ОБЛАСТЬ ИЗОБРЕТЕНИЯ
Данное изобретение относится к частицам имеющего высокий объем пор композиционного материала на основе оксида алюминия, способам их получения, агломератам и полученным из них катализаторам на носителе и способам использования указанных катализаторов.
ПРЕДПОСЫЛКИ СОЗДАНИЯ ИЗОБРЕТЕНИЯ
Область техники, относящаяся к частицам пористого оксида алюминия, полученным из них формованным носителям катализаторов, носителям, пропитанным различными каталитически активными металлами, соединениями металлов и/или промоторами, и различным использованиям таких пропитанных носителей в качестве катализаторов, является широкой и относительно хорошо разработанной.
Хотя в известном уровне техники происходит непрерывная модификация и усовершенствование таких частиц, носителей и катализаторов для повышения их каталитической активности и, хотя в некоторых случаях фактически были достигнуты весьма желательные активности, в промышленности существует сохраняющаяся потребность в улучшенных носителях катализаторов и полученных из них катализаторах, которые обладают повышенной активностью и сроком службы, что определяется желательным балансом морфологических свойств.
Оксид алюминия пригоден для различных применений, включая носители катализаторов и катализаторы для химических процессов, каталитические покрытия (облицовки) для автомобильных глушителей и тому подобное. Во многих указанных применениях может быть желательно добавить к оксиду алюминия каталитические материалы, такие как металлические ионы, тонкоизмельченные металлы, катионы и тому подобное. Уровень и распределение указанных металлов на носителе, а также свойства самого носителя являются ключевыми параметрами, которые оказывают влияние на сложную природу каталитической активности и срок службы.
Оксид алюминия, используемый в каталитических приложениях, получали до сих пор различными способами, такими как водный гидролиз алкоголятов алюминия, осаждение оксида алюминия из квасцов, способы с использованием алюмината натрия и тому подобное. Высокая цена является результатом последних двух способов вследствие того, что количество побочных продуктов, таких как сульфат натрия, фактически превышает количество получаемого требуемого продукта, т.е. бемита. Обычно цена бемита в 4 раза дороже, чем цена активного оксида алюминия.
Вообще говоря, хотя оксид алюминия из указанных источников можно использовать для носителей катализаторов, такое использование связано с некоторыми ограничениями. Это происходит в связи с тем фактом, что для катализаторов на носителях, используемых в химических реакциях, морфологические свойства носителя, такие как площадь поверхности, объем пор и распределение пор по размерам, которые включают общий объем пор, являются очень важными. Такие свойства являются инструментальными свойствами для влияния на природу и концентрацию активных центров катализатора, диффузию реагентов к активным центрам катализатора, диффузию продуктов из активных центров и срок службы катализатора.
Кроме того, носитель и его размеры также влияют на механическую прочность, плотность и характеристики заполнения реактора катализатором, все из которых являются важными при коммерческих применениях.
Катализаторы гидропереработки в процессах переработки нефти представляют собой значительную долю катализаторов на носителе из оксида алюминия в коммерческом использовании. Применение гидропереработки охватывает широкий диапазон типов сырья и эксплуатационных условий, но имеют одну или несколько общих целей, а именно удаление гетероатомных примесей (серы, азота, кислорода, металлов), повышение отношения Н/С в продуктах (тем самым снижается содержание ароматических соединений, плотность и/или коксовый остаток) и расщепление углеродных связей для снижения температурных пределов кипения и средней молекулярной массы.
Более конкретно, хорошо известно использование последовательности реакторов с кипящим слоем, содержащих катализатор, имеющий повышенную эффективность и сохранение активности при десульфуризации и деметаллизации потоков металлсодержащих тяжелых углеводородов.
Так как специалисты по нефтепереработке увеличивают долю более тяжелой сырой нефти худшего качества в исходном сырье, которое нужно переработать, возрастает потребность в процессах обработки фракций, содержащих повышенные уровни металлов, асфальтенов и серы.
Широко известно, что в сырых нефтях и других потоках тяжелых нефтяных углеводородов, таких как нефтяной углеводородный остаток, углеводородные потоки, полученные из битуминозных песков, и углеводородные потоки, полученные из угля, присутствуют различные металлорганические соединения и асфальтены. Наиболее распространенными металлами, найденными в таких углеводородных потоках, являются никель, ванадий и железо. Такие металлы являются очень вредными для различных операций переработки нефти, таких как гидрокрекинг, гидродесульфуризация и каталитический крекинг. Металлы и асфальтены вызывают внутреннюю закупорку слоя катализатора и снижают срок службы катализатора. Осаждение различных металлов на катализаторе приводит к отравлению или дезактивации катализатора. Кроме того, асфальтены имеют тенденцию снижать восприимчивость углеводородов к десульфуризации. Если катализатор, такой как катализатор десульфуризации или псевдоожиженный катализатор крекинга, подвергается воздействию углеводородной фракции, которая содержит металлы и асфальтены, катализатор скоро дезактивируется и будет подвергнут преждевременной замене.
Хотя способы гидропереработки тяжелых углеводородных потоков, включая, но не ограничиваясь ими, тяжелые нефти, нефти, из которых отогнаны легкие фракции, и нефтяные углеводородные остатки, известны, использование каталитических способов на неподвижном слое для превращения такого исходного сырья без заметного осаждения асфальтенов и закупорки реактора и с эффективным удалением металлов и других загрязняющих примесей, таких как соединения серы и соединения азота, не является обычным вследствие того, что используемые катализаторы обычно не способны сохранять активность и эффективность.
Способы гидропревращения очень эффективно проводят в системе кипящего слоя. В кипящем слое предварительно нагретый водород и кубовый остаток входят в нижнюю часть реактора, в котором восходящий поток кубового остатка плюс внутренний рециркулирующий продукт суспендируют частицы катализатора в жидкой фазе. Последние усовершенствования заключались в использовании порошкообразного катализатора, который можно суспендировать без необходимости в рециркулирующей жидкости. В данной системе часть катализатора непрерывно или периодически удаляют в нескольких циклонах, а для поддержания активности добавляют свежий катализатор. Каждый день в системе кипящего слоя заменяют приблизительно около 1 масс.% наличного количества катализатора. Таким образом, общая активность системы представляет собой средневзвешенную активность катализатора при его изменении от свежего до очень старого, т.е. дезактивированного.
В целом, желательно разработать катализатор с наиболее высокой возможной площадью поверхности, чтобы обеспечить максимальную концентрацию каталитических центров и активность. Однако площадь поверхности и диаметр пор обратно пропорционально связаны в практических интервалах значений. Для диффузии, когда катализатор стареет и загрязняется, требуются достаточно большие поры, но большие поры имеют более низкую площадь поверхности.
Более конкретно, разработчик катализатора сталкивается с конкурирующими соображениями, которые часто диктуют, чтобы носители или полученные из них катализаторы обладали определенным балансом морфологических свойств, которые предполагается им придать.
Например, обнаружено (см., например, патент США №4497909), что хотя поры, имеющие диаметр ниже 60 ангстрем (в диапазоне, который называют здесь областью микропор), оказывают влияние на повышение числа активных центров некоторых катализаторов гидрогенизации типа диоксид кремния/оксид алюминия, те же самые центры являются центрами, в первую очередь закупориваемыми коксом, что тем самым вызывает снижение в активности. Аналогично этому, далее обнаружено, что когда такие катализаторы имеют более чем 10% общего объема пор, приходящегося на поры, имеющие диаметр больше, чем 600 ангстрем (в диапазоне, который называют здесь обычно областью макропор), механическая прочность по отношению к раздавливанию снижается, как снижается и активность катализатора. Наконец, для некоторых катализаторов типа диоксид кремния/оксид алюминия обнаружено, что максимизация пор, имеющих диаметр пор от 150 до 600 ангстрем (приблизительно в области, называемой здесь областью мезопор), является желательной для приемлемой активности и срока службы катализатора.
Таким образом, хотя повышение площади поверхности катализатора будет повышать число активных центров, такое повышение площади поверхности, естественно, приводит к увеличению доли пор в области микропор. Как указано выше, микропоры легко закупориваются коксом. Короче говоря, увеличение площади поверхности и максимизация мезопор являются антагонистическими свойствами.
Кроме того, не только площадь поверхности должна быть высокой, но она должна также оставаться стабильной при воздействии условий конверсии, таких как высокая температура и влажность. Поэтому происходит непрекращающийся поиск гидротермически стабильного оксида алюминия с высоким объемом пор и высокой площадью поверхности, пригодного в качестве носителя для катализаторов. Настоящее изобретение было создано в ответ на указанный поиск.
Патент США №4981825 относится к композициям частиц неорганического оксида металла (например, SiO2) и глины, в которых частицы оксида по существу отделены друг от друга частицами глины. Подходящие глины включают лапонит®. Описанное отношение оксид металла:глина находится между 1:1 и 20:1 (предпочтительно, от 4:1 до 10:1). Целевую композицию получают из золя неорганического оксида, имеющего размер частиц от 40 до 800 ангстрем (от 0,004 до 0,08 микрон). Размер частиц конечного продукта зависит от размера частиц в исходном золе, хотя конечный размер частиц не указывается. Критическим является то, что частицы оксида металла и глины имеют противоположные заряды, так что они притягиваются друг к другу, так что частицы глины ингибируют агрегацию частиц оксида металла. Таким образом, частицы глины описаны как помещенные между частицами золя. Регулирование зарядов на двух разных типах частиц определяется значением рН золя. Значение рН неорганического оксида регулируют так, чтобы оно было ниже его изоэлектрической точки, добавлением кислоты, тем самым вызывая образование положительного заряда на частицах неорганического оксида. Хотя описанные подходящие неорганические оксиды металлов включают также Al2O3, не представлены примеры проведения изобретения с использованием Al2O3. Следовательно, перенос данной концепции на Al2O3 осуществляется не без трудности. Например, изоэлектрическая точка Al2O3 находится при основном значении рН около 9. Однако соли Al2O3 образуются только при низком значении рН, меньшем, чем приблизительно 5. Если значение рН превышает приблизительно 5, золь Al2O3 будет осаждаться из дисперсии или никогда не образуется в первую очередь. В противоположность этому, золи SiO2 не должны быть кислотными. Следовательно, хотя любая точка ниже изоэлектрической точки приемлема для золей SiO2, то же самое не является верным для золей Al2O3. Скорее следует проводить операции при рН значительно ниже изоэлектрической точки Al2O3, в области значений рН, при которых образуются золи оксида алюминия. Кроме того, в указанном патенте ничего не говорится о свойствах пор образовавшегося композиционного материала и в нем делается упор только на то, чтобы получить высокую площадь поверхности. Как указано выше, площадь поверхности и высокий объем пор для мезопор обычно являются антагонистическими свойствами.
В противоположность этому, настоящее заявляемое изобретение не выходит из золя Al2O3 и образует золь во время повторной гидратации. Значение рН, при котором образуются заявляемые композиционные материалы, являются слишком высокими для того, чтобы золь образовался во время повторной гидратации, а исходные частицы оксида алюминия являются слишком большими для того, чтобы золь образовался вначале.
Другая область технологии, относящаяся к комбинациям различных глин и оксидов металлов, известна как интеркалированные глины. Интеркалированные глины представлены патентами США №№3803026; 3887454 (см. также 3844978); 3892655 (см. также 3844979); 4637992; 4761391 (см. также 4844790) и 4995964. Патенты интеркалированных глин обычно имеют общее требование, состоящее в том, чтобы применять большие отношения глина:золь. В интеркалированных глинах большая часть их площади поверхности обычно приходится на микропоры, если они не высушены замораживанием.
В патенте США 3803026 описывается гидрогель (или суспензия гидрогеля), содержащий воду, фторсодержащий компонент и аморфный когель, содержащий оксиды или гидроксиды кремния и алюминия. Аморфный когель дополнительно содержит оксид или гидроксид, по меньшей мере, одного элемента, выбранного из магния, цинка, бора, олова, титана, циркония, гафния, тория, лантана, церия, празеодима, неодима и фосфора, причем указанный аморфный когель присутствует в гидрогеле или суспензии гидрогеля в количестве от 5 до 50 масс.%. Значение рН суспензии устанавливают от 6 до 10, и условия превращения вызывают образование значительного количества кристаллического алюмосиликатного минерала, предпочтительно, в тесной смеси со значительным количеством непрореагировавшего аморфного когеля. Молярное отношение диоксид кремния/оксид алюминия составляет, по меньшей мере, 3:1, и образовавшийся материал называют синтетическим слоистым кристаллическим алюмосиликатным минералом типа глины, а непрореагировавший аморфный когель существует главным образом в виде SiO2. В колонке 5, строки 39 и последующие, описано, что образовавшийся алюмосиликат можно размельчить в частицы, пульверизовать в порошок, порошок диспергировать в гидрогеле или суспензии гидрогеля, к которой добавляют компоненты, выбранные из соединений-предшественников, помимо прочего оксида алюминия. Образовавшуюся смесь затем сушат и активируют.Тем не менее, вышеуказанное описание не описывает конкретные примеры с использованием смеси диоксида кремния-алюмината с оксидом алюминия. Следовательно, не описан ни исходный оксид алюминия, на конечный оксид алюминия, ни используемые количества каждого материала.
В патенте США №3887454 (и его родовом патенте 3844978) описан диоктаэдрический, подобный глине материал слоистого типа (LDCM), состоящий из диоксида кремния, оксида алюминия и имеющий включенный в его структуру оксид магния в регулируемых количествах. Предпочтительными глинами являются монтмориллонит и каолин. В колонке 6, строки 24 и последующие, описано, что материал глины может быть комбинирован обычно с компонентами из неорганических оксидов, таких как, помимо прочего, аморфный оксид алюминия. В противоположность этому, заявляемый здесь композиционный материал использует кристаллический оксид алюминия бемит. Аналогичные описания обнаружены в патентах США №№3892655 и 3844979, за исключением того, что указанные последние патенты относятся к триоктаэдрическому, подобному глине, материалу слоистого типа, содержащему оксид магния в качестве его компонента (LTCM) и иллюстрируются глиной типа сапонита.
Патент США №4637992 является патентом на интеркалированную глину, в котором используют коллоидную суспензию неорганических оксидов и добавляют к ним набухаемую глину. Хотя конкретные диапазоны, иллюстрирующие отношение глины к неорганическому оксиду, не описаны, оказывается, что конечный материал все же называют субстратом на основе глины, в который включен неорганический оксид. Следовательно, это предполагает, что конечный материал содержит в основном глину, а не преобладающее количество оксида алюминия и очень небольшие количества глины, как в настоящем изобретении. См., например, столбец 5, строки 46 и последующие патента '992.
Патент США №4844790 (раздел патента США №4761391) относится к деламинированной глине, полученной взаимодействием набухаемой глины с образующим столбики агентом, который включает оксид алюминия. Отношение глины к образующему столбики агенту составляет от 1:1 до 10:1, предпочтительно от 1:1 до 2:1. Однако главной основой патента является глина, содержащая оксид алюминия, а не оксид алюминия, содержащий менее чем 10 масс.% глины. Утверждается, что оксиды металлов удерживают пластинки глины порознь и придают им кислотность, которая является ответственной за каталитическую активность деламинированной глины. Предпочтительной глиной является лапонит®.
Патент США №4995964 относится к продукту, полученному включением в набухаемую глину (гекторит, сапонит, монтмориллонит) олигомеров, полученных из солей редкоземельных элементов и, в частности, трехвалентных редкоземельных элементов, и поливалентных катионов, образующих столбики (pillaring) металлов, таких как Al+3. Материалом оксида алюминия является алюминийсодержащий олигомер, который используют для получения столбиков набухших глин. В заявляемом здесь изобретении не используют или не получают олигомеры гидроксиматериалов алюминия.
В патенте США №4375406 описываются композиции, содержащие волокнистые глины и предварительно кальцинированные оксиды, полученные образованием жидкой суспензии глины с предварительно кальцинированным оксидом, перемешиванием суспензии с образованием содисперсии и формованием и сушкой содисперсии. Отношение образованной волокнистой глины к композиции предварительно кальцинированного оксида может варьироваться от 20:1 до 1:5. Эти количества значительно выше количеств глины, используемой в настоящем изобретении. Кроме того, волокнистая глина не входит в объем описанных здесь набухаемых глин.
Ряд патентов относится к различным типам оксида алюминия и способам их получения, а именно Re 29605; SIR H198 и патенты США №№3322495; 3417028; 3773691; 3850849; 3898322; 3974099; 2987155; 4045331; 4069140; 4073718; 4120943; 4175118; 4708945; 5032379 и 5266300.
Более конкретно, патент США 3974099 относится к гидрогелям диоксид кремния/оксид алюминия из когелей силиката натрия и алюмината натрия. Сущность указанного изобретения относится к осаждению Al2O3 на геле диоксид кремния-оксид алюминия, который стабилизирует центры крекинга по отношению к гидротермической дезактивации (столбец 2, строки 43 и последующие). Образующийся материал обычно содержит приблизительно 38,6% оксида алюминия, когда удаляют весь избыток алюмината натрия. В противоположность этому, диоксид кремния, используемый в настоящем изобретении, является добавкой, которая покрывает поверхность частиц композиционного материала оксид алюминия/глина, поскольку ее добавляют после образования композиционного материала.
В патенте США №4073718 описана основа катализатора из оксида алюминия, стабилизированного диоксидом кремния, на которую нанесен кобальтовый или никелевый катализатор.
В патенте США №4708945 описан катализатор крекинга из диоксида кремния, нанесенного на подобную бемиту поверхность формированием частиц пористого бемита и обработкой их водяным паром при температуре выше 500°С, чтобы вызвать взаимодействие диоксида кремния с бемитом. Обычно используют диоксид кремния в количестве 10%, чтобы достичь образования поверхностного монослоя диоксида кремния с целью повышения термической стабильности.
Патент США №5032379 относится к оксиду алюминия, имеющему объем пор больше, чем 0,4 см3/г и диаметр пор в диапазоне от 30 до 200 ангстрем. Оксид алюминия получают смешиванием двух разных типов способных связываться при повторной гидратации оксидов алюминия с получением продукта, имеющего бимодальное распределение пор.
В патенте США №4266300 описывается носитель из оксида алюминия, полученный смешиванием, по меньшей мере, двух тонкоизмельченных оксидов алюминия, каждый из которых характеризуется, по меньшей мере, одним типом пор, по меньшей мере, в одном из диапазонов (i) от 100000 до 10000 ангстрем, (ii) от 10000 до 1000 ангстрем, (iii) от 1000 до 30 ангстрем.
В патенте США №4791090 описан носитель катализатора с бидисперсным распределением размера микропор. В столбце 4, строка 65 описано, что два размера микропор можно получить смешиванием полностью различных материалов, имеющих различные размеры пор, таких как оксид алюминия и диоксид кремния.
Патент США №4497909 относится к носителям типа диоксид кремния/оксид алюминия, имеющим содержание диоксида кремния меньше, чем приблизительно 40% масс. и, по меньшей мере, один компонент, являющийся благородным металлом группы VII периодической таблицы, и где катализатор содержит поры, имеющие диаметр меньше, чем 600 ангстрем, и занимающие, по меньшей мере, 90% общего размера пор, и поры, имеющие диаметр от 150 до 600 ангстрем, и занимающие, по меньшей мере, 40% общего объема пор среди пор, имеющих диаметр меньше, чем 600 ангстрем.
В следующих патентах описаны различные типы глин: патенты США №№3586478; 4049780; 4629712 и публикации РСТ №№WO 93/11069 и WO 94/16996.
В следующих патентах описаны различные типы агломератов, которые можно образовать из оксида алюминия: патенты США №№3392125; 3630888; 3975510; 4124699; 4276201 (см. также 4309278); 4392987 и 5244648.
В патенте США 4276201 описан катализатор гидропереработки, в котором используют агломерированный носитель из оксида алюминия, например шариковый оксид алюминия, и диоксида кремния, где содержание диоксида кремния составляет меньше, чем 10 масс.% носителя. Агломерированный носитель имеет площадь поверхности 350-500 м2/г. Общий объем пор (TPV) составляет от 1,0 до 2,5 см3/г с менее чем 0,20 см3/г TPV, приходящимся на поры, имеющие диаметр больше, чем 400 ангстрем.
В патенте США №5114895 описана композиция слоистой глины, гомогенно диспергированной в матрице из неорганического оксида, так что слои глины полностью окружены матрицей из неорганического оксида. Матрицу из неорганического оксида выбирают из оксида алюминия, диоксида титана, диоксида кремния, диоксида циркония, P2O5 и их смесей. Подходящие глины включают бентонит, сепиолит, лапонит®, вермикулит, монтмориллонит, каолин, палыгорскит (аттапульгит), гекторит, хлорит, бейделлит, сапонит и нонтронит. Для получения глины, гомогенно диспергированной в матрице из неорганического оксида, предшественник неорганического оксида диспергируют в виде золя или гидрозоля и превращают в гель в присутствии глины. Хотя диапазоны содержания глины от 5 до 70 масс.% широко описаны, в примерах используют, по меньшей мере, 30 масс.% глины. Кроме того, не описано ни одно из свойств пор образовавшегося продукта.
В патенте США №4159969 описан способ получения агломератов оксида алюминия контактированием водного геля оксида алюминия с органической жидкостью, не смешиваемой с водой, где количество указанной жидкости является функцией воды в водном геле оксида алюминия. Во время или после образования геля к оксиду алюминия можно добавить некоторое количество глины, такой как бентонит или каолин, достаточное для повышения прочности агломератов. Не описывается конкретное количество глины, и каолин не является набухаемой глиной. Ни в одном из примеров не применяют глину.
В патенте США №3630888 описан катализатор, имеющий структуру, в которой доступные каналы, имеющие диаметры приблизительно от 100 до 1000 единиц, составляют от 10 до 40% общего объема пор, и в которой доступные каналы, имеющие диаметры больше, чем 1000 единиц, составляют приблизительно от 10 до приблизительно 40% общего объема пор, тогда как оставшаяся часть объема пор составляет от 20 до 80% микропор с диаметрами меньше, чем 100.
В следующих патентах описаны различные операции гидропереработки и использование в них катализаторов: патенты США №№3887455; 4657665; 4886594; публикация РСТ № WO 95/31280.
КРАТКОЕ ИЗЛОЖЕНИЕ СУЩНОСТИ ИЗОБРЕТЕНИЯ
Настоящее изобретение основано на том открытии, что когда активный оксид алюминия диспергируют и подвергают процессу повторной гидратации в присутствии регулируемых количеств диспергированной набухаемой глины, образовавшиеся частицы композиционного материала обнаруживают и сохраняют высокую площадь поверхности, в то же время обладая одновременно более высоким объемом пор и долей типа пор, относящихся к области мезопор, относительно варианта с отсутствием глины. Указанные свойства по существу сохраняются в агломератах, например формованных экструдатах, полученных из частиц композиционного материала до и после пропитки каталитически активными металлическими компонентами, такими как компоненты, используемые для операций гидропереработки. Кроме того, включение набухаемой глины повышает гидротермическую стабильность частиц композиционного материала.
Улучшения в гидротермической стабильности повышают общую экономику способа, в котором используют такие частицы композиционного материала, тогда как смещение в сторону больших значений моды пор в области мезопор повышает активность катализатора на носителе, полученном из частиц композиционного материала. Большее значение моды пор повышает доступность для углеводородов и снижает возможность пор закупориваться из-за осаждения кокса или металлов.
Оксиды алюминия с высоким объемом пор часто получают азеотропной перегонкой со спиртами для удаления воды перед сушкой. Спирт используют для снижения поверхностного натяжения воды, что, в свою очередь, снижает сжатие пор во время сушки. Указанный способ является очень дорогим и неблагоприятным для окружающей среды. Оксиды алюминия имеют высокий средний диаметр пор (APD) и их часто получают агломерацией (спеканием) при высокой температуре. Хотя агломерация повышает APD неагломерированного материала, она неизбежно снижает площадь поверхности относительно неагломерированного материала. Таким образом, это заставляет жертвовать площадью поверхности, чтобы достичь более высокого значения APD. Было обнаружено, что не только можно сместить моду пор в области мезопор в сторону пор большего размера до агломерации, но также считается, что будет иметь место меньшее сокращение диаметра пор при воздействии повышенных температур (обычно связанных с агломерацией без глины). Таким образом, поскольку способ получения можно начать с более высокими модами пор, и имеет место меньшее сокращение, связанное с этими более высокими модами пор, то продукт с высокой площадью поверхности, высоким объемом пор можно получить более эффективным по стоимости и благоприятным для окружающей среды способом, например можно исключить азеотропную перегонку со спиртом и можно снизить температуру, до которой оксид алюминия в противном случае нужно нагреть.
В соответствии с этим, в одном аспекте настоящего изобретения предлагаются частицы пористого композиционного материала, включающего компонент оксида алюминия, и компонент набухаемой глины, однородно диспергированной в компоненте оксида алюминия, где в указанных частицах композиционного материала:
(A) компонент оксида алюминия содержит, по меньшей мере, 75 масс.% оксида алюминия, по меньшей мере, причем 5% указанного оксида алюминия находится в форме кристаллического бемита, полученного из кристаллического бемита, гамма-оксида алюминия, или их смесей;
(B) компонент набухаемой глины является диспергируемым перед включением в частицы композиционного материала и присутствует в частицах композиционного материала в количестве, (а) меньшем, чем приблизительно 10 масс.%, в расчете на общую массу компонента оксида алюминия и компонента набухаемой глины, и (b) эффективном для повышения, по меньшей мере, одного показателя из числа гидротермической стабильности, объема пор по азоту и моды пор в области мезопор по азоту у частиц композиционного материала относительно соответствующей гидротермической стабильности, объема пор и моды пор в области мезопоры частиц компонента оксида алюминия в отсутствие указанной набухаемой глины, и
(C) средний диаметр частиц композиционного материала составляет приблизительно от 0,1 до приблизительно 100 микрон.
