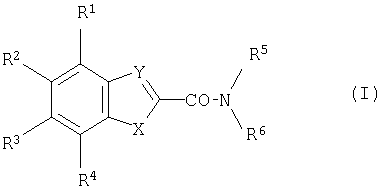
Изобретение относится к новым антагонистам NMDA-рецептора, представляющим собой амидные производные карбоновой кислоты формулы (I)

где
один из R1 R2, R3 и R4 представляет собой ОН или NH2 группу, а другие являются атомами водорода или
две соседние R1, R2, R3 и R4 группы вместе с одним или более идентичными или различными дополнительными гетероатомами и -СН= и/или -СН2- группами образуют 5-6-членное гомо- или гетероциклическое кольцо, предпочтительно пиррольное, пиразольное, имидазольное, оксазольное, оксо-оксазолидиновое или 3-оксо-1,4-оксазиновое кольцо, и две другие R1, R2, R3 и R4 группы являются атомами водорода, R5 и R6 вместе с атомом азота между ними образуют насыщенное или ненасыщенное 4-6-членное гетероциклическое кольцо, которое является замещенным гидроксильной группой, и/или в данном случае фенильной или группами фенокси, фенил-(C1-C4-алкил), фенил-(C1-C4-алкокси), фенокси-(C1-C4-алкил), анилино, фенил-(C1-C4-алкиламино), [фенил-(C1-C4-алкил)]-амино, бензоил, гидроксидифенилметил, C1-C4-алкоксикарбонилфеноксиметил или бензгидрилиденовой группой, необязательно замещенными на ароматическом кольце одним или более атомами галогена, циано или гидроксильной группой, C1-C4-алкильной или C1-C4-алкоксигруппой,
X и Y независимо представляют собой атом кислорода или атом азота или -СН= группу, и к их солям, образованным с кислотами и основаниями.
Поскольку настоящее изобретение относится также к солям соединений формулы (I), образованным с кислотами или основаниями, в особенности солям, образованным с фармацевтически приемлемыми кислотами или основаниями, приведенные значения для соединения формулы (I) относятся либо к свободному соединению, либо к соли, если не оговорено отдельно.
Особенно значительной группой соединений настоящего изобретения являются соединения формулы (Ia),

где значения R1, R2, R3, R4, R5 и R6 те же, что описаны для соединений формулы (I).
В особенности, важными амидными производными карбоновой кислоты формулы (I) являются следующие:
6-(4-бензилпиперидин-1-карбонил)]-1,5-дигидрооксазол[4,5-1]индол-2-он,
6-[4-(4-фторбензилпиперидин-1-карбонил)]-1,5-дигидрооксазол[4,5-f]индол-2-он,
(4-бензилпиперидин-1-ил)-(3,6-дигидропиррол[3,2-е]индазол-7-ил)метанон,
[4-(4-фторбензилпиперидин-1-ил)]-(3,6-дигидропиррол[3,2-е]индазол-7-ил)метанон,
(4-п-толилоксипиперидин-1-ил)]-(3,6-дигидропиррол[3,2-е]индазол-7-ил)метанон,
(4-бензилпиперидин-1-ил)-(3,6-дигидро-имидазо[4,5-е]индол-7-ил)метанон.
Изобретение также относится к фармацевтическим композициям, содержащим в качестве активного ингредиента соединения формулы (I).
Кроме того, объектами настоящего изобретения являются способ получения соединений формулы (I) и химическое и фармацевтическое производство лекарственных средств, содержащих указанные соединения, а также способ лечения с помощью указанных соединений, который заключается во введении млекопитающим, подлежащим лечению, включая человека - эффективного количества/количеств соединений формулы (I) настоящего изобретения как такового или в виде лекарственного средства.
Термин "галоген" в качестве заместителя - как определено ранее - означает фтор, хлор, бром или иод, предпочтительно фтор и хлор. Термин C1-C4-алкильная группа, используемый в настоящем описании, означает метальную, этильную, линейную и изопропильную и различные бутильные группы. Указанные C1-C4-алкильные группы могут быть в составе C1-C4-алкоксигрупп. Термин C1-C4-алканоилоксигруппа означает одновалентную ацилоксигруппу, содержащую атом водорода, также как и C1-C6алкильную группу и карбонилоксигруппу (-СО-O-), присоединенную к ней, предпочтительно формилокси, ацетокси, пропионилокси, различные бутирилокси, валероилокси и капроилоксигруппы.
Изобретение относится также к солям соединений формулы (I), образованным с кислотами или основаниями.
Как органические, так и неорганические кислоты могут быть использованы для формирования кислотно-аддитивных солей. Подходящими неорганическими кислотами могут быть, например, соляная кислота, серная кислота и фосфорная кислота. Представителями одновалентных органических кислот могут быть, например, муравьиная кислота, уксусная кислота, пропионовая кислота и различные масляные кислоты, валериановые кислоты и каприновые кислоты. Представителями двухвалентных органических кислот могут быть, например, щавелевая кислота, малоновая кислота, малеиновая кислота, фумаровая кислота и янтарная кислота. Другие органические кислоты также могут быть использованы, такие как гидроксикислоты, например, лимонная кислота, винная кислота или ароматические карбоновые кислоты, например, бензойная кислота или салициловая кислота, также как алифатические и ароматические сульфоновые кислоты, например, метансульфоновая кислота и п-толуолсульфоновая кислота. Особенно ценной группой кислотно-аддитивных солей является та, в которой сам кислотный компонент не обладает терапевтическим эффектом в применяемой дозе, или она не оказывает неблагоприятного воздействия на эффективность активного компонента. Указанные кислотно-аддитивные соли являются фармацевтически приемлемыми кислотно-аддитивными солями. Причина, почему кислотно-аддитивные соли, которые не принадлежат к фармацевтически приемлемым кислотно-аддитивным солям, но относятся к настоящему изобретению, заключается в том, что при необходимости они могут быть полезными при очистке и выделении желаемого соединения.
Среди солей, образованных с основаниями, особенно важными являются соли, образованные со щелочными металлами, например, натрием, калием, щелочноземельными металлами, например, кальцием и магнием, а также с аммиаком или органическими аминами. Последние основания могут иметь дополнительные заместители, например, гидрокси- или аминогруппы, которые могут влиять, например, на растворимость и обработку продукта.
В соответствии с изобретением соединения формулы (I) получают образованием амидной связи между карбоновой кислотой формулы (II)

где значения R1, R2, R3, R4, Х и Y являются такими, как описано выше для соединения формулы (I) - и амина формулы (III)

где значения R5 и R6 являются такими, как описано выше для соединения формулы (I), и затем полученное амидное производное карбоновой кислоты формулы (I) - где значения R1, R2, R3, R4, R5, R6, Х и Y являются такими, как описано выше для соединения формулы (I) - при необходимости переводят в другое соединение формулы (I) введением новых заместителей и/или модификацией или удалением существующих заместителей, и/или образованием соли и/или выделением соединения из солей, и/или полученные рацематы расщепляют на изомеры с помощью оптически активных кислот или оснований известными способами.
Образование амидной связи предпочтительно осуществляют путем получения активного производного карбоновой кислоты формулы (II), с последующим его взаимодействием с амином формулы (III), предпочтительно в присутствии основания.
В растворе карбоновую кислоту переводят в активное производное in situ во время образования амидной связи в подходящем растворителе (например, диметилформамиде, ацетонитриле, хлорированных углеводородах или углеводородах). Активными производными могут быть хлорангидриды кислот (например, полученные из карбоновой кислоты с тионилхлоридом), смешанные ангидриды (например, полученные из карбоновой кислоты с изобутилхлороформиатом в присутствии основания, например, триэтиламина), активные эфиры (например, полученные из карбоновой кислоты с гидроксибензотриазолом и дициклогексилкарбодиимидом или О-бензотриазол-1-ил-N,N,N',N'-тетраметилуронийгексафторфосфатом (HBTU) в присутствии основания, например, триэтиламина). Активные производные получают при температуре между комнатной и 0°С. К полученному раствору или суспензии добавляют соответствующий амин формулы (III) в виде основания или в виде соли, образованной с неорганической кислотой, так, что основание, например, триэтиламин, необходимый для выделения амина, добавляют в реакционную смесь отдельно. Реакции конденсации отслеживают тонкослойной хроматографией. Необходимое время реакции составляет 6-20 ч. Обработку реакционной смеси проводят различными способами.
Когда реакционная смесь представляет собой суспензию, осадок отфильтровывают и перекристаллизовывают из подходящего растворителя с получением чистого продукта. Если кристаллизация не приводит к чистому продукту, то для его очистки можно затем применить колоночную хроматографию. Колоночную хроматографию проводят либо в нормальной фазе с помощью кизельгеля 60 в качестве адсорбента и в различных системах растворителей, например, толуол/метанол, хлороформ/метанол или толуол/ацетон, в качестве элюентов, либо на обращенной фазе с помощью наполнителей типа Prep-Pak-500/С18 (выпускаемых Waters Associates) и смеси ацетонитрил/вода/трифторуксусная кислота в качестве элюента. Если реакционная смесь в конце реакции ацилирования представляет собой раствор, его концентрируют и остаток кристаллизуют или очищают с помощью хроматографии на колонке, как описано выше. Структуры продуктов определяют с помощью ИК, ЯМР и масс-спектрометрии.
Альтернативно, реакционная смесь может быть очищена с помощью колоночной хроматографии без концентрирования в конце реакции. Фракции, содержащие желаемое соединение, концентрируют, остатки растворяют в диметилсульфоксиде и структуру, чистоту, так же как и концентрацию продукта определяют с помощью ВЭЖХ/МС (колоночной хроматографии высокого давления, с последующей масс-спектрометрией).
Полученные амидные производные карбоновой кислоты формулы (I) - независимо от способа получения - при необходимости могут быть превращены в другое соединение формулы (I) введением других заместителей и/или модификацией и/или удалением существующих заместителей и/или образованием солей с кислотами и/или выделением амидного производного карбоновой кислоты формулы (I) из полученных кислотно-аддитивных солей обработкой основанием, и/или свободное амидное производное карбоновой кислоты формулы (I) может быть превращено в соль обработкой основанием.
Карбоновые кислоты формулы (II) и первичные или вторичные амины формулы (III) являются либо коммерчески доступными, либо могут быть получены различными известными способами. Синтезы некоторых коммерчески недоступных карбоновых кислот формулы (II) описаны в примерах. Следуя указанным способам, могут быть также получены другие коммерчески недоступные карбоновые кислоты формулы (II).
Соединения по изобретению, так же как и их фармацевтически приемлемые соли могут использоваться отдельно или соответственно в форме фармацевтических композиций. Указанные композиции (лекарственные средства) могут находиться в твердой, жидкой или полужидкой форме и могут добавляться фармацевтический адьювант и вспомогательные вещества, которые широко известны в практике, такие как носители, наполнители, разбавители, стабилизаторы, увлажнители или эмульгирующие агенты, вещества, влияющие на значение рН и осмотическое давление, отдушки или ароматизаторы, а также добавки, такие как разрыхлители или наполнители.
