Область изобретения
Изобретение относится к соединениям, которые ингибируют внеклеточное высвобождение воспалительных цитокинов, причем указанные цитокины являются ответственными за одно или несколько патологических состояний человека или высших млекопитающих. Настоящее изобретение относится далее к композициям, включающим указанные соединения, и способу предупреждения, ослабления или регулирования иным путем действия ферментов, которые, как предполагается, являются активными компонентами, ответственными за описываемые в данном описании патологические состояния.
Предпосылки создания изобретения
Интерлейкин-1 (IL-1) и фактор-α некроза опухоли (TNF-α) среди важных биологических веществ известны совместно как «цитокины». Предполагается, что указанные молекулы опосредуют воспалительные реакции, ассоциированные с иммунологическим распознаванием инфекционных агентов.
Предполагается, что указанные провоспалительные цитокины являются важными медиаторами при многих патологических состояниях или синдромах, среди прочего, ревматоидном артрите, остеоартрите, воспалительном заболевании кишечника (IBS), септическом шоке, сердечно-легочной дисфункции, остром респираторном заболевании, кахексии и, следовательно, являются ответственными за развитие и проявление патологических состояний человека.
Поэтому имеется давно ощущаемая потребность в соединениях и фармацевтических композициях, которые включают соединения, которые могут блокировать, ослаблять, регулировать, подавлять или предупреждать высвобождение цитокинов из клеток, которые продуцируют их.
Краткое изложение сущности изобретения
Настоящее изобретение удовлетворяет вышеуказанным потребностям, поскольку неожиданно было обнаружено, что [5,6]- и [5,6,6]-конденсированные пиразолоны и их производные являются эффективными для ингибирования высвобождения воспалительных цитокинов, среди прочих, интерлейкина-1 (IL-1) и фактора некроза опухоли (TNF) из клеток и тем самым предупреждают, ослабляют или регулируют иным путем действия ферментов, которые, как предполагается, являются активными компонентами, ответственными за описанные здесь патологические состояния.
Первый аспект настоящего изобретения относится к соединениям, включающим все их энантиомерные и диастереомерные формы и фармацевтически приемлемые соли, причем соединения имеют формулу:
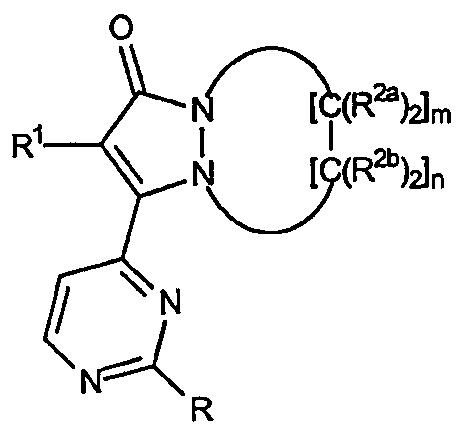
где R представляет собой:
а) водород;
b) -O(CH2)kR3 или
с) -NR4aR4b;
R3 представляет собой замещенный или незамещенный С1-С4алкил, замещенный или незамещенный циклический гидрокарбил, замещенный или незамещенный гетероциклил, замещенный или незамещенный арил, замещенный или незамещенный гетероарил; индекс k равен от 0 до 5;
R4a иR4b представляют собой, каждый независимо:
а) водород или
b) -[С(R5aR5b)]xR6;
каждый R5a иR5b представляет собой независимо водород, -OR7, -N(R7)2, -CO2R7, -CON(R7)2; неразветвленный, разветвленный или циклический С1-С4алкил и их смеси; R6 представляет собой водород, -OR7, -N(R7)2, -CO2R7, -CON(R7)2, замещенный или незамещенный С1-С4алкил, замещенный или незамещенный арил или замещенный или незамещенный гетероарил; R7 представляет собой водород, водорастворимый катион, С1-С4алкил или замещенный или незамещенный арил; индекс х равен от 0 до 5;
R1 представляет собой:
а) замещенный или незамещенный арил или
b) замещенный или незамещенный гетероарил;
звенья R2a и R2b выбраны, каждый независимо, из группы, состоящей из:
а) водорода;
b) -O(CH2)jR8;
c) -(CH2)jN9aR9b;
d) -(CH2)jCO2R10;
e) -(CH2)jOCO2R10;
f) -(CH2)jCON(R10)2;
g) карбонила, когда два звена R2a или два звена R2b у одного и того же атома углерода могут быть взяты вместе с образованием карбонильного звена;
h) двойной связи, когда один R2a и один R2b взяты вместе с образованием двойной связи;
i) кольца, когда один R2a и один R2b взяты вместе с образованием замещенного или незамещенного кольца, включающего от 4 до 8 атомов, причем указанное кольцо выбрано из группы, состоящей из:
i) карбоциклического;
ii) гетероциклического;
iii) арильного;
iv) гетероарильного;
v) бициклического и
vi) гетеробициклического кольца
j) и их смесей;
R8, R9a, R9b и R10 представляют собой, каждый независимо, водород, С1-С4алкил и их смеси; R9a и R9b могут быть взяты вместе с образованием карбоциклического или гетероциклического кольца, включающего от 3 до 7 атомов; два звена R10 могут быть взяты вместе с образованием карбоциклического или гетероциклического кольца, включающего от 3 до 7 атомов; j представляет индекс от 0 до 5; m представляет индекс от 1 до 5; n представляет индекс от 1 до 5, m + n равно от 2 до 6.
Другой аспект настоящего изобретения относится к фармацевтическим композициям, которые могут доставлять соединения настоящего изобретения в организм человека или высшего млекопитающего, причем композиции включают:
а) эффективное количество одного или нескольких соединений настоящего изобретения и
b) один или несколько фармацевтически приемлемых эксципиентов.
Следующий аспект настоящего изобретения относится к способам борьбы с опосредованными одним или несколькими воспалительными цитокинами или модулированными воспалительными цитокинами заболеваниями или состояниями млекопитающих, причем указанный способ включает стадию введения человеку или высшему млекопитающему эффективного количества композиции, включающей одно или несколько соединений настоящего изобретения.
Эти и другие цели, отличительные признаки и преимущества станут очевидны специалисту в данной области после прочтения следующего подробного описания и прилагаемой формулы изобретения. Все проценты, соотношения и пропорции здесь являются массовыми, если не оговорено особо. Все температуры приводятся в градусах Цельсия (°C), если не оговорено особо. Все цитированные документы находятся в соответствующей части и включены здесь в качестве ссылки; цитирование любого документа не должно быть истолковано как допущение, что он является известным уровнем относительно настоящего изобретения.
Подробное описание изобретения
Настоящее изобретение относится к соединениям, которые способны опосредовать, регулировать или ингибировать иным путем внеклеточное высвобождение некоторых цитокинов, особенно воспалительных цитокинов, причем указанные цитокины играют роль в стимуляции, вызывании или проявлении большого числа заболеваний, патологических состояний или синдромов.
Для целей настоящего изобретения термин «гидрокарбил» определяют здесь как любое органическое звено или радикал, который состоит из атомов углерода и водорода. В термин гидрокарбил включены гетероциклы, которые описаны ниже. Примеры различных незамещенных негетероциклических гидрокарбильных звеньев включают пентил, 3-этилоктанил, 1,3-диметилфенил, циклогексил, цис-3-гексил, 7,7-диметилбицикло[2.2.1]гептан-1-ил и нафт-2-ил.
В определение «гидрокарбил» включены ароматические (арильные) и неароматические карбоциклические кольца, неограничивающие примеры которых включают циклопропил, циклобутанил, циклопентанил, циклогексан, циклогексенил, циклогептанил, бицикло[0.1.1]бутанил, бицикло[0.1.2]пентанил, бицикло[0.1.3]гексанил (туйанил), бицикло[0.2.2]гексанил, бицикло[0.1.4]гептанил (каранил), бицикло[2.2.1]гептанил (норборанил), бицикло[0.2.4]октанил (кариофилленил), спиропентанил, дициклопентанспиранил, декалинил, фенил, бензил, нафтил, инденил, 2Н-инденил, азуленил, фенантрил, антрил, флуоренил, аценафтиленил, 1,2,3,4-тетрагидронафталенил и тому подобное.
Термин «гетероцикл» включает как ароматические (гетероарильные), так и неароматические гетероциклические кольца, неограничивающие примеры которых включают: пирролил, 2Н-пирролил, 3Н-пирролил, пиразолил, 2Н-имидазолил, 1,2,3-триазолил, 1,2,4-триазолил, изоксазолил, оксазолил, 1,2,4-оксадиазолил, 2Н-пиранил, 4Н-пиранил, 2Н-пиран-2-онил, пиридинил, пиридазинил, пиримидинил, пиразинил, пиперазинил, симм-триазинил, 4Н-1,2-оксазинил, 2Н-1,3-оксазинил, 1,4-оксазинил, морфолинил, азепинил, оксепинил, 4Н-1,2-диазепинил, инденил, 2Н-инденил, бензофуранил, изобензофуранил, индолил, 3Н-индолил, 1Н-индолил, бензоксазолил, 2Н-1-бензопиранил, хинолинил, изохинолинил, хиназолинил, 2Н-1,4-бензоксазинил, пирролидинил, пирролинил, хиноксалинил, фуранил, тиофенил, бензимидазолил и тому подобное, причем каждый из них может быть замещенным или незамещенным.
Примером звена, определяемого термином «алкиленарил», является бензильное звено, имеющее формулу:
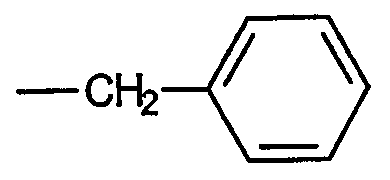
тогда как примером звена, определяемого термином «алкиленгетероарил», является 2-пиколильное звено, имеющее формулу:
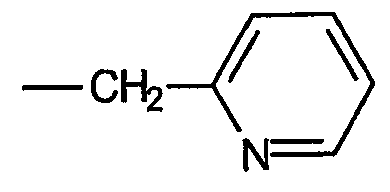
Термин «замещенный» используют на всем протяжении описания. Термин «замещенный» определяют здесь как «охватывающий радикалы или звенья, которые могут заменить атом водорода, два атома водорода или три атома водорода гидрокарбильного радикала. Термин «замещенный» может также включать замену атомов водорода у двух соседних атомов углерода с образованием нового радикала или звена». Например, замещенное звено, которое требует замены одного атома водорода, включает галоген, гидроксил и тому подобное. Замена двух атомов водорода включает карбонил, оксимино и тому подобное. Замена двух атомов водорода у соседних атомов углерода включает эпокси и тому подобное. Замена трех атомов водорода включает циано и тому подобное. Эпоксидное звено является примером замещенного звена, которое требует замены атома водорода у соседних атомов углерода. Термин «замещенный», используемый на всем протяжении настоящего описания, указывает на то, что гидрокарбильный радикал, среди прочего, ароматическое кольцо, алкильная цепь, может иметь один или несколько атомов водорода, замененных заместителем. Когда радикал описан как «замещенный», может быть заменено любое число атомов водорода. Например, 4-гидроксифенил представляет собой «замещенное ароматическое карбоциклическое кольцо», (N,N-диметил-5-амино)октанил представляет собой «замещенное С8алкильное звено, 3-гуанидинопропил представляет собой «замещенное С3алкильное звено» и 2-карбоксипиридинил представляет собой «замещенное гетероарильное звено». Следующие звенья представляют собой неограничивающие примеры звеньев, которые могут служить в качестве замены атомов водорода, когда гидрокарбильное звено описано как «замещенное».
i) -[C(R12)2]p(CH=CH)qR12, где р равно от 0 до 12; q равно от 0 до 12;
ii) -C(Z)R12;
iii) -C(Z)2R12;
iv) -C(Z)CH=CH2;
v) -C(Z)N(R12)2;
vi) -C(Z)NR12N(R12)2;
vii) -CN;
viii) -CNO;
ix) -CF3, -CCl3, -CBr3;
x) -N(R12)2;
xi) -NR12CN;
xii) -NR12C(Z)R12;
xiii) -NR12C(Z)N(R12)2;
xiv) -NHN(R12)2;
xv) -NHOR12;
xvi) -NCS;
xvii) -NO2;
xviii) -OR12;
xix) -OCN;
xx) -OCF3, -OCCl3, -OCBr3;
xxi) -F, -Cl, -Br, -I и их смеси;
xxii) -SCN;
xxiii) -SO3M;
xxiv) -OSO3M;
xxv) -SO2N(R12)2;
xxvi) -SO2R12;
xxvii) -P(O)H2;
xxviii) -PO2;
xxix) -P(O)(OH)2;
xxx) и их смеси,
где R12 представляет собой водород, замещенный или незамещенный неразветвленный, разветвленный или циклический С1-С20алкил, С6-С20арил, С7-С20алкиленарил и их смеси; М представляет собой водород или солеобразующий катион; Z представляет собой =О, =S, =NR12 и их смеси. Подходящие солеобразующие катионы включают натрий, литий, калий, кальций, магний, аммоний и тому подобное.
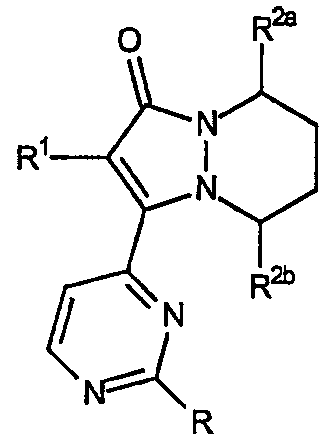
Первый аспект настоящего изобретения, в общем, относится к новым соединениям, подходящим для ингибирования высвобождения воспалительных цитокинов, причем указанные соединения имеют формулу:
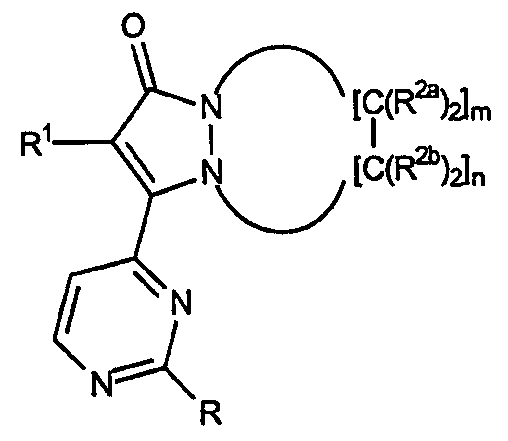
R представляет собой заместитель в положении 2 пиримидин-4-ильной части общей основной структуры, указанное звено R представляет собой:
а) простой эфир, имеющий формулу -О[CH2]kR3 или
b) первичное или вторичное аминозвено, имеющее формулу -NR4aR4b;
где R3 представляет собой замещенный или незамещенный С1-С4алкил, замещенный или незамещенный циклический гидрокарбил, замещенный или незамещенный гетероциклил, замещенный или незамещенный арил, замещенный или незамещенный гетероарил, индекс k равен от 0 до 5.
Ниже указываются различные варианты звеньев R настоящего изобретения, где R представляет собой простой эфир, имеющий формулу -О[CH2]kR3. Однако специалист в данной области не ограничивается иллюстративными вариантами и примерами данного описания.
А) Звенья R включают простые эфиры формулы -OR3 (индекс k равен 0), где R3 представляет собой замещенный или незамещенный арил.
i) Один вариант данного аспекта R включает простые эфиры, имеющие формулу -OR3, где R3 представляет собой замещенный или незамещенный арил. Данный вариант включает следующие неограничивающие примеры R: фенокси, 2-фторфенокси, 3-фторфенокси, 4-фторфенокси, 2,4-дифторфенокси, 3-трифторметилфенокси, 4-трифторметилфенокси, 2,4-дифторметилфенокси и тому подобное.
ii) Другой вариант данного аспекта R включает простые эфиры, имеющие формулу -OR3, где R3 представляет собой замещенный или незамещенный арил. Данный вариант включает следующие неограничивающие примеры: 2-метилфенокси, 3-метилфенокси, 4-метилфенокси, 2,4-диметилфенокси, 2-цианофенокси, 3-цианофенокси, 4-цианофенокси, 4-этилфенокси и тому подобное.
iii) Следующий вариант данного аспекта R включает простые эфиры, имеющие формулу -OR3, где R3 представляет собой замещенный или незамещенный арил. Данный вариант включает следующие неограничивающие примеры: (2-метокси)фенокси, (3-метокси)фенокси, (4-метокси)фенокси, 3-[(N-ацетил)амино]фенокси, 3-бензо[1,3]диоксол-5-ил и тому подобное.
В) Звенья R, включающие простые эфиры, имеющие формулу -OR3 (индекс k равен 0), где R3 представляет собой замещенный или незамещенный гетероарил.
i) Первый вариант данного аспекта R включает простые эфиры, имеющие формулу -OR3, где R3 представляет собой незамещенный гетероарил. Данный вариант включает следующие неограничивающие примеры: пиримидин-2-ил, пиримидин-4-ил, пиридин-2-ил, пиридин-3-ил, пиридин-4-ил и тому подобное.
ii) Второй вариант данного аспекта R включает простые эфиры, имеющие формулу -OR3, где R3 представляет собой замещенный гетероарил. Данный вариант включает следующие неограничивающие примеры: 2-аминопиримидин-4-ил и тому подобное.
С) Звенья R, включающие простые эфиры, имеющие формулу -OCH2R3 (индекс k равен 1), где R3 представляет собой замещенный или незамещенный арил.
i) Первый вариант данного аспекта R включает простые эфиры, имеющие формулу -OCH2R3, где R3 представляет собой замещенный или незамещенный гетероарил. Данный вариант включает следующие неограничивающие примеры: пиримидин-2-ил, пиримидин-4-ил, 2-аминопиримидин-4-ил, 4-аминопиримидин-6-ил, пиридин-2-ил, пиридин-3-ил, пиридин-4-ил и тому подобное.
ii) Второй вариант данного аспекта R включает простой эфир, имеющий формулу -OCH2R3, где R3 представляет собой замещенный или незамещенный алкиленгетероарил. Данный вариант включает следующие неограничивающие примеры: пиридин-3-илэтил, (2-метил-2-пиридин-3-ил)этил и тому подобное.
