ОБЛАСТЬ ИЗОБРЕТЕНИЯ
Данное изобретение касается способа получения негидратируемой кристаллической формы из гидратируемой кристаллической формы 3-бром-1-(3-хлор-2-пиридинил)-N-[4-циано-2-метил-6-[(метиламино)карбонил]фенил]-1H-пиразол-5-карбоксамида.
ПРЕДПОСЫЛКИ ИЗОБРЕТЕНИЯ
PCT патентные публикации WO 04/067528 и WO 06/062978 раскрывают способы получения 3-бром-1-(3-хлор-2-пиридинил)-N-[4-циано-2-метил-6-[(метиламино)карбонил]фенил]-1H-пиразол-5-карбоксамида (соединение 1), а также применение этого соединения как инсектицида. Документ WO 06/062978, кроме того, раскрывает очистку соединения 1 путем рекристаллизации из 1-пропанола.
Из уровня техники хорошо известно, что определенные кристаллические соединения могут существовать как полиморфы. Выражение “полиморф” касается конкретной кристаллической формы химического соединения, которое может кристаллизоваться в разные кристаллические формы, которые имеют разные структуры и/или конформации молекул в кристаллической решетке. Хотя полиморфы могут иметь одинаковый химический состав, они также могут различаться по составу, что обусловлено присутствием или отсутствием сокристаллизованной воды или других молекул, которые могут быть слабо или сильно связаны в решетке. Полиморфы могут отличаться такими химическими, физическими и биологическими свойствами, как форма кристаллов, плотность, твердость, цвет, химическая стабильность, точка плавления, гигроскопичность, суспендируемость, скорость растворения и биологическая доступность.
К настоящему времени не представлялось возможным предусмотреть появление и число кристаллических полиморфов любого отдельного соединения и конкретные физико-химические свойства любого конкретного полиморфа. Важно подчеркнуть, что термодинамическая стабильность и потенциально разное поведение после введения в живые организмы не могут быть определены заранее.
КРАТКОЕ ОПИСАНИЕ ДАННОГО ИЗОБРЕТЕНИЯ
Данное изобретение направлено на способ получения полиморфа A соединения 1, который характеризуется дифракционной рентгенограммой, которая имеет, по меньшей мере, положения максимумов для угла отражения, определенного в градусах 2θ, 6,78, 11,09, 19,94, 20,99, 26,57, 26,98 и 31,52; включающий нагревание при температуре от приблизительно 40°C до точки кипения растворителя смеси, которая содержит растворитель, выбранный из группы, состоящей из воды, н-гептана, 1-хлорбутана, толуола, 1-бутанола и 1-пентанола, и полиморфа B соединения 1, который характеризуется дифракционной рентгенограммой, которая имеет, по меньшей мере, положения максимумов для угла отражения, определенного в градусах 2θ, 7,43, 9,89, 18,68, 19,36, 22,16, 23,09 и 25,70.
КРАТКОЕ ОПИСАНИЕ ГРАФИЧЕСКИХ МАТЕРИАЛОВ
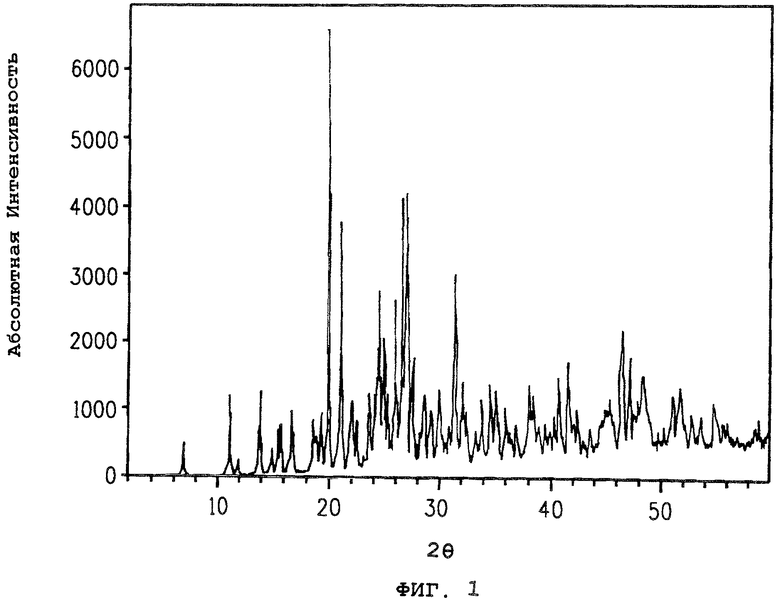
На фиг.1 представлена порошковая дифракционная рентгенограмма полиморфа A соединения 1 и показан подсчет абсолютной интенсивности, нанесенный на график против положения максимумов для угла отражения, определенного в градусах 2θ.