Доза, необходимая для терапевтического действия, может варьироваться в широких пределах и соответствует индивидуальным требованиям в каждом конкретном случае, в зависимости от стадии болезни, состояния и веса тела находящегося на излечении пациента, а также от чувствительности пациента к активному ингредиенту, способа введения и числа обработок в день. Точная используемая доза активного ингредиента может быть благополучно определена лечащим врачом на основании знаний уровня техники, а также - находящегося на излечении пациента.
Фармацевтические композиции, содержащие активный ингредиент в соответствии с настоящим изобретением, обычно содержат от 0.01 до 100 мг активного ингредиента в одной дозированной единице. Конечно, возможно, что количество активного ингредиента в некоторых композициях является большим или меньшим границ, определенных выше.
Твердыми формами фармацевтических композиций могут являться, например, таблетки, драже, капсулы, пилюли или лиофилизованные порошки в ампулах, пригодные для получения инъекций. Жидкими композициями являются инъекционные и инфузионные композиции, жидкие лекарственные средства, запаянные жидкости и капли. Полужидкими композициями могут быть мази, бальзамы, кремы, встряхиваемые микстуры и суппозитории.
Для простого введения желательно, чтобы фармацевтические композиции включали дозированные единицы, содержащие количество активного ингредиента, которое следует ввести однократно или за несколько раз или половину, третью или четвертую часть от них. Такими дозированными единицами являются, например, таблетки, которые можно разделить с помощью канавок, отделяя половину или четверть таблетки для точного введения необходимого количества активного ингредиента.
Таблетки могут быть покрыты оболочкой, растворимой в кислоте, для обеспечения высвобождения содержания активного ингредиента после прохождения желудка. Такие таблетки являются покрытыми энтеросолюбильной оболочкой. Подобное действие может также достигаться инкапсулированием активного ингредиента.
Фармацевтические композиции для орального введения могут содержать, например, лактозу или крахмал в качестве наполнителей, карбоксиметилцеллюлозу натрия, метилцеллюлозу, поливинилпирролидин или крахмальную пасту в качестве связующих веществ или гранулирующих агентов. Добавляют картофельный крахмал или микрокристаллическую целлюлозу в качестве разрыхляющих агентов, а также могут использоваться ультраамилопектин или формальдегидный казеин. В качестве антиадгезивных средств и лубрикантов могут использоваться тальк, коллоидная кремниевая кислота, стеарин, стеарат кальция или магния.
Таблетка может быть получена, например, влажным гранулированием с последующим прессованием. Смешанные активные ингредиенты и наполнители, а также при необходимости часть разрыхляющих агентов гранулируют в водном, спиртовом или водно-спиртовом растворе связующего вещества с использованием подходящего оборудования, затем гранулят высушивают. К сухому грануляту добавляют другие разрыхляющие агенты, лубриканты и антиадгезионные агенты и смесь прессуют в таблетку. В данном случае таблетки изготовляют с канавками, делящими таблетку напополам, для каждого введения.
Таблетки могут быть изготовлены прессованием прямо из смеси активного ингредиента и нужных вспомогательных веществ. При необходимости таблетки могут быть покрыты с помощью слоя из добавок, широко известных в фармацевтической практике, например, таких как стабилизаторы, отдушки, красители, такие как сахар, производные целлюлозы (метил- или этилцеллюлоза, карбоксиметилцеллюлоза натрия и т.д.), поливинилпирролидон, фосфат кальция, карбонат кальция, пищевые красители, пищевые настойки, ароматизаторы, пигменты на основе оксида железа и т.д. В случае капсул смесь активного ингредиента и вспомогательных веществ помещают в капсулы.
Жидкие оральные композиции, например суспензии, сиропы, эликсиры, могут быть изготовлены с помощью воды, гликолей, масел, спиртов, красителей и отдушек.
Для ректального введения композиция заключается в суппозитории или клизмы. Суппозитории кроме активного ингредиента могут содержать носитель, так называемые просуппозитории на основе свиного жира. Носителями могут быть овощные масла, такие как гидрированные овощные масла, триглицериды C12-C18 жирных кислот (предпочтительно носители, имеющие товарное наименование Witepsol). Активный ингредиент гомогенно смешивают с расплавленными просуппозиториями на основе свиного жира и формируют суппозитории.
Для парентерального введения композицию составляют в виде инъекционного раствора. Для получения инъекции раствор активных ингредиентов растворяют в дистиллированной воде и/или в различных органических растворителях, таких как гликолевые эфиры, в данном случае в присутствии растворителей, например, полиоксиэтиленсорбитан-монолаурата, -моноолеата или моностеарата (Tween 20, Tween 60, Tween 80). Инъекционный раствор может также содержать различные вспомогательные вещества, такие как консерванты, например, тетраацетатэтилендиамин, а также агенты, поддерживающие значение рН, и буферы, и в данном случае местное анестезирующее вещество, например лидокаин. Инъекционный раствор, содержащий активный ингредиент по изобретению, фильтруют до его внесения в ампулы и стерилизуют после внесения.
Если активный ингредиент гигроскопичен, он может быть затем стабилизирован лиофилизацией.
Близкие структурные аналоги амидных производных карбоновой кислоты формулы (I) известны из уровня техники.
Замещенные производные индол-2-илкарбонилпиперидина, аналогичные соединениям по изобретению, описаны в WO 9618628 и двух публикациях [J. Med. Chem., 39, 3769 (1996), и J. Med. Chem., 42, 4140 (1999)]. Указанные соединения, обладающие ингибирующей активностью обратной транскриптазы, могут использоваться для лечения пациентов со СПИДом.
Амиды индол-2-карбоновой кислоты являются также известными [Bioorg. Med. Chem. Letters, 10, 483. (2000)], ингибируют рр60c-src тирозинкиназу, и, следовательно, они могут использоваться для лечения пациентов, страдающих онкологическими заболеваниями. Указанные публикации не описывают антагонистического действия на NMDA-рецептор.
Производные бензофуран-2-ил-пиперидина описаны в WO 2000012074. Указанные соединения обладают ингибирующим действием на р38-а киназу и, следовательно, могут использоваться для лечения инфекционных болезней, вызванных грамм-отрицательными бактериями, а также - пациентов с респираторным болезненным синдромом.
Производное метанона, описанное в Protein Sci, 6 (7), 1412 (1997), является ингибитором тромбина. Эти публикации не описывают антагонистического действия на NMDA-рецептор.
Неожиданно было обнаружено, что в противоположность известным соединениям структурно аналогичные соединения - которые, как известно, обладают только различными ингибирующими действиями на фермент - новые амидные производные карбоновой кислоты формулы (I) настоящего изобретения являются высоко эффективными и селективными антагонистами NMDA (N-метил-D-аспартат) рецепторов, и более того, большинство соединений являются селективными антагонистами NR2B подтипа NMDA-рецептора. Эта селективность является особенно важной для снижения нежелательных побочных эффектов соединений.
Антагонисты NMDA-рецепторов могут использоваться во многих заболеваниях, которые сопровождаются избытком высвобождения глутамата, главного возбудительного нейропереносчика в центральной нервной системе. Сверхактивация NMDA-рецепторов глутаматом может приводить к накоплению кальция в клетках. Это может запускать каскад внутриклеточных процессов, которые могут изменять клеточную функцию и даже приводить к гибели нейронов [TINS, 10, 299-302 (1987)].
Распознавание структуры NMDA-рецептора, функции и фармакологии обуславливают последние достижения молекулярной биологии. NMDA-рецепторы представляют собой гетеромерные комплексы, построенные, по крайней мере, из одной NR1-субъединицы и по крайней мере одной из четырех NR2-субъединиц (NR2A-D). Как пространственные конструкции CNS, так и фармакологическая чувствительность NMDA-рецепторов, построенных из различных NR2-субъединиц, являются различными. Особый интерес представляет NR2B-субъединица из-за ее ограниченной конструкции (самая высокая плотность наблюдается в предмозговом и основном желатинообразном веществе спинной связки). Соединения, селективные для этого подтипа, известны [Curr. Pharm. Des. 5, 381-404 (1999)] и, как доказано, являются эффективными на животных моделях с параличом [Stroke 28, 2244-2251 (1997)], травматическим ушибом мозга [Brain Res. 792, 291-298 (1998)], болезнью Паркинсона [Ехр. Neurol. 163, 239-243 (2000)], нейропатической и воспалительной болью [Neuropharmacology 38, 611-623 (1999)]. Подтип селективных антагонистов NMDA-рецепторов, как ожидается, проявляет небольшие или неблагоприятные побочные эффекты, вызываемые действием неселективных антагонистов NMDA-рецепторов на сайт связывания глутамата или в проходе тоннеля.
Заболеваниями, которые, как известно, связаны с NMDA-антагонистами [Drug News Perspect 11, 523-569 (1998) и WO 00/00197], являются церебральная ишемия любого происхождения (например, удар, тепловое воздействие), хронические нейродегенеративные заболевания, такие как болезнь Альцгеймера, болезнь Паркинсона, амиотрофический латеральный склероз (ALS), болезнь Хантингтона, вирус иммунодефицита человека (ВИЧ), связанный с повреждением нейрона, травматический ушиб мозга или спинной связки, боль (например, посттравматическая или послеоперационная) и хронические боли, такие как нейропатическая боль или боль, связанная с раком. Антагонисты NMDA-рецептора могут также использоваться для лечения эпилепсии, беспокойств, депрессии, мигрени, психоза, мышечных спазм, мультиинфарктного слабоумия и слабоумия другого происхождения, гипогликемии, дегенеративных заболеваний сетчатки (например, CMV ретинит), астмы, звона в ушах, потери слуха, вызванной аминогликозидным антибиотиком. NMDA-антагонист может использоваться для снижения толерантности и/или зависимости от опиоидного лечения боли и для лечения синдрома зависимости, например, от алкоголя, опиоидов и кокаина.
Так как заявляемые соединения имеют вышеуказанные биологические активности, объектами настоящего изобретения также являются способ лечения амидными производными карбоновой кислоты формулы (I) или их солями, который предполагает введение млекопитающему, находящемуся на излечении - включая человека - эффективного количества/количеств соединений формулы (I) настоящего изобретения отдельно или в виде лекарственного средства.
Известно, что в течение постнатального развития субъединичная композиция нейронных NMDA-рецепторов изменяется. Подобные изменения обнаружены в клеточных культурах нейронов [Eur. J. Neurosci. 10, 1704-1715 (1998)]. В соответствии с литературными данными и с собственными иммуно-цитохимическими исследованиями нейронных клеток, культурируемых в течение 4-7 дней in vitro, авторами настоящего изобретения, преимущественно экспрессируется NR2B-субъединица вместе с NR1-субъединицей. Такой функциональный тест на NMDA-антагонизм в указанных клетках главным образом отражает действие NR2B-субъединицы, содержащей рецепторы. Так как NMDA-рецепторы, как известно, являются проницаемыми для ионов кальция в процессе возбуждения, авторы охарактеризовали активацию NMDA-рецептора с помощью измерения увеличения концентрации кальция внутри клетки после добавления к клеткам агониста (NMDA).
