Данная заявка имеет притязание на приоритет в соответствии с разделом 35 Кодекса законов США, § 119(е) согласно заявке США с порядковым №60/154594 (реестр №21900-30290.20), поданной 9 мая 2000, и заявке США с порядковым №60/154594, поданной 17 сентября 1999. Приоритет заявлен в соответствии с разделом 35 Кодекса законов США § 120 согласно заявке США с порядковым №09/316761 от 21 мая 1999. Содержание указанных заявок полностью включено в данное описание в качестве ссылок.
Область техники
Данное изобретение относится к лечению различных заболеваний, ассоциированных с повышенной активностью киназы р36-α. Более конкретно, оно касается соединений, которые относятся к производным индольного ряда, связанным с группами пиперазинового или пиперидинового типа, в качестве пригодных в заявляемых способах лечения.
Уровень техники
Было обнаружено, что значительное число хронических и острых состояний связано с возникновением воспалительной реакции. В такой реакции принимает участие огромное количество цитокинов, включая IL-1, IL-6, IL-8 и TNF. Оказалось, что активность таких цитокинов в регулировании воспаления, по крайней мере, частично вызвана активацией фермента в пути передача сигнала клетки, члена семейства MAP киназы, более известного как р38 и, альтернативно, известного как CSBP и RK. Эта киназа активируется двойным фосфорилированием после стимулирование физиохимическим стрессом, обработкой липополисахаридами или провоспалительными цитокинами, такими как IL-1 и TNF. Поэтому ингибиторы активности р38 киназы являются полезными противовоспалительными агентами.
Глазные болезни, связанные с фибропролиферативным состоянием, включают реплантацию сетчатки, сопровождающуюся пролиферативной витреоретинопатией, удаление катаракты с имплантацией внутриглазной линзы и постглаукомный дренаж.
В заявках РСТ WO 98/06715, WO 98/07425 и WO 96/40143, которые включены сюда в качестве ссылок, описана взаимосвязь ингибиторов р38 киназы с различными болезненными состояниями. Как отмечено в данных заявках, ингибиторы р38 киназы пригодны при лечении различных заболеваний, связанных с хроническим воспалением. В данных заявках перечислены ревматоидный артрит, ревматоидный спондилит, остеоартрит, подагрический артрит и другие артритные состояния, сепсис, септический шок, эндотоксический шок, сепсис, вызванный грамотрицательными микроорганизмами, синдром токсического шока, астма, респираторный дистресс-синдром у взрослых, удар, реперфузионные повреждения, повреждения ЦНС, такие как невральная травма и ишемия, псориаз, рестеноз, церебральная малярия, хроническое воспаление легких, силикоз, множественные опухоли в тканях легких, резорбция костей, такая как остеопороз гомологичная болезнь (реакция трансплантат против хозяина), болезнь Крона, язвенный колит, включающий воспаление пищеварительного тракта (IBD) и пирез.
В указанных выше заявках РСТ описаны соединения, которые являются ингибиторами р38 киназы и могут быть пригодны для лечения перечисленных выше болезненных состояний. Такие соединения являются либо имидазолами, либо индолами, замещенными в положениях 3 или 4 пиперазиновым кольцом, присоединенным через карбоксамидную связь. Дополнительные соединения, которое являются конъюгатами пиперазиков с индолами, описаны как инсектициды в WO 97/26252, также включенной сюда в качестве ссылки.
Некоторые ароил/фенил-замещенные пиперазины и пиперидины, которые ингибируют р38-α киназу, описаны в публикации заявки РСТ WO 00/12074, опубликозанной 9 марта 2000. Кроме того, индолилзамещенные пиперидины и пиперазины, которые ингибируют указанный фермент, описаны в публикации заявки PCT WO 99/61426, опубликованной 2 декабря 1999. Карболеновые производные пиперидина и пиперазина, используемые в качестве ингибиторов р38-α киназы, описаны в PCT/US00/07934, поданной 24 марта 2000.
Ни в одном из представленных выше патентов не раскрыты производные индола, описанные в данной заявке, которые ингибируют конкретно р38-α.
Описание изобретения
Данное изобретение относится к способам и соединениям, пригодным при лечении состояний, которые характеризуются повышенной активностью р38-α. Такие состояния включают воспаление, пролифератизные заболевания и некоторые сердечно-сосудистые расстройства, а также болезнь Альцгеймера, описанные ниже.
Обнаружено, что соединения данного изобретения ингибируют р38 киназу, в частности, ее α-изоформу, и, таким образом, могут быть полезны для лечения заболеваний, медиированных активностями указанной киназы. Соединениями данного изобретения являются соединения формулы
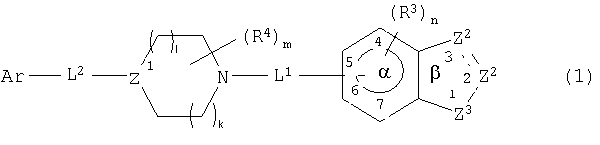
и их фармацевтически приемлемые соли или их фармацевтические композиции, в которых
 представляет одинарную или двойную связь;
представляет одинарную или двойную связь;
один Z2 является СА или CR8A и другой является CR1, CR1 2, NR6 или N, где каждый R1, R6 и R8 независимо являются водородом или не оказывающим влияние заместителем;
А является -Wi-COXjY, где Y является COR2 или его изостером и R2 является водородом или не оказывающим влияние заместителем, каждый из W и Х является разделительным звеном длиной 2-6  и каждый i и j независимо равен 0 или 1;
и каждый i и j независимо равен 0 или 1;
Z3 является NR7 или О;
каждый R3 независимо является не оказывающим влияние заместителем;
n равен 0-3;
каждый из L1-и L2 является связующим звеном;
каждый R4 независимо является не оказывающим влияние заместителем;
m равен 0-4;
Z1 является CR5 или N, где R5 является водородом или не оказывающим влияние заместителем;
каждый из l и k является целым числом от 0 до 2, где сумма l и k равна от 0 до 3;
Ar является арильной группой, замещенной от 0 до 5 не оказывающими влияние заместителями, где два не оказывающих влияние заместителя могут образовывать конденсированное кольцо; и
расстояние между атомом Ar, присоединенным к L2, и центром α кольца составляет 4,5-24 .
Данное изобретение относится к способам лечения воспалительных или пролиферативных состояний с использованием указанных соединений. Данное изобретение также относится к лечению состояний, связанных с сердечной недостаточностью, и болезни Альцгеймера с использованием соединений данного изобретения.
Способы осуществления данного изобретения
Соединения формулы (I) пригодны при лечении состояний, которые характеризуются повышенной активностью р38 киназы, в частности, ее α-изоформы. Состояния «характеризуемые повышенной р38-α активностью» включают такие, при которых указанный фермент присутствует в повышенном количестве, или в которых фермент модифицирован с повышением его обычной активности, или оба случая. Таким образом, «повышенная активность» относится к любому состоянию, при котором эффективность указанных белков нежелательно высока, независимо от причины.
Соединения данного изобретения пригодны при состояниях, при которых р38-α киназа демонстрирует повышенную активность. Такие состояния включают те, в которых фиброз и склероз органов вызваны, или сопровождаются, воспалением, окислительными повреждениями, гипоксией, измененной температурой или внеклеточной осмолярностью, состояния, вызывающие клеточный стресс, апоптоз или некроз. Такие состояния включают ишемические-реперфузионные повреждения, застойную сердечную недостаточность, прогрессирующий легочный и бронхиальный фиброз, гепатит, артрит, воспаление пищеварительного тракта, гломерулярный склероз, межуточный почечный фиброз, хроническое рубцевание глаз, мочевого пузыря и половых путей, дисплазию костного мозга, хронические инфекции или аутоиммунные состояния и травматические или хирургические раны. Эти состояния, конечно, могут лечиться соединениями, которые ингибируют р38-α. Способы лечения соединениями данного изобретения описаны ниже.
Соединения данного изобретения
Соединения, пригодные в данном изобретении, представляют собой производные соединений индольного ряда, содержащие обязательный заместитель А в положении, соответствующем положению 2 или 3 индола. В общем, предпочтительны ядра индольного ряда, хотя альтернативные варианты, входящие в объем данного изобретения, также проиллюстрированы ниже.
В представленном ниже описании, некоторые положения молекулы описаны как возможные «не оказывающие влияния заместители». Данный термин используется, так как заместители в указанных положениях в общем не оказывают влияния на основную активность молекулы, взятой в целом. В этих положениях может быть использовано множество заместителей, и специалист в данной области легко определит, является ли какой-либо определенный произвольный заместитель «оказывающим влияние» или «не оказывающим влияние».
В контексте данного описания термин «не оказывающий влияние заместитель» относится к заместителю, который оставляет способность соединения формулы (I) ингибировать активность р38-α качественно неизменной. Таким образом, заместитель может изменять степень ингибирования р38-α. Однако, поскольку соединение формулы (I) сохраняет способность ингибировать активность р38-α, заместитель может быть определен как «не оказывающий влияние». Результаты множества экспериментов, проводимых для определения способности какого-либо соединения ингибировать активность р38-α, доступны в данной области техники. Предпринятые с этой целью исследования цельной крови представлены ниже: был клонирован ген для р38-α, рекомбинантным методом был приготовлен белок и была проведена оценка его активности, включая оценку способности произвольно выбранного соединения влиять на его активность. Были четко определены основные характеристики молекулы. Положения, которые заняты «не оказывающими влияние заместителями» могут быть замещены обычными органическими группами, как это принято в данной области техники. Для данного изобретения не актуально определение внешних пределов такого замещения. Четкие определения основных характеристик соединений даны ниже. Кроме того, L1 и L2 описаны в данном изобретении как связующие звенья. Природа таких связующих звеньев менее важна, чем расстояние, которое они устанавливают между частями молекулы. Обычные связующие звенья включают алкилен, т.е., (CH2)n-R; алкенилен, т.е., алкиленовую группу, которая содержит двойную связь, включая двойную связь на одном конце. Другие подходящие связующие звенья включают, например, замещенные алкилены или алкенилены, карбонильные группы и подобные.
В контексте данного описания термин «гидрокарбильный остаток» относится к остатку, который содержит только атомы углерода и водорода. Остаток может быть алифатическим или ароматическим, иметь прямую цепь, быть циклическим, разветвленным, насыщенным или ненасыщенным. Гидрокарбильный остаток, так определенный, однако, может содержать гетероатомы в добавление к углеродным и водородным членам остатка заместителя. Таким образом, если определено, что он содержит такие гетероатомы, гидрокарбильный остаток также может содержать карбонильные группы, аминогруппы, гидроксильные группы и подобные, или содержать гетероатомы в «основной цепи» гидрокарбильного остатка.
В контексте данного описания термин «неорганический остаток» относится к остатку, который не содержит углерод. Примеры включают, ко не ограничены ими, галоген, гидрокси, NO2 или NH2.
В контексте данного описания термин «алкил», «алкенил» и «алкинил» включают прямые, разветвленные и циклические одновалентные заместители. Примеры включают метил, этил, изобутил, циклогексил, циклопентилэтил, 2-пропенил, 3-бутинил и подобные. Обычно алкильные, алкенильные и алкинильные заместители содержат 1-10С(алкил) или 2-10С(алкенил или алкинил). Предпочтительно они содержит 1-6С(алкил) или 2-6С(алкенил или алкинил). Гетероалкил, гетероалкенил и гетероалкинил определены так же, но могут содержать 1-2 гетероатома О, S или N или их сочетания в основной цепи остатка.
В контексте данного описания термин «ацил» охватывает определения алкила, алкенила, алкинила и их гетероформ, которые присоединены к дополнительному остатку через карбонильную группу.
«Ароматическая» группа относится к моноциклической или бициклической группе, такой как фенил или нафтил; «гетероароматическая» также относится к моноциклической или конденсированной бициклической кольцевой системе, содержащей один или более гетероатомов, выбранных из О, S и N. Введение гетероатома дает возможность включения 5-членных колец, так же как 6-членных колец. Таким образом, обычные ароматические системы включают пиридил, пиримидил, индолил, бензимидазолил, бензотриазолил, изохинолил, хинолил, бензотиазолил, бензофуранил, тиенил, фурил, пирролил, тиазолил, оксазолил, имидазолил и подобные. Любые моноциклические или конденсированные бициклические кольцевые системы, которые характеризуются ароматичностью в отношении распределения электронов в кольцевой системе, включены в данное определение. Обычно кольцевая система содержит 5-12 атомов в кольце.
Аналогично, «арилалкил» и «гетероалкил» относятся к ароматическим и гетероароматическим системам, которые присоединены к другому остатку через углеродную цепь, включающую замещенные или незамещенные, насыщенные или ненасыщенные углеродные цепи, обычно содержащие 1-6С. Такие углеродные цепи также могут включать карбонильную группу, что делает их способными давать такие заместители, как ацильная группа.
Если соединение формулы 1 содержит один или более хиральных центров, данное изобретение включает оптически чистые формы, а также смеси стереоизомеров или энантиомеров.
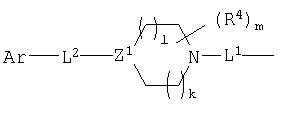
Что касается части соединения между атомом Ar, присоединенным к L2, и кольцом α, L1 и L2 являются связующими звеньями, которые отделяют заместитель Ar от кольца α на расстояние 4,5-24, предпочтительно, 6-20, более предпочтительно, 7,5-10. Расстояние измеряют от центра кольца α до атома Ar, к которому присоединено связующее звено L2. Обычно, но не ограничиваясь ими, L1 и L2 представляют собой СО и их изостеры, или необязательно замещенные изостеры, или формы с более длинной цепью L2, в частности, может быть алкиленом или алкениленом, необязательно замещенным не оказывающими влияния заместителями, или L1 или L2 могут быть, или могут включать гетероатом, такой как N, S или О. Такие заместители включают, но не ограничены ими, группу, выбранную из группы, включающей алкил, алкенил, алкинил, арил, арилалкил, ацил, ароил, гетероарил, гетероалкил, гетероалкенил, гетероалкинил, гетероалкиларил, NH-ароил, галоген, OR, NR2, SR, SOR, SO2R, OCOR, NRCOR, NRCONR2, NRCOOR, OCONR2, RCO, COOR, алкил-OOR, SO3R, CONR2, SO2NR2, NRSO2NR2, CN, CF3, R3Si и NO2, где каждый R независимо является Н, алкилом, алкенилом или арилом или их гетероформами, и где два заместителя на L2 могут быть объединены с образованием неароматического насыщенного или ненасыщенного кольца, которое включает 0-3 гетероатома, таких как О, S и/или N, и которое содержит от 3 до 8 членов, или указанные два заместителя могут быть объединены с образованием карбонильной группы или оксима, простого эфира оксима, сложного эфира оксима или кеталя указанной карбонильной группы.
Изостеры СО и СН2 включают SO, SO2 или СНОН. Предпочтительны СО и СН2. Таким образом, L2 замещен 0-2 заместителями. Там, где это является допустимым, два необязательных заместителя на L2 могут быть объединены с образованием неароматического насыщенного или ненасыщенного гидрокарбильного кольца, которое включает 0-3 гетероатома, таких как О, S и/или N, и которое содержит от 3 до 8 членов. Два необязательных заместителя могут быть объединены с образованием карбонильной группы, которая может быть последовательно превращена в оксим, простой эфир оксима, сложный эфир оксима или кеталь.
Ar является арилом, гетероарилом, включая 6-5 конденсированный гетероарил, циклоалифатическим или циклогетероалифатическим, который может быть необязательно замещен. Предпочтительно Ar является необязательно замещенным фенилом. Каждый заместитель на Ar независимо является гидрокарбильным остатком (1-20С), содержащим 0-5 гетероатомов, выбранных из О, S и N, или неорганическим остатком. Предпочтительные заместители выбирают из группы, состоящей из алкила, алкенила, алкинила, арила, арилалкила, ацила, ароила, гетероарила, гетероалкила, гетероалкенила, гетероалкинила, гетероалкиларила, NH-ароила, галогена, OR, NR2, SR, SOR, SO2R, OCOR, NRCOR, NRCONR2, NRCOOR, OCONR2, RCO, COOR, алкил-OOR, SO3R, CONR2, SO2NR2, NRSO2NR2, CN, CF3, R3Si и NO2, где каждый R независимо является Н, алкилом, алкенилом или арилом или их гетероформами, и где два указанных заместителя могут быть объединены с образованием конденсированного необязательно замещенного ароматического или неароматического, насыщенного или ненасыщенного кольца, которое содержит от 3 до 8 членов. Более предпочтительные заместители включают галоген, алкил (1-4С), и более предпочтительно, фтор, хлор и метил. Указанные заместители могут занимать все доступные положения арильного кольца Ar, предпочтительно, 1-2 положения, наиболее предпочтительно одно положение. Указанные заместители могут быть необязательно замещены заместителями, такими же, как и перечисленные выше. Конечно, некоторые заместители, такие как галоген, не могут быть далее замещены, как известно специалистам в данной области.
Два заместителя на Ar могут быть объединены с образованием конденсированного, необязательно замещенного, ароматического или неароматического, насыщенного или ненасыщенного кольца, которое содержит 3-8 членов.
Между L1 и L2 находится группа пиперидинового ряда следующей формулы:
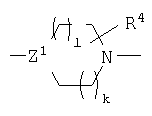
Z1 является CR5 или N, где R5 является Н или не оказывающим влияние заместителем. Каждый из l и k является целым числом от 0 до 2, где сумма l и k равна от 0 до 3. Не оказывающие влияние заместители R5 включают, не ограничиваясь ими, галоген, алкил, алкокси, арил, арилалкил, арилокси, гетероарил, ацил, карбокси или гидрокси. Предпочтительно, R5 является Н, алкилом, OR, NR2, SR или галогеном, где R является Н или алкилом. Кроме того, R5 может быть объединен с R4 заместителем с образованием необязательно замещенного неароматического, насыщенного или ненасыщенного гидрокарбильного кольца, которое содержит 3-8 членов и 0-3 гетероатома, таких как О, N и/или S. Предпочтительные варианты включают соединения, в которых Z1 является СН или N, и такие, в которых оба l и k равны 1.
R4 является не оказывающим влияние заместителем, таким как гидрокарбильный остаток (1-20С), содержащим 0-5 гетероатомов, выбранных из О, S и N. Предпочтительно R4 является алкилом, алкокси, арилом, арилалкилом, арилокси, гетероалкилом, гетероарилом, гетероарилалкилом, RCO, =O, ацилом, галогеном, CN, OR, NRCOR, NR, где R является Н, алкилом (предпочтительно, 1-4C), арилом или их гетероформами. Каждый из подходящих заместителей сам по себе является незамещенным или замещенным 1-3 заместителями. Заместители предпочтительно выбирают из группы, которая включает алкил, алкенил, алкинил, арил, арилалкил, ацил, ароил, гетероарил, гетероалкил, гетероалкенил, гетер оалкинил, гетероалкиларил, NH-ароил, галоген, OR, NR2, SR, SOR, SO2R, OCOR, NRCOR, NRCONR2, NRCOOR, OCONR2, RCO, COOR, алкил-OOR, SO3R, CONR2, SO2NR2, NRSO2NR2, CN, CF3, R3Si и NO2, где каждый R независимо является Н, алкилом, алкенилом или арилом или их гетероформами, и где два заместителя на R4 в соседних положениях могут быть объединены с образованием конденсированного, необязательно замещенного ароматического или неароматического насыщенного или ненасыщенного кольца, которое содержит от 3 до 8 членов, или R4 является =O или его оксимом, простым эфиром оксима, сложным эфиром оксима или кеталем. R4 может содержаться в кольце m раз; m является целым числом от 0 до 4. Предпочтительные варианты R4 включают алкил (1-4С), особенно два алкильных заместителя и карбонил. Наиболее предпочтительно R4 содержит две метильные группы в положениях 2 и 5 или 3 и 6 пиперидинильного или пиперазинильного кольца, или =O предпочтительно в положении 5 кольца. Замещенные формы могут быть хиральными и могут быть предпочтительно отдельным энантиомером.
R3 также является не оказывающим влияние заместителем. Указанные заместители включают гидрокарбильные остатки (1-6С), содержащие 0-2 гетероатома, выбранных из О, S и/или N, и неорганические остатки; n является целым числом от 0 до 3, предпочтительно 0 или 1. Предпочтительно, заместитель, представленный R3, независимо является галогеном, алкилом, гетероалкилом, OCOR, OR, NRCOR, SR или NR2, где R является алкилом, арилом или их гетероформами. Наиболее предпочтительно R3 заместители выбирают из алкила, алкокси или галогена, и наиболее предпочтительно, из метокси, метила и хлора. Наиболее предпочтительно n равно 0 и кольцо α не замещено, за исключением L1, или n равно 1 и R3 является галогеном или метокси.
В кольце, обозначенном как β, Z3 может быть NR7 или О, т.е. соединения могут относиться к индолу или бензофурану. Если Z3 является NR7, предпочтительные варианты R7 включают Н или необязательно замещенный алкил, алкенил, алкинил, арил, арилалкил, ацил, ароил, гетероарил, гетероалкил, гетероалкенил, гетероалкинил, гетероалкиларил, или является SOR, SO2R, RCO, COOR, алкил-OOR, SO3R, CONR2, SO2NR2, CN, CF3, NR2, OR, алкил-SR, алкил-SOR, алкил-SO2R, алкил-OCOR, алкил-COOR, алкил-CN, алкил-CONR2 или R3Si, где каждый R независимо является Н, алкилом, алкенилом или арилом или их гетероформами. Более предпочтительно R7 является водородом или алкилом (1-4С), предпочтительно, метилом, или ацилом (1-4С), или COOR, где R является Н, алкилом, алкенилом или арилом или их гетероформами. R7 также предпочтительно является замещенным алкилом, где предпочтительные заместители либо образуют связи, либо содержат группы сульфиновой или сульфоновой кислоты. Другие предпочтительные заместители включают сульфгидрилзамещенные алкильные заместители. Другие предпочтительные заместители включают CONR2, где R такой, как определен выше.
Предпочтительно, если обозначенная пунктирная линия представляет собой двойную связь; однако соединения, которые содержат насыщенное β кольцо, также включены в объем данного изобретения.
Предпочтительно, обязательный заместитель СА или CR8A находится в положении 3; независимо от положения, которое занимает указанный заместитель, другое положение замещено CR1, CR1 2, NR6 или N. Предпочтительно CR1. Предпочтительные варианты R1 включают алкил, алкенил, алкинил, арил, арилалкил, ацил, ароил, гетероарил, гетероалкил, гетероалкенил, гетероалкинил, гетероалкиларил, NH-ароил, галоген, OR, NR2, SR, SOR, SO2R, OCOR, NRCOR, NRCONR2, NRCOOR, OCONR2, RCO, COOR, алкил-OOR, SO3R, CONR2, SO2NR2, NRSO2NR2, CN, CF3, R3Si и NO2, где каждый R независимо является Н, алкилом, алкенилом или арилом или их гетероформами, и где два заместителя R1 могут быть объединены с образованием конденсированного, необязательно замещенного ароматического или неароматического насыщенного или ненасыщенного кольца, которое содержит от 3 до 8 членов. Наиболее предпочтительно R1 является алкилом, таким как метил, наиболее предпочтительно, кольцо α содержит двойную связь и CR1 является СН или С-алкилом. Другие предпочтительные формы R1 включают Н, алкил, ацил, арил, арилалкил, гетероалкил, гетероарил, галоген, OR, NR2, SR, NRCOR, алкил-OOR, RCO, COOR и CN, где каждый R независимо является Н, алкилом или арилом или их гетероформами.
Хотя в предпочтительном варианте положение, не занятое СА, включает CR1, данное положение также может быть N или NR6. Хотя NR6 наименее предпочтителен (так как в этом случае кольцо, обозначенное β, будет насыщенным), если NR6 присутствует, предпочтительными вариантами R6 являются Н или алкил, алкенил, алкинил, арил, арилалкил, ацил, ароил, гетероарил, гетероалкил, гетероалкенил, гетероалкинил, гетероалкиларил, или SOR, SO2R, RCO, COOR, алкил-COR, SO3R, CONR2, SO2NR2, CN, CF3 или R3Si, где каждый R независимо является Н, алкилом, алкенилом или арилом или их гетероформами.
Предпочтительно, CR8A или СА занимает положение 3 и предпочтительно Z2 в этом положении является СА. Однако, если кольцо β насыщено и присутствует R8, предпочтительными вариантами R8 являются Н, галоген, алкил, алкенил и подобные. Предпочтительно R8 является относительно небольшим заместителем, соответствующим, например, Н или низшему алкилу 1-4С.
А является -Wi-COXjY, где Y является COR2 или его изостером и R2 является не оказывающим влияния заместителем. Каждый из W и Х является разделительным звеном и может быть, например, необязательно замещенным алкилом, алкенилом или алкинилом, каждый из i и j равен 0 или 1. Предпочтительно, W и Х не замещены. Предпочтительно, j равен 0, так, чтобы две карбонильные группы были соседними. Предпочтительно, также, i равен 0, так, чтобы ближайший СО находился рядом с кольцом. Однако соединения, в которых ближайший СО отделен от кольца, могут быть легко получены селективным восстановлением изначально глиоксальзамещенного β кольца. В наиболее предпочтительных вариантах данного изобретения система колец α/β является индолом, содержащим СА в положении 3, где А является COCR2.