В следующем аспекте настоящего изобретения предложен способ получения частиц пористого композиционного материала, включающий:
(А) образование неколлоидной дисперсии, включающей, по меньшей мере, один компонент оксида алюминия, содержащий, по меньшей мере, 75 масс.% активного оксида алюминия, и, по меньшей мере, один компонент набухаемой глины в жидкой диспергирующей среде;
(B) повторную гидратацию активного оксида алюминия из компонента оксида алюминия в присутствии указанной диспергированной набухаемой глины с превращением, по меньшей мере, 5 масс.% активного оксида алюминия в кристаллический бемит и образованием частиц композиционного материала, включающего эффективное количество набухаемой глины, однородно диспергированной в компоненте оксида алюминия, причем указанное эффективное количество набухаемой глины является (i) меньшим, чем 10 масс.%, в расчете на общую массу компонента оксида алюминия и компонента набухаемой глины, и (ii) достаточным для обеспечения повышения значения, по меньшей мере, одного из следующих показателей: гидротермической стабильности, объема пор по азоту и моды пор в области мезопор по азоту у частиц композиционного материала относительно соответствующей гидротермической стабильности, объема пор и типа пор мезопор компонента оксида алюминия в отсутствие указанной набухаемой глины;
(C) выделение частиц композиционного материала из дисперсии и
(D) необязательное прокаливание выделенных частиц композиционного материала при температуре приблизительно от 250 до приблизительно 1000°С в течение времени приблизительно от 0,15 до приблизительно 3 часов.
В другом аспекте настоящего изобретения предложены агломераты вышеуказанных частиц.
В следующем аспекте настоящего изобретения предложены катализаторы на носителе, полученные из вышеуказанных агломератов.
Еще в одном аспекте настоящего изобретения предложен способ гидропереработки нефтяного исходного сырья с использованием вышеуказанных агломератов в качестве носителей для катализаторов гидропереработки.
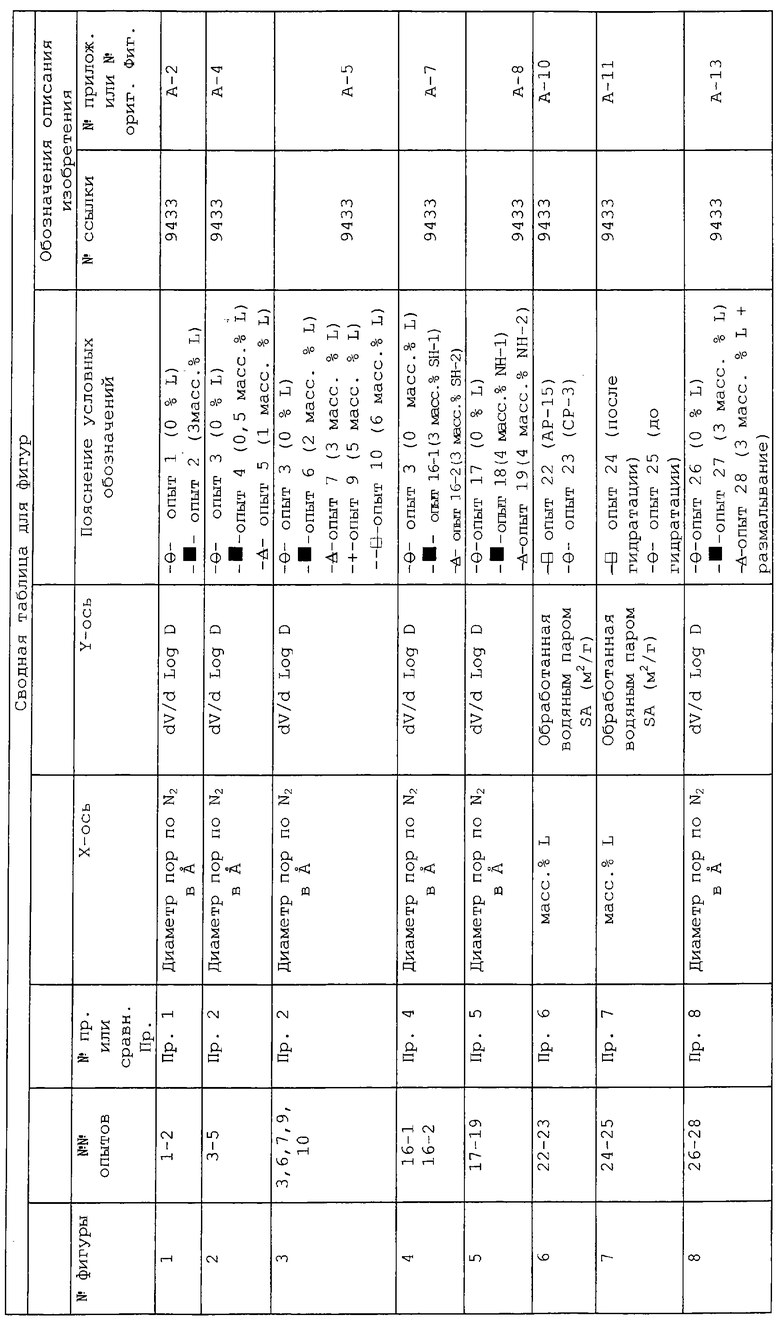
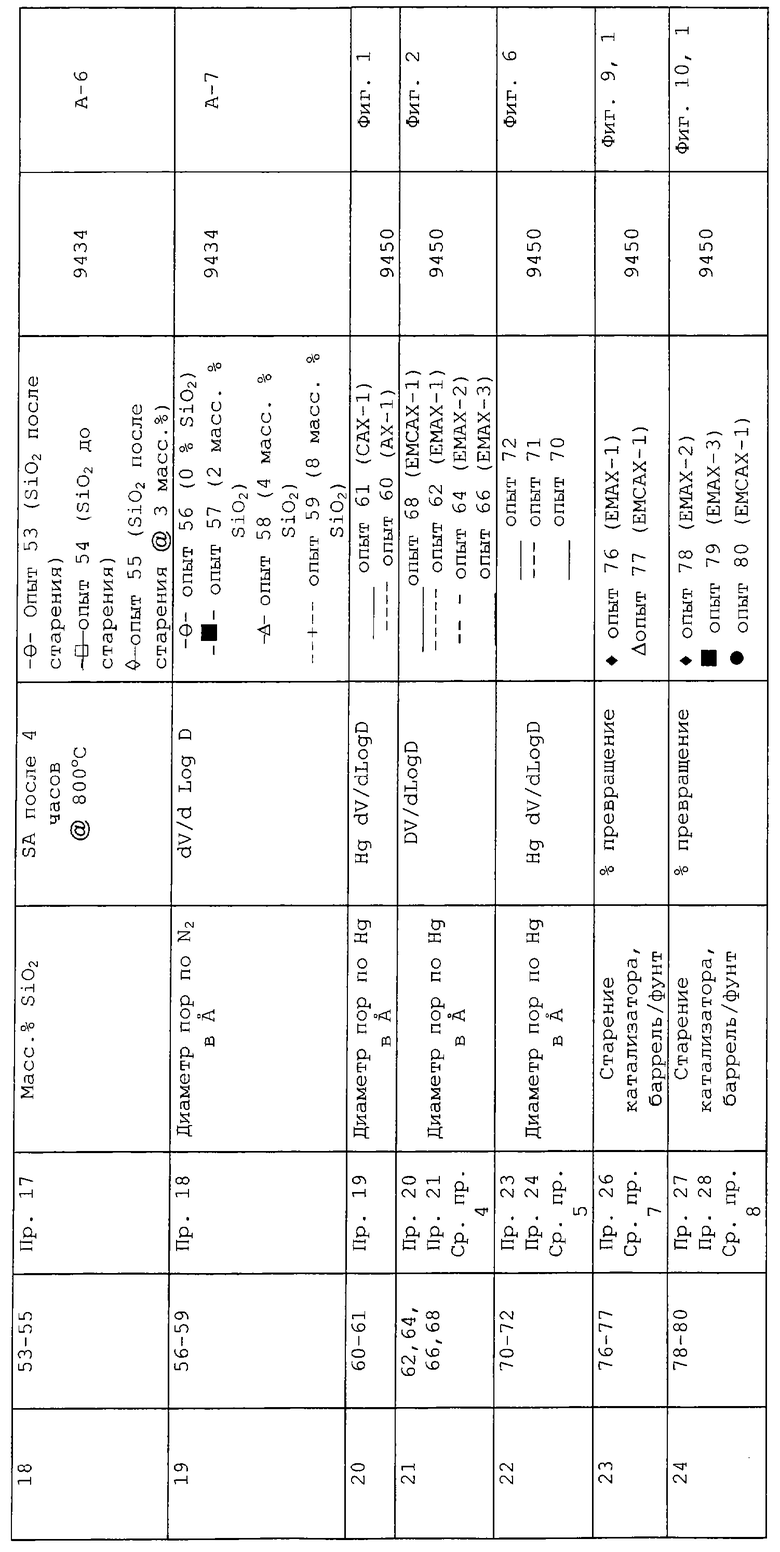
КРАТКОЕ ОПИСАНИЕ ГРАФИЧЕСКОГО МАТЕРИАЛА
В следующей таблице суммированы фигуры от 1 до 24, которые являются графиками, полученными на основе данных из примеров. Соответствующая информация о фигурах, включающая соответствующие номера опытов, номер примера или сравнительного примера, Х-ось, Y-ось и подрисуночные надписи графиков, представлены в следующей таблице:

ОПИСАНИЕ ПРЕДПОЧТИТЕЛЬНЫХ ОСУЩЕСТВЛЕНИЙ
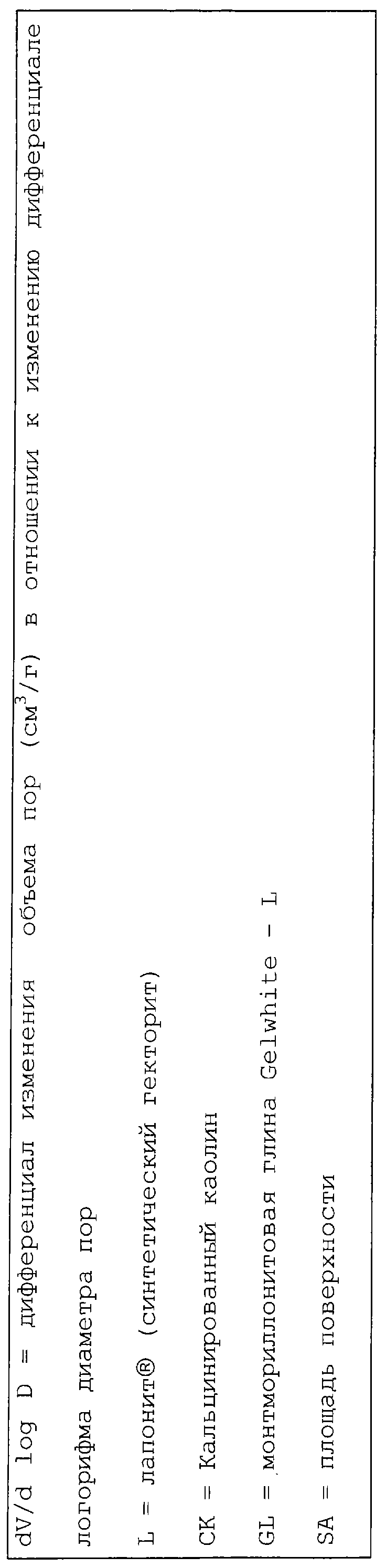
Термин «микропора», используемый здесь, означает поры, имеющие диаметр меньше, чем 100 ангстрем.
Термин «мезопора», используемый здесь, означает поры, имеющие диаметр от 100 до 500 ангстрем.
Термин «макропора», используемый здесь, означает поры, имеющие диаметр больше, чем 500 ангстрем.
Термин «мода пор», используемый здесь, означает диаметр пор, соответствующий максимальному значению (пику) на графике зависимости log дифференциала (приращения) интрузии азота или ртути в см3/г от дифференциала (приращения) log диаметра пор.
Термин «общий объем пор», используемый здесь, означает кумулятивный объем в см3/г всех пор, находимый при определении способами либо по десорбции азота, либо по пенетрации ртути. Более определенно, для частиц оксида алюминия, которые не были агломерированы (например, экструзией), распределение диаметра пор и объем пор вычисляют на основе изотермы десорбции азота (предполагая цилиндрические поры) по методике БЭТ, описанной S. Brunauer, P. Emmett and E. Teller в Journal of American Chemical Society, 60, pp 209-319 (1939).
Что касается частиц оксида алюминия, которые были агломерированы, например, превращены в экструдаты, распределение диаметра пор вычисляют при помощи формулы:
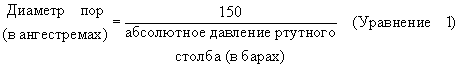
и в соответствии со способом пенетрации ртути (как описано H.L. Ritter and L.C. Drake в Industrial and Engineering Chemistry, Analytical Edition, 17, 787 (1945)) при использовании давлений ртутного столба 1-2000 бар. Площадь поверхности для частиц композиционного материала, а также агломератов определяют, однако, способом десорбции азота.
Общий объем пор образца по N2 является суммой объемов пор по азоту, как определено вышеописанным способом десорбции азота. Аналогично этому, общий объем пор по ртути образца является суммой объемов пор по ртути, как определено вышеописанным способом пенетрации ртути с использованием угла контакта 130°, поверхностного натяжения 485 дин/см и плотности ртути 13,5335 г/см3.
Все морфологические свойства, включая массу, такие как объем пор (см3/г) или площадь поверхности (м2/г), должны быть нормализованы по состоянию без металлов, как определено в соответствии с уравнением 4, описанным в примере 20.
Все площади свежих поверхностей определяли на образцах, которые были высушены и затем кальцинированы на воздухе при 537,8°С в течение 2 часов.
Насыпную плотность измеряли быстрым переносом (за 10 секунд) порошка образца в градуированный цилиндр, который переполняется, когда объем его достигает точно 100 см3. После этого момента порошок больше не добавляют. Скорость добавления порошка предотвращает отстаивание в цилиндре. Массу порошка делят на 100 см3, получая при этом плотность.
Все измерения размеров частиц и распределения размера частиц, описанные здесь, определяют устройством Mastersizer от Malvern, которое действует по принципу лазерной световой лазерной дифракции и является известным всем, знакомым с областью анализа малых частиц.
Компонент оксида алюминия, который смешивают с компонентом из набухаемой глины, включает обычно, по меньшей мере, 75, предпочтительно, по меньшей мере, 80 (например, по меньшей мере, 85), наиболее предпочтительно, по меньшей мере, 90 (например, по меньшей мере, 95) масс.% активного оксида алюминия, такое количество может обычно варьировать от приблизительно 75 до 100, предпочтительно от приблизительно 80 до 100 и наиболее предпочтительно от приблизительно 90 до 100 масс.% активного оксида алюминия. Активный оксид алюминия можно получить различными способами. Например, тригидрат оксида алюминия, осажденный способом Байера, можно измельчить и подвергнуть флэш-кальцинированию. Активный оксид алюминия, в том смысле как он здесь упоминается, характеризуется, тем, что он имеет слабую кристалличность и/или аморфную структуру.
Выражение «слабокристаллическая структура оксида алюминия» в отношении описания предшествующего способа понимается как обозначающее оксид алюминия, который является таким, что рентгеноструктурный анализ дает рентгенограмму, на которой обнаруживают только одну или несколько размытых линий, соответствующих кристаллическим фазам оксидов алюминия с низкотемпературным переходом, и содержит по существу хи-, ро-, эта-, гамма- и псевдогамма-фазы и их смеси.
Выражение «оксид алюминия аморфной структуры» означает оксид алюминия, который является таким, что его рентгеноструктурный анализ не дает никакую из линий, характеристичных для высоко (преимущественно) кристаллической фазы.
Активный оксид алюминия, используемый в настоящей заявке, можно обычно получить быстрой дегидратацией гидроксидов алюминия, таких как байерит, гидраргиллит или гиббсит и нордстрандит, или оксигидроксидов алюминия, таких как бемит и диаспор. Дегидратацию можно проводить в любой подходящей аппаратуре и с использованием горячего газообразного потока. Температура, при которой газы входят в аппаратуру, может обычно варьировать от приблизительно 400 до 1200°С, время контактирования гидроксида или оксигидроксида с горячими газами обычно находится между долей секунды и 4-5 секундами.
Образовавшийся продукт может содержать небольшие, например следовые, количества бемита, гиббсита, гамма, альфа, дельта и других кристаллических структур оксида алюминия.
Образовавшийся активный оксид алюминия обычно проявляет потерю массы приблизительно от 4 до 12 масс.% при нагревании до 538°С в течение 1 часа.
Удельная площадь поверхности активного оксида алюминия, полученного быстрой дегидратацией гидроксидов или оксигидроксидов, измеряемая общепринятым способом ВЕТ, обычно изменяется от приблизительно 50 до 400 м2/г, а диаметр частиц обычно составляет от 0,1 до 300 микрон и, предпочтительно, от 1 до 120 микрон со средним размером частиц обычно больше, чем 1 микрон, предпочтительно, приблизительно от 5 до приблизительно 20, наиболее предпочтительно от приблизительно 5 до приблизительно 15 микрон. Потеря зажигания, измеренная при помощи кальцинирования при 1000°С, обычно изменяется от 3 до 15%, которые соответствуют молярному отношению H2O/Al2O3 приблизительно от 0,17 до 1,0.
В предпочтительном осуществлении используют активный оксид алюминия, полученный быстрой дегидратацией гидрата Байера (гиббсита), который является легко доступным и недорогим промышленным гидроксидом алюминия. Активный оксид алюминия данного типа хорошо известен специалисту в данной области, и способ его получения был описан, например, в патентах США №№2915365; 3222129; 4579839 и, предпочтительно, 4051072, от столбца 3, строка 6, до столбца 4, строка 7, описание данных патентов включается здесь в качестве ссылки.
Применяемый активный оксид алюминия можно использовать как таковой или его можно обработать так, чтобы содержание в нем гидроксида натрия, выраженное как Na2O, было меньше, чем 1000 млн-1.
Более конкретно, частицы композиционного материала, полученные с силикатом или некоторыми глинами, такими как синтетический гекторит, обычно содержат Na2O, который может вызвать спекание оксида алюминия при высоких температурах. Такое спекание будет снижать площадь поверхности. Для исключения такого спекания оксид алюминия, предпочтительно, промывают для удаления Na2O в форме солей. Еще более определенно, оксид алюминия, предпочтительно, суспендируют в воде, содержащей приблизительно 0,05 массовых частей сульфата аммония (А/S), приблизительно 1 массовую часть оксида алюминия и 5 массовых частей воды, в течение 15 минут. Суспензию затем фильтруют, промывают, по меньшей мере, один раз водой для удаления солей и сушат в печи. Указанную промывку можно проводить до или после контактирования с глиной или для любого компонента, который может иметь Na2O. Используемый активный оксид алюминия можно размалывать или не размалывать, но, предпочтительно, его размалывают для облегчения диспергирования в суспензии набухаемой глины, описанной ниже, или с указанной суспензией.
Подходящий порошок активного оксида алюминия, являющийся исходным материалом, коммерчески доступен от Aluminium Company of America с сортами, обозначенными как СР-3, СР-1, СР-5, СР-7 и СР-100. Он доступен также от Porocel (Little Rock, Arkansas) под наименованием АР-15.
Все активные оксиды алюминия, подходящие для использования в компоненте оксида алюминия настоящего изобретения, являются повторно гидратируемыми и образуют гидроксильную связь при контактировании с водой. Настоящее изобретение проводит различие между явлением повторной гидратации, т.е. химическими изменениями, вызванными действием воды и повышенных температур на активный оксид алюминия, и процессом повторной гидратации, т.е. стадиями способа, сопряженными с индуцированием явления повторной гидратации.
Считается, что явление повторной гидратации представляет собой химическое и физическое состояние такого активного оксида алюминия, который был превращен в кристаллический бемит. Однако изменение в состоянии из активного оксида алюминия в бемит не обязательно должно быть связано с утверждением, что процесс повторной гидратации действует на весь образец. Например, в зависимости от условий процесса повторной гидратации, может быть возможно, что только внешняя оболочка частиц активного оксида алюминия или фильтровальной лепешки превращается в бемит, при том, что остальные внутренние части его остаются в виде либо активного оксида алюминия, либо некоторой формы оксида алюминия, иной чем бемит или активный оксид алюминия. Таким образом, хотя «повторно гидратированный оксид алюминия» является химически синонимом бемита, оксид алюминия, полученный повторной гидратацией активного оксида алюминия, включает бемит, активный оксид алюминия и любые побочные продукты оксида алюминия, иные чем бемит, которые могут образоваться во время процесса повторной гидратации. Аналогично этому, процесс повторной гидратации относится к манипулятивным стадиям способа, связанным с добавлением активного оксида алюминия к воде в условиях, например, повышенной температуры, описанных ниже.
Компонент набухаемой глины включает любую из глин с отношением глина:минеральный слоистый силикат 2:1, способных подвергаться набуханию и диспергированию, и их смеси. Набухаемые глины являются увеличивающимися в объеме глинами, пластинки которых удерживаются вместе слабыми ван-дер-ваальсовыми силами и имеют определенную форму или морфологию. Такие глины включают класс глин - смектиты, а также их ионообменные (например, Na+, Li+) производные. В целом, формы с обменом щелочного металла являются предпочтительными вследствие их повышенной способности набухать и диспергироваться. Пригодными являются также диспергируемые слоистые силикаты 2:1, такие как тетракремниевая слюда и тайниолит.
Более конкретно, смектиты являются глинистыми минералами 2:1, которые имеют заряд решетки и типичным образом расширяются при сольватации водой и спиртами, особенно этиленгликолем и глицерином. Указанные минералы включают слои, представленные общей формулой:
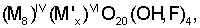
где IV обозначает ион, координированный с четырьмя другими ионами, VI обозначает ион, координированный с шестью другими ионами, и х может быть равен 4 или 6. М обычно представляет Si4+, Al3+и/или Fe3+, но включает также несколько других тетракоординированных ионов, таких как Р5+, В3+, Ge4+, Be2+и тому подобное. М' обычно представляет Al3+ или Mg24, но включает также много возможных гексакоординированных ионов, таких как Fe3+, Fe2+, Ni2+, Co2+, Li4+ и тому подобное. Дефициты зарядов, вызванные различными замещениями в положениях указанных тетра- и гексакоординатных катионов, уравновешиваются одним или несколькими катионами, расположенными между структурными звеньями. Вода также может быть окклюдирована между указанными структурными звеньями, связанными либо с самой структурой, либо с катионами как гидратирующая оболочка. Будучи дегидратированными (дегидроксилированными), вышеуказанные структурные единицы имеют повторяющееся расстояние приблизительно от 9 до 12 ангстрем, при измерении по рентгенограмме. Коммерчески доступные природные смектиты включают монтмориллонит (бентонит), бейделлит, гекторит, сапонит, сауконит и нонтронит. Коммерчески доступными являются также синтетические смектиты, такие как лапонит®, синтетический гекторит, доступный от Laporte Industries Limited.
Смектиты классифицируют на две категории, диоктаэдрические и триоктаэдрические, причем различием их является число октаэдрических центров в центральном слое, которые заняты. Это, в свою очередь, связано с валентностью катиона в центральных слоях.
Диоктаэдрические смектиты имеют центральные катионы, которые являются трехвалентными и соответственно этому занимают только две трети октаэдрических центров, тогда как триоктаэдрические смектиты имеют двухвалентные центральные катионы, где заняты все октаэдрические центры. Диоктаэдрические смектиты включают монтмориллонит, бейделлит и нонтронит, где, например, монтмориллонит имеет октаэдрический катион (М') алюминий, и присутствуют также другие катионы, такие как магний. Триоктаэдрические смектиты, которые являются предпочтительными, включают гекторит и сапонит и их синтетические формы, среди которых, например, гекторит имеет октаэдрический катион (М') магний, и присутствует также литий.
Смектитом, который наиболее выгодно использовать при получении композиций данного изобретения, является триоктаэдрическая смектитовая глина, имеющая полоскообразную морфологию. Однако можно использовать триоктаэдрические смектиты пластинообразной или смешанной полоскообразной и пластинообразной морфологии. Примерами подходящих триоктаэдрических смектитовых глин являются природный сапонит и, что предпочтительно, природный гекторит и синтетический гекторит.
Наиболее предпочтительной набухающей глиной для использования в качестве компонента набухаемой глины являются синтетические гекториты. Процедуры получения синтетических гекторитов хорошо известны и описаны, например, в патентах США №№3803026; 3844979; 3887454; 3892655 и 4049780, описание которых включается здесь в качестве ссылки. Типичным примером синтетического гекторита является лапонит® RD. Глина лапонит® RD является отжатым на фильтре, высушенным на противне и размолотым стержневой мельницей продуктом. Пластины глины лапонит® RD состоят из двух слоев диоксида кремния, окружающих слой магния в октаэдрической координации с замещением литием в данном слое. Глину лапонит® RD и другие лапониты изготовляет и продает Laporte Inorganics, часть Laporte Industries Limited. Типичный анализ и физические свойства глины лапонит® RD представлены ниже, в таблице 1.
Для получения частиц композиционного материала настоящего изобретения неколлоидный активный оксид алюминия, по меньшей мере частично, повторно гидратируют в присутствии диспергированной набухаемой глины.
Повторная гидратация оксида алюминия может, естественно, в конечном счете происходить при комнатной температуре в присутствии воды, но будет занимать продолжительный период времени. Поэтому повторную гидратацию предпочтительно проводят при повышенных температурах, по меньшей мере около 50°С, чтобы ускорить процесс повторной гидратации. Удобно проводить повторную гидратацию простым кипячением с обратным холодильником водной суспензии активного оксида алюминия в течение периода в типичном случае от приблизительно 1 до приблизительно 72, предпочтительно от приблизительно 2 до приблизительно 48 и наиболее предпочтительно от приблизительно 3 до приблизительно 24 часов.