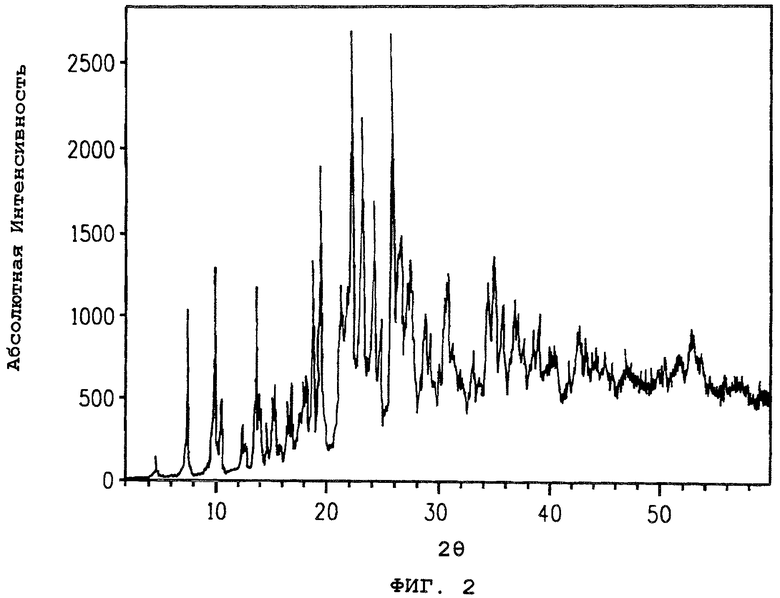
На фиг.2 представлена порошковая дифракционная рентгенограмма полиморфа В соединения 1 и показан подсчет абсолютной интенсивности, нанесенный на график против положения максимумов для угла отражения, определенного в градусах 2θ.
ДЕТАЛЬНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
В контексте данного описания выражения “содержит”, “содержащий”, “включает”, “включающий”, “имеет”, “имеющий” или любая другая их вариация предназначены охватывать неисключительные включения. Например, композиция, процесс, способ, изделие или аппарат, который содержит перечень элементов, не является обязательно ограниченным только теми элементами, но может включать другие элементы, которые специально не приведены или не присущи такой композиции, процессу, способу, изделию или аппарату. Кроме того, если специально не указано противоположное, “или” относится к включающему или, а не к исключающему или. Например, условие A или B удовлетворяет любому одному из следующего: A - истинное (или присутствует), и B - ошибочное (или не присутствует), A - ошибочное (или не присутствует), и B - истинное (или присутствует), и как A, так и B - истинные (или присутствуют).
Также, единственное число элемента или компонента данного изобретения предназначено быть неограничивающим относительно числа случаев (то есть событий) элемента или компонента. Таким образом, единственное число следует читать как такое, которое включает один или, по меньшей мере, один, и форма единственного числа слова элемента или компонента также включает множественное, если только число очевидно не предназначено быть единственным.
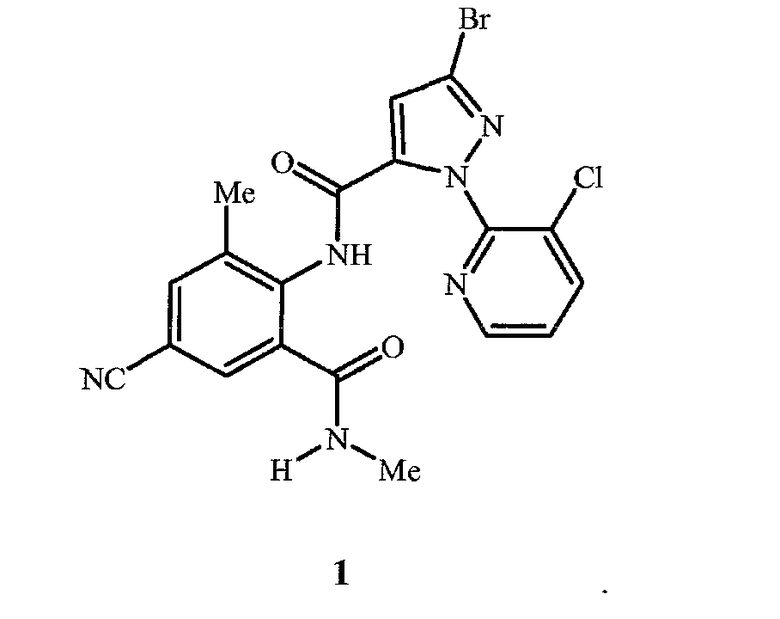
Соединение 1 представляет собой 3-бром-1-(3-хлор-2-пиридинил)-N-[4-циано-2-метил-6-[(метиламино)карбонил]фенил]-1H-пиразол-5-карбоксамид и имеет следующую химическую структуру:
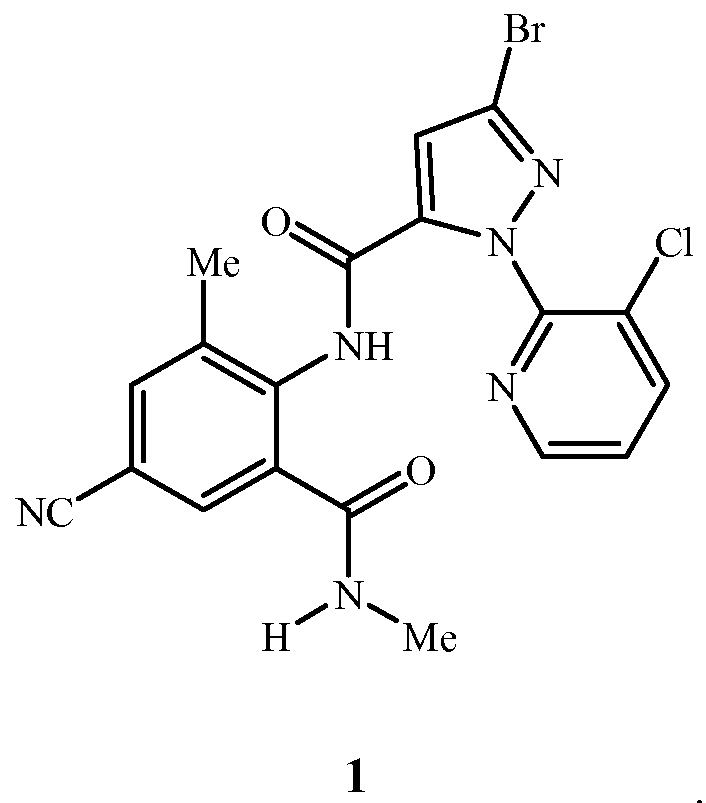
Соединение 1 может существовать в более чем одной кристаллической форме (то есть полиморф). Специалист в данной области оценит, что полиморф соединения 1 может проявлять полезные эффекты (например, пригодность для получения полезных составов, улучшенная биологическая характеристика) относительно другого полиморфа или смеси полиморфов того самого соединения 1. Отличия относительно химической стабильности, фильтруемости, растворимости, гигроскопичности, точки плавления, плотности твердого вещества и текучести могут иметь значительное влияние на разработку способов получения и составов и на качество и эффективность средств обработки растений.
