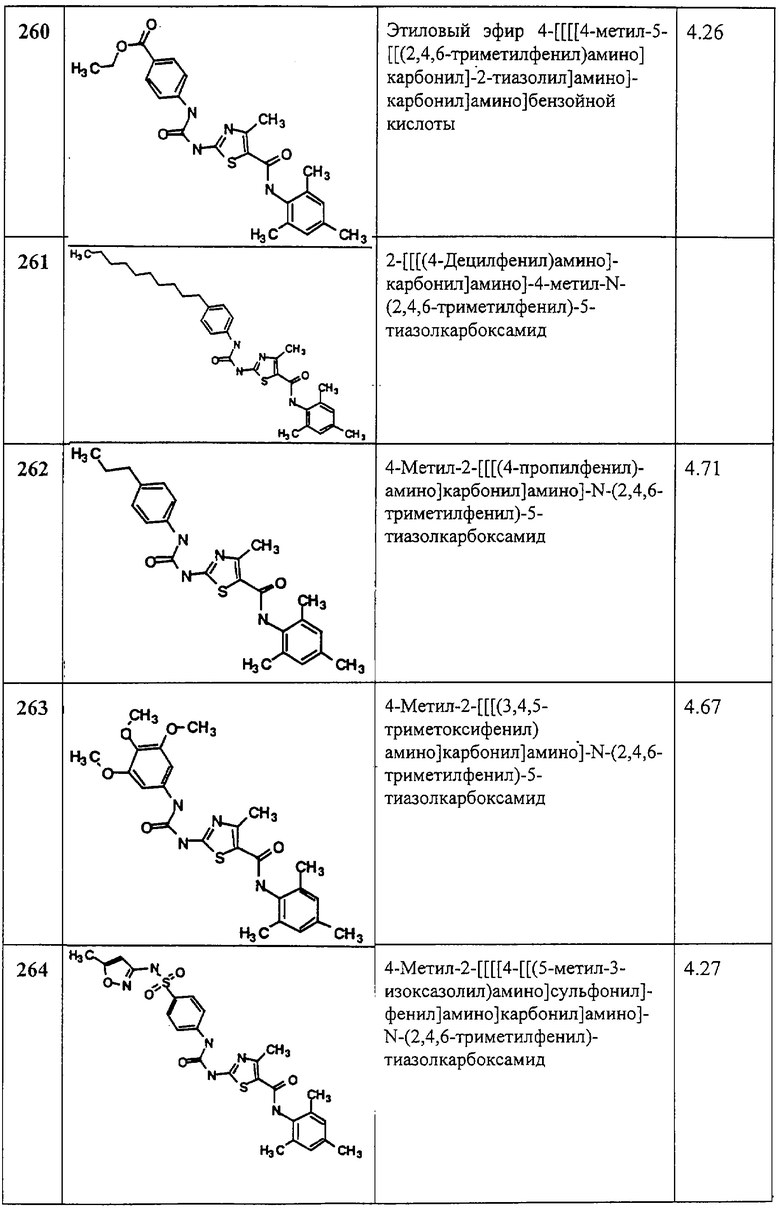
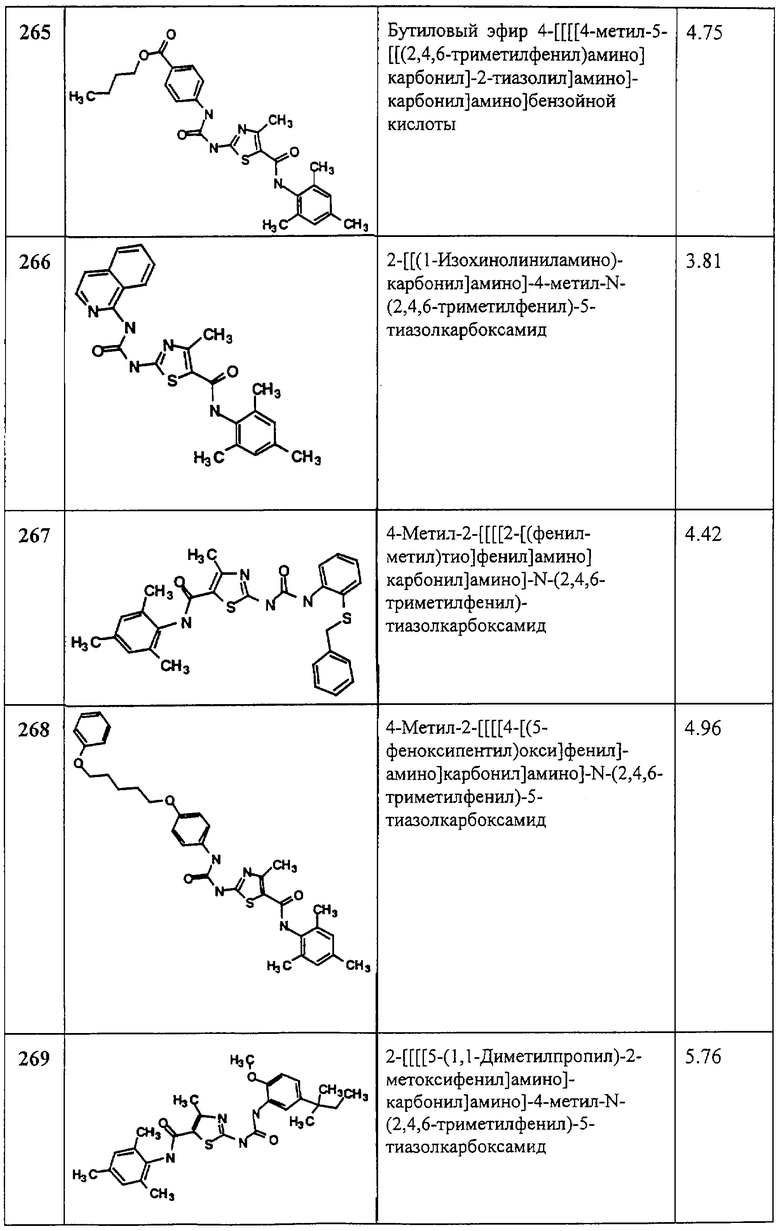
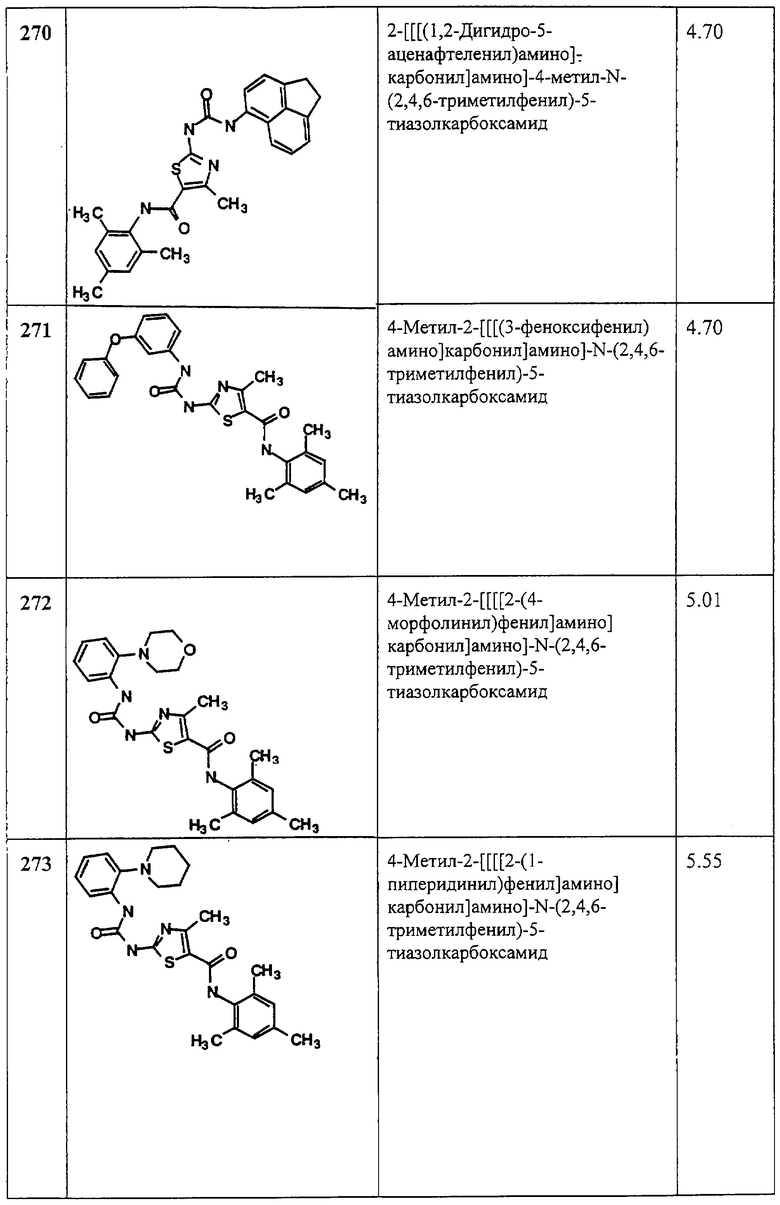
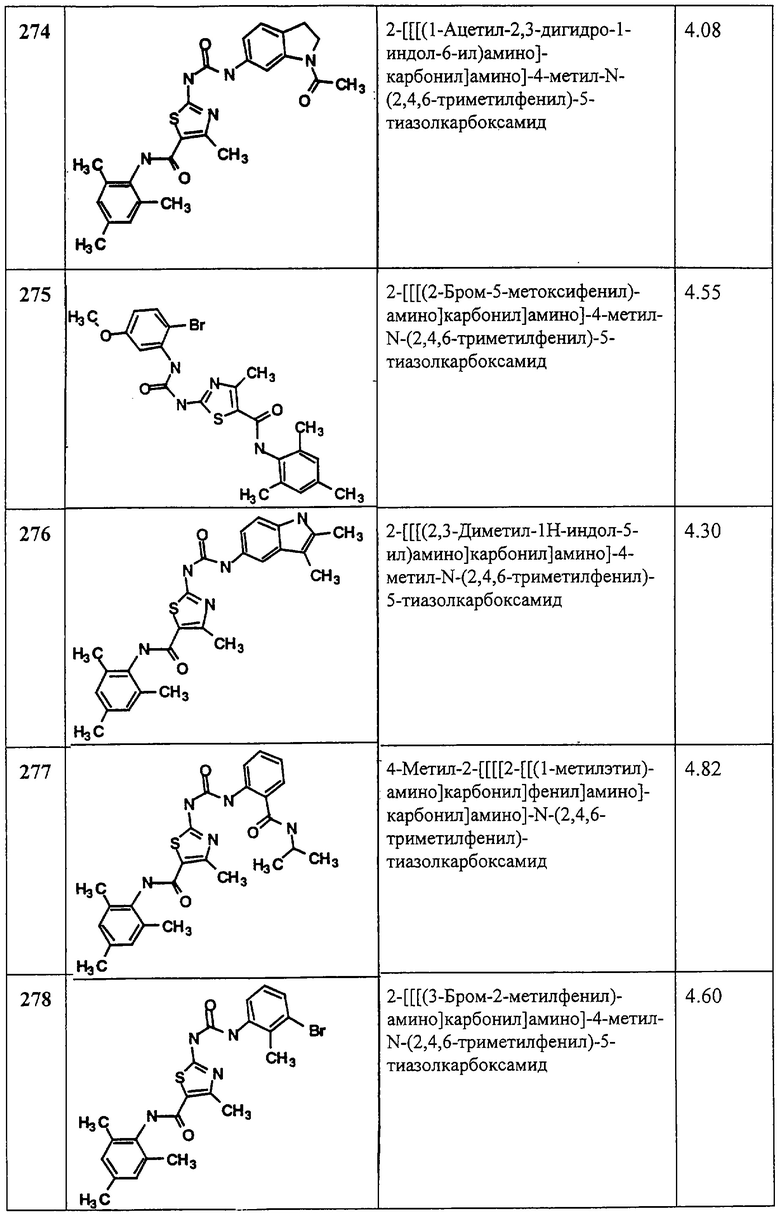
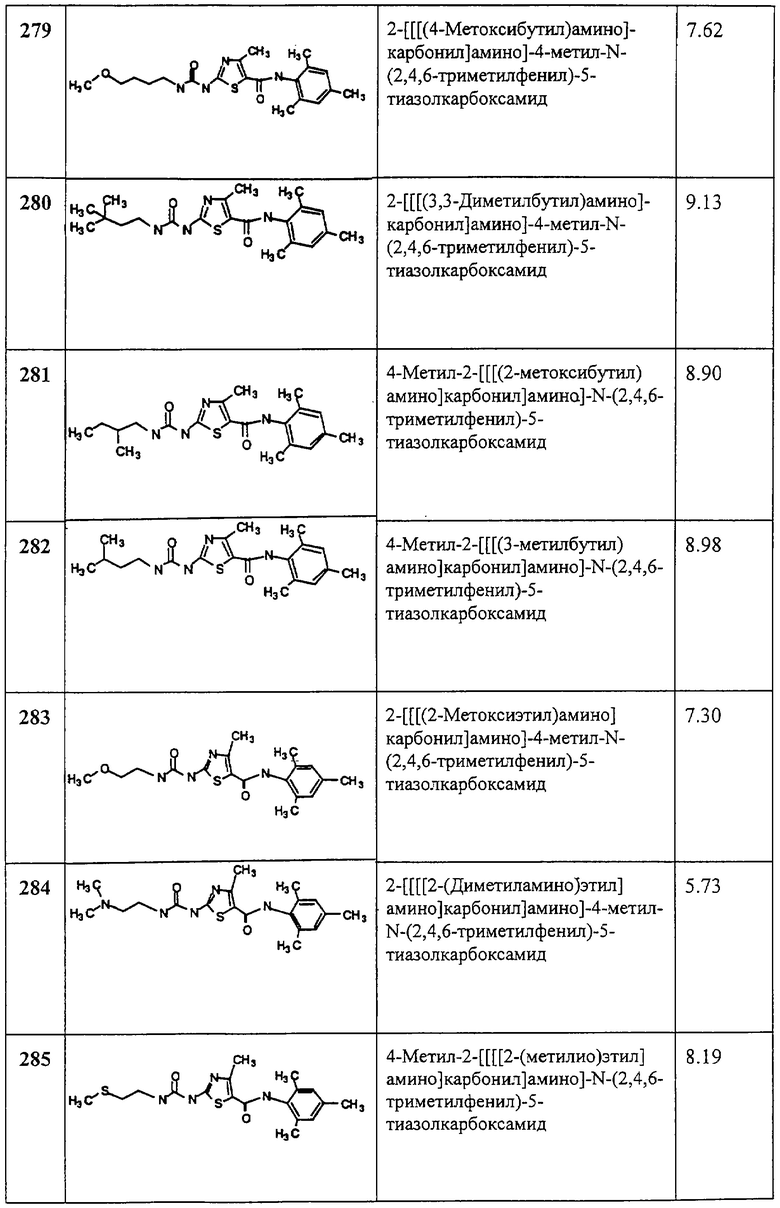
Примеры 286-311.
Общая методика.
Соединения 286-311, за исключением соединения 307, получают по описанной ниже методике.
Раствор хлорангидрида 2-[[(бутиламино)карбонил]амино]-4-метил-5-тиазолкарбоновой кислоты (30 мг, 0,11 ммол), соответствующего анилина (0,12 ммол) в ТГФ (1 мл) обрабатывают диизопропилэтиламином (22,6 мкл, 0,13 ммол). Смесь продувают аргоном и перемешивают механически в ампуле в течение 22 часов, разбавляют хлористым метиленом (4 мл) и промывают 2N HCl aq. (3х). Органический слой отделяют, сушат(Na2SO4), фильтруют и упаривают. Сырой продукт очищают, либо растирают со смесью хлористый метилен - эфир (1:1), либо хроматографией на силикагеле (элюент: 80% EtOAc в смеси гексанов, затем EtOAc), либо автоматической препаративной ВЭЖХ (условия: колонка YMC S5 ODS А 20×100 мм; градиент 10 мин, начиная с 30% растворителя В (90% МеОН, 10% Н2О, 0,1% TFA) и 70% растворителя А (10% МеОН, 90% H2O, 0,1% TFA) до 100% растворителя В, скорость потока 20 мл/мин, λ=220 нм.
Соединение 307 получают по описанной ниже методике. Суспензию 2-[[(бутил-амино)карбонил]амино]-4-метил-5-тиазолкарбоновой кислоты (100 мг, 0,36 ммол) и HATU (170 мг, 0,44 ммол) в ДМФА (3 мл) обрабатывают диизопропилэтиламином (62 мл, 0,44 ммол). Смесь нагревают при 60°С 2 часа, охлаждают, разбавляют хлористым метиленом (12 мл), промывают 8 М раствором мочевины в воде в 2N HCl aq. (6 мл, 3х), 5% КНСО3 aq. (6 мл, 3х), сушат (Na2SO4), фильтруют и упаривают. Остаток растирают со смесью EtOAc - эфир, получая промежуточный смешанный ангидрид (102 мг, 74%) в виде белого вещества. К перемешиваемому раствору 2,6-дихлоранилина (19,6 мг, 0,12 ммол) в ТГФ (1 мл) по каплям добавляют 1М раствор натрий-бис-(триметилсилиламид)а в ТГФ (170 мкл, 0,17 ммол). Через 15 минут смешанный ангидрид - интермедиат (41,3 мг, 0,11 ммол) добавляют сразу (одной порцией). Добавляют несколько капель ДМФА и раствор перемешивают 16 час. Добавляют 1М раствор натрий-бис-(триметилсилиламид)а (110 мкл) и смесь перемешивают еще 2 часа. Смесь разбавляют хлористым метиленом (4 мл) и промывают 2N HCl aq. (2 мл, 3х), насыщ. КНСО3 водн. (3х), сушат (Na2SO4), фильтруют и упаривают. Твердое вещество промывают смесью гексанов (2х) и остаток хроматографируют на колонке с силикагелем. Элюция 80% EtOAc в смеси гексанов, а затем EtOAc дает 307 (12 мг, 27%) в виде легкого рыже-коричневого твердого вещества. “ВЭЖХ время уд.” обозначает время удерживания при следующих условиях: балластная колонка YMC S5 ODS 4,6×50 мм; градиент 4 минуты, начиная от 100% растворителя А (10% МеОН, 90% Н2О, 0,2% Н3PO4) до 100% растворителя В (90% МеОН, 10% Н2O, 0,2% Н3PO4), скорость потока 4 мл/мин, λ=220 нм.
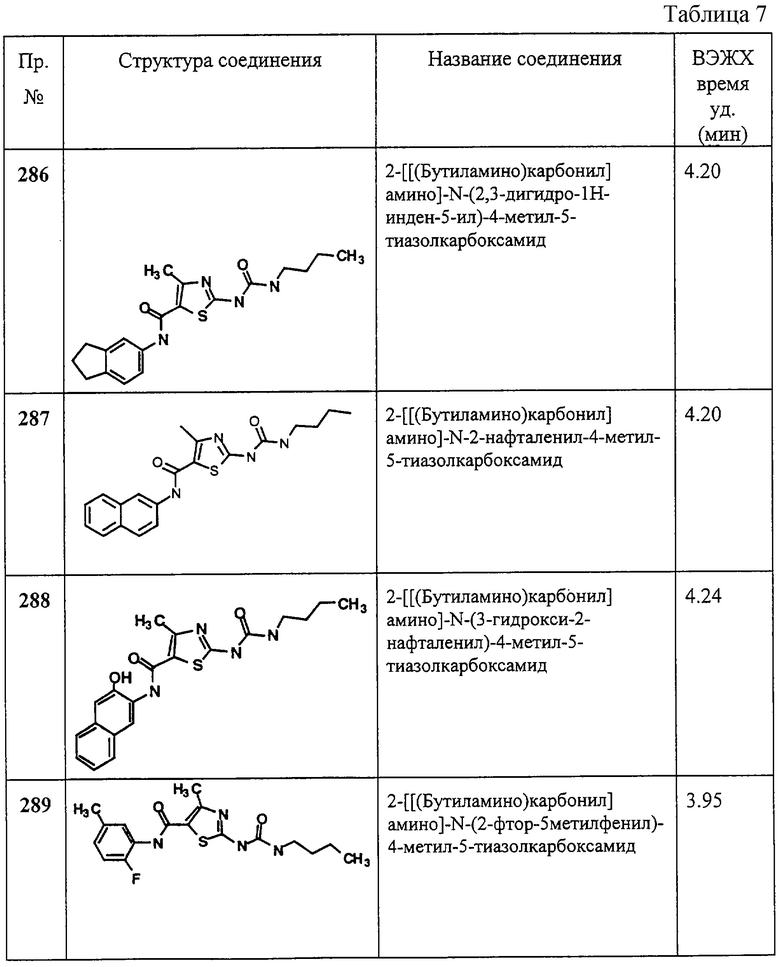
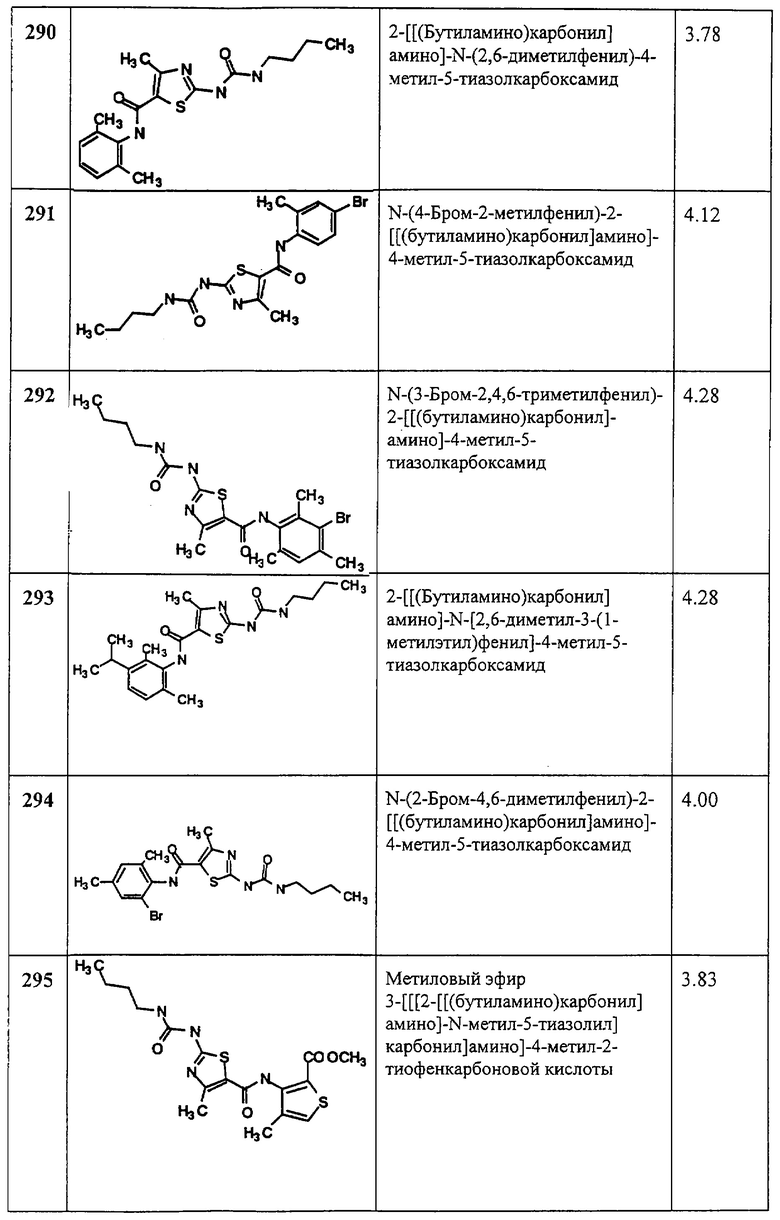
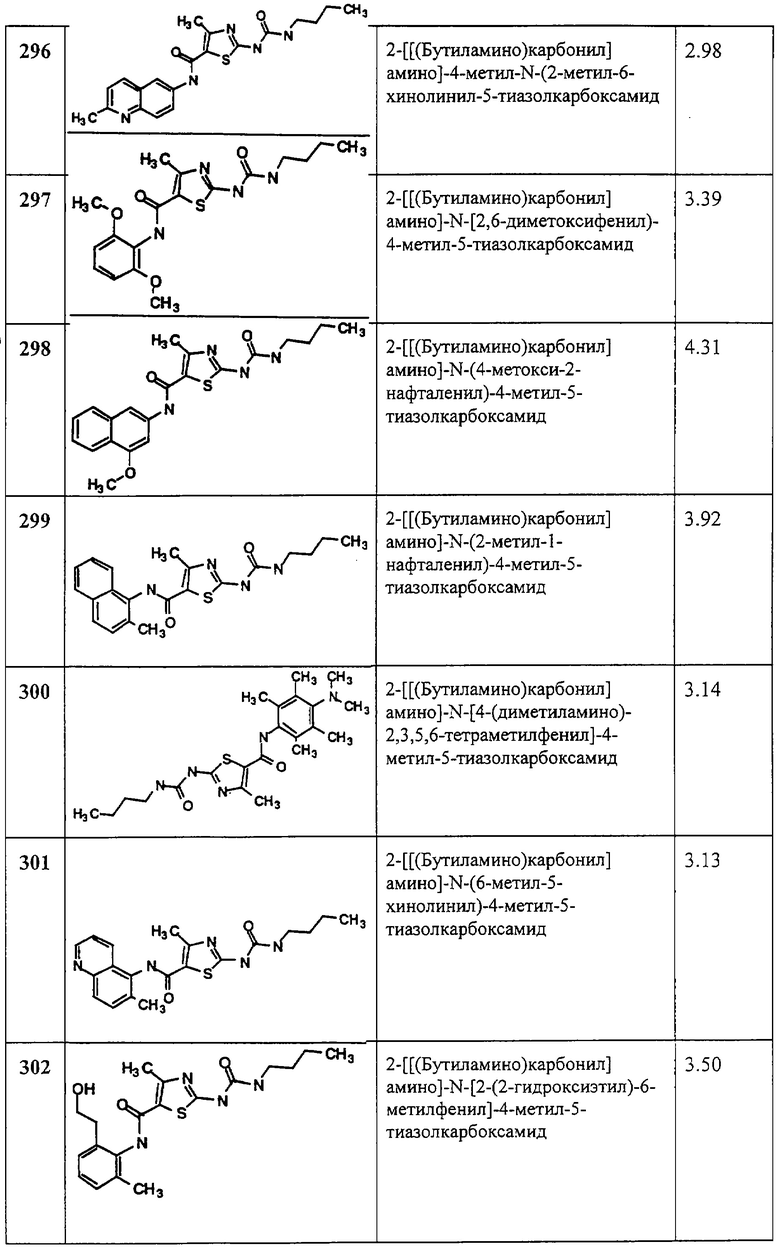
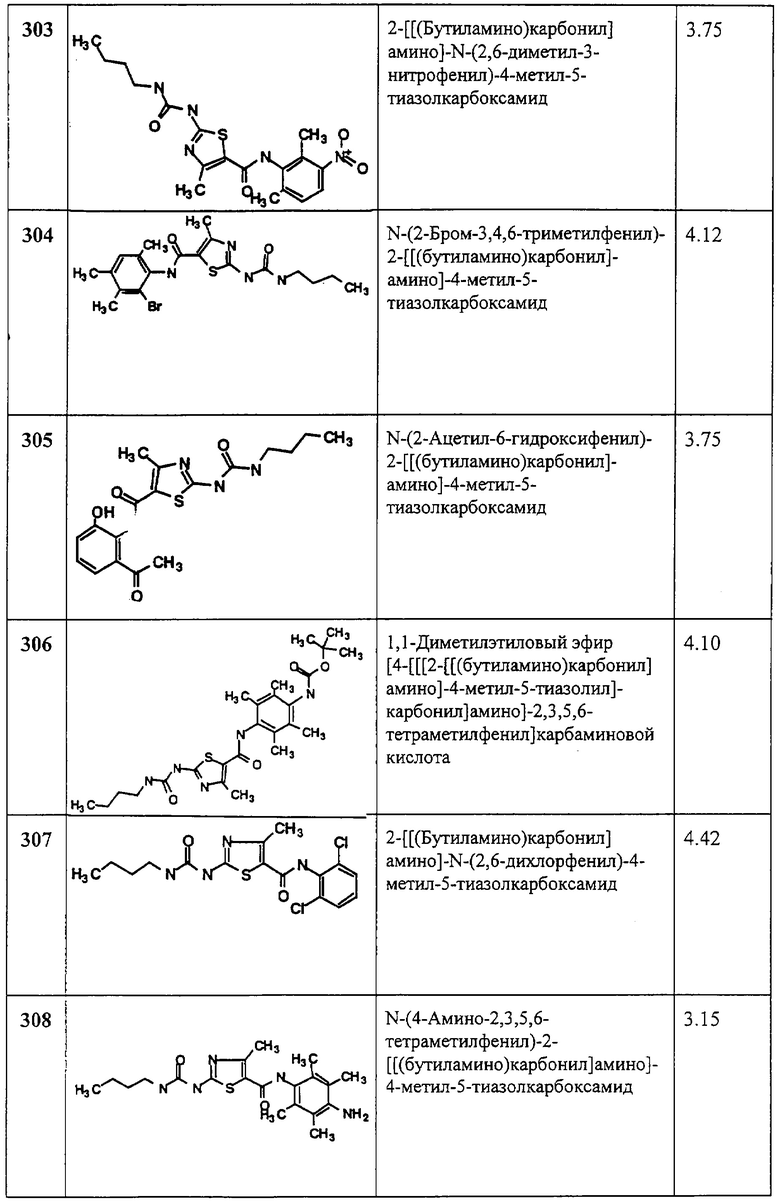
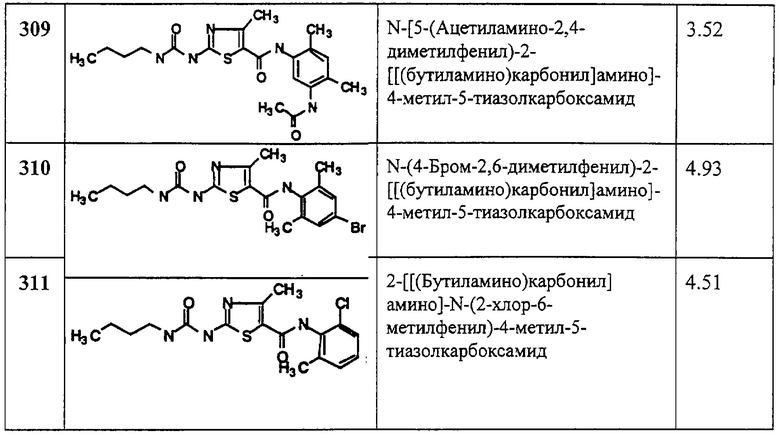
Пример 312.