Не оказывающим влияние заместителем R2, если R2 отличен от Н, является гидрокарбильный остаток (1-20С), содержащий от 0 до 5 гетероатомов, выбранных из О, S и/или N, или неорганический остаток. Предпочтительны варианты, в которых R2 является Н или прямым или разветвленным алкилом, алкенилом, алкинилом, арилом, арилалкилом, гетероалкилом, гетероарилом или гетероарилалкилом, каждый из которых необязательно замещен галогеном, алкилом, гетероалкилом, SR, OR, NR2, OCOR, NRCOR, NRCONR2, NRSO2R, NRSO2NR2, OCONR2, CN, COOR, CONR2, COR или R3Si, где каждый R независимо является Н, алкилом, алкенилом или арилом или их содержащими гетероатом формами, или где R2 является OR, NR2, SR, NRCONR2, OCONR2 или NRSO2NR2, где каждый R независимо является Н, алкилом, алкенилом или арилом или их содержащими гетероатом формами, и где два R, присоединенные к одному и тому же атому, могут образовывать 3-8 членное кольцо, и где указанное кольцо может быть далее замещено алкилом, алкенилом, алкинилом, арилом, арилалкилом, гетероалкилом, гетероарилом или гетероарилалкилом, каждый из которых необязательно замещен галогеном, SR, OR, NR2, OCOR, NRCOR, NRCONR2, NRSO2R, NRSO2NR2, OCONR2, или R3Si, где каждый R независимо является Н, алкилом, алкенилом или арилом или их содержащими гетероатом формами, где два R, присоединенные к одному и тому же атому, могут образовывать 3-6-членное кольцо, замещенное указанными выше заместителями.
Другие предпочтительные варианты R2 включают Н, гетероарилалкил, -NR2, гетероарил, -COOR, -NHRNR2, гетероарил-COOR, гетероарилокси, -OR, гетероарил-NR2, -NROR и алкил. Наиболее предпочтительно R2 является изопропилпиперазинилом, метилпиперазинилом, диметиламином, пиперазинилом, изобутилкарбоксилатом, оксикарбонилэтилом, морфолинилом, аминоэтилдиметиламином, изобутилкарбоксилатпиперазинилом, оксипиперазинилом, этилкарбоксилатпиперазинилом, метокси, этокси, гидрокси, метилом, амино, амкноэтилом, пирролидинилом, аминопропандиолом, пиперидинилом, пирролидинил-пиперидинилом или метилпиперидинилом.
Изостеры COR2, представленные Y, определены следующим образом.
Изостеры имеют переменную липофильность и могут вносить свой вклад в улучшение метаболической стабильности. Таким образом, Y, как показано, может быть замещен изостерами, представленными в таблице 1.
Изотермы кислоты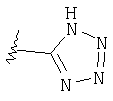
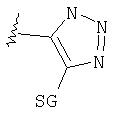
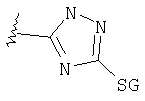
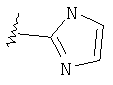
Таким образом, изостеры включают тетразол, 1,2,3-триазол, 1,2,4-триазол и имидазол.
Соединения формулы (I) могут быть в виде их фармацевтически приемлемых кислотно-аддитивных солей, включая соли неорганических кислот, таких как хлористоводородная, серная, бромистоводородная или фосфорная кислоты, или соли органических кислот, таких как уксусная, винная, янтарная, бензойная, салициловая и подобные. Если в соединении формулы (I) присутствует карбоксильная группа, соединение также может иметь форму соли с фармацевтически приемлемым катионом.
Синтез соединений в соответствии с данным изобретением
Следующие схемы реакций иллюстрируют превращение 4-бензилпиперидинилиндол-5-карбоксамида в соединения глиоксаловой кислоты данного изобретения и их производных.
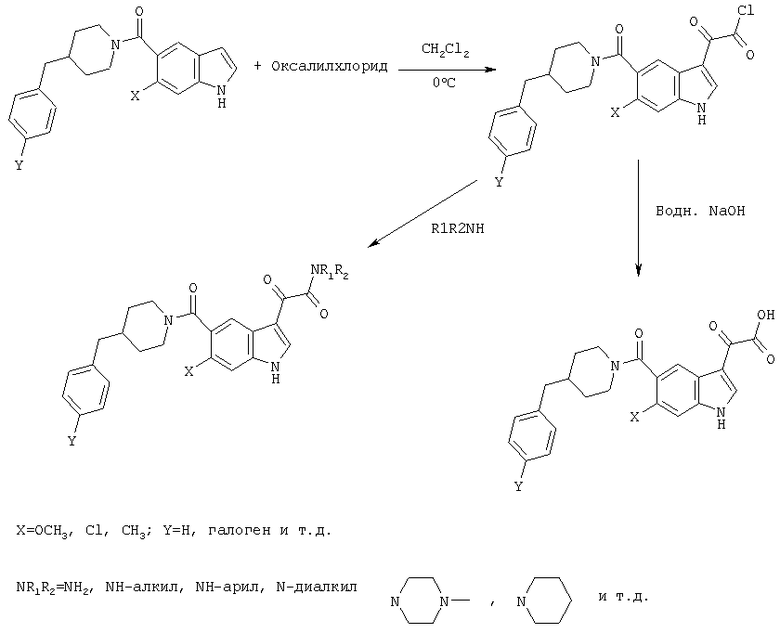
Конечно, иллюстративный 4-бензилпиперидинилкарбонил в положении 5 может быть обобщен как
и заместитель глиоксалевого типа в положении 3 может быть обобщен до WiCOXjY.
Также в указанных схемах могут быть использованы варианты, в которых группа индольного ряда представляет собой
Способы синтеза соединений в соответствии с данным изобретением в основном известны в данной области.
Такие методы могут быть проиллюстрированы следующими общими схемами.
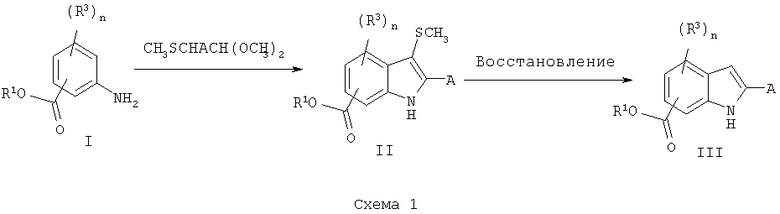
Замещенные сложные эфиры аминобензойной кислоты, такие как I, могут быть обработаны реагентами, такими как диметилацеталь тиометилацетальдегида и N-хлорсукцинамид в метиленхлориде при низкой температуре с последующей обработкой основанием, таким как триэтиламин при температуре кипения с обратным холодильником в метиленхлориде, дихлорэтане или хлороформе с получением индолов II, схема 1. Обработка реагентами, такими как никель Ренея, в подходящем растворителе, таком как этанол, метанол или изопропанол, дает соответствующий сложный эфир карбоновой кислоты, который при гидролизе в основных условиях дает требуемое производное индолкарбоновой кислоты.
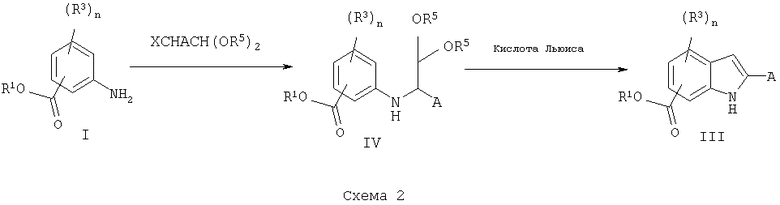
Альтернативно, замещенные сложные эфиры аминобензойной кислоты I могут быть превращены в кетали IV, схема 2, обработкой подходящим альдегидом в условиях восстановительного алкилирования с реагентами, такими как триацетоксиборгидрид натрия в уксусной кислоте в присутствии сульфата натрия. Затем амины могут быть обработаны кислотами Льюиса, такими как хлорид алюминия, хлорид титана, BF3-эфират в дихлорметане или дихлорэтане, при температуре кипения с обратным холодильником с получением соответствующих замещенных сложных метиловых эфиров индола с соответствующими заместителями.
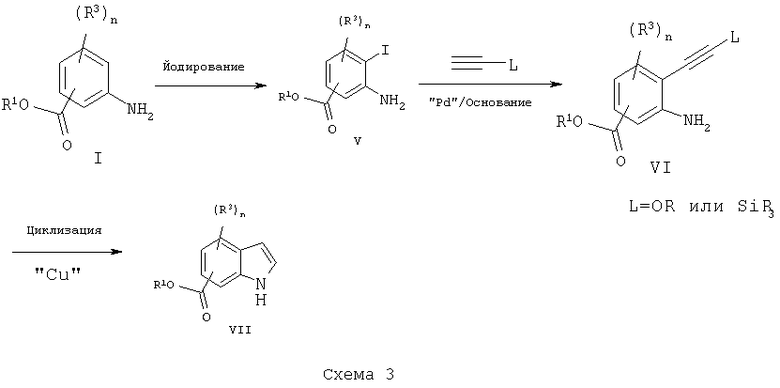
Другой метод может включать обработку замещенных сложных эфиров аминобензойной кислоты I йодом и периодатом натрия в подходящем растворителе, таком как диметилформамид, с получением соответствующих йоданилинов V, схема 3. Они могут быть подвергнуты взаимодействию с ацетиленом, таким как триметилсилилацетилен или этилэтиниловый простой эфир в присутствии подходящего катализатора, такого как палладий и медь, и основания, такого как триэтиламин, с получением силилсвязанных продуктов, таких как VI. Последующая циклизация в растворителе, таком как диметилформамид, и в присутствии катализатора, такого как йодид меди, дает подходящим образом замещенные индолы VII.
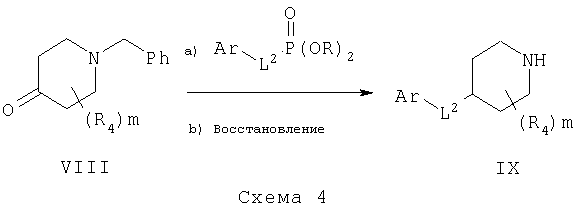
Синтез требуемых пиперидинов может быть проведен обработкой подходящего пиперидона, такого как VIII, схема 4, замещенными сложными эфирами бензилфосфоната в присутствии основания, такого как гидрид натрия, с получением алкенов, которые могут быть восстановлены до соответствующих замещенных 4-бензилпиперидинов, таких как IX. Гидрирование обычно проводят в присутствии каталитических металлов, в растворителях, таких как метанол, этанол и этилацетат.
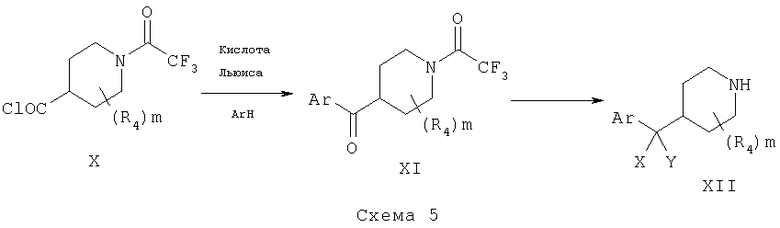
Альтернативный метод может включать использование изонипекотоилхлоридов, таких как X, для ацилирования подходящим образом замещенных бензолов (ArH) в присутствии кислоты Льюиса, такой как хлорид алюминия, с получением кетонов XI, схема 5. Дальнейшие модификации карбонильной группы в XI с помощью методов и способов, хорошо известных в данной области, дают требуемые соединения XII.
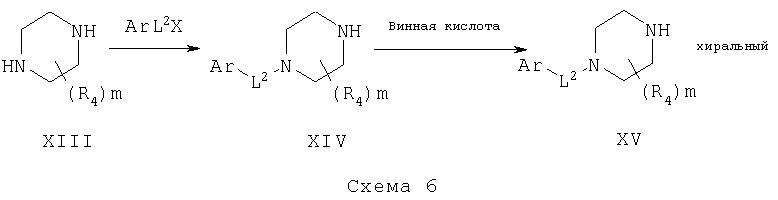
Замещенные пиперазины могут быть подвергнуты взаимодействию с различными подходящими ArL2X в присутствии или отсутствие основания или другого каталитического реагента с получением замещенных пиперазинов XV, схема 6. Они далее могут быть разделены до хиральных компонентов с помощью хирального разделяющего агента, такого как винная кислота, с получением любого энантиомера замещенных пиперазинов XV.
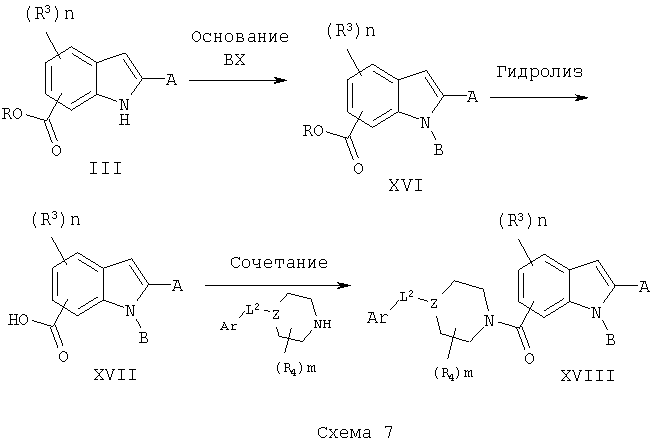
Соединения III могут быть обработаны галогенидами, хлорангидридами или другими электрофилами (ВХ), схема 7, содержащими множество различных заместителей, в присутствии основания, такого как гидрид натрия, во множестве различных растворителей, с получением соединений XVI. Они могут быть превращены в соответствующие кислоты XVII обработкой подходящими реагентами, такими как водное основание. Кислоты затем могут быть подвергнуты взаимодействию с замещенными аминами IX, XII или XV, при использовании конденсирующего агента, такого как EDAC.HCl, в различных растворителях, включая метиленхлорид, диметилформамид, с получением соединений XVIII.
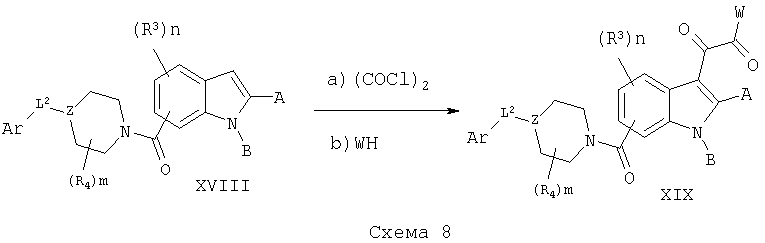
Соединения XVIII могут быть сначала обработаны хлорангидридами, такими как оксалилхлорид, в метиленхлориде в безводных условиях с последующей обработкой различными нуклеофилами WH с получением соединения XIX, схема 8.
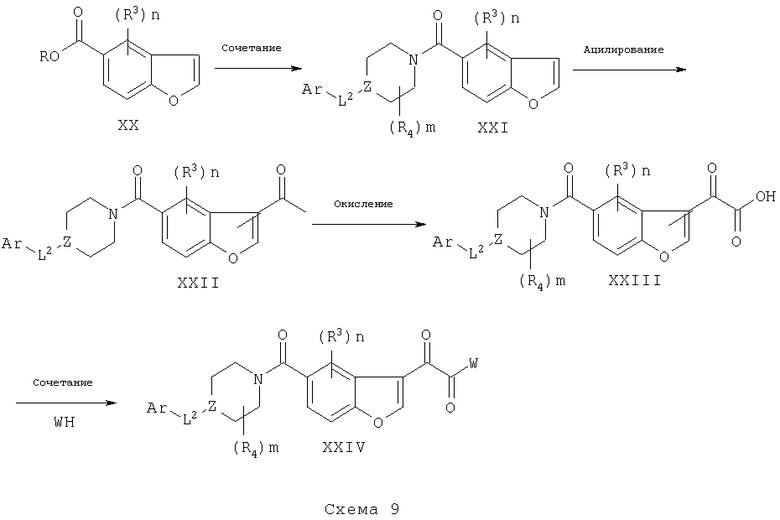
Соединения XXIV могут быть синтезированы, исходя из соответствующим образом замещенных бензофуранов XX посредством взаимодействия их с аминами IX, XII или XV в присутствии соответствующих конденсирующих агентов с получением соединений XXI, схема 9. Дальнейшее ацилирование до XXII может быть проведено с использованием ацилирующего агента, такого как уксусный ангидрид, в присутствии катализатора, такого как Fe(III). Окисление ацетильной группы XXII до группы глиоксалевой кислоты XXIII может быть проведено с использованием окисляющего агента, такого как диоксид селена (ссылка: F Da Settimo et al., Eur. J. Med. Chem (1996), 31, 951-956; М.С.Cook et al., (1975) патент Великобритании 1399089; Е. Campaigne et al., J. Med. Chem (1965), 136-137). Наконец, сочетание кислоты с соответствующим нуклеофилом в WH может быть проведено с использованием любого из множества известных конденсирующих агентов, в различных растворителях с получением соединений XXIV.
Исследования ингибирования р38α киназы
Для каждой из методик исследования, описанных ниже, продуцирование TNF-α коррелируется с активностью р38-α киназы.
А. Исследование ингибирования р38-α киназы с использованием цельной человеческой крови.
Венозную кровь здоровых добровольцев мужского пола собирают в гепаринизированный шприц и используют в течение 2 часов после забора крови. Тестируемые соединения растворяют в 100% ДМСО и 1 мкл аликвот лекарственного средства в концентрациях от 0 до 1 мМ помещают в четырех повторах в ячейки 24-ячеечного титрационного микропланшета (Nunclon Delta SI, Applied Scientific, So. San. Francisco, CA). Добавляют цельную кровь в объеме 1 мл/ячейку и смесь инкубируют в течение 15 минут при постоянном встряхивании (Titer Plate Shaker, Lab-Line Instruments, Inc., Melrose Park, IL) в увлажненной атмосфере 5% CO2 при температуре 37°С. Цельную кровь культивируют либо в неразбавленном виде, либо с конечным разбавлением 1:10 с RPMI 1640 (Gibco 31800 + NaHCO3, Life Technologies, Rockville, MD и Scios, Inc., Sunnyvale, CA). В конце периода инкубации в каждую ячейку добавляют 10 мкл ЛПС [Е.coli 0111:B4, Sigma Chemical Co., St. Louis, МО) до конечной концентрации 1 или 0,1 мкг/мл для неразбавленной или 1:10 разбавленной цельной крови, соответственно. Инкубирование продолжают в течение еще 2 часов. Реакцию останавливают помещением титрационных микропланшетов на ледяную баню, и плазму или не содержащие клеток надсадочные жидкости собирают центрифугированием при 3000 об/мин в течение 10 минут при температуре 4°С. Образцы плазмы хранят при температуре -80°С до начала исследования уровней TNF-α с помощью ELISA, следуя инструкциям, приложенным к набору для исследований Quantikine Human TNF-α assay kit (R&D Systems, Minneapolis, MN).
Значения IC50 рассчитывают, используя концентрации ингибитора, которые дают 50% снижение по сравнению с контролем.
В. Исследование ингибирования р38 киназы в обогащенных одноядерных клетках
При исследовании с использованием обогащенных одноядерных клеток, протокол которого представлен ниже, в качестве исходных используют криоконсервированные одноядерные клетки периферийной крови человека (ОКПКЧ) (Clonetics Corp.), которые промывают и повторно суспендируют в теплой смеси среды для роста клеток. Повторно суспендированные клетки затем просчитывают и высевают при 1×106 клеток/ячейку в 24-ячеечный титрационный микропланшет. Затем планшеты помещают в инкубатор на один час для осаждения клеток в каждой ячейке. После осаждения клеток среду отсасывают и в каждую ячейку титрационного микропланшета добавляют новую среду, содержащую 100 нг/мл стимулирующего цитокин фактора липополисахарида (ЛПС) и тестируемое химическое соединение. Таким образом, каждая ячейка содержит ОКПКЧ, ЛПС и тестируемое химическое соединение. Затем клетки инкубируют в течение 2 часов и количество цитокина Альфа Фактора Некроза Опухоли (TNF-α) измеряют, используя иммуноферментный твердофазный анализ (ELISA). Один из таких ELISA для определения уровней TNF-α коммерчески доступен от R&D Systems. Количество TNF-α, выработанного ОКПКЧ в каждой ячейке, затем сравнивают с контрольной ячейкой для определения, действует ли химическое соединение как ингибитор продуцирования цитокина.
Индуцированный ЛПС синтез цитокина в ОКПКЧ
Криоконсервированные ОКПКЧ (кат №СС-2702 Clonetics Corp.)
Среда LGM-3 (кат №СС-3212 Clonetics Corp.)
Исходный раствор ЛПС 10 г/мл (кат №L 2630 серотип 0111:В4 Sigma)
ELISA для человеческого TNF (R&D Systems)
ДНКаза I (10 мг/мл исходный раствор)
Получение клеток
LGM-3 среду нагревают до 37°С
5 мкл исходного раствора ДНКазы I добавляют в 10 мл среды.
Клетки быстро оттаивают и диспергируют в указанную выше среду
Центрифуга 200хg × 10 мин @ КТ
Осадок после центрифугирования разводят до 10 мл стерильным физиологическим раствором с фосфатным буфером
Центрифуга 200хg × 10 мин @ КТ
Осадок после центрифуги повторно суспендируют в 10 мл LGM-3, затем разбавляют до 50 мл средой LGM-3.
Проводят подсчет клеток
Доводят до 1×106 клеток/ячейку.
Высевают 1 мл/ячейку в 24-ячеечный планшет
Помещают планшет в инкубатор для осаждения в течение 1 часа
Получение инкубационной среды
LGM-3, содержащая 100 нг/мл ЛПС (например, 50 мл среды плюс 0,5 мл исходного раствора ЛПС)
Делят без остатка на 2 мл аликвоты и добавляют 1000Х разбавленный ингибитор
Инкубация
После осаждения клеток среду отсасывают и покрывают 1 мл соответствующей инкубационной среды. Планшет возвращают в инкубатор на 2 часа или 24 часа. После инкубирования надсадочные жидкости переносят в помеченную пробирку и либо сразу проводят ELISA TNF (или другого), либо замораживают для более позднего исследования.
Значения IC50 рассчитывают, используя концентрацию ингибитора, которая дает 50% снижение по сравнению с контролем.
Введение и применение
Соединения данного изобретения пригодны, среди прочих показаний, для лечения состояний, связанных воспалением. Таким образом, соединения формулы (I) или их фармацевтически приемлемые соли используют для производства лекарственных средств для профилактики или терапевтического лечения млекопитающих, включая человека, в случае возникновения состояний, характеризующихся избыточным продуцированием цитокинов и/или неприемлемой или нерегулируемой активностью цитокинов в таких клетках, как кардиомиоциты, кардиофибробласты и макрофаги.
Соединения в соответствии с данным изобретением ингибируют продуцирование цитокинов, таких как TNF, IL-1, IL-6 и IL-8, цитокинов, которые являются важными провоспалительными компонентами при многих различных болезненных состояниях и синдромах. Таким образом, ингибирование указанных цитокинов является важным для контроля и смягчения многих заболеваний. Соединения данного изобретения, как показано в данном описании, ингибируют семейство киназы MAP, называемое также р38 МАРК (или р38), CSBP или SAPK-2. Активация указанного белка, как было показано, сопровождает обострение заболеваний в ответ на причиняемый стресс, вызванный, например, действием липополисахаридов или цитокинов, таких как TNF и IL-1. Ингибирование активности р38, поэтому, обуславливает способность лекарственных средств оказывать благоприятное действие при лечении заболеваний, таких как болезнь Альцгеймера, ишемическая болезнь сердца, застойная сердечная недостаточность, кардиомиопатия, миокардит, васкулит, рестеноз, такой, который может проявиться после пластической операции на коронарных сосудах, атеросклероз, IBD, ревматоидные артриты, ревматоидные спондиллиты, остеоартриты, подагрический артрит и другие артритные состояния, рассеянный склероз, острый респираторный дистресс-синдром (ОРДС), астма, хроническая закупорка легких (ХЗЛ), силикоз, патологическое разрастание легочной ткани, сепсис, септический шок, эндотоксический шок, сепсис, вызванный грамотрицательными микроорганизмами, синдром токсического шока, сердечная и мозговая недостаточность (удар), которые характеризуются ишемией и реперфузионными повреждениями, хирургическим вмешательством, таким как трансплантация и отторжение трансплантата, искусственное кровообращение, коронарное шунтирование, повреждения ЦНС, включая открытые и закрытые травмы головы, воспалительные заболевания глаз, такие как конъюнктивит и увеит, острая почечная недостаточность, гломерулонефрит, воспалительные заболевания пищеварительного тракта, такие как болезнь Крона или язвенный колит, гомологичная болезнь, резорбция костей, такая как остеопороз, диабеты II типа, пирез, псориаз, кахексия, вирусные заболевания, такие как вызываемые ВИЧ, цитомегаловирусом (CMV) и герпесом, и церебральная малярия.