Разработан способ получения негидратируемого полиморфа соединения 1 (полиморф A) из гидратируемого полиморфа соединения 1 (полиморф B), обычно получаемого согласно существующим методикам получения соединения 1. Содержание воды полиморфа B значительно изменяется под действием колебаний атмосферной влажности. В отличие от полиморфа B полиморф A не приобретает или не теряет заметных количеств воды, если подвергается колебаниям атмосферной влажности. Более того, полиморф A обычно не превращается в полиморф B во время долгого хранения. Такая удивительная стабильность облегчает более сравнимый анализ соединения 1. Эти характеристики также делают полиморф A соединения 1 очень подходящим для получения долговечных стабильных твердых составов, которые дают возможность определить стабильное содержание активного ингредиента.
Кроме того, полиморф A имеет физическую форму, обеспечивающую возможность более эффективной фильтрации в сравнении с полиморфом B. Во время промышленного синтеза и выделения замечательная простота отделения полиморфа A может снизить затраты процесса производства.
Порошковая рентгенодифракция применяется для определения кристаллизованных фаз обоих полиморфов A и B соединения 1. Для характеристики полиморфов A и B были получены данные с автоматическим порошковым дифрактометром Philips X'PERT, модель 3040. Образцы при комнатной температуре прогнали в порционном режиме на многопозиционном сменщике проб модели PW 1775 или модели PW 3065. Дифрактометр был оснащен автоматической сменной щелью, ксеноновым пропорциональным счетчиком и графитовым монохроматором. Излучение было Cu (Kα), 45 кВ, 40 мА. Образцы были приготовлены как сухой мазок на низкофоновом стеклянном держателе для образцов. Данные были собраны при 2θ углах от 2 до 60 градусов с применением непрерывного сканирования с эквивалентным размером шага 0,03 градуса и времени подсчитывания 2,0 секунды на шаг. Было применено программное обеспечение MDI/Jade с базой данных Международного комитета для дифракционных данных для идентификации фазы и сравнения дифрактограмм образцов с таковыми справочных материалов.
Порошковая дифракционная рентгенограмма полиморфа A соединения 1 показана на фиг.1. Соответствующие 2θ значения приведены в таблице 1.
Порошковая дифракционная рентгенограмма полиморфа В соединения 1 показана на фиг.2. Соответствующие 2θ значения приведены в таблице 2.
Кристаллические полиморфы соединения 1 также могут быть охарактеризованы ИК спектроскопией. ИК спектры были измерены на ИКПФ (инфракрасная спектроскопия с Фурье преобразованием) спектрометре FTS 3000 (Varian, США) с применением вспомогательного оснащения Golden Gate ATR для твердых веществ. ИК спектры включают следующие максимумы полос, показанные в таблице 3 (полиморф A) и таблице 4 (полиморф B).
Кристаллические полиморфы соединения 1 также могут быть охарактеризованы и отличаться один от другого по Раман-спектроскопии и спектроскопии ближней инфракрасной области.
Варианты осуществления данного изобретения включают:
Вариант осуществления 1. Способ, описанный в кратком описании данного изобретения, где растворителем является н-гептан.
Вариант осуществления 1a. Способ варианта осуществления 1, где температура составляет от приблизительно 40 до приблизительно 100°C.
Вариант осуществления 2. Способ, описанный в кратком описании данного изобретения, где растворителем является толуол.
Вариант осуществления 2a. Способ варианта осуществления 2, где температура составляет от приблизительно 40 до приблизительно 111°C.
Вариант осуществления 3. Способ, описанный в кратком описании данного изобретения, где растворителем является 1-хлорбутан или 1-хлорпентан.
Вариант осуществления 3a. Способ, описанный в кратком описании данного изобретения, где растворителем является 1-хлорбутан.
Вариант осуществления 3b. Способ, описанный в кратком описании данного изобретения, где растворителем является 1-хлорпентан.
Вариант осуществления 3c. Способ варианта осуществления 3a, где температура составляет от приблизительно 40 до приблизительно 77°C.
Вариант осуществления 4. Способ, описанный в кратком описании данного изобретения, где растворителем является 1-бутанол или 1-пентанол.
Вариант осуществления 4a. Способ, описанный в кратком описании данного изобретения, где растворителем является 1-бутанол.
Вариант осуществления 4b. Способ, описанный в кратком описании данного изобретения, где растворителем является 1-пентанол.