D) Звенья R, включающие простые эфиры, имеющие формулу -OR3 (индекс k равен 1), где R3 представляет собой замещенный или незамещенный С1-С4алкил.
i) Первый вариант данного аспекта R представляет собой простой эфир, имеющий формулу -OR3, где R3 представляет собой незамещенный неразветвленный, разветвленный или циклический С1-С4алкил. Данный вариант включает следующие неограничивающие примеры: метил, этил, изопропил, (S)-1-метилпропил и тому подобное.
ii) Второй вариант данного аспекта R представляет собой простой эфир, имеющий формулу -OR3, где R3 представляет собой замещенный неразветвленный, разветвленный или циклический С1-С4алкил. Данный вариант включает следующие неограничивающие примеры: 2-метоксиэтил, (S)-1-метил-3-метилоксипропил и тому подобное.
Ниже указываются различные аспекты звеньев R настоящего изобретения, где R представляет собой амин, имеющий формулу -NR4aR4b, R4a и R4b представляют собой, каждый независимо:
а) водород или
b) -[C(R5aR5b)]xR6;
каждый из R5a и R5b представляет собой, независимо, водород, -OR7, -N(R7)2, -CO2R7; -CON(R7)2; неразветвленный, разветвленный или циклический С1-С4алкил и их смеси; R6 представляет собой водород, -OR7, -N(R7)2, -CO2R7, -CON(R7)2; замещенный или незамещенный С1-С4алкил, замещенный или незамещенный арил или замещенный или незамещенный гетероарил; R7 представляет собой водород, водорастворимый катион, С1-С4алкил или замещенный или незамещенный арил; индекс х равен от 0 до 5. Однако специалист в данной области не ограничивается иллюстративными вариантами и примерами данного описания.
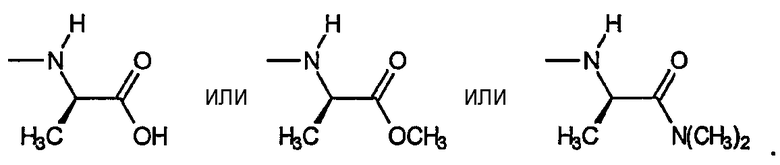
А) Звенья R включают хиральные аминогруппы, где R4a представляет собой водород, R5a представляет собой водород и R5b представляет собой метил, причем указанные звенья имеют формулу:
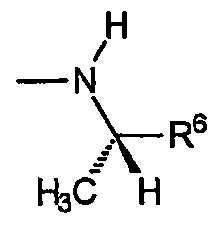
и указанную стереохимию.
i) Первым вариантом данного аспекта R является амин, включающий R6, который представляет собой замещенный или незамещенный фенил. Данный вариант включает следующие неограничивающие примеры: (S)-1-метил-1-фенилметиламино, (S)-1-метил-1-(4-фторфенил)метиламино, (S)-1-метил-1-(4-метилфенил)метиламино, (S)-1-метил-1-(4-метоксифенил)метиламино, (S)-1-метил-1-(2-аминофенил)метиламино, (S)-1-метил-1-(4-аминофенил)метиламино и тому подобное.
ii) Вторым вариантом данного аспекта R является амин, включающий R6, который представляет собой замещенный или незамещенный гетероарил. Данный вариант включает следующие неограничивающие примеры: (S)-1-метил-1-(пиридин-2-ил)метиламино, (S)-1-метил-1-(пиридин-3-ил)метиламино, (S)-1-метил-1-(пиридин-4-ил)метиламино, (S)-1-метил-1-(фуран-2-ил)метиламино, (S)-1-метил-1-(3-бензо[1,3]диоксол-5-ил)метиламино и тому подобное.
iii) Третьим вариантом данного аспекта R является амин, включающий R6, который представляет собой замещенный или незамещенный С1-С4алкил. Данный вариант включает следующие неограничивающие примеры: (S)-1-метилпропиламино, (S)-1-метил-2-(метокси)этиламино.
В) Звенья R включают хиральные аминогруппы, где R4a представляет собой водород, R5a и R5b представляют собой, каждый, С1-С4алкил, причем указанные звенья имеют формулу:
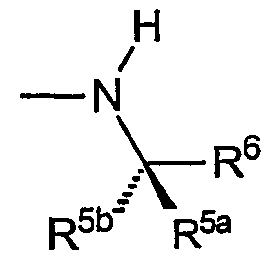
и указанную стереохимию, когда R5a, R5b и R6 не являются одинаковыми.
i) Первым вариантом данного аспекта R является амин, который не имеет хирального центра и неограничивающие примеры которого включают 1,1-диметилэтиламин, 1,1-диметилбензиламин и тому подобное.
ii) Вторым вариантом данного аспекта R является амин, включающий R6, который представляет собой замещенный или незамещенный С1-С4алкил. Данный вариант включает следующие неограничивающие примеры: (S)-1-метил-2-гидрокси-2-метилпропиламин, (S)-1-метил-2-гидрокси-2-метилбутиламин и тому подобное.
С) Звенья R, включающие алкиленариламины, где R4a представляет собой водород, оба R5a и R5b радикала R4b представляют собой водород, R6 представляет собой замещенный или незамещенный арил, причем указанное звено имеет формулу:
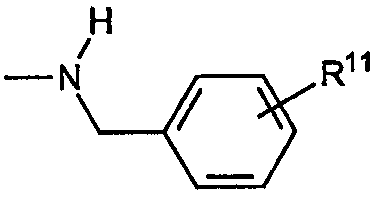
где R11 представляет собой водород или «замещенное звено», как указано выше.
i) Первый вариант данного аспекта включает следующие неограничивающие примеры звеньев R: бензиламино, (2-аминофенил)метиламино; (4-фторфенил)метиламино, (4-метоксифенил)метиламино; (4-пропансульфонилфенил)метиламино и тому подобное.
i) Второй вариант данного аспекта включает следующие неограничивающие примеры звеньев R: (2-метилфенил)метиламино; (3-метилфенил)метиламино, (4-метилфенил)метиламино и тому подобное.
D) Звенья R, включающие амины, где R4a представляет собой водород, R4b включает R5b, равный водороду, и R5b, равный -СО2R7 или -CON(R7)2; причем указанное звено имеет формулу:
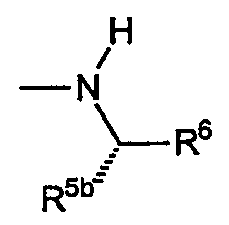
i) Первым вариантом данного аспекта R является амин, включающий R6, который представляет собой замещенный или незамещенный фенил. Данный вариант включает следующие неограничивающие примеры:
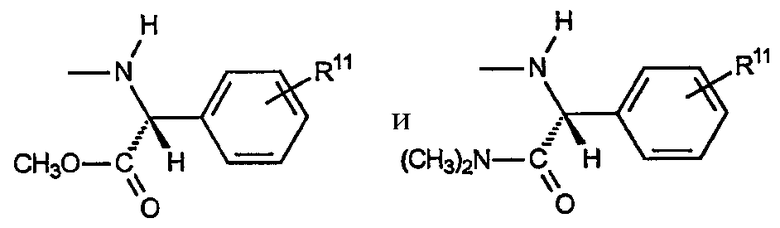
где R11 представляет собой водород или «заместитель», как указано выше.
ii) Вторым вариантом данного аспекта R является амин, включающий R6, который представляет собой замещенный или незамещенный алкил. Данный вариант включает следующие неограничивающие примеры:
Звенья R1 выбраны из:
а) замещенного или незамещенного арила или
b) замещенного или незамещенного гетероарила.
Первый аспект звеньев R1 охватывает галогензамещенные фенильные звенья, неограничивающие примеры которых включают 4-фторфенил, 2,4-дифторфенил, 4-хлорфенил и тому подобное. Звенья R2a и R2b выбраны, каждый независимо, из группы, состоящей из:
а) водорода;
b) -O(CH2)jR8;
c) -(CH2)jN9aR9b;
d) -(CH2)jCO2R10;
e) -(CH2)jOCO2R10;
f) -(CH2)jCON(R10)2;
g) карбонильного звена, когда два R2a или два R2b у одного и того же атома углерода могут быть взяты вместе с образованием карбонильного звена;
h) двойной связи, когда один R2a и один R2b взяты вместе с образованием двойной связи;
i) кольца, когда один R2a и один R2b взяты вместе с образованием замещенного или незамещенного кольца, включающего от 4 до 8 атомов, причем указанное кольцо выбрано из группы, состоящей из:
i) карбоциклического;
ii) гетероциклического;
iii) арильного;
iv) гетероарильного;
v) бициклического и
vi) гетеробициклического кольца;
l) и их смесей;
R8, R9a, R9b и R10 представляют собой, каждый независимо, водород, С1-С4алкил и их смеси; R9a и R9b могут быть взяты вместе с образованием карбоциклического или гетероциклического кольца, включающего от 3 до 7 атомов; два звена R10 могут быть взяты вместе с образованием карбоциклического или гетероциклического кольца, включающего от 3 до 7 атомов; j представляет индекс от 0 до 5.
Системы [5,6]-конденсированных колец
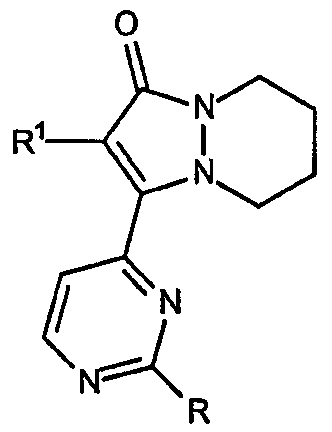
Первый аспект настоящего изобретения относится к циклическим основным структурам, в которых индексы m и n, каждый, равны 2, тем самым включая остовы 2-(R1-замещенный)-3-(2-R-замещенный-пиримидин-4-ил)-5,6,7,8-тетрагидропиразоло[1,2-a]пиридазин-1-она, имеющим формулу:
где звенья R2a и R2b представляют собой, каждый независимо, водород, -(CH2)jCO2R10, -(CH2)jCON(R10)2 и их смеси.
Варианты данной основной структуры включают циклический остов, имеющий формулу:
находящиеся в положении 8 сложные эфиры и амиды 5,6,7,8-тетрагидропиразоло[1,2-a]пиридазин-1-она, имеющие формулу:
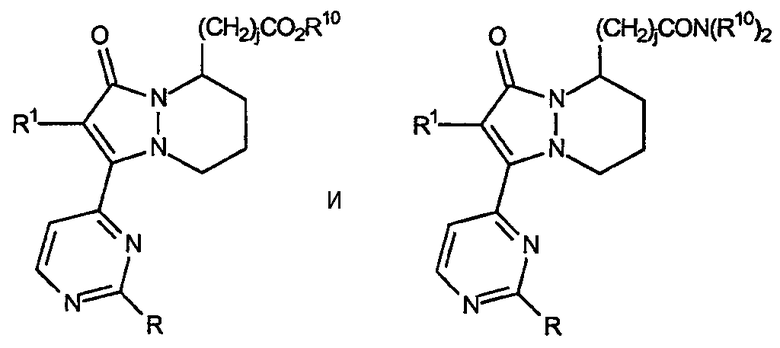
а также находящиеся в положении 5 сложные эфиры и амиды 5,6,7,8-тетрагидропиразоло[1,2-a]пиридазин-1-она, имеющие формулу:
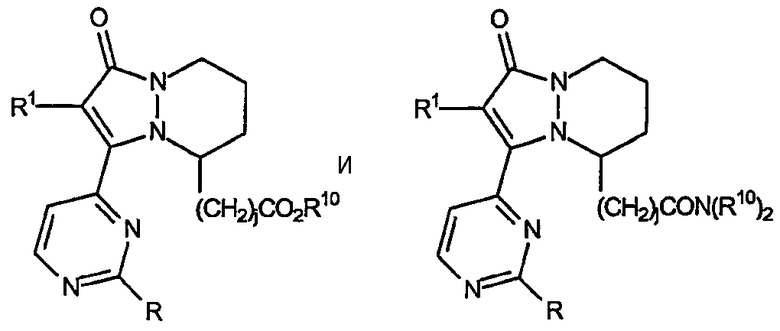
Второй аспект настоящего изобретения, когда он относится к звеньям R2a и R2b, включает остовы 2-(R1-замещенный)-3-(2-R-замещенный-пиримидин-4-ил)-5,6,7,8-тетрагидропиразоло[1,2-a]пиридазин-1-она, имеющие формулу:
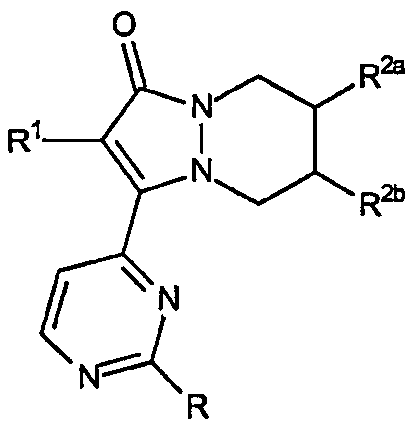
где каждое из звеньев R2a и R2b независимо выбрано из группы, состоящей из:
а) водорода;
b) -O(CH2)jR8 и
с) -(CH2)jNR9aR9b.
Варианты данного аспекта включают 6-гидрокси-2-(R1-замещенный)-3-(2-R-замещенный-пиримидин-4-ил)-5,6,7,8-тетрагидропиразоло[1,2-a]пиридазин-1-оны, 7-гидрокси-2-(R1-замещенный)-3-(2-R-замещенный-пиримидин-4-ил)-5,6,7,8-тетрагидропиразоло[1,2-a]пиридазин-1-оны, 6-(диметиламино)-2-(R1-замещенный)-3-(2-R-замещенный-пиримидин-4-ил)-5,6,7,8-тетрагидропиразоло[1,2-a]пиридазин-1-оны, 6-морфолино-2-(R1-замещенный)-3-(2-R-замещенный-пиримидин-4-ил)-5,6,7,8-тетрагидропиразоло[1,2-a]пиридазин-1-оны.
Третий аспект настоящего изобретения, когда он относится к звеньям R2a и R2b, включает основные структуры, в которых два соседних звена R2a и R2b взяты вместе с образованием двойной связи, например остовы 2-(R1-замещенный)-3-(2-R-замещенный-пиримидин-4-ил)-5,8-дигидропиразоло[1,2-a]пиридазин-1-она, имеющие формулу:
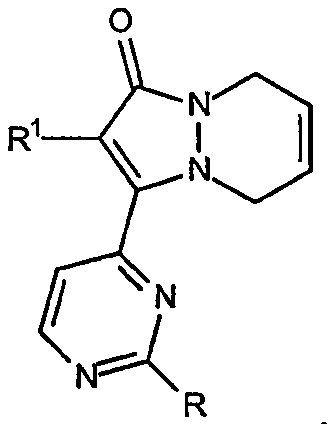
Системы [5,6,X]-конденсированных колец
Настоящее изобретение относится также к [5,6,X]-конденсированным кольцевым системам, где Х представляет собой кольцо, образованное, когда один R2a и один R2b взяты вместе с образованием замещенного или незамещенного кольца, включающего от 4 до 8 атомов. Образованные кольца выбраны из группы, состоящей из:
i) карбоциклического;
ii) гетероциклического;
iii) арильного;
iv) гетероарильного;
v) бициклического и
vi) гетеробициклического кольца.
