Изобретение относится к области неорганической химии, а именно к способам синтеза неорганических фторсодержащих соединений, большое количество которых и разнообразие свойств предопределило их широкое практическое применение в различных областях промышленности, в том числе для получения керамики, эмалей, специальных стекол, лазерных и оптических материалов, фторидных стекол, производства цемента, флюсов и т.д.
Возросший в последнее время интерес к фторидным материалам связан с развитием новых отраслей промышленности, требующих материалов с уникальными свойствами. Так, например, лазерная и оптическая промышленность предъявляет жесткие требования к используемым фторидным материалам по содержанию кислорода, поскольку его присутствие ухудшает химические и физические свойства материала (понижает температуры плавления, стабилизирует промежуточные неустойчивые модификации, приводит к ошибкам при идентификации новых фаз), а например, для отраслей горноперерабатывающей и химической промышленности большей проблемой являются выбор конструкционных материалов и энергоемкость процессов, так как вскрытие руд и неорганический синтез, проводимые с использованием фторсодержащих соединений, чаще осуществляются при высоких температурах.
Метод синтеза фторсодержащих соединений и фторирующие реагенты выбирают в зависимости от требуемых полезных свойств синтезируемых соединений, используя для этого газообразные, жидкие и твердые фторирующие агенты от элементного фтора до сложных комплексных соединений.
Наиболее распространенными являются жидкофазные способы синтеза фторидов с использованием в качестве фторирующих агентов фтористоводородной, фторсульфоновой или кремнефтористоводородной кислот, например, путем их взаимодействия с оксидами, гидроксидами или карбонатами соответствующих металлов с получением гидролитически устойчивых фторидов и бифторидов щелочных и щелочноземельных металлов (п. РФ №2225839, №2226502; п. США №2780523, п. США №4031193).
Однако использование жидких фторирующих агентов приводит к поверхностному и объемному загрязнению получаемых фторидов анионными примесями, гидроксильными группами, удаление которых при прокаливании даже в инертной среде может сопровождаться пирогидролизом, что ведет к снижению качества получаемых продуктов.
Известны способы газофазного фторирования с использованием газообразных фторирующих агентов, например пентафторида хлора для получения фтористого алюминия (а.с. СССР №1100233, опубл. 30.06.84); газообразного фтора или фтористого водорода для получения тетрафторида кремния (п. Великобритании №2079262, п. США №5853685) или фтористого водорода для получения фтористого натрия (а.с. СССР №472900, опубл. 05.06.1975).
Однако работа с газообразными фторидами чрезвычайно опасна и требует особых условий по технике безопасности.
Способы получения фторидных соединений на основе реакций твердофазного синтеза менее разработаны и также обладают рядом ограничений, возникающих как из-за сложного механизма протекания самих реакций, так и из-за условий проведения этих реакций, в том числе высоких температур синтеза, приводящих к загрязнению получаемых соединений материалом аппаратуры.
Известны способы получения фторсодержащих соединений с использованием в качестве фторирующих агентов, например, твердого фторид-бифторида аммония для получения кремнефторида аммония (п. РФ №2226500, опубл. 10.04.2004), трифторида алюминия (п. РФ №2172718, опубл.27.08.2001) или фторсиликатов калия или натрия при переработке титановых руд при температуре 600°-1000°С (п. США №4359449, опубл. 16.11.1982).
Известно использование комплексных фторидов щелочных металлов в аналитической практике для разложения трудновскрываемых минералов. Так, например, для разложения берилла применяют сплавление с 4-х кратным избытком гексафторосиликата натрия при 850°С (Р.Бок. Методы разложения в аналитической химии. М.: Химия, 1984, с. 73).
Наиболее близким к заявляемому является способ синтеза гексафторометаллатов тугоплавких металлов путем спекания силикатов этих металлов с гексафторосиликатом калия, или натрия, или железа. Таким способом получают гексафтороцирконат калия путем спекания циркона с избытком гексафторосиликата калия при температуре 650 - 700°С согласно суммарному уравнению:
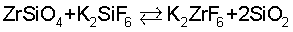
Считается, что процесс протекает с первоначальной диссоциацией K2SiF6 с образованием KF и SiF4, который затем выступает в качестве фторирующего агента для циркона с образованием тетрафторида циркония, взаимодействующего с фторидом калия с образованием гексафтороцирконата калия (Химия и технология редких и рассеянных элементов, ч. 11, М.: Высшая школа, 1976 г., с.322).
