Изобретение относится к получению химических соединений из ряда стероидов и может быть использовано для получения биологически активного вещества, являющегося регулятором роста растений, модулятором абиогенного стресса растений, а именно к улучшенному способу получения 24-эпибрассинолида ((22R,23R,24R)-2α,3α,22,23-тетрагидрокси-В-гомо-7-окса-5α-эргостан-6-она).
Стероидные соединения класса брассиностероидов выполняют многочисленные функции во всех растениях от низших до высших цветковых однодольных и двудольных, выполняя роль управления всеми стадиями развития растений, поэтому их принято относить к так называемым фитогормонам наряду с ауксинами, гиббереллинами, этиленом, абсцизовой кислотой, цитокининами. При этом, брассиностероиды обладают одним чрезвычайно важным преимуществом над остальными фитогормонами - они не обладают сильными негативными эффектами нарушения стадий развития при нарушении дозировок, выполняя роль фитогормона второго уровня - они фактически регулируют действие других физиологически активных молекул в растительных организмах. Поэтому применение брассиностероидов не только безопасно, так как не вызывает сильных отрицательных побочных эффектов даже при случайном нарушении регламентов применения, но и позволяет эффективно управлять развитием растений, смягчать негативные последствия абиогенных стрессов (заморозков, засухи, засоления почв, сильных колебаний температуры среды), что и квалифицируется как адаптогенное антистрессовое воздействие. Применение брассиностероидных препаратов в сельском хозяйстве особенно актуально в условиях драматических изменений климата и деградации агробиоценозов. Использование брассиностероидов в растениеводстве широко изучается во многих странах, многократно признавалось чрезвычайно перспективным, успешно внедрялось на уровне опытных и малых хозяйств, но более широкому применению препятствует сложность синтеза, дороговизна и дефицитность исходных соединений, сложности реализации синтеза в укрупненном масштабе.
Известные брассиностероиды имеют стероидный скелет холестанового типа с боковой цепью в кольце D, два вицинальных диольных фрагмента в кольце А и боковой цепи с гидроксилами в положениях 2, 3, 22, 23 и карбонильной группой в положении 5, с фиксированной стереохимической конфигурацией асимметрических центров. Кольцо В может быть как циклогексановым, так и лактонным семичленным, последний вариант более распространен. Родоначальником класса является брассинолид (1), повсеместно распространенный в растительном царстве, но выделяемый в микро-количествах в основном из пыльцы ряда растений, например, рапса, причем для получения даже миллиграммовых количеств требуется переработка килограммовых количств пыльцы, сбор которой в существенных количествах невозможен. (Grove, М. D. et al. Nature 1979, 281, 216-217). Производство и применение брассинолида не имеет практического значения, как по причине невозможности промышленного синтеза из-за отсутствия необходимого стартового сырья, так и из-за быстрой деактивации в клетках растений при экзогенном применении, хотя по активности в ряде важных физиологических процессах он превосходит все остальные брассиностероиды, что и обусловливает продолжение интереса к возможным новым подходам к синтезу и этого соединения.
Так, Хрипач и сотр. предложили для синтеза брассинолида и его аналогов воспользоваться той же последовательностью стадий, что ведет к эпибрассинолиду (см. ниже), но после формирования функциональности в кольцах А и В стероидного скелета, удалять боковую цепь окислительным расщепление диола, и создавать ее заново в нужной стереохимической конфигурации многостадийным синтезом (I.D. Alshakova, Y.V. Ermolovich, V.N. Zhabinskii, V.A. Khripach Steroids 2015, 97, 72-77).
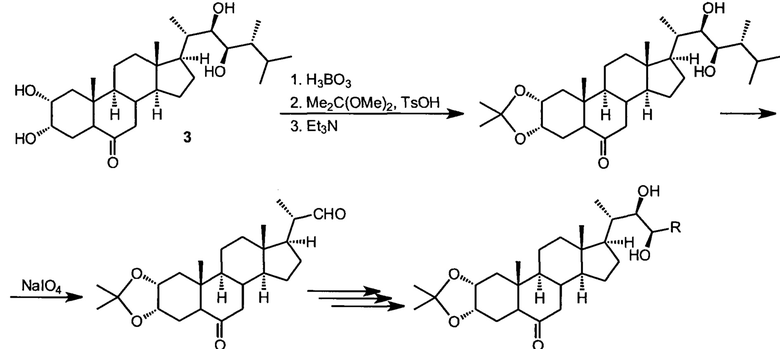
Такой подход открывает возможность наработки исследовательских количеств брассинолида и других ранее недоступных брассиностероидов. Важно то, что в этом подходе требуется наработка значительных количеств эпикастастерона, решаемая способом, описанном в настоящем патенте.
Наибольшее практическое значение имеют три ближайших аналога брассинолида: эпибрассинолид (2), эпикастастерон (3), гомобрассинолид (4), отличающиеся или стереохимической конфигурацией одного стереоцентра, или наличием дополнительной метильной группы в боковой цепи. Эти соединения также являются природными и содержатся в следовых количествах в различных органах растений, но также могут быть синтезированы в значительных количествах. Структурные особенности этих брасиностероидов позволяют а) разрабатывать практически осуществимые в промышленных условиях технологии синтеза из доступного стартового сырья, в основном фитостеролов, являющихся продуктами биотехнологических процессов; б) достичь высокой активности основанных на этих действующих веществах препаратов при экзогенном применении в минимальных количествах. Наиболее высокой ценностью из указанного ряда обладает эпибрассинолид.
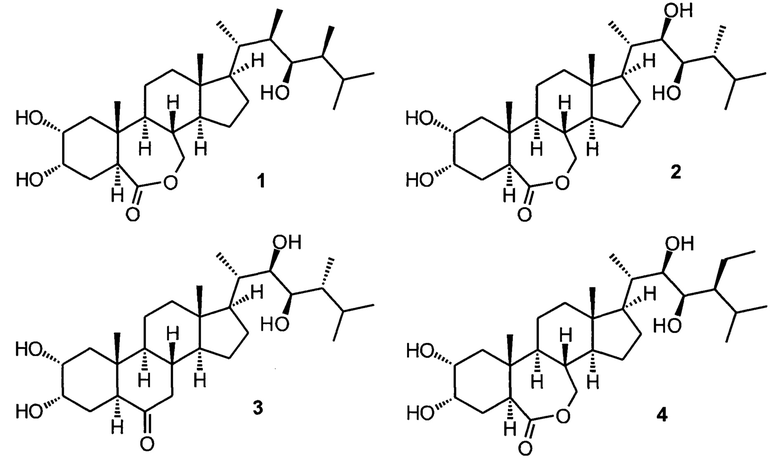
Известно несколько синтетических подходов к указанным брассиностероидам и их ближайшим аналогам, также представляющим практический интерес. В основном, все синтетические подходы используют одни и те же пути синтеза, так как целевые молекулы имеют небольшие различия, вводимые или на отдельных стадиях синтеза с сохранением общности подходов, или через использование исходных с подходящими стереохимическими конфигурациями отдельных центров. Так, брассиностероиды с 24R-конфигурацией (эпибрассинолид, эпикастастерон и их аналоги) получаются с использованием легкодоступного эргостерина, выделяемого из отработанных пивных дрожжей, а брассиностероиды с 24S-конфигурацией (гомобрассинолид и его аналоги) получаются из менее доступных стигматерина и других фитостеринов. Путь синтеза эпикастастерона и эпибрассинолида из эргостерина приобретает еще большую практическую ценность, так как недавно был опубликован биотехнологический подход с использованием генно-модифицированной культуры дрожжей (Yiqi Jiang, et al Nature Communications, 2023,14, 437), позволяющий осуществить производство эпимера эргостерина, (24S)-эпиэргостерина
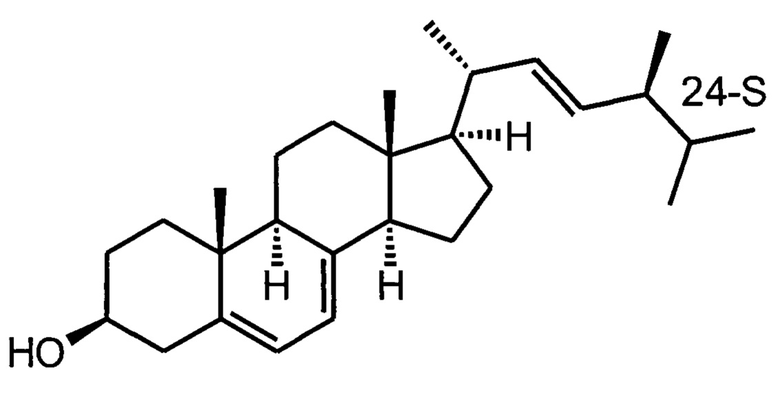
который может стать сырьем для производства брассинолида с использованием той же самой технологии, которая используется для синтеза эпибрассинолида из эргостерина, в том числе той, которая описана в настоящем патенте.
