Изобретение относится к противовоспалительным и обезболивающим агентам, в частности к енольным сложноэфирным и эфирным пролекарствам 3-ацил-2-оксиндол-1-карбоксамидов, составляющих новый класс известных нестероидных противовоспалительных агентов.
Использование оксиндолов в качестве противовоспалительных агентов сообщалось в патенте США 3634453, в котором описываются 1-замещенные 2-оксиндол-3-карбоксамиды. В патенте США 4556672 был описан ряд 3-ацил-2-оксиндол-1-карбоксамидов, которые являются ингибиторами ферментов циклооксигеназы (ЦО) и липоксигеназы (ЛО) и которые могут быть использованы в качестве обезболивающих и противовоспалительных агентов для млекопитающих. В патенте США 5118703 описаны некоторые пролекарства на основе 3-ацил-2-оксиндол-1-карбоксамидов, которые имеют следующую формулу:
где
X и Y каждый представляет собой атом водорода, фтора или хлора;
R1 представляет собой 2-тиенил или бензил;
R представляет собой алканоил, содержащий от 2 до 10 атомов углерода, циклоалкилкарбонил, содержащий 5-7 атомов углерода, фенилалканоил, содержащий 7-10 атомов углерода, хлор-бензоил, метоксибензоил, теноил, ω-алкоксикарбонилалканоил, где вышеуказанная алкоксигруппа содержит от 1 до 3 атомов углерода и вышеуказанная алканоильная группа содержит от 3 до 5 атомов углерода, алкоксикарбонил, содержащий от 2 до 10 атомов углерода, феноксикарбонил, 1-(ацилокси)алкил, где вышеуказанная ацильная группа содержит от 1 до 4 атомов углерода, а вышеуказанная алкильная группа - от 2 до 4 атомов углерода, 1-(алкоксикарбонилокси)алкил, где вышеуказанная алкоксигруппа содержит от 2 до 5 атомов углерода, а вышеуказанная алкильная группа - от 1 до 4 атомов углерода, алкильная группа - от 1 до трех атомов углерода, алкилсульфонильную группу, содержащую от 1 до 3 атомов углерода, метилфенилсульфонильную или диалкилфосфонатную группу, где каждый из вышеуказанных алкилов содержит от 1 до 3 атомов углерода.
Патенты США 5118703 и 4556672 включены в качестве справочного материала.
Изобретение предлагает противовоспалительные эфирные или сложноэфирные пролекарства формулы: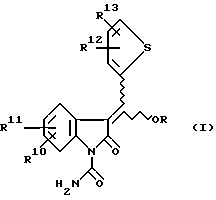
где
R представляют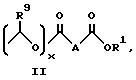
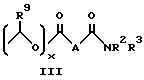
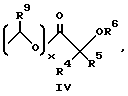
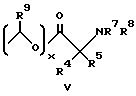
или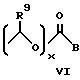
где
x = 0 или 1;
A представляет собой C1-C5-алкилен, необязательно замещенный вплоть до двух C3-C7-циклоалкилами или представляет фениленовую группу;
B представляет собой C2-C6-алкенилфенил, 2-, 3- или 4-пиридил, 2-, 3- или 4-пиперидинил, 2- или 3-пирролидинил, -OCH2CO2R1 или -OCH2CONR2R3;
R1 представляет собой H, C1-C7-алкил, фенил(C1-C4)алкил или (CH2)pCONR2R3;
R2 и R3 независимо друг от друга представляют собой H, C1-C7-алкил или вместе с присоединенным атомом азота представляют собой морфолиновую группу;
R4 и R5 независимо друг от друга представляют собой H, C1-C7-алкил, (CH2)pCONR2R3, (CH2)pNR7R8, (CH2)pOR6 или (CH2)pSR6;
R6 представляет собой H, C1-C6-алкил, C3-C7-циклоалкил, необязательно замещенный вплоть до двух C1-C6-алкильными группами, или взятый вместе с заместителем R4 и присоединенным атомом кислорода образует тетрагидрофурановое кольцо;
R7 и R8 независимо друг от друга представляют собой H, C1-C6-алкил;
R9 представляет собой H или метил;
R10 и R11 представляют галоген;
R12 и R13 независимо представляют водород, галоген;
p = 1 - 3;
при условии, что когда заместитель R представляет собой формулу II, а x = 0, A должен представлять собой группу, отличную от C2-C6-алкиленовой группы.
Предпочтительными являются соединения, где один из заместителей R10 и R11 представляет собой 5-фтор, а другой - 6-хлор.
Другими предпочтительными соединениями являются соединения, где R представляет собой группу формулы: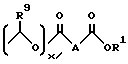
где значения
R1, R9, A указаны выше;
x = 0.
Третью предпочтительную группу соединений формулы I составляют соединения, где R4, R9, R12 и R13 представляют собой атом водорода, R5 представляет собой атом водорода, метил или этил, а R6 представляет собой атом водорода, метил.
Енольные сложные и простые эфиры изобретения не являются енольными кислотами, которыми являются исходные соединения, и обладают способностью в меньшей степени раздражать желудок в сравнении с вышеуказанными исходными соединениями.
Понятие "пролекарство" относится к соединениям, которые являются предшественниками лекарства, которые в свою очередь после применения и абсорбции высвобождают лекарство in vivo в результате метаболизма.
Хотя все обычные способы введения могут быть использованы для соединений изобретения, предпочтительным способом является оральный. После абсорбции в желудочно-кишечном тракте соединения изобретения гидролизуются in vivo до соответствующих соединений формулы I, где заместитель R представляет собой атом водорода, или до их солей. Так как пролекарства изобретения не являются енольными кислотами, то воздействие на желудочно-кишечный тракт кислых исходных соединения сводится до минимума. Кроме того, поскольку желудочно-кишечные осложнения считаются основной отрицательной реакцией на кислотные нестероидные противовоспалительные лекарства (см, например, Del Favero в книге "Side Effects of Drugs Annual 7", Dukes and Elus, Eds. Excerpta Medica, Amsterdam, 1983, p. 104-115), заявляемые соединения формулы I, вероятно, обладают отчетливым преимуществом по сравнению с исходными енольными соединениями.
3-Ацил-2-оксиндол-1-карбоксамиды, превращаемые в соединения формулы I, могут иметь расположение заместителей при экзоциклической двойной связи в положении 3, соответствующее син-, анти-конфигурации, или могут представлять собой смесь изомеров. Следовательно, они представляют собой соединения формул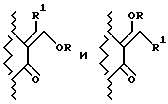
или смесь изомеров, которую представляют в виде формулы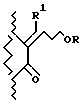
Все формы этих изомеров рассматриваются как часть изобретения.
3-Ацил-2-оксиндол-1-карбоксамиды, которые используются в качестве исходного продукта, могут быть получены по известным методикам, (Пат. США 3634453 и 4556672). Другие исходные реагенты, указанные выше, являются коммерчески доступными или могут быть получены по известным методикам или по методикам, описанным в экспериментальной части данной заявки.
Соединения изобретения могут быть легко получены. Соль соответствующего 3-ацил-2-оксиндол-1-карбоксамида получают в среде инертного растворителя и используют без выделения или после выделения в следующей реакции с хлорангидридом кислоты или α-галогеналкиловым сложным эфиром. Условия этих реакций не являются критическими, температура может изменяться приблизительно от 0 до 50oC, причем предпочтительным температурным интервалом является интервал приблизительно от 0 до 20oC. Время реакции будет изменяться в зависимости от выбранных реагентов и температуры реакции, но предпочтительное время составляет приблизительно от 8 до 90 ч, причем более предпочтительное время составляет приблизительно 20 ч.
Соль 3-ацил-2-оксиндол-1-карбоксамида может представлять собой соль щелочного металла, третичного амина или четвертичную аммонийную соль. Примерами щелочных металлов являются литий, натрий и калий. Третичные амины обычно представляют собой низкомолекулярные алифатические амины, такие как триметиламин, триэтиламин, трибутиламин, и смешанные амины, такие как диизопропилэтиламин, диэтиламинопиридин, а также гетероциклические амины, такие как пиридин-N-метилморфолин. Четвертичные аммонийные соединения могут быть симметричными или смешанными алкиламинами с линейными или разветвленными цепочками. Предпочтительными солями являются натриевые, диизопропилэтиламинные, триэтиламинные и тетрабутиламмониевые соли.
Галогенангидриды кислот могут представлять собой хлорангидриды или бромангидриды, предпочтительными являются хлорангидриды. В качестве α-галогензамещенных сложных эфиров могут использоваться хлор-, бром- или иод-замещенные эфиры, причем предпочтительными являются хлор- и иод-замещенные. Хлорзамещенный сложный эфир предпочтительно используется с иодидом натрия для образования in situ иод-замещенного сложного эфира.
Биологическая активность пролекарств настоящего изобретения измеряется его конверсией в исходное соединение in vivo или его биодоступностью, и определяется в сравнении с исходным соединением.
Например, 3-[гидрокси-2-(тиенил)метилен]-6-хлор-5-фтор- 2,3-дигидро-2-оксо-1H-индол-1-карбоксамид и выбранные пролекарства вводят орально некормленным самцам крыс Sprague-Dawley в дозах 3 мг-экв. 3-[гидрокси-2-(тиенил)метилен]-6-хлор-5-фтор-2,3-дигидро- 2-оксо-1H-индол-1-карбоксамид/кг в виде раствора или суспензии в 0,1%-ной метилцеллюлозе. Дозируемые объемы каждой лекарственной рецептуры сохраняются на уровне 1 мл на 1 кг веса тела. После орального применения отбирают образцы крови путем кровопускания через ретроглазничную пазуху в гепаринизированные пробирки через 1, 3 и 6 ч после введения дозы и сразу же охлаждают. До проведения анализа плазму хранят при -20oC.
Концентрацию 3-[гидрокси-2-(тиенил)метилен] -6-хлор-5-фтор-2,3- дигидро-2-оксо-1H-индол-1-карбоксамида в плазме после введения 3-[гидрокси-2(тиенил)метилен] -6-хлор-5-фтор-2,3-дигидро-2-оксо-1H- индол-1-карбоксамида и пролекарства определяют с помощью высокоэффективной жидкостной хроматографии с ультрафиолетовым детектором при 360 нм. Нижний предел количественного определения 3-[гидрокси-2(тиенил)метилен]-6-хлор-5-фтор-2,3-дигидро-2-оксо-1H-индол- 1-карбоксамида составляет 0,2 мкг/мл.
Площадь под кривой концентрация-время [AUC (0-6 ч)] определяют линейным трапецоидным методом для 3-[гидрокси-2(тиенил)метилен)- 6-хлор-5--фтор-2,3-дигидро-2-оксо-1H-индол-1-карбоксамида после введения 3-[гидрокси-2-(тиенил)метилен)-6-хлор-5-фтор-2,3-дигидро-2-оксо-1H-индол- 1-карбоксамида и каждого пролекарства. Относительная биоактивность 3-[гидрокси-2(тиенил)метилен] -6-хлор-5-фтор-2,3-дигидро-2-оксо-1H- индол-1-карбоксамида после введения каждого пролекарства оценивается путем определения отношения значения площади [AUC(0-6 ч)] 3-[гидрокси-2(тиенил)метилен]-6-хлор-5-фтор-2,3-дигидро-2-оксо-1H- индол-1-карбоксамида после введения пролекарства к значению площади [AUC(0-6 ч)] 3-[гидрокси-2-(тиенил)метилен-6-хлор-5-фтор-2,3- дигидро-2-оксо-1H-индол-1-карбоксамида после введения 3-[гидрокси- 2-(тиенил)метилен-6-хлор-5-фтор-2,3-дигидро-2-оксо-1H-индол-1- карбоксамида.
Данные по "биодоступности" для заявленных соединений приведены ниже.
Соединение примера - Биодоступность%
12 - 95
24 - 53
30 - 90
31 - 143
35 - 58
36 - 86
39 - 62
43 - 80
44 - 100
Пролекарства формулы I оценивались на противовоспалительную и обезболивающую активность по известным методикам, таким как тест на пищевой отек у крыс, тест на вызываемый адъювантами артрит у крыс или тест на вызванные фенилбензохиноном болевые симптомы у мышей, которые ранее использовались для оценки исходных соединений (C.A. Winter. "Progress in Drug Research", ed. E. Jucker. Birkhauser Verlag, Basel. Vol. 10, стр. 139-192 (1966).
Ниже описываются испытания по трем исходным оксиндол карбоксамидам и одному пролекарству, в отношении данных по противовоспалительной активности in vivo.
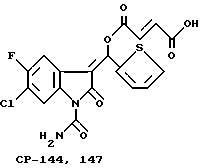
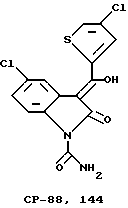
CP-144.477 представляет собой E-1-карбоксамидо-3-[3-карбокси-E- пропеноилокси-(2-тиенил)метилен]-6-хлор-2,3-дигидро-5-фтор-2-оксо- 1H-индол в виде твердого желтого кристаллического вещества при комнатной температуре, имеющего молекулярный вес 436,8.
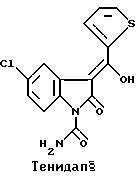
Тенидап является прототипом структурно нового класса противовоспалительных средств - оксиндолов, имеющих биологические характеристики, отличные от характеристик классических нестероидных противовоспалительных лекарственных средств. Так же как и нестероидные противовоспалительные лекарственные средства, тенидап обладает способностью ингибировать доступ CO при метаболизме арахидоновой кислоты и, следовательно in vivo противовоспалительными/обезболивающими свойствами. Однако тенидап обладает несколькими дополнительными видами активности, которыми не обладает класс нестероидных противовоспалительных лекарственных средств.
Тенидап подтвердил свою клиническую активность при лечении как ревматоидного артрита (RA), так и остеоартрита (OA). Промежуточный анализ данных сравнительных испытаний с напроксеном позволяет предположить, что эффективность тенидапа может превысить активность напроксена после длительного приема (в течение месяцев), особенно в случаях с ревматоидным артритом. Кроме того, тенидап (а не напроксен) заметно снижает уровень сыворотки острой фазы белков CRP (C-реактивных) и SAA у пациентов с ревматоидным артритом после нескольких недель терапии. В настоящее время принято считать, что уровень сыворотки CRP связан с тяжестью ревматоидного артрита и длительностью этого заболевания. Такое свойство не присуще нестероидным противовоспалительным лекарственным средствам, в то время как было установлено, что стероиды и золото снижают уровень CRP в такой же степени, как и тенидап. Результаты лабораторных исследований, которые бы конкретно подтверждали свойство тенидапа снижать CRP у пациентов с равметоидным артритом, пока отсутствуют.
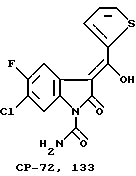
Фармакокинетика CP-144.477 на крысах, собаках и обезьянах
Сравнительная биодоступность CP-72.133 после перорального введения CP-144.477 по сравнению с биодоступностью после перорального введения самого CP-72.133, составила 111%, 62% и 80% у крыс, собак и обезьян соответственно. Концентрация CP-144.477 в крови после перорального введения не подлежала определению у всех видов. Отсутствие систематического воздействия CP-144.477 было неудивительным, поскольку это соединение быстро гидролизовалось in vitro до CP-72.133 в кишечных гомогенатах обезьян и в крысиной, собачьей, обезьяньей и человеческой плазме. Испытания in vitro также показали, что CP-144.477 сохранял стабильность в желудочном соке крыс м обезьян. Все эти данные позволяют предположить, что CP-144.477 сохраняет стабильность в верхней части желудочно-кишечного тракта, но быстро гидролизуется во время и сразу же после пероральной абсорбции.
А. Фармакокинетика CP-72.133 после перорального введения CP-144.477
Фармакокинетика CP-72.133 сравнивалась после перорального введения пролекарства CP-144477 или CP-72.133 крысам, собакам и обезьянам. Концентрация в плазме CP-144.477 и CP-72.133 определялась с использованием жидкостной хроматографии высокого разрешения с ультрафиолетовым определением. Очевидное время полужизни CP-72.133 было таким же после перорального введения CP-144.477 или CP-72.133 крысам и обезьянам, время полужизни CP-72.133 у собак после перорального введения CP144.477 по существу больше, чем после введения CP-72.133, вероятно, благодаря пролонгированной абсорбции пролекарства. Эти испытания также позволили определить относительную биодоступность CP-72.133 после введения CP-144.477 посредством определения отношения AUC CP-72.133 после введения пролекарства или исходного лекарства. После перорального введения CP-144.477 в дозах, равных 3 мг/кг CP-72.133, относительная биодоступность CP-72.133 у крыс, собак и обезьян составила 111%, 62% и 80% соответственно. Важно отметить, что содержание в крови общего количества CP-144.477 было ниже порога количественного определения (< 0,2 мкг/мл) после перорального введения всем видам животных, показывая тем самым, что системное воздействие CP-144.477 было чрезвычайно невелико. Это подтверждалось следующим наблюдением: после перорального введения CP-144.477 в канюлированную воротную вену печени собак, концентрация общего количества пролекарства в воротном и системном кровообращении были ниже порога определения, а системная концентрация CP-72.133 была такой же, как и системная концентрация, полученная после перорального введения CP-72.133 неканюлированным собакам.
Ключевые фармакологические свойства
Первоначально все клеточные эксперименты in vitro показывали, что CP-144.477 имеет так же ингибиторную активность CO. Однако в условиях эксперимента образуется значительное количество CP-72.133, предположительно в результате клеточного гидролиза. В настоящее время проводятся дополнительные эксперименты в среде, не содержащей клеток. В соответствии с фармакокинетическим профилем соединение CP-144.477 показывает, как и ожидается, такой же фармакологический профиль in vivo по отношению к CP-72.133 на модели RFE. Таким образом, ED5- CP-144.477 и CP-72.133 для ингибирования опухания ног составляет 5,5 мг/кг в обоих случаях, а ED50 для ex vivo ингибирования генерации TxB2 составляет менее 0,3 мг/кг в обоих случаях.
А. Активность CP-144.477 in vitro
В клеточной культуре CP-144.477 ингибирует образование простагландина D2 при половине максимальной концентрации, равной концентрации самого CP-72.133 (IC50, 0,02 мкм). Кроме того, CP-144.477 блокирует выработку 5-HETE и LTB4 с такой же эффективностью, как и CP-72.133 (IC50, 1,3 мкм, данные не показаны). Поэтому эти два вещества производят очевидно одинаковое воздействие на метаболизм арахидоновой кислоты с помощью 5-липоксигеназы и циклооксигеназы. Эти эксперименты проводились при таком pH, когда было известно, что CP-144.477 сохраняет стабильность на протяжении периода проведения эксперимента. Однако повторный анализ CP-144.477 после окончания эксперимента показал, что более 40% лекарственного средства в среде присутствовало в качестве соединения, которое хроматографировалось совместно с CP-72.133. Это позволяет предположить, что CP-144.477 конвертируется в CP-72.133 с помощью клеток, и, далее, что результаты, представленные здесь, отражают эту конверсию. Поэтому в настоящее время проводятся эксперименты по определению действия CP-144.477 в качестве ингибитора циклооксигеназы при бесклеточном получении в присутствии ингибиторов протеазы. Предполагается, что эти эксперименты подтвердят тот факт, что CP-144.477 неактивен в качестве ингибитора CO, и его обезболивающее и противоотечное действие как таковое будет объяснено эффективной конверсией в CP-72.133 in vitro.
B. Влияние CP-72.133 и CP-144.477 на отек ног у крыс (RFE)
Инъекция каррагенана в подошву стопы вызывает острую опухолевую реакцию, сопровождающуюся отеком, достигающим своего пика через 3-6 ч после инъекции. Активность на этой модели очень хорошо показывает, каким будет противовоспалительное действие у людей. Фармакологическая активность CP-144.477 и CP-72.133 сравнивалась на модели RFE, при этом измерялось ингибирование CO. Таким образом, у одних и тех же животных определялись ингибирование опухания ног и выработка ex vivo тромбоксана B2 (TxB2) во всей свертывающейся крови. В зависимости от дозы CP-72.133 и CP-144.477 (0,3-10 мг/кг) ингибировали вызванное каррагенаном опухание лап (ED50 5,5 мг/кг и 5,5 мг/кг, соответственно). Оба соединения также ингибировали выработку TxB2 в свертываемой крови, при этом величина ED50 составляла менее 0,3 мг/кг (см. Таблицу А).
Общая фармакология
Результаты испытания in vivo, суммированные ниже, показывают, что острое введение CP-144.477 хорошо переносится без неблагоприятных последствий собаками при дозе 35,4 мг/кг и крысами при дозах 103 мг/кг. Мыши оказались несколько более чувствительными к CP-144.477; при 100 мг/кг наблюдались случаи смерти. Результаты испытаний in vivo показывают, что при высокой концентрации CP-144.477 оказывает некоторое воздействие на подвздошную кишку морских свинок и матку крыс. Однако вышеуказанные данные совпадают с полученными ранее результатами по CP-72.1334 и показывают, что от CP-144.477 не ожидается никакого неожиданного клинического действия.