В течение последних семи лет было показано, что р38 содержит группу MAP киназ, обозначенных как р38-α, р38-β, р38-γ и р38-δ. У Jiang, Y et al, J. Biol Chem (1996) 271:17920-17926 p38-β охарактеризована как белок, содержащий 372-аминокилоты, близко родственный р38-α. При сравнении активности р38-α с активностью р38-β авторы установили, что хотя оба активируются провоспалительными цитокинами и внешним стрессом, р38-β, в основном, активируется такой MAP киназой, как киназа-6 (МКК6) и, в основном, активируется фактором транскрипции 2, таким образом, можно предположить, что разные механизмы действия могут быть связаны этими формами.
У Kumar, S., et al., Biochem Blophys Res Comm (1997) 235:533-538 и Stein, В et al., J. Biol Chem (1997) 272:19509-19517 описана вторая изоформа р38-β, р38-β2, содержащая 364 аминокислоты с 73% идентичностью к р38-α. Из всех указанных ссылок очевидно, что р38-β активируется провоспалительными цитокинами и внешним стрессом, хотя вторая указанная изоформа р38-β, р38-β2, предположительно, в основном экспрессируется в ЦНС, сердце и скелетных мышцах, в отличие от имеющей место практически везде в ткани экспрессии р38-α. Более того, как показали наблюдения, активированный фактор-2 транскрипции (ATF-2) является лучшим субстратом для р38-β2, чем для р38-α, таким образом, полагают, что различные механизмы действия могут быть связаны с этими формами. Физиологическая роль р38-β1 ставилась под вопрос в двух последних исследованиях, так как он не был найден в тканях человека и не демонстрирует значительной активности киназы с субстратами р38-α.
Идентификация р38-γ представлена у Li, Z. et al., Biochem Blophys Res Comm (1996) 228:334-340 и р38-δ у Wang, X. et al., J Biol Chem (1997) 272:23668-23674 и у Kumar, S. et al., Biochem Biophys Res Comm (1997) 235:533-538. Данные позволяют предположить, что две изоформы р38 (γ и δ) представляют собой уникальную субпопуляцию семейства МАРК, основываясь на их формах экспрессии ткани, использовании субстрата, реакции на прямое и косвенное стимулирование и чувствительности к ингибиторам киназы.
Различные результаты, относящиеся к реакции на лекарственные средства, направленные на семейство р38, в отношении р38-α и либо предполагаемого р38-β1 или р38-β2, или обоих представлены у Jiang, Kumar и Stein, указанных выше, а также у Eyers, P. A. et al., Chem and Biol (1995) 5:321-328. В еще одной работе Wang, Y. et al., J Biol Chem (1998) 273:2161-2168 предполагается значимость такого дифференциального действия. Как отмечено у Wang, множество стимулов, таких как инфаркт миокарда, гипертензия, пороки клапанов сердца, вирусный миокардит и расширенная кардиомиопатия приводят к увеличению работы сердца и повышенному механическому давлению на кардиомиоциты. Указано, что это приводит к адаптационному гипертрофическому ответу, который, если его не контролировать, будет иметь несомненно отрицательные последствия. Wang ссылается на более ранние исследования, в которых показано, что при ишемической реперфузии сердца, активность р38 МАРК повышается в связи с гипертрофией и программируемым отмиранием клеток. В приведенной ссылке Wang показывает, что активация действия р-38β приводит к гипертрофии, в то время как активация действия р38-α приводит к апоптозу миоцитов. Таким образом, селективное ингибирование активности р38-α по сравнению с активностью р38-β будет преимущественным при лечении состояний, вызванных сердечной недостаточностью. Такие состояния включают застойную сердечную недостаточность, кардиомиопатию, миокардит, васкулит, рестеноз сосудов, пороки клапанов сердца, состояния, связанные с искусственным кровообращением, коронарным шунтированием, трансплантатами и трансплантатами сосудов. Далее, учитывая то, что α-изоформа является токсичной в других типах мышечных клеток, α-селективные ингибиторы могут быть использованы при состояниях, связанных с кахексией, свойственной TNF или другим состояниям, таким как рак, инфекция или аутоиммунное заболевание.
Таким образом, данное изобретение охватывает применение соединений, которые селективно ингибируют активность р38-α изоформы, для лечения состояний, связанных с активацией р38-α, в частности тех, которые связаны с сердечной гипертрофией, ишемией и другими внешними стрессами, такими как окислительные повреждения, гиперосмолярность или другие агенты или факторы, которые активируют р38-α киназу или сердечную недостаточность, например, застойную сердечную недостаточность, кардиомиопатию и миокардит.
Способ введения и препаративные формы соединений, пригодных в данном изобретении, и родственных им соединений зависят от природы состояния, тяжести состояния, конкретного субъекта, которого лечат, и оценки практикующего врача; препаративная форма зависит от способа введения. Так как соединения в соответствии с данным изобретением представляют собой небольшие молекулы, они удобно вводятся перорально, при смешивании их с подходящими фармацевтическими эксципиентами для получения таблеток, капсул, сиропов и подобных. Подходящие композиции для перорального введения также могут включать вспомогательные компоненты, такие как буферы, вкусовые добавки и подобные. Обычно количество активного ингредиента в композиции составляет от 5% до 95% от общей массы композиции, но в зависимости от носителя могут быть использованы более широкие интервалы. Подходящие носители включают сахарозу, пектин, стеарат магния, лактозу, арахисовое масло, оливковое масло, воду и подобные.
Соединения, пригодные в данном изобретении, также могут вводиться с помощью суппозиториев или других чресслизистых носителей. Обычно такие композиции включают эксципиенты, которые облегчают прохождение соединения через слизистую оболочку, такие как фармацевтически приемлемые детергенты.
Соединения также могут вводиться местно, для местных состояний, таких как псориаз, или в препаративной форме, предназначенной для прохождения через кожу. Такие препаративные формы включают лосьоны, кремы, мази и подобные, которые могут быть получены известными методами.
Соединения также могут вводиться посредством инъекций, включая внутривенные, внутримышечные, подкожные или внутрибрюшинные инъекции. Обычные композиции для такого способа введения включают жидкие препаративные формы в изотонических разбавителях, таких как раствор Ханка или раствор Рингера.
Альтернативные препаративные формы включают интраназальные спреи, липосомные препаративные формы, препаративные формы с замедленным высвобождением и подобные, такие, которые известны в данной области.
Может быть использована любая подходящая препаративная форма. Резюме известных в данной области препаративных форм можно найти в Remington's Pharmaceutical Sciences, последнее издание, Mack Publishing Company, Easton, PA. Ссылка на данное руководство обычна в данной области.
Дозировка соединений данного изобретения зависит от множества факторов, которые варьируются от пациента к пациенту. Однако полагают, что в общем, ежедневная пероральыая доза должна составлять 0,001-100 мг/кг веса тела, предпочтительно от 0,01 до 50 мг/кг и более предпочтительно от около 0,01 мг/кг до 10 мг/кг. Режим дозирования может варьироваться в зависимости от состояния, которые лечат, и оценки практикующего врача.
Необходимо отметить, что соединения формулы (I) могут вводиться как самостоятельные активные ингредиенты, так и в виде смеси нескольких таких соединений формулы I. Кроме того, ингибиторы р38 киназы могут быть использованы как самостоятельные терапевтические агенты или в сочетании с другими терапевтическими агентами. Лекарственные средства, которые могут быть с пользой объединены с такими соединениями, включают природные и синтетические кортикостероиды, особенно преднизон и его производные, моноклональные антитела, направленные на клетки иммунной системы, антитела или растворимые рецепторы или рецептор слитого белка, направленные на иммунные или неиммунные цитокины, и ингибиторы деления клеток, синтеза протеинов или транскрипции или трансляции мРНК с небольшими молекулами, или ингибиторы дифференциации или активации иммунных клеток.
Как сказано выше, хотя соединения данного изобретения могут быть использованы для человека, они также могут быть использованы в ветеринарии для лечения животных.
Следующие примеры предназначены для иллюстрации, но не ограничения данного изобретения, и иллюстрируют использование представленных выше реакционных схем.
Пример 1
6-Метокси-(4-бензилпиперидинил)индол-5-карбоксамид-3-глиоксалевая кислота
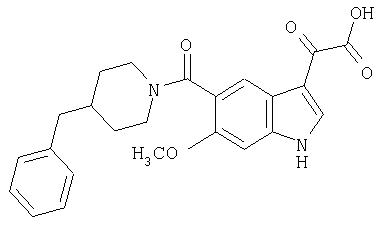
0,348 мг (1 ммоль) 6-метокси-(4-бензилпиперидинил)индол-5-карбоксамида растворяют в 15 мл сухого дихлорметана и охлаждают до температуры 0°С на ледяной бане. С помощью шприца по каплям добавляют 0,6 мл 2-молярного раствора оксалилхлорида в дихлорметане (Aldrich) в инертной атмосфере, и смесь перемешивают при температуре 0°С в течение часа. Ледяную баню удаляют и смесь перемешивают в течение еще одного часа при комнатной температуре. Растворитель выпаривают, и остаток сушат в вакууме в течение 30 минут. Полученное твердое вещество растворяют в смеси ТГФ/воды и подщелачивают 20% водн. NaOH. Растворители удаляют, и остаток растворяют в воде и подкисляют конц. HCl. Выпавшее твердое вещество собирают фильтрацией, сушат и перекристаллизовывают из этанола/воды с получением 350 мг указанного в заголовке соединения.
ЭСМС 421, M+
Пример 2
4-метилпиперазинамид 6-метокси-(4-бензилпиперидинил)-5-карбоксамидоиндол-3-глиоксалевой кислоты
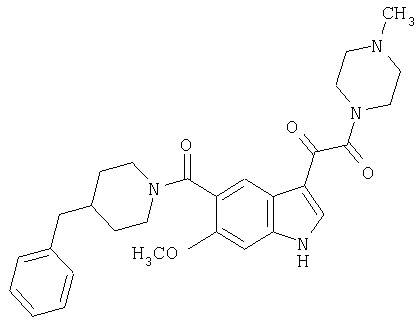
Данное соединение получают с использованием методики, описанной выше для соответствующей кислоты, но заменяя водный NaOH 4-метилпиперазином и проводя реакцию в сухом дихлорметане вместо ТГФ/воды.
ЭСМС 503, М+
Пример 3
1-(2-аминоэтилпирролидин)амид 6-метокси-(4-бензилпиперидинил)-5-карбоксамидоиндол-3-глиоксалевой кислоты
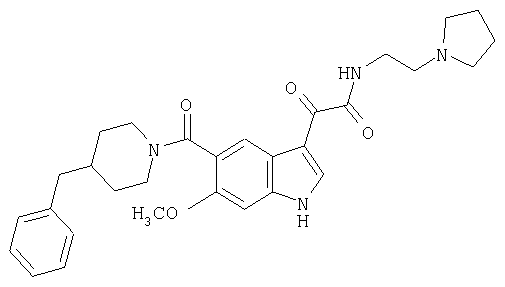
Данное соединение получают с использованием методики, описанной выше, но заменяя 4-метилпиперазин 1-(2-аминоэтил)-пирролидином.
МС М+, 517
Пример 4
амид 4-бензилпиперидинил-5-карбоксамидоиндол-3-глиоксалевой кислоты
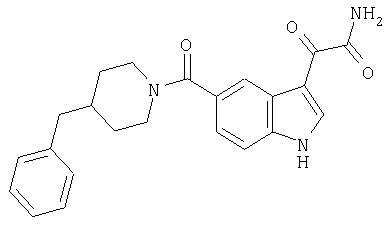
0,318 мг, 1 ммоль, 4-бензилпиперидинилиндол-5-карбоксамида растворяют в сухом дихлорметане и подвергают взаимодействию с 0,6 мл 2-молярного раствора оксалилхлорида при температуре 0°С в течение 1 часа в атмосфере азота. Охлаждающую баню удаляют и смесь перемешивают в течение еще одного часа при комнатной температуре. Растворитель выпаривают, и остаток сушат в вакууме в течение 30 минут. Продукт повторно растворяют в ТГФ и добавляют избыток конц. гидроксида аммония. После перемешивания в течение 1 часа растворитель удаляют и остаток перекристаллизовывают из этилацетата-гексана.
Выход: 220 мг, МС. М+, 389; 345, М+ -CONH2
Пример 5
4-метилпиперазинамид 6-хлор-(4'-фтор-4-бензилпиперидинил)-5-карбоксамидоиндол-3-глиоксалевой кислоты
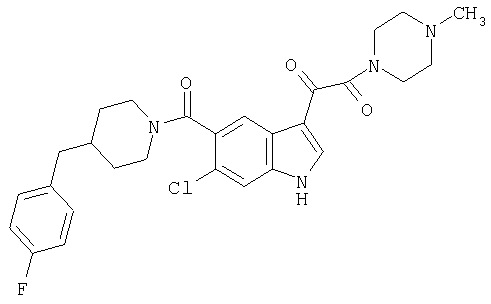
Получают по описанной выше методике.
МС М+, 524
Пример 6
Получение метилового эфира 6-метокси-(4-бензилпиперидинил)индол-5-карбоксамид-3-глиоксалевой кислоты
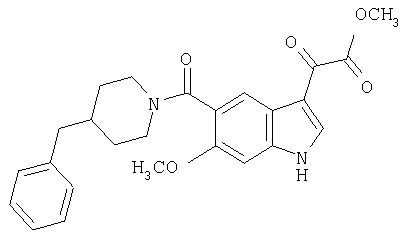
Данное соединение получают по методике, описанной для исходной глиоксалевой кислоты, но заменяя гидроксид натрия метанолом, в ТГФ/воде.
ЭСМС М+, 435
Пример 7
Получение метилового эфира 1-метил-6-метокси-[4'-фтор-(4-бензилпиперидинил)]индол-5-карбоксамид-3-глиоксалевой кислоты
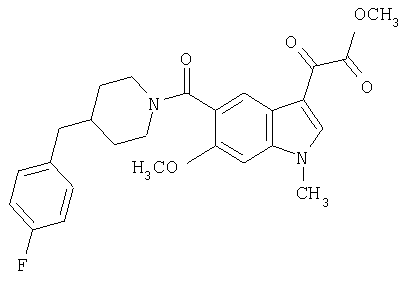
0,435 г метилового эфира 6-метокси-[4'-фтор-(4-бензилпиперидинил)]индол-5-карбоксамид-3-глиоксалевой кислоты растворяют в 10 мл сухого ДМФ и охлаждают до температуры 0°С на ледяной бане. Добавляют 80 мг NaH (60% дисперсия) и смесь перемешивают в течение 15 минут при температуре 0°С и в течение 30 минут при комнатной температуре в инертной атмосфере. Реакционную смесь охлаждают до температуры 0°С и добавляют 200 мкл йодметана. Через 30 минут при температуре 0°С смесь нагревают до комнатной температуры и перемешивание продолжают в течение еще 4 часов. Реакционную смесь выливают в воду и экстрагируют дихлорметаном (3×50 мл). Экстракт сушат, выпаривают и очищают хроматографией на колонке с силикагелем и этилацетат-гексаном (50-90% этилацетат, градиент).
Выход: 70%.
МС М+, 466
Пример 8
Получение 1-метил-6-метокси-[4'фтор-(4-бензилпиперидинил)]индол-5-карбоксамид-3-глиоксалевой кислоты
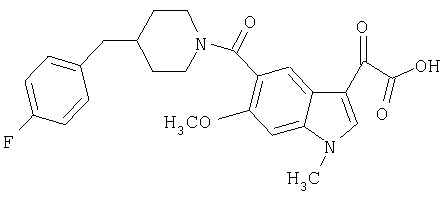
0,24 г (0,51 ммоль) метилового эфира 1-метил-6-метокси-[4'-фтор-(4-бензилпиперидннил)]индол-5-карбоксамид-3-глиоксалевой кислоты растворяют в ТГФ (10 мл), добавляют 1 мл 20% водн. гидроксида натрия и перемешивают в течение 4 ч. Смесь разбавляют водой (5 мл) и перемешивание продолжают в течение 1 часа. ТГФ удаляют при пониженном давлении и оставшийся раствор разбавляют водой и подкисляют конц. HCl, и продукт собирают фильтрацией. Его сушат в вакууме и перекристаллизовывают из этилацетата.
Выход: 180 мг
ЭСМС М+, 453
Пример 9
3-(Метоксибензоил)-(4-бензилпиперидинил)индол-5-карбоксамид
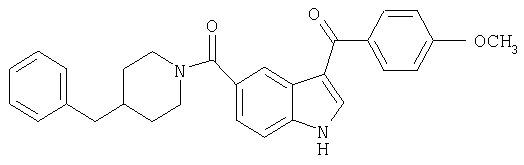
Ацилирование в положении 3 индольного кольца осуществляют по методике С.Х.Yang et al. (Synth. Commun, 27 (12), 2125 (1997). К раствору 0,318 г (1,0 ммоль) 5-(4-бензилпиперидинил)-индолкарбоксамида в дихлорметане добавляют 2,2 мл (2,2 ммоль) 1М раствора ZnCl2 в эфире с последующим добавлением по каплям EtMgBr (1,0 ммоль). Смесь перемешивают в течение 1 часа и добавляют п-анизоилхлорид (180 мг, 1,1 ммоль). Суспензию перемешивают в течение 1 часа и добавляют AlCl3 (0,05 ммоль). Полученную смесь перемешивают в течение 2 часов и гасят насыщ. раствором NB4Cl. Органический слой промывают водн. NaHCO3 и насыщенным раствором соли, сушат над MgSO4 и фильтруют. Растворитель выпаривают и продукт очищают хроматографией на колонке с силикагелем.
МС М+, 452
Пример 10
3-Бензоил-5-(4-бензилпиперидинил)индолкарбоксамид
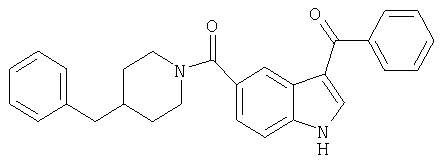
Получают, используя методику, описанную выше, но заменяя п-анизоилхлорид бензоилхлоридом.
МС М+, 422
Пример 11
3-Ацетил-5-(4-бензилпиперидинил)индол-5-карбоксамид
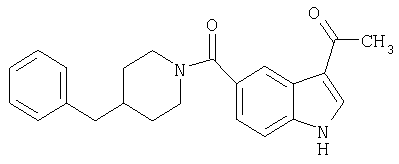
Получают, используя методику, описанную выше, но заменяя п-анизоилхлорид ацетилхлоридом.
МС М+, 360
Пример 12
Получение 3-(2-гидроксиацетил)-5-(4-бензилпиперидинил)-индолкарбоксамида
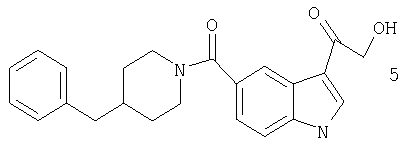
3-(2-Хлорацетил)-5-(4-бензилпиперидинил)индолкарбоксамид получают с использованием методики, описанной выше, но заменяя п-анизоилхлорид хлорацетилхлоридом.
Гидролиз хлорацетильной группы до гидроксиацетильной проводят с использованием опубликованной методики (J. Org. Chem., 1988, 53, 5446). К раствору 50 мг (0,13 ммоль) 3-(2-хлорацетил)-5-(4-бензилпиперидин)индолкарбоксамида в диоксане (3 мл) добавляют 5 мл формамида-воды (10:1). Реакционную смесь нагревают до температуры 110°С в течение 5 часов и охлаждают до комнатной температуры. Реакционную смесь гасят насыщ. раствором NH4Cl, экстрагируют дихлорметаном, сушат (MgSO4) и концентрируют. Остаток очищают препаративной ТСХ, проводимой с дихлорметаном-метанолом (20:1) с получением 20 мг (42%) указанного в заголовке соединения.
МС М+, 376
Пример 13
5-(4-Бензилпиперидинил)карбоксамидоиндол-3-(3'-оксо)-этилпропионат
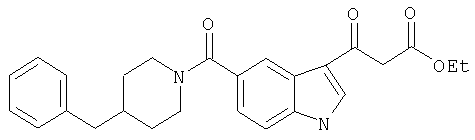
Ацилирование в положении 3 индольного кольца осуществляют по опубликованной методике [Synth. Commun, 1977, 27 (12), 2125]. К раствору 3, 6 мл ZnCl2 (3,6 ммоль, 1М раствор в диэтиловом эфире) в ТГФ по каплям добавляют н-BuLi (2.2 мл, 3,6 ммоль) при температуре 0°С. Во время добавления образуется белая суспензия. Реакционную смесь нагревают до комнатной температуре, перемешивают в течение 1 часа и добавляют раствор 5-(4-бензилпиперидинил)индолкарбоксамида в дихлорметане (10 мл). Полученную смесь перемешивают в течение 1 часа и добавляют этил 3-хлор-3-оксопропионат (585 мг, 3,9 ммоль). Через 1 час реакционную смесь гасят насыщенным NH4Cl, экстрагируют дихлорметаном, сушат (MgSO4) и концентрируют. Остаток очищают хроматографией на колонке с силикагелем с получением 200 мг желаемого продукта.
MC> M+, 431
Соединения 1-55 таблицы 2 получают по той же методике.
Пример 14
Синтез (2S,5R)N-4-Фторбензил-транс-2,5-диметилпиперазина
(+/-)N-Бензил-транс-2,5-диметилпиперазин синтезируют следующим образом
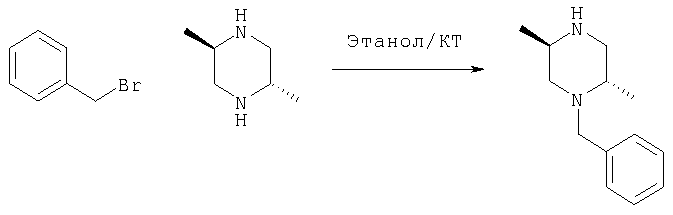
50 г транс-2,5-Диметилпиперазина растворяют в 300 мл этанола и обрабатывают 26,36 мл (1/2 эквивалента) бензилбромида. Смесь перемешивают при комнатной температуре в течение 12 часов и концентрируют. Остаток помещают в этилацетат и промывают 10% водным бикарбонатом натрия и насыщенным хлоридом натрия; сушат над безводным сульфатом магния и концентрируют с получением неочищенного 1-бензил-транс-2,5-диметилпиперазина в виде масла.
Полученный материал подвергают хроматографии, используя ДХМ/МеОН 95/5 для удаления диалкилированного продукта, и затем ДХМ/МеОН/ТЭА 90/10/0,1 для элюирования бензил-транс-2,5-диметилпиперазина. Получают 19,6 г продукта в виде масла.
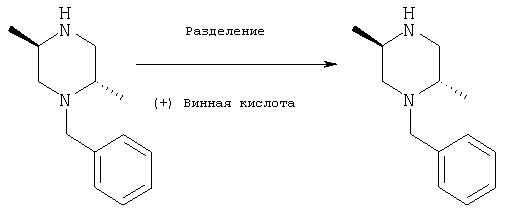
К раствору (+/-)N-бензил-транс-2,5-диметилпиперазина (59 г, 0,29 моль) в метаноле (150 мл) по каплям добавляют раствор (+) винной кислоты (87 г, 0,58 моля) в метаноле (250 мл) на протяжении 5 минут. Кристаллизацию осуществляют при сохранении полученной смеси при температуре на уровне 0°С в течение 48-72 часов. Затирание раствора через 12-16 часов способствует процессу кристаллизации. Смесь фильтруют и промывают холодным метанолом и сушат с получением соли дивинной кислоты (75,9 г) в виде белых кристаллов. Однократная перекристаллизация из метанола, охлаждение до комнатной температуры дает соль в виде белых кристаллов. (58 г) [α]D=+47, (с=1,00, метанол).
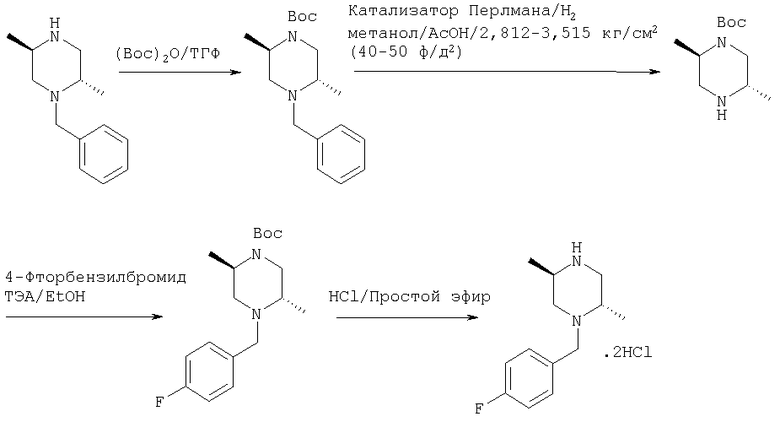
31 г, 15,27 ммоль, диметилбензилпиперазина обрабатывают 43 г, 19,85 ммоль, ди-трет-бутилдикарбоната в 250 мл ТГФ в течение 4 часов. Реакцию контролируют ТСХ и она практически завершается по завершению добавления (BOC)2О. Растворитель удаляют и остаток помещают в этилацетат и промывают 10% водным карбонатом натрия и насыщенным хлоридом натрия, сушат над безводным сульфатом натрия и концентрируют с получением 39,3 г соединения с Вос-защитной группой. Полученное соединение используют на следующей стадии без дальнейшей очистки.