Получение 4-метил-2-[(метилсульфонил)амино]-N-2,4,6-триметилфенил)-5-тиазолкарбоксамида.
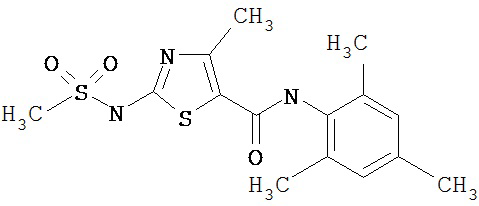
А. Этил-2-[(метилсульфонил)амино]-4-метилтиазол-5-карбоксилат.
Раствор этил-2-амино-4-метилтиазол-5-карбоксилата (558 мг, 3 ммол) в хлористом метилене (15 мл) и пиридине (5 мл) при перемешивании обрабатывают метансульфонилхлоридом (687 мг, 6 ммол) при ~20°С в течение ночи. Раствор разбавляют хлористым метиленом (50 мл) и промывают 2N HCl aq. (15 мл, 3х), сушат (MgSO4), фильтруют и упаривают. Сырой остаток разбавляют эфиром (25 мл) и отфильтровывают твердый продукт, промывают смесью (1:1) эфир - гексан (10 мл, 3х) и сушат в вакууме, получают заглавное соединение (687 мг, 87%) в виде белого твердого вещества.
В. 2-[(Метилсульфонил)амино]-4-метилтиазол-5-карбоновая кислота.
Раствор этил-2-[(метилсульфонил)амино]-4-метилтиазол-5-карбоксилата (300 мг, 1,14 ммол) в метаноле (9 мл) обрабатывают при перемешивании 1N раствором NaOH (28,4 мл, 28,4 ммол). Смесь перемешивают при комнатной температуре. Раствор охлаждают до 0°С и подкисляют 6N HCl aq. до рН 1. Раствор экстрагируют смесью хлористый метилен - хлороформ. Органические вытяжки сушат (MgSO4), фильтруют и упаривают в вакууме, получают заглавное соединение (148 мг, 55%).
С. 4-Метил-2-[(метилсульфонил)амино]-N-(2,4,6-триметилфенил)-5-тиазолкарбоксамид.
Диизопропилэтиламин (87 мкл, 0,5 ммол) добавляют к раствору 312В (99 мг, 0,42 ммол), 2,4,6-триметиланилина (68 мкл, 0,5 ммол) и [O-(7-азабензотриазол-1-ил)-1,1,3,3-тетраметилуроний]гексафторфосфата (HATU, 191 мг, 0,5 ммол) в ДМФА (3 мл). Смесь перемешивают при комнатной температуре в течение ночи, разбавляют EtOAc и промывают 0,5N водн. раствором HCl (15 мл), 10% LiCl водн. (25 мл, 3х), водой (930 мл, 2х), рассолом, сушат (MgSO4), фильтруют и упаривают. Остаток хроматографируют на колонке с силикагелем и элюируют 50% EtOAc в смеси гексанов, а затем 75% EtOAc в смеси гексанов и 2% МеОН в EtOAc, получают заглавное соединение (19 мг, 13%) в виде белого твердого вещества.
Пример 313.
Получение 4-метил-2-[[(фениламино)тиокарбонил]амино]-N-(2,4,6-триметилфенил)-5-тиазолкарбоксамида.
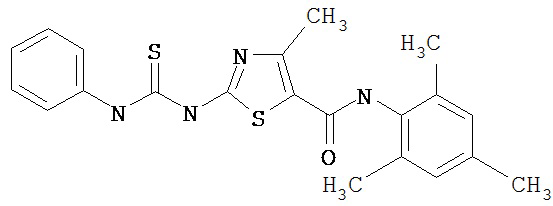
Раствор 2 (45 мг, 0,16 ммол) и фенилизотиоцианата (43 мг, 0,32 ммол) в пиридине (2 мл) нагревают до 80°С в течение 20 часов. Смесь охлаждают, разбавляют смесью хлористый метилен - ТГФ (80 мл, 3:1) и промывают 2N HCl aq. (15 мл, 2х). Органические вытяжки сушат (MgSO4), фильтруют и упаривают. Остаток разбавляют EtOAc (20 мл) и отфильтровывают твердое вещество, промывают эфиром (10 мл, 3х) и сушат в вакууме, получают заглавное соединение (35 мг, 52%) в виде грязно-белого твердого вещества.
Пример 314.
Получение 2-[[(этиламино)карбонил]амино]-4-метил-N-(2,4,6-триметилфенил)-5-тиазолкарбоксамида.
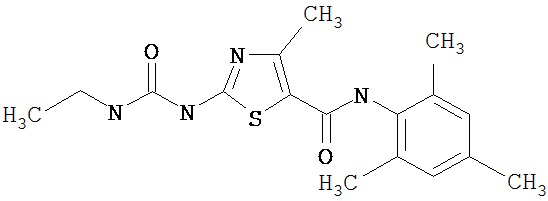
Соединение 314 получают аналогично соединениям 171-180, используя этилизоцианат, получая заглавное соединение 314 в виде белого твердого вещества (65%).
Пример 315.
Получение N-(2-хлор-6-метилфенил)-2-[(циклопропилкарбонил)амино]-5-тиазолкарбоксамида.
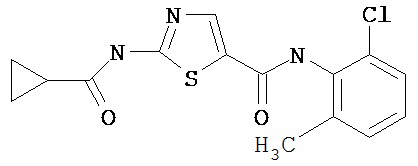
А. Этил-2-трет-бутоксикарбонилоксиамино-4-метилтиазол-5-карбоксилат.
Суспензию этил-2-аминотиазол-5-карбоксилата (972 мг, 6 ммол, В. Plouver, С. Bailly, R. Houssin, j-P. Henlchart Heterocycles 32(4), 693-701, 1991, и H.J. Becker, J.de Jonge Rec. Trav. Chim., 61, 463, 1942), ди-трет-бутилдикарбоната (1,94 г, 9 ммол) и 4-диметиламинопиридина (73 мг, 0,6 ммол) в сухом тетрагидрофуране (75 мл) перемешивают под азотом 24 часа. Растворитель упаривают в вакууме. Остаток суспендируют в эфире (50 мл). Твердое вещество промывают эфиром (10 мл, 3х) и сушат в вакууме, получая заглавное соединение (1,1 г, 70%).
В. 2-трет-Бутоксикарбонилоксиаминотиазол-5-карбоновая кислота.
Раствор этил-2-трет-бутоксикарбонилоксиамино-4-метилтиазол-5-карбоксилата (1,1 г, 4,2 ммол) в смеси тетрагидрофуран - метанол (80 мл, 1:1) обрабатывают 6N NaOH aq. (20 мл, 120 ммол). Смесь перемешивают при комнатной температуре 24 часа. Большую часть ТГФ и метанола отгоняют в вакууме, а водный раствор подкисляют 6N HCl aq. (22 мл). Выпавший осадок отфильтровывают, промывают водой и эфиром, сушат на воздухе, а затем в вакууме, получая заглавную кислоту (940 мг, 96%) в виде грязно-белого твердого вещества.
С. 1,1-Диметилэтиловый эфир [5-[[(2-хлор-6-метилфенил)амино]карбонил]-2-тиазолил]карбаминовой кислоты.
К раствору 2-трет-бутоксикарбонилоксиаминотиазол-5-карбоновой кислоты (234 мг, 1 ммол) в ТГФ (10 мл) и N,N-диметилформамиде (несколько капель) по каплям прибавляют 2М раствор оксалилхлорида в хлористом метилене (1 мл, 2 ммол). Раствор перемешивают при комнатной температуре 4 часа. Растворитель упаривают в вакууме, получая сырой хлоранигидрид кислоты.
К раствору сырого хлорангидрида 2-трет-бутоксикарбонилоксиаминотиазол-5-карбоновой кислоты (1 ммол) в хлористом метилене (10 мл) при 0°С по каплям добавляют 2-хлор-6-метиланилин (212 мг, 1,5 ммол). Добавляют диизопропилэтиламин (516 мг, 4 ммол). Раствор доводят до комнатной температуры и перемешивают 24 часа, разбавляют хлористым метиленом (60 мл) и промывают 2N HCl aq. (15 мл). Органические вытяжки сушат (MgSO4), фильтруют и упаривают. Остаток разбавляют EtOAc - эфиром (25 мл, 1:4) и твердое вещество фильтруют и промывают эфиром (5 мл, 4х) и сушат в вакууме, получая заглавное соединение (175 мг, 48%) в виде рыжевато-коричневого твердого вещества.
D. 2-Амино-N-(2-хлор-6-метилфенил)-5-тиазолкарбоксамид.
Соединение 315D получают аналогично соединению 2, но берут соединение 315С, получая заглавное соединение 315D в виде рыжевато-коричневого твердого вещества.
Е. 2-[(Циклопропилкарбонил)амино]-N-(2-хлор-6-метилфенил)-5-тиазолкарбоксамид.
Раствор 315D (50,6 мг, 0,19 ммол) и ангидрида циклопропанкарбоновой кислоты (302 мг, 1,96 ммол) в диоксане (2 мл) нагревают до 93°С в течение ночи. Смесь упаривают в вакууме, разбавляют EtOAc и промывают насыщенным раствором КНСО3 (2х). Органический слой сушат (Na2SO4), фильтруют и упаривают. Остаток растирают с эфиром и получают заглавное соединение (11 мг, 17%) в виде белого твердого вещества.
Пример 316.
Получение 2-[[[(1,1 - диметилэтил)амино]карбонил]амино]-N-(2-хлор-6-метилфенил)-5-тиазолкарбоксамида.
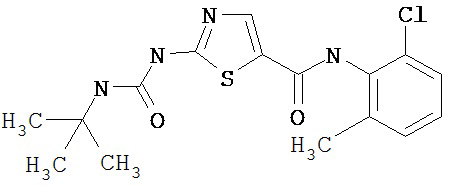
Гидрид натрия (19,2 мг, 0,8 ммол) добавляют к раствору 315D (48,3 мг, 0,18 ммол) и трет-бутилизоцианата (41 мкл, 0,36 ммол) в ТГФ (5 мл) при 0°С. Через 1 час смесь разбавляют EtOAc и промывают холодным насыщенным раствором хлористого аммония. Водный слой отделяют и экстрагируют EtOAc. Вытяжки EtOAc объединяют, сушат (Na2SO4), фильтруют и упаривают. Остаток очищают автоматической препаративной ВЭЖХ (условия: YMC S5 ODS А 20×100 мм колонка; 10 мин градиент, начиная с 10% растворителя В (90% МеОН, 10% H2О, 0,1% TFA) и 90% растворителя А (10% МеОН, 90% H2O, 0,1% TFA) до 100% растворителя В, скорость потока 20 мл/мин, λ=220 нм, получают заглавное соединение (18 мг, 28%) в виде грязно-белой жидкости.
Пример 317.
Получение 2-[[(1,1-диметилэтокси)карбонил]амино]-4-метил-N-(2,4,6-триметилфенил)-5-тиазолацетамида.
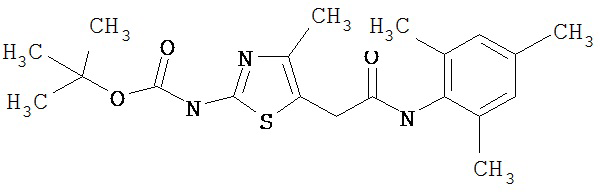
Соединение 317 получают аналогично соединению 1, но берут метил-2-амино-4-метилтиазол-5-ацетат, получая заглавное соединение 317 в виде грязно-белого твердого вещества.
Пример 318.
Получение 2-амино-4-метил-N-(2,4,6-триметилфенил)-5-тиазолацетамида.
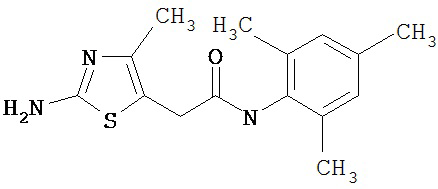
Соединение 318 получают аналогично 2, но берут 317, дающее заглавное соединение 318 в виде легкого твердого вещества коричневого цвета.
Пример 319.
Получение N-(2-хлор-6-метилфенил)-2-[(4,6-диметил-2-пиридинил)амино]-5-тиазолкарбоксамида.
А. 2-Бром-N-(2-хлор-6-метилфенил)-5-тиазолкарбоксамид.
Раствор бромида меди (II) (2,68 г, 12 ммол) в ацетонитриле (50 мл) продувают азотом и охлаждают до 0°С. Добавляют трет-бутил (2 мл, 15 ммол), а затем раствор соединения 315D (2,68 г, 10 ммол) в ацетонитриле (50 мл). Смесь перемешивают при комнатной температуре в течение ночи и упаривают в вакууме. Остаток растворяют в EtOAc, промывают насыщ. раствором NaHCO3 и осадок отделяют фильтрованием. Органический слой сушат (Na2SO4), фильтруют и упаривают. Остаток перекристаллизовывают из смеси EtOAc/эфир/смесь гексанов, получая заглавное соединение (1,68 г, 51%) в виде желтого твердого вещества.
В. N-(2-Хлор-6-метилфенил)-2-[(4,6-диметил-2-пиридинил)амино]-5-тиазолкарбоксамид.
К смеси 319А (25 мг, 0,075 ммол) и 4,6-диметил-2-аминопиридина (37 мг, 0,302 ммол) в ТГФ (1 мл) добавляют 95% гидрида натрия (15 мг). Смесь нагревают при 60°С в течение ночи, охлаждают до комнатной температуры и разбавляют насыщенным водным раствором хлористого аммония. Смесь экстрагируют EtOAc (2х). Органические вытяжки объединяют, промывают водой и сушат (Na2SO4), фильтруют и упаривают. Остаток растирают с эфиром, получают заглавное соединение (17,5 мг, 63%) в виде твердого вещества рыжевато-коричневого цвета.
Пример 320.
Получение N-(2-хлор-6-метилфенил)-2-[(4-этил-2-пиридинил)амино]-5-тиазолкарбоксамида.
Соединение 320 получают аналогично соединению 319В, но берут 4-этил-2-аминопиридин, приводящий к заглавному соединению 320.
Пример 321.
Получение N-(2-хлор-6-метилфенил)-2-[(2,6-диметил-4-пиримидинил)амино]-5-тиазолкарбоксамида.
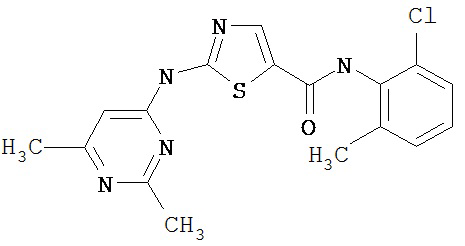
Соединение 321 получают аналогично соединению 319В, но берут 2,6-диметил-4-аминопиримидин, приводящий к заглавному соединению 321.
Пример 322.
Получение N-(2-хлор-6-метилфенил)-2-(3-пиридазиниламино)-5-тиазолкарбоксамида.
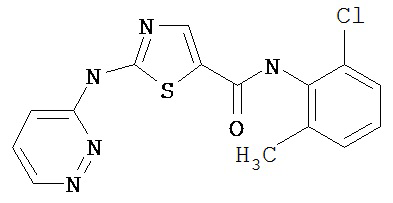
Соединение 322 получают аналогично соединению 319В, но берут 3-аминопиридазин, дающий заглавное соединение 322.
Примеры 323-335.
Общая методика.
Соединения 323-335 получают по описанной ниже методике. К смеси амина 144 (31 мг, 0,11 ммол), соответствующей карбоновой кислоты (0,13 ммол), 1-гидрокси-7-азабензотриазола (19,5 мг, 0,14 ммол) и этил-3-(3-диметиламино)пропилкарбодиимида гидрохлорида (26,8 мг, 0,14 ммол) в ТГФ (0,4 мл) прибавляют диизопропилэтиламин (60 мкл, 0,34 ммол). Смесь нагревают в запаянной трубке под аргоном при 50°С 24 часа. Реакционную смесь разбавляют хлористым метиленом (4 мл) и промывают 1N HCl aq. Раствор в хлористом метилене пропускают через катионообменную колонку Varian Meda Bond Elut SCX, предварительно промытую метанолом и уравновешенную смесью ацетонитрил - метанол (4:1). Колонку элюируют последовательно смесью ацетонитрил - метанол (4:1), метанол - 2М раствор аммиака в метаноле (4:1). Фракции, содержащие продукт, объединяют и упаривают в вакууме. “ВЖЭХ - время уд.” обозначает ВЭЖХ время удерживания в следующих условиях: балластная колонка YMC S5 ODS 4,6×50 мм; 4-минутный градиент, начиная со 100% растворителя А (10% МеОН, 90% Н2О, 0,2% Н3PO4) до 100% растворителя В (90% МеОН, 10% Н2O, 0,2% Н3РО4), скорость потока 4 мл/мин, λ=220 нм.
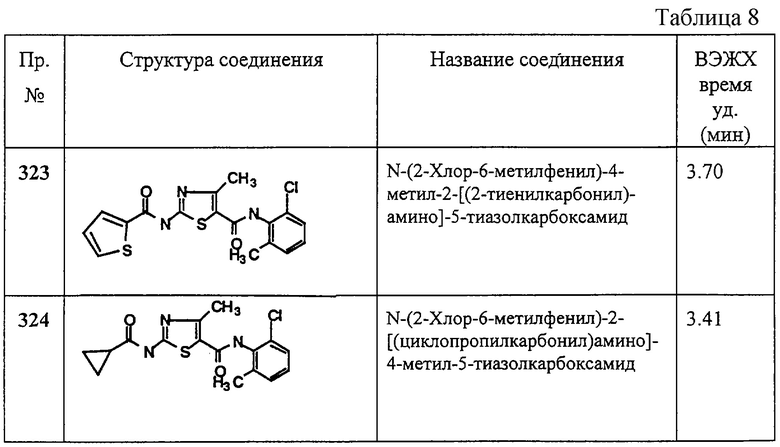
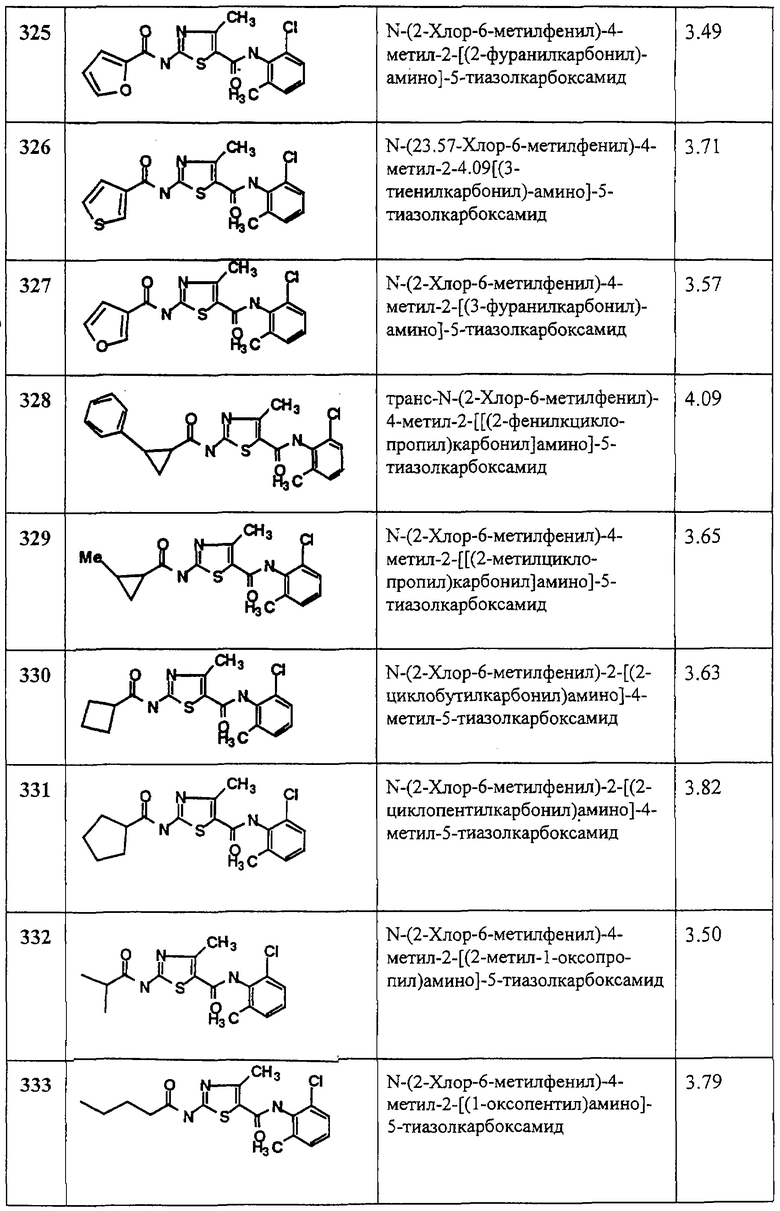
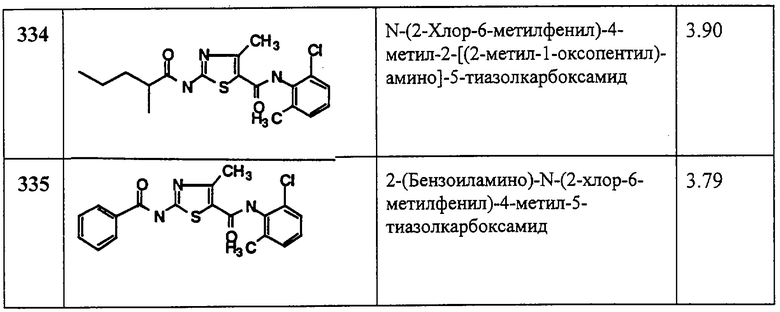
Примеры 336-362.