Вариант осуществления 4c. Способ любого одного из вариантов осуществления 4-4b, где температура составляет от приблизительно 40 до приблизительно 100°C.
Вариант осуществления 5. Способ, описанный в кратком описании данного изобретения, где растворителем является вода.
Вариант осуществления 5a. Способ варианта осуществления 5, где температура составляет от приблизительно 60 до приблизительно 100°C.
Вариант осуществления 5b. Способ варианта осуществления 5a, где температура составляет от приблизительно 70 до приблизительно 100°C.
Вариант осуществления 5c. Способ варианта осуществления 5a, где температура составляет от приблизительно 70 до приблизительно 90°C.
Вариант осуществления 5d. Способ любого одного из вариантов осуществления 5-5c, где смесь нагревают в течение, по меньшей мере, приблизительно 2 часов.
Вариант осуществления 5e. Способ варианта осуществления 5d, где смесь нагревают в течение не более чем приблизительно 48 часов.
Вариант осуществления 5f. Способ варианта осуществления 5e, где смесь нагревают в течение не более чем приблизительно 24 часа.
Вариант осуществления 5g. Способ варианта осуществления 5f, где смесь нагревают в течение не более чем приблизительно 12 часов.
Вариант осуществления 6. Способ любого одного из вариантов осуществления 5-5g, где смесь содержит, по меньшей мере, приблизительно 30% воды по весу.
Вариант осуществления 6a. Способ варианта осуществления 6, где смесь содержит, по меньшей мере, приблизительно 40% воды по весу.
Вариант осуществления 6b. Способ варианта осуществления 6a, где смесь содержит, по меньшей мере, приблизительно 80% воды по весу.
Вариант осуществления 6c. Способ варианта осуществления 6b, где смесь содержит, по меньшей мере, приблизительно 90% воды по весу.
Вариант осуществления 6d. Способ варианта осуществления 6c, где смесь содержит, по меньшей мере, приблизительно 95% воды по весу.
Вариант осуществления 6e. Способ варианта осуществления 6d, где смесь содержит, по меньшей мере, приблизительно 98% воды по весу.
Вариант осуществления 7. Способ, описанный в кратком описании данного изобретения или в любом одном из вариантов осуществления 1-6e, где приблизительно 0,1-10% по весу полиморфа A (соединения 1) относительно веса полиморфа B добавляют к смеси перед нагреванием.
Вариант осуществления 7a. Способ варианта осуществления 7, где приблизительно 0,2-5% по весу полиморфа A (соединения 1) относительно веса полиморфа B добавляют к смеси перед нагреванием.
Полиморф B соединения 1 может быть превращен в полиморф A соединения 1 нагреванием в присутствии жидкой фазы, содержащей растворитель, выбранный из определенных органических растворителей (то есть растворителей, чьи молекулы содержат, по меньшей мере, один атом углерода). Только определенные органические растворители являются пригодными для такого преобразования, и прогнозирование за пределами близких гомологов является невозможным, и, таким образом, определение пригодных классов органических растворителей требует эксперимента. Тем не менее, классы органических растворителей, которые обычно хорошо действуют при преобразовании полиморфа B в полиморф A, как обнаружили, включают C3-C8 н-алкиловые спирты (например, н-пропанол, н-бутанол, н-пентанол), C4-C6 н-алкилхлориды (например, н-бутилхлорид или н-пентилхлорид), C6-C10 алканы (например, н-гексан, гексаны, н-гептан, гептаны), C6-C10 циклоалканы, факультативно замещенные не более чем 2 заместителями, независимо выбранными из C1-C2 алкила (например, циклогексан, метилциклогексан, циклогептан), и бензол, факультативно замещенный не более чем 3 группами, независимо выбранными из C1-C2 алкила (например, бензол, толуол, ксилол). Поскольку полиморф B обычно содержит воду (как воду гидратации и остаточную воду, присутствующие, например, во влажном осадке), а полиморф A является безводным, вода высвобождается во время преобразования. Азеотропная дистилляция часто может быть применена для удаления воды из смеси преобразования полиморфов.
Замечательно, что вода, как обнаружено на данное время, очень хорошо действует как растворитель в нагретой жидкой фазе для преобразования полиморфа B в полиморф A. Это в особенности неожиданно, поскольку полиморф B, который может содержать значительные количества воды в своей кристаллической решетке, может иметь, как ожидают, преимущество над безводным полиморфом A в водной среде. Тем не менее, вода, как обнаружено на данное время, в особенности, пригодна при формировании жидкой фазы для преобразования полиморфа B в полиморф A. Преобразование проходит до приблизительно 100% осуществления и с высокими выходами за коммерчески пригодные периоды времени при температурах, которые не превышают приблизительно 100°C (то есть нормальную точку кипения воды). Не только из-за того, что вода намного дешевле, чем органические растворители, но и потому, что полиморф A имеет небольшую растворимость в воде, его легко можно будет выделить фильтрацией. Альтернативно, если полиморф A находится в высокой концентрации в воде, полиморф A может быть выделен выпариванием воды. В отличие от органических растворителей вода, испаренная из смеси, не требует улавливания.
В одном варианте осуществления данного способа смесь, включающая полиморф B и воду (вместе с возрастающими количествами полиморфа A), содержит твердую фазу, которая включает уменьшающиеся количества полиморфа B и возрастающие количества полиморфа A, вместе с жидкой фазой, которая содержит воду и факультативно другие растворители. Обычно факультативные другие растворители выбраны из органических растворителей, растворимых в воде, хотя органические растворители, которые имеют низкую растворимость в воде, могут также использоваться. Таким образом, обычно жидкая фаза смеси в данном варианте осуществления данного способа содержит, по меньшей мере, приблизительно 50%, и более обычно, по меньшей мере, приблизительно 80%, 90% или 95%, и наиболее обычно, по меньшей мере, приблизительно 98% воды по весу.
