ОБЛАСТЬ ИЗОБРЕТЕНИЯ
Данное изобретение относится к частицам композита оксида алюминия с большим объемом пор и большой площадью поверхности, способам их получения, агломератам и полученным из них нанесенным катализаторам; а также к способам использования указанных катализаторов.
ПРЕДПОСЫЛКИ ИЗОБРЕТЕНИЯ
Уровень техники, относящийся к пористым макрочастицам оксида алюминия, полученным из них сформованным носителям катализаторов, носителям, импрегнированным различными каталитически активными металлами, соединениями металлов и/или промоторами, и к различному использованию таких импрегнированных носителей в качестве катализаторов, является широким и относительно хорошо разработан.
В то время как предшествующий уровень техники показывает непрерывное модифицирование и усовершенствование таких частиц, носителей и катализаторов с целью улучшения их каталитической активности, и в то время как в некоторых случаях весьма желательные активности действительно были достигнуты, в промышленности продолжает существовать необходимость в улучшенных каталитических носителях и полученных из них катализаторах, которые имеют улучшенную активность и долговечность вместе с желательным балансом морфологических свойств.
Оксид алюминия является полезным для различных областей использования, включая носители для катализатора и катализаторы для химических процессов, каталитические насадки для автомобильных глушителей и аналогичное. Во многих из данных областей использования желательно добавить каталитические материалы, такие как ионы металлов, тонкодиспергированные металлы, катионы и т.п., к оксиду алюминия. Уровень и распределение данных металлов на носителе, а также свойства самого носителя являются ключевыми параметрами, которые влияют на сложную природу каталитической активности и долговечность.
Оксид алюминия, полезный для каталитического использования, до сих пор получали различными способами, такими как гидролиз в воде алкоксидов алюминия, осаждение оксида алюминия из квасцов, способы на основе алюмината натрия и аналогичные. Вообще говоря хотя оксид алюминия из данных источников можно использовать для каталитических носителей, такое использование имеет определенные ограничения.
Это объясняется тем фактом, что для используемых в химических реакциях нанесенных катализаторов морфологические свойства носителя, такие как площадь поверхности, объем пор и распределение размеров пор, которые включают в себя общий объем пор, являются очень важными. Такие свойства служат средством, влияющим на природу и концентрацию активных каталитических центров, диффузию реагирующих веществ к активному каталитическому центру, диффузию продуктов от активных центров и время жизни катализатора.
Кроме того, носитель и его размеры также влияют на механическую прочность, плотность и характеристики заполнения реактора, каждая из которых является важной при коммерческом использовании.
Гидрирующие катализаторы для переработки нефти представляют собой огромную долю нанесенных на оксид алюминия катализаторов, находящихся в коммерческом использовании. Применение гидропереработки охватывает широкий диапазон типов сырья и рабочих условий, но имеет одну или более общую цель, а именно удаление содержащих гетероатомы примесей (серы, азота, кислорода, металлов), увеличение Н/С отношения в продуктах (таким образом уменьшая ароматичность, плотность и/или углеродистые остатки) и крекинг углеродных связей для уменьшения диапазона кипения и средней молекулярной массы.
Более конкретно хорошо известно использование серий реакторов с псевдокипящим слоем, содержащих катализатор, имеющий улучшенную эффективность и стабильную активность, при десульфуризации и деметаллировании металлсодержащих тяжелых углеводородных фракций.
При увеличении нефтепереработчиками доли более тяжелой, низкого качества сырой нефти в сырье, которое необходимо перерабатывать, растет необходимость в способах обработки фракций, содержащих все более и более высокие уровни металлов, асфальтенов и серы.
Хорошо известно, что в сырой нефти и других тяжелых нефтяных углеводородных фракциях, таких как нефтяные остатки, углеводородные фракции, полученные из битуминозного песка, и углеводородные фракции, полученные из угля, присутствуют различные металлоорганические соединения и асфальтены. Наиболее обычными (часто встречающимися) металлами, которые можно обнаружить в таких углеводородных потоках, являются никель, ванадий и железо. Такие металлы являются очень вредными для различных операций переработки нефти, таких как гидрокрекинг, гидродесульфуризация и каталитический крекинг. Металлы и асфальтены вызывают закупоривание пор каталитического слоя и уменьшают жизнь катализатора. Различные металлические отложения на катализаторе приводят к отравлению или дезактивации катализатора. Более того асфальтены стремятся уменьшить восприимчивость углеводородов к десульфуризации. Если катализатор, такой как катализатор десульфуризации или псевдоожиженный катализатор для крекинга, подвергается воздействию углеводородной фракции, которая содержит металлы и асфальтены, катализатор быстро дезактивируется и требует преждевременной замены.
Хотя известны способы гидроочистки тяжелых углеводородных фракций, включающих, но не ограничивающихся ими, тяжелую нефть, слабокрекированную нефть и нефтяные углеводородные остатки, использование каталитических процессов с неподвижным слоем для конвертирования такого сырья без заметного осаждения асфальгенов и закупоривания реактора и с эффективным удалением металлов и других загрязняющих примесей, таких как соединения серы и соединения азота, не является обычным, поскольку используемые катализаторы обычно не способны сохранять активность и эксплуатационные характеристики.
Таким образом, определенные процессы гидроконверсии наиболее эффективно проводятся в системе с псевдокипящим слоем. В псевдокипящем слое предварительно нагретый водород и остаток поступают в нижнюю часть реактора, где имеется восходящий поток остатка плюс внутренняя рециркуляция суспендированных в жидкой фазе частиц катализатора. Недавние разработки включали использование порошкообразного катализатора, который может быть суспендирован без необходимости рециркуляции жидкости. В данной системе часть катализатора непрерывно или периодически удаляют в серии циклонов и для поддержания активности добавляют свежий катализатор. Грубо говоря в системе с псевдокипящим слоем каждый день меняют примерно 1 мас. % загрузки катализатора. Таким образом, полная активность системы представляет собой средневзвешенную активность катализатора, изменяющуюся от свежего катализатора до очень старого, т.е. дезактивированного.
В общем желательно разработать катализатор с наиболее высокой возможной площадью поверхности, чтобы предоставить максимум концентрации каталитических центров и активности. Однако площадь поверхности и диаметр пор являются обратносвязанными внутри практических пределов. Когда катализатор стареет и отравляется, для диффузии требуются достаточно большие поры, но большие поры имеют более низкую площадь поверхности.
Более конкретно разработчик столкнулся с конкуренцией соображений, которые часто предписывают баланс морфологических свойств, придаваемых носителям и получаемым из них катализаторам.
Например, признано (см., например, патент США №4497909), что в то время как поры, имеющие диаметр ниже 60 ангстрем (внутри области, которая называется здесь областью микропор), имеют эффект увеличения числа активных центров в определенных гидрирующих катализаторах на основе диоксида кремния/оксида алюминия, эти же самые центры первыми закупориваются коксом, вызывая уменьшение активности. Аналогично далее признано, что когда такие катализаторы имеют более чем 10% от общего объема пор, занятых порами, имеющими диаметр более 600 ангстрем (внутри области, которая называется здесь областью макропор), механическая прочность на раздавливание понижается, так же как и активность катализатора. Наконец признано, что для определенных катализаторов на основе диоксида кремния/оксида алюминия максимизация пор, имеющих диаметр между 150 и 600 ангстрем (примерно внутри области, которая называется здесь областью мезопор), является желательной для подходящей активности и долговечности катализатора.
Таким образом, в то время как увеличение площади поверхности катализатора будет увеличивать количество активных центров, такое увеличение площади поверхности естественно приводит к увеличению доли пор в области микропор. Как показано выше, микропоры легко забиваются коксом. Короче говоря увеличение площади поверхности и максимальное увеличение мезопор являются антагонистическими свойствами.
Более того площадь поверхности должна быть не только высокой, но она также должна оставаться стабильной при воздействии условий конверсии, таких как высокая температура и влажность. Поэтому продолжается поиск гидротермически стабильного, с большим объемом пор и большой площадью поверхности оксида алюминия, подходящего в качестве носителя катализатора.
Один из результатов данного поиска предлагается в имеющей отношение и переуступленной заявке на патент США серийный №09/467742, поданной 21 декабря 1999 года (Doket № W-9433-01). В данной заявке на патент описываются частицы композита, содержащие бемит и способную набухать глину. Такие частицы композита используют активированный оксид алюминия в качестве исходного оксида алюминия, который подвергают повторной гидратации и конвертируют в бемит в присутствии способной набухать глины. В то время как данный способ и получаемый по нему продукт имеет много преимуществ, активированный оксид алюминия является относительно дорогим сырьем. Активированный оксид алюминия можно получить быстрым прокаливанием гиббсита. Поэтому являлось бы еще более благоприятным непосредственно получать бемит с высоким объемом пор, высокой площадью поверхности, используя при этом гиббсит в качестве первичного исходного материала. Одним из препятствий к осуществлению данной цели является стремление гиббсита образовывать большие кристаллиты (например, примерно 500 ангстрем) во время его превращения в бемит. Большие кристаллиты приводят к продукту с низким объемом пор при желательной площади поверхности.
Таким образом, продолжался поиск не только способов получения бемитовых продуктов с высокой площадью поверхности и объемными порами, но также и осуществления этого эффективным образом с точки зрения затрат. Настоящее изобретение было разработано в ответ на данный поиск.
Патент США номер 5728184 нацелен на способ изготовления поликристаллических керамических материалов на основе альфа-оксида алюминия путем получения дисперсии бемита и источника диоксида кремния, гидротермической обработкой дисперсии, конвертированием дисперсии в прекурсор керамического материала на основе альфа-оксида алюминия и обжигом прекурсора. Необязательно в таком способе может быть использован зародышеобразующий материал (иногда называемый материалом затравки) для того, чтобы уменьшить размер кристаллитов альфа-оксида алюминия и увеличить плотность и прочность полученного в результате керамического материала. Описанные зародышеобразующие материалы включают в себя альфа-Al2О3 и альфа-Fe2O3. В столбце 3, строки 25 и последующие описывается, что «зародышеобразующий материал» относится к материалу, который увеличивает превращение промежуточных оксидов алюминия в альфа-оксид алюминия. Таким образом, данный патент использует в качестве сырья бемит и конвертирует бемит в альфа-оксид алюминия. Противоположным образом заявляемое настоящее изобретение основывается на смеси тригидрата оксида алюминия, компонента затравки оксида алюминия, например активированного оксида алюминия, и ингибиторов роста размеров кристаллов (также называемого здесь ИРРК) для получения бемита, имеющего определенные морфологические и кристаллографические свойства, который типично конвертируют в гамма-оксид алюминия при прокаливании. Более того в упомянутом патенте ничего не сказано по поводу свойств пор полученного в результате продукта.
Патент США 4797139 описывает способ получения геля микрокристаллического бемита гидротермической обработкой гиббсита в присутствии затравок бемита при температурах менее 200°С и давлениях менее 200 фунт/кв. дюйм (˜1379 кПа). Указывается, что конверсия гиббсита в микрокристаллический бемит замедляется добавками, такими как фосфаты или фториды, и что следует избегать таких добавок (столбец 2, строки 53 и последующие). Процесс проводят при рН примерно 5 или ниже или альтернативно при рН 8 или выше. Чтобы получить желаемый микрокристаллический бемит, размер затравки должен быть менее 200 ангстрем при количествах, по меньшей мере, 7,5 мас.% относительно прекурсора бемита. Однако если желательно сделать максимальной площадь поверхности, и необходимо использовать бемит при изготовлении пористого гамма-оксида алюминия для каталитического использования, то используемая затравка бемита имеет размер менее 100 ангстрем и предпочтительно менее 50 ангстрем (столбец 3, строки 34 и последующие). Кроме того, если микрокристаллический бемитовый продукт в конечном счете используется для получения керамической массы из альфа-оксида алюминия, затравку альфа-оксида алюминия субмикронного размера для содействия однородности конверсии микрокристаллического бемита в альфа-оксид алюминия желательно смешивать с гиббситовым исходным материалом в течение выдержки в автоклаве (столбец 3, строки 51 и последующие). С бемитом после выдержки в автоклаве или с прекурсором бемита до выдержки в автоклаве могут быть смешаны разнообразные добавки, такие как оксид магния, которые работают как ингибиторы роста кристаллов (столбец 1, строки 47 и последующие). Противоположным образом заявляемое в настоящий момент изобретение опирается на средний размер частиц всех твердых компонентов в дисперсии, которую в конечном счете выдерживают в автоклаве, типично равный примерно от 0,1 до 15 микрон (т.е. от 1000 до примерно 150000 ангстрем), чтобы получить бемитовые композиты с высокой площадью поверхности и высоким объемом пор, которые типично конвертируют в гамма-оксид алюминия прокаливанием.
Патент США 4623364 описывает глиноземные абразивы, получаемые из гелей оксида алюминия, которые формируют частицы альфа-оксида алюминия субмикронного размера (от 0,2 до 0,4 микрометра). Абразивы изготавливают вибропомолом геля с измельчающей средой из оксида алюминия. Предполагается, что измельчение вводит материал из среды размола, представляющей собой оксид алюминия, в гель оксида алюминия, который влияет на образование зародышей кристаллизации альфа-оксида алюминия в течение обжига (столбец 5, строки 55 и последующие). Описываемая помольная среда содержала примерно 90 мас.% альфа-оксида алюминия, содержащего примеси SiO2, MgO и Fe2O3. До или после гелеобразования к оксиду алюминия могут быть добавлены различные добавки, такие как примерно 5 мас.% MgO. Однако MgO присутствует в продукте в виде шпинеля (алюминат магния: MgAl2O4), который окружает непрореагировавший альфа-оксид алюминия (столбец 6, строка 60). К выдерживаемому гелю могут быть добавлены ингибиторы роста зерен, такие как SiO2, Cr2О3, MgO и ZrO2. Целью данного патента можно считать изготовление альфа-оксида алюминия превращением гамма-оксида алюминия из геля оксида алюминия в альфа-оксид алюминия при более низких температурах, например при 1090°С в присутствии затравки относительно 1190°С в отсутствие затравки (столбец 6, строки 40 и последующие).
Противоположным образом цель настоящего изобретения состоит в получении бемита, а не альфа-оксида алюминия, посредством использования затравки активированного оксида алюминия в комбинации, по меньшей мере, с одним ИРРК.
Патент США 4069140 описывает материал носителя, имеющий высокий объем пор, равный, по меньшей мере, 0,8 см3/г с основной частью объема пор, имеющей средний эффективный радиус пор более 100 ангстрем и транспортными порами, имеющими радиусы более 1000 ангстрем. Подходящие носители, описываемые в столбце 6, строки 55 и последующие, включают в себя оксид алюминия, который содержит как бемит, так и аморфный водный оксид алюминия. В столбце 7, строки 15 и последующие далее раскрывается, что носитель может содержать различные наполнители, включая оксид алюминия, диоксид кремния, аморфный диоксид кремния/оксид алюминия, кристаллические алюмосиликаты, уголь, крахмал, волокна целлюлозы и их смеси. В патенте нет описания размера кристаллитов бемита и он, как оказывается, не описывает совместное использование гиббсита и затравки активированного оксида алюминия (или бемита) для получения заявляемого бемитового продукта.
Патент США 4097365 описывает гетерогенные композиты из совместного геля диоксида кремния-оксида алюминия в матрице, состоящей в основном из геля оксида алюминия. Предполагается, что диоксид кремния гетерогенно диспергирован в основе из оксида алюминия в форме обогащенного диоксидом кремния, совместного геля диоксида кремния-оксида алюминия или привитого сополимера, и что основа из оксида алюминия предоставляет матрицу, в которой диспергирован мелкодисперсный композит диоксида кремния-оксида алюминия. Гетерогенно диспергированный, обогащенный диоксидом кремния совместный гель диоксида кремния-оксида алюминия отличается от гомогенного совместного геля. Необходимо заметить, что силикатные ингибиторы роста размера кристаллов, используемые в заявляемом в настоящий момент изобретении, не являются совместными гелями диоксида кремния-оксида алюминия.
Патент США 5178849 описывает способ получения коллоидного бемита, заключающийся в получении суспензии гидрата оксида алюминия, имеющего низкую диспергируемость, подкислении суспензии до рН примерно 3,5 или ниже, чтобы частично растворить гидрат оксида алюминия, но не достаточно для его полного растворения, и затем вываривании подкисленной смеси при температуре примерно от 150 до 200°С при давлении примерно от 5 до 20 атмосфер (например, выдерживая в автоклаве), чтобы получить коллоидный бемит. Предпочтительнее использования бемита в качестве исходного вещества может быть использован тригидрат оксида алюминия (гиббсит) (столбец 2, строка 27). Могут быть добавлены ингибиторы роста зерна, такие как диоксид кремния. К бемиту могут быть добавлены материалы затравки, чтобы увеличить конверсию бемита в альфа-оксид алюминия, или к тригидрату оксида алюминия (прекурсору бемита) для содействия образованию бемита из прекурсора бемита. Материал затравки может быть добавлен до или после гидротермической обработки. Типично материал затравки будет иметь размер частиц менее 1 микрона (столбец 3, строка 55). Материалы затравки для конверсии бемита в альфа-оксид алюминия включают в себя субмикронный альфа-оксид алюминия, а также оксид трехвалентного железа (столбец 3, строка 42). Материалы затравки для конверсии прекурсора бемита в бемит включают в себя субмикронный бемит (столбец 3, строки 46 и последующие). Способ по настоящему изобретению не использует кислотное вываривание такого типа, как описывается в представленном патенте, и не создает коллоидный бемит. Упомянутый патент не описывает комбинацию активированного оксида алюминия и гиббсита, которые конвертируют в кристаллический бемит, имеющий конкретно определенный размер кристаллитов в присутствии ингибитора роста размера кристаллов, такого как силикат, фосфат или сульфат.
Патент США №5114895 описывает композицию слоистой глины, гомогенно диспергированной в матрице неорганического оксида, так что слои глины полностью окружены матрицей неорганического оксида. Матрица неорганического оксида выбрана из оксида алюминия, оксида титана, диоксида кремния, оксида циркония, P2O5 и смесей. Подходящие глины включают в себя бентонит, сепиолит, Laponite™, вермикулит, монтмориллонит, каолин, палыгорскит (аттапульгит), гекторит, хлорит, бейделлит, сапонит и нонтронит. Чтобы получить глину, гомогенно диспергированную в матрице неорганического оксида, прекурсор неорганического оксида диспергируют в виде золя или гидрозоля и проводят гелеобразование в присутствии глины. В то время как широко описывается содержание глины от 5 до 70 масс. %, примеры используют, по меньшей мере, 30 мас. % глины. Кроме того, не описываются ни свойства пор, ни полученный в результате продукт.
Зарегистрированное изобретение № Н-189 суммирует различные способы получения бемита. Более конкретно гиббсит вываривают с кислотой, такой как азотная кислота, и полученный в результате материал нейтрализуют щелочью, а полученную в результате гелеобразную массу затем подвергают искусственному старению и дегидрируют в течение нескольких часов при температурах около 80°С. Затравка альфа-оксида алюминия может быть добавлена до, в течение или после гидролиза без какой-либо разницы в конечном продукте. Противоположным образом активированный оксид алюминия типично существует в "хи"- и "ро"-формах.
Патент США 3392125 нацелен на получение альфа-оксида алюминия частичным прокаливанием, т.е. быстрым прокаливанием тригидрата оксида алюминия (гиббсита) при температуре более 800°С, для получения "хи"- и "ро"-форм, которые также называют активированным оксидом алюминия. Активированный оксид алюминия затем подвергают повторной гидратации и основную его часть конвертируют в фазу оксида алюминия, представляющую собой бемит, псевдобемит или смеси и затем прокаливают при температуре более 1000°С. Дополнительные патенты, которые описывают получение бемита из гиббсита, включают патенты США №4117105; 4344928; 4716029; 4994253 и 5063033.
Патент США 4276201 описывает каталитический носитель, включающий в себя агломераты оксида алюминия и 10% или менее диоксида кремния. Необязательно в носитель могут быть включены меньшие количества других тугоплавких оксидов. Агломераты оксида алюминия готовят посредством контакта водного геля оксида алюминия с органической жидкостью, которая по существу не смешивается с водой при данном отношении органической жидкости к содержащейся в геле воде, так что только часть воды удаляется из водного геля оксида алюминия перед сушкой геля. Затем можно применить ряд методик, чтобы провести агломерирование, таких как помещение геля в роторный пленочный испаритель и выпаривание жидкой фазы при непрерывном перемешивании. Агломерированный оксид алюминия затем прокаливают.
Патент США 4886594 описывает каталитическую композицию для гидроочистки, включающую в себя гидрирующий компонент, состоящий из по существу металлического компонента, где металл выбран из группы VIB, и фосфористого компонента, осаженных на поверхности носителя, содержащего пористый тугоплавкий неорганический оксид и не содержащий цеолит.
Патент США №4981825 относится к композиции частиц неорганического оксида металла (например SiO2) и глины, где частицы оксида по существу отделены друг от друга частицами глины. Подходящие глины включают в себя Laponite™. Описанное отношение оксид металла/глина равно от 1:1 до 20:1 (предпочтительно от 4:1 до 10:1). Композицию, являющуюся предметом изобретения, получают из золя неорганического оксида, имеющего размер частиц от 40 до 800 ангстрем (от 0,004 до 0,08 микрон). Размер частиц в конечном продукте зависит от размера частиц в исходном золе, хотя конечный размер частиц не сообщается. Критично, что частицы оксида металла и глины имеют противоположные заряды, так что они будут притягиваться друг к другу, так что частицы глины ингибируют агрегацию частиц оксида металла. Таким образом, описывается, что частицы глины располагаются между частицами золя. Регулирование зарядов на двух различных типах частиц определяется рН золя. рН неорганического оксида контролируют путем добавления кислоты так, чтобы оно было ниже его изоэлектрической точки, посредством чего индуцируют отрицательный заряд на частицах неорганического оксида. В то время как описывается, что подходящие неорганические оксиды металлов также включают в себя Al2O3, никаких примеров осуществления изобретения с использованием Al2O3 не приведено. Поэтому "перевод" данной концепции на Al2O3 совершается не без трудностей. Например, изоэлектрическая точка Al2O3 находится в щелочном рН, равном примерно 9. Однако золь Al2O3 образуется только при низком рН менее чем примерно 5. Если рН превышает примерно 5, золь Al2O3 будет осаждаться из дисперсии или прежде всего никогда не будет образовываться. Наоборот, золи SiO2 не должны быть кислыми. Поэтому в то время, как любая точка ниже изоэлектрической точки приемлема для золей SiO2, это не является справедливым для золей Al2O3. Наоборот, необходимо работать при рН значительно ниже изоэлектрической точки Al2O3 в области рН, где образуются золи оксида алюминия. Более того, данный патент ничего не сообщает о размере кристаллитов или свойствах пор полученного в результате композита и его суть нацелена только на получение высокой площади поверхности. Как показано выше, площадь поверхности и высокий объем пор являются антагонистическими свойствами.
