ПЕРЕКРЕСТНАЯ ССЫЛКА НА РОДСТВЕННЫЕ ЗАЯВКИ
[001] Настоящая заявка претендует на преимущества Предварительной заявки США №61/676944, поданной 28 июля 2012 г., и Временной заявки США №61/679416, поданной 3 августа 2012 г., все из которых настоящим включены посредством полных ссылок.
ОБЛАСТЬ ИЗОБРЕТЕНИЯ
[002] Изобретение относится к новым соединениям замещенных пиразолонов и их солей, которые полезны в лечении гиперпролиферативных заболеваний, например, раковых образований у млекопитающих. В частности, изобретение касается соединений, ингибирующих активность протеин тирозина киназы, приводящую к ингибированию межклеточной и/или внутриклеточной сигнализации. Настоящее изобретение также касается способа использования таких соединений в лечении гиперпролиферативных болезней у млекопитающих, особенно у людей, и фармацевтических композиций, содержащих такие соединения.
ПРЕДПОСЫЛКИ СОЗДАНИЯ ИЗОБРЕТЕНИЯ
[003] Киназы протеина представляют большую группу протеинов, играющих центральную роль в регулировании большого разнообразия клеточных процессов. Посредством регулирования множества сигнальных путей, киназы протеина управляют метаболизмом клетки, профессией клеточного цикла, пролиферацией клеток и некрозом клеток, дифференцированием и жизнедеятельностью. В человеческом киноме существует более 500 киназ, и предполагается, что более 150 из них вовлекаются в начало и/или развитие различных болезней человека, включая воспалительные заболевания, сердечнососудистые заболевания, нарушения обмена веществ, нейродегенеративные заболевания и рак.
[004] Частичный список таких киназ включает abl, AATK, ALK, Akt, Axl, bmx, bcr-abl, Blk, Brk, Btk, csk, c-kit, c-Met, c-src, c-fins, CDK1, CDK2, CDK3, CDK4, CDK5, CDK6, CDK7, CDK8, CDK9, CDK10, cRafl, CSF1R, CSK, DDR1, DDR2, EPHA, EPHB, EGFR, ErbB2, ErbB3, ErbB4, Erk, Fak, fes, FER, FGFR1, FGFR2, FGFR3, FGFR4, FGFR5, Fgr, fit-1, Fps, Frk, Fyn, GSG2, GSK, Hck, ILK, INSRR, IRAK4, ITK, IGF-1R, INS-R, Jak, KSR1, KDR, LMTK2, LMTK3, LTK, Lck, Lyn, MATK, MEK.T.K, МЛТК, MST1R, MUSK, NPR1, NTRK, MEK, MER, PLK4, PTK, p38, PDGFR, PIK, PKC, PYK2, RET, ROR1, ROR2, RYK, ROS, Ron, SGK493, SRC, SRMS, STYK1, SYK, TEC, TEK, TEX 14, TNK1, TNK2, TNNI3K, TXK, TYK2, Tyro-3, tie, tie2, TRK, Yes и Zap70.
[005] Протеин тирозина киназы являются подклассом киназы протеина. Они также могут быть классифицированы как киназы рецептора фактора роста (например, Axl, VEGFR, c-Met (HGFR), Ron, EGFR, PDGFR и FGFR) или киназы нерецептора (например, c-src и bcr-abl). Киназы тирозина рецептора являются трансмембранными протеинами, обладающими внеклеточной областью связывания для факторов роста, трансмембранной областью и внутриклеточной частью, функционирующей как киназа для фосфорилата, определенного остатка тирозина в протеинах. Неправильное выражение или активность киназ протеина были непосредственно вовлечены в патогенез бесчисленных человеческих раковых образований.
[006] Ангиогенез, формирование новых капилляров из существующих ранее кровеносных сосудов, является необходимым процессом для развития органа во время эмбриогенеза, и критически важен для репродуктивного цикла женщина, воспаления и заживления ран у взрослых. Определенные болезни, как известно, связаны с дерегулированным ангиогенезом, например неоваскуляризация глаз, например ретинопатии (включая диабетическую ретинопатию), возрастная дегенерация желтого пятна, псориаз, гемангиобластома, гемангиома, артериосклероз; воспалительные заболевания, такие как ревматоидные или ревматические воспалительные заболевания, особенно артрит (включая ревматоидный артрит); или другие хронические воспалительные заболевания, например хроническая астма, артериальный или посттрансплационный атеросклероз, эндометриоз, и опухолевые болезни, например так называемые солидные опухоли и жидкие опухоли (например, лейкемии). Солидные опухоли, в частности, зависят от ангиогенеза роста вне определенного критического размера, индуцируя новые капилляры из существующих кровеносных сосудов обеспечивать питание, подавать кислород и выводить токсины. Кроме того, ангиогенез также способствует росту метастаз опухолевых клеток в другие места.
[007] Рост и созревание новых сосудов являются очень сложными и скоординированными процессами, требующими стимулирования многими факторами роста, но сигнализация фактора роста эндотелия сосудов (VEGF) часто представляет критический ограничивающий шаг в физиологическом ангиогенезе и патологическом ангиогенезе. Фактор VEGF связывает и активирует киназу тирозина рецептора, VEGFR. У людей идентифицированы три изоформы VEGFR: VEGFR-1 (Flt-1), VEGFR-2 (KDR/Flk-1) и VEGFR-3 (Flt-4). Изоформа VEGFR-2 добивается большинства клеточных ответов на VEGF, в частности его митогенетические и ангиогенные эффекты. Предположительно, изоформа VEGFR-1 модулирует сигнализацию VEGFR-2, или действует как рецептор плацебо/приманка для секвестирования VEGF вдали от VEGFR-2. Выражение VEGFR-1 также отрегулировано гипоксией, аналогично механизму VEGF, через HIF-1; функции могут варьироваться в зависимости от типа клетки и стадии развития. (Штуттфельд Е, Балмер-Хофер К (сентябрь 2009 г.), «Структура и функция рецепторов VEGF», журнал IUBMB Life 61 (9): 915-22.)
[008] Так как VEGFR-2 является крупным посредником митогенеза и жизнедеятельности сосудистой эндотелиальной клетки (ЭК), а также ангиогенеза и капиллярной проницаемости, ожидается, что прямое ингибирование активности киназы VEGFR-2 приведет к сокращению ангиогенеза и подавлению роста опухоли. Кроме того, ингибирование изоформы VEGFR-2, нацеленной на генетически более стабильные эндотелиальные клетки хозяина, вместо неустойчивых тканей опухоли, может уменьшить шанс развития сопротивления. Несколько агентов, предназначающихся для сигнализации VEGFR, регулируемые либо как единственные агенты, либо в сочетании с химиотерапией, по результатам оказывали положительное влияние на пациентов со злокачественными опухолями поздней стадии ("Таргетная терапия VEGF: механизмы антиопухолевой активности", Nature Reviews Cancer, 2008, 8, 579; "Молекулярное основание для эффективности сунитиниба и будущего клинического развития", Nature Reviews Drug Discovery, 2007, 6, 734; и "Ангиогенез: принцип организации для изобретения лекарства?" Nature Reviews Drug Discovery, 2007, 6, 273).
[009] c-Met, также называемый рецептором фактора роста гепатоцита (HGFR), выражен преимущественно в эпителиальных клетках, но также был идентифицирован в эндотелиальных клетках, миобластах, гематопоэтических клетках и моторных нейронах. Естественный лиганд для c-Met является фактором роста гепатоцита (HGF), также известным как рассеивающий фактор (SF). И в эмбрионах, и у взрослых активированный c-Met продвигает морфогенетическую программу, известную как агрессивный рост, вызывающую распространение клеток, сбой межклеточных контактов и миграцию клеток к их среде ("От Tpr-Met до Met, онкогенез и трубы", Oncogene, 2007, 26, 1276; и "Киназа Тирозина Рецептора Met как Цель Противоопухолевой Таргетной Терапии", Cancer Letter, 2009, 280, 1-14).
[010] Большое разнообразие злокачественных опухолей человека демонстрирует c-Met стимулирование, сверхпродукцию или мутацию, включая карциномы груди, печени, легкого, яичника, почки, щитовидной железы, толстой кишки, глиобластомы, простаты и т.д. c-Met также вовлекается в фиброз легкого и атеросклероз. Агрессивный рост определенных раковых клеток значительно увеличивается стромальными опухолевыми взаимодействиями, включающими путь HGF/c-Met. Таким образом, многочисленные свидетельства, что сигнализация c-Met вовлечена в прогрессию и распространение нескольких видов рака, и понимание роли в болезни, вызвали большой интерес в c-Met как главной цели в поиске лекарства от рака ("Молекулярная терапия рака: оправдаются ли наши ожидания", Euro. J. Cancer, 2008, 44, 641-651; и "Таргетная терапия Пути Сигнализации c-Met при Раке", Clin. Cancer Res., 2006, 12, 3657). Агенты, нацеленные на сигнальный путь c-Met, теперь являются объектом клинических исследований ("Новые Терапевтические Ингибиторы Пути Сигнализации c-Met при Раке", Clinical Cancer Research, 2009, 15, 2207; и "Разработка лекарственного ингибитора МЕТ: таргетная онкогенная зависимость и сложившаяся ситуация", Nature Review Drug Discovery, 2008, 7, 504).
[011] Axl принадлежит к подгруппе киназ тирозина рецептора (К.T.Ks), также включает Tyro3 и Mer (ТАМ). Рецепторы ТАМ характеризуются комбинацией двух областей, подобных иммуноглобулину, и двойных повторений фибронектина типа III во внеклеточной области и цитоплазматической области киназы. Лигандами для рецепторов ТАМ являются Gas6 (ген блокировки роста 6) и протеин S, два протеина, зависимых от витамина K, показывающие 43% аминокислотную идентичность последовательности и имеющие подобные структуры области ("Протеин S фактора антикоагуляции и его связанный ген, Gas6, являются лигандами для группы Tyro 3/Axl киназ тирозина рецептора", Cell, 1995, 80, 661-670; и "Киназа тирозина рецептора Axl, стимулированная протеином, зависимым от витамина K, закодированная геном блокировки роста 6", Nature, 1995, 373, 623-626).
[012] Соответствующие доказательства поддерживают роль системы Gas6/Axl в стимулировании роста и жизнедеятельности клеток в нормальных и раковых клетках ("Киназы тирозина рецептора ТАМ: биологические функции, сигнализация и потенциальное терапевтическое предназначение при раке человека", Adv Cancer Res, 2008, 100, 35-83). Сверхпродукция и сигнализация Axl были вовлечены в несколько видов злокачественных опухолей человека, например, рак толстой кишки, груди, глиомы, щитовидной железы, рак желудка, меланомы, рак легких, и карциномы клеток почечного эпителия (RCC). Более подробная роль биологии Axl была доказана при глиоме, где утрата сигнализации Axl уменьшила рост опухоли глиомы, и при раке молочной железы, при миграции клеток стимулирования Axl, формировании труб, неоваскуляризации и росте опухоли. Axl, как оказалось, играет несколько ролей в онкогенезе, и терапевтические антитела против Axl могут заблокировать функции Axl не только в клетках злокачественных опухолей, но также и в строме опухоли. Дополнительный эффект ингибирования Axl анти-VEGF предполагает, что блокирование функции Axl могло быть эффективным подходом для усиления антиангиогенной терапии ("Axl как потенциальная терапевтическая цель при раке: роль Axl в росте опухоли, метастазе и ангиогенезе", Oncogene, 2009, 28, 3442-3455; и "Киназы Тирозина Рецептора ТАМ: биологические функции, сигнализация и потенциальное терапевтическое предназначение при человеческом раке", Adv Cancer Res., 2008, 100, 35-83).
[013] RON (MST1R, recepteur d'origine nantais), другой член группы MET, является киназой тирозина рецептора для протеина стимулирования макрофага лиганда (MSP, также известный как MST1 и подобный фактору роста гепатоцитов (HGFL)), который связан с лабораторными и естественными рассеиваниями клеток, подвижностью и матричной инвазией - все из которых являются суррогатными маркерами агрессивного фенотипа рака с метастатическим потенциалом. RON добивается опухолеродных фенотипов в раке легких, щитовидной железы, поджелудочной железы, простаты, толстого кишечника и рака молочной железы, и обеспечивает неблагоприятный прогноз при раке молочной железы. Коэкспрессия RON с МЕТ и индукцией выражения RON сигнализацией HGF-MET, была описана при гепатоцеллюлярной карциноме. Кроме того, коэкспрессия МЕТ и RON предвещает худший прогноз при раке яичников, груди и мочевого пузыря. При условии сигнальной избыточности RON и МЕТ, возможно, что сопротивление ингибированию МЕТ установлено сигнализацией RON ("RON (MST1R) - новый прогнозирующий маркер и терапевтическая цель при желудочно-пищеводной аденокарциноме". Cancer Biol Ther. 1 июля 2011 г.; 12 (1): 9-46).
[014] Роли сигнальной оси MSP-RON при патогенезе рака были также экстенсивно изучены в различных системах модели. И в лабораторных и в естественных условиях доказательства показали, что сигнализация MSP-RON важна для агрессивного роста различных типов раковых образований. Нарушенная активация RON, вызванная сверхпродукциями протеина и генерацией опухолеродных изоформ, и обозначенная постоянной активацией мульти-внутриклеточных сигнальных каскадов, происходит в различных типах раковых образований. Сигнализация RON также необходима для роста и жизнедеятельности раковой клетки. Эти особенности делают RON целью медикаментозной раковой терапии ("Сигнализация MSP-RON при раке: патогенез и терапевтический потенциал". Nature Reviews Cancer, 2013, 13, 466-481).
[015] Широко известно, что раковые клетки используют многократные механизмы для уклонения от жестко регулируемых клеточных процессов, например, быстрое увеличение, апоптоз и старение. Таким образом, большинство опухолей может избежать ингибирования любой единственной киназы. Системный анализ опухолей идентифицировал коактивацию киназы тирозина рецептора (К.Т.K) как важный механизм, которым раковые клетки достигают сопротивления к химическому воздействию. Одна из стратегий преодоления коактивации К.Т.K может включать терапевтическую нацеленность на несколько К.Т.K одновременно, чтобы отключить онкогенную сигнализацию К.Т.K и преодолеть компенсационные механизмы. ("Сети Коактивации Киназ Тирозина рецептора при Раке", Cancer Research, 2010, 70, 3857). Подходы к уменьшению опухоли в терапии, нацеленной на сигнализацию VEGFR, с-Met, Ron и/или Axl, могут обойти способность опухолевых клеток преодолеть ингибирование VEGFR, c-Met (HGFR), Ron и/или Axl по отдельности и, таким образом, могут представлять улучшенную терапию рака.
СУЩНОСТЬ ИЗОБРЕТЕНИЯ
[016] В настоящем документе предусматриваются новые соединения и способы для лечения клеточных пролиферативных заболеваний. Соединения, раскрытые в настоящем документе, являются ингибиторами киназ тирозина протеина. Преимущественно, соединения, раскрытые в настоящем документе, являются ингибиторами нескольких функций, способных к ингибированию, например, VEGFR, c-Met (HGFR), Ron и/или Axl. Соответственно, в настоящем документе предусматриваются новые ингибиторы сигнализации рецептора киназы тирозина протеина, например, сигнализации рецептора VEGF, сигнализации рецептора HGF, сигнализации Ron и/или сигнализации Axl.
[017] В частности, обнаружено, что соединения, раскрытые в настоящем документе, и их фармацевтически приемлемые композиции, эффективны как ингибиторы киназ тирозина рецептора, например, как VEGFR, c-Met, Ron и/или Axl.
[018] В одном аспекте, предусмотренном в настоящем документе, соединение, имеющее Формулу (I):
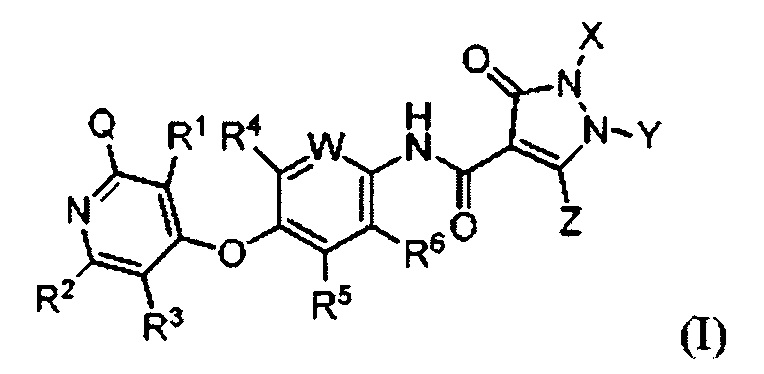
или стереоизомер, геометрический изомер, таутомер, N-оксид, сольват, гидрат, метаболит, фармацевтически приемлемая соль или пропрепарат, в которой каждый из Q, R1, R2, R3, R4, R5, R6, W, X, Y и Z определяется в настоящем документе.
[019] В некоторых воплощениях соединение, раскрытое в настоящем документе, имеет формулу (II):
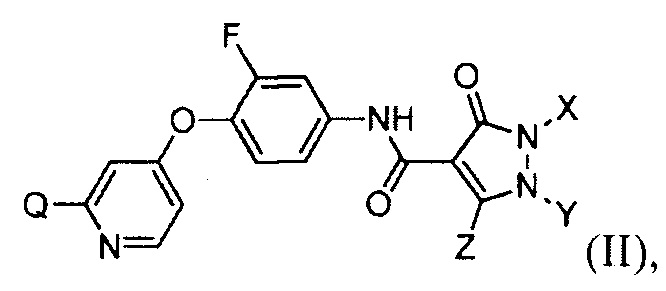
в которой каждый из Q, X, Y и Z определяется в настоящем документе.
[020] В некоторых воплощениях Q является D, -N(Rc)С(=O)NRaRb, -N(Rc)C(=O)Rd, -C(=O)NRaRb, -N(Rc)S(=O)NRaRb, -N(Rc)S(=O)Ra, -N(Rc)S(=O)2NRaRb или - N(Rc)S(=O)2Ra;
W является CR7 или N;
каждый из X, Y и Z независимо H, D, (C1-С6) алкил, (С3-C8) циклоалкил, -(С1-С4) алкилен - (С3-C8) циклоалкил, (С3-С7) гетероциклил, - (С1-С4) алкилен - (С3-С7) гетероциклил, (С6-С10) арил, 5-10-членный гетероарил, включающий 1, 2, 3 или 4 гетероатома, независимо выбранных из О, S и N, - (С1-С4) алкилен - (С6-С10) арил или - (С1-С4) алкилен - (5-10-членный гетероарил), где каждый (С1-С6) алкил, (С3-C8) циклоалкил, - (С1-С4) алкилен - (С3-C8) циклоалкил, (С3-С7) гетероциклил, - (С1-С4) алкилен - (С3-С7) гетероциклил, (С6-С10) арил, 5-10-членный гетероарил, - (С1-С4) алкилен - (С6-С10) арил и - (С1-С4) алкилен - (5-10-членный гетероарил), не замещен или произвольно замещен 1, 2, 3, 4 или 5 заместителей, независимо выбранных из D, F, Cl, Br, CN, (С2-С6) алкенил, (С2-С6) алкинил, ORa, NRaRb, - (C1-C4) алкилен-ORa и - (C1-C4) алкилен-NRaRb;
каждый из R1, R2, R3, R4, R5, R6 и R7 независимо Н, D, F, Cl, Br, CN, N3, ORa, (С1-С6) алкил, (С1-С6) галоалкил, (С2-С6) алкенил или (С2-С6) алкинил;
каждый из Ra, Rb и Rc независимо Н, (С1-С6) алифатический, (С1-С6) галоалкил, (С3-С6) циклоалкил, - (С1-С4) алкилен - (С3-С6) циклоалкил, (С3-С6) гетероциклил, - (С1-С4) алкилен - (С3-С6) гетероциклил, (С6-С10) арил, 5-10-членный гетероарил включающий 1, 2, 3 или 4 гетероатома, независимо выбранных из О, S и N, - (С1-С4) алкилен - (С6-С10) арил или -(С1-С4) алкилен - (5-10-членный гетероарил), где каждый (С1-С6) алифатический, (С1-С6) галоалкил, (С3-С6) циклоалкил, - (С1-С4) алкилен -(С3-С6) циклоалкил, (С3-С6) гетероциклил, - (С1-С4) алкилен - (С3-С6) гетероциклил, (С6-С10) арил, 5-10-членный гетероарил, - (С1-С4) алкилен -(С6-С10) арил и - (С1-С4), алкилен - (5-10-членный гетероарил) не замещается или произвольно замещается 1, 2, 3 или 4 заменителями, независимо выбранными из D, F, Cl, CN, N3, ОН, NH2, (С1-С6) галоалкил, (С1-С6) алкокси и (О-С6) алкиламино; и
Rd является D, (С3-C8) циклоалкил, - (С1-С4) алкилен - (С6-С10) арил, 5-10-членный гетероарил, включающий 1, 2, 3 или 4 гетероатома, независимо выбранных из О, S и N или - (С1-С4) алкилен - (5-10-членный гетероарил), где каждый (С3-C8) циклоалкил, - (С1-C4) алкилен - (С6-С10) арил, 5-10-членный гетероарил и - (С1-С4), алкилен - (5-10-членный гетероарил) не замещается или произвольно замещается 1, 2, 3 или 4 заменителями, независимо выбранными из D, F, Cl, Br, CN, ORa, NRaRb, (С1-С6) алкил, (С2-С6) алкенил, (С2-С6) алкинил, - (С1-С4) алкилен-ORa и - (С1-С4) алкилен-NRaRb.
[021] В других воплощениях каждый из Ra, Rb и Rc независимо Н, (С1-С6) алифатический, (С1-С6) галоалкил, (С3-С6) циклоалкил, (С3-С6) гетероциклил, - (С1-С4) алкилен - (С3-С6) гетероциклил, (С6-С10) арил, 5-10-членный гетероарил, включающий 1, 2, 3 или 4 гетероатома, независимо выбранных из О, S и N, - (С1-С4) алкилен - (С6-С10) арил или -(С1-С4) алкилен - (5-10-членный гетероарил), где каждый (С1-С6) алифатический, (С1-С6) галоалкил, (С3-С6) циклоалкил, (С3-С6) гетероциклил, - (С1-С4) алкилен - (С3-С6) гетероциклил, (С6-С10) арил, 5-10-членный гетероарил, - (С1-С4) алкилен - (С6-С10) арил и - (С1-С4), алкилен - (5-10-членный гетероарил) не замещается или произвольно замещается 1, 2, 3 или 4 заместителями, независимо выбранными из D, F, Cl, CN, N3, ОН, NH2, (С1-С6) галоалкил, (С1-С6) алкокси и (С1-С6) алкиламино; и
Rd является D, (С3-C8) циклоалкил, - (С1-С4) алкилен - (С6-С10) арил, 5-10-членный гетероарил, включающий 1, 2, 3 или 4 гетероатома, независимо выбранных из О, S и N или - (С1-С4) алкилен - (5-10-членный гетероарил), где каждый (С3-C8) циклоалкил, - (С1-С4) алкилен - (С6-С10) арил, 5-10-членный гетероарил и - (С1-С4) алкилен - (5-10-членный гетероарил) не замещается или произвольно замещается 1, 2, 3 или 4 заменителями, независимо выбранными из D, F, Cl, Bra, CN, ORa, NRaRb, (С2-С6) алкенил, (С2-С6) алкинил, - (С1-С4) алкилен-ORa и - (С1-С4) алкилен-NRaRb
[022] В других воплощениях Q является -N (Rc)C(=O)NRaRb, -N(Rc)C(=O)Rd или - С(=O) NRaRb
[023] В других воплощениях каждый из X, Y и Z независимо (С1-С4) алкил, (С3-С6) циклоалкил, - (С1-С2) алкилен - (С3-С6) циклоалкил, (С3-С6) гетероциклил, - (С1-С2) алкилен - (С3-С6) гетероциклил, фенил, 5-10-членный гетероарил, включающий 1, 2, 3 или 4 гетероатома, независимо выбранных из О, S и N, - (С1-С2) фенил алкилен или - (С1-С2) алкилен - (5-10-членный гетероарил), где каждый (С1-С4) алкил, (С3-С6) циклоалкил, -(С1-С2) алкилен - (С3-С6) циклоалкил, (С3-С6) гетероциклил, - (С1-С2) алкилен - (С3-С6) гетероциклил, фенил, 5-10-членный гетероарил, - (С1-С2) фенил алкилен и - (С1-С2) алкилен - (5-10-членный гетероарил) не замещаются или произвольно замещаются 1, 2, 3 или 4 заместителями, независимо выбранными из D, F, Cl, CN, (С2-С4) алкенил, (С2-С4) алкинил, ORa, NRaRb, - (С1-С2) алкилен-ORa и - (С1-С2) алкилен-NRaRb.
[024] В других воплощениях каждый из R1, R2, R3, R4, R5, R6 и R7 независимо Н, D, F или Cl.
[025] В других воплощениях каждый из Ra, Rb и Rc независимо Н, (С1-С4) алкил, (С1-С4) галоалкил, (С3-С6) циклоалкил, - (С1-С2) алкилен - (С3-С6) циклоалкил, (С3-С6) гетероциклил или - (С1-С2) алкилен - (С3-С6) гетероциклил, где каждый (С1-С4) алкил, (С1-С4) галоалкил, (С3-С6) циклоалкил, - (С1-С2) алкилен - (С3-С6) циклоалкил, (С3-С6) гетероциклил и - (С1-С2), алкилен - (С3-С6) гетероциклил не замещается или произвольно замещается 1, 2, 3 или 4 заместителями, независимо выбранными из D, F, Cl, CN, N3, ОН, NH2, (С1-С3) галоалкил, (С1-С3) алкокси и (С1-С3) алкиламино.
[026] В других воплощениях Rd (С3-С6) циклоалкил, где (С3-С6) циклоалкил не замещается или произвольно замещается 1, 2, 3 или 4 заместителями, независимо выбранными из D, F, CN, ORa, NRaRb, (С1-С3) алкил, (С2-С4) алкенил, (С2-С4) алкинил, - (С1-С2) алкилен-ORa и- (С1-С2) алкилен-NRaRb.
[027] В других воплощениях каждый из X, Y и Z независимо Н, D, CH3, метиловая группа замещается 1, 2 или 3 атомами дейтерия, этиловая, пропиловая, изопропиловая, фениловая или фенильная группа замещается 1, 2, 3, 4 или 5 заместителями, независимо выбранными из D, F и Cl.
[028] В других воплощениях Q:
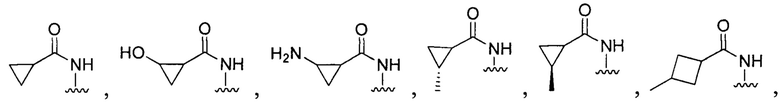
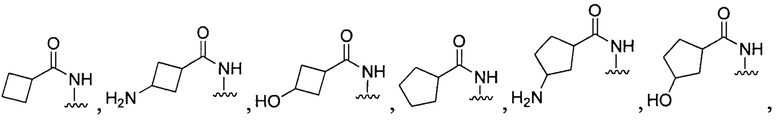
 или
или 
[029] В другом аспекте, предусмотренном в настоящем документе, фармацевтическая композиция, включающая соединение, раскрытое в настоящем документе, является ингибитором киназы тирозина рецептора, или стереоизомером, геометрическим изомером, таутомером, сольватом, метаболитом, фармацевтически приемлемой солью и фармацевтически приемлемым носителем, наполнителем, разжижителем, стимулятором, агентом или комбинацией таких веществ. В некоторых воплощениях фармацевтическая композиция включает соединение, раскрытое в настоящем документе, которое является ингибитором сигнализации рецептора VEGF, сигнализации рецептора HGF, сигнализации Ron и/или сигнализации Axl, или стереоизомером, геометрическим изомером, таутомером, сольватом, метаболитом, фармацевтически приемлемой солью и фармацевтически приемлемым носителем, наполнителем, разжижителем, стимулятором, агентом или комбинацией таких веществ.
[030] В других воплощениях фармацевтическая композиция, раскрытая в настоящем документе далее, включает терапевтический агент.
[031] В других воплощениях терапевтический агент является химиотерапевтическим агентом, антипролиферативным агентом, агентом для лечения атеросклероза, агентом для лечения фиброза легкого или их комбинациями.
[032] В других воплощениях терапевтический агент является хлорамбуцилом, мелфаланом, циклофосфамидом, ифосфамидом, бусульфаном, кармустином, ломустином, стрептозоцином, цисплатином, карбоплатином, лксалиплатином, дакарбазином, темозоломидом, прокарбазином, метотрексатом, фтороурацилом, цитарабином, гемцитабином, меркаптопурином, флударабином, винбластином, винкристином, винорельбином, паклитакселом, доцетакселем, топотеканом, иринотеканом, этопозидом, трабекткдином, дактиномицином, доксорубицином, эпирубицином, даунорубицином, митоксантроном, блеомицином, митомицином, иксабепилоном, тамоксифеном, флутамидом, аналогами гонадорелина, мегестролом, преднидоном, дексаметазоном, метилпреднизолоном, талидомидом, интерфероном альфа, лейковорином, сиролимусом, темсиролимусом, эверолимусом, афатинибом, алисертибом, амуватинибом, апатинибом, акситинибом, бортезомибом, босутинибом, бриванибом, кабозантинибом, цедиранибом, креноланибом, кризотинибом, дабрафенибом, дакомитинибом, данузертибом, дасатинибом, довтинибом, эрлотинибом, форетинибом, ганетеспибом, гефринитибом, ибрутинибом, икотинибом, иматинибом, инипарибом, лапатинибом, ленватинибом, линифанибом, линситинибом, маситинибом, момелотинибом, мотесанибом, нератинибом, нилотинибом, нирапарибом, опрозомибом, олапарибом, пазопанибом, пиктилисибом, понатинибом, квизартинибом, регорафинибом, ригорсертибом, рукапарибом, руксолитинибом, саракатинибом, саридегибом, сорафенибом, сунитинибом, тасоцитинибом, телатенибом, тивантинибом, тивозанибом, тофацинибом, траметинибом, вандетанибом, велипарибом, вемурафенибом, висмодегибом, воласертибом, алемтузумабом, бевацизумабом, брентуксимабом, ведотином, катумаксомабом, цетуксимабом, деносумабом, гемтузумабом, ипилимумабом, нимотузумабом, офатумумабом, панитумумабом, рамуцирумабом, ритуксимабом, тозитумомабом, трастузумабом или комбинацией.
[033] В другом аспекте, предусмотренном в настоящем документе, соединение, раскрытое в настоящем документе, или фармацевтическая композиция, раскрытая в настоящем документе для использования в предотвращении, принятии мер, лечении или уменьшении серьезности пролиферативных расстройств у пациентов.
[034] В другом аспекте, предусмотренном в настоящем документе, способ предотвращения, принятия мер, лечения или уменьшения серьезности пролиферативных расстройств у пациентов путем введения пациентам соединения, раскрытого в настоящем документе.
[035] В другом аспекте, предусмотренном в настоящем документе, способ предотвращения, принятия мер, лечения или уменьшения серьезности пролиферативных расстройств у пациентов путем введения пациентам фармацевтической композиции, раскрытой в настоящем документе.
[036] В некоторых воплощениях пролиферативное расстройство является метастатическим раком. В других воплощениях пролиферативное расстройство является раком толстой кишки, аденокарциномой желудка, раком мочевого пузыря, раком молочной железы, раком почек, раком печени, раком легких, раком кожи, раком щитовидной железы, раком головы и шеи, раком простаты, раком поджелудочной железы, раком ЦНС, глиобластомой или миелопролиферативным расстройством. В дальнейших воплощениях пролиферативное расстройство является фиброзом легкого или атеросклерозом.
[037] В другом аспекте, предусмотренном в настоящем документе, соединение, раскрытое в настоящем документе, или фармацевтическая композиция, раскрытая в настоящем документе для использования при ингибировании или модуляции активности киназы протеина в биологической пробе, включающей связывание биологической пробы с соединением, раскрытым в настоящем документе, или фармацевтической композиции, раскрытой в настоящем документе.
[038] В другом аспекте, предусмотренном в настоящем документе, способ ингибирования или модуляции активности киназы протеина в биологической пробе, включающей связывание биологической пробы с соединением, раскрытым в настоящем документе.
[039] В другом аспекте, предусмотренном в настоящем документе, способ ингибирования или модуляции активности киназы протеина в биологической пробе, включающей связывание биологической пробы с фармацевтической композицией, раскрытой в настоящем документе.
[040] В некоторых воплощениях киназа протеина является киназой тирозина рецептора. В других воплощениях киназа тирозина рецептора является VEGFR, c-Met, Ron, Axl или их комбинацией.
[041] В некоторых воплощениях ингибирование активности киназы протеина рецептора, преимущественно сигнализация рецептора VEGF, сигнализация рецептора HGF, сигнализация Ron, или сигнализация рецептора Axl может быть в клетке или многоклеточном организме. Во многоклеточном организме, способ согласно этому аспекту изобретения включает введение в организм соединения, раскрытого в настоящем документе, или фармацевтической композиции, раскрытой в настоящем документе. В некоторых воплощениях организм является млекопитающим. В других воплощениях человеком. В еще одном воплощении способ далее включает контакт киназы с терапевтическим агентом.
[042] В другом аспекте, предусмотренном в настоящем документе, способы ингибирования пролиферативной активности клетки, способ, включающий контакт клетки с эффективным пролиферативным ингибирующим объемом соединения согласно данному изобретению или его состава. В некоторых воплощениях способ далее включает контакт клетки с терапевтическим агентом.
[043] В другом аспекте, предусмотренном в настоящем документе, способы лечения клеточного пролиферативного заболевания у пациентов, способ, включающий введение пациенту такого эффективного терапевтического объема соединения согласно данному изобретению или его состава. В некоторых воплощениях способ далее включает введение терапевтического агента.
[044] В другом аспекте, предусмотренном в настоящем документе, способы ингибирования роста опухоли у пациента, способ, включающий введение пациенту такого эффективного терапевтического объема соединения согласно данному изобретению или его состава. В некоторых воплощениях способ далее включает введение терапевтического агента.
[045] В другом аспекте, предусмотренном в настоящем документе, способы изготовления, способы отделения и способы очищения составов Формулы (I) или (II).
[046] Предшествующее просто суммирует определенные аспекты изобретения и не имеет ограничивающего характера. Эти аспекты, а также другие аспекты и воплощения более полно описаны далее.
ПОДРОБНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
ОПРЕДЕЛЕНИЯ И ОБЩАЯ ТЕРМИНОЛОГИЯ
[047] Более подробные ссылки относятся к определенным воплощениям изобретения, примеры которого проиллюстрированы в сопутствующих структурах и формулах. Изобретение предназначается для покрытия всех альтернатив, модификаций и эквивалентов, которые могут быть включены в рамках данного изобретения, как определено требованиями. Специалист в данной области признает несколько способов и материалов, подобных или эквивалентных описанным в настоящем документе, которые могут использоваться в практике данного изобретения. Данное изобретение никоим образом не ограничено способами и материалами, описанными в настоящем документе. Если один или несколько упомянутых литературных источников, патентов и подобных материалов отличаются или противоречат настоящей заявке, включая, без ограничения, определенные условия, использование срока, описанные способы и т.п., настоящая заявка является регулирующей.
[048] Как используется в настоящем документе, следующие определения применяются, если иначе не обозначено. В целях настоящего изобретения, химические элементы идентифицируются в соответствии с Периодической таблицей элементов, версии CAS и Руководства химии и физики, 75 издание 1994. Кроме того, общие принципы органической химии описаны в «Органической химии», Томаса Соррелла, University Science Books, Саусалито: 1999, и «Усовершенствованная органическая химия Марча", Майкл Б. Смит и Джерри Марч, John Wiley & Sons, Нью-Йорк: 2007, содержание которых настоящим включено посредством ссылок.
[049] Как описано в настоящем документе, соединения, раскрытые в настоящем документе, могут быть произвольно заменены одним или несколькими заместителями, которые обычно иллюстрируются ниже, или, как иллюстрируется определенными классами, подклассами и разновидностями изобретения. Следует понимать, что фраза «произвольно замещаются» используется наравне с фразой «замещаются или не замещаются». В целом термин «замещенный» относится к замене одного или нескольких водородных радикалов в данной структуре с радикалом указанного заместителя. Если иначе не обозначено, у группы с произвольным замещением может быть заместитель в каждой замещаемой позиции группы. Если более одной позицией в данной структуре можно заменить несколькими заместителями, выбранными из указанной группы, заместитель может быть либо тем же, либо отличающимся в каждой позиции.
[050] Термин «алифатический» или «алифатическая группа» относится к нормальной цепи (т.е. без ветвей) или разветвленной, замещенной или незамещенной углеводородной цепи, полностью насыщенной или содержащей одну или более единиц ненасыщенности. Если иначе не указано, алифатические группы содержат 1-20 атомов углерода. В некоторых воплощениях алифатические группы содержат 1-10 атомов углерода. В других воплощениях алифатические группы содержат 1-8 атомов углерода. В еще других воплощениях алифатические группы содержат 1-6 атомов углерода, и в еще других воплощениях алифатические группы содержат 1-3 атома углерода. Подходящие алифатические группы включают, без ограничений, линейные или разветвленные, замещенные или незамещенные группы алкила, алкенила или алкинила. Например, (С1-С6) алифатические группы включают неразветвленные или разветвленные, незамещенные или произвольно замещенные группы (С1-С6) алкил, (С2-С6) алкенил или (С2-С6) алкинил. Алифатические радикалы произвольно замещаются независимо одним или несколькими заместителями, описанными в настоящем документе.
[051] Термин «алкил» или «алкильная группа» относится к насыщенной линейной или разветвленной цепи одновалентного углеводородного радикала из 1-20 атомов углерода, где алкильный радикал может произвольно замещаться независимо одним или несколькими заместителями, описанными ниже. Если иначе не указано, алкильные группы содержат 1-20 атомов углерода. В некоторых воплощениях алкильные группы содержат 1-10 атомов углерода. В других воплощениях алкильные группы содержат 1-8 атомов углерода. В других воплощениях алкильные группы содержат 1-6 атомов углерода. В еще других воплощениях алкильные группы содержат 1-4 атома углерода, и в еще других воплощениях алкильные группы содержат 1-3 атома углерода.
