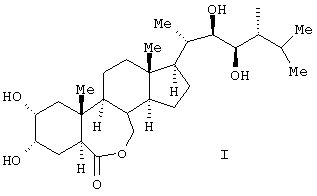
Изобретение относится к получению химического соединения, относящегося к классу стероидных соединений, конкретно к усовершенствованному способу получения 24-эпибрассинолида (2α, 3α, 22R, 23R, 24R)-2,3,22,23-тетрагидрокси-В-гомо-7-окса-5α-холестан-6-она, являющегося биологически активным веществом, фитостимулятором, регулирующего рост растений, ингибирующего транспирацию и увядание, а также средством, усиливающим холодостойкость, ускоряющим рост, утолщение или созревание плодов, корней и корнеплодов, стеблей или луковиц, увеличивающим плодовитость пчел, рыб, отвечающего общей формуле (I):
Известен способ получения стероида изоэргостерина формулы

Изоэргостерин (3α, 5-цикло-24R-метил-5α-холеста-7,22-диен-6β-ол) получают взаимодействием эргостерина с сульфонилгалогенидом - метансульфохлоридом в пиридине (превращают в мезилат) и кипячением полученного сульфопроизводного в водном ацетоне в присутствии бикарбоната калия. Достигают выхода целевого продукта 94%. Данное соединение является промежуточным продуктом в синтезе природного фитогормона - 24-эпибрассинолида (RU 2024541).
Известен способ получения другого стероида 3α, 5-цикло-24S-этил-5α-холест-22-ен-6-она (изоэргостерона) формулы

Способ включает обработку исходного стигмастерина пара-толуолсульфохлоридом, обработку полученного тозилата стигмастерина основанием, окислением 3,5α-цикло-24S-этил-5α-холест-22-ен-6-она хромовой кислотой и выделением целевого продукта, причем в качестве основания используют ацетат калия, и окисление проводят непосредственно в реакционной смеси (SU 1162816, 23.06.1985, кл. С 07 J 9/00). Общий выход продукта 80%. Данное соединение является промежуточным продуктом при получении фитогормона брассинолида и его синтетического аналога (22S, 23S, 24S)-гомобрассинолида, обладающих высокой активностью в качестве стимуляторов роста растений.
Известен способ получения брассиностероидных производных формул (II) и (III).
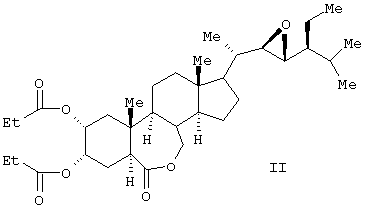
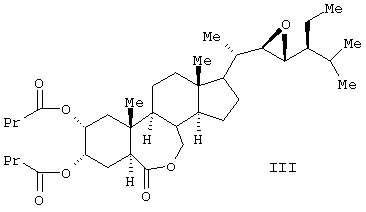
Соединение формулы (II) известного изобретения (называемое далее соединением (II)) и соединение формулы (III) (называемое далее соединением (III)) способны образовываться с большим выходом через дигидроксильные производные, имеющие гидроксигруппы в 2α-положении и в 3α-положении, путем регулирования количества реакционного агента, при осуществлении каталитического гидроксилирования соединения 24S-этил-5α-холеста-2,22Е-диен-6-она, называемого далее соединением (IV) (К.Mori, Agric. Biol. Chem., 44 (50, 1211, 1980) с использованием каталитического количества четырехокиси осмия, в присутствии трет-бутилгидропероксида или N-метилморфолин N-оксида в инертном газе, таком как азот или аргон. Полученные таким образом 2α и 3α-дигидроксисоединения (V) растворяют в пиридине, содержащем 4-диметиламинопиридин для реакции с пропионовым ангидридом или ангидридом масляной кислоты, и таким образом получают (VI) в случае пропионового ангидрида или (VII) в случае ангидрида масляной кислоты. Это соединение (VI) или (VII) растворяют в хлорированном органическом растворителе, который устойчив к окислению, и окисляют органической перекисью, например надбензойной кислотой, м-монохлорнадбензойной кислотой, м-монобромнадбензойной кислотой, мононадфталевой кислотой, трифторнадуксусной кислотой и их натриевой или кальциевой солью таким образом, что из соединения (VI) получается соединение (II) или из соединения (VII) - соединение (III).