Определялось воздействие CP-144.477 на различные системы органов in vivo и in vitro. С помощью метаболизма лекарственного средства было установлено, что исходное соединение не обнаруживается в плазме после перорального введения, благодаря полной конверсии в CP-72.133. Таким образом, используемые экспериментальные дозы 10.3, 51.6 и 103 мг/кг CP-144.477, выбранные для испытаний, являлись свободными от оснований эквивалентами 8, 40 и 80 кг/кг (например, ED50, 5xED50 и 10xED50 для CP-72.133 в испытании по отеку ног у крыс), использованными в предыдущих общих фармакологических испытаниях CP-72.1334. Подобным образом для проведения прямого сравнения между пролекарствами и CP-72.133 в сердечно-сосудистых испытаниях для собак использовалась доза 35.4 мг/кг CP-144.477 (25 мг/кг CP-72.133 - доза используемая ранее).
Название и структура
CP-88.144 представляет собой 5-хлор-2,3-дигидро-3[гидрокси-2-(4-хлортиенил)метилен] -2-оксо- 1H-индол-1-карбоксамид. Присвоенное название отражает преобладание таутомера енола, как определено методами спектроскопии. Молекулярная формула C14H8Cl2O3S определяет молекулярный вес этого соединения - 355,19.
Отек ног у крыс
Инъекции каррагенана в подошву стопы вызывает острую опухолевую реакцию, сопровождающуся отеком, достигающим своего пика через 3-6 ч после инъекции. Активность на этой модели очень хорошо показывает, каким будет противовоспалительное действие у людей.
Как показано в Таблице B, ED50 для CP-88.144 в этой процедуре составляет 4,2 мг/кг, т.е. данное соединение в 5 раз сильнее тенидапа на основании сравнения. Не удивительно, что уровень CP-88.144 в плазме через 4 ч, дозированный на уровне ED50 его RFE, значительно ниже соответствующей величины для тенидапа (1,44±0,76 против 44,8±24,8 мкг/мл). Эти данные согласуются с более высокой активностью CP-88.144, ингибирующей CO in vitro. Подобное сравнение показало, что CP-72.133 в 2,5 раза эффективнее тенидапа в испытании RFE, но это различие не является существенным.
До настоящего времени не проводилось одновременных испытаний CP-88.144 и напроксана или ибупрофена, однако ранее проведенные испытания показали, что тенидап в 4-5 раз эффективнее этих соединений, соответственно. Таким образом, ожидается, что будущие испытания модели RFE покажут, что CP-88.144 эффективнее напроксена и ибупрофена.
Исходя из молярной основы, пролекарства изобретения обычно дозируются на том же уровне и с той же кратностью приема, что и известные 3-ацил-2-оксиндол-1-карбоксамиды, производными которых они являются. Однако неенольная природа настоящих соединений должна в общем случае допускать более высокие переносимые оральные дозы, когда такие высокие дозы необходимы для достижения эффекта обезболивания и лечения воспаления.
Пролекарства изобретения рецептурируются и вводятся аналогично известным исходным соединениям по методикам, которые описаны в процитированных выше работах. Предпочтительным способом введения является оральный, что, таким образом, обеспечивает реализацию преимущества не-енольной природы рассматриваемых соединений.
Пример 1. 6-Хлор-5-фтор-2,3-дигидро-3-[(4-метоксибензоил)окси-(2-тиенил) метилен]-2-оксо-1Н-индол-1-карбоксамид.
Суспендируют 3-[гидрокси-(2-тиенил)метилен] -6-хлор-5-фтор-2,3- дигидро-2-оксо-1Н-индол-1-карбоксамид (5,08 г, 15,0 ммоль) в метиленхлориде и обрабатывают триэтиламином (1,67 г, 165 ммоль). Полученный желтый раствор охлаждают на ледяной бане и обрабатывают 4-метоксибензоилхлоридом (12,8 г, 75,0 ммоль) в одну порцию. Через 18 ч реакционную смесь фильтруют, отделяя желтый осадок. Фильтрат разбавляют дополнительным количеством метиленхлорида, промывают дважды 1 н. соляной кислотой, затем смесью насыщенный раствор NaHCO3/рассол, после чего сушат MgSO4, фильтруют, упаривают, дважды обрабатывают этанолом, полученное твердое вещество растирают со смесью этилацетат/гексан. Продукт объединяют с твердым веществом, выделенным непосредственно из реакционной массы, и перекристаллизовывают из смеси этилацетат/гексан (4: 1) с небольшим количеством ацетона, получают 1,81 г (выход 26%) требуемого продукта в виде желтых кристаллов: т.пл. 220 - 221oC.
Элементный анализ:
Вычислено, %: C 55,88; H 2,98; N 5,92.
C22H14ClFN2O5S.
Найдено, %: C 56,04; H 2,82; N 5,88.
Пример 2. 6-Хлор-5-фтор-2,3-дигидро-3-[(циннамоил)окси-(2-тиенил)метилен]-2- оксо-1Н-индол-1-карбоксамид.
Названное соединение получают по методике примера 1, за исключением того, что используется циннамоилхлорид: т.пл. 214 - 215oC.
Элементный анализ:
Вычислено, %: C 58,91; H 3,01; N 5,97.
C23H14ClFN2O4S.
Найдено, %: C 58,52; H 2,89; N 5,91.
Пример 3. 6-Хлор-5-фтор-2,3-дигидро-3-[(3-метоксибензоил)окси- (2-тиенил)метилен]-2-оксо-1Н-индол-1-карбоксамид.
Названное соединение получают по методике примера 1, за исключением того, что используется 3-метоксибензоилхлорид (4 экв.) и хлороформ в качестве растворителя. Продукт перекристаллизовывают из изопропиловвого спирта: т. пл. 196 - 220oC. Данные спектра 1H ЯМР указывают на то, что продукт содержит как E-, так и Z-изомеры названного соединения в соотношении 17:83.
Элементный анализ:
Вычислено, %: C 55,88; H 2,98; N 5,92.
C22H14ClFN2O5S.
Найдено, %: C 56,00; H 2,82; N 5,78.
Пример 4. 6-Хлор-5-фтор-2,3-дигидро-3-[(2-метоксибензоил)окси- (2-тиенил)метилен]-2-оксо-1Н-индол-1-карбоксамид.
Названное соединение получают по методике примера 1, за исключением того, что используется 2-меткосибензоилхлорид и хлороформ в качестве растворителя. Сырой продукт очищают быстрой хроматографией на силикагеле (элюент хлороформ/метанол, 95:5), после чего перекристаллизовывают из изопропилового спирта: т.пл. 219 - 221oC. Данные спектра 1H ЯМР указывают на то, что продукт содержит только E-изомер названного соединения.
Элементный анализ:
Вычислено, %: C 55,88; H 2,98; N 5,92.
C22H14ClFN2O5S.
Найдено, %: C 55,32; H 3,01; N 5,66.
Пример 5. 6-Хлор-5-фтор-2,3-дигидро-3-[никотиноилокси-(2-тиенил) метилен]-2-оксо-1Н-индол-1-карбоксамид.
Названное соединение получают по методике примера 1, за исключением того, что используется никотиноилхлорид (2,5 экв.) и 3,4 экв. триэтиламина, а также хлороформ в качестве растворителя. Сырой продукт очищают быстрой хроматографией (элюент хлороформ/метанол 83:17), после чего перекристаллизовывают из изопропилового спирта: т.пл. 201 - 202,5oC.
Элементный анализ:
Вычислено, %: C 54,12; H 2,50; N 9,47.
C20H11ClFN3O4S.
Найдено, %: C 54,01; H 2,46; N 9,30.
Пример 6. 6-Хлор-5-фтор-2,3-дигидро-3-[изоникотиноилокси-(2-тиенил)метилен]-2- оксо-1Н-индол-1-карбоксамид
Названное соединение получают по методике примера 1, за исключением того, что используется изоникотиноилхлорид (1,1 экв.) и диизопропилэтиламин (2 экв.). Продукт, полученный непосредственно фильтрацией реакционной смеси, перекристаллизовывают из изопропилового спирта: т.пл. 219,5 - 221oC.
Элементный анализ:
Вычислено, %: C 54,12; H 2,50; N 9,47.
C20H11ClFN3O4S.
Найдено, %: C 54,11; H 2,43; N 9,32.
Пример 7. 6-Хлор-5-фтор-2,3-дигидро-3-[пиколиноилокси-(2-тиенил) метилен]-2-оксо-1Н-индол-1-карбоксамид
Названное соединение получают по методике примера 1, за исключением того, что используются пиколиноилхлорид (1,1 экв.) и диизопропилэтиламин (2 экв. ). Продукт, полученный непосредственно фильтрацией реакционной смеси, перекристаллизовывают из изопропилового спирта: т.пл. 184 - 185oC.
Элементынй анализ:
Вычислено, %: C 54,12; H 2,50; N 9,47.
C20H11ClFN3O4S.
Найдено, %: C 53,73; H 2,34; N 9,31.
Пример 8. 6-Хлор-5-фтор-2,3-дигидро-3-[3-этилоксикарбонил)пропеноилокси- (2-тиенил)метилен]-2-оксо-1Н-индол-1-карбоксамид.
a) 3-(Этилоксикарбонил)пропеноилхлорид получают по методике, описанной Lutz (J.Am. Chem. Soc., 1930, 52, 3430).
b) Названное соединение получают по методике примера 1, за исключением того, что используется 3-(этилоксикарбонил)пропеноилхлорид (2 экв.) и диизопропилэтиламин. Сырой продукт очищают быстрой хроматографией (элюент хлороформ/метанол, 98:2), после чего перекристаллизовывают из изопропанола; т. пл. : 165,5 - 168oC. Данные спектра 1H ЯМР указывают на то, что продукт содержит как E-, так и Z-изомеры названного соединения в соотношении 22:78.
Элементный анализ:
Вычислено, %: C 51,68; H 3,04; N 6,03.
C20H14ClFN2O6S.
Найдено, %: C 51,42; H 3,08; N 5,86.
Пример 9. 6-Хлор-5-фтор-2,3-дигидро-3-[3-метилоксикарбонил) пропеноилокси-(2-тиенил)метилен]-2-оксо-1Н-индол-1-карбоксамид.
a) 3-(Метилоксикарбонил)пропеноилхлорид получают по методике, описанной Lutz (J.Am. Chem. Soc., 1930, 52, 3430).
b) Названное соединение получают по методике примера 1, за исключением того, что используются 3-(метилоксикарбонил)пропеноилхлорид (2 экв.) и диизопропилэтиламин. Сырой продукт очищают быстрой хроматографией (элюент CH2Cl2 / изопропанол, 95:5), после чего перекристаллизовывают из изопропанола: т.пл. 183,5 - 186oC. Данные спектра 1H ЯМР указывают на то, что продукт содержит как E-, так и Z-изомеры названного соединения в соотношении 34: 66.
Элементный анализ:
Вычислено, %: C 50,62; H 2,68; N 6,21.
C19H12ClFN2O6S.
Найдено, %: C 50,56; H 2,84; N 6,04.
Пример 10. 6-Хлор-5-фтор-2,3-дигидро-3-[бензоилоксиацетокси-(2-тиенил)метилен]- 2-оксо-1Н-индол-1-карбоксамид.
Названное соединение получают по методике примера 1, за исключением того, что используются бензилоксиацетилхлорид (2 экв.) и диизопропилэтиламин (2 экв. ). Сырой продукт очищают быстрой хроматографией (элюент хлороформ), после чего перекристаллизовывают из изопропилового спирта: т.пл. 150 - 175oC.
Элементный анализ:
Вычислено, %: C 56,74; H 3,31; N 5,75.
C23H16ClFN2O5S.
Найдено, %: C 58,41; H 3,17; N 5,00.
Пример 11. 6-Хлор-5-фтор-2,3-дигидро-3-[4-(бензилоксикарбонил) бензоилокси-(2-тиенил)метилен]-2-оксо-1Н-индол-1-карбоксамид.
Названное соединение получают по методике примера 1, за исключением того, что используются 4-(бензилоксикарбонил)бензоилхлорид (1,5 экв.) и диизопропилэтиламин (1,5 экв.). Сырой продукт очищают растиранием с хлороформом: т.пл. 212 - 218oC.
Элементный анализ:
Вычислено, %: C 60,37; H 3,14; N 4,85.
C29H18ClFN2O6S.
Найдено, %: C 60,94; H 3,00; N 4,27.
Пример 12. 6-Хлор-5-фтор-2,3-дигидро-3-[3-карбоксипропеноилокси- (2-тиенил)метилен]-2-оксо-1Н-индол-1-карбоксамид.
a) 3-(2-Триметилсилилэтоксикарбонил)пропеноилхлорид получают по модифицированной методике Lutz (J. Am. Chem. Soc., 1930, 52, 3430), в соответствии с которой непрореагировавший фумароилхлорид и ненужный диэфир отделяются с помощью дистилляции в вакууме, а требуемый продукт остается в кубе.
b) Названное соединение получают по методике примера 1, за исключением того, что используются 3-(2-триметилсилилэтилоксикарбонил)пропеноилхлорид (1,3 экв.) и диизопропилэтиламин. Сырой продукт очищают быстрой хроматографией (элюент метиленхлорид; метиленхлорид/метанол, 99:1; а затем метиленхлорид/метанол, 98:2), после чего перекристаллизовывают из ацетонитрила.
c) Комплекс фтористый водород/пиридин (500 г) охлаждают на ледяной бане в полиэтиленовой бутылке, затем в 2-3 порции добавляют 3-(2-триметилсилилэтоксикарбонил)пропеноильное производное (33,84 г, 63 ммоль). После окончания добавления полученную суспензию перемешивают при охлаждении в течение 0,5 ч. Затем реакцию останавливают путем добавления воды. После сушки в вакууме полученный продукт очищают при растирании с горячим этилацетатом. Выход составляет 19,8 г. Аналитический образец получают перекристаллизацией небольшого образца из уксусной кислоты: т.пл. 229 - 232oC. Данные спектра 1H ЯМР указывают на то, что продукт содержит только E-изомер названного соединения.
Элементный анализ:
Вычислено, %: C 49,50; H 2,31; N 6,41.
C18H10ClFN2O6S.
Найдено, %: C 49,23; H 2,23; N 6,37.
Пример 13. 6-Хлор-5-фтор-2,3-дигидро-3-[3-карбоксибензоилокси-(2-тиенил)метилен] -2-оксо-1Н-индол-1-карбоксамид.
f) Раствор изофталоилдихлорида (40,6 г, 200 ммоль) и 2,2,2-трихлорэтанола (30,9 г, 207 ммоль) в толуоле кипятят с обратным холодильником в течение 24 ч. Растворитель упаривают и полученный остаток перегоняют в вакууме. Целевой продукт перегоняется вместе небольшим количеством нежелательного бис(2,2,2-трихлорэтилового)эфира при 125 - 170oC при 0,2 мм рт.ст. Через некоторое время нецелевой продукт отделяется в виде твердого осадка. Целевой 3-(2,2,2-трихлорэтилоксикарбонил)бензоилхлорид (19,0 г) декантируют и используют на следующей стадии.
b) Указанное в названии соединение готовят по методике примера 1 за исключением того, что используются 3-(2,2,2-трихлорэтилоксикарбонил)бензоилхлорид (2 экв. ) и диизопропилэтиламин (2 экв.). Продукт, полученный при фильтровании реакционной смеси, растирают с хлороформом.
c) Раствор 3-(2,2,2-трихлорэтилоксикарбонил)бензоилпропилпроизводного (1,0 г, 1,6 ммоль) в уксусной кислоте (50 мл) обрабатывают порошком цинка 1,0 г, 16 ммоль). Полученную смесь выдерживают на масляной бане при 50oC в течение 18 ч. Нагретую смесь фильтруют (промывая уксусной кислотой) и после охлаждения до комнатной температуры выливают фильтрат в воду (250 мл). 6-Хлор-5-фтор-2,3-дигидро-3-[3-карбоксибензоилокси-(2-тиенил)метилен] -2-оксо-1Н-индол-1-карбоксамид, выделившийся в виде твердого желтого осадка, отфильтровывают и перекристаллизовывают из уксусной кислоты: т.пл. 244 - 248oC. Выход составляет 130 мг.
Элементный анализ:
Вычислено, %: C 50,40; H 3,12; N 6,19.
C19H14ClFN2O6S.
Найдено, %: C 51,21; H 2,96; N 5,99.
Пример 14. 6-Хлор-5-фтор-2,3-дигидро-3-[3-(бутоксикарбонил) пропеноилокси-(2-тиенил)метилен]-2-оксо-1Н-индол-1-карбоксамид.
a) 3-(Бутоксикарбонил)пропеноилхлорид готовят в соответствии с методикой, описанной Lutz (J.Am. Chem. Soc., 1930, 52, 3430).
b) Указанное в названии примера соединение готовят по методике примера 1 за исключением того, что используют 3-(бутоксикарбонил)пропеноилхлорид (2 экв. ) и диизопропилэтиламин. Сырой продукт очищают быстрой хроматографией (используя в качестве элюента метиленхлорид, метиленхлорид-метанол 95:5, а затем метиленхлорид-метанол 90: 10) с последующей перекристаллизацией из ацетонитрила: т.пл. 196 - 197oC. Данные спектра 1H ЯМР указывают на то, что продукт содержит только E-изомер названного соединения.
Элементный анализ:
Вычислено, %:C 53,61; H 3,68; N 5,68.
C22H18ClFN2O6S.
Найдено, %: C 53,53; H 3,55; N 5,78.
Пример 15. 6-Хлор-5-фтор-2,3-дигидро-3-[3-(октилоксикарбонил) пропеноилокси-(2-тиенил)метилен]-2-оксо-1Н-индол-1-карбоксамид.
a) 3-(Октилоксикарбонил)пропеноилхлорид готовят в соответствии с методикой, описанной Lutz (J. Am. Chem. Soc., 1930, 52, 3430).
b) Указанное в названии примера соединение готовят по методике примера 1 за исключением того, что используют 3-(октилоксикарбонил)пропеноилхлорид (2 экв. ) и диизопропилэтиламин. Сырой продукт очищают быстрой хроматографией (используя в качестве элюента метиленхлорид-метанол 99:1) с последующей перекристаллизацией из ацетонитрила: т.пл. 140 - 148oC. Данные спектра 1H ЯМР указывают на то, что продукт содержит как E-, так и Z-изомеры названного соединения в соотношении 33:67.
Элементный анализ:
Вычислено, %: C 56,88; H 4,77; N 5,10.
C26H26ClFN2O6S.
Найдено, %: C 56,81; H 4,62; N 5,07.
Пример 16. 6-Хлор-5-фтор-2,3-дигидро-3-[3-(4-фенилбутилоксикарбонил)пропеноилокси- (2-тиенил)метилен]-2-оксо-1Н-индол-1-карбоксамид.
a) 3-(4-Фенилбутилоксикарбонил)пропеноилхлорид готовят в соответствии с методикой, описанной Lutz (J.Am.Chem. Soc., 1930, 52, 3430).
b) Указанное в названии примера соединение готовят по методике примера 1 за исключением того, что используют 3-(4-фенилбутилоксикарбонил)пропеноилхлорид (2 экв. ) и диизопропилэтиламин. Сырой продукт очищают быстрой хроматографией (используя в качестве элюента метиленхлорид-метанол, 98: 2) с последующей перекристаллизацией из ацетонитрила: Т.пл. 159 - 161oC. Данные спектра 1H ЯМР указывают на то, что продукт содержит как E-, так и Z-изомеры названного соединения в соотношении 27:73.
Элементный анализ:
Вычислено, %: C 59,10; H 3,90; N 4,92.
C28H22ClFN2O6S.
Найдено, %: C 58,89; H 3,71; N 4,72.
Пример 17. 6-Хлор-5-фтор-2,3-дигидро-3-[октаноилокси-(2- тиенил)метилен] -2-оксо-1Н-индол-1-карбоксамид.
Указанное в названии примера соединение готовят по методике примера 1 за исключением того, что используют октаноилхлорид (2 экв.) и диизопропилэтиламин в качестве основания. Сырой продукт очищают быстрой хроматографией на силикагеле (используя в качестве элюента метиленхлорид) с последующей перекристаллизацией из ацетонитрила: т.пл. 149 - 150oC. Данные спектра 1H ЯМР указывают на то, что продукт содержит только Z-изомер названного соединения.
Элементный анализ:
Вычислено, %: C 56,83; H 4,77; N 6,03.