Условия повторной гидратации регулируют так, чтобы получить продукт с высоким объемом пор. В соответствии с этим, условия повторной гидратации регулируют так, чтобы в типичном случае, по меньшей мере, 5, предпочтительно, по меньшей мере, 10 и наиболее предпочтительно, по меньшей мере, 15 масс.% активного оксида алюминия превращено в бемит, и содержание бемита в оксиде алюминия, полученного повторной гидратацией активного оксида алюминия, может составлять в типичном случае приблизительно от 5 до приблизительно 100 (например, от 30 до 100), предпочтительно приблизительно от 10 до приблизительно 100 (например, от 50 до 100), наиболее предпочтительно от приблизительно 15 до приблизительно 100 (например, от 75 до 100) масс.%, в расчете на массу оксида алюминия. Нежелательным побочным продуктом образования бемита является байерит, который является тригидратом оксида алюминия, который образуется, если рН воды превышает приблизительно 10.
С точки зрения первоначального содержания активного оксида алюминия в компоненте из оксида алюминия и степени превращения активного оксида алюминия в кристаллический бемит, компонент из оксида алюминия частиц композиционного материала должен желательно содержать (А) обычно, по меньшей мере, 75, предпочтительно, по меньшей мере, 80 (например, по меньшей мере, 85) и наиболее предпочтительно, по меньшей мере, 90 (например, по меньшей мере, 95) масс.% оксида алюминия, предпочтительно, оксида алюминия, образованного повторной гидратацией активного оксида алюминия, и (В) обычно, по меньшей мере, 3,75 и, предпочтительно, по меньшей мере, 75 и наиболее предпочтительно, по меньшей мере, 10 масс.% компонента оксида алюминия является кристаллическим бемитом, причем количество кристаллического бемита может изменяться обычно приблизительно от 3,75 до приблизительно 100 (например, 40-100), предпочтительно, приблизительно от 7,5 до приблизительно 100 (например, 75-100) и, наиболее предпочтительно, приблизительно от 10 до приблизительно 100 (например, 90-100) масс.%, в расчете на массу компонента оксида алюминия. Аналогично, массовое отношение кристаллического бемита к набухаемой глине в частицах композиционного материала обычно будет изменяться приблизительно от 4:1 до приблизительно 99:1, предпочтительно, приблизительно от 9:1 до приблизительно 50:1 и наиболее предпочтительно, приблизительно от 15:1 до приблизительно 50:1.
Размер кристаллитов (как он определен процедурой, описанной в примере 1) обычно будет меньше, чем приблизительно 110 ангстрем (например, меньше, чем приблизительно 100) и в типичном случае будет составлять приблизительно от 55 до приблизительно 110, предпочтительно, приблизительно от 60 до приблизительно 100 и наиболее предпочтительно, приблизительно от 65 до приблизительно 95 ангстрем.
Образование бемита максимизируется при рН приблизительно 9 (например, от 7 до 10). Так, для стабилизации рН приблизительно при 9 можно добавить буфер, такой как глюконат натрия, но такая добавка может иметь нежелательное влияние на снижение размера кристаллитов бемита, что, в свою очередь, приводит к снижению общего объема пор. Таким образом, предпочтительным является образование бемита без использования буфера. Фактически одним из преимуществ набухаемой глины является то, что она является природным буфером при рН приблизительно 9 и ингибирует повторную гидратацию в байерит.
Как указано выше, повторная гидратация активного оксида алюминия в компоненте из оксида алюминия должна иметь место в присутствии диспергированной набухаемой глины. Без намерения быть связанным с любой конкретной теорией, считают, что высокодиспергированная набухаемая глина захватывается внутрь растущих кристаллов бемита и вызывает образование межкристаллических пустот поддержкой кристаллитов на расстоянии, тем самым повышая объем пор без снижения площади поверхности. По этой причине считают, чем меньше размер частиц набухаемой глины и чем выше степень дисперсности частиц глины в суспензии, тем выше для частиц композиционного материала будет смещение моды пор в область мезопор. Повторную гидратацию оксида алюминия не начинают с золя оксида алюминия и не превращают активный оксид алюминия во время повторной гидратации в золь активного оксида алюминия. Кроме того, если набухаемую глину просто смешивают с предварительно полученным бемитом, а не с бемитом, образуемым, например, повторной гидратацией активного оксида алюминия в присутствии глины, улучшенные свойства пор достигнуты не будут.
В предпочтительном осуществлении компонент оксида алюминия можно предварительно размолоть перед повторной гидратацией активного оксида алюминия, отдельно или в смеси с набухаемой глиной. Предварительное размалывание можно проводить в мельнице мокрого помола, такой как DRAIS, PREMIER или другие типы песочных или галочных барабанных мельниц.
Однако, если предварительное размалывание проводят в отсутствие желательной набухаемой глины, его нужно проводить в условиях, например, достаточно низкой температуры, чтобы избежать преждевременной повторной гидратации оксида алюминия до его контактирования с диспергированной набухаемой глиной.
Предварительное размалывание компонента оксида алюминия обычно проводят при комнатной температуре в течение периода, достаточного для снижения среднего размера частиц, так чтобы он составлял в типичном случае приблизительно от 0,1 до приблизительно 8 (например, от 1 до 8), предпочтительно, приблизительно от 0,1 до приблизительно 5 (например, от 1 до 5) и наиболее предпочтительно, приблизительно от 0,1 до приблизительно 2,5 микрон.
Компонент, являющийся набухаемой глиной, диспергируют в суспензии, обычно в водной суспензии, в условиях, которые предпочтительно будут максимизировать степень дисперсности. Некоторые набухаемые глины являются более легко диспергируемыми, чем другие. Если степень дисперсности, достигаемая во время контактирования с оксидом алюминия, который повторно гидратируется, является низкой, нужное влияние на свойства пор оксида алюминия не может быть достигнуто или максимизировано. В соответствии с этим, может быть необходимо применение стадий для достижения подходящей степени дисперсности, таких как размалывание, регулирование содержания общих летучих компонентов и/или использование вспомогательных диспергирующих средств, таких как тетранатрийпирофосфат (Na4P2O7). Медленное добавление глины к деионизированной воде или воде, содержащей Na4P2O7 для минимизации количества двухвалентных катионов, таких как Са+2 и Mg+2, помогает диспергировать глину. Время, требуемое для диспергирования глины, снижается, если ее добавляют к мешалке с высоким сдвигающим усилием, такой как мешалка COWLES, MYERS или SILVERSON. Удовлетворительную дисперсию можно получить с мешалками типа лопастных, особенно, когда используют емкость с дефлекторами.
Достижение подходящей степени дисперсности трудно оценить количественно, но, в качестве общего правила, чем выше степень прозрачности суспендирующей среды, тем лучше дисперсия, и полностью прозрачная среда наиболее предпочтительна при использовании синтетического гекторита. Это обычно имеет место, когда частицы глины являются преимущественно коллоидными по размеру, например менее чем приблизительно 1 микрон.
В соответствии с этим дисперсию набухаемой глины можно получить смешиванием глины с водой, предпочтительно, в условиях высокого сдвигающего усилия в течение периодов обычно приблизительно от 5 до приблизительно 60 и предпочтительно приблизительно от 10 до приблизительно 30 минут. Температура, при которой образуется дисперсия, не является критической и обычно составляет приблизительно от 10 до приблизительно 60°С. Важно, чтобы вода не содержала другие минералы (например, предпочтительна деионизированная вода), которые могут оказывать влияние на диспергируемость глины.
Степень дисперсности повышается, если исходная глина имеет общее содержание летучих компонентов обычно, по меньшей мере, 6 и, предпочтительно, по меньшей мере, 8 масс.%, и содержание их может составлять обычно приблизительно от 6 до приблизительно 30, предпочтительно, приблизительно от 10 до приблизительно 20 и, наиболее предпочтительно, приблизительно от 12 до приблизительно 18 масс.%.
Количество глины, которое предлагается ввести в конечные частицы композиционного материала, выбирают так, чтобы оно было эффективным для повышения, по меньшей мере, одного из следующих параметров: общий объем пор по азоту, гидротермическая стабильность и/или мода пор по азоту в области мезопор относительно соответствующего объема пор, гидротермическая стабильность (как определено ниже) и мода пор в области мезопор компонента оксида алюминия в отсутствие набухаемой глины. Более конкретно, мода пор в области мезопор обычно повышается по меньшей мере на 10, предпочтительно, по меньшей мере, на 30 и, очень предпочтительно, по меньшей мере, на 50% от соответствующей моды пор в области мезопор, достигаемой в отсутствие набухаемой глины.
Подходящие эффективные количества набухаемой глины обычно меньше, чем приблизительно 10 (например, меньше, чем приблизительно 9), предпочтительно, меньше, чем приблизительно 8, наиболее предпочтительно, меньше, чем приблизительно 6 масс.% и может изменяться обычно приблизительно от 1 до приблизительно от 1 до приблизительно 9 (например, от 1 до приблизительно 8), предпочтительно, приблизительно от 2 до приблизительно 7 и наиболее предпочтительно приблизительно от 2 до приблизительно 5 масс.%, в расчете на общую массу компонента оксида алюминия и компонента смачиваемой глины.
Когда содержание глины частиц композиционного материала повышается выше 1 масс.%, повышается не только объем пор в области мезопор, вплоть до приблизительно 6 масс.% набухаемой глины, после чего он начинает снижаться, но также и площадь поверхности. Кроме того, присутствие глины повышает гидротермическую стабильность частиц композиционного материала вплоть до содержания глины приблизительно 10 масс.%, после чего уровень стабильности прекращает повышаться или снижается.
Из вышеприведенного обсуждения должно быть ясно, что повторную гидратацию оксида алюминия в присутствии диспергированной набухаемой глины можно проводить различными путями.
Например, две отдельно полученные суспензии (дисперсии), содержащие компонент набухаемой глины и компонент неколлоидного оксида алюминия, соответственно, можно объединить или, что предпочтительно, можно получить одну суспензию непосредственно добавлением к воде сначала любого из компонентов или одновременным смешиванием компонентов глины и оксида алюминия с водой.
Однако, если получают две отдельные суспензии, нужно принимать меры, чтобы гарантировать, что повторная гидратация активного оксида алюминия в компоненте оксида алюминия не имеет места преждевременно, до контактирования с диспергированной глиной.
Содержание твердых веществ суспензии, содержащей компонент оксида алюминия и компонент из глины, регулируют так, чтобы оно обычно составляло приблизительно от 2 до приблизительно 30, предпочтительно приблизительно от 4 до приблизительно 25 и наиболее предпочтительно приблизительно от 5 до приблизительно 25 масс.%, в расчете на массу суспензии. Когда содержание твердых веществ снижается в указанных диапазонах, мода пор в области мезопор обычно будет увеличиваться, и наоборот, если содержание глины в масс.% составляет 4 или ниже.
В соответствии с этим, при отсутствии предварительного размалывания глинистого компонента и компонента оксида алюминия для получения суспензии диспергируемого глинистого компонента в диспергированной форме предпочтительно добавить к ней компонент оксида алюминия и затем подвергнуть смесь перемешиванию со сдвигом смеси при повышенной температуре, как описано выше, для однородного диспергирования набухаемой глины и повторной гидратации оксида алюминия.
В одном предпочтительном осуществлении диспергированный глинистый компонент предварительно размалывают в смеси с компонентом из оксида алюминия перед повторной гидратацией оксида алюминия. Таким образом, в данном осуществлении дисперсию набухаемого глинистого компонента получают при перемешивании до полного диспергирования. К дисперсии глины добавляют подходящее количество компонента оксида алюминия и образовавшуюся комбинацию размалывают способом мокрого помола, предпочтительно, тщательно размалывают, при комнатной температуре, например, в мельнице DRAIS, в течение периода обычно приблизительно от 1,0 до приблизительно 3, предпочтительно, приблизительно от 0,5 до приблизительно 2,0 минут. Предварительно размолотую суспензию затем кипятят с обратным холодильником, как описано выше, для повторной гидратации оксида алюминия.
Обнаружено, что предварительное размалывание приводит к повышенной гидротермической стабильности композиции, в то же время приводя к небольшому сдвигу в сторону меньших пор.
Более определенно, гидротермическую стабильность частиц композиционного материала на основе оксида алюминия оценивают сравнением площадей свежих и обработанных водяным паром поверхностей следующим образом.
Площадь поверхности по БЭТ N2 определяют после кальцинирования на воздухе при 537,8°С (1000°F) в течение 2 часов и обозначают как площадь свежей поверхности. Непрокаленный образец затем подвергают действию атмосферы, содержащей приблизительно 20 об. % водяного пара в течение 4 часов при 800°С и автогенном давлении и определяют площадь его поверхности по БЭТ и обозначают как площадь обработанной водяным паром поверхности.
Затем проводят сравнение между площадями свежей и обработанной водяным паром поверхностей. Чем меньше разница между площадями свежей и обработанной водяным паром поверхностей, тем выше гидротермическая стабильность.
После того, как повторная гидратация активного оксида алюминия (в компоненте оксида алюминия) в присутствии набухаемого глинистого компонента завершается, образовавшиеся частицы композиционного материала можно выделить, термическим способом активировать в тех же условиях, как описано ниже для агломератов, или использовать непосредственно для нанесения на них катализатора.
Предпочтительно, частицы композиционного материала выделяют и сушат и, необязательно, сортируют по размерам. Подходящие размеры частиц могут составлять обычно приблизительно от 1 до приблизительно 150 (например, от 1 до приблизительно 100), предпочтительно приблизительно от 2 до приблизительно 60 и наиболее предпочтительно приблизительно от 2 до приблизительно 50 микрон.
Выделение выполняют фильтрованием, упариванием, центрифугированием и тому подобное. Суспензию можно также сушить распылением для осуществления выделения.
Образовавшиеся частицы композиционного материала имеют площадь поверхности БЭТ (на основе, не содержащей металлов) обычно, по меньшей мере, приблизительно 200, предпочтительно, по меньшей мере, приблизительно 240 и наиболее предпочтительно, по меньшей мере, приблизительно 260 м2/г, а площадь поверхности обычно может составлять от приблизительно 200 до приблизительно 400, предпочтительно, приблизительно от 240 до приблизительно 350 и, наиболее предпочтительно, приблизительно от 240 до приблизительно 300 м2/г. Определение площади поверхности проводят на образце, который был высушен при 138°С (280°F) в течение 8 часов и кальцинирован в течение 2 часов при 537,8°С (1000°F).
Средний диаметр пор по азоту частиц композиционного материала обычно составляет приблизительно от 60 до приблизительно 400 (например, от 60 до приблизительно 300), предпочтительно, приблизительно от 70 до приблизительно 275 и, наиболее предпочтительно, приблизительно от 80 до приблизительно 250 ангстрем.
Общий объем пор по азоту частиц композиционного материала (на основе, не содержащей металлов) может изменяться приблизительно от 0,5 до приблизительно 2,0, предпочтительно, приблизительно от 0,6 до приблизительно 1,8 и, наиболее предпочтительно, приблизительно от 0,7 до приблизительно 1,6 см3/г. Перед проверкой диаметра пор и объема пор образцы сушат в печи при 138°С (280°F) и затем кальцинируют в течение 2 часов при 537,8°С (1000°F).
Преимуществом настоящего изобретения является то, что набухаемая глина смещает (сдвигает) моду пор в области мезопор в сторону большего диаметра пор относительно случая ее отсутствия, в то же время при сохранении высокой площади поверхности, как указано выше.
Еще более важно, что настоящее изобретение предлагает механизм регулирования размера моды пор варьированием условий получения, в частности содержания глины в композиционном материале и содержания твердых веществ в суспензии повторной гидратации. Более конкретно, снижение содержания глины от оптимального и/или повышение содержания твердых веществ в суспензии повторной гидратации, будут каждое понижать тип пор.
Таким образом, содержание макропор (т.е. % таких пор в пределах общего объема пор по азоту, который находится в пределах области макропор) частиц композиционного материала обычно составляет не более чем приблизительно 40, предпочтительно, не более чем приблизительно 30, и наиболее предпочтительно, не более чем приблизительно 25% общего объема пор, содержание макропор обычно составляет приблизительно от 5 до приблизительно 50, предпочтительно, приблизительно от 10 до приблизительно 40 и наиболее предпочтительно, приблизительно от 10 до приблизительно 30% общего объема пор.
Содержание мезопор по азоту обычно составляет приблизительно от 20 до приблизительно 90, предпочтительно, приблизительно от 30 до приблизительно 80 и, наиболее предпочтительно, приблизительно от 40 до приблизительно 70% общего объема пор. Кроме того, обычно, по меньшей мере, приблизительно 40, предпочтительно, по меньшей мере, 50 и, наиболее предпочтительно, по меньшей мере, 60% пор в пределах области мезопор имеют диаметры пор обычно приблизительно от 100 до приблизительно 400, предпочтительно приблизительно от 100 до приблизительно 350 и, наиболее предпочтительно, приблизительно от 125 до приблизительно 300 ангстрем.
Мезопоры по азоту таким образом образуемых частиц композиционного материала также желательно имеют тип пор по азоту, предпочтительно, только один тип пор (мономодальный), обычно приблизительно от 60 до приблизительно 400 (например, от 60 до приблизительно 300), предпочтительно, приблизительно от 70 до приблизительно 275 и, очень предпочтительно, приблизительно от 80 до приблизительно 250 ангстрем.
Содержание микропор по азоту для частиц композиционного материала обычно не выше, чем приблизительно 80, предпочтительно, не выше, чем приблизительно 60, и, наиболее предпочтительно, не выше, чем приблизительно 50% общего объема пор, при этом содержание микропор может составлять обычно приблизительно от 80 до приблизительно 5, предпочтительно, приблизительно от 60 до приблизительно 10 и наиболее предпочтительно, приблизительно от 30 до приблизительно 15% общего объема пор.
Кроме того, было обнаружено, что гидротермическую стабильность частиц композиционного материала можно дополнительно повысить включением в них силикатных солей.
Подходящие силикатные соли включают силикаты щелочных и щелочноземельных металлов, наиболее предпочтительно силикат натрия. Менее растворимые силикаты, такие как обнаружены в природных или синтетических глинах или силикагелях, также повышают стабильность. Примерами таких глин являются каолинит, монтмориллонит и гекторит. Кальцинированные глины также дают повышенную гидротермическую стабильность.
Силикат можно добавить к компонентам - оксиду алюминия и набухаемой глине перед повторной гидратацией, но предпочтительным является проведение добавления после повторной гидратации (горячее старение), чтобы максимизировать воздействия, придающие гидротермическую стабильность, и получить высокий объем пор и большой средний диаметр пор. Добавление растворимого силиката перед повторной гидратацией оксида алюминия имеет тенденцию образовывать небольшие поры, которые несколько менее стабильны (т.е. коалесцируют в большие поры при нагревании), чем большие поры, тем самым снижая общий объем пор. Силикат можно добавить через несколько часов горячего старения после того, как установится распределение размера пор.
Количество силиката, эффективное для повышения гидротермической стабильности описанных здесь частиц композиционного материала, может обычно составлять приблизительно от 0,1 до приблизительно 40, приблизительно от 1 до приблизительно 20 и наиболее предпочтительно приблизительно от 2 до приблизительно 10 масс.%, в расчете на общую массу силиката, компонента оксида алюминия и набухаемого глинистого компонента.
Без намерения быть связанными любой конкретной теорией, полагают, что добавленный силикат отличается от силиката в глине тем, что первый, как считается, может свободно мигрировать к оксиду алюминия во время повторной гидратации, тогда как силикат глины остается большей частью незатронутым во время повторной гидратации. Однако некоторую часть наблюдаемого влияния глины на размер пор и стабильность можно отнести к силикату, мигрировавшему из глины к оксиду алюминия во время повторной гидратации.
Хотя частицы оксида алюминия композиционного материала можно использовать непосредственно в качестве носителей, более общепринятым для такой цели является агломерация частиц.
Такие агломераты оксида алюминия можно использовать в качестве катализаторов или носителей катализаторов в любой реакции, которая требует конкретной структуры пор наряду с очень хорошими механическими, термическими и гидротермическими свойствами. Агломераты настоящего изобретения могут, таким образом, найти конкретное применение в качестве носителей катализаторов при обработке выхлопных газов, генерируемых двигателями внутреннего сгорания, и при обработке водородом нефтяных продуктов, такой как гидродесульфуризация, гидродеметаллизация и гидроденитрификация. Их можно также использовать в качестве носителей катализаторов в реакциях для выделения соединений серы (катализаторы Клауса), дегидратации, реформинга, риформинга с водяным паром, дегидрогалогенирования, гидрокрекинга, гидрирования, дегидрирования и дегидроциклизации углеводородов или других органических соединений, а также реакций окисления и восстановления. Их можно также использовать в качестве добавок для флюидизированных катализаторов крекинга, особенно для повышения объема пор и мезо- или макропористости.
Их можно также использовать в качестве катализаторов per se в реакциях, обычно катализируемых оксидами алюминия, таких как реакции гидрокрекинга и изомеризации.
Таким образом, выгодные свойства повышенного содержания мезопор при высокой площади поверхности и гидротермической стабильности частиц композиционного материала переходят к агломератам.
Термин «агломерат» относится к продукту, который объединяет частицы, которые удерживаются вместе различными физическими-химическими силами.
Более конкретно, каждый агломерат состоит из множества соприкасающихся, составляющих агломерат первичных частиц, сортированных по размеру, как описано выше, предпочтительно соединенных и связанных в точках их контактирования.
Таким образом, агломераты настоящего изобретения могут иметь более высокое содержание макропор, чем составляющие их первичные частицы вследствие пустот между составляющими агломераты частицами оксида алюминия композиционного материала.
Тем не менее, агломератные частицы все же сохраняют более высокую моду в области мезопор.
В соответствии с этим, агломераты настоящего изобретения характеризуются как имеющие следующие свойства (на основе, не содержащей металлов) после сушки в течение 8 часов при 121°С (250°F) и кальцинирования в течение 1 часа при 537,8°С (1000°F):
(1) площадь поверхности по азоту, по меньшей мере, приблизительно 100, предпочтительно, по меньшей мере, приблизительно 150 и наиболее предпочтительно, по меньшей мере, 200 м2/г, причем площадь поверхности может составлять обычно приблизительно от 100 до приблизительно 400, предпочтительно, приблизительно от 125 до приблизительно 375, и наиболее предпочтительно, приблизительно от 150 до приблизительно 350 м2/г,
(2) объемную плотность агломератов обычно, по меньшей мере, приблизительно 0,30, предпочтительно, по меньшей мере, приблизительно 0,35, и наиболее предпочтительно, по меньшей мере, приблизительно 0,40 г/мл, объемная плотность может обычно составлять приблизительно от 0,30 до приблизительно 1, предпочтительно, приблизительно от 0,35 до приблизительно 0,95, и наиболее предпочтительно, приблизительно от 0,40 до приблизительно 0,90 г/мл,
(3) общий объем пор по ртути приблизительно от 0,40 до приблизительно 2,0, предпочтительно, приблизительно от 0,5 до приблизительно 1,8, и наиболее предпочтительно, приблизительно от 0,6 до приблизительно 1,5 см3/г,
(4) содержание макропор (т.е. тех пор в пределах общего объема пор, которые находятся в пределах области макропор) обычно не выше, чем приблизительно 40, предпочтительно, не выше, чем приблизительно 30, и наиболее предпочтительно, не выше, чем приблизительно 20% общего объема пор, при этом содержание макропор обычно составляет приблизительно от 5 до приблизительно 40, предпочтительно, приблизительно от 10 до приблизительно 35, и наиболее предпочтительно приблизительно от 15 до приблизительно 30% общего объема пор.
(5) содержание мезопор обычно приблизительно от 15 до приблизительно 95, предпочтительно, приблизительно от 20 до приблизительно 90, и наиболее предпочтительно, приблизительно от 30 до приблизительно 80% общего объема пор. Кроме того, обычно, по меньшей мере, приблизительно 30, предпочтительно, по меньшей мере, приблизительно 40, и наиболее предпочтительно, по меньшей мере, приблизительно 50% пор в пределах области мезопор имеют диаметры пор обычно приблизительно от 80 до приблизительно 400 (например, от 100 до 400), предпочтительно, приблизительно от 90 до приблизительно 350 (например, от 100 до 350), и наиболее предпочтительно, приблизительно от 105 до приблизительно 300 ангстрем.
(6) средний диаметр частиц агломератов обычно приблизительно от 0,5 до приблизительно 5, предпочтительно, приблизительно от 0,6 до приблизительно 2, и наиболее предпочтительно, приблизительно от 0,8 до 1,5 мм.
Мезопоры частиц агломератов, которые кальцинированы, как указано также желательно имеют моду пор, обычно в области мезопор приблизительно от 60 до приблизительно 400, например, от 60 до приблизительно 300, предпочтительно, приблизительно от 65 до приблизительно 275, и наиболее предпочтительно, приблизительно от 70 до приблизительно 250 ангстрем.
Кроме того, агломераты можно смешать с другими стандартными оксидами алюминия для получения носителей, имеющих распределение размера пор с двумя или более модами в области мезопор. Каждый оксид алюминия дает моды в области мезопор в их конкретном характеристическом положении. Предполагаются также смеси двух и более оксидов алюминия, полученных с набухаемыми глинами, имеющими различные моды пор.
Агломерацию композиционного материала оксида алюминия проводят в соответствии со способами, хорошо известными в данной области и, в частности, такими способами, как грануляция, экструзия, формование в шарики во вращающемся барабане для нанесения покрытий и тому подобное. Можно также использовать способ гранулирования, при помощи которого частицы композиционного материала, имеющие диаметр не больше, чем приблизительно 0,1 мм, агломерируют в частицы с диаметром, по меньшей мере, приблизительно 1 мм при помощи жидкости для грануляции.
Как известно специалистам в данной области, агломерацию можно, необязательно, проводить в присутствии дополнительных аморфных или кристаллических связующих, а также к смеси, которую нужно агломерировать, можно добавить порообразующие агенты. Общепринятые связующие включают другие формы оксида алюминия, диоксид кремния-оксид алюминия, глины, диоксид циркония, диоксид кремния-диоксид циркония, оксид магния и диоксид кремния-оксид бора. Общепринятые порообразующие агенты, которые можно использовать, в частности, включают древесную муку, древесный уголь, целлюлозу, крахмалы, нафталин и, в целом, все органические соединения, способные удаляться при кальцинировании. Добавление порообразующих агентов, однако, не является необходимым или желательным.