Общая методика.
Соединения 336-362 получают аналогично соединениям 323-335, но берут 315D вместо 144. Сырые продукты очищают автоматической препаративной ВЭЖХ (условия: колонка YMC S5 ODS А 20х100 мм; 10-минутный градиент, начиная с 10% растворителя В (90% МеОН, 10% H2O, 0,1% TFA) и 90% растворителя А (10% МеОН, 90% H2O, 0,1% TFA) до 100% растворителя В, скорость потока 20 мл/мин, λ=220 нм) и получают заглавные соединения 336-362. “ВЖЭХ - время уд.” обозначает ВЭЖХ время удерживания в следующих условиях: балластная колонка YMC S5 ODS 4,6×50 мм; градиент 4 мин, начиная со 100% растворителя А (10% МеОН, 90% Н2О, 0,2% Н3PO4) до 100% растворителя В (90% МеОН, 10% H2О, 0,2% Н3PO4), скорость потока 4 мл/мин, λ=220 нм.
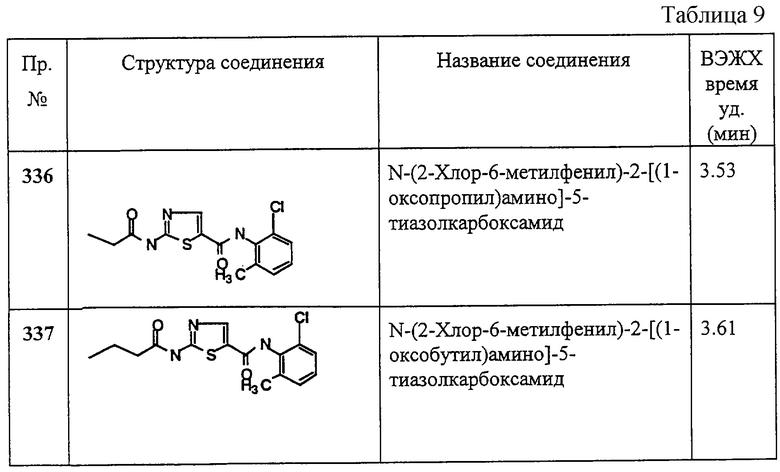
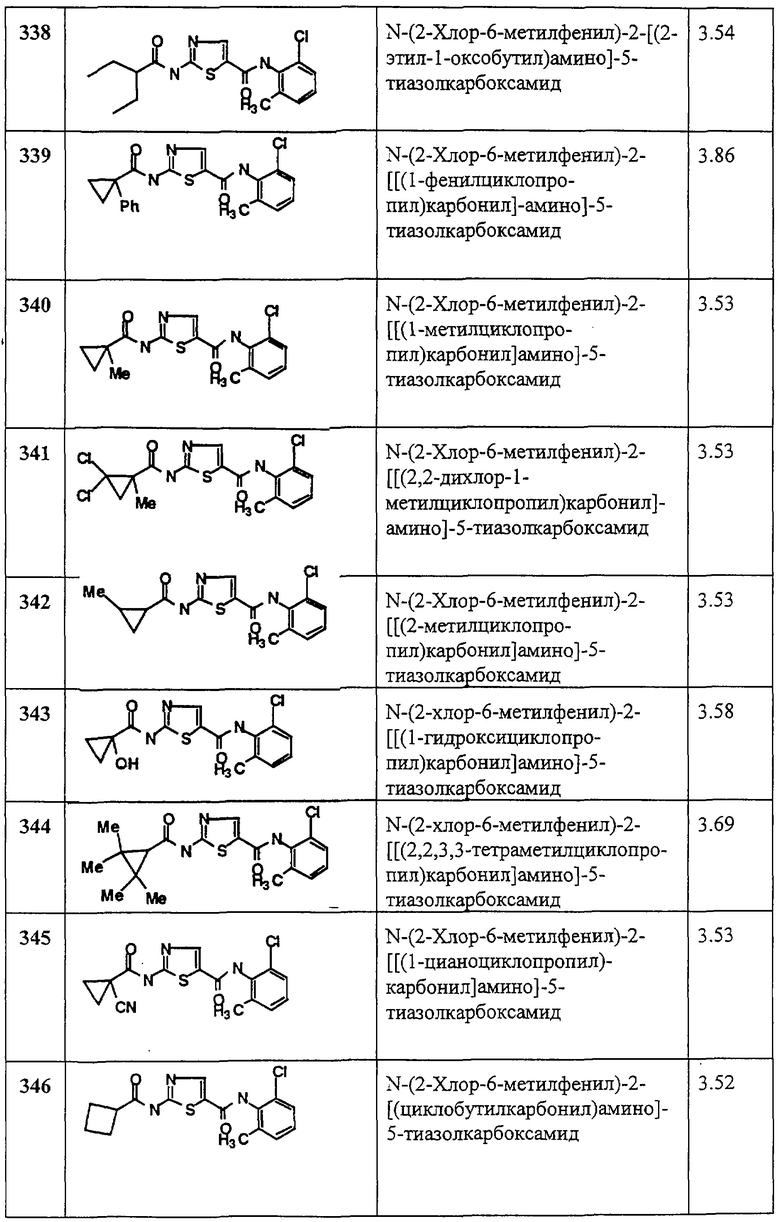
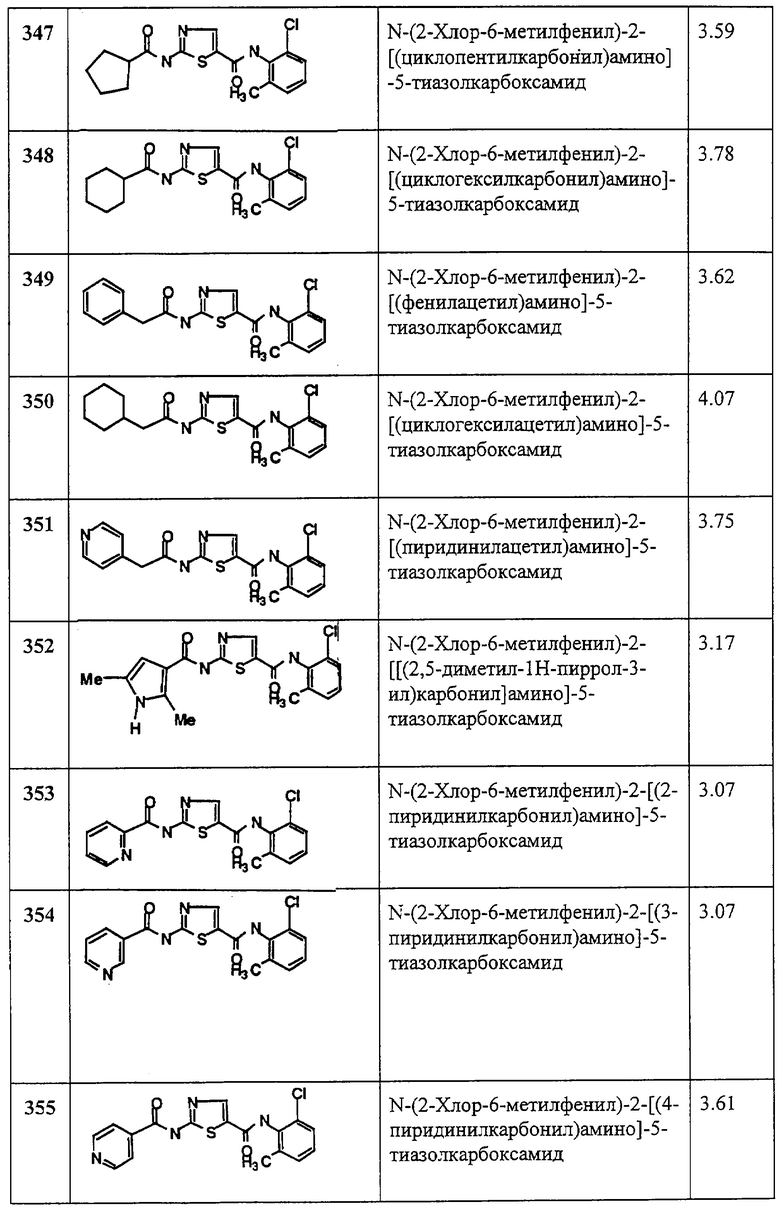
Пример 363.
Получение 2-[(циклопропилкарбонил)амино]-N-(2,6-диметилфенил)-5-тиазолкарбоксамида.
Соединение 363 получают аналогично соединению 315, но берут 2,6-диметиланилин, приводящий к заглавному соединению 363.
Пример 364.
Получение 2-[(циклопропилкарбонил)амино]-N-(2,4,6-триметилфенил)-5-тиазолкарбоксамида.
Соединение 364 получают аналогично соединению 315, но берут 2,4,6-триметиланилин, дающий заглавное соединение 364.
Пример 365.
Получение N-(2-хлор-4,6-диметилфенил)-2-[(циклопропилкарбонил)амино]-5-тиазолкарбоксамида.
Соединение 365 получают аналогично соединению 315, но для получения заглавного соединения 365 берут 2-хлор-4,6-диметиланилин.
Пример 366.
Получение 1,1-диметилэтилового эфира (4-[2-оксо-2-[(2,4,6-триметилфенил)амино]этил]-2-тиазолил]карбаминовой кислоты.
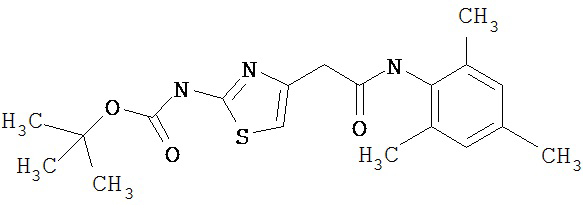
Соединение 366 получают аналогично соединению 1, но берут 2-трет-бутоксикарбониламинотиазол-4-уксусную кислоту, получая заглавное соединение 366 в виде белого твердого вещества.
Пример 367.
Получение 2-амино-N-(2,4,6-триметилфенил)-4-тиазолацетамида.
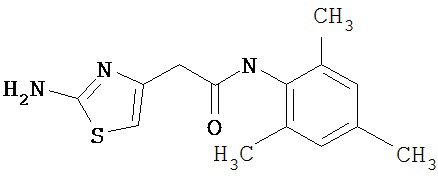
Соединение 367 получают аналогично соединению 4, но берут 365, получая заглавное соединение 367 в виде белого твердого вещества.
Пример 368.
Получение 2-метил-5-нитро-N-(2,4,6-триметилфенил)бензамида.
Соединение 368 получают аналогично соединению 3, но берут 2-метил-5-нитробензойную кислоту, получая заглавное соединение 368 в виде белого твердого вещества.
Пример 369.
Получение 5-амино-2-метил-N-(2,4,6-триметилфенил)бензамида.
К раствору 368 (149 мг, 0,5 ммол) в EtOAc (50 мл) при перемешивании добавляют 10% палладий на угле (30 мг). Реакционная колба соединена с водородным баллоном через трехходовой кран. Воздух из колбы откачивают в вакууме и заполняют ее водородом из баллона. Через 4 часа катализатор фильтруют, промывают EtOAc (5 мл, 5х). Фильтрат упаривают, получая заглавное соединение (133 мг, 99%) в виде белого твердого вещества.
Пример 370.
Получение 2-амино-5-хлор-N-(2,4,6-триметилфенил)-4-пиримидинкарбоксамида.
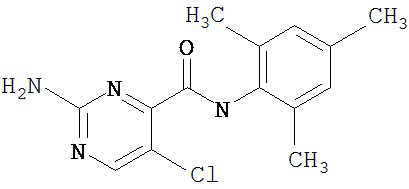
Соединение 370 получают аналогично соединению 3, но берут 2-амино-5-хлорпиримидин-4-карбоновую кислоту, получая заглавное соединение 370 в виде белого твердого вещества.
Пример 371.
Получение 1,1-диметилэтилового эфира [4-метил-5-[[(2,4,6-триметилфенил)амино]карбонил]-2-оксазолил]карбаминовой кислоты.
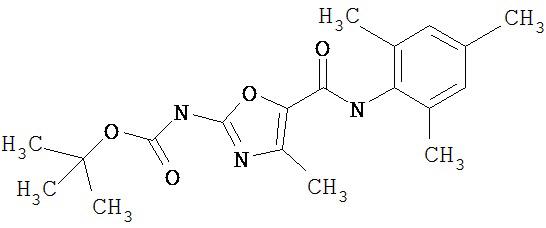
Соединение 371 получают аналогично соединению 1, но берут 2-трет-бутоксикарбонилоксиамино-4-метил-5-оксазолкарбоновую кислоту, получая заглавное соединение 371 в виде легкой белой пены.
Пример 372.
Получение 2-амино-4-(метил)-N-(2,4,6-триметилфенил)-5-оксазолкарбоксамида, трифторацетата (1:1).
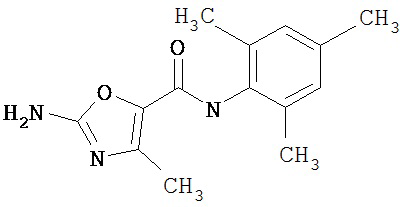
Соединение 372 получают аналогично соединению 4, но берут соединение 369, получая
заглавное соединение 372 в виде белого твердого вещества.
Пример 373.
Получение 2-амино-N-(2,4,6-триметилфенил)-5-пиридинкарбоксамида.
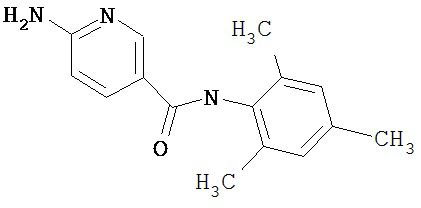
Соединение 373 получают аналогично соединению 3, но используют 6-аминоникотиновую кислоту, получая заглавное соединение 373 в виде белого твердого вещества.
Пример 374.
Получение 3-амино-N-(2,4,6-триметилфенил)-4-пиридинкарбоксамида.
Соединение 374 получают аналогично соединению 3, но используют 3-амино-4-пиридинкарбоновую кислоту, получая заглавное соединение 374 в виде белого твердого вещества.
Пример 375.
Получение 2-амино-4-метил-N-(2,4,6-триметилфенил)-5-пиримидинкарбоксамида.
Соединение 375 получают аналогично соединению 3, но используя 2-амино-4-метил-5-пиримидинкарбоновую кислоту, получают заглавное соединение 375 в виде белого твердого вещества.
Пример 376.
Получение N-(2-хлор-6-метилфенил)-2-[(4-метил-2-пиридинил)амино]-5-тиазолкарбоксамида.
Соединение 376 получают аналогично соединению 319В, но используя 2-амино-4-метилпиридин, и получают заглавное соединение 376 в виде грязно-белого твердого вещества.
Пример 377.
Получение 2-[(6-амино-2-пиридинил)амино]-N-(2-хлор-6-метилфенил)-5-тиазолкарбоксамида.
Соединение 377 получают аналогично соединению 319В, но используя 2,6-диаминопиридин, и получают заглавное соединение 377 в виде легкого коричневого твердого вещества.
Пример 378.
Получение N-(2-хлор-6-метилфенил)-2-[(6-пропил-2-пиридинил)амино]-5-тиазолкарбоксамида.
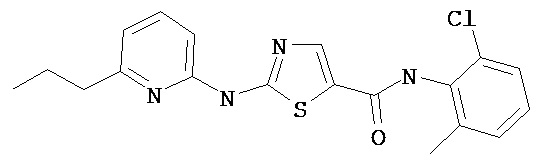
Соединение 378 получают аналогично соединению 319В, но используя 2-амино-6-пропилпиридин, получая заглавное соединение 378 в виде твердого вещества грязно-белого цвета.
Пример 379.
Получение N-(2-хлор-6-метилфенил)-2-[(6-этил-4-пиримидинил)амино]-5-тиазолкарбоксамида.
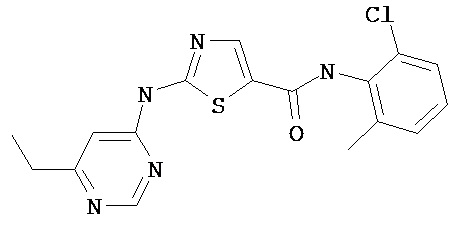
Соединение 379 получают по аналогии с соединением 319В, но используя 2-амино-6-этилпиримидин, получают заглавное соединение 379 в виде твердого вещества белого цвета.
Примеры 380-409.
Общая методика.
Соединения 380-409 получают аналогично соединению 319В. Для нижеследующих примеров 380-527 “ВЖЭХ -время уд.” обозначает время удерживания ВЭЖХ в следующих условиях: балластная колонка YMC S5 ODS 4,6×50 мм; градиент 4 мин, от 100% растворителя А (10% МеОН, 90% Н2О, 0,2% Н3PO4) до 100% растворителя В (90% МеОН, 10% Н2O, 0,2% Н3PO4), скорость потока 4 мл/мин, λ=220 нм. Если употребляется “ВЖЭХ - время уд. В”, это означает ВЭЖХ время удерживания при следующих условиях: колонка YMC S5 ODS 4,6×33 мм Turbo; 2-минутный градиент от 100% растворителя А (10% МеОН, 90% Н2O, 0,1% TFA) до 100% растворителя В (90% МеОН, 10% H2O, 0,1% TFA) при 1 минуте при 100% растворителя В, скорость потока 4 мл/мин, λ=220 нм.
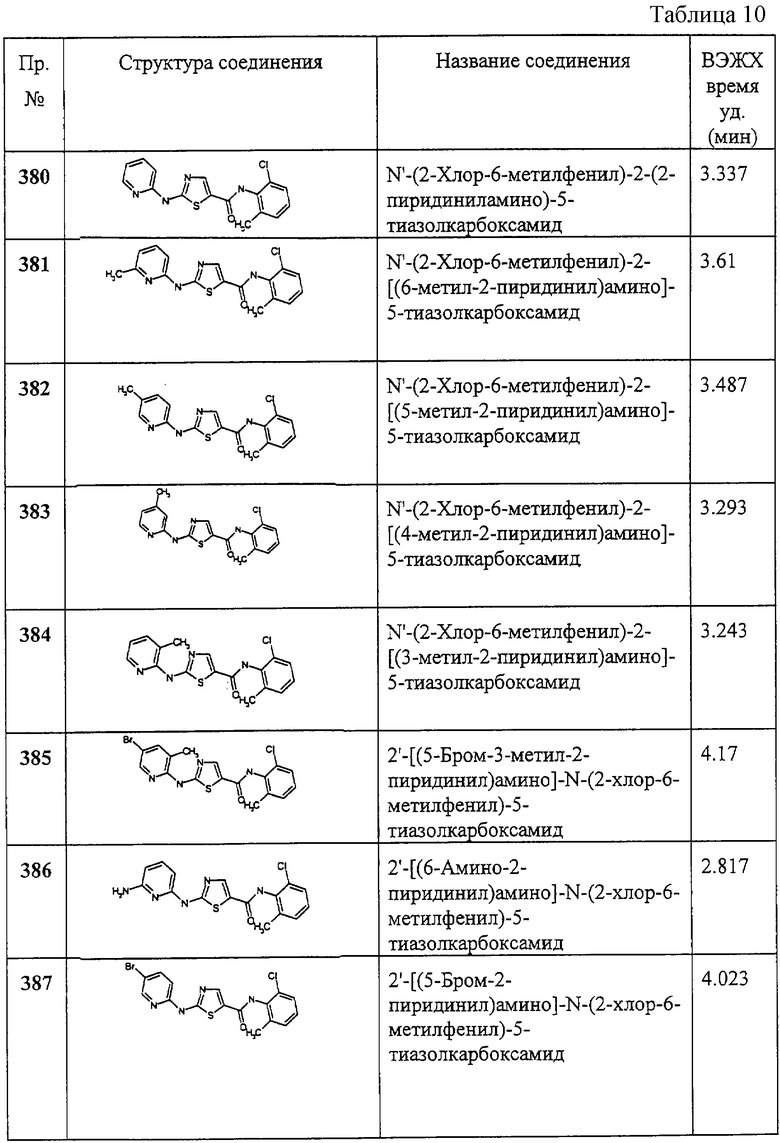
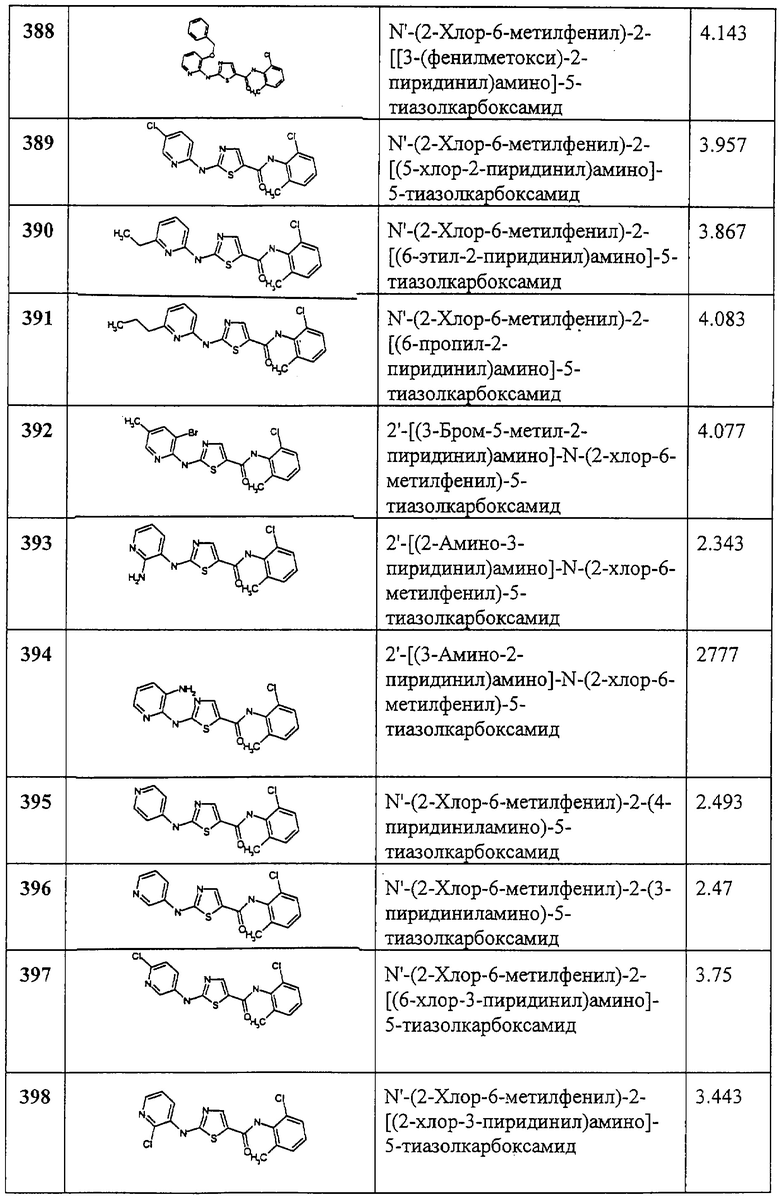
Пример 410.
Получение N'-(2,6-диметилфенил)-2-(фениламино)-5-тиазолкарбоксамида.
A. 1,1-Диметилэтиловый эфир [5-[[(2,6-диметилфенил)амино]карбонил]-2-тиазолил]карбаминовой кислоты.
Соединение 410А получают аналогично соединению 315С, но используют 2,6-диметиланилин.