[052] Некоторые неограничивающие примеры алкильных групп включают, без ограничений, метил (Ме, -СН3), этил (Et, -CH2CH3), 1 пропил (n-Pr, n-пропил, -СН2СН2СН3), 2-пропил (i-Pr, i-пропил,- СН(СН3)2), 1 бутил (n-Bu, n-бутил, -СН2СН2СН2СН3), 2-метил-1-пропил (i-Bu, i-бутил, -СН2СН(СН3)2), 2 бутил (S-bu, S-бутил, -СН(СН3)СН2СН3), 2-метил-2-пропил (t-Bu, t-бутил, -С(СН3)3), 1 пентил (n-пентил, -СН2СН2СН2СН2СН3), 2-пентил (-СН(СН3)СН2СН2СН3), 3 пентил (-СН(СН2СН3)2), 2 метил 2 бутил (-С(СН3)2СН2СН3), 3-метил-2-бутил (-СН(СН3)СН(СН3)2), 3 метил 1 бутил (-СН2СН2СН(СН3)2), 2 метил 1 бутил (-СН2СН(СН3)СН2СН3), 1 гексил (-СН2СН2СН2СН2СН2СН3), 2 гексили (-СН(СН3)СН2СН2СН2СН3), 3 гексил (-СН(СН2СН3)(СН2СН2СН3)), 2 метил 2 пентил (-С(СН3)2СН2СН2СН3), 3-метил-2-пентил (-СН(СН3)СН(СН3)СН2СН3), 4 метил 2 пентил (-СН(СН3)СН2СН(СН3)2), 3-метил-3-пентил (-С(СН3)(СН2СН3)2), 2 метила 3 пентил (-СН(СН2СН3)СН(СН3)2), 2,3-диметил-2-бутил (-С(СН3)2СН(СН3)2), 3,3-диметил-2 бутил (-СН(СН3)С(СН3)3, 1-гептил, 1 октил и т.п.
[053] Термин «алкил» и префиксы «алк-» указывают на содержание нормальной цепи и разветвленной насыщенной углеродной цепи.
[54] Термин «алкилен» относится к влажной двухвалентной группе углеводорода, полученного из насыщенного углеводорода нормальной или разветвленной цепи удалением двух атомов водорода. Если иначе не указано, группы алкилена содержат 1-10 атомов углерода. В некоторых воплощениях группы алкилена содержат 1-6 атомов углерода. В других воплощениях группы алкилена содержат 1-4 атома углерода. В еще других воплощениях группы алкилена содержат 1-2 атома углерода, и иллюстрируется метиленом (-СН2-), этиленом (-СН2СН2-), изопропиленом (-СН(СН3)СН2-) и т.п.
[055] Термин «алкенил» относится к линейной или разветвленной цепи одновалентного углеводородного радикала из 2-12 атомов углерода, по крайней мере, с одним участком ненасыщенности, т.е. углерод-углерод, двойная связь sp2, где радикал алкенил а может произвольно замещаться независимо одним или несколькими заместителями, описанными в настоящем документе, и включает радикалы, имеющие ориентации «cis» и «trans», или альтернативно, ориентации «Е» и «Z». Преимущественно, алкенильная группа содержит 2-8 атомов углерода, и более предпочтительно, 2-6 атомов углерода. Примеры включают, без ограничений, этиленил или винил (-СН=СН2), аллил (-СН2СН=СН2) и т.п.
[056] Термин «алкинил» относится к линейному или отделенному одновалентному углеводородному радикалу из 2-12 атомов углерода, по крайней мере, с одним участком ненасыщенности, т.е. углерод-углерод, тройная связь SP, где радикал алкинила может произвольно замещаться независимо одним или несколькими заместителями, описанными в настоящем документе. Преимущественно, алкинильная группа содержит 2-8 атомов углерода, и более преимущественно 2-6 атомов углерода. Примеры включают, без ограничений, этинил (-С=СН), пропинил (пропаргил, -СН2С=СН), -С=С-СН3 и т.п.
[057] Термин «алкокси» относится к алкильной группе, как ранее определено, присоединенной к основному атому углерода через атом кислорода. Если иначе не указано, алкокси группы содержат 1-20 атомов углерода. В некоторых воплощениях алкокси группы содержат 1-10 атомов углерода. В других воплощениях алкокси группы содержат 1-8 атомов углерода. В еще других воплощениях алкокси группы содержат 1-6 атомов углерода. В еще других воплощениях алкокси группы содержат 1-4 атома углерода. В дальнейших воплощениях алкокси группы содержат 1-3 атома углерода. Алкокси радикалы произвольно замещаются независимо одним или несколькими заместителями, описанными в настоящем документе.
[058] Некоторые неограничивающие примеры алкокси групп включают, без ограничений, метокси (МеО, -ОСН3), этокси (EtO, -OCH2CH3), 1-пропокси (n-PrO, n-пропокси, -ОСН2СН2СН3), 2-пропокси (i-PrO, i-пропокси, -ОСН(СН3)2), 1-бутокси (n-BuO, n-бутокси, -ОСН2СН2СН2СН3), 2 метил 1 пропокси (i-BuO, i-бутокси, -ОСН2СН(СН3)2), 2-бутокси (s-BuO, s-бутокси, -ОСН(СН3)СН2СН3), 2 метил 2 пропокси (t-BuO, t-бутокси, -ОС(СН3)3), 1-пентокси (n-пентокси, -ОСН2СН2СН2СН2СН3), 2-пентокси (-OCH(СН3)СН2СН2СН3), 3-пентокси (-ОСН(СН2СН3)2), 2 метил 2 бутокси (-ОС(СН3)2СН2СН3), 3 метил 2 бутокси (-ОСН(СН3)СН(СН3)2), 3 метил 1 бутокси (-ОСН2СН2СН(СН3)2), 2 метил 1 бутокси (-ОСН2СН(СН3)СН2СН3) и т.п.
[059] Термины «галоалкил», «галоалкенил» или «галоалкокси» относится к алкилу, алкенилу или алкокси, в зависимости от обстоятельств, замещенных одним или несколькими атомами галогена.
[060] Термин «углеродный цикл», «карбоциклил», «карбоциклическое кольцо» или «циклоалифатический» относятся к одновалентному или многовалентному неароматическому, насыщенному или частично ненасыщенному кольцу, имеющему 3-12 атомов углерода как система моноциклического, бициклического или трициклического кольца. Подходящие циклоалифатические группы включают, без ограничений, циклоалкил, циклоалкенил и циклоалкинил. Дальнейшие примеры циклоалифатических групп включают циклопропил, циклобутил, циклопентил, 1 циклопент 1 энил, 1 циклопент 2 энил, 1 циклопент 3 энил, циклогексил, 1-циклогекс-1-энил, 1 циклогекс 2 энил, 1 циклогекс 3 энил, циклогексадиэнил и т.п.
[061] Термин «циклоалкил» относится к одновалентному или многовалентному насыщенному кольцу, имеющему 3-12 атомов углерода как система моноциклического, бициклического или трициклического кольца. Бициклическая кольцевая система включает спиро бициклил или конденсированный бициклил. В некоторых воплощениях циклоалкил содержит 3-10 атомов углерода. В еще других воплощениях циклоалкил содержит 3-8 атомов углерода, и в еще других воплощениях циклоалкил содержит 3-6 атомов углерода. Радикалы циклоалкила произвольно замещаются независимо одним или несколькими заместителями, описанными в настоящем документе.
[062] Термин «гетероцикл», «гетероциклил», или «гетероциклический», как используется взаимозаменяемо в настоящем документе, относится к системе моноциклического, бициклического или трициклического кольца, в которой один или несколько участников кольца независимо выбраны из гетероатомов, и является полностью насыщенным, или содержит одну или несколько единиц ненасыщенности, но которое не является ароматическим, которое имеет одну точку крепления к остальной части молекулы. Бициклическая кольцевая система включает спиро бициклил или конденсированный бициклил, и одно из колец может быть монокрбоциклическим или моногетероциклическим. Один или несколько атомов кольца произвольно замещаются независимо одним или несколькими заместителями, описанными в настоящем документе. В некоторых воплощениях «гетероцикл», «гетероциклил» или «гетероциклическая» группа является моноциклом, имеющим 3-7 участников кольца (2-6 атомов углерода и 1-3 гетероатома, выбранных из N, О, Р, и S, где S или Р произвольно замещаются одним или несколькими оксо для обеспечения группы SO или SO2, РО или PO2). В других воплощениях «гетероцикл», «гетероциклил» или «гетероциклическая» группа является моноциклом, имеющим 3-6 участников кольца (2-5 атомов углерода и 1-3 гетероатома, выбранных из N, О, Р, и S, где S или Р произвольно замещаются одним или несколькими оксо для обеспечения группы SO или SO2, РО или РО2); или бициклом, имеющим 7-10 участников кольца (4-9 атомов углерода и 1-3 гетероатома, выбранных из N, О, Р, и S, где S или Р произвольно замещаются одним или несколькими оксо для обеспечения группы SO или SO2, РО или PO2).
[063] Гетероциклил может быть углеродным радикалом или радикалом гетероатома. Примеры гетероциклических колец включают, без ограничений, пирролидинил, тетрагидрофуранил, дигидрофуранил,тетрагидротиэнил, тетрагидропиранил,
дигидропиранил, тетрагидротиопиранил, пиперидино, морфолино, тиоморфолино, тиоксанил, пиперазинил, гомо-пиперазинил, азетидинил, оксетанил, тиэтанил, гомопиперидинил, оксепанил, тиэпанил, оксазепинил, диазепинил, тиазепинил, 2-пирролинил, 3-пирролинил, индолинил, 2Н-пиранил, 4Н-пиранил, диоксанил, 1,3-диоксоланил, пиразолинил, дитианил, дитиоланил, дигидропиранил, дигидротиэнил, дигидрофуранил, пиразолидинилимидазолинил, имидазолидинил, 1,2,3,4-тетрагидроизо-хинолинил. Примеры гетероциклической группы, где 2 кольцевые атомы углерода замещаются оксо (=O) агентами являются пиримидиндионил и 1,1-диоксо-тиоморфолинил.
[064] Термин «гетероатом» относится к одной или нескольким атомам кислорода, серы, азота, фосфора или кремния, включая любую окисленную форму азота, серы или фосфора; кватернизованной форме любого основного азота; или замещенного азота гетероциклического кольца, например N (как в 3,4 дигидро 2Н пирролиле), NH (как в пирролидиниле) или NR (как в N-замещенном пирролидиниле).
[065] Термин «галоген» относится к F, Cl, Br или I.
[066] Термин «Н» относится к одинарному атому водорода. Этот радикал может быть присоединен, например, к атому кислорода для формирования гидроксильного радикала.
[067] Термин «D» или «2Н» относится к одинарному атому дейтерия. Один из этих радикалов может быть присоединен, например, к метиловой группе для формирования монодейтеризованной метиловой группы (-CDH2), два атомв дейтерия могут присоединяться к метиловой группе для формирования ди-дейтерированного метила (-CD2H), и три атома дейтерия могут присоединяться к метиловой группе для формирования метиловой группы, дейтеризованной тримараном (-CD3).
[068] Термин «N3» относится к агенту азида. Этот радикал может быть присоединен, например, к метиловой группе для формирования азидометана (азид метила, MeN3); или присоединяться к фенильной группе для формирования азида фенила (PhN3).
[069] Термин «арил», используемый отдельно или как часть большего агента, как в «аралкил», «аралкокси» или «арилоксиалкил», относится к моноциклическим, бициклическим, и трициклическим карбоциклическим кольцевым системам, имеющим в общей сложности 6-14 кольцевых участников, предпочтительно, 6-12 кольцевых участников, и более предпочтительно 6-10 кольцевых участников, где, по крайней мере, одно кольцо в системе является ароматическим, где каждое кольцо в системе, содержит 3-7 кольцевых участников, и которое имеет одну точку крепления к остальной части молекулы. Термин «арил» может быть использован наравне с термином «арильное кольцо». Примеры арильных колец включали бы фенил, нафтил и антрацен. Радикалы арила произвольно замещаются независимо одним или несколькими заместителями, описанными в настоящем документе.
[070] Термин «гетероарил», используемый в отдельности или как часть большего агента, как в «гетероаралкил» или «гетероарилалкокси», относится к системам моноциклического, бициклического и трициклического кольца, имеющего в общей сложности 5-14 кольцевых участников, предпочтительно, 5-12 кольцевых участников, и более предпочтительно 5-10 кольцевых участников, где, по крайней мере, одно кольцо в системе является ароматическим, по крайней мере, одно кольцо в системе содержит один или несколько гетероатомов, где каждое кольцо в системе содержит 5-7 кольцевых участников, и имеет одну точку крепления к остальной части молекулы. Термин «гетероарил» может быть использован наравне с термином «гетероарильное кольцо» или термин «гетероароматический». Радикалы гетероарил а произвольно замещаются независимо одним или несколькими заместителями, описанными в настоящем документе.
[071] Некоторые неограничивающие примеры колец гетероарила включают следующие моноциклы: 2-фуранил, 3-фуранил, N-имидазолил, 2-имидазолил, 4-имидазолил, 5-имидазолил, 3-изоксазолил, 4-изоксазолил, 5-изоксазолил, 2-оксазолил, 4-оксазолил, 5-оксазолил, N-пирролил, 2-пирролил, 3-пирролил, 2-пиридил, 3-пиридил, 4-пиридил, 2-пиримидинил, 4-пиримидинил, 5-пиримидинил, пиридазинил (например, 3-пиридазинил), 2-тиазолил, 4-тиазолил, 5-тиазолил, тетразолил (например, 5-тетразолил), триазолил (например, 2-триазолил и 5-триазолил), с 2 тиенилам, 3-тиенил, пиразолил (например, 2-пиразолил), изотиазолил, 1,2,3-оксадиазолил, 1,2,5-оксадиазолил, 1,2,4-оксадиазолил, 1,2,3-триазолил, 1,2,3-тиадиазолил, 1,3,4-тиадиазолил, 1,2,5-тиадиазолил, пиразинил, 1,3,5-триазинил и следующие бициклы: бензимидазолил, бензофурил, бензотиофенил, индолил (например, 2-индолил), пуринил, хинолинил (например, 2-хинолинил, 3-хинолинил, 4-хинолинил), и изохинолинил (например, 1-изохинолинил, 3-изохинолинил или 4-изохинолинил).
[072] Термины «карбокси» или «карбоксил», в отдельности или с другими терминами, например, «карбоксиалкил», относится к -СО2Н. Термин «карбонил», в отдельности или с другими терминами, например, «аминокарбонил», означает -(С=O)-.
[073] Термин «алкиламино» охватывает «N-алкиламино» и «N, N-диалкиламино», где аминогруппы независимо замещаются одним алкильным радикалом или двумя алкильными радикалами, соответственно. Некоторыми неограничивающими примерами алкиламино радикалов является «низшие алкиламино» радикалы, имеющие один или два алкильных радикала из одного - шести атомов углерода, присоединенных к атому азота. Подходящие алкиламино радикалы могут быть моно или диалкиламино, например N-метиламино, N-этиламино, N,N-диметиламино, N,N-диэтиламино и т.п.
[074] Термин «ариламино» относится к аминогруппам, замещенных одним или двумя арильными радикалами, например N-фениламино. Ариламино радикалы могут далее замещаться в арильной части кольца радикала.
[075] Термин «аминоалкил» относится к линейным или разветвленным алкильным радикалам, имеющим один к приблизительно десяти атомам углерода, любой из которых можно заместить одним или несколькими радикалами аминопласта. Более предпочтительные аминоалкил радикалы являются «низшими аминоалкильными» радикалами, имеющими один-шесть атомов углерода и один или несколько радикалов аминопласта. Примеры таких радикалов включают аминометил, аминоэтил, аминопропил, аминобутил и аминогексил.
[076] Термин «ненасыщенный» относится к агенту, имеющему одну или несколько единиц ненасыщенности.
[077] Термин «включающий» означает открытый конец, включая обозначенный компонент, но не исключая другие элементы.
[078] Если не указано иное, структуры, изображенные в настоящем документе, также предназначены для включения всех изомерных форм структуры (например, энантиомерные, диастеререомерные и геометрические (или конформационные)); например, конфигурации R и S для каждого центра асимметрии, изомеры двойной связи (Z) и (Е), и конформационные изомеры (Z) и (Е). Поэтому единственные стереохимические изомеры, а также энантиомерные, диастеререомерные и геометрические (или конформационные) смеси существующих соединений в рамках изобретения.
[079] Термин «таутомер» или «таутомерная форма» относится к структурным изомерам различных энергий, которые являются взаимозаменяемыми через низкоэнергетический барьер. Например, протонные таутомеры (также известные как прототропные таутомеры) включают взаимные преобразования через миграцию протона, например изомеризация имина-энамина и кето-энола. Таутомеры валентности включают взаимные преобразования путем реорганизации некоторых связующих электронов.
[080] Если не указано иное, все таутомерные формы соединений, раскрытых в настоящем документе, в рамках изобретения. Кроме того, если не указано иное, структуры, изображенные в настоящем документе, также включают соединения, отличающиеся только в присутствии одного или нескольких изотопически обогащенных атомов.
[081] Термин «пропрепарат» относится к соединению, преобразовывающемуся в естественных условиях в соединение по формуле (I) или (II). На такую трансформацию может влиять, например, гидролиз в крови или ферментативная трансформация формы пропрепарата в родительскую форму в крови или ткани. Пропрепаратами соединений, раскрытых в настоящем документе, могут быть, например, сложные эфиры. Сложные эфиры, которые могут быть использованы как пропрепараты в данном изобретении, являются сложными фениловыми эфирами, алифатическими сложными эфирами (С1-С24), ациклоксиметильными сложными эфирами, карбонатами, карбаматами и сложными эфирами аминокислот.Например, соединение, раскрытое в настоящем документе, содержит группу ОН, может быть ацилировано в этой позиции в форме пропрепарата. Другие формы пропрепарата включают фосфаты, например, фосфаты, возникающие из фосфонирования группы ОН на родительском соединении. Полное обсуждение пропрепаратов приводится в работе Т. Хигачи и В. Стелла, Пропрепарты как новые системы доставки, Издание 14 A.C.S. Symposium Series, Эдвард Б. Рош, редактор, Биообратимые носители в составлении препарата, American Pharmaceutical Association and Pergaamon Press, 1987, X. Раутио и др., Пропрепараты: Составление и клинические применения, Nature Review Drug Discovery, 2008, 7, 255-270, и С. Дж. Хекер и др., Пропрепараты фосфатов и фосфонатов, Journal of Medicinal Chemistry, 2008, 51, 2328-2345, каждый из которых включен в настоящий документ ссылкой.
[082] «Метаболит» относится к продукту, произведенному через метаболизм в организме указанного соединения или его соли. Метаболиты соединения могут быть идентифицированы с помощью известных обычных способов, и их активность определяется с помощью тестов, например, описанные в настоящем документе. Такие продукты могут возникать, например, в результате окисления, сокращения, гидролиза, амидирования, дезамидирования, эстерификации, деэстерификации, ферментативного расщепления и т.п., вводимого соединения. Соответственно, изобретение включает метаболиты соединений, раскрытых в настоящем документе, включая соединения, произведенные процессом, включающим контакт с соединением, раскрытым в настоящем документе, с млекопитающим в течение времени, достаточного для получения метаболического продукта.
[083] Стереохимические определения и соглашения, используемые в настоящем документе, обычно упоминаются в словаре под ред. С.П. Паркера, Словарь Химических терминов МакГроу-Хилл (1984) McGraw-Hill Book Company, Нью-Йорк; и Э. Элиэля и С. Вилен, «Стереохимия органических соединений», John Wiley & Sons, Inc., Нью-Йорк, 1994. Соединения, раскрытые в настоящем документе, могут содержать асимметричные или хиральные центры, и поэтому существовать в различных стереоизомерных формах. Предполагается, что все стереоизомерные формы соединений, раскрытых в настоящем документе, включая, без ограничений, диастереомеры, энантиомеры и атропоизомеры, а также их смеси, например рацемические смеси, являются частью данного изобретения. Многие органические соединения существуют в оптически активных формах, т.е. у них есть способность вращать плоскость плоскополяризованного света. В описании оптически активного соединения префиксы D и L или R и S используются для обозначения абсолютной конфигурации молекулы относительно ее хирального центра (оси). Префиксы d и 1 или (+) и (-) используются для обозначения признака вращения плоскополяризованного света соединением, при этом (-) или 1 означает, что соединение является левовращающим. Соединение, снабженное префиксом (+) или d, является правовращающим. Для данной химической структуры эти стереоизомеры идентичны за исключением того, что они являются зеркальным отображением друг друга. Определенный стереоизомер может также упоминаться как энантиомер, и смесь таких изомеров часто называют энантиомерной смесью. Смесь энантиомеров 50:50 упоминается как рацемическая смесь или рацемат, который может произойти, если не было стереовыбора или стереоспецифичности в химической реакции или процессе. Термины «рацемическая смесь» и «рацемат» относятся к эквимолекулярной смеси двух энантиомерных разновидностей, лишенных оптической активности.
[084] «Фармацевтически приемлемая соль» относится к органическим или неорганическим солям соединения, раскрытого в настоящем документе. Фармацевтически приемлемые соли известны в области применения. Например, С.М. Бердж и др. подробно описывают фармацевтически приемлемые соли в J. Pharmaceutical Sciences, 66: 1-19, 1977, включенный в настоящий документ ссылкой. Примеры фармацевтически приемлемых, нетоксичных солей включают, без ограничений, соли аминогруппы, сформированные с неорганическими кислотами, такими как соляная кислота, бромистоводородная кислота, фосфорная кислота, серная кислота и перхлорная кислота; или с органическими кислотами, такими как уксусная кислота, щавелевая кислота, малеиновая кислота, винная кислота, лимонная кислота, янтарная кислота или малоновая кислота; или при помощи других способов, используемых в области, например, ионный обмен. Другие фармацевтически приемлемые соли включают адипат, альгинат, аскорбат, аспартат, бенолсульфонат, бензоат, бисульфат, борат, бутират, камфорат, камфорсульфонат, цитрат, циклопентанпропионат, диглюконат, додецилсульфат, этансульфонат, формиат, фумарат, глюкогептонат, глицерофосфат, глюконат, гемисульфат, гептаноат, гексаноат, йодгидрат, 2 гидрокси этансульфонат, лактобионат, лактат, лаурат, лауриловый сульфат, малат, малеат, малонат, метансульфонат, 2-нафталенсульфонат, никотинат, нитрат, олеат, оксалат, пальмитат, памоат, пектинат, персульфат, 3-фенилпропионат, фосфат, пикрат, пивалат, пропионат, стеарат, сукцинат, сульфат, тартрат, роданид, р-толуенсульфонат, ундеканоат, соли валерата и т.п. Соли, полученные из надлежащих основ, включают щелочной металл, щелочноземельный металл, аммоний и соли N+ (С1-4 алкил)4. Настоящее изобретение также предполагает образование четвертичного основания любых основных групп соединений, содержащих азот, раскрытых в настоящем документе. Вода или маслорастворимые или растворимые продукты могут быть получены таким образованием четвертичного основания. Представительные соли щелочных или щелочноземельного металла включают натрий, литий, калий, кальций, магний и т.п. Далее фармацевтически приемлемые соли включают, в надлежащих случаях, нетоксичный аммоний, четвертичный аммоний и катионы амина, сформированные с помощью противоионов, таких как галид, гидроокись, карбоксилат, сульфат, фосфат, нитрат, С1-8 сульфонат и арилзамещенный сульфонат.
[085] «Сольват» относится к ассоциации или комплексу одной или нескольких растворимых молекул и соединения, раскрытого в настоящем документе. Примеры растворителей, формирующих сольваты, включают, без ограничений, воду, изопропиловый спирт, этанол, метанол, DMSO, этилацетат, уксусную кислоту и этаноламин. Термин «гидрат» относится к комплексу, где растворимая молекула является водой.
[086] Термин «блокирующая группа» или «БГ» относится к заместителю, обычно использующемуся для блокировки или защиты определенной функциональности при реакции других функциональных групп соединения. Например, «блокирующая группа аминопласта» является заместителем, присоединенным к аминогруппе, блокирующей или защищающей функциональность аминопласта в соединении. Подходящие блокирующие группы аминопласта включают ацетил, трифторацетил, t-бутокси-карбонил (ВОС, Boc), бензилоксикарбонил (CBZ, Cbz) и 9-флуорэнилметиленоксикарбонил (Fmoc). Точно так же «блокирующая группа гидрокси» относится к заместителю гидроксильной группы, блокирующей или защищающей функциональность гидрокси. Подходящие блокирующие группы включают ацетил и силил. «Блокирующая группа карбокси» относится к заместителю карбоксильной группы, блокирующей или защищающей функциональность карбокси. Общие блокирующие группы карбокси включают -CH2CH2SO2Ph, цианоэтил, 2-(триметилсилил)этил, 2-(триметилсилил)этокси-метил-1, 2-(р-толуенсульфонил) этил, 2-(р-нитрофенилсульфенил)-этил, 2-(дифенилфосфорино)-этил, нитроэтил и т.п. Для общего описания блокирующих групп и их использования, см. Т.В. Грин, Блокирующие группы в органическом синтезе, John Wiley & Sons, Нью-Йорк, 1991; и П.Й. Коциенский, Блокирующие группы, Thieme, Штутгарт, 2005.
ОПИСАНИЕ СОЕДИНЕНИЙ ИЗОБРЕТЕНИЯ
[087] Данное изобретение обеспечивает соединения замещенных пиразолонов, соли и их фармацевтические композиции, которые потенциально полезны в лечении заболеваний, состояний и расстройств, смоделированных киназами тирозина рецептора, особенно рецептора VEGFR, c-Met, Ron и/или Axl. В частности, данное изобретение обеспечивает соединения по Формуле (I):
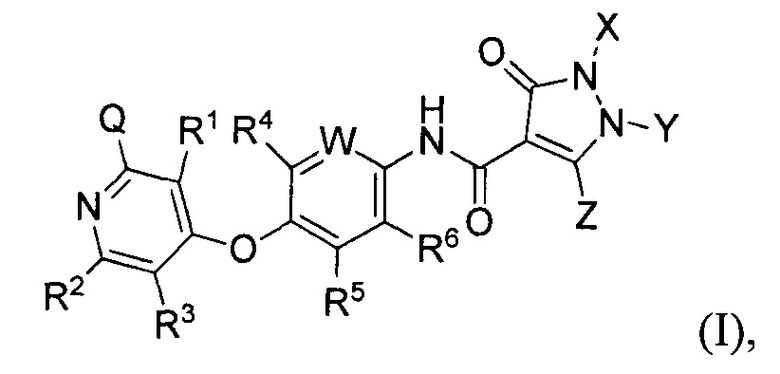
или стереоизомер, геометрический изомер, таутомер, N-оксид, сольват, гидрат, метаболит, фармацевтически приемлемую соль или пропрепарат, где каждый Q, R1, R2, R3, R4, R5, R6, W, X, Y и Z определяется в настоящем документе.
[088] В определенных воплощениях Q в формуле (I) является D, -N(Rc)C(=O)NRaRb, -N(Rc)C(=O)Rd, -C(=O)NRaRb, -N(Rc)S(=O)NRaRb, -N(Rc)S(=O)Ra, -N(Rc)S(=O)2NRaRb или -N(Rc)S(=O)2Ra;
W в формуле (I) является CR7 или N;
каждый из X, Y и Z в формуле (I) независимо Н, D, (С1-С6) алкил, (С3-C8) циклоалкил, - (С1-С4) алкилен - (С3-C8) циклоалкил, (С3-С7) гетероциклил, -(С1-С4) алкилен - (С3-С7) гетероциклил, (С6-С10) арил, 5-10-членный гетероарил, включающий 1, 2, 3 или 4 гетероатома, независимо выбранных из О, S и N, - (С1-С4) алкилен - (С6-С10) арил или - (С1-С4) алкилен - (5-10-членный гетероарил), где каждый (С1-С6) алкил, (С3-C8) циклоалкил, - (С1-С4) алкилен - (С3-C8) циклоалкил, (С3-С7) гетероциклил, - (С1-С4) алкилен -(С3-С7) гетероциклил, (С6-С10) арил, 5-10-членный гетероарил, - (С1-С4) алкилен - (С6-C10) арил и - (С1-С4) алкилен - (5-10-членный гетероарил), не замещен или произвольно замещен 1, 2, 3, 4 или 5 заместителей, независимо выбранных из D, F, Cl, Br, CN, (С2-С6) алкенил, (С2-С6) алкинил, ORa, NRaRb, - (С1-С4) алкилен-ORa и - (С1-С4) алкилен-NRaRb;
каждый из R1, R2, R3, R4, R5, R6 и R7 в формуле (I) независимо Н, D, F, Cl, Br, CN, N3, ORa, (С1-С6) алкил, (С1-С6) галоалкил, (С2-С6) алкенил или (С2-С6) алкинил;
каждый из Ra, Rb и Rc в формуле (I) независимо Н, (С1-С6) алифатический, (С1-С6) галоалкил, (С3-С6) циклоалкил, - (С1-С4) алкилен - (С3-С6) циклоалкил, (С3-С6) гетероциклил, - (С1-С4) алкилен - (С3-С6) гетероциклил, (С6-С10) арил, 5-10-членный гетероарил включающий 1, 2, 3 или 4 гетероатома, независимо выбранных из О, S и N, - (С1-С4) алкилен -(С6-С10) арил или - (С1-С4) алкилен - (5-10-членный гетероарил), где каждый (С1-С6) алифатический, (С1-С6) галоалкил, (С3-С6) циклоалкил, -(С1-С4) алкилен - (С3-С6) циклоалкил, (С3-С6) гетероциклил, - (С1-С4) алкилен - (С3-С6) гетероциклил, (С6-С10) арил, 5-10-членный гетероарил, -(С1-С4) алкилен - (С6-С10) арил и - (С1-С4), алкилен - (5-10-членный гетероарил) не замещается или произвольно замещается 1, 2, 3 или 4 заменителями, независимо выбранными из D, F, Cl, CN, N3, ОН, NH2, (С1-С6) галоалкил, (С1-С6) алкокси и (С1-С6) алкиламино; и
Rd в формуле (I) является D, (С3-C8) циклоалкил, - (С1-С4) алкилен - (С6-С10) арил, 5-10-членный гетероарил, включающий 1, 2, 3 или 4 гетероатома, независимо выбранных из О, S и N или - (С1-С4) алкилен - (5-10-членный гетероарил), где каждый (С3-C8) циклоалкил, - (С1-С4) алкилен - (С6-С10) арил, 5-10-членный гетероарил и - (С1-С4), алкилен - (5-10-членный гетероарил) не замещается или произвольно замещается 1, 2, 3 или 4 заменителями, независимо выбранными из D, F, Cl, Br, CN, ORa, NRaRb, (С1-С6) алкил, (С2-С6) алкенил, (С2-С6) алкинил, - (С1-С4) алкилен-ORa и - (С1-С4) алкилен-NRaRb.
[089] В других воплощениях каждый из Ra, Rb и Rc в формуле (I) независимо Н, (С1-С6) алифатический, (С1-С6) галоалкил, (С3-С6) циклоалкил, (С3-С6) гетероциклил, - (С1-С4) алкилен - (С3-С6) гетероциклил, (С6-С10) арил, 5-10-членный гетероарил, включающий 1, 2, 3 или 4 гетероатома, независимо выбранных из О, S и N, - (С1-С4) алкилен -(С6-С10) арил или - (С1-С4) алкилен - (5-10-членный гетероарил), где каждый (С1-С6) алифатический, (С1-С6) галоалкил, (С3-С6) циклоалкил, (С3-С6) гетероциклил, - (С1-С4) алкилен - (С3-С6) гетероциклил, (С6-С10) арил, 5-10-членный гетероарил, - (С1-С4) алкилен - (С6-С10) арил и - (С1-С4), алкилен - (5-10-членный гетероарил) не замещается или произвольно замещается 1, 2, 3 или 4 заместителями, независимо выбранными из D, F, Cl, CN, N3, ОН, NH2, (С1-С6) галоалкил, (С1-C6) алкокси и (С1-C6) алкиламино; и
Rd в формуле (I) является D, (C3-C8) циклоалкил, - (С1-С4) алкилен - (С6-С10) арил, 5-10-членный гетероарил, включающий 1, 2, 3 или 4 гетероатома, независимо выбранных из О, S и N или - (С1-С4) алкилен - (5-10-членный гетероарил), где каждый (С3-C8) циклоалкил, - (С1-С4) алкилен - (С6-С10) арил, 5-10-членный гетероарил и - (С1-С4) алкилен - (5-10-членный гетероарил) не замещается или произвольно замещается 1, 2, 3 или 4 заменителями, независимо выбранными из D, F, Cl, Bra, CN, ORa, NRaRb, (C2-C6) алкенил, (С2-С6) алкинил, - (С1-С4) алкилен-ORa и - (С1-С4) алкилен-NRaRb.
[090] В других воплощениях Q в формуле (I) является -N(Rc)C(=O)NRaRb, -N(Rc)C(=O)Rd или - С(=O)NRaRb
[091] В других воплощениях каждый из X, Y и Z в формуле (I) независимо (С1-С4) алкил, (С3-С6) циклоалкил, - (C1-C2) алкилен - (С3-С6) циклоалкил, (С3-С6) гетероциклил, - (С1-C2) алкилен - (С3-С6) гетероциклил, фенил, 5-10-членный гетероарил, включающий 1, 2, 3 или 4 гетероатома, независимо выбранных из О, S и N, - (С1-С2) фенил алкилен или - (С1-С2) алкилен - (5-10-членный гетероарил), где каждый (С1-С4) алкил, (С3-С6) циклоалкил, - (С1-С2) алкилен - (С3-С6) циклоалкил, (C3-С6) гетероциклил, -(С1-С2) алкилен - (С3-С6) гетероциклил, фенил, 5-10-членный гетероарил, -(С1-С2) фенил алкилен и - (С1-C2) алкилен - (5-10-членный гетероарил) не замещаются или произвольно замещаются 1, 2, 3 или 4 заместителями, независимо выбранными из D, F, Cl, CN, (С2-С4) алкенил, (С2-С4) алкинил, ORa, NRaRb, - (С1-С2) алкилен-ORa и - (C1-C2) алкилен-NRaRb.
[092] В других воплощениях каждый из R1, R2, R3, R4, R5, R6 и R7 в формуле (I) независимо Н, D, F или Cl.
[093] В других воплощениях каждый из Ra, Rb и Rc в формуле (I) независимо Н, (С1-С4) алкил, (С1-С4) галоалкил, (С3-С6) циклоалкил, - (С1-С2) алкилен - (С3-С6) циклоалкил, (С3-С6) гетероциклил или - (С1-С2) алкилен - (С3-С6) гетероциклил, где каждый (С1-С4) алкил, (С1-С4) галоалкил, (С3-С6) циклоалкил, - (С1-С2) алкилен - (С3-С6) циклоалкил, (С3-С6) гетероциклил и - (С1-С2), алкилен - (С3-С6) гетероциклил не замещается или произвольно замещается 1, 2, 3 или 4 заместителями, независимо выбранными из D, F, Cl, CN, N3, ОН, NH2, (С1-C3) галоалкил, (С1-С3) алкокси и (С1-С3) алкиламино.
[094] В других воплощениях Rd в формуле (I) (С3-С6) циклоалкил, где (С3-С6) циклоалкил не замещается или произвольно замещается 1, 2, 3 или 4 заместителями, независимо выбранными из D, F, CN, ORa, NRaRb, (С1-C3) алкил, (С2-С4) алкенил, (С2-С4) алкинил, - (С1-С2) алкилен-ORa и- (С1-С2) алкилен-NRaRb.
[095] В других воплощениях каждый из X, Y и Z в формуле (I) независимо Н, D, СН3, метиловая группа замещается 1, 2 или 3 атомами дейтерия, этиловая, пропиловая, изопропиловая, фениловая или фенильная группа замещается 1, 2, 3, 4 или 5 заместителями, независимо выбранными из D, F и Cl.
[096] В других воплощениях Q в формуле (I):
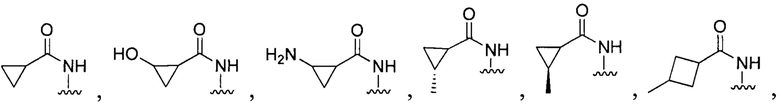
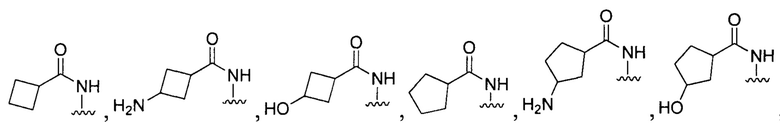
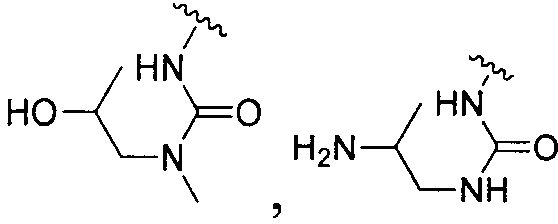 или
или 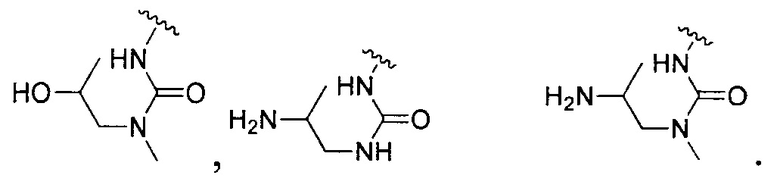
[097] В другом воплощении изобретение обеспечивает соединения, имеющие формулу (II):
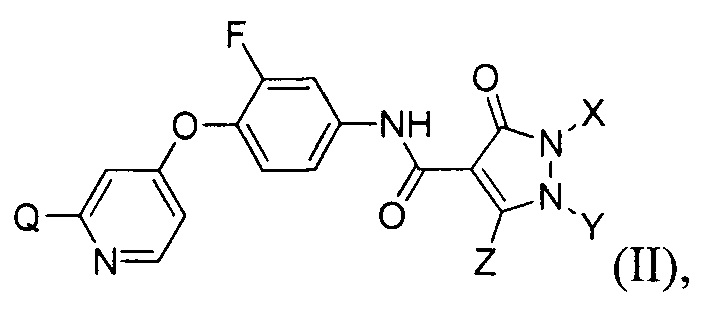
где каждый из Q, X, Y и Z определяется в настоящем документе.
