Настоящее изобретение относится к способу пространственной упаковки химически синтезированных полипептидов. Кроме того, настоящее изобретение относится к способу получения биологически активных белков.
В течение последнего десятилетия отмечается рост потребности в синтетических белках, получаемых в ходе успешного химического синтеза полностью активной ВИЧ-протеазы, фермента, состоящего из 99 остатков, который получают в результате осуществления высокооптимизированных методов твердофазного пептидного синтеза (ТФПС) на основе применения стандартного подхода с Boc/Bzl защитой.
Синтез в 1994 году кристаллического убихитина, небольшого белка, состоящего из 76 остатков, дополнительно продемонстрировал тот факт, что высокочистые белки могут быть синтезированы методами ТФПС на основе процедуры Fmoc/t-Bu, метода, который проще в осуществлении и химически представляет собой менее сложный процесс, чем процедура Boc/Bzl.
К 2000 году накопилось достаточно экспериментальных доказательств того, что белки с одним доменом, содержащие от 60 до 100 аминокислотных остатков, могут быть получены быстро, надежно и экономически эффективно с помощью химического синтеза при использовании пептидного синтезатора в количествах, достаточных для исследования структурных свойств и функциональной активности.
Белки, содержащие дисульфидные мостики, получаемые посредством химического синтеза, при сворачивании имеют такие же свойства, как натуральные формы и формы, полученные генно-инженерными методами. Дисульфидные мостики белков образуют единичные или множественные внутри- и/или межцепочечные циклические структуры, которые придают конформации молекул существенное ограничение, что оказывает решающее воздействие на стабилизацию биологически активной конформации.
Свернутые белки известной структуры с единичным доменом могут быть получены с помощью региоселективного спаривания цистеиновых остатков. В сочетании с обычно используемыми процедурами защиты были разработаны различные комбинации защитных агентов для цистеиновых групп, совместимые с имеющимися способами, которые позволяют осуществлять ступенчатое и попарное удаление защитных групп и/или соокисление цистеиновых остатков с соблюдением полной селективности.
Однако проведенный в последнее время синтез инсулиноподобного пептида - человеческого релаксина - показал, насколько сложен и в этой связи требователен химизм, лежащий в основе региоселективного спаривания цистеиновых остатков, в белках, содержащих множественные цистиновые остатки. Синтез предшественника цепи А был проведен в рамках методики ТФПС с использованием Fmoc/t-Bu-подхода и смолы на основе п-алкоксибензилового спирта, а предшественник В-цепи был получен с использованием смолы ПАМ (сложного эфира 4-карбоксиамидометилбензила, присоединенного к смоле на основе полистирола) с последующим проведением Boc/Bzl-процедуры. Из четырех цистеиновых остатков предшественника А-цепи защита двух из них проводилась в виде производных S-Trt (S-трифенилметила), тогда как для других были выбраны варианты защиты в форме S-Acm (S-ацетамидометила) и S-Meb (S-п-метилбензила) соответственно. Защиту двух цистеинов в предшественнике В-цепи осуществляли с помощью защитных групп S-Acm и S-Meb. Внутримолекулярный S-S-мостик А-цепи получали вначале при окислении йодом в АсОН. Затем в ходе двух стадий получали два межмолекулярных дисульфидных мостика, соединяющих цепи А и В: на первой стадии свободный тиол предшественника цепи А, получаемый при HF-деблокировании защитной группировки S-Meb, подвергали реакции с активированным Cys(Npys) (S-3-нитро-2-пиридинсульфенил) остатком В-цепи (осуществляя направленное образование межмолекулярного гетеродисульфида), а на второй стадии получали оставшийся S-S-мостик путем соокислительного удаления групп S-Acm с помощью йода.
Процедуры ТФПС дают возможность получать с помощью химического синтеза множество полипептидов, содержащих цистеиновые остатки, защищенные одинаковыми блокирующими (защитными) группами. После удаления защитных групп с использованием множества известных окислителей образуются непосредственно дисульфидные связи. Полипептиды и/или белки, содержащие цистеины, защищенные с помощью S-Trt или S-Acm, могут быть достаточно эффективно свернуты посредством обработки йодом, N-йодсукцинимидом и цианогениодидом в строго контролируемых условиях, включая выбор растворителя, значения рН и времени реакции, что позволяет свести к минимуму модификацию чувствительных к окислению остатков Tyr, Met и Trp и избежать избыточного окисления цистеин-тиолов до соответствующих сульфоновых кислот.
Трифторацетат таллия(III) может замещать указанные выше окислители с достижением иногда даже лучших выходов дисульфидных связей. Основными ограничениями для указанного реагента являются его токсичность, трудность удаления таллия из искомого полипептида и необходимость защиты остатков Met и Trp в процессе окисления.
Для прямого окисления с образованием дисульфидных связей полипептидных предшественников, содержащих S-Acm, S-But, S-Meb и S-Mob (S-п-метоксибензил) производные цистеиновых остатков, успешно используются окислители, содержащие смесь сульфоксидных/силильных соединений и трифторуксусной кислоты. Однако основным ограничением для применения указанной смеси является необходимость защиты индольного кольца Trp с помощью формила с целью предотвращения его хлорирования в окислительных условиях.
Способы окислительного сворачивания линейных синтезированных политиоловых предшественников (восстановленных полипептидных форм) являются наиболее известными и применяются чаще всего. В случае самого простого метода соответствующие дисульфидные связи могут быть спонтанно образованы в присутствии воздуха или каких-то других мягких окислителей. Кроме того, сворачивание и спаривание цистеиновых остатков достигается в присутствии как восстановленной (RSH), так и окисленной (R-S-S-R) форм низкомолекулярного сульфгидрильного соединения.
В синтетических полипептидах и небольших белках, состоящих из единичного домена, термодинамическая движущая сила, осуществляющая сворачивание, которое представляет собой результат объединения Н-связывания, ионного спаривания и гидрофобного эффектов, очевидно является вполне достаточной для спонтанного образования нативных изомеров в случайных условиях окислительной ренатурации.
В результате исследования окислительного сворачивания небольших белков, содержащих множественные цистеины, таких как ингибиторы ферментов, токсины или гормоны, была получена полезная информация об определенных структурных мотивах, например цистеин-стабилизированном β-витке, цистеин-стабилизированной поли(Pro)-II спиральной складчатости и цистеин-стабилизированной α-β-структурной складчатости, стабилизация которых является основной движущей силой для корректного образования дисульфидных мостиков даже в относительно малых пептидных молекулах. И если уделять достаточно внимания выбору буферов, температуры и добавок, способных стабилизировать вторичные структурные мотивы, то in vitro может быть достигнуто даже полностью правильное сворачивание частично свернутых или неправильно свернутых белков.
Было разработано множество процедур для сворачивания политиоловых полипептидов с целью минимизации случаев неправильного внутримолекулярного спаривания цистеинов, которое ведет к получению отличных от нативных, неправильно свернутых изомеров, а также с целью избежать, насколько это возможно, образования случайных межмолекулярных дисульфидных связей, которое ускоряет агрегацию и осаждение.
Так, окисление воздухом обычно проводят при высоком разбавлении политиоловой формы предшественника (1 мг/мл или ниже) в условиях нейтральной или слегка щелочной среды. При этом обычно требуется длительный период времени и происходит образование в ходе реакции безвредного побочного продукта в виде воды. Однако окисление воздухом трудно контролировать в связи с тем, что следовые количества ионов металлов очень сильно влияют на скорость этих реакций. Более важно то, что основные и гидрофобные молекулы предшественников имеют склонность к агрегации и осаждению из раствора в условиях, близких к их основным или нейтральным изоэлектрическим точкам, в ходе процесса сворачивания. Более того, в процессе сворачивания образуются побочные продукты, связанные с окислением метионина. Хотя количество химических операций, необходимых для сворачивания политиоловых предшественников, сведено к минимуму, образование дисульфидных мостиков, которому способствует молекулярный кислород воздуха, приводит во многих случаях к низкому выходу, а иногда нужный продукт вовсе не образуется.
В качестве окислителей также использовались ДМСО и феррицианид калия. Однако феррицианид калия необходимо использовать в темноте, и если в полипептидной цепи присутствуют метионин и триптофан, то в ходе процесса сворачивания накапливаются побочные продукты окисления. Использование ДМСО зачастую дает лучшие результаты в связи с тем, что окислительное сворачивание может быть осуществлено в кислых условиях с достаточно высокой скоростью и без образования в процессе реакции вредных продуктов. Указанный метод особенно подходит для сворачивания основных и гидрофобных полипептидных предшественников в связи с более высокими показателями растворимости тех их типов, которые подвергаются окислению в кислых буферах. Однако зачастую сообщается о возникновении проблем, связанных с удалением ДМСО из готового продукта и со снижением селективности образования дисульфидных мостиков. Кроме того, нарушение упорядоченности дисульфидных мостиков ведет к появлению неправильных изомеров, при этом невозможно избежать процесса олигомеризации даже при тщательном контроле экспериментальных условий.
Высокие уровни правильного спаривания цистеинов и сворачивания политиоловых предшественников малых белков чаще всего достигаются при использовании окислительно-восстановительных буферов, таких как окисленный (GSSG) и восстановленный (GSH) глутатион и цистин/цистеин (Cys/Cys).
