Область техники, к которой относится изобретение
Изобретение относится к области медицинской химии. В частности, изобретение относится к арилзамещенным пиразолам, триазолам и тетразолам, а также к открытию противосудорожного действия этих соединений и их действия в качестве блокаторов натриевых (Na+) каналов.
Уровень техники
Показано, что несколько классов используемых в терапии лекарственных средств, включая обезболивающие средства местного назначения, такие как лидокаин и бупивакаин, противоаритмические средства, такие как пропафенон и амиокларон, и противосудорожные средства, такие как ламотригин, фенитоин и карбамазепин, характеризуются общим механизмом действия путем блокирования или модулирования активности Na+ каналов (см. статью Catterall W.A., Trends Pharmacol. Sci., 8:57-65 (1987)). Предполагают, что каждый из этих агентов действует, препятствуя быстрому притоку ионов Na+.
Недавно показано, что другие блокаторы Na+ каналов, такие как BW619C89 и лифаризин, обладают нейропротективным действием на моделях животных с общей или очаговой ишемией и в настоящее время находятся на стадии клинических испытаний (см. статьи Graham и соавт., J. Pharmacol. Exp. Ther., 269: 854-859 (1994); Brown и соавт., British J. Pharmacol., 115: 1425-1432 (1995)).
Нейропротективная активность блокаторов натриевых (Na+) каналов связана с их эффективностью в снижении внеклеточной концентрации глутамата в процессе ишемии посредством ингибирования высвобождения этого экситотоксичного (т.е. индуцирующего токсичность) аминокислотного нейромедиатора (или иначе, подавления возбудимости нервной ткани). Исследования показали, что в отличие от антагонистов глутаматного рецептора, блокаторы Na+ каналов предотвращают гипоксическое повреждение белого вещества мозга млекопитающих (см. статью Stys и соавт., J. Neurosci., 12: 430-439 (1992)). Таким образом, эти блокаторы могут представлять преимущество при лечении некоторых типов инсультов или нейрональных травм в случаях преобладания повреждений каналов белого вещества.
Другим примером клинического использования блокаторов Na+ каналов является рилузол. Показано, что это лекарственное средство продлевает жизнь в подгруппе пациентов с боковым амиотрофическим склерозом (БАС) (см. статью Bensim и соавт., New Engl. J. Med., 330: 585-591 (1994)) и позднее это лекарственное средство было утверждено Управлением по контролю пищевых продуктов и лекарственных средств (FDA) для лечения БАС. Кроме упомянутого выше клинического использования карбамазепин, лидокаин и фенитоин используют для лечения невропатической боли, такой как невралгия тройничного нерва, диабетическая невропатия и другие формы повреждения нервной ткани (см. статью Taylor and Meldrum, Trends Pharmacol. Sci., 16: 309-316 (1995)), а карбамазепин и ламотригин используют при лечении маниакальной депрессии (см. статью Denicott и соавт., J. Clin. Psychiatry, 55: 70-76 (1994). Более того, на основании ряда сходных признаков между хронической болью и звоном в ушах (см. статьи Moller A.R., Am. J. Otol., 18: 577-585 (1997); Tonndorf, J. Hear. Res., 28: 271-275 (1987)) предполагают, что шум в ушах можно рассматривать как форму ощущения хронической боли (см. статью Simpson J.J. и Davies E.W., Tip., 20: 12-18 (1999)). Действительно, показано, что лигнокаин и карбамазепин эффективны при лечении шума в ушах (см. статьи Majumdar В. и соавт., Clin. Otolaryngol., 8: 175-180 (1983); Donaldson I., Laryngol. Otol., 95: 947-951 (1981)).
Показано также, что существует по крайней мере пять-шесть участков связывания на потенциалозависимых Na+ каналах, которые специфически связываются с нейротоксинами (см. статью Catterall W.A., Science, 242: 50-61 (1988)). Дальнейшие исследования показали, что терапевтические противоаритмические средства, противосудорожные средства и обезболивающие средства местного назначения, действие которых опосредовано Na+ каналами, проявляют свою активность путем взаимодействия с внутриклеточным участком Na+ канала и аллостерического ингибирования взаимодействия с участком 2 рецептора нейротоксина (см. статью Catteral W.A., Ann. Rev. Pharmacol. Toxicol., 10: 15-43 (1980)).
Ряд авторов (см. статью Соссо М.Т., Maccioni A., Plumitallo A., Farmaco Ed. Sci., 40: 272-284 (1985)) описали два следующих соединения:
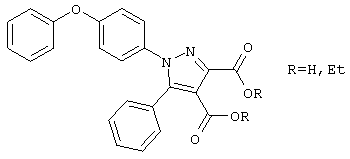
Описаны также другие арилзамещенные гетероциклы в качестве противогрибковых агентов (см. статью Stefancich G. и соавт., Arch. Pharm., (Weinheim Ger.), 323: 273-280, 1990).
До настоящего времени соединения формулы I не использовали для лечения нарушений, восприимчивых к блокированию Na+ каналов у млекопитающих.
Сущность изобретения
Настоящее изобретение относится к открытию действия арилзамещенных пиразолов, триазолов и тетразолов, представленных формулой I, в качестве блокаторов Na+ каналов.
Изобретение относится также к лечению нарушений, восприимчивых к блокированию Na+ каналов, у млекопитающих, страдающих избыточной активностью упомянутых каналов, путем введения эффективного количества соединения формулы I, как описано в данном контексте.
Другим объектом настоящего изобретения является способ лечения, профилактики или снижения степени нейрональных потерь, являющихся результатом общей и очаговой ишемии; лечения, профилактики или уменьшения интенсивности болей, включая острые и хронические боли, а также невропатические боли; лечения, профилактики или уменьшения интенсивности судорог и нейродегенеративных состояний; лечения, профилактики или уменьшения интенсивности маниакальной депрессии с использованием в качестве обезболивающих средств местного действия и противоаритмических средств, а также способа лечения шума в ушах путем введения соединения формулы I млекопитающему, нуждающемуся в таком лечении.
Другим аспектом настоящего изобретения является использование соединений формулы I в качестве блокаторов Na+ каналов.
Настоящее изобретение относится также к использованию соединения формулы I для лечения нейрональных повреждений, образовавшихся в результате общей и очаговой ишемии, и для лечения или профилактики нейродегенеративных состояний, таких как БАС, для лечения шума в ушах в качестве антиманиакальных депрессантов, обезболивающих средств местного действия, противоаритмических средств, противосудорожных средств и для лечения или профилактики диабетической невропатии и для лечения болей, включая острые и хронические боли и мигрени.
Другим объектом настоящего изобретения является фармацевтическая композиция, используемая для лечения нарушений, восприимчивых к блокированию Na+ каналов, причем композиция содержит эффективное количество соединения формулы I в смеси с по меньшей мере одним фармацевтически приемлемым носителем или разбавителем.
Ряд соединений, использованных в настоящем изобретении, до настоящего времени не опубликован. Таким образом, настоящее изобретение относится также к новым арилзамещенным пиразолам, триазолам и тетразолам формулы I.
Кроме того, настоящее изобретение относится к соединениям формулы I, содержащим радиоактивную метку 3H и 14С, и их использованию в качестве радиоактивных лигандов при их связывании с участками связывания на Na+ каналах.
Дополнительные варианты воплощения изобретения и его преимущества будут представлены ниже в подробном описании изобретения, а частично они представляются очевидными из описания или практических рекомендаций изобретения. Варианты воплощения и преимущества изобретения могут быть реализованы и получены с использованием элементов или комбинаций, которые подробно представлены в прилагаемых пунктах формулы изобретения.
Следует понимать, что представленные ниже общее описание изобретения, подробное описание и примеры приведены только в качестве иллюстрации и не ограничивают объем притязаний изобретения, как он заявлен в пунктах формулы изобретения.
На чертеже представлены кривые импульса напряжения
А - Кривые ток-напряжение (IV-кривые);
С - Устойчивая инактивация;
В - Кинетика повторной активации;
D - Период связывания.
Сведения, подтверждающие возможность осуществления изобретения
Настоящее изобретение относится к открытию действия арилзамещенных пиразолов, триазолов и тетразолов формулы I в качестве блокаторов натриевых Na+ каналов. В связи с этим открытием соединения формулы I используют для лечения нарушений, восприимчивых к блокированию каналов ионов натрия.