Противоположным образом заявляемое в настоящий момент изобретение не использует в качестве исходного золь Al2O3 (из которого плохо образуется или вовсе не образуется бемит) и не формирует золь в течение гидротермической обработки. рН, при которой образуются заявляемые в настоящем изобретении композиты, является слишком высоким, чтобы золь образовался в течение гидротермической обработки, а частицы исходного оксида алюминия являются слишком большими, чтобы получился однородный золь.
Другая область технологии, связанная с комбинациями различных глин и оксидов металлов, известна как интеркалированные глины. Интеркалированные глины представлены патентами США №3803026; 3887454 (см. также 3844978); 3892655 (см. также 3844979); 4637992; 4761391 (см. также 4844790); и 4995964. Интеркалированные глины или описывающие их патенты типично содержат требование, заключающееся в том, что применяются большие соотношения глина:золь, и образуется, по меньшей мере, некоторое количество маленьких пор (<25 ангстрем).
Патент США 3803026 описывает гидрогель или суспензию гидрогеля, включающие в себя воду, фторсодержащий компонент, и аморфный совместный гель, включающий оксиды или гидроксиды кремния и алюминия. Аморфный совместный гель далее включает в себя оксид или гидроксид, по меньшей мере, одного элемента, выбранного из магния, цинка, бора, олова, титана, циркония, гафния, тория, лантана, церия, празеодима, неодима и фосфора, причем указанный аморфный совместный гель присутствует в гидрогеле или суспензии гидрогеля в количестве от 5 до 50 мас.%. рН суспензии делают равным от 6 до 10, и условия конверсии создают значительное количество кристаллического алюмосиликатного минерала, предпочтительно в однородной смеси со значительным количеством непрореагировавшего аморфного совместного геля. Молярное отношение диоксид кремния/оксид алюминия, по меньшей мере, равно 3:1, и полученный в результате материал называют синтетическим слоистым кристаллическим глиноподобным алюмосиликатным минералом. В столбце 5, строки 39 и последующие описывается, что полученный в результате алюмосиликат можно измельчить на частицы, растереть в порошок, порошок диспергировать в гидрогель или суспензию гидрогеля, к которой добавляют компоненты, выбранные из исходных соединений, помимо прочего оксида алюминия. Полученную в результате смесь затем сушат и активируют. Несмотря на указанное выше описание, никакие конкретные примеры применения смеси диоксида кремния-оксида алюминия с оксидом алюминия не приводятся. Поэтому исходный оксид алюминия, конечный оксид алюминия и количества каждого из применяемых материалов не описываются.
Патент США №3887454 (и его основной патент 3844978) описывает слоистый, диоктаэдрического типа глиноподобный материал (LDCM), состоящий из диоксида кремния, оксида алюминия и содержащий оксид магния, включенный в его структуру, в контролируемых количествах. Предпочтительными глинами являются монтмориллонит и каолин. В столбце 6, строки 24 и последующие описывается, что материал глины обычно можно объединить с компонентами неорганического оксида, такими как, помимо прочего, аморфный оксид алюминия. Наоборот, заявляемый в настоящее время композит использует кристаллический оксид алюминия, представляющий собой бемит. Аналогичные описания обнаружены в патентах США №3892655 и 3844979, за исключением того, что данные более поздние патенты относятся к глиноподобному минералу слоистого типа и триоктаэдрической структуры, содержащему оксид магния в качестве своего компонента (LTCM), и иллюстрируются глиной типа сапонита.
Патент США №4637992 представляет собой патент, относящийся к интеркалированной глине, который использует коллоидную суспензию неорганических оксидов и добавляет к ней способную набухать глину. В то время как конкретные диапазоны, иллюстрирующие отношение глины к неорганическому оксиду, не раскрываются, оказывается, конечный материал все еще называют субстратом на основе глины, в который включен неорганический оксид. Поэтому это наводит на мысль, что конечный материал содержит главным образом глину, а не преобладающее количество оксида алюминия и очень незначительные количества глины, как в случае настоящего изобретения. См., например, столбец 5, строки 46 и последующие патента '992.
Патент США №4844790 (выделенный из патента США №4761391) относится к расслаивающейся глине, полученной реакцией, способной набухать глины с агентом, способствующим образованию столбчатой структуры, который включает в себя оксид алюминия. Отношение глины к агенту, способствующему образованию столбчатой структуры, равно от 0,1:10 до 10:1, предпочтительно между 1:1 и 2:1. Однако сутью патента, прежде всего, является глина, содержащая оксид алюминия, а не оксид алюминия, содержащий менее 10 мас.% глины. Обсуждается, что оксиды металлов поддерживают по отдельности пластинки глины и кроме того придают кислотность, которая отвечает за каталитическую активность расслаивающейся глины. Предпочтительной глиной является Laponite™.
Патент США №4995964 нацелен на продукт, полученный интеркалированием в способную расширяться глину (гекторит, сапонит, монтмориллонит) олигомеров, полученных из солей редкоземельных металлов и, в частности, трехвалентных редкоземельных металлов, и поливалентных катионов металлов, способствующих образованию столбчатой структуры, таких как Al3+. Материал из оксида алюминия представляет собой алюминийсодержащий олигомер, который используется для придания слоисто-столбчатой структуры глинам. Заявляемое изобретение не использует и не изготавливает олигомеры материалов из гидроксидов алюминия.
Патент США №4375406 описывает композиции, содержащие волокнистые глины и предварительно прокаленные оксиды, приготовленные получением жидкой суспензии глины с предварительно прокаленным оксидом, перемешиванием суспензии для образования совместной дисперсии и формованием и сушкой совместной дисперсии. Отношение волокнистой полученной глины к композиции предварительно прокаленного оксида может изменяться от 20:1 до 1:5. Данные количества значительно выше количеств глины, используемых в заявляемом в настоящий момент изобретении. Более того волокнистые глины, такие как сепиолит или аттапульгит, не попадают в рамки способных набухать глин, описываемых здесь.
Ряд патентов нацелен на различные типы оксида алюминия и способы его получения, а именно Re 29605; SIR H198; и патенты США №3322495; 3417028; 3773691; 3850849; 3898322; 3974099; 3987155; 4045331; 4069140; 4073718; 4120943; 4175118; 4708945; 5032379 и 5266300.
Более конкретно патент США 3974099 направлен на гидрогель из диоксида кремния/оксида алюминия из совместных гелей силиката натрия и алюмината натрия. Суть данного изобретения нацелена на осаждение Al2O3 на гель диоксида кремния/оксида алюминия, который стабилизирует центры крекинга по отношению к гидротермической дезактивации (Столбец 2, строки 43 и последующие). Полученный в результате материал типично имеет примерно 38,6% оксида алюминия, когда удаляют весь избыток алюмината натрия.
Патент США №4073718 описывает каталитическую основу из оксида алюминия, стабилизированного диоксидом кремния, на которую осажден кобальтовый или никелевый катализатор.
Патент США №4708945 описывает катализатор для крекинга из диоксида кремния, нанесенного на подобную бемиту поверхность соединением частиц пористого бемита и обработкой их паром при температуре более 500°С, чтобы вызвать реакцию диоксида кремния с бемитом. Обычно используется 10% диоксида кремния для достижения поверхностного монослоя диоксида кремния для улучшения термической стабильности.
Патент США №5032379 нацелен на оксид алюминия, имеющий объем пор более 0,4 см3/г и диаметр пор в диапазоне от 30 до 200 ангстрем. Оксид алюминия готовят, смешивая два различных типа способных связываться при повторной гидратации оксидов алюминия, чтобы получить продукт, имеющий бимодальное распределение пор.
Патент США №4266300 описывает носитель из оксида алюминия, полученный смешиванием, по меньшей мере, двух мелко дисперсных оксидов алюминия, каждый из которых характеризуется, по меньшей мере, одной модой пор, по меньшей мере, в одном из диапазонов (i) от 100000 до 10000 ангстрем, (ii) от 10000 до 1000 ангстрем, (iii) от 1000 до 30 ангстрем.
Патент США №4791090 описывает носитель катализатора с бидиспергированным распределением размеров микропор. Столбец 4, строка 65 описывает, что можно сделать два размера микропор, смешивая совершенно различные материалы, имеющие различные размеры пор, такие как оксид алюминия и диоксид кремния.
Патент США №4497909 нацелен на носители из диоксида кремния/оксида алюминия, имеющие содержание диоксида кремния примерно менее 40% по массе и, по меньшей мере, один компонент благородного металла из группы VII Периодической таблицы и, где катализатор содержит поры, имеющие диаметр менее 600 ангстрем, занимающие, по меньшей мере, 90% общего объема пор, и поры, имеющие диаметр от 150 до 600 ангстрем, занимающие, по меньшей мере, примерно 40% общего объема пор сформированного из пор, имеющих диаметр менее 600 ангстрем.
Патент США №4159969 описывает способ изготовления агломератов из оксида алюминия посредством контакта водного геля оксида алюминия с органической жидкостью, несмешивающейся с водой, где количество указанной жидкости является функцией воды в водном геле оксида алюминия. В течение или после гелеобразования к оксиду алюминия можно добавить некоторое количество глины, такой как бентонит или каолин, достаточное для увеличения прочности агломератов. Никакое конкретное количество глины не описывается и каолин не является способной набухать глиной. Ни в одном из примеров глина не применяется.
Следующие патенты описывают различные типы глин: патенты США №3586478; 4049780; 4629712 и публикации РСТ № WO 93/11069 и WO 94/16996.
Следующие патенты описывают различные типы агломератов, которые можно получить из оксида алюминия: патенты США №3392125; 3630888; 3975510; 4124699; 4276201 (см. также 4309278); 4392987 и 5244648.
Патент США №3630888 описывает катализатор, имеющий структуру, в которой доступность каналов, имеющих диаметры примерно между 100 и 1000 ангстрем единиц, составляет от 10 до 40% от общего объема пор и в котором доступность каналов, имеющих диаметры более 1000 ангстрем единиц, составляет примерно между 10 и 40% от общего объема пор, в то время как остающийся объем пор равен примерно от 20 до 80% микропор с диаметром менее 100 ангстрем.
Следующие патенты описывают различные операции гидрирования и использования в них катализаторов: патенты США №3887455; 4657665; 4886594; публикация РСТ № WO 95/31280.
КРАТКОЕ ИЗЛОЖЕНИЕ ИЗОБРЕТЕНИЯ
Настоящее изобретение основывается на открытии, что при диспергировании и гидротермической обработке тригидрата оксида алюминия в присутствии контролируемых количеств компонента затравки, представляющей собой диспергированный активированный оксид алюминия, и, по меньшей мере, одного дополнительного компонента, представляющего собой ингибитор роста размера кристаллов, полученные в результате частицы композита, содержащие бемит, обладают маленьким размером кристаллитов, который дает высокую площадь поверхности, в то же время одновременно обладая более высоким объемом пор относительно случая отсутствия затравки и дополнительных компонентов. Данные свойства по существу сохраняются в агломератах, например в формованных экструдатах (extrudates), полученных из частиц композита до и после импрегнирования (пропитки) каталитически активными металлическими компонентами, такими как компоненты, используемые для операций гидропереработки. Получение оксида алюминия в форме с высоким объемом пор и высоким средним диаметром пор делает ненужным прокаливание перед добавлением металлов для увеличения среднего диаметра пор. Также становится ненужным использование органических растворителей для азеотропного удаления воды, которое является дорогим и наносит вред окружающей среде.
Соответственно в одном аспекте по настоящему изобретению предложены пористые частицы композита, включающие в себя компонент оксида алюминия и, по меньшей мере, остаток одного дополнительного компонента, представляющего собой ингибитор роста размеров кристаллов, однородно диспергированный в компоненте оксида алюминия, где указанные частицы композита (после прокаливания при 537,8°С в течение 2 часов) имеют:
(A) удельную площадь поверхности равную, по меньшей мере, примерно 80 м2/г;
(B) средний диаметр пор по азоту примерно 60 от до 1000 ангстрем;
(C) общий объем пор по десорбции азота примерно от 0,2 до 2,5 см3/г; и
(D) средний диаметр частиц примерно от 1 до 15 микрон/ и где в указанных частицах композита:
(i) компонент оксида алюминия включает в себя, по меньшей мере, 70 мас.% (а) кристаллического бемита, имеющего средний размер кристаллитов примерно от 20 до 200 ангстрем; (b) гамма-оксида алюминия, полученного из указанного кристаллического бемита; или (с) их смеси;
(ii) остаток дополнительного компонента получен, по меньшей мере, из одного ионного соединения, имеющего катион и анион, где катион выбран из группы, состоящей из аммония, катиона щелочного металла, катиона щелочноземельного металла и их смесей, а анион выбран из группы, состоящей из гидроксила, силиката, фосфата, сульфата и их смесей, и присутствует в частицах композита в количестве примерно от 0,5 до 10 мас.% относительно объединенного веса компонента оксида алюминия и дополнительного компонента.
В дальнейшем аспекте по настоящему изобретению предлагается способ получения пористых частиц композита, включающий:
(А) смешение (i) тригидрата оксида алюминия, (ii) жидкой среды, способной растворять, по меньшей мере, часть тригидрата оксида алюминия при условиях гидротермической обработки стадии В, (iii) по меньшей мере, одного компонента затравки, представляющей собой активированный оксид алюминия, и (iv) по меньшей мере, одного дополнительного компонента, выбранного из группы (а) по меньшей мере, одного гидроксида, силиката, фосфата или сульфата щелочного или щелочноземельного металла или аммония, и (b) способной набухать глины, и их смесей, таким путем и при условиях, достаточных для диспергирования тригидрата оксида алюминия и компонента затравки оксида алюминия в виде частиц в жидкой среде;
(B) гидротермическую обработку дисперсии, полученной по стадии А при температуре и времени, достаточных для превращения активированного оксида алюминия и тригидрата оксида алюминия в кристаллический бемит, имеющий средний размер кристаллитов примерно от 20 до 200 ангстрем, и для получения частиц композита, включающих в себя остаток указанного дополнительного компонента, однородно диспергированный в указанном кристаллическом бемите, суспендированном в жидкой среде;
(C) отделение жидкой среды от частиц композита, полученных по стадии В.
В дальнейшем аспекте по настоящему изобретению предлагаются нанесенные катализаторы, полученные из вышеуказанных агломератов.
В еще одном дополнительном аспекте по настоящему изобретению предложен способ гидропереработки нефтяного сырья с использованием вышеуказанных агломератов в качестве носителей для гидрирующих катализаторов.
ОПИСАНИЕ ПРЕДПОЧТИТЕЛЬНЫХ ВАРИАНТОВ ОСУЩЕСТВЛЕНИЯ
Используемый здесь термин «микропора» обозначает поры, имеющие диаметр менее чем 100 ангстрем.
Используемый здесь термин «мезопора» обозначает поры, имеющие диаметр между 100 и 500 ангстрем.
Используемый здесь термин «макропора» обозначает поры, имеющие диаметр более чем 500 ангстрем.
Используемый здесь термин «мода поры» обозначает диаметр пор, соответствующий максимуму пика, где логарифм дифференциала интрузии (вхождения) азота или ртути в см3/г нанесен как функция дифференциала диаметра пор.
Используемый здесь термин «общий объем пор» обозначает совокупный объем в см3/г всех пор, определяемых либо методом по десорбции азота, либо методом по проницаемости ртути. Более конкретно для частиц оксида алюминия, которые не были агломерированы (например, экструзией), распределение диаметров пор и объем пор вычисляют со ссылкой к изотерме десорбции азота (предполагая цилиндрические поры) методом БЭТ, описываемым в работе S. Brunauer, P. Emmett and E. Teller, Journal of American Chemical Society, 60, pp. 209-319 (1939).
Относительно частиц оксида алюминия, которые были агломерированы, например сформованы в экструдаты, распределение диаметров пор вычисляют по формуле:
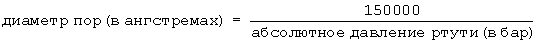
и в соответствии с методом по проницаемости ртути (описываемым в работе H.L. Ritter and L.C. Drake, Industrial and Engineering Chemistry, Analytical Edition 17, 787 (1945)), используя давление ртути 1-2000 бар. Проницаемость ртути является методом, используемым в том случае, когда количество пор с диаметром <60 ангстрем является небольшим, как в случае агломератов.
Общий объем пор образца по N2 представляет собой сумму объемов пор по десорбции азота, определяемую описанным выше методом по десорбции азота. Аналогично общий объем пор образца по ртути представляет собой сумму объемов пор по ртути, определяемую описанным выше методом по проницаемости ртути с использованием контактного угла 140°С.
Термин "площадь поверхности" относится здесь к удельной площади поверхности, определяемой адсорбцией азота с использованием метода БЭТ, как описывается выше, независимо от того, находится ли образец в порошкообразной или агломерированной форме.
Все свежие площади поверхности и измерения пор (например, объем пор и размер пор) определяют на образцах, которые были осушены (при 142°С), подвергнуты катионному обмену и осушены при 142°С, если применялась стадия обмена, и затем прокалены при 537,8°С (1000°F) в течение 2 часов.
Все измерения размеров частиц и распределения размеров частиц, описываемые здесь, определяют на приборе Mastersizer компании Malvern, который работает на принципе дифракции лазерного луча и известен специалистам в области анализа частиц малого размера.
Все морфологические свойства, включающие массу, такие как объем пор (ОП, см3/г) или площадь поверхности (ПП, м2/г) необходимо нормализовать к не содержащей металл основе (НМО).
Образцы нормализуют здесь к не содержащей металл основе в соответствии со следующим уравнением:

где Х обозначает данное свойство пор, такое как ОП (в см3/г) или ПП (м2/г);
W = мас.% металлических оксидов, представляющих собой каталитический промотор, таких как оксиды Ni, Co и Мо, на катализаторе относительно массы пористых составляющих катализатора. Масса непористых составляющих, например непористых разбавителей, прессованного катализатора не включаются в вычисления мас.%;
и НМО = не содержащая металл основа.
Как показано выше, настоящее изобретение относится к композиционным частицам бемита, полученным из гидротермически обработанной смеси компонента затравки оксида алюминия, тригидрата оксида алюминия и, по меньшей мере, одного дополнительного компонента, представляющего собой ингибитор роста размеров кристаллов.
Компонент затравки оксида алюминия представляет собой активированный оксид алюминия, который можно приготовить различными способами. Например, тригидрат оксида алюминия, осажденного в процессе Баера, можно измельчить и быстро прокалить. Активированный оксид алюминия, как указывается здесь, характеризуется плохой кристаллической и/или аморфной структурой.
Предполагается, что выражение, оксид алюминия с плохой кристаллической структурой» для целей вышеуказанного способа означает оксид алюминия, который является таким, что его рентгеноструктурный анализ дает рентгенограмму, которая показывает только одну или несколько диффузных линий, соответствующих кристаллическим фазам оксидов алюминия низкотемпературного перехода, и содержит по существу "хи", "ро", "эта", "гамма" и "псевдогамма" фазы и их смеси.
Выражением «оксид алюминия аморфной структуры» обозначается оксид алюминия, который является таким, что его рентгеноструктурный анализ не дает каких-либо линий, характерных для высоко (преимущественно) кристаллической фазы.
Используемый здесь активированный оксид алюминия обычно можно получить быстрым дегидрированием гидроксидов алюминия, таких как байерит, гидраргиллит или гиббсит, и нордстрандит, или оксигидроксидов алюминия, таких как бемит и диаспор. Дегидрирование можно проводить в любом соответствующем аппарате и с использованием горячего газообразного пара. Температура, при которой газы поступают в аппарат, как правило, может различаться примерно от 400° до 1200°С, а время контакта гидроксида или оксигидроксида с горячими газами обычно равно между долей секунды и 4-5 секундами.
Полученный в результате продукт может содержать незначительные, например, следовые количества бемита, хи, гамма, альфа и других кристаллических структур оксида алюминия, а также остатки гиббсита.
Полученный в результате активированный оксид алюминия типично будет показывать потерю массы при нагревании до 538°С в течение 1 часа, равную примерно от 4 до 12 мас.%.
Удельная площадь поверхности активированного оксида алюминия, полученного быстрой дегидратацией гидроксидов или оксигидроксидов, измеренная традиционным методом БЭТ, как правило, различается между примерно 50 и 400 м2/г, а диаметр частиц, как правило, равен между 0,1 и 300 микронами и предпочтительно между 1 и 120 микронами со средним размером частиц типично более чем 1 микрон, предпочтительно примерно между 5 и 20, наиболее предпочтительно примерно между 5 и 15 микронами. Потеря на обзоливание, измеренная прокаливанием при 1000°С, как правило, различается между 3 и 15%, что соответствует молярному отношению Н2О/ Al2O3 примерно между 0,17 и 0,85.
В предпочтительном варианте осуществления применяют активированный оксид алюминия, получаемый быстрой дегидратацией Bayer гидрата (гидраргиллита), который является легкодоступным и недорогим промышленным гидроксидом алюминия. Активированный оксид алюминия данного типа хорошо известен специалистам в данной области, а способ его получения был описан, например, в патентах США №2915365; 3222129 и предпочтительно 4051072, со столбца 3, строка 6 до столбца 4, строка 7, и описание данных патентов включено здесь в качестве ссылки.
Используемый активированный оксид алюминия может применяться как таковой или может быть обработан так, чтобы содержание в нем гидроксида натрия, выраженное как Na2O, было бы менее 1000 частей на миллион (ЧНМ).
Подходящий порошкообразный исходный материал, представляющий собой активированный оксид алюминия, продается Aluminum Company of America под торговыми марками СР-3, СР-2, СР-1, СР-5, СР-7 или СР-100. Он также продается компанией Porocel (Little Rock, Arkansas) под названием АР-15.