Так, в ходе окислительного сворачивания рибонуклеазы А (R.R. Hantgan et al., Biochemistry 13, 613, 1974), ядерного домена гирудина, состоящего из 49 аминокислот (B. Chatrenet and J.Y. Chang, J. Biol. Chem. 267, 3038, 1992), и ингибитора бычьего панкреатического трипсина (BPTI) (T.E. Creighton, Methods Enzymol. 131, 83, 1986), индуцированного с помощью GSSG/GSH или Cys/Cys, постоянно происходит образование и преобразование свободных сульфгидрильных и дисульфидных групп в течение всего процесса сворачивания. Общие скорости и выходы реакций в указанном случае обычно выше, чем при окислительном сворачивании на воздухе, поскольку тиол/дисульфидный обмен, происходящий с участием промежуточных производных тиолята, облегчает перетасовку ненативного дисульфида в его натуральные формы. Как и в случае окислительного сворачивания на воздухе необходимо высокое разбавление политиолового предшественника для устранения агрегации, образования олигомеров и полимеров и для достижения максимальных выходов целевых белков.
На первой стадии сворачивания гирудина1-49in vitro сворачивание происходит последовательно и необратимо, начиная с несвернутой восстановленной формы (политиола), до образования равновесной смеси изомеров, содержащих один и два дисульфидных мостика, и до образования равновесной смеси продуктов, содержащих три дисульфидных связи (неупорядоченные изомеры) (J.Y. Chang, Biochem. J. 300, 643, 1994). Были идентифицированы почти все 75 возможных видов белка, включая нативную форму: 15 изомеров с одним S-S мостиком, 45 изомеров с двумя S-S мостиками и 15 изомеров с тремя S-S мостиками. На второй стадии сворачивания неупорядоченные типы белка подвергаются перестройке за счет перетасовки ненативных дисульфидов с образованием нативных белковых форм. Образование дисульфида ускоряется в основном окисленным глутатионом или цистином, тогда как для перетасовки дисульфида требуется тиоловый катализатор, например восстановленный глутатион, или цистеин, или меркаптоэтанол.
Очевидно, что эффективность тиоловых реагентов в плане облегчения перетасовки связана с их окислительно-восстановительным потенциалом, и каждый из катализаторов демонстрирует оптимум концентрации. В процессе накопления неупорядоченных гирудинов цистин/цистеин имеет примерно в 10 раз более сильное действие, чем GSSG/GSH. Такое различие может быть объяснено относительным значением окислительно-восстановительного потенциала пары GSSG/GSH (-0,24 в) и пары Cys-Cys/Cys (-0,22 в). При выборе оптимального сочетания условий (температура, буфер, соли и окислительно-восстановительная смесь) процесс сворачивания гирудина1-49 ускоряется до такой степени, что оно полностью завершается в течение 15 минут.
В целом, нативная конформация синтетического белка, содержащего дисульфидные связи, должна образовываться спонтанно в условиях, оптимальных для сворачивания политиоловых форм. Однако во многих случаях, даже в оптимизированных условиях при окислительном сворачивании при использовании указанных выше окислительно-восстановительных буферов, образуется значительное количество побочных продуктов и форм с неверным спариванием. В особенности это справедливо в случае тех белков, которые имеют склонность к образованию нативной конформации только на поверхности специфических мембран или при помощи специфического молекулярного шаперона (S. Sakakibara, Biopolymers, Peptide Science 51, 279, 1999).
Кроме того, несмотря на широкое использование, большинство процессов окислительного сворачивания политиоловых предшественников под воздействием воздуха или окислительно-восстановительных пар GSSG/GSH и цистин/цистеин осуществляется методом проб и ошибок, что было четко продемонстрировано в экспериментах по сворачиванию синтетического хемокина и аналогов хемокина. Фактически в то время как нативные хемокины и множество их аналогов сворачиваются легко, и их свернутые структуры стабилизируются двумя или тремя дисульфидными мостиками, некоторые аналоги не дают хорошего сворачивания в тех же условиях, что использовались в случае соответствующих нативных молекул, образуя частично свернутые формы. Указанные наблюдения дают веские основания полагать, что изменения в первичной структуре политиоловых предшественников могут оказать неблагоприятный эффект на индукцию правильного образования локальных складчатых форм (α-витки, полипролиновые спиральные мотивы и др.) в сворачиваемых полипептидных цепях. В этой связи способность к сворачиванию у многих тиоловых предшественников представляет собой, прежде всего, внутреннее свойство полипептидной цепи, а не функцию специфической окислительной системы, действующей на молекулы.
Об усилении образования выбранных дисульфидных пар при добавлении спиртов, ацетонитрила и ДМСО к буферам при низкой ионной силе также сообщается в литературе. Указанная стратегия основана на усилении образования определенных дисульфидных связей путем корректировки электростатических факторов в среде, способствующей нужному расположению соседних противоположно заряженных аминокислот, которые граничат с выбранными цистеиновыми остатками.
Ферменты, такие как пептидилдисульфидизомераза (ПДИ) и пролилизомераза (ПИ), также были использованы в качестве добавок для катализа и модуляции дисульфидного обмена. Время, необходимое для сворачивания гирудина in vitro, может быть сокращено с 10 часов до 30 секунд, если в буфер для сворачивания добавить ПДИ. В этом случае эффективность сворачивания in vitro существенно не отличается от ситуации, наблюдаемой in vivo.
Политиоловые полипептидные предшественники непосредственно получают при ацидолитическом расщеплении комплекса полипептид-смола в том случае, когда цистеиновые остатки защищены кислотолабильными группами, например, Trt. Альтернативно и предпочтительно, полипептиды, в которых все цистеины защищены кислотоустойчивой группой, например ацетамидометильной группой (Acm), вначале выделяют в виде S-цистеиновых производных путем ацидолитического расщепления комплекса пептид-смола, после чего Acm-группу удаляют путем обработки Hg(AcO)2 в уксусной кислоте с последующим удалением ионов Hg путем гель-фильтрации в присутствии большого избытка меркаптоэтанола.
Сообщается, однако, что в обоих случаях на цистеиновых и триптофановых остатках имеют место некоторые побочные реакции. Индольное кольцо триптофана может быть модифицировано меркаптоэтанолом, а в случае цистеина имеет место множество побочных реакций, наиболее важные из которых представляют собой окисление и алкилирование катионами трет-бутила в ходе ацидолитического удаления полипептидной цепи со смолы.
Таким образом, в связи с недостатками имеющихся методик существует потребность в более эффективных и более простых способах для сворачивания химически синтезированных полипептидов и получения биологически активных белков путем химического синтеза. На этом основании целью настоящего изобретения является разработка эффективного, простого и быстрого способа сворачивания полипептидов и/или белков, при осуществлении которого, в том числе, сводится к минимуму образование изомеров, содержащих неправильные дисульфидные мостики, нет необходимости использовать дорогостоящие реагенты для перетасовки дисульфидных связей, такие как глутатион или ферменты, причем указанный способ является воспроизводимым, простым и масштабируемым. Указанные и другие цели станут очевидными для специалистов в данной области.
Указанная цель достигается в настоящем изобретении за счет разработки способа сворачивания химически синтезированных полипептидов, включающего в себя обработку полипептида, содержащего два или более дериватизированных цистеиновых остатка, восстанавливающим агентом в буфере для сворачивания, имеющем заданный рН и температуру.
Кроме того, предлагается способ получения биологически активных белков, включающий в себя
(а) химический синтез полипептида, содержащего два или более дериватизированных цистеиновых остатков;
(b) обработку указанного полипептида восстанавливающим агентом в буфере для сворачивания, имеющем заданное значение рН и температуры; и
(с) очистку полученных свернутых белков.
Предпочтительно дериватизированный цистеиновый остаток соответствует остатку S-бутилтиоцистеина (S-t-Bu). Так, в соответствии с настоящим изобретением авторы неожиданно обнаружили, что S-t-Bu-производные цистеинов могут быть подвергнуты депротекции, то есть они могут потерять S-t-Bu-группировку с образованием дисульфидных мостиков с другими цистеинами в случае инкубирования в соответствующем буфере для сворачивания при подходящих значениях температуры и рН.
Согласно настоящему изобретению восстанавливающий агент представляет собой предпочтительно свободный цистеин. Избыток цистеина может быть добавлен к буферу (как, например, показано в примерах 1-5) или цистеин может быть получен из полипептида (как показано в примере 6).
В предпочтительном варианте осуществления настоящего изобретения буфер для сворачивания включает в себя одну или более хаотропных солей с целью приведения полипептида в равновесное состояние, которое способствует естественной пространственной упаковке. Указанная цель может быть достигнута, например, при помещении полипептида и/или белка в условия полной денатурации, например, за счет высокой концентрации хаотропных солей с последующим разбавлением хаотропной соли до низкой концентрации для осуществления сворачивания. Хаотропные соли предпочтительно выбирают из группы, состоящей из хлорида гуанидиния и мочевины, и предпочтительно они присутствуют в концентрации 0,1-1 М в процессе сворачивания.
Предпочтительно температура буфера для сворачивания заключена в диапазоне от 25 до 40°С, более предпочтительно - от 27 до 38°С, с тем, чтобы уменьшить изменения при деградации пептида, но при этом соответствовать естественным температурам тела. Наиболее предпочтительно температура в процессе сворачивания составляет примерно 37°С.