Способ варианта осуществления, описанный выше, обеспечивает средства преобразования полиморфа B соединения 1 в полиморф A соединения 1 путем нагревания смеси, которая содержит полиморф B соединения 1 и воду. Обычно, смесь твердого полиморфа B соединения 1 и воды в форме суспензии или взвеси размещают внутри сосуда пригодного размера, оснащенного средствами смешивания и нагревания смеси. Затем смесь нагревают при смешивании в течение периода времени, достаточно длительного для осуществления преобразования полиморфа B в полиморф A. Способы смешивания могут быть внутренними (например, магнитная мешалка или верхнеприводная мешалка) или внешними (например, реакционный сосуд, который вращается или встряхивается). Обычно выгодно добавлять затравку кристаллов полиморфа A к смеси, которая содержит полиморф B, перед нагреванием. Добавление затравки кристаллов уменьшает общее время преобразования и в некоторых случаях уменьшает температуру, необходимую для осуществления преобразования. После преобразования полиморфа B в полиморф A смесь охлаждают, а продукт выделяют. В зависимости от относительных количеств твердой и жидкой фаз выделение продукта может включать дополнительную сушку взвеси, или, если смесь является суспензией, фильтрацию с последующим факультативным промыванием, а затем сушку.
Количество воды в смеси может варьировать для приспособления к разному технологическому оснащению. Например, применение большого излишка воды (то есть, где вода представляет собой жидкую фазу, в которой суспендированы кристаллы полиморфа B) обеспечивает облегчение перемешивания с традиционным оснащением, таким как верхнеприводная мешалка. Такая суспензия, тем не менее, требует значительной энергии для нагревания до желательной температуры. После осуществления преобразования в полиморфа A соединения 1 суспензия может быть отфильтрована для выделения твердого продукта. Этот влажный твердый продукт или влажный осадок может быть дополнительно высушен для получения кристаллического продукта, пригодного для приготовления состава композиций, не включающих воду, или применен непосредственно для приготовления композиций водного состава (например, концентраты водных суспензий).
Преобладающий вариант осуществления данного способа включает изготовление смеси полиморфа B соединения 1 и воды в виде взвеси, которая содержит только количество воды, необходимое для облегчения смешивания. Преимущественно применять меньше воды, поскольку меньше энергии необходимо для нагревания взвеси до желательной температуры. Вдобавок, отдельный этап фильтрации для выделения кристаллов полиморфа A не является необходимым, поскольку кристаллы полиморфа A могут быть легко выделены сушкой взвеси. В зависимости от конфигурации сосуда, который применяют для преобразования полиморфа B в полиморф A, можно преимущественно осуществить этот процесс сушки непосредственно в самом сосуде. В промышленных коммерческих способах исключение потребности переноса твердого вещества из одного контейнера в другой приводит к значительной экономии средств. Альтернативно, кристаллы полиморфа A могут быть перенесены в другой сосуд, пригодный для дальнейшей сушки.
Таким образом, в преобладающем варианте осуществления данного изобретения кристаллы полиморфа B соединения 1, объединенные с водой для формирования взвеси, которая обычно содержит приблизительно 20-60% по весу содержания воды, более обычно 30-50% по весу содержания воды и наиболее обычно приблизительно 40% по весу содержания воды.
Без дополнительного уточнения считается, что специалист в данной области с применением предшествующего описания может использовать данное изобретение в полной мере. Следующие примеры, таким образом, следует толковать только как иллюстративные, а не ограничивающие раскрытие каким бы то ни было образом. Необязательно, чтобы исходный материал для любого примера был получен таким же путем получения. Проценты являются весовыми, за исключением случаев, когда указано другое.
Специфические примеры преобразования полиморфа B соединения 1 в полиморф A соединения 1 приведены ниже.