Важнейшим источником бемита в конечном продукте является тригидрат оксида алюминия. Подходит любая форма тригидрата оксида алюминия, хотя гиббсит, его альфа-форма является предпочтительной.
Дополнительный компонент, который смешивают с компонентами активированного оксида алюминия и тригидрата оксида алюминия, работает как ингибитор роста размеров кристаллов в течение гидротермической обработки. Не привязываясь к какой-либо конкретной теории, считается, что активированный оксид алюминия формирует очень маленькие затравочные кристаллы бемита при повторной гидратации под влиянием ингибитора роста размеров кристаллов. В то же время считается, что тригидрат оксида алюминия частично растворим в жидкой среде, и полагают, что при условиях гидротермической обработки существует равновесие между растворенным тригидратом оксида алюминия и суспендированным нерастворимым тригидратом оксида алюминия. Таким образом, далее считается, что бемит с маленьким размером кристаллитов, полученный из активированного оксида алюминия, служит в качестве маленьких затравочных кристаллов, на поверхности которых кристаллизуется бемит из растворенного тригидрата алюминия. Большие кристаллиты в бемите приводят к получению продукта с низким объемом пор при площади поверхности примерно от 80 до 200 м2/г. Было обнаружено, что определенные ингибиторы роста размеров кристаллов работают в комбинации с активированным оксидом алюминия при определенных условиях для уменьшения размера окончательных кристаллитов бемита.
Подходящие дополнительные компоненты, представляющие собой ингибитор роста размеров кристаллов, выбирают из группы, состоящей из гидроксидов, силикатов, фосфатов и сульфатов щелочных и щелочноземельных металлов или аммония, а также способных набухать глин.
Типичные примеры щелочных металлов или щелочноземельных металлов, подходящих для использования в качестве ингибитора роста размеров кристаллов, включают литий, натрий, калий, цезий, магний, кальций, стронций и барий. Среди щелочных металлов и щелочноземельных металлов, указанных выше, предпочтительными являются литий, натрий, калий, магний и кальций. Наиболее предпочтительным металлом является натрий.
Типичные примеры подходящих гидроксидов щелочных металлов или щелочноземельных металлов включают гидроксид натрия, гидроксид калия, гидроксид кальция, гидроксид лития и гидроксид магния.
Типичные примеры подходящих дополнительных компонентов, являющихся ингибиторами роста размеров кристаллов, среди силикатов щелочных металлов или щелочноземельных металлов включают моно-, ди-, три- и тетразамещенные силикаты щелочных металлов, включая силикат натрия, силикат калия, силикат магния, ортосиликат натрия (Na2SiO3), метасиликат натрия (Na2SiO3), метасиликат калия и жидкое стекло (которое представляет собой жидкую смесь различных силикатов натрия).
Типичные примеры подходящих дополнительных компонентов, являющихся ингибиторами роста размеров кристаллов, среди фосфатов щелочных металлов или щелочноземельных металлов включают натрий фосфорнокислый двухзамещенный, калий фосфорнокислый двухзамещенный, натрий фосфорнокислый трехзамещенный, кальций фосфорнокислый двухзамещенный (орто), кальций фосфорнокислый трехзамещенный, полиметафосфат кальция, полиметафосфат натрия. Предпочтительными фосфатами являются полифосфатные соли, такие как пирофосфаты и триполифосфаты, включая двухзамещенные пирофосфатные соли щелочных металлов и четырехзамещенные пирофосфатные соли щелочных металлов и их смеси, например такие как натрий фосфорнокислый (пиро) двухзамещенный (Na2H2P2O7), натрий фосфорнокислый (пиро) четырехзамещенный (Na4P2O7), калий фосфорнокислый (пиро) четырехзамещенный (К4Р2O7), натрий фосфорнокислый (пиро) однозамещенный, натрий фосфорнокислый (пиро) трехзамещенный и их смеси. Предпочтительные пирофосфатные соли включают натрий фосфорнокислый (пиро) однозамещенный, натрий фосфорнокислый (пиро) двухзамещенный, натрий фосфорнокислый (пиро) четырехзамещенный, калий фосфорнокислый (пиро) четырехзамещенный и их смеси. Наиболее предпочтительной фосфатной солью является натрий фосфорнокислый (пиро) четырехзамещенный.
Также можно использовать соли аммония с любым вышеуказанным анионом.
Типичные примеры подходящих дополнительных компонентов, являющихся ингибиторами роста размеров кристаллов, среди сульфатов щелочных металлов или щелочноземельных металлов включают сульфат магния, сульфат калия, сульфат натрия, сульфат лития и их смеси.
Ингибитор роста размера кристаллов, представляющий собой способную набухать глину, включает любую природную или синтетическую слоистую силикатную глину с отношением 2:1 глина: минерал, способную подвергаться набуханию и дисперсии, и их смеси. Набухающие глины являются глинами, способными увеличиваться в объеме, чьи пластинки удерживаются вместе слабыми Ван-дер-ваальсовыми силами. Природные набухающие глины (в противоположность синтетическим набухающим глинам) типично имеют особую форму или морфологию, так что они показывают отношение длины к ширине типично более чем примерно 2,0, предпочтительно более чем примерно 5,0, и отношение длины к толщине более чем примерно 5,0, предпочтительно более чем примерно 7,0. Как правило, типично более чем примерно 20%, предпочтительно более чем примерно 40% и наиболее предпочтительно более чем примерно 50% частиц природной глины будут иметь обсужденные выше отношения длины к ширине и длины к толщине. При определении данных отношений для частиц с нерегулярной формой длина представляет собой расстояние по прямой линии между двумя точками на частице, которые наиболее далеко расположены друг от друга, в то время как ширина представляет собой расстояние между двумя точками, которые наиболее близко расположены друг к другу. Такие глины включают смектитовый класс глин, а также их производные, подвергнутые ионному обмену (например, Na+, Li+). Обычно формы, подвергнутые обмену со щелочными металлами, являются предпочтительными благодаря их увеличенной способности набухать и диспергироваться. Кроме того, полезными являются диспергируемые 2:1 слоистые силикаты, такие как вермикулит, четырехкремниевая слюда и тайниолит. Синтетические глины, такие как Laponite™, могут показывать более сферическую форму.
Более конкретно смектиты представляют собой 2:1 глинистый минерал, который несет заряд решетки и характерно расширяется при сольватации водой и спиртами, наиболее заметно этиленгликолем и глицерином. Данные минералы включают в себя слои, представленные общей формулой:
(M8)IV(M'x)VIO20(OH, F)4
где IV показывает ион, координированный с четырьмя другими ионами, VI показывает ион, координированный с шестью другими ионами, и х может равняться от 4 до 6. М обычно обозначает Si4+, Al3+ и/или Fe3+, но также включает некоторые другие четырехкоординированные ионы, такие как Р5+, В3+, Ge4+, Be2+ и аналогичные. М' обычно представляет собой Al3+ или Mg2+, но также включает многие шестикоординированные ионы, такие как Fe3+, Fe2+, Ni2+, Co2+, Li2+ и аналогичные. Дефициты заряда, создаваемые различными заместителями в данных положениях четырех и шести координированных катионов, уравниваются одним или несколькими катионами, размещенными между структурными единицами. Между данными структурными единицами также может быть окклюдирована (т.е. захвачена или закупорена) вода, связанная либо с самой структурой, либо с катионами в виде гидратной оболочки. При дегидратации (дегидроксилировании) вышеуказанные структурные единицы имеют повторяющееся расстояние примерно от 9 до 12 ангстрем, измеренное рентгеноструктурным анализом. Имеющиеся в продаже природные смектиты включают монтмориллонит (бентонит), бейделлит, гекторит, сапонит, сауконит и нонтронит. Кроме того, в продаже имеются синтетические смектиты, такие как Laponite™, синтетический гекторит, поставляемый Laporte Industries Limited.
Смектиты классифицируют на две категории, диоктаэдрическую и триоктаэдрическую, причем разница заключается в числе октаэдрических узлов в центральном слое, которые являются занятыми. Это, в свою очередь, связано с валентностью катиона в центральных слоях.
Диоктаэдрические смектиты имеют центральные катионы, которые являются трехвалентными, и заместители, которые являются двухвалентными, тогда как триоктаэдрические смектиты имеют двухвалентные центральные катионы с одновалентными заместителями. Диоктаэдрические смектиты включают монтмориллонит, бейделлит и нонтронит, где, например, монтмориллонит имеет в качестве октаэдрического катиона (М') алюминий и магний в качестве заместителя. Триоктаэдрические смектиты, которые являются предпочтительными, включают гекторит и сапонит и их синтетические формы, где, например, гекторит имеет в качестве октаэдрического катиона M') магний и литий в качестве заместителя.
Смектит, наиболее благоприятно используемый в качестве ингибитора роста размеров кристаллов, представляет собой триоктаэдрическую смектитовую глину, имеющую морфологию лейстовидной (т.е. в форме рейки или планки) или сферической формы. Однако можно использовать триоктаэдрические смектиты с пластинчатой или смешанной лейстовидной и пластинчатой морфологией. Примерами подходящих глин из триоктаэдрических смектитов являются природный сапонит и предпочтительно природный гекторит и синтетический гекторит.
Кроме обсужденной выше формы наиболее предпочтительным является, чтобы частицы исходной глины включали в себя агрегаты случайно ориентированных пластинок. Другими словами, агрегаты, которые образуют частицы глины, предпочтительно должны содержать пластинки, ориентированные гранью к канту (г/к) и кантом к канту (к/к) в добавлении к пластинкам, ориентированным грань к грани (г/г), что является основным способом агрегации пластинок в монтмориллоните. Примеры набухающих глин, которые имеют пластинки с хорошо упорядоченными г/г сочленениями и, следовательно, являются менее предпочтительными, являются природные монтмориллониты и природные гекториты. Встречающиеся в природе монтмориллонит и гекторит состоят из хорошо ориентированных блинообразных пластинок, и такая форма благоприятствует ориентации г/г агрегатов пластинок при воздушной сушке.
Наиболее предпочтительной способной набухать глиной для использования в качестве ингибитора роста размеров кристаллов является синтетические гекториты. Процедуры получения синтетических гекторитов хорошо известны и описываются, например, в патентах США №3803026; 3844979; 3887454; 3892655 и 4049780, описание которых включено здесь в качестве ссылки.
Типичным примером синтетического гекторита является Laponite™ RD. Глина Laponite™ RD представляет собой обработанный на фильтр-прессе, просушенный в лотковой сушилке и измельченный в разбивном барабане продукт. Пластинки глины Laponite™ RD состоят из двух слоев диоксида кремния, окруженных слоем магния в октаэдрической конфигурации. Глину Laponite™ RD и другие Laponite(ы) производит и продает Laport Inorganics, часть компании Laport Industries Limited.
Было обнаружено, что является важным размер частиц гидрата оксида алюминия, когда он находится в смеси с активированным оксидом алюминия непосредственно перед гидротермической обработкой.
Имеющийся в продаже тригидрат оксида алюминия, например гиббсит, типично будет состоять из больших частиц, имеющих средний размер, равный 100 микрон или более.
Чтобы быть эффективным в способе по настоящему изобретению, важно, чтобы средний размер частиц тригидрата оксида алюминия и активированного оксида алюминия, которые подвергаются гидротермической обработке по отдельности и/или вместе, был равен типично примерно от 0,1 до 15,0 (например, от 1 до 15), предпочтительно примерно от 0,1 до 10,0 (например, от 1 до 10) и наиболее предпочтительно примерно от 0,3 до 8,0 (например, от 1 до 8) микрон.
Это можно осуществить раздельным измельчением тригидрата оксида алюминия и активированного оксида алюминия и объединением измельченных материалов, но предпочтительно смешивать тригидрат оксида алюминия и активированный оксид алюминия с образованием суспензии и измельчать суспензию в песчаной мельнице для достижения желаемого среднего размера частиц. Дополнительный компонент, представляющий собой ингибитор роста размера кристаллов, можно добавить до или после измельчения, хотя если ингибитор не полностью растворим в жидкой среде, предпочтительно включить его в операцию измельчения, например, когда ингибитор роста размеров кристаллов представляет собой способную набухать глину.
Наиболее предпочтительным является использовать песчаную мельницу DRAIS и пропускать суспензию через мельницу множество раз. При первом проходе типично применяют мягкие условия, чтобы уменьшить средний размер частиц компонентов из оксида алюминия до промежуточного уровня, равного примерно от 5 до 20 микрон. При втором проходе условия измельчения регулируют так, чтобы они были более жесткими посредством уменьшения скорости, с которой суспензия проходит через мельницу. Измельчение типично проводят при комнатной температуре. Преждевременная регидратация активированного оксида алюминия в бемит до гидротермической обработки не происходит в течение измельчения из-за коротких времен измельчения, равных примерно от 0,1 до 2,0 часов, и низких температур измельчения, равных примерно от 20 до 35°С.
Как только достигнут желательный размер частиц активных ингредиентов, содержащих алюминий, т.е. активированного оксида алюминия и тригидрата оксида алюминия, готовят суспензию в жидкой среде из всех активных ингредиентов (т.е. активированного оксида алюминия, тригидрата оксида алюминия и дополнительного компонента, представляющего собой ингибитор роста размеров кристаллов). Жидкая среда должна быть способна растворять, по меньшей мере, часть тригидрата оксида алюминия при условиях гидротермической обработки. Предпочтительной жидкой средой является вода, предпочтительно в основном вода (например, от 50 до 100 мас.%), наиболее предпочтительно деионизированная вода, хотя может использоваться жидкая органическая среда, например, смешиваемая с водой или не смешиваемая с водой, и/или смеси воды и органической среды, такой как метанол, этанол или диметилсульфоксид.
Количество жидкой среды, используемой для получения суспензии, обычно выбирают так, чтобы получить содержание твердых веществ активных ингредиентов, равное примерно от 5 до 30 мас.% относительно массы жидкой среды и массы активного ингредиента. Если количество жидкости слишком мало, вязкость суспензии будет слишком высокой, в результате чего такие операции, как перемешивание, становятся трудноосуществимыми. С другой стороны, если количество жидкости слишком большое, будет растрачиваться излишнее количество тепловой энергии при гидротермической обработке, что не является экономичным.
В общем жидкую среду и активные ингредиенты однородно смешивают либо совместным измельчением, как обсуждено выше, либо, если ранее не проведено совместное измельчение, любым обычным методом с использованием, например, шаровой мельницы, воздушного перемешивающего устройства, ультразвукового смесителя, шнекового смесителя непрерывного типа или шнекового бура. Шаровая мельница может содержать любую подходящую размалывающую среду, такую как размалывающую среду из альфа-оксида алюминия или размалывающую среду из оксида циркония.
Если дополнительный компонент, являющийся ингибитором роста размеров кристаллов, представляет собой способную набухать глину, его диспергируют в суспензии при условиях, которые предпочтительно увеличивают до предела степень дисперсности. Некоторые способные набухать глины более легко диспергируются, чем другие. Если степень дисперсности, достигнутая при контакте с активированным оксидом алюминия и тригидратом оксида алюминия, является плохой, желаемое воздействие на свойства пор оксида алюминия не может быть достигнуто или увеличено до предела. Соответственно могут являться необходимыми шаги для приведения к соответствующей степени дисперсности, такие как измельчение (предпочтительно совместное измельчение с другими активными ингредиентами), регулирование общего содержания летучих компонентов и/или использование диспергаторов, таких как натрий фосфорнокислый (пиро) черырехзамещенный (tetrasodium pyrophosphate TSPP или Na4P2O7), который, как оказывается, также работает как ингибитор роста размеров кристаллов.
Способную диспергироваться глину можно предварительно диспергировать в воде, используя перемешивающее устройство с высоким сдвигом (например, Silverson) или другое перемешивающее устройство, такое как растворитель Cowles. Еще можно использовать мешалки лопастного типа (например, перемешивающее устройство Lightening) с более длинными временами перемешивания и/или резервуар с перегородками для увеличения сдвига.
Достижение соответствующей степени дисперсности способной набухать глины трудно количественно оценивать, но обычно чем больше степень прозрачности суспендирующей среды, тем лучше дисперсность, и полностью прозрачная среда (когда присутствует только глина) является наиболее предпочтительной. Типично это будет иметь место, когда частицы глины преимущественно являются коллоидными по размеру, например менее чем примерно 1 микрон. Наиболее общим способом уменьшения частиц глины до коллоидного размера является мокрое измельчение, сухое измельчение или оба способа с использованием обычного помольного оборудования.
Соответственно в отсутствии совместного измельчения диспергирование способной набухать глины можно выполнить, смешивая глину с водой, предпочтительно при условиях с высоким сдвигом в течение периодов времени типично примерно от 5 до 60 и предпочтительно примерно от 10 до 30 минут. Температура, при которой получают дисперсию, не является критичной и типично будет находиться в диапазоне примерно от 20 до 40°С. Важно, что вода не содержит других минералов, например предпочтительной является деионизированная вода, что влияет на способность глины диспергироваться. Вода, которая содержит значительное количество солей щелочноземельных металлов или других катионов с большим зарядом, может потребовать TSPP для получения хорошей дисперсии глины.
Степень дисперсии увеличивается, если исходная глина имеет общее содержание летучих компонентов типично, по меньшей мере, 8 и предпочтительно, по меньшей мере, 10 мас.% и может находиться в диапазоне типично примерно от 8 до 25, предпочтительно примерно от 10 до 20 и наиболее предпочтительно примерно от 13 до 18 мас.%.
Отношение тригидрат оксида алюминия: активированный оксид алюминия в суспензии контролируют, чтобы оно типично было равно примерно от 0,6:1 до 19:1, предпочтительно примерно от 1:1 до 9:1 и наиболее предпочтительно примерно от 1,5:1 до 17:1.
Количество ингибитора роста размеров кристаллов (ИРРК) зависит от желаемых свойств бемита. Например, увеличение уровня ИРРК будет уменьшать размер кристаллитов бемита, увеличивать площадь поверхности и объем пор. Таким образом, количество ИРРК типично контролируют, чтобы получить массовое отношение активированный оксид алюминия: ингибитор в суспензии, типично равное примерно от 100:1 до 2:1, предпочтительно примерно от 50:1 до 5:1 и наиболее предпочтительно примерно от 20:1 до 5:1. Количество ингибитора роста размеров кристаллов альтернативно можно выразить как типично изменяющееся примерно от 0,1 до 10, предпочтительно примерно от 0,2 до 8 и наиболее предпочтительно примерно от 0,4 до 5 мас.% относительно массы активных ингредиентов суспензии, т.е. компонентов тригидрата оксида алюминия, активированного оксида алюминия и ИРРК.
Более конкретно, если ИРРК является силикатом, он будет присутствовать в суспензии в количествах типично примерно от 0,2 до 8, предпочтительно примерно от 0,4 до 6 и наиболее предпочтительно примерно от 0,5 до 5 мас.% относительно массы активных ингредиентов в суспензии.
Если ИРРК является гидроксидом, он будет присутствовать в количествах типично примерно от 0,5 до 10, предпочтительно примерно от 1 до 8 и наиболее предпочтительно примерно от 2 до 6 мас.% относительно массы активных ингредиентов в суспензии.
Если ИРРК является фосфатом, он будет присутствовать в количествах (включая воду гидратации) типично примерно от 0,1 до 10, предпочтительно примерно от 0,2 до 8 и наиболее предпочтительно примерно от 0,4 до 6 мас.% относительно массы активных ингредиентов в суспензии.
Если ИРРК является сульфатом, он будет присутствовать в количествах типично примерно от 0,5 до 10, предпочтительно примерно от 1 до 8 и наиболее предпочтительно примерно от 2 до 6 мас.% относительно массы активных ингредиентов в суспензии.
Если ИРРК является способной набухать глиной, он будет присутствовать в суспензии в количествах типично примерно от 0,5 до 8, предпочтительно примерно от 1 до 6 и наиболее предпочтительно примерно от 2 до 5 мас.% относительно массы активных ингредиентов в суспензии.
Если применяются комбинации ИРРК, вышеуказанные проценты все еще отражают подходящие количества каждого компонента в комбинации, однако наиболее предпочтительный диапазон будет слегка уменьшен, поскольку ИРРК до некоторой степени будут действовать согласованно с целью уменьшения размера кристаллитов бемита.
Вышеуказанные количества ИРРК в суспензии, выраженные в виде массового процента активных ингредиентов, переносятся в частицы композита, в которые они встраиваются.
Гидротермическую обработку проводят, подвергая содержащую активные ингредиенты суспензию, действию давления, превышающего атмосферное, температуры и времени, достаточному для конвертирования как тригидрата оксида алюминия, так и активированного оксида алюминия в стабильную кристаллическую фазу бемита. Из данных рентгеноструктурного анализа оказывается, что доля оксида алюминия полностью конвертирована в бемит.
Таким образом, температуру типично будут регулировать примерно до 150°С или выше в течение гидротермической обработки, поскольку образование бемита типично не будет происходить при температуре ниже примерно 150°С. Если применяемая температура слишком высокая, например выше примерно 350°С, фаза бемита может превратиться в фазу α-оксида алюминия в течение продолжительного периода времени, что является нежелательным. Соответственно предпочтительно, чтобы температура гидротермической обработки поддерживалась типично между примерно 150°С и 350°С и предпочтительно примерно между 180° и 250°С. Внутри такого температурного диапазона более высокие температуры вызывают более высокую скорость образования фазы бемита. Более того гидротермическая обработка при давлениях, превышающих несколько сотен атмосфер, может привести скорее к фазе диаспора, чем к фазе бемита. Нижний предел давления не является критичным, пока достигаются целевые температуры. Время и температуру регулируют так, чтобы получить полную конверсию гиббсита в бемит.
Удобно проводить гидротермическую обработку в герметично закрытом резервуаре, таком как автоклав.
С точки зрения вышеизложенного гидротермическую обработку типично будут проводить при температурах, которые типично могут изменяться примерно от 150 до 250, предпочтительно примерно от 170 до 225 и наиболее предпочтительно примерно от 190 до 210°С в течение периодов времени типично примерно от 0,1 до 0,4, предпочтительно примерно от 0,5 до 3 и наиболее предпочтительно примерно от 1 до 2 часов. Источник тепла не является критичным и может включать пар, микроволновое излучение, печь с конвективным обогревом, электрический нагрев и аналогичное.