Согласно другому предпочтительному варианту осуществления настоящего изобретения буфер для сворачивания имеет слегка щелочной показатель рН. Предпочтительно значение рН заключено в диапазоне от 7 до 9, более предпочтительно - от 7 до 8,5, с тем чтобы облегчить процесс сворачивания. Как видно из предыдущего описания, сворачивание белка зависит от сложного комплекса взаимодействий. Так, например, реакция с цистеином не идет при кислом рН, а более высокие значения рН увеличивают риск деградации полипептида.
После завершения сворачивания целевые белки могут быть очищены хорошо известными в технике методами, включая анионо- и катионообменную хроматографию, хроматографию, основанную на гидрофобном взаимодействии, хроматографию с обращенной фазой, аффинную хроматографию, хроматографию, основанную на гидрофильном взаимодействии/катионообмене (HILIC/CEC), вытеснительную хроматографию (ВХ) и вытеснительную хроматографию образца (ВХО). Наиболее предпочтительно использование высокоэффективной хроматографии (с обращенной фазой) с элюцией, а также вытеснительной хроматографии.
В предпочтительном варианте осуществления изобретения для получения биологически активных белков указанный способ включает в себя стадии
(а) сборки S-трет-бутил-тиоцистеинового полипептида на нерастворимой полимерной подложке посредством пошаговой элонгации цепи;
(b) отщепления указанной S-трет-бутил-тиоцистеиновой полипептидной цепи с указанной подложки посредством ацидолизиса;
(с) очистки полученного S-трет-бутил-тиоцистеинового полипептида;
(d) сворачивания очищенного S-трет-бутил-тиоцистеинового полипептида посредством обработки указанного полипептида молярным избытком цистеина в буфере для сворачивания, включающем в себя хаотропную соль, предпочтительно хлорид гуанидиния, и имеющем щелочное значение рН и температуру примерно 37°С; и
(е) очистки полученных свернутых белков посредством высокоэффективной жидкостной хроматографии с обращенной фазой.
В удачном варианте осуществления способа согласно настоящему изобретению полимерная подложка представляет собой полиамид или смолу на основе полистирола, функционализированную кислотолабильным линкером в виде гидроксиметилфеноксиуксусной кислоты, поскольку указанные подложки могут быть успешно использованы для полностью автоматизированных синтезаторов пептидов и в основном позволяют осуществлять синтез длинных полипептидных цепей.
Как отмечалось выше, настоящее изобретение основано на последовательной твердофазной сборке S-трет-бутил-тиоцистеиновых полипептидов и на обнаружении того факта, что значительные количества нативных биологически активных белков могут быть получены посредством проведения сворачивания указанных полипептидов в присутствии восстанавливающего агента, предпочтительно цистеина, при значении рН несколько выше нейтрального и при температуре около 37°С.
Было неожиданно обнаружено, что образование биологически активных белков достигается с использованием высокого молярного избытка цистеина при проведении процедуры удаления S-трет-бутиловой защитной группы, которая сопровождается образованием дисульфидного мостика. Такая процедура является более простой и эффективной, чем описанные на достигнутом уровне техники процедуры, применяемые для сворачивания цистеин-содержащих полипептидов, получаемых с помощью химического синтеза. Таким образом, удаление S-трет-бутила осуществляется на той же стадии, что и сворачивание полипептида.
Альтернативно, тот же свернутый материал может быть получен при использовании сочетания S-трет-бутил-тиоцистеина и цистеина, защищенного соответствующими кислотно-лабильными группами в выбранных положениях полипептидной цепи с целью усиления образования соответствующих дисульфидных связей без необходимости чрезмерного добавления восстанавливающего агента к буферу для сворачивания. В указанном случае кислотно-лабильные группы удаляются, когда пептид отщепляют от смолы при кислом рН. При этом образуются свободные цистеины, которые, в свою очередь, действуют как внутримолекулярный "восстанавливающий агент", обеспечивающий удаление S-трет-бутила и образование дисульфида (как показано в примере 6).
Суть настоящего изобретения основана на
- быстрой сборке S-тио-трет-бутилированных полипептидных цепей на полимерной подложке;
- катализируемом цистеином тиол-дисульфидном обмене производных в слегка щелочных условиях, который ведет к получению цистеинилированных полипептидов, представляющих собой макромолекулярные окисленные формы (белок-S-S-цистеин; полипептид-S-S-цистеин) классической окислительно-восстановительной пары цистин/цистеин;
- поддержании концентрации окисленной макромолекулярной формы на постоянно низком уровне в течение всего процесса сворачивания, так что сводится к минимуму межмолекулярный дисульфидный обмен; и
- отсутствии агрегации неправильно свернутых промежуточных продуктов благодаря быстрому и предпочтительному образованию структур с правильным спариванием цистеина (нативных структур).
Согласно способу сворачивания полипептидов согласно настоящему изобретению, например, на первой стадии 10 мг S-трет-бутильного производного растворяют при комнатной температуре в 1 мл буфера с рН 8,0, включающего в себя 6 М хлорида гуанидиния, 10 мМ Трис и 0,1 М Na2HPO4, и полученный раствор выдерживают при комнатной температуре в течение примерно 20 минут. На второй стадии раствор вначале разбавляют в 10 раз водой до рН 7,2 (0,6 М хлорида гуанидиния, 1,0 мМ Трис, 10 мМ Na2HPO4 при конечной концентрации производного полипептида 1 мг/мл), и затем добавляют при перемешивании большой молярный избыток цистеина (примерно в 100 раз выше концентрации производного полипептида или белка). Температуру смеси постепенно повышают до 37°С и поддерживают постоянной в течение примерно 24 часов для того, чтобы прошло сворачивание полипептида.
Способ сворачивания согласно настоящему изобретению приводит к получению высокогомогенного продукта и применим с небольшими модификациями к любому полипептиду, получаемому путем твердофазного химического синтеза в виде тио-трет-бутилового производного цистеина. Кроме того, способ согласно настоящему изобретению имеет множество других дополнительных преимуществ в сравнении со способами, имеющимися на достигнутом уровне техники, которые применяют политиоловые формы предшественника, такие как
- цистеиновые остатки цепи не алкилируются в процессе ацидолитического расщепления комплекса полипептид-смола;
- не происходит ни переокисления цистеина до сульфоновой кислоты, ни окисления, ведущего к образованию межмолекулярного дисульфидного мостика;
- исключается риск модификации индольного кольца триптофана меркаптоэтанолом, что необходимо для удаления загрязняющих ионов Hg, образующихся при деблокировании Acm с помощью Hg(AcO)2. Фактически тиолят цистеина в смеси для сворачивания согласно настоящему изобретению совсем не модифицирует триптофан;
- чувствительные к окислению остатки метионина, триптофана и тирозина не модифицируются в процессе сворачивания;
- стоимость получения готовых свернутых продуктов в основном ниже в сравнении со стоимостью способов, в которых используются политиоловые полипептиды и окислительно-восстановительные буферы.
Специалистам в данной области очевидно, что несмотря на то, что целевые белки в основном удается получить согласно способу настоящего изобретения с высоким выходом, в ряде случаев, например, в случае сложных белков с множественными дисульфидными связями, в растворе в равновесном состоянии будет оставаться определенная доля промежуточных форм, которые не подверглись переходу в нативную структуру (неправильно свернутые структуры). Такие неправильно упакованные пространственные структуры могут быть легко отделены от правильно упакованных структур посредством ВЭЖХ-ОФ и снова помещены в условия сворачивания согласно настоящему изобретению для повышения общего выхода процесса.
Согласно настоящему изобретению термин полипептид относится к полимеру, состоящему из аминокислот, связанных вместе амидными связями. Термин белок относится к полипептидной структуре в ее трехмерной форме, которая имеет место в клетках и биологических жидкостях живых организмов. Белки могут, например, состоять из единичных свернутых полипептидных цепей или могут представлять собой сложные структуры, состоящие из многих свернутых полипептидных цепей.
Приведенные ниже примеры и графики даны для иллюстрации настоящего изобретения и не направлены на ограничение изобретения сверх тех ограничений, которые накладывает на него формула изобретения.