Первый вариант осуществления данного аспекта относится к кольцевым системам, в которых один R2a и один R2b взяты вместе с образованием 6-членного арильного кольца, среди прочего, системе [5,6,6]-конденсированных колец; 2-(R1-замещенный)-3-(2-R-замещенный-пиримидин-4-ил)-5,10-дигидропиразоло[1,2-b]фталазин-1-ону, имеющему формулу:
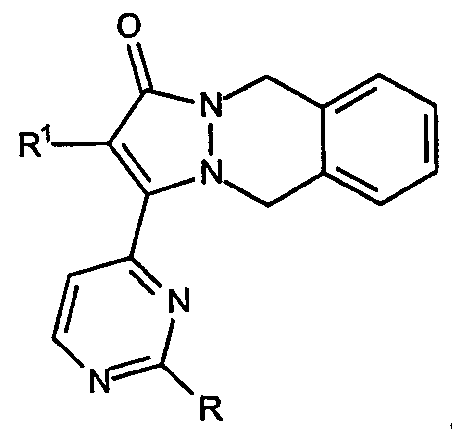
Варианты данного аспекта включают аналоги, которые замещены в С-кольце, например соединения, имеющие формулу:
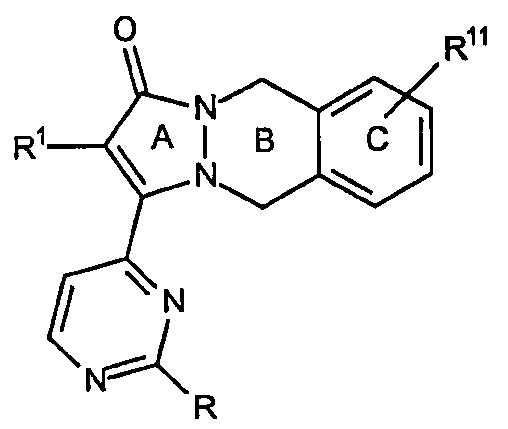
где R12 представляет собой заместитель, как указано выше. Неограничивающими примерами [5,6,6]-циклических основных структур настоящего изобретения являются циклические остовы 2-(R1-замещенный)-3-(2-R-замещенный-пиримидин-4-ил)-5,10-дигидропиразоло[1,2-b]фталазин-1-она, например, соединение, имеющее формулу:
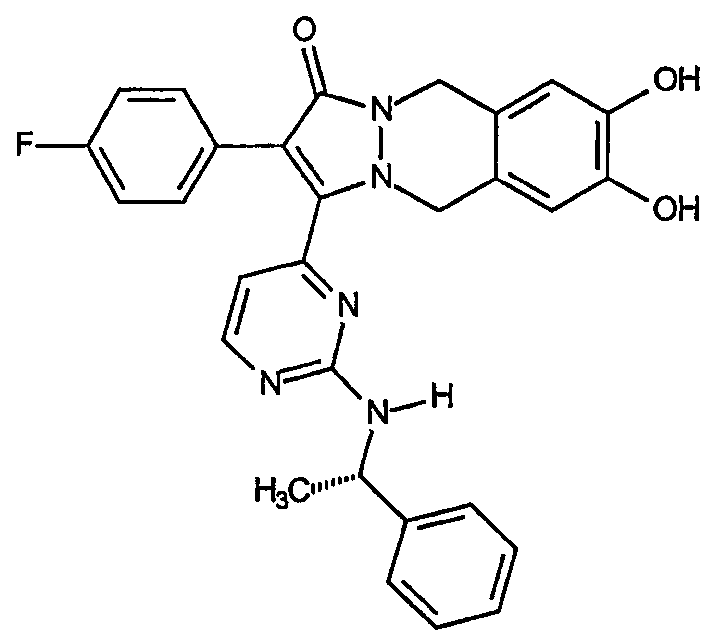
Первый аспект аналогов категории 1, способных ингибировать высвобождение воспалительных цитокинов настоящего изобретения, относится к соединениям, включающим остов 5,10-дигидропиразоло[1,2-b]фталазин-1-она, имеющим формулу:
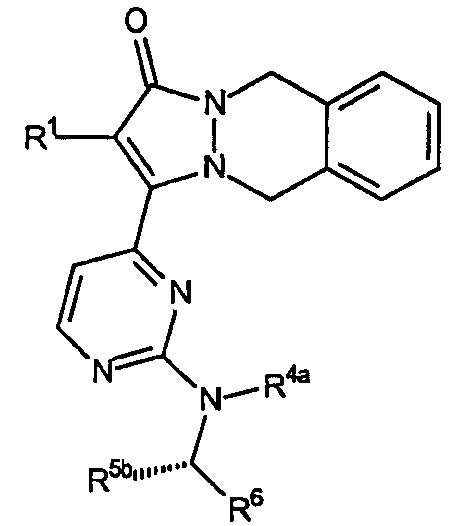
где звенья R представляют собой амины, имеющие формулу -NH[CHR5]R6, и R1, R4a, R5 и R6 имеют значения, указанные ниже в таблице I. Стереохимия R5b является конфигурацией, показанной, когда R5b не является водородом.
Ниже представлена схема получения соединений, относящихся к первому аспекту категории I настоящего изобретения. Первая стадия включает использование промежуточных соединений типа I, например промежуточного соединения 3 для введения выбранного звена R1, в соединяющуюся основную структуру.
Общая схема для получения промежуточного соединения типа I
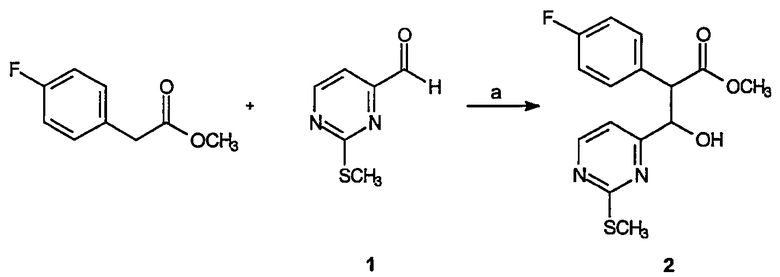
Реагенты и условия: (а) LDA, ТГФ, -78°C, 45 мин.
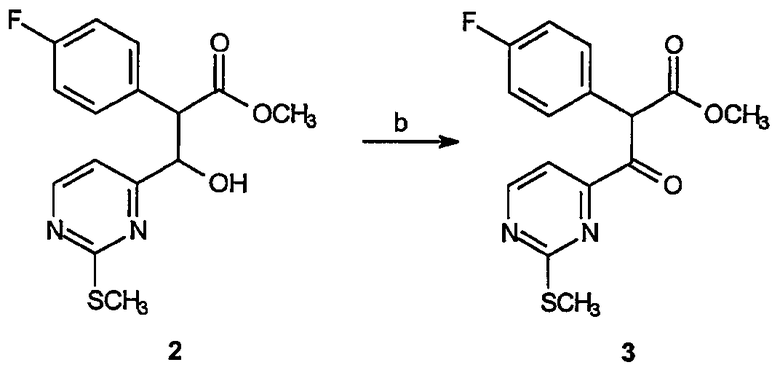
Реагенты и условия: (b) CrO3, CH2Cl2, комнатная температура, 16 час.
ПРИМЕР 1
Метиловый эфир 2-(4-фторфенил)-3-(2-метилсульфанилпиримидин-4-ил)-3-оксопропионовой кислоты (3)
Ниже представлена методика получения 2-метилсульфанилпиримидин-4-карбальдегида, 1, адаптированная из методики H.Bredereck et al., Chem. Ber., 97, pp 3407-3417 (1964), включенной в данное описание в качестве ссылки.
В 3-горлую колбу объемом 12 л в инертной атмосфере загружают диметилацетил-N,N-диметилформамид (801 г) и диметилацеталь пировиноградного альдегида (779 г). Смесь нагревают при кипячении с обратным холодильником в течение 18 часов, в течение этого времени температура понижается приблизительно от 109°C до приблизительно 80°C. Раствор охлаждают и добавляют метанол (4 л) для растворения неочищенного остатка. Раствор затем охлаждают до 20°C и добавляют тиомочевину (892 г, 11,7 моль). После перемешивания смеси в течение приблизительно 15 минут добавляют метоксид натрия (741 г, 13,7 моль) 4 равными порциями на протяжении 1 часа, поддерживая температуру раствора в диапазоне 18-28°C. Смесь перемешивают в течение 5 часов при комнатной температуре, охлаждают до 20°C, затем на протяжении 1,25 часа добавляют метилиодид (2 кг), поддерживая температуру реакции в диапазоне 17-29°C. Перемешивание продолжают в течение 18 часов при комнатной температуре. Метанол и непрореагировавший метилиодид удаляют нагреванием раствора при 35°C и 40 мм рт. ст., получая приблизительно 4,46 кг темного остатка, который распределяют между 14 л воды и 5 л этилацетата. Водную фракцию экстрагируют второй раз этилацетатом, органические слои объединяют и концентрируют в вакууме с получением 685 г масла, которое очищают на диоксиде кремния, получая 522 г 4-диметоксиметил-2-метилсульфанилпиримидина.
Полученный выше диметилацеталь затем гидролизуют в свободный альдегид нагреванием до 60°C в течение 3 часов в 1М HCl. Нейтрализация и экстракция продукта с использованием этилацетата дает 347 г неочищенного продукта, который очищают на диоксиде кремния, получая 401 г 2-метилсульфанилпиримидин-4-карбальдегида, 1.
Получение метилового эфира 2-(4-фторфенил)-3-(2-метилсульфанилпиримидин-4-ил)-3-гидроксипропионовой кислоты (2): К холодному (-78°C) раствору диизопропиламида лития (21,4 мл 2М раствора в ТГФ, 42,8 ммоль) в ТГФ (70 мл) по каплям добавляют раствор метил 4-фторфенилацетата (6,0 г, 35,7 ммоль) в ТГФ (30 мл). Раствор перемешивают в течение 1 часа при -78°C, после чего к реакционной смеси по каплям добавляют раствор 2-метилсульфанилпиримидин-4-карбальдегида, 1 (6,0 г, 39,3 ммоль) в ТГФ (30 мл). Перемешивание продолжают в течение 45 минут при -78°C, затем реакционную смесь гасят, выливая реакционный раствор в водный насыщенный NH4Cl. Водную фазу экстрагируют этилацетатом. Органические фазы объединяют, сушат (MgSO4), фильтруют и концентрируют в вакууме. Неочищенный остаток очищают на диоксиде кремния (33% EtOAc/гексаны), получая 8,7 г (76%) требуемого продукта в виде смеси (1:1) диастереомеров.
Получение метилового эфира 2-(4-фторфенил)-3-(2-метилсульфанилпиримидин-4-ил)-3-оксопропионовой кислоты (3): К суспензии CrO3 в CH2Cl2 (300 мл) добавляют пиридин. Смесь энергично перемешивают в течение 1 часа при комнатной температуре. К хромсодержащей суспензии по каплям добавляют раствор неочищенного метилового эфира 2-(4-фторфенил)-3-(2-метилсульфанилпиримидин-4-ил)-3-гидроксипропионовой кислоты, 2, полученного выше, в CH2Cl2 (50 мл). Реакционную смесь перемешивают при комнатной температуре в течение 16 часов, разбавляют эфиром (1 л) и фильтруют через слой целита. Фильтрат концентрируют в вакууме и образовавшийся остаток очищают на диоксиде кремния (25% EtOAc/гексаны), получая 3,7 г (выход 43%) требуемого продукта в виде желтого твердого вещества.
Следующий пример относится к образованию 6,7-дигидро-5Н-пиразоло[1,2-a]пиразол-1-оновых кольцевых систем с использованием пиразолидина, однако специалист в данной области может заменить его другими циклическими гидразиновыми реагентами для получения других циклических систем настоящего изобретения, среди прочего, указывается использование гексагидропиридазина для получения 5,6,7,8-тетрагидропиразоло[1,2-a]пиридазин-1-онов. В примере, приведенном ниже, промежуточное соединение 3, полученное способом, описанным выше, используют для введения в качестве R1 4-фторфенильного звена, однако замещение для такого звена может быть выполнено во время получения промежуточного сложного β-кетоэфира.
Следующая схема иллюстрирует получение промежуточного соединения типа II, например промежуточного соединения 5, которое включает кольца В и С основной структуры.
Общая схема получения промежуточного соединения типа II
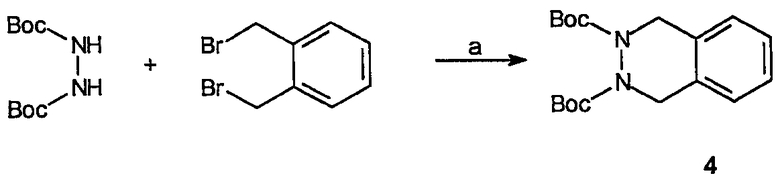
Реагенты и условия: (а) NaH, ДМФ, 90°C, 3 часа.
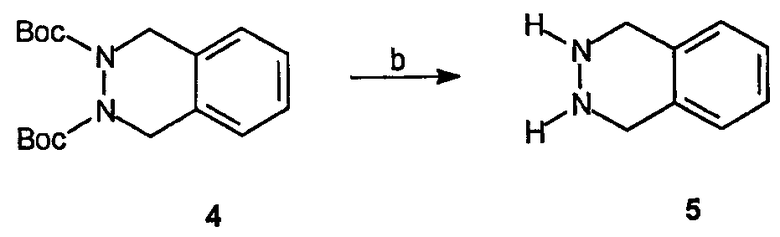
Реагенты и условия: (b) SOCl2, МеОН, комнатная температура, 72 часа.
ПРИМЕР 2
1,2,3,4-Тетрагидрофталазин (5)
Получение ди-трет-бутилового эфира 1,4-дигидрофталазин-2,3-дикарбоновой кислоты (4): К раствору ди-трет-бутилгидразодиформиата (3,0 г, 13,0 ммоль) в ДМФ (20 мл) при комнатной температуре добавляют NaH (0,5 г, 13,0 ммоль). После перемешивания 1 час при комнатной температуре к реакционной смеси добавляют 1,2-бисбромметилбензол (3,4 г, 13,0 ммоль). После перемешивания 1 час при комнатной температуре к реакционной смеси добавляют другую порцию NaH (0,5 г, 13,0 ммоль). Смесь затем нагревают до 90°C в течение 3 часов, дают ей охладиться до комнатной температуры и перемешивание продолжают при комнатной температуре в течение 15 часов. Реакционную смесь можно затем погасить, выливая реакционный раствор в водный насыщенный NH4Cl. Водную фазу экстрагируют эфиром, органическую фазу сушат (MgSO4), фильтруют и концентрируют в вакууме. Неочищенный остаток очищают на диоксиде кремния (5% EtOAc/гексаны), получая 1,0 г (выход 23%) требуемого продукта в виде прозрачного масла.
Получение 1,2,3,4-тетрагидрофталазина (5): Ди-трет-бутиловый эфир 1,4-дигидрофталазин-2,3-дикарбоновой кислоты, 4, (1,0 г, 3 ммоль) растворяют в МеОН (20 мл) и по каплям добавляют SOCl2 (0,5 мл). После перемешивания при комнатной температуре в течение 72 часов растворитель удаляют в вакууме, получая 0,6 г требуемого продукта в виде белого твердого вещества.
Следующая схема иллюстрирует «сборку» остова 3-пиримидин-4-ил-5,10-дигидропиразоло[1,2-b]фталазин-1-она конвергентной стадией, в которой конденсируют промежуточные соединения 3 и 5. Образовавшееся промежуточное соединение затем превращают в конечное соединение, имеющее выбранное звено R.
Реагенты и условия: (с) пиридин, кипячение с обратным холодильником 16 час.
Реагенты и условия: (d) OXONE®, ТГФ/МеОН, комнатная температура, 2 часа.
Реагенты и условия: (е) толуол, 140°C, 12 час.
ПРИМЕР 3
2-(4-Фторфенил)-3-[2-(S)-(1-фенилэтиламино)пиримидин-4-ил]-5,10-дигидропиразоло[1,2-b]фталазин-1-он (8)
Получение 1-(4-фторфенил)-3-(2-метилсульфанилпиримидин-4-ил)-5,10-дигидропиразоло[1,2-b]фталазин-1-она (6): К раствору 1,2,3,4-тетрагидрофталазина, 5 (0,3 г, 1,4 ммоль), в пиридине (5 мл) добавляют метиловый эфир 2-(4-фторфенил)-3-(2-метилсульфанилпиримидин-4-ил)-3-оксопропионовой кислоты, 3 (0,4 г, 1,4 ммоль). Реакционную смесь затем нагревают при кипячении с обратным холодильником в течение 16 часов. Растворитель удаляют в вакууме и образовавшийся остаток очищают препаративной ВЭЖХ, получая 0,2 г (выход 45%) требуемого продукта в виде рыжевато-коричневого твердого вещества.
Получение 2-(4-фторфенил)-3-(2-метансульфонилпиримидин-4-ил)-5,10-дигидропиразоло[1,2-b]фталазин-1-она (7): К раствору 1-(4-фторфенил)-3-(2-метилсульфанилпиримидин-4-ил)-5,10-дигидропиразоло[1,2-b]фталазин-1-она, 6 (2,4 г, 6,8 ммоль) в смеси ТГФ:МеОН (80 мл смеси 1:1) по каплям добавляют раствор OXONE® (16,8 г, 27,2 ммоль) в Н2О (80 мл). После перемешивания в течение 2 часов при комнатной температуре реакционную смесь разбавляют водным насыщенным NaHCO3 и экстрагируют три раза этилацетатом. Объединенные органические фазы сушат (Na2SO4), фильтруют и концентрируют в вакууме, получая 1,5 г (выход 58%) требуемого продукта в виде желтого твердого вещества.
Получение 2-(4-фторфенил)-3-[2-(S)-(1-фенилэтиламино)пиримидин-4-ил]-5,10-дигидропиразоло[1,2-b]фталазин-1-она (8): 2-(4-Фторфенил)-3-(2-метансульфонилпиримидин-4-ил)-5,10-дигидропиразоло[1,2-b]фталазин-1-он, 7 (0,9 г, 2,3 ммоль) растворяют в толуоле (18 мл) вместе с (S)-(-)-α-метилбензиламином (10,5 мл, 81,6 ммоль). Образовавшуюся смесь нагревают до 140°C в течение 12 часов, охлаждают до комнатной температуры и растворитель удаляют в вакууме. Образовавшийся остаток очищают на диоксиде кремния (EtOAc/гексаны, 1:1), получая 0,8 г (выход 80%) требуемого продукта в виде красного липкого твердого вещества.
Первый аспект аналогов категории II настоящего изобретения, способных ингибировать высвобождение воспалительных цитокинов, относится к соединениям, включающим остов 5,8-дигидропиразоло[1,2-a]пиридазин-1-она, где R2a и R2b, взятые вместе, образуют двойную связь, причем указанный остов имеет формулу:
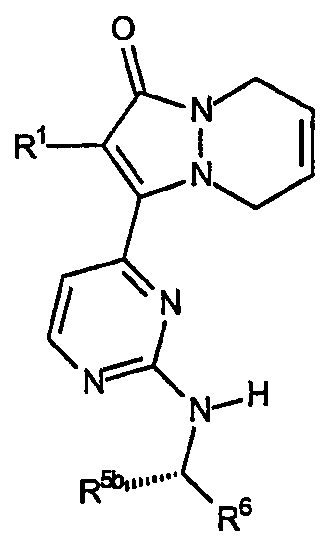
где R1, R5b и R6 описаны в таблице II. Стереохимия R5b является конфигурацией, показанной, когда R5b или R6 не является водородом.
Соединения, которые включают аналоги первого аспекта категории II, могут быть получены синтезом, приведенным ниже на следующей схеме.