В. 2-Амино-N-(2,6-диметилфенил)-5-тиазолкарбоксамид.
Соединение 410В получают аналогично соединению 315D, но используют соединение 410А.
С. Заглавное соединение.
Заглавное соединение получают аналогично соединению 319В, но используют соединение 410В и анилин. ВЭЖХ - время удерживания 3,69 мин.
Примеры 411-427.
Общая методика.
Соединения 411-427 получают аналогично соединению 319В.
Пример 428. Получение 2'-(2-пиридиниламино)-N-(2,4,6-триметилфенил)-5-тиазолкарбоксамида.
А. 1,1-Диметилэтиловый эфир [5-[[(2,4,6-триметилфенил)амино]карбонил1-2-тиазолил]карбаминовой кислоты.
Соединение 428А получают по аналогии с соединением 315С, но используют 2,4,6-триметиланилин.
В. 2-Амино-N-(2,6-диметилфенил)-5-тиазолкарбоксамид.
Соединение 428В получают аналогично соединению 315D, но используют соединение 428А и 2-аминопиридин. Время удерживания при ВЭЖХ 3,66 мин.
Примеры 429-443.
Общая методика.
Соединения 429-443 получают аналогично соединению 319В.
Пример 444.
Получение N'-(2-хлор-6-метилфенил)-2-[[2-метил-6-[[2-(4-морфолинил)этил]амино]-4-пиримидинил]амино]-5-тиазолкарбоксамида.
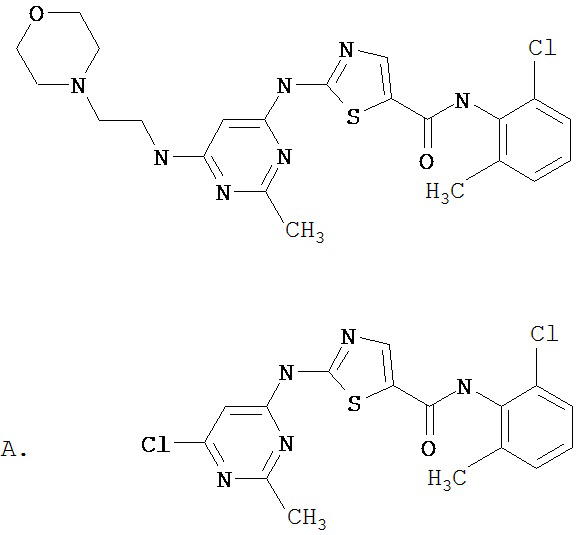
К суспензии NaH (148 мг, 6,17 ммол) в ТГФ (20 мл) добавляют раствор соединения 315D (551 мг, 2,06 ммол) в ТГФ (10 мл) и перемешивают при комнатной температуре 0,5 час.
Раствор 4,6-дихлор-2-метилпиримидина (671,6 мг, 4,12 ммол) в ТГФ (10 мл) перемешивают при комнатной температуре в течение ночи. К реакционной смеси добавляют уксусную кислоту и растворитель удаляют в вакууме. К остатку добавляют воду и нас. раствор NaHCO3 и экстрагируют CH2Cl2. Органический слой удаляют в вакууме и сырой материал очищают хроматографией на колонке, получая 444А (494 мг).
В. Заглавное соединение.
К соединению 444А (30 мг) добавляют N-(2-аминоэтил)морфолин (300 мкл) и смесь нагревают при 80°С в течение 2 час. Воду добавляют к реакционной смеси и продукт собирают фильтрованием. ВЭЖХ - время удерживания 2,357 мин.
Примеры 445-461.
Общая методика.
Соединения 445-461 получают аналогично 444В заменой на соответствующий амин.
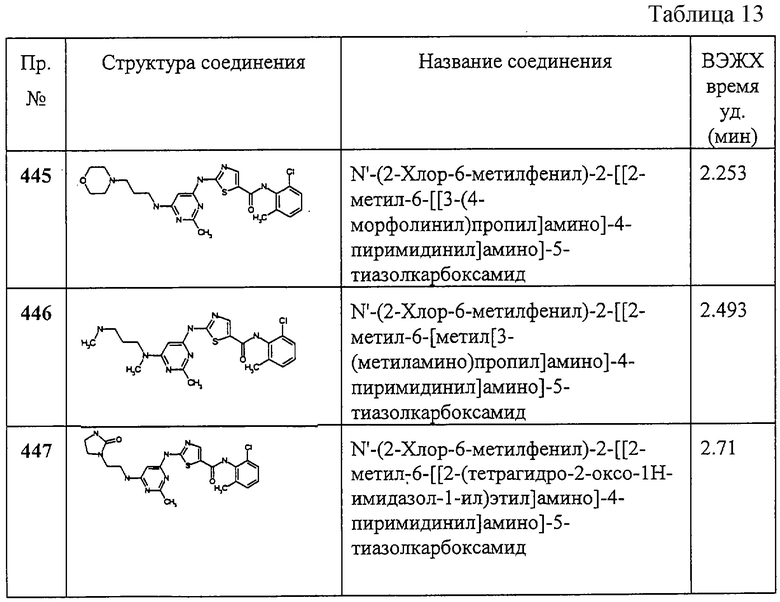
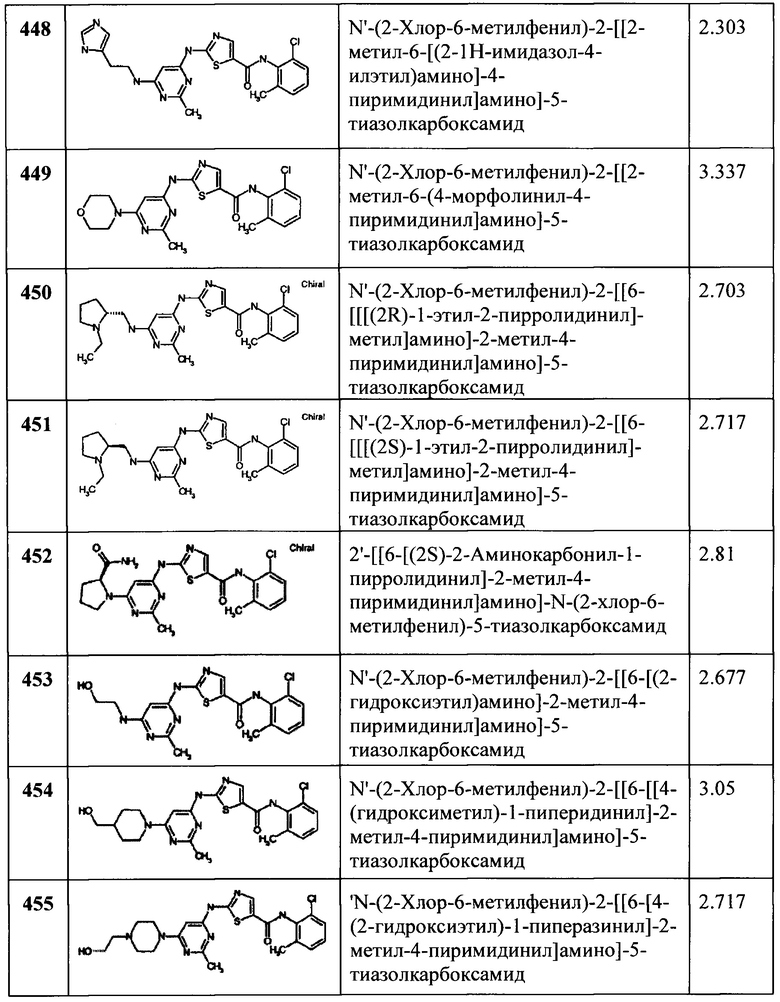
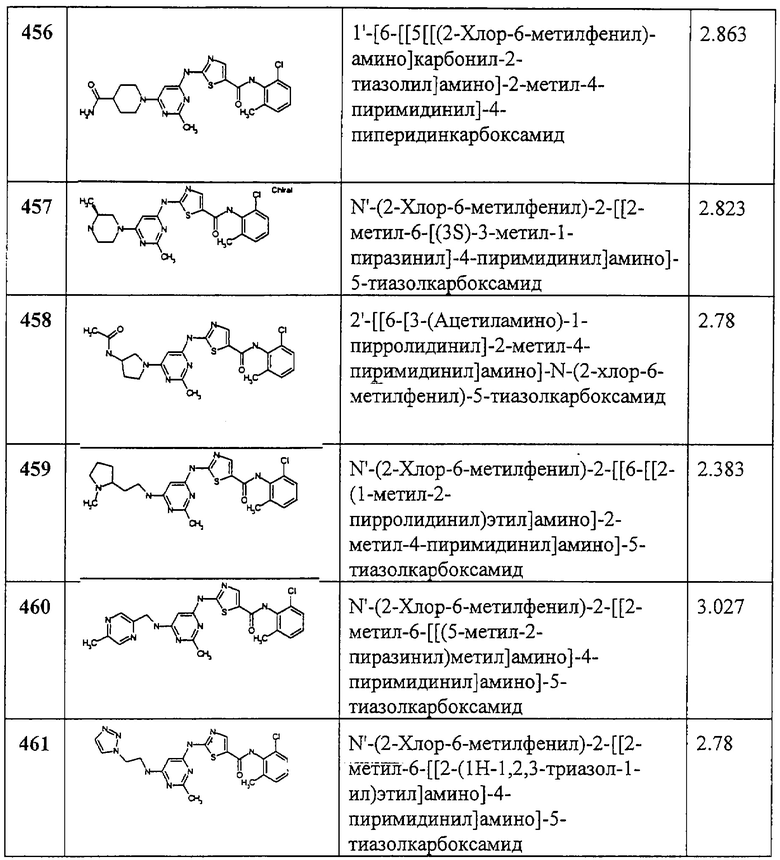
Пример 462.
Получение N'-(2-хлор-6-метилфенил)-2-[[6-[[2-(4-морфолинил)этил]амино]-4-пиримидинил]амино]-5-тиазолкарбоксамида.

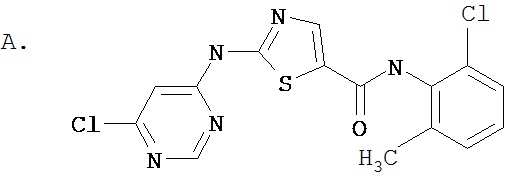
Соединение 462А получают аналогично 444А, но берут 4,6-дихлорпиримидин.
В. Заглавное соединение.
Заглавное соединение получают аналогично 444В, но вместо соединения 444А берут
соединение 462А. Время удерживания при ВЭЖХ 2,553 мин.
Примеры 463-472.
Общая методика.
Соединения 463-472 получают аналогично 444В, заменяя на соответствующий амин. “ВЖЭХ - время уд. В”, обозначает ВЭЖХ - время удерживания при следующих условиях: колонка YMC S5 ODS 4,6×33 мм Turbo; 2-минутный градиент, начиная от 100% растворителя А (10% МеОН, 90% H2O, 0,1% TFA) до 100% растворителя В (90% МеОН, 10% Н2О, 0,1% TFA) при 1 минуте при 100% растворителя В, скорость потока 4 мл/мин, λ=220 нм.
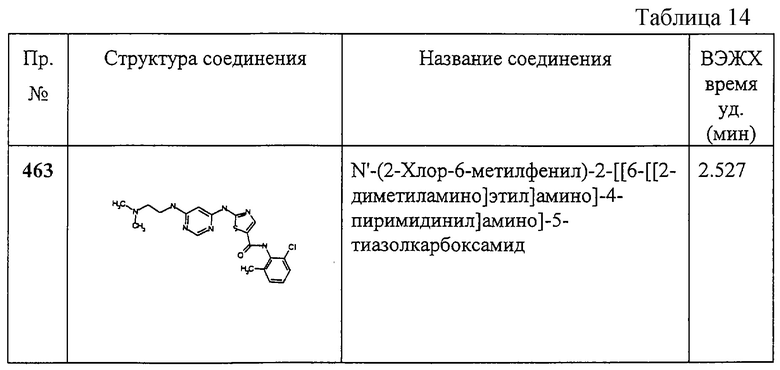
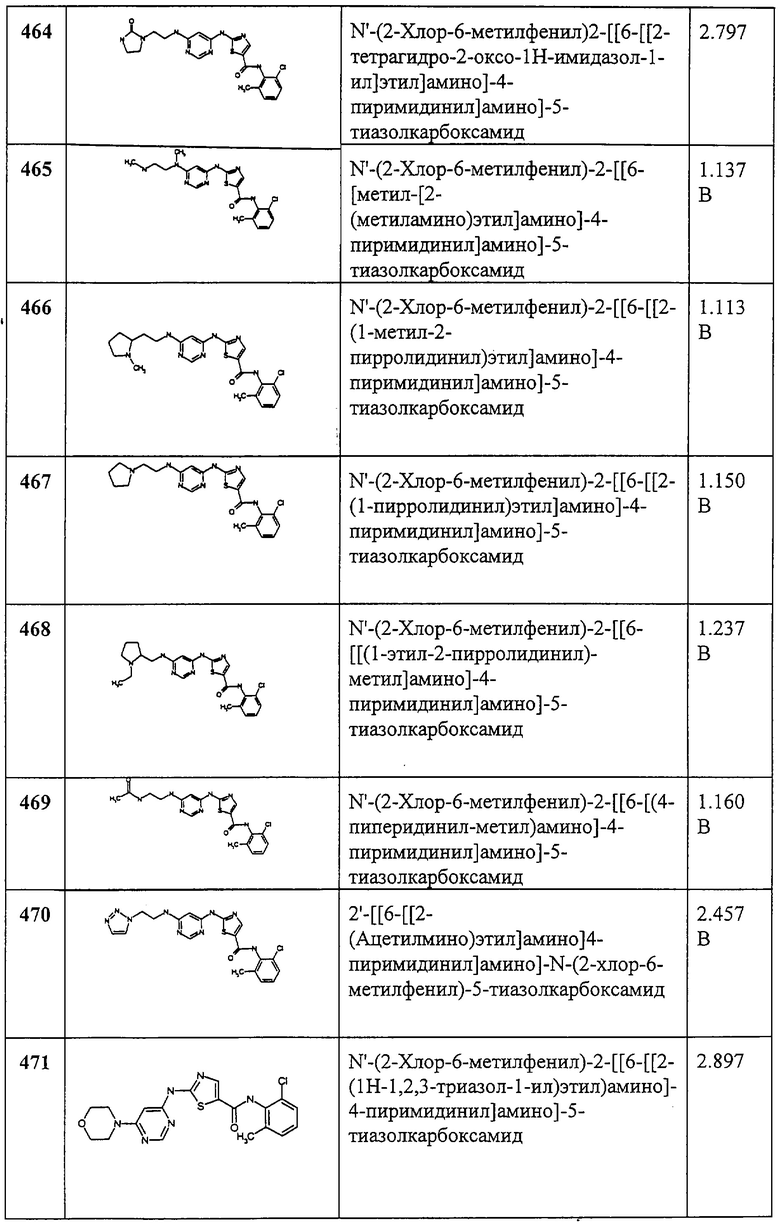
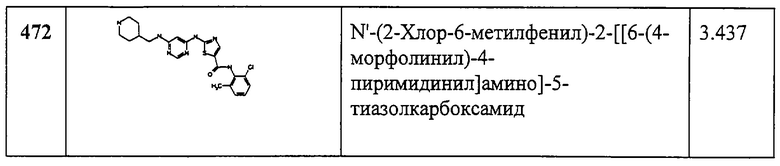
Пример 473.
Получение N'-(2-хлор-6-метилфенил)-2-[[6-[[2-(4-морфолинил)этил]амино]-2-пиридинил]амино]-5-тиазолкарбоксамида.
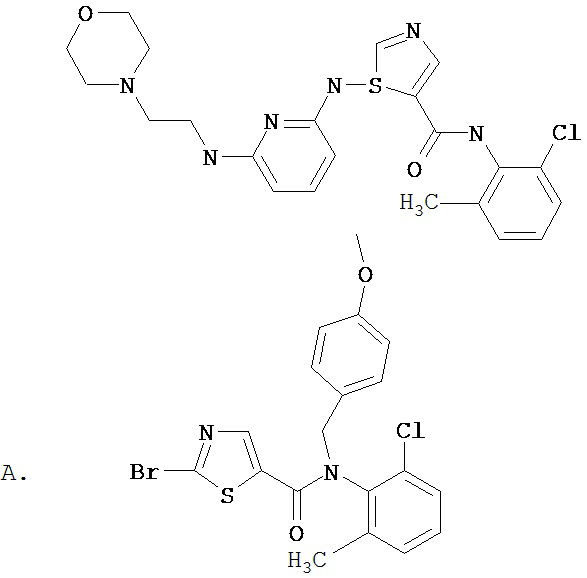
К суспензии NaH (2,83 г, 118 ммол) в ДМФА (350 мл), охлажденной до 0°С, добавляют соединение 319А (31 г, 93,5 ммол). Смесь перемешивают 45 минут при 0°С, затем добавляют Bu4NJ (6,9 г, 18,4 ммол) и далее добавляют 4-метоксибензилхлорид (18 г, 115 ммол). Реакционную смесь доводят до комнатной температуры. После перемешивания в течение ночи при комнатной температуре к реакционной смеси медленно прибавляют уксусную кислоту, затем растворитель удаляют в вакууме. К остатку добавляют воду и нейтрализуют насыщенным водным раствором NaHCO3. Смесь экстрагируют 3 раза EtOAc и объединенные вытяжки (органический слой) промывают водой, а затем насыщенным раствором NaCl. Вытяжки EtOAc упаривают в вакууме и остаток очищают колоночной хроматографией, получая 473А (35 г).
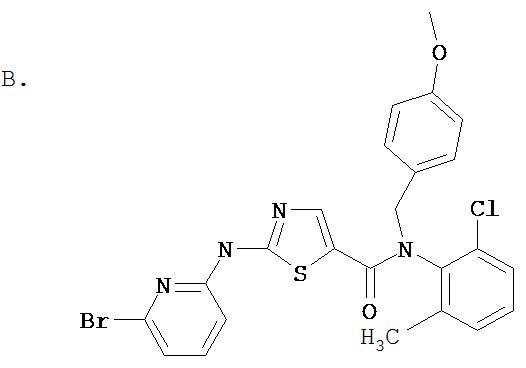
К соединению 473А (0,5 г, 1,1 ммол), растворенному в ТГФ (50 мл), постепенно добавляют NaH (0,13 г, 5,5 ммол), а затем 2-бром-6-аминопиридин (0,76 г, 4,4 ммол). Реакционную смесь нагревают при кипении 2 часа, затем охлаждают до комнатной температуры и “гасят” уксусной кислотой. Растворитель удаляют в вакууме, затем добавляют воду и гексан и перемешивают при комнатной температуре. Твердый осадок собирают фильтрованием и промывают водой и Et2O, получают 473В (0,48 г).
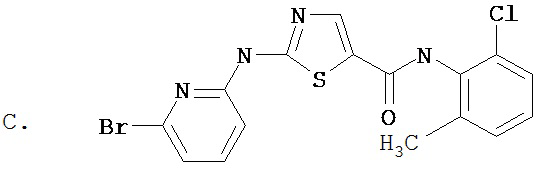
К соединению 473В (0,48 г), растворенному в TFA (5 мл), добавляют анизол (2 мл), а затем трифторметилсульфокислоту (triflic acid) (1 мл). Реакционную смесь перемешивают при комнатной температуре 3 часа, затем постепенно добавляют к быстро перемешиваемой смеси льда, насыщ. NaHCO3, Et2O и CH2Cl2. Смесь перемешивают на холоде 1 час, затем отфильтровывают (твердый) осадок и промывают водой и смесью Et2O/CH2Cl2, получая 473С (0,344 г). Время уд. ВЭЖХ 3,85 мин.
D. Заглавное соединение.
Заглавное соединение получают аналогично 444В, но вместо соединения 444А берут
соединение 473С. ВЭЖХ время уд. 2,80 мин.
Примеры 474-480.
Общая методика.
Соединения 474-480 получают аналогично 443D, заменяя на соответствующий амин.
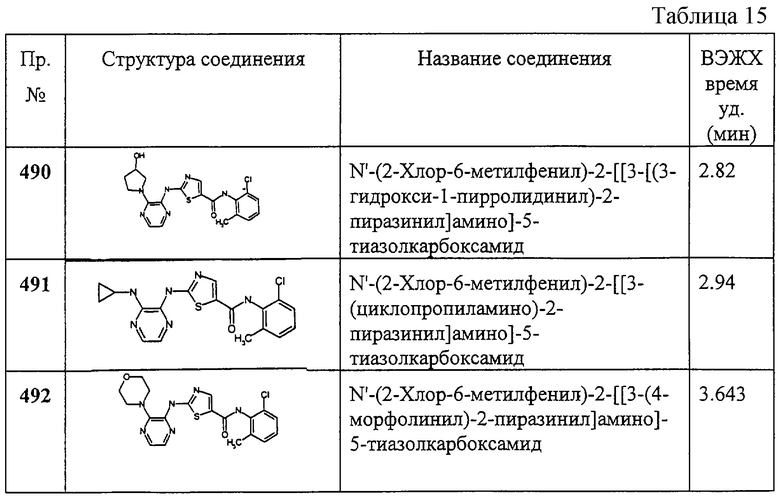
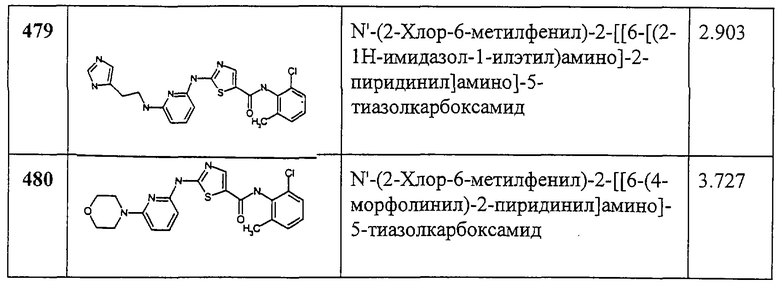
Пример 481.
Получение N'-(2-хлор-6-метилфенил)-2-[[6-[[2-(4-морфолинил)этил]амино]-2-пиразинил]амино]-5-тиазолкарбоксамида.
Соединение 481А получают аналогично 473В, но вместо 2-бром-6-аминопиридина используют 2-хлор-6-аминопиразин.
В. Альтернативный синтез соединения 406.
Соединение 406 получают аналогично 473С, но вместо соединения 473В используют соединение 481А.
С. Заглавное соединение.
Заглавное соединение получают аналогично 444В, но вместо соединения 444А
используют соединение 406. ВЭЖХ - время уд. 2,69 мин.
Примеры 482-486.
Общая методика.
Соединения 482-486 получают аналогично 481С, заменяя на соответствующий амин.
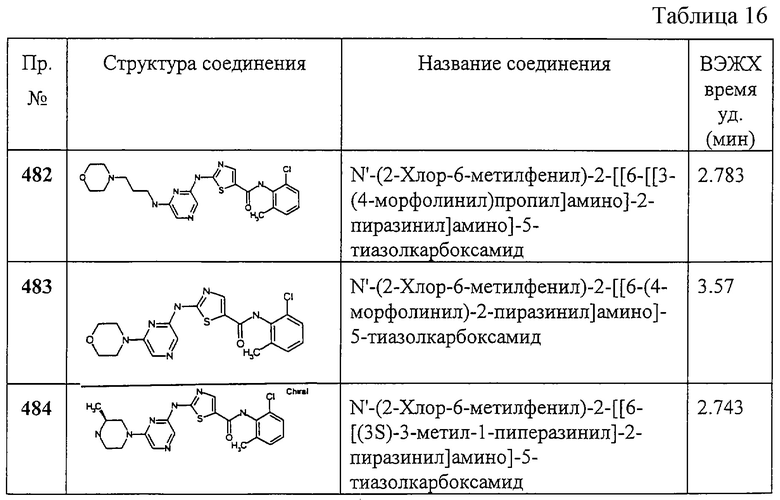
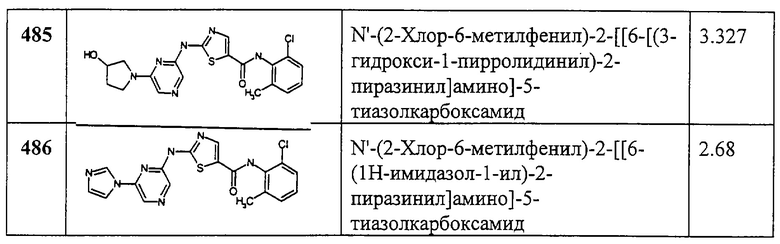
Пример 487.
Получение N'-(2-хлор-6-метилфенил)-2-[[6-(3-гидрокси-1-пирролидинил)-3-пиридазинил]амино]-5-тиазолкарбоксамида.