39,3 мг, 131 ммоль, бензилпиперазина с Вос-защитной группой обрабатывают 3,93 г катализатора Перлмана в 150 мл метанола с 3 мл уксусной кислоты в течение 4 часов при давлении водорода 2,812 кг/см2 (40 ф/д2) в смесителе Парра. Реакционную смесь фильтруют через целит и концентрируют с получением остатка, который сушат в высоком вакууме и затем растворяют в 250 мл сухого этанола и обрабатывают 1,2 эквивалентами, 158 ммоль, 4-фторбензилбромида и 2 эквивалентами, 262 ммоль, триэтиламина в течение 5 часов. Реакционную смесь контролируют ТСХ, которая показывает завершение реакции к этому времени. Растворитель удаляют и остаток помещают в этилацетат и промывают 10% водным карбонатом натрия и насыщенным хлоридом натрия, сушат над безводным сульфатом натрия и концентрируют с получением 47 г неочищенного продукта. Неочищенный продукт подвергают хроматографии на колонке с силикагелем, используя гексан/этилацетат 95/5 для элюирования продукта. После хроматографии получают 29 г 4-фторбензил-транс-2,5-диметил-пиперазина. 20 г 1-(4-Фтор)бензил-транс-2,5-диметил-пиперазина, 62 ммоль, обрабатывают смесью 200 мл 4N HCl/Диоксан/2М HCl/эфир (1:3) в течение 1 часа. Масс-спектроскопия с электронным ударом подтверждает образование продукта и исчезновение исходных материалов. Реакционную смесь концентрируют с получением твердого вещества. Этот материал повторно промывают гексаном и простым эфиром для удаления остаточного диоксана и затем длительно сушат в вакууме перед использованием. Получают 17,6 г конечного продукта в виде белого пушистого твердого вещества.
Пример 15
Синтез хиральных соединений
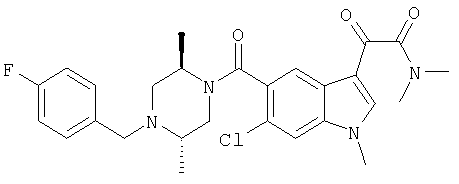

6-хлориндол-5-карбоновую кислоту (1,56 г, 7,44 ммоль) растворяют в сухом метиленхлориде, 60 мл, к полученному раствору добавляют EDAC.HCl (1,57 г, 8,18 ммоль) и ДМАП (10% моль). После перемешивания в атмосфере азота в течение 10 мин добавляют амин (2,19 г, 7,5 ммоль) с последующим добавлением триэтиламина (3 мл, 21,52 ммоль). После выстаивания в течение ночи при комнатной температуре реакционную смесь концентрируют и остаток помещают в этилацетат и промывают 10% водн. карбонатом натрия, насыщенным хлоридом натрия, сушат над безводным сульфатом натрия и фильтруют. Концентрация дает неочищенный продукт, который подвергают хроматоррафии на колонке с силикагелем, используя градиент EtOAc/гексан 2/8-6/4. ТСХ Rf 0,435 (EtOAc:гексан, 1:1), ЭУМС М+ 413.
Продукт с предыдущей стадии с 1,02 г растворяют в 30 мл сухого ДХМ. Реакционную смесь продувают азотом и помещают на ледяную баню. К полученной смеси добавляют 4 мл 2М оксалилхлорида в ДХМ. Реакционную смесь перемешивают при комнатной температуре в течение 1 часа и затем при комнатной температуре в течение 2 часов. Реакционную смесь концентрируют на роторном испарителе. После сушки в вакууме в течение 15 мин остаток (желтое твердое вещество) растворяют в ДХМ, 30 мл, к которому добавляют 4 мл 2М раствора диметиламина в ТГФ. Через 30 минут реакционную смесь концентрируют и остаток помещают в этилацетат и промывают 10% водн. карбонатом натрия, насыщенным хлоридом натрия, сушат над безводным сульфатом натрия и фильтруют. Концентрирование дает неочищенный продукт, который подвергают хроматографии на колонке с силикагелем с использованием градиента EtOAc 100% - EtOAc/MeOH 9:1. ТСХ Rf 0/5 (EtOAc:MeOH, 9:1), ЭУМС М+ 513.
Белое твердое вещество с предыдущей стадии растворяют в 10 мл сухого ДХМ. К полученному раствору добавляют 2М HCl в эфире до выпадения осадка. Затем смесь концентрируют на роторном испарителе досуха и затем сушат в течение ночи в высоком вакууме с получением конечного продукта (1,08 г).
Дополнительные примеры
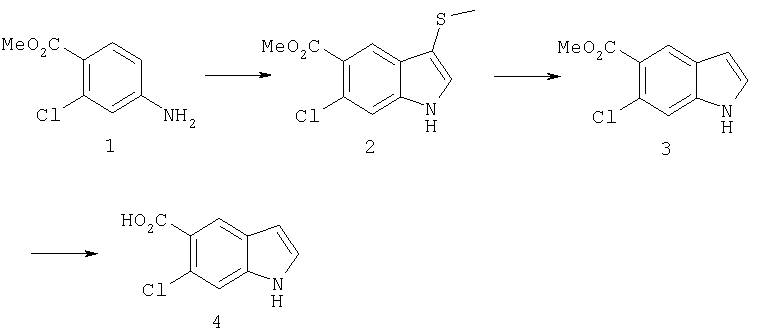
Синтез соединения 2: Метил 4-амино-2-хлорбензоат (1) (18,5 г) растворяют в дихлорметане (350 мл) и добавляют диметилацеталь метилтиоацетальдегида (13,6 г). Смесь охлаждают дс температуры -45°С (баня сухой лед/ацетонитрил). По каплям добавляют N-хлорсукцинимид (16,0 г) в 350 мл дихлорметана на протяжении 1 часа 30 минут, сохраняя температуру бани на уровне -45°С. Реакционную смесь перемешивают еще 1 час, затем по каплям добавляют триэтиламин (16 мл, 100 ммоль) в 30 мл дихлорметана на протяжении 5 минут, реакционную смесь нагревают до комнатной температуры, затем кипятят с обратным холодильником в течение 16 часов. Растворитель удаляют, и остаток помещают в 500 мл четыреххлористого углерода, гидрохлорид триэтиламина кислоту удаляют фильтрацией, фильтрат нагревают при температуре кипения с обратным холодильником в течение 2 часов. Растворитель удаляют на роторном испарителе.
Остаток растворяют в 250 мл тетрагидрофурака и добавляют 250 мл 10% хлористоводородной кислоты. Смесь перемешивают в течение ночи при комнатной температуре до полного исчезновения исходного материала. Растворитель удаляют в вакууме, кислый водный раствор экстрагируют этилацетатом (3×125 мл). Объединенные экстракты этилацетата промывают 10% хлористоводородной кислотой, водой и сушат над безводным сульфатом натрия. Растворитель удаляют в вакууме. Смесь неочищенного продукта очищают на колонке с силикагелем, элюируя этилацетатом:гексаном (15:85) с получением 6,4 г желаемого продукта 2.
Синтез соединения 3: Метил 6-хлор-3-тиометил-5-индолкарбоксилат (5,2 г) растворяют в 150 мл этанола:тетрагидрофурана (9:3) и обрабатывают никелем Ренея. Реакцию контролирует масс-спектрическим анализом с 30-минутными интервалами, с последующим добавлением никеля Ренея до завершения реакции. После завершения реакции реакционную смесь осторожно фильтруют через целит, целит промывают метанолом несколько раз и фильтрат выпаривают. Остаток помещают в этилацетат, промывают водой, сушат над безводным сульфатом натрия. Растворитель удаляют с получением соединения 3 (3,2 г).
Синтез соединения 4: Метиловый эфир, 1,5 г, растворяют в 30 мл метанола/воды 50:50. Реакционную смесь нагревают до температуры 50°С в течение 2 часов с 4 молярными эквивалентами гидроксида натрия. Реакционную смесь охлаждают на ледяной бане, подкисляют до рН 3 5М хлористоводородной кислотой. Этанол удаляют на роторном испарителе, и остаток экстрагируют этилацетатом. Экстракт промывают насыщенным хлоридом натрия и сушат над безводным сульфатом натрия. Выпаривание растворителя дает желаемую кислоту 4 (1/48 г).
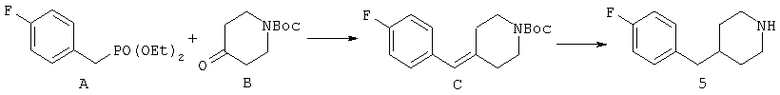
Синтез соединения 5:
СТАДИЯ А: Фосфонат А (38,4 г) и пиперидон В (35,4 г) растворяют в безводном диметилформамиде (400 мл). К полученному раствору порциями добавляют гидрид натрия (60% суспензия в масле), сохраняя температуру реакционной смеси при 0°С. После завершения добавления гидрида натрия реакционную смесь перемешивают в течение 30 минут и затем ледяную баню удаляют, реакционную смесь перемешивают в течение 6 часов и медленно нагревают до температуры окружающей среды. Реакционную смесь снова охлаждают на ледяной бане и гасят метанолом. К реакционной смеси добавляют воду и продукт экстрагируют этилацетатсм. Слой этилацетата промывают насыщенным хлоридом натрия и сушат над безводным сульфатом магния. Растворитель удаляют с получением неочищенного алкена, который очищают хроматографией на колонке, элюируя этилацетатом/гексаном (1:9) с получением 21/8 г желаемого продукта С.
СТАДИЯ В: 10,1 г соединения С растворяют в 50 мл метанола. После продувания раствора азотом добавляют катализатор 5% палладий на углероде (1 г) с последующим добавлением 1 мл уксусной кислоты. Реакционную смесь гидрируют в аппарате Парра в течение 4 часов при 2,812-3,515 кг/см2 (40-50 ф/д2). Реакционную смесь фильтруют через целит и концентрируют. Остаток обрабатывают 2 М хлористоводородной кислотой в эфире для получения соли с хлористоводородной кислотой. Полученное белое твердое вещество продолжительно сушат в вакууме с получением 7,8 г соединения 5 в виде гидрохлорида.
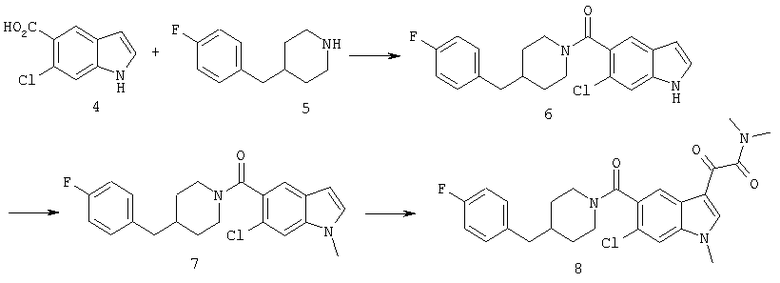
Синтез соединения 6: Смесь 6-хлориндол-5-карбоковой кислоты (1,95 г), гидрохлорида 4-фторбензилпиперидина (2,76 г) помещают в 50 мл сухого дихлорметана и обрабатывают триэтиламином (1,7 мл). Смесь перемешивают до получения прозрачного раствора. Добавляют 1-этил-3-(3-диметиламинопропил)карбодиимид (2,3 г) и диметиламинопиридин (0,25 г) и смесь перемешивают в течение 20 часов при температуре окружающей среды. Смесь выливают в воду и органический слой отделяют. Водный раствор затем дважды экстрагируют дихлорметаном. Объединенный экстракт промывают 10% хлористоводородной кислотой. Органический слой сушат над сульфатом магния и выпаривают. Продукт очищают хроматографией на колонке с силикагелем, элюируя этилацетатом:гексаном (3:7) с получением соединения 6 (2,78 г).
Синтез соединения 7: 8,61 г 6-хлор-(4-F-бензилпиперидинил)индол-5-карбоксамида (6) растворяют в примерно 100 мл сухого ДМФ и раствор охлаждают на ледяной бане. К холодному раствору добавляют 30 мл 1М раствора бис-(триметилсилил)амида натрия в тетрагидрофуране в инертной атмосфере. Реакционную смесь перемешивают при температуре 0°С в течение 15 минут и при температуре окружающей среды в течение еще 30 минут. Реакционную смесь снова охлаждают на ледяной бане и добавляют 2,5 мл йодметана. После перемешивания в течение 30 минут при температуре 0°С смесь нагревают до температуры окружающей среды и перемешивание продолжают в течение 18 часов при температуре окружающей среды. Смесь разбавляют водой (или насыщенным раствором соли) и продукт экстрагируют этилацетатом (4×75 мл). Объединенный экстракт промывают водой и сушат над безв. MgSO4. Растворитель удаляют и продукт очищают хроматографией на колонке с силикагелем, элюируя этилацетатом:гексаном (1:4) с получением 8,0 г желаемого продукта 7.
Синтез соединения 8: 8 г соединения 7 растворяют в примерно 100 мл безводного дихлорметана и охлаждают на ледяной бане. К полученному раствору медленно через шприц добавляют 2М раствор оксалилхлорида (20,8 мл) в дихлорметане, и смесь перемешивают при температуре 0°С в течение 1 часа. Ледяную баню убирают и перемешивание продолжают в течение еще 2 часов при температуре окружающей среды. Растворитель удаляют при пониженном давлении, и остаток откачивают в течение 15 минут для удаления избыточно присутствующего оксалилхлорида. Продукт сразу же повторно растворяют в безв. дихлорметане (150 мл), охлаждают на ледяной бане, быстро через шприц добавляют 30 мл 2М раствора диметиламина в тетрагидрофуране. Через 15 минут перемешивания, перемешивание продолжают в течение еще 15 минут при температуре окружающей среды. Раствор промывают водой для удаления соли и сушат. После упаривания остаток очищают на колонке с силикагелем, элюируя хлороформом:метанолом (99:1) с получением 9,3 г соединения 8.
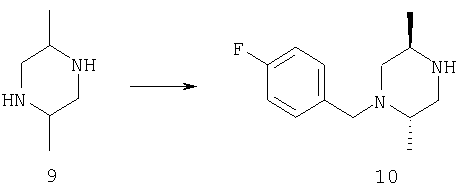
Синтез соединения 10:
СТАДИЯ А: К раствору диметилпиперазина 9 (25 г) в 300 мл абсолютного этанола добавляют 400 мл 2N хлористого водорода в диэтиловом эфире. Раствор нагревают до температуры 70°С на масляной бане в течение 20 минут. Затем раствор охлаждают до комнатной температуры и выстаивают в течение ночи при температуре 6°С. Получают твердое вещество, которое собирают фильтрацией. Выход 39,8 г (дигидрохлорид транс-2,5-диметилпиперазина) после сушки в течение ночи в высоком вакууме.
СТАДИЯ В: Этанольный раствор 42,9 г дигидрохлорида диметилпиперазина со СТАДИИ А и 26,1 г транс-2,5-диметилпиперазина энергично перемешивают на масляной бане при температуре 80°С до растворения всех исходных соединений. Температуру масляной бани понижают до 65°С и добавляют 33,1 г 4-фторбензилхлорида. После перемешивания при этой температуре в течение 30 минут раствор помещают на ночь в холодильник с температурой 6°С. Твердое вещество удаляют из раствора фильтрацией и к фильтрату добавляют 2N хлористый водород в диэтиловом эфире. Фильтрат хранят при температуре 6°С в течение ночи и твердое вещество собирают. Твердое вещество суспендируют в 5% водном растворе гидроксида натрия и три раза экстрагируют этилацетатом. Органический слой сушат над сульфатом натрия и высушивают с получением желтого масла.
СТАДИЯ С: Раствор 50,7 г (L)-винной кислоты в 130 мл кипящего метанола добавляют к 70 мл горячего метанольного раствора 37,5 г продукта со СТАДИИ В. Раствор выстаивают при температуре 6°С в течение 96 часов, затем белые кристаллы собирают фильтрацией. Полученное соединение перекристаллизовывают из кипящего метанола. Продукт собирают фильтрацией после выстаивания при температуре 6°С в течение ночи. Выход 30,5 г соли дивинной кислоты ([α]=+43,2°, с=1).
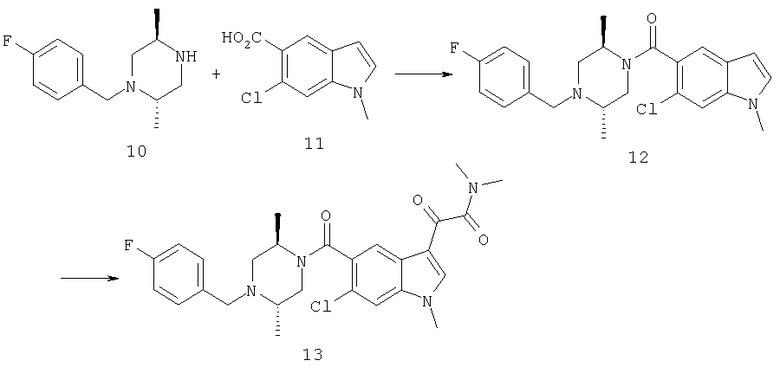
Синтез соединения 11: Сложный эфир индола 3 (0,526 г) растворяют в 10 мл ацетона (сухого) и помещают на ледяную баню. К полученному раствору добавляют измельченный гидроксид калия (0,7 г, 12,5 ммоль), после перемешивания в течение 5 минут при температуре 0°С к реакционной смеси добавляют метилйодид (400 мкл, 6,272 ммоль). Реакционную смесь перемешивают при температуре 0°С в течение 10 минут и затем при комнатной температуре в течение 30 минут. После удаления растворителя остаток помещают в этилацетат и промывают насыщенным хлоридом натрия. После сушки над безводным сульфатом натрия, фильтрации и выпаривания на роторном испарителе получают твердое вещество, 0,7 г. Хроматография на колонке с силикагелем с этилацетатом:гексаном (2:8) дает метиловый эфир соединения 11 в виде белого твердого вещества. 0,52 г Полученного продукта растворяют в 50 мл метанола и обрабатывают 5 мл 10N гидроксида натрия, реакционную смесь нагревают до температуры 50°С в течение 2 часов. Реакционную смесь охлаждают до комнатной температуры и концентрируют до твердого вещества на роторном испарителе. Остаток помещают в 50 мл воды, промывают простым эфиром и помещают на ледяную баню. Основной раствор подкисляют 10% хлористоводородной кислотой до рН 2. Осадок экстрагируют этилацетатом и слой этилацетата промывают насыщенным раствором хлорида натрия. Сушка над безводным сульфатом натрия, фильтрация и концентрация на роторном испарителе дают соединение 11 (0,48 г) в виде белого твердого вещества.
Синтез соединения 12: 1,56 г кислоты (11) растворяют в сухом метиленхлориде, 10 мл, и к полученному раствору добавляют 1-этил-3-(3-диметиламинопропил)карбодиимид (1,57 г) и диметиламинопиридин (каталитический). После перемешивания в атмосфере азота в течение 10 мин добавляют 2,19 г амина (10) с последующим добавлением триэтиламина (3 мл). После перемешивания в течение ночи при комнатной температуре реакционную смесь концентрируют и остаток помещают в этилацетат и промывают 10% водн. карбонатом натрия, водн. насыщенным хлоридом натрия, сушат над безводным сульфатом натрия и фильтруют. Концентрирование дает неочищенный продукт, который подвергают хроматографии на колонке с силикагелем, используя этилацетат:гексан (4:6) в качестве элюента с получением 1,02 г желаемого продукта.
Синтез соединения 13: 1,02 г соединения 12 с предыдущей стадии растворяют в 30 мл сухого дихлорметана. Реакционную смесь продувают азотом и помещают на ледяную баню. К полученной смеси добавляют 4 мл 2М оксалилхлорида в дихлорметане. Реакционную смесь перемешивают при температуре 0°С в течение 1 часа и затем при комнатной температуре в течение 2 часов. Реакционную смесь концентрируют на роторном испарителе. После сушки в вакууме в течение 15 минут остаток (желтое твердое вещество) растворяют в сухом дихлорметане (30 мл), к которому добавляют 4 мл 2М раствора диметиламина в тетрагидрофуране. Через 30 минут реакционную смесь концентрируют и остаток помещают в этилацетат и промывают 10% водн. карбонатом натрия, насыщенным хлоридом натрия, сушат над безводным сульфатом натрия и фильтруют. Концентрирование дает неочищенный продукт, который подвергают хроматографии на колонке с силикагелем с этилацетатом:метанолом (9:1). Полученное белое вещество растворяют в 10 мл сухого дихлорметана. К полученному раствору добавляют достаточное количество 2М хлористоводородной кислоты до тех пор, пока не образуется осадок. Затем смесь концентрируют на роторном испарителе досуха и далее сушат в течение ночи в высоком вакууме с получением соединения 13 (1,08 г).
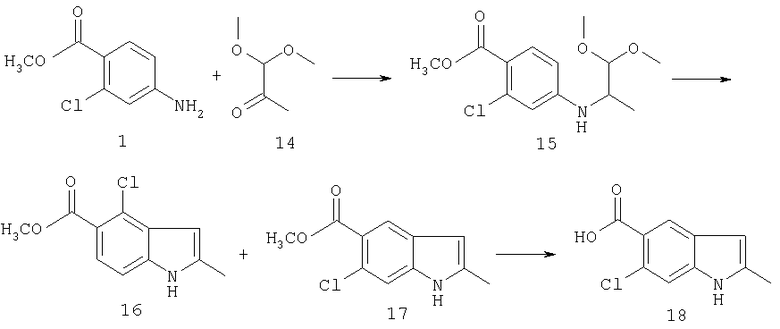
Синтез соединения 15: К раствору анилина 1 (9,25 г, 0,05 моль) и диметилацеталя пировиноградного альдегида 14 (11,8 г, 0,1 моль) в 200 мл ледяной уксусной кислоты добавляют безводный сульфат натрия (71,0 г, 0,5 моль) и смесь перемешивают в течение 30 минут. Затем порциями добавляют порошок триацетоксиборгидрида натрия (31,8 г, 0,15 моль) в течение 5 минут. Реакционную смесь перемешивают в течение еще 2 часов. Уксусную кислоту удаляют при пониженном давлении и остаток подщелачивают добавлением достаточного количества насыщенного раствора бикарбоната натрия. Затем продукт экстрагируют этилацетатом, сушат сульфатом натрия и выпаривают с получением масла. Полученное масло подвергают хроматографии на колонке с силикагелем, используя этилацетат:гексан (3:7) с получением соединения 15 (14 г) в виде бесцветного масла.
Синтез соединений 16 и 17: К суспензии свежего хлорида алюминия (18,5 г) в 200 мл сухого хлороформа при температуре 0°С медленно добавляют раствор кеталя 15 (13.3 г) в 100 мл хлороформа, и смесь нагревают до комнатной температуры и перемешивают в течение ночи. Хлорид алюминия осторожно гасят добавлением ледяной воды и органический слой отделяют и промывают раствором бикарбоната натрия, сушат и выпаривают с получением белого твердого вещества. Изомеры разделяют, используя хроматографию на колонке с силикагелем, используя этилацетат:гексан (1:9). Сначала элюируют 6-хлориндол 17 (2,0 г), затем 4-хлор изомер 16 (3,8 г).
Синтез соединения 18: К раствору 1,3 г индола 17 в 15 мл метанола добавляют раствор 0,9 г гидроксида натрия в 20 мл воды. Реакционную смесь нагревают при температуре 50°С в течение 4 часов до получения прозрачного раствора. Смесь охлаждают, и метанол выпаривают, остаток разбавляют водой и подкисляют 10% хлористоводородной кислотой. Продукт экстрагируют этилацетатом. Органический слой сушат над сульфатом натрия, фильтруют и выпаривают с получением кислоты индола 18 (1,2 г) в виде белого твердого вещества.
2-Метил-6-метоксииндол-5-карбоновую кислоту синтезируют по описанной выше методике.
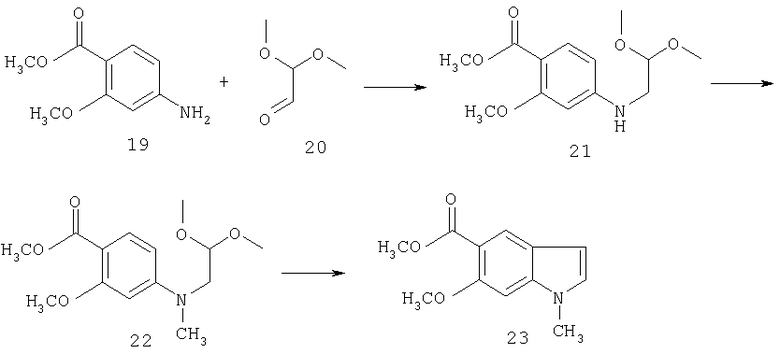
Синтез соединения 21: К раствору метил 4-амино-6-метокси 5-бензоата 19 (6,0 г, 0,033 моль) и диметилацеталя 20 (7,0 г, 0,066 моль) в 150 мл ледяной уксусной кислоты добавляют безводный сульфат натрия (47,0 г, 0,33 моль) и смесь перемешивают в течение 30 минут. Порциями добавляют порошок триацетоксиборгидрида натрия (20,1 г, 0,099 моль) в течение 5 минут. Реакционную смесь перемешивают в течение еще 2 часов. Уксусную кислоту удаляют при пониженном давлении и остаток подщелачивают добавлением достаточного количества насыщенного раствора бикарбоната натрия. Затем продукт экстрагируют этилацетатом, промывают насыщенным хлоридом натрия, сушат над сульфатом натрия и выпаривают с получением масла. Полученное масло подвергают хроматографии на колонке с силикагелем, используя этилацетат:гексан (3:7) в качестве элюента, и получают желаемый продукт 21 (5,2 г) в виде масла.