Если необходимо, затем проводят старение, сушку и/или кальцинирование агломератов.
Агломераты после того, как они образованы, затем обычно подвергают обработке термической активацией при температуре в диапазоне обычно приблизительно от 250 до приблизительно 1000, предпочтительно, приблизительно от 350 до приблизительно 900, и наиболее предпочтительно, приблизительно от 400 до приблизительно 800°С, в течение периодов обычно приблизительно от 0,15 до приблизительно 3,0, предпочтительно приблизительно от 0,33 до приблизительно 2,0 и, очень предпочтительно, приблизительно от 0,5 до приблизительно 1 часа. Атмосферой активации обычно является воздух, но атмосфера может включать инертные газы, такие как азот, или водяной пар.
Обработку активацией можно проводить в несколько стадий, если это необходимо, или она может быть частью обработки для агломерации. В зависимости от определенной температуры активации и используемого времени, агломераты оксида алюминия преимущественно обладают кристаллической структурой, характерной для бемита или гамма-оксида алюминия или их смесей.
Более конкретно, при температурах кальцинирования и времени, увеличивающихся сверх приблизительно 300°С и одного часа, бемит будет превращаться, во все возрастающем количестве, в гамма-оксид алюминия. Однако гамма-оксид алюминия будет иметь свойства пор бемита, из которого он образован. Кроме того, при предпочтительных температурах и времени кальцинирования по существу весь кристаллический бемит превращается в гамма-оксид алюминия. Поэтому сумма содержания кристаллического бемита (масс.%), обсуждаемого выше, плюс содержание гамма-оксида алюминия, являющееся результатом кальцинирования бемита, обычно не превышает первоначального содержания бемита, полученного повторной гидратацией активного оксида алюминия. Данный вывод относится в равной степени к частицам композиционного материала, который активируют и используют непосредственно в форме частиц композиционного материала без агломерации.
Процент γ-Al2О3 (гамма-оксид алюминия) определяют следующим образом:
(1) 100% γ-Al2O3 определяют как интегрированную интенсивность
(площадь под пиком) пика (440) стандарта γ-Al2О3.
(2) Интенсивность пика (101) кварцевой пластины используют в качестве монитора интенсивности рентгеновских лучей.
(3) Сбор данных проводят на автоматическом дифрактометре Philips® 3720, снабженном графитным монохроматором с дифрагированным лучом и запаянной трубкой Cu-рентгеновского излучения. Генератор рентгеновского излучения действует при 45 кВ и 40 мА.
(4) Полную ширину у полумаксимума (FWHM) и интегрированную интенсивность (площадь под пиком) пика (440) γ-Al2О3 получают обработкой кривой по точкам. В случае, когда один пик не может дать хорошую подгонку пика, используют два пика. В случае, когда для подгонки кривой используют два пика, получают два размера кристаллита с использованием уравнения 3, а процент γ-Al2О3 двух размеров кристаллита - с использованием уравнения 2.
(5) Процент γ-Al2О3 образца определяют следующим уравнением:
%γAl203=(Iобразец·Iкварц,с)/(Iстандарт·Iкварц,s) (уравнение 2)
где:
Iобразец - интегрированная интенсивность пика (440) образца;
Iкварц,с - интенсивность пика (101) кварца, измеренная во время измерения стандартного γ-Al2О3;
Iстандарт - интегрированная интенсивность (440) пика стандартного γ-Al2О3 и
Iкварц,s - интенсивность пика (101) кварца, измеренная во время измерения образца.
Размер (L) кристаллита γ-Al2О3 определяют следующей процедурой. Образец размалывают вручную с использованием ступки и пестика. Ровный слой образца помещают на 3,5 г поливинилового спирта (ПВС) и прессуют в течение 10 секунд при 3000 фунт/кв. дюйм с получением гранулы. Гранулу затем сканируют Cu-К-альфа-излучением и строят дифрактограмму между 63 и 73 градусами (2θ). Пик при 66,8 градусов (2θ) используют для вычисления размера кристаллита с использованием уравнения 3 и измеренной ширины пика у половины высоты
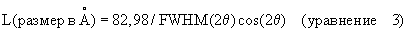
FWHM - полная ширина на половине высоты максимума и
θ - угол дифракции между пучком рентгеновских лучей и плоской поверхностью, на которой установлен образец.
Процент бемита определяют, как описано в примере 1. Большой средний диаметр пор и высокий объем пор делают композиционные материалы оксида алюминия настоящего изобретения пригодными для обработки сырья с высокой молекулярной массой, высококипящего сырья, где не все сырье можно на практике испарить, в операциях как флюид-каталитического крекинга (F.C.C.), так и гидропереработки; для операций крекинга с малым временем контакта, где большие поры могут минимизировать сопротивление диффузии; гидрокрекинга, гидрообработки, гидродесульфуризации и гидроденитрогенирования; переработки экстрактов смолистого песка, битуминозного песка или жидкостей из угля; носителей катализаторов на основе металлов, причем высокий объем пор и диаметр пор обеспечивают повышенную дисперсию металла; отделения соединений с высокой молекулярной массой в растворителе от соединений с низкой молекулярной массой; и применений, требующих небольшого размера частиц оксидов алюминия при низких значениях рН, как например в суспендирующих агентах и полирующих агентах.
Частицы композиционного материала оксида алюминия особенно приспособлены для использования в качестве носителей для различных каталитических систем, использующих в качестве каталитического компонента тяжелые металлы. Следовательно, металлические компоненты таких катализаторов следует добавить и ввести в композиционный материал оксида алюминия. Термическую активацию обычно проводят после образования агломератов, а не до этого.
Такие добавления можно произвести смешиванием каталитических материалов с оксидом алюминия во время получения композиционного материала оксида алюминия, но после его повторной гидратации, во время получения агломератов, например, экструдатов или гранул и тому подобное, пропиткой агломератов оксида алюминия, таких как экструдаты и гранулы, каталитическим материалом посредством погружения в растворы, содержащие каталитический материал, и тому подобное. Способ «сухой» пропитки является другой подходящей альтернативой, где частицы композиционного материала или агломераты контактируют с некоторым количеством пропитывающей жидкости, объем которой соответствует объему пор носителя. Иные и дополнительные способы модификации оксида алюминия могут оказаться желательными для специалистов в данной области.
Пористые оксиды алюминия композиционного материала настоящего изобретения особенно пригодны при использовании в качестве носителей для каталитически активных компонентов гидрирования, таких как металлы группы VIB и группы VIII. Указанные каталитически активные материалы можно подходящим способом применять в операциях гидропереработки.
Более определенно, «гидропереработка», в смысле используемого здесь термина, означает способы очистки нефти и нефтепродуктов реакцией нефтяного сырья (сложных смесей углеводородов, присутствующих в нефти, которые являются жидкими в условиях стандартной температуры и давления) с водородом под давлением в присутствии катализатора для снижения: (а) концентрации, по меньшей мере, одного из следующих компонентов: серы, загрязняющих примесей металлов, азота и углерода по Конрадсону, присутствующих в указанном сырье, и (b) по меньшей мере одного показателя: вязкости, температуры потери текучести и плотности сырья. Гидропереработка включает процессы гидрокрекинга, изомеризации/депарафинизации, окончательной гидрообработки и гидрообработки, которые различаются количеством прореагировавшего водорода и природой обрабатываемого нефтяного сырья.
Обычно подразумевается, что окончательная гидрообработка заключается в гидрообработке углеводородного масла, содержащего преимущественно (по массе) углеводородные соединения в диапазоне кипения смазочного масла («сырье»), где сырье приводят в контакт с твердым катализатором на носителе в условиях повышенной температуры и давления с целью насыщения ароматических и олефиновых соединений и удаления соединений азота, серы и кислорода, присутствующих в сырье, и для улучшения запаха, цвета и свойств термической, окислительной и УФ-стабильности сырья.
Обычно подразумевается, что гидрокрекинг заключается в гидропереработке преимущественно углеводородных соединений, содержащих, по меньшей мере, пять (5) атомов углерода на молекулу («сырье), которую проводят (а) при парциальном давлении водорода выше атмосферного; (b) при температурах обычно ниже 593,3°С (1100°F); (с) с общим суммарным химическим поглощением водорода; (d) в присутствии твердого катализатора на носителе, содержащего, по меньшей мере, один (1) компонент гидрирования, и (е) где указанное сырье дает выход, обычно выше ста тридцати (130) молей (включительно) углеводородов, содержащих, по меньшей мере, три (3) атома углерода на молекулу для каждых ста (100) молей исходного сырья, содержащего, по меньшей мере, пять (5) атомов углерода на молекулу.
Обычно подразумевается, что гидрообработка заключается в гидропереработке преимущественно углеводородных соединений, содержащих, по меньшей мере, пять атомов углерода на молекулу «исходное сырье», для десульфуризации и/или денитрификации указанного исходного сырья, где процесс проводят (а) при парциальном давлении водорода выше атмосферного; (b) при температурах обычно ниже 593,3°С (1100°F); (с) с общим результирующим химическим расходом водорода; (d) в присутствии твердого катализатора на носителе, содержащего, по меньшей мере, один компонент гидрирования, и (е) где (i) исходное сырье дает выход обычно приблизительно от 100 до приблизительно 130 молей (включительно) углеводородов, содержащих, по меньшей мере, три атома углерода на молекулу, для каждых 100 молей исходного сырья, или (ii) сырье включает, по меньшей мере, 50 объемных процентов жидкости недеасфальтированного остатка, обычно кипящего приблизительно выше 565, 6°С (1050°F), как определено перегонкой по ASTM D-1160, и где первой функцией гидропереработки является десульфуризация указанного сырья, или (iii) сырье является продуктом операции получения синтетического масла.
Обычно подразумевается, что изомеризация/депарафинизация заключаются в гидропереработке преимущественно углеводородного масла, имеющего индекс вязкости (VI) и диапазон выкипания, подходящий для смазочного масла («сырье»), где указанное сырье приводят в контакт с твердым катализатором, который содержит в качестве активного компонента микропористое кристаллическое молекулярное сито, в условиях повышенного давления и температуры и в присутствии водорода для получения продукта, у которого свойства текучести на холоду значительно улучшены относительно указанного сырья и интервал кипения которого, по существу, находится внутри интервала кипения сырья.
Более конкретно, компоненты хорошо известных катализаторов гидропереработки обычно включают, по меньшей мере, один компонент-металл, выбранный из группы, состоящей из металлов группы VIII, включая металлы платиновой группы группы VIII, особенно, платину и палладий, металлы группы железа группы VIII, особенно кобальт и никель, металлы группы VIB, особенно молибден и вольфрам, и их смеси. Если сырье имеет достаточно низкое содержание серы, например, ниже, чем приблизительно 1 массовый процент и, предпочтительно, ниже, чем приблизительно 0,5 массового процента, можно использовать металлы платиновой группы группы VIII в качестве компонента гидрирования. В данном осуществлении металл платиновой группы группы VIII, предпочтительно, присутствует в количестве в диапазоне от приблизительно 0,01 массового процента до приблизительно 5 массовых процентов всего катализатора, в пересчете на элементный металл платиновой группы. Когда обрабатываемое сырье содержит более чем приблизительно 0,1 массового процента серы, компонентом гидрирования, предпочтительно, является комбинация, по меньшей мере, одного металла группы железа группы VIII и, по меньшей мере, одного металла группы VIB. Не являющиеся благородными металлами компоненты гидрирования, предпочтительно, присутствуют в конечной композиции катализатора в виде оксидов или сульфидов, более предпочтительно, в виде сульфидов. Предпочтительные композиции катализаторов в целом содержат, по меньшей мере, приблизительно 2, предпочтительно, приблизительно от 5 до приблизительно 40, масс.% металла группы VIB, более предпочтительно, молибдена и/или вольфрама, и обычно, по меньшей мере, приблизительно 0,5 и, предпочтительно, приблизительно от 1 до приблизительно 15 масс.% элементов группы VIII периодической таблицы элементов, более предпочтительно, никеля и/или кобальта, определенных как соответствующие оксиды. Сульфидная форма указанных металлов является более предпочтительной вследствие ее более высокой активности, селективности и сохранения активности.
Компоненты катализатора, например, компоненты катализатора гидропереработки, можно включить в общую композицию катализатора с помощью любой одной из описываемых многочисленных процедур.
Хотя неблагородные металлические компоненты можно объединять в катализатор в виде сульфидов, данный подход не является предпочтительным. Такие компоненты обычно объединяют в виде соли металла, которую можно термически превратить в соответствующий оксид в окисляющей атмосфере или восстановить водородом или другим восстановительным агентом. Композицию можно затем сульфидировать взаимодействием с сернистым соединением, таким как дисульфид углерода, сульфид водорода, углеводородтиолы, элементная сера и тому подобное.
Компоненты катализатора можно ввести в оксид алюминия композиционного материала на любой одной из различных стадий при получении катализатора. Например, соединения металлов, такие как сульфиды, оксиды или водорастворимые соли, такие как гептамолибдат аммония, вольфрамат аммония, нитрат никеля, сульфат кобальта и тому подобное, можно добавить посредством совместного размалывания, пропитки или осаждения, после повторной гидратации, но до того, как композиционный материал, окончательно агломерируют. В альтернативном варианте указанные компоненты можно добавить к композиционному материалу после агломерации пропиткой водным, спиртовым, углеводородным раствором растворимых соединений или предшественников.
Следующее осуществление настоящего изобретения относится к способу гидрообработки углеводородного сырья, по меньшей мере, в одной реакционной зоне с кипящим слоем. Более конкретно, углеводородное сырье приводят контакт с водородом в одной или серии реакционных зон с кипящим слоем в присутствии катализатора гидропереработки, включающего компонент гидрирования в виде каталитических металлов и их производных, как описано выше, осажденный на агломераты композиционного материала оксида алюминия, описанного здесь.
Как хорошо известно, указанное сырье содержит никель, ванадий и асфальтены, например, приблизительно от 40 млн-1 до более чем 1000 млн-1 от общего количества никеля и ванадия и приблизительно до 25 масс.% асфальтенов. Кроме того, для экономики указанных способов желательно получение более легких продуктов, а также деметаллированного остаточного побочного продукта. Данный способ особенно полезен при обработке исходного сырья со значительным количеством металлов, содержащего 150 млн-1 или более никеля и ванадия и имеющего содержание серы в диапазоне приблизительно от 1 масс.% до приблизительно 10 масс.%. Типичное сырье, которое можно удовлетворительно обработать по способу настоящего изобретения, содержит значительное количество (например, приблизительно 90%) компонентов, которые кипят значительно выше 537,8°С (1000°F). Примерами типичного сырья являются сырые нефти, отбензиненные сырые нефти, нефтяные углеводородные остатки, остатки от перегонки нефти как при атмосферном давлении, так и в вакууме, масла, полученные из смолистых песков, и остатки, полученные из масла смолистых песков, и углеводородные потоки продуктов, полученных из угля. Такие углеводородные потоки содержат металлорганические загрязняющие примеси, которые оказывают неблагоприятное влияние в различных способах очистки, в которых используют катализаторы при превращении обрабатываемого данного углеводородного потока. Загрязняющие металлические примеси, которые обнаружены в таком сырье, включают, но не ограничиваются ими, железо, ванадий и никель.
Хотя металлические загрязняющие примеси, такие как ванадий, никель и железо, часть присутствуют в различных углеводородных потоках, в отдельном углеводородном потоке присутствуют также другие металлы. Такие металлы существуют в виде оксидов или сульфидов определенного металла или в виде растворимой соли определенного металла, или в виде металлорганических соединений с высокой молекулярной массой, включая нафтенаты металлов и порфирины металлов и их производные.
Другим характерным явлением гидрообработки тяжелых углеводородов является осаждение нерастворимых углеродистых веществ из асфальтеновой фракции сырья, которое вызывает проблемы работоспособности процесса. Количество таких нерастворимых образованных веществ повышается с увеличением количества материала, кипящего выше 537,8°С (1000°F), которое превращают, или с повышением используемой температуры реакции. Указанные нерастворимые вещества, известные также как твердые вещества горячего фильтрования Шелл, вызывают трудности в работоспособности установки гидропревращения и тем самым ограничивают температуру и сырье, с которыми может работать установка. Другими словами, количество твердых веществ ограничивает превращение данного сырья. Трудности в отношении работоспособности, описываемые выше, могут начать проявлять себя при уровнях твердых веществ уже 0,1 масс.%. Чтобы предотвратить неполадки оборудования для процесса, обычно рекомендуют уровни ниже 0,5 масс.%. Описание испытания нефтепродуктов на фильтруемость в горячем состоянии по Шелл можно найти в публикации A.J.J., Journal of Inst. Of Petroleum (1951) 37, рр. 596-604, Van Kerkvoort, W.J. and Nieuwstad, A.J.J., которая включена здесь в качестве ссылки.
Были сделаны предположения что такие нерастворимые углеродистые вещества образуются, когда тяжелые углеводороды превращаются в установке гидропревращения, что тем самым делает их плохими растворителями для непревращенной асфальтеновой фракции и, следовательно, вызывая образование нерастворимых углеродистых веществ. Образование таких нерастворимых веществ может понижаться из-за того, что имеется некоторая часть площади поверхности в катализаторе гидроконверсии, доступная благодаря очень большим порам, так что большая часть поверхности катализатора является доступной для больших асфальтеновых молекул. Кроме того, большие поры облегчают осаждение никеля и ванадия в катализаторе гидрообработки без закупоривания пор.
Было обнаружено, что использование пористых композиционных материалов в качестве носителей при получении катализаторов, особенно катализаторов гидропереработки, обеспечивает более высокую начальную активность, чем катализаторы, нанесенные на стандартный оксид алюминия.
Хотя преимущество более высокой начальной активности является менее значительным в операции с неподвижным слоем, оно является особенно важным в системе с кипящим слоем. Более конкретно, в системе с кипящим слоем повышение начальной активности имеет большой смысл, поскольку имеется периодическое или непрерывное добавление катализатора для повышения и поддержания общей активности системы. Поскольку общая активность системы с кипящим слоем является средневзвешенной активностью всего присутствующего катализатора, от свежего до дезактивированного, то общую активность повышают постоянным или периодическим добавлением катализатора, обладающего относительно более высокой начальной активностью.
Операции гидрообработки обычно проводят в одном реакторе или последовательном ряде реакторов с кипящим слоем. Как ранее объяснялось, кипящий слой является слоем, в котором частицы твердого катализатора поддерживают в беспорядочном движении посредством подачи направленного вверх потока жидкости и газа. Кипящий слой обычно имеет большой объем, по меньшей мере на 10 процентов больше и вплоть до значения на 70% больше, чем его слой в уплотненном состоянии. Требуемое равновесие частиц катализатора поддерживают введением жидкого сырья, включающего рецикл, если он имеется, в реакционную зону при линейных скоростях, составляющих приблизительно от 0,02 до приблизительно 0,4 фута в секунду и, предпочтительно, приблизительно от 0,05 до приблизительно 0,20 фута в секунду.
Рабочие условия для гидрообработки потоков тяжелых углеводородных потоков, таких как нефтяные углеводородные остатки и так далее, хорошо известны в данной области и включают давление в диапазоне приблизительно от 1000 фунт/кв. дюйм абс. (68 атм) до 3000 фунт/кв. дюйм абс. (204 атм), среднюю температуру слоя катализатора в диапазоне приблизительно от 700°F (371°C) до приблизительно 850°F (454°С), часовую объемную скорость жидкости (LHSV) в диапазоне приблизительно от 0,1 объема углеводорода в час на объем катализатора до приблизительно 5 объемов углеводорода в час на объем катализатора и скорость рецикла водорода или скорости добавления водорода в диапазоне приблизительно от 2000 стандартных кубических футов на баррель (SCFB) (356 м3/м3) до приблизительно 15000 SCFB (2671 м3/м3). Предпочтительно рабочие условия включают общее давление в диапазоне приблизительно от 1200 фунт/кв. дюйм абс. до приблизительно 2000 фунт/кв. дюйм абс. (81-136 атм); среднюю температуру слоя катализатора в диапазоне приблизительно от 730°F (378°C) до приблизительно 820°F (437°C) и LHSV в диапазоне приблизительно от 0,1 до приблизительно 4,0 и скорость рецикла водорода или скорость добавления водорода в диапазоне приблизительно от 5000 SCFB (890 м3/м3) до приблизительно 10000 SCFB (1781 м3/м3). Температуру и объемную скорость способа обычно выбирают так, чтобы, по меньшей мере, 30 об. % фракции сырья, кипящей выше 1000°F, превратилось в продукт, кипящий ниже 1000°F, и более предпочтительно, так, чтобы, по меньшей мере, 70 об.% рассматриваемой фракции превратилось в продукт, кипящий ниже 1000°F.
Для обработки углеводородных дистиллятов рабочие условия обычно включают парциальное давление водорода в диапазоне от приблизительно 200 фунт/кв. дюйм абс. (13 атм) до приблизительно 3000 фунт/кв. дюйм абс. (204 атм), среднюю температуру слоя катализатора в диапазоне приблизительно от 600°F (315°C) до приблизительно 800°F (426°C), LHSV в диапазоне приблизительно от 0,4 объема углеводорода в час на объем катализатора до приблизительно 6 объемов углеводорода в час на объем катализатора и скорость рецикла водорода или скорости добавления водорода в диапазоне приблизительно от 1000 SCFB (178 м3/м3) до приблизительно 10000 SCFB (1381 м3/м3). Предпочтительные рабочие условия для гидрообработки углеводородных дистиллятов включают давление в диапазоне приблизительно от 200 фунт/кв. дюйм абс. (13 атм) до 1200 фунт/кв. дюйм абс. (81 атм); среднюю температуру слоя катализатора в диапазоне приблизительно от 600°F (315°C) до приблизительно 750°F (398°С), LHSV в диапазоне приблизительно от 0,5 объема углеводорода в час на объем катализатора до приблизительно 4 объемов углеводорода в час на объем катализатора и скорость рецикла водорода или скорость добавления водорода в диапазоне приблизительно от 1000 SCFB (178 м3/м3) до приблизительно 6000 SCFB (1068 м3/м3).
Наиболее желательные условия для превращения определенного сырья в заданный продукт, однако, можно лучше всего получить превращением сырья при нескольких различных температурах, давлениях, объемных скоростях и скоростях добавления водорода, устанавливая влияние каждого из указанных параметров и выбирая - лучший компромисс между общим превращением и селективностью.
Все ссылки в данной заявке на элементы или металлы, принадлежащие к определенной группе, относятся к периодической таблице элементов и Hawley's Condensed Chemical Dictionary, 12th Edition. Кроме того, любые ссылки на группу или группы являются ссылками на группу или группы, как отражено в этой периодической таблице элементов, с использованием CAS-системы для нумерации групп.
Все ссылки в формуле изобретения на морфологические свойства, определенные в отношении к массе, такие как площадь поверхности и объем пор, должны быть интерпретированы как относящиеся к системе без металлов, как определено в уравнении 6, например нормализованы так, чтобы вносить поправку на любое влияние каталитического оксида металла (если он присутствует) на массу анализируемого материала. Если не оговорено особо, все композиционные материалы в форме порошка (не агрегированные) в примерах фильтруют после повторной гидратации и затем подвергают ионообмену до низкого содержания натрия с помощью A/S-обмена, как описано выше, перед кальцинированием. Ни один из экструдированных образцов не был подвергнут A/S-обмену.
Следующие примеры приводятся как конкретные иллюстрации заявленного изобретения. Должно быть понятно, однако, что изобретение не ограничивается конкретными деталями, приведенными в примерах. Все части и проценты в примерах, а также в остальной части описания даются по массе, если не оговорено особо. Если не оговорено особо, все определения или перечисления значений площадей поверхности и свойств пор в описании и формуле изобретения должны быть истолкованы, как сделанные на образцах, которые были высушены в печи при 138°С (280°F) и затем кальцинированы при 537,8°С (1000°F) в течение 2 часов при атмосферном давлении на воздухе.
Кроме того, подразумевается, что любой диапазон показателей, перечисленных в описании или формуле изобретения, таких как показатели, представляющие определенный набор свойств, условий физических состояний или процентов, недвусмысленным образом включает любое число, попадающее в такой диапазон, в том числе любую подсерию чисел в указанном таким образом диапазоне.
ПРИМЕР 1
К 1843 г Н2О добавляют 14,4 г в расчете на сухое вещество лапонита® RD - синтетической глины типа гекторит, выпускаемой фирмой LaPorte Industries, Ltd. Образовавшуюся смесь быстро перемешивают в течение 20 минут для диспергирования глины. Образуется очень слабо мутный раствор, при этом почти прозрачная вода указывает на то, что глина является очень хорошо диспергированной и мелкоизмельченной. К дисперсии лапонита® добавляют 23,5 г 10% водного раствора глюконата натрия, затем 465,6 г кальцинированного активного оксида алюминия СР-3, от ALCOA. Суспензию кипятят с обратным холодильником в течение 24 часов. Суспензию фильтруют и сушат в течение ночи при 137,8°С (280°F). Образовавшиеся частицы композиционного материала затем подвергают сухому кальцинированию в течение 2 часов при 537,8°С (этот материал назван здесь свежим) или кальцинируют в течение 4 часов при 800°С в атмосфере 20 об. % водяного пара (такой материал назван здесь обработанным водяным паром) и площадь поверхности измеряют для свежих и обработанных водяным паром образцов.
Степень превращения образца оксида алюминия в кристаллический бемит определяют следующим образом:
(1) 100% бемита определяют как интегральную интенсивность (площадь под пиком) пика (020) оксида алюминия Catapal.
(2) Интенсивность пика (101) кварцевой пластины используют в качестве монитора интенсивности рентгеновских лучей.
(3) Данные получают на автоматическом дифрактометре Philips®, снабженном графитным монохроматором с дифрагированным лучом и запаянной трубкой Cu-рентгеновского излучения. Генератор рентгеновского излучения действует при 45 кВ и 40 мА.
(4) Полную ширину на полувысоте пика максимума (FWHM) и интегральную интенсивность (площадь под пиком) пика (020) бемита получают построением кривой по точкам. В случае, когда один пик не может дать хорошую подгонку пика, используют два пика. В случае, когда для построения кривой по точкам используют два пика, получают два размера кристаллитов с использованием уравнения 5. Процент бемита с двумя размерами кристаллитов получают с использованием уравнения 4.