[098] В определенных воплощениях Q в формуле (II) является -N(Rc)C(=O)NRaRb, -N(Rc)C(=O)Rd, -N(Rc)S(=O)NRaRb, -N(Rc)S(=O)Ra, -N(Rc)S(=O)2NRaRb, -N(Rc)S(=O)2Ra или -C(=O)NRaRb;
каждый из X, Y и Z в формуле (II) независимо Н, D, (С1-С6) алкил, (С3-C8) циклоалкил, (С3-С7) гетероциклил, (С6-С10) арил, 5-10-членный гетероарил, включающий 1, 2, 3 или 4 гетероатома, независимо выбранных из О, S и N, - (С1-С4) алкилен - (С3-C8) циклоалкил, - (С1-С4) алкилен - (С3-С7) гетероциклил, - (С1-С4) алкилен - (С6-С10) арил или - (С1-C4) алкилен - (5-10-членный гетероарил), где каждый алкил (С1-С6), (С3-C8) циклоалкил, (С3-С7) гетероциклил, (С6-С10) арил, 5-10-членный гетероарил, - (С1-С4) алкилен - (С3-C8) циклоалкил, - (С1-С4) алкилен - (С3-С7) гетероциклил, -(С1-С4) алкилен - (С6-С10) арил и - (С1-С4), алкиленом - (5-10-членный гетероарил) произвольно замещается 1, 2, 3, 4 или 5 заместителями, независимо выбранными из D, F, Cl, Br, CN, (С2-С6) алкенил, (С2-С6) алкинил, ORa, NRaRb, - (С1-С4) алкилен-ORa и - (С1-С4) алкилен-NRaRb;
каждый из Ra, Rb и Rc в формуле (II) независимо Н, (С1-С6) алкил, (С3-С6) циклоалкил, (С3-С6) гетероциклил, (С6-С10) арил, 5-10-членный гетероарил, включающий 1, 2, 3 или 4 гетероатомов, независимо выбранных из О, S и N, - (С1-С4) алкилен - (С3-С6) циклоалкил, - (С1-С4) алкилен - (С3-С6) гетероциклил, - (С1-С4) алкилен - (С6-С10) арил или - (С1-С4) алкилен - (5-10-членный гетероарил), где каждый алкил (С1-С6), (С3-С6) циклоалкил, (С3-С6) гетероциклил, (С6-С10) арил, 5-10-членный гетероарил, - (С1-С4) алкилен - (С3-С6) гетероциклил, - (С1-С4) алкилен - (С6-С10) арил и - (С1-С4), алкилен - (5-10-членный гетероарил)произвольно замещается 1, 2, 3 или 4 заместителями, независимо выбранными из D, F, Cl, CN, N3, ОН, NH2, (C1-С6) галоалкил, (С1-С6) алкокси и (С1-С6) алкиламино; и
Rd в формуле (II) (С3-C8) циклоалкил, где (С3-C8) циклоалкил произвольно замещается 1, 2, 3 или 4 заместителями, независимо выбранными из D, F, Cl, ОН, NH2, (С1-С6) алкил, (С1-С6) алкокси и (С1-С6) алкиламино.
[099] В другом воплощении Q в формуле (II) -N(Rc)С(=O)NRaRb, -N(Rc)С(=O)Rd, -N(Rc)S(=O)NRaRb, -N(Rc)S(=O)2NRaRb, -N(Rc)S(=O)2Ra или -С(=O)NRaRb; и
Rd в формуле (II) (С3-C8) циклоалкил, где (С3-C8) циклоалкил произвольно замещается 1, 2, 3 или 4 заместителями, независимо выбранными из D, F, Cl, ОН, NH2, (С1-С6) алкокси и (С1-С6) алкиламино.
[0100] В другом воплощении Q в формуле (II) -N(Rc)С(=O)NRaRb, -N(Rc)С(=O)Rd или -С(=O)NRaRb.
[0101] В другом воплощении каждый из X, Y и Z в формуле (II) независимо Н, D, (С1-С4) алкил или фенил, где каждый (С1-С4) алкил и фенил произвольно замещается 1, 2, 3, 4 или 5 заместителями, независимо выбранными из D, F и Cl.
[0102] В другом воплощении каждый из Ra, Rb и Rc в формуле (II) независимо Н, (С1-С4) алкил, (С3-С6) циклоалкил, (С3-С6) гетероциклил, -(С1-С2) алкилен - (С3-С6) циклоалкил или - (С1-С2) алкилен - (С3-С6) гетероциклил, где каждый (С1-С4) алкил, (С3-С6) циклоалкил, (С3-С6) гетероциклил, - (С1-С2) алкилен - (С3-С6) циклоалкил и - (С1-С2) алкилен -(С3-С6) гетероциклил произвольно замещается 1, 2, 3 или 4 заместителями, независимо выбранными из D, F, Cl, CN, N3, ОН, NH2, (С]-С6) галоалкил, (С1-С6) алкокси и (С1-С6) алкиламино.
[0103] В другом воплощении каждый из X, Y и Z в формуле (II) независимо Н, D, Me, CH2D, CHD2, CD3, этил, пропил, изопропил, фенил или фенильная группа, которые произвольно замещаются 1, 2, 3, 4 или 5 заместителями, независимо выбранными из D, F и Cl.
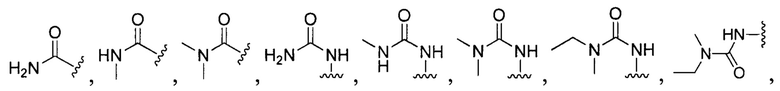
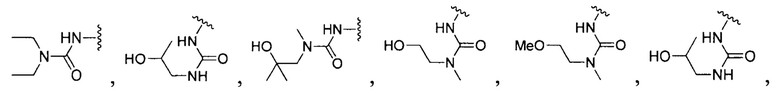
[0104] В другом воплощении Q в формуле (II):
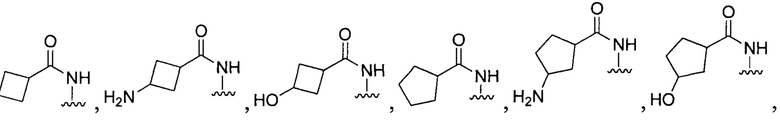
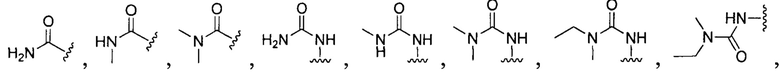
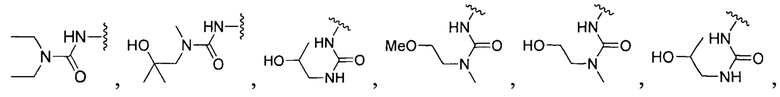
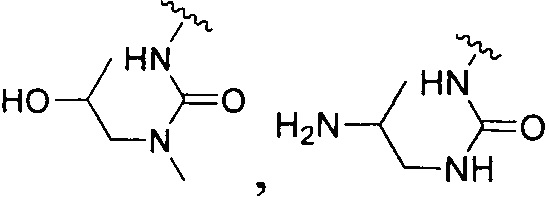 или
или 
[0105] В некоторых воплощениях неограничивающие примеры соединений, раскрытых в настоящем документе, и их фармацевтически приемлемые соли и сольваты, показаны далее:

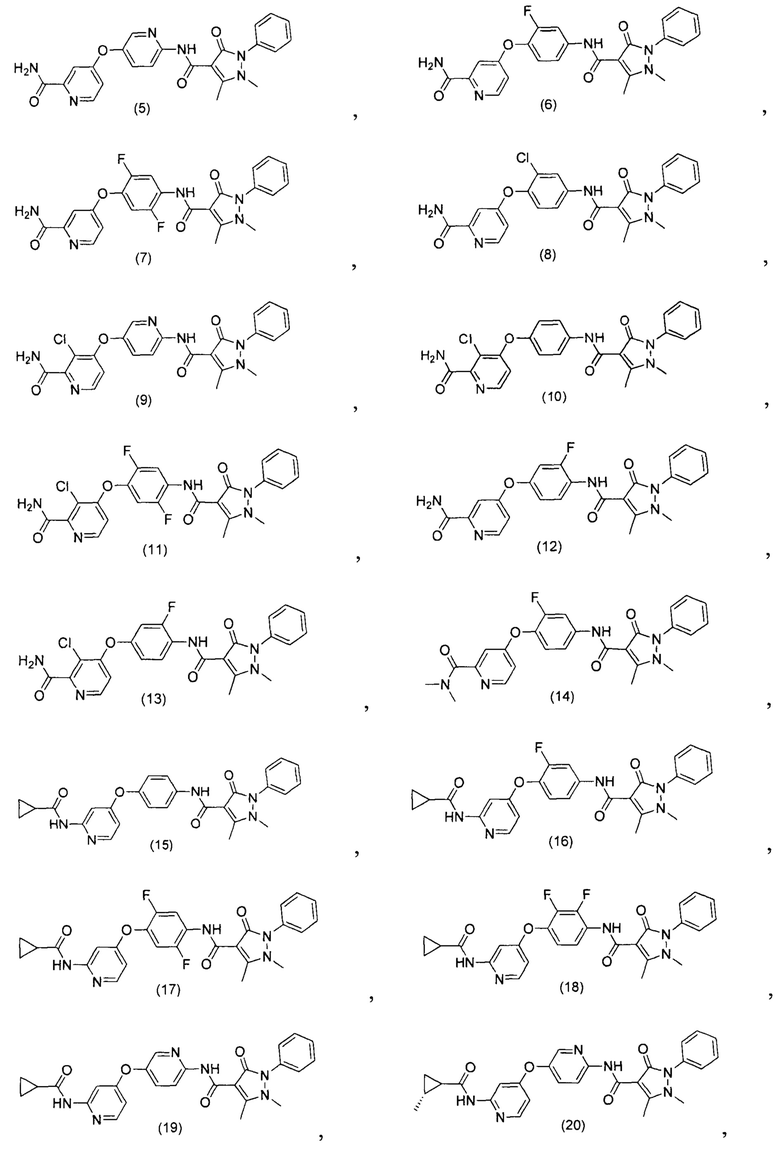
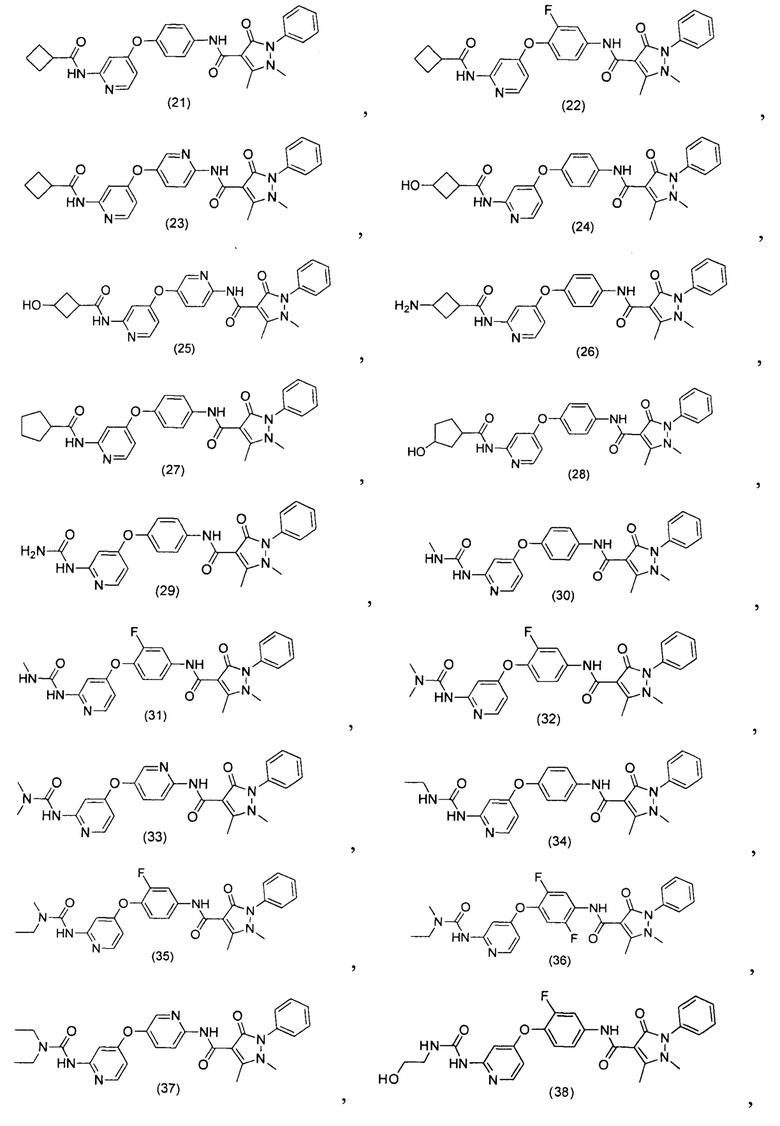
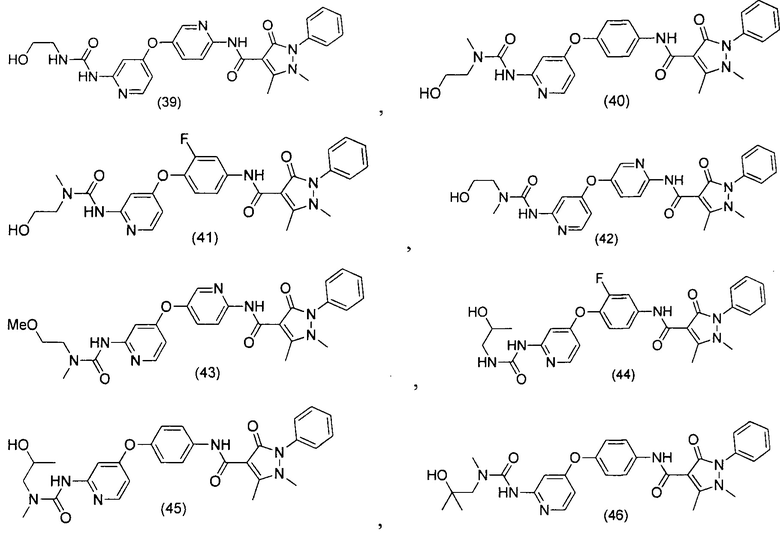
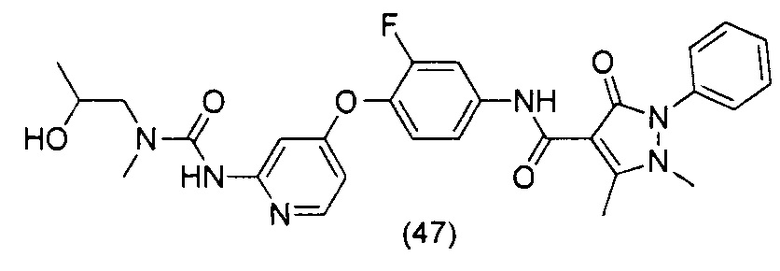 или
или 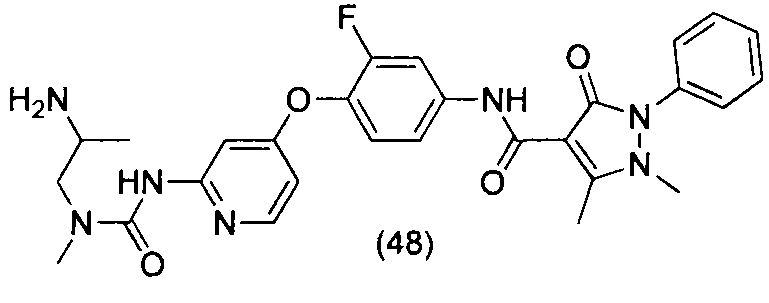
[0106] Данное изобретение также включает использование соединения, раскрытого в настоящем документе, или его фармацевтически приемлемую соль, в изготовлении лекарственных препаратов для лечения, остро или хронически, гиперпролиферативного состояния и/или ангиогенеза, включая, как описано выше. Соединения, раскрытые в настоящем документе, используются при изготовлении лекарственных препаратов для лечения рака. Соединения, раскрытые в настоящем документе, также используются при изготовлении лекарственных препаратов, чтобы уменьшить или предотвратить расстройства посредством ингибирования киназ протеина. Данное изобретение включает фармацевтическую композицию, включающую терапевтически эффективный объем соединения по Формуле (I) или (II) совместно минимум с одним фармацевтически приемлемым носителем, вспомогательным агентом или разжижителем.
[0107] Данное изобретение также включает способ лечения гиперпролиферативного состояния и ангиогенеза у субъекта, у которого присутствуют или который склонен к возникновению таких расстройств, способ, включающий лечение субъекта терапевтически эффективным объемом соединения по Формуле (I) или (II).
[0108] Если не указано иное, все стереоизомеры, геометрические изомеры, таутомеры, сольваты, гидраты, метаболиты, соли и фармацевтически приемлемые пропрепараты соединений, раскрытых в настоящем документе, находятся в рамках изобретения.
[0109] В определенных воплощениях соль является фармацевтически приемлемой солью. Фраза, «фармацевтически приемлемая» указывает, что вещество или соединение должны быть совместимыми химически и/или токсикологическим образом с другими компонентами, включающими формулу изобретения, и/или с млекопитающим, подвергнутым лечению.
[0110] Соединения, раскрытые в настоящем документе также, включают соли таких соединений, которые не обязательно являются фармацевтически приемлемыми солями, и которые могут быть полезны как промежуточные звенья для подготовки и/или очищения соединений по Формуле (I) или (II) и/или для отделения энантиомеров соединений по Формуле (I) или (II).
[0111] Желаемая соль может быть подготовлена любым подходящим способом, доступным в области, например, обработкой свободной основы неорганической кислотой, такой как соляная кислота, бромистоводородная кислота, серная кислота, азотная кислота, фосфорная кислота и т.п., или органической кислотой, такой как уксусная кислота, малеиновая кислота, янтарная кислота, миндальная кислота, фумаровая кислота, малоновая кислота, пировиноградная кислота, щавелевая кислота, гликолевая кислота, салициловая кислота; пиранозидильной кислотой, такой как глюкуроновая кислотная или галактуроновая кислота; альфа-оксикислотой, такой как лимонная кислота или винная кислота; аминокислотой, такой как аспарагиновая кислота или глутаминовая кислота; ароматической кислотой, такой как бензойная кислота или коричная кислота; сульфокислотой, такой как р-толуенсульфоновая кислота или этансульфоновая кислота и т.п.
СОСТАВ, ФОРМУЛЫ И ВВЕДЕНИЕ СОЕДИНЕНИЙ, РАСКРЫТЫХ В НАСТОЯЩЕМ ДОКУМЕНТЕ
[0112] В одном аспекте, показанном в настоящем документе, фармацевтические композиции, включающие соединение по формуле (I) или (II) или соединение, перечисленное в Таблице 1; и фармацевтически приемлемый носитель, вспомогательный агент или средство. Объем соединения в фармацевтических композициях, раскрытых в настоящем документе, является эффективным для обнаружимого ингибирования киназы протеина в биологической пробе или у пациента.
[0113] Следует также отметить, что определенные соединения, раскрытые в настоящем документе, могут существовать в свободной форме для лечения или по мере необходимости как фармацевтически приемлемые производные. Некоторые неограничивающие примеры фармацевтически приемлемых производных включают фармацевтически приемлемые пропрепараты, соли, сложные эфиры, соли таких сложных эфиров или любой другой аддукт или дериватив, который после введения пациенту обеспечивает, прямо или косвенно, соединение, как описано в настоящем документе, или его метаболит или остаток.
[0114] Как описано выше, фармацевтические композиции или фармацевтически приемлемые композиции, раскрытые в настоящем документе, дополнительно, включают фармацевтически приемлемый носитель, вспомогательный или средство, который, как используется в настоящем документе, включает все растворители, разжижители, или другое жидкие агенты, средства распыления или взвести, поверхностно-активные вещества, изотонические агенты, сгущающие или эмульгирующие вещества, консерванты, твердые связующие материалы, смазки и т.п., в зависимости от определенной желаемой формы дозировки. Ремингтон: Наука и практика фармации, 21 выпуск, 2005 г., редактор Д.Б. Трой, Lippincott Williams & Wilkins, Филадельфия, и Энциклопедия фармацевтической технологии, редакторы Дж. Сварбрик и Дж. К. Бойлан, 1988-1999, Marcel Dekker, Нью-Йорк, содержание которых включено посредством ссылок в настоящий документ, описывают различные носители, используемые в формуле изобретения фармацевтически приемлемых композиций и известных способов для их изготовления. В той мере, в которой любая стандартная среда носителя несовместима с соединениями, раскрытыми в настоящем документе, например путем любого нежелательного биологического эффекта или иного вредного взаимодействия с любым другим компонентом фармацевтически приемлемой композиции, его использование находится в рамках данного изобретения.
[0115] Некоторые неограничивающие примеры материалов, которые могут служить фармацевтически приемлемыми носителями, включают иониты, глинозем, стеарат алюминия, лецитин, протеины сыворотки, такие как человеческий сывороточный альбумин; буфферизированные вещества, такие как фосфаты, глицин, сорбиновая кислота, или калиевый сорбат, частичные глицеридные смеси насыщенных растительных жирных кислот, вода, соли или электролиты, такие как сульфат протамина, вторичный кислый фосфорнокислый натрий, вторичный кислый фосфорнокислый калий, хлорид натрия, соли цинка, коллоидная окись кремния, трисиликат магния, поливинила пирролидон, полиакрилаты, воски, полимеры полиэтилен-полиоксипропилен-блок, шерстяной жир; сахар, например, лактоза, глюкоза и сахароза; крахмалы, такие как кукурузный крахмал и картофельный крахмал; целлюлоза и ее производные, такие как карбоксиметилцеллюлоза натрия, этилцеллюлоза и ацетат целлюлозы; порошкообразный трагакант; солод; желатин; тальк; наполнители, такие как масло какао и парафин; масла, такие как арахисовое масло, хлопковое масло, сафлоровое масло, кунжутное масло, оливковое масло, кукурузное масло и соевое масло; гликоли, такие как пропиленгликоль или полиэтиленгликоль; сложные эфиры, такие как этилолеат и лаурат этила; агар; буферные вещества, такие как гидроксид магния и гидроксид алюминия; альгиновая кислота; вода без пирогенов; изотонический раствор; раствор Рингера; этиловый спирт, и буферные растворы фосфата; а также другие нетоксичные совместимые смазки, такие как сульфат лаурила натрия и стеарат магния; а также красящие агенты, разделительные агенты, покрывающие агенты, подсластители, ароматизаторы и отдушки, консерванты и антиокислители могут также присутствовать в составе, по решению разработчика рецептуры.
[0116] Соединения, раскрытые в настоящем документе, могут вводиться перорально, парентерально, ингаляцинным спреем, местно, ректально, через нос, щечно, вагинально или через имплантированный резервуар. Термин «парентеральный», как используется в настоящем документе, включает подкожную, внутривенную, внутримышечную, внутрисуставную, внутрисиновиальную, внутригрудинную, подоболочечную, внутриглазную, внутрипеченочную, внтуриочаговую и внутричерепную инъекцию или инфузию. Преимущественно, соединения вводятся перорально, внутрибрюшинно или внутривенно. Стерильные инъекционные формы соединений, раскрытых в настоящем документе, могут быть водной или маслянистой взвесью. Эти взвеси могут быть сформулированы согласно способам, известным в области, с использованием подходящеих распыливающих или смачивающих средств и суспендирующих агентов. Стерильный инъекционный препарат может также быть стерильным инъекционным растворм или взвесью в нетоксичном парентерально приемлемом разжижителе или растворителе, например, раствор в 1,3-бутандиоле. В качестве приемлемых носителей и растворителей, которые могут использоваться, выступает вода, раствор Рингера и изотонический раствор хлорида натрия. Кроме того, стерильные нелетучие масла традиционно используются как растворимая или суспендирующая среда.
[0117] С этой целью может использоваться любое мягкое нелетучее масло, включая синтетические моно- или диглицериды. Жирные кислоты, такие как олеиновая кислота и ее деривативы глицерида полезны при изготовлении инъекций, также как натуральные фармацевтически приемлемые масла, такие как оливковое масло или касторовое масло, особенно их полиоксиэтилированные версии. Эти масляные растворы или взвеси могут также содержать длинноцепочечный спиртовой разжижитель или дисперсант, например, карбоксиметилцеллюлоза или аналогичные диспергирующие вещества, обычно использующиеся в составлении фармацевтически приемлемых форм дозировки, включая эмульсии и суспензии. Другие обычно используемые сурфактанты, такие как твины, спаны и другие эмульгирующие вещества или усилители биоаккумулирования, обычно использующиеся в изготовлении фармацевтически приемлемых твердых, жидких или других форм дозировки, могут также использоваться в целях составления.
[0118] Фармацевтически приемлемыми композициями, раскрытыми в настоящем документе, могут перорально вводиться в любой приемлемой форме пероральной дозировки, включая, без ограничений, капсулы, таблетки, водные суспензии или растворы. В случае таблеток для перорального применения, обычно используемые носители включают кукурузный крахмал и лактозу. Смазывающие вещества, такие как стеарат магния, также обычно добавляются. Для перорального приема в форме капсулы используемые разжижители включают лактозу и высушенный кукурузный крахмал. Когда для перорального применения требуются водные суспензии, активный ингредиент объединяется с эмульгирующими и суспендирующими агентами. При желании добавляется определенный подсластитель, ароматизатор или краситель.
[0119] Также фармацевтически приемлемые композиции, раскрытые в настоящем документе, могут применяться в форме свечей для ректального введения. Они могут быть изготовлены путем смешивания агента с подходящим нераздражающим наполнителем, который является твердым при комнатной температуре, но становится жидким при ректальной температуре и поэтому будет таять в прямой кишке для высвобождения препарата. Такие материалы включают масло какао, воск и полиэтиленгликоли.
[0120] Фармацевтически приемлемые композиции, раскрытые в настоящем документе, могут также вводиться местно, особенно когда цель лечения включает области или органы, легкодоступные для местного применения, включая заболевания глаза, кожи или нижнего кишечного тракта. Подходящие актуальные составы легко изготовить для каждой из этих областей или органов.
[0121] Местное применение для нижнего кишечного тракта может осуществляться в виде ректального суппозитория (см. выше), или в виде раствора для промывания желудка. Также могут использоваться кожные пластыри для местного применения. Для местного применения фармацевтически приемлемые композиции могут быть выполнены в виде мази, содержащей активный компонент, взвешенный или растворенный в одном или нескольких носителях. Носители для местного применения соединений, раскрытых в настоящем документе, включают, без ограничений, масло, вазелиновое масло, белый вазелин, пропиленгликоль, полиэтиленоксид, полиоксипропилен, эмульгирующий воск и воду. Также фармацевтически приемлемые композиции могут быть выполнены в виде лосьона или крема, содержащего активные компоненты, суспендированные или растворенные в одном или нескольких фармацевтически приемлемых носителях. Подходящие носители включают, без ограничений, масло, сорбитанмоностеарат, полисорбат 60, цетиловый воск сложных эфиров, цетеариловый спирт, 2-октилдодеканол, бензиловый спирт и воду.
[0122] Для глазного применения фармацевтически приемлемые композиции могут быть выполнены, например, в виде микронизированных суспензий в изотоническом растворе, стерильном физрастворе с отрегулированным рН или другом водном растворе. Или, преимущественно, как раствор в изотоническом растворе, стерильном физрастворе с отрегулированным рН или другом водном растворе или с или без консервантов, таких как хлорид бензилалконий. Также для глазного применения, фармацевтически приемлемые составы могут быть выполнены в виде мази, такой как вазелин. Фармацевтически приемлемые композиции, раскрытые в настоящем документе, могут также вводиться в виде носового аэрозоля или ингаляция. Такие составы изготовляются согласно способам, известным в области фармацевтического составления, и могут быть изготовлены как растворы в физрастворе, используя бензиловый спирт или другие подходящие консерванты, поглощающие вещества для улучшения бионакопления, фторуглероды и/или другие стандартные растворы или диспергирующие вещества.
[0123] Жидкие формы для перорального применения включают, без ограничений, фармацевтически приемлемые эмульсии, микроэмульсии, растворы, суспензии, сиропы и эликсиры. В дополнение к активным соединениям, жидкие формы могут содержать инертные разжижители, обычно используемые в области, например, воду или другие растворители, растворяющие агенты и эмульгаторы, такие как этиловый спирт, изопропиловый спирт, этиловый эфир угольной кислоты, этилацетат, бензиловый спирт, бензилбензоат, пропиленгликоль, гликоль с 1,3-бутиленом, диметилформамид, масла (в частности хлопковоесемя, арахисовое, кукурузное масло, жмых зародышей кукурузы, оливковое масло, касторовое и кунжутное масло), глицерин, тетрагидрофуриловый спирт, полиэтиленгликоли и сложные эфиры жирных кислот сорбитана и их смеси. Помимо инертных разжижителей, пероральные составы могут также включать вспомогательные агенты, такие как смачивающие агенты, эмульгирующие и суспендирующие агенты, подсластители, ароматизаторы и отдушки.
[0124] Инъекционные препараты, например стерильные инъекционные водные или маслянистые суспензии, могут быть составлены согласно известному способу, с использованием подходящих распыляющих или смачивающих агентов и суспендирующих агентов. Стерильный инъекционный препарат может также быть стерильным инъекционным растворм, суспензией или эмульсией в нетоксичном парентеральным образом приемлемом разжижителе или растворителем, например, как раствор в 1,3-бутандиолом. В качестве приемлемых носителей и растворителей, которые могут использоваться, применяется вода, раствор Рингера, ФСША, и изотонический раствор хлорида натрия. Кроме того, стерильные нелетучие масла традиционно используются как растворимая или суспендирующая среда. С этой целью может использоваться любое мягкое нелетучее масло, включая синтетические моно- или диглицериды. Кроме того, жирные кислоты, такие как олеиновая кислота, используются в подготовке инъекционных препаратов.
[0125] Инъекционные составы могут стерилизоваться, например, фильтрацией через антибактериальный фильтр или путем слияния стерилизующих веществ в форме стерильных твердых составов, которые могут быть растворены или распылены в стерильной воде или в другой стерильной инъекционной среде до использования. Для продления эффекта соединения данного изобретения часто желательно замедлить поглощение соединения из подкожной или внутримышечной инъекции. Это может быть выполнено при помощи жидкой суспензии из кристаллического или аморфного материала с плохой водной растворимостью. В этом случае скорость поглощения соединения зависит от скорости растворения, что, в свою очередь, может зависеть от размера и формы кристаллов. Также растворение или суспендирование соединения в масляном носителе выполняет задержанное поглощение формы соединения, вводимого парентерально.
[0126] Инъекционные формы депо создаются путем формирования микрокапсульных матриц соединения в биоразлагаемых полимерах, таких как полигликолид полилактида. В зависимости от отношения соединения к полимеру и природы определенного используемого полимера, можно управлять скоростью высвобождения соединения. Примеры других биоразлагаемых полимеров включают поли(орто-эфиры) и поли(ангидриды). Инъекционные формы депо также изготавливаются путем удерживания соединения в липосомах или микроэмульсиях, которые совместимы с тканями организма.
[0127] Составы для ректального или вагинального введения предпочтительно являются свечами, которые могут быть изготовлены путем смешивания соединений, раскрытых в настоящем документе, с подходящим нераздражающими наполнителями или носителями, такими как масло какао, полиэтиленгликоль или парафин, которые являются твердыми при температуре окружающей среды, но тают при температуре тела и тают в прямой кишке или вагинальной впадине, и высвобождают активное соединение.
[0128] Тверды формы для перорального приема включают капсулы, таблетки, пилюли, порошки и гранулы. В таких твердых формах активное соединение смешивается, по крайней мере, с одним инертным, фармацевтически приемлемым наполнителем или носителем, например, цитратом натрия или дикальция фосфатом и/или: а) наполнителями или расширителями, такими как крахмалы, лактоза, сахароза, глюкоза, маннит и кремневая кислота; b) связующими веществами, такими как карбоксиметилцеллюлоза, альгинаты, желатин, поливинилпирролидинон, сахароза и камедь; с) увлажнителями, такими как глицерин; d) разрыхлителями, такими как агар-агар, карбонат кальция, картофельный крахмал или крахмал тапиоки, альгиновая кислота, определенные силикаты и карбонат натрия; е) ингибиторами растворов, такими как керосин; f) акселераторыами абсорбции, такими как четвертичные аммониевые основания; g) смачивающими агентами, такими как цетиловый спирт и моностеарат глицерина; h) абсорбентами, такими как каолиновая и бентонитовая глина; и i) смазками, такими как тальк, стеарат кальция, стеарат магния, твердые полиэтиленгликоли, сульфат лаурила натрия, и их смеси. В случае капсул, таблеток и пилюль, форма может также включить буферные вещества.
[0129] Твердые соединения подобного типа могут также использоваться как наполнители в мягких и твердых капсулах из желатина с помощью таких наполнителей как лактоза или молочный сахар, а также высокомолекулярных полиэтиленгликолей и т.п. Твердые формы таблеток, драже, капсул, пилюль и гранул могут быть изготовлены с покрытиями и оболочками, такими как кишечнорастворимые покрытия и другие покрытия, известные в фармацевтической сфере. Они могут произвольно содержать замутняющие агенты и могут также иметь состав, что они высвобождают активный ингредиент только или преимущественно в определенной части кишечного тракта, произвольно, с задержкой. Примеры вложенных составов, которые могут использоваться, включают полимерные вещества и воски. Твердые составы подобного типа могут также использоваться как наполнители в мягких и твердых капсулах из желатина с помощью таких наполнителей в качестве лактозы или молоного сахара, а также высокомолекулярных полиэтилен гликолей и т.п.
[0130] Активные соединения могут также быть в микрокапсульной форме с одним или несколькими наполнителями, как отмечено выше. Тверды формы таблеток, драже, капсул, пилюль и гранул могут быть изготовлены с покрытиями и оболочками, такими как кишечнорастворимые покрытия, покрытия с контролем высвобождения и другие покрытия, известные в фармацевтической области. В таких твердых формах активное соединение можно включать, по крайней мере, один инертный разжижитель, такой как сахароза, лактоза или крахмал. Такие формы могут также включать, как правило, дополнительные вещества кроме инертных разжижителей, например, таблеточные смазки и другие вспомогательные вещества, например, стеарат магния и микрокристаллическая целлюлоза. В случае капсул, таблеток и пилюль, формы выпуска могут также включать буферные вещества. Они могут произвольно содержать восстанавливающие агенты и могут также иметь состав, при котором активный ингредиент высвобождается только, или преимущественно, в определенной части кишечного тракта, произвольно, с задержкой. Примеры вложенных составов, которые могут использоваться, включают полимерные вещества и воски.
[0131] Формы выпуска для местного или трансдермального введения соединения, раскрытого в настоящем документе, включают мази, пасты, кремы, лосьоны, гели, порошки, растворы, спреи, ингаляторы или пластыри. Активный компонент добавляется в стерильных условиях с фармацевтически приемлемым носителем и любыми необходимыми консервантами или буферами, в зависимости от обстоятельств. Глазные формы, ушные капли и глазные капли также рассматриваются в рамках данного изобретения. Кроме того, данное изобретение рассматривает использование трансдермальных пластырей, имеющих дополнительное преимущество контролируемой поставки соединения в организм. Такие формы могут быть изготовлены путем растворения или распределения соединения в надлежащей среде. Усилители абсорции могут также использоваться для увеличения проникновения соединения через кожу. Скорость может контролироваться обеспечением мембраны контроля скорости или путем распределения состава в полимерной матрице или геле.
[0132] Соединения, раскрытые в настоящем документе, преимущественно изготовляются в форме единицы дозировки для простоты введения и однородности дозировки. Выражение «форма единицы дозировки», как используется в настоящем документе, относится к физически дискретной единице агента, подходящего для пациента подвергающегося лечению. Однако следует понимать, что суммарное ежедневное использование соединений и составов, раскрытых в настоящем документе, определяется лечащим врачом в рамках здравого медицинского суждения. Определенный уровень эффективной дозы для любого определенного пациента или организма зависит от множества факторов, включая расстройство и серьезность расстройства; активность определенного используемого соединения; использование определенного соединения; возраст, масса тела, общее состояние здоровья, пол и диета пациента; время введения, пути введения и скорость экскреции определенного используемого состава; продолжительность лечения; препараты, используемые в комбинации или случано принятые с определенным используемым соединением, и аналогичные факторы, известные в медицинской области.
[0133] Объем соединений, раскрытых в настоящем документе, который может быть объединен с материалами носителя для производства состава в единственной форме выпуска, зависит от рассматриваемого носителя, определенного способа введения. Преимущественно, составы должны быть изготовлены так, чтобы дозировка между 0,01-200 мг/кг масса тела/день ингибитора была введена пациенту, получающему такие составы.
[0134] Соединения, раскрытые в настоящем документе, могут вводиться как единственный фармацевтический агент или в сочетании с одним или несколькими другими дополнительными терапевтическими (фармацевтическими) агентами, если комбинация не вызывает недопустимых неблагоприятных воздействий. Это может иметь определенную уместность при лечении гиперпролиферативных болезней, таких как рак. В этом случае соединение, раскрытое в настоящем документе, может быть объединено с известными цитостатическими агентами, ингибиторами сигнальной трансдукции, или с другими противоопухолевым агентами, а также с примесями и их комбинациями. Как используется в настоящем документе, дополнительные терапевтические агенты, обычно вводимые для лечения определенного заболевания или состояния, известны как «подходящие для рассматриваемой болезни или состояния». Как используется в настоящем документе, «дополнительные терапевтические агенты» включают химиотерапевтические агенты и другие антипролиферативные агенты.
[0135] Например, химиотерапевтические агенты или другие антипролиферативные агенты могут быть объединены с соединениями, раскрытыми в настоящем документе, для лечения пролиферативного заболевания или рака. Примеры химиотерапевтических агентов или других антипролиферативных агентов включают ингибиторы HDAC, включая, без ограничений, САА, MS 275, MGO 103 и описанные в WO 2006/010264, WO 03/024448, WO 2004/069823, US 2006/0058298, US 2005/0288282, WO 00/71703, WO 01/38322, WO 01/70675, WO 03/006652, WO 2004/035525, WO 2005/030705, WO 2005/092899; и агенты деметилирования, включая, без ограничений, 5-аза-dC, Виадаза и Децитабин, и описанные в US 6268137, US 5578716, US 5919772, US 6054439, US 6184211, US 6020318, US 6066625, US 6506735, US 6221849, US 6953783, США 11/393380.