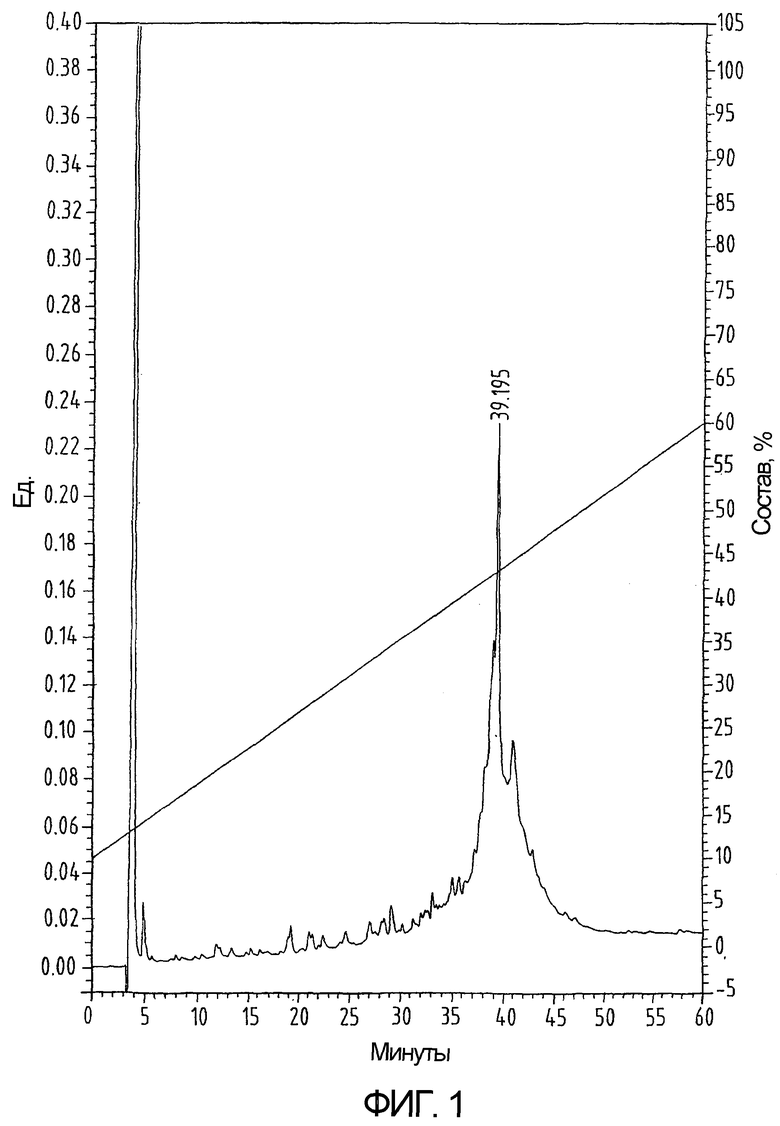
На фиг.1 показан ВЭЖХ-профиль перед удалением S-трет-бутила и сворачиванием hu-I-309 (пример 4).
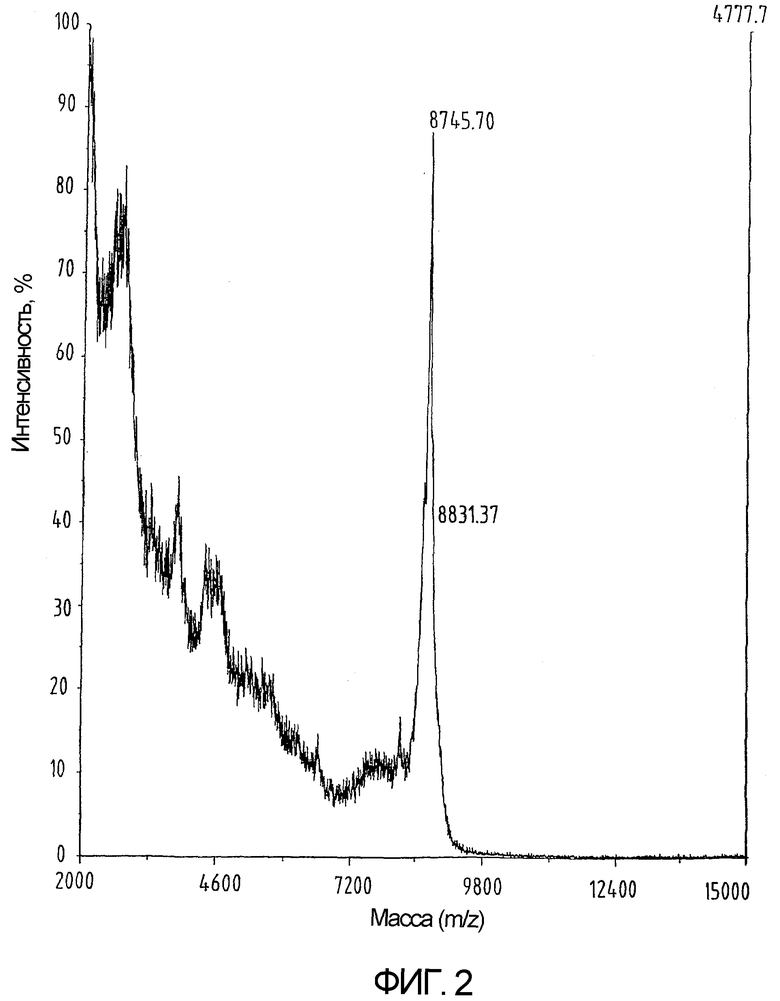
На фиг.2 показан результат определения массы продукта, приведенного на фиг.1.
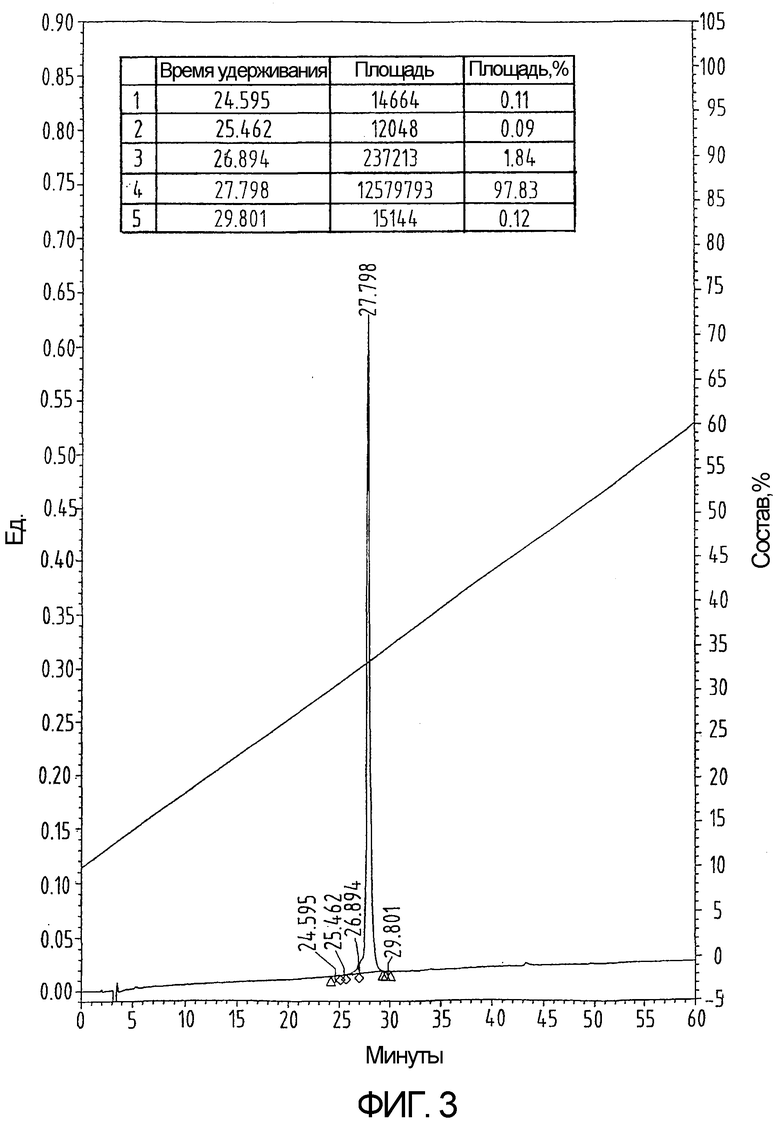
На фиг.3 показан ВЭЖХ-профиль освобожденного от защитной группы свернутого hu-I-309 (пример 4), демонстрирующий более короткое время удерживания.
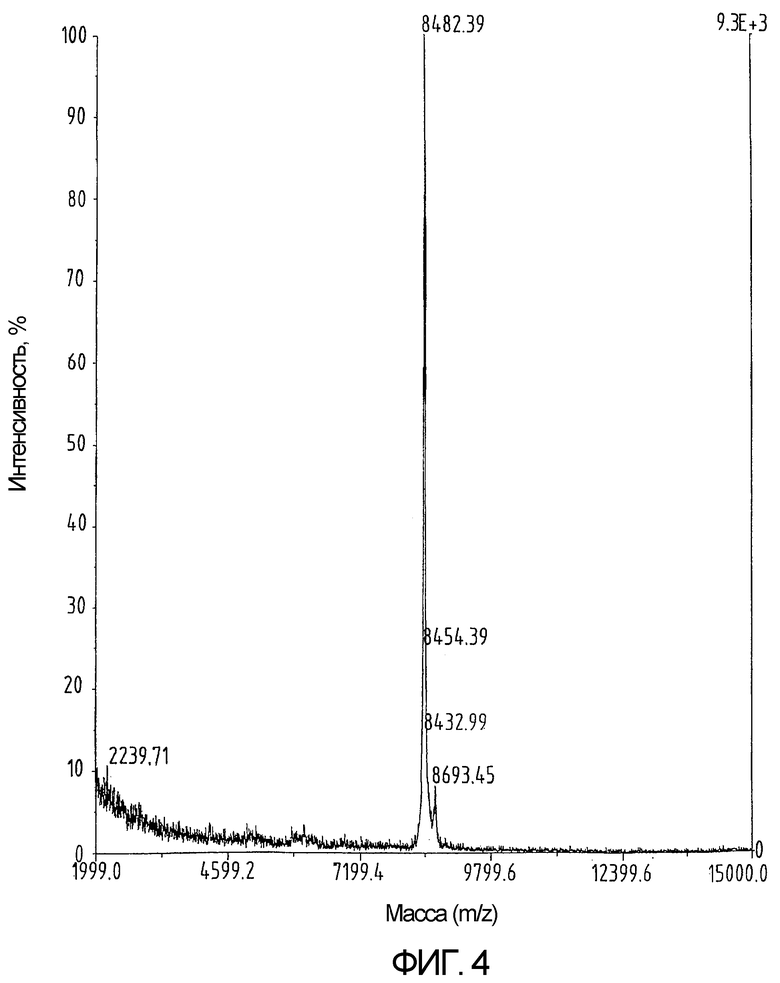
На фиг.4 показан результат определения массы продукта, приведенного на фиг.3.