Реагенты и условия: (а) NaH, ДМФ; от 0°C до 90°C, 4 часа.
Реагенты и условия: (b) SOCl2, MeOH; 0°C, 17 час.
Реагенты и условия: (с) пиридин, 90°C, 16 час.
Реагенты и условия: (d) OXONE®, ТГФ/МеОН/вода, комнатная температура, 2 часа.
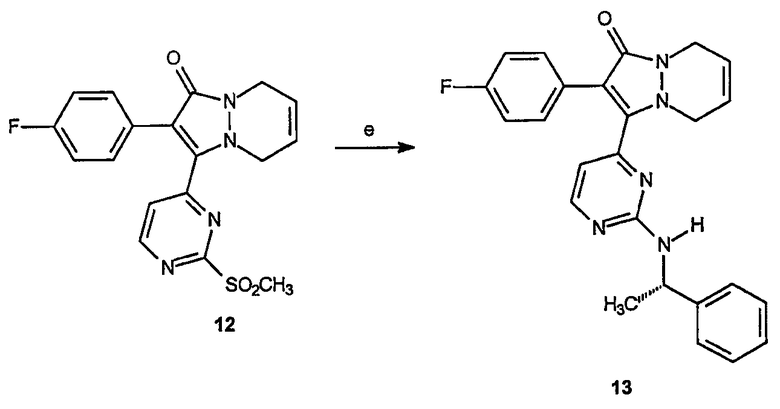
Реагенты и условия: (е) (S)-(-)-α-метилбензиламин, толуол; 140°C в течение 12 час.
ПРИМЕР 4
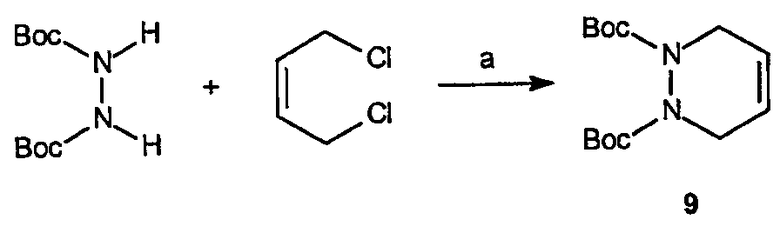
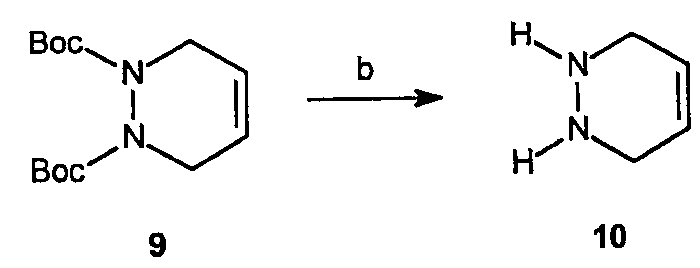
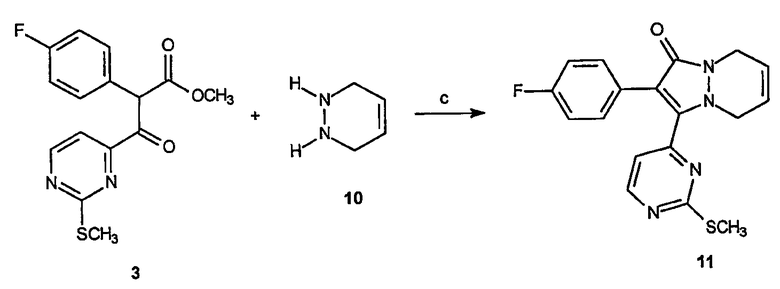
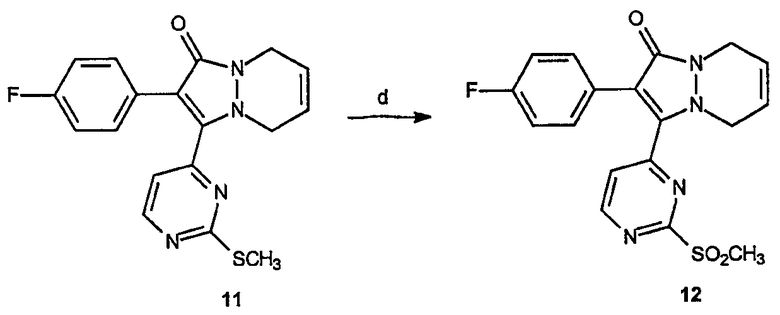
2-(4-Фторфенил)-3-[2-(1-фенилэтиламино)пиримидин-4-ил]-5,8-дигидропиразоло[1,2-a]пиридазин-1-он (13)
Получение ди-трет-бутилового эфира 3,6-дигидропиридазин-1,2-дикарбоновой кислоты (9): К раствору ди-трет-бутилгидразодиформиата (18,6 г, 80,0 ммоль) в ДМФ (220 мл), охлажденному до 0°C, порциями добавляют NaH (8,0 г 60% суспензии в минеральном масле, 200,0 ммоль). После того как раствору дают нагреться и перемешивания 45 минут при комнатной температуре, к реакционной смеси по каплям добавляют цис-1,4-дихлор-2-бутен (8,4 мл, 80,0 ммоль). Смесь затем нагревают при 90°C в течение 4 часов, охлаждают до комнатной температуры и перемешивают дополнительные 15 часов. Реакционную смесь гасят, выливая содержимое реакционного сосуда в ледяную воду. Образовавшуюся водную фазу экстрагируют эфиром, объединенные органические фазы промывают водным насыщенным NaHCO3, сушат, фильтруют и концентрируют в вакууме. Полученный неочищенный продукт растворяют в гексане и образовавшееся твердое вещество выделяют фильтрованием, получая 24 г требуемого продукта в виде белого порошка.
Получение 1,2,3,4-тетрагидропиридазина (10): К раствору ди-трет-бутилового эфира 3,6-дигидропиридазин-1,2-дикарбоновой кислоты, 9 (10,0 г, 35,2 ммоль) в МеОН (140 мл) при 0°C добавляют по каплям SOCl2 (22,0 мл). После постепенного нагревания до комнатной температуры и перемешивания в течение 17 часов растворитель удаляют в вакууме, получая рыжевато-коричневое твердое вещество. Выделенное твердое вещество затем растворяют в МеОН (10 мл) и разбавляют эфиром (250 мл). Образовавшееся белое твердое вещество отфильтровывают, получая 4,3 г (выход 79%) требуемого продукта в виде соли ди-HCl.
Получение 2-(4-фторфенил)-3-(2-метилсульфанилпиримидин-4-ил)-2,3,5,8-тетрагидропиразоло[1,2-a]пиридазин-1-она (11): К раствору 1,2,3,4-тетрагидропиридазина, 5 (5,4 г, 34,2 ммоль) в пиридине (100 мл) добавляют метиловый эфир 2-(4-фторфенил)-3-(2-метилсульфанилпиримидин-4-ил)-3-оксопропионовой кислоты, 3, (7,3 г, 22,8 ммоль). Реакционную смесь нагревают до 90°C в течение 16 часов. Растворитель затем удаляют в вакууме и образовавшийся остаток очищают на диоксиде кремния (100% EtOAc), получая 3,5 г (выход 43%) требуемого продукта в виде желтого твердого вещества.
Получение 2-(4-фторфенил)-3-(2-метансульфонилпиримидин-4-ил)-2,3,5,8-тетрагидропиразоло[1,2-a]пиридазин-1-она (12): К раствору 2-(4-фторфенил)-3-(2-метилсульфанилпиримидин-4-ил)-2,3,5,8-тетрагидропиразоло[1,2-a]пиридазин-1-она, 11 (2,4 г, 6,8 ммоль), в смеси ТГФ/МеОН (80 мл смеси 1:1) добавляют по каплям раствор OXONE® (16,8 г, 27,2 ммоль) в Н2О (80 мл). После перемешивания в течение 2 часов при комнатной температуре реакционную смесь разбавляют водным насыщенным NaHCO3 и экстрагируют EtOAc (3х). Объединенные органические фазы сушат, фильтруют и концентрируют в вакууме, получая 1,5 г (выход 58%) требуемого продукта в виде желтого твердого вещества.
Получение 2-(4-фторфенил)-3-[2-(1-(S)-фенилэтиламино)пиримидин-4-ил]-5,8-дигидропиразоло[1,2-a]пиридазин-1-она (13): 2-(4-Фторфенил)-3-(2-метансульфонилпиримидин-4-ил)-2,3,5,8-тетрагидропиразоло[1,2-a]пиридазин-1-он, 12, (0,9 г, 2,3 ммоль) растворяют в толуоле (18 мл) и к раствору добавляют (S)-(-)-α-метилбензиламин (10,5 мл, 81,6 ммоль). Образовавшуюся смесь нагревают до 140°C в течение 12 часов, охлаждают и растворитель удаляют в вакууме. Образовавшийся неочищенный продукт очищают на диоксиде кремния (EtOAc/гексаны, 1:1), получая 0,8 г (выход 80%) требуемого продукта в виде красного липкого твердого вещества.
Второй аспект аналогов категории II настоящего изобретения, способных ингибировать высвобождение воспалительных цитокинов, относится к соединениям, включающим остов 5,6,7,8-тетрагидропиразоло[1,2-a]пиридазин-1-она и имеющим формулу:
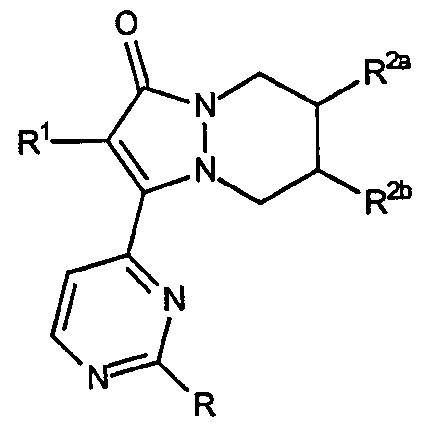
где R, R1, R2a и R2b описаны в таблице III.
Для второго аспекта категории II промежуточные соединения, такие как соединение 13, могут быть использованы для получения аналогов, перечисленных в таблице IV, например соединения 14.
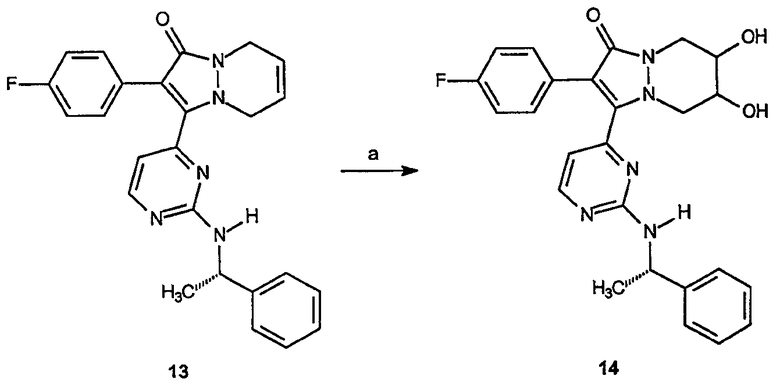
Реагенты и условия: (а) OsO4, K3Fe(CN)6; трет-BuOH:H2O, комнатная температура, 12 час.
ПРИМЕР 5
2-(4-Фторфенил)-6,7-дигидрокси-3-[2-(1-фенилэтиламино)пиримидин-4-ил]-5,6,7,8-тетрагидропиразоло[1,2-a]пиридазин-1-он (14)
Получение 2-(4-фторфенил)-6,7-дигидрокси-3-[2-(1-фенилэтиламино)пиримидин-4-ил]-5,6,7,8-тетрагидропиразоло[1,2-a]пиридазин-1-она (14): К раствору 2-(4-фторфенил)-3-[2-(1-фенилэтиламино)пиримидин-4-ил]-5,8-дигидропиразоло[1,2-a]пиридазин-1-она, 13 (0,8 г, 1,88 ммоль), в смеси трет-BuOH:H2O (24 мл смеси 1:1) добавляют K3Fe(CN)6 (1,9 г, 5,64 ммоль), К2СО3 (0,8 г, 5,6 ммоль) и NaHCO3 (0,5 г, 5,6 ммоль) с последующим добавлением тетраоксида осмия (0,1 г, 0,3 ммоль). Образовавшуюся смесь перемешивают при комнатной температуре в течение 12 часов. Реакционную смесь гасят добавлением водного насыщенного раствора KHSO4 (10 мл). Водную фазу экстрагируют EtOAc (3х) и объединенные органические фазы сушат, фильтруют и концентрируют в вакууме. Образовавшийся неочищенный продукт очищают на диоксиде кремния (100% EtOAc), получая 0,4 г (выход 48%) требуемого продукта.
Кроме того, такое соединение, как 14, может быть само использовано в качестве промежуточного соединения для получения других аналогов, например соединения 15.
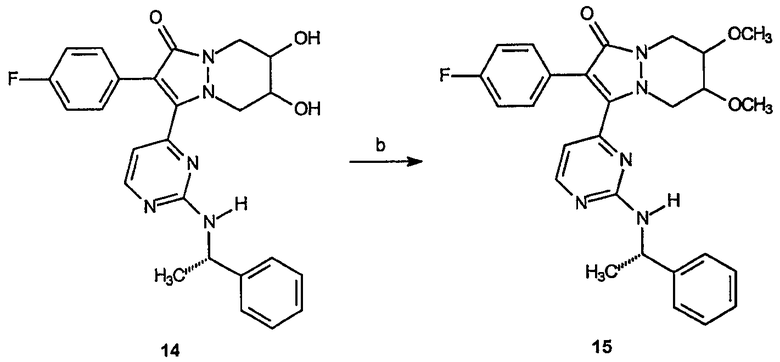
Реагенты и условия: (b) NaH, CH3I, толуол, комнатная температура, 62 часа.
ПРИМЕР 6
2-(4-Фторфенил)-6,7-диметокси-3-[2-(1-(S)-фенилэтиламино)пиримидин-4-ил]-5,6,7,8-тетрагидропиразоло[1,2-a]пиридазин-1-он (15)
Получение 2-(4-фторфенил)-6,7-диметокси-3-[2-(1-(S)-фенилэтиламино)пиримидин-4-ил]-5,6,7,8-тетрагидропиразоло[1,2-a]пиридазин-1-она (15): К раствору 2-(4-фторфенил)-6,7-дигидрокси-3-[2-(1-фенилэтиламино)пиримидин-4-ил]-5,6,7,8-тетрагидропиразоло[1,2-a]пиридазин-1-она, 14 (0,42 г, 0,91 ммоль), в ТГФ (2 мл) добавляют NaH (0,09 г, 2,30 ммоль). После перемешивания при комнатной температуре в течение 1 часа к реакционной смеси по каплям добавляют метилиодид (0,14 г, 2,30 ммоль). После перемешивания в течение 62 часов при комнатной температуре смесь концентрируют в вакууме, растворяют в EtOAc и промывают водным насыщенным NaHCO3. Органическую фазу сушат, фильтруют, концентрируют в вакууме и образовавшийся остаток очищают на диоксиде кремния (100% EtOAc), получая 0,07 г (выход 16%) требуемого продукта в виде желтого твердого вещества.
Первый аспект аналогов категории III настоящего изобретения, способных ингибировать высвобождение воспалительных цитокинов, относится к соединениям, включающим остов 5,6,7,8-тетрагидропиразоло[1,2-a]пиридазин-1-она и имеющим формулу:
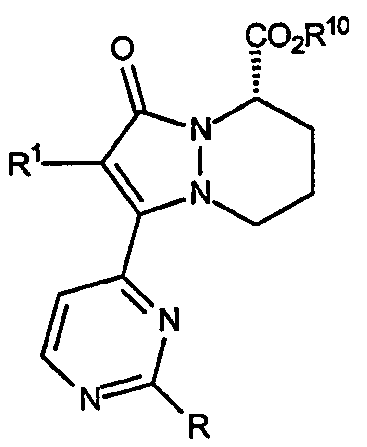
где R представляет собой простой эфир, R, R1 и R10 описаны ниже в таблице IV и аналоги имеют указанную стереохимию.
Соединения, которые включают аналоги первого аспекта категории III, могут быть получены синтезом, приведенным ниже на следующей схеме.
Реагенты и условия: (а) С2О2Cl2, CH2Cl2; комнатная температура, 18 час.
Реагенты и условия: (b) CH2Cl2, комнатная температура, 3 часа.
Реагенты и условия: (с) ТЕА, CH2Cl2; комнатная температура, 18 час.
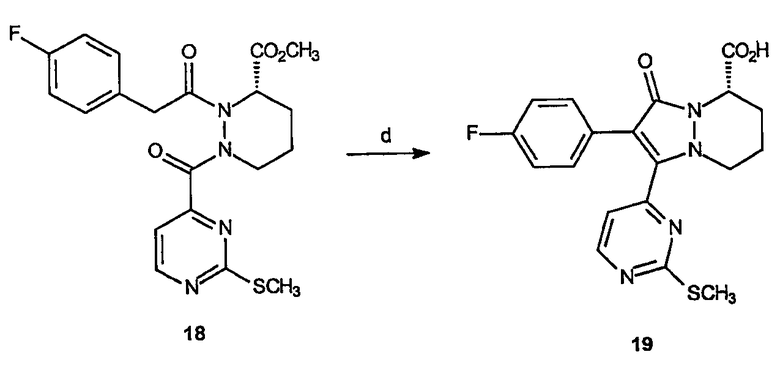
Реагенты и условия: (d) NaOH, МеОН, комнатная температура, 20 мин.