[0136] В другом воплощении данного изобретения, например, химиотерапевтические агенты или другие антипролиферативные агенты могут быть объединены с соединениями, раскрытыми в настоящем документе для лечения пролиферативных заболеваний и рака. Примеры известных химиотерапевтических агентов включают, без ограничений, другие противопухолевые средства или агенты, которые могут использоваться в сочетании с изобретенными противоопухолевыми агентами данного изобретения, и включают хирургию, радиотерапию (например, гамма радиация, нейтронная лучевая радиотерапия, электронно-лучевая радиотерапия, протонная терапия, брахитерапия и системные радиоактивные изотопы), эндокринную терапию, таксаны (TAXOL®, таксотер и т.д.), производные платины, биологические модификаторы реакции (интерфероны, интерлейкины и фактор некроза опухоли (TNF), таргетирование рецептора TRAIL, агенты), гипертермию и криотерапию; агенты для уменьшения любых неблагоприятных воздействий (например, антирвотные средства), и другие утвержденные химиотерапевтические препараты, включая, без ограничений, алкилирующие препараты (мехлоретамин, хлорамбуцил, циклофосфамид, мелфалан, изофамид), антиметаболиты (метотрексат, преметрексед и т.д.), антагонисты пурина и антагонисты пиримидина (6-меркаптопурин, 5-фтороурацил, цитарбил, гемцитабин), веретенные яды (винбластин, винкристин, винорельбин, паклитаксел), подофиллотоксины (этопозид, ириноцетан, топотекан), антибиотики (доксирубицин, блеомицин, митомицин), нитрозомочевина (кармустин, ломустин), неорганические ионы (цисплатин, карбоплатин), ингибиторы клеточного цикла (митотические ингибиторы кинезина KSP, C6NP-E и ингибиторы CDK), ферменты (аспарагиназа) и гормоны (тамоксифен, лейпролид, флутамид и мегестрол), GLEEVEC®, адриамицин, дексаметазон и циклофосфамид. Антиангиогенные агенты (авастин и другие). Моноклональные антитела (белимумаб (BENLYSTA®), брентуксимаб (ADCETRIS®), цетуксимаб (ERBITUX®), гемтузумаб (MYLOTARG®), иплимумаб (YERVOY®), офатумумаб (ARZERR®), панитумумаб (VECTIBIX®), ранибизумаб (LUCENTIS®), ритуксимаб (RITUXAN®), тоситумомаб (BEXXAR®), трастузумаб (HERCEPTIN®)). Ингибиторы киназы (иматиниб (GLEEVEC®), сунитиниб (SUTENT®), сорафениб (NEXAVAR®), сетуксимаб (ERBITUX®), трастузумаб (HERCEPTIN®), эрлотинииб (TARCEVA®), гефинитиб (IRESSA®), дасатиниб (SPRYCEL®), нилотиниб (TASIGNA®), лапатиниб (TYKERB®), кризотиниб (XALKORI®), руксолитиниб (JAKAFI®), вемурафениб (ZELBORAF®), вандетаниб (CAPRELSA®), пазопаниб (VOTRIENT®) и другие). Агенты, подавляющие или активирующие онкологические пути, такие как mTOR, HIF (фактор, индуцированный гипоксией), пути (такие как эверолимус и темисиролимус) и другие. Для всестороннего обсуждения обновленных способов лечения рака, см. http://www.nC1.nih.gov/, список лекарств по онкологии, утвержденный FDA, см. http://www.fda.gov/cder/canC6r/druglist-rame.htm, и руководство Merck, 18-ое изд. 2006 г., содержание которого настоящим включено посредством ссылок.
[0137] В других воплощениях соединения, раскрытые в настоящем документе, могут быть объединены с цитостатическими противоопухолевыми агентами. Примеры таких агентов приведены в 13-м выпуске индекса Merck (2001). Эти агенты включают, без ограничений, аспарагиназы, блеомицин, карбоплатин, кармустин, хлорамбуцил, цисплатин, коласпас, циклофосфамид, цитарабин, дакарбазин, дактиномицин, даунорубицин, доксорубицин (адриамицин), эпирубицин, этопозид, 5-фтороурацил, гексаметилмеламин, гидроксимочевину, ифосфамид, иринотекан, лейковорин, ломустин, мехлоретамин, 6-меркаптопурин, месна, метотрексат, митомицин С, митоксантрон, преднизолон, преднизон, прокарбазин, ралоксифен, стрептозоцин, тамоксифен, тиогуанин, топотекан, винбластин, винкристин и виндезин.
[0138] Другие цитотоксические препараты, подходящие для использования с соединениями, раскрытыми в настоящем документе, включают, без ограничений, составы, которые используются в лечении опухолевых болезней, например, описанные в Фармакологическое основание терапии, Гудмэн и Гилман (Девятый выпуск, 1996, McGraw-Hill). Эти агенты включают, без ограничений, аминоглутэтимид, L-аспарагиназу, имуран, 5-азацидина кладрибин, бусульфан, диэтилстилбэстрола, 2,2-дифтордезоксицитидин, доцетаксел, эритрогидроксинониладеин, этинил эстрадиол, 5-фтородезоксиуридин, 5-фтородезоксиуридина монофосфат, флудабарина фосфат, флуоксиместерон, флутамид, гидроксипрогестерерона капроат, идарубицин, интерферон, медроксипрогестерона ацетат, мегестрола ацетат, мелфалан, митотан, паклитаксел, пентостатин, аспартат N-фосфоноацетил-L-(PALA), пликамицие, семустин, тенипосид, пропионат тестостерона, тиотепа, триметилмеламин, уридин и винорельбин.
[0139] Другие цитостатические противоопухолевые агенты, подходящие для использования в сочетании с соединениями, раскрытыми в настоящем документе также включают недавно обнаруженные цитостатические принципы, такие как оксалипатин, гемцитабин, капецитабин, эпотилон и естественные или синтетические деривативы, темозоламид (Квинн и др., J. Clin. Oncology 2003, 21 (4), 646-651), тоситумомаб (BEXXAR®), трабедектин (Видал и др., Заседание американского общества клинической онкологии, 2004, 23, реферат 3181), и ингибиторы веретенного протеина кинезина Eg5 (Вуд и др., Curr. Opin. Pharmacol. 2001, 1, 370-377).
[0140] В других воплощениях составы, раскрытые в настоящем документе, могут быть объединены с другими ингибиторами сигнальной трансдукции. Примеры таких агентов включают, без ограничений, терапии антителами, такими как трастузумаб (HERCEPTIN®), цетуксимаб (ERBITUX®), ипилимумаб (YERVOY®) и пертузумаб. Примеры таких способов лечения также включают, без ограничений, низкомолекулярные ингибиторы киназы, такие как иматниб (GLEEVEC®), сунитиниб (SUTENT®), сорафениб (NEXAVAR®), эрлотинииб (TARCEVA®), гефинитиб (IRESSA®), дасатиниб (SPRYCEL®), нилотиниб (TASIGNA®), лапатиниб (TYKERB®), кризотиниб (XALKORI®), руксолитиниб (JAKAFI®), вемурафениб (ZELBORAF®), вандетаниб (CAPRELSA®), пазопаниб (VOTRIENT®), афатиниб, алисертиб, амуватиниб, акситиниб, босутиниб, бриваниб, канертиниб, карбозантиниб, цедираниб, креноланиб, дабрафениб, дакомитиниб, данусертиб, довитениб, форетиниб, ганетеспиб, ибрутиниб, инипариб, ленватиниб, линифаниб, линситиниб, маситиниб, момелотиниб, мотесаниб, нератиниб, нирапариб, опрозониб, олапариб, пиктилисиб, понатиниб, квизартиниб, регорафениб, ригосертиб, рукапариб, саракатиниб, саридегиб, тандутиниб, тасоцитиниб, телатиниб, тиватиниб, тивозаниб, тофацитиниб, траметиниб, ваталамиб, велипариб, висмодегиб, воласертиб, BMS-540215, BMS777607, JNJ38877605, TKI258, GDC-0941 (Фолкс, и др., J. Med. Chem., 2008, 51, 5522), BZE235 и другие.
[0141] В других воплощениях соединения, раскрытые в настоящем документе, могут быть объединены с ингибиторами деацетилазы гистона. Примеры таких агентов включают, без ограничения, субероилаилид, гидроксамовую кислоту (SAHA), LAQ-824 (Оттман и др., Заседание американского общества клинической онкологии, 2004, 23, реферат 3024), LBH-589 (Бек и др., Заседание американского общества клинической онкологии, 2004, 23, реферат 3025), MS 275 (Райан и др., Заседание американской ассоциации онкологических исследований, 2004, 45, реферат 2452), FR 901228 (Пикарц и др., Заседания американского общества клинической онкологии, 2004, 23, абстрактные 3028) и MGCDOl 03 (US 6897220).
[0142] В других воплощениях соединения, раскрытые в настоящем документе, могут быть объединены с другими противоопухолевыми агентами, такими как ингибиторы протеасомы и ингибиторы m-TOR. Они включают, без ограничений, бортезомиб и СС1-779 (By и др., Заседание американской ассоциации онкологических исследований, 2004, 45, реферат 3849). Соединения, раскрытые в настоящем документе, могут быть объединены с другими противоопухолевыми агентами, такими как ингибиторы топоизомеразы, включая, без ограничений, камптотецин.
[0143] Такие дополнительные агенты могут вводиться отдельно от состава, содержащего соединение, как часть режима многократной дозировки. Также такие агенты могут быть частью единственной формы дозировки, смешанной вместе с соединением, раскрытым в настоящем документе в единственном составе. При введении как часть режима многократной дозировки, эти два активных вещества могут вводиться одновременно, последовательно или в течение промежутка времени, в зависимости от того, что приведет к желаемой активности агентов.
[0144] Объем соединения и дополнительного терапевтического агента (в соединениях, включающих дополнительный терапевтический агент, как описано выше), который может быть объединен с материалами носителя для производства единственной формы дозировки, зависит от рассматриваемого носителя и определенного способа ведения. Обычно, объем дополнительного терапевтического агента, присутствующего в соединениях, раскрытых в настоящем документе, не превышает объем, обычно вводимый в составе, включающем такой терапевтический агент как единственное активное вещество. Преимущественно объем дополнительного терапевтического агента в раскрываемых составах обычно составляет приблизительно от 50% до 100% от объема, обычно присутствующего в составе, включающем такой агент как единственное терапевтически активное вещество. В составах, включающих дополнительный терапевтический агент, такой дополнительный терапевтический агент и соединение, раскрытое в настоящем документе, могут действовать синергично.
ИСПОЛЬЗОВАНИЕ СОЕДИНЕНИЙ И СОСТАВОВ, РАСКРЫТЫХ В НАСТОЯЩЕМ ДОКУМЕНТЕ
[0145] Изобретение касается фармацевтических композиций, включающих соединение по формуле (I) или (II) или соединения, перечисленные в Таблице 1, и фармацевтически приемлемый носитель, вспомогательное вещество или средство. Объем соединения в составах, раскрытых в настоящем документе, является эффективным для обнаружимого ингибирования киназы протеина, например ингибурующая активность VEGFR, c-Met, Ron или Axl. Соединения, раскрытые в настоящем документе, полезны в терапии как агенты антинеоплазии или для минимизации вредных эффектов сигнализации рецептора VEGFR, с-Met, Rona и/или Axl.
[0146] Соединения, раскрытые в настоящем документе, используются, без ограничений, для профилактики или лечения пролиферативных заболеваний, состояний или расстройств у пациентов путем введения пациенту соединения или состава, раскрытого в настоящем документе, в эффективном объеме. Такие заболевания, состояния или расстройства включают рак, особенно метастатический рак, атеросклероз и фиброз легкого.
[0147] Соединения, раскрытые в настоящем документе, используются для лечения неоплазии, включая рак и метастазы, включая, без ограничений: карцинома, такая как рак мочевого пузыря, груди, толстого кишечника, почки, печени, легкого (включая мелкоклеточный рак легких), пищевода, желчного пузыря, яичников, поджелудочной железы, живота, шейки матки, щитовидной железы, простаты и кожи (включая плоскоклеточную карциному); гематопоэтические опухоли лимфатического происхождения (включая лейкемию, острую лимфоцитарную лейкемию, острую лимфообластную лейкемию, В-клеточную лимфому, Т-клеточную лимфому, лимфому Ходжкина, неходжкинскую лимфому, волосатоклеточную лимфому и лимфому Беркетта); гематопоэтические опухоли миелоидного происхождения (включая острые и хронические миелогенные лейкемии, миелодиспластический синдром и промиелоцитарную лейкемию); опухоли мезенхимального происхождения (включая фибросаркому и рабдомиосаркому, и другие саркомы, например, мягких тканей и кости); опухоли центральной и периферийной нервной системы (включая астроцитому, нейробластому, глиому и шванномы); и другие опухоли (включая меланому, семиному, тератокарциному, остеогенную саркому, пигментную ксеродерму, роговую кератому, фолликулярный рак щитовидной железы и саркому Капоши).
[0148] Соединения, раскрытые в настоящем документе, также используются для лечения офтальмологических состояний, таких как отторжение роговичного трансплантата, неоваскуляризаци глаза, ретинальная неоваскуляризация, включая неоваскуляризацию после ранения или инфекции, диабетическая ретинопатия, ретролентальная фиброплазия и неососудистая глаукома; ретинальная ишемия; кровоизлияние в стекловидное тело; язвенные болезни, такие как язва желудка; патологический, но не злокачественные состояния, такие как гемангиомы, включая инфантильные гемангиомы, ангифибромы носоглотки и аваскулярный некроз кости; и расстройства женской репродуктивной системы, такие как эндометриоз. Соединения также используются для лечения отеков и состояния сосудистой гиперпроницаемости.
[0149] Соединения, раскрытые в настоящем документе, также используются в лечении диабетических состояний, таких как диабетическая ретинопатия и микроангиопатия. Соединения, раскрытые в настоящем документе, также используются для уменьшения кровотока при опухоли у субъекта. Соединения, раскрытые в настоящем документе, также используются для уменьшения роста метастазов опухоли у субъекта.
[0150] Помимо использования для лечения человека, соединения, раскрытые в настоящем документе, также используются для ветеринарного лечения домашних животных, экзотических животных и сельскохозяйственных животных, включая млекопитающих, грызунов и т.п. Более предпочтительные животные включают лошадей, собак и кошек. Как используется в настоящем документе, соединения, раскрытые в настоящем документе, включают их фармацевтически приемлемые производные.
[0151] Если используется сножественное число для обозначения для соединений, солей и т.п., следует понимать также единственное число для обозначения соединения, соли и т.п.
[0152] Способ лечения, включающий введение соединения или состава, раскрытого в настоящем документе, включает введение пациенту дополнительного терапевтического агента (комбинированная терапия), выбранного из следующего: химиотерапевтический или антипролиферативный агент или противовоспалительное средство, где дополнительный терапевтический агент является подходящим для рассматриваемого заболевания, и дополнительный терапевтический агент вводится вместе с соединением или составом, раскрытым в настоящем документе, как единственная форма дозировки или отдельно от соединения или состава, как часть многократной формы дозировки. Дополнительный терапевтический агент можеть вводиться одновременно с соединением, раскрытым в настоящем документе, или в другое время. В последнем случае введение может быть дифференцированым, например, 6 часов, 12 часов, 1 день, 2 дня, 3 дня, 1 неделя, 2 недели, 3 недели, 1 месяц или 2 месяца.
[0153] Изобретение также описывает способ ингибирования роста клетки, продуцирующей VEGFR, c-Met, Ron или Axl, включая контакт клетки с соединением или составом, раскрытым в настоящем документе, таким образом, вызывая ингибирование роста клетки. Примеры клетки, рост которой может быть ингибирован, включают: клетка рака молочной железы, клетка рака ободочной и прямой кишки, клетка рака легких, клетка папиллярной карциномы, клетка рака простаты, клетка лимфомы, клетка рака толстой кишки, клетка рака поджелудочной железы, клетка рака яичника, клетка рака шейки матки, клетка рака центральной нервной системы, клетка остеогенной саркомы, клетка почечной карциномы, клетка гепатоцеллюлярной карциномы, клетка рака мочевого пузыря, клетка рака желудка, плоская клетка карциномы головы и шеи, клетка меланомы или лейкозная клетка.
[0154] Изобретение обеспечивает способ ингибирования активности киназы VEGFR, c-Met, Ron или Axl в биологической пробе, включающем контакт биологической пробы с соединением или составом, раскрытым в настоящем документе. Термин «биологическая проба», как используется в настоящем документе, означает пробу вне живого организма и включает, без ограничения, клеточные культуры или их экстракты; материал биопсии, полученный из млекопитающего или эго экстракты; кровь, слюна, моча, экскременты, сперма, слезы или другие физиологические жидкости или их экстракты. Ингибирование активности киназы, особенно активности киназы VEGFR, c-Met, Ron или Axl, в биологической пробе используется для множества целей, известных в области. Примеры таких целей включают, без ограничений, переливание крови, трансплантация органа, хранение биологического экземпляра и биологическое испытание.
[0155] В определенных воплощениях данного изобретения «эффективный объем» или «эффективная доза» соединения или фармацевтически приемлемой композиции является объемом, эффективным для лечения или уменьшения серьезности одного или нескольких вышеупомянутых расстройств. Соединения и составы, согласно способу данного изобретения, могут быть введены в любом объеме и любым путем введения, эффективным для лечения или уменьшения серьезности расстройства или заболевания. Точный необходимый объем зависит от субъекта, биологического вида, возраста и общего состояния субъекта, серьезности инфекции, определенного агента, способа введения и т.п. Соединение или состав можно также вводить с одним или несколькими другими терапевтическими агентами, как указано выше.
[0156] Соединения, раскрытые в настоящем документе или их фармацевтические композиции, могут также использоваться для покрытия вживляемого медицинского устройства, такого как протезы, искусственные клапаны, сосудистые трансплантаты, стенты и катетеры. Сосудистые стенты, например, используются для преодоления рестеноза (повторного сужения стенки сосуда после ранения). Однако у пациентов, использующих стенты или другие вживляемые устройства, возникает риск формирования тромба или активация тромбоцитов. Эти нежелательные эффекты могут быть предотвращены или минимизированы предварительным покрытием устройства фармацевтически приемлемой композицией, включающей соединение, раскрытое в настоящем документе.
[0157] Подходящие покрытия и общее изготовление покрытых вживляемых устройств описаны в патентах №№ US 6099562, 5886026 и 5304121, содержание каждого из которых включено посредством ссылок в настоящий документ. Покрытия обычно являются биологически совместимыми полимерными материалами, такими как полимер гидрогеля, полиметилдисилоксан, поликапролактон, полиэтиленгликоль, полимолочная кислота, этиленовый винилацетат и их смеси. Покрытия могут быть дополнительно покрыты подходящим верхним слоем фторсиликона, полисахаридами, полиэтиленгликолем, фосфолипидами или их комбинациями для контролированного высвобождения соединения. Вживляемые устройства, покрытые соединением, раскрытым в настоящем документе, являются другим воплощением данного изобретения. Соединения могут также наноситься на вживляемые медицинские устройства, такие как гранулы, или сочетаться с полимерной или другой молекулой, для обеспечения «депо препарата», таким образом, рарешая высвобождение препарата в течение более длинного интервала времени, чем при введении водного раствора препарата.
ОБЩИЕ ПРОЦЕДУРЫ СИНТЕЗА
[0158] Для иллюстрирования изобретения, предусмотрены следующие примеры. Однако следует понимать, что эти примеры не ограничивают изобретение и предназначены только для предложения способа осуществления изобретения.
[0159] Обычно соединения в этом изобретении могут быть подготовлены способами, описанными в настоящем документе, где заместители определяются для формулы (I) или (II) выше, кроме случаев, определенных далее. Следующие неограничивающие схемы и примеры представлены для дальнейшего иллюстрирования изобретения. Специалисты в области признают, что химические реакции, описанные в настоящем документе, могут быть легко адаптированы для изготовления многих других соединений, раскрытых в настоящем документе, и альтернативные способы для изготовления соединений, раскрытых в настоящем документе, находятся в рамках настоящего изобретения. Например, синтез неиллюстрируемых соединений согласно изобретению может быть успешно выполнен при модификации, очевидной для специалистов в области, например, путем надлежащей блокировки мешающих групп, путем использования других подходящих реактивов, известных в области, кроме описанных, и/или путем создания модификации обычных условий реакции. Также другие реакции, раскрытые в настоящем документе или известные в области, признаются применимыми для подготовки других соединений, раскрытых в настоящем документе.
[0160] В примерах, описанных ниже, если иначе не обозначено, все температуры установлены в градусах Цельсия. Реактивы были приобретены у коммерческих поставщиков, таких как Aldrich Chemical Company, Arco Chemical Company и Alfa Chemical Company, Shanghai Medpep Co., Ltd, Aladdin-Shanghai Jinchun Reagents, Ltd, и использовались без дальнейшей очистки, если иначе не обозначено. Общие растворители были приобретены у коммерческих поставщиков, таких как Shantou XiLong Chemical Factory, Guangdong Guanghua Reagent Chemical Factory Co., Ltd., Guangzhou Reagent Chemical Factory, Tainjin YuYu Fine Chemical Ltd., Qindao Tenglong Reagent Chemical Ltd. и Qindao Ocean Chemical Factory.
[0161] Безводный THF, диоксан, толуол и эфир были получены путем флегмирования растворителя с натрием. Безводная CH2Cl2 и CHCl3 были получены путем флегмироания растворителя с СаН2. EtOAc, РЕ, гексаны, DMA и DMF предварительно обработали безводной Na2SO4.
[0162] Реакции, изложенные ниже, обычно проводились под положительным давлением азота или аргона или с сушильной трубой (если не указано иное), в безводных растворителях, и реактивные колбы обычно были оснащены резиновыми септами для введения субстратов и реактивов через шприц. Стеклянную посуду сушили в духовом шкафу и/или теплым воздухом.
[0163] Колоночная хроматография проводилась с помощью колонки с силикагелем. Силикагель (класс крупности 300-400)приобретен у Qindao Ocean Chemical Factory. Спектры 1H NMR были зарегистрированы спектрометром Bruker 400 МГц при температуре окружающей среды. Спектры 1Н NMR были получены как растворы CDCl3, DMSO-d6, CD3OD или ацетон-d6 (в ppm), с помощью TMS (0 ppm) или хлороформа (7,25 ppm) в качестве эталонного стандарта. При пиковой мультиплетности используются следующие сокращения: s (синглет), d (дуплет), t (триплет), m (мультиплет), br (уширенный), dd (двойной дуплет), dt (двойной триплет). Константы связи, в случае необходимости, отображали в герцах (Гц).
[0164] Данные о спектральной массе (MS) с низкой разрешающей способностью обычно определялись на спектрометре LCMS Agilent 1200 (SB-C18 Zorbax, 2.1×30 мм, 4 микрон, 10 минут, расход 0,6 мл/минуты, 5%-95% (муравьиная кислота 0,1% в CH3CN) в (муравьиная кислота 0,1% в H2O)) с обнаружением УФ при 210/254 нм, и режим электроспрея низкого резонанса (ESI).
[0165] Чистоту соединений оценивали высокоэффективной жидкостной хроматографией (ВЭЖХ) Agilent 1100 с обнаружением УФ при 210 нм и 254 нм. Колонка обычно использовалась при 40°С.
[0166] Следующие сокращения используются в настоящей спецификации:
АТР Аденозиновый трифосфат
BINAP 2,2'-bis (дифенилфосфино)-l,1-бинафтил
BBr3 бора трибромид
BSA бычий сывороточный альбумин
ВОС, Boc бутилоксикарбонил
Са(SO3CF3)2 кальция трифторметил сульфонат
Cs2CO3 карбонат цезия
CH2Cl2, DCM метиленхлорид
CHCl3 хлороформ
CDCl3 дейтеризованный хлороформ
Cu медь
Cul йодид меди
DBU l,8-диазабицикло[5.4.0]ундек-7-ен
D2 дейтерий газ
DIBAL диизобутилалюминия гидрид
DIAD диизопропил азодикарбоксилат
DIEA, DIPEA, iPr2Net N,N-диизопропилэтиламин
DEAD диметил азодикарбоксилат
DMF диметилформамид
DMAP 4-диметиламинопиридин
DMSO диметилсульфоксид
DPPA дифенилфосфорил азид
DTT DL-дитиотреитол
EDC, EDCl 1-(3-диметиламинопропил)-3-этилкарбодиимид гидрохлорид
EDTA этилендиаминтетрауксусная кислота
Et3N, TEA триэтиламин
EtOAc, ЕА, этилацетат
Et2O диэтиловый эфир
EtOH этанол
FBS эмбриональная бычья сыворотка
Fe железо
g грамм
h час
HATU 2-(7-аза-1Н-бензотриазол-1-ил)-1,1,3,3-тетраметилуроний гексафторфосфат
HBr бромистоводородная кислота
HCl соляная кислота
НОАс уксусная кислота
НОАТ 1-гидрокси-7-азабензотриазол
H2 водород
HOBt 1-гидроксибензотриазол гидрат
H2O вода
Н2О2 перекись водорода
Н3РО4 ортофосфорная кислота
H2SO4 серная кислота
HNO3 азотная кислота
HCOOK муравьинокислый калий
LiHMDS литий бис (триметилсилил) - амид
LDA литий диизопропиламид
МВР миелиновый основной протеин
МСРВА мета-хлоропербензойная кислота
MeCN, CH3CN ацетонитрил
MgSO4 сульфат магния
МеОН, СН3ОН метанол
Mel метилйодид
MOPS 3-(N-морфолино)пропансульфоновая кислота
2-MeTHF 2-метила тетрагидрофуран
Ml, ml миллилитр
N2 азот
NMP N-метилпирролидинон
NaHCO3 бикарбонат натрия
NaBH4 борогидрид натрия
NaBH3CN цианоборогидрид натрия
NaOtBu терт-бутоксид натрия
NaOH гидроксид натрия
NaClO2 хлорит натрия
NaClO гипохлорит натрия
NaCl хлорид натрия
NaH2PO4 бифосфат натрия
NaH гидрид натрия
NaI йодид натрия
Na2SO4 сульфат натрия
NH3 аммиак
NH4Cl хлорид аммония
Pd/C палладий на углероде
Pd2(dba)3 бис(дибензилиенацетон)палладия
Pd(OAc)2 ацетат палладия
Pd(OH)2 гидроксид палладия
Pd(PPh3)4 палладия тетракис трифенилфосфин
Pd(dppf)Cl2 1,1-бис(дифенилфосфино)ферроцен хлорид палладия
Р(t-Bu)3 три (трет-бутил) фосфин
РЕ нефтяной эфир (60-90°С)
PBS фосфатно-солевой буферный раствор
POCl3 фосфористая хлорокись
PhI(ОАс)2 йодбензол диацетат
K2CO3 карбонат калия
KOH гидроксид калия
К.Т. к.т. комнатная температура
ВУ время удерживания
SOCl2 тионилхлорид
t-BuOK терт-бутанолат калия
TBTU O-бензотриазол-1-ил-N,N,N',N'-тетраметилуроний тетрафторборат
TBS ТРИС-буферизированный физраствор
THF тетрагидрофуран
TFA трифторуксусная кислота
ТЕАС бис(тетра-этиламмония) карбонат
Трис тригидроксиметил аминометан
[0167] Представительные синтетические процедуры для изготовления соединений, раскрытых в настоящем документе, описаны ниже в следующих схемах. Если иначе не обозначено, каждый из X, Y, Z, W, R1, R2, R3, R4, R5, R6 и Rd несут определения, установленные выше в связи с формулой (I) или (II).
Схема 1
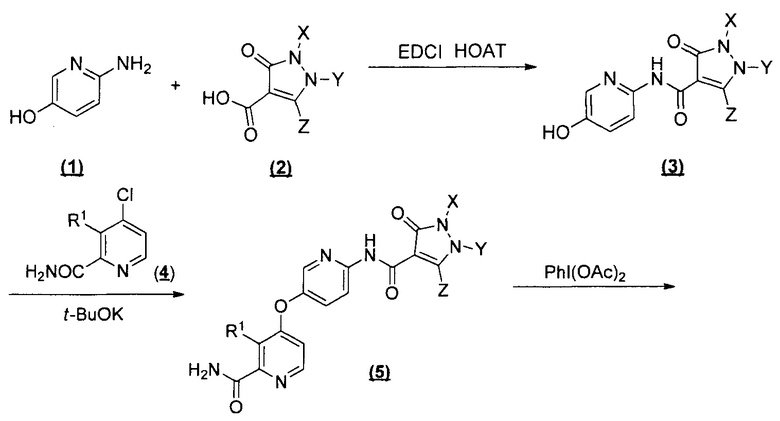
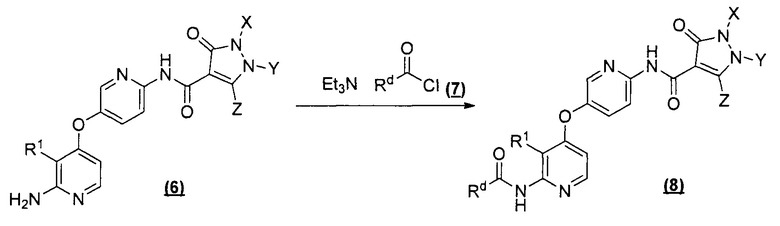
[0168] Соединения, раскрытые в настоящем документе, могут быть изготовлены согласно общим способам синтеза, проиллюстрированным в Схеме 1, и детально описанным в Примерах. Относительно схемы 1, 6 аминопиридин-3-ол (1) сначала сжижается замещенным пиразолоном (2) для обеспечения соединения (3). Связывание пиколинамида (4) с соединением (3) при базовом условии (например, t-BuOK или NaH)при повышенной температуре в полярном растворителе, таком как DMF, обеспечивает получение желаемого амида (5). Перестановка амида в присутствии окислителя, такого как PhI(ОАс)2 или NaClO, приводит к получению аминопиридина (6). Ацилирование аминопиридина (6) с ацильным хлоридом (7) приводит к получению ингибитора киназы (8).
Схема 2
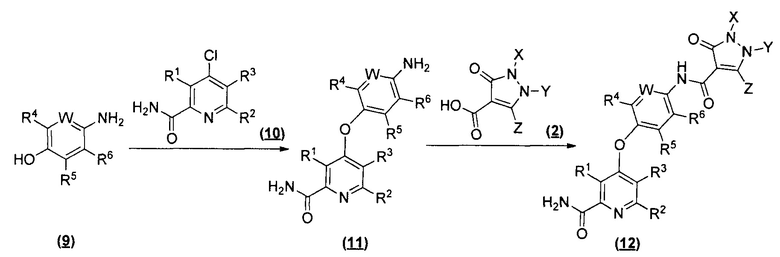
[0169] Также соединения, раскрытые в настоящем документе, могут быть также изготовлены с помощью маршрута синтеза, как показано в Схеме 2. Относительно Схемы 2, арильное соединение (9) (со свободной группой ОН) связывается с замещенным пиридином (10) при повышенной температуре для получения диарилового эфира (11). Конденсирование диарилового эфира (11) с замещенным пиразолоном (2) приводит к получению желаемого ингибитора киназы (12).
Схема 3
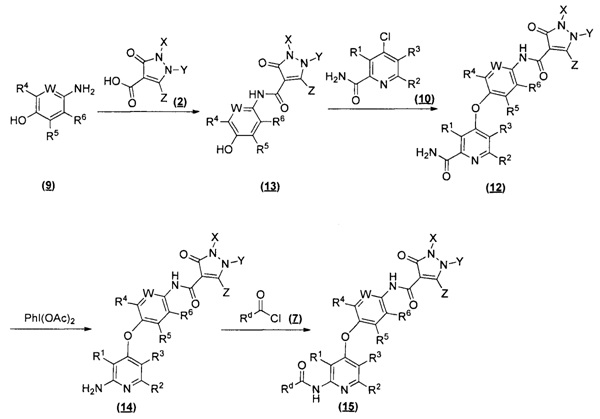
[0170] Желаемый ингибитор (12) и (15) может быть также изготовлен в ходе процесса, проиллюстрированного в Схеме 3. Относительно Схемы 3, сцепление арила (9) (со свободной группой ОН) с кислотой (2) в присутствии реактива связывания, такого как EDCl или HATU, приводит к получению амидного соединния (13). Связывание соединения (13) с замещенным пиридином (10) в присутствии основы, такой как DMAP, iPr2Net, Et3N, t-BuOK, NaH, или CS2CO3 и т.д., приводит к получению амидного соединения (12). Перестановка амидного соединения (12) в присутствии окислителя PhI(ОАс)2 или NaClO приводит к получению аминопиридина (14), который может быть далее преобразован в мочевину (15), в качестве желаемого ингибитора киназы.
ПРИМЕРЫ
Пример 1. 3-хлоро-4-(4-(1,5-диметил-3-оксо-2-фенил-2,3-дигидро-1Н-пиразол-4-карбоксамидо)фенокси)пиколинамид
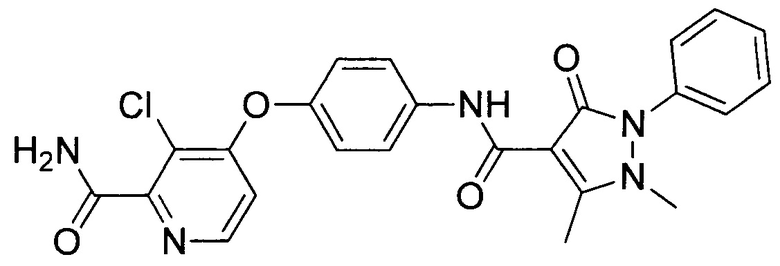
Шаг 1) N-(4-гидроксифенил)-1,5-диметил-3-оксо-2-фенил-2,3-дигидро-1H-пиразол-4-карбоксамид
[0171] К смеси 4-аминофенола (1,09 г, 10 ммоль) и 1,5-диметил-3-оксо-2-фенил-2,3-дигидро-1Н-пиразол-4-карбоновая кислота (2,37 г, 10,2 ммоль) в DCM (30 мл) добавили EDCl (2,3 г, 12 ммоль) и НОАТ (0,27 г, 2 ммоль). Смесь размешивали при 46°С в течение 4 часов, затем охладили до к.т. и разбавили EtOAc (10 мл) и водой (10 мл). Смесь размешивали при к.т. в течение 1 часа, затем отфильтровали до титулованного соединения в виде белой твердой фазы (1,7 г, 52,5%).
MS (ESI, пол. ион) уд.з.: 324.1 [М+Н]+;
1Н NMR (400 МГц, DMSO-d6): δ (ppm) 2.68 (s, 3Н), 3.32 (s, 3Н), 6.72 (d, J=8,8 Гц, 2Н), 7.36-7.42 (m, 4Н), 7.49 (t, J=7,4 Гц, 1Н), 7.57 (t, J=7,6 Гц, 2Н), 9.21 (s, 1Н), 10.46 (s, 1Н).
Шаг 2) 3.4-дихлоропиколинамид
[0172] К смеси 2,2,6,6-тетраметилпиперидин (6,2 мл, 37,2 ммоль) в диэтилэфире (50 мл) добавили n-BuLi в гексане (2.5М, 23 мл, 57,5 ммоль)при 0°С через шприц через 15 минут. Смесь размешивали при 0°С в течение 0,5 часов, затем охладили до -78°С. Раствор 3,4-дихлоропиридина (5,00 г, 33,8 ммоль) в диэтилэфире (20 мл) добавили к смеси через шприц через 15 минут. Реакционный раствор размешивали при -78°С в течение 2 часов, затем добавили изоцианатотриметилсилан (чистота 94%, 6,7 мл, 50,7 ммоль). Смесь нагревали до к.т. и далее вымешивали в течение 2 часов, погасив уксусной кислотой (6,76 г, 112,6 ммоль) в 35 мл воды. Смесь далее быстро размешивали. Титулованный продукт быстро осел в виде белой твердой фазы, собранной путем фильтрации. Больший объем продуктов восстановили из фильтрата. Таким образом, фильтрат экстрагировали этилацетатом (50 мл × 3) и объединенные органические фазы промывали морской водой (50 мл), высушили над безводной Na2SO4 и сконцентрировали в вакууме. Твердую фазу соединили и промывали 35 мл Et2O до титулованного соединения в виде бледно-желтой твердой фазы (2,20 г, 34,0%).
MS (ESI, пол. ион) уд.з.: 191.1 [М+Н]+;
1Н NMR (400 МГц, DMSO-d6): δ (ppm) 8.48 (d, J=5,2 Гц, 1Н), 8.09 (br s, 1Н), 7.82 (s, 1Н), 7.81 (d, J=5,2 Гц, 1Н).
Шаг 3) 3 хлоро-4-(4-(1,5-диметил-3-оксо-2-фенил-2,3-дигидро-1Н-пиразол-4-карбоксамидо)фенокси)пиколинамид
[0173] К смеси N-(4-гидроксифенил)1,5-диметил-3-оксо-2-фенил-2,3-дигидро-1H-пиразол-4-карбоксамид (356 мг, 1,1 ммоль) в DMSO (4 мл) в микроволновом флаконе добавили NaH (88 мг, 2,2 ммоль, 60%, распыленный в минеральном масле) при к.т.
Реакционный раствор размешивали при к.т. в течение 30 минут, затем добавили 3,4-дихлоропиколинамид (191 мг, 1,0 ммоль). Смесь подогревали в мироволновой печи при 160°С в течение 2 часов, затем охладили до к.т., и разбавили водой (10 мл). Полученную смесь экстрагировали этилацетатом (30 мл × 3). Объединенные органические фазы промывали морской водой (30 мл × 3), высушили над безводной Na2SO4 и сконцентрировали в вакууме. Остаток очистили колоночной хроматографией с силикагелем (DCM/MeOH (о/с) = 50/1) до титулованного соединения в виде бледно-желтой твердой фазы (140 мг, 29%).
MS (ESI, пол. ион) уд.з.: 478.1 [М+Н]+;
1Н NMR (400 МГц, DMSO-d6): δ (ppm) 2.71 (s, 3Н), 3.35 (s, 3Н), 6.82 (d, J=5,4 Гц, 1H), 7.19 (d, J=9,0 Гц, 2Н), 7.44 (d, J=7,5 Гц, 2Н), 7.53 (m, 1Н), 7.59 (m, 2Н), 7.74 (m, 3Н), 8.02 (s, 1Н), 8.33 (d, J=5,4 Гц, 1Н), 10.84 (s, 1Н)
Пример 2. 3-хлоро-4-(4-(1,5-диметил-3-оксо-2-фенил-2,3-дигидро-1Н-пиразол-4-карбоксамидо)-2-фторфенокси)пиколинамид
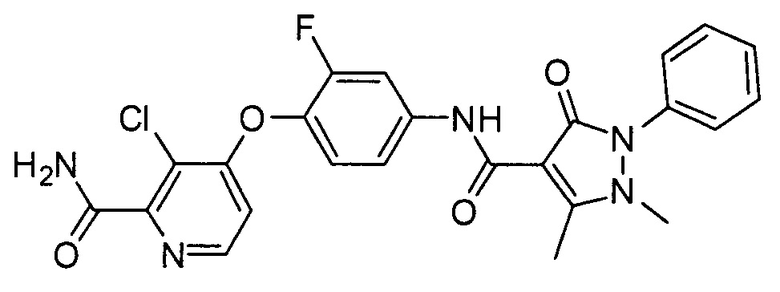
Шаг 1) 4-(4-амино-2-фторфенокси)-3-хлоропиколинамид
[0174] К раствору 4-амино-2-фторфенола (254 мг, 2,0 ммоль) в DMF (5 мл) добавили t-BuOK (359 мг, 3,2 ммоль). Смесь размешивали при к.т. в течение 30 минут. Затем добавили 3,4-дихлоропиколинамид (420 мг, 2,2 ммоль) и смесь подогревали в мироволновой печи при 120°С в течение 2 часов. Смесь охладили до к.т., погасили 25 мл воды. Полученный раствор экстрагировали EtOAc (30 мл × 3) и объединенные органические фазы промывали морской водой (30 мл × 3), высушили над Na2SO4 и сконцентрировали в вакууме. Остаток очистили колоночной хроматографией с силикагелем (EtOAc/PE (о/с) = 2/1) до титулованного соединения в виде бледно-желтой твердой фазы (306 мг, 54,4%).