C22H22ClFN2O4S.
Найдено, %: C 56,85; H 4,49; N 6,03.
Пример 18. 6-Хлор-5-фтор-2,3-дигидро-3-[3-(1- метилпропилоксикарбонил)пропеноилокси-(2-тиенил)метилен]-2- оксо-1Н-индол-1-карбоксамид
a) 3-(1-Метилпропилоксикарбонил)пропеноилхлорид готовят в соответствии с методикой, описанной Lutz (J.Am.Chem. Soc., 1930, 52, 3430).
b) Указанное в названии примера соединение готовят по методике примера 1 за исключением того, что используют 3-(1-метилпропилоксикарбонил)пропеноилхлорид (1,2 экв.) и диизопропилэтиламин. Сырой продукт очищают быстрой хроматографией (используя в качестве элюента метиленхлорид) с последующей перекристаллизацией из ацетонитрила: т.пл. 180 - 186oC. Данные спектра 1Н ЯМР указывают на то, что продукт содержит как E-, так и Z-изомеры названного соединения в соотношении 60:40.
Элементный анализ:
Вычислено, %: C 53,61; H 3,68; N 5,68.
C22H18ClFN2O6S.
Найдено, %: C 53,60; H 3,57; N 5,67.
Пример 19. 6-Хлор-5-фтор-2,3-дигидро-3-[3-(метилоксикарбонил) пропаноилокси-(2-тиенил)метилен]-2-оксо-1H-индол-1-карбоксамид
Указанное в названии примера соединение готовят по методике примера 1 за исключением того, что используют 3-(метилоксикарбонил)пропаноилхлорид (2 экв. ) и диизопропилэтиламин. Сырой продукт очищают быстрой хроматографией (используя в качестве элюента метиленхлорид-метанол, 96:4) с последующей перекристаллизацией из толуола: т. пл. 179 - 183oC. Данные спектра 1H ЯМР указывают на то, что продукт содержит как E-, так и Z-изомеры названного соединения в соотношении 31:69.
Элементный анализ:
Вычислено, %: C 54,27; H 2,48; N 5,75.
C22H12ClFN2O6S.
Найдено, %: C 53,80; H 2,18; N 5,71.
Пример 20. 6-Хлор-5-фтор-2,3-дигидро-3-[3-(этилоксикарбонил) пропаноилокси-(2-тиенил)метилен]-2-оксо-1H-индол-1-карбоксамид
Указанное в названии примера соединение готовят по методике примера 1 за исключением того, что используют 3-(этилоксикарбонил)пропаноилхлорид (2 экв. ) и диизопропилэтиламин (2,2 экв.). Сырой продукт очищают быстрой хроматографией (используя в качестве элюента метиленхлорид-метанол, 97,5:2,5) с последующим растиранием с метиленхлоридом и перекристаллизацией из ацетонитрила: т.пл. 182 - 183oC. Данные спектра 1H ЯМР указывают на то, что продукт содержит только E-изомер названного соединения.
Элементный анализ:
Вычислено, %: C 51,45; H 3,45; N 6,00.
C20H16ClFN2O6S.
Найдено, %: C 51,38; H 3,27; N 5,97.
Пример 21. 6-Хлор-5-фтор-2,3-дигидро-3-[3-[(N,N- диэтилкарбоксамидометил)оксикарбонил] пропаноилокси-(2-тиенил)метилен] - 2-оксо-1H-индол-1-карбоксамид
a) Раствор янтарного ангидрида (16,0 г, 160 ммоль) и бензилового спирта (17,1 г, 158 ммоль) в толуоле (200 мл) кипятят с обратным холодильником в течение 3,5 ч. Затем в вакууме отгоняют растворитель и выделяют 3-(бензилоксикарбонил)пропановую кислоту в виде белого твердого продукта.
b) Раствор 3-(бензилоксикарбонил)пропановой кислоты (15,0 г, 72 ммоль), 2-хлор-N,N-диэтилацетамида (11,9 г, 79,5 ммоль), триэтиламина (11,2 мл, 80,4 ммоль) и иодида натрия (1,1 г, 7,4 ммоль) в этилацетате (280 мл) кипятят с обратным холодильником в течение 3 ч. Полученную смесь фильтруют для удаления хлоргидрида триэтиламина, который промывают большим количеством этилацетата. Фильтрат промывают 1 н. раствором соляной кислоты, насыщенным раствором NaHCO3 и рассолом. После сушки и фильтрования растворитель упаривают в вакууме и выделяют бензиловый эфир 3-[(N,N-диэтилкарбоксамидометил)оксикарбонил] пропановой кислоты в виде вязкого масла (22,2 г).
c) К раствору бензилового эфира 3-[(N,N-диэтилкарбоксамидометил) оксикарбонил] пропановой кислоты (22,2 г, 62,9 ммоль) в этаноле (250 мл) добавляют 10% палладия на угле (1,0 г). Смесь встряхивают в атмосфере водорода (3 атм) при 25oC в течение 18 ч. После удаления катализатора путем фильтрации через диатомовую землю растворитель упаривают и получают 3-[(N,N-диэтилкарбоксамидометил) оксикарбонил] пропановую кислоту в виде белого твердого продукта (10,47 г).
d) Раствор 3-[(N,N-диэтилкарбоксамидометил)оксикарбонил]-пропановой кислоты (5,0 г, 21,6 ммоль) и оксалилхлорида (2,0 мл, 23,5 ммоль) в бензоле (100 мл) кипятят в течение 1 ч. Растворитель упаривают в вакууме и получают 3-[(N,N-диэтилкарбоксамидометил)оксикарбонил] пропаноилхлорид в виде масла.
e) Указанное в названии соединение готовят по методике примера 1 за исключением того, что используют 3-[(N,N-диэтилкарбоксамидометил)оксикарбонил] пропаноилхлорид (2,2 экв.) и диизопропилэтиламин (2,2 экв.). Сырой продукт очищают быстрой хроматографией (используя в качестве элюента метиленхлорид) а затем метиленхлорид/метанол, 97,5:2,5) с последующим растиранием со смесью эфир/метиленхлорид, 1: 1) и перекристаллизацией из толуола: т.пл. 165 - 167oC. Данные спектра 1H ЯМР указывают на то, что продукт содержит только E-изомер названного соединения.
Элементный анализ:
Вычислено, %: C 52,22; H 4,20; N 7,61.
C24H23ClFN3O7S.
Найдено, %: C 52,17; H 4,06; N 7,50.
Пример 22. 5-Хлор-5-фтор-2,3-дигидро-3-[3-карбоксипропеноилокси- (2-тиенил)метилен]-2-оксо-1H-индол-1-карбоксамид.
a) 3-(2-Триметилсилилэтилоксикарбонил)пропеноилхлорид готовят по модифицированной методике, описанной Lutz (J. Am.Chem.Soc., 1930, 52, 3430), в соответствии с которой непрорегаровавший фумароилхлорид и ненужный диэфир отделяются с помощью дистилляции в вакууме, а требуемый продукт остается в кубе.
b) Указанное в названии соединение получают по методике примера 1 за исключением того, что используют 3-(2-триметилсилилэтилоксикарбонил)пропеноилхлорид (1,3 экв. ), 5-хлор-2,3-дигидро-3-[гидрокси-(2-тиенил)метилен] -2-оксо-1H-индол- 1-карбоксамид и диизопропилэтиламин. Сырой продукт очищают быстрой хроматографией (элюент метиленхлорид/этилацетат, 98: 2) с последующей перекристаллизацией из ацетонитрила.
c) Комплекс фтористый водород/пиридин (20 мл) охлаждают на ледяной бане в полиэтиленовой бутылке. Затем добавляют 3-(2-триметилсилилэтилоксикарбонил)пропеноильное производное (0,89 г, 1,71 ммоль). Полученную суспензию перемешивают в течение 0,5 ч. Затем реакцию останавливают путем добавления воды. Продукт отфильтровывают, промывают водой и сушат в вакууме. Выход составляет 0,60 г, т.пл. 198 - 200oC. Данные спектра 1H ЯМР указывают на то, что продукт содержит только E-изомер названного соединения.
Элементный анализ:
Вычислено, %: C 51,62; H 2,65; N 6,69.
C18H11ClN2O6S.
Найдено, %: C 51,18; H 2,62; N 6,51.
Пример 23. 6-Хлор-5-фтор-2,3-дигидро-3-[3-карбоксипропеноилокси- (4-хлор-2-тиенил)метилен]-2-оксо-1H-индол-1-карбоксамид.
a) 3-(2-Триметилсилилэтилоксикарбонил)пропеноилхлорид готовят по модифицированной методике, описанной Lutz (J. Am.Chem.Soc., 1930, 52, 3430), в соответствии с которой непрореагировавший фумароилхлорид и ненужный диэфир отделяются с помощью дистилляции в вакууме, а требуемый продукт остается в кубе.
b) Указанное в названии соединение получают по методике примера 1 за исключением того, что используют 3-(2-триметилсилилэтилоксикарбонил)пропеноилхлорид (1,3 экв.), 6-хлор-5-фтор-2,3-дигидро-3-[гидрокси-(4-хлор-2-тиенил)метилен]- 2-оксо-1H-индол-1-карбоксамид и диизопропилэтиламин. Сырой продукт очищают быстрой хроматографией (элюент гексан/этилацетат, 60: 40) с последующей перекристаллизацией из ацетонитрила.
c) Комплекс фтористый водород/пиридин (20 мл) охлаждают на ледяной бане в полиэтиленовой бутылке. Затем добавляют 3-(2-триметилсилилэтилоксикарбонил)пропеноильное производное (0,82 г, 1,43 ммоль). Полученную суспензию перемешивают в течение 0,5 ч. Затем реакцию останавливают путем добавления воды. Продукт отфильтровывают, промывают водой и сушат в вакууме. Выход составляет 0,50 г, т.пл. 158oC. Данные спектра 1H ЯМР указывают на то, что продукт содержит только E-изомер названного соединения.
Элементный анализ:
Вычислено, %: C 45,88; H 1,92; N 5,94.
C18H9ClFN2O6S.
Найдено, %: C 45,24; H 1,98; N 5,84.
Пример 24. 6-Хлор-5-фтор-2,3-дигидро-3-[этоксикарбонилметоксиацетоксиметокси- (2-тиенил)метилен]-2-оксо-1H-индол-1-карбоксамид.
a) Раствор дигликолевого ангидрида (12 г, 0,103 моль) и этанола (4,74 г, 0,103 моль) в толуоле (100 мл) кипятят с обратным холодильником в течение 16 ч. Затем раствор охлаждают и упаривают, а полученный остаток перегоняют в вакууме. Продукт - моноэтиловый эфир дигликолевой кислоты (9,2 г, выход 55%) собирают при 109 - 135oC при давлении 5 мм рт.ст.
b) К раствору бисульфата тетрабутиламмония (24,6 г, 0,0725 моль) в воде (250 мл) добавляют порциями твердый NaHCO3 (12,2 г, 0,145 моль), а затем добавляют раствор моноэтилового эфира дигликолевой кислоты (11,75 г, 0,0725 моль). Полученную смесь перемешивают в течение 16 ч, а затем экстрагируют метиленхлоридом (3•500 мл). Объединенные органические экстракты сушат, фильтруют и упаривают. Получают соль (21,1 г, 72%), которую используют без дополнительной очистки.
c) Раствор тетрабутиламмониевой соли моноэтилового эфира дигликолевой кислоты (16 г, 0,04 моль) в метиленхлориде (125 мл) добавляют к бромхлорметану (200 мл) в течение 1,5 ч. Полученный раствор перемешивают при комнатной температуре в течение ночи и затем упаривают досуха. Остаток хроматографируют на силикагеле, используя в качестве элюента этилацетат/гексан, 40:60. Фракции, содержащие продукт, объединяют и упаривают, получают этилдихлорметиловый эфир дигликолевой кислоты (0,836 г, выход 10%) в виде бесцветного масла. Согласно данным ЯМР чистота продукта составляет 70%, но продукт используется без очистки на следующей стадии.
d) Хлорметиловый эфир (0,836 г, 3,98 ммоль) со стадии c) растворяют в ацетоне (10 мл) и затем добавляют иодид натрия (1,8 г, 11,9 ммоль) и полученную смесь перемешивают при комнатной температуре в течение 16 ч. Смесь фильтруют и фильтрат упаривают досуха, снова растворяют в метиленхлориде (25 мл) и затем промывают водой (10 мл), раствором тиосульфата натрия (10 мл) и насыщенным раствором хлористого натрия (10 мл). Органическую фазу отделяют, сушат и упаривают. Получают сырой этилиодметиловый эфир дигликолевой кислоты (1 г), который используют на конечной стадии.
e) К суспензии натрий-6-хлор-5-фтор-2,3-дигидро-3-[окси-(2- тиенил)метилен]-2-оксо-1H-индол-1-карбоксамида (1,2 г, 3,3 ммоль) в ацетоне (30 мл) в течение 5 мин добавляют раствор водметилового эфира со стадии d) в ацетоне (10 мл). Смесь перемешивают 16 ч при комнатной температуре. Полученную суспензию разбавляют ацетоном до получения раствора, добавляют двуокись кремния (2 г) и упаривают досуха. Полученную сухую суспензию двуокиси кремния помещают на хроматографическую колонку с двуокисью кремния и далее элюируют смесью этилацетат/гексан, 40:60. Фракции, содержащие продукт, объединяют и упаривают. Получают сырой продукт, который перекристаллизовывают из этанола. Получают названное соединение в виде желтого кристаллического продукта (136 г, т.пл. 129 - 130oC).
Пример 25. 6-Хлор-5-фтор-2,3-дигидро-3-[бензилоксикарбонилметокси- ацетоксиметокси-(2-тиенил)метилен]-2-оксо-1H-индол-1-карбоксамид.
Указанное соединение готовят по методике примера 24 за исключением того, что используют бензиловый спирт вместо этанола на стадии a) и получают монобензиловый эфир дигликолевой кислоты, который используют в остальных методиках. Названное соединение представляет собой желтый кристаллический продукт с т.пл. 122oC.
Пример 26. 6-Хлор-5-фтор-2,3-дигидро-3-[1- (бензилоксикарбонилметоксиацетокси)этокси-(2-тиенил)метилен]-2-оксо- 1H-индол-1-карбоксамид.
а) К раствору монобензилового эфира дигликолевой кислоты (7,28 г, 0,035 ммоль) в метиленхлориде (75 мл) добавляют тионилхлорид (42 г, 0,35 моль) и смесь перемешивают при комнатной температуре в течение 72 ч. Смесь упаривают досуха и сырой хлорангидрид кислоты используют без очистки на следующей стадии.
b) К смеси хлорангидрида кислоты (2,6 г, 0,011 моль) со стадии a) и каталитического количества расплавленного хлорида цинка добавляют ацетальдегид (0,96 г, 0,022 моль) и смесь перемешивают при комнатной температуре в течение 2 ч. Смесь затем упаривают досуха и остаток хроматографируют на SiO2, используя в качестве элюента смесь этилацетат/ гексан, 30 : 70. Фракции, содержащие продукт, объединяют и упаривают, получают бесцветное масло (1,1 г, выход 35%).
c) К суспензии натрий-6-хлор-5-фтор-2,3-дигидро-3-[окси- (2-тиенил)метилен] -2-оксо-1H-индол-1-карбоксамида (0,95 г, 2,62 ммоль) в ацетоне (20 мл) при перемешивании добавляют раствор хлорэтилового эфира со стадии b) в ацетоне (10 мл) в течение 5 мин. Затем добавляют иодид натрия (0,13 г, 0,87 ммоль) и смесь кипятят с обратным холодильником в течение 17 ч. Полученную суспензию охлаждают, разбавляют ацетоном до получения раствора, добавляют двуокись кремния (2 г) и смесь упаривают досуха. Полученную сухую суспензию двуокиси кремния помещают на хроматографическую колонку с двуокисью кремния и элюируют смесью этилацетат/гексан, 40 : 60. Фракции, содержащие продукт, объединяют и упаривают, получают сырой продукт. Перекристаллизацией из эфира получают названное соединение в виде желтого твердого кристаллического продукта (20 мг, т.пл. 89 - 91oC).
Пример 27. 6-Хлор-5-фтор-2,3-дигидро-3-[бензилоксикарбонилметоксиацетокси- (2-тиенил)метилен] -2-оксо-1H-индол-1-индол-1-карбоксамид.
К охлажденному до 0oC раствору 6-хлор-5-фтор-2,3-дигидро-3- [окси-(2-тиенил)метилен] -2-оксо-1H-индол-1-карбоксамида (1 г, 3 ммоль) и триэтиламина (0,33 г, 3,3 ммоль) в метиленхлориде (10 мл) при перемешивании добавляют раствор монобензилового эфира хлорангидрида дигликолевой кислоты (0,79 г, 3,3 ммоль) (см. пример 26, стадия a) в метиленхлориде (10 мл) в течение 5 мин. Смесь перемешивают при комнатной температуре в течение 2 ч, а затем абсорбируют на двуокись кремния и хроматографируют, используя в качестве элюента метиленхлорид/метанол, 15:1. Фракции, содержащие продукт, объединяют и упаривают, сырой продукт перекристаллизовывают из этилацетата, получают названное соединение в виде желтого кристаллического продукта (40 мг, т.пл. 189 - 190oC).
Пример 28. 6-Хлор-5-фтор-2,3-дигидро-3-[N,N-диэтилкарбамо илметоксиацетокси)-(2-тиенил)метилен]-2-оксо-1H-индол-2- карбоксамид
a) К раствору дигликолевого ангидрида (1 г, 8,6 ммоль) в толуоле (10 мл) при перемешивании добавляют диэтиламин (0,628 г, 8,6 ммоль) в течение 10 мин. Наблюдается небольшое выделение тепла. Полученную массу перемешивают при комнатной температуре в течение 16 ч. После упаривания растворителя получают бесцветное, вязкое масло (1,7 г, 100%).
b) К раствору N, N-диэтилкарбамоилметоксиуксусной кислоты (1,5 г, 7,3 ммоль) в метиленхлориде (15 мл) добавляют тионилхлорид (8,7 г, 73 ммоль) и смесь перемешивают при комнатной температуре в течение 16 ч. После упаривания растворителя получают сырой хлорангидрид кислоты, который непосредственно используют на следующей стадии.
c) К раствору 6-хлор-5-фтор-2,3-дигидро-3-[окси-(2-тиенил) метилен]-2-оксо-1H-индол-1-карбоксамида (2,7 г, 8,2 ммоль) и триэтиламина (1 г, 9,8 ммоль) в метиленхлориде (20 мл) добавляют при перемешивании раствор N,N-диэтилкарбамоилметоксиацетилхлорида (1,7 г, 8,2 ммоль) в метиленхлориде (10 мл) в течение 5 мин. Смесь перемешивают при комнатной температуре в течение 72 ч и затем абсорбируют на двуокись кремния и хроматографируют, используя в качестве элюента метиленхлорид/метанол, 25:1. Фракции, содержащие продукт, объединяют и упаривают. Сырой продукт перекристаллизовывают из ацетона, получают названное соединение в виде желтого кристаллического продукта (126 мг, т.пл. 205 - 208oC).
Пример 29. 6-Хлор-5-фтор-2,3-дигидро-3-[(2-(1,1- диметилэтоксикарбониламино)пропаноилокси)метокси-(2-тиенил) метилен] -2-оксо-1H-индол-1-карбоксамид.
Тетрабутиламмонийную соль (ТБА+) N-трет. - бутоксикарбонилаланина (50 ммоль) (полученную три титровании ТБА+-гидроксида в этаноле или при обработке эквивалентом ТБА+HSO
Хлорметиловый эфир N-трет.-бутоксикарбонилаланина (6,20 г, 26,1 ммоль) и иодида натрия (19,5 г, 130 ммоль) смешивают с ацетоном (150 мл) и перемешивают при комнатной температуре в течение 18 ч. Смесь затем фильтруют и фильтрат концентрируют, получают масло, которое растворяют в этилацетате и промывают 10%-ным раствором тиосульфата натрия и водой. После сушки сульфатом магния, фильтрации и упаривания получают 7,40 г (выход 86%) иодметилового эфира N-трет.-бутоксикарбонилаланина в виде масла.
Иодметиловый эфир N-трет.-бутоксикарбонилаланина (7,30 г, 22,2 ммоль) и натриевую соль 3-[гидрокси-(2-тиенил)метилен]-6-хлор-5- фтор-2,3-дигидро-2-оксо-1H-индол-1-карбоксамида (2,67 г, 7,4 ммоль) смешивают с ацетоном (75 мл) и кипятят с обратным холодильником в течение 6 ч. Реакционную массу упаривают и полученный остаток абсорбируют на силикагеле, загружают на колонку и хроматографируют (элюент - метиленхлорид, метанол/метиленхлорид, 5: 95). После объединения и упаривания соответствующих фракций получают сырой продукт, который перекристаллизовывают из смеси этилацетат/гексан. Получают 1,16 г (выход 29%) названного соединения: т.пл. 165 - 170oC.