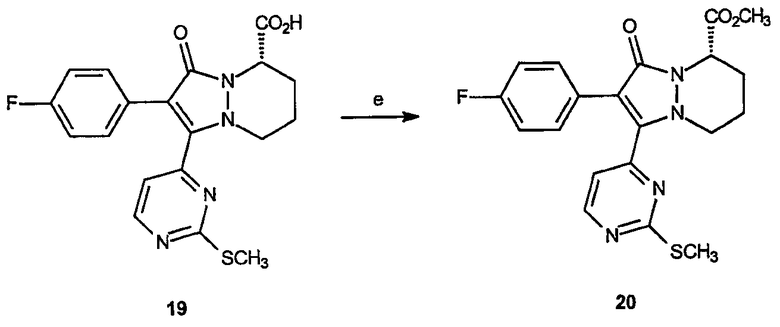
Реагенты и условия: (е) TMS-CHN2, CH2Cl2/МеОН; 20 мин, комнатная температура
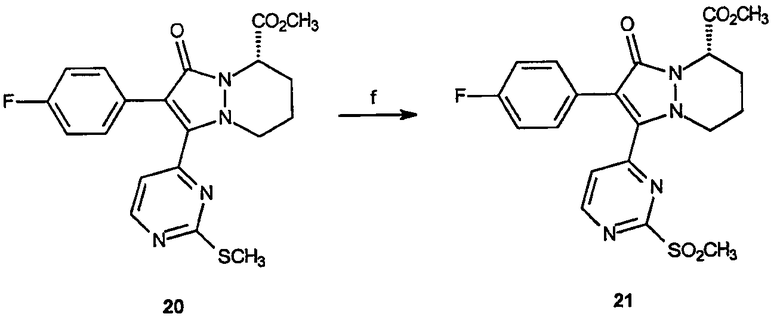
Реагенты и условия: (f) OXONE®, ТГФ/МеОН, комнатная температура, 4 часа.
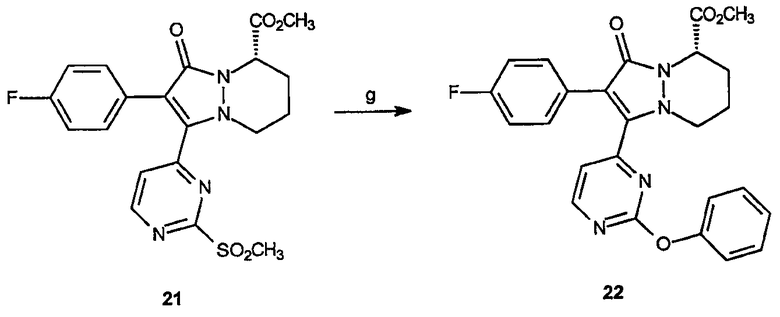
Реагенты и условия: (g) фенол, NaH, ТГФ; комнатная температура, 1 час.
ПРИМЕР 7
Метиловый эфир 2-(4-фторфенил)-3-оксо-1-(3-феноксипиримид-4-ил)-5,6,7,8-тетрагидро-3Н-пиразоло[1,2-а]пиридазин-5-карбоновой кислоты (22)
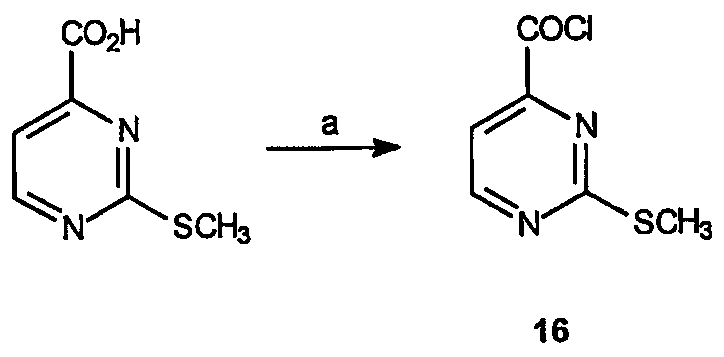
Получение 2-метилсульфанилпиримидин-4-карбонилхлорида (16): К раствору 2-метилсульфанилпиримидин-4-карбоновой кислоты (20 г, 117,7 ммоль) в СН2Cl2 (100 мл) добавляют оксалилхлорид (17,2 г, 197 ммоль) и ДМФ (20 капель). Реакционный раствор перемешивают при комнатной температуре в течение 18 часов, после чего растворитель удаляют в вакууме, получая 21,2 г (выход 95%) требуемого продукта в виде темно-зеленого твердого вещества.
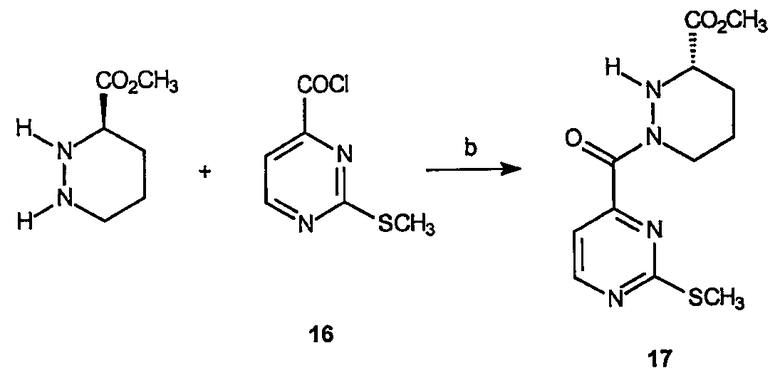
Получение метилового эфира 1-(метилсульфанилпиримидин-4-карбонил)гексагидропиридазин-3-карбоновой кислоты (17): К раствору метилового эфира гексагидропиридазин-3-карбоновой кислоты (1,5 г, 8,3 ммоль) в CH2Cl2 (80 мл) добавляют 2-метилсульфанилпиримидин-4-карбонилхлорид, 16 (1,41 г, 7,5 ммоль), и триэтиламин (1,2 мл, 8,3 ммоль). Смесь перемешивают при комнатной температуре в течение 3 часов. Реакционный раствор затем разбавляют 1 н HCl (100 мл) и органическую фазу декантируют. Водную фазу экстрагируют дополнительным растворителем и органические слои объединяют, сушат и концентрируют в вакууме. Неочищенный продукт очищают на диоксиде кремния (этилацетат/гексан, 1:1), получая 0,9 г (выход 36%) требуемого продукта в виде желтого твердого вещества.
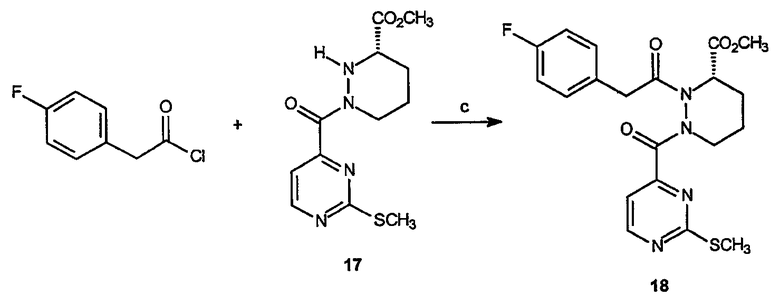
Получение метилового эфира 2-(4-фторбензоил)-1-(2-метилсульфанилпиримидин-4-карбонил)гексагидропиридазин-3-карбоновой кислоты (18): К раствору метилового эфира 1-(метилсульфанилпиримидин-4-карбонил)гексагидропиридазин-3-карбоновой кислоты, 17 (0,9 г, 3 ммоль), в CH2Cl2 (80 мл) добавляют 4-фторфенилацетилхлорид (0,63 мл, 4,6 ммоль) и триэтиламин 0,55 мл, 3,6 ммоль). Реакционный раствор перемешивают при комнатной температуре в течение 18 часов, затем разбавляют 1 н. HCl (50 мл) и органический слой декантируют. Органическую фазу экстрагируют дополнительным растворителем, органические слои объединяют, сушат и концентрируют в вакууме, получая неочищенный продукт. Неочищенное вещество очищают на диоксиде кремния (этилацетат/гексан, 1:1), получая 1,15 г (выход 89%) требуемого продукта в виде желтого твердого вещества.
Получение 2-(4-фторфенил)-1-(3-метилсульфанилпиримидин-4-ил)-3-оксо-5,6,7,8-тетрагидро-3Н-пиразоло[1,2-a]пиридазин-5-карбоновой кислоты (19): К раствору метилового эфира 2-(4-фторбензоил)-1-(2-метилсульфанилпиримидин-4-карбонил)гексагидропиридазин-3-карбоновой кислоты, 18 (1,13 г, 2,62 ммоль), в метаноле (40 мл) добавляют NaOH (1,26 г, 31,4 ммоль). Реакционную смесь перемешивают при комнатной температуре в течение 20 минут, затем разбавляют 1 н. HCl (50 мл). Раствор экстрагируют этилацетатом (3 х 250 мл), органические слои объединяют, сушат и концентрируют в вакууме, получая 0,83 г (выход 79%) масла, которое используют без дополнительной очистки.
Получение метилового эфира 2-(4-фторфенил)-1-(3-метилсульфанилпиримидин-4-ил)-3-оксо-5,6,7,8-тетрагидро-3Н-пиразоло[1,2-a]пиридазин-5-карбоновой кислоты (20): К раствору 2-(4-фторфенил)-1-(3-метилсульфанилфенил)-3-оксо-5,6,7,8-тетрагидро-3Н-пиразоло[1,2-a]пиридазин-5-карбоновой кислоты, 19 (0,83 г, 2,1 ммоль), в метиленхлориде (50 мл) добавляют триметилсилилдиазометан (1,5 мл 2М раствора в гексане, 3 ммоль). Реакционную смесь перемешивают в течение 20 минут при комнатной температуре, затем концентрируют в вакууме с получением неочищенного продукта в виде масла, которое очищают на диоксиде кремния (гексан/этилацетат, 1:4), получая 0,51 г (выход 59%) требуемого продукта в виде желтой пены.
Получение метилового эфира 2-(4-фторфенил)-1-(3-метансульфонилпиримидин-4-ил)-3-оксо-5,6,7,8-тетрагидро-3Н-пиразоло[1,2-a]пиридазин-5-карбоновой кислоты (21): Перемешиваемый раствор метилового эфира 2-(4-фторфенил)-1-(3-метилсульфанилфенил)-3-оксо-5,6,7,8-тетрагидро-3Н-пиразоло[1,2-a]пиридазин-5-карбоновой кислоты, 20 (0,51 г, 1,23 ммоль), в метаноле (30 мл) охлаждают до 0°C. Oxone® (2,27 г, 3,7 ммоль) растворяют в воде (30 мл) и добавляют по каплям к реакционному раствору на протяжении 1 часа. Раствору дают нагреться до комнатной температуры и дополнительно перемешивают 3 часа. Добавляют NaHCO3 (насыщ.) до рН приблизительно 7. Реакционный раствор затем экстрагируют несколько раз этилацетатом, органические фазы объединяют, сушат и концентрируют в вакууме, получая 0,5 г (выход 91%) требуемого продукта в виде желтой пены.
Получение метилового эфира 2-(4-фторфенил)-1-(2-феноксипиримидин-4-ил)-3-оксо-5,6,7,8-тетрагидро-3Н-пиразоло[1,2-a]пиридазин-5-карбоновой кислоты (22): К раствору метилового эфира 2-(4-фторфенил)-1-(3-метансульфонилпиримидин-4-ил)-3-оксо-5,6,7,8-тетрагидро-3Н-пиразоло[1,2-a]пиридазин-5-карбоновой кислоты, 21 (0,033 г, 0,074 ммоль) в ТГФ (3 мл) добавляют фенол и NaH (0,009 г, 0,22 ммоль). Реакционную смесь перемешивают при комнатной температуре в течение 1 часа. Реакционную смесь гасят добавлением 1 н HCl (20 мл) и раствор экстрагируют этилацетатом (3 х 25 мл). Органические фазы объединяют, промывают насыщенным раствором соли, сушат и концентрируют в вакууме, получая неочищенный продукт, который очищают на диоксиде кремния (гексаны/этилацетат, 1:3), получая 0,012 г (выход 35%) требуемого продукта в виде белого твердого вещества. 1Н-ЯМР (300 МГц, CDCl3) δ 8,64 (д, J=4,6 Гц, 1H), 7,59-7,63 (м, 2H), 7,40-7,45 (м, 3H), 7,28-7,30 (м, 1H), 7,18 (д, J=8,4 Гц, 2H), 7,03-7,08 (м, 2H), 4,50-4,56 (м, 1H), 3,99-4,04 (м, 1H), 3,86 (с, 1H), 3,01-3,10 (м, 1H), 2,33-2,41 (м, 1H), 1,86 (уш.с, 2H), 1,64 (уш.с, 3H); ESI/МС: 461 (M+H).
Другие соединения в соответствии с данным аспектом категории III могут быть получены по следующей методике.
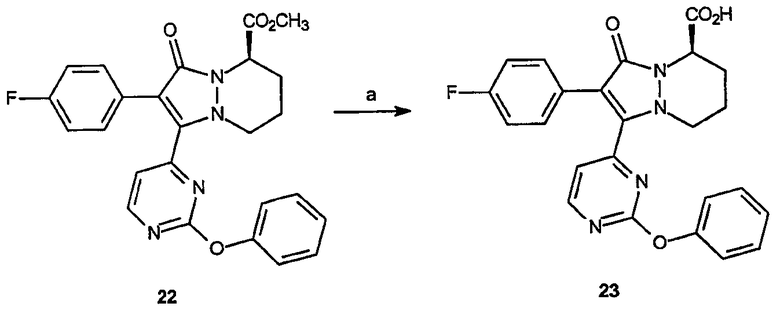
Реагенты и условия: (а) LiOH, МеОН/вода, комнатная температура, 3 часа.
ПРИМЕР 8
Получение 2-(4-фторфенил)-1-(2-феноксипиримидин-4-ил)-3-оксо-5,6,7,8-тетрагидро-3Н-пиразоло[1,2-a]пиридазин-5-карбоновой кислоты (23): К раствору метилового эфира 2-(4-фторфенил)-1-(2-феноксипиримидин-4-ил)-3-оксо-5,6,7,8-тетрагидро-3Н-пиразоло[1,2-a]пиридазин-5-карбоновой кислоты, 22, (0,02 г, 0,0143 ммоль) в метаноле (1 мл) и воде (1 мл) добавляют LiOH (0,016 г, 0,65 ммоль). Реакционный раствор перемешивают при комнатной температуре в течение 3 часов, затем гасят добавлением 1 н. HCl (20 мл). Реакционный раствор экстрагируют этилацетатом (3 х 50 мл), органические слои объединяют, промывают насыщенным раствором соли, сушат и концентрируют в вакууме, получая 0,012 г (выход 63%) требуемого продукта в виде желтого твердого вещества. 1Н-ЯМР (300 МГц, CDCl3) δ 8,45 (дд, J=4,6, 2,1 Гц, 1H), 7,14-7,44 (м, 7H), 6,84-6,,95 (м, 3H), 4,93 (дд, J=11,7, 9,3 Гц, 1H), 4,23 (уш.д, J=12,9 Гц, 1H), 3,04-3,11 (м, 1H), 2,46-2,52 (м, 2H), 1,71-1,93 (м, 2H), APCI/МС: 447 (M +H).
2-(4-Фторфенил)-1-[2-(4-фторфенокси)пиримидин-4-ил]-3-оксо-5,6,7,8-тетрагидро-3Н-пиразоло[1,2-a]пиридазин-5-карбоновая кислота: 1Н-ЯМР (300 МГц, CDCl3) δ 8,50 (д, J=5,1 Гц, 1H), 7,36 (дд, J=8,7, 5,4 Гц, 2H), 7,20-7,31 (м, 4H), 7,02 (т, J=8,7 Гц, 2H), 6,97 (д, J=5,1 Гц, 1H), 5,23-5,25 (м, 1H), 4,24 (д, J=11,4 Гц, 1H), 3,74 (с, 3H), 2,94-2,99 (м, 1H), 2,54-2,59 (м, 1H), 1,82-2,00 (м, 3H); ESI/МС: 479 (M+H).
Второй аспект аналогов категории III настоящего изобретения, способных ингибировать высвобождение воспалительных цитокинов, относится к соединениям, включающим остов 5,6,7,8-тетрагидропиразоло[1,2-a]пиридазин-1-она и имеющим формулу:
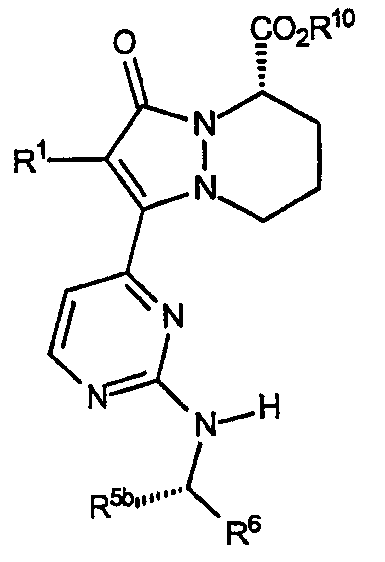
где звенья R являются аминами, имеющими формулу -NH[CHR5b]R6, и R1, R5b, R6 и R10 описаны здесь ниже в таблице V. Стереохимия R5b является конфигурацией, показанной, когда R5b не является водородом.
Соединения, которые включают аналоги второго аспекта категории III, могут быть получены синтезом, приведенным ниже на следующей схеме.