Нагревание предпочтительно проводят при аутогенном давлении, которое обычно достигает примерно от 10 до 20 атмосфер. Конечно, давление можно создать искусственно, если желательно, без изменения сути изобретения. Такое давление может находиться в диапазоне примерно от 5 до 20 атмосфер, но предпочтительно лежит в том же диапазоне, что и давление, создаваемое аутогенно. Используемый здесь термин аутогенное давление относится к давлению, развиваемому в закрытом автоклаве при температуре, но не исключает увеличенное давление, полученное введением пара или газа в автоклав для дальнейшего регулирования общего давления и/или состава в реакции, или уменьшенное давление, полученное стравливанием части пара. Соответственно с точки зрения вышеизложенного давление может типично различаться примерно от 5 до 20, предпочтительно примерно от 10 до 16 и наиболее предпочтительно примерно от 12 до 15 атмосфер.
После завершения гидротермической обработки суспензии дают охладиться до температуры примерно от 20 до 90°С. Охлаждение типично будет происходить с такой же скоростью перемешивания, как при выдержке в автоклаве. После того как охлаждение закончено, жидкость суспензии удаляют обычными способами. Такие методы включают просто сушку суспензии воздухом. Другие подходящие методы включают известные из уровня техники методы удаления свободной жидкости (например, воды) из суспензии и получения сухого продукта. Примеры таких других методов включают центрифугирование или фильтрование. Предпочтительно удаление жидкости проводят, нагревая суспензию для облегчения выпаривания. Более предпочтительно, нагревание проводят в сушильном шкафу с принудительной циркуляцией воздуха при температуре примерно 50-200°С (предпочтительно примерно 100-150°С). Такое нагревание можно выполнить на периодической основе или на непрерывной основе. Стадия удаления жидкости, как правило, удаляет значительную часть жидкой среды из суспензии. Однако в полученном в результате продукте все еще может присутствовать незначительная часть жидкой среды. Суспензию можно осушить другими способами, такими как распылительная сушка или распылительная сушка под вакуумом. Кроме того, суспензию можно использовать без осушки.
Осушенные частицы композита далее можно обработать промывкой для удаления или уменьшения количества солей, например Na2O, прокаливанием, агломерированием и/или импрегнированием. При прокаливании удаляются по существу все летучие вещества, а фаза бемита будет конвертироваться в другие фазы оксида алюминия. При нормальной температуре прокаливания для измерений площади поверхности и объема пор (2 часа при 538°С) оксид алюминия находится в форме гамма-фазы.
При прокаливании, как правило, материал нагревают до температуры типично примерно от 400 до 1000, предпочтительно примерно от 400 до 800 и наиболее предпочтительно примерно от 500 до 750°С и выдерживают при данной температуре, пока не будет удалена свободная вода и предпочтительно, по меньшей мере, примерно 90 мас.% связанных летучих компонентов. Прокаливание можно проводить до или после агломерирования и/или импрегнирования, описанного ниже, или как до, так и после агломерирования и/или импрегнирования. Типично глину будут дегидроксилировать при температурах прокаливания 650-750°С.
Композиционный продукт можно сортировать по размерам любым традиционным способом (например, дроблением или просеиванием). Стадию дробления можно выполнить любым подходящим способом, включая дробление на молотковой мельнице, измельчение в валковой дробилке или измельчение в шаровой мельнице. Можно использовать любой метод измельчения осушенного материала прекурсора. Термин «дробление» используется для включения всех таких методов.
Композиционный продукт в полученном состоянии типично будет включать в себя (а) компонент оксида алюминия, содержащий, по меньшей мере, 70, предпочтительно, по меньшей мере, 85 и наиболее предпочтительно, по меньшей мере, 90 мас.% кристаллического бемита, имеющего диапазон размеров кристаллитов, более детально описываемый в дальнейшем, причем содержание кристаллического бемита в компоненте оксида алюминия может типично находиться в диапазоне примерно от 70 до 100, предпочтительно примерно от 85 до 95 и наиболее предпочтительно примерно от 90 до 95 мас.% относительно массы компонента оксида алюминия, и (b) остаток дополнительного компонента, представляющего собой ингибитор роста размеров кристаллов, однородно диспергированный в компоненте оксида алюминия; причем дополнительный компонент включен или встроен в кристаллиты бемита в процессе их образования. Более конкретно ИРРК, например, катионы щелочноземельного металла, особенно катионы щелочного металла могут вызвать уменьшение объема пор и площади поверхности композиционного продукта при прокаливании, если они присутствуют выше определенных пороговых количеств. Это может являться недостатком для многих областей использования. Такое уменьшение объема пор и площади поверхности типично проявляется, когда содержание катионов щелочного или щелочноземельного металла в композите превышает примерно 0,5 мас.% относительно массы композита. Таким образом, может являться желательным обменять такие катионы на другие катионы, которые не оказывают неблагоприятного влияния на морфологические свойства композита или, по меньшей мере, влияют в значительно меньшей степени. Используемый материал, приводящий к такому результату, называется здесь ионообменной солью.
Типичные примеры катионов, подходящих для использования в ионообменной соли, включают аммоний, катионы, полученные из солей разбавленных кислот, таких как серная, азотная и HCl, переходных металлов, таких как никель, кобальт, молибден или вольфрам, и соли редкоземельных металлов, полученные из редкоземельных элементов, таких как элементы подгруппы церия Периодической таблицы.
Наиболее предпочтительными катионами для осуществления катионного обмена являются катионы аммония. Соответственно предпочтительно промывать первоначально полученный композит бемита водным раствором водорастворимой ионообменной соли.
Типичные примеры анионов, подходящих для ионообменной соли, включают сульфаты, хлориды и нитраты.
Типичные примеры подходящих ионообменных солей включают сульфат аммония, карбонат аммония, нитрат аммония, хлорид аммония, хлорид никеля, сульфат кобальта, нитрат кобальта и аналогичные.
Промывка раствором ионообменной соли типично не будет замещать значительные количества анионов ингибитора роста размеров кристаллов, поскольку они имеют тенденцию удерживаться более сильно, чем катионы, хотя степень анионного обмена не является критичной. Фактически анионы ингибитора роста размеров кристаллов могут показывать свое собственное положительное влияние на приготовленные из них катализаторы. Например, фосфат может помочь диспергировать Ni, Со, Мо или W на импрегнированном катализаторе, а силикат может увеличить активность носителя и его термическую/гидротермическую стабильность.
Катионообменную промывку можно провести суспендированием один или более раз композита бемита в водном растворе, типично содержащем примерно от 0,1 до 10, предпочтительно примерно от 0,2 до 8 и наиболее предпочтительно примерно от 0,4 до 5 мас.% подходящей для обмена соли. Содержание бемита в суспензии типично равно примерно от 10 до 15 мас.% относительно массы суспензии.
Типично бемит суспендируют в разбавленном растворе ионообменной соли в течение примерно от 5 до 30 минут при 65°С с умеренным перемешиванием. рН можно понизить примерно до диапазона от 4,5 до 5,7 кислотой, чтобы помочь удалить Na2O, если он присутствует. Суспензию типично фильтруют и промывают водой для удаления солей. Если уровень сульфата высокий, материал можно повторно суспендировать при рН 8 или выше, используя гидроксид аммония или карбонат аммония для усиления обмена.
Как можно заметить из вышеизложенного, окончательный состав композита бемита относительно дополнительного компонента, представляющего собой ингибитор роста размеров кристаллов, более точно описывается как композиция, полученная из конкретного ингибитора роста кристаллов, используемого в модифицированном виде, если это имеет место, в результате стадии промывки с катионным обменом, если она применяется. Соответственно для удобства окончательный состав компонента ингибитора роста размеров кристаллов, отражающий такие модификации, называется здесь остатком дополнительного компонента, представляющего собой ингибитор роста размеров кристаллов.
В отсутствие стадии промывки с катионным обменом количество и природа остатка дополнительного компонента, представляющего собой ингибитор роста размеров кристаллов, в бемитовом композите будут отражать и по существу являться такими же, как его исходное количество и состав, применяемый в суспензии, которую гидротермически обрабатывают. После стадии промывки с катионным обменом количество первоначального катиона(ов) дополнительного компонента, представляющего собой ингибитор роста размеров кристаллов, остающееся в композите, типично будет равно примерно от 0 до 100, предпочтительно примерно от 0 до 10 и наиболее предпочтительно примерно от 0 до 5 мас.% от количества катиона(ов), первоначально присутствующего до промывки. Аналогично после стадии промывки количество первоначальных анионов ингибитора роста размеров кристаллов, остающееся в композите, типично будет равно примерно от 50 до 100, предпочтительно примерно от 75 до 100 и наиболее предпочтительно примерно от 95 до 100 мас.% от первоначально присутствующего до промывки количества.
Таким образом, любое уменьшение первоначального содержания катиона или аниона ингибитора роста размеров кристаллов в композите будет сопровождаться соответствующим замещением катионами и анионами обменивающейся соли.
Соответственно можно охарактеризовать остаток дополнительного компонента, представляющего собой ингибитор роста размеров кристаллов, в композите как включающий в себя любой из описанных выше первоначальных ингибиторов роста размеров кристаллов вместе с любой обменивающей солью, так что общий остаток типично будет составлять примерно от 0,5 до 10, предпочтительно примерно от 0,5 до 5, наиболее предпочтительно примерно от 0,5 до 3 мас.% относительно массы объединенного компонента оксида алюминия и остатка дополнительного компонента.
Бемитовая часть полученного в результате компонента оксида алюминия в композите будет иметь кристаллическую форму, типично описываемую как обычный бемит, например, в патенте США №4716029 в столбце 1, строки 19 и последующие, но также может включать нетрадиционные формы. При высоких уровнях ингибитора роста размеров кристаллов бемит будет представлен как псевдобемит, т.е. он может иметь очень маленькие кристаллиты. При данных высоких уровнях обычно получают размер кристаллитов от 30 до 60 ангстрем.
Размер кристаллитов в кристаллах бемита компонента оксида алюминия типично равен примерно от 20 до 200 (например, от 100 до 200), предпочтительно примерно от 30 до 150 (например, 120-150) и наиболее предпочтительно примерно от 35 до 100 ангстрем.
Размер кристаллитов бемита можно определить следующей процедурой. Образец размалывают вручную с помощью ступки и пестика. Ровный слой образца помещают на 3,5 г ПВС (поливиниловый спирт) и прессуют в течение 10 секунд при 3000 фунт/кв. дюйм (˜20684 кПа или ˜204 атм), чтобы получить таблетку. Затем таблетку сканируют излучением Cu Кα (альфа) и получают рентгенограмму между 22 и 33 градусами 2 тэта. Пик при 28 градусах 2 тэта используют для вычисления размера кристаллитов с использованием уравнения Шерера (Scherer, +Уравнение 1 ниже) и измеренной ширины пика на половине высоты. Корректировку инструментального уширения пика определяют, применяя такую же процедуру подгонки профиля для сканирования NIST SRM 660 (предоставляет калибровку профиля линии лабораторного гексабората лантана) и затем используя ширину пика в качестве стандарта в виде b в следующем уравнении:
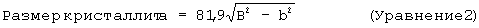
где В = ширина пика образца;
b = ширина пика стандарта.
Обсуждаемый выше угол сканирования 22-33 градуса соответствует образцам, которые не содержат другие кристаллические формы оксида алюминия в дополнение к бемиту, достаточные для маскирования характерных пиков бемита (например, гиббсита). Если такое маскирование наблюдается, можно обратиться к другим немаскированным характерным пикам для определения размера кристаллитов, например находящимся при 14, 28, 38 и 48° 2θ.
Количество кристаллического бемита в оксиде алюминия, содержащем его отличные от бемита формы, можно определить, как описывается в дальнейшем для агломератов.
Образцы сушат в сушильном шкафу при 142°С в течение ночи перед количественной оценкой содержания кристаллического бемита.
Полученные в результате частицы композита можно отделить, термически активировать при тех же условиях, как описывается для агломератов в дальнейшем, или использовать непосредственно для проведения нанесения на него катализатора.
Предпочтительно частицы композита отделяют и сушат и необязательно сортируют по размеру. Подходящий размер частиц типично может находиться в диапазоне примерно от 1 до 150 (например, от 1 до примерно 100), предпочтительно примерно от 2 до 60 и наиболее предпочтительно примерно от 2 до 50 микрон.
Отделение проводят фильтрованием, выпариванием, центрифугированием и аналогичными способами. Суспензию можно также подвергнуть распылительной сушке для эффективного отделения.
Полученные в результате частицы композита имеют площадь поверхности по адсорбции азота типично, по меньшей мере, примерно 80, предпочтительно, по меньшей мере, примерно 150 и наиболее предпочтительно, по меньшей мере, примерно 200 м2/г, причем площадь поверхности типично может находиться в диапазоне примерно от 80 до 500, предпочтительно примерно от 150 до 450 и наиболее предпочтительно примерно от 200 до 400 м2/г.
Средний диаметр пор частиц композита по азоту при парциальном давлении азота р/р0 0,995 типично будет находиться в диапазоне примерно от 60 до 1000, предпочтительно примерно от 80 до 500 и наиболее предпочтительно примерно от 90 до 350 ангстрем.
Общий объем пор, определенный по десорбции азота частицами композита при том же парциальном давлении азота, может различаться примерно от 0,2 до 2,5, предпочтительно примерно от 0,5 до 2,4 и наиболее предпочтительно примерно от 1,0 до 2,3 см3/г.
Преимущество настоящего изобретения заключается в том, что объединенное использование активированного оксида алюминия и ингибитора роста размеров кристаллов увеличивает средний диаметр пор по азоту и общий объем пор, в то же время увеличивая площадь поверхности. Таким образом, варьированием условий синтеза можно контролировать и изменять объем пор и средний диаметр пор, чтобы добиться увеличенной каталитической активности без потери площади поверхности. Катализатор с высоким средним диаметром пор можно приготовить импрегнированием металлами до прокаливания при высокой температуре, уменьшая необходимость предварительной обработки паром для увеличения среднего диаметра пор.
Содержание макропор (т.е. % таких пор внутри общего объема пор по десорбции азота, которые попадают в область макропор) частиц композита типично будет не более примерно 90, предпочтительно не более примерно 75 и наиболее предпочтительно не более примерно 60% от общего объема пор, причем содержание макропор типично будет находиться в диапазоне примерно от 0 до 90, предпочтительно примерно от 5 до 75 и наиболее предпочтительно примерно от 5 до 60% от общего объема пор.
Содержание мезопор по десорбции азота типично будет находиться в диапазоне примерно от 10 до 100, предпочтительно примерно от 15 до 90 и наиболее предпочтительно примерно от 30 до 80% от общего объема пор. Более того, типично, по меньшей мере, 20, предпочтительно, по меньшей мере, 40 и наиболее предпочтительно, по меньшей мере, 60% пор внутри области мезопор будут иметь диаметры пор типично равные примерно от 100 до 400, предпочтительно примерно от 100 до 300 и наиболее предпочтительно примерно от 100 до 250 ангстрем.
Также желательно, чтобы содержание мезопор по азоту в частицах композита, как они получены, обладало бы модой пор, предпочтительно только одной модой пор (мономодальное), типично равной примерно от 60 до 400, предпочтительно примерно от 70 до 300 и наиболее предпочтительно примерно от 80 до 250 ангстрем.
Содержание микропор по азоту в частицах композита типично будет примерно не более 80, предпочтительно не более примерно 60 и наиболее предпочтительно примерно не более 50% от общего объема пор, причем содержание микропор типично может находиться в диапазоне примерно от 0 до 80, предпочтительно примерно от 5 до 60 и наиболее предпочтительно примерно от 10 до 50% от общего объема пор.
Кроме того, агломераты можно смешать с другими традиционными оксидами алюминия, чтобы получить носители, имеющие распределение размеров пор с двумя или более модами в области мезопор. Каждый оксид алюминия вносит вклад в моду мезопор в своем уникальном характерном положении. Также рассматриваются смеси двух и более оксидов алюминия, приготовленные со способными набухать глинами, имеющими различные моды пор.
В то время как композиционные частицы оксида алюминия можно непосредственно использовать в качестве носителей, более удобным и более часто встречающимся является агломерирование частиц для такого использования.
Такие агломераты или агломерированные частицы оксида алюминия можно использовать в качестве катализаторов или носителей для катализаторов в любых реакциях, которые требуют особой структуры пор вместе с хорошими механическими, термическими и гидротермическими свойствами. Агломераты бемита по настоящему изобретению таким образом могут найти конкретное применение в качестве носителей для катализаторов для обработки выхлопных газов, создаваемых в двигателях внутреннего сгорания, и для обработки водородом нефтяных продуктов, например для гидродесульфуризации, гидродеметаллирования и гидроденитрирования. Их также можно использовать в качестве носителей для катализаторов в реакциях удаления соединений серы (катализ Клауса), дегидрирования, реформинга, реформинга паром, дегидрогалогенирования, гидрокрекинга, гидрогенизации, дегидрогенизации и дегидроциклизации углеводородов или других органических соединений, а также в реакциях окисления и восстановления.
Кроме того, они могут быть использованы в качестве катализаторов сами по себе в реакциях, которые типично катализируют оксидами алюминия, таких как гидрокрекинг и реакции изомеризации.
Таким образом, благоприятные свойства, заключающиеся в увеличенном объеме пор при высокой (большой) площади поверхности, хорошей механической прочности и гидротермической стабильности частиц композита, передаются агломератам.
Более конкретно однажды установленные свойства пор агломерата являются термически стабильными и на них по существу не влияет термическая обработка при умеренных температурах, равных 500-700°С, либо до, либо после импрегнирования носителей каталитическими металлами.
Термин "агломерат" относится в данном описании к продукту, который объединяет частицы, которые удерживаются вместе различными физико-химическими силами.
Более конкретно каждый агломерат состоит из множества соприкасающихся, составляющих (составных) первичных частиц, размер которых определен, как описано выше, предпочтительно соединенных и связанных в точках их контакта.
Таким образом, агломераты по настоящему изобретению могут показывать более высокое содержание макропор, чем составляющие их первичные частицы вследствие наличия пустот между составляющими композиционными частицами оксида алюминия.
Тем не менее, агломерированные частицы все еще сохраняют высокие объемы пор в области мезопор.
Соответственно агломераты по настоящему изобретению характеризуются тем, что имеют следующие свойства после прокаливания для превращения в гамма-фазу:
(1) площадь поверхности, равную, по меньшей мере, примерно 100, предпочтительно, по меньшей мере, примерно 150 и наиболее предпочтительно, по меньшей мере, примерно от 200 м2/г, причем площадь поверхности может находиться в диапазоне типично примерно от 100 до 450, предпочтительно примерно от 125 до 425 и наиболее предпочтительно примерно от 150 до 400 м2/г;
(2) средний диаметр пор типично примерно от 50 до 500, предпочтительно примерно от 60 до 400 и наиболее предпочтительно примерно от 70 до 300 ангстрем;
(3) общий объем пор по ртути равный, от 0,2 до 2,5, предпочтительно примерно от 0,5 до 2,4 и наиболее предпочтительно примерно от 1,0 до 2,3 см3/г;
(4) содержание макропор (то есть таких пор внутри общего объема пор, которые попадают внутрь области макропор) типично не более чем примерно 90, предпочтительно не более чем примерно 80 и наиболее предпочтительно не более чем 50% от общего объема пор, причем содержание макропор типично будет находиться в диапазоне примерно от 0 до 90, предпочтительно примерно от 5 до 80 и наиболее предпочтительно примерно от 5 до 50% от общего объема пор;
(5) содержание мезопор типично примерно от 10 до 100, предпочтительно примерно от 15 до 90 и наиболее предпочтительно примерно от 30 до 80% от общего объема пор. Более того типично, по меньшей мере, примерно 20, предпочтительно, по меньшей мере, примерно 40 и наиболее предпочтительно, по меньшей мере, примерно 60% пор внутри области мезопор будут иметь диаметры типично примерно от 100 до 400, предпочтительно примерно от 100 до 300 и наиболее предпочтительно примерно от 100 до 250 ангстрем.
Также желательно, чтобы содержание мезопор в агломерированных частицах, как они образуются, обладало бы модой пор в области меэопор типично примерно от 60 до 400, предпочтительно примерно от 70 до 300 и наиболее предпочтительно примерно от 80 до 250 ангстрем.
Средний диаметр агломерированных частиц типично равен примерно от 0,5 до 5, предпочтительно примерно от 0,6 до 2 и наиболее предпочтительно примерно от 0,8 до 1,5 мм.
Кроме того, агломераты можно смешать с другими традиционными оксидами алюминия, чтобы получить носители, имеющие распределение размеров пор с двумя или более модами в области мезопор. Каждый оксид алюминия вносит вклад моды в области мезопор в своем уникальном характерном положении. Смеси данных оксидов алюминия также можно приготовить в виде агломератов с бимодальным распределением размеров пор. Смеси и полученные в результате моды пор можно изготовить таким образом, чтобы соответствовать размеру/молекулярным массам реагирующих веществ.
Агломерирование композита оксида алюминия выполняют методами, хорошо известными из уровня техники и, в частности, такими методами, как гранулирование, экструзия, формование в гранулы во вращающемся барабане и аналогичными. Также можно использовать методику спекания, где частицы композита, имеющие диаметр примерно не более 0,1 мм, агломерируют в частицы с диаметром, по меньшей мере, примерно 1 мм посредством гранулирующей жидкости.
Как известно специалистам в данной области, агломерацию можно провести при необходимости в присутствии дополнительных аморфных или кристаллических связующих веществ, и к смеси, которую необходимо агломерировать, можно добавить порообразующие реагенты. Традиционные связующие вещества включают в себя другие формы оксида алюминия, диоксид кремния, диоксид кремния-оксид алюминия, глины, оксид циркония, диоксид кремния-оксид циркония, оксид магния и диоксид кремния-оксид бора. Традиционные порообразующие реагенты, которые можно использовать, в частности, включают в себя древесную муку, древесный уголь, целлюлозу, крахмалы, нафталин и в общем все органические соединения, способные удаляться при прокаливании. Однако добавление порообразующих реагентов не является необходимым или желательным.
Если необходимо, затем можно выполнить старение, сушку и/или прокаливание агломератов.