MS (ESI, пол. ион) уд.з.: 282.1 [М+Н]+;
1Н NMR (400 МГц, DMSO-d6): δ (ppm) 8.30 (d, J=5,6 Гц, 1H), 8.03 (s, 1Н), 7.73 (s, 1Н), 7.03 (t, J=9,0 Гц, 1H), 6.72 (dd, J=0,8 Гц, 5,6 Гц, 1Н), 6.53 (dd, J=2,4 Гц, 13,2 Гц, 1Н), 6.44 (dd, J=1,8 Гц, 8,7 Гц, 1H), 5.55 (s, 2Н).
Шаг 2) 3 хлоро-4-(4-(1,5-диметил-3-оксо-2-фенил-2,3-дигидро-1Н-пиразол-4-карбоксамидо)-2-фторфенокси)пиколинамид
[0175] К взвеси 4-(4-амино-2-фторфенокси)-3-хлоропиколинамида (306 мг, 1,40 ммоль) и 1,5-диметил-3-оксо-2-фенил-2,3-дигидро-1Н-пиразол-4-карбоновой кислоты (390 мг, 1,68 ммоль) в DCM (6 мл) добавили EDCl (322 мг, 1,68 ммоль) и HOAT (38 мг, 0,28 ммоль). Смесь размешивали при 45°С в течение 14,5 часов, затем охладили до к.т. и погасили 5 мл воды. Смесь экстрагировали EtOAc (10 мл × 3) и объединенные органические фазы промывали морской водой (10 мл × 3), высушили над Na2SO4 и сконцентрировали в вакууме. Остаток очистили колоночной хроматографией с силикагелем (РЕ/EtOAc (о/с) = 1/4) до титулованного соединения в виде бледно-желтой твердой фазы (647 мг, 93,2%).
MS (ESI, пол. ион) уд.з.: 496.1 [М+Н]+;
1Н NMR (400 МГц, DMSO-d6): δ (ppm) 10.98 (s, 1Н), 8.33 (d, J=5,6 Гц, 1Н), 8.06 (br s, 1Н), 7.99 (dd, J=2,2 Гц, 13,2 Гц, 1Н), 7.75 (br s, 1Н), 7.60 (t, J=7,2 Гц, 2Н), 7.52 (m, 1Н), 7.45 (d, J=5,6 Гц, 2Н), 7.35 (m, 2Н), 6.84 (d, J=5,5 Гц, 1Н), 3.37 (s, 3Н), 2.71 (s, 3Н).
Пример 3. 4-(4-(1,5-диметил-3-оксо-2-фенил-2,3-дигидро-1Н-пиразол-4-карбоксамидо)-2,3-дифторфенокси)пиколинамид
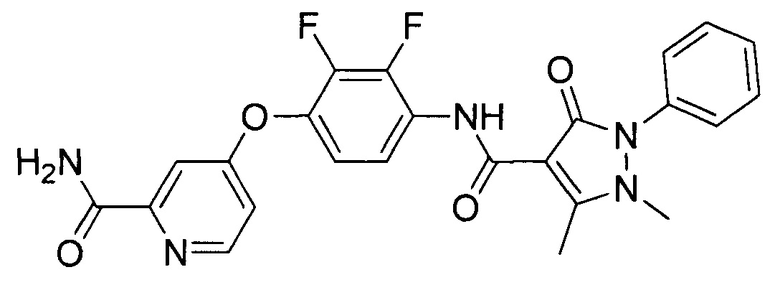
Шаг 1) 1-(бензилокси)-2,3-дифтор-4-нитробензол
[0176] К раствору 2,4,5-трифторнитробензола (5,00 г, 28,2 ммоль) и бензилового спирта (3,07 г, 28,4 ммоль) в DMF (10 мл) добавили K2CO3 (5,87 г, 42,5 ммоль). Реакционный раствор размешивали при к.т. в течение 72 часов, затем разбавили водой (35 мл) и далее быстро вымешивали при 4°С. Осадок собрали через фильтрацию, промывали водой (20 мл) и очищали колоночной хроматографией с силикагелем (EtOAc/PE (о/с) = 1/20) до титулованного соединения в виде бледно-желтой твердой фазы (2,15 г, 28,7%).
1Н NMR (400 МГц, CDC13): δ (ppm) 7.90 (m, 1Н), 7.43 (s, 2Н), 7.42 (s, 2Н), 7.39 (m, 1Н), 6.86 (m, 1Н), 5.27 (s, 2Н).
Шаг 2) 4-амино-2,3-дифторфенол
[0177] К взвеси 1-(бензилокси)-2,3-дифтор-4-нитробензол (1,93 г, 0,73 ммоль) в СН3ОН (45 мл) и THF (9 мл) добавили Pd/C (333 мг, 6% содержание Pd, 53% ~55% содержание воды). Смесь размешивали при 32°С в течение 13 часов под атмосферой Н2. Смесь отфильтровали через CELITE®, промывали 50 мл EtOAc. Фильтрат сконцентрировали в вакууме, промывали 30 мл CH2Cl2 до титулованного соединения в виде темно-коричневой твердой фазы (0,89 г, 84%).
MS (ESI, пол. ион) уд.з.: 146.2 [М+Н]+;
1Н NMR (400 МГц, DMSO-d6): δ (ppm) 6.49 (m, 1Н), 6.38 (m, 1Н), 4.71 (s, 2Н).
Шаг 3) 4-(4-амино-2,3-дифторфенокси)пиколинамид
[0178] К раствору 4-амино-2,3-дифторфенола (208 мг, 1,43 ммоль) в DMF (4 мл) добавили t-BuOK (257 мг, 2,29 ммоль). Реакционный раствор размешивали при к.т. в течение 30 минут, затем добавили 4-хлоропиколинамид (249 мг, 1,59 ммоль). Смесь подогревали в мироволновой печи при 120°С в течение 3 часов, затем охладили до к.т. и разбавили 25 мл воды. Полученную смесь экстрагировали EtOAc (30 мл × 3) и объединенные органические фазы промывали морской водой (30 мл × 3), высушили над безводной Na2SO4 и сконцентрировали в вакууме. Остаток очистили колоночной хроматографией с силикагелем (РЕ/EtOAc (о/с) = 1/2) до титулованного соединения в виде оранжевой твердой фазы (110 мг, 41,5%).
MS (ESI, пол. ион) уд.з.: 266.0 [М+Н]+, 283.2 [M+NH4]+;
1Н NMR (400 МГц, DMSO-d6): δ (ppm) 8.42 (d, J=5,6 Гц, 1Н), 7.84 (br s, 1Н), 7.65 (d, J=2,5 Гц, 1H), 7.03 (m, 1H), 6.77 (m, 1H), 6.56 (m, 1H), 5.56 (br s, 1H), 3.08 (s, 2H).
Шаг 4) 4-(4-(1,5-диметил-3-оксо-2-фенил-2,3-дигидро-1Н-пиразол-4-карбоксамидо)-2,3-дифторфенокси)пиколинамид
[0179] К взвеси 4-(4-амино-2,3-дифторфенокси)пиколинамида (180 мг, 0,68 ммоль) и 1,5-диметил-3-оксо-2-фенил-2,3-дигидро-1Н-пиразол-4-карбоновой кислоты (161 мг, 0,69 ммоль) в DCM (4 мл) добавили EDCl (157 мг, 0,82 ммоль) и HOAT (19 мг, 0,14 ммоль). Смесь размешивали при 45°С в течение 12 часов, затем добавили больше 1,5-диметил-3-оксо-2-фенил-2,3-дигидро-1Н-пиразол-4-карбоновой кислоты (87 мг, 0,37 ммоль) и реакционный раствор далее размешивали при 45°С в течение 5 часов. Смесь охладили до к.т., погасили 5 мл воды и экстрагировали EtOAc (10 мл × 3). Объединенные органические фазы промывали морской водой (10 мл × 3), высушили над Na2SO4 и сконцентрировали в вакууме. Остаток очистили колоночной хроматографией с силикагелем (EtOAc 100%) до титулованного соединения в виде оранжевой твердой фазы (108 мг, 33,2%).
MS (ESI, пол. ион) уд.з.: 480.1 [М+Н]+;
1H NMR (400 МГц, DMSO-d6): δ (ppm) 11.20 (s, 1Н), 8.55 (d, J=5,7 Гц, 1Н), 8.34 (m, 1Н), 8.16 (br s, 1Н), 7.76 (br s, 1H), 7.64 (m, 3H), 7.59 (d, J=7,8 Гц, 1H), 7.46 (m, 3H), 7.28 (m, 1H), 3.38 (s, 3H), 2.71 (s, 3H).
Пример 4. 4-(4-(1,5-диметил-3-оксо-2-фенил-2,3-дигидро-1Н-пиразол-4-карбоксамидо)-2,5-дифторфенокси)пиколинамид
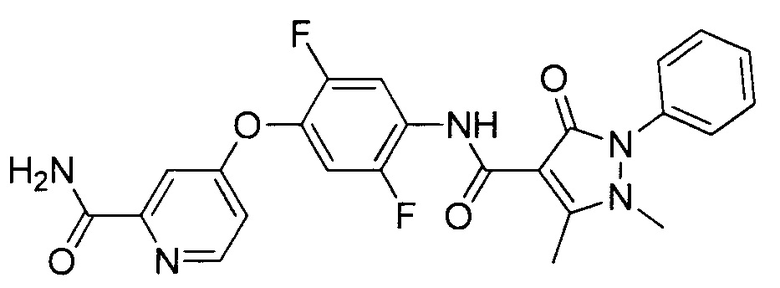
Шаг 1) 1-(бензилокси)-2,5 дифтор-4-нитробензол
[0180] К раствору 2,4,5-трифторнитробензол (5,4 г, 30,5 ммоль) и бензилового спирта (3,2 мл, 30,5 ммоль) в DMF (20 мл) добавили K2CO3 (6,33 г, 46,1 ммоль). Реакционный раствор размешивали при комнатной температуре в течение 72 часов. Воду (60 мл) добавили при 0°С и полученную смесь далее размешивали при 4°С в течение 24 часов. Твердую фазу собрали фильтрацией, промывали 30 мл воды и высушили в вакууме при 45°С для титулованного соединения в виде бледно-желтой твердой фазы (6,0 г, 74%).
1Н NMR (400 МГц, CDC13): δ (ppm) 5.22 (s, 2Н), 6.85-6.89 (m, 1Н), 7.40-7.43 (m, 5Н), 7.89-7.94 (m, 1Н).
Шаг 2) 4-амино-2,5-дифторфенол
[0181] К взвеси 1-(бензилокси)-2,5-дифтор-4-нитробензол (1,06 г, 4 ммоль) в СН3ОН (25 мл) и THF (5 мл) добавили Pd/C (50% содержание Pd, 185 мг). Реакционный раствор размешивали при 32°С под атмосферой Н2 в течение 10 часов. Смесь отфильтровали через CELITE® и фильтрат сконцентрировали в вакууме. Остаток промывали DCM (15 мл) до титулованного соединения в виде темно-коричневой твердой фазы (500 мг, 86%).
MS (ESI, пол. ион) уд.з.: 146.2 [М+Н]+;
1Н NMR (400 МГц, DMSO - <ФК): δ (ppm) 4.68 (s, 2Н), 6.53-6.65 (m, 2Н), 9.06 (br, 1H).
Шаг 3) 4-(4-амино-2,5-дифторфенокси)пиколинамид
[0182] К смеси 4-амино-2,5-дифторфенол (100 мг, 0,64 ммоль) и 4-хлоропиколинамид (110 мг, 0,71 ммоль) в DMF (2 мл) добавили NaH (80 мг, 1,3 ммоль, 60%, распыленный в минеральном масле). Реакционную смесь подогрели в мироволновой печи при 120°С в течение 1,5 час, затем охладили до к.т., разбавили водой (20 мл) и экстрагировали EtOAc (30 мл × 3). Объединенные органические фазы промывали морской водой (80 мл), высшили над Na2SO4 и сконцентрировали в вакууме. Остаток очистили колоночной флэш-хроматографией (EtOAc/PE (о/с) = 4/1) до титулованного соединения в виде коричневой твердой фазы (52 мг, 26%).
MS (ESI, пол. ион) уд.з.: 266.2 [М+Н]+.
Шаг 4) 4-(4-(1,5-диметил-3-оксо-2-фенил-2,3-дигидро-1Н-пиразол-4-карбоксамидо)-2,5-дифторфенокси)пиколинамид
[0183] К раствору 4-(4-амино-2,5-дифторфенокси)пиколинамид (200 мг, 0,76 ммоль) и 1,5-диметил-3-оксо-2-фенил-2,3-дигидро-1Н-пиразол-4-карбоновой кислоты (165 мг, 0,75 ммоль) в DCM (10 мл) добавили EDCl (175 мг, 0,93 ммоль) и HOAT (26 мг, 0,15 ммоль). Реакционный раствор размешивали при 45°С в течение 16 часов, охладили до к.т. и разбавили EtOAc (20 мл). Твердую фазу собрали через фильтрацию, быстро высушили при 45°С в вакууме до титулованного соединения в виде белой твердой фазы (230 мг, 63,7%).
MS (ESI, пол. ион) уд.з.: 480.1 [М+Н]+;
1Н NMR (400 МГц, DMSO-d6): δ (ppm) 11.24 (s, 1Н), 8.53-8.57 (m, 2Н), 8.15 (s, 1Н), 7.75 (s, 1Н), 7.53-7.59 (m, 4Н), 7.44-7.45 (m, 3Н), 7.24-7.25 (d, J=5,2 Гц, 1Н), 3.43 (s, 3Н), 2.70 (s, 3Н).
Пример 5. 3-хлоро-4-(4-(1,5-диметил-3-оксо-2-фенил-2,3-дигидро-1Н-пиразол-4-карбоксамидо)-2,5-дифторфенокси)пиколинамид
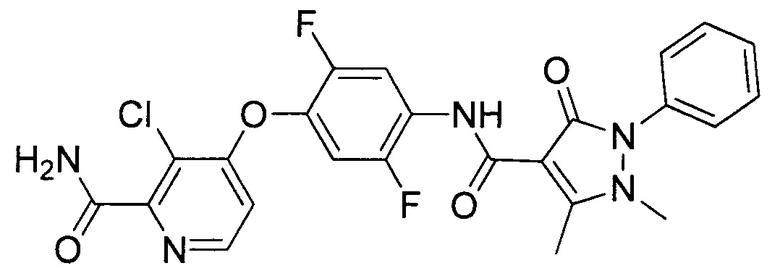
[0184] К раствору N-(2,5-дифтор-4-гидроксифенил)-1,5-диметил-3-оксо-2-фенил-2,3-дигидро-1Н-пиразол-4-карбоксамид (395 мг, 1,1 ммоль) в DMF (5,0 мл) добавили t-BuOK (202 мг, 1,8 ммоль) и смесь размешивали при к.т. в течение 30 минут. Затем добавили 3,4-дихлоропиколинамид (190 мг, 1,0 ммоль) и смесь подогревали в мироволновой печи при 120°С в течение 2 часов, затем охладили до к.т., погасили водой (30 мл) и экстрагировали EtOAc (50 мл × 3). Объединенные органические фазы промывали морской водой (50 мл × 3), высушили над безводной Na2SO4 и сконцентрировали в вакууме. Остаток очистили колоночной хроматографией с силикагелем (MeOH/DCM (о/с) = 1/30) до титулованного соединения в виде бледно-желтой твердой фазы (310 мг, 60%).
MS (ESI, пол. ион) уд.з.: 514.2 [МТН]+;
1Н NMR (400 МГц, DMSO-d6): δ (ppm) 2.70 (s, 3Н), 3.38 (s, 3Н), 6.96 (d, J=5,5 Гц, 1Н), 7.45 (d, J=7,0 Гц, 2Н), 7.51-7.55 (m, 1Н), 7.58-7.66 (m, 3Н), 7.75 (s, 1Н), 8.05 (s, 1Н), 8.34 (d, J=5,5 Гц, 1Н), 8.53-8.58 (m, 1Н), 11.25 (s, 1H)
Пример 6. 4-(3-хлоро-4-(1,5-диметил-3-оксо-2-фенил-2,3-дигидро-1Н-пиразол-4-карбоксамидо) фенокси)пиколинамид
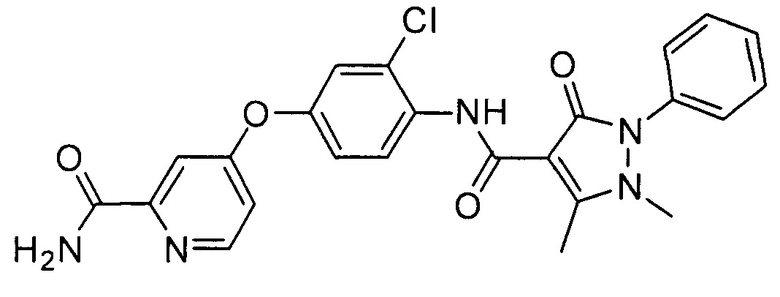
Шаг 1) 4-(4-амино-3-хлорофенокси)пиколинамид
[0185] К смеси 4-амино-2-хлорофенол гидрохлорида (446 мг, 2,4 ммоль) в DMSO (4 мл) добавили NaH (280 мг, 7,0 ммоль, 60%, распыленный в минеральном масле). Реакционный раствор размешивали при к.т. в течение 30 минут с последующим добавлением 4-хлоропиколинамида (345 мг, 2,2 ммоль). Реакционный раствор подогревали в мироволновой печи при 160°С в течение 2 часов, затем охладили до к.т. и разбавили водой (20 мл). Полученную смесь экстрагировали этилацетатом (20 мл × 3) и объединенные органические фазы промывали морской водой (20 мл), высушили над безводной Na2SO4 и сконцентрировали в вакууме. Остаток очистили колоночной хроматографией с силикагелем (EtOAc/РЕ (о/с) = 1/1) до титулованного соединения в виде оранжевой твердой фазы (170 мг, 29%).
MS (ESI, пол. ион) уд.з.: 264.1 [М+Н]+;
1Н NMR (400 МГц, DMSO-d6): δ (ppm) 5.45 (s, 2Н), 6.89 (d, J=8,7 Гц, 1Н), 6.92 (m, 1Н), 7.11 (m, 1Н), 7.16 (d, J=2,6 Гц, 1Н), 7.34 (d, J=2.6 Hz, 1H), 7.68 (s, 1H), 8.10 (s, 1H), 8.47 (d, J=5,6 Гц, 1H).
Шаг 2) 4-(3-хлоро-4-(1,5-диметил-3-оксо-2-фенил-2,3-дигидро-1Н-пиразол-4-карбоксамидо) фенокси)пиколинамид
[0186] К взвеси 4-(4-амино-3-хлорофенокси)пиколинамида (191 мг, 0,72 ммоль) и 1,5-диметил-3-оксо-2-фенил-2,3-дигидро-1Н-пиразол-4-карбоновой кислоты (168 мг, 0,72 ммоль) в DCM (10 мл) добавили EDCl (166 мг, 0,86 ммоль) и HOAT (20 мг, 0,14 ммоль). Реакционный раствор размешивали при 46°С в течение 6 часов с последующим добавлением 1,5-диметил-3-оксо-2-фенил-2,3-дигидро 1Н-пиразол-4-карбоновой кислоты (32 мг, 0,14 ммоль) и EDCl (27 мг, 0,14 ммоль). Смесь далее размешивали при 46°С в течение 13 часов, затем охладили до к.т. и разбавили водой (10 мл). Полученную смесь экстрагировали этилацетатом (10 мл × 3) и объединенные органические фазы промывали морской водой (10 мл), высушили над безводной Na2SO4 и сконцентрировали в вакууме. Остаток очистили колоночной хроматографией с силикагелем (DCM/MeOH (о/с) = 50/1) до титулованного соединения в виде бледно-желтой твердой фазы (160 мг, 46,5%).
MS (ESI, пол. ион) уд.з.: 478.2 [М+Н]+;
1H NMR (400 МГц, DMSO-d6): δ (ppm) 2.71 (s, 3Н), 3.37 (s, 3Н), 7.19 (m, 1Н), 7.23 (m, 1H), 7.43 (m, 3H), 7.50 (m, 2H), 7.60 (m, 2H), 7.72 (s, 1H), 8.13 (s, 1H), 8.52 (d, J=5,6 Гц, 1H), 8.63 (d, J=9,1 Гц, 1H), 11.19 (s, 1H).
Пример 7. 4-((6-(1,5-диметил-3-оксо-2-фенил-2,3-дигидро-1Н-пиразол-4-карбоксамидо)пиридин-3-ил)окси)пиколинамид
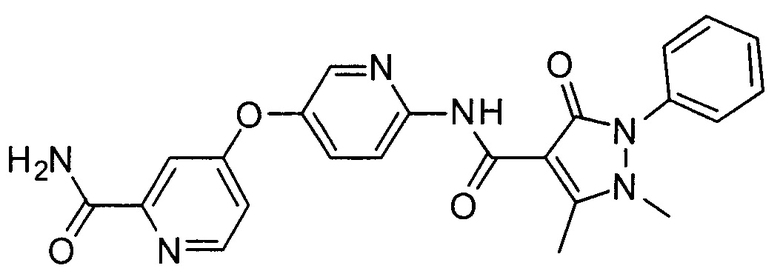
Шаг 1) 4-((6-аминопиридин-3-ил)окси)пиколинамид
[0187] К смеси 6-аминопиридин-3-ол (220 мг, 2 ммоль) и t-BuOK (225 мг, 2,16 ммоль) в DMF (2,5 мл) добавили 4-хлоропиколинамид (315 мг, 2 ммоль). Реакционный раствор нагревали при 80°С в течение 5 часов, затем охладили до к.т. и разбавили EtOAc (50 мл) и H2O (50 мл). Органическую фазу сконцентрировали в вакууме и остаток очистили колоночной хроматографией с силикагелем (DCM/СН3ОН (о/с) = 30/1) до титулованного соединения в виде коричневой твердой фазы (230 мг, 50%).
MS (ESI, пол. ион) уд.з.: 231.1 [М+Н]+;
1Н NMR (400 МГц, CDC13): δ (ppm) 6.09 (s, 2Н), 6.53-6.56 (d, J=8,88 Гц, 1Н), 7.12-7.14 (dd, J=2,64 Гц, 5,6 Гц, 1Н), 7.31-7.34 (dd, J=2,92 Гц, 8,88 Гц, 1Н), 7.35-7.36 (d, J=2,48 Гц, 1Н), 7.70 (s, 1Н), 7.83-7.84 (d, J=2.8 Hz, 1H), 8.11 (s, 1H), 8.46-8.49 (d, J=5.6 Hz, 1H).
Шаг 2) 4-((6-(1,5-диметил-3-оксо-2-фенил-2,3-дигидро-1H-пиразол-4-карбоксамидо)пиридин-3-ил)окси)пиколинамид
[0188] К взвеси 4-((6-аминопиридин-3-ил)окси)пиколинамид (230 мг, 1 ммоль) и 1,5-диметил-3-оксо-2-фенил-2,3-дигидро-1Н-пиразол-карбоновой кислоты 4 (237 мг, 1,02 ммоль) в DCM (5 мл) добавили EDCl (230 мг, 1,2 ммоль) и НОАТ (27 мг, 0,2 ммоль). Реакционный раствор размешивали при 45°С в течение 28 часов, затем охладили до к.т. и разбавили водой (10 мл) и DCM (20 мл). Органическую фазу сконцентрировали в вакууме и остаток очистили колоночной хроматографией с силикагелем (DCM/СН3ОН (о/с) = 40/1) до титулованного соединения в виде светло-серой твердой фазы (111 мг, 25%).
MS (ESI, пол. ион) уд.з.: 445.1 [М+Н]+;
1Н NMR (400 МГц, DMSO-d6): δ (ppm) 2.72 (s, 3Н), 3.33 (s, 3Н), 7.20-7.22 (dd, J=2.64 Hz, 5,64 Гц, 1Н), 7.43-7.46 (m, 3Н), 7.52-7.54 (m, 1Н), 7.58-7.62 (m, 2Н), 7.72 (s, 1Н), 7.75-7.78 (dd, J=2,88 Гц, 8,96 Гц, 1Н), 8.13 (s, 1Н), 8.27-8.28 (d, J=2,68 Гц, 1Н), 8.34-8.36 (d, J=9,08 Гц, 1Н), 8.52-8.54 (d, J=5,6 Гц, 1Н), 11.26 (s, 1Н)
Пример 8. N-(5-((2-(циклопропанкарбоксамидо)пиридин-4-ил)окси)пиридин-2-ил)-1,5-диметил-3-оксо-2-фенил-2,3-дигидро-1Н-пиразол-4-карбоксамид
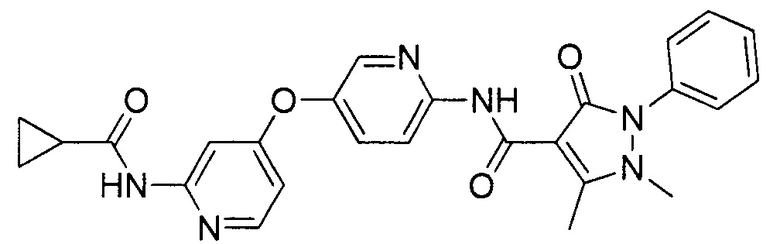
Шаг 1) N-(5-((2-аминопиридин-4-ил)окси)пиридин-2-ил)-1,5-диметил-3-оксо-2-фенил-2,3-дигидро-1Н-пиразол-4-карбоксамид
[0189] К раствору 4-((6-(1,5-диметил-3-оксо-2-фенил-2,3-дигидро-1Н-пиразол-4-карбоксамидо)пиридин-3-ил)окси)пиколинамид (111 мг, 0,25 ммоль) в EtOAc (2 мл), CH3CN (2 мл) и H2O (1 мл) добавили PhI (ОАс)2 (97 мг, 0,3 ммоль). Реакционный раствор размешивали при 0°С в течение 30 минут, затем подогрели до к.т. и далее вымешивали в течение 12 часов. Смесь сконцентрировали в вакууме и остаток очистили колоночной хроматографией с силикагелем (DCM/СН3ОН (о/с) = 40/1) до титулованного соединения в виде светло-бежевой твердой фазы (85 мг, 81,7%).
MS (ESI, пол. ион) уд.з.: 417.2 [М+Н]+;
1Н NMR (400 МГц, DMSO-d6): δ (ppm) 2.71 (s, 3Н), 3.38 (s, 3Н), 5.83 (s, 1Н), 5.98 (s, 2Н), 6.17 (s, 1Н), 7.08-7.10 (m, 2Н), 7.42-7.81 (m, 6Н), 7.80-7.81 (d, J=5.8 Hz, 1Н), 8.17 (s, 1H), 8.29-8.31 (d, J=8,56 Гц, 1H), 11.19 (s, 1H).
Шаг 2) N-(5-((2-(циклопропанкарбоксамидо)пиридин-4-ил)окси)пиридин-2-ил)-1,5-диметил-3-оксо-2-фенил-2,3-дигидро-1Н-пиразол-4-карбоксамид
[0190] К взвеси N-(5-((2-аминопиридин-4-ил)окси)пиридин-2-ил)-1,5-диметил-3-оксо-2-фенил-2,3-дигидро-1Н-пиразол-4-карбоксамид (500 мг, 1,2 ммоль) в DCM (5 мл) и триэтиламин (1 мл) медленно добавили циклопропанкарбонила хлорид (377 мг, 3,6 ммоль)при 0°С. Смесь подогрели до к.т. и вымешивали в течение 4,5 часов, затем разбавили DCM (10 мл) и водой (10 мл). Органическую фазу отделили и сконцентрировали в вакууме. Остаток растворили в МеОН (30 мл) и насыщенном растворе Na2CO3 (30 мл). Полученную смесь размешивали при к.т. в течение 2 часов, затем отфильтровали и фильтрат сконцентрировали в вакууме. Остаток очистили колоночной хроматографией с силикагелем (DCM/MeOH (о/с) = 50/1) до титулованного соединения в виде грязно-белой тердой фазы (350 мг, 60%).
MS (ESI, пол. ион) уд.з.: 485.3 [М+Н]+;
ВЭЖХ: 99,7%;
1Н NMR (600 МГц, CDC13): δ (ppm) 11.25 (s, 1Н), 9.42 (s, 1Н), 8.37 (t, J=12,0 Гц, 1Н), 8.17 (t, J=7,5 Гц, 1Н), 8.10 (t, J=9,8 Гц, 1Н), 7.93 (d, J=2.1 Hz, 1Н), 7.60-7.51 (m, 2Н), 7.51-7.42 (m, 2Н), 7.41-7.36 (m, 2Н), 6.64 (dd, J=6,1 Гц, 2,4 Гц, 1Н), 5.32 (s, 1Н), 3.39 (s, 3Н), 2.81 (s, 3Н), 1.14-1.05 (m, 2Н), 0.97-0.88 (m, 2Н).
Пример 9. 3-хлоро-4-((6-(1,5-диметил-3-оксо-2-фенил-2,3-дигидро-1Н-пиразол-4-карбоксамидо)пиридин-3-ил)окси)пиколинамид
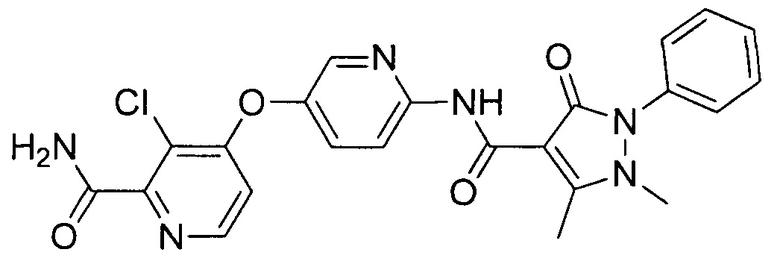
Шаг 1) N-(-5-гидроксипиридин-2-ил)-1,5-диметил-3-оксо-2-фенил-2,3-дигидро-1Н-пиразол-4-карбоксамид
[0191] К взвеси 6-аминопиридин-3-ол (330 мг, 3 ммоль) и 1,5-диметил-3-оксо-2-фенил-2,3-дигидро-1Н-пиразол-4-карбоновой кислоты (710 мг, 306 ммоль) в DMF (10 мл) добавили EDCl (690 мг, 3,6 ммоль) и НОАТ (80 мг, 0,6 ммоль). Реакционный раствор размешивали при 60°С в течение 4 часов, затем охладили до к.т. и разбавили водой (100 мл) и EtOAc (2 мл). Смесь охладили до 0°С и быстр овымешивали. Полученную твердую фазу собрали через фильтрацию до титулованного соединения в виде светло-коричневой твердой фазы (680 мг, 70%).
MS (ESI, пол. ион) уд.з.: 325.1 [М+Н]+;
1H NMR (400 МГц, DMSO-d6): δ (ppm) 2.50 (s, 3H), 3.33 (s, 3H), 7.18-7.20 (dd, J=2,3 Гц, 8,8 Гц, 1H), 7.40-7.42 (d, J=7,5 Гц, 2H), 7.48-7.52 (m, 1H), 7.56-7.60 (m, 2H), 7.81-7.82 (d, J=2,2 Гц, 1H), 7.95 (s, 1H), 8.04-8.06 (d, J=8,8 Гц, 1H), 9.61 (s, 1H), 10.85 (s, 1H).
Шаг 2) 3-хлоро-4-((6-(1,5-диметил-3-оксо-2-фенил-2,3-дигидро-1Н-пиразол-4-карбоксамидо)пиридин-3-ил)окси)пиколинамид
[0192] К смеси 6-аминопиридин-3-ол (324 мг, 1 ммоль) и t-BuOK (135 мг, 1,2 ммоль) в DMF (2 мл) добавили 3,4-дихлоропиколинамид (191 мг, 1 ммоль). Реакционный раствор нагревали при 80°С в течение 15 часов, затем охладили до к.т. и разбавили EtOAc (1 мл) и Н2О (20 мл). Смесь быстро размешивали и полученную твердую фазу собрали через фильтрацию до титулованного соединения в виде коричневой твердой фазы (290 мг, 60,5%).
MS (ESI, пол. ион) уд.з.: 479.2 [М+Н]+;
1Н NMR (400 МГц, CDC13): δ (ppm) 2.72 (s, 3Н), 3.35 (s, 3Н), 6.91-6.92 (d, J=5,5 Гц, 1Н), 6.09 (s, 2Н), 7.43-7.45 (m, 2Н), 7.50-7.54 (m, 1Н), 7.58-7.62 (m, 2Н), 7.73-7.76 (m, 2Н), 8.03 (s, 1Н), 8.26-8.27 (d, J=2,7 Гц, 1Н), 8.33-8.36 (m, 2Н), 11.26 (s, 1Н).
Пример 10. 4-(4-(1,5-диметил-3-оксо-2-фенил-2,3-дигидро-1Н-пиразол-4-карбоксамидо)-2-фторфенокси)пиколинамид
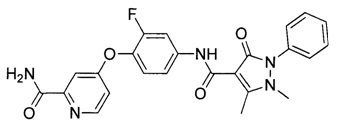
Шаг 1) N-(-3-фтор-4-гидроксифенил)-1,5-диметил-3-оксо-2-фенил-2,3-дигидро-1Н-пиразол-4-карбоксамид
[0193] К взвеси 4-амино-2-фторфенол (2,54 г, 20 ммоль) и 1,5-диметил-3-оксо-2-фенил-2,3-дигидро-1Н-пиразол-4-карбоновой кислоты (4,74 г, 20,4 ммоль) в DCM (60 мл) добавили EDCl (4,6 г, 24 ммоль) и НОАТ (0,54 г, 4 ммоль). Реакционный раствор размешивали при 45°С в течение 12 часов, затем охладили до к.т., погасив H2O (10 мл), и размешивали в течение еще 4 часов. Твердую фазу получили фильтрацией и промывали DCM (20 мл × 3), затем высушили при 60°С в вакууме в течение 12 часов до титулованного соединения в виде бледно-желтой твердой фазы (6,37 г, 93,4%).
MS (ESI, пол. ион) уд.з.: 342.1 [М+Н]+;
1Н NMR (400 МГц, DMSO-d6): δ (ppm) 10.59 (s, 1H), 9.58 (s, 1Н), 7.64 (dd, J=2,4 Гц, 13.5 Hz, 1Н), 7.60 (m, 2Н), 7.50 (m, 1H), 7.42 (m, 2Н), 6.97 (m, 1Н), 6.88 (dd, J=9,6 Гц, 8,8 Гц, 1Н), 3.34 (s, 3Н), 2.70 (s, 3Н).
Шаг 2) 4-(4-(1,5-диметил-3-оксо-2-фенил-2,3-дигидро-1Н-пиразол-4-карбоксамидо)-2-фторфенокси)пиколинамид
[0194] К взвеси N-(3-фтор-4-гидроксифенил)-1,5-диметил-3-оксо-2-фенил-2,3-дигидро-1Н-пиразол-4-карбоксамид (2,2 г, 6,4 ммоль), 4-хлоропиколинамид (1 г, 6,39 ммоль) в DMSO (12 мл) добавили NaH (615 мг, 12,3 ммоль, 50%, распыленный в минеральном масле). Реакционный раствор размешивали при 160°С в течение 20 ч, затем охладили до к.т. и разбавили с H2O (70 мл). Смесь экстрагировали EtOAc (100 мл × 3). Объединенные органические фазы промывали морской водой (100 мл), высушили над Na2SO4 и сконцентрировали в вакууме. Остаток очистили колоночной хроматографией с силикагелем (РЕ/EtOAc (о/с) = 1/4) до титулованного соединения в виде белой твердой фазы (0,85 г, 29%).
MS (ESI, пол. ион) уд.з.: 462.1 [М+Н]+;
1Н NMR (400 МГц, CDC13): δ (ppm) 10.87 (s, 1Н), 8.40-8.41 (d, J=5,6 Гц, 1Н), 7.88-7.92 (dd, J=2,4 Гц, 12,6 Гц, 1Н), 7.82-7.83 (d, J=3,9 Гц, 1H), 7.71-7.71 (d, J=2.5 Гц, 1Н), 7.54-7.58 (m, 2Н), 7.46-7.49 (m, 1Н), 7.35-7.37 (d, J=8,6 Гц, 2Н), 7.07-7.11 (m, 1Н), 6.96-6.98 (dd, J=2,5 Гц, 5,6 Гц, 1Н), 5.56 (s, 1H), 3.37 (s, 3Н), 2.79 (s, 3Н).
Пример 11. N-(4-((2-(циклопропанскарбоксамидо)пиридин-4-ил)окси)-3-фторфенил)-1,5-диметил-3-оксо-2-фенил-2,3-дигидро-1Н-пиразол-4-карбоксамид
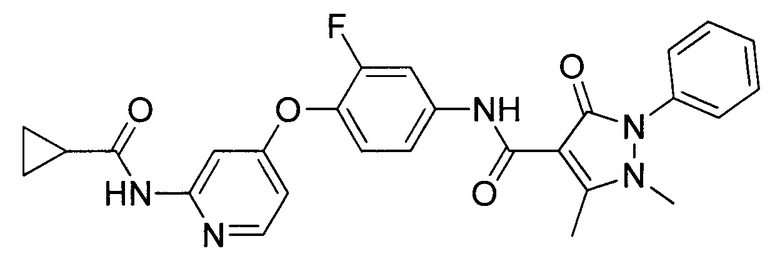
Шаг 1) N-4-((2-аминопиридин-4-ил)окси)-3-фторфенил)-1,5-диметил-3-оксо-2-фенил-2,3-дигидро-1Н-пиразол-4-карбоксамид
[0195] Раствор 4-(4-(1,5-диметил-3-оксо-2-фенил-2,3-дигидро-1Н-пиразол-4-карбоксамид)-2 фторфенокси)пиколинамид (0,4 г, 0,86 ммоль) и PhI (ОАс) 2 (419 мг, 1,5 ммоль) в смеси EtOAc (8 мл), MeCN (8 мл) и H2O (4 мл) охладили до 0°С и размешивали в течение 30 минут. Реакционный раствор затем подогрели до к.т. и размешивали в течение еще 8 часов. Смесь разбавили NaHCO3 (вод., 60 мл) и экстрагировали EtOAc (100 мл × 3). Объединенные органические фазы промывали морской водой, высушили над Na2SO4 и сконцентрировали в вакууме. Остаток очистили колоночной хроматографией с силикагелем (DCM/MeOH (о/с) = 10/1) до титулованного соединения в виде белой твердой фазы (0,21 г, 56%).