ПРИМЕР 1
Получение полиморфа A соединения 1 (с применением взвеси в воде)
В 250 мл плоскодонный цилиндрический реактор с кожухом (внутренний диаметр приблизительно 6 см, Wilmad-LabGlass) загрузили увлажненный водой осадок полиморфа B соединения 1 (67,8 г, полученный по методике примера 15 в PCT патентной публикации WO 06/062978, за исключением того, что выделенный осадок продукта промыли дополнительной водой; увлажненный водой осадок не высушили и применили без дополнительной обработки). Увлажненный водой осадок имел общее содержание влаги приблизительно 40% по весу, включая приблизительно 1% остаточного ацетонитрила. Затем в реактор добавили как затравку кристаллов 2,0 г полиморфа A соединения 1 (полученный нагреванием и азеотропной сушкой взвеси полиморфа B в гептане; 97,4% полиморфа A по анализу в ближней инфракрасной области спектра). Верхнеприводное взбалтывание установили с применением стеклянного четырехлопастного, с 45-градусным наклоном импеллера с общим диаметром 4,5 см и с проекционной высотой лопасти приблизительно 2,2 см. Крышку реактора присоединили и термоэлемент вставили через одно отверстие крышки. Все другие отверстия крышки закупорили для предупреждения испарения влаги из смеси. Взбалтывание начали при приблизительно 21 оборотах в минуту. Горячее масло с рециркуляционного нагревателя/охладителя, установленного на поддержание 83°C, циркулировало через кожух реактора, и содержимому реактора позволили нагреваться и смешиваться в течение 6,25 часов, после чего содержимое реактора охладили и позволили осесть без смешивания в течение ночи. На следующий день снова начали нагревание и смешивание с использованием тех же условий и поддерживали в течение 7,25 часов. Образцы вынимали из реактора во время периодов нагревания после прекращения взбалтывания и снятия реакторной крышки. Перед отбором каждого образца содержимое реактора тщательно перемешивали вручную шпателем для обеспечения однородности. Получили образец весом от 1 до 3 г, а потом поместили в вакуумную печь и сушили в течение ночи при приблизительно 50°C и 17-40 кПа под легким потоком азота. Затем образец анализировали на кристаллическую форму с помощью анализа в ближней инфракрасной области спектра. Результаты анализа кристаллической формы для образцов были следующими:
После нагревания в течение в общем 13,5 часов реактор охладили до 25°C, и содержимое реактора перенесли на чашу для сушки и сушили в течение ночи в вакуумной печи при 50°C и 17-40 кПа под легким потоком азота для получения 28,2 г сухого полиморфа A соединения 1 (92,3% чистоты согласно методу высокоэффективной жидкостной хроматографии (HPLC), 0,1% H2O согласно титрованию по Карлу Фишеру).
ПРИМЕР 2
Получение полиморфа A соединения 1 (с применением суспензии в воде)
В 100 мл круглодонную колбу загрузили полиморф B соединения 1 (5,00 г, приготовленный по методике примера 15 в PCT патентной публикации WO 06/062978 без рекристаллизации из 1-пропанола, 4,2% полиморфа A согласно анализу в ближней инфракрасной области спектра), полиморф A соединения 1 (приготовленный по методике примера 15 в WO 06/062978, включая рекристаллизацию из 1-пропанола, 0,05 г, 97,0% полиморфа A согласно анализу в ближней инфракрасной области спектра) и воду (15 мл). Смесь крутили в течение 4 часов в водяной бане, нагретой до 70°C. После охлаждения до 25°C смесь отфильтровали, промыли несколькими небольшими порциями воды и сушили в вакуумной печи при 60°C и 17-40 кПа для получения полиморфа A соединения 1 (96,8% полиморфа A согласно анализу в ближней инфракрасной области спектра), 4,74 г (93,9% выход), плавление 218-220°C.
ПРИМЕР 3
Получение полиморфа A соединения 1 (с применением суспензии в н-гептане)
В 6 л цилиндрический реактор со стеклянным кожухом, оснащенный верхнеприводным перемешиванием, термоэлементом, погружной трубой для отбора образцов, входом азота, головкой обратного потока дистилляции и дефлегматором, охлажденным замкнутым циркуляционным холодильником, заполненным жидкостью 50:50 гликоль:вода, загрузили полиморфом B соединения 1 (906,1 г увлажненного водой осадка, приблизительно 40% влаги, определенной по потере веса после сушки; приготовленный по методике примера 15 в PCT патентной публикации WO 06/062978 без рекристаллизации из 1-пропанола и без сушки; полиморф B, как определено рентгенодифракцией). Температуру холодильника установили на 5°C. После продувки реактора азотом реактор заполнили 500 мл свежего н-гептана и 2000 мл н-гептанового фильтрата, рециркулированного по идентичным методикам, как описано в данном примере. Реактор снова продули азотом, начали перемешивание и реакционную смесь нагрели до заданного значения кожуха 97,5°C. Реакционная смесь начала кипеть, когда температура смеси достигла приблизительно 80°C при атмосферном давлении, и конденсат (то есть конденсированные испарения) направили из вывода дефлегматора в 1000 мл мерный цилиндр, модифицированный нижним выводом. Конденсат образован двумя отдельными прозрачными жидкими слоями. Нижний слой конденсата, который состоял из воды, периодически удаляли из мерного цилиндра и взвешивали. Приблизительно 350 мл свежего н-гептана добавили назад в реактор для компенсации потери н-гептана, удаленного через цилиндр сбора конденсата. Температура реакционной смеси постепенно росла, поскольку воду удаляли из системы. Когда температура реакционной смеси достигла 90°C, заданное значение кожуха подняли до 110°C и реакционную смесь нагревали для дефлегмации в течение еще приблизительно двух часов. Образцы реакционной смеси периодически отбирали через погружную трубу. Эти образцы отфильтровали, изъяли образованный влажный осадок, высушили в вакуумной печи и анализировали с помощью анализа в ближней инфракрасной области спектра. Результаты анализа кристаллической формы для образцов были следующими:
(b) как определено с помощью анализа в ближней инфракрасной области спектра.
Общий объем водного слоя, удаленного из дистиллята, составлял 363 мл. Реактор охладили до 25°C и выдерживали в течение ночи. Реакционную смесь перемешали за короткое время, чтобы помочь вывести взвесь кристаллов в воронку с крупным стеклянным пористым фильтром, и взвесь фильтровали под вакуумом. Фильтрат рециркулировали и применили для промывания остаточного продукта из реактора на фильтре. Влажный осадок сушили в вакуумной печи в течение ночи при 80°C под легким потоком азота для получения 529,5 г продукта. Высушенным продуктом был полиморф A, как выяснили с помощью анализа в ближней инфракрасной области спектра и рентгенодифракции (97,1% полиморфа A с помощью анализа в ближней инфракрасной области спектра).