Агломераты, как только они получены, затем типично подвергают термической активационной обработке (т.е. прокаливанию) при температуре в диапазоне типично примерно от 300 до 900, предпочтительно примерно от 400 до 800 и наиболее предпочтительно примерно от 450 до 750°С в течение периодов времени типично примерно от 0,1 до 4, предпочтительно примерно от 0,2 до 3 и наиболее предпочтительно примерно от 0,5 до 2 часов. Атмосфера активирования типично представляет собой воздух, но может включать в себя инертные газы, такие как азот. Порошок оксида алюминия, из которого получают агломерат, типично не прокаливают до агломерирования, поскольку может оказаться трудным связать частицы вместе для получения агломерата.
Активационную обработку можно провести в несколько стадий, если это желательно, или она может являться частью обработки агломерата. В зависимости от конкретной температуры активации и применяемого времени агломераты оксида алюминия преимущественно проявляют характеристики кристаллической структуры бемита или гамма-оксида алюминия, или их смесей.
Более конкретно при температурах прокаливания и временах, все в большей степени превышающих примерно 300°С и один час, бемит будет все в большей степени превращаться в гамма-оксид алюминия. Однако гамма-оксид алюминия будет обладать свойствами пор бемита, из которого он получен. Более того, при предпочтительных температурах прокаливания и временах по существу весь кристаллический бемит превращается в гамма-оксид алюминия. Поэтому сумма содержания кристаллического бемита (мас.%), обсуждаемого выше, плюс содержание гамма-оксида алюминия, получаемого в результате прокаливания бемита, типично не будет превышать первоначальное содержание бемита. Данное заключение в равной степени применимо к частицам композита, которые активируют и используют непосредственно в форме частиц композита без агломерирования.
Процент γ-Al2O3 (гамма-оксида алюминия) в образце оксида алюминия определяют следующим образом:
(1) 100% γ-Al2O3 определяют как интегральную интенсивность (площадь под пиком) (440) пика стандарта γ-Al2O3.
(2) Интенсивность (101) пика кварцевой пластины используют в качестве монитора интенсивности рентгеновского излучения.
(3) Сбор данных проводят на автоматическом дифрактометре Philips® 3720, оборудованном графитовым монохроматором дифракционного луча и герметичной Cu трубкой рентгеновских лучей. Генератор рентгеновских лучей работает при 45 кВ и 40 мА.
(4) Полную ширину при половине максимума (FWHM) и интегральную интенсивность (площадь под пиком) (440) пика γ-Al2O3 получают подгонкой эмпирической кривой. В случае, когда один пик не дает хорошего соответствия подгоночных параметров для пика, используют два пика. В случае, когда используют два пика для подгонки эмпирической кривой, получают два размера кристаллитов с использованием уравнения 3. Процент γ-Al2O3 двух размеров кристаллитов получают с использованием уравнения 2.
(5) Процент γ-Al2O3 в образце определяют из следующего уравнения:

где Isample = Интегральная интенсивность (440) пика образца;
Iquartz.c = Интенсивность (101) пика кварца, измеренная во время, когда измеряют стандарт γ-Al2O3;
Istandart = Интегральная интенсивность (440) пика стандарта γ-Al2O3; и
Iquartz.s = Интенсивность (101) пика кварца, измеренная во время, когда измеряют образец.
Размер кристаллитов γ-Al2O3 (L) определяют по следующей процедуре. Образец размалывают вручную с помощью ступки и пестика. Ровный слой образца помещают на 3,5 г ПВС (поливиниловый спирт) и прессуют в течение 10 секунд при 3000 фунт/кв. дюйм, (˜204 атм), чтобы получить таблетку. Затем таблетку сканируют с помощью излучения Cu Кα и получают рентгенограмму между 63 и 73 градусами (2θ). Для вычисления размера кристаллитов с использованием уравнения 4 используют пик при 66,8 градусов (2θ) и измеренную ширину пика при половине высоты.
L(размер в ангстрем)=82,98/FWHM(2θ°)cos(θ°) (Уравнение 4)
где FWHM = Полная ширина при половине максимума; и
θ = Угол дифракции между потоком рентгеновских лучей и планарной поверхностью, на которой находится образец.
Процент бемита, присутствующего в образце оксида алюминия, по отношению к кристаллическому бемиту определяют следующим образом:
(1) 100% бемита определяют как интегральную интенсивность (площадь под пиком) (020) пика оксида алюминия Capatal.
(2) Интенсивность (101) пика кварцевой пластины используют в качестве монитора интенсивности рентгеновского излучения.
(3) Сбор данных проводят на автоматическом дифрактометре Philips® 3720, оборудованном графитовым монохроматором дифракционного луча и герметичной Cu трубкой рентгеновских лучей. Генератор рентгеновских лучей работает при 45 кВ и 40 мА.
(4) Полную ширину при половине максимума (FWHM) и интегральную интенсивность (площадь под пиком) (020) пика бемита получают подгонкой эмпирической кривой. В случае, когда один пик не дает хорошего соответствия подгоночных параметров для пика, используют два пика. В случае, когда используют два пика для подгонки эмпирической кривой, получают два размера кристаллитов с использованием уравнения 6. Процент бемита двух размеров кристаллитов получают с использованием уравнения 5.
(5) Процент бемита в образце определяют следующим уравнением:
%бемита=(Isample·Iquartz.c)/(Icapatal·Iquartz.s) (Уравнение 5)
где Isample = Интегральная интенсивность (020) пика образца;
Iquartz.c = Интенсивность (101) пика кварца, измеренная во время, когда измеряют оксид алюминия Capatal;
Icapatal = Интегральная интенсивность (020) пика оксида алюминия Capatal; и
Iquartz.s = Интенсивность (101) пика кварца, измеренная во время, когда измеряют образец.
Размер кристаллитов бемита (L) определяют по следующей процедуре. Образец размалывают вручную с помощью ступки и пестика. Ровный слой образца помещают на 3,5 г ПВС (поливиниловый спирт) и прессуют в течение 10 секунд при 3000 фунт/кв. дюйм (˜204 атм), чтобы получить таблетку. Затем таблетку сканируют с помощью излучения Cu Кα и получают рентгенограмму между 22 и 33 градусами (2θ). Для вычисления размера кристаллитов с использованием уравнения 6 используют пик при 28 градусах (2θ) и измеренную ширину пика при половине высоты.
L(размер в ангстрем)=82,98/FWHM(2θ°)cos(θ°) (Уравнение 6)
где FWHM = Полная ширина при половине максимума; и
θ = Угол дифракции между потоком рентгеновских лучей и планарной поверхностью, на которой находится образец.
Частицы композита оксида алюминия особенно приспособлены для использования в качестве носителя для различных каталитических систем, использующих тяжелые металлы в качестве каталитического компонента. Поэтому металлические компоненты таких катализаторов должны быть добавлены и введены в композит оксида алюминия.
Такое добавление можно осуществить, смешивая каталитические материалы с оксидом алюминия до или после гидротермической обработки в течение приготовления агломератов, например экструдатов или гранул и т.п., нанося на агломераты оксида алюминия, такие как экструдаты или гранулы, каталитический материал с помощью погружения в раствор, содержащий каталитический материал и т.п. Метод «сухого» импрегнирования представляет собой другую подходящую альтернативу, где частицы композита или агломераты контактируют с некоторым количеством импрегнирующей жидкости, объем которой соответствует объему пор носителя. Другие и дополнительные методы модифицирования оксида алюминия могут оказаться желательными для специалистов в данной области.
Пористые композиты оксидов алюминия по настоящему изобретению являются особенно полезными, когда используются в качестве носителей для каталитически активных компонентов гидрогенизации, таких как металлы VIB группы и VIII группы. Эти каталитически активные материалы могут быть подходящим образом использованы в операциях гидропереработки.
Более конкретно используемый здесь термин "гидропереработка" означает способы переработки нефти с реагированием перерабатываемого сырья (сложных смесей углеводородов, присутствующих в нефти, которые представляют собой жидкость при условиях стандартной температуры и давления) с водородом под давлением в присутствии катализатора для снижения: (а) концентрации, по меньшей мере, одного из веществ, выбранных из серы, примесей металлов, азота и углерода Conradson и присутствующих в указанном перерабатываемом сырье, и (b) по меньшей мере, одного из свойств, включающих вязкость, температуру застывания и плотность перерабатываемого сырья. Гидропереработка включает в себя процессы гидрокрекинга, изомеризации/депарафинизации, рафинирования и гидроочистки, которые отличаются количеством реагирующего водорода и природой обрабатываемого нефтяного сырья.
Типично понимают, что рафинирование включает в себя гидропереработку жидких нефтепродуктов, преимущественно содержащих (по массе) углеводородные соединения в диапазоне кипения смазочного масла ("исходное сырье"), где исходное сырье контактирует с твердым нанесенным катализатором при условиях повышенного давления и температуры с целью насыщения ароматических и олефиновых соединений и удаления присутствующих в исходном сырье соединений азота, серы и кислорода, и с целью улучшения цвета, запаха, термических свойств, устойчивости к окислению, УФ-стабильности исходного сырья.
Типично понимают, что гидрокрекинг включает в себя гидропереработку преимущественно углеводородных соединений, содержащих, по меньшей мере, пять (5) атомов углерода на молекулу ("исходное сырье"), которую проводят: (а) при парциальном давлении водорода выше атмосферного; (b) при температурах типично ниже 593,3°С (1100°F); (с) при полном конечном химическом расходовании водорода; (а) в присутствии твердого нанесенного катализатора, содержащего, по меньшей мере, один (1) гидрирующий компонент; и (е), где указанное перерабатываемое сырье типично дает выход более чем примерно ста тридцати (130) молям углеводородов, содержащих, по меньшей мере, примерно три (3) атома углерода на молекулу, на каждые сто (100) молей перерабатываемого сырья, содержащего, по меньшей мере, пять (5) атомов углерода на молекулу.
Типично понимают, что гидроочистка включает в себя гидропереработку преимущественно углеводородных соединений, содержащих, по меньшей мере, пять атомов углерода на молекулу ("исходное сырье") для десульфуризации и/или денитрификации указанного перерабатываемого исходного сырья, где процесс проводят: (а) при парциальном давлении водорода выше атмосферного; (b) при температурах типично ниже 593,3°С (1100°F); (с) при полном конечном химическом расходовании водорода; (d) в присутствии твердого нанесенного катализатора, содержащего, по меньшей мере, один гидрирующий компонент; и (е), где (i) перерабатываемое сырье типично дает выход примерно от ста до ста тридцати молей углеводородов (включительно), содержащих, по меньшей мере, три атома углерода на молекулу, на каждые 100 молей исходного перерабатываемого сырья; или (ii) исходное сырье включает в себя, по меньшей мере, 50 объемных процентов жидкости недеасфальтированного остатка, типично кипящего примерно при 565,6°С (1050°F), как определяется ASTM D-1160 Дистилляция, и где главная функция гидропереработки заключается в десульфуризации указанного исходного сырья; или (iii) исходное сырье представляет собой продукт операции производства синтетической нефти.
Типично понимают, что изомеризация/депарафинизация включает в себя гидропереработку преимущественно углеводородных нефтепродуктов, имеющих индекс вязкости (VI) и диапазон кипения, соответствующие смазочному маслу ("исходное сырье"), где указанное исходное сырье контактирует с твердым катализатором, который содержит в качестве активного компонента микропористые кристаллические молекулярные сита, при условиях повышенного давления и температуры и в присутствии водорода для получения продукта, чья текучесть на холоде является значительно улучшенной относительно указанного исходного сырья и чей диапазон кипения по существу находится в диапазоне кипения исходного сырья.
Более конкретно хорошо известные компоненты катализатора для гидропереработки типично включают в себя, по меньшей мере, один компонент металла, выбранного из группы, состоящей из металлов VIII группы, включая металлы платиновой группы VIII группы, в частности платину и палладий, металлы группы железа VIII группы, в частности кобальт и никель, металлы группы VI В, в частности молибден и вольфрам и их смеси. Если исходное сырье имеет достаточно низкое содержание серы, например менее примерно 1 мас.% и предпочтительно менее примерно 0,5 мас.%, в качестве гидрирующего компонента можно использовать металлы платиновой группы VIII группы. В данном варианте осуществления металлы платиновой группы VIII группы предпочтительно присутствуют в количестве в диапазоне примерно от 0,01 мас.% до 5 мас.% от общей массы катализатора, в расчете на элементарный металл платиновой группы. Когда перерабатываемое исходное сырье содержит примерно более 1,0 мас.% серы, предпочтительно гидрирующий компонент представляет собой комбинацию, по меньшей мере, одного металла группы железа VIII группы и, по меньшей мере, одного металла группы VIB. Гидрирующие компоненты из неблагородных металлов предпочтительно присутствуют в окончательной каталитической композиции в виде оксидов или сульфидов, более предпочтительно в виде сульфидов. Предпочтительные полные каталитические композиции содержат, по меньшей мере, примерно 2, предпочтительно примерно от 5 до 40 мас.% металла группы VIB, более предпочтительно молибдена и/или вольфрама, и типично, по меньшей мере, 0,5 и предпочтительно примерно от 1 до 15 мас.% металла VIII группы Периодической системы элементов, более предпочтительно никеля и/или кобальта, определенных в виде соответствующих оксидов. Сульфидная форма данных металлов является более предпочтительной из-за более высокой активности, селективности и сохранения активности.
Каталитические компоненты, например каталитические компоненты для гидропереработки, могут быть введены в полную каталитическую композицию с помощью любой из многочисленных описанных процедур.
Хотя компоненты неблагородных металлов можно объединить в катализатор в виде сульфидов, это не является предпочтительным. Такие компоненты обычно объединяют в виде соли металла, которую можно термически конвертировать в соответствующий оксид в окислительной атмосфере или восстановить водородом или другим восстанавливающим агентом. Затем композицию можно сульфидировать реакцией с соединением серы, таким как ди-сероуглерод, сероводород, тиолы углеводородов, элементарная сера и аналогичные.
Каталитические компоненты можно ввести в композит оксида алюминия на любой стадии приготовления катализатора. Например, соединения металлов, такие как сульфиды, оксиды или водорастворимые соли, такие как гептамолибдат аммония, вольфрамат аммония, нитрат никеля, сульфат кобальта и аналогичные, можно добавить совместным измельчением, импрегнированием или осаждением (кристаллизацией) после регидратаций, но до того, как композит окончательно агломерирован. Альтернативно данные компоненты можно добавлять к композиту после агломерирования импрегнированием в водном, спиртовом или углеводородном растворе растворимых соединений или прекурсоров. Импрегнирование является предпочтительным методом.
Дальнейший вариант осуществления настоящего изобретения направлен на способ гидроочистки углеводородного исходного сырья, по меньшей мере, в одной реакционной зоне с псевдокипящим слоем. Более конкретно углеводородное исходное сырье контактирует с водородом в одной или в серии реакционных зон с псевдокипящим слоем в присутствии катализатора для гидропереработки, состоящего из гидрирующего компонента каталитических металлов и производных, как описывается выше, осажденного на описываемых здесь агломератах композита оксида алюминия.
Как хорошо известно, данное исходное сырье содержит никель, ванадий и асфальтены, например, примерно от 40 чнм и до более чем 1000 чнм объединенного общего количества никеля и ванадия, и примерно до 25 мас.% асфальтенов. Далее по экономическим соображениям настоящего процесса желательно производить более легкие продукты, а также полностью деметаллизированный остаточный побочный продукт, который можно использовать для получения кокса анодного качества. Данный способ особенно полезен при очистке исходного сырья со значительными количествами металлов, содержащего 150 чнм и более никеля и ванадия и имеющего содержание серы в диапазоне примерно от 1 до 10 мас.%. Типичное сырье, которое можно удовлетворительно очистить способом по настоящему изобретению, содержит значительные количества компонентов, которые кипят существенно выше 537,8°С (1000°F). Примерами типичного исходного сырья являются сырая нефть, отбензиненная нефть, нефтяные остатки, остатки как атмосферной, так и вакуумной перегонки, нефть, полученная из битуминозного песка, остатки, полученные из нефти битуминозного песка, и углеводородные фракции, полученные из угля. Такие углеводородные фракции содержат металлоорганические примеси, которые создают опасные эффекты в различных процессах переработки нефти, в которых применяются катализаторы для конверсии конкретной перерабатываемой углеводородной фракции. Металлические вредные примеси, которые обнаруживаются в таком исходном сырье, включают в себя, но не ограничиваются ими, железо, ванадий и никель.
В то время как металлические вредные примеси, такие как ванадий, никель и железо, часто присутствуют в различных углеводородных фракциях, другие металлы также часто присутствуют в конкретной углеводородной фракции. Такие металлы существуют в виде оксидов или сульфидов конкретного металла, или в виде растворимой соли конкретного металла, или в виде высокомолекулярных металлоорганических соединений, включая нафтенаты металлов и металлопорфирины, и их производные.
Другим характерным явлением гидроочистки тяжелых углеводородов является осаждение нерастворимых углеродистых веществ из асфальтеновой фракции исходного сырья, которые вызывают производственные проблемы. Количество таких образующихся нерастворимых веществ увеличивается с количеством материала, кипящего выше 537,8°С (1000°F), которое конвертируют, или с увеличением применяемой реакционной температуры. Данные нерастворимые вещества, также известные как твердые вещества горячей фильтрации Shell, создают технологические трудности для установки гидроконверсии и посредством этого ограничивают температуры и сырье, с которыми может работать установка. Другими словами, количество образующихся твердых веществ ограничивает конверсию данного исходного сырья. Технологические трудности, как описывается выше, могут начать проявлять себя при уровнях твердых веществ столь низких, как 0,1 мас.%. Уровни ниже 0,5 мас.%, как правило, рекомендуются для предотвращения загрязнения перерабатывающего оборудования. Описание теста горячего фильтрования Shell имеется в работе A.J.J., Journal of Inst. of Petroleum (1951) 37, pp. 596-604 Van Kerkvoort, W.J. and Nieuwstad, A.J.J., которая включена здесь в качестве ссылки.
Было сделано предположение, что такие нерастворимые углеродосодержащие вещества получаются, когда тяжелые углеводороды конвертируют в установке гидроконверсии, посредством этого предоставляя им более плохой растворитель для неконвертированной асфальтеновой фракции и, следовательно, создавая нерастворимые углеродсодержащие вещества. Образование таких нерастворимых веществ можно уменьшить, имея наиболее высокую площадь поверхности катализатора для гидроконверсии, доступную через очень большие поры, чтобы наибольшая часть поверхности катализатора была бы доступна большим асфальтеновым молекулам. Кроме того, большие поры облегчают осаждение никеля и ванадия в катализаторе для гидроочистки. Таким образом, может являться желательным увеличение содержания макропор в агломератах по настоящему изобретению с помощью методов, хорошо известных из уровня техники для использования при гидроочистке.
Было обнаружено, что использование ИРРК предлагает разработчику средство для регулирования распределения размера пор в области от 400 до 80 ангстрем для того, чтобы приспособиться к изменениям в молекулярной массе перерабатываемого сырья для контролирования процессов диффузии.
Композиты по настоящему изобретению особенно приспособлены для использования в гидроочистке.
Операции гидрирования типично проводят в одном или в серии реакторов с псевдокипящим слоем. Как ранее объяснялось, псевдокипящий слой представляет собой слой, в котором частицы твердого катализатора поддерживаются в случайном (хаотичном) движении восходящим потоком жидкости или газа. Псевдокипящий слой типично имеет большой объем, который превышает, по меньшей мере, от 10% и до 70% объем твердого вещества в осажденном состоянии. Требуемое вскипание частиц катализатора поддерживают введением жидкого сырья, включительно с рециркуляцией, если она имеется, в реакционную зону при линейных скоростях, находящихся в диапазоне примерно от 0,02 до 0,4 футов в секунду (˜0,61-12 см/с) и предпочтительно примерно от 0,05 до 0,20 футов в секунду (˜1,5-6 см/с).
Рабочие условия для гидроочистки тяжелых углеводородных фракций, таких как нефтяные углеводородные остатки и аналогичных, хорошо известны из уровня техники и включают в себя давление в диапазоне примерно от 1000 фунт/кв. дюйм абс. (68 атм) до 3000 фунт/кв. дюйм абс. (204 атм), среднюю температуру слоя катализатора в диапазоне примерно от 700°F (371°С) до 850°F (454°C), часовую объемную скорость жидкости (LHSV) в диапазоне примерно от 0,1 объема углеводорода в час на объем катализатора до 5 объемов углеводорода в час на объем катализатора и скорость рециркуляции водорода или скорость добавления водорода в диапазоне примерно от 2000 стандартных кубических футов на баррель (SCFB) (356 м3/м3) до 15000 SCFB (2671 м3/м3). Предпочтительно рабочие условия включают в себя общее давление в диапазоне примерно от 1200 фунт/кв. дюйм абс. до 2000 фунт/кв. дюйм абс. (81-136 атм); среднюю температуру слоя катализатора в диапазоне примерно от 730°F (387°C) до 820°F (437°С), LHSV в диапазоне примерно от 0,1 до 4,0 и скорость рециркуляции водорода или скорость добавления водорода в диапазоне примерно от 5000 SCFB (890 м3/м3) до 10000 SCFB (1781 м3/м3). Обычно температуры процесса и объемные скорости выбирают так, чтобы, по меньшей мере, 30 об. % подаваемой фракции, кипящей выше 1000°F, конвертировалось в продукт, кипящий ниже 1000°F и более предпочтительно так, чтобы, по меньшей мере, 70 об. % подлежащей переработке фракции конвертировалось в продукт, кипящий ниже 1000°F.
Для обработки углеводородных дистиллятов рабочие условия типично включают в себя парциальное давление водорода в диапазоне примерно от 200 фунт/кв. дюйм абс. (13 атм) до 3000 фунт/кв. дюйм абс. (204 атм), среднюю температуру слоя катализатора в диапазоне примерно от 600°F (315°C) до 800°F (426°С), LHSV в диапазоне примерно от 0,4 объема углеводорода в час на объем катализатора до 6 объемов углеводорода и скорость рециркуляции или скорость добавления водорода в диапазоне примерно от 1000 SCFB (178 м3/м3) до 10000 SCFB (1381 м3/м3). Предпочтительные рабочие условия для гидроочистки углеводородных дистиллятов включают в себя парциальное давление водорода в диапазоне примерно от 200 фунт/кв. дюйм абс. (13 атм) до 1200 фунт/кв. дюйм абс. (81 атм); среднюю температуру слоя катализатора в диапазоне примерно от 600°F (315°C) до 750°F (398°C); LHSV в диапазоне примерно от 0,5 объема углеводорода в час на объем катализатора до 4 объемов углеводорода в час на объем катализатора; и скорость рециркуляции водорода или скорость добавления водорода внутри диапазона примерно от 1000 SCFB (178 м3/м3) до 6000 SCFB (1068 м3/м3).