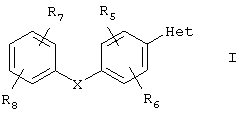
Соединениями, относящимися к данному объекту настоящего изобретения, являются арилзамещенные пиразолы или триазолы, представленные формулой I
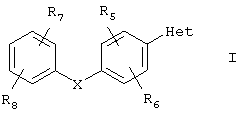
или их фармацевтически приемлемая соль, пролекарство или сольват, где
Х выбирают из группы, включающей в себя О, S;
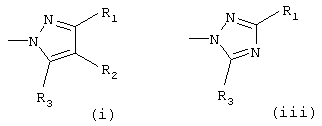
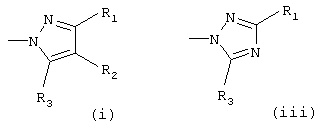
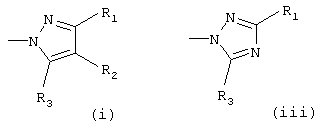
Het означает гетероарил, который выбирают из группы, состоящей из
R1 выбирают из группы, включающей в себя водород, (С1-С6)алкил, C(O)R10;
R2 и R3 независимо выбирают из группы, включающей в себя водород и аминокарбонил;
R5, R6, R7 и R8 независимо выбирают из группы, включающей в себя водород, галоген, галоген(С1-С6)алкил и нитро;
R10 означает OR11;
R11 выбирают из группы, включающей в себя водород и (С1-С6)алкил.
Таким образом, настоящее изобретение относится к разработке способа лечения, профилактики или снижения нейрональных потерь, являющихся результатом общей или очаговой ишемии; лечения, профилактики или снижения интенсивности болей, включая острые и хронические боли, а также невропатические боли; лечения, профилактики или снижения интенсивности судорог или нейродегенеративных состояний; лечения, профилактики или снижения интенсивности маниакальной депрессии; с использованием в качестве обезболивающих средств местного назначения, антиаритмических средств, а также способа лечения звона в ушах путем введения соединения формулы I млекопитающему, нуждающемуся в подобном лечении.
R1 предпочтительно выбирают из группы, включающей в себя водород и C(O)R10;
R10 предпочтительно означает OR11, где R11 определен выше.
R2 и R3 предпочтительно независимо выбирают из группы, включающей в себя водород и аминокарбонил.
Каждая из групп R5-R8 замещает атом водорода, который в ином случае присутствует в любом положении арильного кольца, к которому присоединена группа R.
R5, R6, R7 и R8 предпочтительно независимо выбирают из группы, включающей в себя водород, галоген (предпочтительно, хлор или фтор), галоген(С1-С6)алкил, нитро.
Одна группа предпочтительных соединений, относящихся к формуле I, включает в себя соединения, в которых R1 означает С(O)R10, где R10 определен выше. В этой группе соединений Х более предпочтительно означает О или S и наиболее предпочтительно О.
Наиболее предпочтительными в этой группе являются соединения, где R5 и R6 означают водород; R2 и R3 оба означают Н; a R7 и R8 выбирают из группы, включающей в себя водород, галоген, галоген(С1-С6)алкил, нитро.
Другая группа предпочтительных соединений включает в себя соединения формулы I
или их фармацевтически приемлемую соль, пролекарство или сольват, где
Х означает О или S, предпочтительно О; Het означает гетероарил, который выбирают из группы, состоящей из
предпочтительно (i) или (iii);
R1 предпочтительно означает С(O)R10;
R2 и R3 независимо означают водород; R5 и R6 определены выше и предпочтительно означают водород; R7 и R8 независимо выбирают из группы, включающей в себя водород, галоген, галоген(С1-С6)алкил, нитро.
Примеры предпочтительных соединений, которые могут быть использованы в данном способе по настоящему изобретению, включают без ограничения нижеперечисленным:
1-[4-(4-нитрофенокси)фенил]-1Н-[1,2,4]триазол;
1-[4-(4-фторфенокси)фенил]-3-метилпиразол;
3-метил-1-(4-феноксифенил)пиразол;
1-(4-феноксифенил)-1Н-пиразол-3-карбоксамид;
1-(4-феноксифенил)-1Н-пиразол-5-карбоксамид;
1-[4-(4-фторфенокси)фенил]-1Н-пиразол-3-карбоксамид;
1-[4-(4-нитрофенокси)фенил]-1Н-[1,2,4]триазол-3-карбоксамид и
1-[4-(4-хлор-2-фторфенокси)фенил]-1Н-пиразол-3-карбоксамид.
Другая группа примеров предпочтительных соединений, которая может быть использована по настоящему изобретению, включает в себя 1-[4-(4-фторфенокси)фенил]-5-метилпиразол, 1-(4-феноксифенил)-1Н-пиразол-4-карбоксамид и 4-(4-фторфенокси)фенилпиразол.
Подходящими арильными группами являются С6-С14арил, прежде всего С6-С10арил. Типичные С6-С14арильные группы включают в себя фенил, нафтил, фенантрил, антрацил, инденил, азуленил, бифенил, бифениленил и флуоренильные группы.
Подходящими циклоалкильными группами являются С3-С8циклоалкил. Типичные циклоалкильные группы включают в себя циклопропил, циклобутил, циклопентил, циклогексил и циклогептил.
Термин "гетероарил", использованный в данном описании, относится к группам, содержащим от 5 до 14 атомов в цикле; 6, 10 или 14 п-электронов, объединенных в циклическую систему; и содержащие атомы углерода и 1, 2 или 3 гетероатомов кислорода, азота или серы (где примерами гетероарильных групп являются тиенил, бензо[b]тиенил, нафто[2,3-b]тиенил, тиантренил, фурил, бензофурил, пиранил, изобензофуранил, бензоксазонил, хроменил, ксантенил, феноксантиинил, 2Н-пирролил, пирролил, имидазолил, пиразолил, пиридил, пиразинил, пиримидинил, пиридазинил, индолизинил, изоиндолил, 3Н-индолил, индолил, индазолил, пуринил, 4Н-хинолизинил, изохинолил, хинолил, фталазинил, нафтиридинил, хиназолинил, циннолинил, птеридинил, 4аН-карбазолил, карбазолил, β-карболинил, фенантридинил, акридинил, перимидинил, фенантролинил, феназинил, тиазолил, изотиазолил, фенотиазинил, изоксазолил, фуразанил и феноксазинил.
Подходящие галогеновые группы включают в себя фтор, хлор, бром и иод.
Подходящие алкильные группы включают в себя линейные и разветвленные С1-С10алкильные группы, более предпочтительно С1-С6алкильные группы. Типичные С1-С10алкильные группы включают в себя метил, этил, пропил, изопропил, бутил, втор-бутил, трет-бутил, 3-пентил, гексил и октил. Подходящей является также триметиленовая группа, использованная в качестве заместителя в двух соседних положениях бензольного кольца соединений по настоящему изобретению.
Подходящими алкенильными группами являются С2-С6алкенильные группы, предпочтительно С2-С4алкенил. Типичные С2-С4алкенильные группы включают в себя этенил, пропенил, изопропенил, бутенил и втор-бутенил.
Подходящими алкинильными группами являются С2-С6алкинильные группы, предпочтительно С2-С4алкинил. Типичные С2-С4алкинильные группы включают в себя этинил, пропинил, бутинил и 2-бутинильную группу.
Подходящие арилалкильные группы включают в себя любые из упомянутых выше С1-С10алкильных групп, замещенных любыми упомянутыми выше С6-С14арильными группами. Подходящие значения включают в себя бензил, фенетил и нафтилметил.
Подходящие арилалкенильные группы включают в себя любые из упомянутых выше С2-С4алкенильных групп, замещенных любыми упомянутыми выше С6-С14арильными группами.
Подходящие арилалкинильные группы включают в себя любые из упомянутых выше С2-С4алкинильных групп, замещенных любыми упомянутыми выше С6-С14арильными группами. Подходящие значения включают в себя фенилэтинил и фенилпропинил.
Подходящие гетероарилалкильные группы включают в себя любые из упомянутых выше С1-С10алкильных групп, замещенных любыми упомянутыми выше гетероарильными группами.
Подходящие гетероарилалкенильные группы включают в себя любые из упомянутых выше С2-С4алкенильных групп, замещенных любыми упомянутыми выше гетероарильными группами.
Подходящие гетероарилалкинильные группы включают в себя любые из упомянутых выше С2-С4алкинильных групп, замещенных любыми упомянутыми выше гетероарильными группами.
Подходящие циклоалкилалкильные группы включают в себя любые из упомянутых выше С1-С10алкильных групп, замещенных любыми упомянутыми выше циклоалкильными группами.
Подходящие галогеналкильные группы включают в себя С1-С10алкильные группы, замещенные одним или более атомами фтора, хлора, брома или иода, например фторметил, дифторметил, трифторметил, пентафторэтил, 1,1-дифторэтил и трихлорметил.