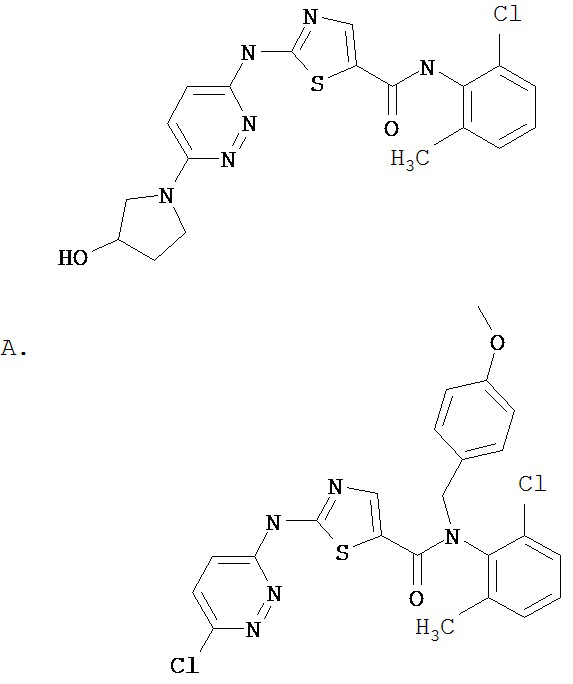
Соединение 487А получают аналогично 473В, но вместо 2-бром-6-аминопиридина используют 2-бром-6-аминопиразин.
Соединение 487В получают аналогично 473С, но вместо соединения 473В используют соединение 487А.
С. Заглавное соединение.
Заглавное соединение получают аналогично 444В, но вместо соединения 444А используют соединение 487В и 3-гидроксипирролидин вместо N-(2-аминоэтил)морфолина. ВЭЖХ - время уд. 2,493 мин.
Пример 488.
Получение N'-(2-хлор-6-метилфенил)-2-[[6-(1Н-имидазол-1-ил)-3-пиридазинил]амино]-5-тиазолкарбоксамида.
Соединение 488 получают аналогично 487С, но вместо 3-гидроксипирролидина используют имидазол. ВЭЖХ - время уд. 2,61 мин.
Пример 489.
Получение N'-(2-хлор-6-метилфенил)-2-[[3-(метиламино)-2-пиразинил]амино]-5-тиазолкарбоксамида.
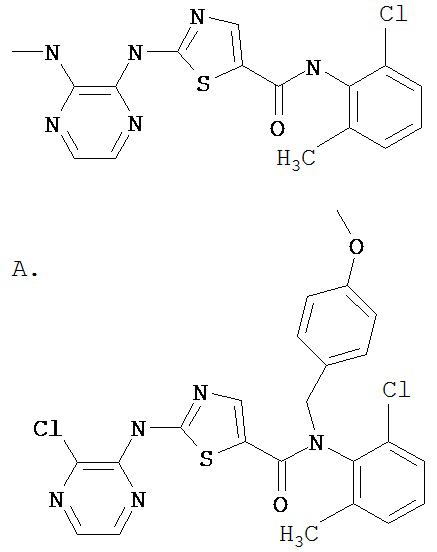
А. Соединение 489А получают аналогично 473В, но вместо 2-бром-6-аминопиридина берут 2-хлор-3-аминопиразин.
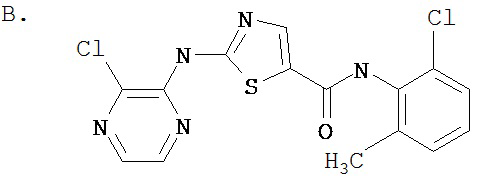
Соединение 489В получают аналогично 473С, но вместо соединения 473В используют
соединение 489А.
С. Заглавное соединение.
Заглавное соединение получают аналогично 444В, но вместо соединения 444А используют соединение 489В и вместо N-(2-аминоэтал)морфолина используют метиламин. ВЭЖХ - время уд. 2,81 мин.
Примеры 490-494.
Общая методика.
Соединения 490-494 получают аналогично 489С, но берут соответствующий амин.
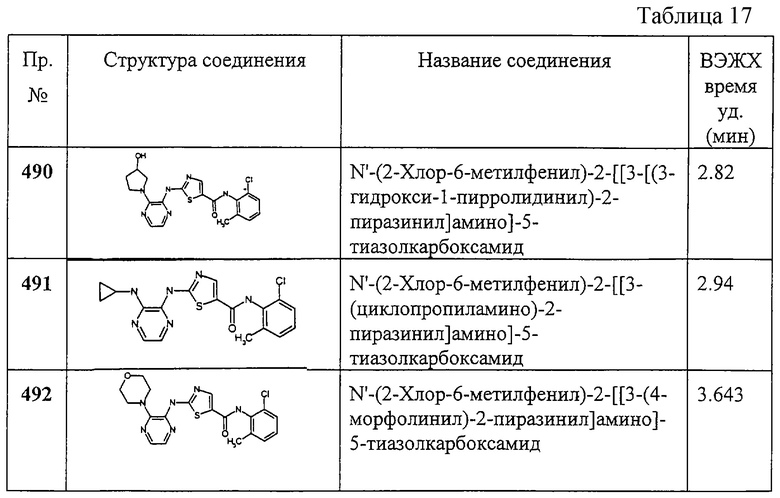
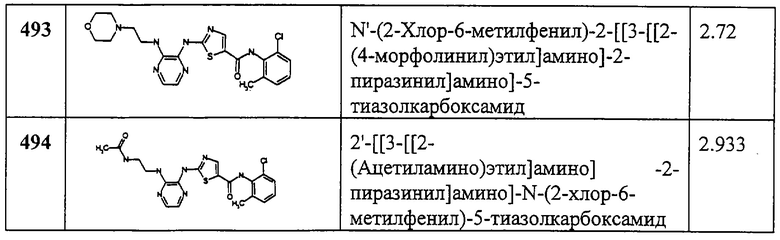
Пример 495.
Получение N'-(2-хлор-6-метилфенил)-2-(циклогексиламино)-5-тиазолкарбоксамида.
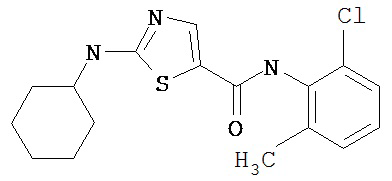
Соединение 495 получают аналогично 444В, но вместо соединения 444А используют соединение 319А и вместо N-(2-аминоэтил)морфолина берут циклогексиламин. ВЭЖХ - время уд. 3,547 мин.
Примеры 496-500.
Общая методика.
Соединения 496-500 получают аналогично 495, но берут соответствующий амин.
Таблица 17а
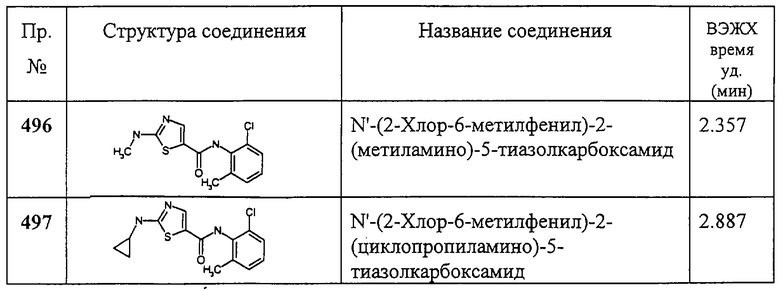
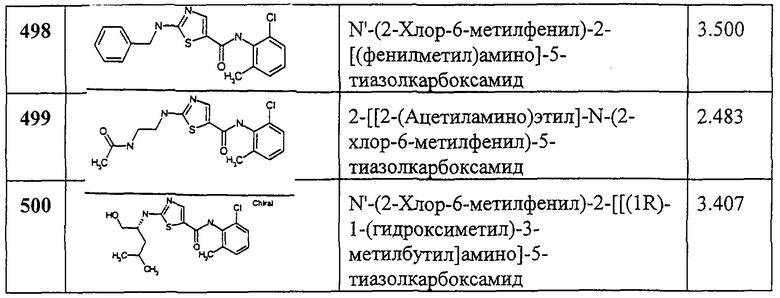
Пример 501.
Получение N'-(2-хлор-6-метилфенил)-2-[[6-(метоксиметил)-4-пиримидинил]амино]-5-тиазолкарбоксамида.
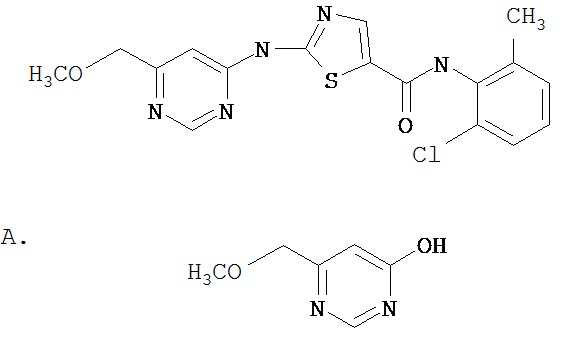
К смеси метил-4-метоксиацетоацетата (метилового эфира 4-метоксиацетоуксусной кислоты) (14,6 г,0,1 мол) и солянокислой соли формамидина (16,1 г, 0,2 мол) в 70 мл сухого МеОН порциями добавляют 25% раствор метоксида натрия (70 мл, 0,3 мол) в МеОН. Сразу же образуется белый осадок. Реакционную смесь перемешивают при комнатной температуре 1,0 час. Добавляют уксусную кислоту (28,6 мл, 0,5 мол) и реакционную смесь упаривают в вакууме. К остатку добавляют воду и смесь (сверх) перенасыщают NaCl и экстрагируют EtOAc (х5). Объединенные экстракты сушат над безводным Na2SO4 и упаривают в вакууме, получая соединение 501А в виде желтого твердого вещества.
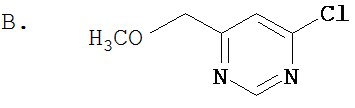
Смесь соединения 501А (5,3 г, 37,8 ммол) и POCl3 (40 мл) нагревают при кипении 2 часа. Упаривают в вакууме и остаток выливают в смесь лед-CH2Cl2. Доводят рН до 6,5-7 концентрированным NH4OH. Смесь экстрагируют СН2Cl2 (×3) и объединенные экстракты сушат над Na2SO4. Упаривание в вакууме с последующей флеш-хроматографией (CH2Cl2 - EtOAc 9:1) на силикагеле дает 5,33 г соединения 501В в виде светло-желтого масла.
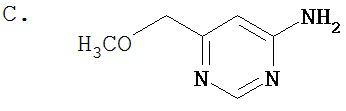
Смесь соединения 501В (3,2 г, 20 ммол) и NH4OH (50 мл) нагревают при 85°С в трубке под давлением 3,0 часа. По охлаждении до комнатной температуры реакционную смесь упаривают в вакууме и остаток растирают с эфиром, получая 2,81 г соединения 501С в виде светло-желтого твердого вещества.
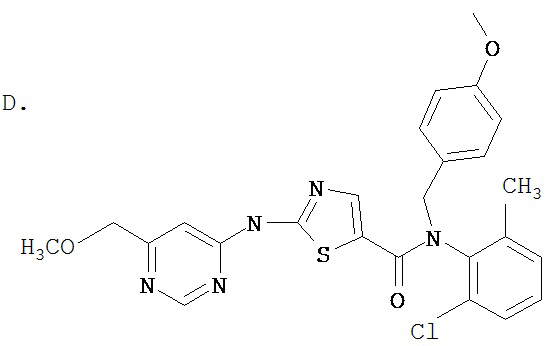
Соединение 501D получают из соединения 501С аналогично соединению 473В.
Е. Заглавное соединение.
Заглавное соединение получают из соединения 501D аналогично соединению 473С.
Время уд. при ВЭЖХ 3,25 мин.
Пример 502.
Получение N'-(2-хлор-6-метилфенил)-2-[[6-(гидроксиметил)-4-пиримидинил]амино]-5-тиазолкарбоксамида.
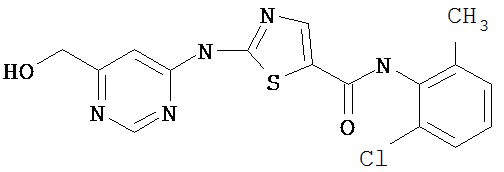
К раствору соединения 501 (56 мг, 0,144 ммол) в сухом СН2Cl2 (3,0 мл), охлажденному до 0°С, добавляют чистый BBr3 (0,054 мл, 0,574 ммол). Смесь перемешивают 1 час при комнатной температуре. Медленно и осторожно при 0°С добавляют МеОН и образующуюся смесь упаривают в вакууме. К остатку добавляют воду и доводят рН до 7 нас.NaHCO3. Белый осадок отфильтровывают, промывают водой/эфиром и сушат в высоком вакууме, получают 52 мг соединения 502 в виде грязно-белого твердого вещества. ВЭЖХ - время удерживания 2,84 мин.
Пример 503.
Получение N'-(2-хлор-6-метилфенил)-2-[[6-(4-морфолинилметил)-4-пиримидинил] амино]-5-тиазолкарбоксамида.
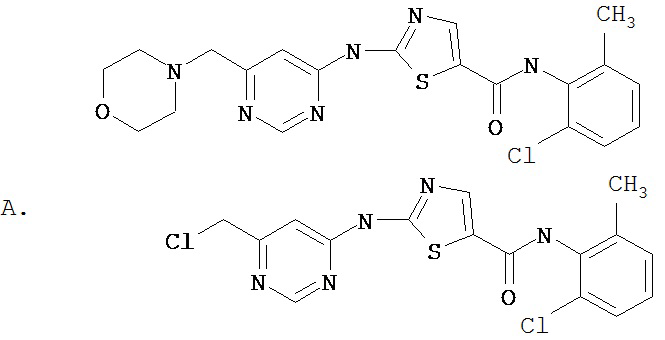
К суспензии соединения 502 (44,2 мг, 0,118 ммол) в 0,5. мл сух. СН2Cl2 добавляют тионилхлорид (0,086 мл, 1,18 ммол). Реакционную смесь перемешивают 5,0 часов. Упаривают в вакууме и остаток после азеотропного упаривания с CH2Cl2 дает 56 мг соединение 503 в виде желтого твердого вещества.
В. Заглавное соединение.
Смесь соединения 503А (20 мг), морфолина (0,014 мл) и диизопропилэтиламина (0,09 мл) в 0,5 мл сухого диоксана нагревают при 85°С в течение 4 час. Упаривание в вакууме с последующей флеш-хроматографией (CH2Cl2-MeOH-NH4OH 95:5:0,5) на силикагеле дает 15 мг заглавного соединения в виде грязно-белого твердого вещества. Время удерживания ВЭЖХ 2,52 мин.
Примеры 504-513.
Общая методика.
Соединения 504-513 получают из соединения 503А аналогично соединению 503. Эти соединения имеют следующее строение.
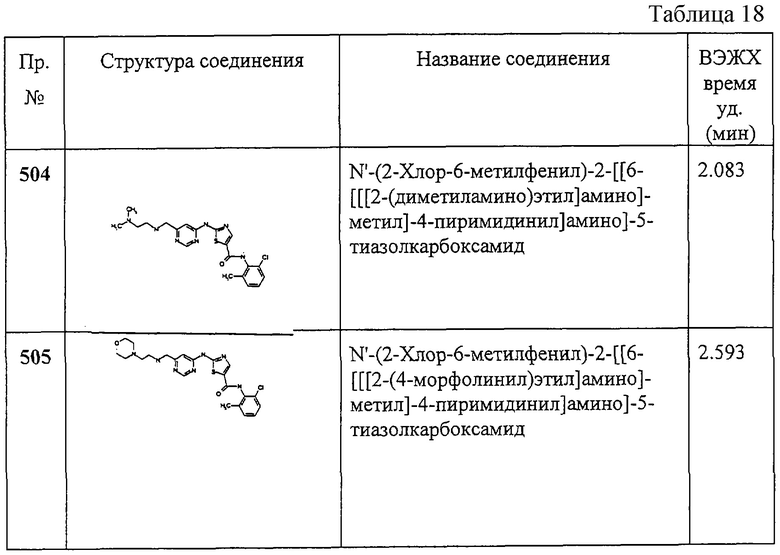
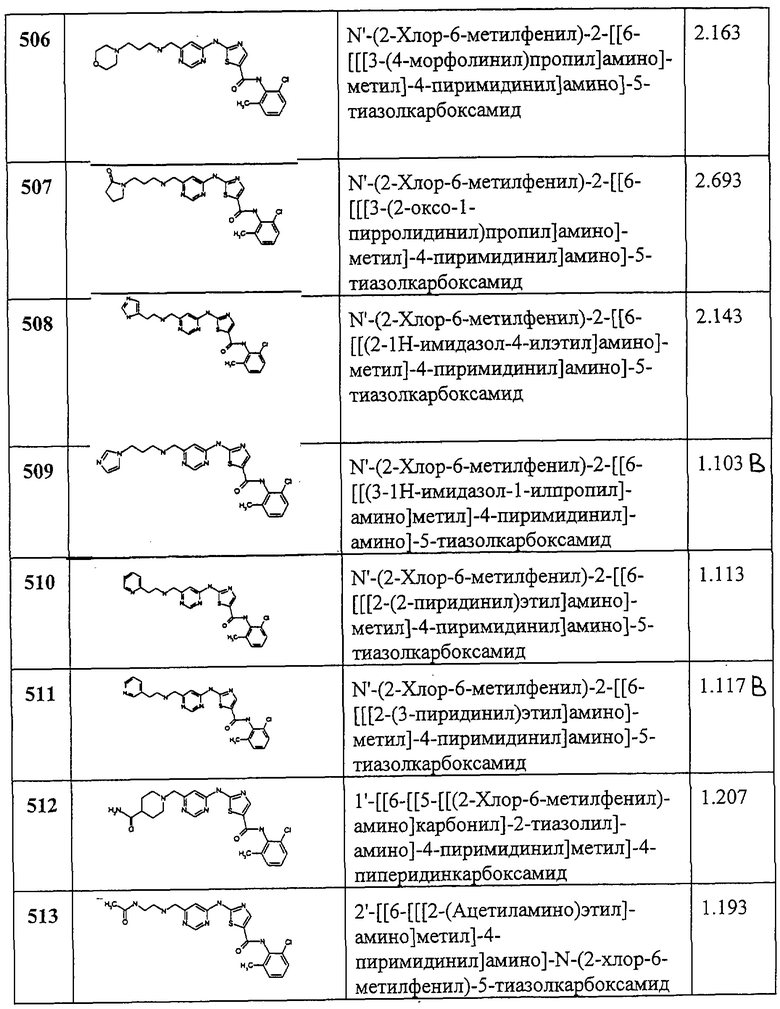
Пример 514.
Получение N'-(2-хлор-6-метилфенил)-2-(2-нафталениламино)-5-тиазолкарбоксамида.
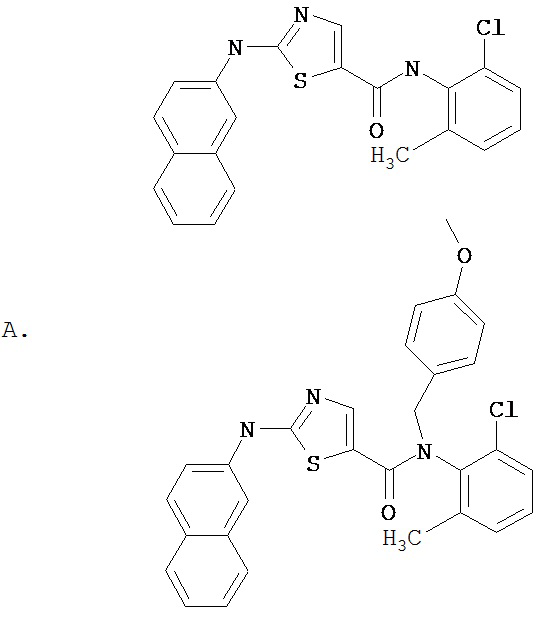
Соединение 514А получают из соединения 473А аналогично 473В, но вместо 2-бром-аминопиридина берут 2-аминонафталин.
В. Заглавное соединение.
Заглавное соединение получают аналогично соединению 473С, но вместо соединения 473В берут соединение 514А. Время удерживания при ВЭЖХ 4,11 мин.
Пример 515.
Получение N'-(2-хлор-6-метилфенил)-2-(2-хинолиниламино)-5-тиазолкарбоксамида.
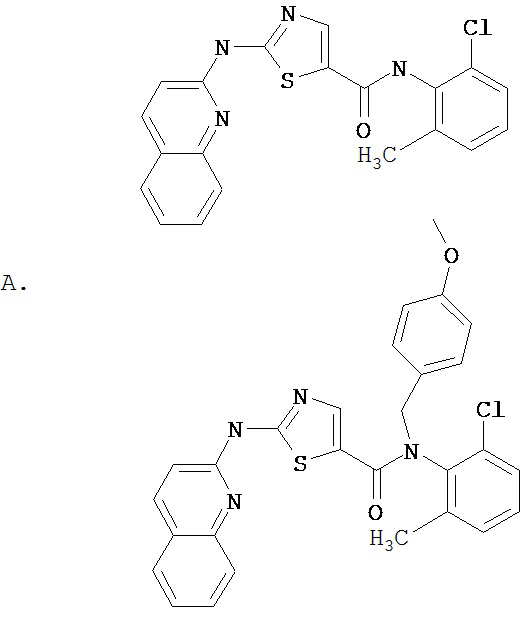
Соединение 515А получают из соединения 473А аналогично соединению 473В, но вместо 2-бромаминопиридина берут 2-аминохинолин.
В. Заглавное соединение.
Заглавное соединение получают аналогично соединению 473С, но вместо соединения 473В берут соединение 515А. ВЭЖХ - время удерживания 3,94 мин.
Пример 516.
Получение N'-(2-хлор-6-метилфенил)-2-(3-изохинолиниламино)-5-тиазолкарбоксамида.
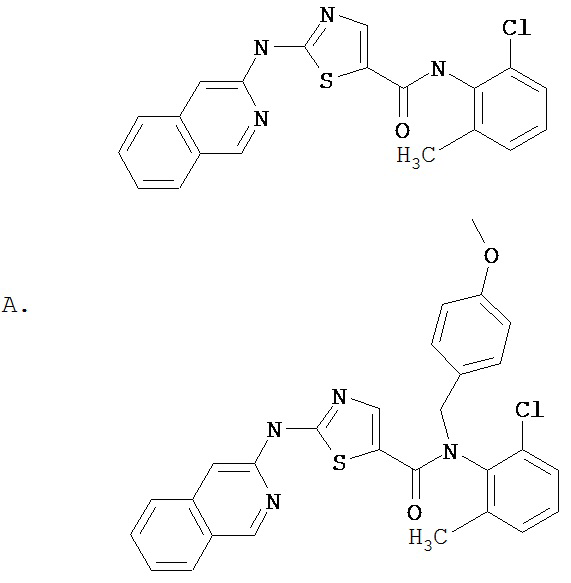
Соединение 516А получают из соединения 473А аналогично соединению 473В, но вместо 2-бром-6-аминопиридина берут 3-аминоизохинолин.
В. Заглавное соединение.
Заглавное соединение получают аналогично соединению 473С, но вместо соединения 473В берут соединение 516А. Время удерживания при ВЭЖХ 3,94 мин.
Пример 517.
Получение N'-(2-хлор-6-метилфенил)-2-(2-хиноксалиниламино)-5-тиазолкарбоксамида.
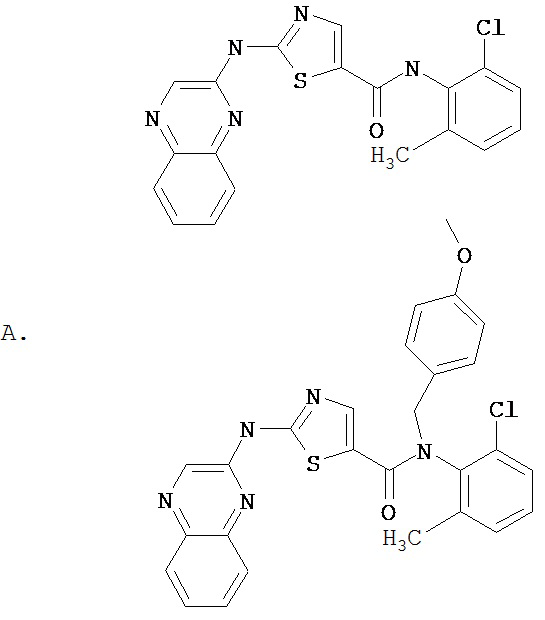
Соединение 517А получают из соединения 473А аналогично соединению 473В, но вместо 2-бром-6-аминопиридина берут 2-аминохиноксалин.
В. Заглавное соединение.
Заглавное соединение получают аналогично соединению 473С, но вместо соединения 473В берут соединение 517А. Время удерживания при ВЭЖХ 3,927 мин.
Пример 518.
Получение N'-(2-хлор-6-метилфенил)-4-метил-2-[[2-метил-6-(4-морфолинил)-4-пиримидинил]амино]-5-тиазолкарбоксамида.
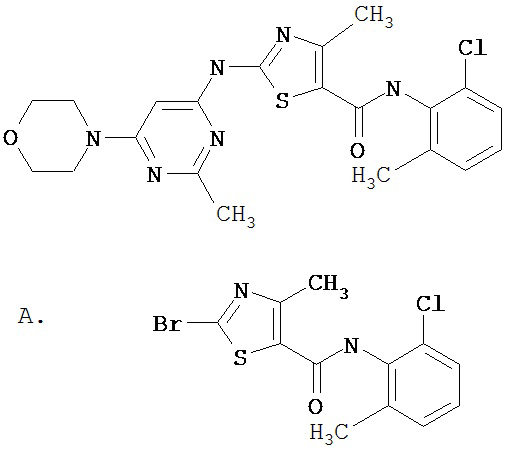
Соединение 518А получают из соединения 144 аналогично соединению 319А.