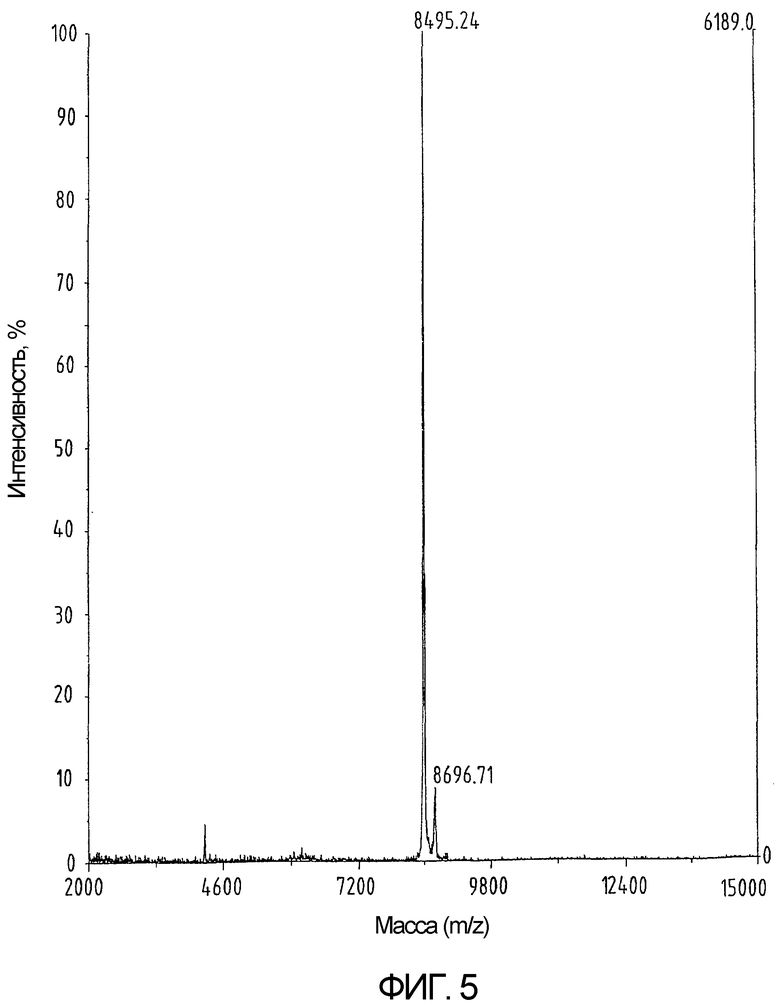
На фиг.5 показан результат определения массы продукта, изображенного на фиг.3, после обработки NEM. В сравнении с фиг.4 не наблюдается изменения массы, что указывает на отсутствие свободных -SH групп.
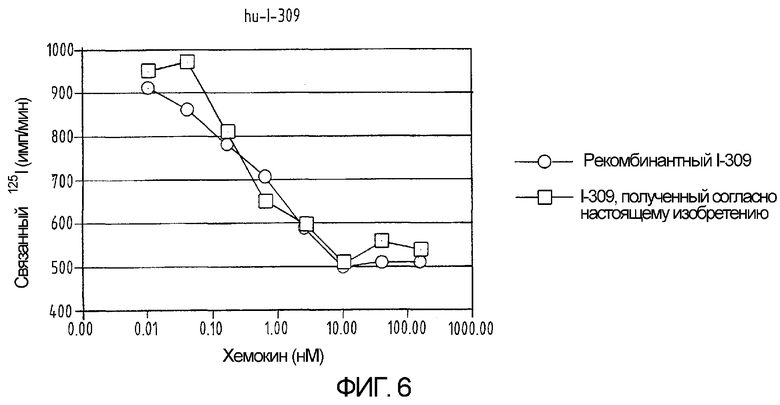
Фиг.6 представляет собой графическую иллюстрацию сравнения биологической активности рекомбинантного I-309 и синтетического I-309, свернутого согласно настоящему изобретению. Биологическую активность оценивают по связыванию хемокина, меченного 125I, с лимфоцитами человека.
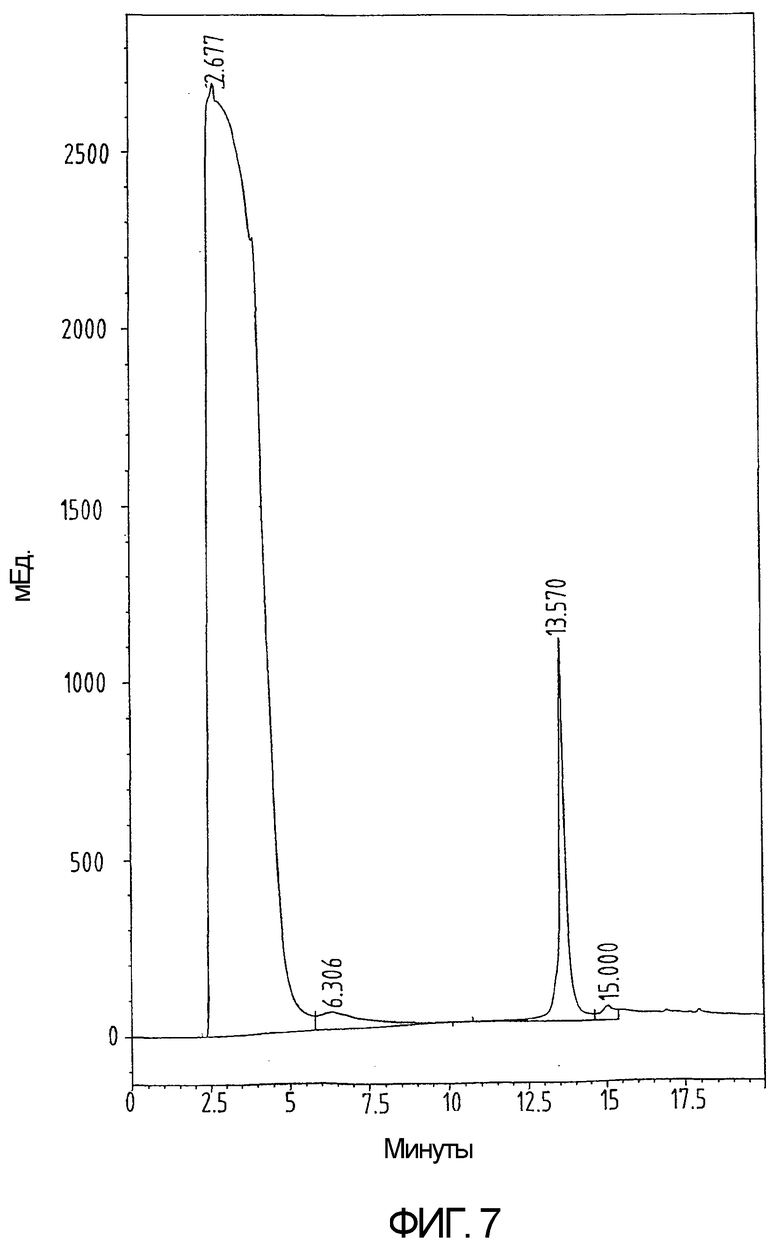
На фиг.7 показан аналитический профиль ВЭЖХ белка из примера 5 после сворачивания.
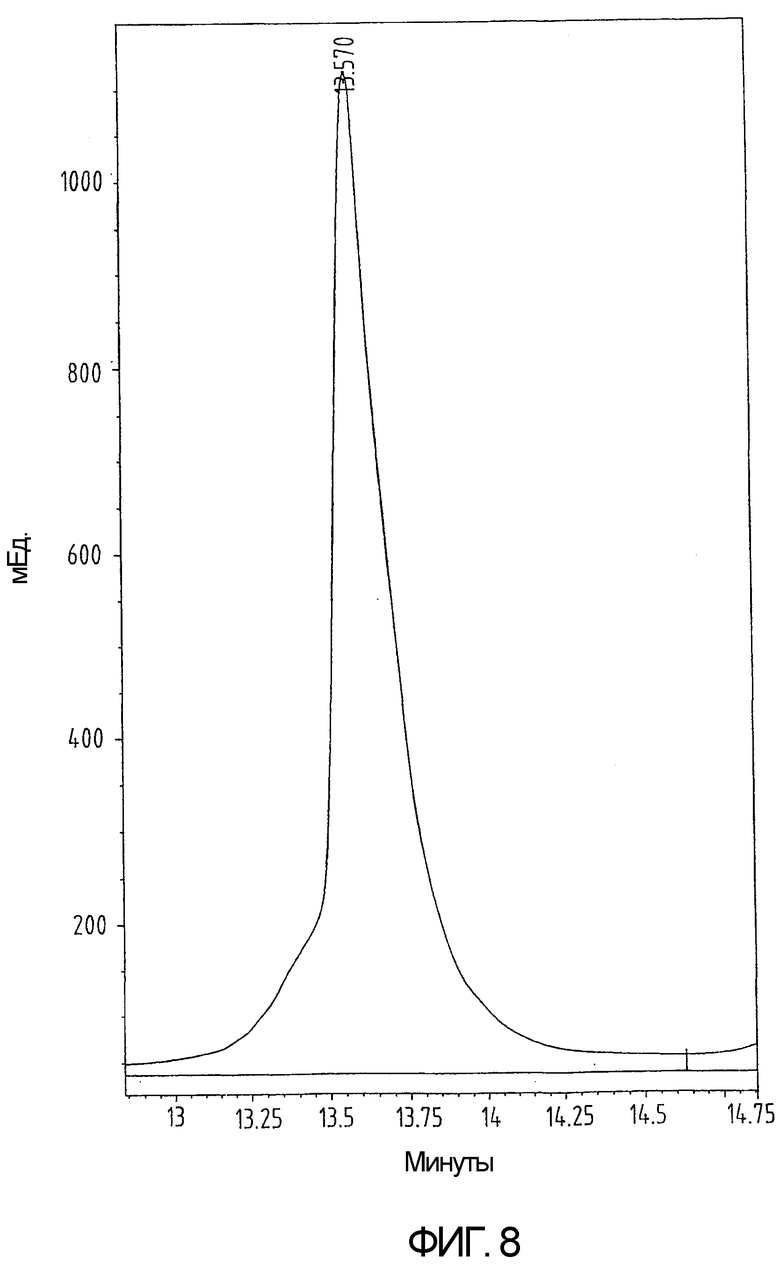
На фиг.8 показан профиль препаративной ВЭЖХ свернутого белка, приведенного в примере 5.