Элементный анализ:
Вычислено, %: C 51,16; H 4,29; N 7,78.
C23H23ClFN3O7S.
Найдено, %: C 51,26; H 4,10; N 7,10.
Соединения примеров 30 - 40 синтезированы в соответствии с методикой примера 29.
Общая формула данных соединений имеет вид:
Физические свойства и значения R представлены в табл. 1.
Пример 41. N-трет.-Бутоксикарбонил-защищенные аминокислотные производные 3-[гидрокси-(2-тиенил)метилен] -6-хлор-5-фтор-2,3-дигидро-2- оксо-1H-индол-1-карбоксамида превращают в соответствующие незащищенные соединения двумя методами.
Метод A. N-трет.-Бутоксикарбонил-защищенное аминокислотное производное растворяют в охлажденной (баня лед-вода) трифротуксусной кислоте (ТФУ) для получения 0,1 M раствора и перемешивают при этой температуре в течение 1 ч. К полученному раствору добавляют один эквивалент моногидрата 4-толуолсульфокислоты (ТСК), после чего ТФУ упаривают в вакууме. Для более полного удаления ТФУ добавляют толуол и упаривают, повторяя эту операцию дважды. Твердый остаток перекристаллизовывают из смеси метанол/этилацетат, получают требуемое незащищенное соединение.
Метод B. N-трет.-Бутоксикарбонил-защищенное аминоксилотное производное растворяют в смеси диоксан-этилацетат 2:1 для получения 0,1 M раствора и насыщают его газообразным HCl при температуре не выше 10oC. Через 3 ч выпавший в осадок продукт отфильтровывают, промывают этилацетатом, перекристаллизовывают, промывают этилацетатом, перекристаллизовывают из смеси этилацетат/этанол, получают требуемое незащищенное соединение.
Соединения примеров 42 - 45-5 получены методом A или B примера 41 (см. табл. 2)
Пример 46. 6-Хлор-5-фтор-2,3-дигидро-3-[1-(4-фенилметоксикарбонил-бензоилокси)- 1-этокси-(2-тиенил)метилен]-2-оксо-1H-индол-1-карбоксамид.
Хлористый цинк (524 мг) плавят в вакууме и после охлаждения обрабатывают 4-(бензилоксикарбонил)бензоилхлоридом (21,48 г, 78,2 ммоль). Через 30 мин при 0oC добавляют ацетальдегид (3,44 г, 78 ммоль). Реакционную смесь перемешивают при комнатной температуре в течение 30 мин и разбавляют метиленхлоридом. Полученную смесь перемешивают еще 30 мин, а затем промывают три раза водой. Органический слой сушат сульфатом магния, упаривают в вакууме и получают серо-коричневый твердый продукт, который подвергают хроматографированию (элюент метиленхлорид/гексан, 25: 75) с получением 7,65 г (выход 31%) 1- хлорэтилового эфира 4-(бензилоксикарбонил) бензойной кислоты в виде белого твердого продукта: т.пл. 79 - 82oC.
Элементный анализ:
Вычислено, %: C 64,06; H 4,74.
C1H15ClO4.
Найдено, %: C 63,88; H 4,44.
Полученный хлорэтиловый эфир (4,00 г, 12,5 ммоль) смешивают с натриевой солью 3-[гидрокси-(2-тиенил)метилен] -6-хлор-5-фтор-2,3-дигидро-2-оксо-1H-индол- 1-карбоксамида (4,51 г, 12,5 ммоль) и иодидом натрия (644 мг, 4,3 ммоль) в ацетоне (40 мл) кипятят с обратным холодильником в течение 12 ч.
Реакционную смесь отфильтровывают и фильтрат упаривают в вакууме. Полученную коричнево-оранжевую смолу хроматографируют (элюент этилацетат/гексан, 25:75; гексан; этилацетат/метиленхлорид, 1:99), получают 1,947 г (выход 25%) оранжевой пены.
Элементный анализ:
Вычислено, %: C 58,81; H 3,72; N 4,43.
C31H22ClFN2O7S • 2/3 H2O.
Найдено, %: C 58,74; H 3,38; N 4,38.
Соединение примера 47 получено в соответствии с аналогичной последовательностью реакций (см.табл.3).
Пример 49. 3-[[[(5-бензилокси)глутарил]метилен]окси-(2-тиенил) метилен] -6-хлор-5-фтор-2,3-дигидро-2-оксо-1H-индол-1-карбоксамид.
Смесь 114 г (0,24 моль) тетрабутиламмонийбензилглутарата (A.R.English, D, Girard, V. J. Jasys, R. J. Martingano, M.S. Kellogg. J.Med.Chem., 1990, 33.344) и 600 мл бромхлорметана перемешивают при 0oC (ледяная баня) при медленном повышении температуры до комнатной в течение 16 ч. Избыток бромхлорметана упаривают в вакууме и полученный остаток растворяют в этилацетате, промывают последовательно водным раствором 1 н. соляной кислоты (2 • 1 л) и насыщенным раствором бикарбоната натрия (1 • 2 л), сушат сульфатом натрия и упаривают, получают 35 г масла. Смесь масла, 55,2 г (0,368 моль) иодида натрия и 150 мл ацетона перемешивают в течение ночи при комнатной температуре. Остаток распределяют между этилацетатом (500 мл) и водой (500 мл). Органический слой отделяют, промывают насыщенным водным раствором тиосульфата натрия (2 • 500 мл), сушат сульфатом натрия и упаривают. Получают 34,7 г желтого масла, которое очищают быстрой хроматографией (элюент этилацетат/гексан, 4:6). Выделяют 10,7 г (выход 32%) [[5-(бензилокси)глутарил]окси] метилиодида в виде масла (Rf-9,55, этилацетат/гексан, 1:1). Спектр ПМР (CDCl3), δ м.д.: 1,92 - 2,20 (2H, м), 2,41 (2H, т., J=7), 2,45 (2H, т, J=7), 5,16 (с, 2H), 5,88 (2H, с), 7,30-7,40 (5H, м).
Смесь 20,0 г (0,59 моль) 3-[гидрокси-(2-тиенил)метилен]-6-хлор-5-фтор-2,3-дигидро-2-оксо-1H-индол-1-карбоксамида, 590 мл (0,059 моль) 1 н. водного раствора гидроксида натрия и 300 мл метанола упаривают в вакууме. Остаток хорошо промывают эфиром и получают 20,5 г (выход 95%) желто-оранжевой натриевой соли.
Суспензию 10,1 г (0,028 моль) описанной выше соли, 10,0 г (0,028 моль) [[5-(бензилокси)глутарил)окси] метилиодида и 300 мл ацетона перемешивают в течение 16 ч при комнатной температуре. Смесь становится гомогенной, после чего ее упаривают в вакууме, а полученный остаток распределяют между 350 мл этилацетата и 350 мл насыщенного водного раствора тиосульфата натрия. Органический слой отделяют, промывают снова 350 мл насыщенного водного раствора тиосульфата натрия, сушат сульфатом магния и упаривают в вакууме. После очистки 15,6 мг полученного твердого продукта оранжевого цвета с помощью быстрой хроматографии (элюент этилацетат/гексан, 3:7) получают 1,8 г (выход 11%) названного соединения в виде твердого продукта желтого цвета (Rf-0,3, этилацетат/гексан, 1: 1). Спектр ПМР (CDCl3), δ м.д.: 1,90-2,06 (2H, м) 2,32-2,56 (4H, м), 5,12 (2H, с), 5,54 (1H, уш.с.), 5,72 (2H, с), 7,24 - 7,27 (1H, м), 7,28 - 7,41 (5H, м), 7,50-7,53 (H, м), 7,75-7,78 (2H,м), 8,37-8,41 (2H, м).
Пример 50. 6-Хлор-5-фтор-3-[[(глутарил)метилен]окси-(2-тиенил)метилен] -6-хлор-5-фтор- 2,3-дигидро-2-оксо-1H-индол-1-карбоксамид
Раствор 1,75 г 3-[[(5-бензилокси)глутарил]метилен]окси-(2-тиенил)метилен]-6-хлор-5-фтор- 2,3-дигидро-2-оксо-1H-индол-1-карбоксамида в 200 мл этилацетата гидрируют при давлении 45 фунтов/кв.дюйм (3,06 атм) в присутствии 900 мг Pd(OH)2 в течение 2,5 ч. Добавляют еще 900 мг катализатора, а затем через несколько часов еще раз добавляют 900 мг катализатора. Гидрирование продолжают в течение ночи. Катализатор отфильтровывают и фильтрат упаривают в вакууме. Получают 2,2 г оранжевого полутвердого продукта. Продукт очищают быстрой хроматографией (элюент метанол/хлороформ с соотношением от 2:98 до 10:90), собирая и упаривая фракции, содержащие соединение с Rf = 0,3 (элюент метанол/хлороформ, 1:9). Остаток растирают с эфиром, получают 66 мг (выход 4%) названного соединения в виде желтого твердого продукта: т.пл. 184 - 186oC. Спектр ПМР (DMCO-d6), δ м.д.: 1,62-1,80 (2H, м), 2,20 (2H, т, J = 7), 2,40 (2H, т, J = 7), 5,75 (2H, с), 7,30 (1H, т.J = 3), 7,65 (1H, д, J = 2), 7,76-7,85 (2H, м), 7,95 (1H, с), 8,05 (1H, д, J = 7), 8,25 (1H, д, J= 7), 8,32 (1H, с). Масс-спектр (m/e): 337 и 339, 295 и 297, 211 и 213 (осн.).
Пример 51. 6-Хлор-5-фтор-2,3-дигидро-3-[(2-фуроил)окси-(2-тиенил)метилен]-2-оксо- 1H-индол-1-карбоксамид.
К суспензии натриевой соли (z)-6-хлор-5-фтор-2,3-дигидро-3- (гидрокси-2-тиенилметилен)-2-оксо-1H-индол-1-карбоксамида (1,4 г, 3,88 ммоль) и иодида натрия (20 г, 0,12 ммоль) в 30 мл метиленхлорида при перемешивании добавляют 2-фуроилхлорид (380 мг, 3,88 ммоль). Полученную суспензию перемешивают в течение 20 ч при комнатной температуре. Суспензию отфильтровывают и промывают метиленхлоридом для удаления непрореагировавшей натриевой соли 6-хлор-5-фтор-2,3-дигидро-3-(гидрокси-2-тиенилметилен)-2-оксо-1H-индол-1- карбоксамида. Фильтрат упаривают в вакууме, получают твердый оранжевый продукт, который хроматографируют на колонке с силикагелем (100 г силикагеля, элюент метиленхлорид), получают названное соединение в виде неочищенного твердого продукта. Твердый продукт перекристаллизовывают из метиленхлорида и гексана и получают желтый твердый продукт, 210 мг (выход 12,9%): т.пл. 215 - 216oC.
Элементный анализ:
Вычислено, %: C 52,73; H 2,33; N 6,47.
C19H10ClFN2O5S.
Найдено, %: C 52,66; H 2,21; N 6,46.
Пример 52. 6-Хлор-5-фтор-2,3-дигидро-3-[(ацетоксиацетоил)окси-(2-тиенил)метилен]-2- оксо-1H-индол-1-карбоксамид.
К суспензии натриевой соли (z)-6-хлор-5-фтор-2,3-дигидро-3-(гидрокси-2-тиенилметилен)-2-оксо-1H-индол- 1-карбоксамида (2,0 г, 5,6 ммоль) и иодида натрия (200 мг, 1,33 ммоль) в 25 мл ацетона при перемешивании добавляют ацетоксиацетилхлорид (770 мг, 5,6 ммоль) и полученную смесь кипятят в течение 18 ч. Полученную суспензию охлаждают до комнатной температуры и фильтруют для удаления непрореагировавшей натриевой соли 6-хлор-5- фтор-2,3-дигидро-3-(гидрокси-2-тиенилметилен) -2-оксо-1H-индол-1-карбоксамида. Фильтрат упаривают в вакууме, получают твердый желтый продукт, который хроматографируют на колонке с силикагелем (75 г силикагеля, элюент гексан/этилацетат, 9:1), получают названное соединение в виде твердого продукта, который перекристаллизовывают из ацетонитрила и получают желтый твердый продукт, 500 мг (выход 20%): т.пл. 188 - 189oC.
Элементный анализ:
Вычислено, %: C 49,27; H 2,76; N 6,38.
C18H12ClFN2O6S.
Найдено, %: C 49,14; H 2,54; N 6,17.
Пример 53. 6-Хлор-5-фтор-2,3-дигидро-3-[(метоксиацетоил)окси- (2-тиенил)метилен]-2-оксо-1H-индол-1-карбоксамид.
Названное соединение получают, используя метоксиацетилхлорид, по методике, описанной в примере 52, за исключением того, что сырой продукт очищают хроматографией (форасил) с этилацетатом в качестве элюента. После кристаллизации из ацетонитрила получают желтый твердый продукт, 550 мг (выход 24,2%): т.пл. 199 - 200oC.
Элементный анализ:
Вычислено, %: H 49,70; H 2,94; N 6,82.
C17H12ClFN2O5S.
Найдено, %: C 49,51; H 2,66; N 6,92.
Пример 54. 6-Хлор-5-фтор-2,3-дигидро-3-[транс-(2-триметилсилиэтил-оксикарбонил)- 2-(циклобутаноил)окси-(2-тиенил)метилен] -2-оксо-1H-индол-1-карбоксамид.
К раствору 6-хлор-5-фтор-2,3-дигидро-3-(гидрокси-2-тиенилметилен- -2-оксо-1H-индол-1-карбоксамид (14,1 г, 0,0416 ммоль) в 120 мл метиленхлорида и диизопропилэтиламина (6,89 г, 0,053 моль) при перемешивании добавляют транс-1-(2-триметилсилилэтилоксикарбонил)-2-циклобутилкарбонилхлорид (14,0 г, 0,053 моль). После перемешивания в течение 18 ч при комнатной температуре отфильтровывают образующийся твердый продукт. Фильтрат упаривают в вакууме и получают твердый продукт желтого цвета. Твердый продукт хроматографируют на колонке с силикагелем, используя в качестве элюента гексан/этилацетат, 3:1. Получают сырой твердый продукт, после кристаллизации которого из ацетона получают желтый твердый продукт, 1,0 г (выход 4,3%): т.пл. 183 - 184oC.
Элементный анализ:
Вычислено,%: C 53,14; H 4,64; N 4,96.
C25H26ClFN2O6SSi.
Найдено,%: C 53,08; H 4,45; N 4,86.
Пример 55. 6-Хлор-5-фтор-2,3-дигидро-3-[транс-1-(2- триметилсилилэтилоксикарбонил)-2-циклопропаноил)окси-(2-тиенил) метилен] -2-оксо-1H-индол-1-карбоксамид, Z-изомер.
Названное соединение готовят с использованием подходящих исходных материалов по методике примера 54, за исключением того, что его отделяют на слое силикагеля (элюент гексан/этилацетат, 4:1), получают сырой продукт. После кристаллизации из хлороформа и гексана получают желтый твердый продукт, 140 мг (выход 3,7%): т.пл. 181 - 182oC.
Элементный анализ:
Вычислено,%: C 52,31; H 4,39; N 5,08.
C24H24ClFN2O6SSi.
Найдено,%: C 52,32; H 4,36; N 5,12.
Пример 56. 6-Хлор-5-фтор-2,3-дигидро-3-[транс-1-(2- триметилсилилэтилоксикарбонил)-2-(циклопропаноил)окси-2-(2-тиенил) метилен]-2-оксо-1H-индол-1-карбоксамид, E-изомер.
Названное соединение готовят с использованием подходящих исходных материалов по методике примера 54. После кристаллизации из хлороформа и гексана получают желтый твердый продукт, 160 мг (выход 4,2%): т.пл. 175 - 176oC.
Элементный анализ:
Вычислено,%: C 52,31; H 4,39; N 5,08.
C24H24ClFN2O6SSi.
Найдено,%: C 52,45; H 4,28; N 4,92.
Пример 57. 6-Хлор-5-фтор-2,3-дигидро-3-[(циклопентаноил) окси-2-(2-тиенил)метилен]-2-оксо-1H-индол-1-карбоксамид.
Названное соединение готовят с использованием подходящих исходных материалов по методике примера 54 за исключением того, что после того, что после хроматографирования на силикагеле (элюент гексан/этилацетат, 7:3), получают сырой продукт, который кристаллизуется из ацетата и гексана. Получают желтый твердый продукт, 260 мг (выход 3,3%): т.пл. 194oC (разл.).
Элементный анализ:
Вычислено,%: C 55,24; H 3,71; N 6,44.
C20H16ClFN2O4SSi.
Найдено,%: C 55,31; H 3,47; N 6,37.
Пример 58. 6-Хлор-5-фтор-2,3-дигидро-3-[(2,2,3,3- тетраметилциклопропаноил)окси-2-(2-тиенил)метилен]-2-оксо-1H-индол-1- карбоксамид.
Названное соединение готовят с использованием соответствующих материалов по методике примера 54, за исключением того, что после очистки на слое силикагеля (элюент гексан/этилацетат, 7: 3), получают сырой продукт, после кристаллизации которого из ацетона получают желтый твердый продукт, 230 мг (выход 2,5%): т.пл. 202oC.
Элементный анализ:
Вычислено,%: C 57,08; H 4,35; N 6,05.
C22H20ClFN2O4S.
Найдено,%: C 57,02; H 4,36; N 5,87.
Пример 59. {1-[(1-Карбамоил-6-хлор-5-фтор-2-оксо-1,2-дигидро-индол -3-илиден)-тиофен-2-илметокси] этоксикарбонилокси} уксусной кислоты метиловый эфир.
Тетрабутиламмониевую соль 3-[гидрокси-(2-тиенил)метилен] -6- хлор-5-фтор-2,3-дигидро-2-оксо-1H-индол-1-карбоксамида (2,98 г) и метиловый эфир (1-индэтокси)карбонилоксиуксусной кислоты (2,18 г) смешивают в ацетоне и полученный раствор перемешивают в течение 20 ч при 20oC. Растворитель упаривают в вакууме и остаток экстрагируют диэтиловым эфиром. Эфирный раствор упаривают в вакууме, получают частично кристаллизующееся при стоянии масло. После фильтрации от оставшегося масла полученный твердый продукт суспендируют в небольшом количестве диэтилового эфира и фильтруют. Получают 495 мг желтого твердого продукта. Дополнительно 295 мг сравнительно чистого названного продукта получают при хроматографировании на силикагеле (элюент хлороформ с 2% ацетона) объединенных эфирных остатков. Продукт идентифицируют с помощью спектров ПМР. Данные спектров ПМР для соединений примеров 59-70 представлены в табл. 4 после примера 70.
Пример 60. {1-[(1-Карбамоил-6-хлор-5-фтор-2-оксо-1,2-дигидро-индол-3-илиден)- тиофен-2-илметокси]этоксикарбонилокси}-N,N-диметилацетамид.
Тетрабутиламмониевую соль 3-[гидрокси-(2-тиенил)метилен]-6- хлор-5-фтор-2,3-дигидро-2-оксо-1H-индол-1-карбоксамида (6,25 г) и N,N-диметил-(1-иодэтокси)карбонилоксиацетамид (5,55 г) смешивают в 60 мл ацетона и полученный раствор перемешивают в течение 90 ч при 20oC. Осадок отфильтровывают и сушат в вакууме. Твердый остаток затем хроматографируют на силикагеле, элюент хлороформ/ацетон, 90:10. Фракции, содержащие названный продукт, смешивают и упаривают. Остаток суспендируют в небольшом количестве диэтилового эфира и фильтруют. Получают 820 мг желтого твердого продукта, т.пл. 194,5 - 195,5oC.
Пример 61. {1-[(2-Карбамоил-6-хлор-5-фтор-2-оксо-1,2-дигидро- индол-3-илиден)тиофен-2-илметокси]этоксикарбонилокси}-N,N- диэтилацетамид.
Тетрабутиламмониевую соль 3-[гидрокси-(2-тиенил)метилен] -6- хлор-5-фтор-2,3-дигидро-2-оксо-1H-индол-1-карбоксамида (13,85 г) и N,N-диэтил-(1-иодэтокси)карбонилоксиацетамид (13,6 г) смешивают в 100 мл ацетона и полученный раствор перемешивают в течение 42 ч при 20oC. Нерастворимые продукты отфильтровывают и перемешивают в хлороформе. Оставшиеся твердые продукты отфильтровывают, а хлороформ упаривают в вакууме. Остаток хроматографируют на силикагеле, элюент хлороформ/ацетон, 90:10. Фракции, содержащие названный продукт, смешивают и упаривают. Остаток суспендируют в небольшом количестве диэтилового эфира и фильтруют. Получают 2,37 г желтого твердого продукта: т. пл. 172 - 174oC.