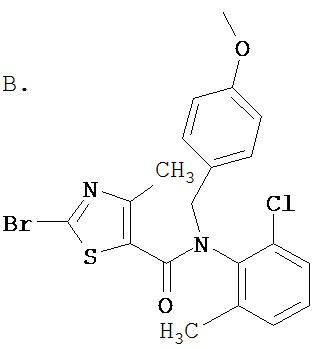
Соединение 518В получают аналогично соединению 473А, но вместо соединения 319А берут соединение 518А.
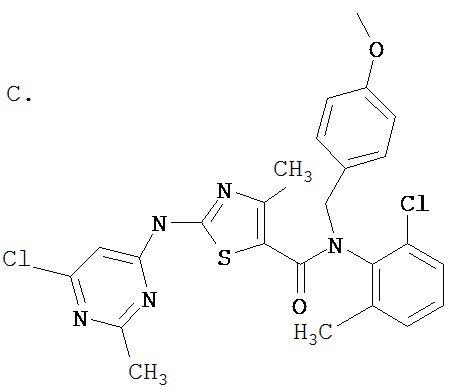
Соединение 518С получают аналогично соединению 473В, но вместо соединения 473А берут соединение 518В и вместо 2-амино-6-бромпиридина берут 4-амино-6-хлор-2-метилпиридин.
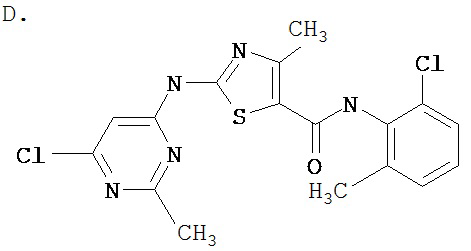
Соединение 518D получают аналогично соединению 473С, но вместо соединения 473В берут соединение 518С.
Е. Заглавное соединение.
Заглавное соединение получают аналогично соединению 444В, но вместо соединения 444А берут соединение 518D и вместо N-(2-аминоэтил)-морфолина берут морфолин. ВЭЖХ - время удерживания 3,397 мин.
Пример 519.
Получение N'-(2-хлор-6-метилфенил)-4-метил-2-[[2-метил-6-[[2-(4-морфолинил)этил]амино]-4-пиримидинил]амино]-5-тиазолкарбоксамида.
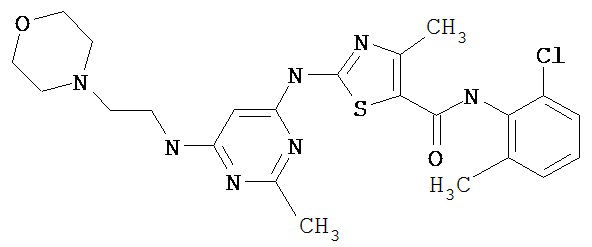
Соединение 519 получают аналогично соединению 518Е, но вместо морфолина берут N-2-аминоэтилморфолин. Время удерживания при ВЭЖХ 2,493 мин.
Пример 520.
Альтернативный способ получения соединения 321.
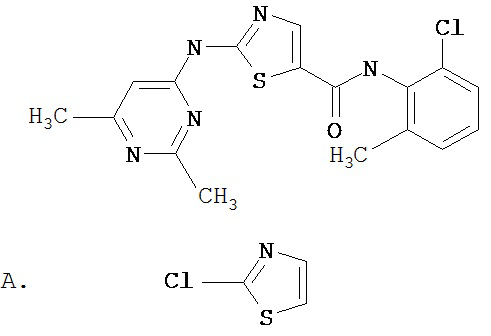
Соединение 520А получают из 2-аминотиазола по методике, описанной в патентной заявке Великобритании GB 2323595 A.
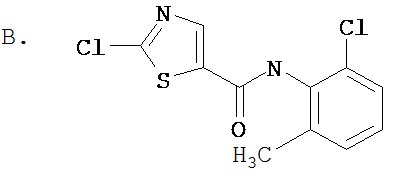
К раствору соединения 520А (480 мг, 4,0 ммол) в сухом ТГФ (10 мл), охлажденному до -78°С, добавляют 2,5М раствор n-BuLi (1,68 мл, 4,2 ммол) в гексане по каплям из шприца при температуре внутри колбы ниже -75°С. По окончании прибавления получают суспензию цвета беж. Реакционную смесь перемешивают 15 минут при -78°С. Добавляют раствор 2-хлор-6-метилфенилизоцианата (0,6 мл, 4,4 ммол) в 5 мл сухого ТГФ и реакционную смесь перемешивают еще 2 часа при -78°С. Добавляют насыщенный раствор NH4Cl (10 мл), смесь распределяют в системе EtOAc-вода и экстрагируют EtOAc (x2). Объединенные вытяжки сушат над Na2SO4 и упаривают в вакууме, получая после перекристаллизации из EtOAc-гексана 0,99 г заглавного соединения в виде бледно-желтого кристаллического вещества.
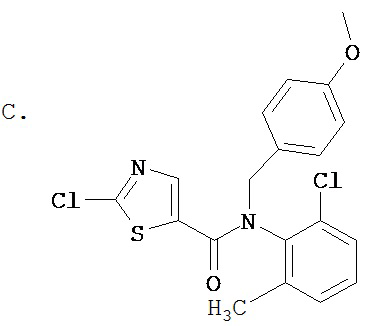
Соединение 520С получают аналогично соединению 473А, но вместо соединения 319А берут соединение 520В.
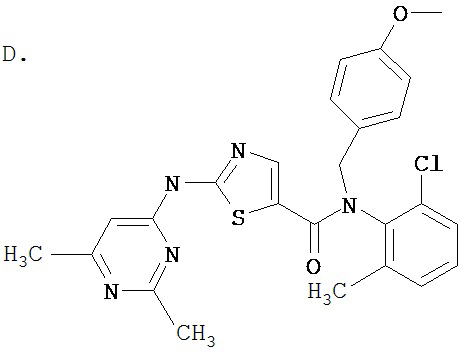
Соединение 520D получают из соединения 520С аналогично соединению 473В.
Е. Заглавное соединение.
Соединение 321 получают аналогично соединению 473С.
Пример 521.
Получение 2'-[[2,6-диметил-4-пиримидинил]амино]-N-фенил-5-тиазолкарбоксамида.
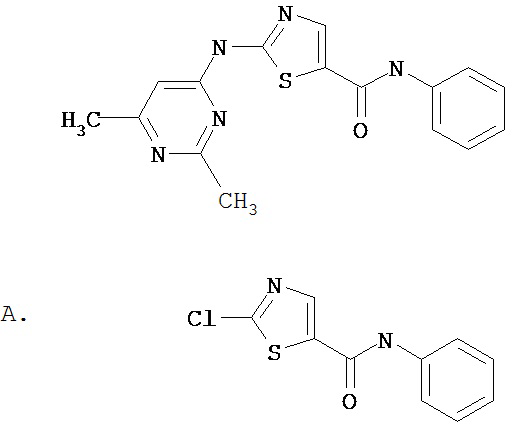
Соединение 521А получают аналогично соединению 520В, но вместо 2-хлор-6-метилфенилизоцианата используют фенилизоцианат.
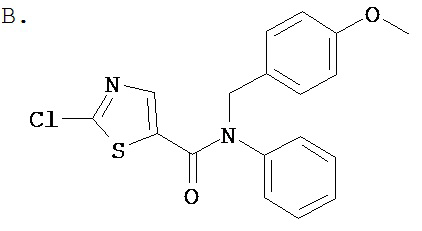
Соединение 521В получают аналогично соединению 473А, применяя соединение 521А вместо соединения 319А.
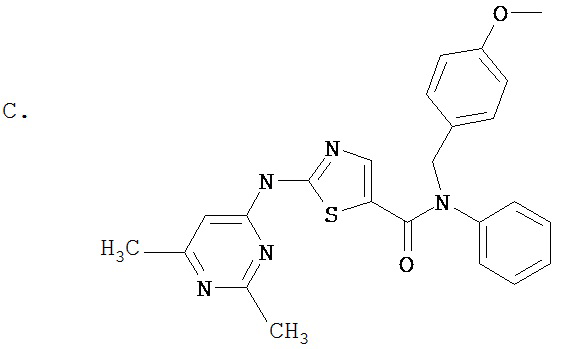
Соединение 521С получают из соединения 521В аналогично соединению 473В.
D. Заглавное соединение.
Заглавное соединение получают аналогично соединению 473С. ВЭЖХ - время удерживания 1,3 мин, метод В.
Пример 522.
Получение 2'-[(2,6-диметил-4-пиримидинил)метиламино]-N-(2-метилфенил)-5-тиазолкарбоксамида.
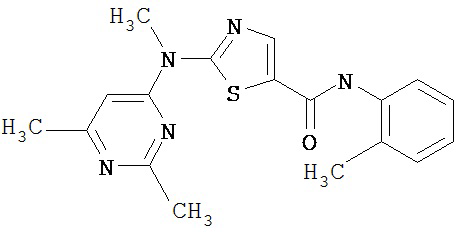
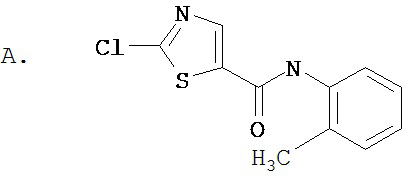
Соединение 522А получают аналогично соединению 520В, но вместо 2-хлор-6-метилфенилизоцианата используют 2-метилфенилизоцианат.
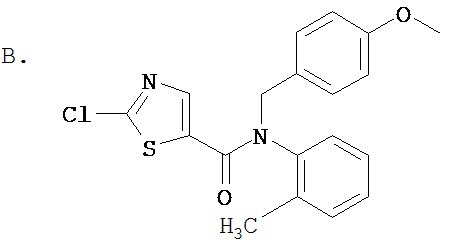
Соединение 522В получают аналогично соединению 473А, но вместо соединения 319А берут соединение 522А.
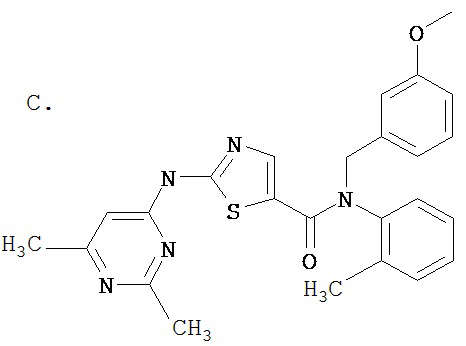
Соединение 522С получают из соединения 522В аналогично соединению 473В.
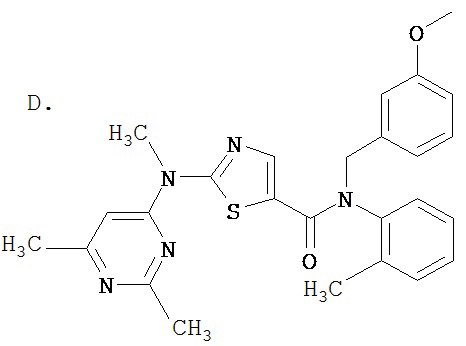
К раствору соединения 522С (280 мг, 0,61 ммол) в 2 мл ДМФА при комнатной температуре добавляют гидрид натрия (60% в масле; 40 мг, 1 ммол). После 30-минутного перемешивания добавляют иодметан (0,2 мл, 3 ммол) и реакционную смесь перемешивают 4 часа. После распределения реакционной смеси в системе этилацетат (50 мл) и вода (50 мл) органический слой промывают водой (2×50 мл) и рассолом (50 мл). Высушивание (MgSO4) и упаривание дают масло, которое хроматографируют на колонке 2,5×15 см с силикагелем, используя 50-75% этилацетат/гексан. Чистые фракции упаривают и остаток перекристаллизовывают из этилацетата/гексана, получая 100 мг 522D в виде легкого желтого твердого вещества
Е. Заглавное соединение.
Заглавное соединение получают аналогично соединению 473С. ВЭЖХ - время
удерживания 1,21 мин, метод В.
Пример 523.
Получение 2'-[(2,6-диметил-4-пиримидинил)амино]-N-(2-метилфенил)-5-тиазолкарбоксамида.
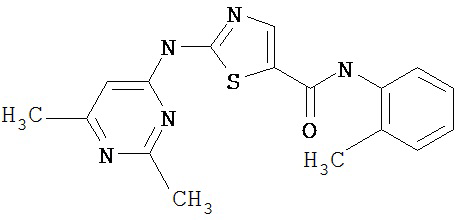
Соединение 523 получают аналогично соединению 473С, но вместо соединения 473В берут соединение 522С. ВЭЖХ - время удерживания 1,24 мин, метод В.
Пример 524.
Получение N'-(3,5-диметоксифенил)-2-[(2,6-диметил-4-пиримидинил)амино]-5-тиазолкарбоксамида.
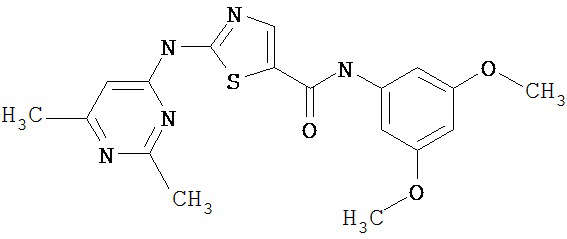
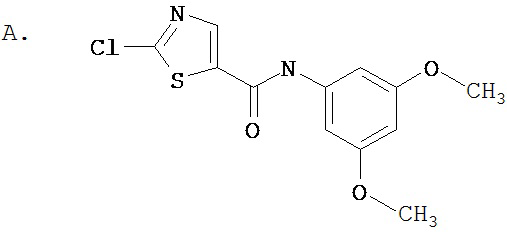
Соединение 524А получают аналогично соединению 520В, но вместо 2-хлор-6-метилфенилизоцианата берут 3,5-диметоксифенилизоцианат.
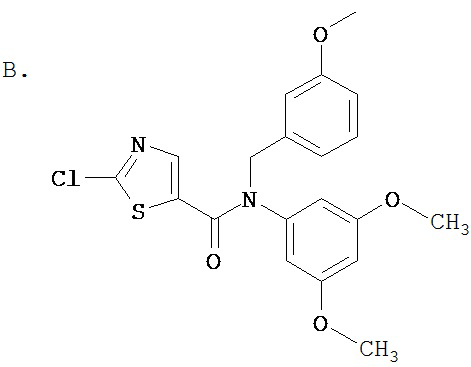
Соединение 524В получают аналогично соединению 473А, применяя соединение 524А вместо соединения 319А.
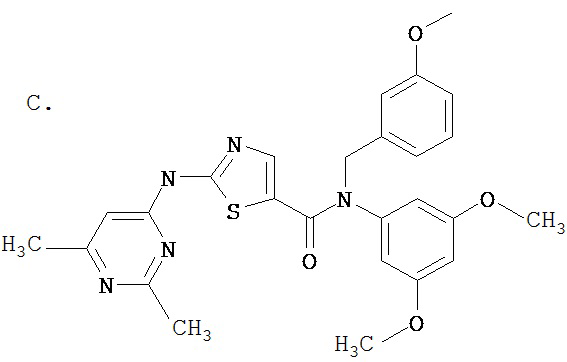
Соединение 524С получают из соединения 524В аналогично соединению 473В.
D. Заглавное соединение.
Заглавное соединение получают аналогично соединению 473С, но вместо соединения
473В берут 524С. Время удерживания ВЭЖХ 1,28 мин, метод В.
Пример 525.
Получение N'-[2,6-бис(1-метилэтил)фенил]-2-[(2,6-диметил-4-пиримидинил)амино]-5-тиазолкарбоксамида.
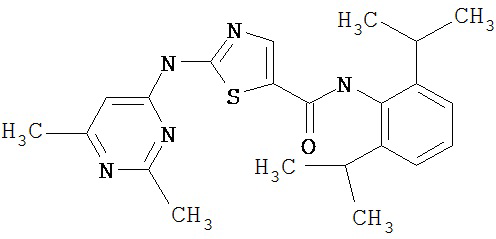
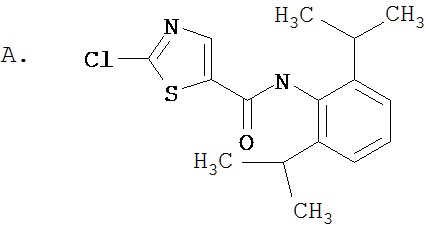
Соединение 525А получают аналогично соединению 520В, но вместо 2-хлор-6-метилфенилизоцианата берут 2,2-диизопропилфенилизоцианат.
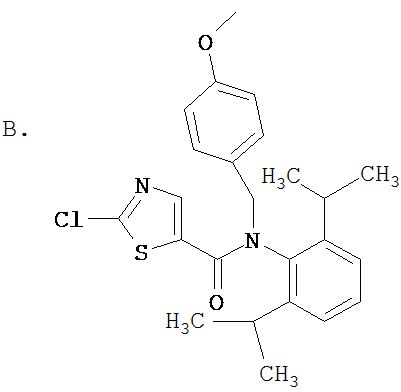
Соединение 525В получают аналогично соединению 473А, применяя вместо соединения 319А соединение 525А.
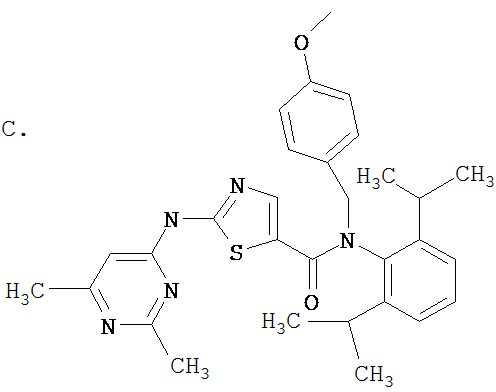
Соединение 525С получают из соединения 525В аналогично соединению 473В.
D. Заглавное соединение.
Заглавное соединение получают аналогично соединению 473С, но вместо соединения 473В берут соединение 525С. ВЭЖХ - время удерживания 1,6 мин, метод В.
Пример 526.
Получение N'-(2-хлор-6-метилфенил)-2-[(2,6-диметил-4-пиримидинил)метиламино]-5-тиазолкарбоксамида.
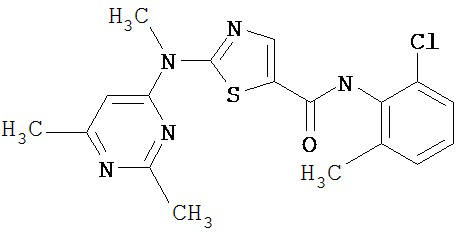
Смесь соединения 321 (110 мг, 0,29 ммол), карбоната калия (138 мг, 1 ммол) и иодметана (0,06 мл, 1 ммол) в ДМФА перемешивают 2 часа при комнатной температуре. После распределения реакционной смеси между этилацетатом (25 мл) и водой (25 мл) органический слой промывают водой (2×25 мл) и рассолом (25 мл). Высушиванием (MgSO4) и упариванием получают масло, которое хроматографируют на колонке 2,5×15 см с силикагелем, элюируя 1-4% МеОН/CHCl3, и собирают фракции, содержащие 526; получают 20 мг продукта. ВЭЖХ - время удерживания 1,3 мин, метод В.
Пример 527.
Получение N'-(2-хлор-6-метилфенил)-2-[(2,6-диметил-4-пиримидинил)амино] -N-метил-5-тиазолкарбоксамида.
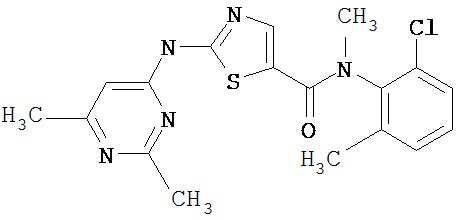
Соединение 527 получают аналогично соединению 526, но собирают фракции, содержащие соединение 527, получая 60 мг продукта. ВЭЖХ - время удерживания 1,23 мин, метод В.
Пример 528.
Получение 2-бром-N,N-(2-хлор-6-метилфенил)-(4-метоксибензил)-5-тиазолкарбоксамида.
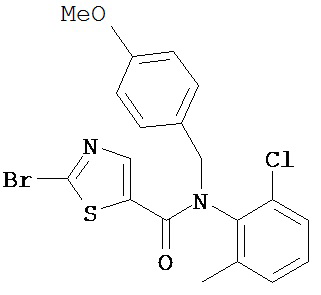
К охлажденному (0°С) раствору 2-хлор-6-метиланилина (2,86 мл, 23,3 ммол, 1,10 экв.) в ТГФ по каплям добавляют 1,0 М раствор литий бис(триметилсилил)- амида (42,2 мл, 42,2 ммол, 2,00 экв.) с помощью шприца. Гомогенный раствор перемешивают 5 минут, а затем добавляют через иглу раствор этил-2-бром-5-тиазолкарбоксилата (5,00 г, 21,1 ммол, 1,00 экв., получен аналогично соединению 319А) в ТГФ. Раствор перемешивают 15 минут, пока ТСХ не покажет отсутствие исходных. Затем к реакционной смеси прибавляют 4-метоксибензилхлорида (7,15 мл, 52,7 ммол, 2,5 экв.), потом каталитическое количество тетрабутиламмонийиодида (1,56 г, 4,22 ммол, 0,20 экв.). Гомогенную смесь перемешивают при комнатной температуре в течение ночи, а затем упаривают в вакууме. Остаток распределяют между этилацетатом и водой, вытяжки промывают рассолом и сушат Na2SO4. После фильтрования и удаления растворителя продукт очищают флеш-хроматографией (10-20% этилацетата в смеси гексанов), получая заглавное соединение в виде рыжевато-коричневого твердого вещества (47%).
Пример 529.
Получение N,N-(2-хлор-6-метилфенил)-(4-метоксибензил)-2-[(6-бром-2-пиридинил)амино]-5-тиазолкарбоксамида.
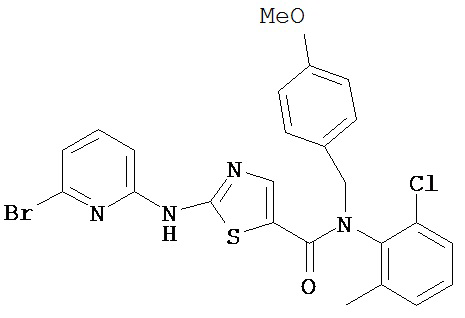
Соединение 529 получают аналогично соединению 319В, но берут в качестве реагентов соединение 528 и 6-бром-2-аминопиридин.
Пример 530.
Получение N-(2-хлор-6-метилфенил)-2-[(6-бром-2-пиридинил)амино] -5-тиазолкарбоксамида.
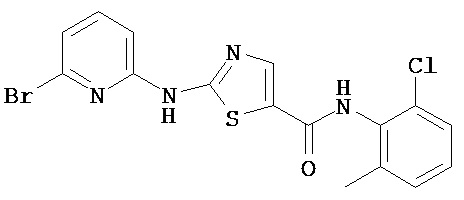
Соединение 529 (0,500 г, 0,919 ммол, 1,00 экв.) растворяют в 5 мл трифторуксусной кислоты и к нему при комнатной температуре добавляют 2 мл анизола, а затем 1 мл трифторметансульфоновой кислоты. Темно-красный гомогенный раствор перемешивают в течение ночи, а затем осторожно “гасят”, выливая раствор в смесь лед/бикарбонат натрия. Белый осадок отфильтровывают и последовательно промывают водой, смесью 1:1 гексан/эфир и эфиром, получая заглавное соединение (41%).
Примеры 531-538.
Общая методика.
Соединения 531-538 получают по описанной ниже общей методике. В ампулу на 1 драм (60 или - 28 грамм) помещают соединение 530 и избыток амина и нагревают при 90°С в течение ночи. Остаток затем очищают обращенно-фазовой ВЭЖХ, получая чистое соединение. Для нижеприведенных соединений 531-555 “ВЖЭХ - время уд.” обозначает время удерживания в процессе ВЭЖХ при следующих условиях: YMC ODS А С 18 S7 3,0×50 мм; 2-минутный градиент, начиная со 100% растворителя А (10% МеОН, 90% Н2О, 0,1% TFA) до 100% растворителя В (90% МеОН, 10% H2O, 0,1% TFA), скорость потока 5 мл/мин, λ=220 нм.