(5) Процент бемита в образце определяют по следующему уравнению:
%бемита=(Iобразец·Iкварц,с)/(Icatapal·Iкварц,s) (уравнение 4)
где:
Iобразец - интегральная интенсивность пика (020) образца;
Iкварц,с - интенсивность пика (101) кварца, измеренная во время измерения оксида алюминия Catapal;
Icatapal - интегрированная интенсивность пика (020) оксида алюминия Catapal и
Iкварц,s - интенсивность пика (101) кварца, измеренная во время измерения образца.
Размер (L) кристаллита бемита определяют по следующей методике. Образец размалывают вручную с применением ступки и пестика. Ровный слой образца помещают на 3,5 г поливинилового спирта (ПВС) и прессуют в течение 10 секунд при 3000 фунт/кв. дюйм с получением гранулы. Гранулу затем сканируют Cu-К-альфа-излучением и строят дифрактограмму от 22 до 33 градусов (2θ). Пик у значения 28 градусов (2θ) используют для вычисления размера кристаллита с использованием уравнения 5 и измеренной ширины пика на половине его высоты
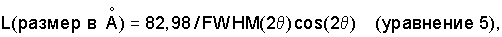
где
FWHM - полная ширина на полу высоте пика максимума и
θ - угол дифракции между пучком рентгеновских лучей и плоской поверхностью, на которой установлен образец.
Полученные в результате анализа свойства приведены в таблице 2 и на фигуре 1 и обозначены как опыт 2. Добавление 3% лапонита® дает повышенные площади свежей и обработанной водяным паром поверхности по сравнению со сравнительным примером 1. Фигура 1 также иллюстрирует большое увеличение общего объема пор по азоту и смещение в сторону большего размера.пор.
СРАВНИТЕЛЬНЫЙ ПРИМЕР 1
Повторяли пример 1, за исключением того, что к образцу не был добавлен лапонит®. Результаты приведены в таблице 2, на фигуре 1 и обозначены как опыт 1.
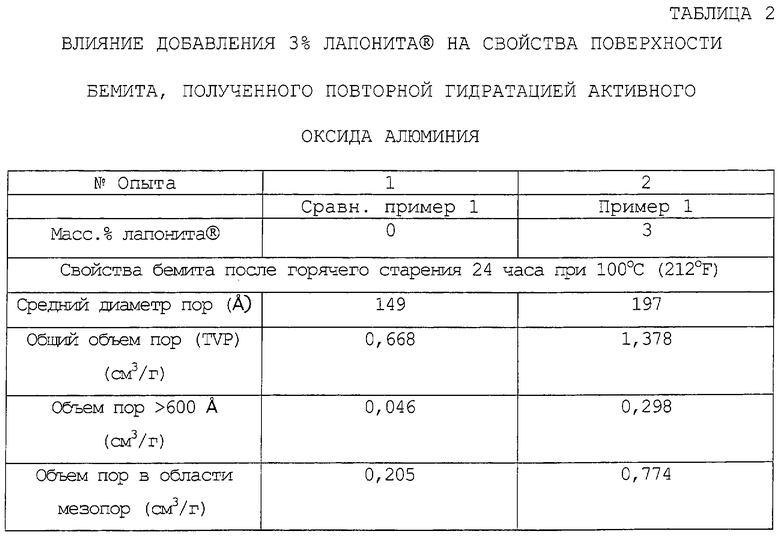
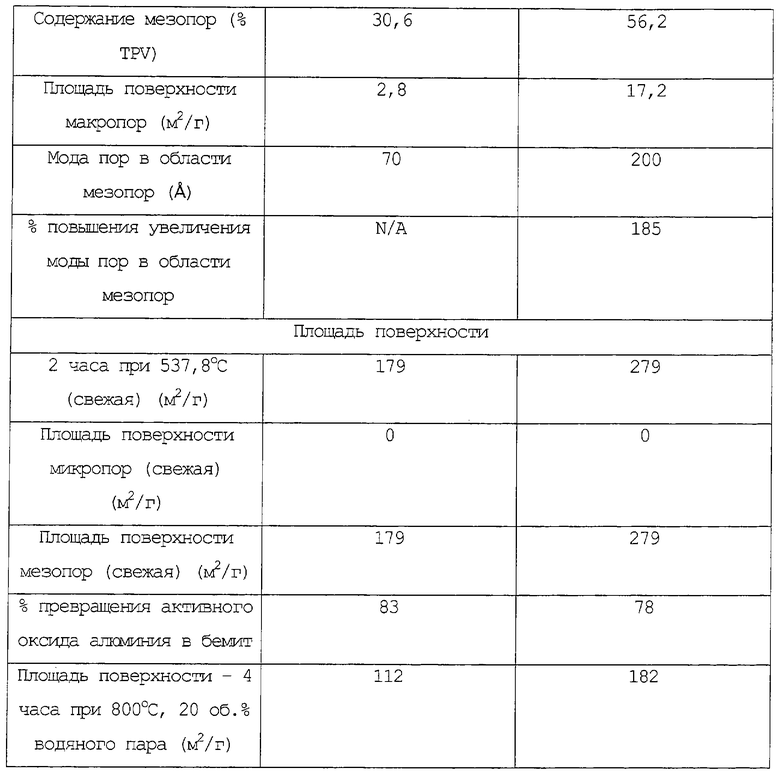
ПРИМЕР 2
Повторяли пример 1 за исключением того, что уровень синтетического гекторита - лапонита® варьировали от 0,1 до 10 масс.% от общего содержания твердых веществ (лапонит® + оксид алюминия) (соответственно опытам 3-12). После 24 часов старения при кипячении с обратным холодильником образцы фильтруют и сушат в течение ночи при 137,8°С (280°F). Измеряют размер кристаллита бемита выбранных образцов, а также площадь поверхности через два часа выдерживания при 537,8°С (1000°F) или через 4 часа в 20 об. % водяном паре при 800°С. Измеряют также показатель диспергируемости (DPI) частиц композиционного материала. В этом тесте измеряют % частиц, имеющих диаметр частицы <1 микрона после диспергирования в воде с измеренным количеством HCl (237 миллиэквивалентов/моль оксида алюминия) и перемешивания. Результаты влияния уровня синтетического гекторита на свойства бемита суммированы в таблице 3.
Можно видеть, что диспергируемая набухаемая глина
(a) повышает общий объем пор по азоту и средний диаметр пор до максимального уровня в диапазоне 3-5 масс.%;
(b) снижает размер кристаллитов бемита;
(c) значительно повышает площадь свежей и обработанной водяным паром поверхности и объем пор по азоту; и
(d) повышает диспергируемость оксида алюминия, когда добавляют 3 масс % или больше глины.
Было также отмечено, что когда масс.% добавленной синтетической глины повышали, твердость высушенного в печи бемита повышалась. При 0 масс.% высушенный в печи материал является мягким порошком, при 3 масс.% глины он является умеренно твердым, тогда как при 5-10 масс.% он является совершенно твердым. Считают, что данный факт означает, что экструдаты/гранулы, изготовленные из частиц композиционного материала, имеющего уровни 3 масс.% и выше, должны иметь высокую прочность на раздавливание. Графики распределения размера пор по азоту при уровнях содержания глины от 0 до 1 масс.% показаны на фигуре 2, а графики для уровней содержания глины от 0 до 6 масс.% показаны на фигуре 3.
Фигура 2 показывает реальное уменьшение моды пор в области мезопор при низких уровнях лапонита®. Фигура 3 показывает стойкий сдвиг в сторону больших мод пор в области мезопор при все более возрастающих концентрациях глины от 2 до 5 масс.%. Таблица 3 показывает наличие пика общего объема пор (TPV), среднего диаметра пор (ADP) и пиков площадей свежей и обработанной водяным паром поверхности (SA) при концентрации глины 5 масс.%.
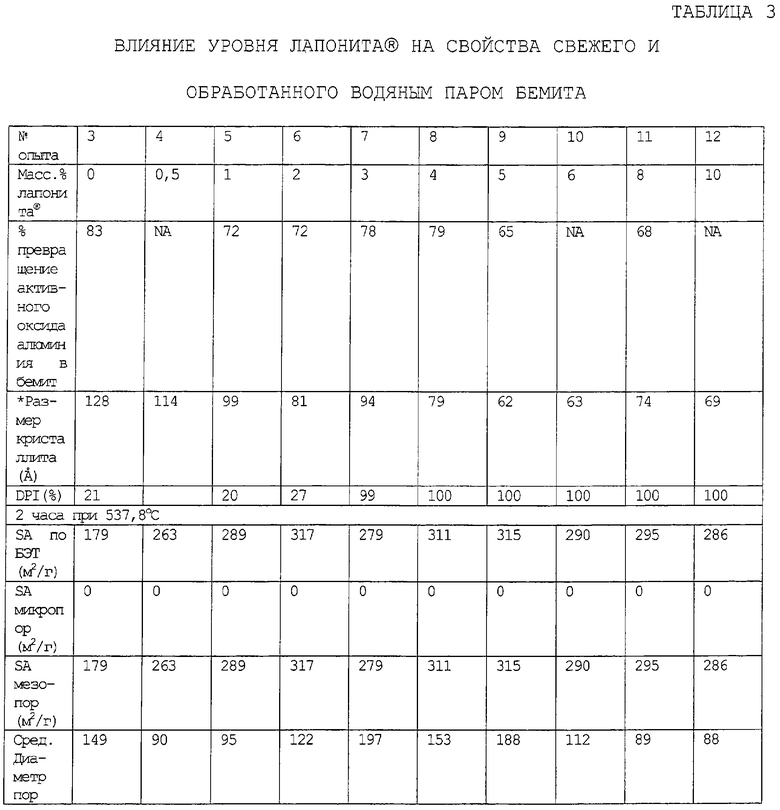


ПРИМЕР 3
Данный пример иллюстрирует влияние условий сушки и общего количества летучих компонентов (TV), измеренного при 954,4°С (1750°F), на диспергируемость синтетического гекторита и, соответственно, на продукт из оксида алюминия.
Порцию синтетического гекторита в автоклаве на два галлона получают в соответствии с примером 2 патента США 4049780.
После выдерживания в автоклаве суспензию геля синтетического гекторита фильтруют, промывают водой и делят на образцы от 1 до 4 следующим образом:
(1) сохраненный в виде фильтровальной лепешки, TV=82,43%;
(2) высушенный в печи в течение ночи при 100°С (212°F), TV=12,83%;
(3) высушенный распылением (S.D.) при 130°С у выпускного отверстия, TV=19,38%;
(4) высушенный распылением при 180°С у выпускного отверстия, TV=15,45%.
Высушенные распылением образцы 3 и 4 получают повторным суспендированием фильтровальной лепешки до приблизительно 2% твердых веществ с последующей сушкой распылением в небольшом настольном сушильном аппарате.
Четыре отдельных композиционных материала оксид алюминия/синтетический гекторит получают в соответствии с примером 2, но с использованием 3 масс.% одного из образцов синтетического гекторита от 1 до 4. Содержание твердых веществ каждой из суспензий глина/оксид алюминия составляет 17 масс.%. Более конкретно, каждый из вышеуказанных образцов синтетического гекторита от 1 до 4 диспергируют в воде быстрым перемешиванием в течение 1/2 часа. Затем добавляют к каждой дисперсии кальцинированный оксид алюминия и кипятят с обратным холодильником в течение 24 часов с хорошим перемешиванием. Влияние синтетического гекторита на объем пор оксида алюминия показано в таблице 4, опыты с 13 по 16. Общий объем пор бемитного продукта повышается при повышении диспергируемости синтетического гекторита. Визуально заметно, что диспергированные в воде синтетические гекториты повышают прозрачность (и, следовательно, диспергируемость) в следующем порядке: фильтровальная лепешка < высушенный в печи < S.D. при 130°С < S.D. при 180°С, что является порядком увеличения объема пор, среднего диаметра пор и показателя диспергируемости оксида алюминия. В соответствии с этим, смещение объема пор можно регулировать с помощью уровня используемой диспергируемой набухающей глины и/или степени дисперсности набухающей глины. Степень дисперсности или размер частиц глины в дисперсии можно регулировать условиями синтеза глины (молярные отношения при подаче реагентов, температура автоклава и т.д.) или условиями сушки. Следует отметить далее, что площадь свежей и обработанной водяным паром поверхности (S.A.) примера 4 все еще выше, чем примера 1, при достижении значительно более высокого TPV.
ВЛИЯНИЕ УСЛОВИЙ СУШКИ СИНТЕТИЧЕСКОГО ГЕКТОРИТА НА СВОЙСТВА ПОВЕРХНОСТИ БЕМИТА ИЗ АКТИВНОГО ОКСИДА АЛЮМИНИЯ И 3% ГЛИНЫ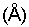
ПРИМЕР 4
Данный пример иллюстрирует влияние диспергируемости на морфологические свойства композиционного материала, опосредуемые температурой реакции образования глины.
Первый образец синтетического гекторита, маркированный SH-1 и обозначенный как опыт 16-1, получали в целом в соответствии с примером 3 при исходном введении 1,49 моль SiO2, 1,0 моль MgO, 0,06 моль Li, 0,08 моль Na посредством добавления 97,9 г кремниевой кислоты (H4SiO4), 58,3 г Mg(ОН)2, 2,55 г LiCl и 4,7 г NaCl к 1,169 г Н2О и кипячением с обратным холодильником в течение 24 час. Второй синтетический гекторит (SH-2), обозначенный как опыт 16-2, получали с использованием начального введения 87,4 г кремниевой кислоты, 58,3 г Mg(ОН)2 и 10,5 г LiCl, добавленных к 1,083 г H2O, и горячего старения в пластиковом сосуде в течение приблизительно 24 часов при 101,7°С (215°F). Оба образца имели рентгенограмму гекторита. Суспензию каждой глины получали смешиванием в воде в течение 2 минут и к данной суспензии добавляли 291 г, в расчете на сухую массу, кальцинированного оксида алюминия СР-3 и 1,5 г глюконата натрия. Отношение синтетическая глина/оксид алюминия составляло 3/97. Данную суспензию кипятили с обратным холодильником в течение 24 часов, фильтровали и сушили в печи. Во время кипячения с обратным холодильником исходных материалов - синтетических гекторитов наблюдали как для SH-1, так и для SH-2, что частицы были крупнозернистыми и не были диспергированы до коллоидного золя.
Результаты распределения размера частиц по азоту суммированы на фигуре 4 вместе с графиком для контрольного опыта 3. Указанные результаты показывают, что данный оксид алюминия, полученный с недиспергируемыми или слабо диспергированными синтетическими гекторитами (SH-1 и SH-2), не имеет такого же сдвига в распределении размера пор даже для контрольного образца без синтетического гекторита. Количества реагентов для SH-1 и SH-2 суммированы в таблице 5.
В целом, чем выше температура реакции образования синтетического гекторита или продолжительнее время реакции, тем выше будет его диспергируемость. Таким образом, предпочтительными являются температуры, по меньшей мере, от 150 до 200°С.Таких температур можно достичь в автоклаве.
ПРИМЕР 5
Данный пример иллюстрирует влияние применения высокоочищенного нефторированного гекторита вместо синтетического гекторита. Два образца высокоочищенного природного гекторита были получены от American Colloid Co. Указанные глины, гекталит 200 (обозначенный отдельно NH-1) и гектабрит DP (обозначенный NH-2), диспергировали в течение 1 минуты в смесителе. К каждой дисперсии добавляли кальцинированный оксид алюминия и глюконат натрия, получая при этом 3% глины и 97% кальцинированного оксида алюминия (СР-3, ALCOA). Уровень глюконата был 0,5 масс.%, в расчете на оксид алюминия. Обе суспензии кипятили с обратным холодильником в течение 24 часов при перемешивании, фильтровали и сушили в печи. Результаты распределения размера пор по азоту приводятся на фигуре 5. Данную процедуру повторяли с использованием образцов глины SH-1 и SH-2 и обозначали как опыты 20 и 21.
Стандартный образец оксида алюминия, обозначенный СЕ-2, также получали в соответствии с примером 5, за исключением того, что не применяли глину.
Сравнение графиков фигур 1 и 3 с графиком фигуры 5 показывает только слабое смещение моды в области мезопор в сторону больших диаметров при использовании природного гекторита по сравнению с синтетическим гекторитом (опыты 20-21). Другие морфологические свойства образцов природного гекторита (опыты 18-19), образца из опыта 7 и сравнительного образца СЕ-2 (опыт 17) также приводятся в таблице 6.
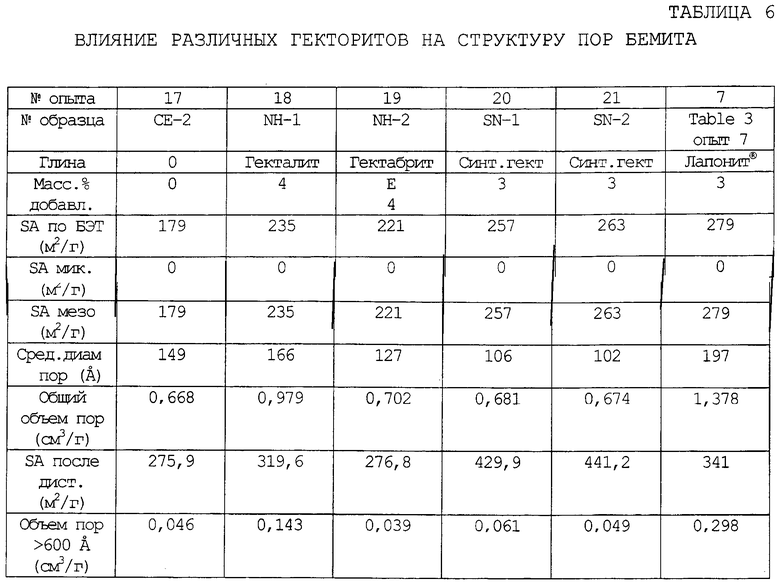
ПРИМЕР 6
Данный пример иллюстрирует влияние синтетического гекторита на гидротермическую стабильность различных кальцинированных оксидов алюминия. Так, кальцинированные оксиды алюминия, выпускаемые Porocel под товарным знаком АР-15, использовали для получения композиционных материалов бемит/лапонит® с использованием процедуры примера 1, за исключением того, что уровни содержания диспергируемого гекторита (лапонит® RD) варьировали как 0, 1,5 и 3 масс.% и не использовали глюконат натрия (опыт 22). Результаты показывают, что получена хорошая гидротермическая стабильность с добавленным глюконата или без добавленного глюконата. Стабильность хорошо сопоставима со стабильностью, полученной с оксидом алюминия СР-3.
Образовавшиеся образцы подвергали старению в водяном паре (20%) в течение 4 часов при 800°С и определяли площадь поверхности по BET в м2/г. Кроме того, образцы композиционного материала получали в соответствии с примером 1 с использованием оксида алюминия СР-3, за исключением того, что содержание лапонита® варьировали как 0, 0,1, 0,2, 0,25, 1,5, 2,3 и 5 масс.%, и образовавшиеся продукты подвергали старению, как описано выше для образцов, полученных из АР-15. Результаты показаны на фигуре 6 как опыт 23.
ПРИМЕР 7
Данный пример иллюстрирует влияние изменения момента добавления синтетического лапонита® до и после повторной гидратации кальцинированного оксида алюминия.
Порцию бемита из кальцинированного оксида алюминия получали следующим образом: к 1888 г Н2О в стеклянном контейнере на 3 л добавляли 24,4 г 10 масс.% раствора глюконата натрия и затем 480 г, в пересчете на сухую основу, кальцинированного оксида алюминия СР-3 от ALCOA. Данную суспензию кипятили с обратным холодильником в течение 24 часов для повторной гидратации оксида алюминия. Повторно гидратированный оксид алюминия затем делили на 5 равных порций. К каждой добавляли изменяющиеся количества лапонита® RD (Laporte) при значениях 0, 2, 4, 6 и 8 масс.%. Площадь поверхности указанных материалов определяли после старения 4 часа в 20% водяном паре, результаты приводятся на фигуре 7 как опыт 24. Вышеуказанную процедуру повторяли, за исключением того, что лапонит® добавляли в количестве 0, 0,25, 1,0, 2,0, 2,25, 3,0, 3,75, 4,0, 5,0, 6,0, 8,0 и 10 масс.% перед кипячением оксида алюминия с обратным холодильником, т.е. перед повторной гидратацией. Образовавшиеся продукты затем также подвергали старению в водяном паре (20%) в течение 4 часов при 800°С, определяли площади поверхности по БЭТ, результаты суммированы на фигуре 7, опыт 25. Как можно видеть на фигуре, добавление диспергируемого гекторита перед повторной гидратацией (опыт 25) дает более гидротермически стабильный продукт по сравнению с добавлением после повторной гидратации (опыт 24). Считается, что это обусловлено более высокой площадью свежей поверхности, объемом пор и средним диаметром пор данного продукта, а также лучшей диспергируемостью глины в оксиде алюминия, когда ее добавляют в начале повторной гидратации.
ПРИМЕР 8
Данный пример иллюстрирует влияние на гидротермическую стабильность предварительного размалывания суспензии, содержащей диспергируемый синтетический гекторит и кальцинированный оксид алюминия (СР-3, ALCOA).
К 8,331 г Н20 добавляют с быстрым перемешиванием 51,9 г лапонита® RD (Laporte, TV=13,26%). Суспензию подвергают старению при комнатной температуре с перемешиванием в течение 20 минут для диспергирования глины. Образуется практически прозрачный раствор, что указывает на хорошую коллоидную дисперсию глины. К дисперсии добавляют 1616,7 г СР-3 (TV=10%) с хорошим перемешиванием. Суспензию, содержащую 3 масс.% лапонита®, затем подвергают размалыванию в жестких условиях (80% среда, 0,75 л/1 мин) в мельнице 4L DRAIS. Размолотую суспензию кипятят с обратным холодильником в течение 24 часов и часть фильтруют и сушат в печи. Образовавшийся образец обозначают как опыт 28. Определение 0,75 л/1 мин относится к введению/выведению 0,75 литра в минуту в мельницу и из мельницы.
Вышеуказанную процедуру повторяют, за исключением того, что не применяют предварительное размалывание. Данный образец обозначают как опыт 27.
Кроме того, получают контрольный образец, где отсутствует лапонит® и не используют предварительное размалывание. Данный образец обозначен как пример 26.
Все образцы затем делят на две части и первую часть подвергают старению в течение 4 часов в 20% водяном паре при 800°С, а вторую часть подвергают старению при 537,8°С в течение 2 часов. Для каждого подвегнутого старению образца определяют площадь поверхности. Результаты гидротермической стабильности суммированы в таблице 7, а свойства пор по азоту приводятся на фигуре 8.
Таблица 7 показывает, что предварительное размалывание приводит к получению продукта со значительно более высокой площадью свежей поверхности и обработанной водяным паром поверхности, чем в случае отсутствия глины или неразмолотого образца с таким же уровнем диспергируемой глины. Результаты распределения размера пор по азоту показывают, что предварительное размалывание дает небольшой сдвиг в сторону большего уровня пор с меньшим диаметром, но более высокий TPV.
ПРИМЕР 9
Данный пример иллюстрирует влияние и степень влияния предварительного диспергирования, достигнутого с лабораторным препаратом синтетического гекторита, высушенного до низкого TV (9,64%).
Синтетический гекторит получают медленным добавлением раствора 75,3 г Na2CO3, растворенного в 289 г H2O, к раствору 183,5 г MgSO4·7H2O+3,4 г LiCl. Затем на протяжении периода получаса добавляют раствор 267,4 силиката (27,11% SiO2), разбавленного 826 г Н2О. Образовавшуюся суспензию геля кипятят в течение 30 минут для удаления карбоната, затем выдерживают в автоклаве в течение 2 часов при 200°С, фильтруют, промывают на фильтре 1 л деионизированной воды при 65,6°С (150°F) и сушат при 135°С.
Части снова суспендируют в воде следующим образом:
Опыт 29 перемешивают при умеренной степени перемешивания в течение 30 минут.
Опыт 30 диспергируют смесителем Silverson в течение 10 минут при 1000 об./мин.
Опыт 31 перемешивают в течение 18 часов стержнем магнитной мешалки.
К каждой из вышеуказанных суспензий добавляют достаточное количество кальцинированного оксида алюминия (СР-3, ALCOA) для получения суспензии с 15% твердых веществ, содержащей 3 масс.% синтетического гекторита и 97% оксида алюминия. Суспензию кипятят с обратным холодильником в течение 24 часов при перемешивании. Суспензию фильтруют и сушат в печи при 137,8°С (280°F). Средний диаметр пор каждого продукта, полученного в опытах 29-31, измеряют после кальцинирования в течение 2 часов при 537,8°С (1000°F). Определяют площадь поверхности и TPV, результаты суммированы в таблице 8.
Средний диаметр пор повышался от 147 до 159 и 176 ангстрем для опытов с 29 по 31, соответственно, указывая на то, что лучшее диспергирование достигается, когда повышают время и/или интенсивность диспергирования. Считается, что данный образец трудно было диспергировать вследствие его относительно низкого общего содержания летучих компонентов - 9,64%. Таким образом, диспергируемость глины можно улучшить регулированием TV, степени дисперсности глины или размера кристаллитов глины, которые, в свою очередь, регулирует средний диаметр пор оксида алюминия.
ВЛИЯНИЕ СТЕПЕНИ ДИСПЕРСНОСТИ СИНТЕТИЧЕСКОГО ГЕКТОРИТА НА ОБЩИЙ ОБЪЕМ ПОР N2 И СРЕДНИЙ ДИАМЕТР ПОР БЕМИТА, ПОЛУЧЕННОГО ПОВТОРНОЙ ГИДРАТАЦИЕЙ АКТИВНОГО ОКСИДА АЛЮМИНИЯ
Общее содержание летучих компонентов синтетического гекторита, образца, используемого для опытов 29-31, составляло только 9,64% и считается, что данную глину трудно полностью диспергировать, независимо от степени перемешивания.