Однако наиболее желательные условия для конверсии конкретного сырья в предопределенный продукт могут быть наилучшим образом получены конвертированием сырья при нескольких различающихся температурах, давлениях, объемных скоростях и скоростях добавления водорода, с корреляцией влияния каждой из данных переменных величин и выбирая наилучший компромисс между полной конверсией и селективностью.
Все сделанные здесь ссылки на элементы или металлы, принадлежащие к определенной группе, относятся к Периодической таблице элементов и Hawley's Condensed Chemical Dictionary, 12th Edition. Кроме того, любые ссылки на группу или группы должны быть ссылками к группе или группам, как отражается в данной Периодической таблице элементов, с использованием системы CAS для нумерации групп.
Следующие ниже примеры даются в качестве конкретных иллюстраций заявляемого изобретения. Однако необходимо понимать, что изобретение не ограничивается конкретными деталями, сформулированными в примерах. Все части и проценты в примерах, а также в оставшейся части описания являются массовыми, если это не оговорено иначе. Если это не оговорено здесь иным образом, все измерения или перечисления площади поверхности и свойств пор в описании и формуле изобретения необходимо истолковывать как сделанные на образцах, которые были прокалены при 537,8°С (1000°F) в течение 2 часов при атмосферном давлении на воздухе.
Что касается таблиц 1А и 1В, то используемые ингибиторы роста размера кристаллов представляли собой силикат в виде силиката натрия, щелочь в виде NaOH, натрий фосфорнокислый (пиро) четырехзамещенный (TSPP, т.е. Na4P2O7), сульфат натрия (Na2SO4) и Laponite™. Массу используемого Laponite™ сообщают по сухой основе и корректируют по общему содержанию летучих компонентов (total volatiles, TV), измеряемому при 954,4°С (1750°F). Источником NaOH является как силикат натрия (3,2 SiO2/Na2O), так и NaOH. TSPP добавляли в виде гидрата с 10 молекулами воды, но воду не включали в сообщенную добавленную массу, т.е. использовали Na4P2O7. Было определено, что TV гиббсита равно 34,65%, измеренное при 954,4°С (1750°F). Когда приготовили измельченную суспензию гиббсита, % твердых веществ определили, сначала высушивая взвешенную часть суспензии при 137,8°С (280°F) и затем прокаливая в течение 1 часа при 954,4°С (1750°F). TV прокаленной затравки оксида алюминия определяли прокаливанием при 954,4°С (1750°F) и измеряя потерю массы.
Более того % твердых веществ, сообщенный в колонке 1 таблицы 1А, представляет общий мас.% твердых веществ в суспензии, которую выдерживают в автоклаве. Колонки 7 и 9 таблицы 1А представляют собой % твердых веществ из колонки 1, которые состоят из затравки оксида алюминия (колонка 7) и ИРРК (колонка 9) соответственно. Колонка 8 из таблицы 1А представляет собой % твердых веществ из колонки 9, которые соответствуют индивидуальным компонентам, составляющим общее количество ИРРК из колонки 9.
Далее предполагается, что любой диапазон чисел в описании или формуле изобретения, например, представляющий конкретный ряд свойств, условий, физических состояний или процентов, определенно включает в себя любое число, попадающее в такой диапазон, включая любое подмножество чисел внутри любого диапазона, перечисленного таким образом.
Сравнительный пример 1
Данный сравнительный пример описывает типичный бемитовый продукт, полученный из гиббсита. Используемые гиббситы имеются в продаже под торговыми марками SUPERFINE, HYDRAL.710 и FRF85, поставляемые ALCAN, ALCOA и ALCAN соответственно. Средний размер частиц (СРЧ) для каждого образца гиббсита в микронах для серий 1-3 был 11, 7 и 4 соответственно. Сведения о СРЧ для гиббсита, использованного в сериях 4-5, в распоряжении нет. Соответственно суспензии данных гиббситов с мелким размером частиц готовят, добавляя каждый образец в воду до примерно 15% твердого вещества. Суспензии выдерживают в автоклаве в течение 1 часа при 200°С при перемешивании и затем сушат в течение ночи при 138°С. Это дает большие кристаллиты бемита с низким объемом пор по азоту, который также сообщается в таблице 1, серии 1-5.
Сравнительный пример 2
Данный пример иллюстрирует влияние изменения количества затравки бемита на морфологические свойства бемита, получаемого из гидротермически обработанного гиббсита. Суспензии гиббсита, измельченного в песчаной мельнице и поставляемого ALCOA под торговой маркой C-30D, добавляют в суспензии воды и различных количеств затравок бемита, имеющих размер кристаллитов 130 ангстрем. Окончательное содержание твердого вещества равно примерно 20 мас.%. Каждую суспензию выдерживают в автоклаве с перемешиванием при 200°С и затем сушат в сушильном шкафу в течение ночи при 138°С. Результаты суммируются в таблице 1, серии 6-9. Таблица 1 показывает, что увеличение уровня затравки имеет лишь относительно малый эффект на уменьшение размера кристаллитов и увеличение объема пор полученного в результате бемитового продукта.
Пример 1
Данный пример иллюстрирует влияние ингибитора роста размеров кристаллов и содержания затравки оксида алюминия на размер кристаллитов бемита и свойства пор. К суспензиям гиббсита (которые характеризуются колонками 2, 3 и 4 из таблицы 1А), продаваемого компанией ALCOA под торговой маркой C-30D, размер частиц которого уменьшают измельчением суспензии при скорости 1500 мл/минуту и вторым проходом при 800 мл/минуту до среднего размера частиц примерно 3 микрона, добавляют суспензии активированного оксида алюминия, продаваемого компанией ALCOA под торговой маркой СР-3, и раствор силиката натрия и гидроксида натрия (который в некоторых случаях, т.е. для серии 12 подвергают старению в течение 18 часов, и для серии 13 в течение 1 часа) в различных количествах, так что содержание полученных в результате суспензий, которые выдерживают в автоклаве, суммируется в таблице 1А, серии. 10-13.
Суспензии затем выдерживают в автоклаве при 200°С в течение 1 или 2 часов, как сообщается в таблице 1А. Характеристики продукта суммируются в таблице 1В, серии 10-13. Таблица 1В иллюстрирует, что при данных реакционных условиях 30% загрузки затравки из серий 12 и 13 дают намного более высокие объемы пор и площадь поверхности, чем 20% загрузки затравки из серий 10 и 12.
Пример 2
Данный пример иллюстрирует влияние измельченного гиббсита на объем пор, полученного в результате бемита. К измельченной суспензии гиббсита, полученного от компании Reynolds Aluminum Со. под торговой маркой RH30, добавляют либо СР-3 (активированный оксид алюминия со СРЧ 3 микрона), либо АР-15 (активированный оксид алюминия со СРЧ 8 микрон) вместе с 2% силикатом натрия при молярном отношении Na2O:SiO2, равном 1,0. Окончательное содержание твердого вещества для обеих суспензий равно примерно 15 мас.%. Индивидуальные количества, выраженные как процент твердого вещества, суммируются в колонках 4, 7 и 9 таблицы 1А. После выдержки в автоклаве в течения 2 часов при 200°С суспензии сушат в течение ночи при 138°С. Результаты анализа продукта суммируются в таблице 1В, серии 14 и 15. Видно, что затравка оксида алюминия с меньшим размером частиц дает бемит с более высоким объемом пор, чем затравка оксида алюминия с большим размером частиц.
Пример 3
Данный пример иллюстрирует влияние используемого метасиликата в качестве ингибитора роста размеров кристаллов. К дважды измельченной на песчаной мельнице суспензии гиббсита C-30D, имеющего СРЧ примерно 100 микрон, добавляют воду, метасиликат натрия и СР-3 затравку, представляющую собой активированный оксид алюминия, имеющий СРЧ примерно 30 микрон. Содержание твердых веществ в полученной в результате суспензии равно примерно 15 мас.% и содержание активных ингредиентов в виде процента твердых веществ равно 68 мас.% гиббсита, 30 мас.% СР-3 и 2 мас.% метасиликата. После выдержки в автоклаве в течение 2 часов при 200°С суспензию сушат в течение ночи при 138°С. Полученный в результате продукт анализируют и результаты суммируются в таблице 1, серия 16. Можно увидеть, что получается оксид алюминия с высоким объемом пор со средним диаметром пор выше 200 ангстрем.
Пример 4
Данный пример иллюстрирует влияние совместного измельчения всех активных ингредиентов на морфологические свойства продукта. К 11446,1 граммам Н2O добавляют 360,0 грамм (12,5 мас.%) силиката натрия (3,2 моля SiO2/NaO2) и 83,9 грамм 50 мас.% раствора гидроксида натрия. Затем к полученному в результате раствору добавляют 2410,1 грамм Н-30 гиббсита и 700 грамм АР-15 активированного оксида алюминия, продаваемого компанией Porocel. Суспензию дважды измельчают в 4 л песчаной мельнице, пока СРЧ гиббсита и активированного оксида алюминия не будет равна примерно 3,0 микрона, выдерживают в автоклаве в течение 2 часов при 200°С, затем сушат в сушильном шкафу в течение ночи при 138°С. Общее содержание твердых веществ в суспензии равно 15,3 мас.%, а содержание в ней активных ингредиентов в виде процента таких твердых веществ равно 68,18 мас.% гиббсита, 29,27 мас.% АР-15, 1,95 мас.% SiO2 и 2,64 мас.% NaOH. Продукт идентифицировали рентгенографически как бемит с размером кристаллитов 95 ангстрем. Данный пример и морфологические свойства описываются в таблице 1В, серия 17.
Пример 5
Данный пример иллюстрирует раздельное измельчение гиббсита без других активных ингредиентов относительно совместного измельчения всех активных ингредиентов. Таким образом, для серии 18 суспензию 25 мас.% гиббсита типа ALCOA С-30 дважды измельчают в 4 л песчаной мельнице, пока СРЧ не становиться равным 3,0 микрона. Из гиббсита, полученного в результате измельчения в песчаной мельнице, делают суспензию в воде вместе с затравкой, представляющей собой оксид алюминия типа СР-3, имеющей СРЧ 3,0 микрона, и метасиликатом натрия в количествах, сообщаемых в таблице 1А, серия 18. Для серии 19 такой же исходный материал гиббсита, как используемый для серии 18, смешивают с таким же оксидом алюминия и метасиликатом натрия в тех же пропорциях, как в серии 18, за исключением того, что содержание метасиликата равно только примерно 1 мас.% против примерно 2 мас.% для серии 18, и совместно измельчают до тех пор, пока не будет получен такой же СРЧ.
Обе суспензии подвергают микроволновой обработке в течение 20 минут при 200°С в герметично закрытом реакторе, охлаждают и сушат в течение ночи при 138°С. Содержание суспензии и анализ продуктов суммируются в таблице 1А и В, серии 18 и 19. В обоих случаях наблюдается примерно 90% конверсия гиббсита в бемит только за 20 минут при 200°С. Несмотря на остаток гиббсита, общий объем пор по азоту намного больше 1 см3/г. Данный пример также иллюстрирует, что микроволновый нагрев может дать бемит с высоким объемом пор из измельченной суспензии.
Пример 6
Данный пример иллюстрирует влияние используемых комбинаций ингибиторов роста размеров кристаллов на размер кристаллитов оксида алюминия и объем пор. Готовят три суспензии, содержащие гиббсит Н-30 и затравку, представляющую активный оксид алюминия АР-15. Природу ингибитора роста размера кристаллов варьируют среди метасиликата натрия, сульфата натрия и натрия фосфорнокислого (пиро) четырехзамещенного (TSPP), как сообщается в таблице 1 А для серий 20-22. Каждую суспензию измельчают совместно, используя два прохода в 4 л песчаной мельнице. Каждую суспензию выдерживают в автоклаве в течение 2 часов при 200°С с перемешиванием при 600 об/мин, охлаждают и сушат в сушильном шкафу в течение ночи при 138°С. Данный пример и морфологические свойства полученных в результате продуктов суммируются в таблицах 1А и 1В, серии 20-22. Можно заметить, что размер кристаллитов значительно уменьшается при добавлении TSPP, площадь поверхности и общий объем пор по азоту значительно увеличивается при добавлении TSPP, а добавление сульфата натрия не дает существенный эффект на размер кристаллитов, но увеличивает общий объем пор по азоту.
Пример 7
Данный пример иллюстрирует, что бемит с очень высоким средним диаметром пор можно получить выдержкой в автоклаве совместно измельченной смеси гиббсита и активированного оксида алюминия с 6% диоксидом кремния в виде метасиликата натрия.
Соответственно суспензию гиббсита (Н-30), активированного оксида алюминия (АР-15), диоксида кремния в виде метасиликата натрия и натрия фосфорнокислого (пиро) четырехзамещенного (TSPP) готовят при примерно 15 мас.% общего содержания твердого вещества. Данную суспензию готовят, растворяя TSPP (Na4P2O7·10Н2О) в деионизированной воде, добавляя водный раствор силиката натрия при мольном соотношении SiO2/Na2O, равном 3,2, раствор гидроксида натрия, активированный оксид алюминия АР-15 и гиббсит Н-30. Состав полученной в результате суспензии, которую выдерживают в автоклаве, суммируется в таблице 1А, серия 23. Все добавления делают при перемешивании с использованием перемешивающего устройства типа Cowles. Суспензию затем измельчают в 4 л мельнице DRAIS с первым проходом примерно при 1,5 л/мин и вторым проходом примерно при 500 мл/мин. Затем суспензию выдерживают в автоклаве в течение 2 часов при 200°С с перемешиванием при 580 об/мин. После охлаждения суспензию сушат в течение ночи примерно при 140°С. Проводят ионный обмен образца, чтобы понизить содержание натрия, повторным суспендированием с раствором сульфатом аммония (A/S), используя 1 г A/S/г образца в течение 15 минут, фильтруют, промывают водой и сушат в сушильном шкафу. Затем образцы прокаливают в течение 2 часов при 538,7°С для измерения площади поверхности. Морфологические свойства продукта суммируются в таблице 1В, серия 23. Данный материал оказывается очень стабильным гидротермически, как свидетельствует площадь поверхности 154 м2/г или сохранение 83% площади поверхности после обработки при 800°С в течении 4 часов в атмосфере 20% пара.
Пример 8
Данный пример иллюстрирует влияние использования гидроксида натрия в качестве ингибитора роста размеров кристаллов. Суспензию с 15% твердого вещества готовят из Н-30 (гиббсит), АР-15 (активированный оксид алюминия) и NaOH, получая суспензию, характеристики которой приводятся в таблице 1А, серия 24. Суспензию готовят, добавляя 162,2 г 50 мас.% водного раствора NaOH к 7158 г воды. Затем к полученному в результате раствору при интенсивном перемешивании добавляют 1258 г (без дополнительной обработки) Н-30 (гиббсит) и 619,9 г (без дополнительной обработки) АР-15 (активированный оксид алюминия). Суспензию дважды совместно измельчают в 4 л мельнице DRAIS с первым проходом при 1,5 л/мин и со вторым проходом при 500 мл/мин, вследствие чего содержание твердых веществ падает примерно от 15 мас.% до 11,5 мас.%. Затем суспензию выдерживают в автоклаве в течение 2 часов при 200°С. Морфологические свойства суммируются в таблице 1В, серия 24.
Пример 9
Повторяют пример 8, за исключением того, что уровень активированного оксида алюминия в твердом веществе понижают с 38,18 до 23,6 мас. %. После совместного измельчения и выдержки в автоклаве как в примере 8 измеряют морфологические свойства, которые приведены в таблице 1В, серия 25. Как видно из серий 24 и 25, увеличение размера кристаллитов уменьшает площадь поверхности и понижает объем пор и СРЧ.
Пример 10
Данный пример иллюстрирует влияние высоких уровней затравки и 3-компонентного ИРРК, т.е. метасиликата натрия, NaOH и TSPP, на площадь поверхности и объем пор. Суспензию из Н-30 (гиббсит), АР-15 (активированный оксид алюминия), силиката, добавленного в виде метасиликата натрия, и 0,0065 молей TSPP/моль оксида алюминия готовят, добавляя 37,7 г ТЗРР·10Н2О к 6848,1 г воды, после чего следует объединение при быстром перемешивании 441,6 г 12,5 мас.% метасиликата натрия (мольное соотношение SiO2/Na2O 3,2), 102,9 г 50 мас.% водного раствора NaOH, 1224,8 г Н-30 и 582,7 г АР-15. Суспензию, имеющую содержание твердых веществ, как сообщается в таблице 1А, серия 26, измельчают двойным проходом по примеру 8. Суспензию разделяют и одну часть выдерживают в автоклаве в течение 2 часов при 200°С, в то время как другую часть выдерживают в автоклаве в течение 1 часа при 200°С. Оба продукта сушат в течение ночи при 140°С. Морфологические свойства двух продуктов суммируются в таблице 1В, серии 26 и 27 соответственно.
Пример 11
Данный пример иллюстрирует использование силиката и TSPP в качестве ИРРК без щелочи. Готовят и обозначают как серии 28 и 29 две суспензии, содержащие Н-30 (гиббсит), АР-15 (активированный оксид алюминия), силикат и TSPP. Содержание каждой суспензии суммируется в таблице 1А. Однако для серии 28 мольное отношение TSPP:общее количество оксида алюминия равно 0,0065, а для серии 29 оно равно 0,00325. Обе суспензии дважды измельчают в 4 л мельнице DRAIS с первым проходом при 1500 мл/мин и со вторым проходом при 500 мл/мин. Обе суспензии выдерживают в автоклаве в течение 2 часов при 200°С, охлаждают и затем сушат в течение ночи при 140°С. Результаты морфологического анализа суммируются в таблице 1В, серии 28-29. Образец из серии 28 испытывают на гидротермическую стабильность, нагревая образец в течение 4 часов при 800°С в атмосфере 20% пара, площадь поверхности тестируют и обнаруживают, что она равна 249 м2/г, что составляет 71% от исходной.
Пример 12
Данный пример иллюстрирует использование и влияние TSPP и NaOH в качестве ИРРК. Данный материал также имеет довольно "острое" распределение размера пор со средним диаметром пор между 150-200 ангстрем. Суспензию готовят из Н-30 (гиббсит), АР-15 (активированный оксид алюминия) и гидроксида натрия, получая 5,35 мас.% NaOH и 0,02 моля TSPP/моль общего оксида алюминия. Суспензию готовят, растворяя 120,7 г натрия фосфорнокислого (пиро) четырехзамещенного (TSPP) в 7034,7 г воды, добавляя 164,2 г 50 мас.% водного раствора гидроксида натрия, 613,3 г АР-15 и 1267 г Н-30. Добавление всех веществ осуществляют при перемешивании в устройстве Cowles. Суспензию совместно измельчают в мельнице DRAIS с первым проходом при 1500 мл/мин и со вторым проходом при 500 мл/мин. Затем суспензию выдерживают в автоклаве в течение 2 часов при 200°С, охлаждают и затем сушат в течение ночи при 140°С. Результаты морфологического анализа суммируются в таблице 1А, серия 30.
Пример 13
Данный пример иллюстрирует влияние добавления NaOH и Laponite™, синтетического гекторита, производимого Laporte Industries в качестве ИРРК. Две суспензии (серии 31 и 32), измельченные в песчаной мельнице по примеру 8, готовят из C30D гиббсита, ALCOA СР-3 активированного оксида алюминия и SiO2 от молярного соотношения 3,22 SiO2/Na2O с добавлением щелочи до молярного отношения Na2O/SiO2, равного 2,0. Отдельно полностью диспергируют глину Laponite в течение 1/2 часа в воде перед добавлением к ней измельченной суспензии для серии 32. После выдержки обеих суспензий в автоклаве в течении двух часов при 200°С продукты сушат в течение ночи при 138°С. Содержание суспензии, которую выдерживают в автоклаве, суммируется в таблице 1А, а результаты морфологического анализа приводятся в таблице 1В, серии 31 и 32. Видно, что Laponite™ уменьшает размер кристаллитов, существенно увеличивает площадь поверхности и существенно увеличивает общий объем пор по азоту.
Пример 14
Данный пример сравнивает одну и ту же суспензию, выдержанную в автоклаве с/без Laponite RDS™. Отметим, что Laponite RDS содержит фосфатную добавку (так что мольное соотношение Р2О5/Al2О3 в конечной суспензии равно 0,018), поэтому ее можно диспергировать в воде при более высокой концентрации. К первой измельченной в два прохода в песчаной мельнице суспензии гиббсита добавляют воду, активированный оксид алюминия СР-3 в качестве затравки, силикат натрия и гидроксид натрия, имеющие молярное отношение SiO2/Na2O, равное 3,2, и полученную в результате суспензию обозначают как серию 33. Серию 33 повторяют и полученную в результате суспензию обозначают как серию 34, за исключением того, что к суспензии из серии 34 добавляют хорошо диспергированную суспензию Laponite RDS™ (физическая смесь Laponite RD™ и TSPP, имеющая состав 6,7 мас.% Na2O; 26,7 мас.% MgO; 1,9 мас.% SO4; 4,5 мас.% Р2O5; 0,76 мас.% LiO2; 59 мас.% SiO2 и ATV 13,77 мас.%) так, чтобы окончательная суспензия содержала 5 мас.% Laponite RDS™ относительно массы твердых веществ. Обе суспензии выдерживают в автоклаве в течение 2 часов при 200°С с перемешиванием при 600 об/мин. Составы суспензий до выдержки в автоклаве суммируются в таблице 1А, серии 33 и 34. После охлаждения суспензии сушат в сушильном шкафу в течение ночи при 138°С. Их морфологические свойства суммируются в таблице 1В, серии 33 и 34. Видно, что добавление Laponite RDSTM дает бемит с более высоким объемом пор по азоту, меньшим размером кристаллитов, более высокой площадью поверхности и значительно увеличенным общим объемом пор.
