Каберголин является производным эрголина, взаимодействующим с рецепторами допамина D2 и наделенным различными видами полезной фармацевтической активности, и используется при лечении гипер-пролактинемии, расстройств центральной нервной системы (ЦНС) и других подобных заболеваний.
Каберголин является родовым названием 1((6-аллилэрголин-8β-ил)-карбонил)-1-(3-диметиламинопропил)-3-этилмочевины, описанным и заявленным в US 4526892. Синтез молекулы каберголина описан также в Eur. J. Med. Chem., 24, 421, (1989) и в GB-2103603-B.
Форма I, подобно каберголину, проявляет значительный ингибирующий эффект в отношении пролактина и имеет терапевтические свойства, позволяющие лечить пациентов, имеющих патологические состояния, связанные с анормальным уровнем пролактина, и поэтому полезна при лечении человека и/или в ветеринарии. Каберголин также эффективен, отдельно или в комбинации, при лечении возвратной обструкции дыхательных путей, для регулирования внутриглазного давления и для лечения глаукомы. Каберголин используется также в области ветеринарии в качестве антипролактинового средства и для резкого сокращения пролиферации у позвоночных животных. Некоторые применения каберголина описаны, например, в WO 99/48484, WO 99/36095, US 5705510, WO 95/05176, EP 040325. Форма I каберголина в особенности полезна при лечении болезни Паркинсона (ПБ), синдрома усталых ног (УНС), лечении заболеваний, подобных прогрессирующему супрануклеарному параличу (ПСП) и мультисистемной атрофии (МСА).
Кристаллическая форма I каберголина, безводная несольватированная форма каберголина, была впервые получена путем кристаллизации из диэтилового эфира, как описано в Il Farmaco, 50 (3), 175-178 (1995).
Другой способ получения кристаллической формы I каберголина через сольватированную толуолом форму V был описан в WO 01/70740. Выход продукта в этом способе обычно составляет около 60%. В целях снижения стоимости материала в высокой степени желательно улучшить выход продукта промышленного получения кристаллической формы I каберголина и облегчить контроль профиля десольватации для формы V в процессе крупномасштабного производства. Поэтому предметом настоящего изобретения является получение высокочистой формы I каберголина с использованием системы органических растворителей, которая ранее никогда не использовалась. Эффективное получение высокочистого каберголина в кристаллической форме I с выходом продукта, превышающим 90%, обладает преимуществами в отношении промышленных затрат и экологии. Кроме того, был обнаружен особый, уникальный и желательный способ десольватирования полученной формы V, позволяющий выделить форму I.
Настоящее изобретение относится к новому способу получения кристаллической формы I каберголина.
Способ по настоящему изобретению включает получение формы V с использованием гептана в качестве осаждающего растворителя и его исключительное превращение в кристаллическую форму I каберголина. Настоящий способ кристаллизации из системы растворителей толуол-гептан для формы V включает "обратное добавление" толуол-каберголинового концентрата к холодному гептану.
Согласно второму аспекту, изобретение относится к новому способу получения сольватированной чистой кристаллической формы V каберголина посредством конверсии фаз исходного аморфного осадка в форму V под кинетическим контролем и, согласно третьему аспекту, изобретение относится к способу получения чистой кристаллической формы I каберголина из сольватированной кристаллической формы V каберголина, который включает использование гептана в качестве приемлемого растворителя для промывания формы V перед десольватированием в печи.
Краткое описание чертежей
ФИГ.1 - порошковая рентгенограмма (ДРЛ), имеющая пики, характерные для кристаллической сольватированной формы V каберголина, полученной в соответствии с Примером 1.