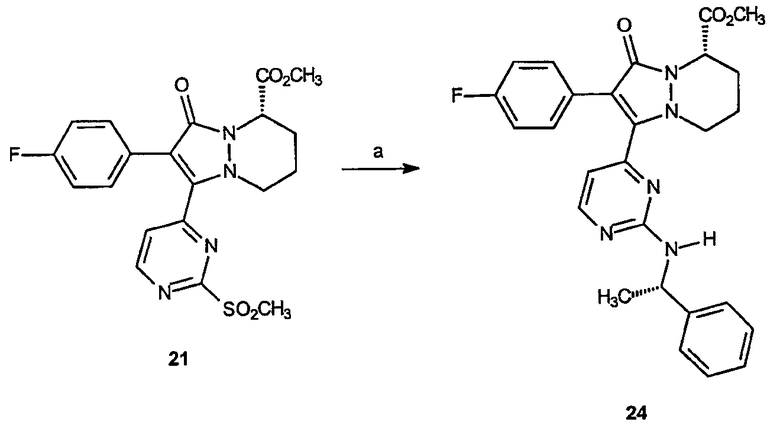
Реагенты и условия: (а) (S)-(-)-α-метилбензиламин, толуол; 100°C, 4 часа.
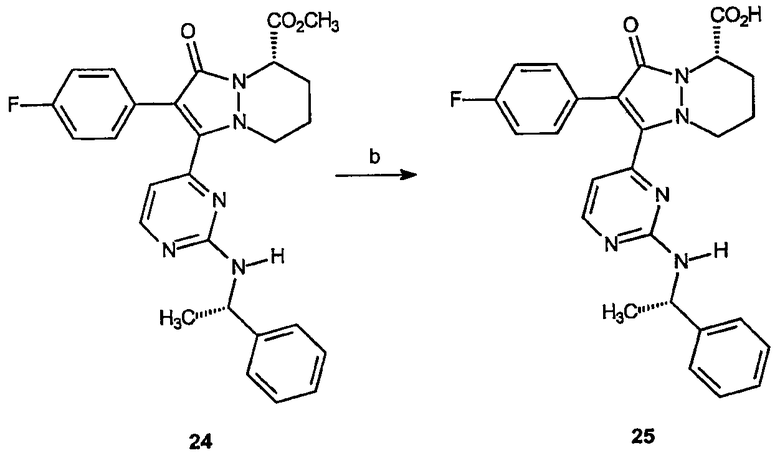
Реагенты и условия: (b) LiOH, МеОН/вода; комнатная температура, 3 часа.
ПРИМЕР 9
Метиловый эфир 2-(4-фторфенил)-3-оксо-1-[2-(1-(S)-(фенилэтиламино)пиримидин-4-ил]-5,6,7,8-тетрагидро-3Н-пиразоло[1,2-a]пиридазин-5-карбоновой кислоты (24)
Получениеметилового эфира 2-(4-фторфенил)-3-оксо-1-[2-(1-(S)-(фенилэтиламино)пиримидин-4-ил]-5,6,7,8-тетрагидро-3Н-пиразоло[1,2-a]пиридазин-5-карбоновой кислоты (24): К раствору метилового эфира 2-(4-фторфенил)-1-(3-метансульфонилпиримидин-4-ил)-3-оксо-5,6,7,8-тетрагидро-3Н-пиразоло[1,2-a]пиридазин-5-карбоновой кислоты, 21, (0,10 г, 0,22 ммоль) в толуоле (1,4 мл) добавляют (S)-(-)-α-метилбензиламин (1,4 мл, 1,12 ммоль). Реакционный раствор нагревают до 100°C в течение 4 часов, после чего реакционную смесь охлаждают и разбавляют 1 н. HCl. Образовавшийся раствор экстрагируют этилацетатом (3 х 25 мл), органические слои объединяют, сушат и концентрируют в вакууме, получая 0,071 г (выход 66%) требуемого продукта в виде желтого твердого вещества. 1Н-ЯМР (300 МГц, CDCl3) δ 8,22 (ддд, J=11,4, 5,1, 2,1 Гц, 1H), 7,22-7,37 (м, 7H), 6,97 (дт, J=8,7, 2,1 Гц, 2H), 6,41 (ддд, J=15,6, 5,1, 2,1 Гц, 1H), 5,72-5,83 (м, 1H), 5,2 (уш.с, 2H), 5,52-5,62 (м, 1H), 3,77 (с, 3H), 3,47 (д, J=2,7 Гц, 1H), 2,47-2,51 (м, 2H), 2,00 (уш.с, 1H); 1,41 (д, J=6,6 Гц, 3H); APCI/МС: 487 (M+H).
ПРИМЕР 10
2-(4-Фторфенил)-3-оксо-1-[2-(1-(S)-(фенилэтиламино)пиримидин-4-ил]-5,6,7,8-тетрагидро-3Н-пиразоло[1,2-a]пиридазин-5-карбоновая кислота (25)
Получение2-(4-фторфенил)-3-оксо-1-[2-(1-(S)-(фенилэтиламино)пиримидин-4-ил]-5,6,7,8-тетрагидро-3Н-пиразоло[1,2-a]пиридазин-5-карбоновой кислоты (25): К раствору метилового эфира 2-(4-фторфенил)-3-оксо-1-[2-(S)-(1-фенилэтиламино)пиримидин-4-ил]-5,6,7,8-тетрагидро-3Н-пиразоло[1,2-a]пиридазин-5-карбоновой кислоты, 24 (0,066 г, 0,14 ммоль), в метаноле (2 мл) и воде (2 мл) добавляют LiOH (0,033 г, 1,36 ммоль). Смесь перемешивают при комнатной температуре в течение 3 часов, затем разбавляют 1 н. HCl (25 мл), после чего раствор экстрагируют этилацетатом (3 х 50 мл). Объединенные органические слои промывают насыщенным раствором соли, сушат и концентрируют в вакууме, получая 0,043 г (выход 65%) требуемого продукта в виде желтого твердого вещества. 1Н-ЯМР (300 МГц, CDCl3) δ 8,13-8,19 (м, 1H), 7,22-7,34 (м, 7H), 6,97 (т, J=8,7 Гц, 2H), 6,34 (дд, J=15,3, 5,1 Гц, 1H), 5,11-5,24 (м, 2H), 3,56 (уш.с, 1H), 2,96 (уш.с, 1H), 2,52-2,64 (м, 2H), 1,79-1,96 (м, 2H), 1,57 (д, J=6,9 Гц, 3H): ESI/МС: 474 (M+H).
Метиловый эфир 2-(4-фторфенил)-3-оксо-1-[2-(1-(S)-метилметоксиэтиламино)пиримидин-4-ил]-5,6,7,8-тетрагидро-3Н-пиразоло[1,2-a]пиридазин-5-карбоновой кислоты: 1Н-ЯМР (300 МГц, CDCl3) δ 8,25 (д, J=5,1 Гц, 1H), 7,43 (дд, J=9,0,7 Гц, 2H), 6,99 (т, J=9,0 Гц, 2H), 6,44 (д, J=5,1 Гц, 1H), 5,50-5,54 (м, 1H), 5,26 (д, J=3,6Гц, 1H), 4,15-4,25 (м, 2H), 3,76 (с, 3H), 3,37-3,47 (м, 4H), 2,95-3,06 (м, 1H), 2,51-2,62 (м, 1H), 1,92-2,02 (м, 3H), 1,23-1,30 (м, 3H); ESI/МС: 456 (M+H).
Третий аспект аналогов категории III настоящего изобретения, способных ингибировать высвобождение воспалительных цитокинов, относится к соединениям, включающим остов 5,6,7,8-тетрагидропиразоло[1,2-a]пиридазин-1-она и имеющим формулу:
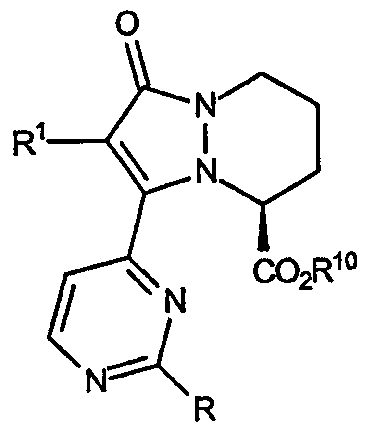
где R представляет собой простую эфирную группу формулы: -OR3. В таблице VI описаны различные значения R, R1 и R10.
Соединения, которые включают третий аспект аналогов категории III, могут быть получены по методике, приведенной на следующей схеме.
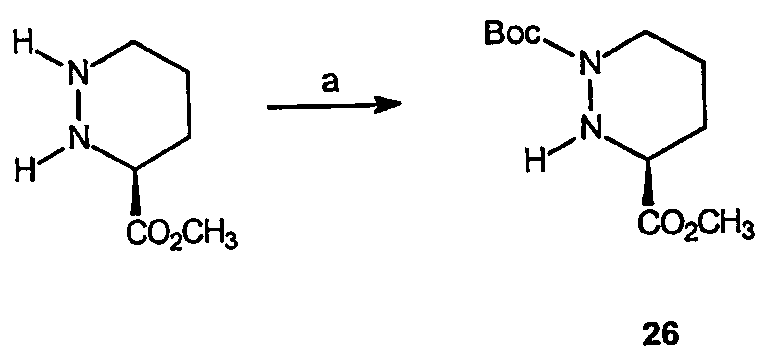
Реагенты и условия: (а) (ВОС)2О, ТЕА, CH2Cl2, комнатная температура, 12 час.
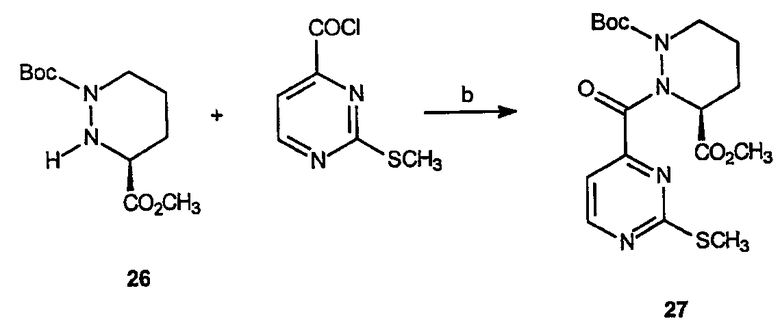
Реагенты и условия: (b) CH2Cl2, ТЕА, комнатная температура, 10 час.
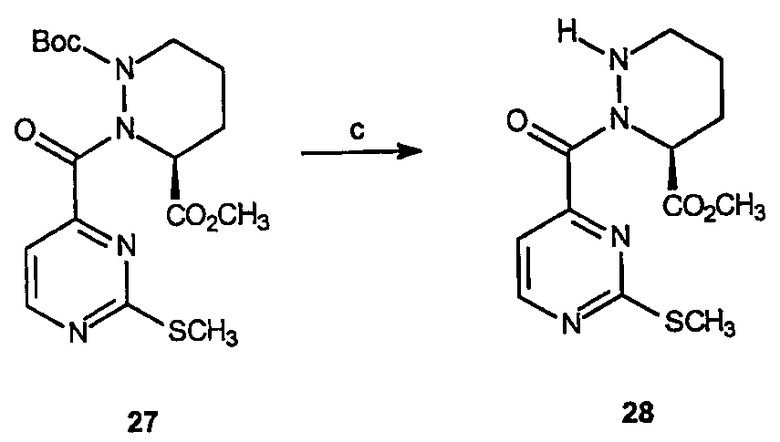
Реагенты и условия: (с) ТФУ, CH2Cl2/вода; 0°C 2 часа, комнатная температура 1 час.
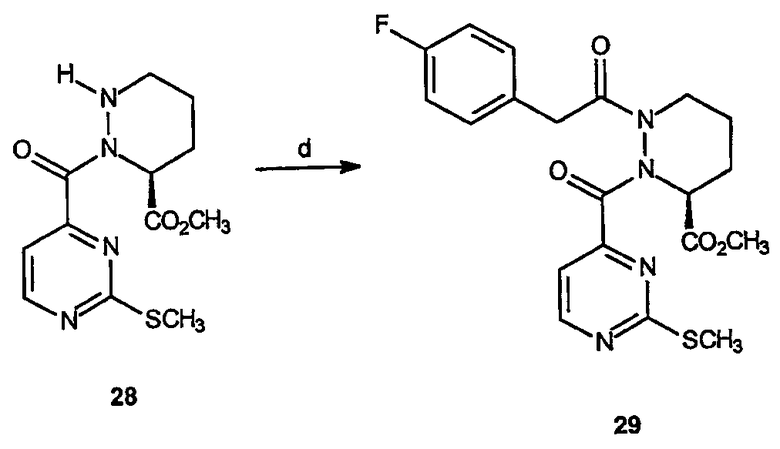
Реагенты и условия: (d) ТЕА, CH2Cl2; комнатная температура 12 час.
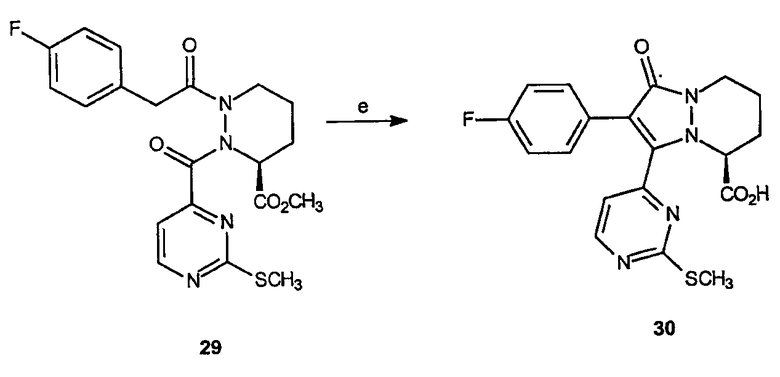
Реагенты и условия: (e) NaOH, МеОН; комнатная температура 15 час.
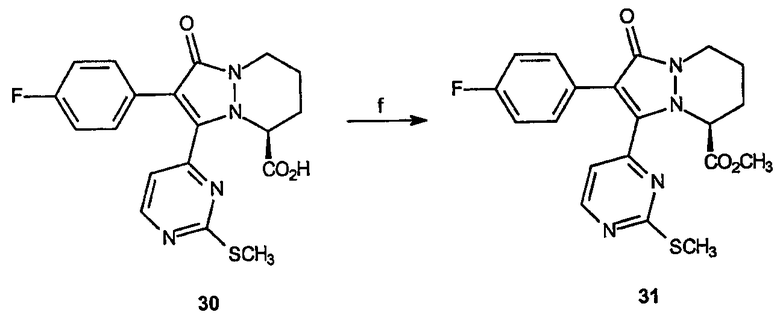
Реагенты и условия: (f) CH2N2, Et2О/EtOAc; комнатная температура 5 мин.
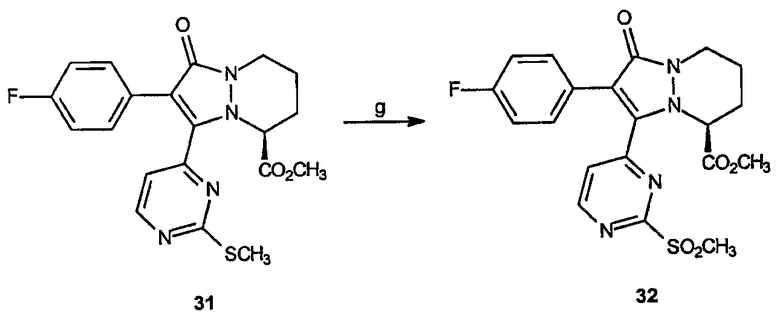
Реагенты и условия: (g) Oxone®, ТГФ/МеОН/вода; комнатная температура 5 час.
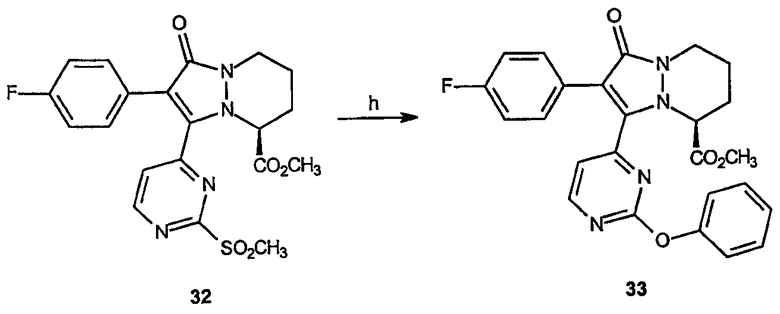
Реагенты и условия: (h) фенол, NaOH, ТГФ; комнатная температура 8 час.
ПРИМЕР 11
Метиловый эфир 2-(4-фторфенил)-1-оксо-3-(2-феноксипиримидин-4-ил)-5,6,7,8-тетрагидро-1Н-пиразоло[1,2-a]пиридазин-5-(S)-карбоновой кислоты (33)
Получение 3-(S)-метилового эфира 1-трет-бутилового эфира тетрагидропиридазин-1,3-дикарбоновой кислоты (26): К метиловому эфиру пиперазиновой кислоты (3,44 г, 19 ммоль) в метиленхлориде (150 мл) добавляют (Вос)2О (4,2 г, 19 ммоль) и триэтиламин (2,65 мл, 19 ммоль). Реакционную смесь перемешивают 12 часов, концентрируют в вакууме с получением желтого масла, которое очищают на диоксиде кремния (этилацетат/гексан, 1:1), получая 4,5 г (выход 98%) требуемого продукта в виде светло-желтого масла.
Получение 3-(S)-метилового эфира 1-трет-бутилового эфира 2-(2-метилсульфанилпиримидин-4-карбонил)тетрагидропиридазин-1,3-дикарбоновой кислоты (27): К раствору 3-(S)-метилового эфира 1-трет-бутилового эфира тетрагидропиридазин-1,3-дикарбоновой кислоты, 26 (3,91 г, 15,9 ммоль), в метиленхлориде (200 мл) добавляют 2-метансульфанилпиримидин-4-карбонилхлорид, 16, (3,32 г, 17,6 ммоль) и триэтиламин (3,5 мл, 25,3 ммоль) так, чтобы рН был приблизительно нейтральным. Образовавшуюся смесь перемешивают в течение 10 часов при комнатной температуре и смесь промывают водой (100 мл), насыщенным раствором соли (100 мл), сушат и концентрируют в вакууме с получением масла, которое очищают на диоксиде кремния (этилацетат/гексан, 1:1), получая 5,22 г (выход 83%) требуемого продукта в виде желтого масла.