Подходящие гидроксиалкильные группы включают в себя С1-С10алкильные группы, замещенные гидроксигруппой, например гидроксиметил, гидроксиэтил, гидроксипропил и гидроксибутил.
Подходящие алкоксигруппы включают в себя кислород, замещенный одной из С1-С10алкильных групп, упомянутых выше.
Подходящие алкилтиогруппы включают в себя серу, замещенную одной из С1-С10алкильных групп, упомянутых выше.
Подходящими ациламиногруппами являются любые С1-С6ацил(алканоил) группы, присоединенные к аминному азоту, например ацетамидо, пропионамидо, бутаноиламидо, пентаноиламидо, гексаноиламидо, а также арилзамещенные С2-С6замещенные ацильные группы.
Подходящими ацилоксигруппам и являются любые ацил(алканоил) группы, присоединенные к окси (-O-) группе, например ацетокси, пропионоилокси, бутаноилокси, пентаноилокси, гексаноилокси и т.п.
Термин "гетероцикл" использован в данном описании для обозначения насыщенной или частично ненасыщенной 3-7-членной моноциклической или 7-10-членной бициклической кольцевой системы, которая состоит из атомов углерода и из от одного до четырех гетероатомов, независимо выбранных из группы, состоящей из О, N и S, где гетероатомы серы и азота могут быть необязательно окислены, а азот может быть необязательно кватернизован, причем упомянутая система включает в себя любую бициклическую систему, в которой определенные выше гетероциклические кольца могут быть конденсироваными с бензольным кольцом, и где бициклическое кольцо может быть замещено по атому углерода или азота, при условии, что полученное соединение является стабильным. Примеры включают в себя без ограничения перечисленным пирролидин, пиперазин, морфолин, имидазолин, пиразолидин, бензодиазепины и т.п.
Подходящие гетероциклоалкильные группы включают в себя любые из упомянутых выше С1-С10алкильных групп, замещенных любыми упомянутыми выше гетероциклическими группами.
Подходящими алкиламино и диалкиламино группами являются -NHR20 и -NR20R21, где R20 и R21 являются С1-С10алкильными группами.
Аминокарбонильной группой является -С(O)NH2.
Подходящими алкиламинокарбонильными группами являются карбонильные группы, замещенные группами -NHR20 и -NR20R21, где R20 и R21 являются С1-С10алкильными группами, как определено выше.
Подходящие алкилтиольные группы включают в себя любые упомянутые выше С1-С10алкильные группы, замещенные -SH группой.
Подходящие алкилсульфинильные группы включают в себя любые упомянутые выше С1-С10алкильные группы, присоединенные к сульфинильной группе (-SO-).
Подходящие алкилсульфонильные группы включают в себя любые упомянутые выше С1-С10алкильные группы, присоединенные к сульфонильной группе (-SO2-).
Карбамоилоксигруппа означает -O-С(O)-NH2.
Карбоксигруппа означает -СООН.
Азидогруппа означает -N3.
Уреидогруппа означает -NH-С(O)-NH2.
Аминогруппа означает -NH2.
Амидная группа означает органический радикал, содержащий в качестве функциональной группы -NHC(О)-.
Изобретение, описанное в контексте данной заявки, охватывает все фармацевтически приемлемые соли описанных соединений. Фармацевтически приемлемые соли включают в себя без ограничения перечисленным соли металлов, такие как соль натрия, соль калия, соль цезия и т.п.; соли щелочноземельных металлов, такие как соль кальция, соль магния и т.п.; соли органических аминов, такие как соль триэтиламина, соль пиридина, соль пиколина, соль этаноламина, соль триэтаноламина, соль дициклогексиламина, соль N,N'-дибензилэтилендиамина и т.п.; соли неорганических кислот, такие как гидрохлорид, гидробромид, сульфат, фосфат и т.п.; соли органичесих кислот, такие как формиат, ацетат, трифторацетат, малеат, тартрат и т.п.; сульфонаты, такие как метансульфонат, бензолсульфонат, п-толуолсульфонат и т.п.; соли аминокислот, такие как аргинат, аспаргинат, глутамат и т.п.
Настоящее изобретение предназначено также для охвата пролекарств описанных соединений. Пролекарства представляют собой любые ковалентно связанные носители, высвобождающие in vivo исходное лекарственное активное средство. Примеры пролекарств включают в себя сложные эфиры или амиды формулы I, где радикалы R2-R11 означают гидроксиалкил или аминоалкил, причем эти соединения могут быть получены при их взаимодействии с ангидридами, такими как ангидрид янтарной кислоты.
Настоящее изобретение охватывает также продукты метаболизма in vivo заявленных соединений. Такие продукты могут образоваться в результате окисления, восстановления, гидролиза, амидирования, этерификации и т.д. вводимого соединения, в основном, вследствие ферментативного процесса. Соответственно изобретение включает в себя соединения, образующиеся в процессе контактирования соединения с организмом в течение времени, достаточного для образования продукта метаболизма. Такие продукты обычно идентифицируют с использованием соединения по настоящему изобретению, содержащего радиоактивную метку, которое вводят в детектируемой дозе парентеральным способом животному, такому как крыса, мышь, морская свинка, обезьяна или человек, выдерживают их в течение достаточного времени для процесса метаболизма, затем выделяют продукты превращения соединения из мочи, крови или других биологических образцов.
Изобретение, описанное в данном контексте, включает в себя соединения, содержащие изотопную метку, т.е. содержащие один или более атомов с другой атомной массой или массовым числом. Примеры изотопов, которые могут быть включены в описываемые соединения, включают в себя изотопы водорода, углерода, азота, кислорода, фосфора, фтора и хлора, такие как 2H, 3H, 13С, 14С, 15N, 18O, 17О, 31P, 32P, 35S, 18F и 36Cl соответственно.
Некоторые из соединений, описанных в данном контексте, могут содержать один или более асимметричных центров, что может приводить к образованию энантиомеров, диастереомеров и других стереоизомерных форм. Настоящее изобретение также включает рацемические смеси, смеси их разделенных форм, также как и индивидуальные энантиомеры, которые могут быть разделены в соответствии с методами, хорошо известными специалистам в данной области техники. Если соединения, описанные в данном контексте, содержат двойные связи или другие центры геометрической асимметрии и если это не оговорено специально, они включают в себя оба геометрических изомера Е и Z. Настоящее изобретение охватывает также все таутомеры.
Использованный в данном контексте термин "стереоизомеры" является общим термином для обозначения всех изомеров индивидуальных молекул, отличающихся только ориентацией их атомов в пространстве. Он включает в себя энантиомеры и изомеры соединений с более чем один хиральным центром, который не является зеркальным отображением другого изомера (диастереомеры).
Термин "хиральный центр" означает атом углерода, к которому присоединены 4 различных группы.
Термин "энантиомер" или "энантиомерный" означает молекулу, которая не может быть совмещена со своим зеркальным отображением и, следовательно, является оптически активной, причем энантиомер вращает плоскость поляризованного света в одном направлении, а его зеркальное отображение вращает плоскость поляризованного света в противоположном направлении.
Термин "рацемический" означает смесь равных частей энантиомеров, которая является оптически неактивной.
Термин "разделение" означает разделение или концентрирование или обеднение одной из двух энантиомерных форм молекулы. Словосочетание "энантиомерный избыток" означает смесь, в которой один из энантиомеров присутствует в большей концентрации по сравнению с молекулой его зеркального отображения.
Поскольку соединения формулы I являются блокаторами натриевых каналов, они могут быть использованы для лечения ряда заболеваний и состояний, опосредованных притоком ионов натрия. Следовательно, изобретение относится к способу лечения, профилактики или снижения нейрональных потерь, связанных с инсультом, общей или очаговой ишемией, травмами центральной нервной системы, гипогликемией или хирургией, травмами спинного мозга, а также к способу лечения и снижению интенсивности нейродегенеративных заболеваний, включающих болезнь Альцгеймера, амиотропный побочный склероз, болезнь Паркинсона, лечение или снижение интенсивности тревожных состояний, судорог, глаукомы, мигреней и мышечных спазмов. Соединения формулы I используют также в качестве лекарственных средств от звона в ушах, противоманиакальных депрессантов, обезболивающих средств местного назначения, противоаритмических средств, а также для лечения, профилактики или снижения интенсивности болей, включая послеоперационные, хронические и невропатические боли. В каждом случае согласно способу по настоящему изобретению требуется введение животному, нуждающемуся в таком лечении, эффективного количества блокатора натриевых каналов по настоящему изобретению, или его фармацевтически приемлемой соли, или пролекарства.