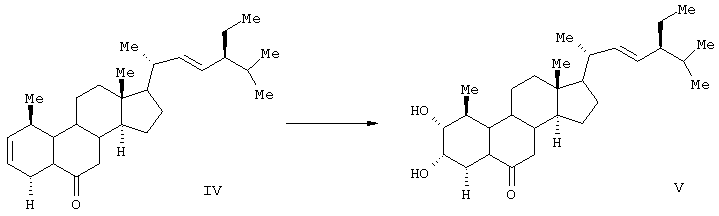
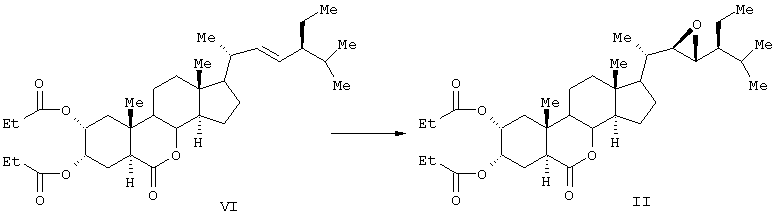
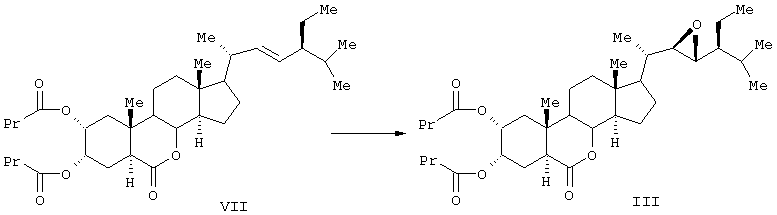
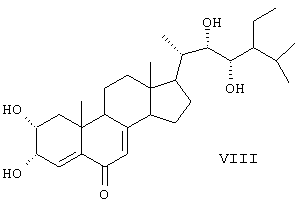
Данные соединения используются в качестве активных ингредиентов. Известен способ получения активного стероида 2α, 3α, 22S, 23S-тетраокси-24S-этидхолест-4-ен-6-она формулы (VIII).
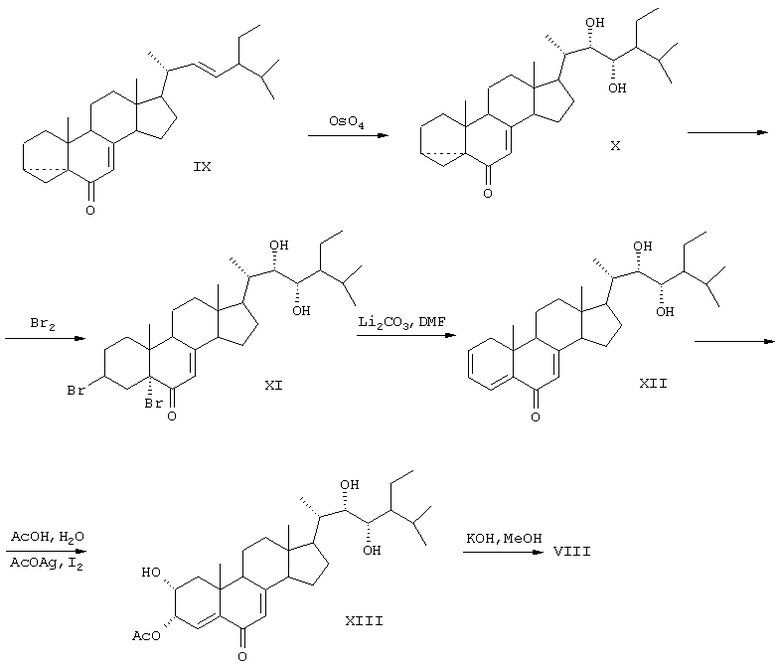
В результате окисления циклокетона (DC) четырехокисью осмия получают диол (X), который обрабатывают бромом. Полученный дибромид (XI) дегидробромируют кипячением в ДМФА в присутствии карбоната лития. Окисляют образовавшийся триенон (XII) в условиях метода Вудворда. На заключительной стадии проводят мягкий щелочной гидролиз и получают 2α, 3α, 22S, 23S-тетраокси-24S-этилхолест-4-ен-6-он (VIII) (SU 1433005 А1, 30.12.1991).
