Настоящее изобретение относится к новым производным пиперидина, используемым в качестве ингибиторов глюкозилцерамидсинтазы (ГЦС, UDP-глюкоза:церамид глюкозилтрансфераза; UDP-глюкоза:N-ацилсфингозин D-глюкозилтрансфераза, ЕС 2.4.1.80), способам их получения и их применению в медицине, в особенности при лечении и профилактике болезненных состояний, вызываемых ГЦС. Соединения нашли применение при лечении заболеваний, обусловленных накоплением гликолипидов, заболеваний, связанных с аккумуляцией гликолипидов, раков с аномальным гликолипидным синтезом, инфекционных заболеваний, вызываемых организмами, которые используют поверхность клетки гликолипида в качестве рецептора, инфекционных заболеваний, при которых синтез глюкозилцерамида является необходимым или важным, заболеваний, при которых происходит избыточный гликолипидный синтез, нейронные расстройства и нейронное поражение. Описаны также их синтез, фармацевтические композиции, включающие соединения, и способы лечения с использованием соединений.
ГЦС представляет собой внутриклеточный фермент, который катализирует соединение уридиндифосфатглюкозы и церамида в гликолипид, глюкозилцерамид. В настоящее время роль ГЦС в биологии является объектом интенсивного интереса фундаментальной и прикладной науки. Например, многие исследователи изучают роль ГЦС в регуляции уровней церамидов, поскольку данная молекула может индуцировать гибель апоптотных клеток (J. Biol. Chem., 2000, Mar 10, 275(10), 7138-43). Подобным образом ведутся активные исследования в отношении роли ГЦС в поддержании холестерин/гликолипидных "переправ", участков на поверхности клеточной мембраны с особой проницаемостью и функциональностью, которые, как считают, вовлечены в целый ряд событий трансдукции сигналов (Nature, 1997, Jun 5, 387(6633), 569-72).
ГЦС является также мишенью при лечении некоторых заболеваний человека. Глюкозилцерамид и структурно родственные гликолипиды накапливаются в лизосомах пациента, что приводит к мутации одного из незаменимых ферментов разрушения гликолипидов (например, болезней Гоше, Тея-Сакса, Сэндхоффа, ганглиозидоза GM1 и болезни Фабри). Накопление гликолипидов наблюдается также как вторичный эффект в некоторых тканях (например, нейронной ткани) при генетических болезнях, обусловленных накоплением, таких как болезнь С Ниманна-Пика, мукополисахаридозы, муколипидоз типа IV (Proc. Natl. Acad. Sci. USA, 1998, May 26, 95(11), 6373-8) и α-маннозидоз (Proc. Natl. Acad. Sci. USA, 1991 Dec 15, 88(24), 11330-4). Было выдвинуто предположение, что ингибиторы ГЦС могут быть применены для снижения скорости синтеза гликолипида в больных клетках так, чтобы присутствовало меньше гликолипида, который будет накапливаться, т.е. для подхода к лечению, называемого депривацией субстрата. Исследования показали, что ингибиторы ГЦС действительно могут быть использованы для снижения аккумуляции гликолипида, наблюдаемой на клеточных и животных моделях накопления гликолипида (Proc. Natl. Acad. Sci. USA, 1999, May 25, 96(11), 6388-93; Science, 1997, Apr 18, 276(5311), 428-31; J. Clin. Invest., 2000, Jun, 105(11), 1563-71). Кроме того, недавний отчет о клинических испытаниях показал, что ингибиторы ГЦС, такие как N-бутилдеоксиножиримицин (NB-DNJ) могут быть использованы при лечении пациентов с болезнью Гоше (Lancet, 2000, Apr 29, 355(9214), 1481-5). Применение иминосахара NB-DNJ в качестве ингибитора ГЦС описано в EP-A-0698012. В EP-A-0536402 и EP-A-0698012 описано, что N-алкилпроизводные деоксигалактоножиримицина, например, N-бутилдеоксигалактоножиримицин (NB-DGJ) также могут быть использованы при лечении расстройств, обусловленных накоплением гликолипида. В ЕР-А-0698012 описано также, что соответствующие N-бутилпроизводные маннозы (NB-DMJ), фукозы (NB-DFJ) и N-ацетилглюкозамин (NB-NAG) не действуют как ингибиторы биосинтеза гликолипида.
Было предложено использовать ингибиторы ГЦС при лечении злокачественных опухолей человека. Опухоли могут синтезировать аномальные количества гликолипидов и/или гликолипиды, не присутствующие в нормальной ткани. Вдобавок, гликолипиды или ганглиозиды в особенности накапливаются клетками опухоли и высвобождаются в межклеточное пространство и в кровоток. И опухолевое депо, и опухолевые ганглиозиды, связанные с поверхностью клетки, могут влиять на взаимодействия опухоль-клетка хозяин, такие как контакты клетка-клетка или адгезия (Methods Enzymol., 2000, 312, 447-58), подвижность клеток (Mol. Chem. Neuropathol., 1995 Feb-Apr, 24(2-3), 121-35), события сигнализации фактора роста (J. Biol. Chem., 2000 Nov 3, 275(44), 34213-23), стимулированный опухолью ангиогенез (Acta Oncol., 1997, 36(4), 383-7) и специфические иммунные реакции опухоли (J. Immunol., 1999 Oct 1, 163(7), 3718-26). Все эти события могут вызвать развитие и прогрессирование опухоли. Известно, что гликолипиды, в частности глюкозилцерамид, накапливаются в устойчивых к мультилекарствам (MDR) опухолевых клетках (Anticancer Res., 1998 Jan-Feb, 18(1B), 475-80), и воздействие in vitro на такие клетки ингибиторами ГЦС может реверсировать MDR фенотип (J. Biol. Chem., 1997, Jan 17, 272(3), 1682-7; Br. J. Cancer, 1999, Oct, 81(3), 423-30).
Гликолипиды поверхности клеток также играют роль в инфекционном заболевании, служа рецепторами для прикрепления патогенных бактерий (APMIS, 1990, Dec, 98(12), 1053-60, Review), грибков (Infect. Immun., 1990, Jul, 58(7), 2085-90) и вирусов (FEBS Lett.,1984 May 7, 170(1), 15-8). Кроме того, гликолипиды на поверхности клеток связываются с бактериальными токсинами (Methods Enzymol., 2000, 312, 459-73), например, с подгруппой В холерного токсина (ганглиозид GM1) и вероцитоксином (глоботриаосилцерамид GB3) (J. Infect. Dis., 2001, supl.70-73, 183).
Применение ингибиторов ГЦС может быть также полезным при ряде других клинических показаний, которые связаны с аномальностью синтеза гликолипидов. При атеросклеротическом поражении аорты человека наблюдается более высокое содержание ганглиозида, чем в неповрежденных областях аорты, и концентрации ганглиозида в сыворотке крови пациентов с атеросклерозом выше, чем у нормальных субъектов (Lipids, 1994, 29(1), 1-5). Ткани, выделенные из почек пациентов с полициститной болезнью почек, содержат более высокие концентрации и глюкозилцерамида, и лактозилцерамида (J. Lipid. Res., 1996, Jun, 37(6), 1334-44). Ренальная гипертрофия на животной модели диабета связана с увеличением синтеза гликолипидов (J. Clin. Invest., 1993, Mar, 91(3), 797-803).
Гликолипидный метаболизм играет также критическую роль при других нейронных заболеваниях, таких как болезнь Альцгеймера и эпилепсия. Например, в нейронах пациентов с болезнью С Ниманна-Пика (NPC) присутствуют клубки, напоминающие морфологию, наблюдаемую при болезни Альцгеймера.
Интересно, что GM1 ганглиозид, связанный с амилоидным бета-протеином, вызывает конформационные изменения, которые поддерживают образование им волокнистых полимеров, и фибриллярное отложение этого протеина показывает раннюю стадию болезни Альцгеймера (Yanagisawa et al. (1995), Nat. Med., 1, 1062-6; Choo-Smith et al. (1997), Biol. Chem., 272, 22987-90). Таким образом, снижение синтеза GM1 такими агентами как NB-DNJ может ингибировать образование волокон, наблюдаемое при болезни Альцгеймера.