Известен метод синтеза 24-эпибрассинолида и 24-эпикастастерона, описанный в статье Мак-Морриса и Патила (Trevor С.McMorris and Prakash A. Patil Improved synthesis of 24-epibrassinolide from ergosterol J. Org. Chem. 1993, 58, 8, 2338-2339), и развитый в работе Хрипача и сотрудников (Khripach VA, Zhabinskii VN, Olkhovik VK, Ivanova Gl, Zhernosek EV, Kotyatkina Al. Improved synthesis of epibrassinolide. Russ J Org Chem 1994, 30,1735-40.), и наиболее приспособленный как основа для разработки промышленного синтеза брассиностероидов. В этом методе технический эргостерин (5) чистотой не менее 85% превращают в сульфонат (6) действием метан- или толуолсульфохлорида в среде сухого пиридина. После тщательного высушивания в вакууме, сульфонат эргостерина подвергают сольволитической изостериновой перегруппировке в среде кипящего водного ацетона в присутствии гидрокарбоната калия с образованием изоэргостерина (7), который далее окисляют комплексом хромового ангидрида с пиридином в среде пиридина в изоэргостерон (8). Ключевой стадией синтеза является сопряженное восстановление двойной связи енона раствором металлического лития в разбавленном растворе в жидком аммиаке с образованием дигидроизоэргостерона (9). Это соединение подвергают ретро-изостериновой перегруппировке в присутствии смеси кислоты Льюиса и протонной кислоты в среде кипящего диметилацетамида в инертной атмосфере. Вторая ключевая стадия синтеза, создающая стереоцентры системы брассиностероида это асимметрическое дигидроксилирование по методу Шарплесса тетраоксидом осмия в присутствии хирального лиганда, п-хлорбензоата дигидрохинидина, а также стехиометрического реокислителя гексацианоферрата (III) калия, добавки метансульфонамида, ускоряющего обращение катализатора, и карбоната калия в двухфазной системе трет-бутанол-вода. Продуктом этой стадии является брассиностероид эпикастастерон (3), который далее может быть превращен в более ценный брассиностероид, эпибрассинолид (2) с помощью реакции Байера-Виллигера с трифторнадуксусной кислотой в хлороформе, причем реакцию можно выполнять как с самим эпикастастероном, так и после защиты гидроксильных групп ацетилированием. Работа с защищенным эпикастастероном дает более высокие выходы целевого эпибрассинолида, но добавляет к пути синтеза стадии ацетилирования и снятия защиты.
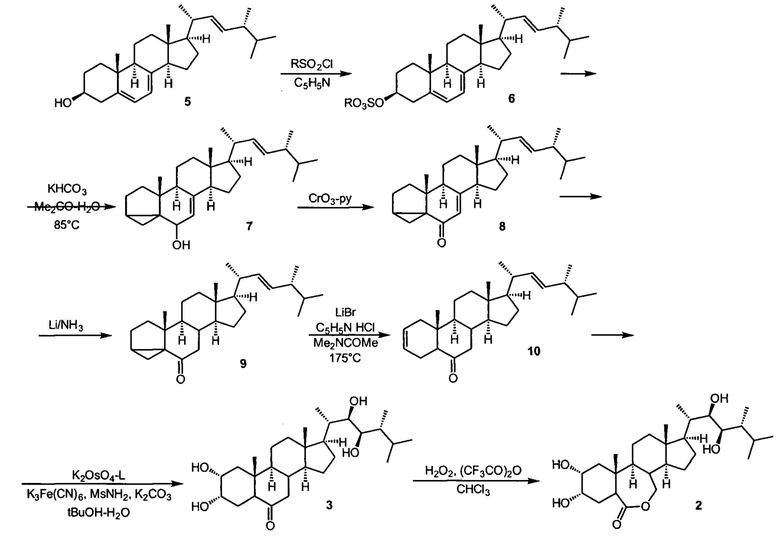
Этот метод позволяет синтезировать эпикастастерон и эпибрассинолид в лабораторном масштабе и масштабе полупромышленных пилотных установок, но обладает несколькими принципиальными недостатками, препятствующими его более широкому использованию. Принципиальная проблема метода - плохая воспроизводимость, низкая чистота целевого продукта, очень маленький и непостоянный суммарный выход, не превышающий 10% в пересчете на продукт технической квалификации. Эти проблемы обусловлены неоптимизированными условиями стадий синтеза, и в основном заложены в стадиях а) сольволитической перегрупппировки сульфоната эргостерина; б) стаадией сопряженного восстановления двойной связи в изоэргостерине; в) недостаточной диастереселективностью стадии асимметрического дигидроксилирования, в которой образуется 24-эпикастастерон. Особенно большие проблемы возникают на стадии сопряженного восстановления, изоэргостерона в дигидроизоэргостерон, которое осуществляется действием разбавленного раствора щелочного металла в жидком аммиаке, и поэтому требует работы в криогенных условиях с неблагоприятным по экономическим и экологическим соображениями соотношением субстрат-жидкий аммиак с очень высоким расходом аммиака на единицу массы полупродукта, что требует или использования установки для рециклизации аммиака, или эффективного оборудования для улавливания аммиака после реакции. Во-вторых, сильнощелочные условия восстановления в данной стадии и в самой реакции и во время послереакционной обработки реакционной смеси являются причиной побочных реакций эпимеризации, в результате которй образуются продукты восстановления с другой стереохимической конфигурацией сочленения колец В и С (цис-сочленение вместо правильного транс-сочленения, присущего всем веществам класса брассиностероидов).