Пример 62. 1-[2-Карбамоил-6-хлор-5-фтор-2-оксо-1,2-дигидро- индол-3-илиден)тиофен-2-илметокси]метиловый эфир N,N- диметиламида янтарной кислоты.
Тетрабутиламмониевую соль 3-[гидрокси-(2-тиенил)метилен] -6- хлор-5-фтор-2,3-дигидро-2-оксо-1H-индол-1-карбоксамида (6,65 г) и иодметиловый эфир N, N-диметиламида янтарной кислоты (3,27 г) смешивают в 100 мл ацетона и полученный раствор перемешивают в течение 65 ч при 20oC. После упаривания растворителя в вакууме твердый остаток экстрагируют три раза хлороформом. Хлороформные экстракты объединяют и упаривают в вакууме. Остаток хроматографируют на силикагеле, элюент хлороформ/ацетон, 75:25. Фракции, содержащие названный продукт, смешивают и упаривают. Получают 189 мг оранжевого масла. Другие фракции, содержащие названный продукт объединяют, упаривают и снова хроматографируют на силикагеле, элюент хлороформ/ацетон, 90:10. Фракции, содержащие продукт, объединяют со 189 мг масла, полученного выше, упаривают и получают 490 мг оранжевого твердого продукта, который суспендируют в небольшом количестве диэтилового эфира и фильтруют. Получают 180 мг твердого оранжевого вещества: т.пл. 183 - 187oC.
Пример 63. 1-[1-Карбамоил-6-хлор-5-фтор-2-оксо-1,2-дигидро- индол-3-илиден)тиофен-2-илметокси]метиловый эфир N,N- диэтиламида янтарной кислоты.
Тетрабутиламмониевую соль 3-[гидрокси-(2-тиенил)метилен] -6-хлор-5-фтор-2,3- дигидро-2-оксо-1H-индол-1-карбоксамида (4,36 г) и иодметиловый эфир N, N-диэтиламида янтарной кислоты (2,36 г) смешивают в 30 мл ацетона и полученный раствор перемешивают в течение 87 ч при 20oC. После упаривания растворителя в вакууме твердый остаток хроматографируют на силикагеле, элюент хлороформ/ацетон, 90:10. Фракции, содержащие названный продукт, смешивают и упаривают. Получают масло, содержащее некоторое количество твердого продукта. Добавляют к полученному маслу диэтиловый эфир и отфильтровывают нерастворившиеся вещества. (Твердый продукт содержит как тетрабутиламмонийиодид, так и продукт C-алкилирования). Эфирный экстракт упаривают и остаток хроматографируют на силикагеле, элюент хлороформ/ацетон, 90:10. Фракции, содержащие продукт, объединяют, упаривают в вакууме. Полученное оранжевое масло растворяют в диэтиловом эфире и выдерживают в течение ночи. Отфильтровывают желтый кристаллический осадок и сушат в вакууме. Получают 265 мг твердого продукта, т.пл. 151 - 152,5oC.
Пример 64. [(1-Карбамоил-6-хлор-5-фтор-2-оксо-1,2-дигидро-индол-3-илиден)- тиофен-2-илметокси]метиловый эфир N,N-дипропиламида янтарной кислоты.
Тетрабутиламмониевую соль 3-[гидрокси(2-тиенил)метилен]-6-хлор-5-фтор-2,3-дигидро-2-оксо-1H-индол- 1-карбоксамида (6,07 г) и иодметиловый эфир N, N-дипропиламида янтарной кислоты (2,6 г) смешивают в 50 мл ацетона и полученный раствор перемешивают в течение 18 ч при 20oC. Растворитель упаривают в вакууме, а полученный остаток перемешивают с диэтиловым эфиром. Образующийся осадок отфильтровывают. Эфирный раствор упаривают в вакууме и получают 6,5 г оранжевого масла. Полученное масло хроматографируют на силикагеле, элюент хлороформ/ацетон, 95:5. Фракции, содержащие названный продукт, смешивают и упаривают. К полученному остатку добавляют диэтиловый эфир и некоторое время выдерживают. Образовавшийся остаток отфильтровывают и получают 210 мг продукта. Другие фракции, содержащие названный продукт, объединяют и упаривают, остаток хроматографируют на силикагеле, элюент этилацетат/гексан, 50:50. Фракции, содержащие продукт, объединяют, упаривают в вакууме. Полученное масло растворяют в диэтиловом эфире и выдерживают в течение 18 ч. Отфильтровывают желтый кристаллический осадок и дополнительно получают 220 мг: т.пл. 131, 5 - 133,5oC.
Пример 65. [(1-Карбамоил-6-хлор-5-фтор-2-оксо-1,2-дигидро-индол- 3-илиден)-тиофен-2-илметокси]метиловый эфир N,N-гексаметиленамид янтарной кислоты.
Тетрабутиламмониевую соль 3-[гидрокси-(2-тиенил)метилен]-6-хлор- 5-фтор-2,3-дигидро-2-оксо-1H-индол-1-карбоксамида (4,4 г) и иодметиловый эфир 4-гомопиперидино-4-оксо-масляной кислоты (11,98 г) смешивают в 30 мл ацетона и полученный раствор перемешивают в течение 17 ч при 20oC. Полученную смесь фильтруют и фильтрат упаривают в вакууме. Остаток хроматографируют на силикагеле, элюент хлороформ/ацетон, 93:7. Фракции, обогащенные названным продуктом, смешивают и упаривают. Полученное оранжевое масло хроматографируют на силикагеле, используя тот же элюент. Фракции, обогащенные названным продуктом, объединяют и упаривают. К полученному остатку добавляют диэтиловый эфир и полученный раствор выдерживают некоторое время. Отфильтровывают образовавшийся желтый осадок, получают 250 мг продукта: т.пл. 149 - 151oC.
Пример 66. [(1-Карбамоил-6-хлор-5-фтор-2-оксо-1,2-дигидро- индол-3-илиден)-тиофен-2-илметокси] метиловый эфир N, N-диметилкарбамоилокси уксусной кислоты.
Тетрабуталаммониевую соль 3[гидрокси-(2-тиенил)метилен]-6-хлор-5-фтор-2,3-дигидро-2-оксо-1H-индол- 1-карбоксамида (3,28 г) и иодметилового эфира N, N-диметиламинокарбонилоксиуксусной кислоты (1,18 г) смешивают в 15 мл ацетона и полученный раствор перемешивают в течение 17 ч при 20oC. Полученную смесь фильтруют и фильтрат упаривают в вакууме. Остаток хроматографируют на силикагеле, элюент хлороформ/ацетон, 95:5. Фракции, обогащенные названным продуктом, смешивают и упаривают. К полученному маслу добавляют диэтиловый эфир и раствор выдерживают некоторое время. Отфильтровывают образовавшийся желтый осадок, сушат, получают 240 мг продукта: т.пл. 185,5 - 187oC.
Пример 67. [(1-Карбамоил-6-хлор-5-фтор-2-оксо-1,2-дигидро- индол-3-илиден)тиофен-2-илметокси] метиловый эфир N,N-диэтилкарбамоил- оксиуксусной кислоты.
Тетрабутиламмониевую соль 3-[гидрокси-(2-тиенил)метилен] -6-хлор-5-фтор-2,3-дигидро-2-оксо-1H- индол-1-карбоксамида (11,6 г) и иодметилового эфира N, N-диэтиламинокарбонилоксиуксусной кислоты (6,36 г) смешивают в 100 мл ацетона и полученный раствор перемешивают в течение 17 ч при 20oC. Полученную смесь фильтруют и упаривают в вакууме, получают полутвердый продукт, к которому добавляют диэтиловый эфир и отфильтровывают нерастворившиеся в эфире вещества. Эфирный раствор оставляют на несколько часов. Отфильтровывают образовавшийся белый осадок (продукт C-алкилирования). После дополнительного выдерживания из эфирного фильтрата выпадает желтый осадок, который отфильтровывают, сушат, получают 990 мг кристаллического продукта: т.пл. 156,8 - 157,8oC.
Пример 68. [(1-Карбамоил-6-хлор-5-фтор-2-оксо-1,2-дигидро- индол-3-илиден)-тиофен-2-илметокси]метиловый эфир N-пивалоилглицина.
Тетрабутиламмониевую соль 3-[гидрокси-(2-тиенил)метилен] -6-хлор-5-фтор-2,3-дигидро-2-оксо-1H-индол-1-карбоксамида (2,80 г) и иодметиловый эфир N-пивалоилглицина (1,45 г) смешивают в 20 мл ацетона и полученный раствор перемешивают в течение 17 ч при 20oC. После упаривания растворителя в вакууме желто-оранжевый остаток перемешивают с диэтиловым эфиром и фильтруют. Эфирный фильтрат упаривают и остаток перемешивают с небольшим количеством диэтилового эфира. Отфильтровывают нерастворимый в эфире осадок и растворяют в этилацетате для хроматографирования на силикагеле, элюент этилацетат. Фракции, обогащенные названным продуктом, объединяют и упаривают. Получают желтый твердый продукт, который суспендируют в небольшом количестве диэтилового эфира и фильтруют. Получают 140 мг: т.пл. 200 - 204oC.
Пример 69. [(1-Карбамоил-6-хлор-5-фтор-2-оксо-1,2-дигидро- индол-3-илоиден)-тиофен-2-илметокси]метиловый эфир N-(2-этилбутирил)глицина.
Тетрабутиламмониевую соль 3-[гидрокси-(2-тиенил)метилен] - 6-хлор-5-фтор-2,3-дигидро-2-оксо-1H-индол-1-карбоксамида (5,13 г) и иодметил N-(2-этилбутирил)глицинат (2,78 г) смешивают в 35 мл ацетона и полученный раствор перемешивают в течение 17 ч при 20oC. После упаривания в вакууме растворителя получают желто-оранжевый остаток, который суспендируют в диэтиловом эфире и фильтруют. Эфирный фильтрат упаривают и остаток перемешивают с небольшим количеством диэтилового эфира. Отфильтровывают желтый осадок (200 мг) и растворяют в этилацетате для хроматографирования на силикагеле, элюент этилацетат. Фракции, обогащенные названным продуктом, объединяют и упаривают. Получают 40 г желтого твердого продукта. Дополнительные 256 мг названного продукта получают при хроматографировании на силикагеле (элюент этилацетат) фракции, нерастворившейся в ацетоне.
Пример 70. 1-[(-Карбамоил-6-хлор-5-фтор-2-оксо-1,2-дигидро-индол-3-илиден)- тиофен-2-илметокси]этиловый эфир изопропилкарбоновой кислоты.
Тетрабутиламмониевую соль 3-[гидрокси-(2-тиенил)метилен] - 6-хлор-5-фтор-2,3-дигидро-2-оксо-1H-индол-1-карбоксамида (13,6 г) и 1-иодэтиловый эфир изопропилкарбоната (Wan-Joo Kim, et ai., The Journal of Antibiotics, (1991), 44, 1086) (6,08 г) смешивают в 90 мл ацетона и полученный раствор перемешивают в течение 17 ч при 20oC. Нерастворимые продукты отфильтровывают, суспендируют в хлороформе и фильтруют. Хлороформный фильтрат смешивают с раствором ацетона и растворители упаривают в вакууме. Остаток хроматографируют на силикагеле, элюент хлороформ/ацетон, 98:2. Фракции, содержащие названный продукт, объединяют и упаривают. Получают оранжевое масло. К полученному маслу при перемешивании добавляют диэтиловый эфир и отфильтровывают желтый кристаллический продукт. Получают 1,10 г: т.пл. 183 - 184oC.
Данные спектров ЯМР соединений примеров 59-70 приведены в табл. 4.
Пример 71. 6-Хлор-5-фтор-2,3-дигидро-2-[1-метоксиацетокси)- этокси-(2-тиенил)метилен]оксо-1H-индол-1-карбоксамид.
a) К метоксиацетилхлориду (10,0 г, 0,092 моль) добавляют каталитическое количество расплавленного хлорида цинка, раствор охлаждают до -20oC и добавляют ацетальдегид (4,06 г, 0,092 моль). Охлаждение снимают и перемешивают в течение 1 часа при комнатной температуре. Перегоняют при 30 мм рт.ст. и собирают фракции, выходящие при 50 - 80oC. Получают 3,3 г масла (выход 22,9%).
b) К суспензии натрий-6-хлор-5-фтор-2,3-дигидро-3-(гидрокси-2- тиенилметилен)-2-оксо-1H-индол-1- карбоксамида (3,5 г, 9,7 ммоль) и йодида натрия (500 мг, 3,3 ммоль) в 30 мл ацетона при перемешивании добавляют 1-хлорэтил метоксиацетил (1,8 г, 9,7 ммоль). Полученную суспензию кипятят с обратным холодильником в течение 7 ч, охлаждают и упаривают в вакууме с получением оранжевого твердого вещества. Твердое вещество хроматографируют на колонке с силикагелем (125 мг силикагеля), используя в качестве элюента гексан/этилацетат, 9: 1, получают сырой продукт. При кристаллизации из ацетона получают желтое твердое вещество, 1,1 г (25%): т.пл. 175 - 176oC.
Элементный анализ:
Вычислено,%: C 50,17; H 3,55; N 6,16.
C19H16ClFN2O6S.
Найдено,%: C 50,31; H 3,32; N 6,32.
Пример 72. 6-Хлор-5-фтор-2,3-дигидро-3-[1-(1-морфолинокарбамоил) -этокси-(2-тиенил)метилен]-2-оксо-1H-индол-1-карбоксамид.
a) К раствору морфолина (8,7 г, 0,1 моль) и триэтиламина (10,1 г, 0,1 моль) в метиленхлориде (120 мл) добавляют 1-хлорэтилхлорформиат (14,3 г, 0,1 моль) с такой скоростью, чтобы не допустить кипения. После перемешивания в течение 10 мин раствор выливают в воду (200 мл). Органический слой отделяют и сушат сульфатом магния, затем упаривают с получением 17,8 г светло-коричневого масла (выход 92%).
b) К суспензии натрий-6-хлор-5-фтор-2,3-дигидро-3-(гидрокси- тиенилметилен)-2-оксо-1H-иендол-1- карбоксамида (10 г, 0,028 моль) и йодида натрия (300 мг, 0,0017 ммоль) в 45 мл ацетона при перемешивании добавляют морфолинокарбамоиловый эфир со стадии a (5,36 г, 0,028 ммоль) и смесь кипятят с обратным холодильником в течение 5,5 ч. Суспензию охлаждают и упаривают досуха, твердое вещество хроматографируют на колонке с силикагелем (4,5 • 42 см), элюента гексан/этилацетат, 7:3, получают сырой продукт. При кристаллизации из ацетона выделяют желтое твердое вещество, 2,37 г (17,3%): т.пл. 124 - 126oC.
Элементный анализ:
Вычислено,%: C 50,86; H 3,86; N 8,47.
C21H19ClFN3O6S.
Найдено,%: C 50,91; H 3,78; N 8,54.
Пример 73. Соль 6-хлор-5-фтор-2,3-дигидро-3-[1-(1-амино-циклопентоилоксиметокси- (2-тиенил)метилен]-2-оксо-1H-индол-1-карбоксамид-метансульфокислоты.
a) 1-N-трет.-бутоксикарбониламино-1-циклопентанкарбоновая кислота
К раствору 1-амино-1-циклопентанкарбоновой кислоты (10 г, 0,0774 моль), триэтиламина (11,75 г, 0,1161 моль), воды (45 мл) и диоксана (45 мл) добавляют BOC-ON (20,9 г, 0,085 моль) при перемешивании в течение 7 ч при комнатной температуре. Смесь экстрагируют этилацетатом и слой этилацетат промывают водой (50 мл). Водный слой подкисляют лимонной кислотой до pH 3,5 и экстрагируют этилацетатом (2 • 100 мл). Слой этилацетата сушат сульфатом магния и упаривают, получают продукт в виде масла, 14 г (79%).
b) 1-N-трет.-BOC-амино-1-хлорметилциклопентаноат.
К гидросульфату тетрабутиламмония (22,5 г, 0,066 моль) в воде (125 мл) медленно добавляют бикарбонат натрия (11,1 г, 0,132 моль) и перемешивают в течение 15 мин. Добавляют соединение a) в хлороформе (250 мл) и перемешивают в течение 3 ч. Органический слой отделяют и сушат сульфатом магния, затем упаривают. Получают масло, к которому добавляют бром-хлорметан (225 мл) и раствор перемешивают в течение ночи при комнатной температуре. Смесь упаривают до вязкого масла, которое растирают с эфиром и фильтруют. Фильтрат упаривают, получают сырой продукт в виде масла 9,2 г (выход 50%).
c) К суспензии натриевой соли 6-хлор-5-фтор-2,3-дигидро-3- (гидрокси-2-тиенилметилен)-2-оксо-1H-индол-1-карбоксамида (2,85 г, 7,92 ммоль) и йодида натрия (285 мг, 1,9 ммоль) в 70 мл ацетона при перемешивании добавляют 1-N-трет. -бутоксиамино-1-хлорметиленциклопентаноат (2,2 г, 7,92 ммоль). Полученную суспензию нагревают с обратным холодильником в течение 7 ч. Реакционную суспензию охлаждают до комнатной температуры и фильтруют для удаления непрореагировавшей натриевой соли 6-хлор-5-фтор-2,3-дигидро-3-(гидрокси-2-тиенилметилен)-2-оксо- 1H-индол-1-карбоксамида. Фильтрат упаривают в вакууме до оранжевого твердого вещества, которое хроматографируют на слое силикагеле (75 г), используя в качестве элюента гексан/этилацетат, 7:3. Получают твердое вещество, которое перекристаллизовывают из ацетонитрила с получением желтого твердого вещества, 320 мг (выход 7,1%): т.пл. 204oC (разл.).
Элементный анализ:
Вычислено,%: C 53,84; H 4,69; N 7,24.
C26H27ClFN3O7S.
Найдено,%: C 53,83; H 4,42; N 7,29.
d) Раствор производного 1-N-трет. - бутоксикарбониламиноциклопентаноильного производного со стадии c) (410 мг, 0,7 ммоль) в 10 трифторуксусной кислоты перемешивают при комнатной температуре в течение 20 мин. Смесь упаривают в вакууме, получают маслянистый твердый продукт, добавляют 25 мл метиленхлорида и метансульфокислоту (67,9 мг, 0,7 ммоль) и упаривают в вакууме с получением желтого твердого вещества, которое перемешивают с 25 мл метиленхлорида в течение 15 мин. Отфильтровывают осадок и получают желтый твердый продукт, 400 мг (99%): т.пл. 230oC (разл.).
Элементный анализ:
Вычислено,%: C 45,87; H 4,02; N 7,30.
C22H23ClFN3O8S2.
Найдено,%: C 45,63; H 3,95; N 7,19.
Пример 74. 6-Хлор-5-фтор-2,3-дигидро-3- [ацетоксиметоксикарбонилметокси-(2-тиенил)метилен]-2-оксо-1H-индол-1- карбоксамид
a) К смеси ацетоксиацетилхлорид (20 г, 0,146 моль) и каталитического количества расплавленного хлорида цинка добавляют параформальдегид (6,2 г, 0,206 моль), смесь нагревают на паровой бане в течение 3,5 ч. После охлаждения реакционную смесь перегоняют и собирают фракции, кипящие при 72-82oC и 1,2 - 1,9 мм рт.ст. Получают 3,2 г масла (выход 13%)
b) Названный продукт получают с использованием промежуточного соединения со стадии a) по методике, аналогичной методике, описанной в примере 73, за исключением того, что используют в качестве элюента гексан/этилацетат, 3:1. Кристаллизация из этилацетата дает желтое твердое вещество, 900 мг (8,9%): т.пл. 176 - 177oC.
Элементный анализ:
Вычислено,%: C 48,68; H 3,01; N 5,98.
C19H14ClFN2O7S.
Найдено,%: C 48,60; H 3,02; N 6,00.
Пример 75. 6-Хлор-5-фтор-2,3-дигидро-3-[бензилметокси-(2- тиенил)метилен]-2-оксо-1H-индол-1-карбоксамид.
a) Хлорметилбензоат получают способом, аналогичным описанному в стадии a), пример 74. Продукт реакции перегоняют при 74 - 76oC при 2,0 мм рт.ст. Получают желтый сырой продукт в виде масла (16,42 г, 50%).
b) Названный продукт получают с использованием промежуточного соединения со стадии a) по методике, аналогичной методике, описанной в примере 73. Кристаллизация из этилацетата дает желтое твердое вещество, 130 мг (5,5%): т. пл. 202 - 203oC.