Напротив, предварительные клинические испытания показали, что нейродегенеративные процессы, наблюдаемые при болезни Паркинсона, ударе и повреждении позвоночника, кажутся усиливающимися при лечении пациентов ганглиозидом GM1 (Alter, (1998), Ann. NY. Acad. Sci., 845, 391-401; Schneider (1998), Ann. NY. Acad. Sci., 845, 363-73; Geisler (1998), Ann. NY. Acad. Sci., 845, 374-81). Возможно, что совместное введение ингибиторов синтеза глюкозилцерамида может обеспечить больший клинический контроль во время этого курса лечения. Ингибиторы, подобные NB-DNJ, должны ограничить специфическую несовместимость пациентов, блокируя их нейронный гликолипидный синтез. Кроме того, ингибирование синтеза глюкозилцерамида должно ограничить метаболизм введенных гликолипидов в другие, возможно, непродуктивные формы. Таким образом, способность модулировать синтез глюкозилцерамида такими ингибиторами, как NB-DNJ, может быть полезной при лечении широкого круга нейронных расстройств.
Было также показано, что иминосахара могут обратимо индуцировать мужскую стерильность и могут поэтому быть использованы в качестве мужских контрацептивов.
Соединение (2S,3R,4R,5S)-1-бутил-2-(гидроксиметил)-3,4,5-пиперидинтриол описано в Anal. Biochem., 2000, 284(1), 136-142 как соединение для сравнения при анализе и фармацевтическая полезность этого соединения не указывается и не предполагается.
WO 01/10429 (опубликованная после даты приоритета данной заявки) описывает соединение N-нонилальтростатин (2S,3S,4R,5S)-1-нонил-2-(гидроксиметил)-3,4,5-пиперидинтриол и его применение при лечении вирусных инфекций.
Tet. Lett., 1990, 31(47), 6777-80 описывает соединение (2S,3R,4R,5S)-1-фенилметил-2-(гидроксиметил)-3,4,5-пиперидинтриол как образующиеся в малых количествах побочные продукты при синтезе (2R,3R,4R,5S)-1-фенилметил-2-гидроксиметил-3,4,5-пиперидинтриол и фармацевтическая полезность этого соединения не указывается и не предполагается. Соединения (2S,3R,4R,5S)-1-фенилметил-2-[(фенилметокси)метил]-3,4,5-трис(фенилметокси)пиперидин и (2S,3R,4R,5S)-1-фенилметил-2-[(ацетилокси)метил]-3,4,5-трис(ацетилокси)пиперидин также описаны как побочные продукты, получаемые при синтезе соответствующих (2R,3R,4R,5S) соединений.
Tetrahedron, 1997, 53(9), 3407-16 описывает соединение пиперидина, а именно, (2S,3S,4R,5S)-1-(фенилметил)-2-[(фенилметокси)метил]-3,4-ди(ацетилокси)-5-(фенилметокси)пиперидин как побочный продукт, полученный при синтезе соответствующего (2R,3S,4R,5S) соединения.
Bioorganic and Medicinal Chemistry, 1996, 4(11), 1857-65 описывает соединение пиперидина (2S,3R,4R,5S)-1-фенилметил-2-[(фенилметокси)метил]-3,4-ди(фенилметокси)-5-(бензоилокси)пиперидин как промежуточное соединение при синтезе (2S,3R,4R,5S)-2-(гидроксиметил)-3,4,5-пиперидинтриола.
Ввиду важности ГЦС в широком спектре интересов фундаментальной и прикладной науки необходимо, чтобы были разработаны новые инструменты, которые обеспечили способы модулировать функцию данного фермента. Для этой цели авторы синтезировали ряд новых соединений, которые полезны для ингибирования каталитической активности ГЦС.
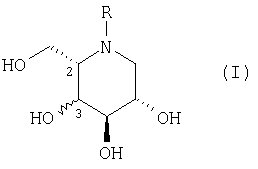
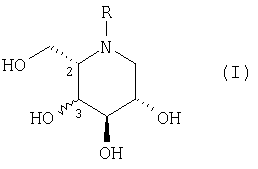
Согласно изобретению предлагается соединение формулы (I), или его фармацевтически приемлемая соль, или его пролекарство:

где:
R представляет С1-16 алкил с прямой или разветвленной цепью, необязательно замещенной С3-7 циклоалкилом и необязательно прерванной -О-, причем кислород отделен от азота кольца по меньшей мере двумя атомами углерода, или С1-10 алкиларил, где арил представляет фенил, пиридил, тиенил или фурил, в котором фенил необязательно замещен одним или несколькими заместителями, выбранными из F, Cl, Br, CF3, OCF3, OR1, и С1-6 алкила с прямой или разветвленной цепью; и
R1 представляет водород или С1-6 алкил с прямой или разветвленной цепью;
при условии, что соединение не представляет собой:
a) (2S,3R,4R,5S)-1-бутил-2-(гидроксиметил)-3,4,5-пиперидинтриол;
b) (2S,3R,4R,5S)-1-фенилметил-2-(гидроксиметил)-3,4,5-пиперидинтриол;
c) (2S,3S,4R,5S)-1-нонил-2-(гидроксиметил)-3,4,5-пиперидинтриол;
d) (2S,3R,4R,5S)-1-додецил-2-(гидроксиметил)-3,4,5-пиперидинтриол; или
e) (2S,3R,4R,5S)-1-(1-фенил)этил-2-(гидроксиметил)-3,4,5-пиперидинтриол.
Гидроксильная группа в положении 3 может быть прикреплена или в R-, или в S-конфигурации. Гидроксильная группа в положении 3 предпочтительно находится в R-конфигурации, т.е. соединение формулы (I) имеет стереохимию (2S,3R,4R,5S).
R предпочтительно представляет С1-16 алкил с прямой или разветвленной цепью или С1-10 алкилфенил, в котором фенил необязательно замещен одним или несколькими заместителями, выбранными из F, Cl, Br, CF3, OCF3, OR1, и С1-6 алкила с прямой или разветвленной цепью. Более предпочтительно, R представляет С1-16 алкил с прямой или разветвленной цепью. Еще более предпочтительно, R представляет С3-10 алкил с прямой цепью, в особенности С4-7 алкил с прямой цепью.
Соединения, используемые в способах по изобретению, предпочтительно имеют молекулярную массу меньше 800, более предпочтительно, меньше 600.
Конкретные соединения по изобретению, которые могут быть упомянуты, включают следующие:
(2S,3R,4R,5S)-1-пропил-2-(гидроксиметил)-3,4,5-пиперидинтриол;
(2S,3R,4R,5S)-1-пентил-2-(гидроксиметил)-3,4,5-пиперидинтриол;
(2S,3R,4R,5S)-1-гептил-2-(гидроксиметил)-3,4,5-пиперидинтриол;
(2S,3S,4R,5S)-1-бутил-2-(гидроксиметил)-3,4,5-пиперидинтриол;
(2S,3R,4R,5S)-1-нонил-2-(гидроксиметил)-3,4,5-пиперидинтриол;
(2S,3R,4R,5S)-1-(1-этил)пропил-2-(гидроксиметил)-3,4,5-пиперидинтриол;
(2S,3R,4R,5S)-1-(3-метил)бутил-2-(гидроксиметил)-3,4,5-пиперидинтриол;
(2S,3R,4R,5S)-1-(2-фенил)этил-2-(гидроксиметил)-3,4,5-пиперидинтриол;
(2S,3R,4R,5S)-1-(3-фенил)пропил-2-(гидроксиметил)-3,4,5-пиперидинтриол;
(2S,3R,4R,5S)-1-(1-этил)гексил-2-(гидроксиметил)-3,4,5-пиперидинтриол;
(2S,3R,4R,5S)-1-(2-этил)бутил-2-(гидроксиметил)-3,4,5-пиперидинтриол;
(2S,3R,4R,5S)-1-[(2R)-(2-метил-2-фенил)этил]-2-(гидроксиметил)-3,4,5-пиперидинтриол;
(2S,3R,4R,5S)-1-[(2S)-(2-метил-2-фенил)этил]-2-(гидроксиметил)-3,4,5-пиперидинтриол;
и их фармацевтически приемлемые соли и пролекарства.
Наиболее предпочтительным соединением является (2S,3R,4R,5S)-1-пентил-2-(гидроксиметил)-3,4,5-пиперидинтриол и его фармацевтически приемлемые соли.