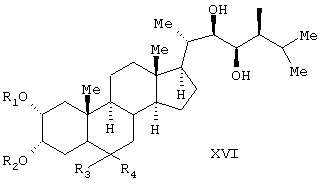
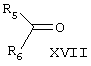
 Известен способ получения брассинолида формулы (XIV) взаимодействием 1 моля стероида формулы (XV), (где R1 и R2 - Н, алкильная или ацильная группа, R5 и R6 - Н, алкил, циклоалкил или замещенный фенил) с 1-5 молями трифторперуксусной кислоты в инертном растворителе при температуре (-20)-(+50)°С. Стероид формулы (XV) получают взаимодействием стероида формулы (XVI), где R1 и R2 - Н, алкильная или ацильная группа; R3 и R4 - карбонил или кеталь) с соединением формулы (XVII), (JP, 62167797, 24.07.1987, С 07 J 3/00).
Известен способ получения брассинолида формулы (XIV) взаимодействием 1 моля стероида формулы (XV), (где R1 и R2 - Н, алкильная или ацильная группа, R5 и R6 - Н, алкил, циклоалкил или замещенный фенил) с 1-5 молями трифторперуксусной кислоты в инертном растворителе при температуре (-20)-(+50)°С. Стероид формулы (XV) получают взаимодействием стероида формулы (XVI), где R1 и R2 - Н, алкильная или ацильная группа; R3 и R4 - карбонил или кеталь) с соединением формулы (XVII), (JP, 62167797, 24.07.1987, С 07 J 3/00).


Технической задачей заявленного изобретения является получение 24-эпибрассинолида более высокого качества, т.е. получение более чистого продукта (с более высокой температурой плавления), что приводит к повышению его биологической активности.
Поставленная техническая задача достигается новым способом получения эпибрассинолида формулы (I),

включающим следующие стадии: а) синтез мезилата эргостерина (XVIII) обработкой природного стерина - эргостерина метансульфохлоридом в пиридине при температуре реакционной среды не более 12°С;
б) синтез изоэргостерина (XIX) кипячением полученного сульфопроизводного - мезилата эргостерина в водном ацетоне в присутствии бикарбоната калия (буферного средства);
в) синтез изоэргостерона (XX) окислением изоэргостерина хромовым ангидридом в пиридине;
г) синтез 7,8-дигидроэргостерона (XXI) восстановлением енона (кетона) изоэргостерина дитионитом натрия в водной микрогетерогенной среде с высокой солюбилизирующей способностью в присутствии солюбилизирующей среды, включающей одноцепочные катионные, анионные или неионные ПАВ (поверхостно-активные вещества) из ряда СnН2n+1X, где n=9-18, Х=NMe3, NEt3, СООН, SO3H, OSO2M, OP(O)(OM)2, где М=металл, полиэтиленгликоль, косолюбилизатор - алифатические спирты С2-С6 или моноэфиры этилен- или диэтиленгликоля, электролит и воду при мольном соотношении их соответственно 1:5-6:100-250;
д) стероидная перегруппировка 7,8-дигидроизоэргостерона при нагревании его в смеси с безводным бромидом лития, гидрохлоридом пиридиния и диметилацетамидом;
е) синтез эпикастастерона (XXII) обработкой (22Е,24R)-5α-эргоста-2,22-диен-6-она метансульфонамидом и карбонатом калия с использованием каталитических количеств гексацианоферрата (III) калия и четырехокиси осмия;
ж) растворение образовавшейся таким образом тетрагидроокиси в хлороформе с последующей обработкой трифторуксусным ангидридом в хлорированном органическом растворителе с последующим окислением перекисью водорода и выделением целевого продукта (I).
В качестве ПАВ в предложенном способе используют, например, следующие катионные, анионные или неионные ПАВ из ряда CnH2n+1X, где n=9-18, X=NMe3, NEt3, СООН, SO3H, OSO2M, ОР(O)(ОМ)2, где М = щелочной металл.
Пример 1. Получение мезилата эргостерина.