Определение активности NMDA-антагониста in vitro с помощью измерения концентрации кальция внутри клетки ячеечным флуориметрическим счетчиком
Измерения кальция внутри клетки проводят на первичных неокортикальных культурах клетки, полученных от эмбрионов Charles River крысы в возрасте 17 дней (для более подробного описания получения неокортикальной культуры клетки смотри Johnson, M.I.; Bunge, R.P. (1992): Primary cell cultures of peripheral and central neurons and glia. In: Protocols for Neural Cell Culture, eds: Fedoroff, S.; Richardson A., The Humana Press Inc., 13-38.) После выделения клетки помещают в стандартные 96-ячеечные микропланшеты и культуры поддерживают в атмосфере 95% воздуха-5% СО2 при 37°С до измерений кальция.
Культуры используют для измерений кальция внутри клетки через 4-7 дней in vitro. До измерения в клетки вводят флуоресцентный Са2+ - чувствительный краситель, Fluo-4/AM (2-2.5 мкМ). Для прекращения введения клетки промывают дважды раствором, использующимся для измерения (140 мМ NaCl, 5 мМ KCl, 2 мМ CaCl2, 5 мМ HEPES, 5 мМ HEPES-Na, 20 мМ глюкозы, 10 мкМ глицина, рН 7.4). После промывания к клеткам добавляют тестируемые соединения в указанном выше растворе (90 мкл/на ячейку). Измерения кальция внутри клетки проводят ячеечным флуориметрическим счетчиком: повышение Fluo-4-флуоресценции и, следовательно, кальция внутри клетки вызывают введением 40 мкМ NMDA. Ингибирующая активность тестируемых соединений оценивается измерением понижения в повышении уровня кальция в присутствии различных концентраций соединений. После измерения используют стандартную калибровочную процедуру с небольшими изменениями для преобразования флуоресцентных данных в значения концентрации кальция [Meth. Cell. Biol. 40, 155-181 (1994)].
Кривые доза - отклик и IC50-величины рассчитывают с помощью данных, полученных по крайней мере из трех независимых экспериментов. Ингибирующая активность соединения в точке единичной концентрации выражается как ингибирование в процентах отклика NMDA. Сигмоидальные кривые концентрация - ингибирование соответствуют данным и определяют IC50 величины как концентрацию, которая проявляет половину от максимального ингибирования, вызванного соединением.
В Таблице 1 представлены IC50 величины для большинства эффективных соединений данного изобретения, измеренные в этом тесте (колонки 1-2), вместе с самыми эффективными исследованными соединениями из уровня техники (колонки 3-4).

Соединениями из уровня техники являются следующие:
Со 101244: 1-[2-(4-гидроксифенокси)этил]-4-гидрокси-4-(4-метилбензил) пиперидин
EMD 95885: 6-[3-(4-фторбензил)пиперидин-1-ил]пропионил]-2,3-дигидробензоксазол-2-он
СР-101,606: (1S,2S)-1-(4-гидроксифенил)-2-(4-гидрокси-4-фенилпиперидин-1-ил)-1-пропанол
Со-111103: 1-[2-(4-гидроксифенокси)этил]-4-(4-фторбензил)пиперидин
Ro 25.6981: R-(R*,S*)-1-(4-гидроксифенил)-2-метил-3-[4-(фенилметил)пиперидин-1-ил]-1-пропанол.
Как показывает Таблица 1, большинство соединений настоящего изобретения, подвергшихся исследованию, превышают по активности соединения, известные из уровня техники.
Тест на субъединичную селективность на клетках, экспрессирующих рекомбинантные NMDA-рецепторы крыс
Для подтверждения селективности NR2B-субъединицы соединений используют клетки, трансфицированные с ДНК крысы NR1a и NR2A или NR2B-субъединицы. Гены, клонированные в соответствии с известными последовательностями [gi508809 (NRIa крысы), gi205738 (NR2B крысы), gi2905805 (NR2A крысы)], встраивают в индуцированные векторы экспрессии млекопитающего, несущие различные устойчивые гены (гигромицин в случае NRIa или неомицин в случае NR2-субъединиц). Конструкции векторов вводят в НЕК293 клетки с помощью катионного липидного способа трансфекции. Экспрессию белка вызывают с помощью 3 мкМ Муристерона А. Клетки поддерживают в присутствии 365 мкМ кетамина в течение 48-72 часов в атмосфере 95% воздух-5% CO2 при 37°С до экспериментов.
Оценка NMDA-антагонистической активности на клетках, трансфицированных NR1a/NR2B-субъединицами флуориметрическим методом
Для введения клеточных клонов, стабильно экспрессирующих NR1a/NR2B-рецепторы, трансфицированные клетки обрабатывают выбранными антибиотиками в течение 4 недель, затем выращивают устойчивые клоны. Экспрессию белка NR2B-субъединицы осуществляют с помощью поточного иммуноцитохимического метода, основанного на цитометрии. Далее положительные клоны тестируют на функциональную активность в экспериментах с фиксированным делением. Лучший клон, продуцирующий самый большой ионный поток, вызванный NMDA, используют для тестирования NMDA антагонизма с помощью измерения NMDA, вызывающего повышение концентрации кальция в цитозоле. Индукция экспрессии протеина и выращивание клеток были такими же, как описано выше.
Клетки помещают в стандартные 96-ячеечные микропланшеты. Для измерения NMDA-антагонизма используют флуориметрические испытания с помощью счетчика на подложке. По существу способ является таким же, как способ, описанный выше для тестирования первичных культур кортикальных нейронов крыс.
Оценка NMDA-антагонистической активности на клетках, трансфицированных NR1a/NR2A-субъединицами, методом с фиксированным делением
В экспериментах с фиксированным пятном используют клетки, быстро экспрессирующие NR1a/NR2A-рецепторы и выращенные под покровными стеклами. Цельноклеточное записывающее устройство с фиксированным делением изготовляют в соответствии со стандартными способами. Клеточные культуры постоянно промывают внеклеточным раствором (140 мМ NaCl, 5 мМ KCl, 5 мМ Hepes, 5 мМ Na-Hepes, 2 мМ CaCl2, 20 мМ глюкозы, 10 мкМ глицина, рН 7.35) при комнатной температуре. Делительные пипетки сопротивлением между 3 и 6 M Ω наполняют внеклеточным раствором (140 мМ CsCl, 11 мМ EGTA и 10 мМ Hepes, pH 7.3). Записывают внутренний поток, вызываемый 100 мкМ NMDA, от клеток при фиксированном напряжении -70 мВ. Соединения пропускают через мультибаррельное выталкивающее устройство, контролируемое электромагнитными клапанами. Первый NMDA вводят повторно до стабилизации откликов, затем вводят тестируемое соединение. Степень ингибирования - выраженную в процентах - рассчитывают из пика потоков, вызванных NMDA, в присутствии и отсутствии тестируемого соединения. Соотношение селективности (NR2B/NR2A) рассчитывают как соотношение тестируемой дозы на NR1/2A трансфицированные клетки и ИК50 величины NMDA антагонизма на NR1/NR2B экспрессированные клетки. Результаты представлены в Таблице 2.
Оценка селективности для NR2B от NR2A субъединицы, содержащей рецепторы
**: Результаты экспериментов с фиксированным делением на NR1a/NR2A быстро трансфицирующих НЕК клетках. Представлена тестируемая концентрация. Приведены значения 3, 6, 2 экспериментов для 4570001461, 45070002260 и СР-101,606 соответственно.
Селективность: соотношение селективности (NR2B/NR2A), рассчитанное как соотношение тестируемой концентрации на NR1/2A трансфицированные клетки и ИК50 величины на NR1/NR2B экспрессирующие клетки.
В соответствии с результатами, представленными в Таблице 2, соединения 4570001461 и 4570002260, так же как и СР-101,606, являются высоко селективными в отношении NR2B-субъединицы, содержащей NMDA-рецепторы. Синтез соединений и фармацевтических композиций в соответствии с изобретением представлен следующими не ограничивающими примерами. Кодовые номера соединений, которые приведены в биологических тестах, указаны после названия соединений, получаемых в Примерах.
Пример 1
6-(4-Бензилпиперидин-1-карбонил-3H-фуро[3',2':4,5]бензо[1,2-d]оксазол-2-он (4514255)
а) Этил 3Н-фуро[2,3-f]бензоксазол-2-он-6-карбоксилат
Смесь 0.9 г (4.2 ммоль) этил 5-гидрокси-6-аминобензофуран-2-карбоксилата [Helv. Chim. Acta 77, 100 (1994)], 60 мл тетрагидрофурана, 3.1 мл 20% водного раствора фосгена в толуоле и 2.0 мл триэтиламина перемешивают при комнатной температуре в течение часа. Тетрагидрофуран отгоняют в вакууме, к остатку добавляют воду и продукт экстрагируют этилацетатом. Объединенные органические слои промывают 5% водным раствором гидрокарбоната натрия, водой, 1 N раствором соляной кислоты и снова водой, сушат над сульфатом натрия и концентрируют, что дает 1.0 г (96%) названного соединения в виде масла.
b) 3Н-Фуро[2,3-f]бензоксазол-2-он-6-карбоновая кислота
Перемешиваемую смесь 1.0 г (4 ммоль) этил 3Н-фуро[2,3-f]бензоксазол-2-он-6-карбоксилата, 100 мл этанола и 0.5 г гидроксида калия нагревают при кипении с обратным холодильником в течение часа. Смесь концентрируют, остаток растворяют в воде и подкисляют 20% водным раствором серной кислоты. Осажденные кристаллы отфильтровывают и промывают водой, что дает 0.84 г (95%) названного соединения. Т.пл.:190-192°С (вода).
c) 6-(4-Бензилпиперидин-1-карбонил)-3Н-фуро[3',2':4,5]бензо[1,2-d]оксазол-2-он
Смесь 0.42 г (1.9 ммоль) 3Н-фуро[2,3-f]бензоксазол-2-он-6-карбоновой кислоты, 0.3 мл (2.1 ммоль) триэтиламина, 0.35 мл (2.0 ммоль) 4-бензилпиперидина, 0.76 г (2.0 ммоль) HBTU (Advanced Chem. Tech.) и 10 мл диметилформамида перемешивают при комнатной температуре в течение 6 часов. Реакционную смесь концентрируют и остаток очищают с помощью хроматографической колонки, используя кизельгель 60 в качестве адсорбента (Merck) и смесь - толуол: ацетон = 2:1 в качестве элюента, затем продукт кристаллизуют из диэтилового эфира, что дает 0.39 г (54%) названного соединения. Т.пл.:205-210°С (диэтиловый эфир).