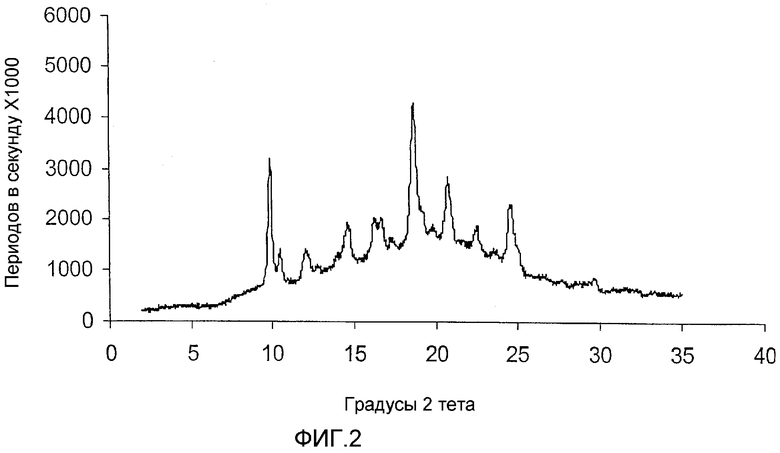
ФИГ.2 - порошковая рентгенограмма (ДРЛ), имеющая пики, характерные для кристаллической формы I каберголина, в соответствии с Примером 2.
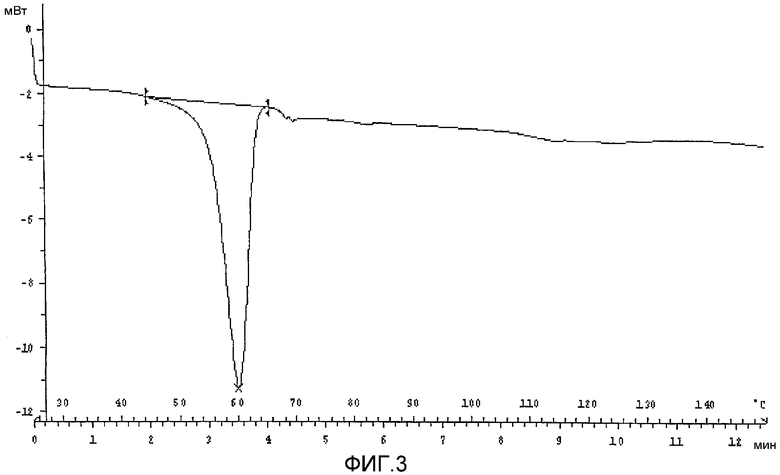
ФИГ.3 - профиль дифференциального сканирующего калориметра (ДСК) формы V, имеющий тепловое событие, связанное с эвтектическим плавлением каберголина с толуолом.
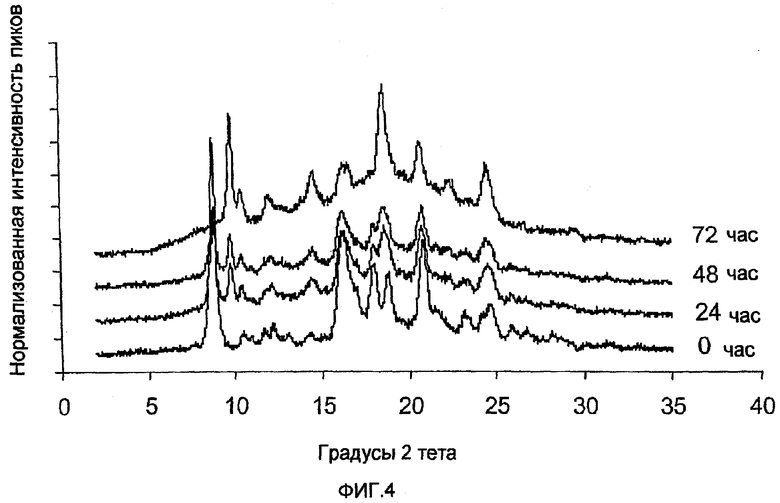
ФИГ.4 - распределенный по времени анализ порошковой рентгенографии процесса десольватирования формы V, проведенный в соответствии с примером 1 в произвольно выбранных условиях.