В двугорлой колбе на 250 мл, снабженной капельной воронкой, термометром и мешалкой, приготовили раствор 10 г эргостерина в 150 мл пиридина (перегнанного над едким натром). Раствор охладили до 10°С и при перемешивании по каплям добавили 9,4 мл метансульфохлорида с такой скоростью, чтобы температура реакционной смеси не поднималась выше 12°С. Смесь перемешивали 1 час при 8-10°С, после чего вылили ее в 800 мл воды. Полученной суспензии дали отстояться в течение одного часа, затем отфильтровали осадок на воронке Бюхнера. Осадок промыли 1000 мл воды, высушили над едким натром в эксикаторе в течение 12 часов. Получили 11 г (92%) продукта в виде белого порошка, использованного без дополнительной очистки.
Пример 2. Получение изоэргостерина.
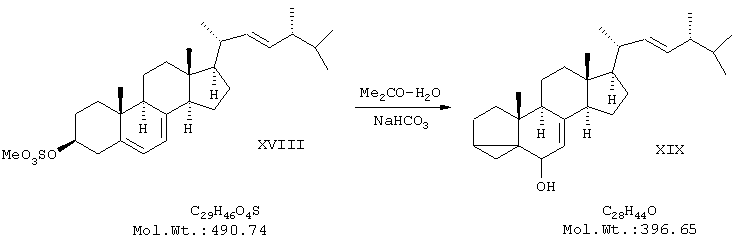
В двугорлой колбе на 2000 мл, снабженной обратным холодильником и мешалкой, смешали 1280 мл ацетона (перегнанного над KMnO4, 320 мл воды и 1,15 г КНСО3. Полученный раствор нагрели до кипения, затем при интенсивном перемешивании быстро засыпали в колбу 5,5 г мелкоизмельченного мезилата эргостерина. Реакционную смесь кипятили в течение 5 минут, после чего заменили обратный холодильник на нисходящий и отогнали 500 мл растворителя. Колбу охладили сначала до комнатной температуры, а потом до 0°С. Выпавший через несколько часов белый хлопьевидный осадок отфильтровали на воронке Бюхнера и промыли 500 мл воды, после чего сушили в эксикаторе над NaOH в течение 12 часов. После высушивания продукт перекристаллизовали из 150 мл ацетона, медленно (в течение 1 часа) охлаждая раствор от комнатной температуры до -20°С. Выпавшие кристаллы отфильтровали на воронке Бюхнера и сушили в вакууме. Получили 3,7 г (80%) продукта в виде бесцветных игольчатых кристаллов с т.пл.130-131°С. ПМР-спектр (CDCl3, 500 МГц): 0,48 (2Н), 0,66 (3Н), 0,85 (6Н), 0,90 (3Н), 1,03 (3Н), 1,10 (3Н), 1,2-2,2 (неразрешенный сложный мультиплет), 3,4 (1Н), 5,2 (2Н), 5,5 (1Н).
Пример 3. Получение изоэргостерона
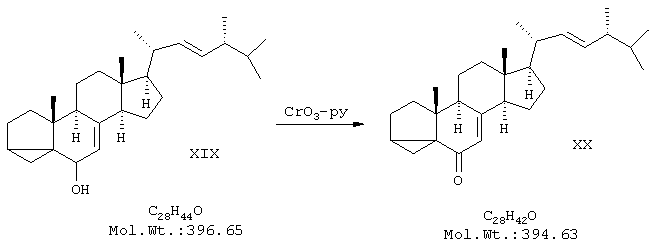
В двугорлую колбу на 500 мл, снабженную капельной воронкой и мешалкой, поместили 70 мл пиридина (перегнанного над NaOH). Колбу охладили до 15°С и при перемешивании добавили 1,15 г хромового ангидрида. Смесь перемешивали 10 мин до полного растворения хромового ангидрида. В течение 50 минут при интенсивном перемешивании добавили еще 5 порций CrO3 по 1,15 г, поддерживая температуру 15-20°С. Получили суспензию комплекса CrO3 с пиридином в пиридине. К суспензии добавили с помощью капельной воронки раствор 6,96 г изоэргостерина в 70 мл пиридина. Реакционную смесь перемешивали 24 ч при 20°С. После этого смесь разбавили 300 мл диэтилового эфира, выпавший осадок отфильтровывали на воронке Бюхнера, промывали 100 мл эфира. Фильтрат упарили в вакууме досуха, твердый остаток перекристаллизовали из 10 мл ацетона, получили 4,63 г продукта в виде бледно-желтых пластинчатых кристаллов с т.пл.169-170°С. Маточный раствор упарили и полученный остаток хроматографировали на колонке с силикагелем, используя в качестве элюента смесь петролейного эфира и этилацетата в соотношении 10:1. Таким образом получили еще 0,84 г продукта с т.пл.168-169°С. Общий выход 5,47 г (79%). ПМР-спектр (CDCl3, 500 МГц): 0,65 (3Н), 0,77 (1Н), 0,84 (6Н), 0,93 (3Н), 1,05 (3Н), 1,2-2,3 (неразрешенный сложный мультиплет), 5,2 (2Н), 5,8 (1Н).