Особую группу соединений по изобретению, которая может быть упомянута, составляют соединения формулы (Ia) или их фармацевтически приемлемые соли:

в которой:
R представляет С1-16 алкил с прямой или разветвленной цепью или С1-10 алкиларил, где арил представляет фенил, пиридил, тиенил или фурил, в котором фенил необязательно замещен одним или несколькими заместителями, выбранными из F, Cl, Br, CF3, OCF3, OR1, и С1-6 алкила с прямой или разветвленной цепью; и
R1 представляет водород или С1-6 алкил с прямой или разветвленной цепью;
при условии, что соединение не представляет собой:
a) (2S,3R,4R,5S)-1-бутил-2-(гидроксиметил)-3,4,5-пиперидинтриол;
b) (2S,3R,4R,5S)-1-фенилметил-2-(гидроксиметил)-3,4,5-пиперидинтриол;
c) (2S,3S,4R,5S)-1-нонил-2-(гидроксиметил)-3,4,5-пиперидинтриол;
d) (2S,3R,4R,5S)-1-додецил-2-(гидроксиметил)-3,4,5-пиперидинтриол; или
e) (2S,3R,4R,5S)-1-(1-фенил)этил-2-(гидроксиметил)-3,4,5-пиперидинтриол.
Как описано здесь, соединения по настоящему изобретению могут быть использованы для ингибирования ГЦС. Следовательно, во втором аспекте настоящее изобретение предлагает применение соединений формулы (I), но без ограничений a), b), d) и е), в медицине. Конкретные соединения, используемые по указанному выше назначению, включают, кроме упомянутых выше, соединение (2S,3R,4R,5S)-1-бутил-2-(гидроксиметил)-3,4,5-пиперидинтриол.
Подходящие фармацевтически приемлемые соли соединений формулы (I) включают, но не ограничиваются этим, соли с неорганическими кислотами, такие как гидрохлорид, сульфат, фосфат, дифосфат, гидробромид и нитрат, или соли с органическими кислотами, такие как малат, малеат, фумарат, тартрат, сукцинат, цитрат, ацетат, лактат, метансульфонат, п-толуолсульфонат, пальмитат, салицилат и стеарат.
Подходящие пролекарства соединений формулы (I) включают, но не ограничиваются этим, фармацевтически приемлемые эфиры, такие как С1-6алкильные эфиры.
Некоторые из соединений по данному изобретению могут быть кристаллизованы или перекристаллизованы из растворителей, таких как водные и органические растворители. В таких случаях могут образоваться сольваты. В объем данного изобретения включены стехиометрические сольваты, включая гидраты, а также соединения, содержащие переменные количества воды, которые могут образоваться в результате таких процессов, как лиофилизация.
Некоторые из соединений формулы (I) могут существовать в форме оптических изомеров, например, диастереоизомеров, и смесей изомеров во всех соотношениях, например, рацемических смесей. Изобретение охватывает все такие формы, в частности, чистые изомерные формы. Различные изомерные формы могут быть отделены или выделены одна от другой обычными способами, или любой заданный изомер может быть получен обычными синтетическими методами или стереоспецифичными или асимметричными синтезами.
Поскольку соединения формулы (I) предназначены для применения в фармацевтических композициях, будет легко понятно, что каждое из них предпочтительно представлено в достаточно чистом виде, например, с чистотой по меньшей мере 60%, более желательно, по меньшей мере 75%, и предпочтительно по меньшей мере 85%, в особенности, с чистотой по меньшей мере 98% (%% массовые). Нечистые препараты соединений могут быть использованы для приготовления более чистых форм, используемых в фармацевтических композициях; такие менее чистые препараты соединений должны содержать по меньшей мере 1%, более желательно, по меньшей мере 5%, и предпочтительно от 10 до 59% соединения формулы (I) или его фармацевтически приемлемого производного.
Термин "алкил", как он использован здесь, как сам по себе или как часть большего термина, например, "алкиларил", включает радикалы и с прямой, и с разветвленной цепью. Термин "алкил" включает также такие радикалы, в которых один или несколько атомов водорода замещены фтором.
Соединения формулы (I) могут быть получены по известным методикам из известных или имеющихся в продаже исходных веществ. Если исходные вещества являются недоступными из промышленных источников, их синтез описан здесь, или они могут быть получены по известным методикам.
Конкретно, соединения формулы (I) могут быть получены способом, включающим:
a) взаимодействие соединения формулы (II):
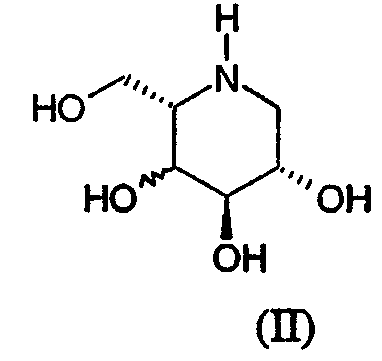
c NaBH3CN и альдегидом формулы R2CHO в смеси уксусная кислота-метанол, или с NaBH(OAc)3 и альдегидом формулы R2CHO в растворителе, таком как дихлорметан; где R2 представляет С1-15 алкил с прямой или разветвленной цепью, необязательно замещенной С3-7циклоалкилом и необязательно прерванной -О-, причем кислород отделен от группы СНО по меньшей мере одним атомом углерода, или С0-9алкиларил, где арил определен в формуле (I); или
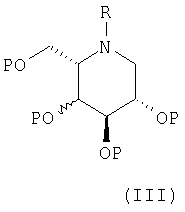
(b) снятием защиты с соединения формулы (III):
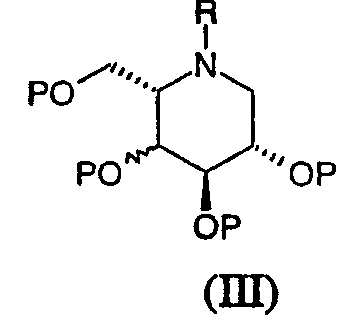
в которой R является таким, как определено в формуле (I), и Р, которые могут быть одинаковыми или различными, представляют гидрокси-защищенные группы, например, бензил (Bn). Если Р представляет CH2Ph, снятие защиты проводят в присутствии газообразного водорода и катализатора, такого как PdCl2 или палладий-на-угле, в подходящем растворителе, таком как спирт, например, этанол. Должно быть понятно, что, если Р представляет CH2Ph и R представляет CH2Ph, группа R также может быть удалена при таких условиях, давая соединения формулы (II); следовательно, соединения формулы (I), где R представляет CH2Ph, предпочтительно получают, используя вышеприведенный способ (а).
Соединения формулы (II) известны, см., например, Carbohydr. Res., 1993, 246, 377-81 (2S3R4R5S) и Tet. Lett., 1997, 38(45), 8009-12 (2S3S4R5S).
Соединения формулы (III) могут быть получены при взаимодействии соединения формулы (IV):
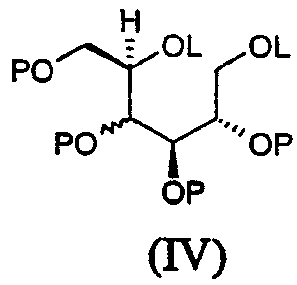
где L, которые могут быть одинаковыми или различными, являются удаляемыми группами, такими как мезил, и Р является таковым, как определено в формуле (III), с амином формулы RNH2, в которой R является таким, как определено в формуле (I), либо в чистом виде, либо в растворителе, таком как тетрагидрофуран.
Соединение (IVa), в котором L представляет мезил и P представляет бензил, известно: V.S.Rao et al., Can. J. Chem. (1981), 59(2), 333-8; P.A.Fowler et al., Carbohydr. Res. (1993), 246, 377-81.
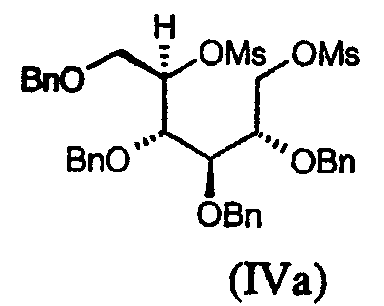
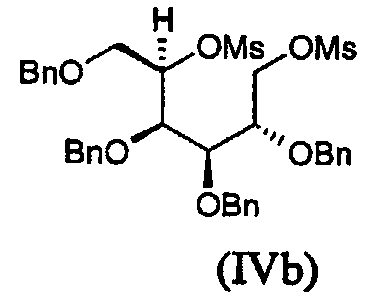
Соединение (IVb), в котором L представляет мезил и P представляет бензил, может быть получено реакцией 2,3,4,6-тетра-О-бензил-D-галактитола с мезилхлоридом в присутствии основания, такого как пиридин.
Любые новые промежуточные соединения, которые описаны здесь, также включены в объем настоящего изобретения.