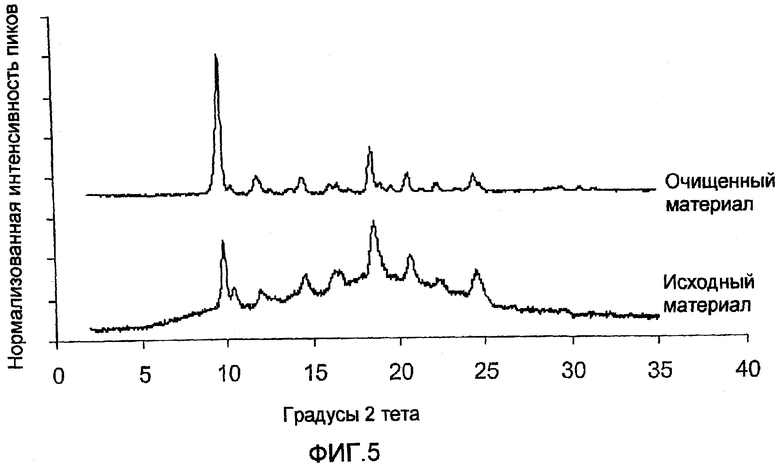
ФИГ.5 - сравнение порошковых рентгенограмм формы I, полученной в примере 3, и формы I, полученной в примере 2.
Подробное описание изобретения
В соответствии с настоящим изобретением, форма I может быть легко получена способом "обратного добавления", исходя из сырья. Механизм этого способа включает осаждение аморфного каберголина с последующей конверсией фаз до формы V в ходе процесса кристаллизации. Результат этого подхода заключается в том, что форма V, полученная посредством обратного добавления, обладает более высокой свободной энергией, чем форма V, полученная из смеси толуол-диэтиловый эфир, описанная в предшествующем уровне техники. Результатом этого является особый процесс десольватирования формы V, полученной посредством этого нового способа, который оказался более благоприятным для контролируемого превращения в форму I. Использование гептана в качестве промывочного растворителя после фильтрации также способствует снижению содержания толуола во влажном фильтрационном остатке, что, в свою очередь, облегчает контролируемое десольватирование формы V в форму I в процессе десольватирования и сушки.
Таким образом, также предусмотрен способ превращения формы V в кристаллическую форму I каберголина.
В ходе кристаллизации посредством "обратного добавления" могут быть получены смеси формы V с аморфным каберголином, поскольку эта процедура включает осаждение твердого вещества, которое затем при переходе фаз превращается под кинетическим контролем в форму V. Содержание аморфного вещества может не снижаться в ходе процесса десольватирования и сушки. Таким образом, также предусмотрен способ снижения содержания аморфного вещества либо в промежуточной форме 5, либо в форме I в случае, если образуются смеси.
Способ получения кристаллической формы I каберголина по настоящему изобретению заключается в кристаллизации, проводимой из смеси толуол/гептан. Вместо гептана также может использоваться гексан. Однако гептан предпочтительнее за счет его токсикологических свойств, которые более подходят для фармацевтического применения.
Способ включает растворение сырого целевого каберголина, полученного в форме масла посредством синтеза, описанного в Eur. J. Med. Chem., 24, 421, (1989), или любой смеси, содержащей кристаллическую форму каберголина, включающей кристаллы формы I, полученной в ходе процедур, описанных в вышеупомянутой ссылке, в подходящем количестве толуола, предпочтительно в количестве от 2,5 до 4,0 грамм толуола на грамм каберголина, более предпочтительно около 3,5 грамм толуола на грамм каберголина, при комнатной температуре.
Полученный концентрат добавляют к холодному гептану при температуре ниже -10°С таким образом, что предпочтительно на грамм каберголина приходится приблизительно от 10 до 20 г гептана. В процессе добавления концентрата каберголина емкость, содержащая гептан при температуре ниже -10°С, выдерживается при перемешивании, а скорость прерывистого добавления концентрата каберголина к холодному гептану контролируется таким образом, что весь концентрат добавляется периодически в течение не менее 2 часов. При добавлении каждой капли концентрата каберголина формируется твердый каберголин.