Однако, как и все известные способы твердофазного синтеза неорганических фторидов, способ является энергозатратным, так как протекает при температурах выше 600°С. Кроме этого, поскольку на выходе получают смесь двух твердых фаз, требуемый продукт загрязняется трудно удаляемым диоксидом кремния.
Задача изобретения состоит в разработке энергосберегающего способа синтеза фторсодержащих неорганических соединений на основе твердофазных реакций и повышение чистоты получаемого соединения, что достигается за счет использования фторирующего агента, не загрязняющего соединение продуктами своего разложения.
Поставленная задача решается способом получения неорганических фторсодержащих соединений путем твердофазного взаимодействия неорганических соединений, выбранных из группы оксидов, фторидов, солей слабых кислот элементов I-IV и VIII групп Периодической системы элементов с гексафторосиликатом аммония при температуре не выше 280°С.
Способ позволяет при минимальных энергетических затратах получать простые и комплексные фториды металлов I-IV и VIII групп, не загрязненных избытком фторирующего реагента и твердыми продуктами его разложения. Чистота конечных продуктов определяется реакцией синтеза и чистотой исходных реагентов и находится в пределах 90-99,9%.
Предложенное использование (NH4)2SiF6 - гексафторосиликата аммония (ГФСА) в качестве фторирующего реагента в реакциях твердофазного синтеза при температурах не выше 280°С основывается на новом неизвестным ранее механизме ступенчатого разложения ГФСА на HF, NH3 и летучий NH4SiF5, при котором при данных температурах фторирующим реагентом выступает продукт разложения ГФСА - фтористый водород, в отличие от известного процесса фторирования солями кремнефтористоводородной кислоты, в которых фторирующим реагентом является SiF4. Таким образом, известное вещество (ГФСА) используется по новому назначению (фторирующий агент при температурах не выше 280°С), которое обусловлено такими ранее неизвестными для него свойствами (механизм разложения при температурах не выше 280°С), которые необходимы для реализации его назначения.
Известно, что ГФСА является твердым продуктом фторирования диоксида кремния и силикатов металлов гидродифторидом аммония, а также он образуется как побочный продукт фторирования кремнийсодержащих концентратов. ГФСА достаточно легко может быть очищен возгонкой, хорошо растворяется в воде и разлагается раствором аммиака до белой сажи - SiO2. ГФСА устойчив до 100°С, однако выше этой температуры начинает терять массу (около 0,2% в час), а при температуре выше 300°С он переходит в газовую фазу без остатка. По одним данным это происходит вследствие сублимации (Рысс И.Г. Химия фтора и его неорганических соединений. - М.: Госхимиздат, 1956. - С.382), а по другим в результате диссоциации при 319°С (Раков Э.Г. Фториды аммония: Итоги науки и техники. Неорганическая химия. Т.15. - М.: ВИНИТИ, 1988. - 154 с.; Химическая энциклопедия. В 5-ти томах. T.1. - М.: Советская энциклопедия, 1988. - С.282).
Вероятно, вышеприведенные сведения явились причиной того, что в известных источниках информации не обнаружилось сведений о возможности использования ГФСА для получения фторсодержащих соединений при температурах не выше 280°С.
Изучение термических свойств ГФСА проводилось нами на лабораторной установке, состоящей из реактора и конденсатора, представляющих собой две сочлененные трубки из платины, в одной из которых (реактор) нагревали вещество, а в другой (конденсатор) конденсировали образовавшийся продукт (возгон). Для полноты сбора легколетучих фракций конденсатор удлиняли фторопластовой трубкой. Образец исследуемого ГФСА в лодочке помещали в реактор, систему продували аргоном, выдерживали образец при необходимой температуре и собирали образовавшийся в разных зонах конденсатора возгон, который затем подвергали исследованию.