Получение 3-(S)-метилового эфира 2-(2-метилсульфанилпиримидин-4-карбонил)тетрагидропиридазин-1,3-дикарбоновой кислоты (28): К раствору 3-(S)-метилового эфира 1-трет-бутилового эфира 2-(2-метилсульфанилпиримидин-4-карбонил)тетрагидропиридазин-1,3-дикарбоновой кислоты, 27 (7 г, 17,6 ммоль), в метиленхлориде (50 мл) добавляют трифторуксусную кислоту (50 мл) при 0°C. Реакционную смесь перемешивают в течение 2 часов при охлаждении, 1 час при комнатной температуре, затем концентрируют в вакууме, получая остаток, который может быть растворен в толуоле и снова концентрирован с получением 7,2 г (выход 100%) требуемого соединения в форме трифторацетатной соли в виде желтого масла, которое используют без дополнительной очистки.
Получение 1-[2-(4-фторфенил)-2-оксоэтил]-2-(2-метилсульфанилпиримидин-4-карбонил)гексагидропиридазин-3-(S)-карбоновой кислоты (29): К раствору 3-(S)-метилового эфира 2-(2-метилсульфанилпиримидин-4-карбонил)тетрагидропиридазин-1,3-дикарбоновой кислоты, 28 (7,2 г, 17,6 ммоль), в метиленхлориде (150 мл) добавляют 4-фторфенилацетилхлорид (3 г, 17,6 ммоль) и триэтиламин (3,65 мл, 26,4 ммоль). Образовавшуюся смесь перемешивают в течение 12 часов, затем концентрируют в вакууме, получая коричневое масло. Неочищенный остаток очищают препаративной ВЭЖХ, получая 5,33 г (выход 70%) требуемого продукта в виде желтого масла.
Получение 2-(4-фторфенил)-3-(2-метилсульфанилпиримидин-4-ил)-1-оксо-5,6,7,8-тетрагидро-1Н-пиразоло[1,2-a]пиридазин-5-(S)-карбоновой кислоты (30): К раствору 1-[2-(4-фторфенил)-2-оксоэтил]-2-(2-метилсульфанилпиримидин-4-карбонил)гексагидропиридазин-3-(S)-карбоновой кислоты, 29 (1 г), в метаноле (170 мл) добавляют NaOH (0,23 г, 5,8 ммоль). Образовавшуюся смесь перемешивают в течение 15 часов и смесь концентрируют в вакууме с получением остатка, который растворяют в воде (150 мл). Раствор подкисляют 3 н. HCl до рН 1 и экстрагируют этилацетатом (300 мл). Органический слой концентрируют в вакууме и образовавшееся неочищенное вещество очищают препаративной ВЭЖХ, получая 7,0 г (выход 76%) требуемого продукта в виде твердого вещества кремового цвета.
Получение метилового эфира 2-(4-фторфенил)-3-(2-метилсульфанилпиримидин-4-ил)-1-оксо-5,6,7,8-тетрагидро-1Н-пиразоло[1,2-a]пиридазин-5-(S)-карбоновой кислоты (31): К раствору 2-(4-фторфенил)-3-(2-метилсульфанилпиримидин-4-ил)-1-оксо-5,6,7,8-тетрагидро-1Н-пиразоло[1,2-a]пиридазин-5-карбоновой кислоты, 30, в смеси диэтиловый эфир/этилацетат (2,5:1, 70 мл) добавляют свежеприготовленный диазометан в диэтиловом эфире (5 мл). Реакционную смесь перемешивают в течение 5 минут, затем гасят добавлением НОАс (0,5 мл). Полученный раствор промывают NaHCO3, насыщенным раствором соли, сушат и концентрируют в вакууме, получая 1 г (выход 98%) требуемого продукта в виде светло-желтого твердого вещества.
Получение метилового эфира 2-(4-фторфенил)-3-(2-метансульфонилпиримидин-4-ил)-1-оксо-5,6,7,8-тетрагидро-1Н-пиразоло[1,2-a]пиридазин-5-(S)-карбоновой кислоты (32): К раствору метилового эфира 2-(4-фторфенил)-3-(2-метилсульфанилпиримидин-4-ил)-1-оксо-5,6,7,8-тетрагидро-1Н-пиразоло[1,2-a]пиридазин-5-карбоновой кислоты, 31 (0,48 г, 1,16 ммоль), в смеси ТГФ/метанол, 1:1, (50 мл) добавляют Oxone® (2,14 г, 3,5 ммоль) в воде (50 мл). Реакционную смесь перемешивают в течение 5 часов при комнатной температуре, уменьшают объем в вакууме приблизительно до 25 мл и добавляют этилацетат (200 мл). Органическую фазу обрабатывают NaHCO3, насыщенным раствором соли, сушат и концентрируют в вакууме, получая 0,5 г требуемого продукта в виде желтого твердого вещества.
Получение метилового эфира 2-(4-фторфенил)-3-(2-феноксипиримидин-4-ил)-1-оксо-5,6,7,8-тетрагидро-1Н-пиразоло[1,2-a]пиридазин-5-(S)-карбоновой кислоты (33): NaOH (0,112 г, 2,8 ммоль) добавляют к раствору фенола (0,316 г, 3,36 ммоль) в ТГФ (100 мл). Метиловый эфир 2-(4-фторфенил)-3-(2-метансульфонилпиримидин-4-ил)-1-оксо-5,6,7,8-тетрагидро-1Н-пиразоло[1,2-a]пиридазин-5-карбоновой кислоты, 32 (0,5 г), растворяют в ТГФ (50 мл) и добавляют по каплям к раствору на протяжении 5 минут. Полученную смесь перемешивают при комнатной температуре в течение 8 часов, после чего добавляют воду (20 мл). Раствор экстрагируют этилацетатом (100 мл), органический слой промывают насыщенным раствором соли (50 мл) и концентрируют в вакууме, получая 0,278 г (выход 54%) требуемого продукта в виде желтого твердого вещества. 1Н-ЯМР (300 МГц, CDCl3) δ 1,75 (м, 2H), 1,97 (м, 1H), 2,42 (д, J=12,8 Гц, 1H), 3,27 (м, 1H), 3,27 (м, 1H), 3,6 (с, 3H), 4,5 (уш.д, J=12,8 Гц, 1H), 5,25 (м, 1H), 6,87 (д, J=5,7 Гц, 1H), 7,05 (м, 2H), 7,23 (м, 2H), 7,35 (м, 3H), 7,52 (м, 2H), 8,42 (д, J=5,7 Гц, 1H): точная масса, вычисленная для C25H21FN4O4, 460,46, МС-ESI (М+1) 461.
Четвертый аспект аналогов категории III настоящего изобретения, способных ингибировать высвобождение воспалительных цитокинов, относится к соединениям, включающим остов 5,6,7,8-тетрагидропиразоло[1,2-a]пиридазин-1-она и имеющим формулу:
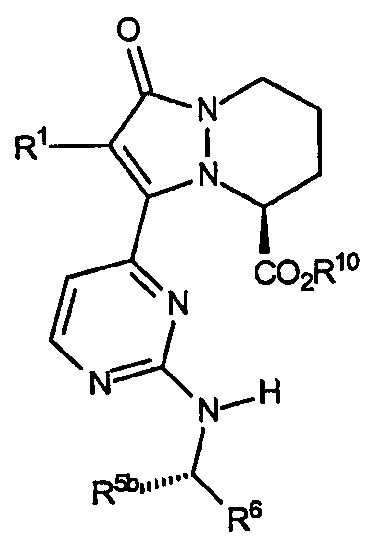
где звенья R представляют собой амины, имеющие формулу -NH[CHR5b]R6, и R1, R5b, R6 и R10 имеют значения, указанные здесь ниже в таблице VII. Стереохимия R5b является конфигурацией, показанной, когда R5b не является водородом.
Соединения, которые включают аналоги четвертого аспекта категории III, могут быть получены синтезом, показанным здесь ниже на следующей схеме, начинающейся с промежуточного соединения 32.
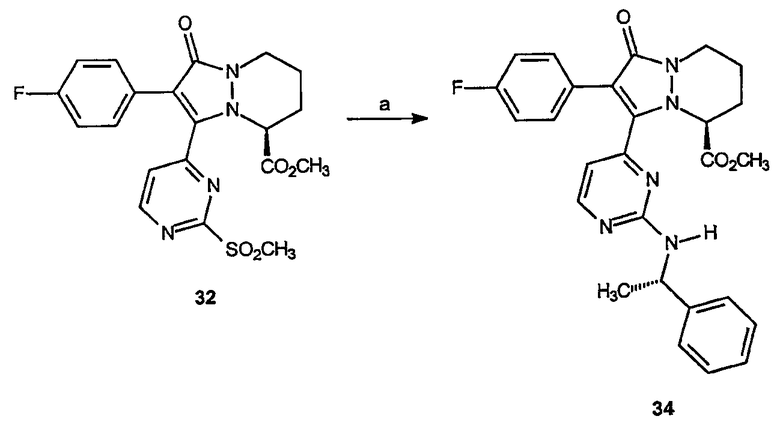
Пятый аспект аналогов категории III настоящего изобретения, способных ингибировать высвобождение воспалительных цитокинов, относится к соединениям, включающим остов 5,6,7,8-тетрагидропиразоло[1,2-a]пиридазин-1-она и имеющим формулу:
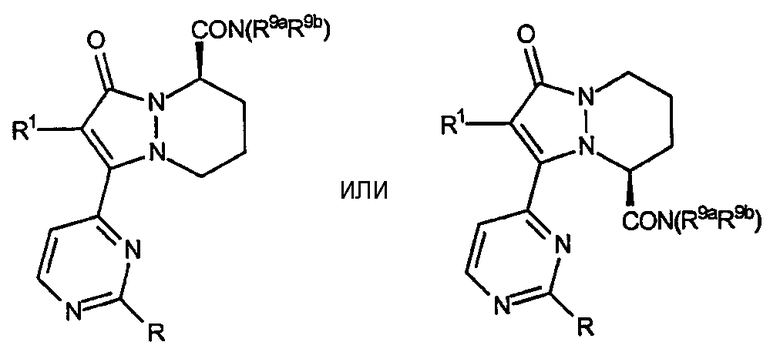
где R, R1, R9a и R9b имеют значения, указанные ниже в таблице VIII.
Другой вариант данного аспекта относится к соединениям, у которых R9a и R9b, взятые вместе, образуют карбоциклическое или гетероциклическое кольцо, включающее от 3 до 7 атомов. В таблице IX описаны соединения, включенные данным вариантом пятого аспекта категории III.
Другие соединения настоящего изобретения включают:
2-(4-фторфенил)-5-(пиперазин-1-карбонил)-3-(2-феноксипиримидин-4-ил)-5,6,7,8-тетрагидро-3Н-пиразоло[1,2-a]пиридазин-1-он;
2-(4-фторфенил)-8-(пиперазин-1-карбонил)-3-(2-феноксипиримидин-4-ил)-5,6,7,8-тетрагидропиразоло[1,2-a]пиридазин-1-он;
2-(4-фторфенил)-8-(морфолин-4-карбонил)-3-(2-феноксипиримидин-4-ил)-5,6,7,8-тетрагидропиразоло[1,2-a]пиридазин-1-он;
2-(4-фторфенил)-5-(морфолин-4-карбонил)-3-[2-(4-фторфенокси)пиримидин-4-ил]-5,6,7,8-тетрагидропиразоло[1,2-a]пиридазин-1-он;
2-(4-фторфенил)-8-(морфолин-4-карбонил)-3-[2-(4-фторфенокси)пиримидин-4-ил]-5,6,7,8-тетрагидропиразоло[1,2-a]пиридазин-1-он;
2-(4-фторфенил)-5-(морфолин-4-карбонил)-3-{2-[1-(S)-(α)-(метил)бензиламино]пиримидин-4-ил}-5,6,7,8-тетрагидропиразоло[1,2-a]пиридазин-1-он;
2-(4-фторфенил)-8-(морфолин-4-карбонил)-3-{2-[1-(S)-(α)-(метил)бензиламино]пиримидин-4-ил}-5,6,7,8-тетрагидропиразоло[1,2-a]пиридазин-1-он.
Аналоги (соединения) настоящего изобретения разделены на несколько категорий, чтобы помочь специалисту в данной области применить рациональную синтетическую стратегию для получения аналогов, которые явно не приводятся в данном описании в качестве примеров. Расположение по категориям не означает повышенную или пониженную эффективность для любой из описываемых и рассматриваемых композиций.
Во многих случаях было найдено, что соединения, перечисленные и описанные здесь выше, проявляют активность (IC50 в описанном ниже анализе на основе клеток или в анализах, которые приводятся в ссылках) при уровне ниже 1 микромолярного (мкМ).
Соединения настоящего изобретения способны эффективно блокировать продуцирование воспалительных цитокинов из клеток, что, таким образом, обеспечивает возможность подавления, ослабления, прекращения, замедления или профилактики развития одного или нескольких патологических состояний или синдромов, которые относятся к внеклеточному высвобождению одного или нескольких цитокинов. Воспалительные патологические состояния включают состояния, которые относятся к следующим неограничивающим примерам молекул:
i) Интерлейкин-1 (IL-1): принимает участие в качестве молекулы, ответственной за большое число патологических состояний, среди прочего, ревматоидного артрита, остеоартрита, а также других патологических состояний, которые относятся к разрушению соединительной ткани.
ii) Циклооксигеназа-2 (СОХ-2): ингибиторы высвобождения цитокина предложены в качестве ингибиторов индуцируемой экспрессии СОХ-2, которая, как было обнаружено, повышается цитокинами. M.K. O'Banion et al., Proc. Natl. Acad. Sci. U.S.A., 89, 4888 (1998).
iii) Фактор-α некроза опухоли (TNF-α): предполагается, что указанный провоспалительный цитокин является важным медиатором во многих патологических состояниях или синдромах, среди прочего, ревматоидном артрите, остеоартрите, воспалительном заболевании кишечника (IBS), септическом шоке, сердечно-легочной дисфункции, остром респираторном заболевании и кахексии.
Каждое из патологических состояний, которое специалисту в данной области требуется вылечить, может требовать свой, отличный от других уровень или количество описанных здесь соединений, для достижения терапевтического уровня. Специалист в данной области может определить такое количество любым из известных способов испытания, известных специалисту в данной области.
Настоящее изобретение далее относится к формам настоящих соединений, которые в обычных для человека или высшего млекопитающего физиологических условиях высвобождают описываемые соединения. Один вариант этого аспекта включает фармацевтически приемлемые соли описываемых аналогов. Для цели совместимости со способом доставки, эксципиентами и тому подобное специалист в данной области может выбрать одну форму соли настоящих аналогов из числа других, поскольку соединения сами являются активными соединениями, которые подавляют описанные в данном описании процессы заболеваний.
К данному аспекту имеют отношение различные «пролекарственные» формы предшественников аналогов настоящего изобретения. Соединения настоящего изобретения могут быть желательно получены в виде химических производных, которые сами не являются активными против описываемой активности цитокинов, но вместо этого являются формами настоящих аналогов, которые при доставке в организм человека или высшего животного будут подвергаться химической реакции, катализируемой нормальной функцией организма, среди прочего, ферментами, присутствующими в желудке, сывороткой крови, причем указанная химическая реакция высвобождает основное активное соединение. Термин «пролекарство» относится к соединениям, которые превращаются in vivo в активное фармацевтическое средство.
КОМПОЗИЦИИ
Настоящее изобретение относится также к композициям или готовым препаративным формам, которые включают соединения настоящего изобретения, ингибирующие высвобождение воспалительных цитокинов. В общем, композиции настоящего изобретения включают:
а) эффективное количество одного или нескольких бициклических пиразолонов и их производных настоящего изобретения, которые являются эффективными для ингибирования высвобождения воспалительных цитокинов, и
b) один или несколько фармацевтически приемлемых эксципиентов.
Для целей настоящего изобретения термин «эксципиент» и «носитель» используют взаимозаменяемым образом на всем протяжении описания настоящего изобретения, и указанные термины определяют в данном описании как «ингредиенты, которые используют в практике приготовления безопасной и эффективной фармацевтической композиции».
Специалист должен понимать, что эксципиенты используют главным образом для того, чтобы они служили для доставки безопасного, стабильного и функционального фармацевтического средства, являясь не только частью общего наполнителя для доставки, но также средством для достижения эффективной абсорбции реципиентом активного ингредиента. Эксципиент может выполнять роль простого и непосредственного инертного наполнителя, или эксципиент, используемый в данном описании, может быть частью стабилизирующей рН системы или покрытия для гарантии безопасной доставки ингредиентов в желудок. Специалист может также воспользоваться тем фактом, что соединения настоящего изобретения имеют повышенную клеточную эффективность, улучшенные фармакокинетические свойства, а также улучшенную пероральную биологическую доступность.
Настоящее изобретение относится также к композициям или готовым препаративным формам, которые включают форму предшественника или «пролекарства» соединений настоящего изобретения, ингибирующих высвобождение воспалительных цитокинов. В общем, такие содержащие предшественники композиции настоящего изобретения включают:
а) эффективное количество одного или нескольких производных бициклических пиразолонов настоящего изобретения, которые действуют, высвобождая in vivo соответствующий аналог, который является эффективным в ингибировании высвобождения воспалительных цитокинов, и
b) один или несколько фармацевтически приемлемых эксципиентов.