Изобретение также направлено на разработку способа лечения состояний, восприимчивых к блокированию натриевых каналов, у животных, подверженных таким заболеваниям. Наиболее предпочтительные варианты воплощения настоящего изобретения заключаются в использовании арилзамещенных гетероарильных соединений в способе по настоящему изобретению, представленных определенной выше формулой I.
Соединения по настоящему изобретению могут быть получены с использованием методов, известных специалистам в данной области техники.
Синтез пиразолов формулы I может быть осуществлен, как показано на схемах Синтез 1 и 2. Конденсацию бороновой кислоты проводят согласно методике, описанной в статье Lam Y.S. и соавт., Tetrahedron Lett., 39: 2941-2944 (1998).
Синтез 1
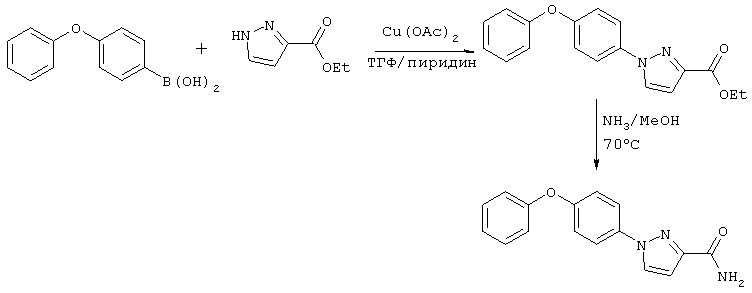
Синтез 2
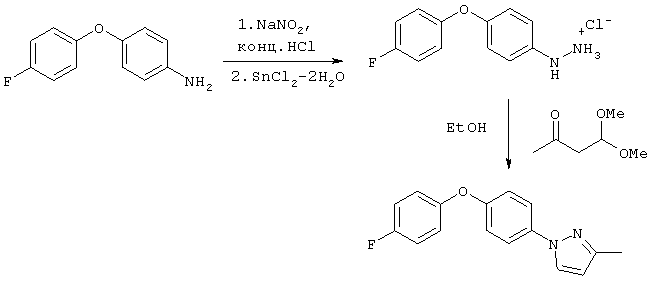
Триазолы формулы I могут быть получены, как показано на схеме Синтез 3, с использованием выпускаемого в промышленности 4-(1,2,4-триазол-1-ил)фенола (производства фирмы Lancaster Synthesis).
Синтез 3
Изобретение относится к соединениям формулы I, содержащим радиоактивную метку 3H и 14С, и к их использованию в качестве радиоактивных лигандов для определения участков связывания в натриевом канале. Например, одним из способов использования меченых соединений по изобретению является характеризация специфического связывания с рецептором. Другим применением меченых соединений по изобретению, наряду с испытанием на моделях животных, является изучение взаимосвязи структуры и активности. Конкурентный анализ связывания с рецепторами проводят при постоянной концентрации меченого соединения формулы I и возрастающих концентрациях тестируемого соединения.
Содержащие тритий соединения формулы I могут быть получены путем введения трития в соединение формулы I, например, методом каталитического дегалогенирования в присутствии трития. Этот способ включает в себя взаимодействие соответствующего галогензамещенного предшественника соединения формулы I в атмосфере газообразного трития в присутствии подходящего катализатора, например Pd/C, в присутствии или отсутствии основания. Другие подходящие методы описаны в книге Filer "Isotopes in the Physical and Biomedical Sciences" (Изотопы в физических и медикобиологических исследованиях) т.1, Labeled Compounds (Меченые соединения) (часть А), глава 6. 14C-меченые соединения могут быть получены с использованием исходного материала, содержащего углерод 14С.
Способность соединений по настоящему изобретению блокировать натриевые каналы исследуют путем электрофизиологического анализа диссоциированных нейронов гиппокампа. Связывание этих соединений с нейронными потенциалозависимыми натриевыми каналами может быть также исследовано с использованием мембран передней доли мозга крысы и [3H]ВХТ-В.
Натриевые каналы представляют собой высокомолекулярные трансмембранные белки, экспрессируемые в различных тканях. Эти потенциалозависимые каналы являются ответственными за быстрое возрастание проницаемости ионов Na+ в ответ на деполяризацию, связанную с потенциалом действия, во многих возбудимых клетках, включая мышечные, нервные клетки и клетки сердечной мышцы.
Аспектом настоящего изобретения является открытие механизма действия соединений, описанных в данном контексте в качестве специфических блокаторов Na+ каналов. На основании открытия такого механизма эти соединения могут быть использованы для лечения или профилактики нейрональных потерь, происходящих в результате очаговой или общей ишемии, и для лечения или профилактики нейродегенеративных нарушений, включая БАС, состояние тревоги и эпилепсию. Предполагается также, что такие соединения будут эффективными при лечении, профилактике или уменьшении интенсивности невропатических болей, послеоперационных болей, хронических болей и шума в ушах. Предполагается также, что соединения будут использованы в качестве противоаритмических средств, обезболивающих средств и противоманиакальных депрессантов.
Настоящее изобретение относится к соединениям формулы I, которые являются блокаторами потенциалозависимых натриевых каналов. Согласно настоящему изобретению такие соединения, обладающие свойствами, предпочтительно блокирующими натриевые каналы, характеризуются величиной IC50 приблизительно 100 мкМ или менее, измеренной методом электрофизиологического анализа, который описан в данном контексте. Величина IC50 соединений по настоящему изобретению предпочтительно составляет 10 мкМ или менее. Наиболее предпочтительно величина IC50 соединений по настоящему изобретению составляет 1,0 мкМ или менее. Замещенные гетероарильные соединения по настоящему изобретению могут быть исследованы для определения их блокирующей Na+ каналы активности с использованием следующих методов: электрофизиологического анализа и анализа связывающей активности.
Электрофизиологический метод анализа
Подготовка клеток: клеточная линия НЕК-293 (NaIIA-B2), стабильно экспрессирующая изоформу rBIIA Na+ каналов, получена в лаборатории. Клетки культивируют по стандартной технологии, как описано ранее (см. статью Verdoom Т.А., Neuron, 4: 919-928 (1990)). Для электрофизиологического анализа клетки наносят на предварительно покрытые поли-D-лизином чашки Петри Cellware размером 35 мм (производства фирмы BIOCOAT, Becton Dickinson) при плотности ˜104 клеток на чашку в день пересеивания из конфлуентной культуры. Эксперименты показали, что клетки пригодны для измерений в течение 2-3 суток после нанесения.
Измерение потенциалозависимых Na+ токов методом фиксации потенциала: измерения методом фиксации потенциала на целых клетках проводят с использованием стандартных методов фиксации потенциала (см. статью HamiII и соавт., Pfluegers Arch., 391: 85-100 (1981)) и усилителя Axopatch 200A amplifier (производства фирмы Axon Instruments, Foster City, CA). В измерительную камеру непрерывно подают внешний раствор (150 мМ NaCl, 5,4 мМ KCI, 1,8 мМ CaCl2, 1 мМ MgCl2, 10 мМ HEPES, 10 мМ глюкозы, рН 7,4 доводят NaOH, величина осмоляльности составляет ˜320 ммоль/кг) со скоростью приблизительно 1 мл/мин. Измерительные пипетки вытягивают из толстостенных капилляров (производства фирмы WPI, Sarasota, FI) и оплавляют в пламени. Интервал сопротивления пипетки, заполненной внутренним раствором (содержащим в мМ: 130 CsF, 20 NaCl, 2 MgCl2, 10 EGTA, 10 HEPES, рН доводят до 7,4 CsOH, осмоляльность составляет ˜320 ммоль/кг), составляет от 1 до 3 МОм. Лекарственные средства и промежуточные промывные растворы вводят через линейный пучок шлангов (производства фирмы Drummond Microcaps, 2 мкл, длина 64 мм). Соединения растворяют в диметилсульфоксиде (ДМСО) и получают исходный 30 мМ раствор, который впоследствии разбавляют внешним раствором до конечной концентрации 0,1-100 мкМ. При самой высокой концентрации (1%) ДМСО лишь незначительно ингибирует величину Na+ тока. Величину тока регистрируют при комнатной температуре (22-25°С), фильтруют при 3 кГц с использованием активного 8-полюсного фильтра Bessel (производства фирмы Frequency Devices, Haverhill, MA), переводят в цифровую форму через интервалы 10-15 мкс и сохраняют с использованием аналого-цифрового интерфейса Digidata 1200 и программного обеспечения Pclamp6/Clampex (фирмы Axon Instruments). При необходимости последовательное сопротивление обычно компенсируют примерно на 75%.
Для оценки эффективности и кинетики ингибирования Na+ каналов соединениями по изобретению используют кривые импульса напряжения, представленные на чертеже.