ПРИМЕР 4
Получение полиморфа A соединения 1 (с применением суспензии в 1-хлорбутане)
Стеклянный флакон с завинчивающейся крышкой заполнили полиморфом B соединения 1 (0,509 г), полиморфом A соединения 1 (0,503 г, приготовленный из полиморфа B с помощью способа, аналогичного примеру 3) и 1-хлорбутаном (5,8 г). Добавили магнитную мешалку и флакон закрыли. Флакон поместили в алюминиевый поддон на верх нагретой плиты магнитной мешалки. Алюминиевый поддон нагрели до 45°C и реакционную смесь перемешивали при этой температуре в течение приблизительно 27 часов. Затем реакционную смесь отфильтровали через воронку Бюхнера с применением вакуума. Отфильтрованный осадок сушили на воздухе в течение приблизительно 30 минут, а потом перенесли в новый стеклянный флакон. Флакон накрыли тканью и поместили в вакуумную печь, которую поддерживали при 60-70°C и 17-40 кПа в течение приблизительно 3 дней. Высушенные твердые вещества анализировали в ближней инфракрасной области спектра и обнаружили 97,4% полиморфа A.
ПРИМЕР 5
Получение полиморфа A соединения 1 (с применением суспензии в толуоле)
1000 мл цилиндрический реактор со стеклянным кожухом, оснащенный верхнеприводным перемешиванием, ловушкой Дина-Старка и дефлегматором, термоэлементом и капельной воронкой, заполнили полиморфом B соединения 1 (100 г, полученный по методике примера 15 в PCT патентной публикации WO 06/062978, за исключением того, что выделенный осадок продукта переработали во взвесь в смеси ацетонитрила с водой, отфильтровали и высушили; полиморф B подтвердили с помощью рентгенодифракции). После продувки реактора азотом реактор заполнили 500 мл толуола и содержимое реактора перемешали для формирования взвеси. Взвесь нагрели путем повышения температуры жидкости кожуха до 120°C. Конденсат, который начал собираться, когда взвесь достигла 102,6°C, собрали в ловушку Дина-Старка. Через приблизительно один час при дефлегмации 4,4 г нижнего (водного) слоя удалили из ловушки. Через еще двадцать минут взвесь оказалась более жидкой и состояла из больших твердых частиц, которые быстро осели на дно реактора, когда перемешивание временно остановили. Через приблизительно два часа общего времени при дефлегмации реакционную смесь охладили до 20°C. Реакционную смесь вынули и отфильтровали с применением вакуума для получения влажного осадка, который имел вид песка. Осадок продукта промыли в общем 150 мл свежего толуола двумя порциями, а затем перенесли в чашку для сушки. Осадок продукта высушивали в вакуумной печи при 100°C и 17-40 кПа с легким потоком азота в течение трех дней. Высушенный продукт определили как полиморф A соединения 1 (92,2 граммов) путем рентгенодифракции; анализ в ближней инфракрасной области спектра показал, что продуктом является 95,6% полиморф A.
ПРИМЕР 6
Получение полиморфа A соединения 1 (с применением суспензии в 1-бутаноле)
Стеклянный флакон с завинчивающейся крышкой заполнили полиморфом B соединения 1 (0,572 г), полиморфом A соединения 1 (0,578 г, приготовленный из полиморфа B с помощью способа, аналогичного примеру 3) и 1-бутанолом (4,0 г). Добавили магнитную мешалку и флакон закрыли. Флакон поместили в алюминиевый поддон на верх нагретой плиты магнитной мешалки. Алюминиевый поддон нагрели до 60°C и реакционную смесь перемешивали при этой температуре в течение приблизительно 24 часов. Затем реакционную смесь отфильтровали через воронку Бюхнера с применением вакуума. Отфильтрованный осадок сушили на воздухе в течение приблизительно 30 минут, а потом перенесли в новый стеклянный флакон. Флакон накрыли тканью и поместили в вакуумную печь, которую поддерживали при 60°C и 17-40 кПа в течение приблизительно 3 дней. Высушенные твердые вещества анализировали в ближней инфракрасной области спектра и обнаружили 96,7% полиморфа A.
ПРИМЕР 7
Получение полиморфа A соединения 1 (с применением суспензии в 1-пентаноле)
Стеклянный флакон с завинчивающейся крышкой заполнили полиморфом B соединения 1 (0,611 г), полиморфом A соединения 1 (0,605 г, изготовленный из полиморфа B с помощью способа, аналогичного примеру 3) и 1-пентанолом (4,0 г). Добавили магнитную мешалку и флакон закрыли. Флакон поместили в алюминиевый поддон на верх нагретой плиты магнитной мешалки. Алюминиевый поддон нагрели до 60°C и реакционную смесь перемешивали при этой температуре в течение приблизительно 24 часов. Затем реакционную смесь отфильтровали через воронку Бюхнера с применением вакуума. Отфильтрованный осадок сушили на воздухе в течение приблизительно 30 минут, а потом перенесли в новый стеклянный флакон. Флакон накрыли тканью и поместили в вакуумную печь, которую поддерживали при 60°C и 17-40 кПа в течение приблизительно 3 дней. Высушенные твердые вещества анализировали в ближней инфракрасной области спектра и обнаружили 97,2% полиморфа A.