Таким образом, согласно следующему аспекту изобретения предложено соединение формулы (III), как оно определено выше, при условии, что соединение не представляет собой:
i) (2S,3R,4R,5S)-1-фенилметил-2-[(фенилметокси)метил]-3,4,5-трис(фенилметокси)пиперидин;
ii) (2S,3R,4R,5S)-1-фенилметил-2-[(ацетилокси)метил]-3,4,5-трис(ацетилокси)пиперидин;
iii) (2S,3S,4R,5S)-1-фенилметил-2-[(фенилметокси)метил]-3,4-ди(ацетилокси)-5-(фенилметокси)пиперидин;
iv) (2S,3S,4R,5S)-1-(метил)-2-[(фенилметокси)метил]-3,4-ди(ацетилокси)-5-(фенилметокси)пиперидин;
v) сложный эфир бутандионовой кислоты (3α, 5α)-холестан-3-ола и (2S,3R,4R,5S)-1-(фенилметил)-2-(гидроксиметил)-3,4,5-трис(фенилметокси)пиперидина или
vi) (2S,3R,4R,5S)-1-(фенилметил)-2-[(фенилкарбонилокси)метил]-3,4-ди(фенилметокси)-5-(фенилкарбонилокси)пиперидин.
Во время синтеза соединений формулы (I) лабильные функциональные группы в промежуточных соединениях, например, гидрокси-, карбокси- и аминогруппы, могут быть защищены. Исчерпывающее обсуждение способов, которыми могут быть защищены различные лабильные группы, и способов разложения полученных защищенных производных приведено, например, в Protective Groups in Organic Chemistry, T.W.Greene and P.G.M.Wuts, (Wiley-Interscience, New York, 2nd edition, 1991).
Дополнительные детали получения соединений формулы (I) можно найти в примерах.
Соединения формулы (I) могут быть получены по отдельности или в виде библиотеки соединений, включающей по меньшей мере 2, например, от 5 до 500 соединений, и, более предпочтительно, от 10 до 100 соединений формулы (I). Библиотеки соединений формулы (I) могут быть получены комбинаторным подходом "раздели и смешай" или множественным параллельным синтезом с использованием либо химии фазы раствора или химии твердой фазы способами, известными специалистам в этой области.
Так, согласно следующему аспекту изобретения предложена библиотека соединений, включающая по меньшей мере 2 соединения формулы (I) или их фармацевтически приемлемые соли.
Фармацевтически эффективные соединения формулы (I) и их фармацевтически приемлемые соли могут быть введены в обычных дозированных формах, приготовленных объединением соединения формулы (I) ("активного ингредиента") со стандартными фармацевтическими носителями или разбавителями согласно обычным методам, хорошо известным из практики. Такие методы могут включать смешение, гранулирование и прессование или растворение ингредиентов, в зависимости от требуемой формы препарата.
Согласно следующему аспекту настоящее изобретение предлагает фармацевтические композиции, включающие одно или несколько соединений формулы (I), но без ограничений a), b), d) и е), вместе с одним или несколькими фармацевтически приемлемыми носителями или наполнителями.
Фармацевтические композиции по изобретению могут быть приготовлены для введения любым способом и включают формы, подходящие для перорального, местного или парентерального введения млекопитающим, включая человека.
Фармацевтические формы могут быть предназначены для введения любым подходящим путем, например, для перорального (включая защечное или подъязычное), ректального, назального, местного (включая защечное, подъязычное или трансдермальное), вагинального или парентерального (включая подкожное, внутримышечное, внутривенное или интрадермальное) введения. Такие формы могут готовиться любым известным в фармацевтической практике способом, например, вводом в сочетание активного ингредиента с носителем (носителями) или наполнителем (наполнителями).
Фармацевтические формы, предназначенные для перорального введения, могут быть представлены в виде дискретных дозированных единиц, таких как капсулы или таблетки; порошков или гранул; растворов или суспензий в водных или неводных жидкостях; съедобных пен или кремов; или эмульсий масло-в-воде или вода-в-масле.
Фармацевтические формы, предназначенные для трансдермального введения, могут быть представлены в виде дискретных наклеек, предназначенных оставаться в тесном контакте с эпидермисом реципиента в течение длительного периода времени. Например, активный ингредиент может доставляться из наклейки, например, ионтофорезом, как в общем случае описано в Pharmaceutical Research, 3(6), 318 (1986).
Фармацевтические формы, предназначенные для местного введения, могут быть составлены как мази, кремы, суспензии, лосьоны, порошки, растворы, пасты, гели, пропитанные повязки, спреи, аэрозоли или масла и могут содержать в мазях и кремах обычные добавки, такие как консерванты, растворители для облегчения проникновения лекарств и эмоленты.
Для нанесения в глаза или на другие внешние ткани, например, на ткани рта или шеи, формы предпочтительно наносят в виде местной мази или крема. Если готовят мазь, то активный ингредиент может быть введен или с парафинной, или со смешивающейся с водой основой мази. Альтернативно, активный ингредиент может быть введен в крем с основой крема масло-в-воде или вода-в-масле.
Фармацевтические формы, предназначенные для местного введения в глаза, включают глазные капли, в которых активный ингредиент растворен или суспендирован в подходящем носителе, предпочтительно, в водном растворителе.
Фармацевтические формы, предназначенные для местного введения в рот, включают лепешки, пастилки и полоскания рта.
Фармацевтические формы, предназначенные для ректального введения, могут быть представлены в виде свечей или клизм.
Фармацевтические формы, предназначенные для назального введения, где носитель представляет собой твердое вещество, включают крупный порошок, имеющий размер частиц, например, в интервале от 20 до 500 микрон, который вводят таким образом, при котором берут понюшку, т.е. быстрым вдыханием через носовой проход из контейнера с порошком, удерживаемого близко к носу. Подходящие формы, в которых носитель является жидким, для введения в виде спрея для носа или капель в нос включают водные или масляные растворы активного ингредиента.
Фармацевтические формы, предназначенные для введения ингаляцией, включают пыли с тонкими частицами или аэрозоли, которые могут быть созданы с применением различных типов нажимаемых аэрозольных контейнеров, распылителей или инсуффляторов.
Фармацевтические формы, предназначенные для вагинального введения, могут быть представлены в виде пессариев, тампонов, кремов, гелей, паст, пен или композиций для распыления.
Фармацевтические формы, предназначенные для парентерального введения, включают водные и неводные стерильные растворы для инъекций, которые могут содержать антиоксиданты, буферы, бактериостатики и растворенное вещество, которое делает композицию изотоничной с кровью предполагаемого реципиента; и водные и неводные стерильные суспензии, которые могут включать суспендирующие агенты и загустители. Формы могут поставляться в однодозовых или многодозовых контейнерах, например, в запаянных ампулах и пузырьках, и могут храниться в высушенном лиофилизацией состоянии, требуя только добавления стерильной жидкости, например, воды для инъекций, непосредственно перед употреблением. Приготовляемые на месте инъекционные растворы и суспензии могут готовиться из стерильных порошков, гранул и таблеток.
Должно быть понятно, что в дополнение к ингредиентам, в частности, указанным выше, композиции могут включать другие агенты, традиционно известные из уровня техники, с учетом типа конкретной композиции, например, те, которые являются подходящими для перорального введения, могут включать вкусовые агенты.
Фармацевтические композиции согласно изобретению предназначены предпочтительно для перорального введения.
Композиции могут содержать также совместимые обычные носители, такие как основы кремов или мазей, и этанол или олеиловый спирт для лосьонов. Такие носители могут составлять от примерно 1% до примерно 98% от формы. Более предпочтительно, они должны составлять до примерно 80% от композиции.
Таблетки и капсулы для перорального введения могут быть в форме единичных доз и могут содержать обычные наполнители, такие как связующие агенты, например, сироп, камедь, желатин, сорбитол, трагакант, или поливинилпирролидон; наполнители, например, лактозу, сахар, кукурузный крахмал, фосфат кальция, сорбитол или глицин; таблетирующие смазывающие агенты, например, стеарат магния, тальк, полиэтиленгликоль или двуокись кремния; разрыхлители, например, картофельный крахмал; или приемлемые смачивающие агенты, такие как лаурилсульфат натрия. На таблетки могут быть нанесены покрытия известными из фармацевтической практики способами. Жидкие препараты для перорального введения могут быть, например, в форме водных или масляных суспензий, растворов, эмульсий, сиропов или эликсиров или могут быть поставлены как сухой продукт для восстановления водой или другим подходящим носителем перед употреблением. Такие жидкие препараты могут содержать обычные добавки, такие как суспендирующие агенты, например, сорбитол, метилцеллюлозу, глюкозный сироп, желатин, гидроксиэтилцеллюлозу, карбоксиметилцеллюлозу, гель стеарата алюминия или гидрогенизированные пищевые жиры, эмульгаторы, например, лецитин, сорбитан моноолеат или камедь; неводные носители (которые могут включать пищевые масла), например, миндальное масло, маслянистые эфиры, такие как эфиры глицерина, пропиленгликоля или этилового спирта; консерванты, например, метил или пропил п-гидроксибензоат или сорбиновую кислоту и, если требуется, обычные вкусовые и окрашивающие агенты.