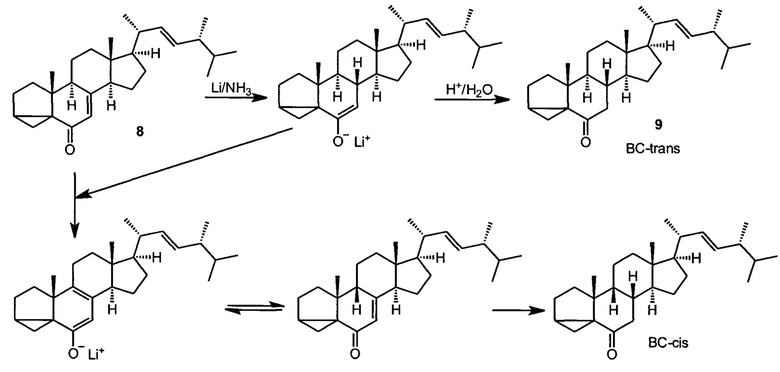
Эти побочные продукты, образующиеся в значительных количествах, необходимо удалять с помощью хроматографии, так как эти примеси сильно снижают качество и активность целевого продукта, и сильно затрудняют все последующие стадии синтеза. По этой причине все стадии синтеза от стадии восстановления дигидроэргостерина должны сопровождаться хроматографической очисткой полупродуктов и целевого продукта, что сильно снижает и общую производительность, и сильно удорожает продукт.
Наиболее близким аналогом, т.е. прототипом, к предлагаемому изобретению является "Способ получения 24-эпибрассинолида" (патент РФ №2272044, авторы Чепраков А.В., Филатов М.А., Лукашев Н.В., Малеванная Н.Н.). В прототипе целевой продукт 24-эпибрассинолид получают из коммерческого сырья, эргостерина в синтезе, включающем следующие стадии с суммарным выходом по эргостерину 23%:
1) получение метансульфоната эргостерина обработкой эргостерина метансульфонилхлоридом в среде пиридина;
2) сольволитическую перегруппировку мезилата эргостерина в среде кипящего ацетона с небольшой добавкой воды и гидрокарбоната калия в качестве буфера;
3) окисление изоэргостерина в изоэргостерон комплексом хромового ангидрида с пиридином в среде сухого пиридина;
4) селективное восстановление сопряженной двойной связи в изоэргостероне дитионитом натрия в среде водной солюбилизующей системы с высокой солюбилизующей емкостью;
5) обратную стероидную перегруппировку дигидроизоэргостерона в среде диметилацетамида, катализируемую безводным бромидом лития;
6) асимметрическое дигидроксилирование по методу Шарплесса с использованием хирального лиганда 4-хлорбензоата дигидрохинидина с образованием 24-эпикастастерона;
7) окислению 24-эпикастастерона по методу Байера-Виллигера трифторнадуксусной кислотой
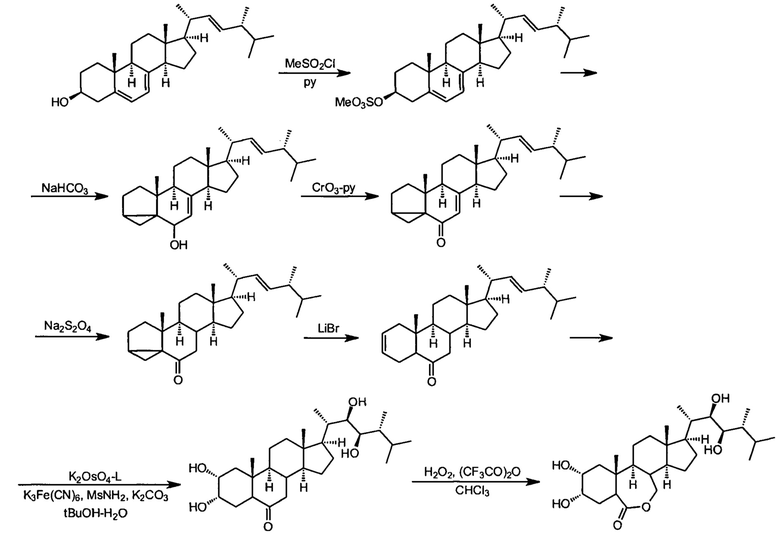
Предложенный в прототипе метод использует ту же общую последовательность стадий, что и метод, описанный в работе Мак-Морриса и Пракаша (Trevor С.McMorris and Prakash A. Patil Improved synthesis of 24-epibrassinolide from ergosterol J. Org. Chem. 1993, 58, 8, 2338-2339), но вводит значительные модификации, которые позволили достичь воспроизводимости, высокого суммарного выхода продукта требуемой чистоты, что было достигнуто за счет ряда изменений на отдельных стадиях, наиболее значительным улучшением является замена ключевой стадии восстановления изоэргостерона в дигидроизоэргостерон. Вместо восстановления разбавленными растворами щелочных металлов в жидком аммиаке метод-прототип использует безопасную и легко масштабируемую методику восстановления техническим дитионитом натрия в среде водных микрогетерогенных систем с высокой солюбилизующей способностью, получаемые смешением поверхностно-активных веществ анионного, катионного или неионного типа, а также косолюбилизаторов (алифатических спиртов или моноэфиров моно или диэтиленгликоля), и водного раствора неорганической соли-электролита в соотношении 1:5-6:100-250. Это изменение позволило не только исключить использование дорого, опасного и токсичного криогенного растворителя, жидкого аммиака, требующего особого оборудования для работы и послереакционной регенерации или утилизации, но и сделало условия реакции восстановления мягкими и исключающими среду высокой щелочности, являющуюся причиной образования побочных продуктов. Восстановление описанным в прототипе способом высокоселективно, исключает эпимеризацию, и поэтому делает ненужным хроматографическую очистку полупродукта этой и последующих стадий.
Тем не менее метод-прототип сохранил многие другие недостатки исходного пути синтеза. Наиболее существенными являются следующие. Сольволитическая перегруппировка (изостероидная перегруппировка) сульфоната эргостерина выполнялось в среде кипящего ацетона, используемого в очень больших количествах относительно загрузок сульфоната эргостерина: на каждый грамм исходного необходимо брать более 200 мл чистого свежеперегнанного ацетона. Такие условия создают высокую степень опасности для исполнителей синтеза - горячий ацетон пожароопасен и токсичен. Расход ацетона для этой стадии чрезвычайно велик. При пересчете на 1 грамм конечного продукта синтеза, 24-эпибрассинолида, с учетом суммарного выхода, расходуется более 2 литров ацетона, причем после реакции происходит разбавление смеси водой и регенерация растворителя из таких отходов практически невозможна, что создает проблему утилизации больших количеств токсичных, летучих и пожароопасных ацетонсодержащих отходов. Именно эта стадия поэтому становится неприемлемой при промышленном масштабировании синтеза и должна быть заменена в приоритетном порядке. Авторы настоящего патента предложили использовать разработанную ими водную микрогетерогенную систему с высокой солюбилизующей способностью, успешно использованную ранее в прототипе на стадии восстановления изоэргостерона, и для стадии сольволитической изомеризации. Разработанная система обладает уникальными характеристиками: она состоит в основном из воды, с небольшими добавками в несколько весовых или объемных процентов простых и доступных, биоразлагаемых ПАВ типа четвертичных солей аммония с длинноцепочечной алкильной группой, а также косолюбилизатора, простого алифатического спирта типа н-бутанола и балансирующего электролита, который одновременно является буфером для поддержания нужного уровня кислотности системы. Использование такой системы в качестве среды реакции снимает проблемы пожароопасности и токсичности, а также регенерации или утилизации. Как показано в нижеописанных примерах, это техническое решение дает и существенное улучшение всей последовательности синтеза, так как приводит к увеличению выходов полупродуктов, упрощению процедур выделения и очистки полупродуктов, сокращению времени синтеза и общей интенсификации процесса. Кроме того, последовательность первых трех стадий в этом методе требует тщательной очистки и сушки каждого из полупродуктов (сульфоната эргостерина, изоэргостерина, изоэргостерона) перед загрузкой с следующую стадию, а использование недоочищенных продуктов приводит к резкому снижению выходов и образованию побочных продуктов, в частности, продукта дегидратации эргостерина вместо изоэргостерона.