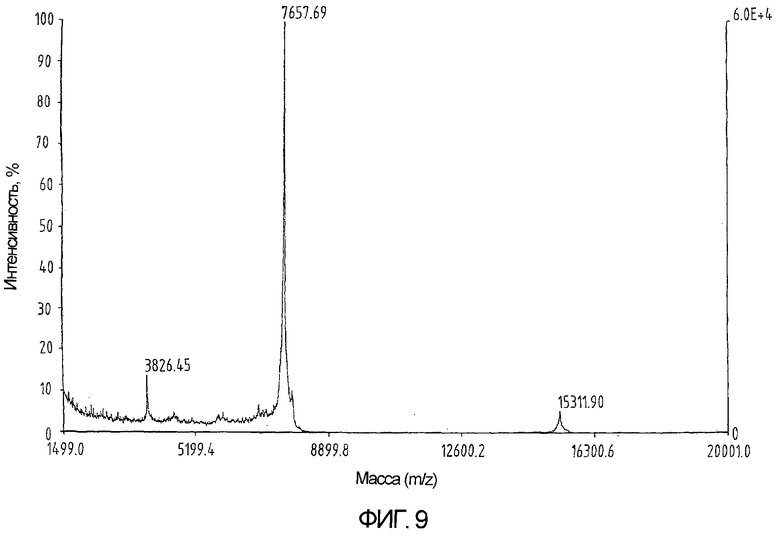
На фиг.9 показан результат определения массы очищенного продукта из примера 5, указывающий на наличие ожидаемой молекулярной массы.
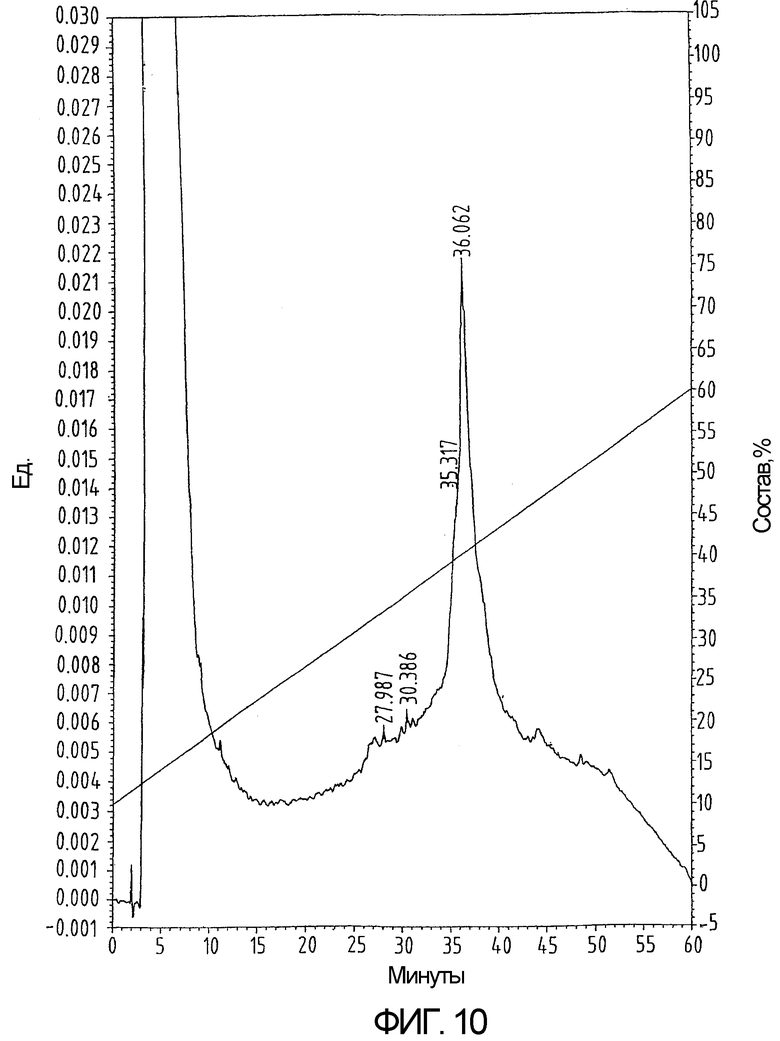
На фиг.10 показан ВЭЖХ-профиль полипептида из примера 6 перед удалением S-трет-бутила и сворачиванием.
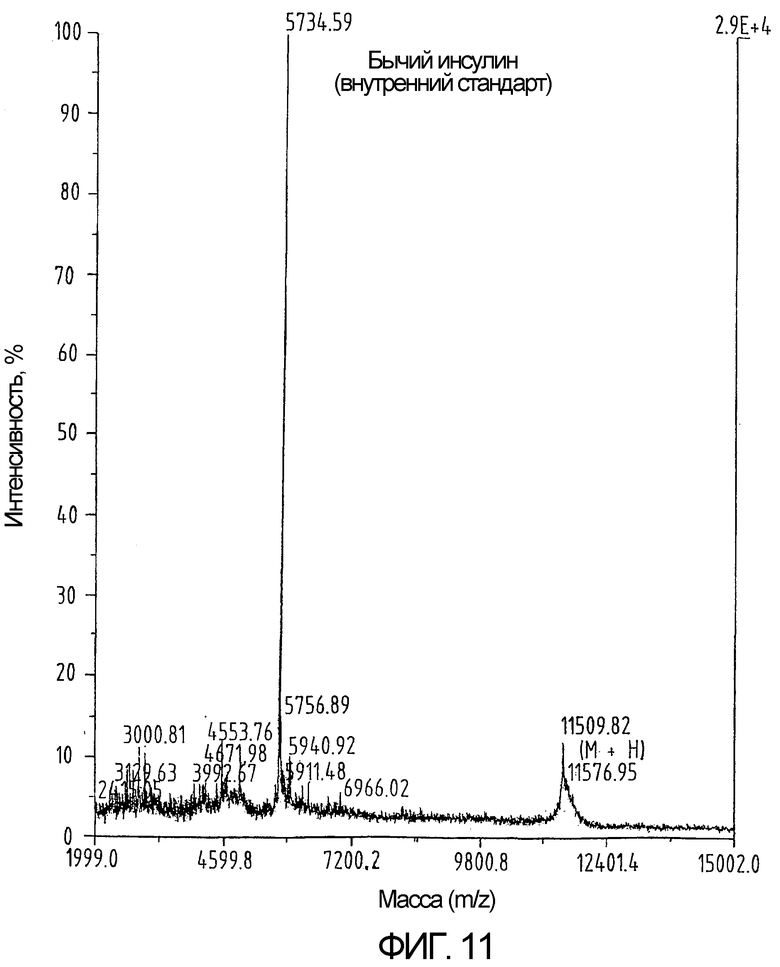
На фиг.11 показан результат определения массы полипептида, приведенного на фиг.10 (М=Н).