Для регистрации напряжения, при котором достигается максимальная величина внутреннего Na+ тока, используют зависимость тока от напряжения (IV-кривые), кривая А. При проведении эксперимента эту величину используют в качестве испытательного напряжения, Vt. Кривую устойчивой инактивации (или наличия), кривая С, используют для регистрации напряжения, при котором наблюдается практически полная (≥95%) инактивация Na+ каналов, эту величину используют на протяжении всего эксперимента в качестве кондиционирующего предварительного импульса Vc. На кривой В показано, как быстро каналы восстанавливаются после инактивации при гиперполяризованном напряжении. Эта кривая позволяет установить продолжительность интервала гиперполяризации, который используют при измерении кинетики связывания соединений с инактивированными Na+ каналами (кривая D). Повторная активация каналов в контрольных условиях происходит быстро (≥90%-ное восстановление в течение первых 5-10 мс). Если лекарственное средство значительно замедляет процесс повторной активации, то становится возможным (кривая D) точно измерить кинетику связывания ингибитора с инактивированными каналами, а также устойчивое сродство (k+ и Кi). Для определения величины k+ строят зависимость уменьшения пиков тока в последовательных экспериментах при варьировании интервала предварительного импульса от продолжительности предварительного импульса и измеряют постоянную времени (т) методом моноэкспоненциального приближения. Зависимость 1/т от концентрации антагониста позволяет рассчитать макроскопические скорости связывания антагонистов. Для определения величины Ki кривые частичного ингибирования, измеренные при фракционных откликах в устойчивом состоянии, приводили в соответствие с логистическим уравнением
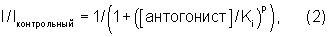
где Iконтрольный означает максимальный Na+ ток в отсутствие антагонста, [антагонист] означает концентрацию лекарственного средства, Ki означает концентрацию антагониста, при которой наблюдается половина максимального ингибирования и р означает величину наклона.
Анализ связывания in vitro
Способность соединений по настоящему изобретению модулировать участок 1 и участок 2 Na+ каналов определяют с использованием следующих операций, подробно описанных в статьях Yasushi, J. Biol. Chem., 261: 6149-6152 (1986) и Creveling, Mol. Pharmacol., 23: 350-358 (1983) соответственно. В качестве источника белков Na+ каналов используют мембраны передней доли мозга крысы. Анализ связывания проводят в 130 мкМ холинхлориде при 37°С и при инкубировании в течение 60 мин в присутствии [3H]сакситоксина и [3Н]батрахотоксина в качестве радиоактивно меченных лигандов для участка 1 и участка 2 соответственно.
Фармакологические исследования in vivo
Соединения по настоящему изобретению могут быть испытаны in vivo на противосудорожную активность после введения внутривенным, пероральным или внутрибрюшинным способом с использованием ряда методов противосудорожных испытаний на мышах, включая испытание на максимальный эпилептический припадок, индуцированный электрошоком (МЭП). Максимальные припадки индуцируют методом электрошока у самцов мышей NSA весом 15-20 г и самцов крыс Sprague-Dawley весом 200-225 г путем приложения тока (для мышей: 50 мА, 60 импульс/с, продолжительность импульса 0,8 мс, продолжительность опыта 1 с, прямой ток; для крыс: 99 мА, 125 импульс/с, продолжительность импульса 0,8 мс, продолжительность опыта 2 с, прямой ток) с использованием прибора Ugo Basile ЕСТ (модель 7801). Мышь удерживают путем захвата дряблой кожи на поверхности спины, и к роговице обоих глаз легким касанием прикладывают два роговичных электрода, покрытых солевым раствором. Крысы свободно передвигаются по поверхности стола с прикрепленными к ушам электродами. Прикладывают ток, и за животными наблюдают вплоть до 30 с, регистрируя отклик в виде тонических судорог разгибающей мышцы задней конечности. Тонический приступ определяют как избыточное вытяжение задней конечности на 90° от плоскости тела. Результаты обрабатывают количественным методом.
Соединения могут быть испытаны на антиноцицептивную активность с использованием формалиновой модели, как описано в статье Hunskaar S., Fasmer O.B. и Hole К., J. Neurosci. Methods, 14: 69-76 (1985). Во всех экспериментах используют самцов мышей Swiss Webster NIH (20-30 г, Marian, San Diego, CA). В день эксперимента мышей не кормят. Мышей помещают в банки из оргстекла на период по крайней мере 1 ч для привыкания к окружающей среде. Затем после привыкания мышей взвешивают и вводят исследуемое соединение внутрибрюшинным или пероральным способом или соответствующий объем носителя (10%-ный Твин-80). Через 15 мин после внутрибрюшинного введения дозы и через 30 мин после перорального введения дозы мышам вводят формалин (20 мкл 5%-ного раствора формалина в солевом растворе) в спинную поверхность правой задней лапы. Мышей переносят в банки из оргстекла и регистрируют период времени, в который наблюдается зализывание или покусывание лапы. Периоды зализывания и покусывания регистрируют через 5-минутные интервалы в течение 1 ч после инъекции формалина. Все эксперименты проводят с зашориванием глаз в течение светового цикла. Раннюю стадию отклика на формалин измеряют как зализывание/покусывание в течение 0-5 мин, позднюю стадию измеряют как зализывание/покусывание в течение 15-50 мин. Различие между группами животных, обработанных лекарственным средством и носителем, анализировали методом однофакторного дисперсионного анализа (ANOVA). Величину Р≤0,05 принимают за значимую величину. Соединения, обладающие блокирующей активностью в отношении острой и второй фазы индуцированной формалином активности зализывания лапы, рассматриваются как эффективные в отношении острых и хронических болей.
Соединения могут быть испытаны на их способность лечения хронических болей (антиаллодиническая и антигипералгезическая активности) на модели периферической невропатии Чанга. Самцов крыс Sprague-Dawley весом 200-225 г анестезируют галотаном (1-3% в смеси 70% воздуха и 30% кислорода) и измеряют их температуру тела в процессе анестезии с использованием гомотермического одеяла. На спине на уровне L5 и L6 делают средний надрез размером 2 см и паравертебральные группы мышц оттягивают билатерально. Затем обнажают спинные нервы L5 и L6, их отделяют и прочно сшивают шелковой нитью 6-0. В качестве отрицательного контроля проводят фиктивную операцию, обнажая спинные нервы L5 и L6 на противоположной стороне тела.
Тактильная аллодиния: крыс переносят в подвешенную экспериментальную клетку с решетчатым проволочным полом и выдерживают животных в течение 5-10 мин для привыкания к данным условиям. На подошвенную часть задней ноги накладывают ряд моноволокон Семмес-Вейнштайна для определения порога отдергивания. Первым используют волокно с массой изгиба 9,1 г (величина log 0,96), причем используют его до 5 раз, чтобы определить, вызовет ли оно отклик в виде отдергивания лапы. Если у животного наблюдается отклик в виде отдергивания, то следующее, более легкое волокно применяют до пяти раз, определяя, может ли оно вызвать отклик. Эту процедуру повторяют, используя все более легкие волокна, до тех пор, пока отклика не наблюдается, и регистрируют самое легкое волокно, при котором наблюдается ответ. Если не наблюдается ответного отдергивания при использовании первого волокна массой 9,1 г, то применяют волокна с большей массой до тех пор, пока волокно не вызовет отклика, и регистрируют, при использовании какого волокна наблюдается ответ. Для каждого животного в каждой временной точке проводят по три измерения, чтобы определить средний порог отдергивания. Измерения проводят до введения лекарственного средства и через 1, 2, 4 и 24 ч после его введения. Одновременно проводят тестирование на тактильную аллодинию и механическую гиперальгезию.
Механическая гиперальгезия. Крыс помещают в подвешенную клетку с проволочным решетчатым полом и выдерживают их в течение 5 мин для привыкания к условиям эксперимента. Слегка притупленной иглой касаются подошвенной части задней лапы таким образом, чтобы игла слегка вдавливалась в кожу, не проникая через нее. Прикосновение иглы к контрольным лапам обычно вызывает быструю реакцию отдергивания, слишком короткую, чтобы зафиксировать ее секундомером, и время отдергивания произвольно считают равным 0,5 с. У животных с невропатией на лапе с оперированной стороны в ответ на прикосновение затупленной иглы наблюдается чрезмерно сильное отдергивание. Максимальное время 10 с принимают за предельную величину. Время отдергивания для обеих лап животных в каждой временной точке измеряют по три раза с пятиминутным периодом между прикосновениями для восстановления в исходное состояние. Для расчета среднего значения времени отдергивания для каждой временной точки используют три измерения. Одновременно проводят тестирование на тактильную аллодинию и механическую гиперальгезию.