СРАВНИТЕЛЬНЫЙ ПРИМЕР 2
Данный пример иллюстрирует влияние ненабухаемых глин, таких как каолин или кальцинированный каолин, на структуру пор.
Пример 1 повторяли, за исключением того, что используемой глиной был каолин или кальцинированный каолин в количествах, указанных в таблице 9. Образовавшиеся композиционные материалы обозначают как опыт 33 (каолин), опыт 34 (кальцинированный каолин при 6 масс.%) и опыт 35 (кальцинированный каолин при 12 масс.%). Кальцинирование каолина проводили при 900°С в течение 0,66 часа. Свойства поверхности указанных материалов измеряли для случая площади свежей поверхности, результаты приводятся в таблице 9. Применяли также контрольный оксид алюминия без глины, полученный таким же способом и обозначенный как опыт 32. Количества каолина, присутствующего в частицах композиционного материала, также приводятся в таблице 9. Данные десорбции азота для опытов с 32 по 35 также приводятся на фигуре 9. Как можно видеть из данных таблицы 9 и фигуры 9, каолин действительно вызывал уменьшение среднего диаметра пор (APD) и общего объема пор относительно контроля, но происходило повышение площади поверхности. Каолин является ненабухаемой глиной.
ПРИМЕР 10
Данный пример иллюстрирует влияние использования менее предпочтительной монтмориллонитной глины на свойства пор.
Повторяли сравнительный пример 2, за исключением того, что монтмориллонитная глина, выпускаемая фирмой Southern Clay Products под товарным знаком Gelwhite L, заменяла каолиновую глину при 6 масс.% (опыт 36) и 12 масс.% (опыт 37). Контроль при 0 масс.% глины обозначали как пример 32. Результаты суммированы в таблице 9 и на фигуре 10. Как можно видеть их них, свойства пор и площадь поверхности оксида алюминия выше при 6 масс., чем при 12 масс.% глины. Кроме того, оказывается, что мода пор в области мезопор не смещается вправо и распределение размера пор оказывается «размазанным» (растянутым по оси X). Считается, что это можно приписать тому факту, что монтмориллонит сложно хорошо диспергировать без некоторой другой обработки, такой как значительное размалывание для снижения размера частиц, ионообмен или использование диспергаторов, таких как тетранатрийпирофосфат.
ПРИМЕР 11
Повторяли сравнительный пример 2 за исключением того, что каолиновую глину заменяли силикатом натрия при 0,5 масс.% (опыт 39) и 1 масс.% (опыт 38). Контроль без силиката представлен опытом 32.
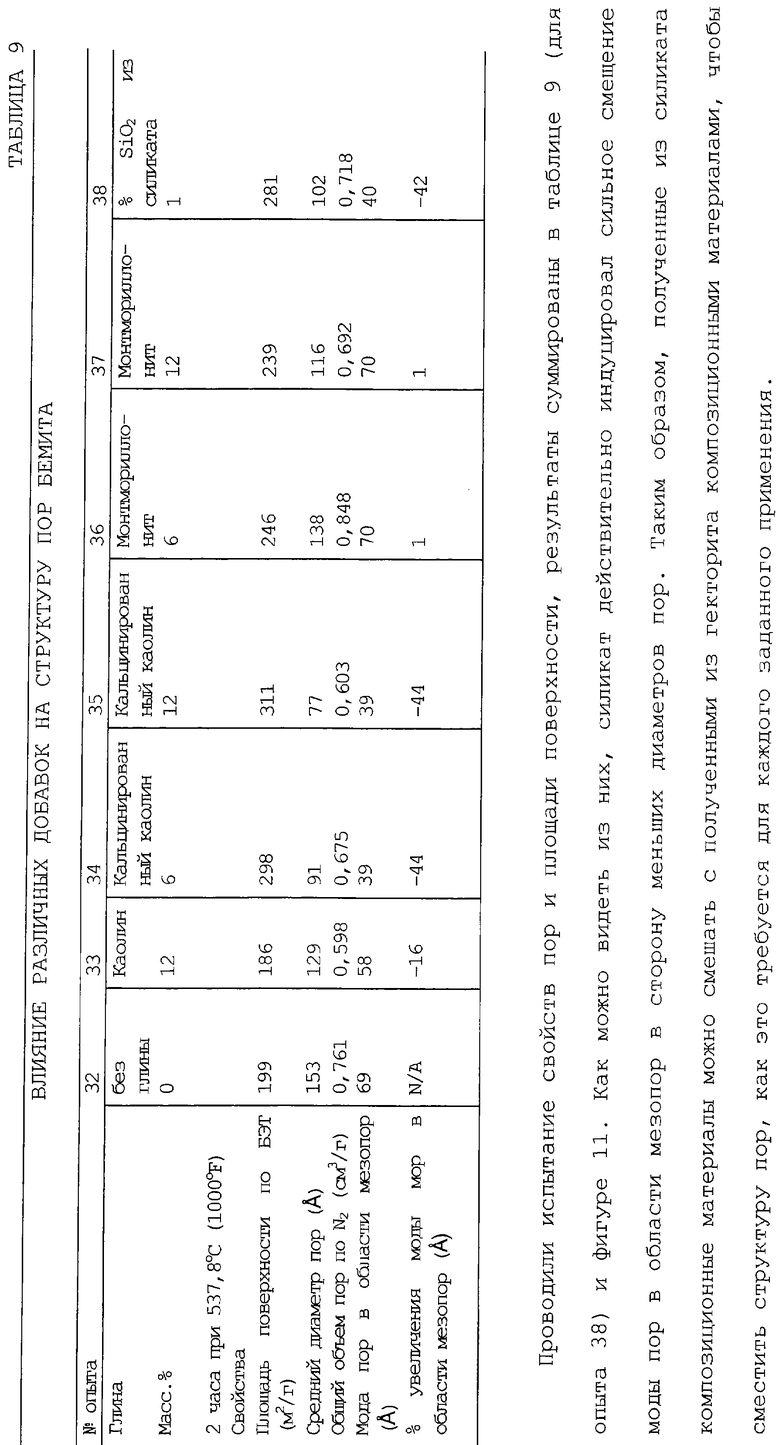
Пример 12
Данный пример иллюстрирует влияние на гидротермическую стабильность размалывания кальцинированного оксида алюминия перед повторной гидратацией в присутствии набухаемой глины.
Так, повторяли пример 8, опыт 28, и объединенную суспензию лапонита® (при 3 масс.%) и кальцинированного оксида алюминия размалывали перед повторной гидратацией и обозначали как опыт 42. После кипячения с обратным холодильником в течение 24 часов при 100°С (212°F) бемит фильтровали и сушили в печи при 140°С в течение 6 часов. Порции кальцинировали при 800°С в атмосфере приблизительно 20% водяного пара при варьировании времени и затем измеряли площадь поверхности. Вышеуказанную процедуру повторяли, за исключением того, что опускали стадию размалывания, и продукт обозначали как опыт 41. Был также получен контрольный образец, не содержащий лапонит® и не прошедший стадию размалывания, обозначен как опыт 40 и подвергнут такой же обработке водяным паром и определениям площади поверхности. Результаты суммированы на фигуре 12. На фигуре 13 данные каждого опыта фигуры 12 выражены в виде процента площади поверхности, полученной на свежем образце, нагретом в течение 2 часов при 537,8°С (1000°F) (т.е. нагретой в водяном паре в течение 0 часов). Данный процент обозначен как % сохранения площади поверхности. Как можно видеть из данных фигуры, стабильность площади поверхности повышается в следующем порядке: 0% лапонита® (опыт 40)<3% лапонита® (опыт 41)<3% лапонита® и размалывание (опыт 42).
ПРИМЕР 13
Данный пример иллюстрирует влияние добавления силиката Na к бемиту, полученному из повторно гидратированного, кальцинированного оксида алюминия, после выполнения синтеза.
Образец кальцинированного оксида алюминия, выпускаемого фирмой Porcel под торговой маркой СР-3 (опыт 43), и образец кальцинированного оксида алюминия, выпускаемого Porcel под торговой маркой АР-15 (опыт 44), каждый отдельно, суспендировали в воде, содержащей 0,5 масс.% (в расчете на оксид алюминия) глюконата натрия, при содержании твердых компонентов 17 масс.% и подвергали горячему старению в течение 24 часов при кипячении с обратным холодильником. Обе порции затем разделяли и к ним добавляли варьируемые количества силиката натрия, подвергали старению в течение 30 минут при 21°С, рН доводили до 9,0 при помощи 4% H2SO4, фильтровали, снова суспендировали с сульфатом аммония для удаления Na20, фильтровали, промывали водой и сушили. Каждый образец обрабатывали водяным паром в течение 4 часов при 800°С в атмосфере приблизительно 20 об. % водяного пара и измеряли его площадь поверхности.
Результаты по продуктам каждого опыта суммированы на фигуре 14. Как можно из видеть из ее данных, силикат значительно улучшает гидротермическую стабильность образцов бемита.
ПРИМЕР 14
Данный пример иллюстрирует влияние добавления силиката к композиционному материалу настоящего изобретения после его образования.
Две порции бемита получали с использованием 3 масс.% (опыт 45) и 5 масс.% (опыт 46) лапонита® RD (Laporte) в качестве источника диспергируемой глины. Суспензии лапонита® получали добавлением глины к воде с быстрым смешиванием и перешиванием в течение 20 минут. Глюконат алюминия добавляли в количестве 0,5 масс.% (в расчете на оксид алюминия) с последующим добавлением СР-3, т.е. кальцинированного оксида алюминия от ALCOA. Каждую суспензию кипятили с обратным холодильником в течение 24 часов с перемешиванием, фильтровали и сушили в печи в течение ночи при 137,8°С (280°F). Порции каждого продукта снова диспергировали в воде, добавляли силикат натрия и смесь подвергали старению 30 минут при 21°С. рН регулировали до 9,0 при помощи 4% H2SO4, фильтровали, подвергали ионообмену в виде суспензии с сульфатом аммония для удаления Na2O, фильтровали, промывали водой и сушили в печи при 137,8°С (280°F). Каждый образец затем подвергали контакту с атмосферой с 20 масс.% водяного пара в течение 4 часов при 800°С и определяли его площадь поверхности. График зависимости масс.% SiO2 от площади обработанной водяным паром поверхности представлен на фигуре 15. Как можно видеть из фигуры 15, получили очень высокие площади поверхности. Кроме того, сравнение указанных площадей обработанных водяным паром поверхностей с образцами без глины примера 13 (фигура 14) представлено на фигуре 16. Как видно из ее данных, повышенные площади поверхности получали комбинированием добавления диспергируемой глины с добавлением силиката после синтеза. Считают, что частью причин для более высокой площади обработанной водяным паром поверхности оксидов алюминия с диспергируемой глиной является более высокая площадь свежей поверхности, более высокий объем пор и более высокий средний диаметр пор.
ПРИМЕР 15
Данный пример иллюстрирует влияние добавления силиката к оксиду алюминия, полученному со слабо диспергированным синтетическим гекторитом, полученным только при 100°С (212°F) (более низкая температура получения, что вызывает значительно более низкую степень диспергируемости), после проведения синтеза.
Порцию синтетического гекторита получали добавлением 97,9 г кремниевой кислоты (H2SiO4) к 1169 г Н2О в полимерном реакторе на 3 л при перемешивании. В реактор добавляли при перемешивании 58,3 г Mg(ОН)2, 2,55 г LiCl и 4,7 г NaCl. Суспензию кипятили с обратным холодильником в течение 24 часов, фильтровали, промывали три раза водой при 65,6°С (150°F) и сушили при 137,8°С (280°F). Была получена рентгенограмма, очень похожая на рентгенограмму лапонита® RD. Достаточное количество данного высушенного материала смешивали в 800 мл Н20 в течение 2 минут с получением при этом 3% конечной массы оксида алюминия и с диспергированием его в максимально возможной степени. В противоположность лапониту® RD, получали мутную суспензию, что является показателем низкой степени дисперсности. Данную суспензию добавляли в полимерный реактор на 3 л вместе с 0,5 масс.% глюконата натрия (в расчете на оксид алюминия) и дополнительной водой с получением суспензии с 17% твердых веществ. Затем добавляли СР-3 (кальцинированный оксид алюминия от ALCOA). Данную суспензию кипятили с обратным холодильником в течение 24 часов, фильтровали и сушили при 137,8°С (280°F). Части повторно суспендировали в воде и добавляли изменяемые количества силиката, как показано на фигуре 17, опыт 49. Каждый образец обрабатывали водяным паром в течение 4 часов при 800°С приблизительно в 20% водяном паре и определяли его площадь поверхности.
Вышеуказанную процедуру повторяли, за исключением того, что высокодиспергированный лапонит® RD заменяли на синтетический гекторит, изготовленный и использованный в опыте 49. Каждый образец образующегося продукта анализировали на площадь поверхности и результаты обозначали как опыт 48. Эти результаты приведены на фигуре 17.
Контрольный образец, также получали по процедурам опыта 49, за исключением того, что не использовали глину. Площади поверхности нанесены на фигуру 17 и обозначены как опыт 47.
Как можно видеть из данных фигуры 17, стабильность площади поверхности повышается в следующем порядке: без глины + диоксид кремния < слабо диспергированная глина+диоксид кремния < хорошо диспергированная глина + диоксид кремния.
ПРИМЕР 16
Данный пример иллюстрирует влияние предварительного размалывания суспензии, содержащей кальцинированный оксид алюминия и диспергируемую глину, перед повторной гидратацией и с послесинтетическим добавлением силиката натрия.
Суспензию получают диспергированием 51,9 г лапонита® RD (TV=13,26%) в 8331 г Н2O с быстрым перемешиванием в течение 20 минут с последующим добавлением 1616,7 г СР-3 (кальцинированный оксид алюминия от ALCOA, TV=10%). Образовавшуюся суспензию размалывают в мельнице DRAIS на 4 л при скорости приблизительно 1 л/минуту с загрузкой приблизительно 60% стеклянной среды. Суспензию подвергают горячему старению в течение 24 часов при кипячении с обратным холодильником, фильтруют и сушат в печи. Образцы данного оксида алюминия типа бемита снова диспергируют в воде с варьированием количеств силиката натрия, как указано в таблице 10, подвергают старению 1/4 часа при 21°С, рН доводят до 9,0 с с помощью 4% H2SO4, фильтруют и обозначают как опыт 52. Вышеуказанную процедуру повторяют, за исключением того, что стадию размалывания опускают и образовавшиеся образцы обозначают как опыт 51. Контроль получают так же, за исключением того, что опускают стадию размалывания и добавление диоксида кремния и глины. Контроль обозначают опытом 50. Каждый из образцов опытов от 50 до 52 снова диспергируют в воде, содержащей сульфат аммония, в течение 1/4 часа, фильтруют, промывают водой и сушат в печи. Такой ионообмен проводят для снижения Na2O до низкого (<0,25 масс.%) уровня. Образцы затем кальцинируют в течение 4 часов при 800°С в атмосфере 20 об. % водяного пара. Влияние уровня силиката, а также размалывания/отсутствия размалывания и присутствия диспергируемого гекторита на площадь обработанной водяным паром поверхности суммировано в таблице 10. Размалывание дает самую высокую площадь поверхности с добавлением силиката или без его добавления.
ПРИМЕР 17
Данный пример иллюстрирует влияние добавления силиката натрия перед или после повторной гидратации (горячее старение) суспензии кальцинированного оксида алюминия.
Суспензии кальцинированного оксида алюминия (СР-3, ALCOA) получают с варьированием уровней силиката и затем подвергают горячему старению в течение 24 часов при кипячении с обратным холодильником. Образовавшиеся образцы группируют как опыт 54. Суспензии получают также с использованием только кальцинированного оксида алюминия (обозначен как опыт 53) или кальцинированного оксида алюминия, к которому перед горячим старением добавляют 3% диспергируемого гекторита (лапонит® RD) (обозначено как опыт 55). Последние два препарата обрабатывают различными количествами силиката после горячего старения, изменяемые количества показаны на фигуре 8. Все образцы подвергают обмену с сульфатом аммония для снижения Na2O до низкого уровня (<0,25 масс.%) перед обработкой водяным паром при 800°С в течение 4 часов в атмосфере приблизительно 20% водяного пара. Площади поверхности образовавшихся продуктов для опытов с 53 по 55 указываются на фигуре 18. Результаты площади поверхности указывают, что добавление силиката улучшает гидротермическую стабильность всех образцов. Однако площади поверхности следуют обычно в следующем порядке: Al2O3+3% лапонита® > добавление силиката после старения > добавление силиката перед старением. Добавление силиката до горячего старения также снижает объем пор/диаметр пор бемита.
ПРИМЕР 18
Данный пример иллюстрирует влияние добавления силиката после повторной гидратации на структуру обработанных паром пор композиционного материала бемитный оксид алюминия/лапонит®.
Образцы бемита, содержащего 3% лапонит®, с изменением уровней силиката: 0 масс.% (опыт 56), 2 масс.% (опыт 57), 4 масс.% (опыт 58) и 8 масс.% (опыт 59) получали, как описано в примере 14. После 4 часов обработки при 800°С в 20% водяном паре измеряли распределение размера пор по азоту. Результаты приводятся на фигуре 19. Фигура 19 показывает только небольшие изменения в распределении пор для 0, 2, 4% добавленного силиката, однако, при 8% силиката поры смещаются к более низкому среднему диаметру пор.
ПРИМЕР 19
Данный пример иллюстрирует получение композиционного материала оксид алюминия/набухаемая глина, который используют в следующем примере для получения из него агломератов. 7014 г ОВ (первоначальная основа, не скорректированная по TV) (3% по массе, в пересчете на общую массу оксида алюминия и глины) синтетического гекторита лапонит® суспендируют в 350 галлонах водопроводной воды при температуре окружающей среды. Суспензию перемешивают в открытом танке мешалкой с 4 лопастями в течение 30 минут при максимальном перемешивании (приблизительно 300 об./мин) для обеспечения хорошей дисперсности. Затем к суспендированному лапониту® добавляют 234 кг (515 фунтов) ОВ активированного оксида алюминия Alcoa CP-3. После добавления всего CP-3 суспензию нагревают до 93,3°С (200°F), при такой температуре ее выдерживают в течение 24 часов. Суспензию фильтруют и промывают водопроводной водой при 65,6-71,1°С (150-160°F) на ленточном фильтре с тремя зонами для промывок Eimco. Фильтровальную лепешку сушат распылением при температуре входа 371,1°С (700°F) и выхода 121,1°С (250°F).
Образовавшийся продукт обозначают как образец № АХ-1.
Сумма свойств АХ-1 представлена в таблице 11, а график его распределения размера пор представлен на фигуре 20.
Данные по АХ-1 на фигуре 2 обозначены как опыт 60.
СРАВНИТЕЛЬНЫЙ ПРИМЕР 3
Контрольный образец бемитного оксида алюминия синтезировали следующим образом:
К 4950 объемным частям воды, нагретой в реакторе до 63,3°С (146°F), при постоянном перемешивании добавляют 150 объемных частей 7,0 масс.% раствора сульфата аммония как Al2O3 и образовавшуюся смесь перемешивают в течение четырех минут. В реактор затем одновременно вводят два отдельных раствора. Первый раствор является 7,0 масс.% раствором сульфата алюминия (в пересчете на Al2O3) в воде и второй раствор является 20 масс.% раствором алюмината натрия (в пересчете на Al2O3) в воде. После завершения добавления массовое отношение сульфат алюминия : сульфат алюминия-натрия в реакторе составляет 5:3. Скорости потоков во время добавления регулируют так, чтобы обеспечить рН 7,6. Когда рН соответствует требуемому значению 7,6, добавление сульфата алюминия заканчивают, а добавление алюмината натрия продолжают до достижения рН 9,3. Добавление алюмината натрия затем заканчивают и содержимое реактора подвергают старению в течение 2 часов при 66°С (150°F). Осажденный продукт затем фильтруют, промывают и сушат распылением при температуре ввода 371°С (700°F) и температуре вывода 135°С (275°F) с образованием алюминиевого порошка, который сортируют по размеру для получения частиц размером 10-200 микрон. Образовавшийся продукт обозначают САХ-1. Свойства его суммированы в таблице 11 и на фигуре 20. Данные САХ-1 на фигуре 20 обозначены опытом 61.
Как можно видеть из таблицы 11 и фигуры 20, АХ-1 (опыт 60) и САХ-1 (опыт 61) имеют сходные площади поверхности, но АХ-1 имеет приблизительно на 20 об. % больший объем пор, чем САХ-1, и мода пор в области мезопор для АХ-1 составляет приблизительно 150 ангстрем по сравнению с 50-70 ангстремами для САХ-1.
ПРИМЕР 20
Часть А
Данный пример иллюстрирует получение АХ-1 с предварительной пропиткой перед экструзией.
13,6 кг (30 фунтов) ОВ оксида алюминия АХ-1 смешивают в смесителе Эйриха с 10,5 кг водопроводной воды, 6,2 кг раствора молибдата аммония и 2,0 кг раствора нитрата никеля товарного сорта (15% Ni). Раствор молибдата аммония получают растворением 2,2 кг продажных кристаллов димолибдата аммония в 4,0 кг деионизированной воды. Смесь экструдируют в пилотной установке экструдера 4'' Bonnot, получая при этом экструдаты с диаметром 0,04'' с использованием стандартных условий экструзии. Экструдаты сушат при 121,1°С (250°F) в течение 4 часов и кальцинируют при 648,9°С (1200°F) в течение 1 часа. Образовавшийся экструдат обозначают ЕМАХ-1 (опыт 62).
Часть В
Результаты по свойствам пор части А примера 20 нормализуют по основе, не содержащей металлов, результаты обозначены как опыт 63. Образцы нормализуют по основе, не содержащей металла, в соответствии со следующим уравнением:
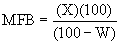
Уравнение 6
где Х обозначает соответствующую характеристику пор, такую как PV (в см3/г) или SA (м2/г);
W - масс.% являющихся каталитическими промоторами оксидов металлов, таких как оксид Ni, Co и Мо, на катализаторе, в расчете на массу пористых компонентов катализатора. Масса непористых компонентов экструдата катализатора, например непористых разбавителей, не включена в расчет масс.%,
MFB - Основа без металлов.
СРАВНИТЕЛЬНЫЙ ПРИМЕР 4
Часть А
Пример 20, часть А, повторяют, за исключением того, что образец АХ-1 из примера 19 заменяют образцом САХ-1 сравнительного примера 3. Образовавшийся экструдатный продукт обозначают ЕМСАХ-1 (опыт 68).
Часть В
Результаты по свойствам пор части A сравнительного примера 4 нормализовали по основе, не содержащей металлов, и результаты обозначили как опыт 69.
Физические и композиционные свойства образцов пропитанных металлом катализаторов ЕМАХ-1 и ЕМСАХ-1 представлены в таблице 12, а распределение размера пор по ртути и другие свойства указанных образцов приводятся в таблицах 13А и В.
Отмечают также более высокое содержание SiO2 вследствие природы диспергируемой глины, содержащейся в АХ-1. График распределения размера пор по ртути для каждого катализатора показан на фигуре 21.
Особенно следует отметить, что SA и TPV как опыта 62, так и опыта 68 являются сходными, но мода пор для опыта 62 составляет 145 ангстрем по сравнению только с 65 ангстрем для опыта 68. Указанные моды являются очень близкими таковым исходных оксидов алюминия, что является характеристикой стабилизирующего действия предварительной пропитки металлами на свойства оксида алюминия. Как можно далее увидеть из данных таблиц 13 A и В, диаметры пор смещены от <100 ангстрем в опыте 68 к области 100-250 ангстрем и, более преимущественно, к области 130-250 ангстрем для опыта 62. Несмотря на смещение и увеличение моды пор, общая площадь поверхности и общий объем в опыте 62 подобны опыту 68.
ПРЕДВАРИТЕЛЬНО ПРОПИТАННЫЕ МЕТАЛЛОМ ЭКСТРУДАТЫ
CBD - объемная плотность в слежавшемся состоянии
ПРИМЕР 21
Часть А
Образец оксида алюминия АХ-1, полученный в соответствии с примером 19, использовали для получения пропитанного металлом экструдата в соответствии с примером 20, часть Е, за исключением того, что смесь содержала на 300 г больше воды, чтобы повысить уровень пористости в порах больше, чем 250 ангстрем в диаметре. Образовавшиеся экструдаты обозначали ЕМАХ-2 (опыт 64).
Часть В
Результаты для свойств пор части A данного примера нормализовали по основе, не содержащей металла, и результаты обозначали как опыт 54 и приводили в таблицах 13А и 13В.