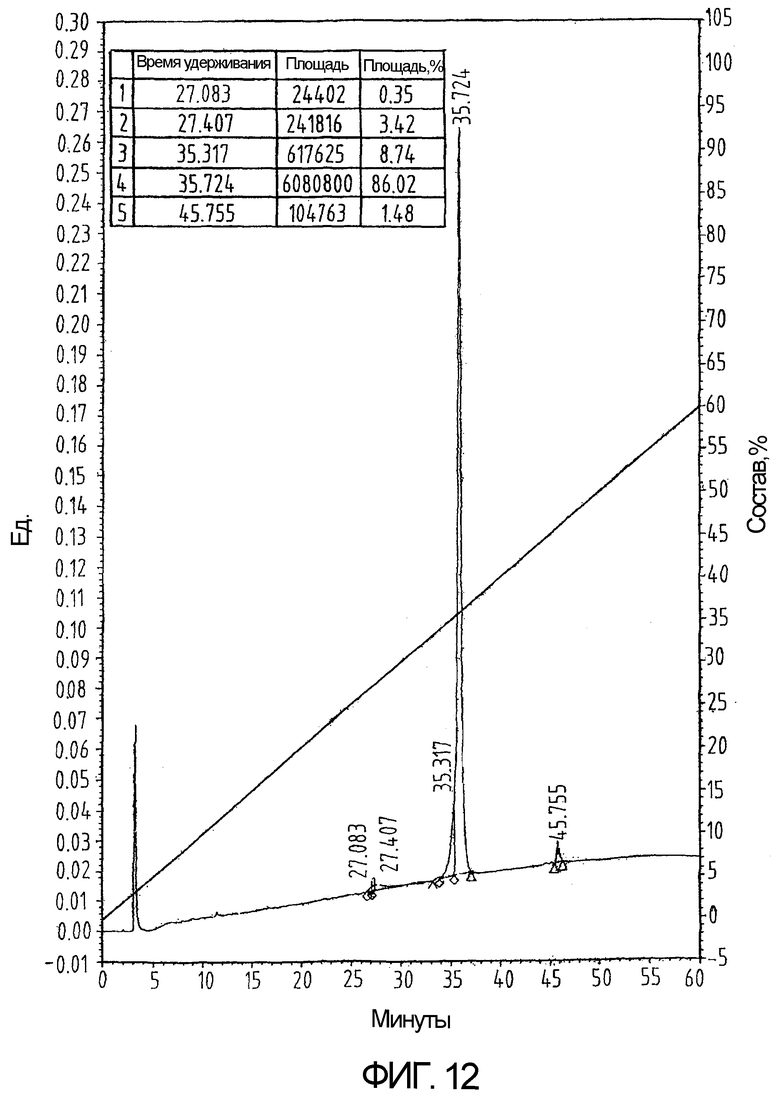
На фиг.12 показан ВЭЖХ-профиль белка из примера 6 после сворачивания, демонстрирующий более короткое время удерживания.
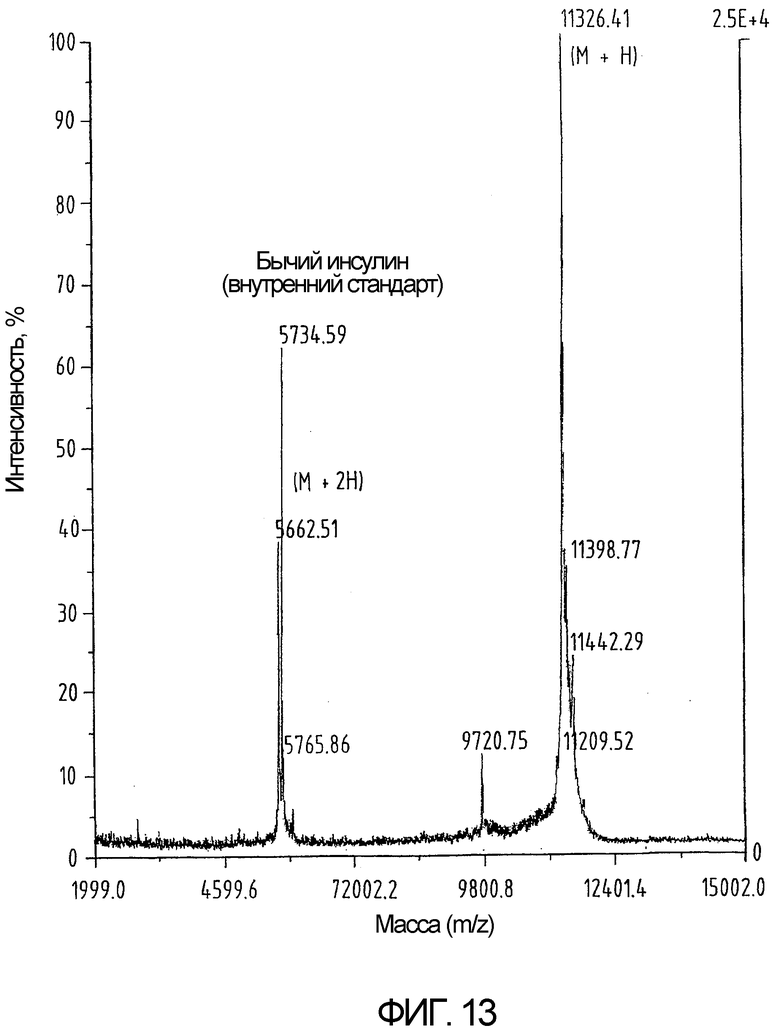
На фиг.13 показан результат определения массы белка, показанного на фиг.12 (М=Н), указывающий на наличие ожидаемой молекулярной массы.
Примеры
Пример 1
Синтез и сворачивание Cys10,11,34,50(S-t-Bu)-hu-TARC
(тимус и активация регулируемого хемокина)
Производное хемокина, состоящее из 71 аминокислотного остатка, подвергают сборке на пептидном синтезаторе 433 А (Perkin Elmer/ABI) с использованием технологии Fmoc/t-Bu и смолы на основе полистирола, содержащей функциональные группы, полученные при обработке кислотолабильным линкером на основе гидроксиметилфеноксиуксусной кислоты (смола Ванга; Wang resin), к которой Fmoc-Ser(t-Bu) присоединен посредством реакции этерификации, катализируемой ДМАП (4-диметиламинопиридином). Степень замещения составляет 0,57 ммоль/г. Синтез проводят по шкале с шагом 0,27 ммоль, используя пятикратный избыток Fmoc-аминокислот и реагентов, активированных смесью DCI (N,N'-диизопропилкарбодиимид)/HOBt (1-гидроксибензотриазол), в ДМФ. Время связывания составляет примерно 60 минут с проведением спектрофотометрического мониторинга депротекции Fmoc.
Четыре цистеиновых тиола защищают с помощью S-трет-бутильных групп и используют схему максимальной защиты для всех других боковых цепей: Ser(t-Bu), Thr(t-Bu), Tyr(t-Bu), Asp(O-t-Bu), Glu(O-t-Bu), Lys(Boc), Trp(Boc), Asn(Trt), Gln(Trt) и Arg(Pmc). После каждого связывания проводят покрытие уксусным ангидридом и ДИЭА в ДМФ.
Полученный комплекс полипептид-смола обрабатывают при комнатной температуре свежеприготовленной смесью ТФУ/вода/ТИС (триизопропилсилан)/фенол (78:5:12:5, объем/объем/объем/вес, 10 мл/г смолы) в течение 2,5-3,0 часов. Отщепленное полипептидное производное осаждают путем прямой фильтрации расщепленной смеси в холодном метил-трет-бутиловом эфире (МТБЭ) и осадок отделяют путем центрифугирования, дважды промывают эфиром и сушат на воздухе.
Затем неочищенный продукт растворяют в разбавленной уксусной кислоте, лиофилизируют, перерастворяют в 50% уксусной кислоте и наносят на колонку с Сефадекс G-50 (70х25 см), с использованием 50% уксусной кислоты в качестве подвижной фазы. Собранные фракции анализируют посредством MALDI-TOF-масс-спектрометрии и те фракции, которые содержат нужные полипептидные производные (М.в. 8436,9 Да), объединяют и лиофилизируют после разбавления водой.
Объединенные фракции вновь растворяют в 50% уксусной кислоте и дальше очищают путем нанесения на полупрепаративную колонку Vydac C4 размером 250х10 мм. Образцы элюируют при скорости тока 3 мл/мин с использованием линейного градиента 20-80% B в течение 60 минут, где В представляет собой 0,1% ТФУ в ацетонитриле, а А представляет собой 0,1% ТФУ в воде. Обнаружение проводят при длине волны 280 нм, и только фракции, содержащие целевой полипептид, объединяют и лиофилизируют перед проведением в дальнейшем процесса сворачивания.
Сворачивание производного хемокина, очищенного методом ВЭЖХ-ОФ, проводят вначале посредством растворения 10 мг продукта в 1 мл 6М GnHCl, 0,1M Na2HPO4 и 10 мМ Трис при рН 8,0 и при комнатной температуре. Через 20 минут раствор разбавляют при добавлении 10 мл воды до конечной концентрации 0,6М GnHCl, 10mM Na2HPO4, 1mM Трис, рН 7,2 и концентрации пептида 1 мг/мл. Сворачивание инициируют добавлением цистеина в концентрации примерно 20мМ (примерно 100-кратный молярный избыток относительно концентрации пептида) и постепенно повышают температуру до 37°С.
Реакцию сворачивания, протекающую при постоянной температуре 37°С на воздухе, отслеживают посредством проведения анализа методом ВЭЖХ-ОФ с использованием аликвот по 25 мкл раствора, погашенного уксусной кислотой, на приборе Waters 2690 Separation Module, снабженном фотодиодным детектором Waters 996, с использованием аналитической колонки Vydac C4 и 20-60% градиента ацетонитрила в смеси 0,1% ТФУ/вода в течение 40 минут при скорости тока 1,0 мл/мин. По 1 микролитру из каждого пика ВЭЖХ (соответствующего промежуточным продуктам тиол-дисульфидного обмена в процессе сворачивания) собирают, смешивают с 1 микролитром насыщенного раствора синаповой кислоты в смеси ацетонитрил/1% ТФУ (1:2) в воде, сушат в вакууме и анализируют с помощью MALDI-TOF масс-спектрометрии с использованием спектрометра Voyager-DE (Perseptive Biosystem, Framingham, MA), оснащенного азотным лазером. Через 24 часа образуется 78% свернутого полипептида. Пик, М.в. которого соответствует молекулярному весу свернутого продукта, далее проверяют в реакции с N-этилмалеидом (NEM) для обнаружения присутствия свободных тиоловых групп (+125 Да для каждой SH-группы).