Свечи должны содержать обычные основы для свечей, например, масло какао или другие глицериды.
Для парентерального введения готовят жидкие единичные дозы, используя соединение и стерильный носитель, причем предпочтительным носителем является вода. Соединение может быть или суспендировано или растворено в носителе в зависимости от носителя и используемой концентрации. При приготовлении растворов соединение может быть растворено в воде для инъекций и стерилизовано фильтрацией перед заполнением подходящего флакона или ампулы и запаиванием.
Благоприятно, когда в носителе могут быть растворены такие агенты, как местные обезболивающие, консерванты и буферные агенты. Для улучшения стабильности композиция может быть заморожена после заполнения сосуда, и вода может быть удалена под вакуумом. Затем сухой лиофилизированный порошок запаивают в сосуде и для восстановления жидкости перед использованием можно приложить сопутствующий сосуд с водой для инъекций. Парентеральные суспензии готовят практически таким же образом за исключением того, что соединение суспендируют в носителе вместо того, чтобы его растворять, и стерилизация не может быть осуществлена фильтрацией. Соединение может быть стерилизовано выдержкой в этиленоксиде перед суспендированием в стерильном носителе. Благоприятно, чтобы в композицию был включен ПАВ или смачивающий агент для того, чтобы обеспечить равномерное распределение соединения.
Композиции могут содержать от 0,1% мас., предпочтительно от 10-60% мас. активного вещества в зависимости от способа введения.
Фармацевтические формы могут быть представлены в виде единичных дозированных форм, содержащих заранее определенное количество активного ингредиента на дозу. Такая единица может содержать, например, до 1 г, обычно от 10 мг до 600 мг, предпочтительно от 50 мг до 300 мг, и более предпочтительно от 50 мг до 150 мг в зависимости от состояния, которое подвергается лечению, способа введения и возраста, веса и состояния пациента. Предпочтительными формами дозированных единиц являются те, которые содержат суточную дозу активного ингредиента, или поддозу, как указывалось здесь выше, или нужную ее часть.
Специалисту должно быть понятно, что оптимальное количество и интервалы приема индивидуальных доз соединения формулы (I) должны определяться природой и степенью состояния, которое должно подвергаться лечению, формой, способом и местом введения и тем, какое именно млекопитающее подвергается лечению, и что такой оптимум может быть определен обычными способами. Специалисту должно быть также ясно, что оптимальный курс, т.е. число доз соединения формулы (I), принимаемых за сутки в течение определенного числа суток, может быть выяснено специалистами при использовании обычного курса тестов определения лечения.
При введении соединения формулы (I) или его фармацевтически приемлемого производного в вышеуказанном интервале дозировок не обнаружены токсикологические эффекты.
Соединения по настоящему изобретению полезны тем, что они способны ингибировать глюкозилцерамидсинтазу. Так, соединения по изобретению могут быть использованы при лечении различных заболеваний, обусловленных накоплением гликолипидов, таких как болезнь Гоше, болезнь Сэндхоффа, болезнь Тея-Сакса, болезнь Фабри, GM1 ганглиозидозис, и т.п. Кроме того, соединения согласно изобретению могут также найти применение при лечении заболеваний, при которых происходит аккумуляция гликолипидов, таких как болезнь Ниманна-Пика, мукополисахаридозы (MPS I, MPS IIIA, MPS IIIB, MPS VI и MPS VII), муколипидоз типа IV и α-маннозидоз.
Соединения по настоящему изобретению могут быть также использованы при лечении раковых заболеваний, при которых гликолипидный синтез является аномальным, таких как опухоли мозга, нейробластома, злокачественная меланома, почечная аденокарцинома, и раки, устойчивые к мультилекарствам, в целом.
Соединения по настоящему изобретению могут быть также использованы при лечении заболеваний, вызываемых инфекционными организмами, которые используют гликолипиды на поверхности клеток в качестве рецепторов для инфекционного организма или вырабатываемого инфекционным организмом токсина.
Соединения по настоящему изобретению могут быть также использованы при лечении заболеваний, вызываемых инфекционными организмами, для которых синтез глюкозилцерамида является необходимым или важным процессом, таких как патогенный грибок Cryptococcus neoformans.
Соединения по настоящему изобретению могут быть также использованы при лечении заболеваний, при которых происходит избыточный синтез гликолипида, таких как, но не ограниченных этим, атеросклероз, полициститная болезнь почек и диабетическая почечная гипертрофия.
Соединения по настоящему изобретению могут быть также использованы при лечении нейронных заболеваний, таких как болезнь Альцгеймера и эпилепсия, и нейронных дегенеративных заболеваний, таких как болезнь Паркинсона.
Соединения по настоящему изобретению могут быть также использованы при лечении нейронных повреждений, таких как повреждение спинного мозга или удар.
Соединения по настоящему изобретению могут быть также использованы при лечении тучности.
Поэтому в дополнительных аспектах настоящее изобретение предлагает:
(i) применение соединения формулы (I), но без ограничений а)-е), в качестве ингибитора глюкозилцерамидсинтазы;
(ii) применение соединения формулы (I), но без ограничений а)-е), при изготовлении лекарства для лечения заболеваний, обусловленных накоплением гликолипидов. Примерами заболеваний, обусловленных накоплением гликолипидов, которые могут подвергаться лечению, являются, но не ограничиваются этим, болезнь Гоше, болезнь Сэндхоффа, болезнь Тея-Сакса, болезнь Фабри или GM1 ганглиозидоз;
(iii) применение соединения формулы (I), но без ограничений а)-е), при изготовлении лекарства для лечения болезни Ниманна-Пика типов А и С;
(iv) применение соединения формулы (I), но без ограничений а)-е), при изготовлении лекарства для лечения мукополисахаридоза типа I, мукополисахаридоза типа IIID, мукополисахаридоза типа IIIA, мукополисахаридоза типа VI или мукополисахаридоза типа VII;
(v) применение соединения формулы (I), но без ограничений а)-е), при изготовлении лекарства для лечения α-маннозидоза или муколипидоза типа IV;
(vi) применение соединения формулы (I), но без ограничений а)-е), при изготовлении лекарства для лечения раковых заболеваний, при которых гликолипидный синтез является аномальным, включающих, но не ограниченных этим, нейронные раки, включая нейробластому, рак мозга, почечную аденокарциному, злокачественную меланому, множественную миелому и раки, устойчивые к мультилекарствам;
(vii) применение соединения формулы (I), но без ограничений а)-е), при изготовлении лекарства для применения при лечении болезни Альцгеймера, эпилепсии или удара;
(viii) применение соединения формулы (I), но без ограничений а)-е), при изготовлении лекарства для применения при лечении болезни Паркинсона;
(ix) применение соединения формулы (I), но без ограничений а)-е), при изготовлении лекарства для лечения повреждения позвоночника;
(x) применение соединения формулы (I), но без ограничений а)-е), при изготовлении лекарства для использования при лечении заболеваний, вызываемых инфекционными микроорганизмами, которые используют гликолипиды на поверхности клеток в качестве рецепторов для самого организма или вырабатываемых организмом токсинов;
(xi) применение соединения формулы (I), но без ограничений а)-е), при изготовлении лекарства для использования при лечении заболеваний, вызываемых инфекционными организмами, для которых синтез глюкозилцерамида является необходимым или важным процессом, таких как, но не ограниченных этим, патологии, связанные с заражением патогенным грибком Cryptococcus neoformans;