Пример 4. Получение 7,8-дигидроизоэргостерона.

Реакцию ведут в водной микрогетерогенной среде с высокой солюбилизующей способностью (далее - солюбилизующей среде). Для получения таких сред используется смесь поверхностно-активного солюбилизатора, косолюбилизатора, воды и электролита в соотношении, обеспечивающем достижение оптимального гидрофильно-липофильного баланса системы. Полученные среды термодинамически стабильны, обладают визуальной гомогенностью (прозрачны и не расслаиваются при произвольно долгом выдерживании в диапазоне температур от комнатной до 100°С), способны солюбилизовать без внешних признаков образования новой фазы значительные количества гидрофобных водонерастворимых органических соединений (например, углеводородов, стероидов) до молярных концентраций. В качестве солюбилизатора используются одноцепочечные катионные, анионные или неионные ПАВ из ряда CnH2n+1X, где n=9-18; Х=NMe3, NEt3, СООМ, SO3М, OSO2M, ОР(O)(ОМ)2, ПЭГ.
В качестве косолюбилизатора используются алифатические спирты нормального или разветвленного строения с числом атомов углерода от 2 до 6, а также моноэфиры моно- или диэтиленгликоля.
Состав солюбилизующей среды - солюбилизатор-косолюбилизатор-вода в мольном соотношении 1:5-6:100-250. Компоненты смешиваются до получения однородного раствора, используемого далее в качестве среды реакции. В двугорлую колбу на 250 мл, снабженную мешалкой и обратным холодильником, поместили 65 мл солюбилизующей среды (в качестве примера среду приготовили из 3,6 мл н-бутанола, 3,6 г цетилтриметиламмонийбромида и 60 мл воды. Смесь перемешивали при 20°С до тех пор, пока не образовался прозрачный раствор (около 10 мин). К полученному раствору при перемешивании прилили раствор 1,16 г енона в 6 мл толуола и 2,1 г гидрокарбоната натрия. Через образовавшуюся смесь пропускали при интенсивном перемешивании ток азота или аргона в течение 10 мин, после чего смесь нагрели до кипения и добавили 1,2 г дитионита натрия. Кипятили 1,5 ч, добавив в течение этого времени еще 2 порции дитионита натрия по 250 мг. После этого охладили до 20°С, разбавили хлористым метиленом (100 мл) и вылили в 300 мл насыщенного раствора NaCl. Отделили органический слой, промыли его водой, (2×200 мл), насыщенным раствором NaCl (1×200 мл) и высушили над Na2SO4. Раствор упарили в вакууме, твердый остаток растворили в 50 мл диэтилового эфира и пропустили через короткую колонку (5 см) с силикагелем, раствор упарили в вакууме. Остаток хроматографировали на силикагеле (циклогексан/этилацетат 80/1). Получили 0,93 г (80%) продукта в виде белого порошка с т.пл.110-111°С. ПМР-спектр (CDCl3, 500 МГц): 0,70 (3Н), 0,81 (6Н), 0,92 (3Н), 1,02 (6Н), 1,1-2,1 (неразрешенный сложный мультиплет), 5,2 (2Н).
Пример 5. Получение (22Е, 24R)-5α-эргоста-2,22-диен-6-она.

В двугорлой колбе на 50 мл, снабженной обратным холодильником, смешали 1 г дигидроизоэргостерона, 0,11 г безводного бромида лития, 0,06 г гидрохлорида пиридиния, 10 мл диметилацетамида. В колбу впустили ток азота, после чего ее нагрели до 160°С и выдержали 3 ч при этой т-ре. Смесь охладили до 20°С и вылили ее в 200 мл воды. Осадок отфильтровали на воронке Бюхнера и высушили в эксикаторе над NaOH. Полученное твердое вещество перекристаллизовали из 30 мл метанола, получили 0,82 г продукта в виде белого порошка с т.пл.123-4°С (выход 82%). ПМР-спектр (CDCl3, 500 МГц): 0,67 (3Н), 0,70 (3Н), 0,82 (6Н), 0,91 (3Н), 1,02 (3Н), 1,2-2,4 (неразрешенный сложный мультиплет), 5,2 (2Н), 5,60 (1Н), 5,68 (1H).