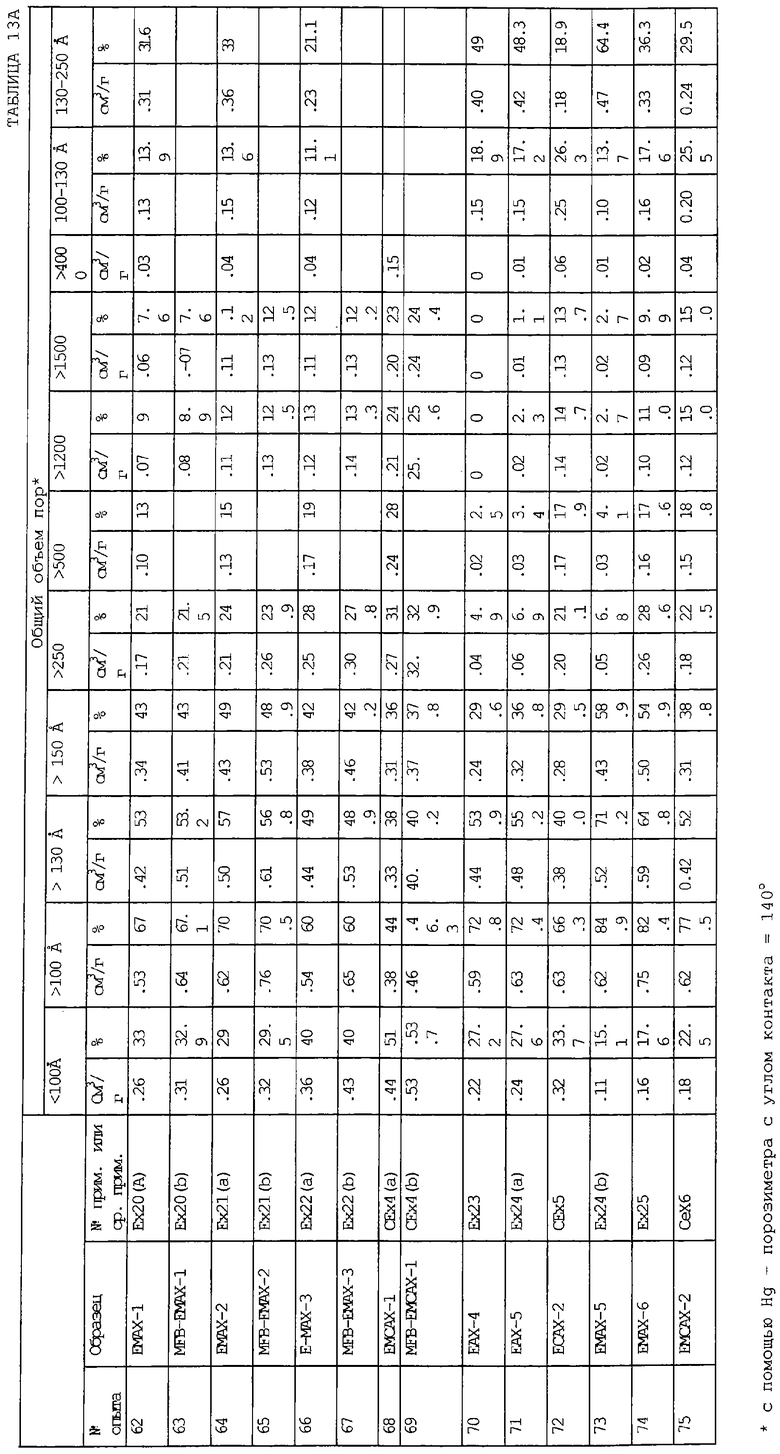

MFB - основа без металла
Пример 22
Часть А
Катализатор получали таким же путем, как ЕМАХ-1 (опыт 62), за исключением того, что источником оксида алюминия была физическая смесь порошков 9,09 кг (20 фунтов) исходной порции (0 В) АХ-1 (опыт 60) и 4,5 кг (10 фунтов) OB CAX-1 (опыт 61). Оксид алюминия CAX-1 добавляли для повышения макропористости (поры >250 ) в катализаторе. Образовавшийся продукт обозначали ЕМАХ-3 (опыт 66) и его свойства суммированы в таблицах 12, 13А и В и на фигуре 21. Как можно видеть из них, добавление CAX-1 делает поры в области <100 ангстрем и области <250 ангстрем бимодальными.
Часть В
Свойства пор образца части А нормализовали по основе, не содержащей металлов, и результаты обозначали как пример 67 и приводили в таблицах 13А и В.
ПРИМЕР 23
Данный пример описывает процедуру получения (только) основы катализатора в виде оксида алюминия, которую можно в конечном счете превратить в катализатор окончательной обработкой посредством способа «последующей пропитки». Основу для последующим способом пропитанных катализаторов, получаемых последующей пропиткой, получают эструзией/прокаливанием оксида алюминия в отсутствие металлов-промоторов (Ni и Мо в данном случае).
30 фунтов ОВ оксида алюминия АХ-1 смешивали с 31 фунтами водопроводной воды в смесителе Эйриха пилотного масштаба. Смесь экструдировали с использованием экструдера 4'' Bonnot с образованием экструдатов с диаметром 0,04''. Экструдаты сушили при 121,1° (250°F) в течение 4 часов и затем кальцинировали при 732,2°С (1350°F) в течение 1 часа.
Образовавшийся продукт обозначили как ЕАХ-4 (пример 70), свойства его пор по ртути показаны в таблицах 13А и В.
СРАВНИТЕЛЬНЫЙ ПРИМЕР 5
Пример 22 повторяли, за исключением того, что исходным оксидом алюминия был САХ-1. Образовавшийся продукт обозначили как ЕСАХ-2 (опыт 72). Свойства пор по ртути показаны в таблицах 13А и В.
При сравнении опыта 70 с 72 можно увидеть, что опыт 70 имеет почти 70% от TPV в области 100-250 ангстрем и основную часть (49%) в области 130-250 ангстрем. Можно далее отметить, что в опыте 72 мода пор исходного оксида алюминия САХ-1 (опыт 61) смещается от 65 ангстрем до 100 ангстрем в экструдате. То же самое не является верным для опыта 70 (фигура 22) по сравнению с АХ-1 опыта 60 (фигура 20), которые оба имеют моду пор приблизительно 145 ангстрем. Кроме того, в опыте 70 имеет место приблизительно такая же мода пор, как и в 62 и 64 (фигура 21). Таким образом, оказывается, что оксид алюминия АХ-1 настоящего изобретения имеет одинаковую моду пор, независимо от того, пропитывают ли его до или после обработки. Следует далее отметить, что опыт 70 по существу не имеет пористости >250 ангстрем в отличие от его предварительно пропитанного аналога опыта 62.
ПРИМЕР 24
Часть А
Пример 23 повторяли, за исключением того, что 14,5 кг (32 фунта) воды (на 1 фунт больше, чем в примере 23) применяли в смеси для повышения общей пористости в порах >250 ангстрем. Образовавшийся экструдат обозначили ЕАХ-5 (опыт 71).
Композиционные свойства, диаметр частиц и прочность на раздавливание в опытах с 70 по 72 суммированы в таблице 14 и распределение пор по ртути образцов опытов 70-72 суммировано в таблицах 13А и В. Распределение пор по ртути опытов 70-72 показано также на фигуре 22.
Часть В
Образец экструдата, не содержащий металла, ЕАХ-5 (опыт 71) части А подвергали последующей пропитке следующим образом:
Раствор молибдата аммония в количестве 313 г, рН которого доводили до рН 5,2-5,4, смешивали с 120 г нитрата никеля. Добавляли воду для получения общего раствора 440 см3. Весь раствор переносили на 550 г основы ЕАХ-5. Пропитку проводили способом начальной влажности в пластиковом мешке. Пропитанный материал сушили в течение ночи при 121,1°С (250°F) и кальцинировали при 537,8°С (1000°F) в течение одного часа. Образовавшийся катализатор обозначили ЕМАХ-5 (опыт 73), свойства его пор по ртути показаны в таблицах 13А и В.
СРАВНИТЕЛЬНЫЙ ПРИМЕР 6
Пример 24, часть В, повторяли, за исключением того, что образец ЕАХ-5 заменяли на контрольный образец экструдата ЕСАХ-2 опыта 72. Образовавшийся образец экструдата, пропитанного металлами, обозначили ЕМСАХ-2 (опыт 75), его свойства пор по ртути показаны в таблицах 13А и В.
ПРИМЕР 25
Пример 22 повторяли, за исключением того, что смесь была составлена из равных количеств АХ-1 и САХ-1, по 6,8 кг (15 фунтов) каждого. Смесь перемешивали с 31 фунтами воды в смесителе Эйриха, пропитывали никелем и молибденом с использованием раствора металлов, полученного в соответствии с примером 24, часть В, за исключением того, что общий объем раствора был 550 см3 вследствие более высокого объема пор настоящего образца.
Образовавшийся пропитанный металлом экструдат обозначили ЕМАХ-6 (опыт 74), его свойства пор по ртути показаны в таблице 13А и В.
Композиционные свойства, диаметр частиц, объемная плотность и прочность на раздавливание для опытов от 73 до 75 суммированы в таблице 15.
ПРИМЕРЫ ОСНОВЫ, НЕ СОДЕРЖАЩЕЙ МЕТАЛЛОВ
ТАБЛИЦА 15
ПРИМЕРЫ С ПОСЛЕДУЮЩЕЙ ПРОПИТКОЙ
ПРИМЕР 26
Вакуумные остатки со дна колонны, полученные из арабской средней сырой нефти, имеющей свойства, суммированные в таблице 16, выбраны в качестве сырья для пилотной установки гидрообработки кубовых остатков в неподвижном слое. Эксплуатационные условия пилотной установки суммированы в таблице 17.
Катализатор опыта 62 (ЕМАХ-1) помещают в пилотную установку и испытывают, как описано ниже. Пилотная установка имеет четыре независимых реактора, расположенных в обычной песочной бане. Песочная баня поддерживает четыре реактора приблизительно при одной и той же температуре. В каждый реактор загружают 75 см3 испытуемого катализатора. Инертные стеклянные шарики загружают выше и ниже слоя катализатора для предварительного нагрева реагентов до требуемых условий и заполнения любого дополнительного пространства. Водород и сырье - кубовый остаток входят в нижнюю часть реактора и протекают вместе через слой катализатора и выходят в верхней части реактора. Продукты поступают в емкость разделения газа и жидкости, которая расположена вслед за реактором. Газообразные продукты выходят из системы через клапан регулирования давления, который используют для регулирования давления в реакторе. Жидкие продукты выходят из емкости разделения через клапан регулирования уровня в емкость для жидкого продукта, которая аккумулирует продукт до тех пор, пока его не удаляют из системы. Скорость подачи водорода в реактор регулируют регулятором расхода массы. Скорость подачи кубового остатка в реактор регулируют питающим насосом. Температуру реакторов регулируют изменением температуры песочной бани.
Катализатор испытывали при стандартных условиях: LHSV (часовая объемная скорость) 1,0 (75 см3/час сырья и 75 см3 объема слоя катализатора), температура реактора 426,7°С (800°F) и избыточное давление H2 2000 фунт/кв. дюйм. Скорость потока водорода поддерживают на уровне 75 нормальных литров (NL)/час (примечание: NL измеряют при 0°С и давлении 1 атмосфера). Такие условия обеспечивают приблизительно такой же уровень конверсии, а также удаление серы и кокса по Конрадсону, какие можно было бы ожидать для общепринятых катализаторов в коммерческой установке гидрокрекинга в кипящем слое. Процентное превращение материалов, кипящих выше 537,8°С (1000°F), в материалы, кипящие ниже 537,8°С, измеряли как функцию времени, выраженную в виде баррелей переработанного сырья на фунт загруженного катализатора.
Результаты показаны на фигуре 23 и обозначены как опыт 76.
СРАВНИТЕЛЬНЫЙ ПРИМЕР 7
Пример 26 повторяли за исключением того, что катализатор опыта 68 использовали вместо катализатора опыта 62.
Результаты превращения также суммированы на фигуре 23 как опыт 77. Как можно видеть на фигуре 23, полученный из АХ-1 образец опыта 76 проявляет более высокую активность в крекинге высококипящего (1000°F) кубового материала в более легкие продукты, чем полученный из САХ-1 образец опыта 77. В приведенные данные внесли поправки на стандартные эксплуатационные условия для исключения любых отклонений, вызванных изменениями в реальных эксплуатационных условиях. Полученный из АХ-1 катализатор опыта 76 также имеет преимущества в активности по насыщению (повышение API продукта), десульфуризации и удалению кокса по Конрадсону. Теоретически считается, что более высокий крекинг и гидроактивность данного катализатора имеют место вследствие модифицированного распределения размера пор и, возможно, химического состава основы, обусловленных исходными материалами АХ-1.
Удаление осадка и металлов в опыте 76 незначительно уступает контрольному образцу в опыте 77. Предполагается, что количество осадка повышается при более высоком превращении кубового остатка. Образец опыте 76 также имеет более низкую пористость в макрообласти, что также может оказывать влияние на эксплуатационные характеристики по осадку и металлам на данном сырье.
СВОЙСТВА ИСХОДНОГО СЫРЬЯ
ПРИМЕР 27
Пример 24 повторяли, за исключением того, что катализатором опыта 64 (ЕМАХ-2) заменяли катализатор стандартного примера и исходным сырьем был другой средний вакуумный кубовый остаток арабской нефти, свойства которого суммированы в таблице 16. Результаты сгруппированы как опыт 78 и эффективность превращения суммирована на фигуре 24.
ПРИМЕР 28
Пример 27 повторяли с использованием катализатора опыта 66 (ЕМАХ-3), результаты сгруппированы как опыт 79 и изображены на фигуре 24.
СРАВНИТЕЛЬНЫЙ ПРИМЕР 8
Пример 27 повторяли с использованием контрольного катализатора опыта 68 (ЕМСАХ-1). Результаты сгруппированы как опыт 80 и изображены на фигуре 24.
Как можно видеть на фигуре 24, опыты 78 и 79 значительно превосходят контроль по конверсии.
Кроме того, в следующих аспектах:
(a) API общего жидкого продукта - контроль уступал опыту 79, но превосходил опыт 78;
(b) в снижении содержания серы - контроль уступал опыту 79, но превосходил опыт 78;
(c) в снижении остатка кокса (CCR) в масс.% по Конрадсону - контроль уступал опыту 79, но превосходил опыт 78;
(d) в снижении осадка - контроль превосходил опыты 78 и 79, причем опыт 79 был лучше, чем опыт 78.
(e) в снижении содержания ванадия - контроль превосходил опыты 78 и 79, причем опыт 79 был лучше, чем опыт 78.
(f) в снижении содержания никеля - контроль уступал опыту 79, но превосходил опыт 78.
Следует отметить, что CCR представляет собой остаточный углеродистый материал, образующийся после того, как были отогнаны все легкие углеводороды. Его измеряют стандартным испытанием на деструктивную дистилляцию (ASTM(D-189)). Испытание проводят на исходном сырье и на продуктах. Разница между двумя числами является снижением CCR.
Принципы, предпочтительные варианты осуществления и рабочие режимы настоящего изобретения были описаны в предшествующем описании. Изобретение не должно, однако, истолковываться как ограниченное описанными конкретными формами, т.к. они должны рассматриваться как иллюстративные, а не ограничивающие. Специалистом в данной области могут быть сделаны варианты и изменения, не выходящие за пределы сущности изобретения.