Однако начальное состояние этих твердых веществ является по природе аморфным и в данном изобретении оно определяется как твердая форма, не содержащая дальний порядок в трех измерениях, характерный для кристаллов. Отсутствие этого дальнего порядка наилучшим образом выявляется при анализе порошковой рентгенографии. В то время как анализ порошковой рентгенографии наилучшим образом может подходить для того, чтобы охарактеризовать кристаллическую фазу и обнаруживать небольшие количества аморфного твердого вещества, смешанного с кристаллическим материалом, микроскопия с поляризованным светом также может быть использована специалистами в известном уровне техники для того, чтобы быстро определить, является ли образец аморфным или кристаллическим.
Суспензия аморфного каберголина перемешивается при температуре ниже -10°С в течение не более чем трех дней для превращения твердого вещества в кристаллическую форму V предпочтительно в течение, как минимум, 48 часов.
В этих условиях получается форма V, которая может быть выделена при помощи обычных процедур, например путем фильтрации под низким давлением или фильтрации центрифугированием, с последующим промыванием твердого вещества чистым гептаном, предпочтительно по 5 мл на каждый грамм каберголина, для того, чтобы удалить остаточный маточный раствор, содержащий значительное количество избыточного толуола сверх молярного содержания формы V, сольватированной толуолом. Это облегчает последующие процессы десольватирования и сушки с целью получения формы I.
Кристаллы формы I получают путем проведения процессов десольватирования и сушки кристаллов формы V для превращения фаз и для снижения остаточного толуола до уровней, приемлемых для фармацевтического использования. Это можно осуществить при помощи любых подходящих методов, не ограничивающими примерами которых являются нагрев твердого вещества, снижение внешнего давления, окружающего твердое вещество, или их сочетаний. Давление в процессе сушки и время сушки не являются строго критическими. Давление в процессе сушки поддерживается предпочтительно около 101 кПа или меньше. Однако в то время как давление в процессе сушки снижается, температура, при которой сушка может быть проведена, и/или время сушки также снижаются. В частности, для твердых веществ, смоченных высококипящими растворителями, таких как толуол, сушка под вакуумом позволит использовать более низкие температуры сушки. Оптимальное сочетание давления и температуры обычно определяют по диаграмме зависимости давления пара от температуры для толуола и исходя из технологических факторов, относящихся к конструкции сушилки. Время сушки должно быть достаточным только для того, чтобы осуществилось фазовое превращение формы V в форму I, и для снижения уровня толуола до фармацевтически приемлемого уровня. Когда твердое вещество нагревают с целью удаления растворителя, например, в печи, выбирается такая температура, которая предпочтительно не превышает приблизительно 150°С.
Как было упомянуто выше, кристаллы формы V, полученные посредством процесса обратного добавления, и кристаллы формы I, в последствии полученные по завершении процесса сушки, могут содержать некоторое количество аморфного каберголина. Это количество может быть уменьшено до уровня, находящегося ниже обычного предела обнаружения методом порошковой рентгенографии, путем суспендирования кристаллов формы V или формы I при умеренном перемешивании в чистом гептане (предпочтительно 20 г гептана на грамм каберголина) при температуре от 45° до 60°С в течение приблизительно от 4 до 20 часов, предпочтительно в течение приблизительно 24 часов при 45°С. Для дальнейшего ускорения превращения аморфного каберголина в кристаллический каберголин к суспензии также может быть добавлено некоторое количество толуола.
Снижение содержания аморфной формы также может быть достигнуто при помощи других "основанных на испарении" методов, хорошо известных из уровня техники.
Кристаллы формы I каберголина, полученные в соответствии со способом по настоящему изобретению, предпочтительно имеют чистоту полиморфа >95%, более предпочтительно >98% при выходе в избытке 90 мас.%, в сравнении с приблизительно 60% для способа, описанного в WO 01/70740.