Предлагаемое в настоящем патенте изобретение позволяет решить следующие задачи:
1. Увеличение суммарного выхода 24-эпибрассинолида в расчете на введенный в реакцию эргостерин с 23% в прототипе до 38% в заявляемом методе, при сохранении высоких качественных параметров получаемого продукта, оцениваемых по совокупности физико-химических параметров (тмпература плавления, удельный угол вращения плоскости поляризованного света, спектры ЯМР). Увеличение суммарного выхода дает значительную экономию как исходного эргостерина, дорогостоящего импортного сырья, так и реагентов и вспомогательных материалов и растворителей на единицу массы конечного продукта.
2. В предлагаемом методе синтеза три стадии из семи выполняются в экологически и технологически безопасных, рециклизуемых, биоразлагаемых системах, или на основе солюбилизующих систем на водной основе, или водно-спиртовой среде, практически не использующих летучих органических загрязнителей (VOC). Эти стадии соответствуют большинству критериев устойчивого развития и экологической безопасности. В методе прототипе и других аналогах именно эти стадии представляли наиболее значительные технологические и экологические опасности из-за использования больших объемов токсичных и летучих растворителей, нуждающихся в предреакционной очистке и послереакционной утилизации, что значительно увеличивало общую энерго- и трудоемкость последовательности синтеза.
3. Улучшение выхода и степени превращения на стадии асимметрического дигидроксилирования за счет использования более эффективного хирального лиганда DHQD-PHAL, в сравнении с методом-прототипом, в котором на этой стадии используется более простой, но менее эффективный лиганд дигидрохинуклидина 4-хлорбензоат, что приводит к более высокой степени превращения менее реакционноспособной двойной связи в боковой цепи, и соответствующее улучшение выхода 24-эпикастастерона.
4. Усовершенствование методов выделения и очистки полупродуктов, отказ от хроматографической очистки на всех стадиях дали дополнительное улучшение выходов на пятой и заключительной стадиях синтеза.
Технический результат заключается в следующих изобретениях:
1. Проведение второй стадии синтеза, сольволитической перегруппировки мезилата эргостерина в водной солюбилизующей системе высокой емкости на основе катионного ПАВ цетилтриметиламмонийбромида и косолюбилизатора, алифатического спирта в количестве не более 5% по массе от общей массы реагирующей системы, основным компонентом в которой является вода. Это позволило заменить стадию перегруппировки в разбавленном растворе кипящего ацетона, пожароопасного и токсичного растворителя, полностью отказаться от использования этого растворителя, а также более чем вдвое увеличить производительность, заменить более дорогой и дефицитный бикарбонат калия на легкодоступный дешевый бикарбонат натрия (пищевую соду).
2. Использование водных микрогетерогенных систем высокой емкости для осуществления изостероидной перегруппировки в ряду стероидов ранее известно не было и является изобретением авторов настоящей заявки. Прототипом этого метода является осуществление восстановления двойной связи в сопряженном непредельном кетоне стероидного строения, описанного в патенте-прототипе настоящей заявки.
3. Три первые стадии синтеза выполняются без очистки промежуточных продуктов, что упрощает технологию, снижает расход токсичных растворителей, увеличивает суммарный выход по трем стадиям с 58% в прототипе до 70% в предлагаемом методе, что и является основой высокого суммарного выхода.
4. Изменена техника выполнения четвертой стадии - восстановитель вводится в виде водного раствора, что снижает время реакции, упрощает инструментальное оформление процесса (заменяет технически сложное устройство для ввода порошкообразного реагента на обычную капельную воронку или шприцевой дозатор), уменьшает вдвое расход восстановителя, увеличивает конверсию полупродукта и выход дигидроизоэргостерона за счет улучшения селективности процесса.
Указанные технические результаты достигаются за счет использования следующий последовательности синтеза 24-эпибрассинолида:
1) синтез мезилата (метансульфоната) эргостерина реакцией природного эргостерина, доступного коммерческого сырья, с метансульфонилхлоридом в сухом пиридине при температуре реакционной среды не выше +10°С;
2) синтез изоэргостерина за счет сольволитической изостероидной перегруппировки контролируемым гидролизом мезилата эргостерина в водной солюбилизующей среде высокой емкости (микроэмульсионной системе третьего типа, состоящей из воды с добавлением катионного ПАВ из ряда н-CnH2n+lNR3X (где n=11-15, X=Cl, Br; R=метил, этил) и косолюбилизатора, алифатического (С2-С5)-спирта, в количестве не более 5% по объемной доле) в присутствии бикарбоната натрия в качестве буферного средства и балансирующего электролита;
3) синтез изоэргостерона окислением изоэргостерина комплексом хромового ангидрида с пиридином в пиридине;
4) синтез 7,8-дигидроизоэргостерона восстановлением изоэргостерона техническим дитионитом натрия в водной солюбилизующей среде высокой емкости (микроэмульсионной системе третьего типа, состоящей из воды с добавлением катионного ПАВ из ряда н-CnH2n+lNR3X (где n=11-15, X=Cl,Br; R=метил, этил) и косолюбилизатора, алифатического (С2-С5)-спирта, в количестве не более 5% по объемной доле) в присутствии бикарбоната натрия в качестве буферного средства и балансирующего электролита;
5) обратную стероидную (циклопропилметил-гомоаллильную) перегруппировку 7,8-дигидроизоэргостерона с образованием (22Е, 24R)-5α-эргоста-2,22-диен-6-она нагреванием с каталитическими количествами безводного бромида лития и гидрохлорида пиридиния в сухом диметилацетамиде;
6) синтез 24-эпикастастерона асимметрическим дигидроксилированием (22Е, 24R)-5α-эргоста-2,22-диен-6-она в присутствии каталитической системы осмат калия - лиганды DHQD-PHAL (гидрохинидина 1,4-фталазиндиил диэфир) и гидрохинидина 4-хлорбензоат, стехиометрического окислителя гексацианоферрата(III) калия, метансульфонамида, карбоната калия в двухфазной среде трет-бутанол-вода
7) окислением 24-эпикастастерона трифторнадуксусной кислотой, образовавшейся при смешении трифторуксусного ангидрида и 50%-ной перекиси водорода в среде сухого хлороформа, с выделением и очисткой целевого продукта.