Нейропротективная активность соединений может быть исследована после индуцирования очаговой и общей ишемии у крыс или песчанок по методике, описанной в статьях Buchan и соавт. (Stroke, Suppl., 148-152 (1993)), Sheardown и соавт. (Eur. J. Pharmacol., 236: 347-353 (1993)) и Graham и соавт. (J. Pharmacol. Exp. Therap., 276: 1-4 (1996)).
Нейропротективная активность соединений может быть исследована после травматического повреждения спинного мозга согласно методике, описанной в статьях Wrathall и соавт. (Exp. Neurology, 137: 119-126 (1996)) и Iwasaki и соавт. (J. Neurosci., 134: 21-25 (1995)).
Композиции по настоящему изобретению включают в себя все композиции, в которых соединения по настоящему изобретению содержатся в количествах, эффективных для достижения соответствующей цели. В связи с тем, что индивидуальные потребности индивидуумов различны, определение оптимального диапазона эффективных количеств каждого компонента зависит от специалистов в данной области техники. В типичном случае соединения или эквивалентные количества их фармакологически приемлемых солей могут быть введены млекопитающим, например человеку, пероральным способом в дозе от 0,0025 до 50 мг в сутки на килограмм веса тела млекопитающего, нуждающегося в лечении эпилепсии, нейродегенеративных заболеваний, аритмии, маниакальной депрессии, боли и в анестезии. При внутримышечных инъекциях доза обычно составляет примерно половину пероральной дозы.
В случае лечения или предупреждения нейрональной потери при очаговой или общей ишемии, травме головного и спинного мозга, гипоксии, гипогликемии, при состоянии эпилепсии и при хирургических операциях соединение можно вводить внутривенно в дозе от приблизительно 0,025 до приблизительно 10 мг/кг.
Однократная доза для перорального приема может составлять от приблизительно 0,01 до приблизительно 50 мг, предпочтительно приблизительно от 0,1 до приблизительно 10 мг соединения. Однократную дозу можно вводить один или несколько раз в день в виде одной или более таблеток, каждая из которых содержит от приблизительно 0,1 до приблизительно 10, наиболее удобно от приблизительно 0,25 до приблизительно 50 мг соединения или его сольватов.
Кроме введения соединения в необработанном виде, соединения по изобретению можно вводить в составе фармацевтического препарата, содержащего подходящие фармацевтически приемлемые носители, включающие наполнители и вспомогательные материалы, облегчающие процесс обработки соединений в препараты, которые могут быть использованы в фармацевтике. Препараты, прежде всего те, которые могут быть введены перорально и которые предпочтительны для определенного способа введения, такого как таблетки, драже и капсулы, а также препараты, которые могут быть введены ректально, такие как суппозитории, и также подходящие растворы для введения в виде инъекций или перорального приема предпочтительно содержат от приблизительно 0,01 до 99%, предпочтительно от приблизительно 0,25 до 75% активного вещества (активных веществ), включая наполнитель.
В объем притязаний настоящего изобретения включены также нетоксичные фармацевтически приемлемые соли соединений по настоящему изобретению. Кислотно-аддитивные соли получают путем смешивания раствора конкретного гетероарильного соединения по настоящему изобретению с раствором фармацевтически приемлемой нетоксичной кислоты, такой как соляная кислота, фумаровая кислота, малеиновая кислота, янтарная кислота, уксусная кислота, лимонная кислота, винная кислота, угольная кислота, фосфорная кислота, щавелевая кислота, дихлоруксусная кислота и тому подобные. Соли с основаниями получают путем смешивания раствора гетероарильного соединения по настоящему изобретению с раствором фармацевтически приемлемого нетоксичного основания, такого как гидроксид натрия, гидроксид калия, гидроксидхолина, карбонат натрия и им подобные.
Фармацевтические композиции по настоящему изобретению могут быть введены любому животному, которое может ощутить благотворное действие соединений по изобретению. Наиболее предпочтительными среди таких животных являются млекопитающие, например человек, хотя изобретение не предназначено для такого ограничения.
Фармацевтические композиции по настоящему изобретению могут быть введены любым способом, при котором достигается предназначенное действие. Например, их можно вводить парентерально, подкожно, внутривенно, внутримышечно, внутрибрюшинно, чрескожно или трансбуккально (защечный способ). Альтернативным или одновременным способом приема является пероральный прием. Вводимая доза зависит от возраста, состояния здоровья, веса принимающего препарат, вида сопутствующего лечения, если таковое имеется, частоты приема и природы требуемого эффекта.
Фармацевтические препараты по настоящему изобретению получают широко распространенным методом, например путем обычного смешивания, гранулирования, получения драже, процессов растворения и лиофилизации. Таким образом, фармацевтические препараты для перорального приема могут быть получены путем смешивания активных компонентов с твердыми носителями, необязательно путем измельчения полученной смеси и при необходимости путем обработки смеси гранул после добавления подходящих вспомогательных веществ для изготовления таблеток или ядер драже.
Подходящими наполнителями являются, прежде всего, такие наполнители, как сахариды, например лактоза или сахароза, маннит или сорбит, препараты целлюлозы и/или фосфаты кальция, например трехзамещенный фосфат кальция или двухзамещенный фосфат кальция, а также такие связующие агенты, как крахмальная паста с использованием, например, кукурузного крахмала, пшеничного крахмала, рисового крахмала, картофельного крахмала, желатина, трагаканта, метилцеллюлозы, гидроксипропилметилцеллюлозы, натрийкарбоксиметилцеллюлозы и/или поливинилпирролидона. При необходимости могут быть добавлены дезинтегрирующие агенты, такие как вышеупомянутые крахмалы и также карбоксиметилкрахмал, поперечно-сшитый поливинилпирролидон, агар или альгиновая кислота или ее соли, например альгинат натрия. Вспомогательными веществами являются, прежде всего, агенты, регулирующие текучесть, и замасливатели, например диоксид кремния, тальк, стеариновая кислота или ее соли, такие как стеарат магния или стеарат кальция и/или полиэтиленгликоль. Ядра драже покрывают соответствующей оболочкой, которая при необходимости является устойчивой к желудочным сокам. Для этой цели могут быть использованы концентрированные растворы сахаридов, которые необязательно могут содержать аравийскую камедь, тальк, поливинилпирролидон, полиэтиленгликоль и/или диоксид титана, лакирующие растворы и приемлемые органические растворители или смеси растворителей. Для получения оболочек, устойчивых к желудочным сокам, используют растворы подходящих препаратов целлюлозы, таких как фталат ацетилцеллюлозы или фталат гидроксипропилметилцеллюлозы. В оболочки таблеток или драже, например, для их идентификации или обозначения сочетаний доз активных соединений могут быть добавлены пигменты или красители.
Другие фармацевтические препараты, которые могут быть введены перорально, включают в себя штампованные капсулы из желатина, а также мягкие запаянные капсулы из желатина и пластификатора, такого как глицерин или сорбит. Штампованные капсулы могут содержать активные соединения в виде гранул, которые могут быть смешаны с наполнителями, такими как лактоза, связывающими агентами, такими как крахмалы, и/или замасливателями, такими как тальк или стеарат магния, и необязательно стабилизаторами. В мягких капсулах активные соединения предпочтительно растворены или суспендированы в подходящих жидкостях, таких как нелетучие масла или жидкий парафин. Кроме того, могут быть добавлены стабилизаторы.
Возможные фармацевтические препараты, которые могут быть использованы ректально, включают в себя, например, суппозитории, содержащие комбинацию одного или более активных веществ с основой суппозитория. Приемлемыми основами для суппозиториев являются, например, природные или синтетические триглицериды или насыщенные углеводороды. Кроме этого, можно использовать желатиновые ректальные капсулы, содержащие комбинацию активных соединений с основой. Возможные материалы основы включают в себя, например, жидкие триглицериды, полиэтиленгликоли или насыщенные углеводороды.
Приемлемые составы для парентерального введения включают в себя водные растворы активных соединений в водорастворимой форме, например водорастворимые соли и щелочные растворы. Кроме того, могут быть использованы суспензии активных соединений в виде соответствующих масляных инъекционных суспензий. Подходящие липофильные растворители или носители включают в себя нелетучие масла, например кунжутное масло, или синтетические эфиры жирных кислот, например этиловый эфир олеиновой кислоты, или триглицериды, или полиэтиленгликоль 400 (соединения растворимы в ПЭГ 400). Водные инъекционные суспензии могут содержать соединения, повышающие вязкость суспензии, и включать в себя, например, натрийкарбоксиметилцеллюлозу, сорбит и/или декстран. По выбору суспензия может также содержать стабилизаторы.