Характеристика
Порошковая рентгенография (ДРЛ) была использована для характеристики сольватированной формы V и формы I каберголина.
Анализ порошковой рентгенографии
Порошковая рентгенография проводилась с использованием либо порошкового диффрактометра Siemens D5000, либо многоцелевого диффрактометра Inel. В случае диффрактометра Siemens D5000 грубые данные были получены для величин 2(два тета) от 2 до 50, с шагами 0,020 и периодами шагов две секунды. В случае многоцелевого диффрактометра Inel образцы помещались в алюминиевый держатель образца и грубые данные были получены в течение одной тысячи секунд при всех величинах 2 одновременно.
Следует заметить, что в то время как пиковые позиции в порошковой рентгенограмме отражают длительный порядок в трех измерениях в кристаллической форме, определенной параметрами ее кристаллической решетки, и должны быть одинаковыми для данной твердой формы, относительные пиковые интенсивности отражают не только внутреннюю упорядоченность или структуру. На относительные интенсивности могут влиять такие параметры как различие во внешней форме кристаллов той же самой формы, которые, в свою очередь, могут меняться в зависимости от условий процесса, подходящих для кристаллизации данной формы. Более того, приготовление образца перед анализом порошковой рентгенографии также может привести к различиям в относительных интенсивностях для той же самой твердой формы.
Порошковая рентгенограмма для формы I каберголина (Фиг. 1), приготовленной в соответствии с примером 1 и прошедшей через многоцелевой диффрактометр Inel, демонстрирует кристаллическую структуру с отчетливыми пиками, отображенными в следующей таблице 1. Процентные пиковые интенсивности в таблице 1 вычислены после корректировки максимума (отражающего некоторое количество аморфного каберголина, смешанного с формой I) на основной линии порошковой рентгенограммы формы I, показанной на фигуре 1.
Данные по рентгенографии, форма I
%
Порошковая рентгенограмма для известной сольватированной толуолом формы V каберголина, полученной в соответствии с примером 2 (Фиг. 2) и также описанной в WO 01/70740, имеет кристаллическую структуру с отчетливыми пиками, отображенными в следующей таблице 2. Процентные пиковые интенсивности в таблице 2 вычислены после корректировки максимума (отражающего некоторое количество аморфного каберголина, смешанного с формой V) на основной линии порошковой рентгенограммы формы V на фиг. 2.
Данные по рентгенографии, форма V
%
Способ десольватирования и фазового превращения формы V, полученной в соответствии с примером 1, в форму I изучался путем помещения 1,50 г образца формы V в кристаллизатор в вакуумной печи, работающей при 43°С и вакууме 94,8 кПа в течение 48 часов. За этой стадией сушки последовали 24 часа при 57°С и вакууме 94,8 кПа. Образцы забирались каждые 24 часа для анализа порошковой рентгенографии. На фиг. 4 показан распределенный по времени процесс при этих произвольно выбранных условиях. Данные показывают, что форма V, полученная в соответствии с примером 1, начала превращаться в форму I (характеризующаяся 9,870 и 18,707 градусами 2 пиков) в течение 24 часов, и превращение было завершено в течение 72 часов.
Анализ порошковой рентгенографии также был использован для оценки эффективности, описанной в примере 3 процедуры снижения аморфного содержания формы I, которая может быть получена при помощи процедур, описанных в примерах 1 и 2. На фиг. 5 изображены результаты анализа порошковой рентгенографии, проведенной до и после обработки формы I в ходе процедуры, описанной в примере 3.
Анализ дифференциальной сканирующей калориметрии (ДСК)
Профили дифференциальной сканирующей калориметрии были получены с помощью дифференциального сканирующего калориметра Mettler-Toledo 822e. Данные были собраны между 25 и 150°С при снижении нагрева 10°С/мин. Использовались сорока-микролитровые герметично запечатанные алюминиевые колбы с игольным отверстием в крышке.