Оценку биологической активности hu-TARC, полученного по методике согласно настоящему изобретению, проводят по методу Имаи (T. Imai et al., J. Biol. Chem., 271, 21514, 1996).
Т-клеточные линии человека Hut 78, Hut 102 и Jurkat, а также свежие моноциты, нейтрофилы и лимфоциты оценивают по их способности мигрировать через поликарбонатный фильтр в ответ на воздействие TARC. У моноцитов или нейтрофилов не отмечалось хемотаксической реакции ни под действием TARC, полученного химическим синтезом, ни под действием рекомбинантного TARC. В Т-клеточных линиях Hut 78 и Hut 102 синтетический TARC, равно как и рекомбинантный TARC, индуцирует миграцию с достижением типичной колоколообразной кривой, и максимальный эффект при этом наблюдается при концентрации 100 нг/мл.
Пример 2
Синтез и сворачивание Cys10,34,50(S-t-Bu)-hu-TARC и
Cys11,34,50(S-t-Bu)-hu-TARC
Синтез, очистку и сворачивание производных Cys10,34,50(S-t-Bu)-hu-TARC и Cys11,34,50(S-t-Bu)-hu-TARC проводят в тех же условиях, что были приняты для Cys10,11,34,50(S-t-Bu)-hu-TARC (пример 1), единственное отличие заключается в том, что создают Trt-защиту для Cys10 и Cys11 соответственно, которую удаляют одновременно с отщеплением полипептидных предшественников от смолы. Выходы готовых свернутых хемокинов составляют 80% и 79% соответственно.
Пример 3
Синтез и сворачивание Cys34,50(S-Bu)-hu-TARC
Синтез, очистку и сворачивание производного Cys34,50(S-Bu)-hu-TARC проводят в тех же условиях, что были использованы для производных в примерах 1 и 2, за исключением того, что оба остатка Cys10 и Cys11 защищают Trt, который удаляют в процессе конечного отщепления от смолы с помощью ТФУ. Выход свернутого продукта составляет 75%.
Пример 4
Синтез и сворачивание Cys10,11,26,34,50,68(S-t-Bu)-hu-I-309
Синтез hu-I-309, содержащего 6 цистеинов, защищенных (S-t-Bu), проводят по шкале с шагом 0,12 ммоль в тех же условиях, что были описаны в примере 1, с использованием Fmoc-Lys(Boc) смолы Ванга (Wang resin) (степень замещения составляет 0,61 ммоль/г). Полученный комплекс полипептид-смола обрабатывают, как было описано в примере 1, и очищенный на G-50 материал далее очищают путем нанесения на колонку Vydac C18 размером 250х10 мм (как показано на фиг.1 и 2).
Сворачивание производного хемокина, очищенного методом ВЭЖХ-ОФ, проводят посредством растворения 65 мг продукта в 60 мл 0,6М GuHCl, 10мM Na2HPO4 и 1 мМ Трис при рН 8,0 и добавления цистеина в концентрации, достигающей 100-кратного молярного избытка относительно концентрации пептида. Полипептидный раствор выдерживают при температуре 37°С в течение 4 дней. После подкисления с помощью ТФУ свернутый материал выделяют методом ВЭЖХ-ОФ с использованием колонки Vydac C18 размером 250х10 мм (как показано на фиг.3 и 4). Полноту спаривания цистеина проверяют с помощью масс-спектрометрии после реакции с N-этилмалеимидом (NEM). Никакого увеличения мол. веса при этом не наблюдается, что указывает на отсутствие свободных тиоловых групп (как показано на фиг.5). Выход готового свернутого хемокина составляет почти 25%. Синтетический свернутый hu-I-309 демонстрирует биологическую активность, эквивалентную активности рекомбинантного белка (фиг.6).
Проводят аналитическое хроматографирование с использованием следующих условий:
Колонка: С18 размером 250х4,6 мм (Vydac#238TP54)
Подвижная фаза: А = 100% Н2О / 0,1% ТФУ
В = 100% CH3CN / 0,1% ТФУ
Градиент: композиция В% описана на хроматограмме
Детектор: 214 нм
Пример 5
Синтез и сворачивание C-концевого фрагмента
Plasmodium vivax
Синтез и очистку циркумспорозоитного белка Plasmodium vivax (PvCS 303-372), содержащего 4 цистеина, защищенных (S-t-Bu), проводят в тех же условиях, что были описаны в примере 1.
Сворачивание проводят посредством добавления 27 мг пептида в 2,7 мл 6М GuHCl в 0,1М Трис-буфере с рН 8,5. Полученный раствор перемешивают в течение 10 минут. Затем добавляют 13,5 мл 1мМ ЭДТА, 0,2М NaCl, забуференного при рН 8,8 в 0,2М Трис-буфере. В конце добавляют 10,8 мл 35мМ цистеина в 1мМ ЭДТА, 0,2М NaCl, забуференного при рН 8,8 в 0,2М Трис-буфере. Реакционную смесь доводят до температуры 37°С. Реакцию свертывания проверяют на полноту завершения методом ВЭЖХ с обращенной фазой (3-6 часов) (фиг.7) и останавливают реакцию охлаждением в течение 5 минут при 4°С с последующим добавлением 10% ТФУ при 4°С до достижения конечной концентрации 1% ТФУ (3 мл 10% ТФУ). Впоследствии продукт очищают методом ВЭЖХ с обращенной фазой (фиг.8) и определяют массу готового продукта (фиг.9). Выход готового окисленного продукта составляет 70-80%.
Проводят аналитическое хроматографирование с использованием следующих условий:
Колонка: С4 размером 250х4,6 мм (Vydac#214TP54)
Подвижная фаза: А = 100% Н2О / 0,1% ТФУ
В = 100% CH3CN / 0,1% ТФУ
Градиент: композиция В% описана на хроматограмме
Детектор: 214 нм
Пример 6
Крупномасштабный синтез и сворачивание
C-терминального фрагмента Plasmodium falciparum
Крупномасштабный синтез и очистку циркумспорозоитного белка Plasmodium falciparum (PfCS 282-383), содержащего только 2 цистеина, защищенных (S-t-Bu), из 4, проводят в тех же условиях, что были описаны в примере 1 (как показано на фиг.10 и 11), за исключением следующих изменений.
Сворачивание проводят посредством растворения 1,1 г частично очищенного пептида в 1,0 л 0,1М CH3COONH4, рН 8,0 без добавления свободного цистеина в буфер для сворачивания. Реакционную смесь выдерживают при температуре 32°С в течение 18 часов. Полученный продукт очищают методом ВЭЖХ с обращенной фазой (фиг.12 и 13). Выход готового окисленного продукта составляет почти 37%.
Проводят аналитическое хроматографирование с использованием следующих условий:
Колонка: С18 размером 250х4,6 мм (Vydac#238TP54)
Подвижная фаза: А = 100% Н2О / 0,1% ТФУ
В = 100% CH3CN / 0,1% ТФУ
Градиент: композиция В% описана на хроматограмме
Детектор: 214 нм
Настоящее изобретение относится к способу пространственной упаковки (сворачивания) химически синтезированных полипептидов, включающему в себя обработку полипептида и/или белка, который содержит два или более S-бутил-тиоцистеиновых остатка, цистеином, в буфере для сворачивания, который имеет среднее значение рН от 7 до 9 и температуру в интервале от 25°С до 40°С. 3 н. и 16 з.п. ф-лы, 13 ил.
(а) химический синтез полипептида, содержащего два или более S-бутил-тиоцистеиновых остатка;
(b) обработку указанного полипептида цистеином в буфере для сворачивания, который имеет среднее значение рН от 7 до 9 и температуру в интервале от 25 до 40°С; и
(c) очистку полученных свернутых полипептидов и/или белков.
(a) сборки S-трет-бутил-тиоцистеинового полипептида на нерастворимой полимерной подложке посредством пошаговой элонгации цепи;
(b) отщепления указанной S-трет-бутил-тиоцистеиновой полипептидной цепи от указанной подложки посредством ацидолизиса;
(c) очистки полученного S-трет-бутил-тиоцистеинового полипептида;
(d) сворачивания очищенного S-трет-бутил-тиоцистеинового полипептида посредством обработки указанных полипептидных производных молярным избытком цистеина в буфере для сворачивания, содержащем хаотропную соль и имеющем щелочное значение рН и температуру примерно 37°С; и
(e) очистки полученных свернутых белков посредством высокоэффективной жидкостной хроматографии с обращенной фазой.
| КОВАЛЕНТНОЕ МОДИФИЦИРОВАНИЕ ПОЛИПЕПТИДОВ ПОЛИЭТИЛЕНГЛИКОЛЯМИ | 1992 |
|
RU2148586C1 |
| WO 9108762 A1, 27.06.1991 | |||
| Узел ввода | 1976 |
|
SU628573A1 |
| WO 9506064 A1, 02.03.1995. | |||