Следующие примеры приведены для иллюстрации способа и композиций по настоящему изобретению без ограничения перечисленным. Другие подходящие модификации и использование ряда условий и параметров, обычно используемых в клинической терапии и очевидных для специалистов в данной области, включены в объем притязаний и сущность настоящего изобретения.
1-[4-(4-Фторфенокси)фенил]-3-метилпиразол
а) Гидрохлорид 4-(4-фторфенокси)фенилгидразина. Суспензию мелко измельченного порошкообразного 4-фтор-4'-аминодифенилового эфира (2,00 г, 9,84 ммоль) в 10 мл воды охлаждают на бане вода/лед и добавляют по каплям через капельную воронку 19,4 мл концентрированной HCl. Полученную смесь охлаждают до -5°С на бане лед/ацетон и к реакционной смеси по каплям добавляют раствор нитрита натрия (кристаллический; 0,714 г, 10,3 ммоль) в 8 мл холодной воды с такой скоростью, чтобы температура реакционной смеси оставалась в интервале от -5 до 0°С. Раствор SnCl2-2H2O (6,66 г, 29,5 ммоль) в 20 мл конц. HCl при -20°С обрабатывают порциями реакционной смеси, поддерживая температуру реакции ниже -10°С. При этом образуется осадок серого цвета и полученную реакционную смесь перемешивают при -20°С в течение 90 мин. Твердое вещество выделяют фильтрованием и промывают холодным EtOH (2×10 мл). Неочищенный гидразин, 2,36 г, используют на следующих стадиях без очистки.
б) 1-[4-(4-Фторфенокси)фенил]-3-метилпиразол. Суспензию гидразина (500 мг, 2,05 ммоль) в 5,5 мл смеси EtOH/вода, 1:1 обрабатывают 300 мкл (299 мг, 2,03 ммоль) 90%-ного диметилацеталя ацетилацетальдегида и полученную смесь нагревают в воздушно-струйной сушилке в течение 2 мин. Реакционную смесь охлаждают и экстрагируют гексаном (4×10 мл). Объединенные органические слои промывают солевым раствором, сушат (Na2SO4) и концентрируют. Осадок подвергают колоночной хроматографии (силикагель, 10% EtOAC/гексан), при этом получают 134 мг (24%) соединения, указанного в заголовке, в виде твердого вещества белого цвета, Тпл 80-81°С. 1H ЯМР (CDCl3): δ 7,74 (s, 1H), 7,58 (d, J=8,4 Гц, 2Н), 7,06-6,95 (m, 6H), 6,23 (s, 1H), 2,37 (s, 3Н).
1-(4-Феноксифенил)-1Н-пиразол-3-карбоксамид и 1-(4-феноксифенил)-1Н-пиразол-5-карбоксамид
а) 3-Этоксикарбонил-1-(4-феноксифенил)-1Н-пиразол и 5-этоксикарбонил-1-(4-феноксифенил)-1Н-пиразол. К суспензии 4-феноксифенилбороновой кислоты (1,70 г, 7,85 ммоль), этилового эфира 3-пиразолкарбоновой кислоты (0,55 г, 3,92 ммоль), ацетата меди (II) (1,1 г, 5,89 ммоль) и молекулярных сит 4Å (измельченных и нагретых перед использованием при 200°С в течение 2 ч) в 30 мл безводного ТГФ добавляют 0,6 мл пиридина. Реакционную смесь перемешивают на воздухе при комнатной температуре в течение 2 сут и затем фильтруют, фильтрат концентрируют досуха. Неочищенный продукт очищают экспресс-хроматографией, элюируют смесью 15% EtOAC/гексан, при этом получают 5-этоксикарбонил-1-(4-феноксифенил)-1Н-пиразол (Rf=0,6, 55 мг, 4,6%) и 3-этоксикарбонил-1-(4-феноксифенил)-1Н-пиразол (Rf=0,5, 125 мг, 10,3%).
б) 1-(4-Феноксифенил)-1Н-пиразол-3-карбоксамид. Раствор 3-этоксикарбонил-1-(4-феноксифенил)-1Н-пиразола (120 мг, 0,39 ммоль) в 5 мл 2н. раствора аммиака в МеОН перемешивают при комнатной температуре в течение 4 сут. По данным ТСХ реакция не завершена, и раствор переносят в закрытую пробирку, нагревают при 70°С в течение ночи. Реакционную смесь концентрируют и очищают препаративной ТСХ, элюируют 50% смесью EtOAC/гексан, при этом получают 1-(4-феноксифенил)-1Н-пиразол-3-карбоксамид (Rf=0,26, 56 мг, 52%), Тпл=165-167°С. 1H ЯМР (300 МГц, ДМСО-d6): δ 8,50 (d, J=2,7 Гц, 1Н, пиразол), 7,92 (d, J=9,0 Гц, 2Н, фенил), 7,71 (ушир, 1Н, NH2), 7,43 (m, 2Н, фенокси), 7,39 (ушир., 1Н, NH2), 7,18 (m, 1Н, фенокси), 7,17 (d, J=9,0 Гц, 2Н, фенил), 7,07 (m, 2Н, фенокси), 6,87 (d, J=2,7 Гц, 1Н, пиразол).
По методике, описанной выше, из 5-этоксикарбонил-1-(4-феноксифенил)-1Н-пиразола (50 мг, 0,16 ммоль) получают 1-(4-феноксифенил)-1Н-пиразол-5-карбоксамид (25 мг, 55%), Тпл=142-144°С. 1 H ЯМР (300 МГц, ДМСО-d6): δ 8,03 (ушир.s, 1Н, NH2), 7,70 (d, J=1,8 Гц, 1Н, пиразол), 7,54 (ушир.s, 1Н, NH2), 7,42 (m, 2Н, фенокси), 7,39 (d, J=9,0 Гц, 2Н, фенил), 7,20 (m, 1H, фенокси), 7,08 (m, 2Н, фенокси), 7,06 (d, J=9,0 Гц, 2Н, фенил), 6,90 (d, J=1,8 Гц, 1H, пиразол).
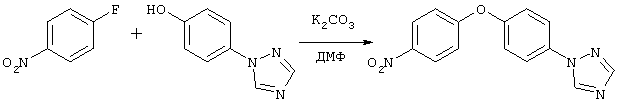
1-[4-(4-Нитрофенокси)фенил]-1Н-[1,2,4]триазол
Смесь 1-фтор-4-нитробензола (0,17 мл, 1,6 ммоль), 4-{[1,2,4]триазол-1-ил}фенола (0,26 г, 1,58 ммоль) и карбоната калия (1,69 г, 12,2 ммоль) в ДМФ кипятят с обратным холодильником в течение ночи. Реакционную смесь охлаждают до комнатной температуры, затем распределяют между водой и этилацетатом. Водный слой экстрагируют этилацетатом. Объединенные органические слои промывают водным раствором гидроксида натрия (2н.), водой (2 раза), сушат над сульфатом натрия, фильтруют и упаривают при пониженном давлении, при этом получают осадок желтого цвета. Продукт очищают колоночной хроматографией (силикагель; смесь гексан/этилацетат, 1:1) и перекристаллизовывают из смеси хлороформ/гексан, при этом получают 165 мг (37%) соединения, указанного в заголовке, в виде твердого вещества желтого цвета, Тпл 131-132°С. 1H ЯМР (CDCl3): δ 8,55 (s, 1H), 8,25 (d, J=9 Гц, 2Н), 8,12 (s, 1H), 7,75 (d, J=9,0 Гц, 2Н), 7,24 (d, J=9 Гц, 2Н), 7,08 (d, J=9 Гц, 2Н).
Противосудорожная активность соединений по настоящему изобретению
Способность соединений по настоящему изобретению блокировать МЭП определяют, как описано ранее.
Соединение по настоящему изобретению вводят мышам перорально за 30 мин до эксперимента. Соединение оказывает защитное действие против МЭП, причем величина ED50 (доза, при которой наблюдается защитное действие у 50% животных), предпочтительно составляет менее 10 мг/кг.
Активность соединения в качестве противосудорожного средства определяли на модели МЭП у мышей: ED50 для 1-(4-феноксифенил)-1Н-пиразол-3-карбоксамида после внутривенного введения составляет 0,7 мг/кг; ED50 для 1-[4-(4-нитрофенокси)фенил]-1Н-[1,2,4]тризола после перорального введения составляет 6,6 мг/кг; ED50 для 1-(4-феноксифенил)-1Н-пиразол-5-карбоксамида после внутривенного введения составляет 10,0 мг/кг; ED50 для 4-[4-(4-триметилфенокси)фенил]-пиримидин-2-карбоксамида после перорального введения составляет 2,5 мг/мл, а после внутривенного введения - 0,5 мг/мл.