Пример 6. Получение 24-эпикастастерона.

В колбе на 100 мл, снабженной мешалкой, смешали 3 г эргоста-2,22-диен-6-она, 1,497 г гексацианоферрата(III) калия, 0,629 г карбоната калия, 0,144 г метансульфонамида, 0,011 г осмата калия, 0,071 г 4-хлорбензоата дигидрохинидина, 15 мл трет-бутанола и 15 мл воды. Смесь перемешивали при 20°С в течение 6 суток, после чего добавили 0,6 г сульфита натрия и перемешивали еще 18 часов. Трет-бутанол отогнали в вакууме, остаток экстрагировали этилацетатом (6×20 мл). Остаток промыли 1 раз водой (20 мл), 0,25 М серной кислотой (3×20 мл), насыщенным раствором NaCl (1×20 мл). Этилацетат упарили и полученный остаток хроматографировали на силикагеле, используя в качестве элюента смесь хлороформа и этилацетат (9:1). Фракции, содержащие эпикастастерон упарили, получили 0,23 г продукта в виде белого порошка с т.пл.247-248°С (Выход 77%). ПМР-спектр (CDCl3, 500 МГц): 0,67 (3Н), 0,77 (3Н), 0,86 (6Н), 0,93 (3Н), 1,0 (3Н), 1,1-2,2 (неразрешенный сложный мультиплет), 2,25 (2Н), 2,66 (1Н), 3,41 (1Н), 3,7 (1Н), 3,8 (1Н), 4,1 (1Н).
Пример 7. Получение эпибрассинолида

В двугорлой колбе на 50 мл, снабженной капельной воронкой, мешалкой, хлоркальциевой трубкой, смешали 3,4 мл хлороформа, 2,6 мл трифторуксусного ангидрида и 0,415 мл 30% р-ра перекиси водорода. Полученный раствор охладили до 0°С и по каплям при перемешивании добавили раствор 0,17 г эпикастастерона в 10 мл хлороформа. Смесь перемешивали 2 ч при 20°С, после этого разбавили 7 мл хлороформа. Полученную смесь промыли водой (1×10 мл), водным раствором карбоната натрия (2×10 мл), водным раствором гидросульфита натрия (2×10 мл), насыщенным раствором NaCl, а затем высушили над сульфатом натрия. Растворитель упарили в вакууме и остаток перекристаллизовали из 20 мл этилацетата, получив 0,14 г эпибрассинолида в виде белого порошка с т.пл.256-258°С. Получено 0,14 г (80% от теории) ПМР-спектр (CDCl3, 500 МГц): 0,70 (3Н), 0,85 (6Н), 0,92 (6Н), 1,05 (3Н), 1,2-2,3 (неразрешенный сложный мультиплет), 3,10 (1Н), 3,45 (1Н), 3,7 (2Н), 4,01 (1Н), 4,2 (1Н).
Преимуществами заявленного способа являются:
а) использование широкого круга дешевых простых взаимозаменимых ПАВ, в том числе выпускаемых отечественной промышленностью вместо уникального импортного межфазного переносчика, не имеющего доступных аналогов;
б) солюбилизующая среда является однофазной термодинамически устойчивой жидкостью, вследствие чего реакционная смесь не требует особых режимов высокоэффективного перемешивания (специальных мешалок, контроля скорости вращения и т.п.). Могут с успехом применяться обычные мешалки (магнитные или механические), и результаты процесса практически не зависят от условий перемешивания;
в) результаты реакции воспроизводимы при соблюдении основных параметров процесса, указанных в методике;
г) процесс не зависит от малоэффективного межфазного переноса аниона восстановителя, что позволяет использовать меньшие количества дитионита натрия;
д) выделение продукта осуществляется с помощью стандартной процедуры экстракции и не требует использования больших количеств дорогого хроматографического силикагеля;
е) продукт более высокого качества (более высокая т.пл.), так как не содержит примесей межфазного переносчика.