Проведенные нами термодинамические расчеты и экспериментальные исследования термических свойств ГФСА с анализом образующихся возгонов показали, что, во-первых, изменение энергии Гиббса для реакции разложения (NH4)2SiF6 на SiF4, 2NH3 и 2HF для стандартных условий составляет +222,2 кДж/моль, при 319°С - +7,4, и значение, равное нулю, энергия Гиббса принимает только при 329°С, что поставило под сомнение справочные данные о термическом разложении ГФСА при 319°С на SiF4, NH3 и HF, во-вторых, на термограмме возгона (фиг.1, где приведены кривые ДТГ при скорости нагревания 2,5 град/мин): (а) - ГФСА, (б) - его возгона при 300°С и (в) - после повторной перегонки возгона ГФСА при 240°С) присутствует не один эффект потери массы, а два, и, в-третьих, обнаружены различия в растворимости реактивного ГФСА и ГФСА из возгона. При этом нами обнаружено, что основная масса возгона, осаждающаяся в примыкающей к реактору зоне конденсатора, представляет собой кубическую модификацию ГФСА с примесью гексагональной модификации. В то же время в возгоне, собранном в дальней более холодной зоне конденсатора, по данным рентгенофазового анализа кубическая модификация ГФСА отсутствует, а имеется смесь ГФСА гексагональной модификации и оксофторосиликата аммония переменного состава, появление которого связано с высокой чувствительностью образующегося по реакции NH4SiF5 к следовым количествам влаги в зоне реакции. В таблице приведены результаты расчета фазового состава возгонов, собранных в наиболее холодной части конденсатора, по данным химического анализа, подтверждающие присутствие оксофторосиликатов аммония.
Подтверждением присутствия в возгоне кислородсодержащей фазы служат и результаты ИК спектроскопического исследования. На фиг.2 приведены ИК-спектры возгонов (NH4)2SiF6 после первой (2) и второй перегонки (3), из которых хорошо видно, как увеличивается интенсивность полос поглощения в области колебаний связи Si-O при 1080-1150 см-1. Для сравнения приведены спектры оптического кварца (4) и реактива (NH4)2SiF6 (1).
Обнаружение в конденсаторе тонкодисперсного, с малой плотностью и на ощупь слегка маслянистого продукта, содержащего ГФСА гексагональной модификации, структурно уплотненного всего на 74%, служит доказательством частичной, не полностью обратимой термической диссоциации исходной кубической модификации до промежуточного гидролитически неустойчивого соединения NH4SiF5.
По литературным данным анион SiF5 -1 с координационным числом "5" в ИК спектре должен быть активен при 785 и 874 см-1 (Эннан А.А., Кац Б.М. Успехи химии. 1974, т.43, в. 7, с.1186). Однако из-за частичной замены фтора кислородом в этом анионе и присутствия сильной полосы колебаний октаэдрического иона SiF6 2- при 740 см-1 наблюдается только нарушение симметричности последней, что свидетельствует о присутствии в возгоне второго компонента.
Таким образом, при нагревании ГФСА до температур, не превышающих 280°С, когда еще не происходит сублимация исходной соли, а ее распад на аммиак, фтористый водород и тетрафторид кремния еще термодинамически невозможен, наблюдается разложение ГФСА на NH3, HF и пентафторосиликат аммония, переходящих в газовую фазу. При конденсации паров образуется смесь из ГФСА гексагональной структуры и частично гидролизованного NH4SiF5, который обнаруживается в виде оксофторосиликатов аммония переменного состава. Именно обнаруженный заявителем механизм термического разложения ГФСА при невысоких температурах с образованием фтористого водорода позволил использовать ГФСА в качестве активного реагента для получения неорганических фторсодержащих соединений твердофазным путем при температурах не выше 280°С, что ранее не было известно.
Для подтверждения заявляемого способа были проведены твердофазные синтезы фторсодержащих соединений с использованием в качестве исходных соединений различных классов неорганических соединений, выбранных из группы оксидов, фторидов металлов I-IV и VIII групп Периодической системы и их солей слабых кислот, например угольной, уксусной, щавелевой, а в качестве фторирующего агента - (NH4)2SiF6.
Способ осуществляли с препаратами квалификации "ч". В качестве объектов фторирования были выбраны следующие образцы: оксиды - железа, алюминия, кальция; карбонаты - кальция и натрия; фториды - циркония и алюминия.