Пример 2
6-(4-Бензилоксипиперидин-1-карбонил)-3Н-фуро[3',2':4,5]бензо[1,2-d]оксазол-2-он (4514254)
Названное соединение получают из 3Н-фуро[2,3-f]бензоксазол-2-он-6-карбоновой кислоты и 4-бензилоксипиперидина в соответствии со способом, описанным в примере 1/с. Т.пл.: 217-219°С (диэтиловый эфир).
Пример 3
1-(4-Бензилпиперидин-1-ил)-1-(1,6-дигидро-1,6-диаза-аз-индацен-2-ил)метанон (4514305)
a) Метил (2)-2-азидо-3-(1Н-индол-5-ил)акрилат
В атмосфере азота к раствору метоксида натрия (полученного из 15 мл метанола и 0.66 г (29 ммоль) натрия) добавляют по каплям при температуре 0°С смесь 1.02 г (7 ммоль) индол-5-карбальдегида [Helv. Chim. Acta, 1616 (1968)], 3.34 г (29 ммоль) метил азидо-ацетат и 7 мл метанола и таким образом полученную смесь перемешивают при той же самой температуре в течение 5 часов. Затем реакционную смесь разбавляют 50 мл воды и экстрагируют трижды 50 мл хлороформа. Объединенные органические слои промывают 20 мл воды, отфильтровывают с помощью разделяющей фазы пористой бумаги и концентрируют, что дает 1.3 г (77%) названного соединения. Т.пл.: 130-133°С (хлороформ).
b) Метиловый эфир 1.6-дигидро-1,6-диаза-аз-индацен-2-карбоновой кислоты
К кипящему раствору 36 мл ксилола добавляют небольшими дозами 1.09 г (4.5 ммоль) метилового эфира (2)-2-азидо-3-(1Н-индол-5-ил)акриловой кислоты. Реакционную смесь нагревают при кипении с обратным холодильником, пока не закончится выделение азота, затем концентрируют и остаток кристаллизуют гексаном, продукт отфильтровывают и промывают гексаном, что дает 0.6 г (62%) названного соединения. Т.пл.: 183-184°С (гексан).
с) 1,6-Дигидро-1,6-диаза-аз-индацен-2-карбоновая кислота
Смесь 0.53 г (2.5 ммоль) метилового эфира 1,6-дигидро-1,6-диаза-аз-индацен-2-карбоновой кислоты, 0.36 г (2.5 ммоль) триметилсиланолята калия (Aldrich) и 6.0 мл тетрагидрофурана нагревают при кипении с обратным холодильником в течение часа, потом добавляют еще 0.18 г (1.25 ммоль) триметилсиланолята калия и после 5 часов нагревания при кипении с обратным холодильником реакционную смесь концентрируют. Остаток перемешивают с 20 мл воды, нерастворенный остаток отфильтровывают, к фильтрату добавляют 0.32 мл соляной кислоты, осажденный сырой продукт отфильтровывают и очищают с помощью хроматографической колонки, используя кизелыель 60 (Merck) в качестве адсорбента и смесь - хлороформ: метанол = 9:1 в качестве элюента. Продукт кристаллизуют из диэтилового эфира, что дает 0.22 г (44%) названного соединения. Т.пл.: 248-250°С (диэтиловый эфир).
д) 1-(4-Бензилпиперидин-1-ил)-1-(1,6-дигидро-1,6-диаза-аз-индацен-2-ил)метанон
Названное соединение получают из 1,6-дигидро-1,6-диаза-аз-индацен-2-карбоновой кислоты и 4-бензилпиперидина в соответствии со способом, описанным в примере 1/с. Т.пл.:186-188°С (диэтиловый эфир).
Пример 4
(4-Бензилпиперидин-1-ил)-(2-пропил-8Н-оксазол[5,4-g]индол-7-ил)метанон (4570001079)
a) 1-(4-Бензилпиперидин-1-ил)-1-(6-гидрокси-1Н-индол-2-ил)метанон
Смесь 5.0 г (28.2 ммоль) 6-гидроксииндол-2-карбоновой кислоты [J. Chem. Soc. 1605-1608 (1948)], 4.4 мл (31,6 ммоль) триэтиламина, 5.0 г (28.5 ммоль) 4-бензилпиперидина, 12.0 г (31,6 ммоль) HBTU (Advanced Chem. Tech.) и 50 мл диметилформамида перемешивают при комнатной температуре в течение 6 часов. Выпавший в осадок продукт отфильтровывают и перекристаллизовывают из этанола, что дает 6.75 г (71%) названного соединения. Т.пл.: 214-215°С (этанол).
b) (4-Бензилпиперидин-1-ил)-(2-пропил-8Н-оказол[5,4-g]индол-7-ил)метанон
В атмосфере аргона к раствору 0.5 г (1.49 ммоль) 1-(4-бензилпиперидин-1-ил)-1-(6-гидрокси-1Н-индол-2-ил)метанона и 0.14 г (0.2 ммоль) н-бутиламина в 100 мл диметилового эфира этиленгликоля добавляют порциями по каплям при температуре 0°С 10 г (115 ммоль) диоксида марганца. После перемешивания в течение часа реакционную смесь отфильтровывают, фильтрат концентрируют и остаток очищают с помощью хроматографической колонки, используя кизельгель 60 в качестве адсорбента (Merck) и смесь - гексан: этилацетат = 2:1 - в качестве элюента, что дает 0.1 г (17.3%) названного соединения. Т.пл.: 195-196°С (гексан - этилацетат).
Пример 5
6-(4-Бензилпиперидин-1-карбонил)-1,5-дигидро-оказол[4,5-f]индол-2-он (4570001461)
a) Метиловый эфир (2)-2-азидо-3-(4-бензилокси-3-нитрофенил)акриловой кислоты
В атмосфере аргона к раствору метоксида натрия (полученного из 2.5 г (108.7 ммоль) натрия и 70 мл метанола) добавляют при температуре 0°С смесь 6.1 г (23.7 ммоль) 4-бензилокси-3-нитробензальдегида и 11.2 г (97.3 ммоль) метилазидоацетата в 100 мл метанола. Смесь перемешивают при 0°С в течение 5 часов, затем разбавляют 350 мл воды, осажденные кристаллы отфильтровывают, промывают водой и сушат, что дает 4.93 г (59%) названного соединения. Т.пл.: 95-96°С (вода).
b) Метиловый эфир 6-бензилокси-5-нитро-индол-2-карбоновой кислоты
К перемешиваемому раствору 200 мл кипящего ксилола 4.93 г ((13.9 ммоль) добавляют небольшими дозами метиловый эфир (2)-2-азидо-3-(4-бензилокси-3-нитрофенил)акриловой кислоты. После завершения добавления реакционную смесь нагревают при кипении с обратным холодильником, пока не закончится образование газа (приблизительно 0.5 часа), затем ее охлаждают до комнатной температуры, выпавший в осадок продукт отфильтровывают и промывают н-гексаном, что дает 0.67 г (15%) названного соединения. Т.пл.: 184-187°С (ксилол).
c) Метиловый эфир 5-амино-6-гидроксииндол-2-карбоновой кислоты
Смесь 0.67 г (2.0 ммоль) метилового эфира 6-бензилокси-5-нитро-индол-2-карбоновой кислоты, 60 мл тетрагидрофурана гидрируют в течение 5 часов с помощью 0.1 г 10% Pd/C. Катализатор отфильтровывают и фильтрат, содержащей названное соединение, используют непосредственно на следующем этапе.
д) Метиловый эфир 2-оксо-1,5-дигидро-2Н-оказол[4,5-f]индол-6-карбоновая кислота
Названное соединение получают из метилового эфира 5-амино-6-гидроксииндол-2-карбоновой кислоты в соответствии со способом, описанным в примере I/a. Т.пл.: 267-277°С (вода).
е) 2-Оксо-1,5-дигидро-2Н-оказол[4,5-f]индол-6-карбоновая кислота
Названное соединение получают из метилового эфира 2-оксо-1,5-дигидро-2Н-оказол[4,5-f]индол-6-карбоновой кислоты в соответствии со способом, описанным в примере 1/b. Т.пл.: 288-290°С (вода).
г) 6-(4-Бензилпиперидин-1-карбонил)-1,5-дигидрооказол[4,5-f]индол-2-он
Названное соединение получают из 2-оксо-1,5-дигидро-2Н-оказол[4,5-f]индол-6-карбоновой кислоты и 4-бензилпиперидина в соответствии со способом, описанным в примере 1/с. Т.пл.: 270-271°С (диэтиловый эфир).
Пример 6
6-(4-Бензилоксипиперидин-1-карбонил)-1,5-дигидрооксазол[4,5-f]индол-2-он (4570001462)
Названное соединение получают из 2-оксо-1,5-дигидро-2Н-оказол[4,5-f]индол-6-карбоновой кислоты и 4-бензилоксипиперидина [Тетраhedron 54, 13981, (1998)] в соответствии со способом, описанным в примере 1/с. Т.пл.: 258-261°С (диэтиловый эфир).
Пример 7
6-[4-(4-Фторбензилпиперидин-1-карбонил1-1,5-дигидрооксазол[4,5-]индол-2-он (4570001484)
Названное соединение получают из 2-оксо-1,5-дигидро-2Н-оказол[4,5-f]индол-6-карбоновой кислоты и 4-(4-фторбензил)пиперидина [J. Med. Chem., 35, 4903, (1992)] в соответствии со способом, описанным в примере 1/с. Т.пл.: 244-247°С (диэтиловый эфир).
Пример 8
[4-(4-Фторбензил)пиперидин-1-ил]-(1,6-дигидропиррол[2,3-g]индазол-7-ил)-метанон (другая таутомерная форма соединения - [4-(4-фторбензил)пиперидин-1-ил]-(3≤6-дигидропиррол[2,3-g]индазол-7-ил)метанон) (4570001661)
а) Этиловый эфир 2-[(1Н-индазол-6-ил)-гидразоно]-пропионовой кислоты (другая таутомерная форма соединения - этиловый эфир 2-[(3Н-индазол-6-ил)-гидразоно]-пропионовой кислоты)
К перемешиваемой смеси 6.66 г (50 ммоль) 6-аминоиндазола, 40 мл воды и 25 мл концентрированного раствора соляной кислоты добавляют по каплям при температуре 0°С 3.5 г нитрит натрия в 10 мл воды и перемешивание продолжают при той же температуре в течение 0.5 часов. Затем таким образом полученный раствор добавляют к следующей перемешиваемой смеси: 86 мл воды, 15 г гидроксида калия, 15 г ацетата натрия, 60 мл этанола и 8 мл этилового эфира 2-метилацетоуксусной кислоты (чистота 90%). После добавления реакционную смесь перемешивают при температуре 0°С в течение часа, выпавший в осадок продукт отфильтровывают, промывают водой и сушат, что дает 8.16 г (66%) названного соединения. Т.пл.: 210-211°С (этанол).
b) Этиловый эфир 1,6-дигидро-пиррол[2.3-g]индазол-7-карбоновой кислоты (другая таутомерная форма соединения - этиловый эфир 3.6-дигидропиррол[2,3-g]индазол-7-карбоновой кислоты)
Смесь 4.0 г (16.2 ммоль) этилового эфира 2-[(1Н-индазол-6-ил)-гидразоно]-пропионовой кислоты и 20 г полифосфорной кислоты медленно нагревают до 120°С и выдерживают при этой температуре в течение 0.5 часов. Затем реакционную смесь охлаждают до комнатной температуры, добавляют 30 мл воды и 15 мл концентрированной соляной кислоты. Таким образом полученную смесь экстрагируют этилацетатом, сушат над сульфатом натрия и концентрируют, что дает 1,6 г (43%) названного соединения. Т.пл.: 120-121°С (этилацетат).