СПОСОБ ПРИМЕНЕНИЯ
Настоящее изобретение относится также к способу регулирования уровня одного или нескольких индуцирующих воспаление цитокинов, среди прочих, интерлейкина-1 (IL-1), фактора-α некроза опухоли (TNF-α), интерлейкина-6 (IL-6) и интерлейкина-8 (IL-8) и, таким образом, способу лечения или профилактики, модулирования или ослабления патологических состояний, на которые влияют уровни внеклеточных воспалительных цитокинов. Настоящий способ включает стадию введения человеку или высшему млекопитающему эффективного количества композиции, включающей один или несколько ингибиторов воспалительных цитокинов настоящего изобретения.
Поскольку ингибиторы воспалительных цитокинов настоящего изобретения могут быть доставлены способом, по которому может быть достигнут более чем один сайт регуляции, в одно и то же время может быть модулировано более чем одно патологическое состояние. Нелимитирующие примеры заболеваний, на которые воздействуют, регулированием или ингибированием, ингибиторы воспалительных цитокинов, посредством чего модулируется избыточная активность цитокинов, включают остеоартрит, ревматоидный артрит, диабет, заболевание, вызванное вирусом иммунодефицита человека (ВИЧ).
МЕТОДИКИ
Оценка эффективности соединений настоящего изобретения может быть проведена, например, измерением констант ингибирования цитокинов и величины IC50, любым методом по выбору специалиста.
Нелимитирующие примеры подходящих анализов включают:
i) Ферментативный анализ с субстратом в УФ-видимом диапазоне спектра, как описано L. AI Reiter, Int. J. Peptide Protein Res., 43, 87-96 (1994).
ii) Ферментативный анализ с флуоресцентным субстратом, как описано Thornberry et al., Nature, 356, 768-774 (1992).
iii) Анализ с использованием клеток МКПК (РМСВ), как описано в патенте США 6204261 В1, Batchelor et al., выданном 20 марта 2001.
Каждый из перечисленных выше материалов включен в данное описание в качестве ссылки.
Кроме того, ингибирование фактора некроза опухоли, TNF-α, может быть измерено с использованием стимулированных липополисахаридом (LPS) моноцитарных клеток человека, как описано в:
i) K.M. Mohler et al., "Protection Against a Lethal Dose of Endotoxin by an Inhibitor of Tumour Necrosis Factor Processing", Nature, 370, pp 218-220 (1994).
ii) патенте США 6297381 B1, Cirillo et al., выданном 2 октября 2001, включенном в качестве ссылки и воспроизведенном в данном описании ниже в соответствующей его части.
Ингибирование продуцирования цитокинов может быть обнаружено измерением ингибирования TNF-α в стимулированных липополисахаридом клетках ТНР. Все клетки и реагенты растворяют в RPMI 1640 с Феноловым красным и L-глутамином, дополненным дополнительным L-глутамином (всего: 4 мМ), пенициллином и стрептомицином (каждый 50 единиц/мл) и фетальной бычьей сывороткой (3% FBS) (GIBCO, все концентрации указываются конечные). Анализ проводят в стерильных условиях, только препарат испытуемого соединения не является стерильным. Исходные растворы готовят в ДМСО с последующим разведением в RPMI 1640 в 2 раза выше, чем требуемая конечная концентрация для анализа. Конфлюэнтные клетки ТНР.1 (2 х 106 клеток/мл, конечная концентрация; American Type Culture Company, Rockville, Md) добавляют в 96-луночные полипропиленовые круглодонные культуральные планшеты (Costar 3790; стерильные), содержащие 125 мкл испытуемого соединения (2-кратно концентрированного) или наполнитель ДМСО (контроли, слепые опыты). Концентрация ДМСО не должна превышать 0,2% конечной. Клеточную смесь прединкубируют в течение 30 минут при 37°C, при 5% СО2 перед стимуляцией липополисахаридом (LPS, 1 мкг/мл в конце; Sigma L-2630, из серотипа 0111.В4 Е. Coli; сохраняют в виде исходного раствора концентрации 1 мг/мл в защищенном от эндотоксина, разбавленном Н2О наполнителе при -80°C). Слепые опыты (нестимулированные) получают носитель Н2О; конечный объем инкубации составляет 250 мкл. Инкубация (4 часа) происходит, как описано выше. Анализ должен быть закончен центрифугированием планшетов в течение 5 минут при комнатной температуре, 1600 об./мин (4033 g); супернатант затем переносят в прозрачные 96-луночные планшеты и сохраняют при -80°C до анализа TNF-α человека коммерчески доступным набором для ТИФА (Biosource #КНС3015, Camarillo, Ca). Вычисленной величиной IC50 является концентрация испытуемого соединения, которая вызывает 50% снижение при максимальном продуцировании TNF-α.
Хотя конкретные варианты осуществления настоящего изобретения были иллюстрированы и описаны, для специалистов в данной области должно быть очевидно, что могут быть сделаны различные другие изменения и модификации без выхода за пределы сущности и объема изобретения. Следовательно, прилагаемая формула изобретения охватывает все такие изменения и модификации, которые входят в объем настоящего изобретения.
| название | год | авторы | номер документа |
|---|---|---|---|
| 6,7-ДИГИДРО-5H-ПИРАЗОЛО[1,2-А]ПИРАЗОЛ-1-ОНЫ, РЕГУЛИРУЮЩИЕ ВОСПАЛИТЕЛЬНЫЕ ЦИТОКИНЫ (ВАРИАНТЫ), И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 2003 |
|
RU2289584C2 |
| 6,7-ДИГИДРО-5H-ПИРАЗОЛО[1,2-a]ПИРАЗОЛ-1-ОНЫ (ВАРИАНТЫ), ИНГИБИРУЮЩИЕ ВЫСВОБОЖДЕНИЕ ВОСПАЛИТЕЛЬНЫХ ЦИТОКИНОВ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ И СПОСОБ ИНГИБИРОВАНИЯ | 2002 |
|
RU2299885C2 |
| СПИРО-6,7-ДИГИДРО-5Н-ПИРАЗОЛО[1,2а]ПИРАЗОЛ-1-ОНЫ, КОТОРЫЕ РЕГУЛИРУЮТ ВОСПАЛИТЕЛЬНЫЕ ЦИТОКИНЫ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ И СПОСОБ ИНГИБИРОВАНИЯ | 2002 |
|
RU2272040C2 |
| СОЕДИНЕНИЕ НА ОСНОВЕ ДИГИДРОНАФТИРИДИНОНА, И СПОСОБ ЕГО ПОЛУЧЕНИЯ, И ЕГО ПРИМЕНЕНИЕ В МЕДИЦИНЕ | 2021 |
|
RU2809869C1 |
| КОНДЕНСИРОВАННЫЕ ПИРРОЛДИКАРБОКСАМИДЫ И ИХ ПРИМЕНЕНИЕ В КАЧЕСТВЕ ФАРМАЦЕВТИЧЕСКИХ СРЕДСТВ | 2013 |
|
RU2650111C2 |
| ЗАМЕЩЕННЫЕ 4-(АРИЛАМИНО) СЕЛЕНОФЕНОПИРИМИДИНОВЫЕ СОЕДИНЕНИЯ И СПОСОБЫ ИХ ПРИМЕНЕНИЯ | 2011 |
|
RU2566293C2 |
| КОНДЕНСИРОВАННЫЕ ТРИАЗОЛАМИНЫ В КАЧЕСТВЕ МОДУЛЯТОРОВ Р2Х7 | 2010 |
|
RU2533122C2 |
| БИЦИКЛИЧЕСКИЕ ГЕТЕРОЦИКЛИЧЕСКИЕ ПРОИЗВОДНЫЕ, ИХ ПОЛУЧЕНИЕ И ПРИМЕНЕНИЕ | 2013 |
|
RU2627269C2 |
| ГЕТЕРОЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ | 2005 |
|
RU2390522C2 |
| ПРОИЗВОДНЫЕ ЦИКЛОАЛКАНО-ПИРИДИНА, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ИХ ОСНОВЕ И ПРОМЕЖУТОЧНЫЕ СОЕДИНЕНИЯ | 1997 |
|
RU2194698C2 |
Настоящее изобретение относится к соединению, в том числе ко всем их энантиомерным и диастереомерным формам и фармацевтически приемлемым солям, которые способны предотвращать внеклеточное высвобождение воспалительных цитокинов. Предлагаемые соединения имеют формулу (I), где R представляет собой: a) -OR3 или b) -NR4aR4b; R3 представляет собой замещенный или незамещенный фенил, где заместителями являются: i) галоген; ii) С1-С6 алкил; iii) трифторметил; iv) трихлорметил; v) трибромметил; vi) циано; и vii) C1-С6 алкокси; R4a и R4b представляют собой, каждый независимо: а) водород или b) -[C(R5aR5b)]xR6; индекс х равен от 0 до 5; каждый R5a и R5b представляет собой, независимо, водород, неразветвленный или разветвленный C1-C4алкил, С3-С7 циклический алкил; R6 представляет собой -OR7 или C1-C4алкил; R1 представляет собой водород или C1-C4алкил; R1 представляет собой галогензамещенный фенил; звенья R2a и R2b выбраны, каждый независимо, из группы, состоящей из: а) водорода; b) -O(CH2)jR8; с) -(CH2)jCO2R10; d) -(CH2)jCON(R10)2; e) двойной связи, когда один R2a и один R2b взяты вместе с образованием двойной связи; f) кольца, когда один R2a и один R2b взяты вместе с образованием кольца, причем указанное кольцо выбрано из группы, состоящей из: i) бензольного и ii) диоксоланового; R8 и R10 представляют собой, каждый независимо, водород или С1-С4алкил; j представляет индекс от 0 до 5; m представляет индекс от 1 до 3; n представляет индекс от 1 до 3; m+n равно 4. Объектами изобретения также являются фармацевтическая композиция на основе вышеописанных соединений, ингибирующая внеклеточное высвобождение воспалительных цитокинов, и способ регулирования внеклеточного высвобождения воспалительных цитокинов. 3 н. и 7 з.п. ф-лы, 9 табл.
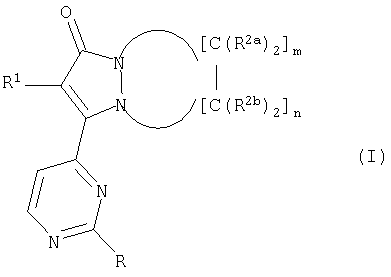
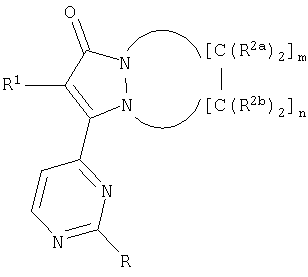
где R представляет собой
a) -OR3 или
b) -NR4aR4b;
R3 представляет собой замещенный или незамещенный фенил, где заместителями являются
i) галоген;
ii) C1-С6 алкил;
iii) трифторметил;
iv) трихлорметил;
v) трибромметил;
vi) циано и
vii) C1-С6 алкокси;
R4a и R4b представляют собой каждый независимо
a) водород или
b) -[C(R5aR5b)]xR6;
индекс х равен от 0 до 5;
каждый R5a и R5b представляет собой, независимо, водород, неразветвленный или разветвленный C1-C4 алкил, С3-С7 циклический алкил;
R6 представляет собой -OR7 или С1-C4 алкил;
R7 представляет собой водород или C1-C4 алкил;
R1 представляет собой галогензамещенный фенил;
звенья R2a и R2b выбраны каждый независимо из группы, состоящей из
a) водорода;
b) -O(CH2)jR8;
c) -(CH2)jCO2R10;
d) -(CH2)jCON(R10)2;
e) двойной связи, когда один R2a и один R2b взяты вместе с образованием двойной связи;
f) кольца, когда один R2a и один R2b взяты вместе с образованием кольца, причем указанное кольцо выбрано из группы, состоящей из
i) бензольного и
ii) диоксоланового;
R8 и R10 представляют собой каждый независимо водород или С1-С4алкил;
j представляет индекс от 0 до 5;
m представляет индекс от 1 до 3;
n представляет индекс от 1 до 3;
m+n равно 4.
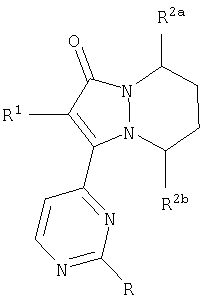
где R представляет собой
a) -OR3 или
b) -NR4aR4b;
R3 представляет собой замещенный или незамещенный фенил;
R4a и R4b каждый независимо представляют собой
a) водород или
b) -[C(R5aR5b)]xR6;
индекс х равен от 0 до 5;
каждый R5a и R5b представляет собой независимо водород, неразветвленный или разветвленный С1-С4 алкил, С3-С7 циклический алкил;
R6 представляет собой С1-С4алкил;
R7 представляет собой водород или С1-С1алкил;
R1 выбран из группы, состоящей из 4-фторфенила, 2,4-дифторфенила и 4-хлорфенила;
каждое звено R2a или R2b независимо выбрано из группы, состоящей из
a) водорода и
b) -O(CH2)jR8;
j равно 0.
R1 представляет собой 4-фторфенил;
R представляет собой простое эфирное звено, выбранное из группы, состоящей из фенокси, 2-фторфенокси, 3-фторфенокси, 4-фторфенокси, 2,6-дифторфенокси, 2-цианофенокси, 3-цианофенокси, 2-трифторметилфенокси, 4-трифторметилфенокси, 2-метилфенокси, 4-метилфенокси, 2,4-диметилфенокси, 2-метоксифенокси, 4-метоксифенокси, или
R представляет собой аминозвено, выбранное из группы, состоящей из метиламино, этиламино, пропиламино, циклопропиламино, циклопропилметил-амино, трет-бутиламино и 1-(S)-(циклопропил)этиламино.
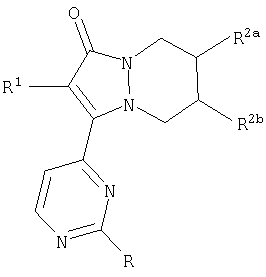
где R представляет собой
а) простой эфир, имеющий формулу -OR3 или
b) -NR4aR4b;
R3 представляет собой замещенный или незамещенный фенил;
R4a и R4b каждый независимо представляет собой
а) водород или
b) -[С(R5aR5b)]хR6;
индекс х равен от 0 до 5;
каждый R5a и R5b представляет собой независимо водород, неразветвленный или разветвленный C1-C4 алкил, С3-С7 циклический алкил;
R6 представляет собой -OR7 или C1-C4 алкил;
R7 представляет собой водород или С1-С1алкил;
R1 выбран из группы, состоящей из 4-фторфенила, 2,4-дифторфенила и 4-хлорфенила;
каждое звено R2a или R2b независимо выбрано из группы, состоящей из
a) водорода;
b) кольца, когда один R2a и один R2b взяты вместе с образованием кольца, причем указанное кольцо выбрано из группы, состоящей из
i) бензольного;
ii) диоксоланового;
с) двойной связи, когда один R2a и один R2b взяты вместе с образованием двойной связи;
j равно 0.
R1 представляет собой 4-фторфенил;
R представляет собой простое эфирное звено, выбранное из группы, состоящей из фенокси, 2-фторфенокси, 3-фторфенокси, 4-фторфенокси, 2,6-дифторфенокси, 2-цианофенокси, 3-цианофенокси, 2-трифторметилфенокси, 4-трифторметил-фенокси, 2-метилфенокси, 4-метилфенокси, 2,4-диметилфенокси, 2-метокси-фенокси, 4-метоксифенокси, или
R представляет собой аминозвено, выбранное из группы, состоящей из метиламино, этиламино, пропиламино, циклопропиламино, циклопропилметил-амино, трет-бутиламино и 1-(S)-(циклопропил)этиламино.
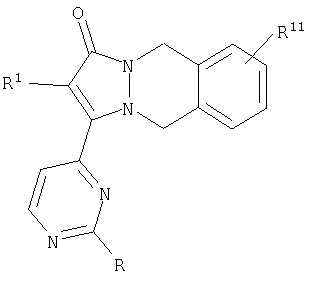
где R представляет собой
a) простой эфир, имеющий формулу -OR3, или
b) амин, имеющий формулу -NR4aR4b;
R3 представляет собой замещенный или незамещенный фенил;
R4a и R4b каждый независимо представляет собой
a) водород или
b) -[C(R5aR5b)]xR6;
индекс х равен 0;
каждый R5a и R5b представляет собой независимо водород, неразветвленный или разветвленный С1-С4алкил, циклический С3-С7алкил;
R6 представляет собой -OR7 или С1-С4алкил;
R7 представляет собой водород или С1-С1алкил;
R1 выбран из группы, состоящей из 4-фторфенила, 2,4-дифторфенила и 4-хлорфенила;
R11 представляет собой водород.
R1 представляет собой 4-фторфенил;
R представляет собой простое эфирное звено, выбранное из группы, состоящей из фенокси, 2-фторфенокси, 3-фторфенокси, 4-фторфенокси, 2,6-дифторфенокси, 2-цианофенокси, 3-цианофенокси, 2-трифторметилфенокси, 4-трифторметилфенокси, 2-метилфенокси, 4-метилфенокси, 2,4-диметилфенокси, 2-метоксифенокси и 4-метоксифенокси, или
R представляет собой аминозвено, выбранное из группы, состоящей метиламино, этиламино, пропиламино, циклопропиламино, циклопропилметил-амино, трет-бутиламино и 1-(S)-(циклопропил)этиламино.
a) эффективное количество одного или нескольких соединений по п.1 и
b) один или несколько фармацевтически приемлемых эксципиентов.
а) эффективное количество одного или нескольких соединений по п.1 и b) один или несколько фармацевтически приемлемых эксципиентов.
| ВПТБ | 0 |
|
SU375848A1 |
| US 3222366 А, 07.12.1965 | |||
| US 5366973 А, 22.11.1994. | |||