Синтез соединения 22: К раствору соединения 21 (3,6 г) и йодметана (5,7 г) в 50 мл безводного диметилформамида добавляют трет-бутокскд калия (1,0 М в тетрагидрофуране, 20 мл) при температуре окружающей среды. Реакционную смесь перемешивают при температуре окружающей среды в течение 0,5 часа и выливают в 250 мл этилацетата, промывают водой (4×100 мл), насыщенным раствором соли (50 мл) и сушат над сульфатом магния. Выпаривание растворителя дает 3/26 г соединения 22. Продукт используют на следующей стадии без очистки.
Синтез соединения 23: К суспензии безводного хлорида алюминия (0,71 г) в 20 мл безводного 1,2-дихлорэтана добавляют по каплям раствор соединения 22 (1 г) в 10 мл 1,2-дихлорэтана при перемешивании. Реакционную смесь нагревают до температуры 80°С в течение 0,5 часа. По истечении указанного времени реакционную смесь гасят метанолом, растворители выпаривают, затем добавляют этилацетат (100 мл). Органическую фазу промывают водой, водн. раствором бикарбоната натрия и насыщенным раствором соли и концентрируют. Неочищенный продукт очищают хроматографией на колонке с силикагелем, используя этилацетат:гексан (3:7), с получением соединения 23 (0,22 г).
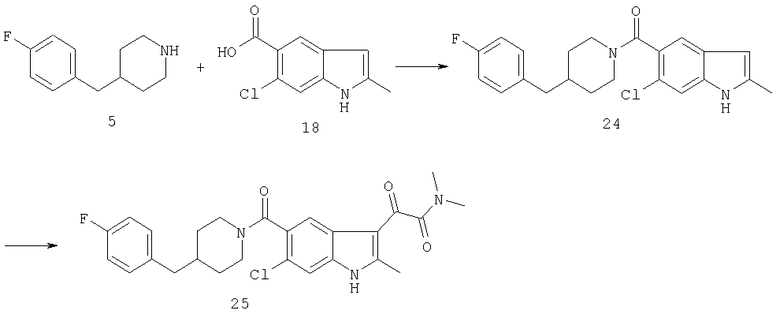
Синтез соединения 24: К суспензии 1,2 г кислоты индола 18 и 1,6 г соединения 5 в 30 мл дихлорметана добавляют 0,7 г триэтиламина с последующим добавлением 1,4 г 1-этил-3-(3-диметиламинопропил)карбодиимида. Полученный прозрачный раствор перемешивают в течение 4 часов. Растворитель выпаривают, и остаток помещают в этилацетат и промывают водой, разб. хлористоводородной кислотой и насыщенным раствором соли. Органический слой сушат сульфатом натрия и выпаривают. Продукт выделяют (1,3 г) в виде белого твердого вещества после хроматографии на колонке с силикагелем с использованием этилацетата:гексана (1:4).
Синтез соединения 25: Раствор 1,3 г соединения 24 в 20 мл дихлорметана охлаждают до температуры 0°С и добавляют 2 М раствор оксалилхлорида в дихлорметане (3,4 мл, 6,8 ммоль), реакционную смесь перемешивают при температуре 0°С в течение 1 часа. Температуру повышают до комнатной температуры и перемешивание продолжают в течение еще 1 часа. Растворитель выпаривают и остаток сушат в вакууме. Хлорангидрид растворяют в дихлорметане (25 мл) и добавляют сразу весь раствор 6,8 мл (13,6 ммоль) 2 М N,N-диметиламина в тетрагидрофуране. Растворитель удаляют и продукт очищают хроматографией на колонке с силикагелем, используя метанол:хлороформ (2:98). Продукт получают в виде белого твердого вещества (1,4 г).
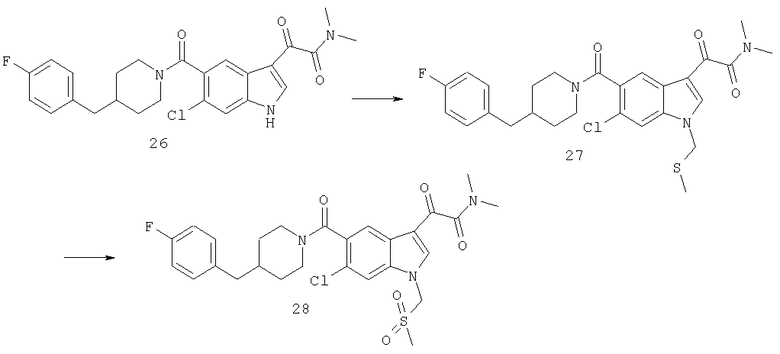
Синтез соединения 27: Раствор соединения 26 (130 мг) в безводном диметилформамиде (5 мл) перемешивают при температуре 0°С в атмосфере азота. Добавляют гидрид натрия (25 мг, 60% дисперсия в масле) и перемешивают в течение 5 минут, затем при комнатной температуре в течение 30 минут. Смесь охлаждают до температуры 0°С и добавляют хлорметил метилсульфид (70 мкл). Реакционную смесь перемешивают в течение 20 часов при комнатной температуре и затем экстрагируют этилацетатом. Органический слой сушат над сульфатом натрия, концентрируют в вакууме. Остаток счищают на хроматотроне с использованием метанола: хлороформа (2:98) с получением 100 мг соединения 27.
Синтез соединения 28: Раствор оксона (120 мг) в воде (1 мл) добавляют к суспензии соединения 27 (100 мг) в ледяной уксусной кислоте (4 мл). Реакционную смесь перемешивают при комнатной температуре в течение 18 часов, промывают водой и концентрируют в вакууме. Остаток очищают на хроматотроне, используя метанол: хлороформ (2:98) с получением 60 мг соединения 28.
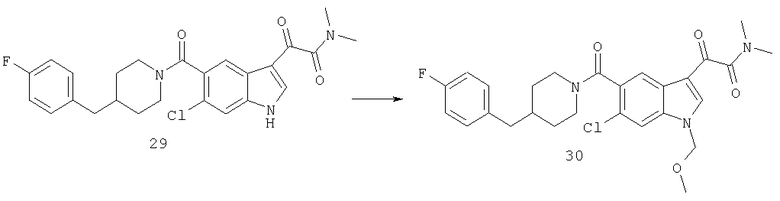
Синтез соединения 30: К раствору соединения 29 (200 мг) в тетрагидрофуране (20 мл) по каплям добавляют бис(триметилсилил)амид натрия (0,51 мл, 0,51 ммоль) при температуре 0°С. Смесь, превратившуюся в светло-желтый раствор, медленно нагревают до температуры окружающей среды и перемешивают в течение 30 минут. Затем добавляют метоксиметилхлорид и реакционную смесь перемешивают в течение ночи. Реакционную смесь гасят хлоридом аммония и экстрагируют дихлорметаном. Объединенный органический слой промывают насыщенным раствором соли, сушат и концентрируют. Остаток очищают хроматографией на колонке с силикагелем, элюируя метанолом:дихлорметаыом (5:95) с получением 190 мг соединения 30.
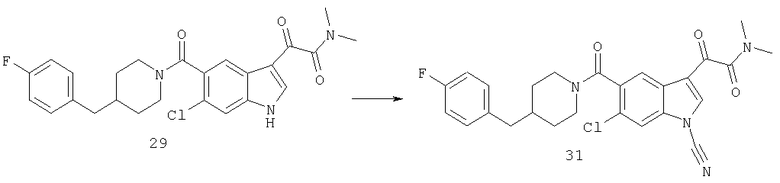
Синтез соединения 31: к раствору соединения 29 (200 мг) в тетрагидрофураке (10 мл) по каплям добавляют бис (триметилсилил) амид натрия (0,56 мл, 0,56 ммоль) при температуре 0°С. Смесь медленно нагревают до температуры окружающей среды, перемешивают в течение 30 минут и затем добавляют к раствору тозилцианида (115 мг) в тетрагидрофуране (10 мл). Полученную смесь перемешивают в течение 2 часов при комнатной температуре, гасят хлоридом аммония и экстрагируют дихлорметаном. Объединенный органический слой промывают насыщенным раствором соли, сушат и концентрируют. Остаток очищают хроматографией на колонке с силикагелем, элюируя метанолом:дихлорметаном (5:95) с получением 90 мг соединения 31.
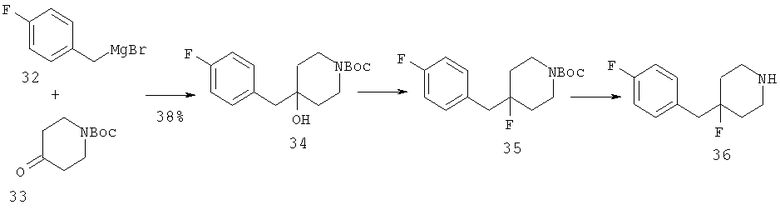
Синтез соединения 34: Пиперидон 33 медленно добавляют к раствору 4-фторбензил хлорида магния (32) (33 мл, 8,3 ммоль, 0,25 М в этиловом эфире) при температуре 0°С. Реакционную смесь нагревают до температуры окружающей среды и затем при температуре кипения с обратным холодильником в течение 6 часов. Полученный мутный раствор обрабатывают хлоридом амиония (насыщенным) и экстрагируют простым эфиром. Объединенный органический слой промывают насыщенным раствором соли, сушат и концентрируют. Остаток очищают хроматографией на колонке с силикагелем, элюируя гексаном:этилацетатом (4:1) с получением 990 мг соединения 34.
Синтез соединения 35: Спирт 34 (670 мг) в дихлорметаке (10 мл) добавляют к раствору трифторида диэтиламиносеры (0,5 мл, 4/34 ммоль) в дихлорметане (20 мл) при температуре -78°С. Реакционную смесь медленно нагревают до температуры окружающей среды и перемешивают в течение 2 часов, и затем обрабатывают карбонатом натрия (насыщенным) и экстрагируют дихлорметаном. Объединенный органический слой промывают насыщенным растворов соли, сушат и концентрируют. Остаток очищают хроматографией на колонке с силикагелем, элюируя гексаном:этилацетатом (8:1) с получением 300 мг соединения 35.
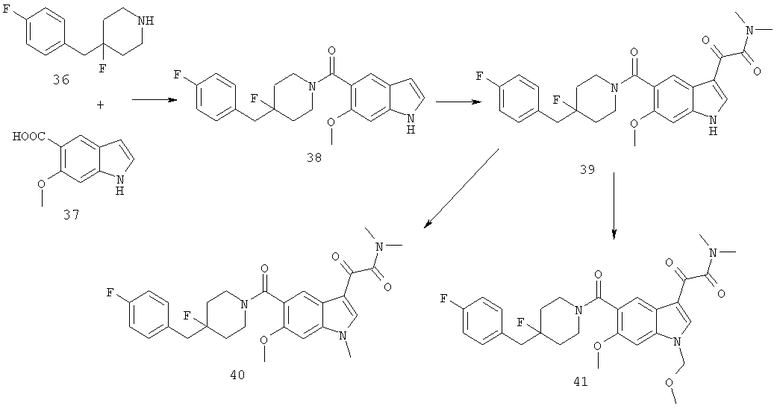
Синтез соединения 36: Смесь соединения 35 (456 мг) и 4 М хлористоводородной кислоты в диоксане (10 мл) перемешивают в течение 4 часов. Реакционную смесь нейтрализуют карбонатом натрия, экстрагируют этилацетатом. Объединенный органический слой промывают насыщенным раствором соли, сушат и концентрируют. Неочищенный продукт используют в следующей реакции без дальнейшей очистки.
Синтез соединения 38: К суспензии индолкарбоновой кислоты 37 (475 мг) в безводном дихлорметане (20 мл) добавляют пиперидин 36 (350 мг, 1,66 ммоль). Смесь перемешивают в течение 10 минут и затем добавляют 1-[3-(диметиламинопропил)1-3-этилкарбодиимид гидрохлорид (475 мг) и диметиламинопиридин (202 мг). Реакционная смесь становится прозрачной, и ее непрерывно перемешивают в течение ночи. Затем реакционную смесь обрабатывают 10% раствором хлористоводородной кислоты и экстрагируют дихлорметаном. Объединенные органические экстракты промывают бикарбонатом натрия, насыщенным раствором соли, сушат и концентрируют. Остаток очищают хроматографией на колонке с силикагелем, элюируя дихлорметаном:этилацетатом (9:1) с получением 300 мг соединения 38 в виде белой пены.
Синтез соединения 39: К суспензии соединения 38 (200 мг) по каплям добавляют оксалилхлорид (0,52 мл, 1,04 ммоль, 2М в дихлорметане) при температуре 0°С. Реакционную смесь перемешивают при температуре 0°С в течение 30 минут, затем нагревают до комнатной температуры и перемешивают в течение 5 часов. Образуется желтая суспензия. Растворитель и избыток оксалилхлорида удаляют при пониженном давлении. Образовавшееся желтое твердое вещество сушат в вакууме и растворяют в дихлорметане, затем охлаждают на ледяной бане. Добавляют диметиламин (1,04 мл, 2,08 ммоль, 2 М в тетрагидрофуране). Через 30 минут реакционную смесь обрабатывают водой и экстрагируют дихлорметаном. Органические экстракты промывают водой, насыщенным раствором соли, сушат и концентрируют. Остаток очищают хроматографией на колонке с силикагелем, элюируя 3% МеОН в дихлорметане с получением 190 мг соединения 39 в виде белого твердого вещества.
Синтез соединения 40: К раствору соединения 39 (75 мг) в тетрагидрофуране (10 мл) по каплям добавляют KHMDS (0,46 мл, 0,23 ммоль, 0,5 М в толуоле) при температуре 0°С. Смесь медленно нагревают до комнатной температуры и перемешивают в течение 30 минут. К реакционной смеси медленно добавляют метилйодид (33 мг, 0,23 ммоль), затем перемешивают в течение 2 часов. Реакционную смесь гасят хлоридом аммония и экстрагируют дихлорметаном. Объединенные органические экстракты промывают насыщенным раствором соли, сушат и концентрируют. Остаток очищают хроматографией на колонке с силикагелем, элюируя метанолом:дихлорметаном (2:98) с получением 50 мг соединения 40.
Синтез соединения 41: К раствору соединения 40 (75 мг) в тетрагидрофуране (10 мл) по каплям добавляют KHMDS (0,46 мл, 0,23 ммоль, 0,5 М в толуоле) при температуре 0°С. Смесь медленно нагревают до комнатной температуры и перемешивают в течение 30 минут. К реакционной смеси медленно добавляют метоксиметилхлорид (18 мг, 0,23 ммоль), затем перемешивают в течение ночи. Реакционную смесь гасят хлоридом аммония и экстрагируют дихлорметаном. Объединенные органические экстракты промывают насыщенным раствором соли, сушат и концентрируют. Остаток очищают хроматографией на колонке с силикагелем, элюируя метанолом:дихлорметаном (2:98) с получением 65 мг соединения 41.
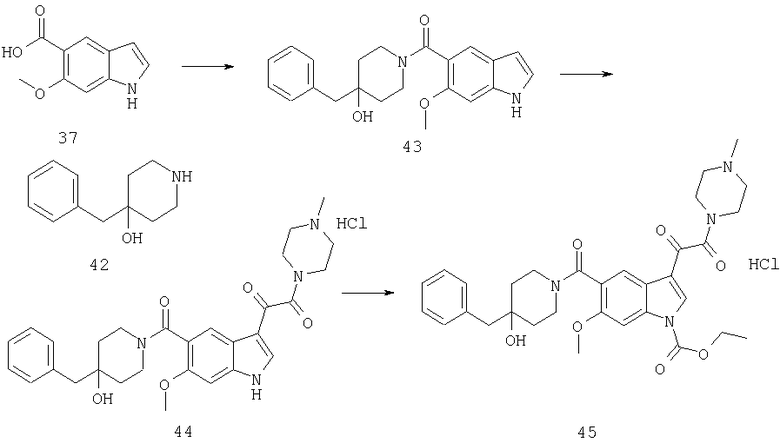
Синтез соединения 43: К суспензии 6-метокси-5-индолкарбоновой кислоты (37) (200 мг) в дихлорметаке (5 мл) одной порцией добавляют гидрохлорид 1-[3-(диметиламинопропил)]-3-этилкарбодиимида (258 мг, 1,35 ммоль). Реакционную смесь перемешивают до растворения всех твердых веществ. Добавляют 4-бензил-4-гидроксипиперидин (42) (258 мг), полученный удалением защитной группы из соединения 34 по методике синтеза соединения 36, и полученную смесь перемешивают при температуре окружающей среды. Реакционная смесь становится мутной. Добавляют каталитическое количество диметиламинопиридина (10 мг) и перемешивают при температуре окружающей среды в течение ночи. Реакционную смесь обрабатывают водой и экстрагируют дихлорметаном. Объединенный органический слой промывают 10% раствором хлористоводородной кислоты, бикарбонатом натрия (насыщенным) и насыщенным раствором соли, затем сушат и концентрируют с получением 300 мг (82%) соединения 43 в виде желтой пены.
Синтез соединения 44: Оксалилхлорид (0,41 мл, 1,65 ммоль, 2 М в дихлорметане) по каплям добавляют к раствору 6-метокси-(4-бензил-4-гидроксипиперидинил)индол-5-карбоксамида (300 мг) в дихлорметане (20 мл) при температуре 0°С. Реакционную смесь перемешивают при температуре 0°С в течение 30 минут и затем медленно нагревают до температуры окружающей среды. Через 2 часа образуется желтый осадок. Реакционную смесь концентрируют при пониженном давлении и сушат в течение 1 часа при пониженном давлении. Полученное желтое твердое вещество суспендируют в дихлорметане (20 мл). При комнатной температуре добавляют метилпиперазин (0,2 мл) и диизопропилэтиламин (0,2 мл). Смесь перемешивают в течение 1 часа, обрабатывают водой и экстрагируют дихлорметаном. Остаток очищают хроматографией на колонке с силикагелем, элюируя дихлорметаном:метанолом (10:1) с получением 180 мг белого твердого вещества. 20 мг полученного твердого вещества растворяют в метаноле (1 мл). К полученному раствору по каплям добавляют насыщенный раствор хлористоводородной кислоты до достижения рН около 3. Затем растворители удаляют, и продукт сушат с получением соединения 44.
Синтез соединения 45: К раствору соединения 44 (65 мг) в безводном тетрагидрофуране (10 мл) по каплям добавляют бис(триметилсилил)амид натрия (0,25 мл, 0,25 ммоль, 1,0 М раствор в тетрагидрофуране) при температуре окружающей среды. Смесь перемешивают при температуре окружающей среды в течение 30 минут и затем к реакционной смеси добавляют этилхлорформиат (0,024 мл, 0,25 ммоль). Через 1 час реакционную смесь гасят хлоридом аммония (насыщенным) и экстрагируют этилацетатом. Объединенный органический слой промывают насыщенным раствором соли, сушат (сульфат магния) и концентрируют при пониженном давлении. Остаток очищают хроматографией на колонке с силикагелем, элюируя дихлорметаном: метанолом (10:1) с получением 15 мг указанного в заголовке соединения 45 в виде белого твердого вещества. Полученное твердое вещество растворяют в метаноле (1 мл) и к полученному раствору по каплям добавляют насыщенный раствор хлористоводородный кислоты в метаноле до тех пор, пока рН не станет около 3. Затем растворитель удаляют, и продукт сушат с получением гидрохлорида соединения 45.
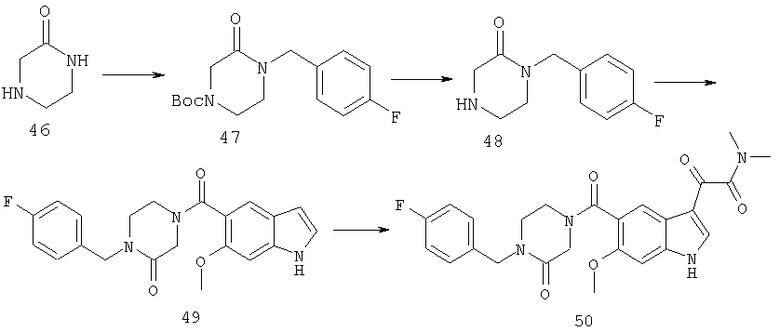
Синтез соединения 47:
СТАДИЯ А: Пиперазинон 46 (5,0 г) растворяют в 15 мл тетрагидрофурана, к полученному раствору добавляют раствор ди-трет-бутилдикарбоната (12,04 г в 50 мл тетрагидрофурана). После перемешивания при комнатной температуре в течение 4 часов реакционную смесь концентрируют досуха на роторном испарителе и полученное твердое вещество используют в следующей реакции без дальнейшей очистки.
СТАДИЯ В: 10 г продукта со стадии А растворяют в безводном ацетоне и реакционную смесь охлаждают на ледяной баке. К полученному раствору добавляют измельченный гидроксид калия (14,02 г). После перемешивания при температуре 0°С в течение 10 минут добавляют 4-фторбензилбромид (23,5 г) и реакционную смесь перемешивают при температуре 0°С в течение 20 минут и при комнатной температуре в течение 1 часа. После удаления растворителя на роторном испарителе остаток помещают в этилацетат и промывают 10% водным карбонатом натрия, насыщенным раствором соли и сушат над безводным сульфатом натрия. Фильтрация и концентрирование дают неочищенный продукт, который очищают хроматографией на колонке с силикагелем, используя этилацетат:гексан, с получением 14 г соединения 47.
Синтез соединения 48: 14 г соединения 47 растворяют в 100 мл метиленхлорида и обрабатывают 100 мл 2 М хлористоводородной кислоты в эфире в течение 2 часов. Растворитель удаляют и полученное твердое вещество промывают простым эфиром и гексаном и сушат в высоком вакууме с получением соединения 48 (11/3 г) в виде соли гидрохлорида.
Синтез соединения 49: 1 г соединения 48 растворяют в 20 мл метиленхлорида и обрабатывают 6-метокси-5-индолкарбоновой кислотой (37) (0,87 г), 1-этил-3-(3-диметиламинопропил)-карбодиимидом (0,95 г), триэтиламином (700 мкл) и диметил-аминопиридином (каталитическим) и реакционную смесь перемешивают в течение ночи. Реакционную смесь концентрируют и остаток помещают в этилацетат, промывают 10% водным карбонатом натрия, насыщенным раствором соли и сушат над безводным сульфатом натрия. Фильтрование и концентрирование дают неочищенный материал, который очищают хроматографией на колонке с силикагелем, используя этилацетат/гексан, с получением соединения 49 (1,6 г).
Синтез соединения 50: 1,6 г соединения 49 растворяют в 25 мл сухого дихлорметана. Реакционную смесь продувают азотом и помещают на ледяную баню. К полученному раствору добавляют 5 мл 2М оксалилхлорида в дихлорметане. Реакционную смесь перемешивают при комнатной температуре в течение 1 часа и затем при комнатной температуре в течение 2 часов. Реакционную смесь концентрируют на роторном испарителе и затем сушат в вакууме в течение 15 минут, остаток (желтое твердое вещество) растворяют в сухом дихлорметане. К полученному остатку добавляют 2М раствор диметиламина в ТГФ (5 мл) и реакционную смесь перемешивают в течение 0,5 часа. Затем реакционную смесь концентрируют, и остаток помещают в этилацетат и промывают 10% водн. карбонатом натрия, насыщенным хлоридом натрия, сушат над безводным сульфатом натрия и фильтруют. Концентрирование дает неочищенный продукт, который подвергают хроматографии на колонке с силикагелем, используя этилацетат/гексан, с получением соединения 50 (1,45 г).
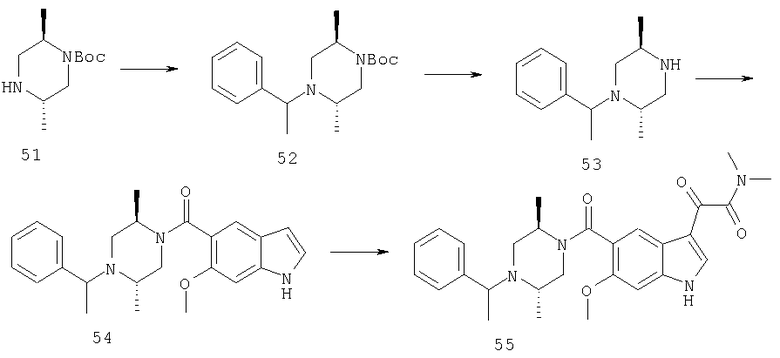
Синтез соединения 52: 2R,5S-трансдиметилпиперазин 51 (5,0 г) растворяют в 15 мл этанола, к полученному раствору добавляют 2-бромэтилбензол (4,4 г). Полученную смесь перемешивают при температуре 40°С в течение 30 минут, охлаждают до комнатной температуры и концентрируют досуха на роторном испарителе. Остаток помещают в этилацетат и промывают 10% водным карбонатов натрия, насыщенным раствором соли и сушат над безводным сульфатом натрия. Фильтрация и концентрирование дает неочищенный материал, который очищают хроматографией на колонке с силикагелем, используя этилацетат/гексан, с получением соединения 52 (5,4 г).