(xii) применение соединения формулы (I), но без ограничений а)-е), при изготовлении лекарства для использования при лечении заболеваний, связанных с аномальным гликолипидным синтезом, таких как, но не ограниченных этим, полициститная болезнь почек, диабетическая почечная гипертрофия и атеросклероз;
(xiii) применение соединения формулы (I), но без ограничений а)-е), при изготовлении лекарства для лечения состояний, излечиваемых введением ганглиозида, такого как ганглиозид GM1. Примерами таких состояний являются болезнь Паркинсона, удар и повреждения спинного мозга;
(xiv) применение соединения формулы (I), но без ограничений а)-е), при изготовлении лекарства для обратимого вызывания бесплодия у самцов млекопитающих;
(xv) применение соединения формулы (I), но без ограничений а)-е), при изготовлении лекарства для лечения тучности, например, в качестве агента, подавляющего аппетит;
(xvi) способ лечения заболеваний, обусловленных накоплением гликолипидов, например, болезни Гоше, болезни Сэндхоффа, болезни Тея-Сакса, болезни Фабри или GM1 ганглиозидоза, который включает стадию введения пациенту эффективного количества соединения формулы (I), но без ограничений а)-е);
(xvii) способ лечения болезни Ниманна-Пика типов А и С, который включает стадию введения пациенту эффективного количества соединения формулы (I), но без ограничений а)-е);
(xviii) способ лечения мукополисахаридоза типа I, мукополисахаридоза типа IIID, мукополисахаридоза типа IIIA, мукополисахаридоза типа VI или мукополисахаридоза типа VII, который включает стадию введения пациенту эффективного количества соединения формулы (I), но без ограничений а)-е);
(xix) способ лечения α-маннозидоза или муколипидоза типа IV, который включает стадию введения пациенту эффективного количества соединения формулы (I), но без ограничений а)-е);
(xx) способ лечения раковых заболеваний, при которых гликолипидный синтез является аномальным, включающих, но не ограниченных этим, нейронные раки, включая нейробластому, рак мозга, почечную аденокарциному, злокачественную меланому, множественную миелому и раки, устойчивые к мультилекарствам, который включает стадию введения пациенту эффективного количества соединения формулы (I), но без ограничений а)-е);
(xxi) способ лечения болезни Альцгеймера, эпилепсии или удара, который включает стадию введения пациенту эффективного количества соединения формулы (I), но без ограничений а)-е);
(xxii) способ лечения болезни Паркинсона, который включает стадию введения пациенту эффективного количества соединения формулы (I), но без ограничений а)-е);
(xxiii) способ лечения повреждения позвоночника, который включает стадию введения пациенту эффективного количества соединения формулы (I), но без ограничений а)-е);
(xxiv) способ лечения заболеваний, вызываемых инфекционными микроорганизмами, которые используют гликолипиды на поверхности клеток в качестве рецепторов для самого организма или вырабатываемых организмом токсинов, который включает стадию введения пациенту эффективного количества соединения формулы (I), но без ограничений а)-е);
(xxv) способ лечения заболеваний, вызываемых инфекционными организмами, для которых синтез глюкозилцерамида является необходимым или важным процессом, таких как, но не ограниченных этим, патологии, связанные с заражением патогенным грибком Cryptococcus neoformans, который включает стадию введения пациенту эффективного количества соединения формулы (I), но без ограничений а)-е);
(xxvi) способ лечения заболеваний, связанных с аномальным гликолипдным синтезом, таких как, но не ограниченных этим, полициститная болезнь почек, диабетическая почечная гипертрофия и атеросклероз, который включает стадию введения пациенту эффективного количества соединения формулы (I), но без ограничений а)-е);
(xxvii) способ лечения состояний, излечиваемых введением ганглиозида, такого как ганглиозид GM1, который включает стадию введения пациенту эффективного количества соединения формулы (I), но без ограничений а)-е). Примерами таких состояний являются болезнь Паркинсона, удар и повреждения спинного мозга;
(xxviii) способ обратимого вызывания бесплодия у самцов млекопитающих, который включает стадию введения указанному самцу млекопитающего эффективного количества соединения формулы (I), но без ограничений а)-е);
(xxix) способ лечения тучности, который включает стадию введения пациенту эффективного количества соединения формулы (I), но без ограничений а)-е).
Изобретение также относится к применению соединения формулы (I), но без ограничений а)-е), для лечения вышеупомянутых заболеваний и состояний.
Фигура 1 показывает пути метаболизма гликолипидов в клетках млекопитающего. Показаны реакция, катализируемая глюкозилцерамидсинтазой, соединение уридиндифосфатглюкозы и церамида в глюкозилцерамид. Представлены также ферментные пути, приводящие к заболеваниям накопления гликолипидов у человека, а также реакция глюкозилцерамидсинтазы, ингибированной N-бутилдеоксиножиримицином (NB-DNJ).
Сокращения: UDP-Glc - уридиндифосфоглюкоза; Cer - церамид; Sial - сиаловая кислота; Gal - галактоза; GalNAc - N-ацетилгалактозамин; Glc - глюкоза; и
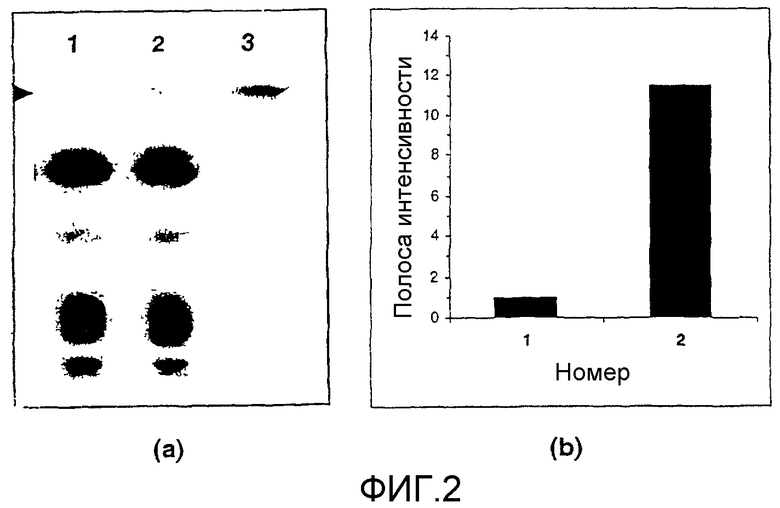
Фигура 2 показывает (а) хроматограмму тонкослойной хроматографии (ТСХ) неполярной липидной фракции, извлеченной из клеток карциномы груди MCF-7, обработанных в течение 7 суток 50 мкМ соединения примера 2 (1), клеток карциномы груди MCF-7 (2) и (3) представляет стандарт глюкозилцерамида, и (b) представляет измерение интенсивности полосы глюкозилцерамида из ТСХ хроматограммы относительно базовой линии образца, обработанного соединением примера 2 (1) и необработанного контроля (2).
Все публикации, включая, но не ограничиваясь этим, патенты и патентные заявки, процитированные в описании, включены в качестве ссылки таким образом, как если бы было полностью сказано, что каждая индивидуальная публикация отдельно и конкретно введена в качестве ссылки.
Изобретение подтверждено следующими примерами, которые являются просто иллюстративными и не должны рассматриваться как ограничение объема настоящего изобретения.
Пример 1. (2S,3R,4R,5S)-1-пропил-2-(гидроксиметил)-3,4,5-пиперидинтриол
a) 2,3,4,6-Тетра-О-бензил-1,5-ди-О-мезил-D-глюцитол
2,3,4,6-Тетра-О-бензил-D-глюцитол (45 г) растворяли в пиридине (200 мл) и добавляли в течение свыше 30 мин к раствору мезилхлорида (15 мл) в пиридине (100 мл) при 0°С. Прозрачный раствор хранили в течение ночи при 4°С, после чего анализ ТСХ показывал завершение реакции. Реакционную смесь распределяли между этилацетатом и водой со льдом. Органические фракции промывали 5% хлористоводородной кислотой и затем насыщенным водным раствором бикарбоната натрия, сушили (Na2SO4) и концентрировали до желто-оранжевого масла. Масло подвергали азеотропной осушке с толуолом и напрямую использовали на следующей стадии.
b) (2S,3R,4R,5S)-1-пропил-2-[(фенилметокси)метил]-3,4,5-трис(фенилметокси)пиперидин
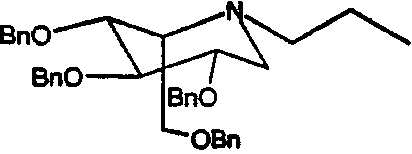
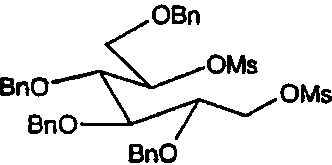
Сырой 2,3,4,6-тетра-О-бензил-1,5-ди-О-мезил-D-глюцитол (998 мг) растворяли в н-пропиламине (10 мл) и перемешивали при 55°С в течение 4 суток. ТСХ анализ показал, что реакция дошла до завершения. Реакционную смесь концентрировали и полученное сырое масло очищали флэш-хроматографией (градиентное элюирование 0→16% этилацетат/петролейный эфир), получая (2S,3R,4R,5S)-1-пропил-2-[(фенилметокси)метил]-3,4,5-трис(фенилметокси)пиперидин (610 мг, 73%).