| название | год | авторы | номер документа |
|---|---|---|---|
| Способ получения 24-эпибрассинолида | 2023 |
|
RU2839811C2 |
| СПОСОБ ПОЛУЧЕНИЯ α 5-ЦИКЛО-24R-МЕТИЛ- a -ХОЛЕСТ-7,22-ДИЕН -6β -ОЛА | 1991 |
|
RU2024541C1 |
| СПОСОБ ВОССТАНОВЛЕНИЯ НЕНАСЫЩЕННЫХ КЕТОНОВ В НАСЫЩЕННЫЕ КЕТОНЫ | 2005 |
|
RU2293720C1 |
| СПОСОБ ПОЛУЧЕНИЯ НОВЫХ 3β-АЦЕТОКСИ-17α-ГИДРОПЕРОКСИ-16α-МЕТИЛПРЕГНАНОВ ИЗ Δ-20-КЕТОСТЕРОИДОВ И СПОСОБ ПОЛУЧЕНИЯ 3β-АЦЕТОКСИ-17α-ГИДРОКСИ-16α-МЕТИЛПРЕГНАНОВ С ИСПОЛЬЗОВАНИЕМ 3β-АЦЕТОКСИ-17α-ГИДРОПЕРОКСИ-16α-МЕТИЛПРЕГНАНОВ | 2009 |
|
RU2418805C1 |
| СПОСОБ ПОЛУЧЕНИЯ 6-ДЕГИДРО-6-МЕТИЛГИДРОКОРТИЗОНА ИЛИ ЕГО ЭФИРОВ ИЗ 21-АЦЕТАТА ГИДРОКОРТИЗОНА | 2017 |
|
RU2663893C1 |
| СПОСОБ ПОЛУЧЕНИЯ 6α-МЕТИЛГИДРОКОРТИЗОНА | 2006 |
|
RU2297423C1 |
| СПОСОБ ПОЛУЧЕНИЯ 6-МЕТИЛЕНГИДРОКОРТИЗОНА ИЛИ ЕГО ЭФИРОВ ИЗ 21-АЦЕТАТА ГИДРОКОРТИЗОНА | 2017 |
|
RU2664101C1 |
| СПОСОБ ПОЛУЧЕНИЯ 6α-МЕТИЛГИДРОКОРТИЗОНА ИЛИ ЕГО ЭФИРОВ ИЗ 21-АЦЕТАТА ГИДРОКОРТИЗОНА | 2017 |
|
RU2663484C1 |
| СПОСОБ ПОЛУЧЕНИЯ 6-(N-МЕТИЛ-N-ФЕНИЛ)АМИНОМЕТИЛ-ГИДРОКОРТИЗОНА ИЛИ ЕГО ЭФИРОВ ИЗ 21-АЦЕТАТА ГИДРОКОРТИЗОНА | 2017 |
|
RU2663483C1 |
| ТРИФЕНИЛФОСФОНИЕВЫЕ СОЛИ ЛУПАНОВЫХ ТРИТЕРПЕНОИДОВ, СПОСОБ ПОЛУЧЕНИЯ И ПРИМЕНЕНИЕ В КАЧЕСТВЕ ПРОТИВООПУХОЛЕВЫХ ВЕЩЕСТВ | 2012 |
|
RU2551647C2 |
Изобретение относится к улучшенному способу получения химических соединений из ряда стероидов и конкретно касается получения эпибрассинолида [(22R, 23R, 24R)-2α, 3α, 22, 23-тетрагидрокси-В-гомо-7-окса-5α-эргостан-6-она, относящегося к биологически активному веществу - фитостимулятору, регулирующему рост растений. Способ заключается в последовательном осуществлении следующих стадий: а) синтез мезилата эргостерина обработкой эргостерина метансульфохлоридом в пиридине; б) синтез изоэргостерина кипячением мезилата эргостерина в водном ацетоне в присутствии бикарбоната калия (натрия); в) синтез изоэргостерона окислением изоэргостерина хромовым ангидридом в пиридине; г) синтез 7,8-дигидроэргостерона восстановлением изоэргостерона дитионитом натрия в присутствии солюбилизирующей среды, содержащей катионные, анионные или неионные ПАВ (поверхостно-активные вещества) из ряда СnН2n+1X, где n=9-18, Х=NMe3, NEt3, COOH, SO3Н, OSO2M, OP(O)(OM)2, где М = щелочной металл, полиэтиленгликоль, косолюбилизатор - алифатические спирты С2-С6 или моноэфиры этилен- или диэтиленгликоля, электролит и воду при мольном соотношении их соответственно 1:5-6:100-250; д) стероидную перегруппировку 7,8-дигидроизоэргостерона; е) образование 24-эпикастастерона, обработкой (22Е, 24R)-5 α-эргоста-2,22-диен-6-она метансульфонамидом и карбонатом калия с использованием каталитических количеств гексацианоферрата (III) калия и четырехокиси осмия; ж) растворение образовавшегося таким образом 24-эпикастастерона в хлороформе с последующей обработкой трифторнадуксусной кислотой, получающейся при смешении трифторуксусного ангидрида и перекиси водорода в хлорированном органическом растворителе, и выделением целевого продукта (I) с высоким выходом.