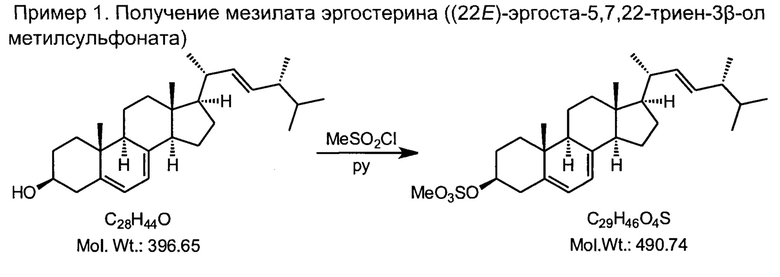
В трехгорлую колбу на 2000 мл, снабженную капельной воронкой, термометром, механической мешалкой, загружали 80 г (0.2 моль) эргостерина и 1.2 л пиридина, свежеперегнанного над едким натром, и перемешивали до полного растворения осадка. Раствор охлаждали до +5°С с помощью охлаждающей бани со льдом, и при интенсивном перемешивании прикапывали 30 мл метансульфохлорида (2-х кратный избыток) с такой скоростью, чтобы температура не повышалась выше +10°С. После завершения прибавления метансульфохлорида смесь перемешивали еще один час при температуре не выше +10°С, и выливали на 2 кг льда в полиэтиленовом сосуде емкостью 5 литров. Как только лед растает, полученную суспензию фильтровали с отсасыванием на воронке Бюхнера с бумажным фильтром или на воронке со стеклянным фильтрующим элементом средней пористости, вещество промывают одним литром ледяной воды, хорошо отжимают, и немедленно используют в следующей стадии без досушки.
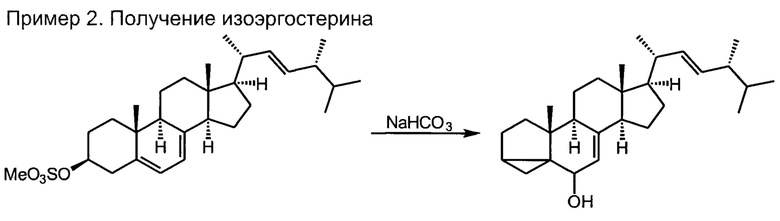
Продукт, полученный на первой стадии, имеет переменную влажность, его взвешивали и делили по массе на 4 равные части. Каждую из частей подвергали процедуре, описанной далее. При невозможности ввести продукт первой стадии в реакцию в течение суток после выделения, его следует хранить в морозильной камере холодильника не более 3 месяцев, в противном случае качество продукта второй стадии будут ухудшаться.
В высокий химический стакан емкостью 2 л, установленный на магнитной мешалке с регулируемым нагревом, помещали 40 г цетилтриметиламмоний бромида, 50 мл н-бутанола, 50 мл толуола и при непрерывном перемешивании приливали 1 литр водного раствора 20 г гидрокарбоната натрия. Смесь нагревали до 80°С, причем раствор должен стать визуально гомогенным. При интенсивном перемешивании в раствор максимально быстро вносят небольшими порциями влажный продукт первой стадии, следя чтобы он быстро распространялся по объему раствора, возможные комки разбивают шпателем. В момент внесения особенно первых порций возможно сильное вспенивание. Полученный раствор перемешивали 30 минут при температуре 75-90°С, охлаждали, переносили в экстрактор жидкость-жидкость и экстрагировали 300 мл этилацетата. Процедуру повторяли стремя другими порциями. Объединенные экстракты упаривали на роторном испарителе до начала выпадения кристаллического осадка, дополнительно охлаждали смесь в морозильной камере и фильтровали на воронке Бюхнера с отсасыванием. Белая кристаллическая масса, которую необходимо использовать в третьей стадии синтеза не позднее 10 дней, до использования хранить в морозильной камере.
Пример 3. Получение изоэргостерона
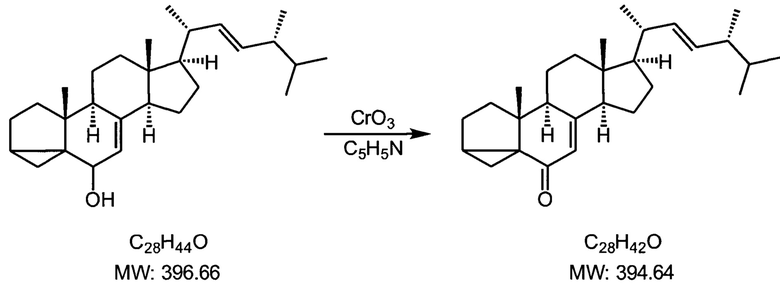
Вещество, полученное на второй стадии, делят на две равные порции, каждую обрабатывают по описанной процедуре.
В трехгорлую колбу на 2000 мл, снабженную капельной воронкой, мешалкой и термометром, защищенную через воздушный холодильник трубкой с пеллетами КОН, помещали 600 мл пиридина, свежеперегнанного над едким натром. Колбу охлаждали до +5 -+10°С, и при интенсивном перемешивании добавляли шестью порциями по 5 грамм 30 г хромового ангидрида, каждый раз перемешивая до полного растворения ангидрида, при этом ближе к концу прибавления начинает выпадать оранжевый комплекс хромового ангидрида с пиридином. Во время прибавления температура смеси не должна превышать 20°С. К суспензии прикапывали раствор одной порции продукта второй стадии в 600 мл пиридина, и оставляли перемешиваться при комнатной температуре на 1 сутки. Далее смесь разбавляли равным объемом диэтилового эфира, перемешивали 0.5 часа и отфильтровывали выпавший темно-зеленый осадок на воронке Бюхнера. Осадок промывали порциями 300 мл диэтилового эфира, тщательно перемешивая осадок на фильтре. Объединенный раствор упаривали на роторном испарителе досуха, остаток перекристаллизовывали из ацетона. Маточник после фильтрования оставляли и прибавляли при перекристаллизации к следующей порции. Процедуру повторяли с второй порцией продукта второй стадии.
Суммарный выход по двум порциям загрузки - 56 г, что соответствует выходу в 70% по трем первым стадиям. Белые пластинки, т.пл. 168-169°С. Спектр 1H ЯМР (CDCl3, 400 МГц): 5.8 (с, 1Н), 5.2 (м, 2Н), 1.2-2.3 (перекрывающиеся мультиплеты, 23Н), 1.05 (с, 3Н), 0.93 (с, 3Н), 0.84 (с, 6Н), 0.77 (с, 1Н), 0.65 (с, 3Н).
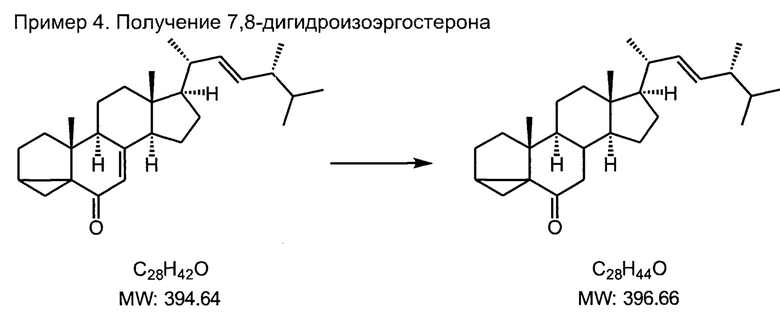
В трехгорлую колбу на 2 л, снабженную эффективным обратным холодильником высотой не менее 50 см, капельной воронкой с компенсатором давления, термометром или датчиком температуры и эффективной мешалкой загружали 45 г цетилтриметиламмонийбромида, 20 грамм изоэргостерона, 40 мл н-бутанола, 40 мл толуола. Включали перемешивание и нагрев и вливали 1 литр кипящей воды. Смесь интенсивно перемешивали и добавляли раствор 20 г гидрокарбоната натрия в 400 мл горячей воды. Смесь нагревали до интенсивного кипения и начинал прикапывать раствор 10 г технического дитионита (гидросульфита) натрия в 200 мл холодной воды. Успех этой стадии зависит от качества дитионита натрия, это должен быть свежий (по дате изготовления) реагент от поставщика с известной репутацией; использование старого реагента с просроченным сроком хранения не допускается. Раствор дитионита в холодной воде готовится непосредственно перед загрузкой в капельную воронку, допускается задержка не более 10 минут. Прикапывание раствора осуществляли медленно, с постоянной скоростью, в течение часа, при этом раствор должен непрерывно интенсивно кипеть.