1Н ЯМР (CDCl3): δ 0,9(3H, т), 1,4(2H, м), 2,45(2H, м), 2,6(1H, м), 2,8(1H, дд, J=5, 11 Гц), 3,3(1H, м), 3,5(2H, м), 3,6(2H, м), 3,7(1H, дд), 4,4-4,8(8H, м, OCH2Ph), 7,2-7,4(20H, м, ArH).
с) (2S,3R,4R,5S)-1-пропил-2-(гидроксиметил)-3,4,5-пиперидинтриол
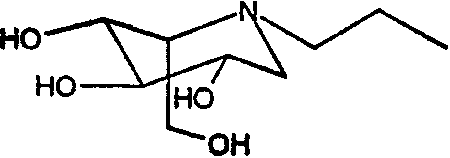
(2S,3R,4R,5S)-1-Пропил-2-[(фенилметокси)метил]-3,4,5-трис(фенилметокси)пиперидин (610 мг) растворяли в МеОН (10 мл) и перемешивали в течение ночи в атмосфере водорода в присутствии PdCl2 (300 мг). ТСХ показала завершение реакции. Реакционную смесь фильтровали через целит (с последующей промывкой метанолом/водой) и фильтрат концентрировали. Раствор разбавляли водой (10 мл) и медленно загружали на 5 г смолы Dowex 50X4-200, предварительно промытой хлористоводородной кислотой. Смолу промывали водой и затем элюировали смесью 1:7 концентрированный водный аммиак:вода. Продуктовые фракции концентрировали, получая (2S,3R,4R,5S)-1-пропил-2-(гидроксиметил)-3,4,5-пиперидинтриол (200 мг, 90%) в виде резиноподобного твердого вещества.
1Н ЯМР (D2О): δ 0,75(3H, т), 1,35(2H, м), 2,45(3H, м), 2,75(1H, дд, J=5, 12,5 Гц), 3,0(1H, дд, J=4, 9 Гц), 3,3(1H, т), 3,45(1H, м), 3,6(1H, дд, J=5, 10 Гц), 3,7(2H, м).
Пример 2. (2S,3R,4R,5S)-1-бутил-2-(гидроксиметил)-3,4,5-пиперидинтриол
a) (2S,3R,4R,5S)-1-бутил-2-[(фенилметокси)метил]-3,4,5-трис(фенилметокси)пиперидин
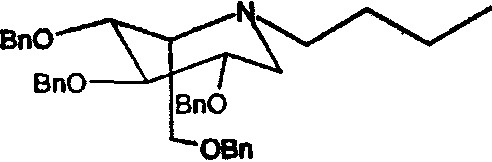
Сырой 2,3,4,6-Тетра-О-бензил-1,5-ди-О-мезил-D-глюцитол (пример 1а, 30 г) растворяли в н-бутиламине (200 мл) и перемешивали при 50°С в течение 4 суток. ТСХ анализ показал, что реакция дошла до завершения. Реакционную смесь концентрировали и полученное сырое масло очищали флэш-хроматографией (градиентное элюирование 0→16% этилацетат/петролейный эфир), получая (2S,3R,4R,5S)-1-бутил-2-[(фенилметокси)метил]-3,4,5-трис(фенилметокси)пиперидин (23 г, 90%).
1Н ЯМР (CDCl3): δ 0,9(3H, т), 1,2(2H, м), 1,4(2H, м), 2,5(2H, м), 2,7(1H, м), 2,9(1H, дд, J=6, 12 Гц), 3,4(1H, м), 3,5(1H, AB квартет, J=10 Гц), 3,55(1H, м), 3,65(1H, м), 3,7(1H, дд, J=2, 13 Гц), 3,8(1H, дд, J=6, 10 Гц), 4,4-4,9(8H, м, OCH2Ph), 7,2-7,4(20H, м, ArH).
b) (2S,3R,4R,5S)-1-бутил-2-(гидроксиметил)-3,4,5-пиперидинтриол
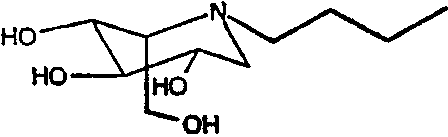
(2S,3R,4R,5S)-1-бутил-2-[(фенилметокси)метил]-3,4,5-трис(фенилметокси)пиперидин (15 г) растворяли в МеОН (300 мл) и перемешивали в течение ночи в атмосфере водорода в присутствии PdCl2 (5 г). ТСХ показала завершение реакции. Реакционную смесь фильтровали через целит (с последующей промывкой метанолом/водой) и фильтрат концентрировали до остаточного объема 50 мл. Раствор медленно загружали на 70 г смолы Dowex 50X12-200, которая была предварительно промыта хлористоводородной кислотой. Смолу промывали водой и затем элюировали смесью 1:7 концентрированный водный аммиак:вода. Продуктовые фракции концентрировали, получая (2S,3R,4R,5S)-1-бутил-2-(гидроксиметил)-3,4,5-пиперидинтриол (4,8 г, 85%) в виде бесцветного масла.
1Н ЯМР (D2О): δ 0,90(3H, т), 1,31(2H, м), 1,49(2H, м), 2,53(1H, дд), 2,63(1H, ддд), 2,72(1H, ддд), 2,87(1H, дд), 3,14(1H, кв), 3,44(1H, т), 3,61(1H, ддд), 3,75(1H, дд), 3,85(1H, дд), 3,89(1H, дд).
Пример 3. (2S,3R,4R,5S)-1-пентил-2-(гидроксиметил)-3,4,5-пиперидинтриол
a) (2S,3R,4R,5S)-1-пентил-2-[(фенилметокси)метил]-3,4,5-трис(фенилметокси)пиперидин
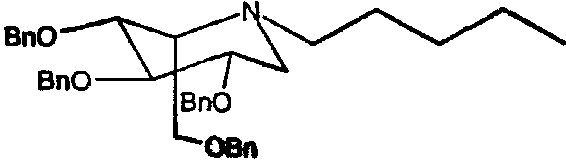
Сырой 2,3,4,6-тетра-О-бензил-1,5-ди-О-мезил-D-глюцитол (пример 1а, 1 г) растворяли в н-пентиламине (10 мл) и перемешивали при 55°С в течение 4 суток. ТСХ анализ показал, что реакция дошла до завершения. Реакционную смесь концентрировали и полученное сырое масло очищали флэш-хроматографией (градиентное элюирование 0→12% этилацетат/петролейный эфир), получая (2S,3R,4R,5S)-1-пентил-2-[(фенилметокси)метил]-3,4,5-трис(фенилметокси)пиперидин (680 мг, 76%).
1Н ЯМР (CDCl3): δ 1,0(3H, т), 1,2(2H, м), 1,4(4H, м), 1,6(2H, м), 2,7(2H, м), 2,85(1H, м), 3,05(1H, дд, J=5, 10,5 Гц), 3,55(1H, м), 3,7(2H, м), 3,85(2H, м), 3,95 (1H, дд, J=5, 9 Гц), 4,6-5,05(8H, м, OCH2Ph), 7,4-7,5(20H, м, ArH).
b) (2S,3R,4R,5S)-1-пентил-2-(гидроксиметил)-3,4,5-пиперидинтриол
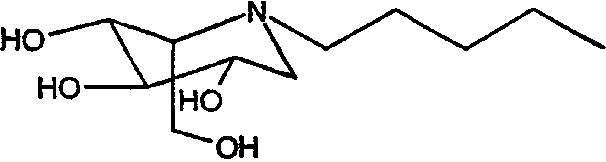
(2S,3R,4R,5S)-1-пентил-2-[(фенилметокси)метил]-3,4,5-трис(фенилметокси)пиперидин (680 мг) растворяли в МеОН (10 мл) и перемешивали в течение ночи в атмосфере водорода в присутствии PdCl2 (300 мг). ТСХ показала завершение реакции. Реакционную смесь фильтровали через целит (с последующей промывкой метанолом/водой) и фильтрат концентрировали. Концентрат разбавляли водой и медленно загружали на 5 г смолы Dowex 50X4-200, предварительно промытой хлористоводородной кислотой. Смолу промывали водой и затем элюировали смесью 1:7 концентрированный водный аммиак:вода. Продуктовые фракции концентрировали, получая (2S,3R,4R,5S)-1-пентил-2-(гидроксиметил)-3,4,5-пиперидинтриол (240 мг, 90%) в виде резиноподобного твердого вещества.