Профиль дифференциальной сканирующей калориметрии для формы V (Фиг. 3) демонстрирует единственное эндотермическое тепловое событие, сосредоточенное вокруг 62°С. Это тепловое событие соответствует эвтектическому плавлению формы V в толуоле. В соответствии с настоящим изобретением эвтектическое плавление определено как переход растворителя, содержащего твердое вещество, в гомогенный жидкий раствор без какой-либо значительной потери растворителя, связанного с твердым веществом. Калориметрия в растворе была проведена с использованием калориметра с раствором Parr 1455 для того, чтобы получить данные по энтальпии раствора и понять различия между формой V, полученной с помощью процесса обратного добавления, описанного здесь, и процедурой получения формы V, которая была описана в WO 01/70740. Измерения были проведены два раза при приблизительно 21°С путем растворения приблизительно 0,3 г образца формы V, полученного в каждом процессе, в приблизительно 100 мл чистого толуола.
Форма V, полученная в ходе процедуры обратного добавления, описанной здесь, имела среднее значение энтальпии раствора 23,93 килоджоуля/моль, в то время как форма V, полученная в ходе процедуры, описанной в WO 01/70740, имела среднее значение 25,56 килоджоуля/моль. Более низкие значения для формы V, полученной в ходе процедуры обратного добавления, говорят о том, что она экзотермически превращается в кристаллы формы V, полученной в ходе процедуры, описанной в WO 01/70740. Одной из причин для более низкой энтальпии раствора для формы V, полученной в ходе процесса "обратного добавления", является "сниженная молекулярная упорядоченность", возможно являющаяся результатом небольшого количества аморфного каберголина, смешанного с формой V. Поскольку тот факт, что в процессе "обратного добавления" кристаллизуется форма V посредством фазового превращения аморфного каберголина, может привести к небольшим количествам аморфного каберголина, остающимся даже после фазового превращения, предполагается, что форма V, по-видимому, является завершенной в суспензии. Различия в энтальпиях раствора для формы V, полученной различными способами, также могут иметь благоприятные последствия для процесса десольватирования, который завершается получением формы I.
Примеры
Следующие примеры содержат подробные описания способов получения кристаллических форм каберголина, описанных в данном изобретении. Эти подробные описания входят в объем изобретения и иллюстрируют изобретение, никоим образом не ограничивая этот объем. Все процентные соотношения являются массовыми, если не указано обратное.
Пример 1. Получение кристаллической формы V каберголина
2,0 г каберголина растворяли в 7,01 г толуола в 25 мл сцинтилляционном пузырьке путем перемешивания с магнитной мешалкой. В 125 мл реакторе с рубашкой, оснащенном верхней системой перемешивания, охлаждали 30 г гептана при -18°С для того, чтобы температура в реакторе достигла -15°С. Затем концентрат каберголина в толуоле порционно добавляли к холодному гептану в течение более 2 часов при перемешивании в реакторе, установленном на 203 оборота в минуту. После завершения загрузки концентрата перемешивание было снижено до 175 оборотов в минуту. Твердое вещество образовывалось при добавлении каждой капли концентрата. Микроскопия с поляризованным светом подтвердила, что это исходное твердое вещество является аморфным. Суспензия перемешивалась в течение 48 часов при -15°С после завершения загрузки концентрата каберголина для того, чтобы превратить с переходом фаз аморфный каберголин в кристаллическую форму V каберголина. По истечении 48 часов суспензия выгружалась в фильтрационную колбу, работающую при пониженном давлении. Фильтрационный остаток промывался 10 мл гептана для удаления маточного раствора и вымывания избыточного толуола из твердого вещества. Твердое вещество было оставлено на фильтре в течение 25 минут под давлением.
Оно было идентифицировано как форма V при помощи ДРЛ с получением данных, отображенных на фиг. 1 и в таблице 1. Выход составил около 100 мас.% в расчете на содержание чистого "свободного от толуола" каберголина.