Синтез соединения 53: 5 г соединения 52 растворяют в 60 мл метиленхлорида и обрабатывают 60 мл 2 М хлористоводородной кислоты в эфире в течение 3 часов. Растворитель удаляют, и полученное твердое вещество промывают простым эфиром и гексаном и сушат в высоком вакууме с получением соединения 53 в виде соли гидрохлорида (3,6 г).
Синтез соединения 54: соединение 53 (1,32 г) растворяют в 20 мл метиленхлорида и обрабатывают 6-метокси-5-индолкарбоновой кислотой (37) (0,9 г) в присутствии 1-этил-3-(3-диметиламинопропил) карбодиимида (1,04 г), 800 мкл триэтиламина и каталитического ДМАП, в течение ночи. Реакционную смесь концентрируют и остаток помещают в этилацетат, промывают 10% водным карбонатом натрия, насыщенным раствором соли и сушат над безводным сульфатом натрия. Фильтрование и концентрирование дают неочищенный материал, который очищают хроматографией на колонке с силикагелем, используя этилацетат/гексан, с получением соединения 54 (0,95 г).
Синтез соединения 55: 0,6 г соединения 54 растворяют в 20 мл метиленхлорида. Реакционную смесь продувают азотом и помещают на ледяную баню. К полученному раствору добавляют 2 мл 2М оксалилхлорида в метиленхлориде. Реакционную смесь перемешивают при комнатной температуре в течение 1 часа и затем при комнатной температуре в течение 2 часов. Реакционную смесь концентрируют на роторном испарителе и затем сушат в вакууме в течение 15 минут. Остаток (желтое твердое вещество) растворяют в 15 мл сухого метиленхлорида, к полученному раствору добавляют 2 мл 2М раствора диметиламина в тетрагидрофуране. Через 30 минут реакционную смесь концентрируют и остаток помещают в этилацетат и промывают 10% водн. карбонатом натрия, насыщенным хлоридом натрия, сушат над безводным сульфатом натрия и фильтруют. Концентрирование дает неочищенный продукт, который очищают хроматографией на колонке с силикагелем, используя этилацетат/гексан, с получением соединения 55 (0,78 г).
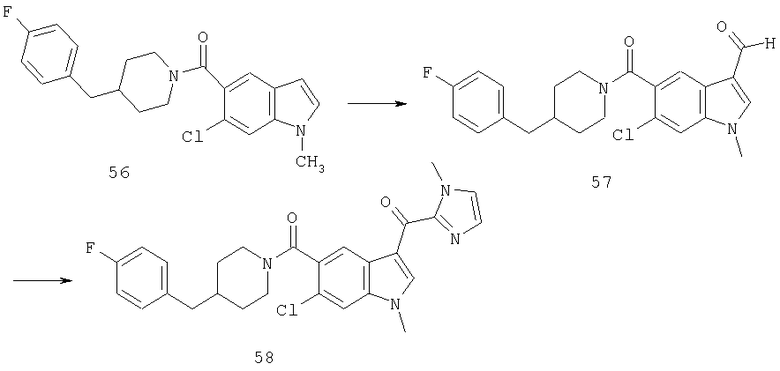
Синтез соединения 57: 7,65 г (19,97 ммоль) индола 56 растворяют в 80 мл безводного диметилформамида и охлаждают на ледяной бане. В инертной атмосфере по каплям добавляют 40 мл (2М раствора в дихлорметане) оксалилхлорида на протяжении 20 минут. После перемешивания в течение 15 минут при температуре 0°С реакционную смесь нагревают до температуры окружающей среды и перемешивание продолжают в течение 1 часа. Реакционную смесь выливают в воду и экстрагируют этилацетатом (3×100 мл). Объединенный экстракт промывают водой, сушат над сульфатом натрия и упаривают. Продукт затем очищают хроматографией на колонке с силикагелем, элюируя этилацетатом:гексаном (30:70) с получением 8 г (97,5%) альдегида 57.
Синтез соединения 58: К раствору 1-метилимидазола (149 мг) в тетрагидрофуране (10 мл) по каплям добавляют н-бутиллитий (1/14 мл, 1,82 ммоль, 1,6 М в гексане) при температуре -78°С. Реакционную смесь перемешивают при температуре -40°С в течение 30 минут. Добавляют соединение 57 (500 мг) в тетрагидрофуране (10 мл). Реакционную смесь нагревают до температуры окружающей среды и перемешивают в течение ночи. Реакционную смесь гасят хлоридом аммония (насыщенным), экстрагируют этилацетатом. Объединенный органический слой промывают насыщенным раствором соли, сушат и концентрируют. Неочищенную смесь кипятят с обратным холодильником с фенилгидразином (0,6 мл) в этаноле (10 мл) для удаления соединения 57, которое не израсходовалось в процессе реакции. После удаления растворителя остаток очищают хроматографией на колонке с силикагелем, элюируя этилацетатом:гексаном (4:1) с получением 120 мг соединения 58.
Используя описанные выше методики, получают соединения из таблиц 2 и 3, которые тестируют для определения способности ингибировать р38-α киназу. Было обнаружено, что соединения в таблицах 2 и 3 имеют значения IC50 для ингибирования р38-α в интервале от 0,1 до 1,5 мкммоль.
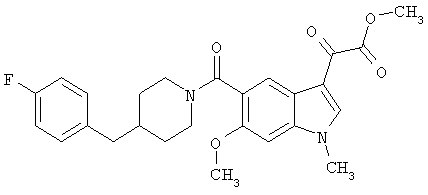
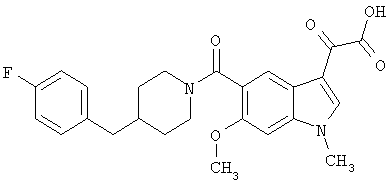
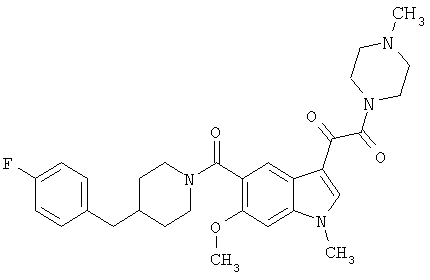
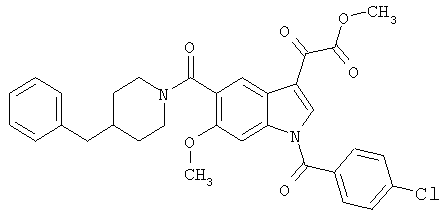
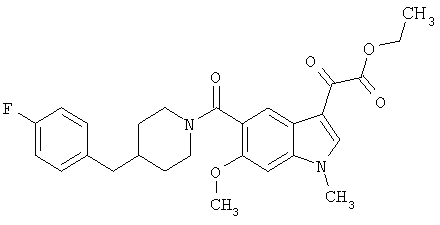
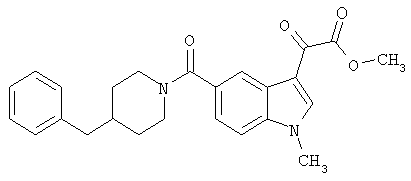
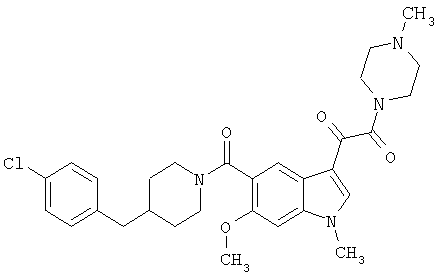
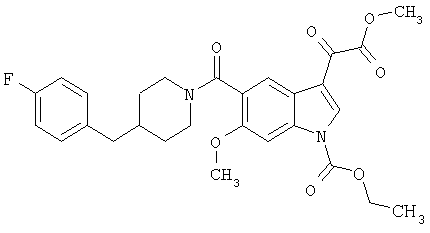
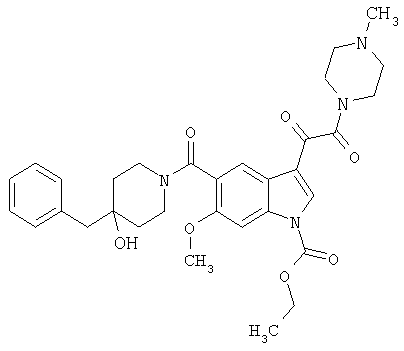
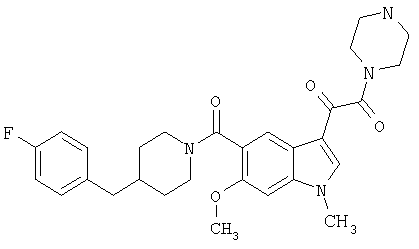
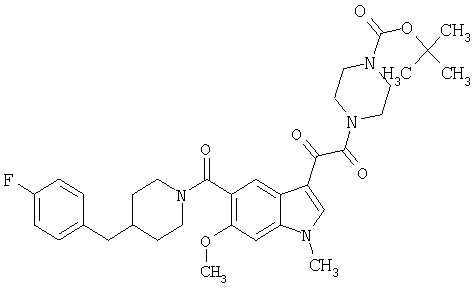
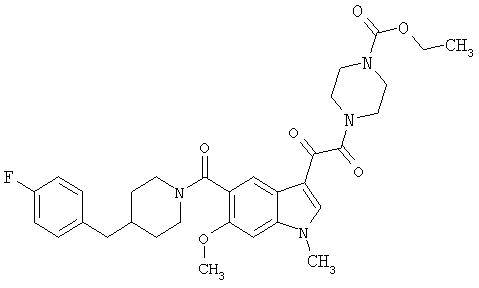
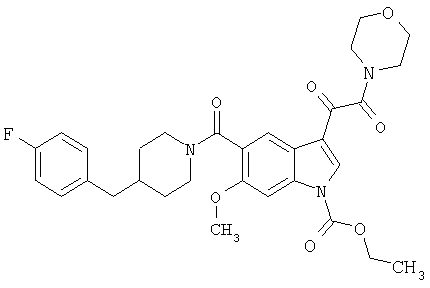

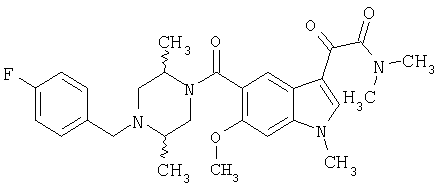

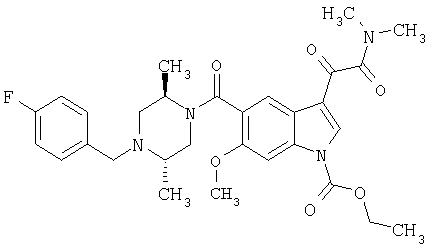
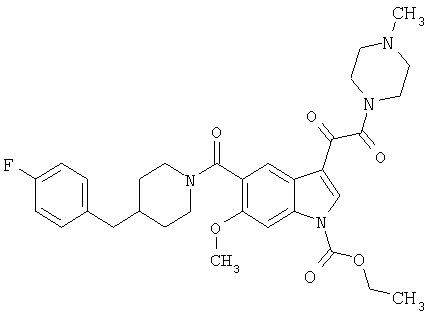
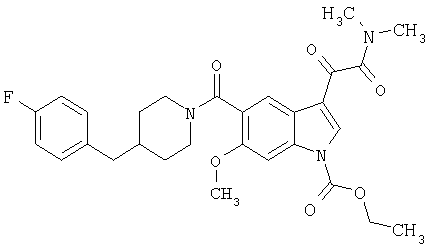
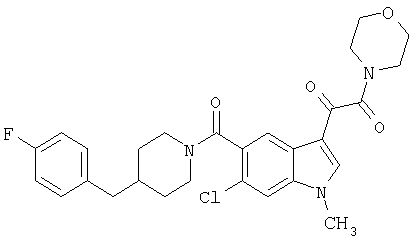
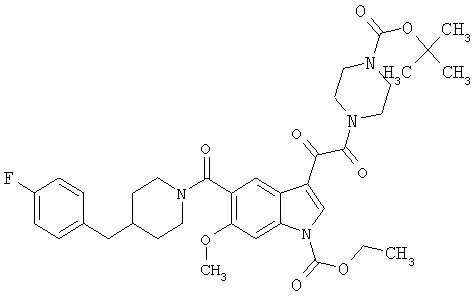
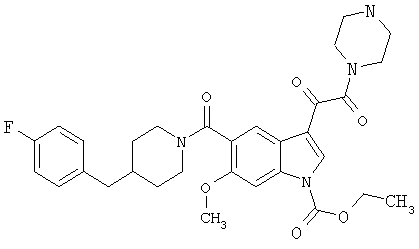
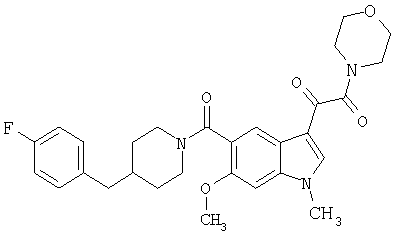
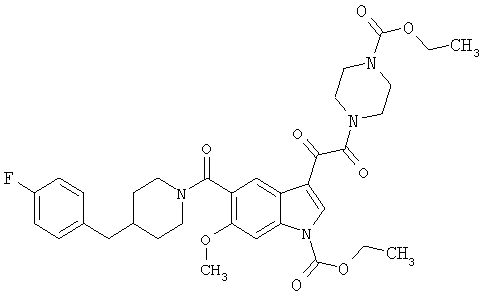
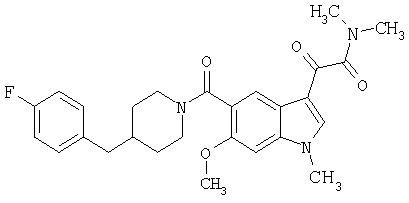
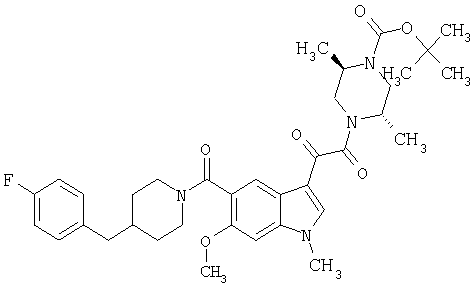
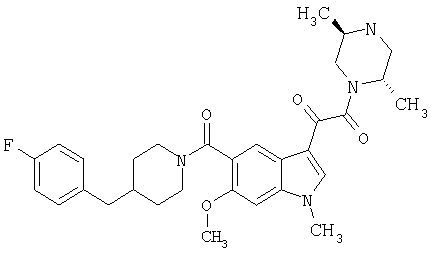
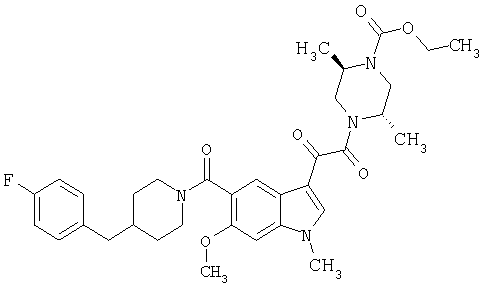

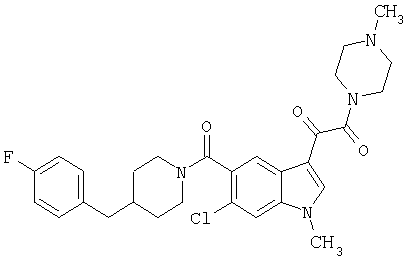
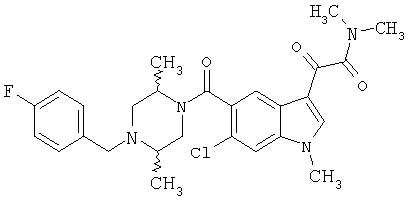
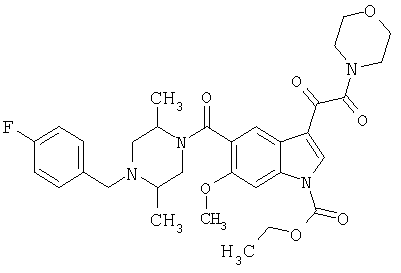
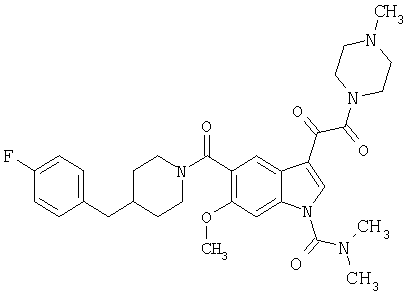
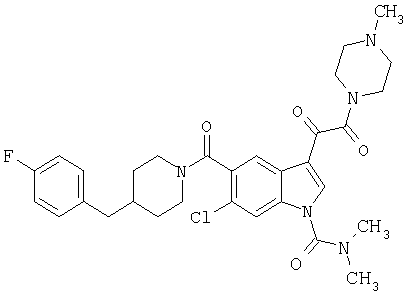
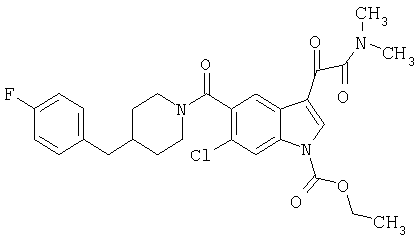
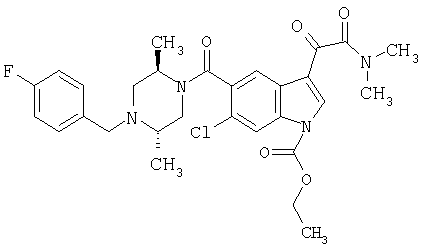
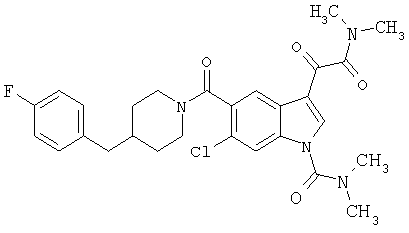
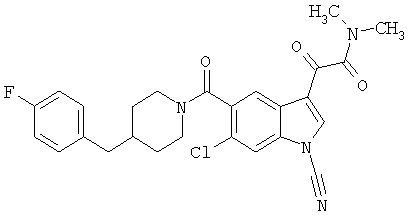
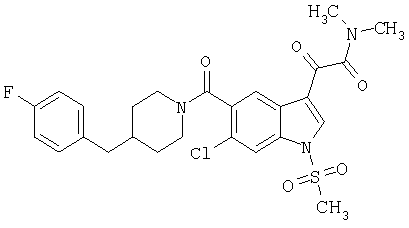
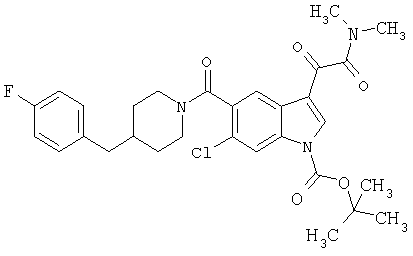
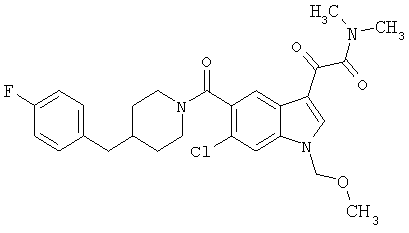
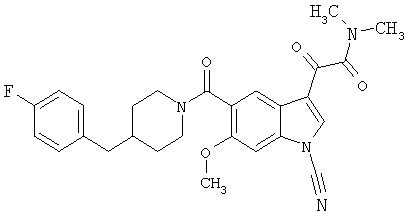
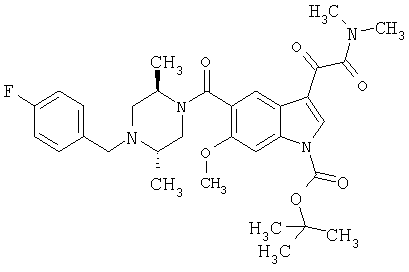
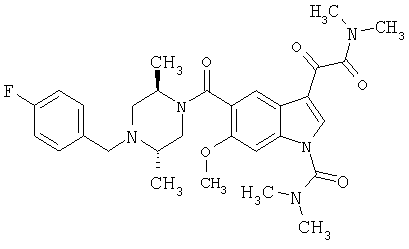
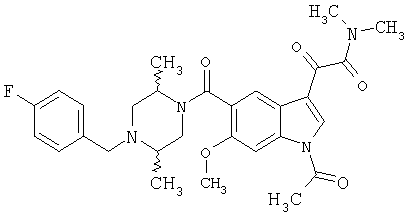
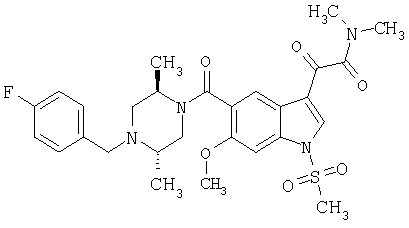
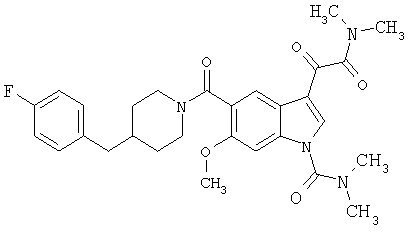

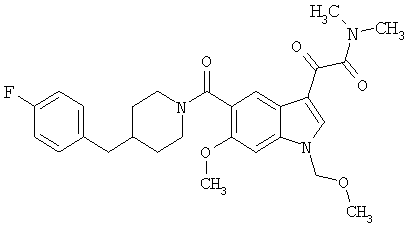
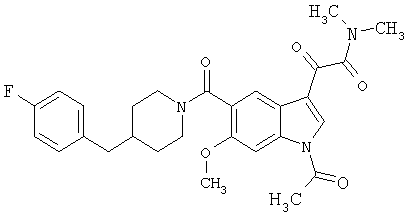
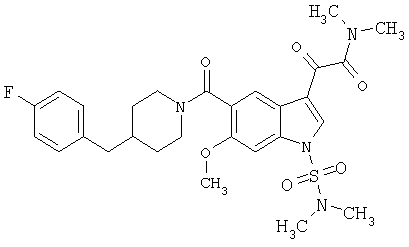
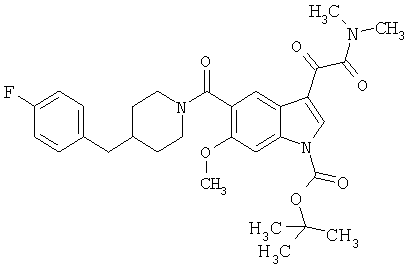
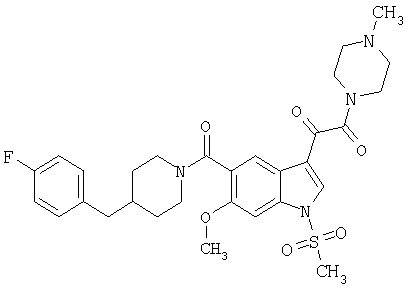
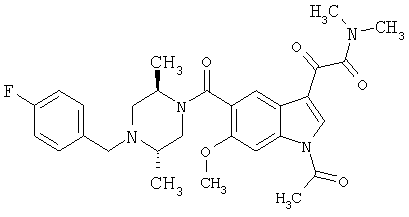
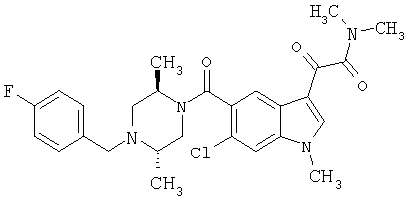
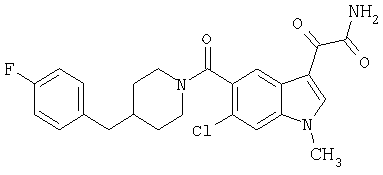
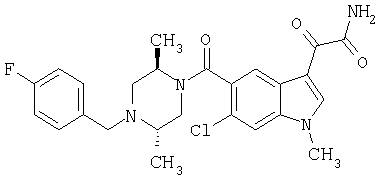
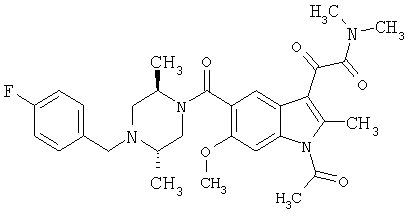
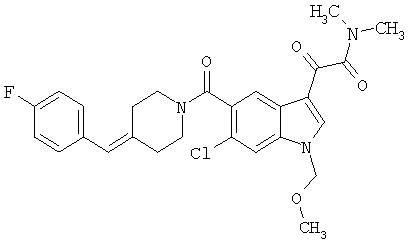
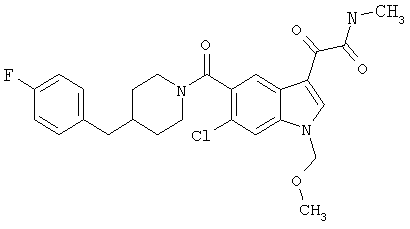
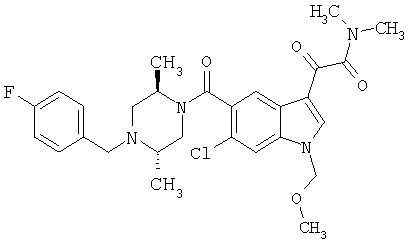
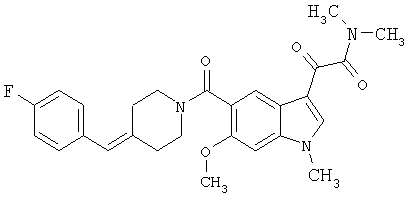
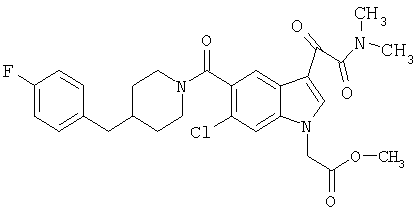
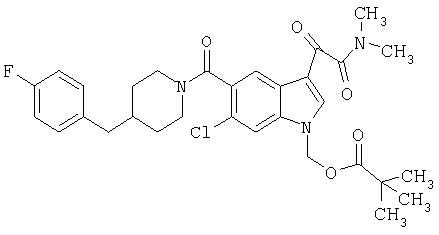
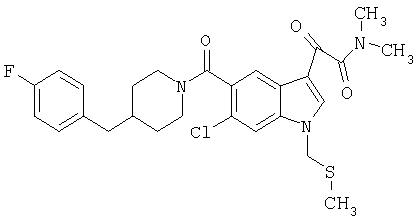
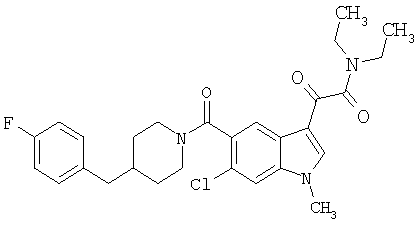
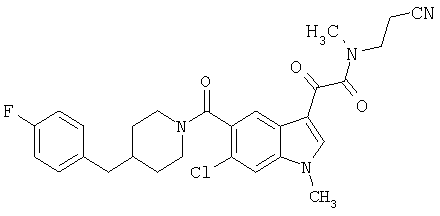
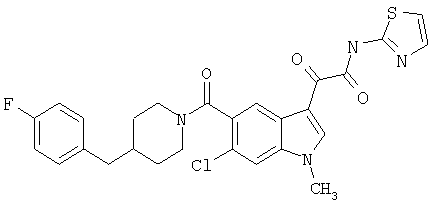
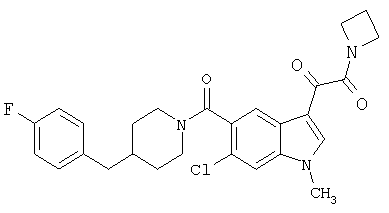
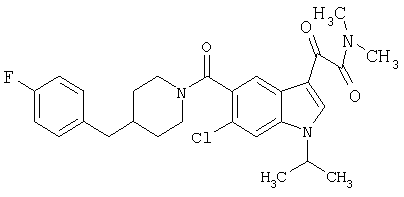
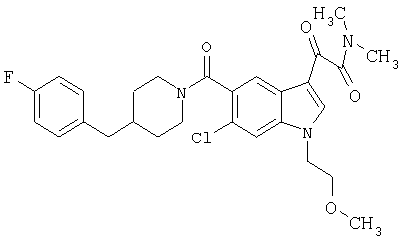
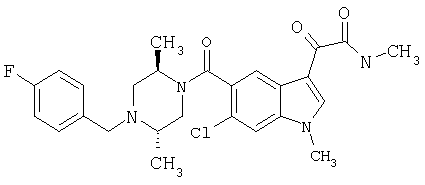
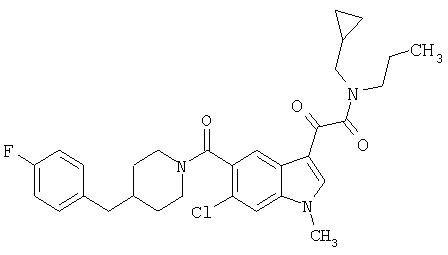
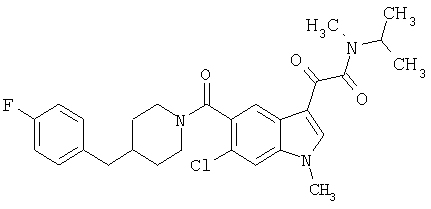
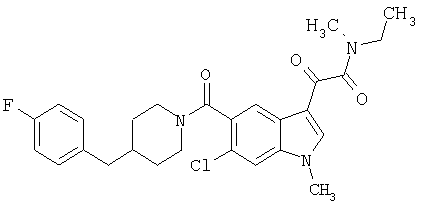
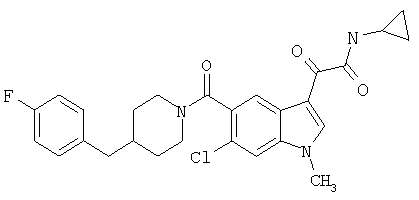
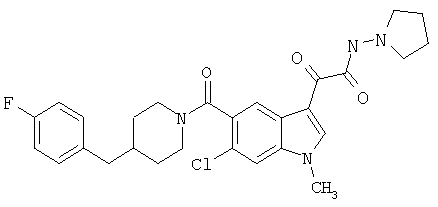
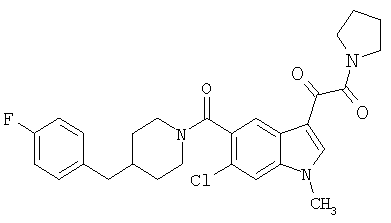
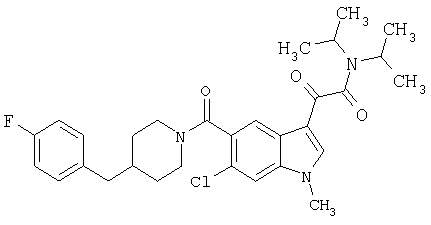
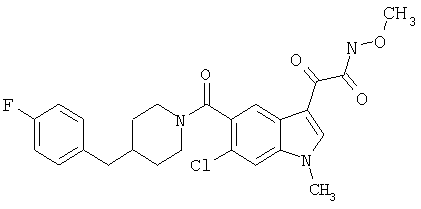
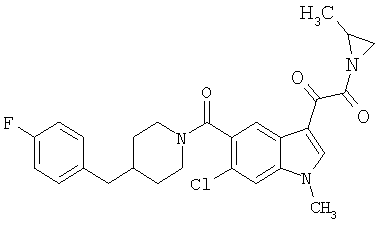
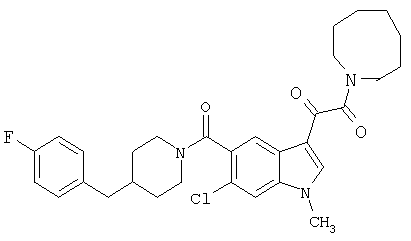
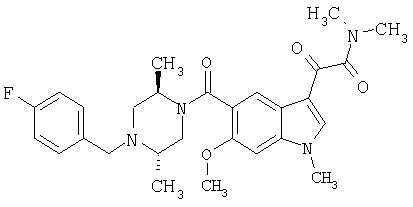
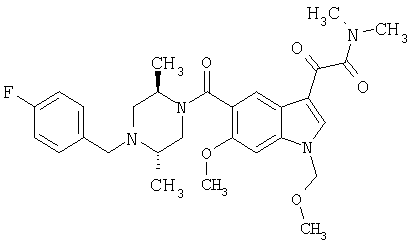
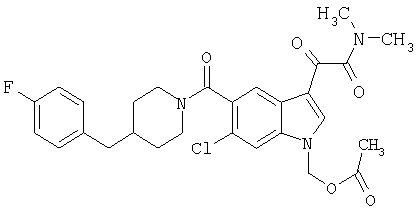
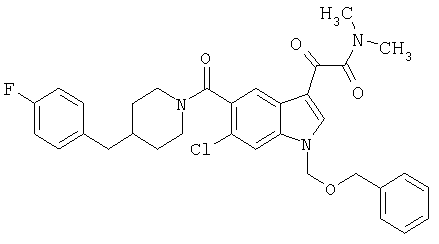
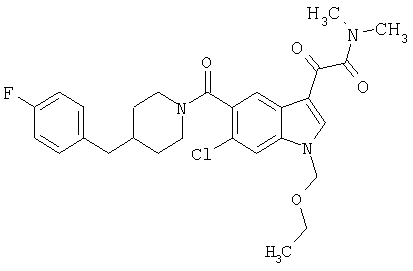
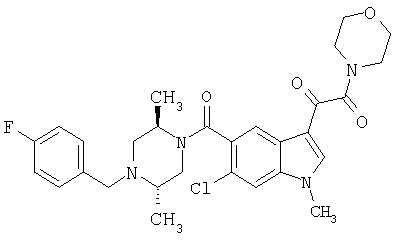
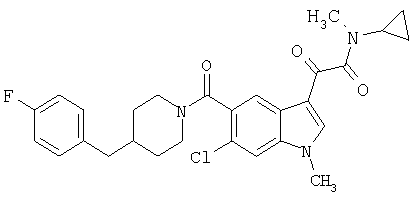
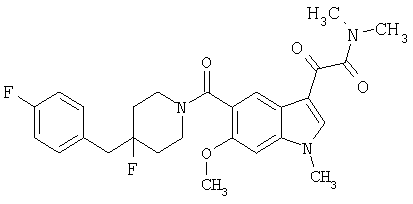
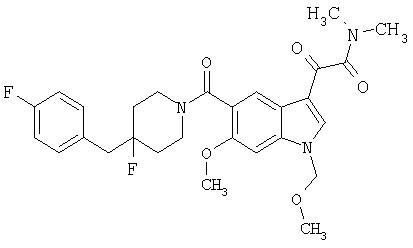
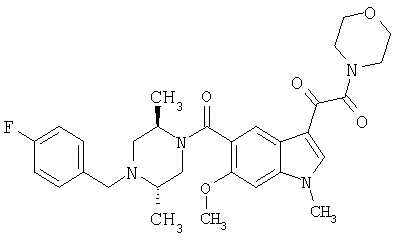
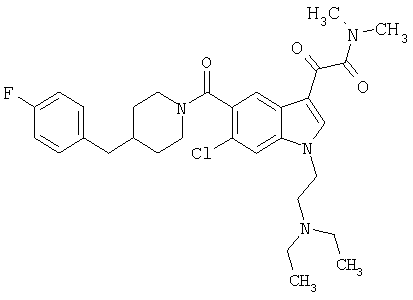
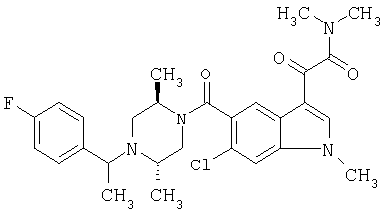
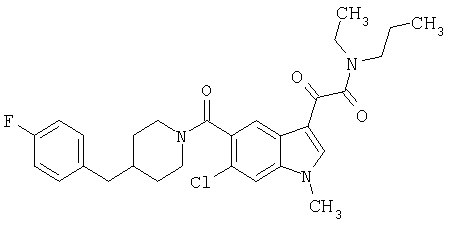
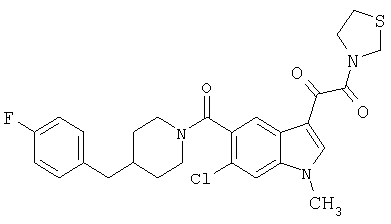
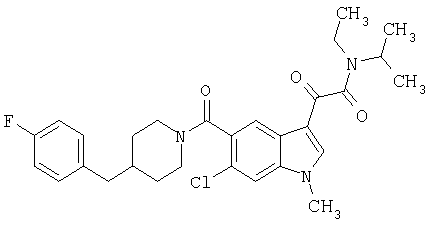
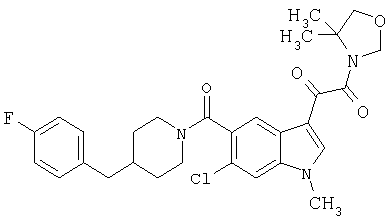
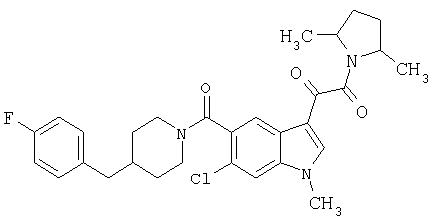
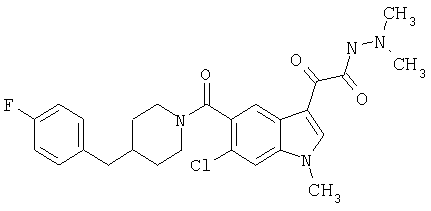
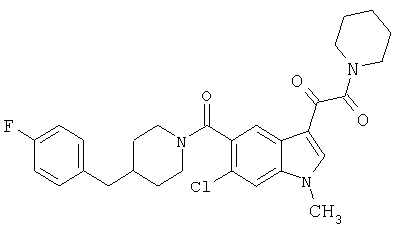
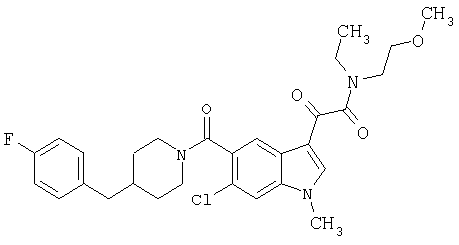