Для анализа полученных продуктов использовали методы химического, термического, рентгенофазового анализа и ИК спектроскопии.
Содержание фтора и аммония в образцах определяли путем отгонки H2SiF6 и NH3 с последующим титрованием полученных растворов нитратом тория и серной кислотой соответственно.
Рентгенофазовый анализ (РФА) выполнялся на дифрактометре ДРОН-2 (Си Кα-излучение, скорость сканирования 4 град/мин) и рентгеновском автоматическом дифрактометре Д-8 с последующей идентификацией полученного продукта по ASTM (X-ray diffraction data cards).
ИК спектры образцов снимали на спектрометре SHIMADZU в виде суспензии в вазелиновом масле.
Пример 1. Взаимодействие (NH4)2SiF6 с СаО.
Навески 0,5 г оксида кальция и 3,2 г (NH4)2SiF6 (мольное отношение 1:2) смешивают и растирают в агатовой ступке. Реакция начинается сразу, что фиксируется по резкому запаху выделяющегося аммиака. Однако полностью реакция заканчивается при последующем нагревании смеси при 250°С в течение 10 мин. Образуется 0,69 г CaF2, что составляет 99,1% от теоретического.
СаО+2(NH4)2SiF6=CaF2↓+H2O↑+2NH3↑+2NH4SiF5↑
Идентификацию образовавшегося CaF2 проводят методом РФА (карточка ASTM No. 4-864). Содержание основного вещества (CaF2) составляет 95%.
При мольном отношении исходных компонентов в шихте ниже 1:2, наряду с CaF2 образуется SiO2, который может быть легко отделен от CaF2 промыванием слабым раствором фтористоводородной кислоты.
Пример 2. Взаимодействие (NH4)2SiF6 с Fe2O3.
1 г Fe1О3 смешивают с 3,3 г (NH4)2SiF6, растирают в стеклоуглеродном тигле и нагревают в изотермическом режиме при 260°С в течение часа. Образовавшийся продукт извлекают из тигля. Вес продукта 1,73 г, что составляет 94,1% от теоретического. Твердый продукт представляет собой NH4FeF4, что подтверждается РФА (карточка ASTM No. 20-503). Содержание основного вещества составляет 98%.
Fe2O3+3(NH4)2SiF6=2NH4FeF4↓+SiF4↑+2NH3↑+Н2O↑+2NH4SiOF3↑
Пример 3. Взаимодействие (NH4)2SiF6 с ZrO2
1 г ZrO2 смешивают с 2,9 г (NH4)2SiF6 (мольное отношение 1:2) и нагревают в стеклоуглеродном тигле при 250°С в течение часа. Получают 1,5 г продукта (выход 90,0%), который по данным РАФ относится к NH4ZrF5 (карточка ASTM No. 20-1460)
ZrO2+2(NH4)2SiF6=αNH4ZrF5↓+(NH4)2SiF6SiO2↑+NH3↑+HF↑
По данным ИК-спектра твердого продукта в нем отсутствует SiO2, что объяснимо с учетом химического транспорта за счет образования летучих соединений (NH4)2SiF6 SiO2 или 2NH4SiOF3. Содержание основного вещества составляет 95%.
Пример 4. Взаимодействие (NH4)2SiF6 с Al2О3
1 г Al2O3 смешивают с 3,5 г (NH4)2SiF6, растирают и нагревают в изотермическом режиме при 280°С в течение 1 часа. Образовавшийся продукт извлекают из тигля. Вес продукта 2,30 г. Выход 97,0%. Состав продукта доказан методом РФА (карточка ASTM No. 2077). Содержание основного вещества составляет 98%.