Элементный анализ:
Вычислено,%: C 55,88; H 2,98; N 5,92.
C22H14ClFN2O5S.
Найдено,%: C 55,69; H 2,69; N 5,86.
Пример 76. 6-Хлор-5-фтор-2,3-дигидро-3-[1-уксусная кислота-1- циклопентилацетоксиметокси-(2-тиенил)метилен]-2-оксо-1H-индол-1- карбоксамид.
1-(4-Метоксибензилоксикарбонилметил)-1-циклопентануксусная кислота.
a) Смесь 3,3-тетраметиленглутарового ангидрида (10,0 г, 5,95 ммоль), 4-метоксибензилового спирта (8,2 г, 5,95 ммоль) и пиридина (4,7 г, 5,95 ммоль) соединяют и нагревают на паровой бане в течение 1,5 ч. Охлаждают, подкисляют, добавляя по каплям 6 н. HCl, и экстрагируют эфиром и этилацетатом, 1: 1 (3 • 150 мл). Объединенные органические слои промывают насыщенным раствором бикарбоната натрия (3 • 150 мл). Водный основной слой подкисляют 6 н. HCl, экстрагируют этилацетатом и эфиром (200 мл) и сушат сульфатом магния. Упаривают в вакууме и получают сырое масло, 11,6 г (64%).
b) 1-(4-Метоксибензилоксикарбонилметил)-1- хлорметилоксикарбонилциклопентан получают по описанию примера 73, стадия b). После упаривания получают сырой продукт в виде масла, 6,5 г (48%), которое используют на стадии c).
c) 4-Метоксибензилокси производное названного соединения получают способом, аналогичным описанному в примере 73 c). Получают 300 мг (1,6%) красного вязкого масла.
Элементный анализ:
Вычислено,%: C 58,49; H 4,6; N 4,26.
C32H30ClFN2O8S.
Найдено,%: C 58,89; H 4,32; N 4,33.
d) 4-Метоксибензилпроизводное со стадии c) (400 мг, 0,61 ммоль) в 15 мл трифторуксусной кислоты перемешивают в течение 45 мин, упаривают в вакууме, получают желтое твердое вещество. Добавляют 60 мл метиленхлорида и упаривают. Полученное желтое вещество перекристаллизовывают из метанола, выделяют 600 мг продукта (выход 15%): т.пл. 187 - 188oC.
Элементный анализ:
Вычислено,%: C 53,68; H 4,13; N 5,22.
C24H22ClFN2O7S.
Найдено,%: C 53,94; H 4,04; N 5,02.
Пример 77. 6-Хлор-5-фтор-2,3-дигидро-3-[L-пропилметокси-(2-тиенил)метилен]-2-оксо- 1H-индол-1-карбоксамид соль метансульфокислоты.
a) Хлорметиловый эфир N-трет.-бутоксикарбонил-L-пролина получают по методике, описанной в примере 73, стадия b). Сырой продукт выделяют в виде масла. 35,4 г (83%).
b) Производное N-трет.-бутоксикарбонил-L-пролина получают способом, аналогичным описанному в примере 73, стадия c). Кристаллизация из эфира и гексана дает желтое твердое вещество, 2,65 г (9,36%); т.пл. 168 - 169oC.
Элементный анализ:
Вычислено,%: C 52,96; H 4,62; N 7,41.
C25H26ClFN3O7S.
Найдено,%: C 52,85; H 4,46; N 7,43.
c) Названное соединение получают способом, аналогичным описанному в примере 73, стадия d). Кристаллизация из 2-пропанола дает желтое твердое вещество, 333 мг (34%): т.пл. 90 - 120oC (аморфное вещество).
Элементный анализ:
Вычислено,%: C 44,52; H 3,83; N 7,42.
C20H17ClFN3O5S•CH3SO3H•1/4 H2O.
Найдено,%: C 44,35; H 3,92; N 7,23.
Пример 78. 6-Хлор-5-фтор-2,3-дигидро-3-[1-карбоновая кислота-3-пиперидилкарбонилметокси- (2-тиенил)метилен] -2-оксо-1H-индол-1-карбоксамид, соль метансульфокислоты.
a) Получение N-трет.бутоксикарбонилпиперидин-3-карбоновой кислоты проводят по описанию примера 73, стадия a). Получают сырое твердое вещество (19 г). Кристаллизация из этилацетата дает белое твердое вещество, 11,1 г (48%): т.пл. 162 - 163oC.
b) Получение 1-N-трет.-бутоксикарбонил-пиперидин-3-хлорметилового эфира проводят способом, аналогичным описанному в примере 73, стадия b). После упаривания получают белое влажное твердое вещество, 13,5 г (78%), которое непосредственно используется на следующей стадии.
c) 3-хлорметиловый эфир 1-N-трет.-бутоксикарбонил-пиперидина используют для получения бутоксикарбонильных производных названного соединения способом, аналогичным описанному в примере 73. После кристаллизации из эфира и гексана получают 580 мг желтого твердого вещества (2,5%): т.пл. 145 - 146oC.
Элементный анализ:
Вычислено,%: C 53,84; H 4,69; N 7,24.
C26H27ClFN3O7S.
Найдено,%: C 53,83; H 4,57; N 7,16.
d) Названное соединение получают способом, аналогичным описанному в примере 73, стадия d). Кристаллизация из ацетона дает желтое твердое вещество, 280 мг (68,7%): т.пл. 184 - 185oC
Элементный анализ:
Вычислено,%: C 45,87; H 4,02; N 7,30.
C22H23ClFN3O8S2.
Найдено,%: C 45,75; H 3,85; N 7,17.
CP-156,011.
Пример 79. 6-Хлор-5-фтор-2,3-дигидро-3-[1-карбокси-3-этил- 3-метил-пентаноилметокси-(2-тиенил)метилен]-2-оксо-1H-индол-1-карбоксамид.
Названное соединение получают способом, аналогичным описанному в примере 76. Кристаллизация из ацетона и гексана дает желтое твердое вещество, 380 мг (27%): т.пл. 124 - 125oC.
Элементный анализ:
Вычислено,%: C 52,62; H 4,22; N 5,34.
C23H22ClFN2O7S.
Найдено,%: C 52,83; H 3,99; N 5,20.
Пример 80. 6-Хлор-5-фтор-2,3-дигидро-3-[транс-1-карбокси- 2-циклогексилкарбонилметокси-(2-тиенил)метилен]-2-оксо- 1H-индол-1-карбоксамид.
Названное соединение получают способом, аналогичным описанному в примере 76. Кристаллизация из ацетона дает желтое твердое вещество, 590 мг (28,3%): т.пл. 214 - 215oC.
Элементный анализ:
Вычислено,%: C 52,83; H 3,85; N 5,26.
C23H20ClFN2O7S.
Найдено,%: C 53,17; H 3,72; N 5,10.
Пример 81. 6-Хлор-5-фтор-2,3-дигидро-3-[транс-1-карбокси- 2-циклопропилкарбонилметокси-(2-тиенил)метилен]-2-оксо- 1H-индол-1-карбоксамид, соль метансульфокислоты.
a) 1-N-трет.бутоксикарбонилциклопропаноильное производное названного соединения получают способом, аналогичным описанному в примере 73, стадия c). Кристаллизация из эфира дает желтое твердое вещество, 780 мг (4,4%): т.пл. 179 - 180oC.
Элементный анализ:
Вычислено,%: C 52,22; H 4,20; N 7,61.
C24H23ClFN3O7S.
Найдено,%: C 52,30; H 4,03; N 7,67.
b) Названное соединение получают способом, аналогичным описанному в примере 73. Кристаллизация из метанола дает желтое твердое вещество, 235 мг (78%): т.пл. 175 - 182oC (аморфное твердое вещество).
Элементный анализ:
Вычислено,%: C 43,13; H 3,62; N 7,54.
C19H15ClFN3O5S•CH3SO3H •1/2H2O.
Найдено,%: C 43,17; H 3,33; N 7,46.
Пример 82. 6-Хлор-5-фтор-2,3-дигидро-3-[транс-1-карбокси-2-циклопропилкарбонилметокси- (2-тиенил)метилен]-2-оксо-1H-индол-1-карбоксамид.
a) К раствору гидроксида натрия (12,0 г, 0,3 моль) в воде (200 мл) добавляют диэтиловый эфир 1,2-циклопропандикарбоновой кислоты (18,6 г, 0,10 моль) и перемешивают при 80oC в течение 5 ч. Раствор упаривают до твердого вещества при охлаждении (0 - 5oC). Медленно при охлаждении добавляют концентрированную соляную кислоту (50 мл), затем нагревают на паровой бане, отфильтровывают нерастворимые соединения и охлаждают в бане с ледяной водой. Полученные кристаллы отбирают и растворяют в ацетоне и упаривают с получением транс-1,2-циклопропандикарбоновой кислоты, 6,2 г (47,6%), т.пл. 176 - 177oC. Лит.данные: JACS, 4994-4999 (1957) т.пл. 176 - 172oC.
b) Смесь транс-1,2-циклопропандикарбоновой кислоты (3,0 г, 0,0231 моль) и тионилхлорида (20 мл) нагревают с обратным холодильником в течение 1,5 ч, затем охлаждают и упаривают. Полученное масло растворяют в бензоле (20 мл) и медленно добавляют 2-(триметилсилил)этанол (2,13 г, 0,018 моль) и затем перемешивают в течение ночи при комнатной температуре. Реакционную смесь фильтруют и упаривают с получением сырого продукта в виде светло-коричневого масла, 3,96 г (42%).
c) (Z)-изомер 6-хлор-5-фтор-2,3-дигидро-2-[транс-1-(2- триметилсилоксикарбонил)-2-(циклопропаноил)окси-(2-тиенил)метилен] - 2-оксно-1H-индол-1-карбоксамида получают способом, аналогичным описанному в примере 54, за исключением того, что соединение отделяют на слое силикагеля и в качестве элюента используют гексан/этилацетат, 4: 1. Получают сырой продукт, кристаллизация которого из смеси хлороформа и гексана дает желтое вещество. 140 мг (3,7%): т.пл. 181 - 182oC.
Элементный анализ:
Вычислено,%: C 52,31; H 4,39; N 5,08.
C24H24ClFN2O6SSi.
Найдено,%: C 52,32; H 4,36; N 5,12.
d) E-изомер выделяют способом, аналогичным описанному в стадии c). Кристаллизация из хлороформа и гексана дает желтое твердое вещество, 160 мг (4,2%): т.пл. 175 - 176oC. CP-152,775.
Элементный анализ:
Вычислено,%: C 52,31; H 4,39; N 5,08.
C24H24ClFN2O6SSi.
Найдено,%: C 52,45; H 4,28; N 4,92.
e) Суспензию триметилсилоксициклопропил-производного названного соединения (800 мг, 1,45 ммоль) в фтористоводородном комплексе пиридина (5 мл) при -20oC перемешивают в течение 30 мин, добавляют 20 мл воды и отфильтровывают сырое твердое вещество. Кристаллизация из эфира дает желтое твердое вещество, 412 мг (61,7%): т.пл. 195oC (разл.).
Элементный анализ:
Вычислено,%: C 49,63; H 2,85; N 6,09.
C19H12ClFN2O6S•1/2H2O.
Найдено, %: C 49,81; H 2,62; N 6,06.
Пример 83. 6-Хлор-5-фтор-2,3-дигидро-3-[1,3-диоксан-5-карбонилметокси- (2-тиенил)метилен]-2-оксо-1H-индол-1-карбоксамид.
a) 1,3-диоксан-5-карбоновую кислоту синтезируют по методике, описанной в Synthesis Communication, 24, (1974).
b) Хлорметиловый эфир получают способом, аналогичным описанному в примере 73, стадия c). Получают 4,3 г светло-желтого масла (48%).
c) Производное 1,3-диоксана получают способом, аналогичным описанному в примере 73. Продукт выделяют на колонке с силикагелем (42х5,5 см), элюент гексан/этилацетат, 7:1. Затем упаривают в вакууме и получают 150 мг желтого твердого вещества (1,3%): т.пл. 199 - 200oC.
Элементный анализ:
Вычислено,%: C 49,75; H 3,34; N 5,80.
C20H16ClFN2O7S.
Найдено,%: C 49,84; H 3,17; N 5,8.
Пример 84. 6-Хлор-5-фтор-2,3-дигидро-3-[1-(4-метоксибензоксикарбонил)- 5-(3-метил)пентоилметокси-(2-тиенил)метилен]-2-оксо-1H-индол-1-карбоксамид.
a) Хлорметиловый эфир получают способом аналогичным описанному в примере 73, стадия b). Продукт выделяют в виде масла, 9,92 г (26%). Используют непосредственно на следующей стадии.
Названное соединение получают способом, аналогичным описанному в примере 76. После кристаллизации из эфира получают 150 мг (7,6%) твердого оранжевого вещества: т.пл. 113 - 114oC.
Элементный анализ:
Вычислено,%: C 56,45; H 4,25; N 4,54.
C29H26ClFN2O8S.
Найдено,%: C 56,32; H 4,13; N 4,56.
Методика 1. Метил-(1-хлорэтокси)карбонилоксиацетат.
В охлаждаемый на бане с ледяной водой раствор метил гликолята (13,5 мл) и N,N-диизопропилэтиламина (30,46 мл) в тетрагидрофуране (180 мл) добавляют по каплям при перемешивании 25,0 г 1-хлорэтилхлороформиата. Охлаждение прекращают и смесь перемешивают в течение 3 ч. После фильтрования для удаления нерастворимого гидрохлорида амина раствор в ТГФ упаривают при пониженном давлении. Остаток растворяют в смеси этилацатета и воды. Слой этилацетата отделяют, сушат безводным сульфатом натрия и упаривают при пониженном давлении. Получают 31,1 г красноватого масла ЯМР.
Методика 2. Метил-(1-йодоэтокси)карбонилоксиацетат.
Йодид натрия (13,19 г), ацетон (35 мл) и метил -(1-хлорэтокси) карбонилоксиацетата (8,65 г) кипятят с обратным холодильником в течение 2,5 ч. После охлаждения смесь фильтруют и фильтрат упаривают в вакууме, получают коричневое масло, N-метилморфолин (1,2 мл) и 5,8 мл коричневатого масла растворяют в 50 мл метиленхлорида и полученную смесь перемешивают при комнатной температуре в течение 45 мин. После фильтрования раствор в метиленхлориде упаривают и к остатку добавляют этилацетат и воду. Слой этилацетата отделяют, сушат безводным сульфатом натрия и упаривают, получают 2,18 г сырого продукта, содержащего примерно 68% метил-(1-йодоэтокси)карбонилоксиацетата и 32% метил (1-хлорэтокси) карбонилоксиацетата. Полученный сырой продукт используют без дополнительной очистки.
Методика 3. N,N-диметиламид гликолевой кислоты.
К раствору диметиламина (2,52 г) в 150 мл метанола добавляют 14,8 г метилового эфира гликолевой кислоты и смесь выдерживают при 20oC в течение 18 ч. Избыток диметиламина и метанола упаривают при пониженном давлении и добавляют диэтиловый эфир для кристаллизации. После фильтрования и сушки получают белые кристаллы, 13, 15 г: т.пл. 44 - 46oC.
Методика 4. N,N -диметил -(1-хлорэтокси)карбонилоксиацетамид.
Охлаждаемый в бане с ледяной водой раствор N,N-диметиламида гликолевой кислоты (15,64 г) и N,N-диизопропилэтиламина (26,4 мл) в тетрагидрофуране (100 мл) по каплям при перемешивании добавляют 16,37 мл 1-хлорэтилхлорформиата. Охлаждение прекращают и смесь перемешивают еще в течение 4 ч. После фильтрования для удаления нерастворимого гидрохлорида амина раствор в ТГФ упаривают при пониженном давлении. Получают 25,5 г коричневого твердого продукта. Сырой продукт растирают в петролейном эфире, фильтруют и сушат в вакууме, получают практически белый твердый продукт, 19,98 г: т.пл. 59-60,8oC.
Методика 5. N, N-диметил-(1-иодэтокси)карбонилоксиацетамид.
Йодид натрия (17,16 г), ацетон (35 мл) и N,N-диметил(1-хлорэтокси)карбонилоксиацетамид (12,0 г кипятят с обратным холодильником в течение 1,5 ч. После охлаждения смесь фильтруют и фильтрат упаривают. К остатку добавляют этилацетат и отфильтровывают нерастворимые вещества. После упаривания этилацетата добавляют хлороформ и отфильтровывают нерастворимые вещества. После упаривания раствора в хлороформе получают 13,0 г красноватого масла. К остатку добавляют диэтиловый эфир и растворимую в эфире часть декантируют от нерастворимой. Упаривание эфирного раствора дает 4,0 желто-оранжевого масла. К желто-оранжевому маслу добавляют метиленхлорид (30 мл) и 0,42 мл N-метилморфолина, смесь перемешивают при комнатной температуре в течение 45 мин, затем концентрируют в вакууме. Остаток распределяют между этилацетатом и водой, слой этилацетата отделяют, сушат сульфатом натрия, упаривают при пониженном давлении, получают 2,55 г желтого твердого вещества, которое, как показывают данные ЯМР, является смесью примерно 66% N, N-диметил-(1-йодоэтокси) карбонилоксиацетамида и примерно 33% N,N-диметил-(1-хлорэтокси)карбонилоксиацетамида. Полученный сырой продукт используют без дополнительной очистки.
Методика 6. N,N-диэтиламид гликолевой кислоты.
К охлажденному до 0oC раствору диэтиламина (189 мл) и триэтиламина (25,5 мл) в 500 мл метиленхлорида по каплям при перемешивании добавляют 25 г ацетоксиацетилхлорида. Ледяную баню убирают и смесь оставляют нагреваться до 20oC при перемешивании в течение еще 2 ч. После фильтрования с целью удаления гидрохлоридов аминов метиленхлоридный раствор упаривают при пониженном давлении. Получают 29,1 г сырого N,N-диэтилацетоксиацетамида в виде желтого масла. Желтое масло растворяют в 125 мл метанола и добавляют 168 мл 1 гидроксида натрия. Наблюдается слабый экзотермический эффект. Смесь оставляют перемешиваться в течение 2 ч без нагревания. Растворители упаривают в вакууме до 1/4 объема и оставшийся водный раствор экстрагируют этилацетатом (200 мл). Этилацетатный экстракт сушат безводным сульфатом натрия и упаривают при пониженном давлении, получают 14,0 г N,N-диэтиогликольамида в виде желтого масла.
Методика 7. N,N-диэтил-(1-хлорэтокси)карбонилоксиацетамид.
В охлаждаемый на бане с ледяной водой раствор N,N-диэтиламида гликолевой кислоты (19,25 г) и N,N-диизопропилэтиламина (26,2 мл) в тетрагидрофуране (100 мл) при перемешивании по каплям добавляют 15,8 мл 1-хлорэтилхлорформиата. Охлаждение ледяной баней прекращают и смесь перемешивают еще в течение 4 ч. После фильтрования для удаления нерастворимого гидрохлорида амина раствор в ТГФ упаривают при пониженном давлении. Твердый остаток растворяют в этилацетате и раствор промывают водой, сушат безводным сульфатом натрия и упаривают до твердого остатка. После растирания в диэтиловом эфире твердое вещество отфильтровывают и сушат, получают 18,4 г практически белых кристаллов: т.пл. 76-78oC.
Методика 8. N,N-диэтил-(1-йодэтокси)карбонилоксиацетамид.
Йодид натрия (15,14 г) ацетон (35 мл) и N, N-диэтил-(1-хлорэтокси)карбонилоксиацетамида (12,0 г) кипятят с обратным холодильником в течение 1,5 ч. После охлаждения смесь фильтруют и фильтрат упаривают. К остатку при перемешивании добавляют этилацетат и воду. Слой этилацетата отделяют, сушат безводным сульфатом натрия и упаривают в вакууме, получают 13,6 г красноватого масла, которое, как показывают данные ЯМР, является смесью примерно 58% N,N-диэтил-(1-йодоэтокси)карбонилоксиацетамида и примерно 42% N,N-диэтил-(1-хлорэтокси)карбоанилоксиацетамида. Этот материал используют без дополнительной очистки.
Методика 9. Диметиламмониевая соль N, N-диметиламида янтарной кислоты.
Газообразный диметиламин медленно в течение 1 ч пропускают через предварительно охлажденную (0oC) смесь ангидрида янтарной кислоты (10 г) и тетрагидрофурана (75 мл). Охлажденную баню удаляют и реакционную смесь перемешивают еще в течение 30 мин, затем упаривают при пониженном давлении, получают 17,57 г прозрачного масла.