с) 1,6-Дигидропиррол[2,3-г]индазол-7-карбоновая кислота (другая таутомерная форма соединения - 3,6-дигидропиррол[2,3-г]индазол-7-карбоновая кислота)
Названное соединение получают из этилового эфира 1,6-дигидропиррол[2,3-г]индазол-7-карбоновой кислоты в соответствии со способом, описанным в примере 1/b. Т.пл.: 270-275°С (вода).
d) [4-(4-Фторбензил)пиперидин-1-ил)-(1,6-дигидропиррол[2,3-g]индазол-7-ил)-метанон (другая таутомерная форма соединения - [4-(4-фторбензил)пиперидин-1-ил)-(3,6-дигидропиррол[2,3-g]индазол-7-ил)-метанон)
Названное соединение получают из 1,6-дигидропиррол[2,3-g]индазол-7-карбоновой кислоты и 4-(4-фторбензил)пиперидина в соответствии со способом, описанным в примере 1/с. Т.пл.: 162-165°С (диэтиловый эфир).
Пример 9
(4-Бензилпиперидин-1-ил)-(1,6-дигидропиррол[2.3-g]индазол-7-ил)-метанон (другая таутомерная форма соединения - (4-бензилпиперидин-1-ил)-(3,6-дигидропиррол[2.3-g]индазол-7-ил)-метанон) (4570001662)
Названное соединение получают из 1,6-дигидропиррол[2,3-g]индазол-7-карбоновой кислоты и 4-бензилпиперидина в соответствии со способом, описанным в примере 1/с. Т.пл.: 209-210°С (диэтиловый эфир).
Пример 10
[4-(4-Фторбензил)пиперидин-1-ил]-(3.6-дигидроимидазо[4,5-е]индол-7-ил)метанон (другая таутомерная форма соединения - [4-(4-фторбензил)пиперидин-1-ил]-(1,6-дигидроимидазо[4,5-е]индол-7-ил)метанон) (4570001688)
а) Этиловый эфир 2-[(1Н-бензимидазол-5-ил)-гидразоно]-пропионовой кислоты (другая таутомерная форма соединения - этиловый эфир 2-[(3Н-бензимидазол-5-ил)-гидразоно]-пропионовой кислоты)
Названное соединение получают из 5-амино-бензимидазола [Helv. China. Acta, 32, 135 (1949)] в соответствии со способом, описанным в Примере 8/а. Т.пл.: 121-1276С. (вода).
b) Этиловый эфир 3,6-дигидро-имидазо[4,5-е]индол-7-карбоновой кислоты (другая таутомерная форма соединения - этиловый эфир 1,6-дигидроимидазо[4,5-е]индол-7-карбоновой кислоты)
Названное соединение получают из этилового эфира 2-[(1Н-бензимидазол-5-ил)-гидразоно]-пропионовой кислоты в соответствии со способом, описанным в примере 8/b. Т.пл.: пена
с) 3,6-Дигидроимидазо[4,5-е]индол-7-карбоновая кислота (другая таутомерная форма соединения - 1,6-дигидроимидазо[4,5-е]индол-7-карбоновая кислота)
Названное соединение получают из этилового эфира 3,6-дигидроимидазо[4,5-е]индол-7-карбоновой кислоты в соответствии со способом, описанным в примере 1/b. Т.пл.: 185-190°С (вода).
д) [4-(4-Фторбензил)пиперидин-1-ил]-(3,6-дигидроимидазо[4,5-е]индол-7-ил)метанон (другая таутомерная форма соединения - [4-(4-фторбензил)пиперидин-1-ил-(1,6-дигидро-имидазо[4,5-е]индол-7-ил)метанон]
Названное соединение получают из 3,6-дигидро-имидазо[4,5-е]индол-7-карбоновой кислоты и 4-(4-фторбензил)пиперидина в соответствии со способом, описанным в примере 1/с. Т.пл.: 283-287°С (диэтиловый эфир).
Пример 11
(4-Бензилпиперидин-1-ил)-(3,6-дигидропиррол[3,2-е]индазол-7-ил)метанон (другая таутомерная форма соединения - (4-бензилпиперидин-1-ил)-(1,6-дигидропиррол[3,2-е]индазол-7-ил)метанон) (4570001689)
а) Этиловый эфир 2-[(1Н-индазол-5-ил)-гидразоно]-пропионовой кислоты (другая таутомерная форма соединения - этиловый эфир 2-[(3Н-индазол-5-ил)-гидразоно-1-пропионовой кислоты)
Названное соединение получают из 5-аминоиндазола в соответствии со способом, описанным в примере 8/а. Т.пл.: 111-113°С (вода).
b) Этиловый эфир 3,6-дигидропиррол[3,2-е]индазол-7-карбоновой кислоты (другая таутомерная форма соединения - этиловый эфир 1,6-дигидропиррол[3,2-е]индазол-7-карбоновой кислоты)
Названное соединение получают из этилового эфира 2-[(1Н-индазол-5-ил)-гидразоно]-пропионовой кислоты в соответствии со способом, описанным в примере 8/b. Т.пл.: 220-221°С (метанол).
с) 3.6-Дигидропиррол[3,2-е]индазол-7-карбоновая кислота (другая таутомерная форма соединения - 1,6-дигидропиррол[3,2-е]индазол-7-карбоновая кислота)
Названное соединение получают из этилового эфира 3,6-дигидропиррол[3,2-е]индазол-7-карбоновой кислоты в соответствии со способом, описанным в примере 1/b. Т.пл.: 183-189°С (вода).
д) (4-Бензилпиперидин-1-ил)-(3,6-дигидро-пиррол[3,2-е]индазол-7-ил)метанон (другая таутомерная форма соединения - (4-бензилпиперидин-1-ил)-(1,6-дигидропиррол[3,2-е]индазол-7-ил)метанон)
Названное соединение получают из 3,6-дигидропиррол[3,2-е]индазол-7-карбоновой кислоты и 4-бензилпиперидина в соответствии со способом, описанным в примере 1/с. Т.пл.: 205-207°С (диэтиловый эфир).
Пример 12
[4-(4-Фторбензил)пиперидин-1-ил]-(3,6-дигидропиррол[3,2-е]индазол-7-ил)метанон (другая таутомерная форма соединения - [4-(4-фторбензил)пиперидин-1-ил]-(1,6-дигидропиррол [3,2-e]индазол-7-ил)метанон) (4570001690)
Названное соединение получают из 3,6-дигидро-пиррол[3,2-е]индазол-7-карбоновой кислоты и 4-(4-фторбензил)пиперидина в соответствии со способом, описанным в примере 1/с. Т.пл.: 169-173°С (диэтиловый эфир).
Пример 13
(4-Бензилпиперидин-1-ил)-(3,6-дигидроимидазо[4,5-е]индол-7-ил)метанон (другая таутомерная форма соединения - (4-бензилпиперидин-1-ил)-(1,6-дигидроимидазо[4,5-е]индол-7-ил)метанон) (4570001779)
Названное соединение получают из 3,6-дигидро-имидазо[4,5-е]индол-7-карбоновой кислоты и 4-бензилпиперидина в соответствии со способом, описанным в примере 1/с. Т.пл.: 256-257°С (диэтиловый эфир).
Пример 14
7-(4-Бензилпиперидин-1-карбонил)-1,6-дигидро-3-окса-1,6,8-триаза-аз-индацен-2-он другая таутомерная форма соединения - 7-(4-бензилпиперидин-1-карбонил)-1,8-дигидро-3-окса-1,6,8-триаза-аз-индацен-2-он) (4570001971)
a) N-Бутил-N'-(4-метокси-2-нитрофенил)-оксаламид
К суспензии 44.0 г (164 ммоль) этилового эфира N-(4-метокси-2-нитрофенил)-оксаламовой кислоты [J. Med. Chem., 18, 926 (1975)] в 330 мл толуола добавляют по каплям, поддерживая температуру ниже 20°С, 16.8 мл (170 ммоль) н-бутиламина, затем смесь перемешивают при комнатной температуре в течение ночи. Реакционную смесь концентрируют и добавляют к остатку 200 мл диэтилового эфира. Таким образом полученную суспензию отфильтровывают, промывают диэтиловым эфиром и сушат, что дает 45.3 г (93%) названного соединения. Т.пл.: 127-128°С (диэтиловый эфир).
b) N-(2-Амино-4-метоксифенил)-N'-бутил-оксаламид
Смесь 27.0 г (91 ммоль) N-бутил-N'-(4-метокси-2-нитрофенил)-оксаламида, 1200 мл метанола и 7.3 г 10% Pd/C катализатора гидрируют в течение 3 часов. Катализатор отфильтровывают, промывают ацетоном, фильтрат концентрируют и остаток обрабатывают 100 мл диэтилового эфира. Полученный продукт отфильтровывают, промывают диэтиловым эфиром и сушат, что дает 21.8 г (90%) названного соединения. Т.пл.: 180-181°С (диэтиловый эфир).
с) Бутиламид 6-метокси-1Н-бензимидазол-2-карбоновой кислоты (другая таутомерная форма соединения - бутиламид 5-метокси-1Н-бензимидазол-2-карбоновой кислоты)
В атмосфере азота 41.0 г (154 ммоль) N-(2-амино-4-метоксифенил)-N'-бутилоксаламида нагревают до 240 С в течение 10 минут. После охлаждения остаток обрабатывают 300 мл ацетона, отфильтровывают и фильтрат концентрируют. Таким образом полученный остаток кристаллизуют из 150 мл гексана, отфильтровывают и сушат, что дает 26.5 г (69.5%) названного соединения. Т.пл.: 125-126°С (гексан).
d) 6-Гидрокси-1Н-бензимидазол-2-карбоновая кислота (другая таутомерная форма соединения - 5-гидрокси-1Н-бензимидазол-2-карбоновая кислота)
Раствор 26.0 г (105 ммоль) бутиламида 6-метокси-1Н-бензимидазол-2-карбоновой кислоты в 780 мл 48% бромистого водорода перемешивают при 90°С в течение 12 часов, затем при 125°С в течение 12 часов. Реакционную смесь охлаждают, выпавший в осадок продукт отфильтровывают, промывают водой и сушат, что дает 14.4 г (76%) названного соединения. Т.пл.: 206-207°С (вода).