Al2O3+2(NH4)2SiF6=2NH4AlF4↓+2SiOF2↑+2NH3↑+H2O↑
Пример 5. Взаимодействие (NH4)2SiF6 с Na2CO3
1 г кальцинированной соды Na2CO3 с 1,7 г (NH4)2SiF6 смешивают, растирают и нагревают до температуры 280°С в тигле из нержавеющей стали в течение 30 мин. Продукт извлекают из тигля. Вес продукта составляет 1,2 г. Согласно уравнению реакции:
2Na2CO3+2(NH4)2SiF6=Na2SiF6↓+2NaF↓+SiF4↑+4NH3↑+2CO2↑+Н2O↑
образовавшийся твердый продукт представляет собой смесь двух соединений Na2SiF6 и NaF, что подтверждается данными РФА (карточки ASTM No. 8-36 (для Na2SiF6) и No. 4-793 (для NaF). При необходимсти эта смесь легко переводится в NaF известными способами, например простым прокаливанием при температуре 620°С.
Пример 6. Взаимодействие (NH4)2SiF6 с СаСО3
1 г СаСО3 растирают с 1,7 г (NH4)2SiF6 смешивают и растирают в агатовой ступке. Смесь помещают в стеклоуглеродный тигель, нагревают при 250°С 12 мин. Образуется 0,77 г CaF2, что подтверждается методом РФА (карточка ASTM No. 4-864). Выход продукта составил 98,7%. Содержание основного вещества составляет 99%.
СаСО3+(NH4)2SiF6=CaF2↓+CO2↑+2NH3↑+SiF4↑+Н2O↑
Пример 7. Взаимодействие (NH4)2SiF6 с ZrF4
1 г ZrF4 смешивают с 1,1 г (NH4)2SiF6 и нагревают в стеклоуглеродном тигле при 270°С в течение 45 мин. Образуется 1,43 г (NH4)2ZiF6, что составляет 99,3% от теоретического. Содержание основного вещества составляет 99,5%.
ZrF4+(NH4)2SiF6=(NH4)2ZrF6↓+SiF4↑
Идентификацию полученного вещества провели методом РФА по данным ASTM (карточка №20-1458).
Пример 8. Взаимодействие (NH4)2SiF6 с AlF3
1 г AlF3 смешивают с 4,2 г (NH4)2SiF6 и нагревают в стеклоуглеродном тигле при 275°С в течение 45 мин. Образуется 2,1 г (NH4)2AlF6, что составляет 91,3% от теоретического. По данным ASTM (карточка No. 22-1036), твердый продукт представляет собой (NH4)2AlF6. Содержание основного вещества составляет 98%.
AlF3+2(NH4)2SiF6=(NH4)3AlF6↓+NH4SiF5↑+SiF4↑
| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБ ПОЛУЧЕНИЯ АМОРФНОГО ДИОКСИДА КРЕМНИЯ | 2005 |
|
RU2280614C1 |
| СПОСОБ ОБЕСКРЕМНИВАНИЯ МИНЕРАЛЬНОГО СЫРЬЯ | 2005 |
|
RU2317252C2 |
| СПОСОБ ПЕРЕРАБОТКИ БОРОСИЛИКАТНЫХ КОНЦЕНТРАТОВ | 2008 |