Методика 10. Хлорметиловый эфир N,N-диметиламида янтарной кислоты.
К 50 мл метиленхлорида при перемешивании добавляют 10 мл воды, 31,4 г гидросульфата тетрабутиламмония, 92,5 мл 1 н. раствора гидроксида натрия и 17,57 г диметиламмониевой соли N,N-диметиламида янтарной кислоты в 50 мл метиленхлорида. После перемешивания в течение 2,25 ч слой метиленхлорида отделяют, сушат безводным сульфатом натрия и упаривают в вакууме, получают 26,8 г масла. Масло растворяют в 100 мл бромхлорметана и перемешивают при 20oC в течение 17 ч. После упаривания бромхлорметана в вакууме остаток масла экстрагируют несколько раз диэтиловым эфиром. Эфирные экстракты объединяют и упаривают, получают 5,4 г масла. Сырой продукт очищают хроматографированием на силикагеле с использованием в качестве элюента смеси хлороформ/этилацетат, 70:30. Фракции, содержащие требуемый продукт, объединяют и концентрируют, получают 2,3 прозрачного масла.
Методика 11. Йодометиловый эфир N,N-диметиламида янтарной кислоты.
Йодид натрия (3,57 добавляют к раствору хлорметилового эфира N,N-диметиламида янтарной кислоты (2,3 г) в 10 мл ацетона и смесь перемешивают при 20oC в течение 2 ч. После упаривания растворителя в вакууме остаток растворяют в смеси метиленхлорида и воды. Слой метиленхлорида отделяют, сушат безводным сульфатом натрия и упаривают, получают 3,27 г коричневого масла, которое используют без дополнительной очистки.
Методика 12. Хлорметиловый эфир N,N-диэтиламида янтарной кислоты.
Гидросульфат тетрабутиламмония (19,6 г) N,N-диэтиламида янтарной кислоты (10,0 г), гидрокарбонат натрия (4,85 г) и 1 н. раствор гидроксида натрия (58 мл) смешивают в 400 мл метиленхлорида и 80 мл воды. Через 1,5 ч фазы разделяют. Слой метиленхлорида сушат безводным сульфатом натрия и упаривают при пониженном давлении, получают 9,6 г масла. Водный слой экстрагируют метиленхлоридом и после сушки и упаривания растворителя получают еще 4,2 г масла. Часть (4,2 г) маслянистой соли тетрабутиламмония растворяют в 100 мл бромхлорметана и раствор перемешивают при 20oC в течение 40 ч. После упаривания бромхлорметана в вакууме остаток повторно экстрагируют диэтиловым эфиром. Объединенные эфирные экстракты упаривают до 2,3 г прозрачного масла. Масло очищают хроматографированием на силикагеле, в качестве элюента используют смеси хлороформ/этилацетат, 80-20, получают 1,62 г масла.
Методика 13. Йодметиловый эфир N,N-диэтиламида янтарной кислоты.
Йодид натрия (2,19 г) и хлорметилового эфира N,N-диэтиламида янтарной кислоты (1,62 г) смешивают с 15 мл ацетона и смесь перемешивают при 20oC в течение 3 ч. Растворитель упаривают в вакууме и остаток распределяют между метиленхлоридом и водой. Слой метиленхлорида отделяют, сушат безводным сульфатом натрия и упаривают при пониженном давлении, получают 1,81 г масла, которое используют без дополнительной очистки.
Методика 14. Дипропиламмониевая соль N,N-дипропиламида янтарной кислоты.
Ангидрид янтарной кислоты (15,0 г) добавляют к предварительно охлажденному (0oC) раствору дипропиламина (41 мл) в 200 мл метанола. Охлаждение прекращают и смесь перемешивают в течение 30 мин, затем растворитель отгоняют, получают 42,8 г бледно-желтого масла.
Методика 15. Хлорметиловый эфир N,N-дипропиламида янтарной кислоты.
Смесь, содержащую дипропиламмоний N,N-дипропилсукцинаната (41,8 г), гидросульфат тетрабутиламмония (48,05 г), метиленхлорид 250 мл, воду (80 мл) и 1 н. раствора гидроксида натрия (142 мл) перемешивают в течение 1,5 ч. Слой метиленхлорида отделяют, сушат безводным сульфатом натрия и упаривают в вакууме, получают 44,12 г масла. Двадцать граммов масла растворяют в 100 мл бромхлорметана и раствор перемешивают в течение 4,5 ч. Бромхлорметан упаривают в вакууме. Остаток экстрагируют несколько раз диэтиловым эфиром, экстракты объединяют и упаривают, получают 6,5 г желтого масла, которое используют без дополнительной очистки.
Методика 16. Йодметиловый эфир N,N-дипропиламида янтарной кислоты.
Йодид натрия (7,8 г) добавляют к раствору хлорметилового эфира N,N-дипроламида янтарной кислоты (6,5 г) в 30 мл ацетона и полученную смесь перемешивают в течение 2,5 ч. После упаривания растворителя в вакууме полученный остаток растворяют в смеси метиленхлорида и воды. Слой метиленхлорида отделяют, сушат безводным сульфатом натрия и упаривают, получают 8,8 г оранжевого масла, которое используют без дополнительной очистки.
Методика 17. Гомопиперидиний 4-гомопиперидино-4-оксобутират.
Ангидрид янтарной кислоты (15,0 г) осторожно добавляют к раствору гомопиперидина (33,78 мл) в терагидрофуране (100 мл) и полученную смесь перемешивают в течение 30 мин. Растворитель упаривают в вакууме, получают 33,16 г масла.
Методика 18. Хлорметил 4-гомопиперидино-4-оксобутират.
Смесь гомопиперидиний 4-гомопиперидино-4-оксобутирата (33,16 г), гидросульфата тетрабутиламмония (49,74 г), метиленхлорида (200 мл), воды (50 мл) и 1 н. раствора гидроксида натрия (146 мл) перемешивают в течение 1,5 ч. Фазу метиленхлорида отделяют, сушат безводным сульфатом натрия и упаривают в вакууме, получают 22,0 г прозрачного масла. Масло растворяют в 100 мл бромхлорметана и перемешивают в течение 17 ч при 20oC. После упаривания в вакууме бромхлорметана остаток экстрагируют диэтиловым эфиром. Эфирный раствор сушат сульфатом натрия и упаривают в вакууме, получают 10,5 г масла. Масло очищают хроматографией на силикагеле с использованием в качестве элюента смеси хлороформ/этилацетат, 85: 15. Фракции, содержащие требуемый хлорметиловый эфир, объединяют и упаривают, получают 4,36 г прозрачного масла.
Методика 19. Йодметил 4-гомопиперидино-4-оксобутират.
Йодид натрия (5,28 г) добавляют к раствору хлорметил 4-гомопиперидино-4-оксобутирата (4,36 г) в 20 мл ацетона и смесь перемешивают в течение 3 ч. Растворитель упаривают в вакууме и остаток распределяют между метиленхлоридом и водой. Слой метиленхлорида отделяют, сушат безводным сульфатом натрия и упаривают в вакууме, получают 4,4 г масла, которое используют без дополнительной очистки.
Методика 20. Метил-N,N-диметиламинокарбонилоксиацетат.
Пиридин (67 мл), метиловый эфир гликолевой кислоты (12,85 мл) и диметилкарбамоилхлорид (15,33 мл) смешивают при 0oC, затем охлаждающую баню удаляют и раствор перемешивают при 20oC в течение 17 ч. Затем реакционную смесь нагревают до 65oC в течение 4 ч, после чего охлаждают, добавляют воду и этилацетат и полученную смесь подкисляют добавлением 1 н. соляной кислоты. Слой этилацетата отделяют, промывают рассолом, сушат безводным сульфатом натрия и концентрируют в вакууме, получают 7,1 г желтого масла. Остаток хроматографируют на силикагеле, элюент хлороформ. Приемники, как установлено, содержат желаемый продукт, их содержимое объединяют и концентрируют, получают 2,5 г масла.
Методика 21. Натриевая соль N,N-диметиламинокарбонилокси уксусной кислоты.
Вышеописанный эфир (2,5 г) растворяют в 10 мл метанола, добавляют 1 н. раствор гидроксида натрия (15,5 мл) и полученный раствор перемешивают при 20oC в течение 1 ч. После упаривания реакционной смеси при пониженном давлении получают белый твердый продукт, который используют без дополнительной очистки.
Методика 22. Хлорметил N,N-диметиламинокарбонилокси-ацетат.
Натриевую соль N, N-диметиламинокарбонилоксиуксусной кислоты (2,62 г) смешивают со 100 мл метиленхлорида, 30 мл воды 5,27 г гидросульфата тетрабутиламмония и 1,3 г гидрокарбоната натрия и перемешивают в течение 1,5 ч. Слой метиленхлорида отделяют, сушат безводным сульфатом натрия и упаривают в вакууме, получают 2,8 г масла. Масло растворяют в 80 мл бромхлорметана и перемешивают при 20oC в течение 18 ч. Метиленхлорид упаривают в вакууме и остаток несколько раз экстрагируют диэтиловым эфиром. Экстракты объединяют и упаривают, получают 0,95 г масла, которое используют без дополнительной очистки.
Методика 23. Йодметил N,N-диметиламинокарбонилокси-ацетат.
Йодид натрия (1,46 г) и хлорметил N,N-диметиламинокарбонилоксиацетат (0,95 г) смешивают с 30 мл ацетона и перемешивают при 20oC в течение 19 ч. Ацетон упаривают в вакууме, получают 1,18 г масла, которое используют без дополнительной очистки.
Методика 24. Метил N,N-диэтиламинокарбонилоксиацетат.
Пиридин (54 мл), метиловый эфир гликолевой кислоты (10,3 мл) и диэтилкарбамоилхлорид (16,9 мл) смешивают и выдерживают при 65oC в течение 40 ч, затем еще в течение 24 ч при 85oC и дополнительно в течение 24 ч при 95oC. После охлаждения реакционную смесь распределяют между этилацетатом и водой. Слой этилацетата отделяют, промывают 1 н. HCl, водой и рассолом, сушат над безводным сульфатом натрия и концентрируют в вакууме, получают 15,0 г масла, которое используют без дополнительной очистки.
Методика 25. Натриевая соль N,N-диэтиламинокарбонилоксиуксусной кислоты.
Вышеописанный эфир (15 г) растворяют в 100 мл метанола, добавляют 1 н. раствор гидроксида натрия (79,3 мл) и полученный раствор перемешивают при 20oC в течение 1,5 ч. Упаривание реакционной смеси при пониженном давлении дает белое твердое вещество, 15,3 г (после сушки над P2O5), которое используют без дополнительной очистки.
Методика 26. Йодметил N,N-диэтиламинокарбонилоксиацетат.
Натриевую соль N,N-диэтиламинокарбонилоксиуксусной кислоты (15,3 г) смешивают с 800 мл метиленхлорида, 240 мл воды, 26,35 г гидросульфата тетрабутиламмония и 6,52 г гидрокарбоната натрия и перемешивают в течение 1,5 ч. Слой метиленхлорида отделяют, сушат безводным сульфатом натрия и упаривают в вакууме, остаток несколько раз экстрагируют диэтиловым эфиром. Экстракты объединяют и упаривают, получают 10,63 г масла. Сырой хлорметиловый эфир смешивают с йодидом натрия (14,25 г) в 100 мл ацетона и перемешивают при 20oC в течение 3,5 ч. Растворитель упаривают в вакууме и остаток распределяют между хлороформом и водой. Слой хлороформа сушат безводным сульфатом натрия и упаривают в вакууме, получают 11,56 г светло-желтого масла, которое используют без дополнительной очистки.
Методика 27. Этиловый эфир N-пивалоилглицина.
В охлаждаемую ледяной баней колбу, содержащую 150 мл метиленхлорида, при перемешивании добавляют гидрохлорид этилового эфира глицина (25,0 г), пивалоилхлорид (20,9 мл), а затем медленно добавляют 62,4 мл N,N-диизопропилэтиламина. После удаления ледяной бани реакционную смесь перемешивают в течение 4 ч. Растворитель упаривают в вакууме и оставшееся масло распределяют между этилацетатом и водой. Слой этилацетата промывают смесью 1:1 насыщенного водного раствора гидрокарбоната натрия и воды, сушат безводным сульфатом натрия и упаривают в вакууме, получают 11,3 г масла.
Методика 28. Натриевая соль N-пивалоилглицина.
К раствору этилового эфира N-пивалоилглицина (11,3 г) в 50 мл метанола добавляют 69,2 мл 1 н. раствор гидроксида натрия и полученный раствор перемешивают в течение 2 ч. Растворитель упаривают в вакууме. Добавляют свежий метанол и еще раз упаривают, получают 12,45 г практически белого масла.
Методика 29. Хлорметиловый эфир N-пивалоилглицина.
К раствору натриевой соли N-пивалоилглицина (12,45 г) и гидросульфата тетрабутиламмония (21,8 г) в 100 мл метиленхлорида и 50 мл воды при перемешивании добавляют 64,3 мл 1 н. раствора гидроксида натрия и полученную смесь перемешивают в течение 3 ч. Слой метиленхлорида отделяют, сушат сульфатом натрия и упаривают в вакууме, получают 20,9 г масла. Масло растворяют в 50 мл бромхлорметана и перемешивают в течение 17 ч при 20oC. После упаривания бромхлорметана остаток обрабатывают несколько раз диэтиловым эфиром. Объединенные экстракты упаривают в вакууме, получают 2,2 г масла. После хроматографирования на силикагеле, элюент этилацетат /хлороформ, получают 1,2 г хлорметилового эфира.
Методика 30. Йодметиловый эфир N-пивалоилглицина
Смесь йодида натрия (1,71 г), хлорметилового эфира N-пивалоилглицина (1,2 г) и ацетона (20 мл) перемешивают при 20oC в течение 19 ч. Растворитель упаривают в вакууме и остаток распределяют между хлороформом и водой. Фазы разделяют и водный слой повторно экстрагируют хлороформом. Слои хлороформа объединяют, сушат безводным сульфатом натрия и упаривают в вакууме, получают 1,45 г масла. Продукт используют без дополнительной очистки.
Методика 31. Этиловый эфир N-(2-этилбутирил)глицина.
В охлаждаемую ледяной баней колбу, содержащую 200 мл метиленхлорида, при перемешивании добавляют гидрохлорид этилового эфира глицина (25,0 г), ангидрид 2-этилмасляной кислоты (39,3 мл), а затем медленно добавляют 54,8 мл триэтиламина. После удаления ледяной бани реакционную смесь перемешивают в течение 17 ч. Растворитель упаривают в вакууме и остаток распределяют между этилацетатом и водой. Слой этилацетата промывают смесью насыщенного водного раствора гидрокарбоната натрия и воды, 1:1, сушат безводным сульфатом натрия и упаривают в вакууме, получают 33,75 г белого рыхлого твердого вещества.
Методика 32. Натриевая соль N-(2-этилбутирил) глицина.
К раствору этилового эфира N-(2-этилбутирил) глицина (33,75 г) в 100 мл метанола добавляют 167,6 мл 1 н. раствора гидроксида натрия и раствор перемешивают в течение 1,5 ч. Растворители упаривают в вакууме, добавляют свежий метанол и затем упаривают, получают 32,6 г светло-желтого масла.
Методика 33. Хлорметиловый эфир N-(2-этилбутирил) глицина.
К раствору натриевой соли N-(2-этилбутирил) глицина (32,6 г) и гидросульфата тетрабутиламмония (53,3 г) в 100 мл метиленхлорида и 100 мл воды при перемешивании добавляют 157 мл 1 н. раствора гидроксида натрия и смесь перемешивают в течение 2,5 ч. Фазы разделяют и водный слой повторно экстрагируют 400 мл метиленхлорида. Органические слои объединяют, сушат сульфатом натрия и упаривают в вакууме, получают 59,8 г масла. Масло растворяют в 75 мл бромхлорметана и перемешивают в течение 15 ч при 20oC. После упаривания бромхлорметана остаток обрабатывают несколько раз диэтиловым эфиром. Объединенные экстракты упаривают в вакууме, получают 7,84 г масла. После хроматографирования на силикагеле, элюент этилацетат/хлороформ, 3:1, получают 2,33 г хлорметилового эфира.
Методика 34. Йодметиловый эфир N-(2-этилбутирил)глицина.
Смесь йодида натрия (3,15 г), хлорметиловый эфир N-(2-этилбутирил)глицина (2,33 г) и ацетона (20 мл) перемешивают при 20oC в течение 17 ч. Растворитель упаривают в вакууме и остаток распределяют между метиленхлоридом и водой. Фазы разделяют и водный слой дважды экстрагируют метиленхлоридом. Органические экстракты объединяют, сушат безводным сульфатом натрия и упаривают в вакууме, получают 2,78 г твердого вещества. Продукт используют без дополнительной очистки.
Методика 35. 1-Йодэтил-изопропилкарбонат.
1-Хлорметилхлороформиат переводят в 1-йодэтил-изопропилкарбонат с использованием известного способа (Wan-Joo Kim, et al., The Journal of Antibiotics, (1991) 44, pg. 1086).
Галоидалкильные сложноэфирные полупродукт приведены в табл.5.