Пример 15
Данный пример иллюстрирует влияние добавления небольших количеств натрия фосфорнокислого (пиро) четырехзамещенного (TSPP) на размер кристаллитов и объем пор продукта из оксида алюминия, выдержанного в автоклаве, изготовленного с Laponite™. Готовят две идентичные суспензии и обозначают их как серии 35 и 36 соответственно, за исключением того, что к одной (серия 36) добавляют 0,00234 молей TSPP/моль оксида алюминия. Суспензии готовят диспергированием 3,0 г Laponite RD™ (способная набухать глина, характеризующаяся тем, что содержит 59-60 мас.% SiO2, 27-29 мас.% MgO, 0,7-0,9 мас.% LiO и 2,2-3,5 мас.% Na2O) в 596,6 г Н2O. Затем TSPP добавляют к суспензии Laponite™ из серии 36 и не добавляют к суспензии из серии 35. Затем к каждой суспензии Laponite™ добавляют 14,4 г раствора силиката натрия (12,5 мас.% SiO2, молярное соотношение SiO2/Na2O = 3,2) и 8,1 г 50 мас.% водного раствора NaOH. Затем к каждой суспензии добавляют 522,8 г измельченного за два прохода в песчаной мельнице гиббсита (Н-30 от Kaiser, общее содержание летучих компонентов (TV)=75,9 мас.%) вместе с 55,1 г активированного оксида алюминия СР-3 (TV=10,0 мас.%). Обе суспензии выдерживают в автоклаве в течение 2 часов при 200°С, и продукт из оксида алюминия сушат в течение ночи при 138°С. Состав суспензии, которую выдерживают в автоклаве, суммируется в таблице 1А, и результаты морфологического анализа даются в таблице 1, серии 35 и 36. Видно, что добавление небольших количеств фосфатной соли существенно уменьшает размер кристаллитов и увеличивает объем пор.
Пример 16
Данный пример иллюстрирует влияние совместного измельчения всех исходных материалов вместо измельчения только гиббсита на свойства выдержанного в автоклаве бемита в системе, содержащей Laponite™. Готовят две суспензии, обозначаемые сериями 37 и 38, содержащие примерно 68 мас.% гиббсита, примерно 27 мас.% АР-15 (активированный оксид алюминия), примерно 2 мас.% SiO2 (молярное отношение Na2O/SiO2=1,0) и примерно 3 мас.% Laponite™, как показано в таблице 1А. Для серии 37 только гиббсит был измельчен в песчаной мельнице в виде суспензии, в то время как для серии 38 вся суспензия, входящая в автоклав, была совместно измельчена. Обе суспензии измельчили двойным пропусканием с умеренным первым проходом и сильным вторым. Морфологический анализ дается в таблице 1В, серии 37 и 38. Видно, что совместное измельчение значительно уменьшает размер кристаллитов бемита и увеличивает объем пор по азоту.
Как и во всех случаях определения площади поверхности и свойств пор, такие свойства, как сообщаемые в сериях 37 и 38, получают после прокаливания при 537,8°С в течение 2 часов. Однако часть непрокаленного образца из серии 38 прокалили в течение 4 часов при 800°С в атмосфере 15 мас.% пара, после этого определяют морфологические свойства и они сообщаются как серия 39 в таблице 1В. Среди заслуживающих внимания черт распределения размера пор продукта из серий 38 и 39 можно указать очень маленький объем пор в порах <100 ангстрем, моду пор в области мезопор при примерно 250 ангстрем и превосходную гидротермическую стабильность с сохранением 95% площади поверхности после обработки паром.
Пример 17
Данный пример иллюстрирует влияние Laponite™ на две различные совместно измельченные суспензии, приготовленные с 0 и 5 мас.% Laponite™ и обозначенные как серии 40 и 41 соответственно. Входные данные и анализ продуктов после выдержки в автоклаве в течение 2 часов при 200°С и затем сушки в течение ночи суммируются в таблице 1А. Таким образом, процедура совместного измельчения такая же, как используемая для примера 16, и составы суспензий суммируются в таблице 1А, серии 40 и 41. Добавка 5 мас.% Laponite™ дает оксид алюминия с меньшим размером кристаллитов, более высокой площадью поверхности и намного более высоким объемом пор, чем в образце, выдержанном в автоклаве без Laponite™. Распределение размеров пор по азоту для бемита с 5% добавленного Laponite™ (серия 41) подтверждает высокую долю пор с диаметром между 300 и 1000 ангстрем с модой пор в области макропор при 780 ангстрем.
Пример 18
Данный пример иллюстрирует влияние измельчения на получение очень высоких объемов пор. Таким образом, серию 41 повторяют два раза (и обозначают как серии 42 и 43), за исключением того, что совместное измельчение изменяют контролированием числа проходов через песчаную мельницу. Также изменяют размер частиц исходного гиббсита. Более конкретно в сериях от 42 до 43 используют 1 и 0 проходов через песчаную мельницу соответственно. Размер частиц исходного гиббсита до совместного измельчения равен 8 микрон для серий 42 и 43 относительно 100 микрон для серии 41. Размер частиц, равный 8 микрон, получают предварительным измельчением гиббсита. Все измельченные суспензии измельчают со всеми присутствующими ингредиентами. Входные уровни и результаты суммируются в таблицах 1А и В, серии 42 и 43. Из сравнения серий 41-43 видно, что объем пор увеличивается, а размер кристаллитов уменьшается с усилением степени измельчения. Распределение размера пор по азоту подтверждает данное увеличение объема пор при увеличении степени измельчения.
Пример 19
Данный пример иллюстрирует влияние использования TSPP в качестве ИРРК при различных количествах. Таким образом, готовят три одинаковые суспензии (обозначаемые сериями 44-46), содержащие гиббсит, затравку оксида алюминия (АР-15), силикат натрия, гидроксид натрия и Laponite™ в количествах, сообщаемых в таблице 1А, серии 44-46. Также к суспензиям добавляют натрий фосфорнокислый (пиро) четырехзамещенный (TSPP) в количествах 0,0; 0,00256 или 0,00511 моль TSPP на моль Al2O3, соответственно для серий 44-46. Все суспензии измельчают, дважды пропуская через песчаную мельницу после того, как добавлены все ингредиенты, и затем выдерживают в автоклаве в течение 2 часов при 200°С с перемешиванием при 600 об/мин. Каждый продукт затем сушат в сушильном шкафу в течение ночи при 138°С. Результаты морфологического анализа суммируются в таблице 1В, серии 44-46. Видно, что добавление TSPP уменьшает размер кристаллитов и увеличивает объем пор оксида алюминия в форме бемита. Однако будет наблюдаться, что если количество TSPP является слишком высоким, как в серии 46, это будет ингибировать конверсию гиббсита в бемит. Соответственно серия 46 рассматривается как сравнительный пример 3. Высокие уровни TSPP можно использовать без ингибирования конверсии гиббсита путем изменения реакционных условий, например, уменьшая уровень силиката или увеличивая уровень внесения затравки оксида алюминия.
Пример 20
Данный пример иллюстрирует эффект использования природной глины Polargel®T (смесь примерно 10 мас.% природной гекторитовой и примерно 90 мас.% монтмориллонитовой глин) от American Colloid Co. в качестве ИРРК. Можно охарактеризовать, что Polargel™ содержит 2,35 мас.% Na2O, 14,43 мас.% Al2O3, 75,35 мас.% SiO2, 3,11 мас.% MgO, 1,78 мас.% CaO, 0,84 мас.% К2O, 0,067 мас.% Li2O и TV при 954°С, равное 11,68 мас.%. Две суспензии, обозначаемые как серии 47 и 48, готовят с 1627,4 г гиббсита Н-30, 469,2 г активированного оксида алюминия АР-15, 250,2 г (12,5% SiO2) силиката натрия (молярное отношение SiO2/Na2O=3,2), 58,3 г 50% раствора NaOH и 6741 г H2O. Суспензия из серии 47 содержит 1 г Laponite™ RD (TV=13,26 мас.%), а суспензия для серии 48 содержит 54,1 г Polargel™ Т (TV=11,68 мас.%). Каждую суспензию готовят диспергированием соответствующей глины в воде в течение 1/2 часа при быстром перемешивании. К каждой суспензии затем добавляют силикат и щелочь, а затем активированный оксид алюминия и гиббсит. Обе суспензии измельчают двойным проходом в 4 л песчаной мельнице, выдерживают в автоклаве в течение 2 часов при 200°С, охлаждают и затем сушат в сушильном шкафу в течение ночи при 138°С. Результаты морфологического анализа даются в таблице 1В, серии 47 и 48. Видно, что оксиды алюминия с близким высоким объемом пор получают с синтетическим гекторитом или со смешанной природной глиной.
Пример 21
Данный пример иллюстрирует действие неизмельченного гиббсита в системе, содержащей TSPP/Laponite™ и различные количества TSPP. Готовят три суспензии, обозначенные как серии 49-51, используя 190,1 г гиббсита (непрокаленного АР-15 от Porocel), 50,0 г активированного оксида алюминия АР-15, 14,4 г (12,5%) силиката натрия (3,2 SiO2/Na2O), 3,4 г 50% гидроксида натрия, 10,4 г Laponite RD™ и 0,00256, и 0,0039 молей TSPP/моль оксида алюминия для серий 49-51 соответственно. Все суспензии перемешивают, выдерживают в автоклаве в течение 2 часов при 200°С и сушат в сушильном шкафу в течение ночи при 138°С. Морфологические аналитические результаты сообщаются в таблице 1В, серии 49-51. Видно, что без TSPP и без измельчения можно получить оксид алюминия с умеренно высоким объемом пор, и добавление TSPP уменьшает размер кристаллитов и дополнительно увеличивает объем пор.