Способ получения 24-эпибрассинолида (22R,23R,24R)-2α,3α,22,23-тетрагидрокси-В-гомо-7-окса-5α-эргостан-6-она общей формулы (I)
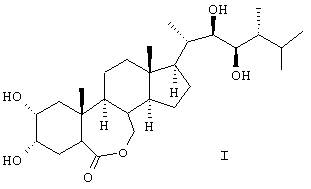
включающий следующие стадии:
а) синтез мезилата эргостерина обработкой природного стерина - эргостерина метансульфохлоридом в пиридине при температуре реакционной среды не более 12°С;
б) синтез изоэргостерина кипячением полученного мезилата эргостерина в водном ацетоне в присутствии бикарбоната калия (натрия) в качестве буферного средства;
в) синтез изоэргостерона окислением изоэргостерина хромовым ангидридом в пиридине;
г) синтез 7,8-дигидроэргостерона восстановлением изоэргостерона дитионитом натрия в водной микрогетерогенной среде с высокой солюбилизирующей способностью в присутствии солюбилизирующей среды, включающей одноцепочные катионные, анионные или неионные ПАВ (поверхостно-активные вещества) из ряда CnH2n+1X, где n=9-18, X=NMe3, NEt3, COOH, SO3H, OSO2M, ОР(O)(ОМ)2, где М = щелочной металл, полиэтиленгликоль, ко-солюбилизатор - алифатические спирты C2-С6 или моноэфиры этилен- или диэтиленгликоля, электролит и воду при мольном соотношении их соответственно 1:5-6:100-250;
д) стероидную перегруппировку 7,8-дигидроизоэргостерона при нагревании его в смеси с безводным бромидом лития, гидрохлоридом пиридиния и диметилацетамидом;
е) синтез 24-эпикастастерона, обработкой (22Е,24R)-5α-эргоста-2,22-диен-6-она метансульфонамидом и карбонатом калия с использованием каталитических количеств четырехокиси осмия и стехиометрического реокислителя гексацианоферрата (III) калия в присутствии соли оптически активного амина 4-хлорбензоата дигидрохинидина;
ж) растворение образовавшегося таким образом 24-эпикастастерона в хлороформе с последующей обработкой трифторнадуксусной кислотой, получающейся при смешении трифторуксусного ангидрида и перекиси водорода в хлорированном органическом растворителе, с последующим выделением целевого продукта (I).
| СПОСОБ ПОЛУЧЕНИЯ α 5-ЦИКЛО-24R-МЕТИЛ- a -ХОЛЕСТ-7,22-ДИЕН -6β -ОЛА | 1991 |
|
RU2024541C1 |
| Способ получения 3 @ ,5-цикло-24 @ -этил-5 @ -холест-22-ен-6-она | 1984 |
|
SU1162816A1 |
| K.Mori, Agric | |||
| Biol | |||
| Chem | |||
| Приспособление для плетения проволочного каркаса для железобетонных пустотелых камней | 1920 |
|
SU44A1 |
| 2 @ ,3 @ ,22S,23S-Тетраокси-24S-этилхолест-4-ен-он, обладающий фиторостостимулирующей активностью | 1987 |
|
SU1433005A1 |
| JP 62167797, A, 24.07.1987 | |||
| УСТРОЙСТВО для ЗАМЕТА И ВЫБОРКИ КОШЕЛЬКОВОГО НЕВОДА НА СУДАХ С КОРМОВЫМ СЛИПОМ | 0 |
|
SU178607A1 |