Смесь продолжали перемешивать, охлаждали до 50°С, добавляли 300 мл воды и 200 мл этилацетата. Слои делили с помощью делительной воронки, и еще 4 раза экстрагировали этилацетатом по 100 мл.
Экстракт упаривали на роторном испарителе досуха, остаток перекристаллизовали из ацетона, охлаждая кристаллизующуюся массу в морозильной камере. Белые мелкие кристаллы фильтровали на стеклянном пористом фильтре средней пористости, промывали небольшим количеством холодного ацетона. Маточники упаривают, и остаток присоединяют к следующей загрузке этой стадии.
Выход 16.3 г (81%), белые кристаллы, т.пл. 110-111С
Спектр 1Н ЯМР (CDCl3, 400 МГц, м.д. ТМС): 5.2 (м, 2Н), 1.2-2.1 (перекрывающиеся мультиплеты, 24Н), 1.02 (с, 6Н), 0.92 (с, 3Н), 0.81 (с, 6Н), 0.70 (с, 3Н).
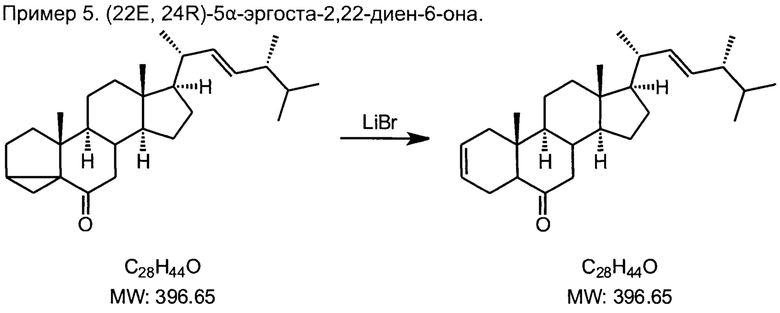
В двухгорлую колбу на 500 мл, снабженную обратным холодильником с верхним вводом аргона через силиконовую прокладку, и термометром помещали 16.3 г дигидроизоэргостерона, 1.8 г безводного бромида лития (вещество гигроскопично, взвешивать в закрытом бюксе), 1 г гидрохлорида пиридиния (вещество гигроскопично, взвешивать в закрытом бюксе), 150 мл сухого диметилацетамида. Смесь перемешивали при температуре кипения растворителя (165°С) в слабом токе аргона 1 час. После охлаждения до температуры менее 100°С смесь выливали в 1 литр воды с льдом в стакане на 2 л, при перемешивании шпателем так, чтобы в сосуде не образовывалось непромешанных зон, при этом выпадает обильный серый всплывающий осадок. Осадок фильтровали на воронке Бюхнера, тщательно отжимали, и оставляли сушиться на воздухе 2-3 суток, постепенно растирая материал в грубый порошок. Вещество перекристаллизовывали из 200 мл этанола, дополнительно охлаждая маточник льдом, маточник после фильтрования кристаллов упаривали досуха и присоединяли к следующей порции продукта. Сушили на воздухе. Белый мелкокристаллический порошок, 14.2 г (86%), т.пл. 123-123.5°С.
Спектр 1Н ЯМР (CDCl3, 400 МГц, м.д. ТМС): 5.68 (м, 1Н), 5.60 (м, 1Н), 5.2 (м, 2Н), 1.2-2.4 (перекрывающиеся мультиплеты, 22Н), 1.02 (с, 3Н), 0.91 (с, 6Н), 0.82 (с, 6Н), 0.70 (с, 3Н), 0.67(с, 3Н).
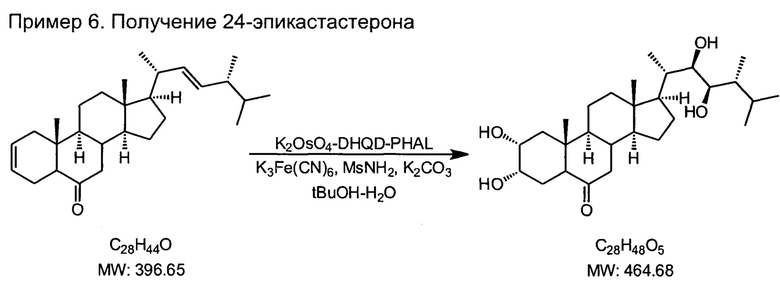
В одногорлую колбу на 1 л, снабженную эффективной мешалкой, помещали 14.2 г 5а-эргоста-2,22-диен-6-она, 8 г гексацианоферрата(III) калия, 3 г карбоната калия, 0.7 г метансульфонамида, 0.05 г осмата калия, 0.5 г лиганда DHQD-PHAL, 0.5 г п-хлорбензоата дигидрохинидина, 300 мл трет-бутанола и 300 мл воды. Колбу экранировали от прямого света алюминиевой фольгой и перемешивали при комнатной температуре 5 суток, добавляли еще 0.5 г лиганда DHQD-PHAL, и перемешивали еще 5 суток. К смеси добавляли 3 г сульфита натрия перемешивали еще 12 часов. Слои разделяли на делительной воронке, водный слой экстрагировали три раза по 100 мл этилацетата. Органический слой промывали водой, 3%-ным раствором серной кислоты (2 раза или больше, последняя промывка должна не иметь зеленоватой окраски), снова водой. Органический слой не сушили, а упаривали на роторном испарителе досуха, растворяли остаток в 100 мл хлороформа и фильтровали через слой силикагеля для колоночной хроматографии толщиной 50-100 мм на фильтре с пористой пластиной диаметром 5-8 см. Силикагель несколько раз промывали дихлорметаном порциями 100 мл, каждый раз прокачивая раствор с помощью вакуумного насоса до высушивания слоя. Объединенные растворы упаривали досуха. Остаток сушили на воздухе 3-5 суток до постоянной массы.
Белый или слегка кремовый тонкий порошок, т.пл. 246-248°С, 14.6 г (выход 88%). Спектр 1Н ЯМР (CDCl3, 400 МГц, м.д. ТМС): 4.1 (с, 1Н), 3.8 (уш. с, 1Н), 3.7 (уш. с, 1Н), 3.41 (м, 1Н), 2.66 (с, 1Н), 2.25 (м, 2Н), 1.2-2.1 (перекрывающиеся мультиплеты, 22Н), 1.0 (с, 3Н), 0.93 (с, 3Н), 0.86 (с, 6Н), 0.77 (с, 3Н), 0.67(с, 3Н).