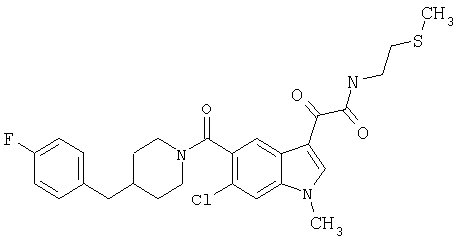
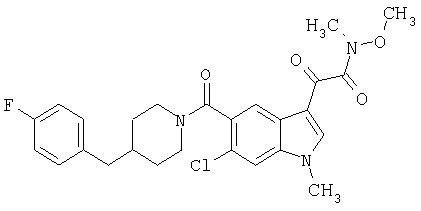
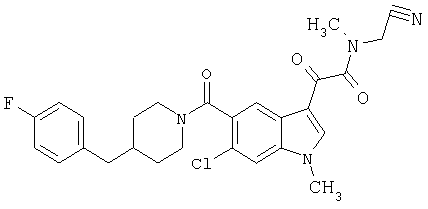
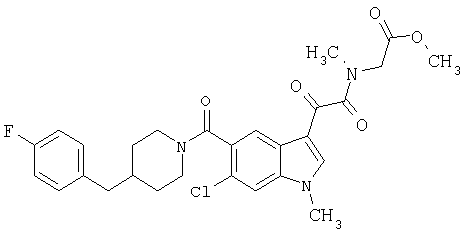
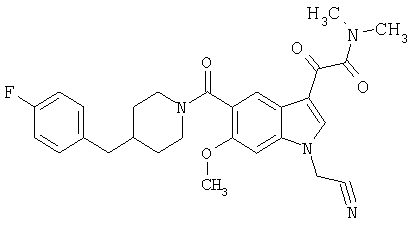
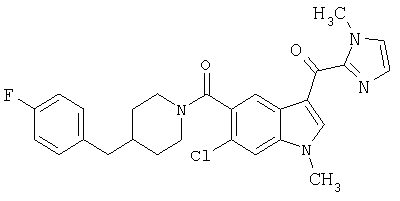
Таблица 3
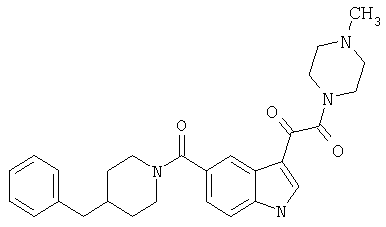
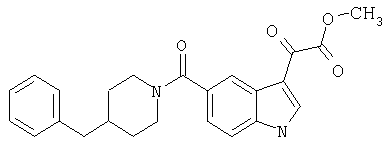
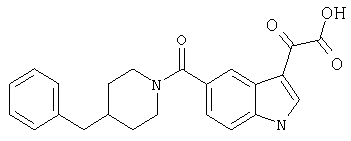
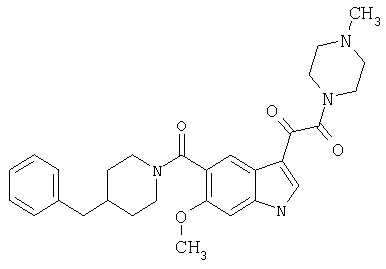
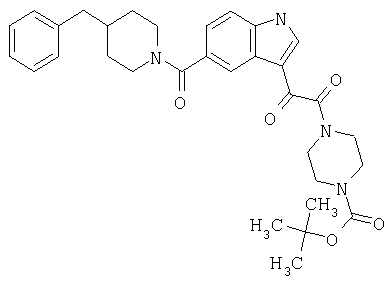
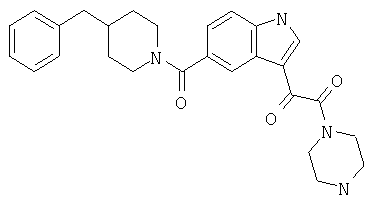

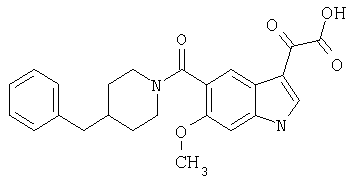
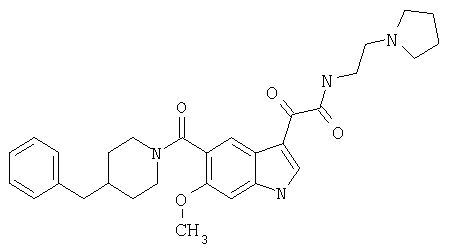
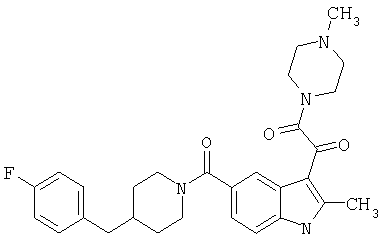
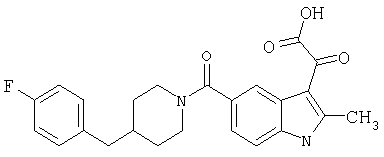
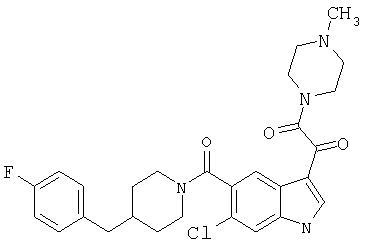
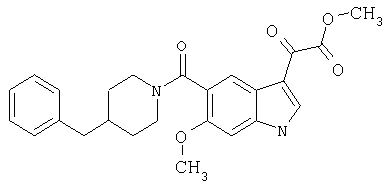
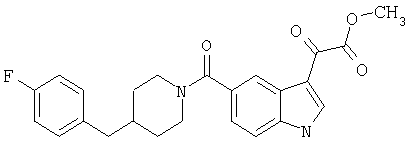
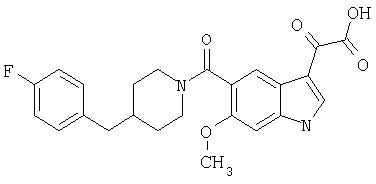
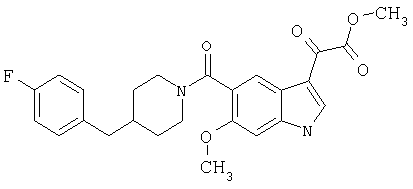
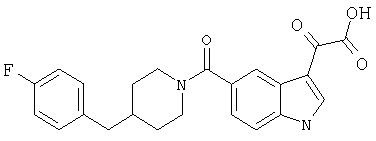
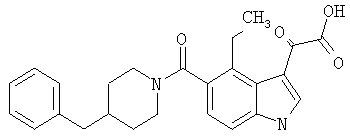
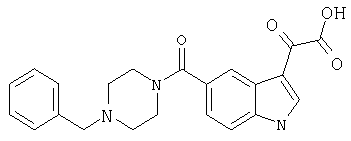
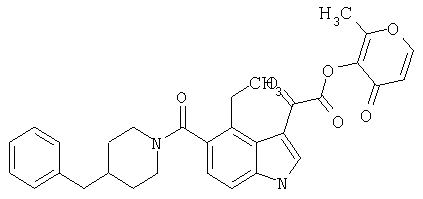
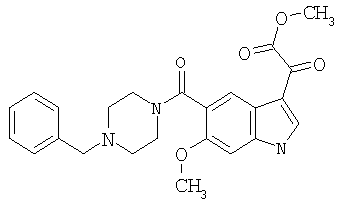
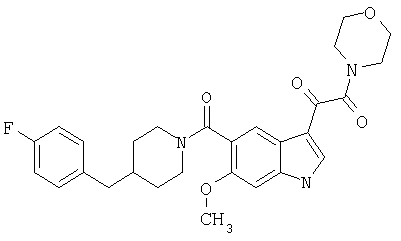
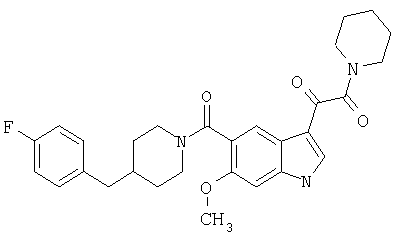
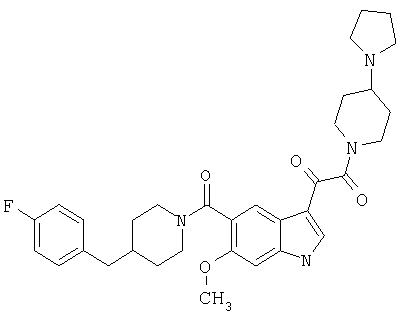
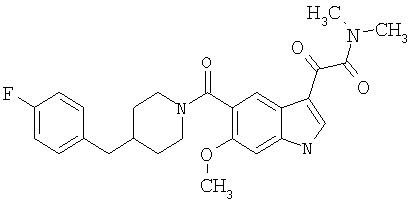
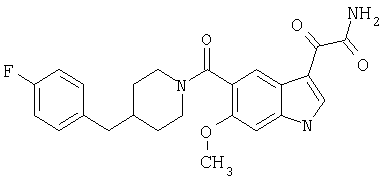
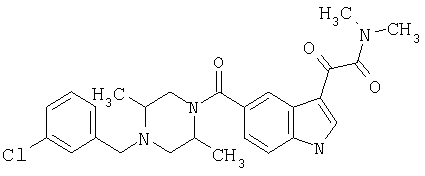
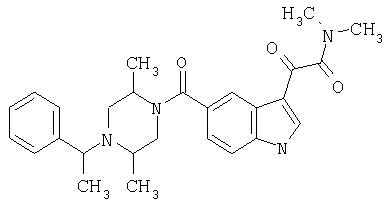
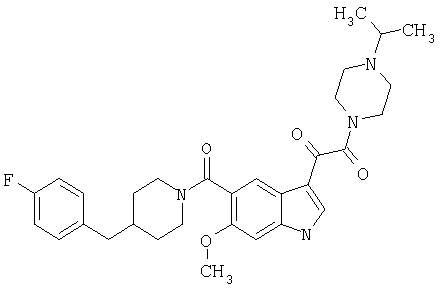
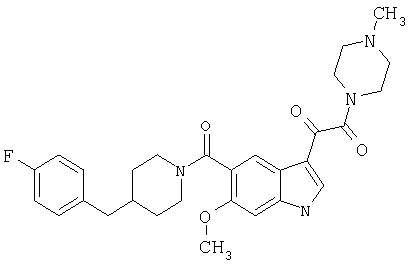
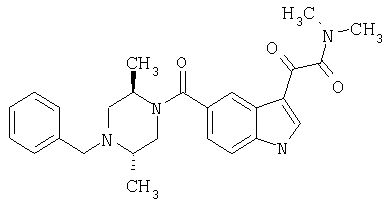
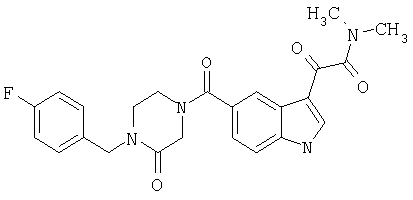
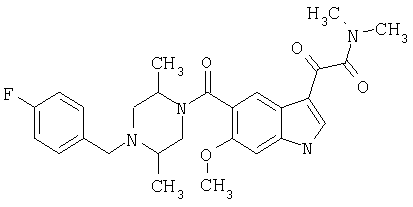
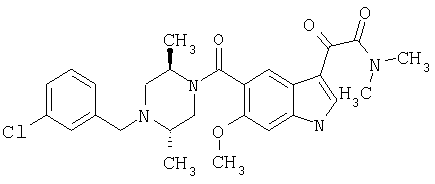
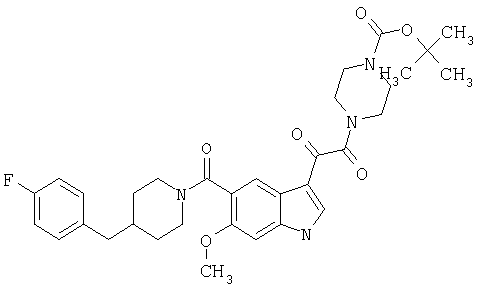
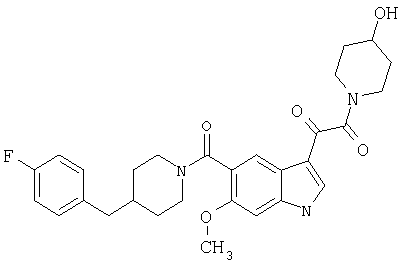
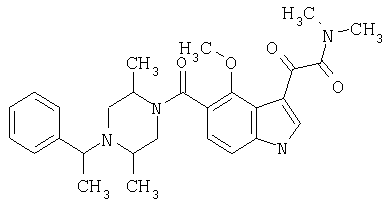
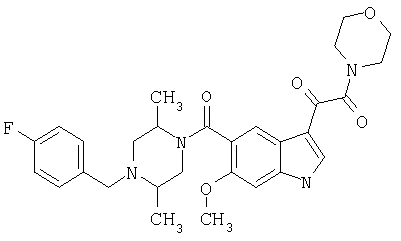
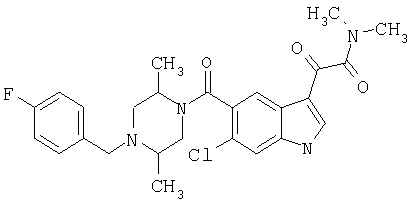
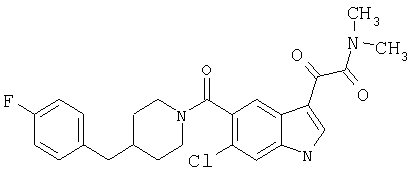
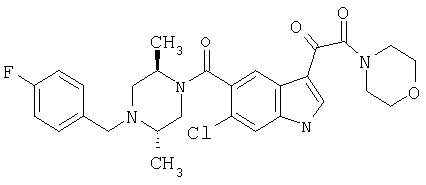
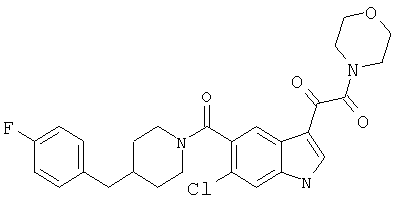
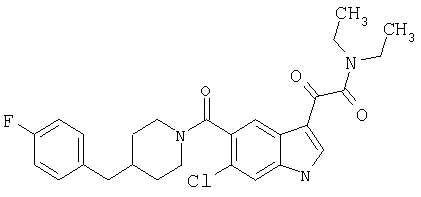
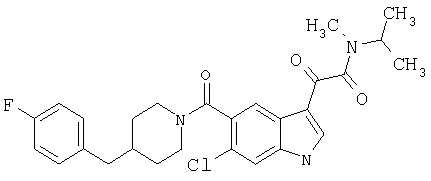
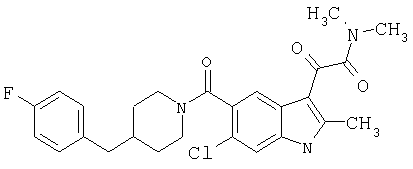
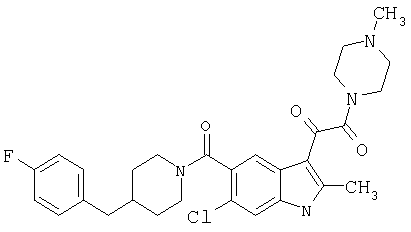
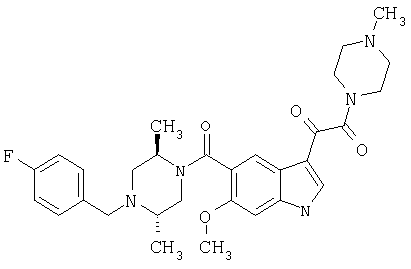
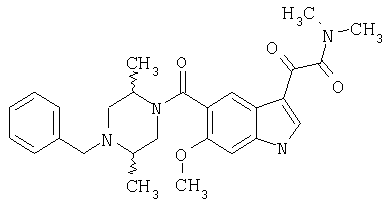
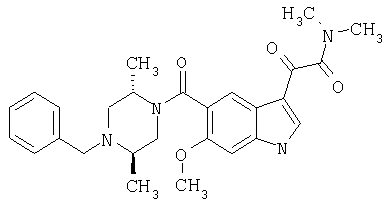
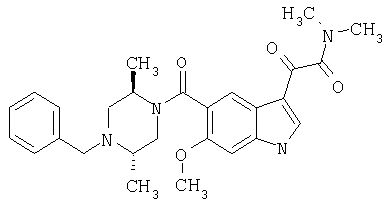
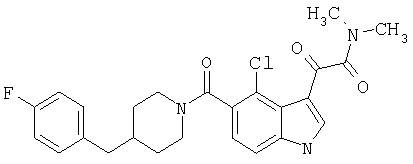
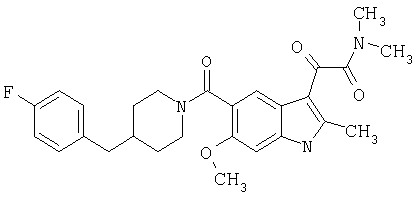
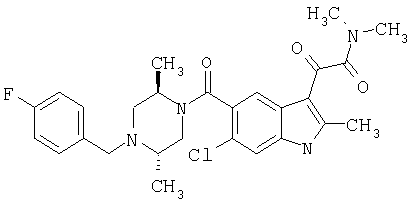
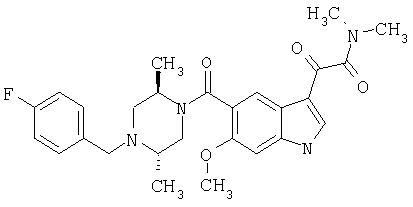
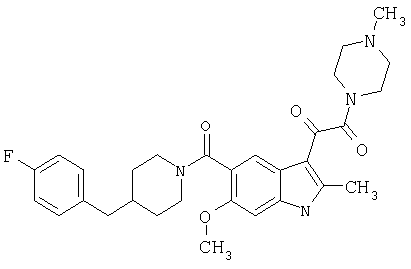
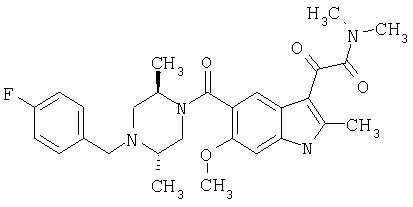

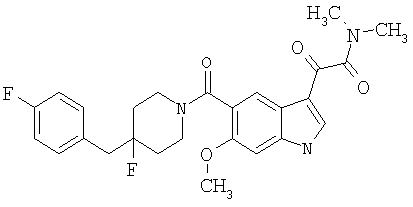
| название | год | авторы | номер документа |
|---|---|---|---|
| ГЕТЕРОЦИКЛИЧЕСКОЕ СОЕДИНЕНИЕ | 1999 |
|
RU2270833C2 |
| АМИДНЫЕ ПРОИЗВОДНЫЕ КАРБОНОВОЙ КИСЛОТЫ, СПОСОБ ИХ ПОЛУЧЕНИЯ И ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ НА ИХ ОСНОВЕ | 2001 |
|
RU2271361C2 |
| СОЕДИНЕНИЯ ПИРРОЛО- И ТИАЗОЛОПИРИДИНА (ВАРИАНТЫ) И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ИХ ОСНОВЕ | 2007 |
|
RU2461557C2 |
| 2-АМИНОБЕНЗОКСАЗОЛКАРБОКСАМИДЫ В КАЧЕСТВЕ МОДУЛЯТОРОВ 5-НТ3 | 2007 |
|
RU2448105C2 |
| АМИДНЫЕ ПРОИЗВОДНЫЕ 2,3,4,5-ТЕТРАГИДРОБЕНЗО [f]-[1,4] ОКСАЗЕПИН-5-КАРБОНОВОЙ КИСЛОТЫ В КАЧЕСТВЕ ИНГИБИТОРОВ ГАММА-СЕКРЕТАЗЫ, ПРЕДНАЗНАЧЕННЫХ ДЛЯ ЛЕЧЕНИЯ БОЛЕЗНИ АЛЬЦГЕЙМЕРА | 2004 |
|
RU2332408C2 |
| НОВЫЕ ПРОИЗВОДНЫЕ ГЕТЕРОЦИКЛИЧЕСКИХ СОЕДИНЕНИЙ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ, СПОСОБ МОДУЛЯЦИИ АУТОИММУННЫХ ЗАБОЛЕВАНИЙ, СПОСОБ ПОЛУЧЕНИЯ ЭТИХ СОЕДИНЕНИЙ | 2000 |
|
RU2255937C2 |
| N-ГЕТЕРОЦИКЛИЧЕСКИЕ ПРОИЗВОДНЫЕ И ИХ ПРИМЕНЕНИЕ В КАЧЕСТВЕ ИНГИБИТОРОВ СИНТАЗЫ ОКСИДА АЗОТА | 1998 |
|
RU2241708C2 |
| ЦИТОТОКСИЧЕСКИЕ И АНТИМИТОТИЧЕСКИЕ СОЕДИНЕНИЯ И СПОСОБЫ ИХ ПРИМЕНЕНИЯ | 2015 |
|
RU2723651C2 |
| ПРОИЗВОДНЫЕ КАРБОНОВЫХ КИСЛОТ (ВАРИАНТЫ), ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ И СПОСОБ СЕЛЕКТИВНОГО ИНГИБИРОВАНИЯ СВЯЗЫВАНИЯ αβ ИНТЕГРИНА У МЛЕКОПИТАЮЩЕГО | 2000 |
|
RU2263109C2 |
| ЗАМЕЩЕННЫЕ ТИОФЕНКАРБОКСАМИДЫ И АНАЛОГИ В КАЧЕСТВЕ АНТИБАКТЕРИАЛЬНЫХ СРЕДСТВ | 2019 |
|
RU2797316C2 |
Изобретение относится к новым производным индола формулы
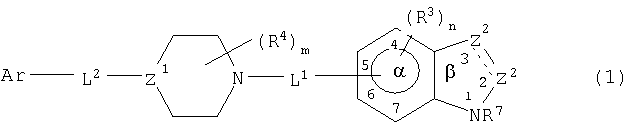
и их фармацевтически приемлемые соли,
где:
 представляет двойную связь;
представляет двойную связь;
Z1 представляет N, CR5, где R5 представляет Н, C1-С6 алкил, ОН, C1-С6 алкокси или галоген; Z2 в положении 2 представляет CR1 и Z2 в положении 3 представляет СА; где каждый R1 независимо означает C1-С6 алкил; А является -Wi-COXjY, где Y является COR2, где R2 является OR, NR2, NRNR2 или NROR, где каждый R независимо представляет Н, C1-С6 алкил, или C5-С6 гетероарил, содержащий в кольце один или два гетероатома, выбранных из N, О и S, каждый из которых необязательно замещен одной или несколькими группами, выбранными из NR'2, OR', COOR', C1-С6 алкила, CN, =O и SR', где каждый R' представляет Н или C1-С6 алкил и где два R или R', присоединенные к одному и тому же атому N, могут образовывать 3-8-членное кольцо, выбранное из группы, включающей пиперазиновое кольцо, морфолиновое кольцо, тиазолидиновое кольцо, оксазолидиновое кольцо, пирролидиновое кольцо, пиперидиновое кольцо, азациклопропановое кольцо, азациклобутановое кольцо и азациклооктановое кольцо; и где указанное кольцо может быть дополнительно замещено C1-С6 алкилом или COO-C1-С6 алкилом, и Х является незамещенным алкиленом (1-6С), или Y является имидазолом, замещенным метилом, i равен 0, j равен 0 или 1; R7 является Н или C1-С6 алкилом, SOR, SO2R, RCO, COOR, C1-С6 алкил-COR, CONR2, SO2NR2, CN, OR, C1-С6 алкил-SR, C1-С6 алкил-OCOR, C1-С6 алкил-COOR, C1-С6 алкил-CN или C1-С6 алкил-CONR2, где каждый R независимо представляет Н, C1-С6 алкил или арил, который необязательно замещен галогеном, C1-C4 алкилом или C1-C4 алкокси, или R7 является метоксиметилом, метоксиэтилом, этоксиметилом, бензилоксиметилом или 2-метоксиэтилоксиметилом; каждый R3 независимо является галогеном, C1-С6 алкилом, OR, SR или NR2, где R представляет Н или C1-С6 алкил; n равен 0-3; L1 является СО; L2 является алкиленом (1-4С), необязательно замещенным одним или двумя группами С1-С4 алкила; каждый R4 независимо выбирают из группы, включающей C1-С6 алкил, галоген, OR, NR2, SR, SOR, SO2R, RCO, COOR, CONR2, SO2NR2, где каждый R независимо представляет Н или C1-С6 алкил, или R4 представляет =O; m равен 0-4; Ar является арильной группой, замещенной от 0 до 5 заместителями, выбранными из группы, включающей C1-С6 алкил, галоген, OR, NR2, SR, SOR, SO2R, RCO, COOR, CONR2 и SO2NR2, где каждый R независимо представляет Н или C1-С6 алкил. Соединения 1 обладают ингибирующей активностью в отношении р38-α киназы, что позволяет использовать их в фармацевтической композиции.
3 н. и 60 з.п. ф-лы, 3 табл.
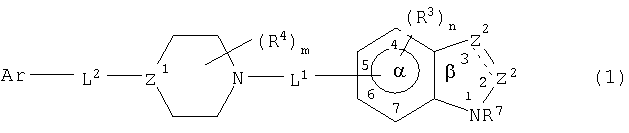
и их фармацевтически приемлемые соли,
где  представляет двойную связь;
представляет двойную связь;
Z1 представляет N или CR5, где R5 представляет собой H, C1-С6 алкил, гидрокси, C1-С6 алкокси или галоген;
Z2 в положении 2 представляет CR1 и Z2 в положении 3 представляет СА,
где каждый R1 независимо является водородом или C1-С6 алкилом;
А является -Wi-COXjY, где Y является COR2, где R2 является OR, NR2, NRNR2 или NROR, где каждый R независимо представляет Н, C1-С6 алкил, или С5-С6 гетероарил, содержащий в кольце один или два гетероатома, выбранных из N, О и S, каждый из которых необязательно замещен одной или несколькими группами, выбранными из NR'2, OR', COOR', C1-С6 алкила, CN, =O и SR', где каждый R' представляет Н или C1-С6 алкил, и где два R или R', присоединенные к одному и тому же атому N, могут образовывать 3-8-членное кольцо, выбранное из группы, включающей пиперазиновое кольцо, морфолиновое кольцо, тиазолидиновое кольцо, оксазолидиновое кольцо, пирролидиновое кольцо, пиперидиновое кольцо, азациклопропановое кольцо, азациклобутановое кольцо и азациклооктановое кольцо; и где указанное кольцо может быть дополнительно замещено C1-С6 алкилом или COO-C1-С6 алкилом, и Х является незамещенным алкиленом (1-6С), или Y является имидазолом, замещенным метилом, i равен 0, j равен 0 или 1;