е) 6-Гидрокси-7-нитро-1-Н-бензимидазол-2-карбоновая кислота (другая таутомерная форма соединения - 5-гидрокси-4-нитро-1Н-бензимидазол-2-карбоновая кислота)
К раствору 2.0 г (11.2 ммоль) 6-гидрокси-1-Н-бензимидазол-2-карбоновой кислоты в 25 мл трифторуксусной кислоте добавляют при температуре ниже 20°С 1.0 г (11.7 ммоль) нитрата натрия и смесь перемешивают при 20°С в течение 2 часов. Реакционную смесь выливают в ледяную воду, выпавший в осадок продукт отфильтровывают, промывают водой и сушат, что дает 1.7 г (68%) названного соединения. Т.пл.: 218°С (вода).
f) (4-Бензилпиперидин-1-ил)-(6-гидрокси-7-нитро-1Н-бензимидазол-2-ил)метанон (другая таутомерная форма соединения - (4-бензилпиперидин-1-ил)-(5-гидрокси-4-нитро-1Н-бензимидазол-2-ил)метанон)
Названное соединение получают из 6-гидрокси-7-нитро-1Н-бензимидазол-2-карбоновой кислоты и 4-бензилпиперидина в соответствии со способом, описанным в примере 1/с. Т.пл.: 102°С (диэтиловый эфир).
g) (7-Амино-6-гидрокси-1Н-бензимидазол-2-ил)-(4-бензилпиперидин-1-ил)метанон (другая таутомерная форма соединения - (4-амино-5-гидрокси-1Н-бензимидазол-2-ил)-(4-бензилпиперидин-1-ил)метанон
Смесь 1.0 г (4-бензилпиперидин-1-ил)-(6-гидрокси-7-нитро-1Н-бензимидазол-2-ил)метанона, 30 мл метанола гидрируют в течение 2 часов с помощью 0.4 г 10% Pd/C катализатора. Катализатор отфильтровывают и фильтрат концентрируют. Остаток обрабатывают диэтиловым эфиром, кристаллический продукт отфильтровывают, промывают диэтиловым эфиром и сушат, что дает 0.5 г (54%) названного соединения. Т.пл.: 108°С (диэтиловый эфир).
h) 7-(4-Бензилпиперидин-1-карбонил)-1,6-дигидро-3-окса-1,6,8-триаза-аз-индацен-2-он (другая таутомерная форма соединения - 7-(4-бензилпиперидин-1-карбонил)-1,8-дигидро-3-окса-1,6,8-триаза-аз-индацен-2-он)
К раствору 0.45 г (1.28 ммоль) (7-амино-6-гидрокси-1Н-бензимидазол-2-ил)-(4-бензилпиперидин-1-ил)метанона в 5 мл тетрагидрофурана добавляют при температуре 20°С 0.2 г (1.37 ммоль) 1,1'-карбонилдиимидазола. Реакционную смесь перемешивают при комнатной температуре в течение 2 часов, затем концентрируют. Остаток очищают с помощью хроматографической колонки, используя кизельгель 60 в качестве адсорбента (Merck) и смесь - толуол: метанол = 4:1 в качестве элюента. Таким образом полученный продукт кристаллизуют из изопропанола, отфильтровывают и сушат, что дает 0.45 г (93.5%) названного соединения. Т.пл.: >270°С (изопропанол).
Пример 15
6-(4-Бензилпиперидин-1-карбонил)-3,5-дигидроимидазо[4',5';4,5]бензо[1,2-d]оксазол-2-он (другая таутомерная форма соединения - 6-(4-бензилпиперидин-1-карбонил)-3,7-дигидро-имидазо [4',5';4,5]бензо[1,2-d]оксазол-2-он) (4570001972)
a)6-Амино-5-нитро-3Н-бензоксазол-2-он
К раствору 2.0 г (13.3 ммоль) 6-амино-3Н-бензоксазол-2-она [J. Chem. Soc., 321 (1938)] в 20 мл трифторуксусной кислоты добавляют при температуре ниже 20°С 1.2 г (14.1 ммоль) нитрата натрия. Реакционную смесь перемешивают при комнатной температуре в течение ночи, затем концентрируют. Остаток очищают с помощью хроматографической колонки, используя кизельгель 60 в качестве адсорбента (Merck) и смесь - толуол: метанол = 4:1 в качестве элюента, что дает 2.50 г (96%) названного соединения. Т.пл.: 198°С (толуол - метанол).
b) Этиловый эфир (4-бензилпиперидин-1-ил)оксоуксусной кислоты
К раствору 10 г (57 ммоль) 4-бензилпиперидина и 10 мл (57.4 ммоль) N-этиловый эфир диизопропиламина в 100 мл дихлорметана добавляют по каплям при температуре 0°С 7.05 мл (63.1 ммоль) этилоксалилхлорида, затем смесь перемешивают при той же температуре в течение 30 минут. Реакционную смесь разбавляют водой, разделяют, органический слой сушат и концентрируют, что дает 15.5 г (99%) названного соединения в виде масла.
c) (4-Бензилпиперидин-1-ил)оксоуксусная кислота
Смесь 15.5 г (56 ммоль) этилового эфира (4-бензилпиперидин-1-ил)оксоуксусной кислоты, 5 г (79.4 ммоль) гидроксида калия и 250 мл метанола перемешивают при комнатной температуре в течение 6 часов. Затем реакционную смесь концентрируют, остаток переносят в воду, подкисляют 1N соляной кислотой, выпавший в осадок продукт отфильтровывают, промывают водой и сушат, что дает 11.95 г (85%) названного соединения. Т.пл.: 115°С (вода).
д) (4-Бензилпиперидин-1-ил)оксоацетилхлорид
Смесь 26.2 г (106 ммоль) (4-бензилпиперидин-1-ил)оксоуксусной кислоты и 50 мл тионилхлорида нагревают при кипении с обратным холодильником в течение 2 часов, затем охлаждают и концентрируют, что дает 28.0 г (99.5%) названного соединения в виде масла.
e) 2-(4-Бензилпиперидин-1-ил)-N-(5-нитро-2-оксо-2,3-дигидро-бензоксазол-6-ил)-2-оксо-ацетамид
К раствору 3.23 г (16.56 ммоль) 6-амино-5-нитро-3Н-бензоксазол-2-она (этап а), 2.58 мл (18.5 ммоль) триэтиламина и 100 мл хлороформа добавляют по каплям при температуре 20°С 5.25 г (19.75 ммоль) (4-бензилпиперидин-1-ил)оксоацетилхлорида в 20 мл хлороформа, затем смесь перемешивают при комнатной температуре в течение 2 часов. Смесь промывают водой, органический слой сушат и концентрируют. Остаток очищают с помощью хроматографической колонки, используя кизельгель 60 в качестве адсорбента (Merck) и смесь - толуол: метанол = 4:1 в качестве элюента, что дает 3.3 г (47%) названного соединения. Т.пл.: пена.
f) N-(5-Амино-2-оксо-2,3-дигидробензоксазол-6-ил)-2-(4-бензилпиперидин-1-ил-2-оксо-ацетамид
Смесь 3.3 г (7.7 ммоль) 2-(4-бензилпиперидин-1-ил)-N-(5-нитро-2-оксо-2,3-дигидробензоксазол-6-ил)-2-оксо-ацетамида, 100 мл метанола гидрируют в течение 8 часов с помощью 0.3 г 10% Pd/C катализатора. Катализатор отфильтровывают и фильтрат концентрируют. Остаток очищают с помощью хроматографической колонки, используя кизельгель 60 в качестве адсорбента (Merck) и смесь - толуол: ацетон = 2:1 в качестве элюента, что дает 1.06 г (34.5%) названного соединения. Т.пл.: 296°С (толуол - ацетон).
g) 6-Бензилпиперидин-1-карбонил)-3,5-дигидроимидазо[4',5';4,5]бензо[1,2-d]оксазол-2-он (другая таутомерная форма соединения - 6-(4-бензилпиперидин-1-карбонил)-3,7-дигидроимидазо[4',5':4,5]бензо[1,2-d]оксазол-2-он)
1.0 г (2.5 ммоль) N-(5-амино-2-оксо-2,3-дигидро-бензоксазол-6-ил)-2-(4-бензилпиперидин-1-ил)-2-оксо-ацетамида нагревают до 240°С в течение 10 минут, затем охлаждают. Полученную смесь очищают с помощью хроматографической колонки, используя кизельгель 60 в качестве адсорбента (Merck) и смесь - толуол: ацетон = 1:1 в качестве элюента, что дает 0.16 г (17%) названного соединения. Т.пл.: >290°С (толуол - ацетон).
Пример 16
6-(4-Бензилпиперидин-1-карбонил)-3,5-дигидро-1Н-имидазо[4,5-f]индол-2-он (4570002045)
a) Этиловый эфир-[(2-оксо-2,3-дигидро-1Н-бензимидазол-5-ил)гидразоно]-пропионовой кислоты
Названное соединение получают из 5-амино-1,3-дигидро-бензимидазол-2-она [J. Am. Chem. Soc., 80, 1657 (1958)] в соответствии со способом, описанным в примере 8/а. Т.пл.: 220°С (вода).
b) Этиловый эфир 2-оксо-1,2.3,5-тетрагидроимидазо[4,5-f]индол-6-карбоновой кислоты
Названное соединение получают из этилового эфира 2-[(2-оксо-2,3-дигидро-1Н-бензимидазол-5-ил)-гидразоно]-пропионовой кислоты в соответствии со способом, описанным в примере 8/b. Т.пл.: 196-197°С (этилацетат).
c) 2-Оксо-1,2,3,5-тетрагидро-имидазо[4,5-1]индол-6-карбоновая кислота
Названное соединение получают из этилового эфира 2-оксо-1,2,3,5-тетрагидро-имидазо[4,5-f]индол-6-карбоновой кислоты в соответствии со способом, описанным в примере 1/b. Т.пл.: >270°С (вода).
д) 6-(4-Бензилпиперидин-1-карбонил)-3,5-дигидро-1Н-имидазо[4,5-г]индол-2-он
Названное соединение получают из 2-оксо-1,2,3,5-тетрагидроимидазо[4,5-f]индол-6-карбоновой кислоты и 4-бензилпиперидина в соответствии со способом, описанным в примере 1/с. Т.пл.: >270°С (ацетонитрил).