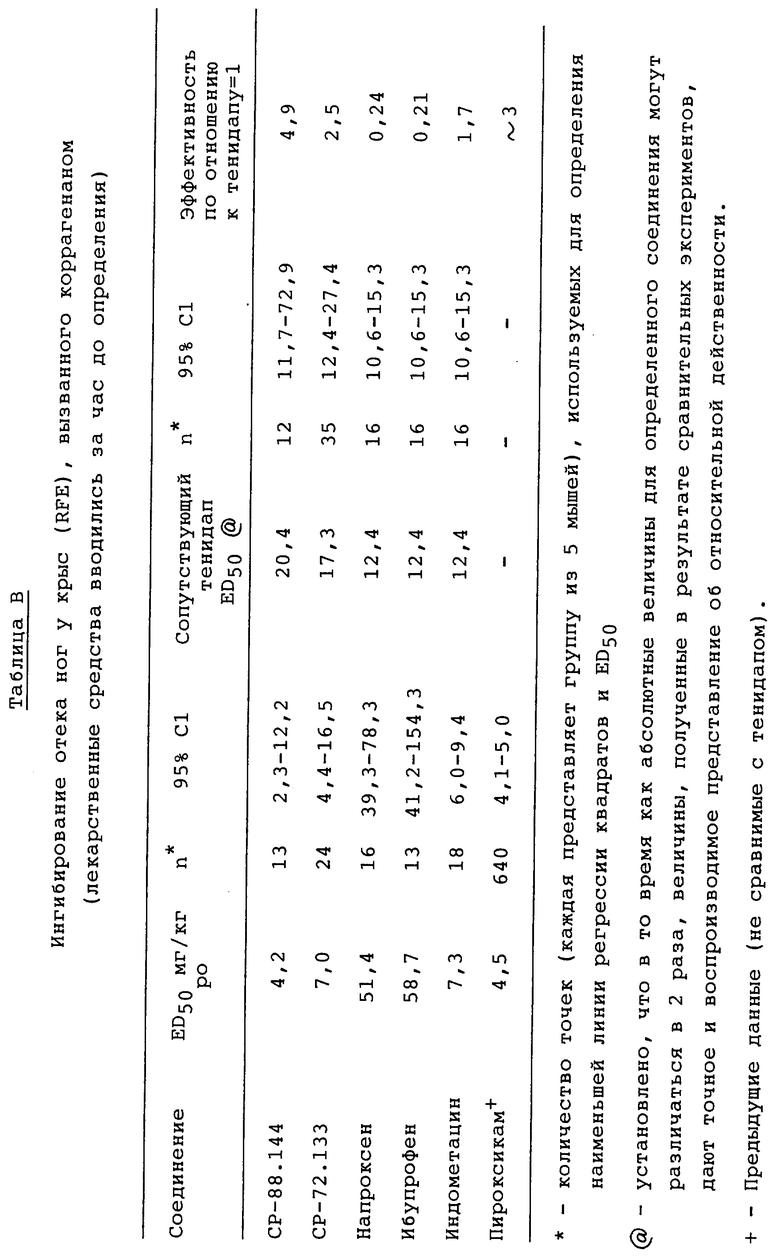
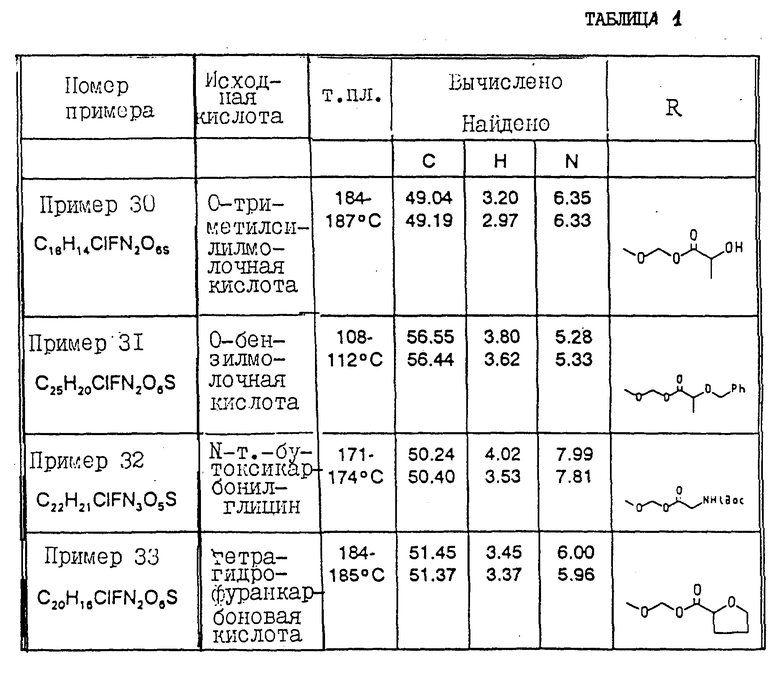
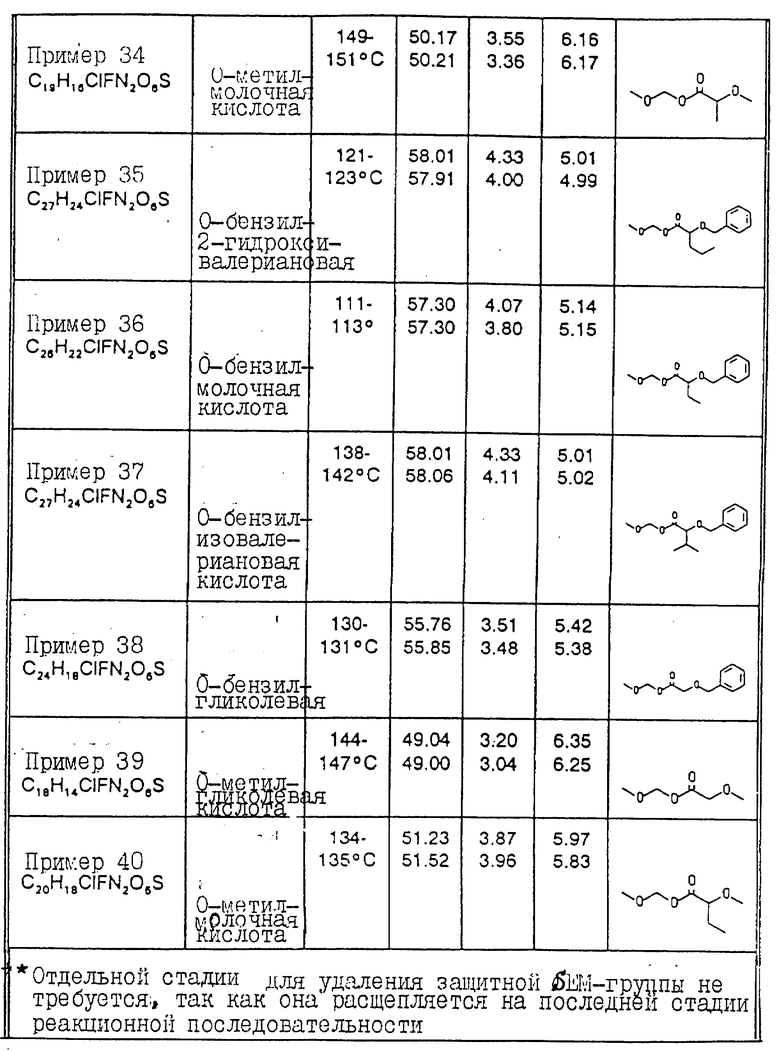
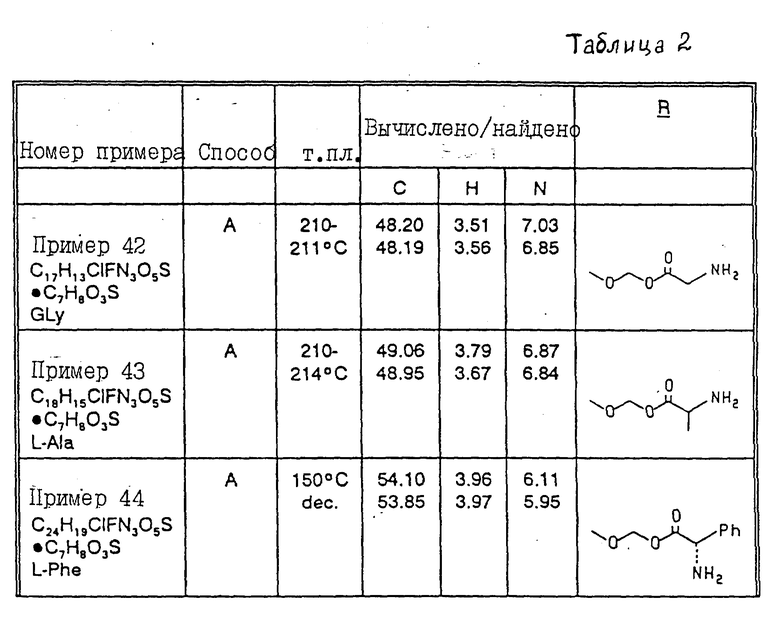
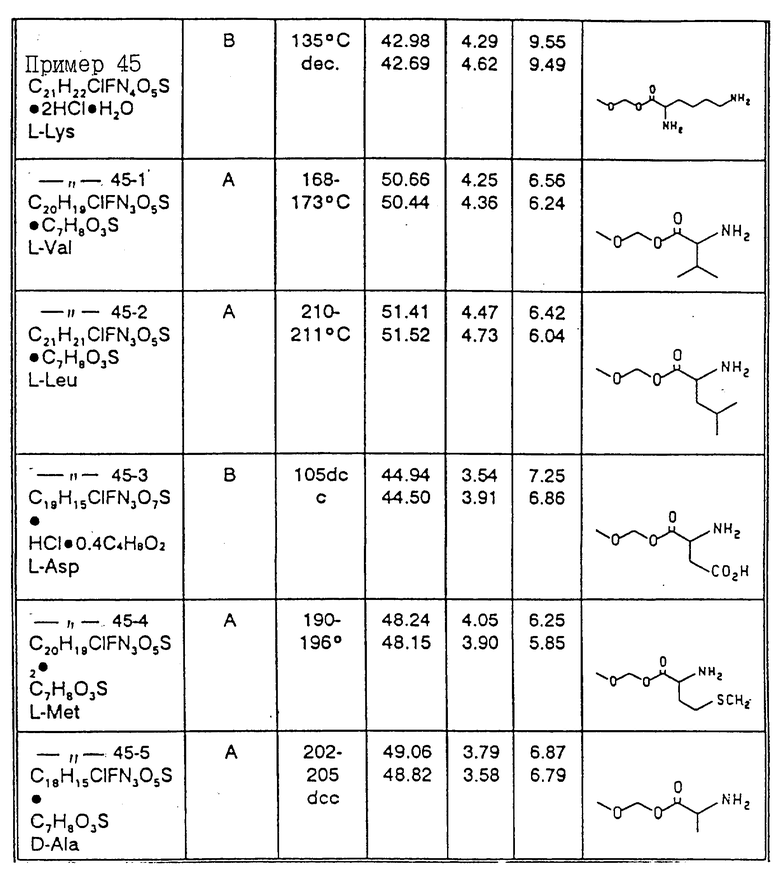

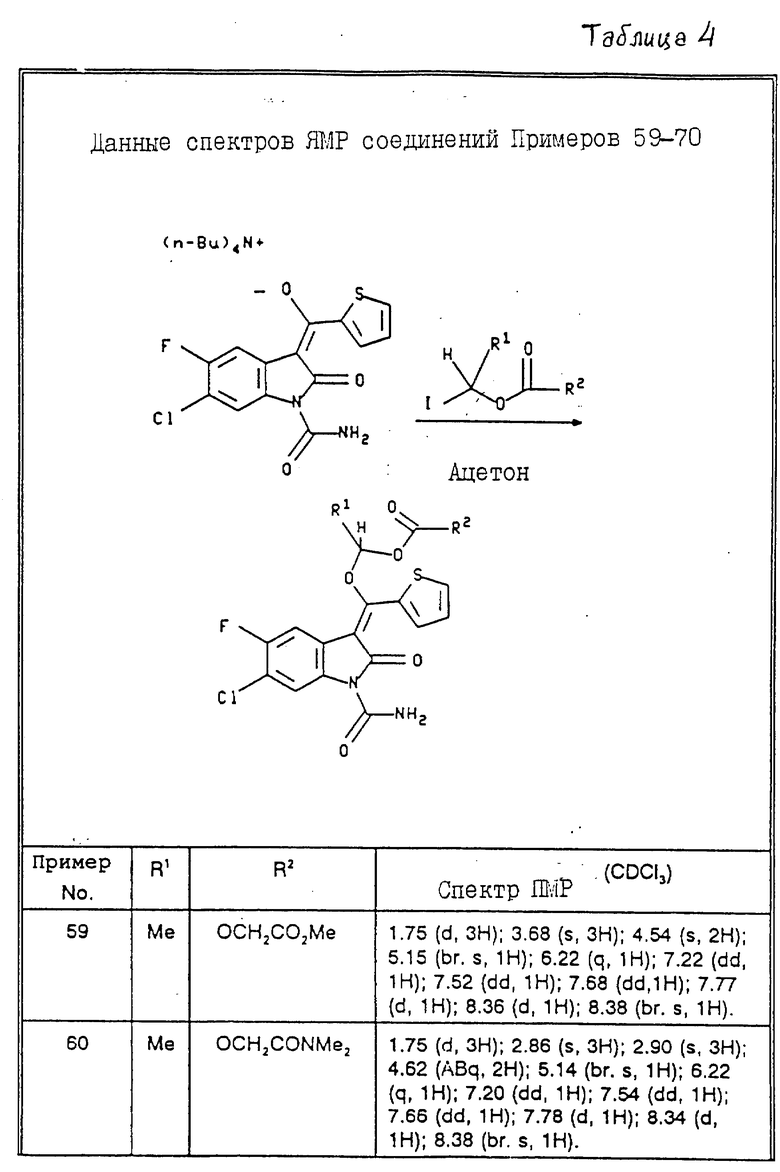
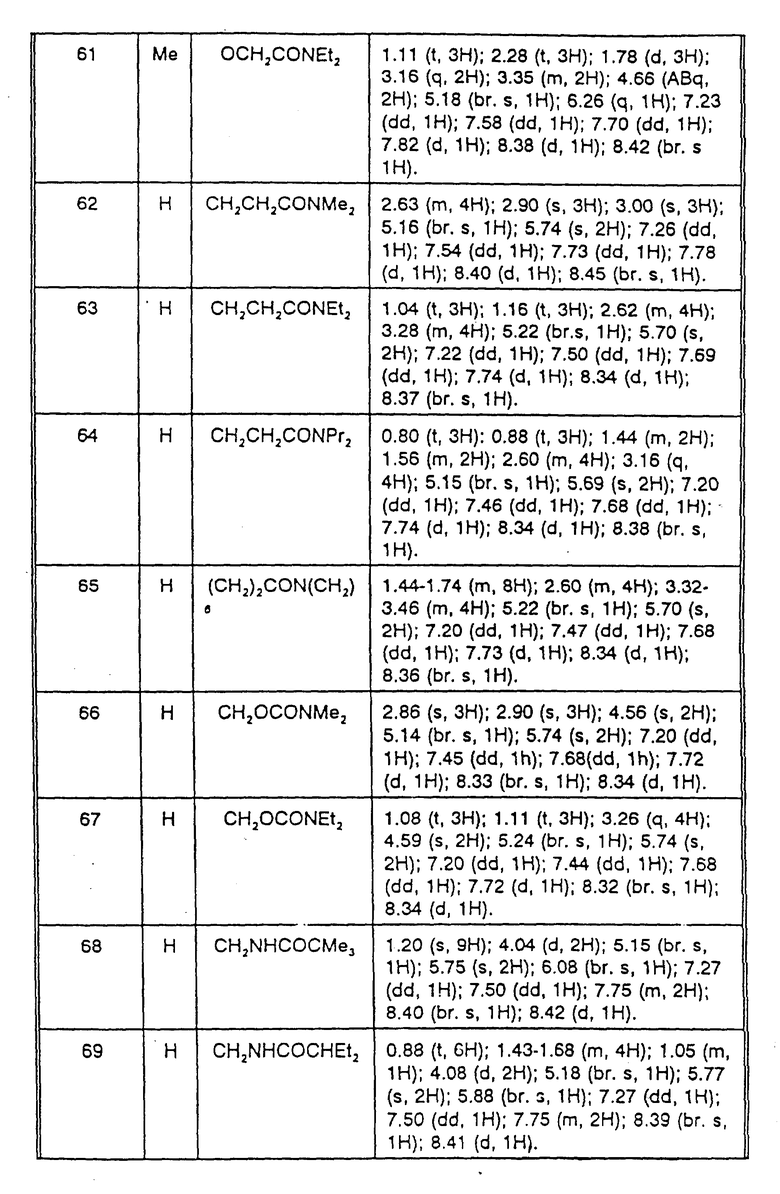

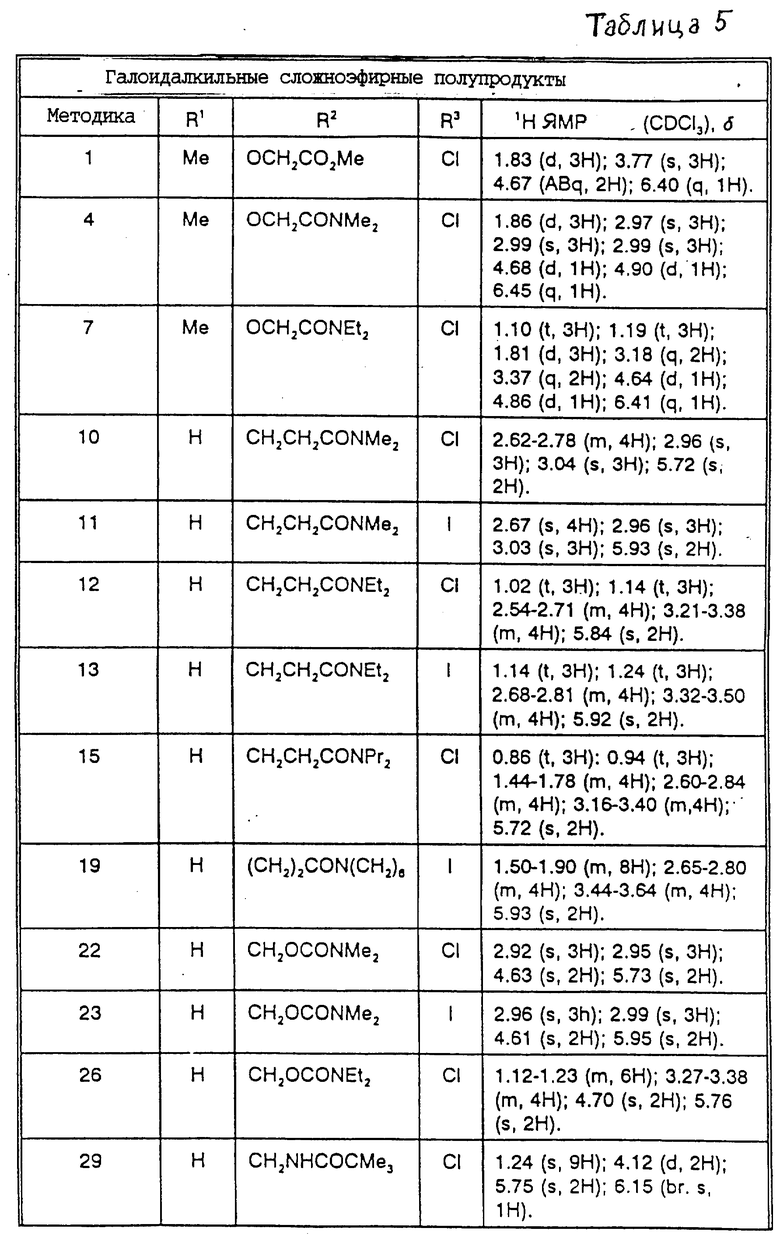
Пролекарства-3-ацил-2-оксииндол-1-карбоксамида формулы I, где R представляет собой фрагменты формулы II-VI, x - O или 1, A - C1-5-алкилен, необязательно замещенный 1-2 C3-7-циклоалкилами или фениленовая группа; B - C2-5-алкенилфенил, пиридил, пиперидинил, пирролидинил, -OCH2CO2 R1 или -OCH2CONR2R3; R1 - H, алкил, фенил низший алкил или (CH2)pCONR2R3; R2 и R3 - H, алкил или вместе с присоединенным атомом азота образуют морфолиновую группу; R4 и R5 - H, алкил, (CH2)pCO-NR4R3, (CH2)pNR7R8, (CH2)pOR6 или (CH2)pSR6; R6 - H, алкил, C3-7-циклоалкил, необязательно замещенный 1-2 алкильными группами, или взятые вместе с заместителем и присоединенным атомом кислорода образуют тетрагидрофурановое кольцо; R7 и R8 - H, алкил; R9 - H или метил; R10 и R11 - галоген; R12 и R13 - H, галоген, p = 1 - 3. Соединения I относятся к фенольным сложноэфирным и эфирнм пролекарствам оксииндола, обладающим обезболивающим и противовоспалительным действием. 3 с. и 12 з.п.ф-лы, 7 табл.
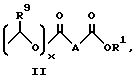
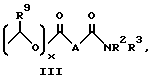
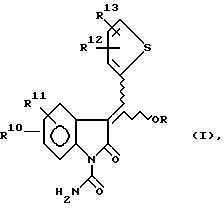
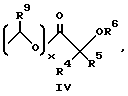
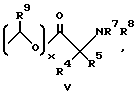
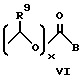

где R представляет собой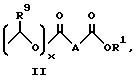
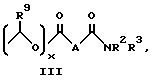
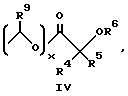
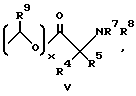
или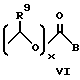
где x принимает значения 0 или 1;
А представляет собой С1-С5-алкилен, необязательно замещенный вплоть до двух С3-С7-циклоалкилами, или представляет собой фениленовую группу;
В представляет собой С2-С6-алкенилфенил, 2-, 3- или 4-пиридил, 2-, 3- или 4-пиперидинил, 2- или 3-пирролидинил, -OCH2CO2R1 или -OCH2CONR2R3;
заместитель R1 представляет собой H, С1-С7-алкил, фенил (С1-С4)алкил или (CH2)p CONR2R3;
заместители R2 и R3 независимо друг от друга представляют собой H, С1-С7-алкил, или вместе с присоединенным атомом азота представляют собой морфолиновую группу;
R4 и R5 независимо друг от друга представляют собой H, С1-С7-алкил, (CH2)p CONR2R3, (CH2)p NR7R8, (CH2)p OR6 или (CH2)pSR6;
R6 представляет собой H, С1-С6-алкил, С3-С7-циклоалкил, необязательно замещенный вплоть до двух С1-С6-алкильными группами, или, взятый вместе с заместителем R4 и присоединенным атомом кислорода, образует тетрагидрофурановое кольцо;
R7 и R8 независимо друг от друга представляют собой H, С1-С6-алкил;
R9 представляет собой H или метил;
R10 и R11 представляют собой галоген;
R12 и R13 независимо представляют водород, галоген;
p принимает значения 1 - 3;
при условии, что когда заместитель R представляет собой формулу II, а x равен 0, А должен представлять собой группу, отличную от С2-С6-алкиленовой группы.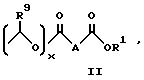
где значения R1, R9, А и x указаны выше.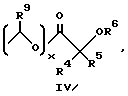
где R4, R5, R6 и R9 имеют значения, указанные выше, и x принимает значение 1.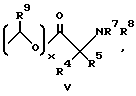
где R4, R5, R7, R8 и R9 имеют значения, указанные выше, и x принимает значение 1.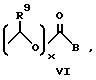
где R9 и В имеют значения, указанные выше, и x принимает значение 1.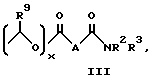
где R2, R3, R9, А и x имеют значения, указанные выше.
| US 5036099, 1991 | |||
| US 5118703, 1992 | |||
| Машковский М.Д | |||
| Лекарственные средства | |||
| - М.: Медицина, ч.1, 1988, с.190-192. |