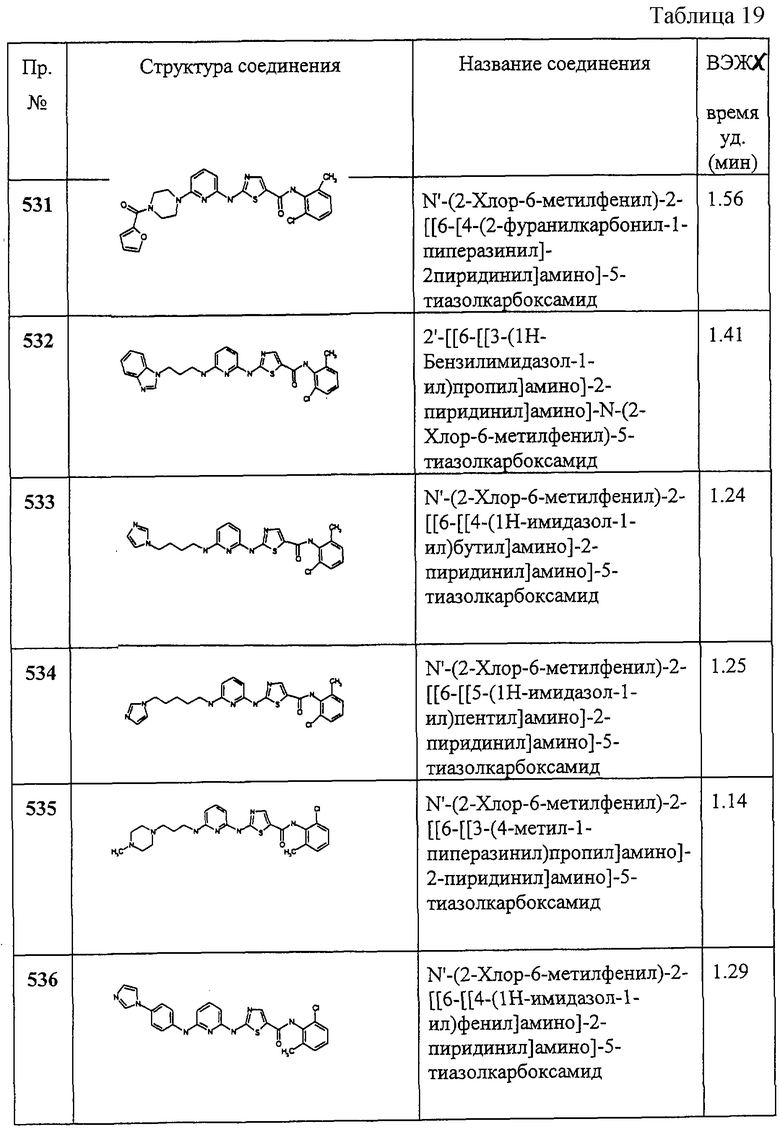
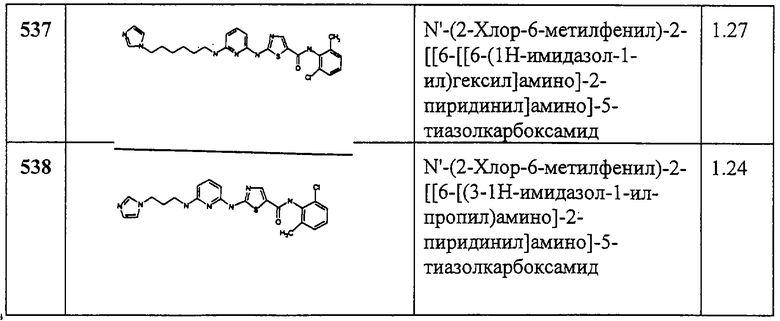
Пример 539.
Получение этил-2-[(6-бром-2-пиридинил)амино]-5-тиазолкарбоксилата.
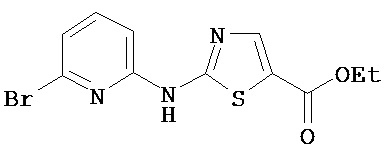
Соединение 539 получают аналогично 319В, но используют в качестве реагентов этил-2-бром-5-тиазолкарбоксилат и 6-бром-2-аминопиридин.
Примеры 540-550.
Общая методика.
Соединения 540-550 получают по нижеописанной общей методике. Соединение 539 конденсируется с соответствующим анилином по методике, описанной в примере 528, при этом получают соответствующий N-(4-метоксибензил)амид. Промежуточный бромпиридин затем реагирует с N-(3-аминопропил)имидазолом по методике, предложенной для примеров 531-538, с образованием соответствующего диаминопиридина. Снятие 4-метоксибензольной группы по методике, описанной в Примере 530, с последующей очисткой обращенно-фазовой препаративной ВЭЖХ дает соединения 540-550.
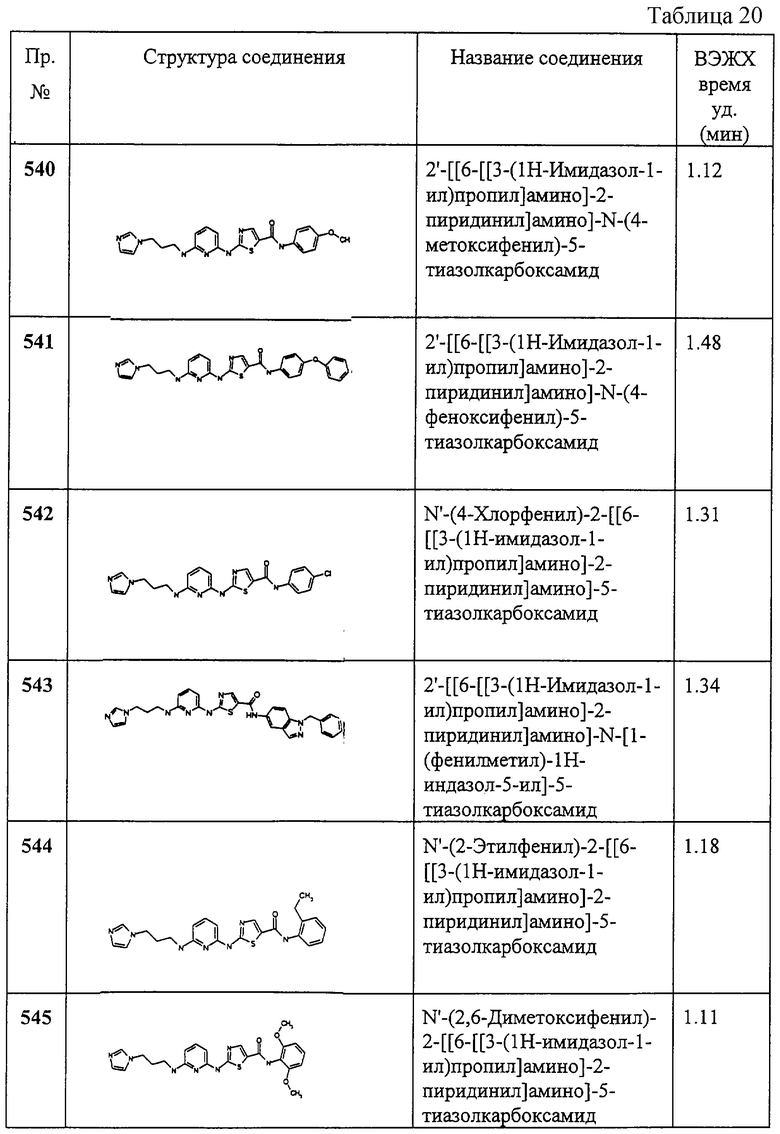
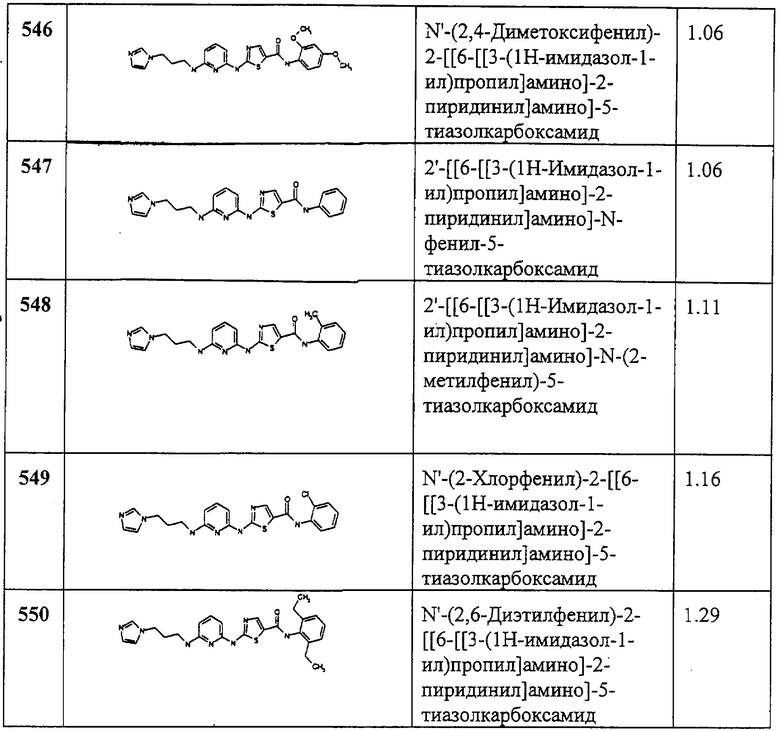
Пример 551.
Получение этил-2-[(6-бром-2-пиридинил)амино]-4-метил-5-тиазолкарбоксилата.
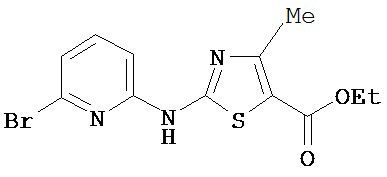
Соединение 551 получают аналогично 319В, но в качестве реагентов используют 2-бром-4-метил-5-тиазолкарбоксилат и 6-бром-2-аминопиридин.
Примеры 552 и 553.
Соединения 552 и 553 получают по методике, аналогичной методике получения соединений 540-550, но в качестве исходного используют соединение 551.
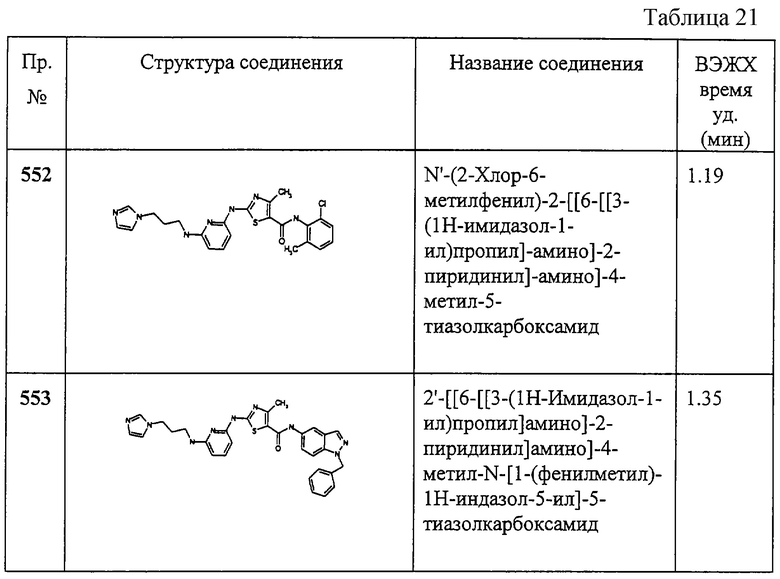
Пример 554.
Получение N'-(2-хлор-6-метилфенил)-2-[[3-[[3-(1Н-имидазол-1-ил)пропил]-амино]фенил]амино]-5-тиазолкарбоксамида.
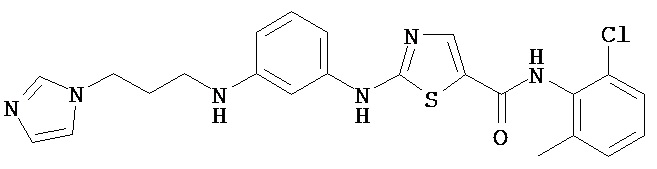
Раствор 528 (0,127 г, 0,281 ммол, 1,00 экв.) и 3-[N,N-(трет-бутоксикарбонил)-(3-аминопропил)-имидазолил]-1,3-фенилендиамина (0,178 г, 0,563 ммол, 2,00 экв.) в 0,200 мл ДМСО нагревают при 120°С в запаянной ампуле в течение ночи. Очистка обращенно-фазовой препаративной ВЭЖХ с последующим снятием защиты по методике, описанной для соединения 530, дает заглавное соединение.
Пример 555.
Получение N'-(2-хлор-6-метилфенил)-2-[[5-[[3-(1Н-имидазол-1-ил)пропил]-амино]-2-нитрофенил]амино]-5-тиазолкарбоксамида.
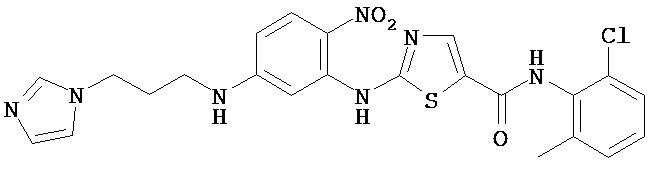
В раствор 2,4-дифторнитробензола (0,400 мл, 3,65 ммол, 1,00 экв.) в ацетонитриле помещают К2СО3 (0,605 г, 4,38 ммол, 1,20 экв.), а затем этил-2-амино-5-тиазолкарбоксилат (0,628 г, 3,65 ммол, 1,00 экв.) в виде твердого вещества. Гетерогенную смесь запаивают и нагревают при 120°С в течение ночи. Раствор отфильтровывают и затем упаривают в вакууме. Очистка флеш-хроматографией дает этил-2-[(3-фтор-6-нитро-1-фенил)амино]-5-тиазолкарбоксилат в виде желтого твердого вещества (9%). Это промежуточное соединение реагирует с 2-хлор-6-метиланилином по методике, описанной для соединения 528, давая N'-(2-хлор-6-метилфенил)-2-[3-(фтор-6-нитро-1-фенил)амино]-5-тиазолкарбоксамид (21%). Заглавное соединение синтезируют реакцией интермедиата с избытком N-(3-аминопропил)-имидазола при 80°С с последующей очисткой обращенно-фазовой препаративной ВЭЖХ.
Примеры 556-566.
Общая методика.
Соединения 556-566 получают по нижеописанной общей методике. Смесь 2-бром-N-[2-хлор-6-метилфенил]-5-тиазолкарбоксамида 319А, анилина (1 экв.), 1,0 N HCl aq. (0,5 экв.) в н-BuOH нагревают в течение ночи при 120°С в запаянной ампуле, разбавляют метанолом и продукт выделяют препаративной ВЭЖХ (YMC S5 ODS 30×100 мм колонка; элюция в градиенте смеси двух растворителей (смесь А: 10% МеОН, 90% воды и 0,1% TFA, смесь В: 90% МеОН, 10% воды и 0,1% TFA). В случае анилинов, замещенных на карбоксильную кислотную группу, реакционную смесь обрабатывают 1N водн. NaOH (5 экв.) в течение ночи перед заключительной очисткой продукта с помощью ВЭЖХ. “ВЖЭХ - время уд.” обозначает время удерживания в процессе ВЭЖХ в следующих условиях: YMC S5 ODS 4,6×30 мм (для 556-560) или YMC S7 ODS 3×50 мм колонке (для 561-566); 2-минутный градиент, начиная от 100% растворителя А (10% МеОН, 90% H2O, 0,1% TFA) до 100% растворителя В (90% МеОН, 10% H2O, 0,1% TFA), скорость потока 5 мл/мин, λ=220 нм.
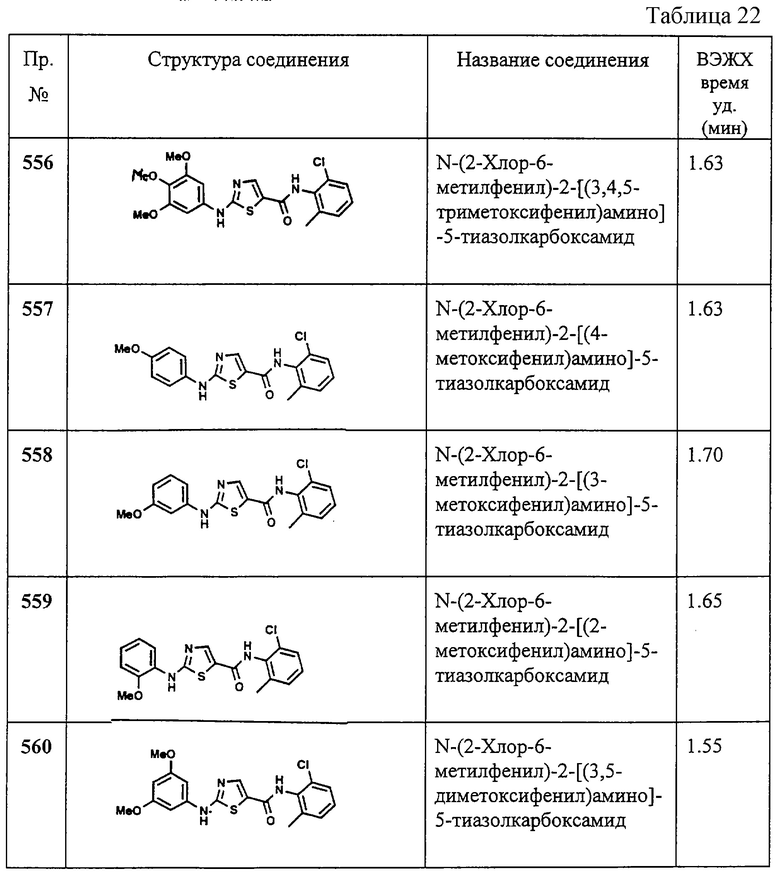
Пример 567.
N-(2-Хлор-6-метилфенил)-2-[[1-[3-(1Н-имидазол-1-ил)пропил]-1Н-бензимидазол-4-ил]амино]-5-тиазолкарбоксамид.
Смесь 1-бром-3-хлорпропана (10 мл, 0,10 ммол), имидазола (6,81 г, 0,10 ммол) в этанольном NaOEt (41,3 мл, 21 вес.%, 1,1 мол) нагревают при кипении 1 час. По охлаждении до комнатной температуры фильтруют и осадок на фильтре промывают EtOH. Растворитель упаривают из фильтрата и получают сырой 3-хлор-1-(имидазо-1-ил)-пропан в виде масла. Часть сырого хлорида (1,07 г, 7,40 ммол) добавляют к смеси 4-нитро-бензимидазола (1,09 г, 6,66 ммол) и NaH (293 мг, 60% в масле, 8,14 ммол) в ДМФА (15 мл). После нагревания в течение ночи при 60°С и затем 3 часа при 75°С растворитель удаляют. Остаток распределяют между водой и смесью 10% МеОН в ДСМ. Органическую фазу отделяют, сушат (Na2SO4) и растворитель отгоняют. Радиальная хроматография (4 мм пластинка с силикагелем, которую элюируют в ступенчатом градиенте ДСМ, содержащем 2, 3, 4,…10% МеОН) дает основной продукт 1-[3-имидазо-1-илпропил]-4-нитробензимидазол в виде твердого вещества (518 мг, 28%). Смесь этого вещества (250 мг) и 10% палладия на угле (200 мг) в EtOH (10 мл) в атмосфере водорода (баллон) энергично перемешивают 1 час. Катализатор отфильтровывают и растворитель упаривают под давлением, в остатке получают сырой 4-амино-1-[3-имидазо-1-илпропил]-бензимидазол в виде твердого вещества. Часть этого вещества (46 мг, 0,191 ммол) добавляют к смеси 319А (63 мг, 1,0 экв.), водного раствора HCl (0,24 мл, 1,0 М, 1,25 экв.) и н-BuOH (1 мл). Нагревают в запаянной ампуле при 120°С в течение 44 часов. По охлаждении до комнатной температуры препаративной ВЭЖХ выделяют соединение 567 (время удерживания ВЭЖХ (YMC S5 ODS 4,6×30 мм) 1,20 мин).
Пример 568.
N-(2-Хлор-6-метилфенил)-2-[[1-[2-(1Н-имидазол-1-ил)этил]-1Н-индазол-6-ил]амино]-5-тиазолкарбоксамид.
Смесь 1-бром-2-хлорэтана (4,6 мл, 0,055 мол), имидазола (3,40 г, 0,050 мол) в этанольном NaOEt (19 мл, 21 вес.%, 1 мол) нагревают при кипячении 2 час. По охлаждении до комнатной температуры реакционную смесь отфильтровывают и осадок на фильтре промывают EtOH. Растворитель отгоняют из фильтрата и получают сырой 2-хлор-1-(имидазо-1-ил)-этан. Часть сырого хлорида (2,24 г, 17,2 ммол) прибавляют к смеси 6-нитроиндазола (1,63 г, 10,0 ммол), К2СО3 (1,50 мг, 1,1 экв.) и KJ (1,70 г,1,1 экв.) в ДМФА (15 мл). После нагревания при 70°С в течение ночи, а затем при 90°С в течение 4 часов растворитель удаляют. Остаток распределяют между водой и смесью 5% МеОН в ДСМ. Органическую фазу отделяют, сушат (Na2SO4) и растворители удаляют. Радиальная хроматография (4 мм пластины с силикагелем, которые элюируют в ступенчатом градиенте ДСМ, содержащем 0, 1, 2% МеОН) дает 659 мг 1-[2-имидазо-1-илэтил]-6-нитроиндазола и 450 мг изомерного 2-[2-имидазо-1-илэтил]-6-нитроиндазола. Смесь 1-[2-имидазо-1-илэтил]-6-нитроиндазола (650 мг) и 10% палладия на угле (600 мг) в EtOH (10 мл) в атмосфере водорода (баллон) энергично перемешивают в течение ночи. Катализатор отфильтровывают, растворитель удаляют в вакууме, остается сырой 6-амино-1-[2-имидазо-1-илэтил]-индазол в виде твердого вещества. Часть этого вещества (68,1 мг, 1,5 экв.) прибавляют к смеси 556 (99,3 мг, 0,300 ммол), водного раствора HCl (0,45 мл, 1,0 М, 1,5 экв.) и н-BuOH (1,5 мл). Нагревают в запаянной ампуле при 120°С в течение 44 часов. По охлаждении до комнатной температуры препаративной хроматографией выделяют соединение 568 (ВЭЖХ - время удерживания (YMC ODS S7 3×50 мм) 1,31 мин).
Пример 569.
N-(2-Хлор-6-метилфенил)-2-[[2-[2-(1H-имидазол-1-ил)этил]-2Н-индазол-6-ил]амино]-5-тиазолкарбоксамид.
Исходя из изомерного 2-[2-имидазо-1-илэтил]-6-нитроиндазола, соединение 569 (ВЭЖХ - время удерживания (YMC ODS S7 3×50 мм) 1,28 мин) получают аналогично соединению 568.
Пример 570 и 571.
N-(2-Хлор-6-метилфенил)-2-[(1-метил-1Н-бензимидазол-6-ил)амино]-5-тиазолкарбоксамид.
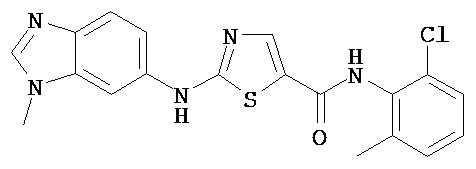
N-(2-Хлор-6-метилфенил)-2-[(1-метил-1Н-бензимидазол-5-ил)амино] -5-тиазолкарбоксамид.
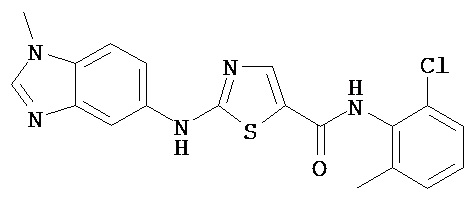
Исходя из 5-нитробензимидазола и иодистого метила соединение 570 (ВЭЖХ - время удерживания (YMC ODS S7 3×50 мм) 1,23 мин) и соединение 571 (ВЭЖХ - время удерживания (YMC ODS S7 3×50 мм) 1,23 мин) получают аналогично соединениям 557 и 558.
Пример 572.
N-(2-Хлор-6-метилфенил)-2-[[2-[3-(1Н-имидазол-1-ил)пропил]амино-1Н-бензимидазол-5-ил]амино]-5-тиазолкарбоксамид.
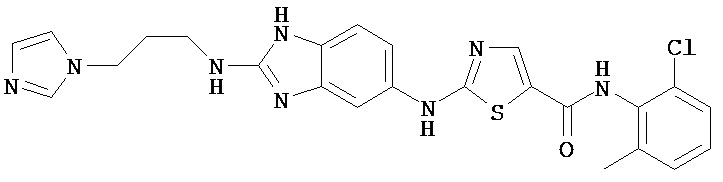
Смесь 2-хлор-5-нитробензимидазола (985 мг, 5,0 ммол) и 1-(3-аминопропил)-имидазола (1,8 мл, 3 экв.) в толуоле (15 мл) нагревают при кипении 5 час. Реакционную смесь распределяют между EtOAc и рассолом, получают осадок, который отфильтровывают. Флеш-хроматография его (силикагель; элюция в ступенчатом градиенте смесями, содержащими ДСМ и 1, 2, 3…10% МеОН) дает 2-[3-[имидазо-1-ил]пропиламино]-5-нитробензимидазол (550 мг) в виде твердого вещества. Это соединение соединяют с 10% Pd на угле (500 мг), взвешенным в EtOH, и перемешивают в атмосфере водорода (баллон) в течение ночи. Отфильтровывают катализатор, растворитель отгоняют в вакууме, остается сырой 5-амино-2-[3-имидазо-1-илпропиламино]-5-бензимидазол в виде твердого вещества. Часть его (77 мг, 0,30 ммол) прибавляют к смеси соединения 319А (99 мг, 1,0 экв.), водного раствора HCl (0,60 мл, 1,0 М, 2 экв.) и н-BuOH (1,5 мл). Нагревают в запаянной ампуле при 120°С 20 часов. По охлаждении до комнатной температуры выделяют препаративной хроматографией соединение 572. (ВЭЖХ - время удерживания (YMC ODS S7 3×50 мм) 1,20 мин).
Пример 573.
N-(2-Хлор-6-метилфенил)-2-[[2-(4-морфолинилметил)-1Н-бензимидазол-5-ил]амино]-5-тиазолкарбоксамид.