Пример 17
2-(4-Бензилпиперидин-1-карбонил)-1,5-дигидро-8-окса-1,5-диаза-циклопента[b]нафталин-6-он (4570002185)
а) Метиловый эфир 6-оксо-1,5,6,7-тетрагидро-8-окса-1,5-диаза-циклопента[b]нафталин-2-карбоновой кислоты
К раствору 2.0 г (9.7 ммоль) метилового эфира 5-амино-6-гидроксииндол-2-карбоновой кислоты (Пример 5/с) в 300 мл тетрагидрофурана добавляют 2.45 г (29.2 ммоль) гидрокарбоната натрия и 1.3 мл (16.3 ммоль) хлорацетилхлорида и смесь перемешивают при комнатной температуре в течение ночи. Реакционную смесь концентрируют и к остатку добавляют 100 мл воды. Осажденные кристаллы отфильтровывают, промывают водой и суспендируют в 120 мл ацетонитрила. К суспензии добавляют 4.05 г (29.3 ммоль) карбоната калия и полученную смесь перемешивают при комнатной температуре в течение ночи. Реакционную смесь концентрируют и к остатку добавляют 100 мл воды. Продукт отфильтровывают, промывают водой и сушат, что дает 1.7 г (71%) названного соединения. Т.пл.: 188-196°С (вода).
b) 6-Оксо-1,5,6,7-тетрагидро-8-окса-1,5-диаза-циклопента[b]нафталин-2-карбоновая кислота
Названное соединение получают из метилового эфира 6-оксо-1,5,6,7-тетрагидро-8-окса-1,5-диаза-циклопента[b]нафталин-2-карбоновой кислоты в соответствии со способом, описанным в примере 8/b. Т.пл.:231-237°С (вода).
с) 2-(4-Бензилпиперидин-1-карбонил)-1,5-дигидро-8-окса-1,5-диаза-циклопента[b]нафталин-6-он
Названное соединение получают из 6-оксо-1,5,6,7-тетрагидро-8-окса-1,5-диаза-циклопента[b]нафталин-2-карбоновой кислоты и 4-бензилпиперидина в соответствии со способом, описанным в примере 1/с. Т.пл.: 225-231°С (диэтиловый эфир).
Пример 18
2-[4-(4-Фторбензил)пиперидин-1-карбонил]-1,5-дигидро-8-окса-1,5-диазациклопента[b]нафталин-6-он (4570002193)
Названное соединение получают из 6-оксо-1,5,6,7-тетрагидро-8-окса-1,5-диаза-циклопента[b]нафталин-2-карбоновой кислоты и 4-(4-фторбензил)пиперидина в соответствии со способом, описанным в примере 1/с. Т.пл.: 219-226°С (диэтиловый эфир).
Пример 19
(3,6-Дигидро-пиррол[3,2-е]индазол-7-ил)-(4-п-толилоксипиперидин-1-ил)метанон (другая таутомерная форма соединения - (1,6-дигидропиррол[3,2-е]индазол-7-ил)-(4-п-толилоксипиперидин-1-ил)метанон (4570002260)
Названное соединение получают из 3,6-дигидропиррол[3,2-е]имидазол-7-карбоновой кислоты (Пример 11/с) и 4-п-толилоксипиперидина [J. Med. Chem., 21, 309 (1978)] в соответствии со способом, описанным в примере 1/с. Т.пл.: 218-222°С (диэтиловый эфир).
Пример 20
(3,6-Дигидро-пиррол[3,2-е]индазол-7-ил)-[4-(4-метилбензил)пиперидин-1-ил]метанон (другая таутомерная форма соединения - (1,6-дигидро-пиррол[3,2-е]индазол-7-ил)-[4-(4-метилбензил)пиперидин-1-ил]метанон (4570002340)
Названное соединение получают из 3,6-дигидропиррол[3,2-е]индазол-7-карбоновой кислоты (Пример 11/с) и 4-(4-метилбензил)пиперидина [J. Org. Chem., 64, 3763 (1999)] в соответствии со способом, описанным в примере 1/с. Т.пл.: 253-255°С (диэтиловый эфир).
Пример 21
1-(4-Бензилпиперидин-1-ил)-1-(6-гидрокси-1Н-индол-2-ил)метанон (4513579)
Смесь 5.0 г (28.2 ммоль) 6-гидроксииндол-2-карбоновой кислоты [J. Chem. Soc. 1605-1608 (1948)], 4.4 мл (31.6 ммоль) триэтиламина, 5.0 г (28.5 ммоль) 4-бензилпиперидина, 12.0 г (31.6 ммоль) HBTU (Advanced Chem. Tech.) и 50 мл диметилформамида перемешивают при комнатной температуре в течение 6 часов. Полученный продукт отфильтровывают и перекристаллизовывают из этанола, чтобы получить 6.75 г (71%) названного соединения. Т.пл.: 214-215°С (этанол).
Пример 22
1-[4-(4-Фторбензил)пиперидин-1-ил]-1-(6-гидрокси-1Н-индол-2-ил)метанон (4513848)
Названное соединение получают из 4-(4-фторбензил)пиперидина [J. Med. Chem., 35, 4903 (1992)] и 6-гидрокси-1Н-индол-2-карбоновой кислоты в ацетонитриле при комнатной температуре. Реакционную смесь концентрируют и остаток очищают с помощью хроматографии на колонке, используя Кизельгель 60 в качестве адсорбента (Merck) и смесь - толуол: метанол = 4:1 в качестве элюента. Т.пл.: 180-182°С (толуол).
Пример 23
1-(4-Бензилпиперидин-1-ил)-1-(5-нитро-1Н-индол-2-ил)метанон (4514205)
Названное соединение получают из 5-нитроиндол-2-карбоновой кислоты (J. Am. Chem. Soc., 4621 (1958)] и 4-бензилпиперидина в соответствии со способом, описанным в примере 1/с. Т.пл. 220-224°С (диэтиловый эфир).
Пример 24
1-(5-Амино-1Н-индол-2-ил)-1-(4-бензилпиперидин-1-ил)метанон (4514244)
Смесь 0.5 г (1.38 ммоль) 1-(4-бензилпиперидин-1-ил)-1-(5-нитро-1Н-индол-2-ил)метанона, 20 мл метанола и 0.1 г 10% Pd/C катализатора гидрируют в течение 5 часов.
Катализатор отфильтровывают, промывают метанолом и фильтрат концентрируют. Остаток обрабатывают диэтиловым эфиром и выпавшие в осадок кристаллы отфильтровывают, чтобы получить 0.27 г (59%) названного соединения. Т.пл.: 175-180°С (диэтиловый эфир).
Пример 25
(4-Бензилпиперидин-1-ил)-(6-гидрокси-1Н-бензоимидазол-2-ил)-метанон (4570001103)
a) N-Бутил-N'-(4-метокси-2-нитрофенил)оксаламид
К суспензии 44,0 г (164 ммоль) этилового эфира N-(4-метокси-2-нитрофенил)оксаламовой кислоты [J. Med. Chem., 18, 926 (1975)] и 330 мл толуола добавляют 16.8 мл (170 ммоль) н-бутиламина при температуре около 20°С. Реакционную смесь перемешивают при комнатной температуре в течение 10 часов, затем концентрируют и остаток кристаллизуют из диэтилового эфира, выпавший в осадок продукт отфильтровывают, промывают диэтиловым эфиром и сушат, чтобы получить 45.3 г (93.3%) названного соединения. Т.пл.: 127-128°С (диэтиловый эфир).
b) N-(2-Амино-4-метоксифенил)-N'-бутил-оксаламид
Смесь 27.0 г (91 ммоль) N-Бутил-N'-(4-метокси-2-нитрофенил)оксаламида, 1200 мл метанола и 7.3 г 5% Pd/C катализатора гидрируют в течение 3 часов. К реакционной смеси добавляют 600 мл ацетона. Катализатор отфильтровывают, промывают ацетоном, фильтрат концентрируют и остаток кристаллизуют из диэтилового эфира, чтобы получить 21.8 г (90.1%) названного соединения. Т.пл.: 180-181°С (диэтиловый эфир).
c) Бутиламид 6-метокси-1Н-бензоимидазол-2-карбоновой кислоты
В атмосфере азота при температуре 240 С в течение 10 минут перемешивают 41.0 г (154 ммоль) N-(2-амино-4-метоксифенил)-N'-бутилоксаламида. Смесь охлаждают до комнатной температуры, затем добавляют 300 мл ацетона и перемешивают в течение часа. Полученный продукт отфильтровывают. Фильтрат концентрируют и остаток смешивают с 150 мл н-гексана. Полученный продукт отфильтровывают, промывают гексаном и сушат, чтобы получить 26.5 г (69.5%) названного соединения. Т.пл.: 125-126°С (н-гексан).
d) 6-Гидрокси-1Н-бензоимидазол-2-карбоновая кислота
Смесь 26.0 г (105 ммоль) 6-метокси-1Н-бензоимидазол-2-карбоновой кислоты бутиламида и 780 мл 48% водного раствора бромистоводородной кислоты перемешивают при температуре 110°С в течение 8 часов, затем нагревают при кипении с обратным холодильником в течение 12 часов. Смесь охлаждают до комнатной температуры, выпавший в осадок продукт отфильтровывают, промывают водой до тех пор, пока значение рН не будет нейтральным, и сушат, чтобы получить 14.3 г (76.2%) названного соединения. Т.пл.: 206-207°С (вода).
e) (4-Бензилпиперидин-1-ил)-(6-гидрокси-1Н-бензоимидазол-2-ил)-метанон (4570001103)
Смесь 3.0 г (16.75 ммоль) 6-гидрокси-1H-бензоимидазол-2 карбоновой кислоты, 2.4 мл (17.2 ммоль) триэтиламина, 3.0 г (17.1 ммоль) 4-бензилпиперидина, 7.0 г (18.5 ммоль) HBTU и 100 мл диметилформамида перемешивают при комнатной температуре в течение 16 часов. Реакционную смесь концентрируют и остаток очищают с помощью хроматографической колонки, используя Кизельгель 60 в качестве адсорбента (Merck) и смесь - толуол: метанол = 4:1 - в качестве элюента, затем продукт перекристаллизовывают из толуола, чтобы получить 3.58 г (63.5%) названного соединения. Т.пл.: 186°С (толуол).
Пример 26
(6-Гидрокси-1Н-бензоимидазол-2-ил'[4-(4-метилбензил)-пиперидин-1-ил]-метанон (4570001378)
Названное соединение получают из 6-гидрокси-1H-бензоимидазол-2-карбоновой кислоты [пример 25 d] и 4-(4-метилбензил)пиперидина [J.Org. Chem., 64, 3763 (1999)] в соответствии со способом, описанным в примере 25. Т.пл.: 93°С (диизопропиловый эфир).