MS (ESI, пол. ион) уд.з.: 434.2 [М+Н]+;
1Н NMR (400 МГц, CDC13): δ (ppm) 10.83 (s, 1Н), 7.91-7.92 (d, J=5,9 Гц, 1Н), 7.83-7.86 (dd, J=2,4 Гц, 10,1 Гц, 1Н), 7.56-7.58 (m, 2Н), 7.46-7.52 (d, J=5,9 Гц, 2Н), 7.35-7.37 (d, J=8,6 Гц, 2Н), 7.04-7.09 (m, 1Н), 6.29-6.31 (m, 1Н), 5.92-5.93 (d, J=2,1 Гц, 1Н), 4.45 (s, 2Н), 3.37 (s, 3Н), 2.79 (s, 3Н).
Шаг 2) N-(4-((2-циклопропанкарбоксамидо)пиридин-4-ил)окси)-3-фторфенил)-1,5-диметил-3-оксо-2-фенил-2,3-дигидро-1Н-пиразол-4-карбоксамид
[0196] К раствору N-(4-((2-аминопиридин-4-ил)окси)-3-фторфенил)-1,5-диметил-3-оксо-2-фенил-2,3-дигидро-1Н-пиразол-4-карбоксамид (217 мг, 0,5 ммоль) в DCM (5 мл) добавили Et3N (253 мг, 2,5 ммоль) и циклопропанкарбонила хлорид (125 мг, 1,2 ммоль)при 0°С. Реакционную смесь размешивали при к.т. в течение 4 часов, погасили с H2O (10 мл) и экстрагировали DCM (50 мл × 3). Объединенные органические фазы промывали морской водой (50 мл × 3), высушили над безводной Na2SO4H и сконцентрировали в вакууме. Остаток растворили в СН3ОН (20 мл) и раствор Na2CO3 (106 мг, 1,0 ммоль) в H2O (1 мл). Полученную смесь размешивали при к.т. в течение 1 часа и сконцентрировали в вакууме. Остаток очистили колоночной хроматографией с силикагелем (чистый DCM) до титулованного соединения в виде бежевой твердой фазы (158 мг, 63,2%).
MS (ESI, пол. ион) уд.з.: 502.3 [М+Н]+;
1Н NMR (600 МГц, CDCl3): δ (ppm) 10.93 (s, 1H), 10.73 (s, 1Н), 8.03 (d, J=6,0 Гц, 2Н), 7.96 (dd, J=12,5 Гц, 2,3 Гц, 1Н), 7.59 (t, J=7,8 Гц, 2Н), 7.51 (d, J=7,5 Гц, 1H), 7.38 (d, J=7,4 Гц, 2H), 7.30 (d, J=9,2 Гц, 1H), 7.12 (t, J=8,7 Гц, 1H), 6.74 (dd, J=6,3 Гц, 1,9 Гц, 1H), 3.40 (s, 3H), 2.81 (s, 3H), 1.79 (m, 1H), 1.12 (m, 2H), 0.97 (m, 2H).
Пример 12. 4-(5-хлоро-4-(1,5-диметил-3-оксо-2-фенил-2,3-дигидро-1Н-пиразол-4-карбоксамидо)-2-фторфенокси)пиколинамид
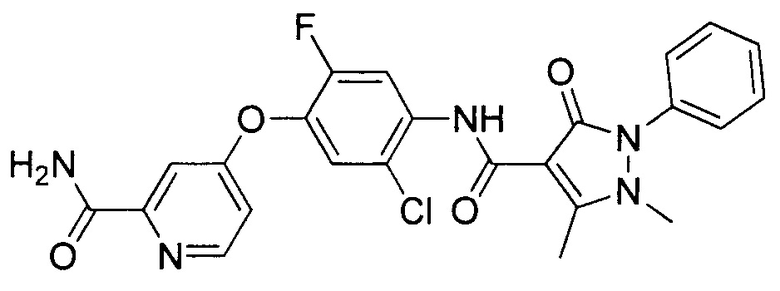
Шаг 1) 1-хлоро-4,5-дифтор-2-нитробензол
[0197] В колбу добавили 4-хлоро-1,2-дифторбензол (8,97 г, 60,4 ммоль) с последующим добавлением 98% кон. H2SO4 (16,1 мл, 302 ммоль) и 65% кон. HNO3 (5,0 мл, 66,4 ммоль)при 0°С. Смесь размешивали при к.т. в течение 5 часов, затем влили в воду со льдом (500 мл). Полученную смесь экстрагировали этилацетатом (200 мл × 3). Объединенную органическую фазу промывали насыщенным водным раствором NaHCO3 (200 мл × 2) и морской водой (200 мл), высушили над безводной Na2SO4 и сконцентрировали в вакууме до титулованного соединения в виде желтой жидкости (11,31 г, 96,7%).
1Н NMR (400 МГц, CDC13): δ (ppm) 7.41-7.45 (m, 1Н), 7.86-7.90 (m, 1Н).
Шаг 2) калий 5-хлоро-2-фтор-4-нитрофенолат
[0198] Смесь 1-хлоро-4,5-дифтор-2-нитробензол (5,12 г, 26,5 ммоль) и 15% водный раствор KOH (19,9 г) размешивали с обратным холодильником в течение 3 часов, затем охладили до к.т. и отфильтровали до титулованного соединения в виде желтых кристаллоидов (5.67 г, 93,3%).
1Н NMR (400 МГц, DMSO-d6): δ (ppm) 6.20 (d, J=13,2 Гц, 1Н), 7.70 (d, J=8,6 Гц, 1Н).
Шаг 3) 4-амино-5-хлоро-2-фторфенол
[0199] К раствору калия 5-хлоро-2-фтор-4-нитрофенолата (1,0 г, 4,35 ммоль, Yuxiang) в 95% EtOH (22 мл) и H2O (8 мл) добавили Fe (0,97 г, 17,4 ммоль) и NH4Cl (1,86 г, 34,8 ммоль). Смесь размешивали при к.т. в течение 10 часов, затем разбавили метанолом (100 мл) и этилацетатом (100 мл). Отфильтровали, а фильтрат сконцентрировали в вакууме. Остаток разбавили в воде (50 мл) и этилацетате (50 мл). Органическую фазу отделили и водную фазу экстрагировали этилацетатом (50 мл × 2). Объединенные органические уровни промывали морской водой (50 мл × 3), высушили над безводной Na2SO4 и сконцентрировали в вакууме до титулованного соединения в виде бледной твердой фазы (0.6 г, 85,3%).
1Н NMR (400 МГц, DMSO-d6): δ (ppm) 4.90 (s, 2Н), 6.60 (d, J=12,9 Гц, 1Н), 6.79 (d, J=8,8 Гц, 1Н), 9.11 (s, 1Н).
Шаг 4) N-(2 хлоро-5-фтор-4-гидроксифенил)-1,5-диметил-3-оксо-2-фенил-2,3-дигидро-1Н-пиразол-4-карбоксамид
[0200] К взвеси 4 амино-5-хлоро-2-фторфенол (0,97 г, 6 ммоль) и 1,5-диметил-3-оксо-2-фенил-2,3-дигидро-1Н-пиразол-4-карбоновая кислота (1,42 г, 6,12 ммоль) в DMF (20 мл) добавили EDCl (0,38 мг, 7,2 ммоль) и HOAT (0,16 г, 1,2 ммоль). Смесь была разогрета до 80°С и вымешивалась в течение 24 часов. Затем добавили H2O (200 мл) и EtOAc (2 мл). Полученную смесь размешивали при 0°С в течение 2 часов, затем отфильтровали до титулованного соединения в виде светло-коричневой твердой фазы (1,2 г, 53,2%).
MS (ESI, пол. ион) уд.з.: 376.1 [М+Н]+;
1Н NMR (400 МГц, DMSO-d6): δ (ppm) 2.68 (s, 3Н), 3.34 (s, 3Н), 7.02-7.04 (d, J=8,8 Гц, 1Н), 7.41-7.43 (m, 2Н), 7.48-7.52 (m, 1Н), 7.56-7.60 (m, 2Н), 829-8.33 (d, J=13,8 Гц, 1Н), 10.08 (s, 1Н), 10.95 (s, 1Н).
Шаг 5) 4-(5-хлоро-4-(1,5-диметил-3-оксо-2-фенил-2,3-дигидро-1Н-пиразол-4-карбоксамидо)-2-фторфенокси)пиколинамид
[0201] К взвеси N-(2-хлоро-5-фтор-4-гидроксифенил)-1,5-диметил-3-оксо-2-фенил-2,3-дигидро-1Н-пиразол-4-карбоксамид (751,6 мг, 2 ммоль) и t-BuOK (224,4 мг, 2 ммоль) в DMF (4 мл) добавили 4-хлоропиколинамид (313,2 мг, 2 ммоль). Реакцию подогревали до 120°С и вымешивали в течение 15 часов. После охлаждения смеси до к.т., добавили H2O (40 мл) и полученную смесь быстро размешивали при к.т. Отфильтрованный и отжатый осадок очистили колоночной хроматографией с силикагелем (DCM/СН3ОН (о/с) = 50/1) до титулованного соединения в виде светло-коричневой твердой фазы (290 мг, 60,5%).
MS (ESI, пол. ион) уд.з.: 496.2 [М+Н]+;
1NMR (400 МГц, DMSO-d6): δ (ppm) 11.37 (s, 1Н), 8.69-8.66 (d, J=13,4 Гц, 1Н), 8.55-8.54 (d, J=5,6 Гц, 1H), 8.15 (s, 1Н), 7.78-7.75 (m, 2Н), 7.62-7.58 (m, 2Н), 7.55-7.51 (m, 1Н), 7.46-7.43 (m, 3Н), 7.26-7.24 (dd, J=5,6 Гц, 2,6 Гц, 1Н), 3.38 (s, 3Н), 2.72 (s, 3Н).
Пример 13. 4-(2-хлоро-4-(1,5-диметил-3-оксо-2-фенил-2,3-дигидро-1H-пиразол-4-карбоксамидо)фенокси)пиколинамид
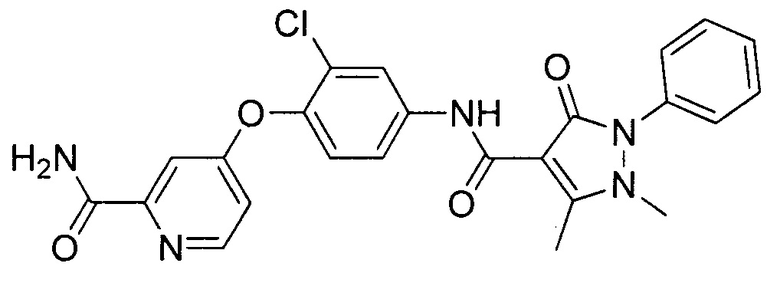
Шаг 1) N-(3-хлоро-4-гидроксифенил)-1,5-диметил-3-оксо-2-фенил-2,3-дигидро-1Н-пиразол-4-карбоксамид
[0202] К взвеси 4-амино-2-хлорофенол (4,0 г, 28,00 ммоль) и 1,5-диметил-3-оксо-2-фенил-2,3-дигидро-1Н-пиразол-4-карбоновая кислота (7,4 г, 30,11 ммоль) в DCM (70 мл) добавили EDCl (6,65 г, 30,11 ммоль) и HOAT (0,76 г, 5,68 ммоль). Смесь размешивали при 45°С в течение 20 часов, затем охлаждали до к.т. и отфильтровали. Отжатый осадок промывали DCM (20 мл × 3) и быстро высушили при 50°С в вакуумной духовке до титулованного соединения в виде серой твердой фазы (7,1 г, 72,1%).
MS (ESI, пол. ион) уд.з.: 358.1 [М+Н]+;
1HNMR (400 МГц, DMSO-d6): δ (ppm) 10.56 (s, 1Н), 9.92 (s, 1Н), 7.59 (m, 2Н), 7.50 (m, 1Н), 7.42 (m, 2Н), 7.83 (dd, J=2,6 Гц, 8,7 Гц, 1Н), 6.90 (d, J=8,7 Гц, 1H), 6.88 (dd, J=9,6 Гц, 8,8 Гц, 1Н), 3.33 (s, 3Н), 2.68 (s, 3Н).
Шаг 2) 4-(2-хлоро-4(1,5-диметил-3-оксо-2-фенил-2,3-дигидро-1Н-пиразол-4-карбоксамидо)фенокси)пиколинамид
[0203] К взвеси N-(3 хлоро-4 гидроксифенил)-1,5-диметил-3-оксо-2-фенил-2,3-дигидро-1Н-пиразол-4-карбоксамид (1,074 г, 3,0 ммоль) в DMF (12 мл) добавили t-BuOK (539 мг, 4,8 ммоль). Смесь размешивали при к.т. в течение 30 минут, затем добавили 4-хлоропиколинамид (517 мг, 3,3 ммоль). Смесь размешивали при 120°С в течение 36 часов, затем охладили до к.т., погасив 50 мл воды и экстрагировав EtOAc (50 мл × 3).
Объединенные органические фазы промыли морской водой (50 мл × 3), высушили над безводной Na2SO4 и концентрировали в вакууме. Остаток очистили колоночной хроматографией с силикагелем (EtOAc/РЕ (о/с) = 3/1) до титулованного соединения в виде белой твердой фазы (580 мг, 40,4%).
MS (ESI, пол. ион) уд.з.: 478.0 [М+Н]+;
1HNMR (400 МГц, DMSO-d6): δ (ppm) 10.95 (s, 1Н), 8.53 (d, J=5,6 Гц, 1Н), 8.16 (d, J=2,4 Гц, 1Н), 8.13 (br s, 1Н), 7.72 (br s, 1H), 7.60 (m, 2H), 7.51 (m, 2H), 7.44 (d, J=7.3 Hz, 2H), 7.38 (d, J=8,8 Гц, 1H), 7.32 (d, J=2,6 Гц, 1H), 7.17 (dd, J=2,6 Гц, 5,6 Гц, 1H), 3.16 (d, J=5,2 Гц, 3Н), 2.70 (s, 3Н)
Пример 14. 4-(4-(1,5-диметил-3-оксо-2-фенил-2,3-дигидро-1Н-пиразол-4-карбоксамидо)-3-фторфенокси)пиколинамид
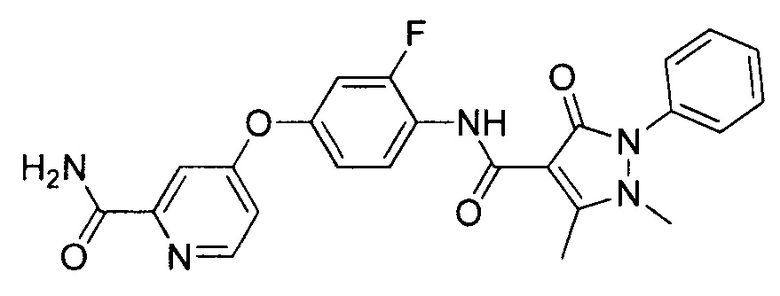
Шаг 1) 4-амино-3-фторфенол
[0204] Взвесь 3-фтор-4-нитрофенола (2,0 г, 12,73 ммоль), 10%-й Pd/C (0,4 г) и HCOOK (8,75 г, 101,85 ммоль) в НСООК (70 мл / 20 мл) размешивали в 50°С в течение 5 часов, затем охладили до к.т. и профильтровали через CELITE®. Фильтрат разбавили водой (30 мл) и экстрагировали THF (50 мл). Органический уровень высушили над Na2SO4 и сконцентрировали в вакууме. Остаток разбавили водой (50 мл) и экстрагировали DCM (50 мл). Органический уровень высушили над безводным Na2SO4 и сконцентрировали в вакууме до титулованного соединения в виде коричневой твердой фазы (1,17 г, 72,3%).
MS (ESI, пол., ион) уд.з.: 128.1 [М+Н]+, К.т. = 0,204 мин.
Шаг 2) N-(2-фтор-4-гидроксифенил)-1,5-диметил-3-оксо-2-фенил-2,3-дигидро-1Н-пиразол-4-карбоксамид
[0205] К взвеси 4-амино-3-фторфенола (1,0 г, 7,87 ммоль) и 1,5-диметил-3-оксо-2-фенил-2,3-дигидро-1Н-пиразол-4-карбоновая кислота (2,19 г, 9,44 ммоль) в CH2Cl2 (20 мл) добавили EDCl (3,02 г, 15,7 ммоль) и HOAT (0,21 г, 1,57 ммоль). Реакционную смесь дефлегмировали в течение 20 часов и затем охладили до к.т.
В смесь добавили воду (10 мл), быстро размешивали при к.т., затем отфильтровали и отжатый осадок промывали водой (5 мл) с последующим очищением колоночной хроматографией с силикагелем (CH2Cl2/МеОН (о/с) = 70/1) до титулованного соединения в виде желтоватой твердой фазы (1,25 г, 46,6%).
MS (ESI, пол. ион) уд.з.: 342.1 [М+Н]+, К.т. = 2,712 минуты.
Шаг 3) 4-(4-(1,5-диметил-3-оксо-2-фенил-2,3-дигидро-1Н-пиразол-4-карбоксамидоУ3-фторфенокси)пиколинамид
[0206] К смеси N-(2-фтор-4-гидроксифенил)-l,5-диметил-3-оксо-2-фенил-2,3-дигидро-1Н-пиразол-4-карбоксамида (300 мг, 0,879 ммоль) и t-BuOK (118 мг, 1,05 ммоль) добавили DMF (2,5 мл). Полученную смесь размешивали при к.т. в течение 30 минут, затем добавили 4-хлоропиколинамид (165 мг, 1,05 ммоль). Смесь нагревали при 120°С в течение 5 часов, затем охладили до к.т. и добавили H2O (50 мл) и EtOAc (2 мл). Полученную смесь быстро размешивали при к.т.
Отфильтрованный осадок промывали водой (5 мл) до титулованного соединения в виде темно-коричневой твердой фазы (370 мг, 91,2%).
MS (ESI, пол. ион) уд.з.: 462.2 [М+Н]+, К.т. = 3,012 мин.
Пример 15. 3-хлоро-4-(4-(1,5-диметил-3-оксо-2-фенил-2,3-дигидро-1Н-пиразол-4-карбоксамидо)-3-фторфенокси)пиколинамид
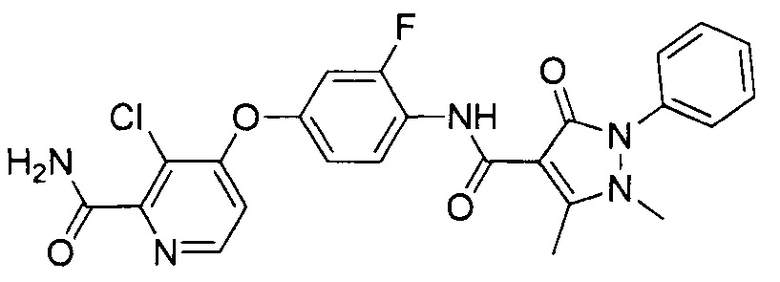
[0207] Смесь N-(2-фтор-4-гидроксифенил)-1,5-диметил-3-оксо-2-фенил-2,3-дигидро-1Н-пиразол-4-карбоксамид (300 мг, 0,879 ммоль) и t-BuOK (118 мг, 1,5 ммоль) в DMF (3 мл) размешивали при к.т. в течение 30 минут, затем добавили 3,4-дихлоропиколинамид (201 мг, 1,05 ммоль). Реакционную смесь нагревали до 120°С и вымешивали в течение 12 часов. Добавили EtOAc (1 мл) и Н2О (20 мл) и полученную смесь быстро размешивали при к.т.
Отфильтровали до титулованного соединения в виде коричневой твердой фазы (379 мг, 87,0%).
MS (ESI, пол. ион) уд.з.: 496.0 [М+Н]+, К.т. = 2.642 мин.
Пример 16. N-(4-((2-(циклопропанкарбоксамидо)пиридин-4-ил)окси)фенил)-1,5-диметил-3-оксо-2-фенил-2,3-дигидро-1Н-пиразол-4-карбоксамид
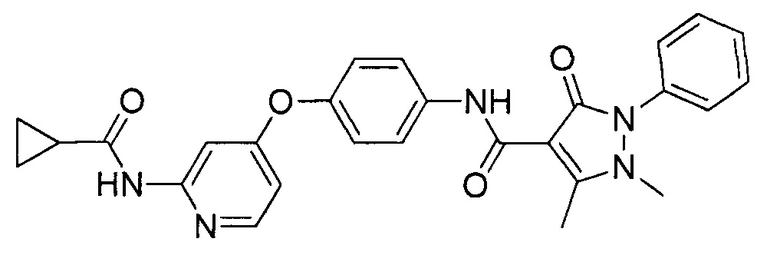
Шаг 1) 4-аминофенол
[0208] К смеси 4-нитрофенола (7 г, 50,3 ммоль) и HCOOK (1,8 г, 21,48 ммоль) в THF (210 мл) и H2O (70 мл) добавили Pd/C (110 мг, 10% Pd содержание, 53% ~55% содержание воды). Реакционную смесь размешивали при 50°С в течение 24 часов и затем сконцентрировали в вакууме. Остаток развели DCM (100 мл) и профильтровали через CELITE®. Фильтрат сконцентрировали в вакууме до титулованного соединения в виде бледно-оранжевой твердой фазы (3,28 г, 60%).
MS (ESI, пол. ион) уд.з.: 110.1 [М+Н]+.
Шаг 2) 4-(4-амнофенокси)пиридин-2-амин
[0209] К смеси 4-аминофенола (218 мг, 2 ммоль) и 4-хлоропиридин-2-амин (256 мг, 2 ммоль) в DMSO (2,5 мл) добавили NaOCH3 (216 мг, 4 ммоль). Реакционный раствор подогрели в мироволновой печи при 180°С в течение 40 минут, затем охладили до к.т. и погасили водой (10 мл). Смесь экстрагировали EtOAc (10 мл × 3). Объединенные органические фазы высушили над безводной Na2SO4 и сконцентрировали в вакууме. Остаток очистили колоночной хроматографией с силикагелем (DCM/СН3ОН (о/с) = 30/1) до титулованного соединения в виде коричневой твердой фазы (103 мг, 26%).
MS (ESI, пол. ион) уд.з.: 202.2 [М+Н]+;
1Н NMR (400 МГц, CDC13): δ (ppm) 3.65 (s, 2Н), 4.37 (s, 1Н), 5.89-5.90 (d, J=2,04 Гц, 1Н), 6.25-6.27 (dd, J=2,08 Гц, 5,88 Гц, 1H), 6.68-6.71 (m, 2Н), 6.86-6.89 (m, 2Н), 7.88-7.89 (d, 7=5.88 Гц, 1Н).
Шаг 3) N-(4-((2аминопиридин-4-ил)окси)фенил)-1,5-диметил-3-оксо-2-фенил-2,3-дигидро-1Н-пиразол-4-карбоксамид
[0210] К смеси 4-(4-аминофенокси)пиридин-2-амин (101 мг, 0,5 ммоль) и 1,5-диметил-3-оксо-2-фенил-2,3-дигидро-1Н-пиразол-4-карбоновая кислота (118 мг, 0,51 ммоль) в DCM (5 мл) добавили EDCl (115 мг, 0,6 ммоль) и НОАТ (13,6 мг, 0,1 ммоль). Реакционный раствор размешивали при 45°С в течение 3 часов и затем погасили водой (20 мл). Органическую фазу отделили и сконцентрировали в вакууме. Остаток очистили колоночной хроматографией с силикагелем (DCM/СН3ОН (о/с) = 20 л) до титулованного соединения в виде светло-серой твердой фазы (110 мг, 49,2%).
MS (ESI, пол. ион) уд.з.: 416.4 [М+Н]+;
lH NMR (400 МГц, DMSO-d6): δ (ppm) 2.71 (s, 3Н), 3.36 (s, 3Н), 5.80-5.81 (d, J=2,16 Гц, 1Н), 5.92 (s, 2Н), 6.12-6.14 (dd, J=2,24 Гц, 5,8 Гц, 1Н), 7.08-7.10 (m, 2Н), 7.42-7.45 (m, 2Н), 7.51-7.53 (m, 1Н), 7.57-7.61 (m, 2Н), 7.65-7.67 (m, 2Н), 7.77-7.79 (d, J=5,8 Гц, 1Н)
Шаг 4) N-(4-((2-(циклопропанкарбоксамидо)пиридин-4-ил)окси)фенил)фенил-1,5-диметил-3-оксо 2 2,3-дигидро-1Н-пиразол-4-карбоксамид
[0211] К раствору N-(4-((2-аминопиридин-4-ил)кислород)фенил)--l,5-диметил-3-оксо-2-фенил-2,3 дигидро-1Н-пиразол-4-карбоксамид (200 мг, 0,48 ммоль) в DCM (5 мл) добавили триэтиламин (1 мл), затем добавили путем ления циклопропанкарбонил хлорида (104,5 мг, 0,96 ммоль)при 0°С. Реакционную смесь подогрели до к.т. и вымешивали в течение 3 часов, затем погасили водой (10 мл). В отделенный органический слой добавили насыщенную вод. Na2C03 (10 мл) и EtOH (30 мл). Смесь размешивали при к.т. в течение 30 минут и сконцентрировали в вакууме. Остаток очистили колоночной хроматографией с силикагелем (DCM/MeOH (о/с) = 80/1) до титулованного соединения в виде белой твердой фазы (130 мг, 56%).
ВЭЖХ: 99,5%;
MS (ESI, пол. ион) уд.з.: 484.3 [М+Н]+;
1HNMR (600 МГц, CDC13): δ (ppm) 10.77 (s, 1Н), 9.45 (s, 1Н), 8.05 (d, J=5,8 Гц, 1Н), 7.90 (s, 1Н), 7.74 (d, J=8,9 Гц, 2Н), 7.57 (t, J=7,8 Гц, 2Н), 7.48 (t, J=7,5 Гц, 1Н), 7.38 (d, J=7,4 Гц, 2Н), 7.05 (d, J=8,9 Гц, 2Н), 6.62 (dd, J=6,0 Гц, 2,0 Гц, 1H), 3.37 (s, 3Н), 2.81 (s, 3Н), 1.65 (td, J=7,7 Гц, 4,0 Гц, 1Н), 1.26 (d, J=12,8 Гц, 2Н), 1.10 (dt, J=7,9 Гц, 4,0 Гц, 2Н).
Пример 17. N-(5-((2-(3-(2-гидроксиэтил)пиридин-4-ил)окси)пиридин-2-ил)-1,5-диметил-3-оксо-2-фенил-2,3-дигидро-1Н-пиразол-4-карбоксамид
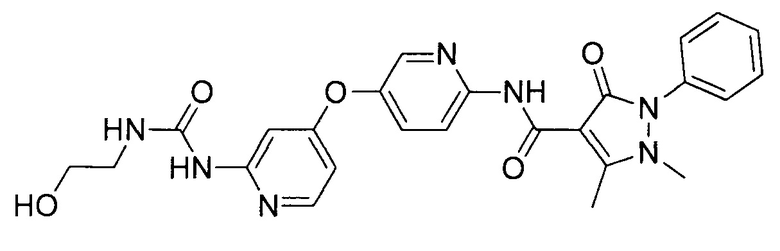
Шаг 1) 4-хлоропиколинамид
[0212] К раствору 4-хлоропиколиновой кислоты (100 мг, 0,64 ммоль) в DCM (10 мл) добавили одну каплю DMF и SOCl2 (0,14 мл, 1,92 ммоль). Реакционную смесь быстро размешивали при к.т., затем сконцентрировали в вакууме. Полученный остаток растворили в THF (10 мл), далее добавили Et3N (0,2 мл, 0,13 ммоль). Смесь размешивали в атмосфере аммиачного газа в течение 1 часа, затем сконцентрировали в вакууме, развели водой (20 мл) и экстрагировали этилацетатом (10 мл × 3). Объединенные органические фазы вымыли морской водой (20 мл), высушили над безводной Na2SO4, сконцентрировали в вакууме и остаток очистили колоночной хроматографией с силикагелем (РЕ/EtOAc (о/с) = 5/1) до титулованного соединения в виде желтой твердой фазы (70 мг 70%).
MS (ESI, пол. ион) уд.з.: 157.0 [М+Н]+;
1Н NMR (300 МГц, CD3OD): δ (ppm) 8.48 (d, J=5,15 Гц, 1Н), 8.22 (d, J=2,10 Гц, 1Н), 7.79 (br s, 1H), 7.46 (dd, J=5,28 Гц, 2,13 Гц, 1H), 5.80 (br s, 1H).
Шаг 2) N-(5-гидроксипиридин-2-ил)-2.5-диметил-3-оксо-1-фенил-2,3-дигидро-1Н-пиразол-4-карбоксамид
[0213] Взвесь 2-амино-5-гидроксипиридин гидрохлорида (500 мг, 3,41 ммоль) и Et3N (0,77 мл, 5,5 ммоль) в DMF (8 мл) размешивали при к.т. в течение 1 часа. Одновременно, к раствору l,5-диметил-3-оксо-2-фенил-2,3-дигидро-1Н-пиразол-4-карбоновой кислоты (0,64 г, 2,75 ммоль), HOAT (449 мг, 3,3 ммоль) и EDCl (0,63 г, 3,3 ммоль) в DMF (8 мл) добавили каплю Et3N (0,77 мл, 5,5 ммоль). Смесь размешивали при к.т. в течение 1 часа, далее добавили вышеупомянутую подготовленную смесь. Реакционный раствор нагевали при 60°С в течение 5 часов, затем охладили до к.т., разбавили водой (150 мл) и довели до рН фактора=6 при помощи НС1 (1 М). Смесь экстрагировали EtOAc (20 мл × 4) и объединенные органические фазы промывали морской водой (20 мл), сушили над безводной Na2SO4 и сконцентрировали в вакууме. Остаток очистили колоночной хроматографией с силикагелем (DCM/MeOH (о/с) = 10/1) до титулованного соединения в виде бледно-желтой твердой фазы (699 мг, 78%).
MS (ESI, пол. ион) уд.з.: 325.1 [М+Н]+.
Шаг 3) 4-((6-(1,5-диметил-3-оксо-2-фенил-2,3-дигидро-lH-пиразол-4-карбоксамидо)пиридин-3-ил)окси)пиколинамид
[0214] К раствору N-(5 гидроксипиридин-2-ил)-l,5-диметил-3-оксо-2-фенил-2,3-дигидро-1Н-пиразол-4-карбоксамида (468 мг, 1,4 ммоль) в DMF (30 мл) добавили калиевый терт-бутоксид (486 мг, 4,33 ммоль). Раствор размешивали при к.т. в течение 20 минут, далее добавили 4-хлоропиколинамид (293 мг, 1,87 ммоль). Реакционный раствор нагревали при 120°С в течение 2 часов, затем охладили до к.т., развели водой (15 мл) и экстрагироввали EtOAc (20 мл × 3). Объединенные органические фазы промывали морской водой (20 мл) и сконцентрировали в вакууме. Остаток очистили колоночной хроматографией с силикагелем (DCM/MeOH (о/с) = 50/1) до титулованного соединения в виде грязно-белой твердой фазы (285 мг, 44%).
MS (ESI, пол. ион) уд.з.: 444.8 [М+Н]+.
Шаг 4) N-(5-((2-аминопиридин-4-ил)оксилпиридин-2-ил)-l,5-диметил-3-оксо-2-фенил-2,3-дигидро-1Н-пиразол-4-карбоксамид
[0215] К раствору 4-((6-(l,5-диметил-3-оксо-2-фенил-2,3-дигидро-1Н-пиразол-4-карбоксамидо)пиридин-3-ил)окси)пиколинамид (60 мг, 0,13 ммоль) в EtOAc/MeCN/H2O (2 мл / 2 мл / 1 мл) добавили йодбензола диацетат (54 мг, 0,16 ммоль)при 0°С. Реакционный раствор размешивали при к.т. в течение 2 часов, затем сконцентрировали в вакууме. Полученный остаток растворили в EtOAc (10 мл), промыли насыщеной вод. NaHCO3 и затем высушили над безводной Na2SO4. Органический раствор сконцентрировали в вакууме до титулованного соединения в виде желтой твердой фазы (43 мг, 77%).
MS (ESI, пол. ион) уд.з.: 416.9 [М+Н]+;
1Н NMR (300 МГц, CDC13): δ (ppm) 11.21 (s, 1H), 8.31 (d, J=9,0 Гц, 1Н), 8.13 (d, J=2,8 Гц, 1Н), 7.92 (d, J=5,9 Гц, 1H), 7.57-7.27 (m, 6Н), 6.29 (dd, J=5,91 Гц, 2,13 Гц, 1Н), 5.94 (d, J=2,19 Гц, 1H), 4.51 (br s, 2Н), 3.37 (s, 3Н), 2.79 (s, 3Н).
Шаг 5) фенил (4-((6-(1,5-диметил-3-оксо-2 фенил-2,3-дигидро-1Н-пиразол-4-карбоксамидо)пиридин-3-ил)окси)пиридин-2-ил) карбамат
[0216] К взвеси N-(5-((2-аминопиридин-4-ил)окси)пиридин-2-ил)-l,5-диметил-3-оксо-2-фенил-2,3-дигидро-lH-пиразол-4-карбоксамид (0,25 г, 0,6 ммоль) в DCM (20 мл) добавили пиридин (0,65 мл, 6 ммоль). Взвесь охладили до 0°С, далее медленно добавили раствор фенилкарбонохлоридата (98 мг, 0,63 ммоль) в DCM (5 мл). Реакционный раствор размешивали при к.т. в течение 2 часов, разбавили DCM (20 мл) и затем промывали водой (25 мл × 2) с последующим промыванием морской водой (25 мл). Органический раствор высушили над безводной Na2SO4, затем сконцентрировали в вакууме и остаток очистили колоночной хроматографией с силикагелем (DCM/MeOH (о/с) = 100/1) до титулованного соединения в виде белой твердой фазы (134 мг, 42%).
MS (ESI, пол. ион) уд.з.: 536.8 [М+Н]+.
Шаг 6) N-(5-((2-(3-(2-гидроксиэтил)уреидо)пиридин-4-ил)окси)пиридин-2-ил)-1,5-диметил-3-оксо-2-фенил-2,3 дигидро-1Н-пиразол-4-карбоксамид
[0217] Смесь фенила (4-((6-(1,5-диметил-3-оксо-2-фенил-2,3-дигидро-1Н-пиразол-4-карбоксамидо)пиридин-3-ил)окси)пиридин-2-ил), карбамата (0,1 г, 0,187 ммоль), NMP (10 мл) с 2-аминоэтанолом (14,0 мг, 0,224 ммоль) нагревали при 40°С в течение 2 часов. Растворитель испарили под низким давлением при 40°С и затем растворили EtOAc (20 мл). Осадок собрали фильтрацией и промыли EtOAc (3 мл) до титулованного соединения в виде белой твердой фазы (92 мг, 98%).
MS (ESI, пол. ион) уд.з.: 504.2 [М+Н]+;
1Н NMR (300 МГц, DMSO-d6): δ (ppm) 11.23 (s, 1Н), 9.14 (s, 1Н), 8.33 (d, J=9,0 Гц, 1Н), 8.21 (d, J=2,9 Гц, 1Н), 8.08 (d, J=5,8 Гц, 1Н), 8.04 (br s, 1Н), 7.69 (dd, J=8,9 Гц, 2,9 Гц, 1H), 7.63-7.43 (m, 5H), 6.97 (d, J=2,19 Гц, 1H), 6.58 (d, J=5,8 Гц, 1H), 4.73 (t, J=5,2 Гц, 2H), 3.43 (q, J=5,7 Гц, 2H), 3.37 (s, 3H), 3.32 (s, 3H), 3.18 (q, J=5.6 Hz, 2H), 2.72 (s, 3H)
Пример 18. N-(5-((2-(3-(2-гидроксиэтил)- 3-метилуреидо)пиридин-4-ил)окси)пиридин-2-ил)-1,5 диметил-3-оксо-2-фенил-2,3-дигидро-1Н-пиразол-4-карбоксамид
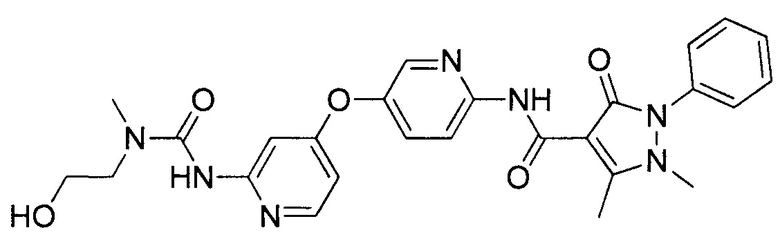
[0218] Смесь фенила (4-((6-(1,5-диметил-3-оксо-2-фенил-2,3-дигидро-1Н-пиразол-4-карбоксамидо)пиридин-3-ил)окси)пиридин-2-ил) карбамат (30 мг, 0,05 ммоль), NMP (5 мл) и 2-(метиламино) этанола (5,0 мг, 0,06 ммоль) нагревали при 40°С в течение 1,5 часов. Смесь охладили до к.т. и разбавили EtOAc (15 мл) и водой (10 мл). Отделенный органический слой промыли водой (10 мл × 3), далее промыли морской водой (10 мл), высушили над безводной MgSO4 и сконцентрировали в вакууме. Остаток очистили колоночной хроматографией с силикагелем (DCM/MeOH (о/с) = 20/1) и затем промыли эфиром (5 мл) до титулованного соединения в виде белой твердой фазы (17 мг, 59%).
MS (ESI, пол. ион) уд.з.: 518.2 [М+Н]+;
1Н NMR (300 МГц, CD3OD): δ (ppm) 8.44 (d, J=9,0 Гц, 1Н), 8.24 (d, J=2,85 Гц, 1Н), 8.20 (d, J=7,14 Гц, 1Н), 7.73 (dd, J=9,0 Гц, 2,88 Гц, 1Н), 7.64-7.56 (m, 3Н), 7.46-7.43 (m, 2Н), 3.72 (t, J=5,2 Гц, 2Н), 3.53 (t, J=5,2 Гц, 2Н), 3.43 (s, 3Н), 3.08 (s, 3Н), 2.80 (s, 3Н)
Пример 19. N-(5-((2-(циклобутанкарбоксамидо)пиридин-4-ил)окси)пиридин-2-ил)-1,5-диметил-3-оксо-2-фенил-2,3-дигидро-lH-пиразол-4-карбоксамид

[0219] К взвеси N-(5-((2-аминопиридин-4-ил)окси)пиридин-2-ил)-1,5-диметил-3-оксо-2-фенил-2,3-дигидро-lH-пиразол-4-карбоксамид (50 мг, 0,12 ммоль) в THF (13 мл) добавили Et3N (0,034 мл, 0,24 ммоль). Взвесь охладили до 0°С, затем раствор циклобутанкарбонил хлорида (28,5 мг, 0,24 ммоль) в THF (3 мл) медленно добавили к смеси. Реакционный раствор размешивали при к.т. в течение 1,25 часов, затем разбавили EtOAc (15 мл). Смесь промывали водой (15 мл × 2) с последующим промыванием морской водой (15 мл), высушили над безводной Na2SO4 и сконцентрировали в вакууме. Остаток очистили колоночной хроматографией с силикагелем (DCM/MeOH (о/с) = 60/1) до титулованного соединения в виде белой твердой фазы (25 мг, 41%).