Активность соединения по настоящему изобретению в качестве блокатора натриевых каналов
Соединения по настоящему изобретению испытывают с использованием электрофизиологического анализа и анализа связывания, описанных выше, и получают дозозависимое ингибирование управляемых потенциалом токов ионов натрия, регистрируемых на клетках НЕК-293, стабильно экспрессирующих изоформу rBIIA натриевых каналов. Блокирующий эффект, оказываемый предпочтительными соединениями на токи Na+, является высокочувствительным к фиксируемому напряжению, что свидетельствует о связывании соединения с потенциалозависимыми Na+ каналами в инактивированном состоянии и что соединение имеет низкую активность по отношению к Na+-каналам в состоянии покоя (см. статьи Ragsdale и соавт., Mol. Pharmacol., 40: 756-765 (1991); Kuo и Bean, Mol. Pharmacol., 46: 716-725 (1994)). Константа диссоциации антагониста (Кd) предпочтительных соединений для инактивированнных натриевых каналов приблизительно составляет менее 400 нМ. 1-(4-Феноксифенил)-1Н-пиразол-3-карбоксамид исследуют с использованием изоформы rBIIA натриевых каналов, значение Кi, составляет 0,35 мкМ; а для 4-[4-(4-триметилфенокси)фенил]-пиримидин-2-карбоксамида она составляет 0,21 мкМ.
| название | год | авторы | номер документа |
|---|---|---|---|
| ЗАМЕЩЕННЫЕ НИКОТИНИМИДНЫЕ ИНГИБИТОРЫ ВТК, ИХ ПОЛУЧЕНИЕ И ПРИМЕНЕНИЕ В ТЕРАПИИ РАКОВЫХ, ВОСПАЛИТЕЛЬНЫХ И АУТОИММУННЫХ ЗАБОЛЕВАНИЙ | 2014 |
|
RU2677884C2 |
| ПИРАЗОЛОПИРИДИНОВЫЕ ПРОИЗВОДНЫЕ КАК ИНГИБИТОРЫ НАДФН-ОКСИДАЗЫ | 2009 |
|
RU2548022C2 |
| АРИЛ- И ГЕТЕРОАРИЛЗАМЕЩЕННЫЕ ГЕТЕРОЦИКЛИЧЕСКИЕ ПРОИЗВОДНЫЕ МОЧЕВИНЫ, СПОСОБ ИНГИБИРОВАНИЯ КИНАЗЫ RAF И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 1998 |
|
RU2265597C2 |
| СОЕДИНЕНИЯ ЗАМЕЩЕННЫХ ПИРАЗОЛОНОВ И СПОСОБЫ ИСПОЛЬЗОВАНИЯ | 2013 |
|
RU2650895C2 |
| ЗАМЕЩЕННЫЕ ТРИАЗОЛЫ В КАЧЕСТВЕ БЛОКАТОРОВ НАТРИЕВЫХ КАНАЛОВ | 2004 |
|
RU2372339C2 |
| АМИДОПРОИЗВОДНЫЕ КАК БЛОКАТОРЫ TTX-S | 2013 |
|
RU2632899C2 |
| Соединения 6, 7-дигидро-5H-пиразоло[5,1-b][1,3]оксазин-2-карбоксамида | 2017 |
|
RU2719599C2 |
| ПИРАЗОЛОПИРИДИНОВЫЕ ПРОИЗВОДНЫЕ КАК ИНГИБИТОРЫ НАДФН-ОКСИДАЗЫ | 2009 |
|
RU2532161C2 |
| СПОСОБ ИНГИБИРОВАНИЯ ПРОДУЦИРОВАНИЯ ОСТАТОЧНЫХ ЛИПОПРОТЕИНОВ | 2004 |
|
RU2330682C2 |
| ЭМУЛЬГИРУЕМЫЙ КОНЦЕНТРАТ, ВКЛЮЧАЮЩИЙ ПЕСТИЦИД, ЖИРНЫЙ АМИД И ЛАКТАМИД | 2015 |
|
RU2688819C2 |
Изобретение относится к арилзамещенным пиразолам или триазолам общей формулы I
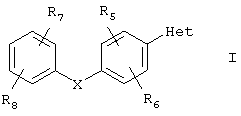
их фармацевтически приемлемым солям, пролекарствам или сольватам, где Х - O или S; Het - одна из групп
R1-Н, С1-С6алкил, аминокарбонил, С(O)R10; R2 и R3 независимо - Н и аминокарбонил, R5, R6, R7 и R8 независимо - Н, галоген, галоген C1-С6алкил, NO2; R10-OR11, где R11 - H, C1-С6алкил. Соединения I пригодны для лечения нейрональных повреждений, наблюдающихся в результате общей и очаговой ишемии, для лечения или профилактики нейродегенеративных состояний, таких как боковой амиотрофический склероз (БАС), и для лечения, профилактики или снижения интенсивности острых или хронических болей, в качестве агентов против шума в ушах, противосудорожных средств, а также в качестве противоманиакальных депрессантов, в качестве обезболивающих средств местного назначения, в качестве противоаритмических средств и для лечения или профилактики диабетической невропатии. 4 н. и 9 з.п. ф-лы, 1 ил.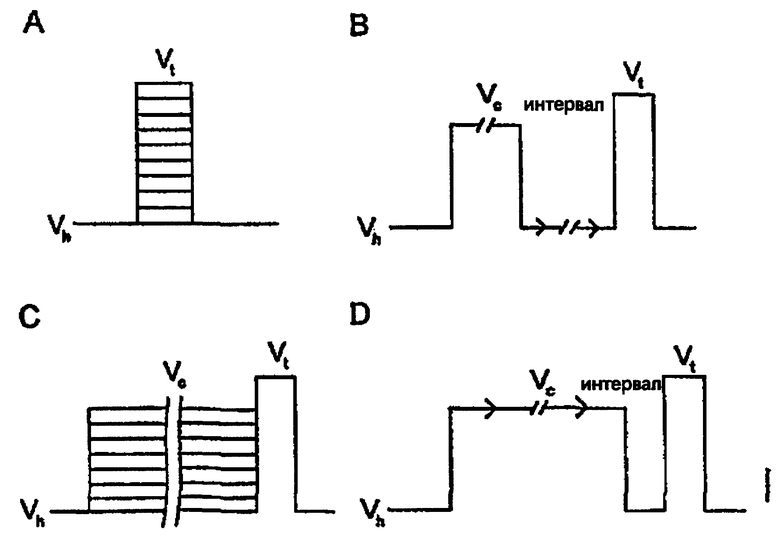
или их фармацевтически приемлемые соли, пролекарства или сольваты, где
Х означает одну из групп О, S;
Het означает гетероарил, выбранный из группы, состоящей из
R1 выбран из группы, включающей в себя водород, (С1-С6)алкил, аминокарбонил, C(O)R10;
R2 и R3 независимо выбраны из группы, включающей в себя водород и аминокарбонил;
R5, R6, R7 и R8 независимо выбраны из группы, включающей в себя водород, галоген, галоген(С1-С6)алкил и нитро;
R10 означает OR11;
R11 выбран из группы, включающей в себя водород и (С1-С6)алкил; если Het означает (iii), и Х означает О, то R1 не может быть C(O)R10.
R3 и R2 оба означают Н и
R7 и R8 выбраны из группы, включающей в себя водород, галоген и нитро.
R1 означает водород, (С1-С6)алкил или аминокарбонил;
Х означает О;
R2 и R3 означают водород;
R5 и R6 означают водород;
R7 и R8 независимо выбраны из группы, включающей в себя водород, галоген, галогенС1-С6алкил и нитро.
или его фармацевтически приемлемой, соли, пролекарства или сольвата, где Х означает О или S;
Het означает гетероарил, выбранный из группы, состоящей из
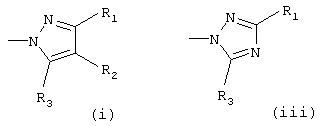
R1 выбирают из группы, включающей в себя водород, (С1-С6)алкил, аминокарбонил и C(O)R10;
R2 и R3 независимо выбирают из группы, включающей в себя водород, и аминокарбонил;
R5, R6, R7 и R8 независимо выбирают из группы, включающей в себя водород, галоген, галоген(С1-С6)алкил и нитро;
R10 означает OR11;
R11 означает водород или (С1-С6)алкил.
| Способ получения конденсированных производных пиррола | 1987 |
|
SU1577698A3 |
| WO 9611911 A, 25.04.1996 | |||
| DE 1936760 A, 22.01.1970. | |||