Пример 22
Данный пример иллюстрирует эффект проведения гидротермической обработки с микроволновым излучением. К 54,9 г Н2O добавляют 0,87 г Laponite RD™ и перемешивают в течение 1/2 часа, чтобы диспергировать глину. Затем к дисперсии добавляют 1,2 г 12,5% раствора SiO2 (SiO2/Na2O молярное отношение=3,2) вместе с 0,28г 50% раствора гидроксида натрия. Затем добавляют 4,2 г активированного оксида алюминия СР-3 и 42,8 г измельченного двойным проходом через песчаную мельницу гиббсита Н-30, приготовленного при содержании твердого вещества примерно 25%, и суспензию обрабатывают микроволновым излучением в герметично закрытом контейнере в течение 20 минут при 200°С. После охлаждения суспензию сушат в течение ночи при 138°С. Морфологические аналитические результаты суммируются в таблице 1В, серия 52. Видно, что несмотря на то, что конверсия в бемит менее 100%, общий объем пор по азоту выше 1 см3/г.
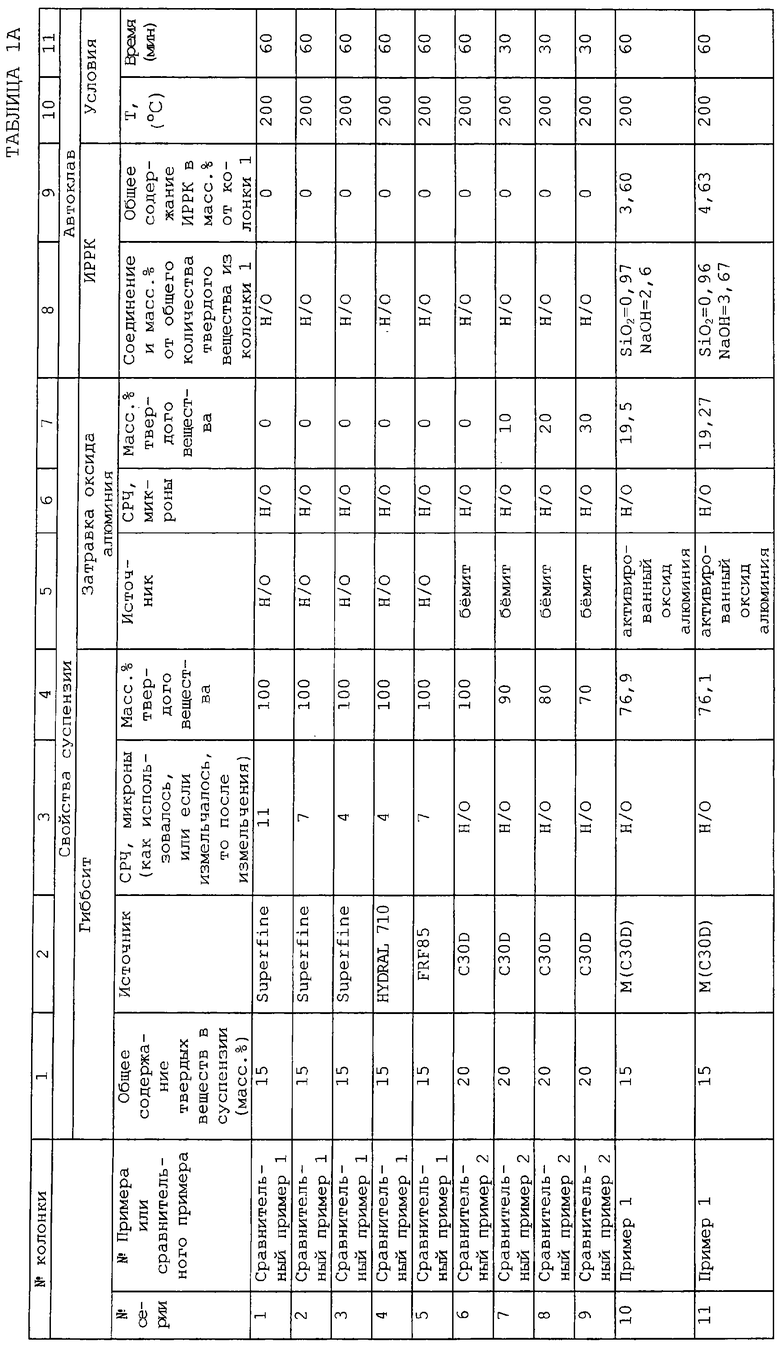

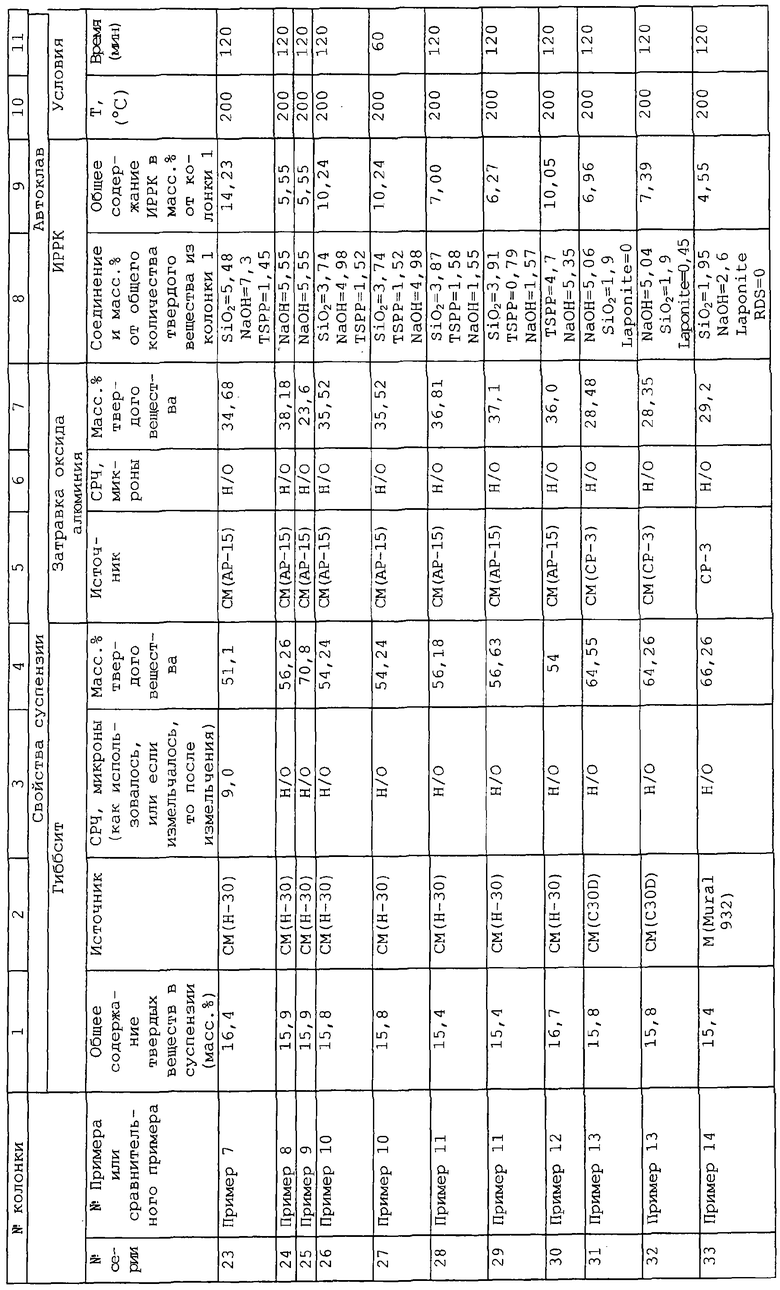
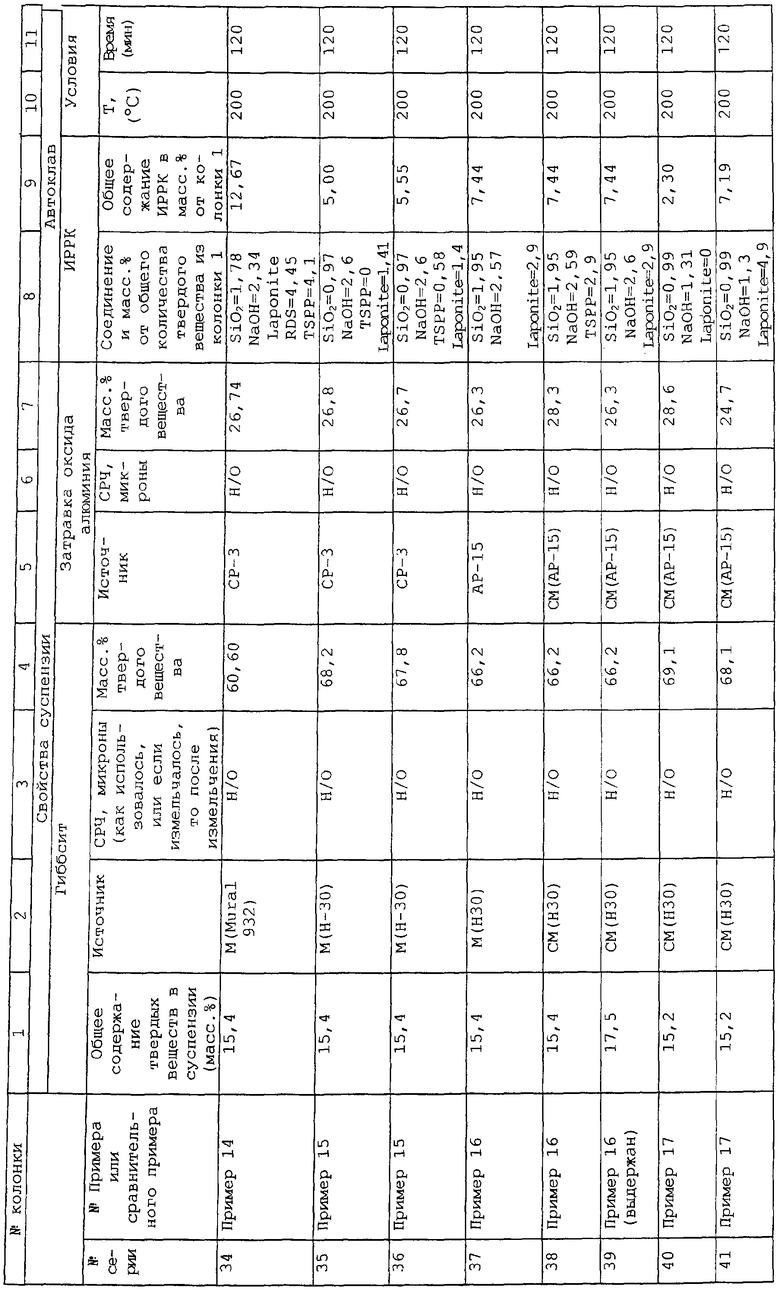
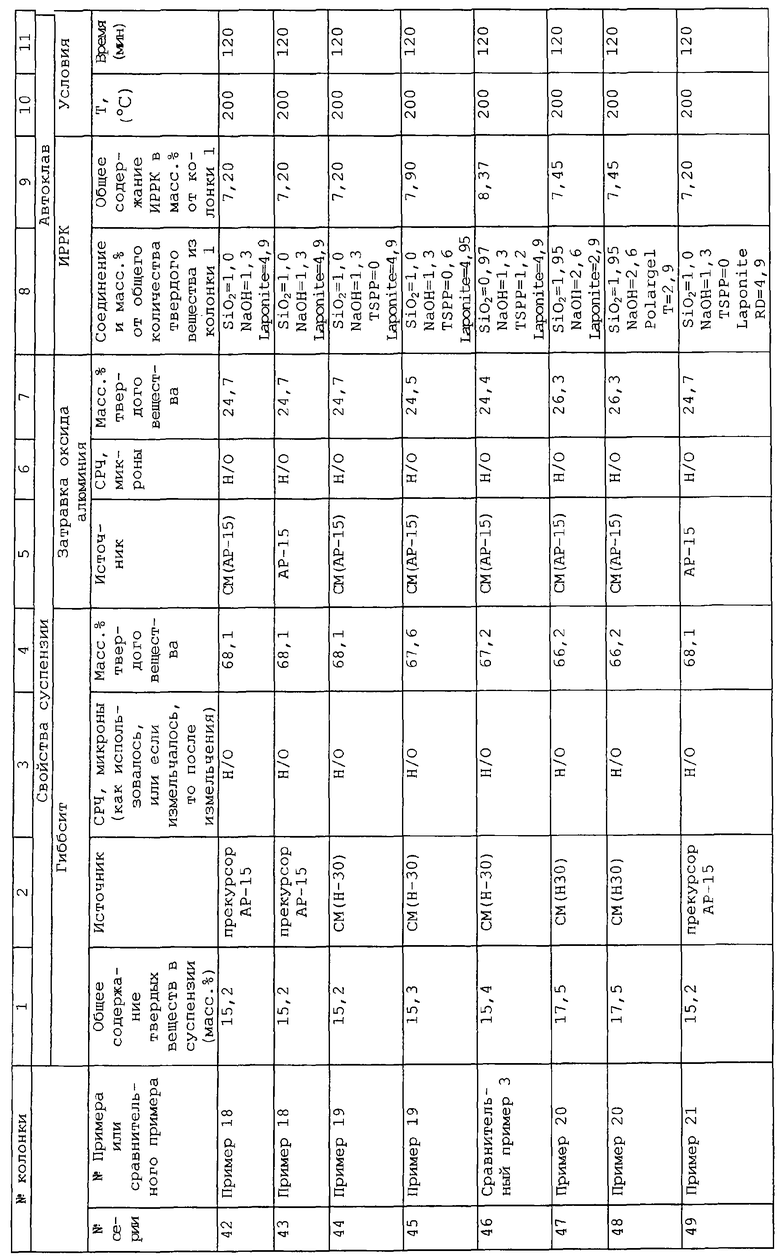
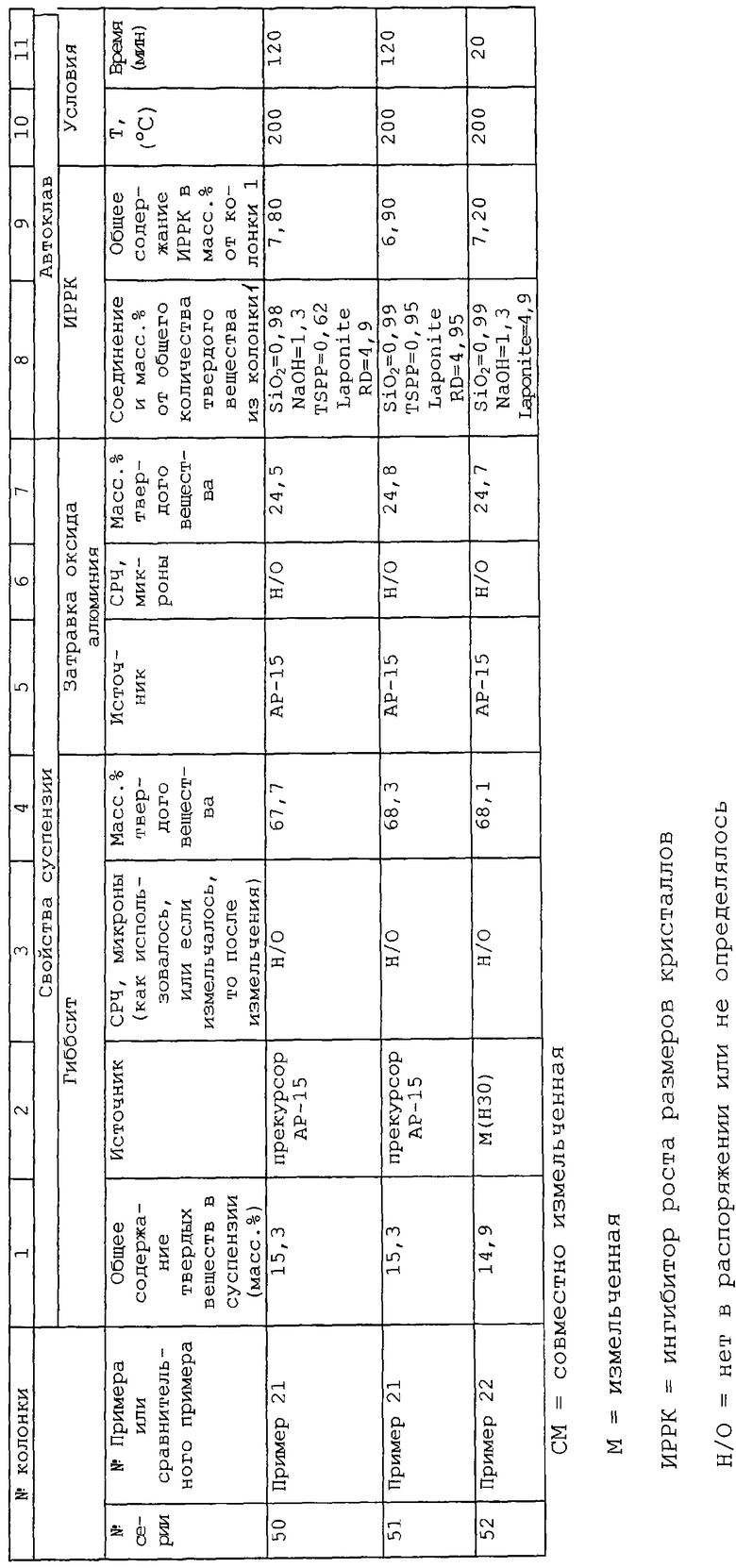
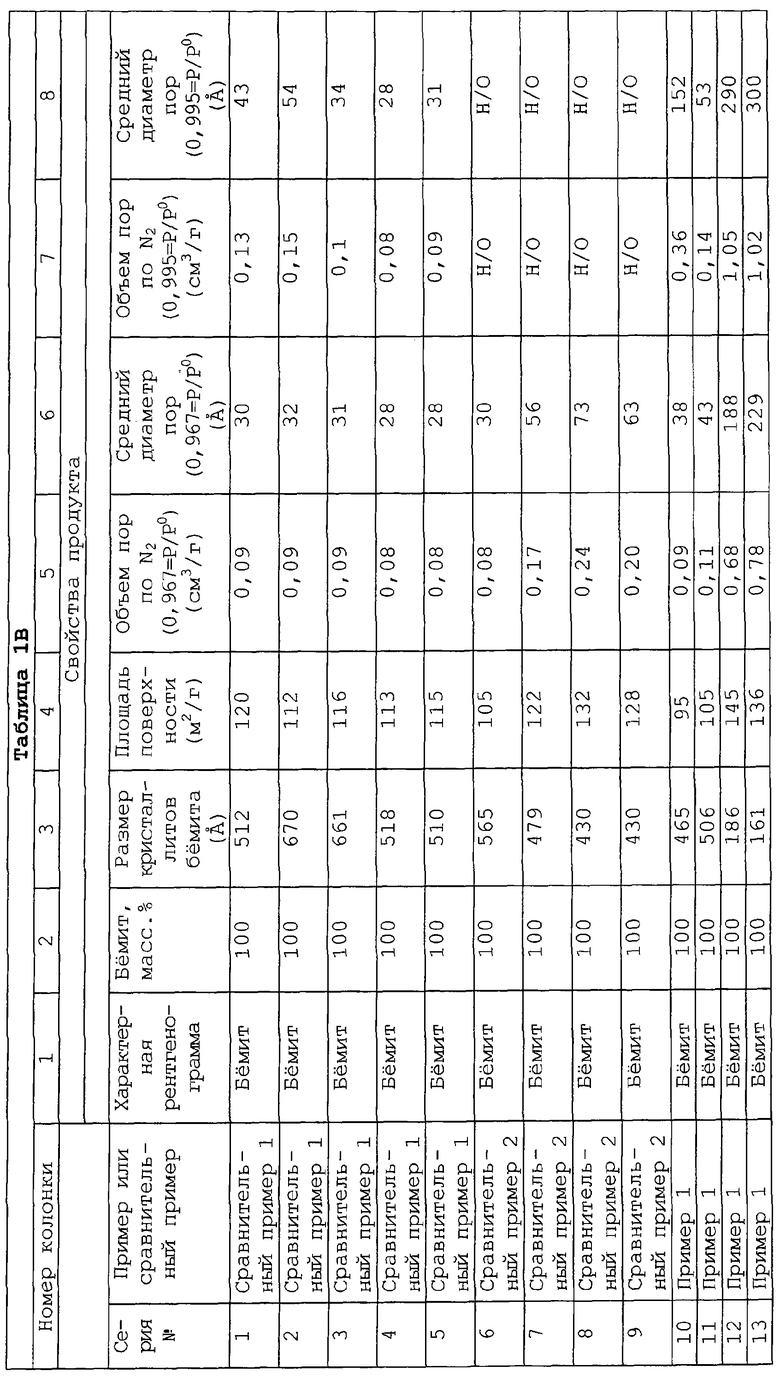
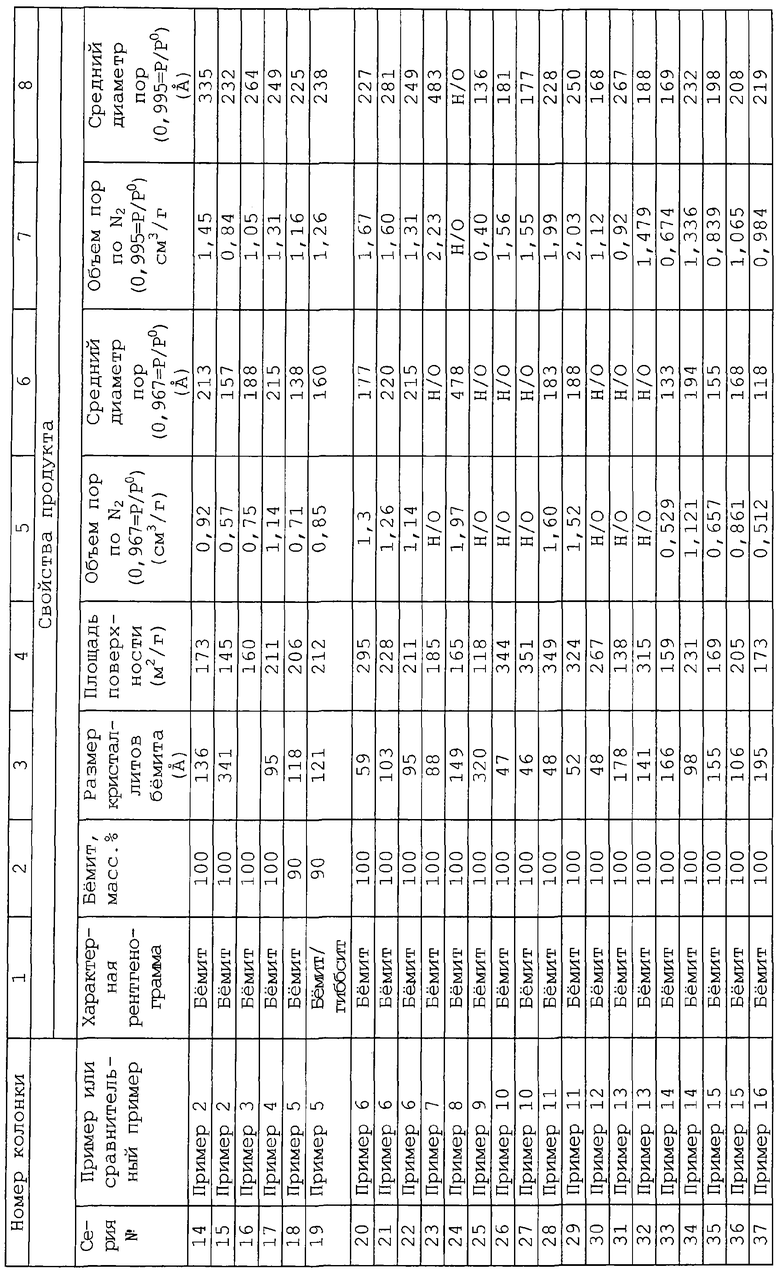
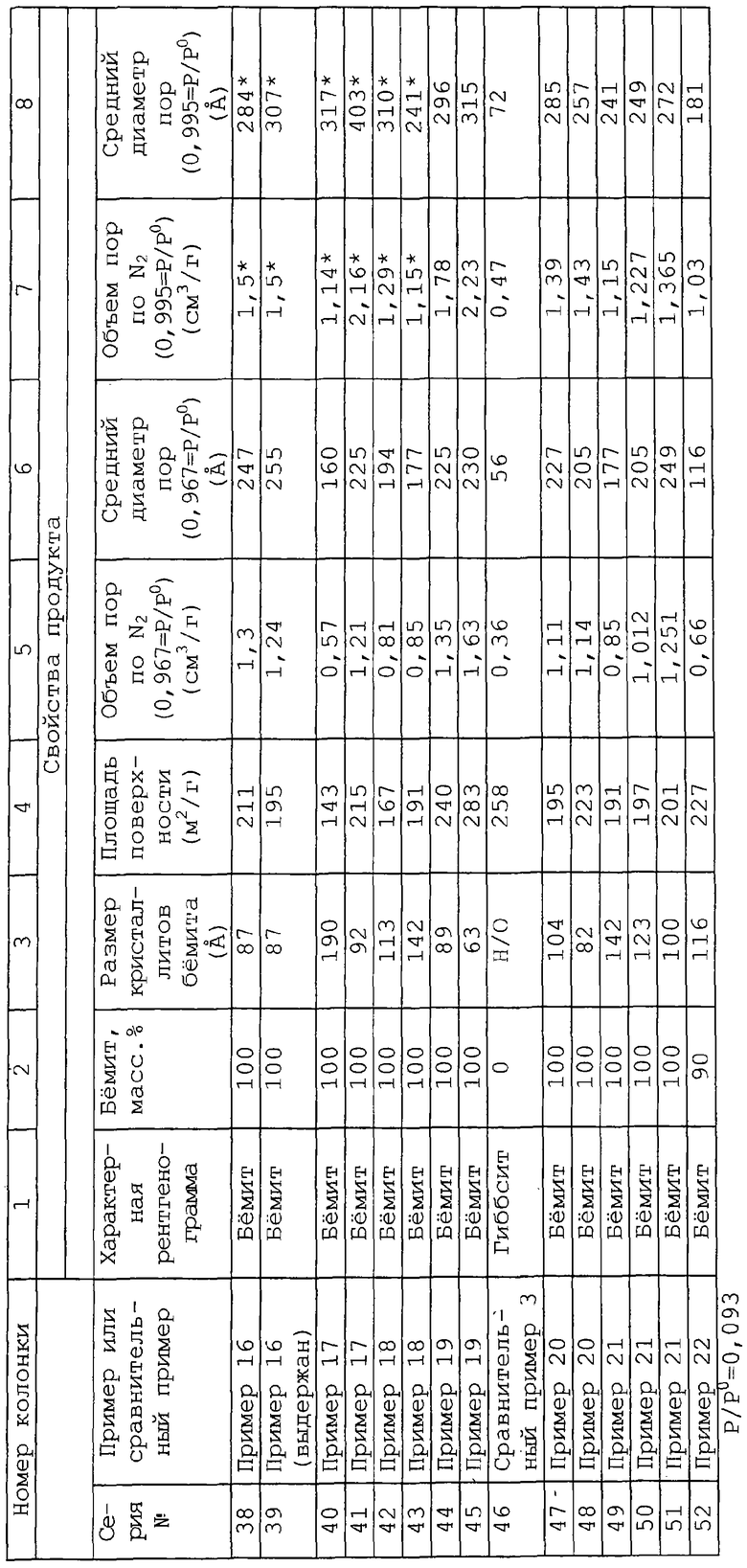
Принципы, предпочтительные варианты осуществления и режимы работы по настоящему изобретению были описаны в предшествующем описании. Однако изобретение, которое намереваются здесь защищать, не следует рассматривать как ограниченное конкретными описанными формами, поскольку их следует рассматривать скорее как иллюстративные, чем ограничивающие. Специалисты в данной области могут сделать вариации и изменения без отклонения от духа изобретения.
Пористые частицы композита, содержащие компонент оксида алюминия и остаток по меньшей мере одного дополнительного компонента ингибитора роста размеров кристаллов, диспергированного внутри компонента оксида алюминия, где указанные частицы композита имеют:
(A) удельную площадь поверхности, равную по меньшей мере 80 м2/г;
(B) средний диаметр пор по азоту от 60 до 1000 ангстрем;
(C) общий объем пор по азоту от 0,2 до 2,5 см3/г; и
(D) средний диаметр частиц от 1 до 15 микрон, и где в указанных частицах композита:
(i) компонент оксида алюминия содержит по меньшей мере 70 мас.% (а) кристаллического бемита, имеющего средний размер кристаллитов от 20 до 200 ангстрем; (b) гамма-оксида алюминия, полученного из указанного кристаллического бемита;
или (с) их смеси;
(ii) остаток дополнительного компонента получен по меньшей мере из одного ионного соединения, имеющего катион и анион, где катион выбран из группы, содержащей аммоний, катион щелочного металла, катион щелочноземельного металла и их смеси, а анион выбран из группы, содержащей гидроксил, силикат, фосфат, сульфат и их смеси, и присутствует в частицах композита в количестве от 0,5 до 10 мас.% относительлно объединенного веса компонента оксида алюминия и остатка дополнительного компонента. Также предлагается способ получения частиц композита, изготовленные из них агломерированные частицы и способ гидропереработки нефтяного исходного сырья с использованием указанных агломератов. Получение оксида алюминия в форме с высоким объемом пор и высоким средним диаметром пор делает ненужным прокаливание перед добавлением металлов для увеличения среднего диаметра пор. Также становится ненужным использование органических растворителей для азеотропного удаления воды. 5 н. и 31 з.п., 2 табл.
(A) удельную площадь поверхности по меньшей мере 80 м2/г;
(B) средний диаметр пор по азоту от 60 до 1000 Å;
(C) общий объем пор по азоту от 0,2 до 2,5 см3/г; и
(D) средний диаметр частиц от 1 до 15 мкм, и при этом в указанных частицах композита
(i) компонент оксида алюминия содержит по меньшей мере 70 мас.% (а) кристаллического бемита, имеющего средний размер кристаллитов от 20 до 200 Å; (b) гамма-оксида алюминия, полученного из указанного кристаллического бемита; или (с) их смеси;
(ii) остаток дополнительного компонента получен из по меньшей мере одного ионного соединения, имеющего катион и анион, где катион выбран из группы, содержащей аммоний, катион щелочного металла, катион щелочноземельного металла и их смеси, а анион выбран из группы, содержащей гидроксил, силикат, фосфат, сульфат и их смеси, и присутствует в частицах композита в количестве от 0,5 до 10 мас.% относительно объединенного веса компонента оксида алюминия и остатка дополнительного компонента.
(i) содержание макропор не более 75% от общего объема пор;
(ii) содержание мезопор от 15 до 90% от общего объема пор по азоту, и где по меньшей мере 20% пор в области мезопор имеют диаметр от 100 до 400Å; и
(iii) содержание микропор не более 80% от общего объема пор по азоту.
(А) смешивают (i) тригидрат оксида алюминия, (ii) жидкую среду, способную растворять по меньшей мере часть тригидрата оксида алюминия при условиях гидротермической обработки стадии (В), (iii) по меньшей мере один компонент затравки активированного оксида алюминия, и (iv) по меньшей мере один дополнительный компонент, выбранный из группы (а) по меньшей мере одного гидроксида, силиката, фосфата или сульфата щелочного или щелочноземельного металла или аммония, (b) способной набухать глины и (с) их смесей, таким образом и при таких условиях, которые достаточны для диспергирования тригидрата оксида алюминия и компонента затравки оксида алюминия в виде частиц в жидкой среде, при этом тригидрат оксида алюминия и компонент затравки активированного оксида алюминия вместе и/или по отдельности обладают средним размером частиц от 0,1 до 15 мкм;
(B) проводят гидротермическую обработку дисперсии, полученной по стадии А, при температуре и в течение времени, достаточных для превращения активированного оксида алюминия и тригидрата оксида алюминия в кристаллический бемит, имеющий средний размер кристаллитов от 20 до 200Å, и для получения частиц композита, содержащих остаток указанного дополнительного компонента, диспергированный в указанном кристаллическом бемите, который суспендирован в жидкой среде;
(C) отделяют жидкую среду от частиц композита, полученных по стадии В.
(i) удельной площадью поверхности по меньшей мере 100 м2/г;
(ii) средним диаметром пор от 50 до 500Å; и
(iii) общим объемом пор по ртути от 0,2 до 2,5 см3/г.
(i) удельной площадью поверхности по меньшей мере 100 м2/г;
(ii) средним диаметром пор от 50 до 500Å; и
(iii) общим объемом пор по ртути от 0,2 до 2,5 см3/г.
(i) удельную площадь поверхности по меньшей мере 80 м2/г;
(ii) средний диаметр пор по азоту от 60 до 1000Å; и (iii) общий объем пор по азоту от 0,2 до 2,5 см2/г; и полученные способом, включающим в себя
(A) смешение (i) тригидрата оксида алюминия, (ii) жидкой среды, способной растворять по меньшей мере часть тригидрата оксида алюминия при условиях гидротермической обработки, (iii) по меньшей мере одного компонента затравки активированного оксида алюминия, и (iv) по меньшей мере одного дополнительного компонента, выбранного из группы, состоящей из (а) гидроксида, силиката, фосфата или сульфата щелочного или щелочноземельного металла и (b) способной набухать глины, таким путем и при условиях, достаточных для диспергирования тригидрата оксида алюминия и компонента затравки оксида алюминия в виде частиц, имеющих средний размер частиц от 1 до 15 мкм, в жидкой среде;
(B) гидротермическую обработку дисперсии, полученной по стадии А, при температуре и в течение времени, достаточных для превращения активированного оксида алюминия и тригидрата оксида алюминия в кристаллический бемит, имеющий средний размер кристаллитов от 20 до 200 Å, и для получения частиц композита, содержащих остаток указанного дополнительного компонента, диспергированный в указанном кристаллическом бемите, который суспендирован в жидкой среде;
(С) отделение жидкой среды от частиц композита, полученных по стадии В.
| US 4797139 А, 10.01.1989 | |||
| US 4301037 А, 17.11.1981 | |||
| US 5194243 А, 16.03.1993 | |||
| US 5114895 А, 19.05.1992 | |||
| US 5178849 А, 12.01.1993 | |||
| Способ получения бемита | 1985 |
|
SU1289819A1 |
| ЧАСТИЧНО КРИСТАЛЛИЧЕСКИЙ ПЕРЕХОДНОЙ ОКСИД АЛЮМИНИЯ, СПОСОБ ЕГО ПОЛУЧЕНИЯ И СПОСОБ ПОЛУЧЕНИЯ ФОРМОВАННОГО γ -ОКСИДА АЛЮМИНИЯ ИЗ НЕГО | 1992 |
|
RU2078043C1 |
| US 4657665 А, 14.04.1987 | |||
| Прибор для очистки паром от сажи дымогарных трубок в паровозных котлах | 1913 |
|
SU95A1 |