MS (ESI, пол. ион) уд.з.: 499.2 [М+Н]+;
1Н NMR (300 МГц, CDC13): δ (ppm) 11.21 (s, 1Н), 8.35 (d, J=9,0 Гц, 1Н), 8.14 (d, J=2.6 Hz, 1Н), 8.13-8.08 (m, 2H), 7.89-7.88 (m, 1H), 7.55-7.35 (m, 1H), 6.56 (d, J=3,0 Гц, 1H), 3.36 (s, 3H), 3.22-3.11 (m, 1H), 2.78 (s, 3H), 2.43-2.29 (m, 2H), 2.26-2.15 (m, 2H), 2.4-1.87 (m, 2H).
Пример 20. N-(5-((2-(3-гидроксициклобутанкарбоксамидо)пиридин-4-ил)окси)пиридин-2-ил)-1,5 диметил-3-оксо-2-фенил-2,3-дигидро-lH-пиразол-4-карбоксамид
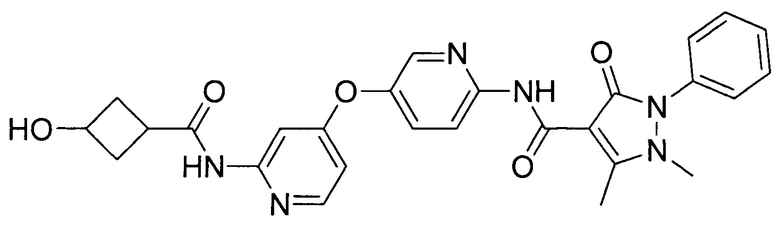
Шаг 1) 3-ацетоксициклобутанкарбоновая кислота
[0220] К раствору 3-гидроксициклобутанкарбоновой кислоты (100 мг, 0,86 ммоль) в DCM (10 мл) добавили DMAP (1,0 мг, 0,086 ммоль). Расвор охладили до 0°С, далее добавили ацетилхлорид (0,14 мл, 2,6 ммоль). Реакционный раствор нагревали при 45°С в течение 4 часов, затем охладили до к.т. и разбавили DCM (20 мл). Смесь промыли водой (10 мл), далее промывали морской водой (10 мл), высушили над безводной Na2SO4, отфильтровали и сконцентрировали в вакууме к полученному сырому продукту, использованному в следующем шаге без дальнейшей очистки.
Шаг 2) 3-((4-((6-(15-диметил-3-оксо-2-фенил-2,3-дигидро-1Н-пиразол-4-карбоксамидо)пиридин-3-ил)окси)пиридин-2-ил)карбамоил)циклобутил ацетат
[0221] К раствору N-(5-((2-аминопиридин-4-ил)окси)пиридин-2-ил)-1,5-диметил-3 оксо-2-фенил-2,3-дигидро-1Н-пиразол-4-карбоксамид (50 мг, 0,12 ммоль), HOAT (19 мг, 0,24 ммоль), EDCl (28 мг, 0,14 ммоль) и DIPEA (0,1 мл, 0,6 ммоль) в DMF (5 мл) добавили раствор 3-ацетоксициклобутанкарбоновой кислоты в DMF (5 мл)при 0°С. Реакционный раствор нагревали при 120°С в течение 5 часов, затем охладили до к.т., разбавили водой (50 мл) и экстрагировали EtOAc (30 мл × 3). Объединенные органические фазы промывали морской водой (10 мл), высушили над безводной Na2SO4, отфильтровали и сконцентрировали в вакууме. Полученный остаток очистили колоночной хроматографией с силикагелем (DCM/EtOAc (о/с) = 1/2) до титулованного соединения в виде белой твердой фазы (20 мг, 30%).
MS (ESI, пол. ион) уд.з.: 556.9 [М+Н]+.
Шаг 3) N-(5-((2-(3-гидроксициклобутанкарбоксамидо)пиридин-4-ил)окси)пиридин-2-ил)-1,5-диметил-3-оксо-2-фенил-2,3-дигидро-1H-пиразол-4-карбоксамид
[0222] Раствор 3-((4-((6-(1,5-диметил-3-оксо-2-фенил-2,3-дигидро-1Н-пиразол-4-карбоксамидо)пиридин-3-ил)кислород)пиридин-2-ил) карбамоил), циклобутил ацетат (40 мг, 0,072 ммоль) в МеОН (2,5 мл) охладили до 0°С, затем к смеси добавили NaOH (6 мг, 0,144 ммоль). Реакционный раствор размешивали при к.т. в течение 1 часа, затем сконцентрировали в вакууме. Остаток промывали водой (5 мл) и эфиром (5 мл), затем отфильтровали до титулованного соединения в виде белой твердой фазы (17 мг, 42%).
MS (ESI, пол. ион) уд.з.: 515.0 [М+Н]+;
1Н NMR (300 МГц, DMSO-d6): δ (ppm) 11.24 (s, 1Н), 10.48 (s, 1Н), 8.33 (d, J=9,0 Гц, 1Н), 8.23 (d, J=2,88 Гц, 1Н), 8.19 (d, J=5,7 Гц, 1Н), 7.73-7.69 (m, 2Н), 7.63-7.5 (m, 3Н), 7.46-7.43 (m, 2Н), 6.72 (dd, J=2,4 Гц, 5,7 Гц, 1Н), 5.09 (d, J=7,3 Гц, 1Н), 3.96 (m, 1Н), 3.37 (s, 3Н), 2.72 (s, 3Н), 2.77-2.66 (m, 1H), 2.34-2.26 (m, 2Н), 2.04-1.91 (m, 2Н).
Пример 21. N-(4-((2-(циклобутанкарбоксамидо)пиридин-4-ил)окси)3-фторфенил)-1,5-диметил-3-оксо-2-фенил-2,3-дигидро-1Н-пиразол-4-карбоксамид

Шаг 1) 4 амино-2-фторфенол
[0223] К раствору 2-фтор-4-нитрофенола (5,0 г, 31,8 ммоль) в метаноле (200 мл) добавили Pd/C (1,0 г, 10%). Реакционный раствор быстро размешивали при к.т. в атмосфере Н2. В смесь отфильтровали через CELITE®, затем промыли МеОН (5 мл). Фильтрат сконцентрировали в вакууме до титулованного соединения в виде коричневой твердой фазы (4,02 г, >99%).
MS (ESI, пол. ион) уд.з.: 128.1 [М+Н]+.
Шаг 2) N-(3-фтор-4-гидрооксифенил)-1,5-диметил-3-оксо-2-фенил-2,3-дигидро-1Н-пиразол-4-карбоксамид
[0224] К раствору l,5-диметил-3-оксо-2-фенил-2,3-дигидро-1Н-пиразол-4-карбоновой кислоты (6,12 г, 26,3 ммоль), HOAT (4,3 г, 31,6 ммоль) и EDCl (6,03 г, 31,6 ммоль) в DMF (200 мл) добавили Et3N (9,2 мл, 65,8 ммоль)по капле. Раствор размешивали при к.т. в течение 20 минут, затем раствор 4-амино-2-фторфенола (4,02 г, 31,6 ммоль) в DMF (20 мл) добавили к системе. Реакционный раствор нагревали при 70°С в течение 4 часов, затем сконцентрировали до 2 мл в вакууме и разбавили Н2О/EtOAc (150 мл / 50 мл). Осадок получили фильтрацией и затем промыли EtOAc (10 мл), высушили до коричневой твердой фазы (5,49 г); фильтрат отделили и органическую фазу сконцентрировали в вакууме. Полученный остаток очистили колоночной хроматографией с силикагелем (DCM/EtOAc (о/с) = 10/1) до желтой твердой фазы (1,692 г). Титулованное соединение получили путем объединения осадка и желтой твердой фазы для 80% продукта.
MS (ESI, пол. ион) уд.з.: 342.0 [М+Н]+;
1Н NMR (300 МГц, CDC13): δ (ppm) 10.58 (s, 1Н), 7.72 (dd, J=2,4 Гц, 12,69 Гц, 1Н), 7.58-7.32 (m, 5Н), 7.09-7.04 (m, 1Н), 6.89 (t, J=9,3 Гц, 1Н), 3.34 (s, 3Н), 2.78 (s, 3Н).
Шаг 3) 4-(4-(1,5-диметил-3-оксо-2-фенил-2,3-дигидро-1Н-пиразол-4-карбоксамидо)-2-фторфенокси)пиколинамид
[0225] К раствору N-(3-фтор-4-гидроксифенил)-l,5-диметил-3-оксо-2-фенил-2,3-дигидро-1Н-пиразол-4-карбоксамид (7,18 г, 21,1 ммоль) в DMF (200 мл) добавили калиевый терт-бутоксид (7,08 г, 63,2 ммоль). Раствор размешивали при к.т. в течение 20 минут с последующим добавлением 4-хлоропиколинамида (4,27 г, 25,2 ммоль). Реакционный раствор нагревали при 120°С в течение 3 часов, затем концентрировали до 2 мл в вакууме и разводили водой (60 мл). Осадок получали фильтрацией и затем промывали водой (10 мл). Сырой продукт очистили колоночной хроматографией с силикагелем (DCM/EtOAc/MeOH (o/cN) = 1/1/0.05) до титулованного соединения в виде желтой твердой фазы (4,15 г, 43% продукта).
MS (ESI, пол. ион) уд.з.: 462.0 [М+Н]+;
1H NMR (300 МГц, DMSO-d6): δ (ppm) 10.97 (s, 1Н), 8.53 (d, J=6,0 Гц, 1Н), 8.12 (br s, 1Н), 7.97 (dd, J=2,0 Гц, 13,23 Гц, 1Н), 7.72 (br s, 1Н), 7.63-7.42 (m, 5Н), 7.42-7.32 (m, 3Н), 7.21 (dd, J=2,7 Гц, 5,7 Гц, 1Н), 3.37 (s, 3Н), 2.71 (s, 3Н)
Шаг 4) N-(4-((2-аминопиридин-4ил)окси)-3-фторфенил)-1,5-диметил-3-оксо-2-фенил-2,3-дигидро-1Н-пиразол-4-карбоксамид
[0226] К раствору 4-(4-(1,5-диметил-3-оксо-2-фенил-2,3-дигидро-1Н-пиразол-4-карбоксамидо)-2-фторфенокси)пиколинамид (2,5 г, 6,0 ммоль) в EtOAc/MeCN/H20 (60 мл / 60 мл / 30 мл) добавили йодбензол диацетат (2,9 г, 9,0 ммоль) в 0°С. Реакционный раствор размешивали при к.т. в течение 2 часов, затем сконцентрировали в вакууме. Полученный остаток разбавили EtOAc (150 мл), промыли насыщенным вод. NaHCO3 (80 мл), высушили над безводной Na2SO4 и сконцентрировали в вакууме. Остаток промывали DCM/эфиром (1/10, 22 мл) до титулованного соединения в виде желтой твердой фазы (2,09 г, 81%-й доход).
MS (ESI, пол. ион) уд.з.: 434.0 [М+Н]+;
1HNMR (300 МГц, CDC13): δ (ppm) 10.83 (s, 1Н), 7.92 (d, J=5,8 Гц, 1Н), 7.84 (dd, J=2.4 Hz, 12,5 Гц, 1Н), 7.60-7.33 (m, 5Н), 7.26-7.22 (m, 1Н), 7.06 (t, J=8,7 Гц, 1Н), 6.29 (dd, J=2,2 Гц, 5,9 Гц, 1Н), 5.92 (d, J=2,19 Гц, 1Н), 4.40 (br s, 2Н), 3.37 (s, 3Н), 2.79 (s, 3Н).
Шаг 5) N-(4-((2-(циклобутанкарбоксамидо)пиридин-4-ил)окси)-3-фторфенил)-1,5-диметил-3-оксо-2-фенил-2,3-дигидро-1Н-пиразол-4-карбоксамид
[0227] К взвеси N-(4-((2-аминопиридин-4-ил)окси)-3-фторфенил)-l,5-диметил-3-оксо-2-фенил-2,3-дигидро-1Н-пиразол-4-карбоксамид (100 мг, 0,231 ммоль) в THF/DMF (15 мл / 0,5 мл) добавили Et3N (0,058 мл, 0,462 ммоль). Взвесь охладили до 0°С, затем раствор циклобутанкарбонил хлорида (30 мг, 0,254 ммоль) в THF (5 мл) добавили к смеси по капле через 1 час. Реакционный раствор размешивали при к.т. в течение 2 часов, затем разбавили EtOAc (20 мл). Смесь промывали водой (25 мл × 3), с последующим промыванием морской водой (25 мл), высушили над безводной Na2SO4, отфильтровали и сконцентрировали в вакууме. Остаток очистили колоночной хроматографией с силикагелем (DCM/EtOAc/MeOH (o/cN) = 100/20/1) до титулованного соединения в виде белой твердой фазы (51 мг, 44%).
MS (ESI, пол. ион) уд.з.: 516.0 [М+Н]+;
1Н NMR (300 МГц, CDC13): δ (ppm) 10.84 (s, 1H), 8.06 (d, J=5,8 Гц, 1Н), 7.91-7.86 (m, 3Н), 7.59-7.35 (m, 5Н), 7.28-7.24 (m, 1H), 7.09 (t, J=8,7 Гц, 1Н), 6.56 (dd, J=2,9 Гц, 5,8 Гц, 1Н), 3.68 (s, 3Н), 3.16 (m, 1Н), 2.79 (s, 3Н), 2.43-2.29 (m, 2Н), 2.26-2.15 (m, 2Н), 2.06-1.84 (m, 2Н).
Пример 22. N-(3-фтор-4-((2-(3-(2-гидроксиэтил)уреидо)пиридин-4-ил)окси)фенил)-1,5-диметил-3-оксо-2-фенил-2,3-дигидро-1Н-пиразол-4-карбоксамид
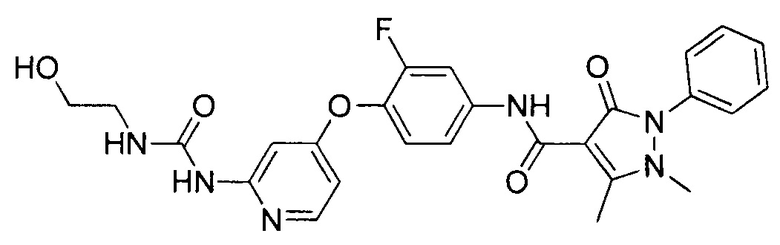
Шаг 1) фенил (4-(4-(1,5-диметил-3-оксо-2-фенил-2,3-дигидро-1Н-пиразол-4-карбоксамидо)-2-фторфенокси)пиридин-2-ил)карбамат
[0228] Взвесь N-(4-((2-аминопиридин-4-ил)окси)-3-фторфенил)-l,5-диметил-3-оксо-2-фенил-2,3-дигидро-1Н-пиразол-4-карбоксамид (500 мг, 1,10 ммоль) в DCM/пиридине (50 мл / 25 мл) охладили до 0°С, затем раствор фенилкарбонохлоридата (450 мг, 2,90 ммоль) в DCM (5 мл) медленно добавили к смеси через 0,5 часа. Реакционный раствор размешивали при к.т. в течение 2 часов, затем разбавили DCM (250 мл). Смесь промыли водой (150 мл × 3) с последующим промыванием морской водой (150 мл), высушили над безводной Na2SO4, отфильтровали и сконцентрировали в вакууме. Остаток очистили колоночной хроматографией с силикагелем (DCM/MeOH (о/с) = 150/1) до титулованного соединения в виде белой твердой фазы (352 мг, 46%).
MS (ESI, пол. ион) уд.з.: 554.0 [М+Н]+.
Шаг 2) N-(3-фтор-4-((2-(3-(2-гидроксиэтил)уреидо)пиридин-4-ил)оксилфенил)-1,5-диметил-3-оксо-2-фенил-2,3-дигидро-1Н-пиразол-4-карбоксамид
[0229] Смесь фенила (4-(4-(1,5-диметил-3-оксо-2-фенил-2,3-дигидро-1Н-пиразол-4-карбоксамидо)-2-фторфенокси)пиридин-2-ил)карбамат (100 мг, 0,18 ммоль), NMP (5 мл) и 2-аминоэтанола (3,0 мг, 0,22 ммоль) нагревали при 40°С в течение 2 часов. Растворитель испарили под низким давлением в 40°С и разбавили EtOAc (20 мл). Осадок собрали фильтрацией и затем промывали EtOAc (3 мл) до титулованного соединения в виде белой твердой фазы (65 мг, 69%).
MS (ESI, пол. ион) уд.з.: 521.0 [М+Н]+;
1Н NMR (300 МГц, DMSO-d6): δ (ppm) 10.94 (s, 1Н), 9.14 (s, 1Н), 8.06 (d, J=5,9 Гц, 1Н), 8.00 (br s, 1Н), 7.94 (dd, J=1,50 Гц, 12,84 Гц, 1Н), 7.63-7.42 (m, 5Н), 7.31-7.29 (m, 2Н), 6.97 (d, J=2,04 Гц, 1Н), 6.56 (dd, J=2,4 Гц, 5,9 Гц, 1Н), 4.72 (t, J=5,1 Гц, 1Н), 3.42 (q, J=5,4 Гц, 2Н), 3.37 (s, 3Н), 3.17 (q, J=5,6 Гц, 2Н), 2.70 (s, 3Н).
Пример 23. N-(3-фтор-4-((2-(3-(2-гидроксипропил)-3-метилуреидо)пиридин-4-ил)окси)фенил)-1,5-диметил-3-оксо-2-фенил-2,3-дигидро-lH-пиразол-4-карбоксамид

[0230] Смесь фенила (4-(4-(l,5-диметил-3-оксо-2-фенил-2,3-дигидро-lH-пиразол-4-карбоксамидо)-2-фторфенокси)пиридин-2-ил) карбамат (100 мг, 0,18 ммоль), NMP (5 мл) и l-аминопропан-2-ол (16 мг, 0,2 ммоль) нагревали при 40°С в течение 2 часов. Растворитель испарили под низким давлением при 40°С и затем разбавили EtOAc (20 мл). Осадок собрали фильтрацией до титулованного соединения в виде белой твердой фазы (58 мг, 60%).
MS (ESI, пол. ион) уд.з.: 535.0 [М+Н]+;
1Н NMR (300 МГц, DMSO-d6): δ (ppm) 10.94 (s, 1Н), 9.14 (s, 1Н), 8.06 (d, J=5,8 Гц, 1Н), 7.98 (br s, 1Н), 7.93 (dd, J=1,5 Гц, 12,8 Гц, 1H), 7.63-7.42 (m, 5H), 7.31-7.29 (m, 2H), 6.97 (d, J=2,1 Гц, 1H), 6.56 (dd, J=2,4 Гц, 5,8 Гц, 1H), 4.73 (d, J=4,8 Гц, 1H), 3.69-3.61 (m, 1H), 3.37 (s, 3H), 3.18-3.10 (m, 1H), 3.00-2.92 (m, 1H), 2.70 (s, 3H), 1.02 (d, J=6.2 Гц, 3Н).
Пример 24. N-(4-((2-(3,3-диметилуреидо)пиридин-4-ил)окси)-3-фторфенил)-1,5-диметил-3-оксо-2-фенил-2,3-дигидро-1Н-пиразол-4-карбоксамид
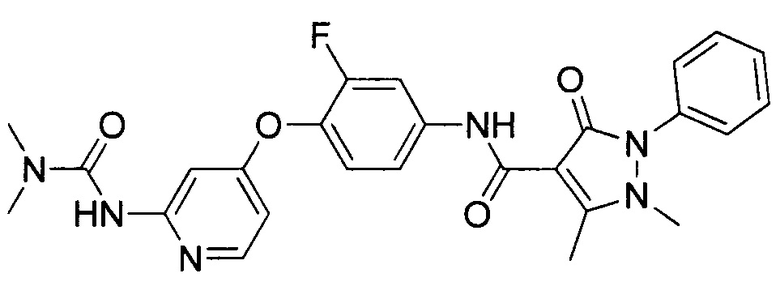
[0231] Смесь фенила (4-(4-(1,5-диметил-3-оксо-2-фенил-2,3-дигидро-1Н-пиразол-4-карбоксамидо)-2-фторфенокси)пиридин-2-ил)карбамат (100 мг, 0,18 ммоль), NMP (5 мл) и водный диметиламин (0,045 мл, 33%) нагревали при 40°С в течение 2 часов. Растворитель испарили под низким давлением в 40°С и затем разбавили EtOAc (20 мл). Осадок собрали фильтрацией до титулованного соединения в виде белой твердой фазы (25 мг, 27%).
MS (ESI, пол. ион) уд.з.: 505.0 [М+Н]+;
1Н NMR (300 МГц, CD3OD): δ (ppm) 8.07 (d, J=5,8 Гц, 1Н), 7.88 (dd, /=2,4 Гц, 12,9 Гц, 1H), 7.66-7.43 (m, 5Н), 7.31-7.27 (m, 1Н), 7.21 (t, J=8,7 Гц, 1H), 6.59 (dd, J=2,4 Гц, 5,8 Гц, 1Н), 4.84 (s, 3Н), 3.41 (s, 3Н), 3.00 (s, 6Н), 2.76 (s, 3Н).
Пример 25. N-(4-((2-(3-этил-3-метилуреидо)пиридин-4-ил)окси)-3-фторфенил)-1,5-диметил-3-оксо-2-фенил-2,3-дигидро-1Н-пиразол-4-карбоксамид
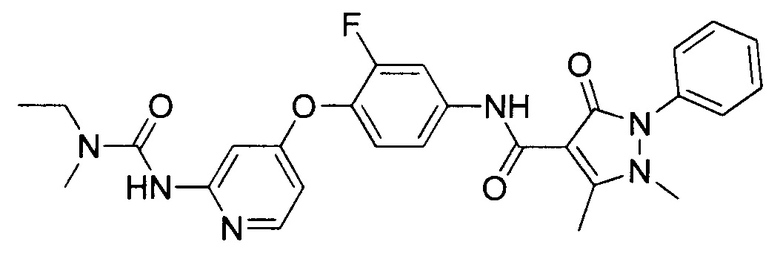
[0232] Смесь фенила (4-(4-(l,5-диметил-3-оксо-2-фенил-2,3-дигидро-1Н-пиразол-4-карбоксамидо)-2-фторфенокси)пиридин-2-ил)карбамат (100 мг, 0,18 ммоль), NMP (5 мл) и N-метилэтанамин (12 мг, 0,2 ммоль) нагревали в 40°С в течение 2 часов. Растворитель испарили под низким давлением при 40°С и затем разбавили водой (10 мл). Осадок собрали фильтрацией и затем очистили колоночной хроматографией с силикагелем (DCM/MeOH (о/с) = 60/1) до титулованного соединения в виде белой твердой фазы (55 мг, 62%).
MS (ESI, пол. ион) уд.з.: 519.3 [М+Н]+;
1H NMR (300 МГц, CD3OD): δ (ppm) 8.07 (d, J=5,8 Гц, 1H), 7.88 (dd, J=2,4 Гц, 12,9 Гц, 1Н), 7.66-7.43 (m, 5Н), 7.31-7.27 (m, 1Н), 7.21 (t, J=8,6 Гц, 1Н), 6.59 (dd, J=2,4 Гц, 5,8 Гц, 1Н), 3.41 (s, 3Н), 3.40 (q, J=7,2 Гц, 2Н), 2.99 (s, 3Н), 1.76 (s, 3Н), 1.16 (t, J=7.2, 3Н).
Пример 26. N-(4-((2-(циклобутанкарбоксамидо)пиридин-4-ил)окси)фенил)-1,5-диметил-3-оксо-2-фенил-2,3-дигидро-1Н-пиразол-4-карбоксамид
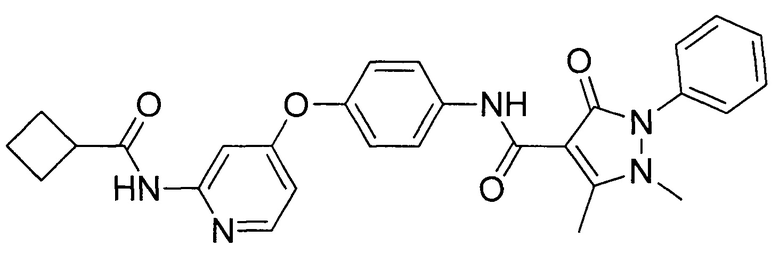
Шаг 1) 4-аминофенол
[0233] К раствору 4-нитрофенола (2 г, 14,4 ммоль) в EtOH (80 мл) добавили Pd/C (10%, 300 мг). Реакционный раствор размешивали при к.т. в течение 5 часов в атмосфере Н2, затем отфильтровали через CELITE®, промыли EtOH (10 мл). Фильтрат сконцентрировали в вакууме до титулованного соединения в виде коричневой твердой фазы (1,58 г, >99%).
MS (ESI, пол. ион) уд.з.: 110.1 [М+Н]+.
Шаг 2) N-[-(4-гидроксифенил)-1,5-диметил-3-оксо-2-фенил-2,3-дигидро-1Н-пиразол-4-карбоксамид
[0234] К раствору l,5-диметил-3-оксо-2-фенил-2,3-дигидро-1Н-пиразол-4-карбоновой кислоты (1,70 г, 7,35 ммоль), НОАТ (1,2 г, 8,82 ммоль) и EDC1 (1,68 г, 8,82 ммоль) в DMF (40 мл) добавили Et2N (2,6 мл, 18,38 ммоль)по капле. Раствор размешивали при к.т. в течение 20 минут, затем раствор 4-аминофенола (994 мг, 9,11 ммоль) в DMF (6 мл) добавили по капле к реакционной системе. Реакционный раствор нагревали при 70°С в течение 5 часов, затем сконцентрировали до 2 мл в вакууме и разбавили водой (50 мл). Осадок собрали фильтрацией, промыли водой (5 мл) и высушили. Сырой продукт промывали DCM (20 мл) до титулованного соединения в виде коричневой твердой фазы (2,02 г, 85%).
MS (ESI, пол. ион) уд.з.: 324.0 [М+Н]+;
1Н NMR (300 МГц, DMSO-d6): δ (ppm) 10.46 (s, 1Н), 9.17 (s, 1Н), 7.61-7.55 (m, 2Н), 7.52-7.46 (m, 1Н), 7.43-7.34 (m, 4Н), 6.73-6.68 (m, 2Н), 3.33 (s, 3Н), 2.69 (s, 3Н).
Шаг 3) 4-(4-(1,5-диметил-3-оксо-2-фенил-2,3-дигидро-lH-пиразол-4-карбоксамидо)фенокси)пиколинамид
[0235] К раствору N-(4-гидроксифенил)-1,5-диметил-3-оксо-2-фенил-2,3-дигидро-1Н-пиразол-4 карбоксамида (1,80 г, 5,57 ммоль) в DMF (35 мл) добавили калиевый терт-бутоксид (1,87 г, 16,7 ммоль). Раствор размешивали при к.т. в течение 20 минут с последующим добавлением 4-хлоропиколинамида (1,046 г, 6,68 ммоль). Реакционный раствор нагревали при 120°С в течение 3 часов, затем концентрировали до 2 мл в вакууме и разбавили водой (60 мл). Осадок получили фильтрацией, промывали водой (5 мл) и высушивали. Сырой продукт промывали DCM (20 мл) до титулованного соединения в виде коричневой твердой фазы (1,8 г, 73%).
MS (ESI, пол. ион) уд.з.: 444.2 [М+Н]+;
1Н NMR (300 МГц, DMSO-d6): δ (ppm) 10.84 (s, 1Н), 8.51 (d, J=5,64 Гц, 1Н), 8.11 (d, J=2,64 Гц, 1Н), 7.75-7.69 (m, 3Н), 7.63-7.56 (m, 2Н), 7.54-7.48 (m, 1H), 7.46-7.39 (m, 3Н), 7.22-7.15 (m, 3Н), 3.36 (s, 3Н), 2.71 (s, 3Н).
Шаг 4) N-(4-((2-аминопиридин-4-ил)окси)фенил)-1,5-диметил-3-оксо-2-фенил-2,3-дигидро-1Н-пиразол-4-карбоксамид
[0236] К раствору 4-(4-(l,5-диметил-3-оксо-2-фенил-2,3-дигидро-1Н-пиразол-4-карбоксамидо) фенокси)пиколанимид (1,773 г, 3,998 ммоль) в EtOAc/MeCN/H20 (20 мл / 20 мл / 10 мл) добавили йодобензол диацетат (1,6 г, 4,96 ммоль) в 0°С. Реакционный раствор быстро размешивали при к.т., затем сконцентрировали в вакууме и разбавили EtOAc (100 мл). Смесь промывали насыщенной вод. NaHCO3 (60 мл), высушили над Na2SO4 и сконцентрировали в вакууме. Остаток очистили колоночной хроматографией с силикагелем (DCM/MeOH (о/с) = 30/1) до титулованного соединения в виде желтой твердой фазы (799 мг, 48%).
MS (ESI, пол. ион) уд.з.: 416.1 [М+Н]+;
1Н NMR (300 МГц, DMSO-d6): δ (ppm) 10.77 (s, 1Н), 7.78 (d, J=9,8 Гц, 1Н), 7.68-7.63 (m, 2Н), 7.62-7.56 (m, 2Н), 7.54-7.48 (m, 1Н), 7.45-7.41 (m, 2Н), 6.13 (dd, J=2,3 Гц, 9,8 Гц, 1Н), 5.90 (br s, 2Н), 5.82 (d, J=2,2 Гц, 1Н), 3.36 (s, 3Н), 2.71 (s, 3Н).
Шаг 5) N-(4-((2-(циклобутанкарбоксамидо)пиридин-4-ил)окси)фенил)-1,5-диметил-3-оксо-2 фенил 2,3 дигидро-1Н-пиразол-4-карбоксамид
[0237] К взвеси N-(4-((2-аминопиридин-4-ил)окси)фенил)-l,5-диметил-3-оксо-2-фенил-2,3-дигидро-1Н-пиразол-4-карбоксамид (100 мг, 0,241 ммоль) в THF/DMF (15 мл / 1 мл) добавили Et3N (0,06 мл, 0,482 ммоль). Взвесь охладили до 0°С, затем раствор циклобутанкарбонил хлорида (31,0 мг, 0,265 ммоль) в THF (5 мл) добавили к реакционной смеси по капле, через 1 час. Реакционный раствор размешивали при к.т. в течение 5 часов и затем разбавили EtOAc (20 мл). Полученную смесь промыли водой (25 мл × 3) с последующим промыванием морской водой (25 мл), высушили над безводной Na2SO4, отфильтровали и сконцентрировали в вакууме. Остаток очистили колоночной хроматографией с силикагелем (DCM/EtOAc/MeOH (о/с) = 100/25/1) до титулованного соединения в виде белой твердой фазы (30 мг, 25%).
MS (ESI, пол. ион) уд.з.: 498.0 [М+Н]+;
1Н NMR (300 МГц, CDC13): δ (ppm) 10.73 (s, 1Н), 8.05 (d, J=5,8 Гц, 1Н), 7.87-7.86 (m, 2Н), 7.74-7.70 (m, 2Н), 7.58-7.36 (m, 5Н), 7.07-7.03 (m, 2Н), 6.54 (dd, J=2,3 Гц, 5,8 Гц, 1H), 3.35 (s, 3Н), 3.22-3.10 (m, 1Н), 2.80 (s, 3Н), 2.43-2.31 (m, 2Н), 2.26-2.15 (m, 2Н), 2.08-1.86 (m, 2Н).
Пример 27. N-(4-((2-(циклопентанкарбоксамидо)пиридин-4-ил)окси)фенил)-1,5-диметил-3-оксо-2-фенил-2,3-дигидро-1Н-пиразол-4-карбоксамид
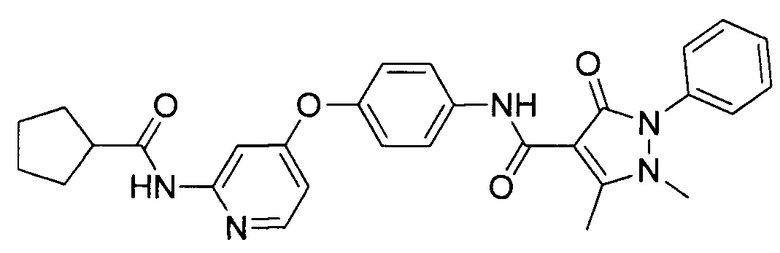
[0238] К взвеси N-(4-((2-аминопиридин-4-ил)окси)фенил)-l,5-диметил-3-оксо-2-фенил-2,3-дигидро-1Н-пиразол-4-карбоксамида (100 мг, 0,241 ммоль) в THF/DMF (20 мл / 1 мл) добавили Et3N (0,06 мл, 0,481 ммоль). Взвесь охладили до 0°С, раствор циклопентанкарбонил хлорида (64,0 мг, 0,481 ммоль) в THF (5 мл) добавили к реакционной смеси по капле, через 2 часа. Реакционный раствор размешивали при к.т. в течение 1,5 часов, затем разбавили EtOAc (25 мл). Полученную смесь промыли водой (25 мл × 3) с последующим промыванием морской водой (25 мл), высушили над безводной Na2SO4, отфильтровали и сконцентрировали в вакууме. Остаток очистили колоночной хроматографией с силикагелем (DCM/EtOAc/MeOH (о/с) = 80/15/1) до титулованного соединения в виде белой твердой фазы (39 мг, 32%).
MS (ESI, пол. ион) уд.з.: 512.2 [М+Н]+;
1Н NMR (300 МГц, CDCl3): δ (ppm) 10.73 (s, 1Н), 8.05 (d, J=5,7 Гц, 1H), 8.01 (br s, 1Н), 7.73-7.68 (m, 2Н), 7.58-7.34 (m, 5Н), 7.06-7.01 (m, 2Н), 6.54 (dd, J=2,4 Гц, 5,7 Гц, 1Н), 3.35 (s, 3Н), 2.79 (s, 3Н), 2.71-2.63 (m, 1Н), 1.95-1.57 (m, 8Н).
Пример 28. N-(4-((2-(3-(2 гидроксиэтил)-3-метилуреидо)пиридин-4-ил)окси)фенил)-1,5-диметил-3-оксо-2-фенил-2,3-дигидро-1Н-пиразол-4-карбоксамид
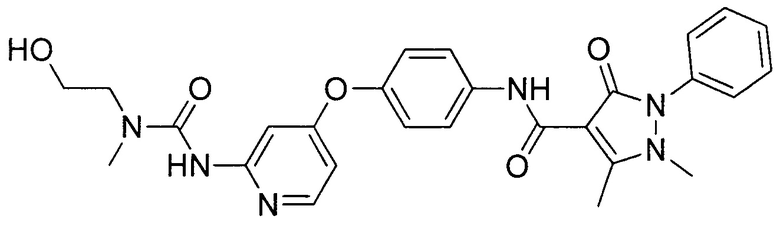
Шаг 1) фенил (4-(4-(1,5-диметил-3-оксо-2-фенил-2,3-дигидро-1H-пиразол-4-карбоксамидо)фенокси)пиридин-2-ил) карбамат
[0239] Взвесь N-(4-((2-аминопиридин-4-ил)окси)фенил)-1,5-диметил-3-оксо-2-фенил-2,3-дигидро-1Н-пиразол-4-карбоксамид (600 мг, 1,44 ммоль) в DCM/пиридине (30 мл / 20 мл) охладили до 0°С, затем раствор фенилкарбонохлоридата (565 мг, 3,61 ммоль) в DCM (10 мл) медленно добавили к смеси через 1 час. Реакционный раствор размешивали при к.т. в течение 2 часов, затем разбавили DCM (200 мл). Смесь промыли водой (140 мл × 4) с последующим промыванием морской водой (150 мл), высушили над безводной Na2SO4, отфильтровали и сконцентрировали в вакууме. Остаток очистили колоночной хроматографией с силикагелем (DCM/EtOAc (о/с) = 2/1) до титулованного соединения в виде белой твердой фазы (327 мг, 42%).
MS (ESI, пол. ион) уд.з.: 536.0 [М+Н]+.
Шаг 2) N-(4-((2-(3-(2-гидроксиэтил)-3метилуреидо)пиридин-4-ил)окси)фенил)-1,5-диметил-3-оксо-2-фенил-2,3-дигидро-1Н-пиразол-4-карбоксамид
[0240] Смесь фенила (4-(4-(1,5-диметил-3-оксо-2-фенил-2,3-дигидро-1Н-пиразол-4-карбоксамидо) фенокси)пиридин-2-ил) карбамат (50 мг, 0,093 ммоль), NMP (3 мл) и 2-(метиламино) этанол (8,4 мг, 0,112 ммоль) нагревали при 40°С в течение 2 часов. Растворитель испарили под низким давлением при 40°С и затем разбавили EtOAc (10 мл). Полученную смесь промыли водой (7 мл × 3) с последующим промыванием морской водой (10 мл), высушили над безводной Na2SO4, отфильтровали и сконцентрировали в вакууме. Остаток промывали EtOAc (2 мл) до титулованного соединения в виде белой твердой фазы (25 мг, 52%).
MS (ESI, пол. ион) уд.з.: 517.0 [М+Н]+;
1Н NMR (300 МГц, DMSO-d6): δ (ppm) 10.80 (s, 1Н), 9.04 (s, 1Н), 8.06 (d, J=5.7 Гц, 1Н), 7.70-7.65 (m, 2Н), 7.62-7.41 (m, 5Н), 7.36 (d, J=2,2 Гц, 1Н), 7.14-7.09 (m, 2Н), 6.54 (dd, J=2,3 Гц, 5,7 Гц, 1Н), 5.31 (br s, 1Н), 3.56 (q, J=4.8 Гц, 2H), 3.36 (s, 3Н), 3.36-3.33 (m, 2H), 2.88 (s, 3Н), 2.71 (s, 3Н).