Пример 7. Получение 24-эпибрассинолида
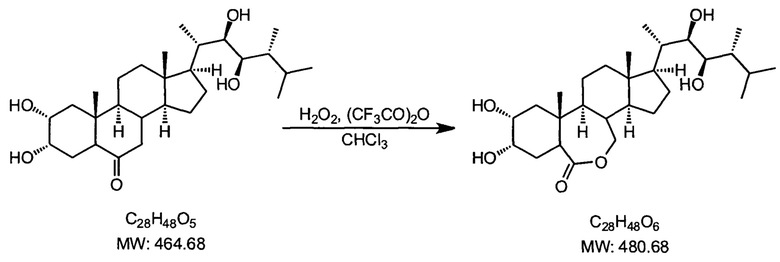
В трехгорлую колбу, снабженную капельной воронкой с компенсацией давления, обратным холодильников, защищенным хлоркальциевой трубкой, термометром помещали 300 мл сухого хлороформа и 220 мл трифторуксусного ангидрида, и при охлаждении льдом медленно прикапывали 30 мл 50%-ной перекиси водорода, следя за тем, чтобы температура не превышала 15°С (при выходе температуры из этого предела возможно быстрое саморазогревание смеси с разложением и выбросом). К полученному раствору при охлаждении медленно прикапывают раствор 14.6 г эпикастастерона в 800 мл сухого хлороформа, выдерживая температуру смеси не выше +10°С. После окончания прикапывания перемешивали еще час при комнатной температуре (охлаждение нужно убрать), после чего приливали 100 мл 10%-ного раствора фосфата калия (K3PO4) вместо раствора соды, что позволяет избежать вспенивания, способствует хорошему разделению слоев, подавляет эмульгирование, в разы ускоряет последующую обработку реакционной смеси. Смесь промывали путем перемешивания в колбе с последующим делением слоев в делительной воронке на 2 л растворами сульфита натрия (10 г в 100 мл воды), водой (2 раза по 200 мл), насыщенным раствором хлорида натрия. Органический слой не сушили, а сразу упаривали на роторном испарителе до объема 100 мл, фильтровали через слой силикагеля на фильтре с пористой пластиной, силикагель промывали одной порцией хлороформа (100 мл). Раствор упаривали на роторном испарителе до прекращения конденсации растворителя, добавляли 50 мл этанола, упаривали досуха, повторяли упаривание с этанолом до того, когда при полном упаривании остаток застывает в стеклообразную хрупкую пузырящуюся массу, которую извлекали из колбы стальным шпателем, растирали в фарфоровой чашке в грубый порошок и сушили на воздухе до постоянной массы, продолжая измельчать до состояния тонкого сыпучего порошка.
Белый порошок (допускается слабый желтоватый или кремовый оттенок), 13.4 г (89%), т.пл. 256-258°С.
Спектр 1Н ЯМР (CDCl3, 400 МГц, м.д. ТМС): 4.2 (м, 1Н), 4.0 (уш. с, 1Н), 3.7 (уш. с, 2Н), 3.45 (м, 1Н), 3.1 (м, 1Н), 1.2-2.2 (перекрывающиеся мультиплеты, 22Н), 1.05 (с, 3Н), 0.92 (с, 6Н), 0.85 (с, 6Н), 0.70 (с, 3Н).
| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБ ПОЛУЧЕНИЯ 24-ЭПИБРАССИНОЛИДА | 2004 |
|
RU2272044C1 |
| СПОСОБ ПОЛУЧЕНИЯ α 5-ЦИКЛО-24R-МЕТИЛ- a -ХОЛЕСТ-7,22-ДИЕН -6β -ОЛА | 1991 |
|
RU2024541C1 |
| Способ получения 3 @ -окси-24R-метилхолест-5-ена | 1988 |
|
SU1594181A1 |
| (22R)-20- 3′ ИЗОПРОПИЛИЗОКСАЗОЛИН-5-ИЛ) 3α ЦИКЛО 5α ПРЕГНАН-6-ОН, ПРОЯВЛЯЮЩИЙ ФИТОРОСТОСТИМУЛИРУЮЩУЮ АКТИВНОСТЬ, И СПОСОБ ЕГО ПОЛУЧЕНИЯ | 1990 |
|
SU1786807A1 |
| (22ζ)-6b-МЕТОКСИ-3a,5-ЦИКЛО-5a-ХОЛЕСТАН-24-ОН-22-ОЛ В КАЧЕСТВЕ ПОЛУПРОДУКТА В СИНТЕЗЕ (22R,23R)-3b-АЦЕТОКСИ-22,23-ИЗОПРОПИЛИДЕНДИОКСИ-24-МЕТИЛХОЛЕСТ-5-ЕНА | 1991 |
|
RU2024540C1 |
| 2β,3α,5α-ТРИГИДРОКСИ-АНДРОСТ-6-ОН, СПОСОБЫ ЕГО ПОЛУЧЕНИЯ И ПРИМЕНЕНИЯ | 2014 |
|
RU2629929C2 |
| СПОСОБ ПОЛУЧЕНИЯ 2'-ФТОР-2'-АЛКИЛЗАМЕЩЕННЫХ ИЛИ ДРУГИХ ЗАМЕЩЕННЫХ РИБОФУРАНОЗИЛПИРИМИДИНОВ И ПУРИНОВ И ИХ ПРОИЗВОДНЫХ | 2005 |
|
RU2433124C2 |
| ПРОИЗВОДНЫЕ ЭПОКСИЦИКЛОГЕКСАНА И РЕГУЛЯТОРЫ РОСТА РАСТЕНИЙ | 1995 |
|
RU2126396C1 |
| СПОСОБ СИНТЕЗА ПРОИЗВОДНЫХ 9-АЛЛИЛКАМПТОТЕЦИНА | 2014 |
|
RU2658017C2 |
| АНТАГОНИСТЫ ПРОГЕСТЕРОНА | 2012 |
|
RU2608521C2 |
Изобретение относится к способу получения 24-эпибрассинолида (22R,23R,24R)-2α,3α,22,23-тетрагидрокси-В-гомо-7-окса-5α-эргостан-6-она, соответствующего указанной ниже формуле, который является регулятором роста растений и модулятором абиогенного стресса растений. Предлагаемый способ включает стадии 1)-7). На стадии 1) осуществляют синтез мезилата (метансульфоната) эргостерина реакцией природного стерина, доступного коммерческого сырья, с метансульфонилхлоридом в сухом пиридине при температуре реакционной среды не выше +10°С. На стадии 2) проводят синтез изоэргостерина за счет сольволитической изостероидной перегруппировки контролируемым гидролизом мезилата в водной солюбилизующей среде высокой емкости (микроэмульсионной системе третьего типа, состоящей из воды с добавлением катионного ПАВ, представляющего собой цетилтриметиламмоний бромид C16H33N(CH3)3Br, и косолюбилизатора, алифатического (С2-С5)-спирта, в количестве не более 5% по объемной доле) в присутствии бикарбоната натрия в качестве буферного средства и балансирующего электролита. На стадии 3) осуществляют синтез изоэргостерона окислением изоэргостерина комплексом хромового ангидрида с пиридином в пиридине. На стадии 4) выполняют синтез 7,8-дигидроизоэргостерона восстановлением изоэргостерона техническим дитионитом натрия в водной солюбилизующей среде высокой емкости (микроэмульсионной системе третьего типа, состоящей из воды с добавлением катионного ПАВ, представляющего собой цетилтриметиламмоний бромид C16H33N(CH3)3Br, и косолюбилизатора, алифатического (С2-С5)-спирта, в количестве не более 5% по объемной доле) в присутствии бикарбоната натрия в качестве буферного средства и балансирующего электролита. На стадии 5) осуществляют обратную стероидную (циклопропилметил-гомоаллильную) перегруппировку 7,8-дигидроизоэргостерона с образованием (22Е,24R)-5α-эргоста-2,22-диен-6-она нагреванием с каталитическими количествами безводного бромида лития и гидрохлорида пиридиния в сухом диметилацетамиде. На стадии 6) выполняют синтез 24-эпикастастерона асимметрическим дигидроксилированием (22E,24R)-5α-эргоста-2,22-диен-6-она в присутствии каталитической системы осмат калия - лиганды DHQD-PHAL (гидрохинидина 1,4-фталазиндиил диэфир) и гидрохинидина 4-хлорбензоат, стехиометрического окислителя гексацианоферрата(III) калия, метансульфонамида, карбоната калия в двухфазной среде трет-бутанол-вода. На стадии 7) выполняют окисление 24-эпикастастерона трифторнадуксусной кислотой, образовавшейся при смешении трифторуксусного ангидрида и 50%-ной перекиси водорода в среде сухого хлороформа, с выделением и очисткой целевого продукта. Предлагаемый способ позволяет получать 24-эпибрассинолид с высоким суммарным выходом по усовершенствованной методике. 7 пр.