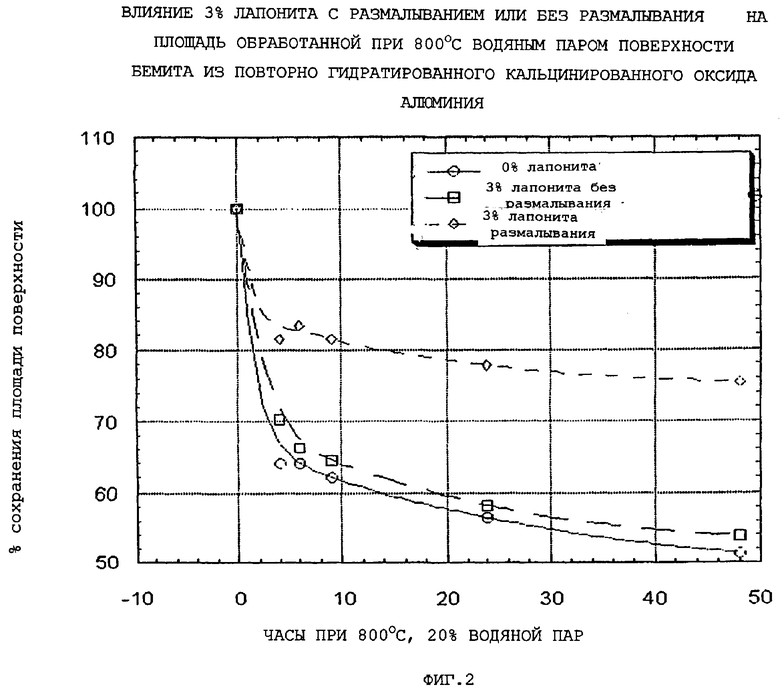

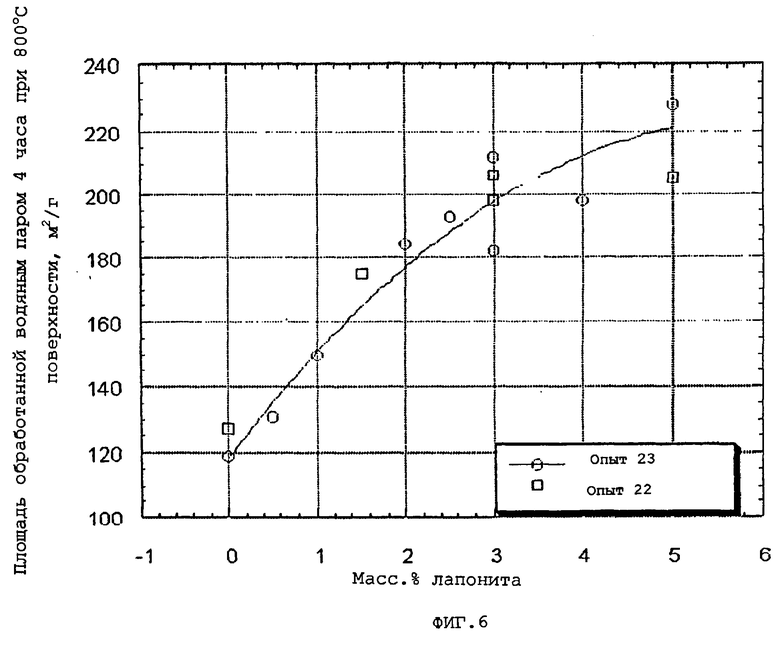
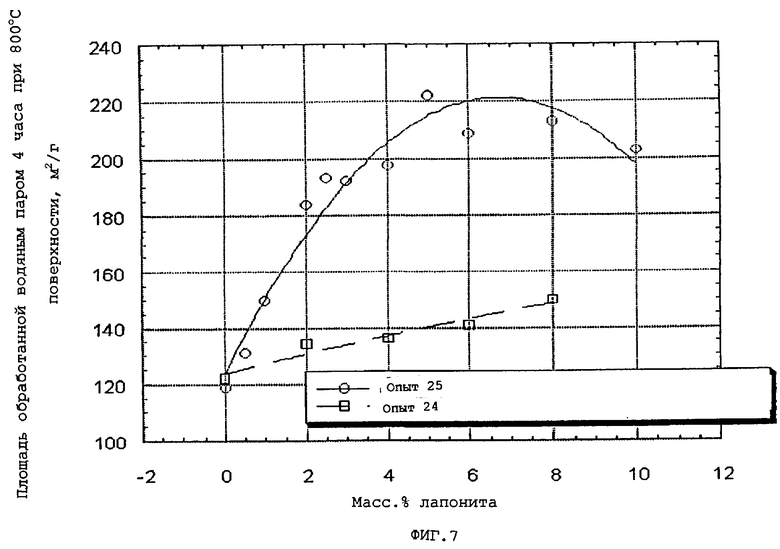
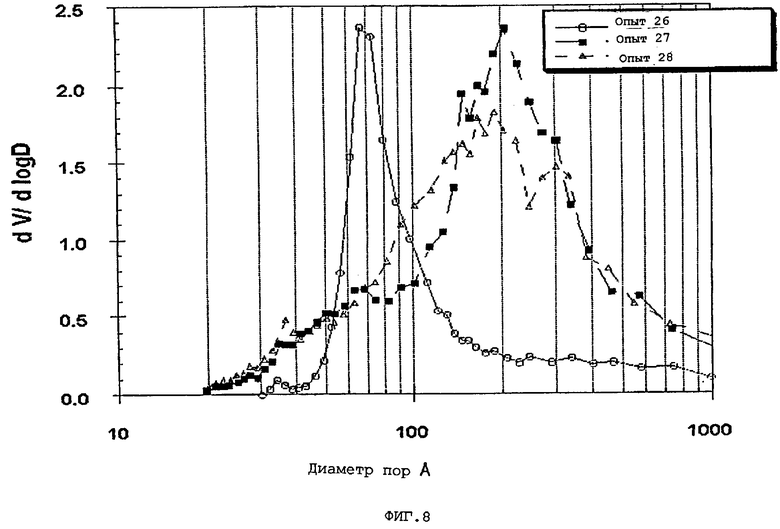
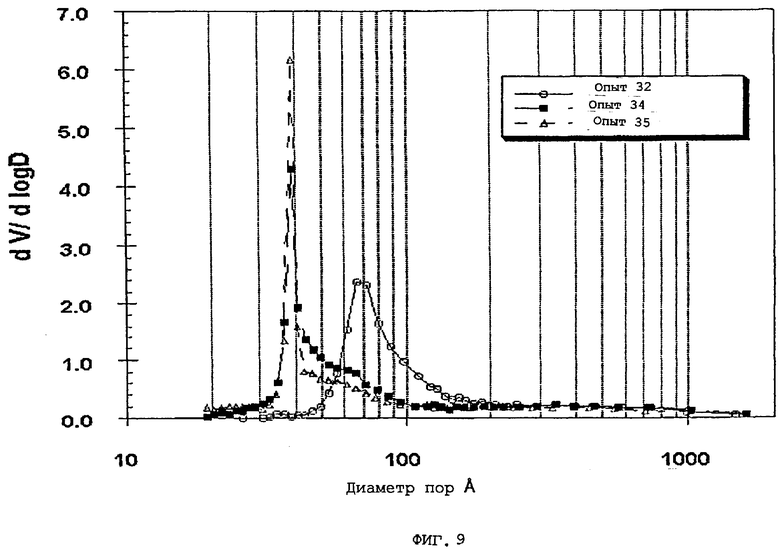
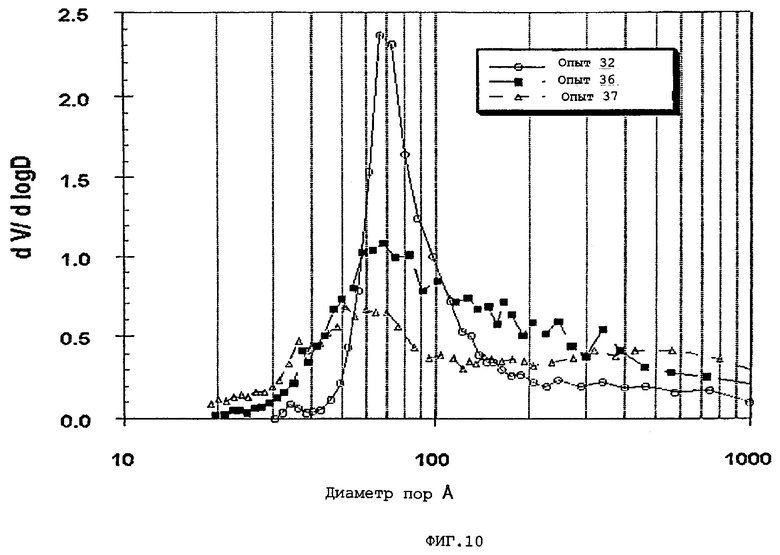
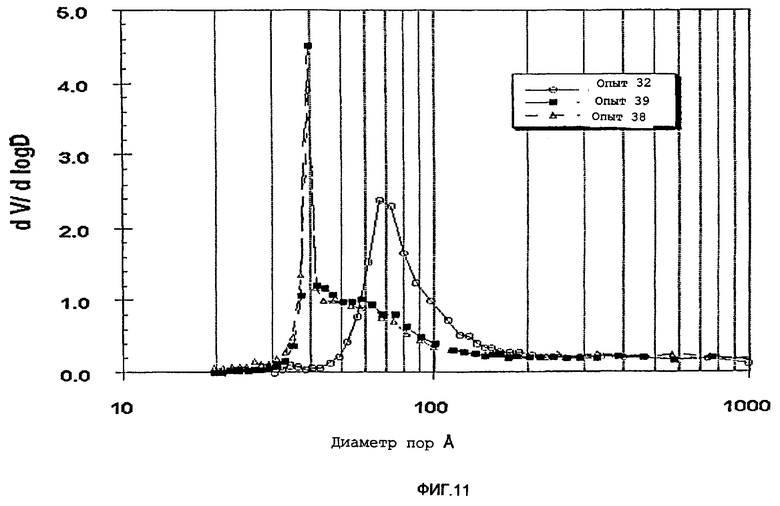
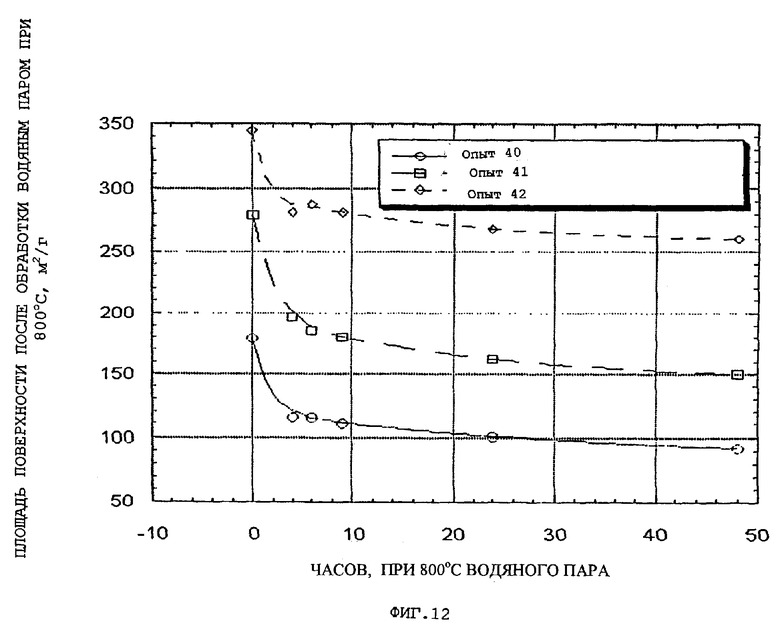
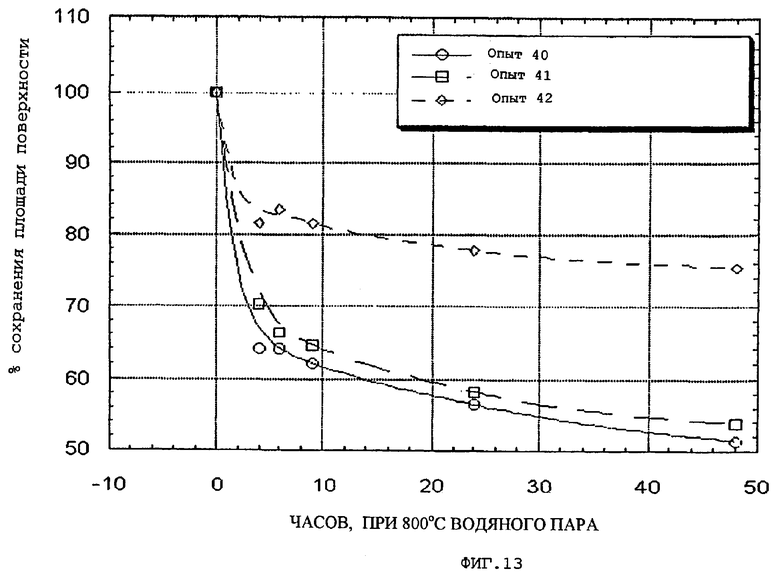

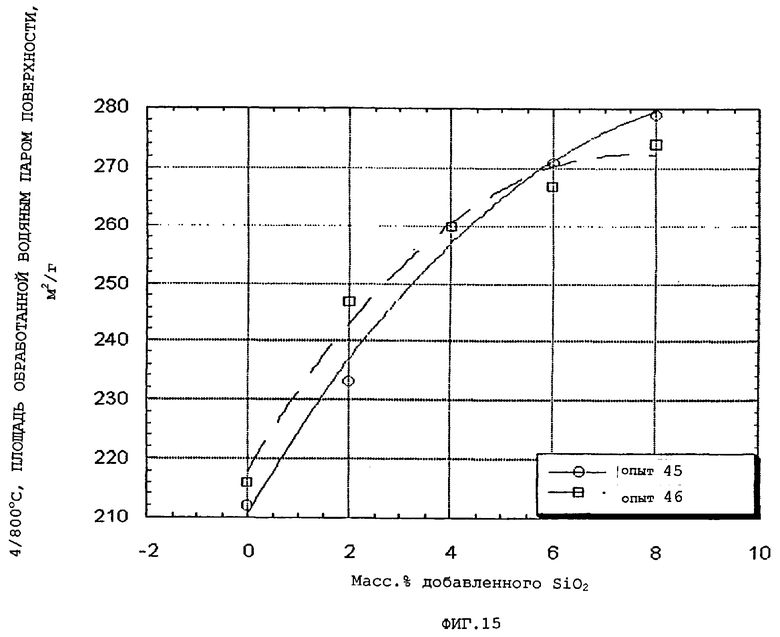
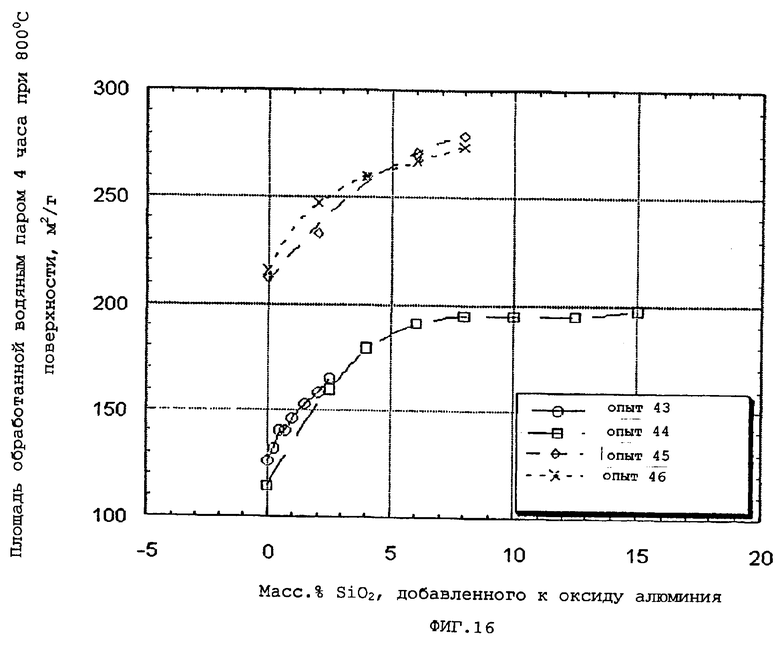
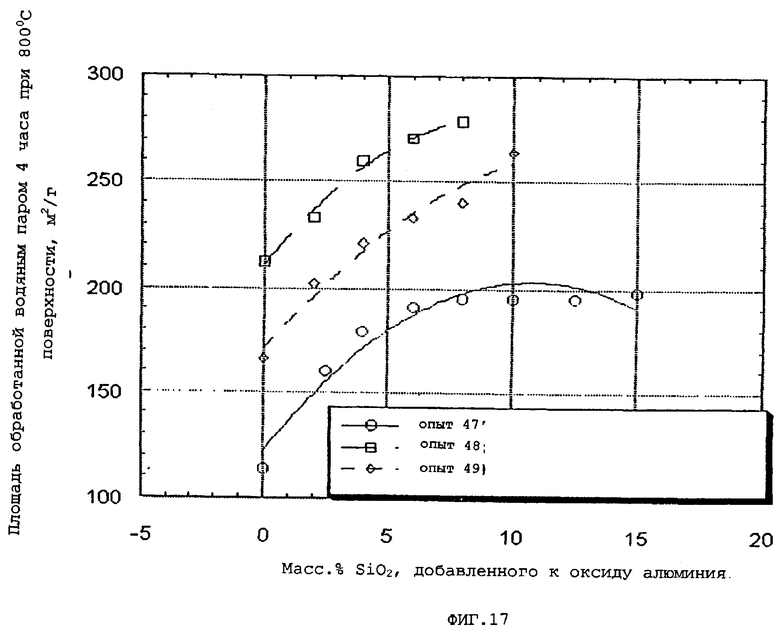
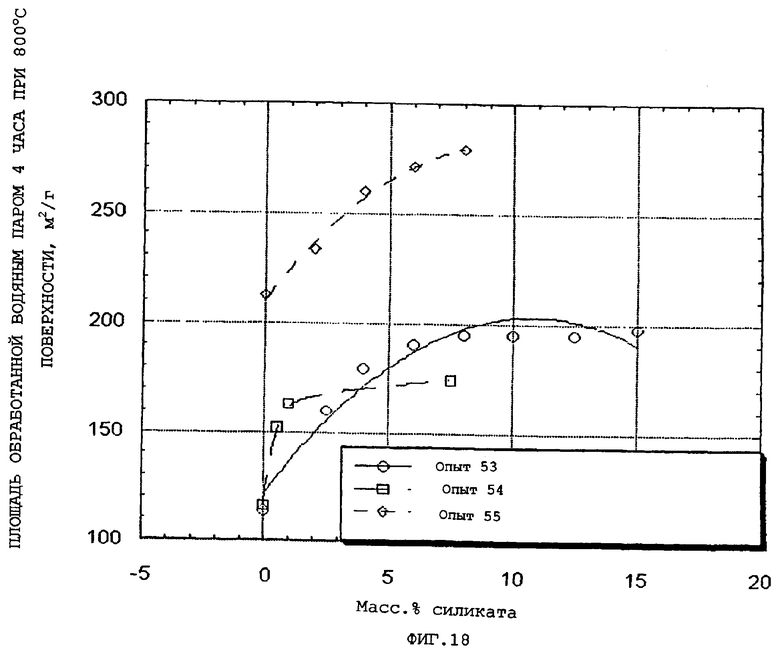
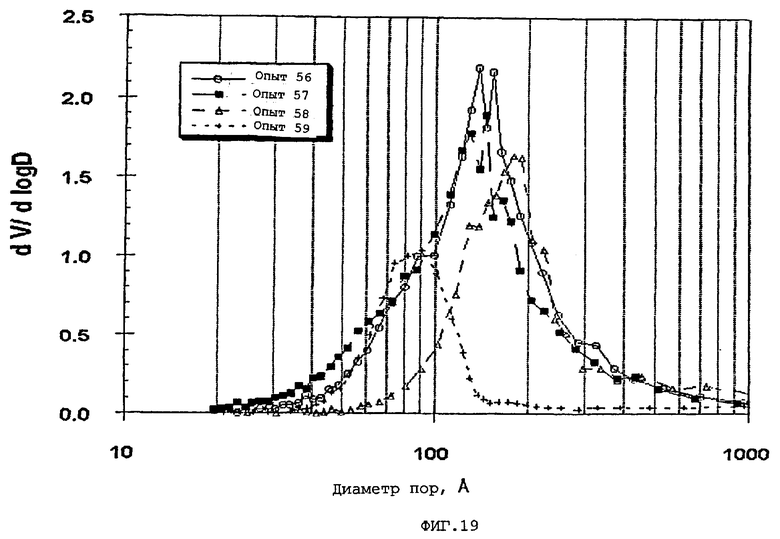
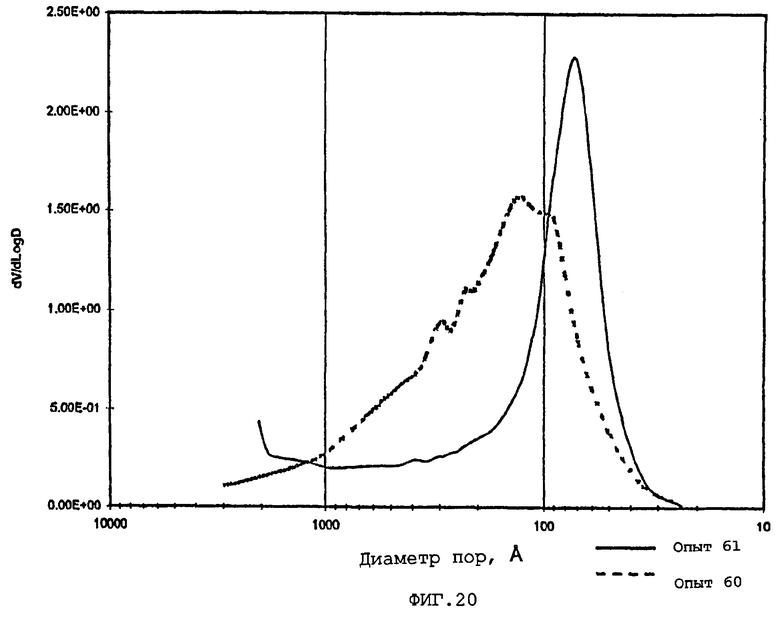

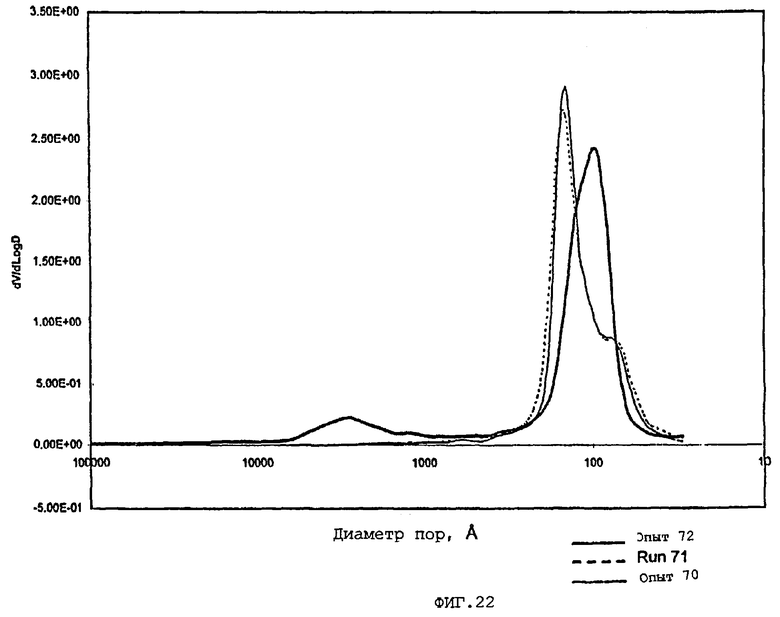
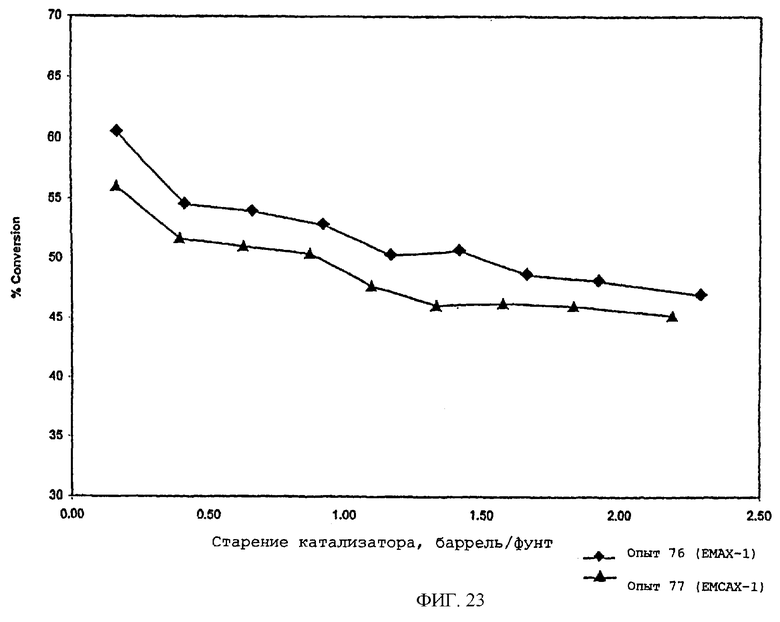
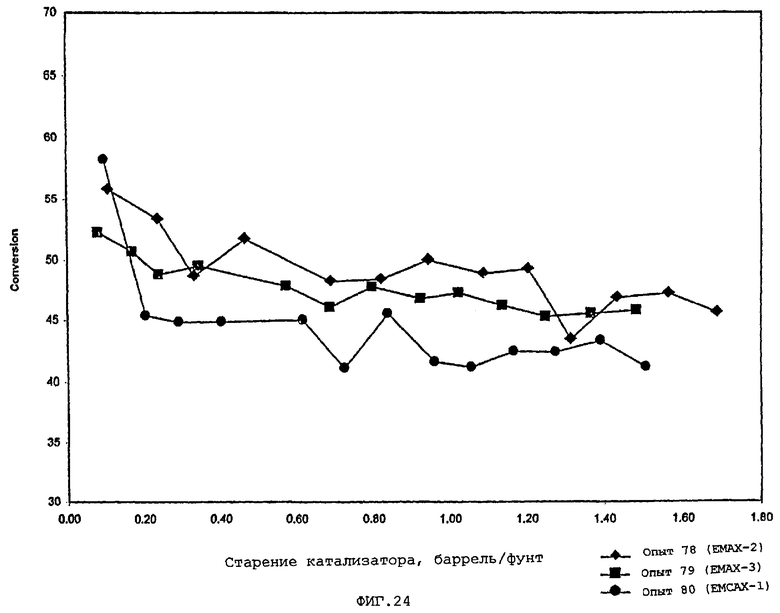
Изобретение относится к области носителей катализаторов и способам их получения. Предложены пористые частицы композиционного материала, которые включают компонент оксида алюминия и компонент набухаемой глины, тонко диспергированный в компоненте оксида алюминия в количестве, эффективном для повышения гидротермической стабильности, объема пор и/или моды пор в области мезопор у частиц композиционного материала относительно случая отсутствия набухаемой глины. Предложены также способы получения частиц композиционного материала, частиц агломератов, полученных из них, и способ гидропереработки нефтяного исходного сырья с использованием агломератов в качестве носителя для катализатора гидропереработки. Технический результат: данный носитель катализатора обладает повышенной гидротермической стабильностью, объемом пор. 6 н. и 38 з.п. ф-лы, 17 табл., 24 ил.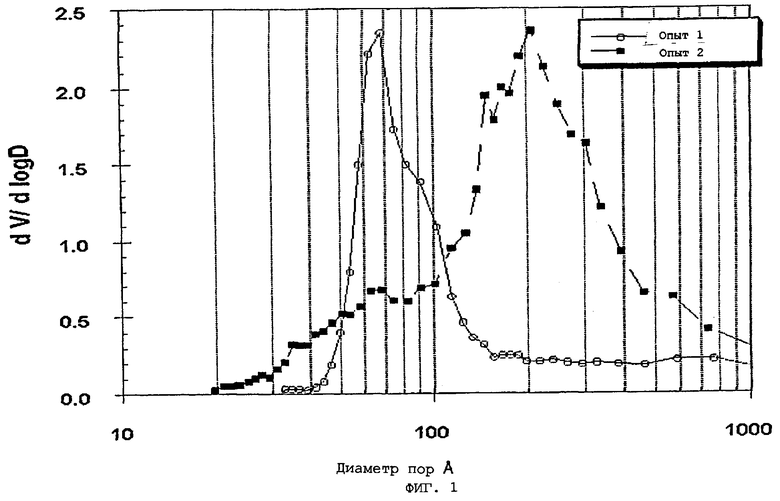
(A) компонент оксида алюминия включает, по меньшей мере, 75 мас.% оксида алюминия, при этом, по меньшей мере, 5% указанного оксида алюминия находится в форме кристаллического бемита, гамма-оксида алюминия, полученного из кристаллического бемита, или их смесей;
(B) компонент набухаемой глины является диспергируемым до включения в частицу композиционного материала и присутствует в частицах композиционного материала в количестве, (а) меньшем, чем 10 мас.%, в расчете на суммарную массу компонента оксида алюминия и компонента набухаемой глины, и (b) эффективном для повышения, по меньшей мере, одной из следующих характеристик: гидротермической стабильности, объема пор по азоту и моды пор в области мезопор по азоту у частиц композиционного материала относительно соответствующей гидротермической стабильности, объема пор и моды пор в области мезопор у компонента оксида алюминия в отсутствие указанной набухаемой глины,
(С) средний диаметр частиц композиционного материала составляет приблизительно от 1 до приблизительно 150 мкм,
и где указанные частицы композиционного материала при кальцинировании при 537,8°С в течение 2 ч имеют:
(i) удельную площадь поверхности, по меньшей мере, приблизительно 200 м2/г;
(ii) средний диаметр пор по азоту приблизительно от 60 до 400 Å и
(iii) средний объем пор по азоту приблизительно от 0,5 до приблизительно 2,0 см3/г.
(A) удельную площадь поверхности, по меньшей мере, приблизительно 200 м2/г;
(B) средний диаметр пор по азоту от приблизительно 60 до 300 Å;
(C) общий объем пор по азоту приблизительно от 0,5 до приблизительно 2,0 см3/г и отличаются тем, что имеют
(i) содержание макропор не выше, чем приблизительно 40% общего объема пор;
(ii) содержание мезопор приблизительно от 20 до приблизительно 90% общего объема пор по азоту, где, по меньшей мере, приблизительно 40% пор в области мезопор имеют диаметр приблизительно от 100 до приблизительно 400 Å, и
(iii) содержание микропор не выше, чем приблизительно 80% общего объема по азоту,
и где в указанных частицах композиционного материала
(i) компонент оксида алюминия включает, по меньшей мере, 75 мас.% оксида алюминия, при этом, по меньшей мере, 5 мас.% указанного оксида алюминия находится в форме кристаллического бемита, гамма-оксида алюминия, полученного из кристаллического бемита или их смесей;
(ii) компонент набухаемой глины является диспергируемым в воде до включения в частицу композиционного материала и присутствует в частице композиционного материала в количестве (а) меньшем, чем 10 мас.%, в расчете на суммарную массу компонента оксида алюминия и компонента набухаемой глины, и (b) эффективном для повышения, по меньшей мере, одной из следующих характеристик: гидротермической стабильности, объема пор по азоту и моды пор в области мезопор по азоту у частиц композиционного материала относительно соответствующей гидротермической стабильности, объема пор и моды пор в области мезопор у компонента оксида алюминия в отсутствие указанной набухаемой глины, и
(iii) средний диаметр пор частиц композиционного материала составляет приблизительно от 1 до приблизительно 150 мкм.
(A) образование неколлоидной дисперсии, включающей, по меньшей мере, один компонент оксида алюминия, содержащий, по меньшей мере, 75 мас.% активного оксида алюминия, и, по меньшей мере, один компонент набухаемой глины в жидкой диспергирующей среде;
(B) повторную гидратацию активного оксида алюминия из компонента оксида алюминия в присутствии указанной диспергированной набухаемой глины с превращением, по меньшей мере, 5 мас.% активного оксида алюминия в кристаллический бемит и образованием частиц композиционного материала, содержащих эффективное количество набухаемой глины, тонко диспергированной в компоненте из оксида алюминия, причем указанное эффективное количество набухаемой глины является (i) меньшим, чем 10 мас.%, в расчете на суммарную массу компонента оксида алюминия и компонента набухаемой глины, и (ii) достаточным для обеспечения повышения, по меньшей мере, одной из следующих характеристик: гидротермической стабильности, объема пор по азоту и моды пор в области мезопор по азоту у частиц композиционного материала относительно соответствующей гидротермической стабильности, объема пор и моды пор в области мезопор компонента оксида алюминия в отсутствие указанной набухаемой глины, и
(С) выделение частиц композиционного материала из дисперсии,
и при этом компонент оксида алюминия в (А) включает, по меньшей мере, 90 мас.% оксида алюминия, полученного повторной гидратацией активного оксида алюминия, компонент набухаемой глины включает, по меньшей мере, одну смектитовую глину типа смектита, присутствующую в дисперсии в количестве приблизительно от 1 до приблизительно 8 мас.%, в расчете на суммарную массу компонента оксида алюминия и компонента набухаемой глины, повторную гидратацию регулируют таким образом, чтобы превратить приблизительно от 30 до приблизительно 100 мас.% активного оксида алюминия в кристаллический бемит, имеющий размер кристаллита меньше, чем приблизительно 110 Å, и жидкой диспергирующей средой является вода.
(A) размер частиц агломератов составляет приблизительно от 0,5 до приблизительно 5 мм;
(B) компонент оксида алюминия содержит, по меньшей мере, 75 мас.% повторно гидратированного активного оксида алюминия, причем, по меньшей мере, 3,75 мас.% указанного компонента из оксида алюминия находится в форме кристаллического бемита, гамма-оксида алюминия, полученного из кристаллического бемита, или их смеси,
(C) компонент набухаемой глины присутствует в компоненте оксида алюминия в количестве (i) меньшем, чем 10 мас.%, в расчете на суммарную массу компонентов оксида алюминия и набухаемой глины, и (ii) эффективном для повышения, по меньшей мере, одной из следующих характеристик: гидротермической стабильности, объема пор по ртути и моды пор в области мезопор по ртути у частиц агломератов относительно соответствующей гидротермической стабильности, объема пор и моды пор в области мезопор у частиц агломератов в отсутствие набухаемой глины,
и где указанные частицы агломератов носителя при кальцинировании при 537,8°С в течение 2 ч имеют:
(i) удельную площадь поверхности, по меньшей мере, приблизительно 200 м2/г;
(ii) моду пор в области мезопор приблизительно от 60 до приблизительно 400 Å и
(iii) общий объем пор по ртути приблизительно от 0,6 до приблизительно 1,5 см3/г.
(A) размер частиц агломератов составляет приблизительно от 0,5 до приблизительно 5 мм;
(B) компонент оксида алюминия включает, по меньшей мере, 75 мас.% оксида алюминия, причем по меньшей мере, 3,75 мас.% указанного компонента из оксида алюминия находится в форме кристаллического бемита, гамма-оксида алюминия, полученного из кристаллического бемита или их смесей,
(C) компонент набухаемой глины присутствует в компоненте оксида алюминия в количестве (i) меньшем, чем 10 мас.%, в расчете на общую массу компонентов оксида алюминия и набухаемой глины, и (ii) эффективном для повышения, по меньшей мере, одной из следующих характеристик: гидротермической стабильности, объема пор по ртути и моды пор в области мезопор по ртути у частиц агломератов относительно соответствующей гидротермической стабильности, объемов пор и моды пор в области мезопор у частиц агломератов в отсутствие указанной набухаемой глины, и где частицы агломератов носителя имеют:
(i) удельную площадь поверхности, по меньшей мере, приблизительно 200 м2/г,
(ii) моду пор в области мезопор по ртути от приблизительно 60 до приблизительно 400 Å и
(iii) общий объем пор по ртути приблизительно от 0,5 до приблизительно 1,8 см3/г.
(А) удельную площадь поверхности, по меньшей мере, приблизительно 200 м2/г;
(В) средний диаметр пор по азоту приблизительно от 60 до 300 Å и
(iii) общий объем пор по азоту приблизительно от 0,5 до приблизительно 2,0 см3/г, и которые получены способом, включающим
(i) образование неколлоидной дисперсии, включающей, по меньшей мере, один компонент оксида алюминия, содержащий, по меньшей мере, 75 мас.% активного оксида алюминия и, по меньшей мере, один компонент набухаемой глины в жидкой диспергирующей среде;
(ii) повторную гидратацию активного оксида алюминия из компонента оксида алюминия в присутствии указанной диспергированной набухаемой глины с превращением, по меньшей мере, 5 мас.% активного оксида алюминия в кристаллический бемит и с образованием частиц композиционного материала, содержащих эффективное количество набухаемой глины, тонко диспергированной в компоненте оксида алюминия, причем указанное эффективное количество набухаемой глины является (i) меньшим, чем приблизительно 10 мас.%, в расчете на суммарную массу компонента оксида алюминия и компонента набухаемой глины, и (b) достаточном для обеспечения повышения, по меньшей мере, одной из следующих характеристик: гидротермической стабильности, объема пор по азоту и моды пор в области мезопор по азоту у частиц композиционного материала по сравнению с соответствующей гидротермической стабильностью, объемом пор и модой пор в области мезопор у компонента из оксида алюминия в отсутствие указанной набухаемой глины;
(iii) выделение частиц композиционного материала из дисперсии.
| US 5114895 A1, 19.05.1992 | |||
| КАТАЛИЗАТОР ДЛЯ ОБРАБОТКИ ГАЗОВ, СОДЕРЖАЩИХ СЕРНИСТЫЕ СОЕДИНЕНИЯ, И СПОСОБ ОБРАБОТКИ УКАЗАННЫХ ГАЗОВ | 1995 |
|
RU2112595C1 |