Пример 29. 1,5-диметил-N-(4-((2-(3-метилуреидо)пиридин-4-ил)окси)фенил)-3-оксо-2-фенил-2,3-дигидро-1Н-пиразол-4-карбоксамид
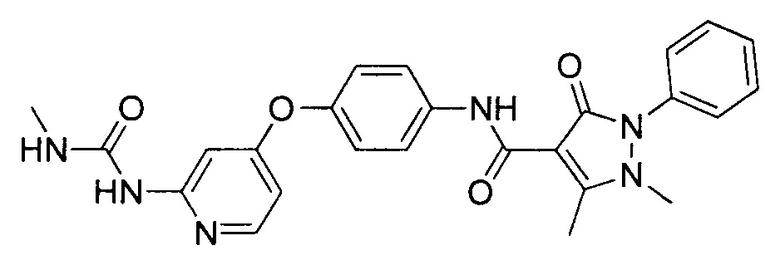
[0241] Смесь фенила (4-(4-(1,5-диметил-3-оксо-2-фенил-2,3-дигидро-1Н-пиразол-4-карбоксамидо) фенокси)пиридин-2-ил) карбамат (100 мг, 0,187 ммоль), NMP (3 мл) и раствор метиламина (25%, 0,2 мл) быстро нагревали при 40°С. Растворитель испарили под низким давлением при 40°С и затем разбавили водой (10 мл). Осадок собрали фильтрацией и затем очистили колоночной хроматографией с силикагелем (DCM/EtOAc/MeOH (o/cN) = 1/1/0.01) до титулованного соединения в виде белой твердой фазы (46 мг, 52%).
MS (ESI, пол. ион) уд.з.: 473.1 [М+Н]+;
1Н NMR (300 МГц, DMSO-d6): δ (ppm) 10.81 (s, 1Н), 9.13 (s, 1Н), 8.04 (d, J=5,9 Гц, 1Н), 7.99-7.94 (m, 1Н), 7.71-7.65 (m, 2Н), 7.62-7.42 (m, 5Н), 7.15-7.09 (m, 2Н), 6.87 (d, J=2,28 Гц, 1Н), 6.51 (dd, J=2,3 Гц, 5,9 Гц, 1Н), 3.36 (s, 3Н), 2.71 (s, 3Н), 2.68 (d, J=4.6 Hz, 3Н).
Пример 30. N-(4-((2-(3-этилуреидо)пиридин-4-ил)окси)фенил)-1,5-диметил-3-оксо-2-фенил-2,3 дигидро-1Н-пиразол-4-карбоксамид
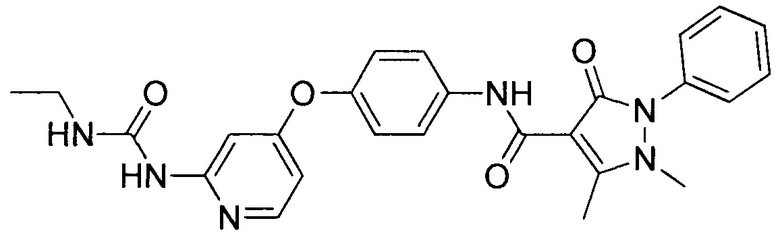
[0242] К раствору гидрохлорида этиламина (29 мг, 0,356 ммоль) в NMP (3 мл) добавили Et3N (0,1 мл). Раствор размешивали при к.т. в течение 10 минут с последующим добавлением фенила (4-(4-(1,5-диметил-3-оксо-2-фенил-2,3-дигидро-1Н-пиразол-4-карбоксамидо) фенокси)пиридин-2-ил) карбамат (100 мг, 0,187 ммоль). Реакционный раствор быстро размешивали при 40°С, затем сконцентрировали в вакууме при 40°С и разбавили водой (10 мл). Осадок собрали фильтрацией и затем очистили колоночной хроматографией с силикагелем (DCM/EtOAc/MeOH (о/с) = 80/40/1) до титулованного соединения в виде белой твердой фазы (46 мг, 50%).
MS (ESI, пол. ион) уд.з.: 487.0 [М+Н]+;
1Н NMR (300 МГц, DMSO-d6): δ (ppm) 10.81 (s, 1H), 9.13 (s, 1Н), 8.04 (d, J=5,9 Гц, 1H), 7.99-7.94 (m, 1Н), 7.71-7.65 (m, 2Н), 7.62-7.42 (m, 5Н), 7.15-7.09 (m, 2Н), 6.87 (d, J=2,3 Гц, 1Н), 6.51 (dd, J=2,3 Гц, 5,9 Гц, 1Н), 3.36 (s, 3Н), 2.71 (s, 3Н), 2.68 (d, J=4.6 Hz, 3Н).
БИОЛОГИЧЕСКОЕ ТЕСТИРОВАНИЕ
[0243] Система LC/MS/MS, используемая в анализе, состоит из вакуумного сепаратора Agilent 1200, двойного насоса, автоматического дозатора с луночным планшетом, терморегулируемой колонки, масс-спектрометра Agilent G6430 Triple Quadrupole с источником ионизации электрораспылением (ESI) Количественный анализ был выполнен с помощью режима MRM. Параметры для переходов MRM привдены в Таблице А.

[0244] Колонка Agilent XDB-C18, 2.1×30 мм, 3.5 μМ использовалась для анализа. 5 μЛ образцов были введены. Условие анализа: подвижная фаза - муравьиная кислота 0,1% в воде (А) и муравьиная кислота кислота в метаноле 0,1% (В). Расход составлял 0,4 мл/минуты. Градиент подвижной фазы приведен в Таблице В.
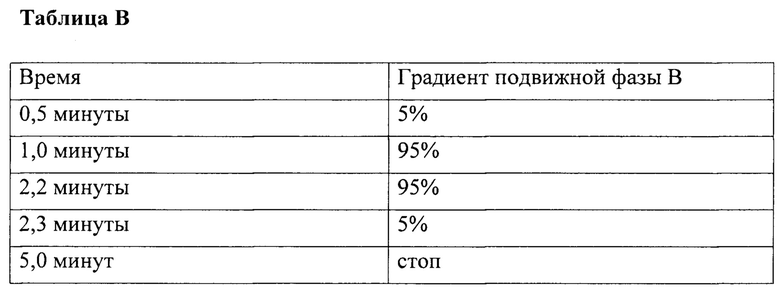
[0245] Также спектрометр Agilent 6330 LC/MS/MS, оборудованный двойными насосами G1312A, автоматическим дозатором G1367A и датчиком G1314C UV, использовался в анализе. Источник ESI использовался на спектрометре LC/MS/MS. Анализ проводили в режиме положительного иона, при необходимости, и переход MRM для каждого аналита был оптимизирован с помощью титрованного раствора. Колонка МР-С18 Capcell 100×4,6 мм I.D., 5 μM (Phenomenex, Торранс, Калифорния, США) использовалась во время анализа. Подвижная фаза - 5-мм ацетат аммиака, МеОН 0,1% в воде (А): 5-мм ацетат аммиака, МеОН 0,1% в ацетонитриле (В) (70:30, о/с). Расход составлял 0,6 мл/минуту. Колонка сохранялась при температуре окружающей среды. 20 μЛ образцов были введены.
Пример А: стабильность соединения в микросомах печени человека и крысы
[0246] Инкубации микросом печени крысы и человека проводились в двойном экземпляре в трубах из полипропилена. Стандартные смеси инкубации состояли из микросом печени крысы или человека (0,5 мг протеин/мл), соединения (5 μМ) и NADPH (1,0 мм) в суммарном объеме буфера фосфата калия на 200 μЛ (PBS, 100 мм, рН фактор 7.4). Соединения растворили в DMSO и разбавили PBS, таким образом, чтобы заключительная концентрация DMSO составляла 0,05%. Ферментативные реакции начинали с добавлением протеина после преинкубации в течение 3 минут и инкубации на водяной бане, открытой для воздуха при 37°С. Реакции были завершены в различные моментв времени (0, 5, 10, 15, 30, 60 минут)путем добавления равного объема ледяного ацетонитрила. Образцы были сохранены при -80°С до испытания LC/MS/MS.
[0247] Концентрации соединений в смесях инкубации микросом печени крысы или человека были определены способом LC/MS/MS. Диапазоны линейности в диапазоне концентрации были определены для каждого проверяемого соединения.
[0248] Параллельная инкубация выполнялась с помощью денатурированных микросом в качестве отрицательного контроля, и реакции были завершены в различные моменты времени (0, 15, 60 минут)после инкубации при 37°С.
[0249] Декстрометорфан (70 μМ) был выбран как положительный контроль, и реакции были завершены в различные моменты времени (0, 5, 10, 15, 30, 60 минут)после инкубации при 37°С. Образцы положительного и отрицательного контроля были включены в каждое испытание для обеспечения целостности системы микросомальной инкубации.
Анализ данных
[0250] Концентрации соединений при инкубации микросом печени крысы или человека принимались как процент от соответствующего нулевого момента времени для каждой реакции. В естественных условиях CLint экстраполировались (см. Наритоми И., Терашита С, Кимура С, Сузуки А., Кагайама А., Суджияма И. Прогнозирование печеночного клиренса человека в естественных экспериментах на животных и в лабораторных метаболических исследованиях с микросомами печени животных и человека. Drug Metabolism and Disposition 2001, 29: 1316-1324.)
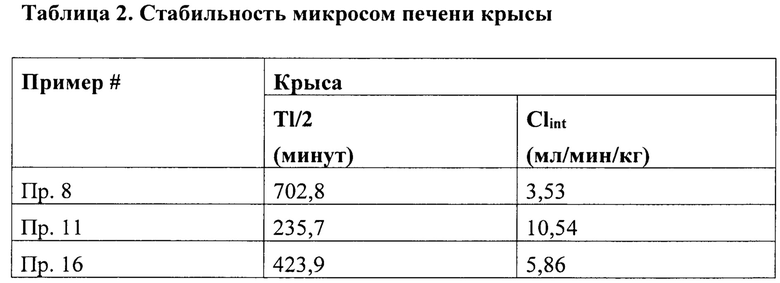
[0251] Соединения, раскрытые в настоящем документе, очень стабильны в микросомах печени крысы. Например, Примеры 8, 11 и 16 были мало усвоены, когда они были выведены в микросомах печени крысы при экспериментальном условии, таким образом, демонстрируя продленное время Т1/2.
Пример В: оценка фармакокинетики после внутривенного введения и перорального приема соединений, раскрытых в настоящем документе, у мышей, крыс, собак и обезьян
[0252] Соединения, раскрытые в настоящем документе, оценены в фармакокинетических исследованиях у мышей, крыс, собак или обезьян. Соединения вводились как водный раствор, 2%-й НРМС + 1%-й TWEEN®80 в водном растворе, 5% DMSO + 5% солутол в физиологическом растворе, взвеси 4% МС или в капсуле. Для внутривенного введения животные обычно получали дозу 1 или 2 мг/кг. Для перорального приема (п.о) мышам и крысам обычно давали дозу 5 или 10 мг/кг и собакам и обезьянам обычно давали дозу 10 мг/кг. Образцы крови (0,3 мл) были взяты через 0,25, 0.5, 1.0, 2.0, 3.0, 4.0, 6.0, 8.0, 12 и 24 ч или через 0.083, 0.25, 0.5, 1.0, 2.0, 4.0, 6.0, 8,0 и 24 ч и центрифугировались на скорости 3000 или 4000 об/мин в течение 2-10 минут. Плазменные растворы были собраны, сохранены при -20°С или -70°С, пока не были проанализированы на LC/MS/MS, как описано выше.
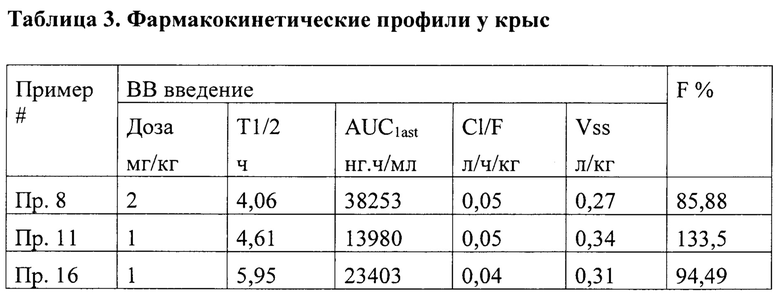
[0253] Соединения, раскрытые в настоящем документе, продемонстрировали оптимизированные фармакокинетические свойства с желательным клиренсом (О), периодом полураспада (Т1/2) и превосходным пероральным биоаккумулированием, при введении соединения внутривенно или перорально.
[0254] Эффективность соединений, раскрытых в настоящем документе, как ингибиторов киназ тирозина рецептора, например c-Met, VEGFR, Ron и Axl, и в качестве противопухолевых агентов в моделях ксенотрансплантатов животных могут быть оценены следующим образом. Результаты испытания могут продемонстрировать, что определенные соединения, раскрытые в настоящем документе, потенциально ингибируют c-Met, VEGF-R2, Rona и фосфорилирование Axl и демонстрируют потенциальную противоопухолевую акивность, зависимую от дозы в определенных моделях ксенотрансплантатов.
Анализ киназы
[0255] Анализ киназы может быть выполнен измерением объединения γ - 33Р АТР в иммобилизованный миелиновый основной протеин (МБР). Высокосвязывающие белые 384 луночные планшеты (Greiner)покрыты МБР (Сигма #М-1891) инкубацией 60 μл/лунка 20 μг/мл МВР в ТРИС-буфферизированном физрастворе (TBS; 50 мм рН ТРИС 8.0, 138-мм NaCl, 2.7-мм KCl) в течение 24 часов при 4°С. Пластины промывали 3×100 μл TBS. Реакции киназы выполняются в суммарном объеме буфера киназы 34 μЛ (5-мм рН Hepes 7.6, 15-мм NaCl, бычий гамма-глобулин 0,01% (Sigma №1-5506), 10-мм MgCh, 1-мм DTT, TritonX-100 0,02%). Разведение соединения выполняли в DMSO и добавляли в аналитические лунки к заключительной концентрации DMSO 1%. Каждая точка данных измеряется дважды, и, по крайней мере, два двойных анализа выполняется для определения каждого отдельного соединения. Фермент добавляется к заключительным концентрациям 10 нм или 20 нм, например. Смесь немаркированного АТР и γ - 33Р АТР добавляется для начала реакции (2×106 cpm в минуту γ - 33Р АТР на лунку (3000 С1/ммоль) и 10 μМ немаркированных АТР. Реакции выполняются в течение 1 часа при комнатной температуре с перемешиванием. Планшеты промываются 7х TBS с последующим добавлением сверкающей жидкости 50 μл/лунка (Wallac). Планшеты считываются с помощью счетчика Wallac Trilux. Это только один формат такого испытания; возможны различные другие форматы, известные специалисту в области.
[0256] Вышеупомянутая процедура анализа может использоваться для определения IC50 для ингибирования и/или постоянного ингибирования, Кi. IC50 определяется как концентрация соединения, необходимая для сокращения активности фермента на 50% при условии анализа. Значение IC50 оценивается путем подготовки кривой на 10 точек с помощью ряда разбавлений на 1/2 (например, типичная кривая может быть подготовлена с помощью следующих концентраций соединения: 100 μМ, 30 μМ, 10 μМ, 3 μМ, 1 μМ, 0.3 μМ, 0.1 μМ, 0.03 μМ, 0.01 μМ и 0 μМ).
Анализ c-Met (h)
[0257] Met (h) инкубируется при 8-мм рН MOPS 7.0, 0,2-мм EDTA, 250 μМ KKKSPGEYVNIEFG, 10-мм MgAcetate и [γ-33Р-АТР] (определенная активность приблизительно 500 cpm/пмоль, концентрация по мере необходимости). Реакция инициируется добавлением соединения MgATP. После инкубации в течение 40 минут при комнатной температуре реакция останавливается добавлением 3% раствора фосфорной кислоты. 10 μЛ реакционного раствора затем наносится на фильтр Р30 и промывается три раза в течение 5 минут 75-мм фосфорной кислоты и один раз метанолом до высыхания и сцинтилляционного подсчета.
Анализ KDR (h) (VEGF-R2 (h)):
[0258] KDR (h) инкубируется прис 8-мм рН MOPS 7.0, 0,2-мм EDTA, 0,33 мг/мл миелиновым основным протеином, 10-мм MgAcetate и [γ-33Р-АТР] (определенная активность приблизительно 500 cpm/пмоль, концентрация по мере необходимости). Реакция инициируется добавлением соединения MgATP. После инкубации в течение 40 минут при комнатной температуре реакция останавливается добавлением 3% раствора фосфорной кислоты. 10 μЛ реакционного раствора затем наносится на фильтр Р30 и промывается три раза в течение 5 минут 75-мм фосфорной кислоты и один раз метанолом до высыхания и сцинтилляционного подсчета.
Анализ Axl (h)
[0259] Axl (h) инкубируется при 8-мм рН MOPS 7.0, 0,2-мм EDTA, 250 μМ KKSRGDYMTMQIG, 10-мм MgAqeTate и [γ-33Р-АТР] (определенная активность приблизительно 500 cpm/пмоль, концентрация по мере необходимости). Реакция инициируется добавлением соединения MgATP. После инкубации в течение 40 минут при комнатной температуре реакция останавливается добавлением 3% раствора фосфорной кислоты. 10 μЛ реакционного раствора затем наносится на фильтр Р30 и промывается три раза в течение 5 минут 75-мм фосфорной кислоты и один раз метанолом до высыхания и сцинтилляционного подсчета.
[0260] Анализ киназы, описанный в настоящем документе, может быть выполнен в Millipore UK Ltd, Технологическом парке Данди, Данди DD2 1SW, Великобритания.
[0261] Также активность киназы соединений можно измерить с помощью KINOMEscaw™, основывающегося на анализе связывания, который количественно измеряет способность соединения конкурировать с иммобилизованным активным лигандом. Испытание выполнялось путем объединения трех компонентов: ДНК-маркированной киназы; иммобилизованным лигандом; и испытуемым соединением. Способность испытуемого соединения конкурировать с иммобилизованным лигандом измерялась количественным PCR ДНК-маркера.
[0262] Для большей части анализов маркируемые киназой штаммы фага Т7 были подготовлены на носителе Е. coli, полученного из штамма BL21. Бактерии Е. coli были выращены к лог-фазе и заражены фагом Т7, и инкубированы с перемешиванием при 32°С до лизиса. Лизаты центрифугировались и фильтровались для удаления обломков клетки. Оставшиеся киназы производились в клетках HEK-293 и впоследствии маркировались ДНК для обнаружения qPCR. Покрытые стрептавидином магнитные бусинки рассматривали с низкомолекулярными биотинилированными лигандами в течение 30 минут при комнатной температуре для генерации родственных смол для испытания киназы. Бусинки лигандов заблокированы избыточным биотином и промыты блокирующим буфером (SEABLOCK™ (Pierce), 1% BSA, TWEEN®20 0,05%, 1-мм DTT), чтобы удалить несвязанный лиганд и уменьшить неопределенное связывание. Реакции связывания были собраны путем объединения киназ, бусинок родственных лигандов и испытуемых соединений в Гх связывающем буфере (20%) SEABLOCK™, 0.17х PBS, TWEEN®20 0,05%, 6-мм DTT). Все реакции выполнялись в полистирольных 96-луночных планшетах в заключительном объеме 0.135 мл. Аналитические планшеты были инкубированы при комнатной температуре с переешиванием в течение 1 часа, и родсвенные бусинки были промыты буфером (1x PBS, TWEEN®20 0,05%). Бусинки затем повторно взвешены в буфере (1x PBS, TWEEN®20 0,05%, 0.5 дМ небиотинилированный родственный лиганд) и инкубированы при комнатной температуре с перемешиванием в течение 30 минут.Концентрация киназы в элюатах была измерена qPCR.
[0263] Анализ киназы, описанный в настоящем документе, выполнялся с помощью KINOMEscaw™ Профильная услуга в DiscoveRx Corporation, 42 501 Олбрэ-Ст.Фремонт, Калифорния 94538, США, и выбранные результаты перечислены в Таблице 4.
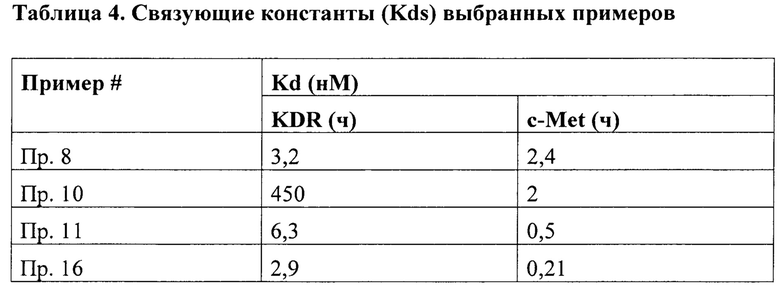
[264] Соединения, раскрытые в настоящем документе, продемонстрировали потенциальную активность в анализах c-Met (h) и KDR (h).
Анализ клеточного фосфорилирования
[0265] Обычно клетки предварительно инкубируются испыттуемыми составами для обеспечения полного таргетного связывания. Уровень автофосфорилирования определен способом Sandwich-ELISA. Значения IC50 определяются путем испытания 8 концентраций соединения на полулогарифмических этапах (каждая концентрация дублируется). Анализ клеточного фосфорилирования, описанный в настоящем документе, может быть выполнен в ProQinase GmbH, Брайзахер Штрассе 117 D-79106, Фрайбург, Германия.
Анализ фосфорилирования c-Met
[0266] Известно, что клеточная линия аденокарциномы желудка человека MKN45 сверхпродуцирует c-Met. Сверхпродукция c-Met приводит к конститутивному, независимому от лиганда автофосфорилированию киназы. Путем добавления SU 11274 уровни фосфинового МЕТ в основном уменьшены, и, таким образом, достигнуто динамическое поведение, чтобы определить ингибирующие потенциалы соединений. Сигнал фосфинового МЕТ впоследствии определяется количественно способом Sandwich-ELISA. Анализ валидируется на основе известных ингибиторов активности киназы МЕТ.
Анализ фосфорилирования VEGF-R2
[0267] Известно, что иммортализованные эндотелиальные клетки пупочной вены человека (HUE) сверхпродуцируют человеческий рецептор VEGF-R2. Стимулирование этих клеток с физиологическим лигандом VEGF-A приводит к прочному автофосфорилированию рецептора. Соединения предварительно инкубируются перед стимулированием клетки, чтобы обеспечить полное таргетное связывание. Условия стимулирования оптимизированы для определения связанного с дозой ингибирования сигнала фосфинового VEGF-R2, впоследствии определяющегося количественно способом sandwich-ELISA. Анализ валидируется на основе известных ингибиторов активности киназы VEGF-R2.
Анализ фосфорилирования Axl
[0268] Анализ клеточного фосфорилирования AXL был сгенерирован на фоне эмбрионального фибробласта мыши (MEF). Клетки были трансфицированны для продукции непроцессированного протеина AXL. После клонового выбора была получена преобразованная клеточная линия с высоким уровнем автофосфорилированного AXL. Путем добавления староспорина фосфиновые-AXL уровни в основном уменьшены, и, таким образом, достигнуто динамическое поведение, чтобы опрелить ингибирующие потенциалы соединений. Фосфиновые-AXL уровни определяются количественно способом Sandwich-ELISA.
Модели ксенотрансплантата опухоли
[0269] Эффективность соединений, раскрытых в настоящем документе, оценена в стандартной модели онкогенеза у крыс.Опухолевые клетки человека (клетки глиобластомы U87MG от АТСС) были использованы в культуре, собраны и введены подкожно на задний фланг 6-7-недельных бестимусных самок мышей (BALB/cA, nu/nu Hunan SLAC Laboratory Animal, Co.) (n=6-10 для группы средства и для каждой группы дозирования). Когда опухоли достигли объема 100-250 мм3, животные были несистемно разделены на контрольные группы (например, 5% % DMSO+70 Captisol® (30%), 7% НС1 (рН1), 18% Captisol® (30%); или 7% DMSO, 7% НС1 (pHl), 70% Captisol® (30%), 16% Captisol® (30%) и т.п.) и группы соединения. Последующее введение соединения перорально начинает в любой момент, начиная с 0 до 15 дня после возникновения опухолевой клетки, и обычно продолжается один раз в день на период эксперимента.
Анализ ингибирования роста опухоли (TGI)
[0270] Прогрессия роста опухоли оценена объемами опухоли и регистрируется как функция времени. Длинные (Д) и короткие (Ш) оси подкожных опухолей измеряли кронциркулем два раза в неделю, и объем опухоли (TV) рассчитывали как (Д × Ш2)/2). TGI рассчитывали от разницы между средними объемами опухоли мышей контрольной и испытательной группы, выраженной как процент от среднего объема опухоли в испытательной и контрольной группе, следующим отношением:

Начальный статистический анализ выполнен повторным дисперсионным анализом отличий (RMANOVA) с последующим испытанием Scheffe psot hoc на многократные сравнения. Только контроль (5%-й % DMSO + 70 Captisol® (30%), 7% HCl (pHl), 18% Captisol® (30%); или 7% DMSO, 7% HC1 (pHl), 70% Captisol® (30%), 16% Captisol® (30%) и т.п.), отрицательный контроль.
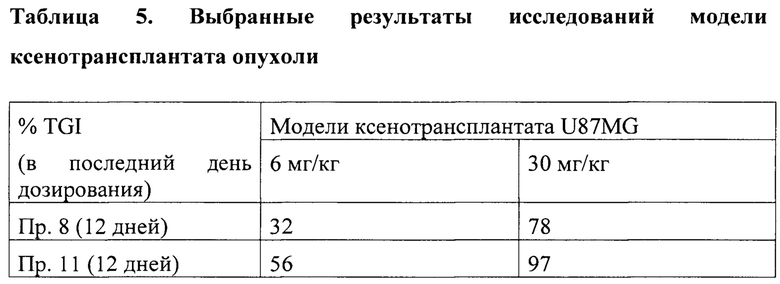
[0271] Наконец, нужно отметить, что существуют альтернативные способы реализовать данное изобретение. Соответственно, существующие воплощения считаются иллюстративными и не ограничивающими, и изобретение не должно быть ограничено подробной информацией, предоставленной в настоящем документе, но может быть изменено в объеме и эквивалентах приложенных пунктов формулы изобретения. Все публикации и патенты, процитированные в настоящем документе, включены посредством ссылок.
| название | год | авторы | номер документа |
|---|---|---|---|
| ТРИЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ И ИХ ИСПОЛЬЗОВАНИЕ | 2016 |
|
RU2724100C2 |
| ФАРМАЦЕВТИЧЕСКИЕ СОЕДИНЕНИЯ | 2020 |
|
RU2840815C2 |
| БИЦИКЛИЧЕСКИЕ ЛАКТАМЫ И СПОСОБЫ ИХ ПРИМЕНЕНИЯ | 2016 |
|
RU2827714C1 |
| ФАРМАЦЕВТИЧЕСКОЕ СРЕДСТВО, СОДЕРЖАЩЕЕ ИНГИБИТОР НАТРИЙЗАВИСИМОГО ПЕРЕНОСЧИКА ФОСФАТА | 2015 |
|
RU2811864C1 |
| ФАРМАЦЕВТИЧЕСКОЕ СРЕДСТВО, СОДЕРЖАЩЕЕ ИНГИБИТОР НАТРИЙЗАВИСИМОГО ПЕРЕНОСЧИКА ФОСФАТА | 2015 |
|
RU2740008C2 |
| НОВЫЕ АНТИГЕЛЬМИНТНЫЕ СОЕДИНЕНИЯ | 2019 |
|
RU2828007C2 |
| НОВЫЕ ПИРАЗОЛПИРИМИДИНОВЫЕ ПРОИЗВОДНЫЕ | 2017 |
|
RU2769448C2 |
| МОДУЛЯТОРЫ ПРОТЕОЛИЗА И СООТВЕТСТВУЮЩИЕ СПОСОБЫ ПРИМЕНЕНИЯ | 2019 |
|
RU2805511C2 |
| НОВЫЕ АКТИВАТОРЫ РАСТВОРИМОЙ ГУАНИЛАТЦИКЛАЗЫ И ИНГИБИТОРЫ ФОСФОДИЭСТЕРАЗЫ С ДВОЙНЫМ МЕХАНИЗМОМ ДЕЙСТВИЯ И ИХ ПРИМЕНЕНИЕ | 2018 |
|
RU2758373C2 |
| ПОЛИЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ И СПОСОБЫ ЦЕЛЕНАПРАВЛЕННОЙ ДЕГРАДАЦИИ ПОЛИПЕПТИДОВ БЫСТРО УСКОРЕННОЙ ФИБРОСАРКОМЫ | 2019 |
|
RU2830173C2 |
Изобретение раскрывает новые соединения замещенных пиразолонов формулы (I) и их фармацевтически приемлемые соли, которые ингибируют активность протеин тирозинкиназы и модулируют клеточную активность, например быстрое увеличение, дифференцирование, апоптоз, миграцию и инвазию. В соединениях формулы (I) Q представляет собой -N(Rc)C(=O)Rd; W является CR7 или N; X представляет собой (С6-С10)арил; каждый из Y и Z представляет собой независимо (С1-С6)алкил; каждый из R1, R2, R3, R4, R5, R6 и R7 представляет собой независимо Н, D, F или Cl; каждый из Ra, Rb и Rc представляет собой независимо Н; и Rd представляет собой (С3-C8) циклоалкил, где (С3-С8)циклоалкил не замещается или необязательно замещается 1 или 2 заместителями, независимо выбранными из D, F, Cl, Br, -CN, -ORa-, -NRaRb и (С1-С6)алкила. Изобретение также раскрывает фармацевтически приемлемые композиции, включающие такие соединения, и способы использования композиций и соединений формулы (I) в лечении гиперпролиферативных расстройств у млекопитающих, в частности у людей. 5 н. и 10 з.п. ф-лы, 7 табл., 30 пр.
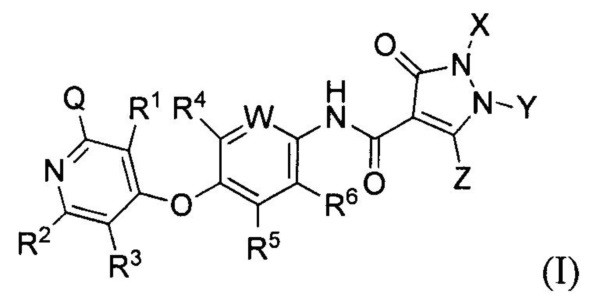
1. Соединение формулы (I):
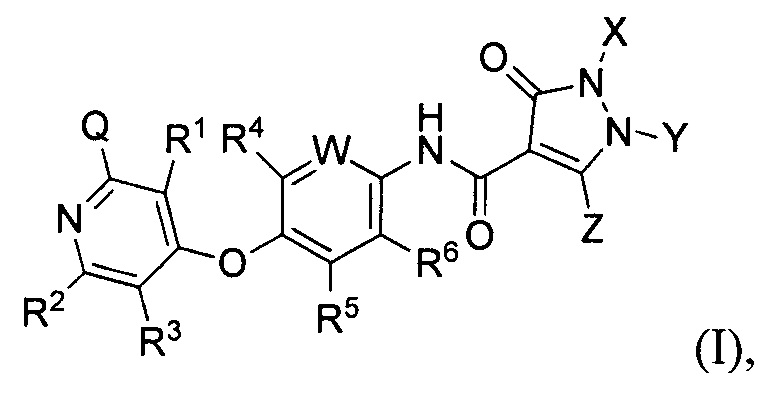
или фармацевтически приемлемая соль,
где Q представляет собой -N(Rc)C(=O)Rd;
W является CR7 или N;
X представляет собой (С6-С10)арил;
каждый из Y и Z представляет собой независимо (С1-С6)алкил;
каждый из R1, R2, R3, R4, R5, R6 и R7 представляет собой независимо Н, D, F или Cl;
каждый из Ra, Rb и Rc представляет собой независимо Н; и
Rd представляет собой (С3-C8) циклоалкил, где (С3-С8)циклоалкил не замещается или необязательно замещается 1 или 2 заместителями, независимо выбранными из D, F, Cl, Br, -CN, -ORa-, -NRaRb и (С1-С6)алкила.
2. Соединение по п. 1, где Rd представляет собой (С3-С6)циклоалкил, где (С3-С6) циклоалкил не замещается или необязательно замещается 1 или 2 заместителями, независимо выбранными из D, F, CN, ORa, NRaRb и (С1-С3)алкила.
3. Соединение по п. 1, где каждый из X представляет собой фенил; каждый из Y и Z независимо представляет собой СН3, этил, пропил или изопропил.
4. Соединение по п. 1, где Q представляет собой:
 или
или
5. Соединение по п. 1, имеющее Формулу (II):
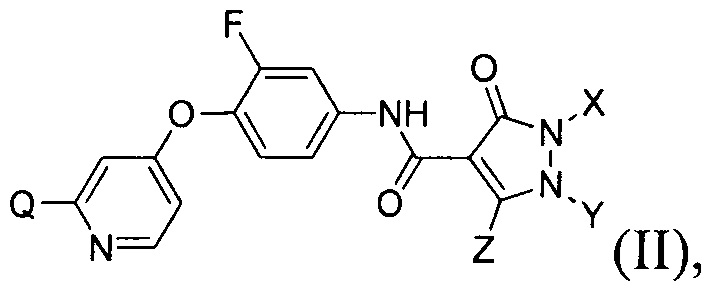
где Q является -N(Rc)C(=O)Rd;
X представляет собой (С6-С10)арил;
каждый из Y и Z независимо представляет собой (С1-С6)алкил;
каждый из Ra, Rb и Rc независимо представляет собой Н; и
Rd представляет собой (С3-С8)циклоалкил, где (С3-C8) циклоалкил необязательно замещается 1 или 2 заместителями, независимо выбранными из D, F, Cl, ОН, NH2 и (С1-С6)алкила.
6. Соединение по п. 5, где X представляет собой фенил; каждый из Y и Z независимо представляет собой (С1-С4)алкил.
7. Соединение по п. 5, где X представляет собой фенил; каждый из Y и Z независимо представляет собой Me, этил, пропил или изопропил.
8. Соединение по п. 5, где Q является:
 или
или
9. Соединение по п. 1, имеющее одну из следующих структур:
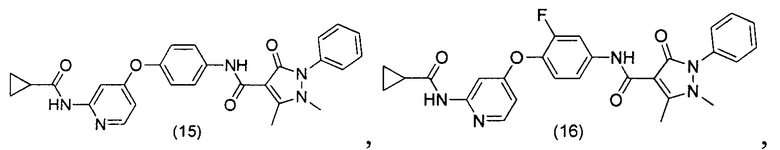
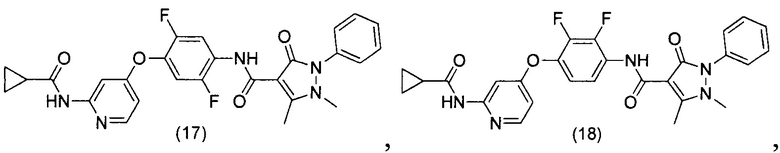
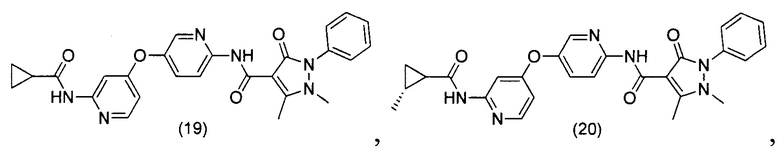
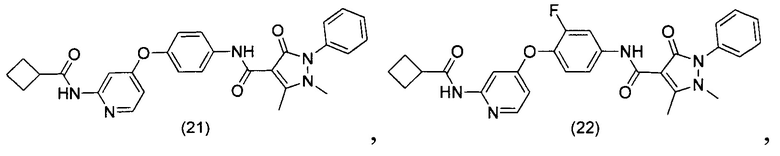
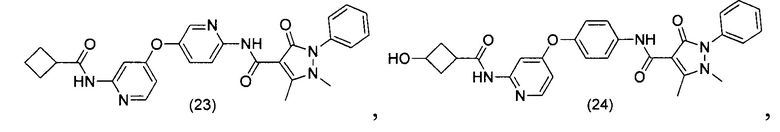

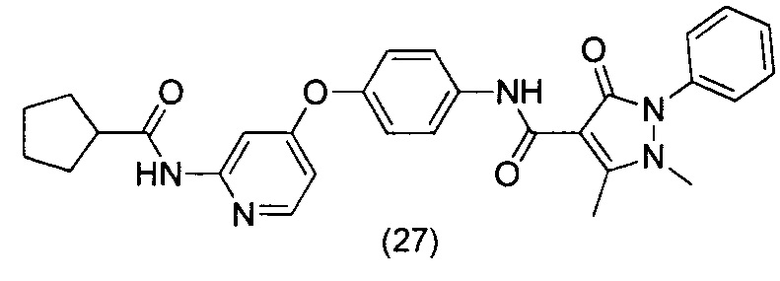 или
или 
10. Соединение по любому из пп. 1-9 для использования в предотвращении, контроле, лечении или уменьшении серьезности пролиферативных расстройств у пациента.
11. Фармацевтическая композиция, включающая соединение по любому из пп. 1-9, и фармацевтически приемлемый носитель, наполнитель, разжижитель, стимулятор, агент или комбинацию таких веществ для использования в предотвращении, контроле, лечении или уменьшении серьезности пролиферативных расстройств у пациента.
12. Соединение по п. 10, где пролиферативное расстройство является метастатическим раком, раком толстой кишки, аденокарциномой желудка, раком мочевого пузыря, раком молочной железы, раком почек, раком печени, раком легких, раком кожи, раком щитовидной железы, раком головы и шеи, раком простаты, раком поджелудочной железы, раком ЦНС, глиобластомой или миелопролиферативным расстройством, атеросклерозом или фиброзом легкого.
13. Фармацевтическая композиция по п. 11, где пролиферативное расстройство является метастатическим раком, раком толстой кишки, аденокарциномой желудка, раком мочевого пузыря, раком молочной железы, раком почек, раком печени, раком легких, раком кожи, раком щитовидной железы, раком головы и шеи, раком простаты, раком поджелудочной железы, раком ЦНС, глиобластомой или миелопролиферативным расстройством, атеросклерозом или фиброзом легкого.
14. Соединение по любому из пп. 1-9 для использования в ингибировании или модуляции активности протеинкиназы в биологической пробе, включая контакт биологической пробы с соединением;
где протеинкиназа является рецепторной тирозинкиназой,
при этом рецепторная тирозинкиназа является VEGFR, C-met, Ron, A×L или их комбинацией.
15. Фармацевтическая композиция, включающая соединение по любому из пп. 1-9, и фармацевтический приемлемый носитель, наполнитель, разжижитель, стимулятор, агент или комбинация таких веществ для использования в ингибировании или модуляции активности протеинкиназы в биологической пробе, включая контакт биологической пробы с фармацевтической композицией,
где протеинкиназа является рецепторной тирозинкиназой;
а также рецепторная тирозинкиназа является VEGFR, c-Met, Ron, A×l или их комбинацией.
| Устройство для установки на железной крыше мачт для радиосетей | 1928 |
|
SU13231A1 |
| US 7714138 B2, 11.05.2010 | |||
| ПЛАСТИНЧАТЫЙ МНОГОКАНАЛЬНЫЙ КАВИТАЦИОННЫЙ РЕАКТОР | 2006 |
|
RU2336123C1 |