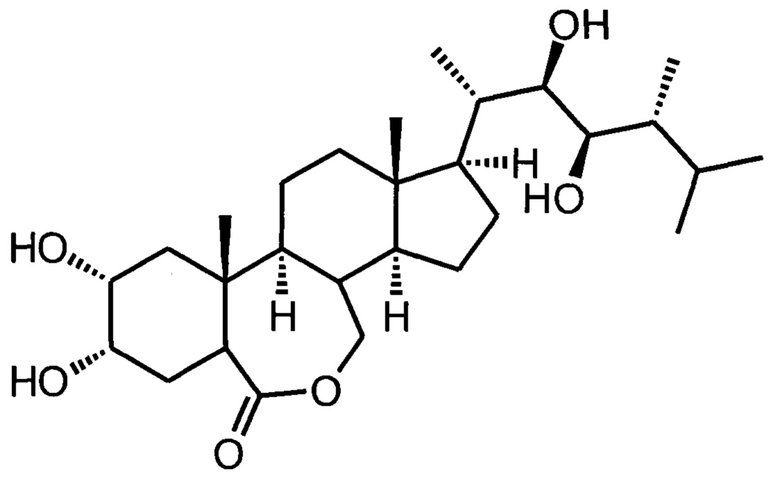
Способ получения 24-эпибрассинолида (22R,23R,24R)-2α,3α,22,23-тетрагидрокси-В-гомо-7-окса-5α-эргостан-6-она, соответствующего формуле
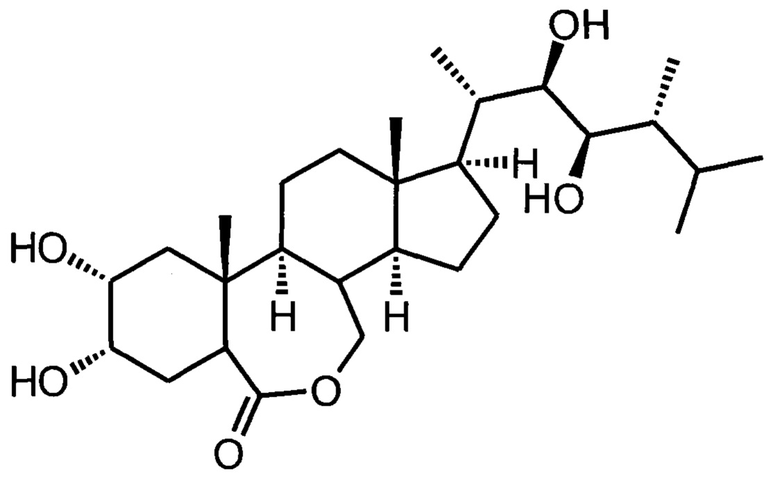 ,
,
включающий следующие стадии:
1) синтез мезилата (метансульфоната) эргостерина реакцией природного стерина, доступного коммерческого сырья, с метансульфонилхлоридом в сухом пиридине при температуре реакционной среды не выше +10°С;
2) синтез изоэргостерина за счет сольволитической изостероидной перегруппировки контролируемым гидролизом мезилата в водной солюбилизующей среде высокой емкости (микроэмульсионной системе третьего типа, состоящей из воды с добавлением катионного ПАВ, представляющего собой цетилтриметиламмоний бромид C16H33N(CH3)3Br, и косолюбилизатора, алифатического (С2-С5)-спирта, в количестве не более 5% по объемной доле) в присутствии бикарбоната натрия в качестве буферного средства и балансирующего электролита;
3) синтез изоэргостерона окислением изоэргостерина комплексом хромового ангидрида с пиридином в пиридине;
4) синтез 7,8-дигидроизоэргостерона восстановлением изоэргостерона техническим дитионитом натрия в водной солюбилизующей среде высокой емкости (микроэмульсионной системе третьего типа, состоящей из воды с добавлением катионного ПАВ, представляющего собой цетилтриметиламмоний бромид C16H33N(CH3)3Br, и косолюбилизатора, алифатического (С2-С5)-спирта, в количестве не более 5% по объемной доле) в присутствии бикарбоната натрия в качестве буферного средства и балансирующего электролита;
5) обратную стероидную (циклопропилметил-гомоаллильную) перегруппировку 7,8-дигидроизоэргостерона с образованием (22Е,24R)-5α-эргоста-2,22-диен-6-она нагреванием с каталитическими количествами безводного бромида лития и гидрохлорида пиридиния в сухом диметилацетамиде;
6) синтез 24-эпикастастерона асимметрическим дигидроксилированием (22E,24R)-5α-эргоста-2,22-диен-6-она в присутствии каталитической системы осмат калия - лиганды DHQD-PHAL (гидрохинидина 1,4-фталазиндиил диэфир) и гидрохинидина 4-хлорбензоат, стехиометрического окислителя гексацианоферрата(III) калия, метансульфонамида, карбоната калия в двухфазной среде трет-бутанол-вода;
7) окисление 24-эпикастастерона трифторнадуксусной кислотой, образовавшейся при смешении трифторуксусного ангидрида и 50%-ной перекиси водорода в среде сухого хлороформа, с выделением и очисткой целевого продукта.
| СПОСОБ ПОЛУЧЕНИЯ 24-ЭПИБРАССИНОЛИДА | 2004 |
|
RU2272044C1 |
| В.А | |||
| ХРИПАЧ И ДР | |||
| Усовершенствованный синтез эпибрассинолида, ЖУРНАЛ ОРГАНИЧЕСКОЙ ХИМИИ, 1994, Т | |||
| Способ обработки медных солей нафтеновых кислот | 1923 |
|
SU30A1 |
| Походная разборная печь для варки пищи и печения хлеба | 1920 |
|
SU11A1 |
| Электрический плавкий предохранитель с автоматической заменой перегоревшей нити | 1924 |
|
SU1650A1 |
| CN 111004303 A, 14.04.2020. | |||