Пример 2. Получение кристаллической формы I каберголина
Сольватированная толуолом форма V, полученная в примере 1, помещалась в вакуумную печь при температуре 43°С и вакууме 94,8 кПа на 48 часов, затем на 6 часов при 55°С. После сушки суммарный выход продукта составил около 93% в расчете на начальное содержание чистого каберголина, и полученная твердая форма была идентифицирована при помощи ДРЛ как форма I. Образец имел все характеристические пики, приведенные в таблице 2, однако, он также имел небольшой "максимум" на основной линии порошковой рентгенограммы, указывающий на наличие некоторого количества аморфного материала, смешанного с формой I (фиг. 2 и образец, помеченный как "исходный материал" на фиг. 5).
Пример 3. Снижение содержания аморфного вещества в кристаллической форме I каберголина
В 12 мл пузырек, оснащенный магнитной мешалкой, добавляли 100 мг аморфного вещества, содержащего форму I, полученную в примере 2. Затем добавили 2,0 г гептана. Полученная суспензия перемешивалась в течение 24 часов на магнитной плитке при 45°С. Затем суспензия выгружалась в фильтрационную колбу, работающую под давлением. Фильтрационный остаток промывался 1,0 мл гептана и высушивался на воздухе в течение тридцати минут. Твердое вещество анализировалось при помощи порошковой рентгенографии. Оно было идентифицировано как твердое вещество формы I с аморфным каберголином ниже предела обнаружения для прибора порошковой рентгенографии (см. образец "очищенного материала" на фиг. 5).
Изобретение описывает улучшенный способ получения кристаллической формы I каберголина, включающий получение формы V с использованием гептана в качестве осаждающего растворителя при исключительном превращении в кристаллическую форму I каберголина. Способ включает стадию кристаллизации из толуол-гептановой системы растворителей формы V при "обратном добавлении" толуол-каберголинового концентрата к холодному гептану. Способ позволяет получить высоко чистый продукт с выходом ˜90%. 3 н. и 8 з.п. ф-лы, 2 табл., 5 ил.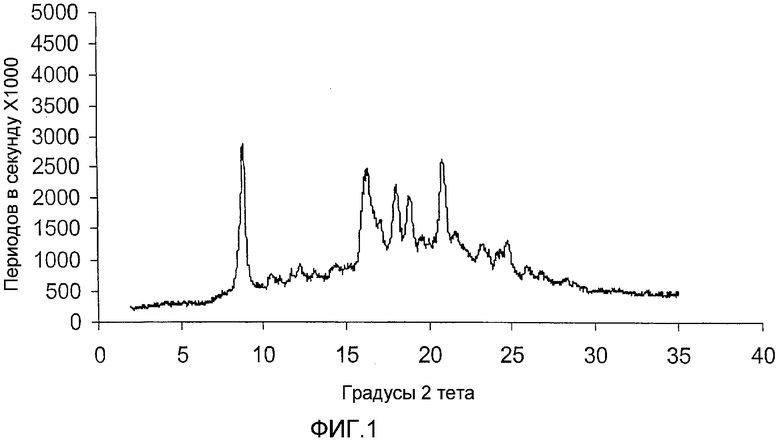
Приоритет по пунктам и признакам:
| US 4526892 A, 02.07.1985 | |||
| Печь для непрерывного получения сернистого натрия | 1921 |
|
SU1A1 |
| US 5705510 A, 06.01.1991 | |||
| СПОСОБ РАБОТЫ БЛОЧНОЙ ГАЗОВОЙ ГОРЕЛКИ И БЛОЧНАЯ ГАЗОВАЯ ГОРЕЛКА | 1995 |
|
RU2103603C1 |
| Прибор для очистки паром от сажи дымогарных трубок в паровозных котлах | 1913 |
|
SU95A1 |
| Прибор, замыкающий сигнальную цепь при повышении температуры | 1918 |
|
SU99A1 |