R7 является Н или C1-С6 алкилом, SOR, SO2R, RCO, COOR, C1-С6 алкил-COR, CONR2, SO2NR2, CN, OR, C1-С6 алкил-SR, C1-С6 алкил-OCOR, C1-С6 алкил-COOR, C1-С6 алкил-CN или C1-С6 алкил-CONR2, где каждый R независимо представляет Н, C1-С6 алкил или арил, который необязательно замещен галогеном, С1-С4 алкилом или C1-C4 алкокси, или R7 является метоксиметилом, метоксиэтилом, этоксиметилом, бензилоксиметилом или 2-метоксиэтилоксиметилом;
каждый R3 независимо является галогеном, C1-С6 алкилом, OR, SR или NR2, где R представляет Н или C1-С6 алкил;
n равен 0-3;
L1 является СО;
L2 является алкиленом (1-4С), необязательно замещенным одним или двумя группами C1-C4 алкила;
каждый R4 независимо выбирают из группы, включающей C1-С6 алкил, галоген, OR, NR2, SR, SOR, SO2R, RCO, COOR, CONR2, SO2NR2, где каждый R независимо представляет Н или C1-С6 алкил,
или R4 представляет =O;
m равен 0-4;
Ar является арильной группой, замещенной от 0 до 5 заместителями, выбранными из группы, включающей C1-С6 алкил, галоген, OR, NR2, SR, SOR, SO2R, RCO, COOR, CONR2 и SO2NR2, где каждый R независимо представляет Н или C1-С6 алкил.
метоксиоксалилиндол-1-карбоновой кислоты этиловый эфир;
и их фармацевтически приемлемые соли.
6-метокси-[4'-фтор-(4-бензилпиперидинил)]индол-5-карбоксамид-3-глиоксиловой кислоты N,N-диметиламид (соединение 144);
1-метил-6-хлор-[4'-фтор-(4-бензилпиперидинил)]индол-5-карбоксамид-3-глиоксиловой кислоты N,N-диметиламид (соединение 58);
1-метил-6-метокси-[4'-фтор-(4-бензилпиперидинил)]индол-5-карбоксамид-3-глиоксиловой кислоты N,N-диметиламид (соединение 25);
1-метил-6-хлор-[4'-фтор-(4-бензилпиперидинил)]индол-5-карбоксамид-3-глиоксиловой кислоты морфолинамид (соединение 20);
1-метил-6-метокси-[4'-фтор-(4-бензилпиперидинил)]индол-5-карбоксамид-3-глиоксиловой кислоты морфолинамид (соединение 23);
6-метокси-[4'-фтор-(4-бензилпиперидинил)]индол-5-карбоксамид-3-глиоксиловой кислоты морфолинамид (соединение 141);
6-хлор-[4'-фтор-(4-бензилпиперидинил)]индол-5-карбоксамид-3-глиоксиловой кислоты морфолинамид (соединение 165);
6-метокси-[4'-фтор-(4-бензилпиперидинил)]индол-5-карбоксамид-3-амид глиоксиловой кислоты (соединение 145);
6-хлор-[4'-фтор-(4-бензилпиперидинил)]индол-5-карбоксамид-3-глиоксиловой кислоты N,N-диметиламид (соединение 163),
и их фармацевтически приемлемые соли.
1-метил-6-метокси-[4'-фтор-(4-бензил-2,5-диметилпиперазин)]индол-5-карбоксамид-3-глиоксиловой кислоты N,N-диметиламид (соединение 15);
1-метил-6-хлор-[4'-фтор-(4-бензил-2,5-диметилпиперазин)]индол-5-карбоксамид-3-глиоксиловой кислоты N,N-диметиламид (соединение 33);
1-метил-6-хлор-[4'-фтор-(4-бензил-2R,5S-диметилпиперазин)]индол-5-карбоксамид-3-глиоксиловой кислоты N,N-диметиламид (соединение 57);
1-метил-6-хлор-[4'-фтор-(4-бензил-2R,5S-диметилпиперазин)]индол-5-карбоксамид-3-амид глиоксиловой кислоты (соединение 59);
1-метил-6-хлор-[4'-фтор-(4-бензил-2R,5S-диметилпиперазин)]индол-5-карбоксамид-3-глиоксиловой кислоты N-метиламид (соединение 77);
1-метил-6-метокси-[4'-фтор-(4-бензил-2R,5S-диметилпиперазин)]индол-5-карбоксамид-3-глиоксиловой кислоты N,N-диметиламид (соединение 89);
1-метил-6-хлор-[4'-фтор-(4-бензил-2R,5S-диметилпиперазин)]индол-5-карбоксамид-3-глиоксиловой кислоты морфолинамид (соединение 96);
1-метил-6-метокси-[4'-фтор-(4-бензил-2R,5S-диметилпиперазин)]индол-
5-карбоксамид-3-глиоксиловой кислоты морфолинамид (соединение 100),
и их фармацевтически приемлемые соли.
и их фармацевтически приемлемые соли,
где
представляет двойную связь;
Z1 представляет N, СН, СОН или CF;
Z2 в положении 2 является CR, где R является Н или алкилом (С1-6);
Z2 в положении 3 является C-C(=O)-C(=O)-OR или C-C(=O)-C(=O)-NR2, где каждый R независимо является Н или алкилом(С1-6), или где NR2 представляет морфолиновое, пиперидиновое или пиперазиновое кольцо, каждое из которых может быть замещено вплоть до 3 группами, выбранными из R' и -OR', где R' является алкилом (С1-6);
R7 является Н или является C1-С6 алкилом, SO2R, RCO, COOR, C1-С6алкил-COR, CONR2, SO2NR2, CN, CF3, NR2, OR, C1-C6 алкил-SR, C1-С6 алкил-OR, C1-С6 алкил-COOR или C1-С6 алкил-CN, где каждый R независимо является Н или C1-С6 алкилом, или R7 является метоксиметилом, метоксиэтилом, этоксиметилом;
каждый R3 независимо является галогеном, C1-С6 алкилом, OR, SR или N(R)2, где R является Н или C1-С6 алкилом;
n равен 0-3;
m равен 0-4;
L1 является СО;
L2 является алкиленом (1-4С), необязательно замещенным одной или двумя группами C1-C4 алкила;
каждый R4 независимо выбран из группы, включающей C1-С6 алкил, галоген, OR, SR, SOR, SO2R, RCO, COOR, CONR2 и SO2NR2,
где каждый R независимо является Н или C1-С6 алкилом;
Ar является арильной группой, замещенной 0-5 заместителями, выбранными из группы, включающей C1-C6 алкил, галоген, OR, SR, SOR, SO2R, RCO, COOR, CONR2 и SO2NR2, где каждый R независимо является Н или C1-С6 алкилом.
и его фармацевтически приемлемые соли,
где представляет двойную связь;
Z1 представляет N или CR5, где R5 представляет собой Н, C1-С6 алкил, гидрокси, C1-С6 алкокси или галоген;
Z2 в положении 2 представляет CR1 и Z2 в положении 3 представляет СА;
где каждый R1 независимо является водородом или C1-С6 алкилом;
А является -Wi-COXjY, где Y является COR2,
где R2 является OR, NR2, NRNR2 или NROR, где каждый R независимо представляет Н, C1-С6 алкил, или С5-С6 гетероарил, содержащий в кольце один или два гетероатома, выбранных из N, О и S, каждый из которых необязательно замещен одной или несколькими группами, выбранными из NR'2, OR', COOR', C1-С6 алкила, CN, =O и SR', где каждый R' представляет Н или C1-С6 алкил, и где два R или R', присоединенные к одному и тому же атому N, могут образовывать 3-8-членное кольцо, выбранное из группы, включающей пиперазиновое кольцо, морфолиновое кольцо, тиазолидиновое кольцо, оксазолидиновое кольцо, пирролидиновое кольцо, пиперидиновое кольцо, азациклопропановое кольцо, азациклобутановое кольцо и азациклооктановое кольцо; и где указанное кольцо может быть дополнительно замещено C1-С6 алкилом или COO-C1-C6 алкилом, и Х является незамещенным алкиленом (1-6С), или Y является имидазолом, замещенным метилом, i равен 0, j равен 0 или 1;
R7 является Н или C1-С6 алкилом, SOR, SO2R, RCO, COOR, C1-С6 алкил-COR, CONR2, SO2NR2, CN, OR, C1-С6 алкил-SR, C1-С6 алкил-OCOR, C1-С6 алкил-COOR, C1-С6 алкил-CN или C1-С6 алкил-CONR2, где каждый R независимо представляет Н, C1-С6 алкил или арил, который необязательно замещен галогеном, C1-C4 алкилом, или C1-C4 алкокси, или R7 является метоксиметилом, метоксиэтилом, этоксиметилом, бензилоксиметилом или 2-метоксиэтилоксиметилом;
каждый R3 независимо является галогеном, C1-С6 алкилом, OR, SR или NR2, где R представляет Н или C1-С6 алкил;
n равен 0-3;
L1 является СО;
L2 является алкиленом (1-4С), необязательно замещенным одним или двумя группами С1-С4 алкила;
каждый R4 независимо выбирают из группы, включающей C1-С6 алкил, галоген, OR, NR2, SR, SOR, SO2R, RCO, COOR, CONR2, SO2NR2, где каждый R независимо представляет Н или C1-С6 алкил, или R4 представляет =O;
m равен 0-4;
Ar является арильной группой, замещенной от 0 до 5 заместителями, выбранными из группы, включающей C1-С6 алкил, галоген, OR, NR2, SR, SOR, SO2R, RCO, COOR, CONR2 и SO2NR2, где каждый R независимо представляет Н или C1-С6 алкил.
Приоритет по пунктам и признакам:
| Прибор, замыкающий сигнальную цепь при повышении температуры | 1918 |
|
SU99A1 |
| Дорожная спиртовая кухня | 1918 |
|
SU98A1 |
| Приспособление в пере для письма с целью увеличения на нем запаса чернил и уменьшения скорости их высыхания | 1917 |
|
SU96A1 |
| Дорожная спиртовая кухня | 1918 |
|
SU98A1 |
| ПРОИЗВОДНЫЕ ПИПЕРИДИНА И ПИПЕРАЗИНА, СПОСОБ ИХ ПОЛУЧЕНИЯ, СОДЕРЖАЩИЕ ИХ ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ И СПОСОБ ИХ ПОЛУЧЕНИЯ | 1994 |
|
RU2132848C1 |