Пример 27
Получение фармацевтических композиций:
a) Таблетки:
0.01-50% активного ингредиента, 15-50% лактозы, 15-50% картофельного крахмала, 5-15% поливинилпирролидона, 1-5% талька, 0.01-3% стеарата магния, 1-3% коллоидного диоксида кремния и 2-7% ультраамилопектина перемешивают, затем гранулируют с помощью влажной грануляции и прессуют в таблетки.
b) Драже, таблетки, покрытые пленкой:
Таблетки, изготовленные в соответствии со способом, описанным выше, покрывают слоем, состоящим из энтеро- или гастрорастворяющей пленки или из сахара и талька. Драже шлифуют с помощью смеси из пчелиного воска и карнаубского воска.
c) Капсулы:
0.01-50% активного ингредиента, 1-5% лаурилсульфата натрия, 15-50% крахмала, 15-50% лактозы, 1-3% коллоидного диоксида кремния и 0.01-3% стеарата магния тщательно перемешивают, смесь пропускают через сито и наполняют ею твердые желатиновые капсулы.
d) Суспензии;
Ингредиенты: 0.01-15% активного компонента, 0.1-2% гидроксида натрия, 0.1-3% лимонной кислоты, 0.05-0.2% нипагина (метил 4-гидроксибензоат натрия), 0.005-0.02% нипазола, 0.01-0.5% карбопола (полиакриловая кислота), 0.1-5% этилового спирта 96%, 0.1-1% отдушки, 20-70% сорбита (70% водный раствор) и 30-50% дистиллированной воды.
К раствору нипагина и лимонной кислоты в 20 мл дистиллированной воды добавляют небольшими дозами карбопол при энергичном перемешивании и раствор оставляют стоять в течение 10-12 часов. Затем при перемешивании добавляют гидроксид натрия в 1 мл дистиллированной воды, водный раствор сорбита, а затем экстракт с привкусом малины. Активный ингредиент добавляют к полученному носителю небольшими дозами и суспендируют с погружением в гомогенизатор. Под конец суспензию разбавляют до требуемого окончательного объема дистиллированной водой и суспензию в виде сиропа пропускают через коллоидное мельничное оборудование.
е) Свечи:
Для каждой свечи 0.01-15% активного компонента и 1-20% лактозы тщательно перемешивают, затем расплавляют 50-95% адепса на свечу (например, Witepsol 4), охлаждают до 35°С и смесь из активного компонента и лактозы перемешивают с помощью гомогенизатора. Полученную смесь формуют в охлаждаемых формах.
f) Лиофилизованные порошковые композиции в ампулах:
Готовят 5% раствор маннитола или лактозы с бидистиллированной водой для инъекционного применения, после чего раствор отфильтровывают так, чтобы сделать его стерильным. Готовят 0.01-5% раствор активного компонента также с бидистиллированной водой для применения в инъекции и полученный раствор отфильтровывают так, чтобы сделать его стерильным. Полученные два раствора смешивают в асептических условиях, наполняют в ампулы дозой 1 мл, содержимое ампул лиофилизуют и ампулы запаивают в атмосфере азота. Содержание ампул растворяют в стерильной воде или 0.9% (физиологическом) стерильном водном растворе хлорида натрия перед введением.
| название | год | авторы | номер документа |
|---|---|---|---|
| ГЕТЕРОАРИЛЬНЫЕ СОЕДИНЕНИЯ, ИХ КОМПОЗИЦИИ И ИХ ПРИМЕНЕНИЕ В КАЧЕСТВЕ ИНГИБИТОРОВ ПРОТЕИНКИНАЗЫ | 2007 |
|
RU2474582C2 |
| ПРОЛЕКАРСТВА 3-АЦИЛ-2-ОКСИНДОЛ-1-КАРБОКСАМИДОВ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 1993 |
|
RU2124514C1 |
| ПРОИЗВОДНЫЕ ГЕТЕРОАРИЛПИРРОЛИДИНИЛ- И ПИПЕРИДИНИЛКЕТОНА | 2007 |
|
RU2479575C2 |
| ПРОИЗВОДНЫЕ ИНДОЛА И ИНДАЗОЛА, ОБЛАДАЮЩИЕ КОНСЕРВИРУЮЩИМ ДЕЙСТВИЕМ ПО ОТНОШЕНИЮ К КЛЕТКАМ, ТКАНЯМ И ОРГАНАМ | 2009 |
|
RU2460525C2 |
| ПРОИЗВОДНЫЕ ИНДАЗОЛА КАК ИНГИБИТОРЫ ГОРМОНЧУВСТВИТЕЛЬНЫХ ЛИПАЗ | 2005 |
|
RU2370491C2 |
| ПРОИЗВОДНЫЕ ПИПЕРИДИНА И ФАРМАЦЕВТИЧЕСКИЙ ПРЕПАРАТ НА ИХ ОСНОВЕ | 1997 |
|
RU2184112C2 |
| Способ получения производных пиридо/1,2-а/пиримидинов или их солей,или их оптически активных изомеров | 1978 |
|
SU906379A3 |
| ПРОИЗВОДНЫЕ БЕНЗИЛПИПЕРИДИНА, СПОСОБ ИХ ПОЛУЧЕНИЯ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ И СПОСОБ ЕЕ ПОЛУЧЕНИЯ | 1995 |
|
RU2160259C2 |
| СОЕДИНЕНИЯ ДЛЯ ЛЕЧЕНИЯ РАКА | 2009 |
|
RU2514427C2 |
| НОВЫЕ ПРОИЗВОДНЫЕ ВИНИЛОГЕННЫХ КИСЛОТ | 2006 |
|
RU2425830C2 |
Изобретение относится к новым амидным производным карбоновой кислоты, являющимся антагонистами NMDA-рецептора формулы (I):

где один из R1, R2, R3 и R4 представляет собой ОН или NH2-группу, а другие являются атомами водорода или две соседние R1, R2, R3 и R4 группы в данном случае вместе с одним или более идентичными или различными дополнительными гетероатомами и -СН= и/или -СН2- группами образуют 5-6-членное гомо- или гетероциклическое кольцо, предпочтительно пиррольное, пиразольное, имидазольное, оксазольное, оксо-оксазолидиновое или 3-оксо-1,4-оксазиновое кольцо, и другие две R1, R2, R3 и R4-группы являются атомами водорода, R5 и R6 вместе с атомом азота, находящимся между ними, образуют насыщенное или ненасыщенное 4-6-членное гетероциклическое кольцо, которое является замещенным группами фенокси, фенил-(С1-С4-алкокси), фенокси-(С1-С4-алкил), бензоил группой, необязательно замещенной на ароматическом кольце одним или более атомами галогена, С1-С4-алкильной или C1-C4-алкоксигруппой, Х и Y независимо являются атомом кислорода, азота или -СН= группой, и к их солям, образованным с кислотами и основаниями. Изобретение также относится к способу получения соединений формулы (I) и к фармацевтическим композициям, обладающим активностью в качестве селективных антагонистов NR2B-рецептора на основе этих соединений. Технический результат - получение новых соединений и фармацевтических композиций на их основе в целях лечения следующих заболеваний: хронические нейродегенеративные заболевания, хронические болевые состояния, бактериальные и вирусные инфекции. 5 н. и 6 з.п. ф-лы, 2 табл.

где один из R1, R2, R3 и R4 представляет собой ОН или NH2 группу, а другие являются атомами водорода или две соседние R1, R2, R3 и R4 группы, в данном случае, вместе с одним или более идентичными или различными дополнительными гетероатомами и -СН= и/или -СН2- группами образуют 5-6 членное гомо- или гетероциклическое кольцо, предпочтительно пиррольное, пиразольное, имидазольное, оксазольное, оксооксазолидиновое или 3-оксо-1,4-оксазиновое кольцо, и другие две R1, R2, R3 и R4 группы являются атомами водорода,
R5 и R6 вместе с атомом азота, находящимся между ними, образуют насыщенное или ненасыщенное, 4-6 - членное гетероциклическое кольцо, которое является замещенным группами фенокси, фенил-(C1-C4-алкокси), фенокси-(C1-C4-алкил), бензоил группой, необязательно замещенной на ароматическом кольце одним или более атомами галогена, C1-C4-алкильной или C1-C4-алкоксигруппой,
Х и Y независимо являются атомом кислорода, азота или -СН= группой,
и их соли, образованные с кислотами и основаниями.

где значения R1, R2, R3, R4, R5 и R6 те же, что определены в п.1, и их соли, образованные с кислотами и основаниями.
6-(4-бензилпиперидин-1-карбонил)-1,5-дигидро-оказол[4,5-f]индол-2-он,
6-[4-(4-фторбензилпиперидин-1-карбонил)]-1,5-дигидро-оказол[4,5-f]индол-2-он,
(4-бензилпиперидин-1-ил)-(3,б-дигидропиррол[3,2-е]индазол-7-ил)метанон,
[4-(4-фторбензилпиперидин-1-ил)]-(3,6-дигидропиррол[3,2-е]индазол-7-ил)метанон,
(4-п-толилоксипиперидин-1-ил)-(3,6-дигидропиррол[3,2-е]индазол-7-ил)метанон,
(4-бензилпиперидин-1-ил)-(3,6-дигидроимидазо[4,5-е]индол-7-ил)метанон.
производного карбоновой кислоты формулы (I):
где значения R1, R2, R3, R4, R5, R6, Х и Y те же, что определены в п.1, и/или их фармацевтически приемлемые соли, образованные с кислотами или основаниями, в качестве активного ингредиента, и носители, наполнители и им подобные добавки, обычно применяемые в фармацевтике.

где значения R1, R2, R3, R4, R5, R6, Х и Y те же, что определены в п.1,
и их соли, образованные с кислотами и основаниями, заключающийся в образовании
амидной связи между карбоновой кислотой формулы (II)

где значения R1, R2, R3, R4, X и Y те же, что описаны в п.1, и амином формулы (III)

где значения R5 и R6 те же, что приведены в п.1, после чего полученное таким образом амидное производное карбоновой кислоты формулы (I), где значения R1, R2, R3, R4, R5, R6, Х и Y те же, что определены в п.1, при необходимости, превращают в другие соединения формулы (I) с помощью введения новых заместителей и/или модифицируя или удаляя существующие, и/или формируя соль, и/или высвобождая соединение из соли с помощью известных способов.
где значения R1, R2, R3, R4, R5, R6, Х и Y те же, что определены в п.1, и/или его фармацевтически приемлемых солей, образованных с кислотами или основаниями, с носителями, наполнителями и им подобными добавками, обычно применяемыми в фармацевтике.
где значения R1, R2, R3, R4, R5, R6, Х и Y те же, что определены в п.1, и/или его фармацевтически приемлемых солей, образованных с кислотами или основаниями млекопитающим, включая человека, нуждающимся в лечении, в виде соединения как такового или соответственно в форме фармацевтической композиции, полученной на известном вспомогательном оборудовании, обычно применяемом в фармацевтике.
| RU 97120624 А, 10.09.1999 | |||
| ТРИЦИКЛИЧЕСКИЕ ДИКАРБОНИЛЬНЫЕ ПРОИЗВОДНЫЕ И ЛЕКАРСТВЕННЫЙ ПРЕПАРАТ НА ИХ ОСНОВЕ | 1995 |
|
RU2145606C1 |
| Поляризованное электронное реле | 1934 |
|
SU42213A1 |
| WO 9814427 A1, 09.04.1998 | |||
| WO 9526346 A1, 05.10.1995. | |||