|
RU2375305C1 |
| Способ переработки датолитового концентрата | 2020 |
|
RU2748972C1 |
| СПОСОБ ОПРЕДЕЛЕНИЯ ПРИМЕСЕЙ В КРЕМНИЙСОДЕРЖАЩИХ МАТЕРИАЛАХ | 2006 |
|
RU2306546C1 |
| СПОСОБ ПЕРЕРАБОТКИ ТИТАН-КРЕМНИЙСОДЕРЖАЩЕГО СЫРЬЯ | 2008 |
|
RU2377332C2 |
| СПОСОБ ПОЛУЧЕНИЯ ОСОБО ЧИСТОГО КРЕМНИЯ | 2008 |
|
RU2355634C1 |
| СПОСОБ ОПРЕДЕЛЕНИЯ КРЕМНИЯ | 1999 |
|
RU2157523C1 |
| СПОСОБ ГИДРОХИМИЧЕСКОГО ПОЛУЧЕНИЯ ВЫСОКОДИСПЕРСНОГО ДИОКСИДА КРЕМНИЯ ИЗ ТЕХНОГЕННОГО КРЕМНИЙСОДЕРЖАЩЕГО СЫРЬЯ | 2004 |
|
RU2261841C1 |
| Способ переработки титансодержащего минерального сырья | 2019 |
|
RU2717418C1 |

Изобретение относится к области неорганической химии, а именно к способам синтеза неорганических фторсодержащих соединений. Неорганические фторсодержащие соединения синтезируют твердофазным взаимодействием неорганического соединения с солью кремнефтористоводородной кислоты при нагревании. Взаимодействие осуществляют при температуре не выше 280°С. Неорганическое соединение выбирают из группы оксидов, фторидов или солей слабых кислот элементов I-IV и VIII групп Периодической системы элементов. В качестве соли кремнефтористоводородной кислоты используют гексафторсиликат аммония. Технический результат заключается в разработке энергосберегающего способа синтеза фторсодержащих соединений на основе твердофазных реакций и повышении чистоты получаемого соединения. 2 ил., 1 табл.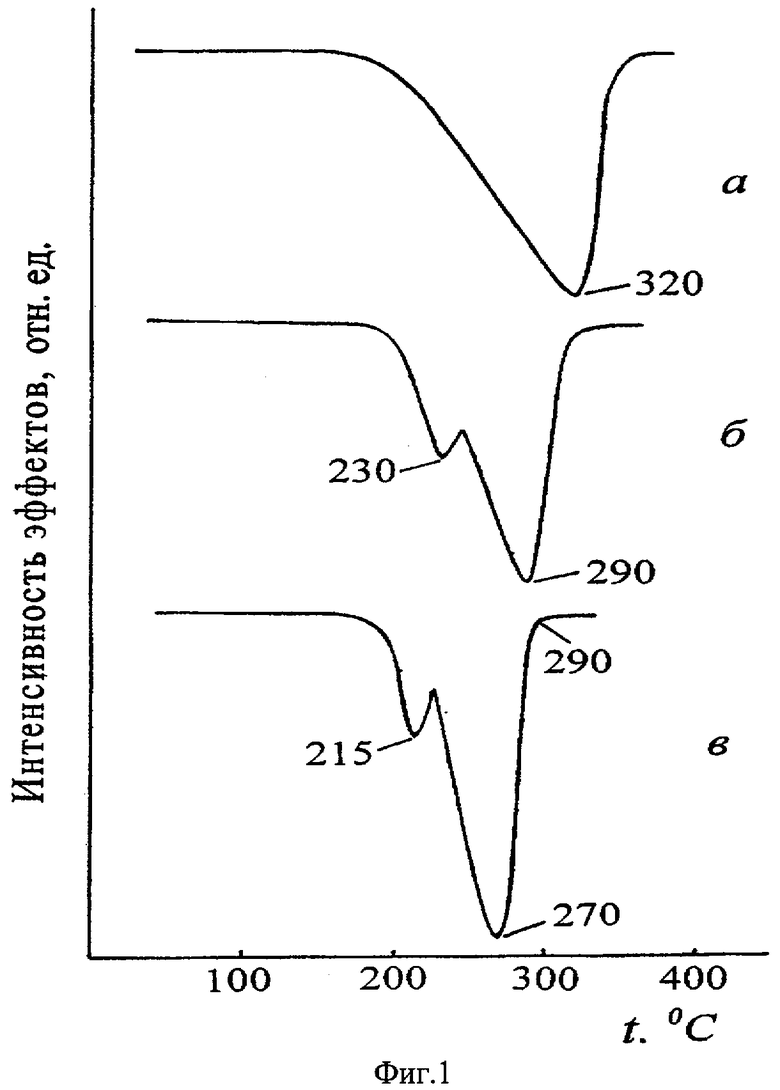
Способ синтеза неорганических фторсодержащих соединений путем реакции твердофазного взаимодействия неорганического соединения с солью кремнефтористоводородной кислоты при нагревании, отличающийся тем, что взаимодействие осуществляют при температуре не выше 280°С, при этом неорганическое соединение выбирают из группы оксидов, фторидов или солей слабых кислот элементов I-IV и VIII групп Периодической системы элементов, а в качестве соли кремнефтористоводородной кислоты используют гексафторсиликат аммония.
| Способ крепления стен котлована | 1981 |
|
SU990966A1 |
| RU 2058408 C1, 20.04.1996 | |||
| JP 2003253460 А, 10.09.2003 | |||
| US 6171706 A, 09.01.2001. | |||