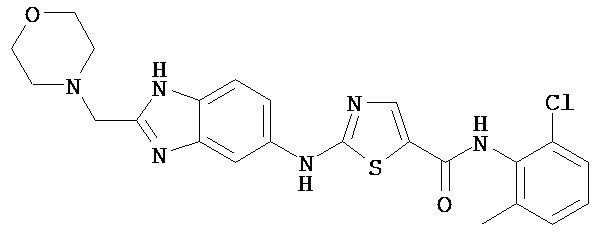
Смесь 3,4-диаминонитробензола (15,3 г, 0,10 мол) и хлоруксусной кислоты (14,18 г, 1,5 экв.) в 5,0 N водной HCl (80 мл) кипятят 1 час. По охлаждении до комнатной температуры реакционную смесь фильтруют через целит и фильтрат выдерживают при 0°С 2 дня. Образовавшиеся кристаллы собирают и перекристаллизовывают из смеси EtOH и воды, получают 7,2 г солянокислой соли 2-хлорметил-5-нитробензимидазола. Часть этой соли (528 мг, 2,13 ммол) и морфолин (1,31 мл, 7 экв.) кипятят в толуоле (15 мл) 4 часа. По охлаждении до комнатной температуры реакционную смесь фильтруют и осадок на фильтре промывают толуолом. Растворитель удаляют из фильтрата и получают сырой 2-[N-морфолинилметил]-5-нитробензимидазол в виде масла. Часть его (657 мг) и 10% палладий на угле (650 мг) в EtOH (10 мл) перемешивают в течение ночи в атмосфере водорода (баллон). Катализатор отфильтровывают, удаляют растворитель, остается сырой 5-амино-2-[N-морфолинилметил]-бензимидазол в виде масла. Часть его конденсируют с 556, как описано для 570, получают соединение 573 (ВЭЖХ - время удерживания (YMC ODS S7 3×50 мм) 0,92 мин).
Пример 574.
N-(2-Хлор-6-метилфенил)-2-[[2-(1Н-имидазол-1-илметил)-1Н-бензимидазол-5-ил]амино]-5-тиазолкарбоксамид.
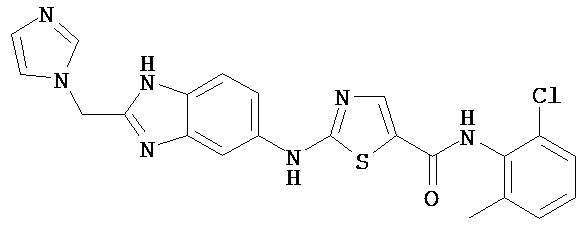
Исходя из имидазола и 2-хлорметил-5-нитробензимидазола соединение 574 (ВЭЖХ - время удерживания (YMC ODS S7 3×50 мм) 1,17 мин) получают аналогично соединению 570.
Пример 575.
N-(2-Хлор-6-метилфенил)-2-[[3-[[5-(1Н-имидазол-1-ил)-2-пиридинил]амино]фенил]амино]-5-тиазолкарбоксамид.
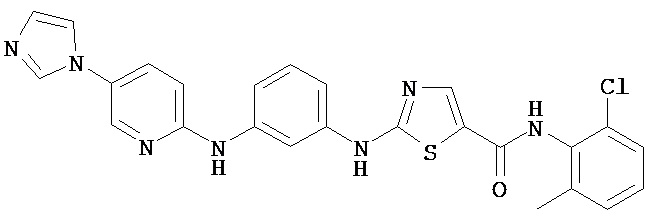
Смесь 3-нитроанилина (2,91 г, 21,1 ммол) и 2,5-дибромпиридина (5,0 г, 1 экв.) нагревают при 185°С 1 час. После охлаждения до комнатной температуры твердое вещество измельчают и обрабатывают смесью насыщ. NaOH aq. и 10% МеОН в ДСМ. Суспендированное твердое вещество отфильтровывают и промывают малым количеством 10% МеОН в ДСМ, а затем водой, после сушки получают 3,72 г сырого N-[5-бромпиридин-2-ил]-5-нитроанилина. Часть его (500 мг, 1,70 ммол) смешивают с имидазолом (116 мг, 1 экв.), CuJ (81 мг, 0,25 экв.) и K2СО3 (235 мг, 1 экв.) в ДМФА (2 мл) и смесь в течение 2 дней нагревают при 130°С. По охлаждении до комнатной температуры растворитель удаляют и остаток распределяют между водой и смесью 20% МеОН в ДСМ. Органический слой отделяют, сушат (Na2SO4) и удаляют растворители, остается твердый N-[5-имидазо-1-ил]-пиридин-2-ил]-5-нитроанилин. Его обрабатывают 10% палладием на угле (650 мг) в EtOH в атмосфере водорода 1,5 часа. После удаления катализатора и затем растворителя остается сырой N-[5-имидазо-1-ил]-пиридин-2-ил]-5-аминоанилин. Его очищают радиальной хроматографией (пластину с силикагелем 4 мм элюируют в ступенчатом градиенте ДСМ, содержащем 1, 2, 3…6% МеОН). Затем анилин конденсируют с 319А, как описано для 570, получая соединение 575 (ВЭЖХ - время удерживания (YMC ODS S5 4,6×30 мм) 1,42 мин).
Пример 576 и 577.
N-(2-Хлор-6-метилфенил)-2-[[3-[3-(1Н-имидазол-1-ил)пропокси]фенил]амино]-5-тиазолкарбоксамид.
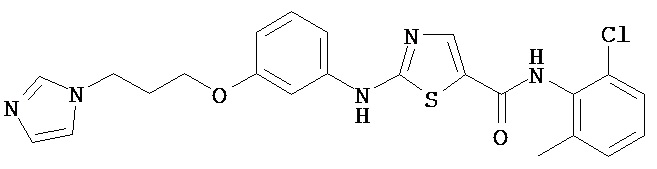
N-(2-Хлор-6-метилфенил)-2-[[4-[3-(1Н-имидазол-1-ил)пропокси]фенил]амино]-5-тиазолкарбоксамид.
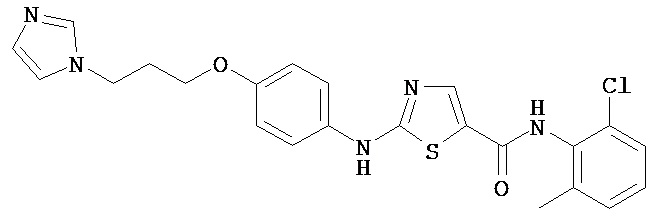
Суспензию 3-нитрофенола (837 мг, 6,02 ммол), 1-хлор-3-[имидазо-1-ил]-пропана (811 мг, 1 экв.), К2СО3 (3,3 г, 4 экв.) и NaJ (1,0 г, 1,1 экв.) в ДМФ нагревают при 120°С в течение 6 часов. По охлаждении до комнатной температуры реакционную смесь фильтруют и осадок промывают ДМФА. Растворитель удаляют и остаток хроматографируют (радиальная хроматография; пластину с силикагелем 4 мм элюируют в ступенчатом градиенте ДСМ, содержащем 0; 1; 2,5; 5; 7,5% МеОН), получают 400 мг 3-[3-имидазо-1-илпропилокси]] -нитробензола. Его обрабатывают 10% палладием на угле (400 мг) в EtOH в атмосфере водорода 4 часа. После удаления катализатора и растворителя остается 3-[3-имидазо-1-илпропилокси]]-анилин, который конденсируют с 319А, как описано для 570, получают соединение 576 (ВЭЖХ - время удерживания (YMC ODS S5 4,6×30 мм) 1,33 мин). Исходя из 4-нитрофенола и 1-хлор-3-[имидазо-1-ил]-пропана получают соединение 577 (ВЭЖХ - время удерживания (YMC ODS S5 4,6×30 мм): 1,42 мин) аналогично соединению 576.
Пример 578.
N-(2-Хлор-6-метилфенил)-2-[[4-[2-(1Н-имидазол-1-ил)этокси]-3-метоксифенил]амино]-5-тиазолкарбоксамид.
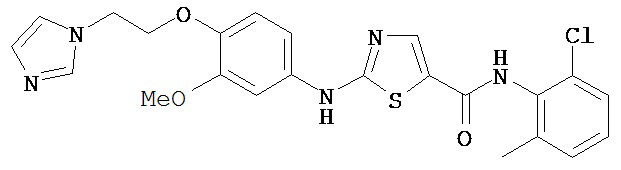
Исходя из 2-метокси-4-нитрофенола и 1-хлор-3-[имидазо-1-ил]-этана, соединение 578 (ВЭЖХ - время удерживания (YMC ODS S5 4,6×30 мм) 1,35 мин) получают аналогично соединению 576.
Пример 579 и 580.
N-(2-Хлор-6-метилфенил)-2-[[3-[[[3-(1Н-имидазол-1-ил)пропил]амино]сульфонил]фенил]амино]-5-тиазолкарбоксамид.
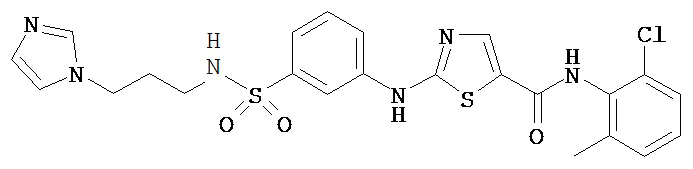
N-(2-Хлор-6-метилфенил)-2-[[4-[[[3-(1Н-имидазол-1-ил)пропил]амино]сульфонил]фенил]амино]-5-тиазолкарбоксамид.
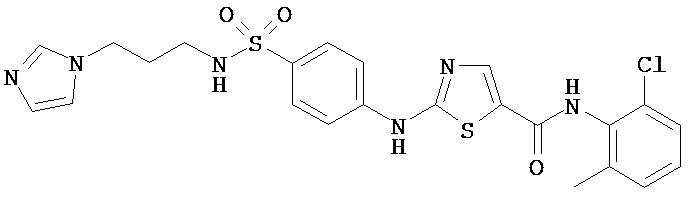
3-Имидазо-1-илпропиламин (2,04 мл, 2,5 экв.) прибавляют к раствору 3-нитробензолсульфонилхлорида (1,5 г, 6,77 ммол) в ТГФ (20 мл) при комнатной температуре. Через 1 час растворитель удаляют и остаток распределяют между водой и смесью 10% МеОН в ДСМ. Органический слой отделяют, промывают водой и сушат (Na2SO4). Сырой продукт N-[3-имидазо-1-ил]-пропил]-3-нитробензолсульфонамид обрабатывают 10% палладием на угле (2 г) в ТГФ (60 мл) в атмосфере водорода в течение ночи. После удаления катализатора и растворителя получают сырой 3-амино-N-[3-[имидазо-1-ил]-пропил]-бензолсульфонамид, который затем реагирует с 319А, как описано для 570, давая соединение 579 (ВЭЖХ – время удерживания (YMC ODS S7 3×50 мм) 1,22 мин). Исходя из 4-нитробензолсульфонилхлорида и 3-[имидазо-1-ил]-пропиламина получают соединение 580 (ВЭЖХ - время удерживания (YMC ODS S7 3×50 мм) 1,21 мин) аналогично соединению 579.
Пример 581
Из представленных выше соединений N-(2-хлор-6-метилфенил)-2-[[6-[4-(2-гидроксиэтил)-1-пиперазинил]-2-метил-4-пиримидинил]амино]-5-тиазолкарбоксамид, фигурирующий в примерах под №455 (далее для краткости “соединение 455”) было идентифицировано как ингибитор Srs/Abl киназ с исключительной антипролиферативной активностью против гематологических клеточных линий и клеточных линий солидных опухолей. Соединение 1 было активным при пероральном введении на модельном К562 ксенотрансплантате хронического миелогенного лейкоза (CML), демонстрируя полную регрессию опухолей и низкую токсичность при уровне многократных доз.
Целевая терапия реально осуществляется при проведении преклинического и клинического онкологического исследования. Применение иматиниба (Gleevec™) для лечения хронического миелогенного лейкоза (CML) служит подтверждением концепции, что терапевтические агенты, нацеленные на специфические для ракового заболевания метаболические пути (каскады реакций), могут привести к значительному улучшению по сравнению с обычными химиотерапевтическими агентами. CML является миелопролиферативным нарушением, которое характеризуется гиперпролиферацией стволовых клеток с последующей дифференцировкой в клетки периферической белой крови (лейкоциты). Присутствие хромосомы Philadelphia, образующейся при транслокации домена Abl киназы на хромосому 9 со специфической областью точки разрыва (bcr) на хромосоме 22, является характерным для CML. Генным продуктом этой транслокации является конститутивно-активированная тирозинкиназа, известная как Bcr-Abl, которая стимулирует пролиферацию стволовых клеток в костном мозге и в результате вызывает патологию заболевания. Нацеливая тирозинкиназную активность Bcr-Abl, иматиниб нормализует число клеток периферической белой крови (лимфоцитов) и значительно снижает позитивный в отношении хромосомы Филадельфия клон стволовых клеток в костном мозге, вызывая в клинике эффективный гематологический и цитогенетический ответ.
Протоонкоген с-Src играет основную роль в развитии, росте и образовании метастазов в случае широкого ряда разновидностей рака человека. Активация Src в форме повышенного уровня киназной активности и/или экспрессии белка продемонстрирована на нескольких основных типах рака, включая рак толстой кишки, молочной железы, поджелудочной железы и мозга. Src киназа модулирует сигнальную трансдукцию через множество онкогенных путей, включая EGFR, Her2/neu, PDGFR, FGFR и VEGFR. Таким образом, можно ожидать, что блокирование передачи сигнала с помощью ингибирования киназной активности Src явится эффективным способом модулирования аберрантных путей, которые стимулируют онкологическую трансформацию клеток.
Соединение 455 получали с общим выходом 61% из хлортиазола по оптимизированной синтетической последовательности, подробно изображенной на Схеме XII. Конденсация литиевого аниона соединения 1 с 2-хлор-6-метилфенилизоцианатом в ТГФ протекает гладко, при этом образуется соответствующий карбоксамид, который защищают в виде 4-метоксибензильного производного 2. Замена 2-хлорзамещенного на натриевую соль 4-амино-6-хлор-2-метилпиримидина при кипячении с последующим снятием защитной 4-метоксибензильной группы в TfOH/ТФК дают хлорпиримидин 3. Наконец, реакция 3 с 1-(2-гидроксиэтил)пиперазином в диоксане при кипячении дает соединение 455, которое затем под действием HCl в эфире превращается в соответствующий гидрохлорид. Оказалось, что масштабы этой реакции можно увеличивать и все стадии можно проводить в количествах 50 г или более со сравнимыми выходами.
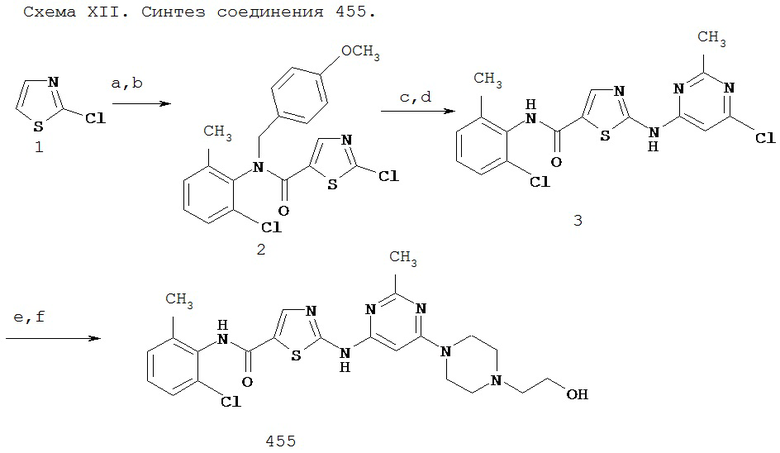
(а) н-BuLi, ТГФ, 2-хлор-6-метилфенилизоцианат, -78°С, 86%; (b) NaH, кипячение, 4-метоксибензилхлорид, ТГФ, 95:; (c)NaH, ТГФ, 4-амино-6-хлор-2-метилпиримидин, кипячение, 83%; (d) TfOH, ТФК/СН2Cl2 (1:1), 99%; (е) 1-(2-гидроксиэтил)пиперазин, 1,4-диоксан, кипячение; (f) HCl, Et2O, MeOH, 91% (две стадии).
Для лучшего понимания фермент-ингибиторных свойств соединения 455, это соединение изучали на панели внутренней киназной селективности и определили значения Кi, как для Src, так и для Bcr-Abl киназы. Было подтверждено, что соединение является высокоактивным ингибитором как для Src, так и для Bcr-Abl, измеренные значения Кi, составляют 16±1.0 пМ и 30±22 пМ, соответственно. Данные относительно селективности соединения суммированы в таблице 1. Было найдено, что соединение эффективно ингибирует других представителей Src-семейства. Соединение также демонстрирует заметную активность против с-набора и PDGFRβ. В отношении всех других киназ-мишеней на панели наблюдалась более чем 1000-кратная селективность.
Профиль селективности соединения 455 в отношении киназ.
IC50, nM
IC50, nM
В фармакокинетическом исследовании на крысах соединение 455 было охарактеризовано как соединение с высоким объемом распределения (Vss) с системным клиренсом (Cl) около 40% печеночного кровотока. Пероральная доза 10 мг/кг, вводимая в виде раствора в смеси 1:1 пропиленгликоль : вода, быстро всасывалась и демонстрировала подходящий период полужизни (t1/2) и среднее резидентное время (MRT). Найденная пероральная биодоступность (Fpo) в этом исследовании составляла 27%. Совместно с данными 4-часовой пероральной экспозиции, полученными на мышах, эти данные позволили сделать заключение, что имеет фармакокинетический профиль, подходящий для успешного перехода к изучению in vivo эффективности. Параметры РК, полученные в исследовании на крысах, суммированы в таблице 2.
Фармакокинетические свойства соединения, полученные на крысах Sprague-Dawley
In vivo активность соединения 455 изучали анализом К562 ксенотрансплантата на моделях голых мышей. Опухолевые клетки имплантируют подкожно и выращивают примерно до 300 мг и проводят 2 цикла лечения перорально по схеме: 5 дней принимают, перерыв 2 дня. При однократном ежедневном приеме доз соединения 13 либо по 5, либо по 50 мг/кг наблюдалось уменьшение (регрессия) после одного цикла лечения и полное исчезновение опухолевой массы к концу лечения лекарственным веществом. Ни в одной группе животных не наблюдалось никакой токсичности (смерти животных, отсутствие прибавки в весе). Таким образом, установлено, что соединение 455 обладает высокой in vivo активностью и высокой границей безопасности на этой животной модели CML.
Соединение 455 было испытано на токсичность в нескольких тестах. Оно было негативным в тесте Эймса на мутагенность. Оно было негативным также при анализе человеческих гепатоцитов в иммортализованных (>40 мкМ) и в примированных клеточных линиях (до 16 мкМ). Оно проявило также минимальную кардиотоксичность.
| название | год | авторы | номер документа |
|---|---|---|---|
| ИНГИБИТОРЫ ДИПЕПТИДИЛПЕПТИДАЗЫ IV НА ОСНОВЕ КОНДЕНСИРОВАННЫХ ЦИКЛОПРОПИЛПИРРОЛИДИНОВ И СПОСОБ ИХ ПРИМЕНЕНИЯ | 2001 |
|
RU2286986C2 |
| ПРОИЗВОДНЫЕ ПИРРОЛО [2,3-D] ПИРИМИДИНОВ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ, ПИРРОЛО [2,3-D] ПИРИМИДИНЫ, ПРОИЗВОДНЫЕ ПИРРОЛА | 1993 |
|
RU2124015C1 |
| ПРОИЗВОДНЫЕ ПИРАЗОЛА И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ, СОДЕРЖАЩАЯ ИХ | 1993 |
|
RU2142946C1 |
| ПИРАЗОЛО- И ПИРРОЛОПИРИДИНЫ | 1995 |
|
RU2135498C1 |
| 1,4-ЗАМЕЩЕННЫЕ ПИПЕРАЗИНЫ ИЛИ 4-АЛКИЛИДЕНИЛПИПЕРИДИНЫ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ИХ ОСНОВЕ И СПОСОБ ЛЕЧЕНИЯ | 2000 |
|
RU2241707C2 |
| О-АРИЛГЛЮКОЗИДНЫЕ ИНГИБИТОРЫ SGLT2 И СПОСОБ ИХ ПРИМЕНЕНИЯ | 2001 |
|
RU2269540C2 |
| ЦИКЛИЧЕСКИЕ ИНГИБИТОРЫ ПРОТЕИНТИРОЗИНКИНАЗ | 2000 |
|
RU2312860C2 |
| ЗАМЕЩЕННЫЕ ДИАЛКИЛ (ОКСИДО)-Λ-СУЛЬФАНИЛИДЕН НИКОТИНАМИД ПРОИЗВОДНЫЕ В КАЧЕСТВЕ ИНГИБИТОРОВ КИНАЗЫ | 2014 |
|
RU2711749C2 |
| ПИРАЗОЛОПИРИМИДИНЫ И ИХ ФАРМАЦЕВТИЧЕСКИ ПРИЕМЛЕМЫЕ СОЛИ | 1993 |
|
RU2124016C1 |
| ИНГИБИТОРЫ СЕРИН/ТРЕОНИН КИНАЗЫ ДЛЯ ЛЕЧЕНИЯ ГИПЕРПРОЛИФЕРАТИВНЫХ ЗАБОЛЕВАНИЙ | 2013 |
|
RU2644947C2 |
Изобретение относится к N-(2-хлор-6-метилфенил)-2-[[6-[4-(2-гидроксиэтил)-1-пиперазинил]-2-метил-4-пиримидинил]амино]-5-тиазолкарбоксамиду формулы
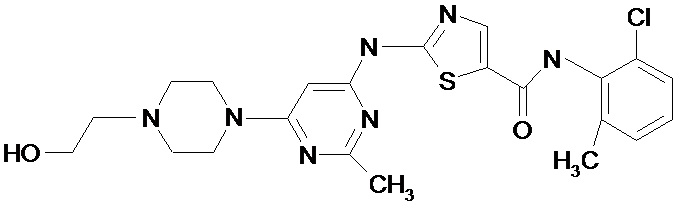
и к его фармацевтически приемлемым солям. Также описана фармацевтическая композиция, ингибирующая протеинтирозинкиназы и содержащая указанное соединение, способ лечения обусловленного протеинтирозинкиназами нарушения, такого как иммунологическое расстройство и онкологическое заболевание, и способ лечения рака. 4 н. и 1 з.п. ф-лы, 25 табл.
1. 'N-(2-Хлор-6-метилфенил)-2-[[6-[4-(2-гидроксиэтил)-1-пиперазинил]-2-метил-4-пиримидинил]амино]-5-тиазолкарбоксамид
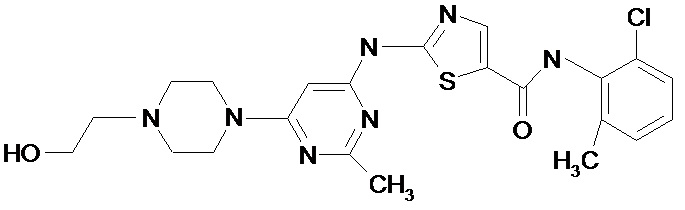
или его фармацевтически приемлемые соли.
2. Способ лечения рака, включающий введение пациенту эффективного количества соединения по п. 1.
3. Фармацевтическая композиция, ингибирующая протеинтирозинкиназы и содержащая соединение по п. 1 и фармацевтически приемлемый носитель или разбавитель.
4. Способ лечения обусловленного протеинтирозинкиназами нарушения, включающий введение пациенту эффективного количества соединения по п. 1 или фармацевтической композиции по п. 3.
5. Способ по п. 4, отличающийся тем, что пациенту вводят, по меньшей мере, один дополнительный терапевтический агент, выбранный из циклоспорина A; CTLA4-Ig; антител, выбираемых из анти-ICAM-3, рецептора анти-IL-2 (Anti-Tac), анти-СD45RВ, анти-СD2, анти-СD3 (ОКТ-3), анти-СD4, анти-CD80, анти-СD86 и моноклонального антитела ОКТ3; агентов, блокирующих взаимодействие между CD40 и gp39; слитых белков, созданных из CD40 и gp39; ингибиторов NF-каппа В-функции; нестероидных противовоспалительных средств (НПВС); стероидов; соединений золота; антипролиферативных агентов; FK506, микофенолата мофетила; цитотоксических лекарственных средств; ингибиторов TNF-α; антител против TNF или растворимого рецептора TNF; рапамицина; лефлюнимида; ингибиторов циклооксигеназы-2; паклитаксела; цисплатина, карбоплатина, доксорубицина, карминомицина, даунорубицина, аминоптерина, метотрексата, метоптерина, митомицина С, эктеинасцидина 743, порфиромицина, 5-флуороурацила, 6-меркаптопурина, гемцитабина, цитозин арабинозида, подофиллотоксина, этопозида, этопозида фосфата, тенипозида, мелфелана, винбластина, винкристина, лейрозидина, эпотилона, виндезина или лейрозина.
| Дорожная спиртовая кухня | 1918 |
|
SU98A1 |
| US 3547917 A , 15.12.1970 | |||
| Способ получения аминотиазоловили иХ КиСлОТНО-АддиТиВНыХ СОлЕй | 1979 |
|
SU843746A3 |