| название | год | авторы | номер документа |
|---|---|---|---|
| Способ получения полиморфа гидрохлорида 2-[4-(метиламинометил)фенил]-5-фтор-бензофуран-7-карбоксамида | 2018 |
|
RU2783418C1 |
| КРИСТАЛЛИЧЕСКИЕ ФОРМЫ ИНГИБИТОРА LTA4H | 2019 |
|
RU2808992C2 |
| КРИСТАЛЛИЧЕСКАЯ ФОРМА И СПОСОБ ЕЕ ОЧИСТКИ | 2011 |
|
RU2604734C2 |
| СПОСОБ ПОЛУЧЕНИЯ АГОНИСТА A-АДЕНОЗИНОВОГО РЕЦЕПТОРА И ЕГО ПОЛИМОРФОВ | 2007 |
|
RU2447081C2 |
| НОВАЯ ПОЛИМОРФНАЯ ФОРМА КАЛЬЦИМИМЕТИЧЕСКОГО СОЕДИНЕНИЯ | 2011 |
|
RU2599788C2 |
| ТВЕРДЫЕ ФОРМЫ, СОДЕРЖАЩИЕ (-)-О-ДЕСМЕТИЛВЕНЛАФАКСИН, И ИХ ПРИМЕНЕНИЯ | 2008 |
|
RU2477269C2 |
| СПОСОБ ПОЛУЧЕНИЯ ПОЛИМОРФНОЙ ФОРМЫ 3-[5-АМИНО-4-(3-ЦИАНОБЕНЗОИЛ)ПИРАЗОЛ-1-ИЛ]-N-ЦИКЛОПРОПИЛ-4-МЕТИЛБЕНЗАМИДА | 2017 |
|
RU2765719C2 |
| КРИСТАЛЛИЧЕСКИЕ ФОРМЫ ИНГИБИТОРА TLR7/TLR8 | 2019 |
|
RU2792005C2 |
| КРИСТАЛЛИЧЕСКИЕ СОЛИ И ПОЛИМОРФЫ АНТАГОНИСТА P2X3 | 2017 |
|
RU2782068C2 |
| КРИСТАЛЛИЧЕСКИЕ ФОРМЫ ИНГИБИТОРА BTK | 2020 |
|
RU2828460C2 |
Изобретение относится к способу получения негидратируемой кристаллической формы (полиморфа А) 3-бром-1-(3-хлор-2-пиридинил)-N-[4-циано-2-метил-6-[(метиламино)карбонил]фенил]-1Н-пиразол-5-карбоксамида (соединение 1), которая характеризуется дифракционной рентгенограммой с пиками углов отражения, указанными в формуле изобретения. Согласно изобретению способ включает нагревание гидратируемой кристаллической формы соединения 1 (полиморфа В), имеющей характеристики рентгеновской дифракции, указанные в формуле изобретения, при температуре от приблизительно 40°С до точки кипения растворителя смеси, содержащей растворитель, выбранный из группы, состоящей из воды, н-гептана, 1-хлорбутана, толуола, 1-бутанола и 1-пентанола. Технический результат - получение стабильной полиморфной формы А соединения 1, что позволяет получать стабильные твердые инсектицидные составы. 14 з.п. ф-лы, 2 ил., 6 табл., 7 пр.
1. Способ получения полиморфа А 3-бром-1-(3-хлор-2-пиридинил)-N-[4-циано-2-метил-6-[(метиламино)карбонил]фенил]-1Н-пиразол-5-карбоксамида, характеризующегося дифракционной рентгенограммой, которая имеет, по меньшей мере, положения максимумов для угла отражения, определенного в градусах 2θ,
включающий нагревание при температуре от приблизительно 40°С до точки кипения растворителя смеси, содержащей растворитель, выбранный из группы, состоящей из воды, н-гептана, 1-хлорбутана, толуола, 1-бутанола и 1-пентанола, и полиморф В 3-бром-1-(3-хлор-2-пиридинил)-N-[4-циано-2-метил-6-[(метиламино)карбонил]фенил]-1Н-пиразол-5-карбоксамида, характеризующегося дифракционной рентгенограммой, которая имеет, по меньшей мере, положения максимумов для угла отражения, определенного в градусах 2θ,
2. Способ по п.1, где растворителем является н-гептан.
3. Способ по п.1, где растворителем является толуол.
4. Способ по п.1, где растворителем является 1-хлорбутан.
5. Способ по п.1, где растворителем является 1-бутанол или 1-пентанол.
6. Способ по п.1, где растворителем является вода.
7. Способ по п.6, где температура составляет от приблизительно 60 до приблизительно 100°С.
8. Способ по п.7, где температура составляет от приблизительно 70 до приблизительно 100°С.
9. Способ по п.8, где температура составляет от приблизительно 70 до приблизительно 90°С.
10. Способ по п.6, где смесь нагревают в течение, по меньшей мере, приблизительно 2 ч.
11. Способ по п.10, где смесь нагревают в течение не более чем приблизительно 48 ч.
12. Способ по п.11, где смесь нагревают в течение не более чем приблизительно 24 ч.
13. Способ по п.12, где смесь нагревают в течение не более чем приблизительно 12 ч.
14. Способ по п.6, где приблизительно 0,1-10 мас.% полиморфа А относительно массы полиморфа В добавляют к смеси перед нагреванием.
15. Способ по п.14, где приблизительно 0,2-5 мас.% полиморфа А относительно массы полиморфа В добавляют к смеси перед нагреванием.
| Пломбировальные щипцы | 1923 |
|
SU2006A1 |
| RU 2005127049 A, 27.01.2006. | |||