1Н ЯМР (D2О): δ 0,75(3H, т), 1,15(4H, м), 1,35(2H, м), 2,35(1H, дд, J=10, 12,5 Гц), 2,5(2H, м), 2,7(1H, дд, J=5, 12 Гц), 3,0(1H, дд, J=4, 9 Гц), 3,25(1H, т), 3,45(1H, м), 3,6(1H, дд, J=5, 10 Гц), 3,75(2H, м).
Пример 4. (2S,3R,4R,5S)-1-гептил-2-(гидроксиметил)-3,4,5-пиперидинтриол
a) (2S,3R,4R,5S)-1-гептил-2-[(фенилметокси)метил]-3,4,5-трис(фенилметокси)пиперидин
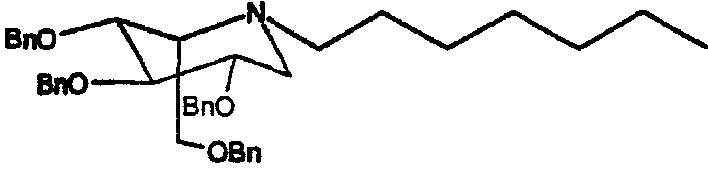
Сырой 2,3,4,6-тетра-О-бензил-1,5-ди-О-мезил-D-глюцитол (пример 1а, 1 г) растворяли в н-гептиламине (10 мл) и перемешивали при 55°С в течение 4 суток. ТСХ анализ показал, что реакция дошла до завершения. Реакционную смесь концентрировали и полученное сырое масло очищали флэш-хроматографией (градиентное элюирование 0→25% диэтиловый эфир/петролейный эфир), получая (2S,3R,4R,5S)-1-гептил-2-[(фенилметокси)метил]-3,4,5-трис(фенилметокси)пиперидин (690 мг, 76%).
1Н ЯМР (CDCl3): δ 0,9(3H, т), 1,3(8H, м), 1,4(2H, м), 2,5(2H, м), 2,7(1H, м), 2,9(1H, дд, J=5, 11 Гц), 3,4(1H, м), 3,55(2H, м), 3,7(2H, м), 3,8(1H, дд, J=6, 13 Гц), 4,4-4,9(8H, м, OCH2Ph), 7,2-7,4(20H, м, ArH).
b) (2S,3R,4R,5S)-1-гептил-2-(гидроксиметил)-3,4,5-пиперидинтриол
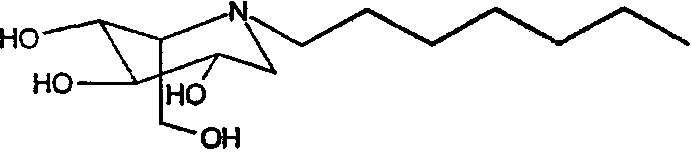
(2S,3R,4R,5S)-1-гептил-2-[(фенилметокси)метил]-3,4,5-трис(фенилметокси)пиперидин (690 мг) растворяли в МеОН (10 мл) и перемешивали в течение ночи в атмосфере водорода в присутствии PdCl2 (350 мг). ТСХ показала завершение реакции. Реакционную смесь фильтровали через целит (с последующей промывкой метанолом/водой) и фильтрат концентрировали. Концентрат разбавляли водой (5 мл) и медленно загружали на 5 г смолы Dowex 50X4-200, предварительно промытой хлористоводородной кислотой. Смолу промывали водой и затем элюировали смесью 1:7 концентрированный водный аммиак:вода. Продуктовые фракции концентрировали, получая (2S,3R,4R,5S)-1-гептил-2-(гидроксиметил)-3,4,5-пиперидинтриол (260 мг, 90%) в виде резиноподобного твердого вещества.
1Н ЯМР (D2О): δ 0,7(3H, т), 1,1(8H, м), 1,3(2H, м), 2,45(3H, м), 2,7(1H, дд, J=5, 10 Гц), 2,95(1H, дд, J=4, 9 Гц), 3,25(1H, т), 3,4(1H, м), 3,55(1H, дд, J=5,5, 9,5 Гц), 3,65(2H, м).
Пример 5. (2S,3S,4R,5S)-1-бутил-2-(гидроксиметил)-3,4,5-пиперидинтриол
а) 2,3,4,6-Тетра-О-бензил-D-галактитол
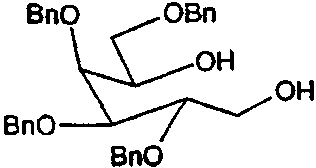
2,3,4,6-Тетра-О-бензил-D-галактопиранозу (107 г) растворяли в этаноле (0,6 л) и при перемешивании при 0°С добавляли боргидрид натрия (31 г). После перемешивания в течение ночи ТСХ анализ показал завершение реакции. Этанольный раствор распределяли между водой (3 л) и простым эфиром (1,5 л). Органическую фазу сушили (Na2SO4), фильтровали и концентрировали. Полученное масло очищали флэш-хроматографией (градиентное элюирование с использованием 20→50% этилацетат/петролейный эфир) и затем кристаллизовали из смеси этилацетат/петролейный эфир, получая 2,3,4,6-тетра-О-бензил-D-галактитол (97 г, 91%) в виде белого твердого вещества.
1Н ЯМР (CDCl3): δ 2,4(1H, уш.с), 3,35(1H, уш.с), 3,55(2H, м), 3,8(3H, м), 3,9(2H, м), 4,1(1H, м), 4,4-4,8(8H, м, ОСН2Pb), 7,2-7,4(20H, м, ArH).
Масс-спектр: m/z 543 (M+H)+ 565 (M+Na)+.
b) 2,3,4,6-Тетра-О-бензил-1,5-ди-О-мезил-D-галактитол
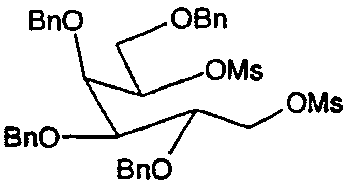
2,3,4,6-Тетра-О-бензил-D-галактитол (7,6 г) перемешивали при 0°С в пиридине (20 мл) и добавляли раствор мезилхлорида (2,5 мл) в пиридине (20 мл). Раствор хранили в течение ночи при 4°С. Анализ ТСХ показывал завершение реакции. Реакционную смесь распределяли между этилацетатом и водой со льдом. Органические фракции промывали 5% хлористоводородной кислотой и затем насыщенным водным раствором бикарбоната натрия, сушили (Na2SO4) и концентрировали, получая бесцветное масло, которое напрямую использовали на следующей стадии.
c) (2S,3S,4R,5S)-1-бутил-2-[(фенилметокси)метил]-3,4,5-трис(фенилметокси)пиперидин
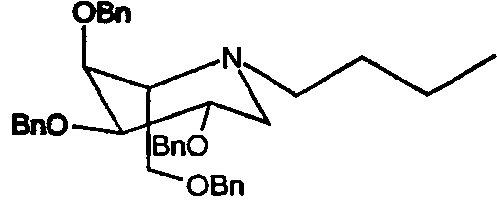
Сырой 2,3,4,6-тетра-О-бензил-1,5-ди-О-мезил-D-галактитол растворяли в н-бутиламине (50 мл) и перемешивали при 55°С в течение 5 суток. Реакционную смесь концентрировали и сырое масло очищали флэш-хроматографией (градиентное элюирование 5→16% этилацетат/петролейный эфир), получая (2S,3S,4R,5S)-1-бутил-2-[(фенилметокси)метил]-3,4,5-трис(фенилметокси)пиперидин (4,8 г, 59% от 2,3,4,6-тетра-О-бензил-1,5-ди-О-мезил-D-галактитола) в виде бесцветного масла.
1Н ЯМР (CDCl3): δ 0,9(т, 3H, J=6 Гц), 1,25(м, 2H), 1,4(м, 2H), 2,6(м, 3H), 2,8(м, 1H), 3,0(м, 1H), 3,4(м, 1H), 3,55(2H, м), 3,75(1H, м), 3,8(1H, м), 4,3-4,6(8H, м, OCH2Ph), 7,15-7,3(20H, м, ArH).
d) (2S,3S,4R,5S)-1-бутил-2-(гидроксиметил)-3,4,5-пиперидинтриол
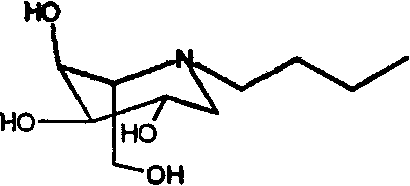
(2S,3S,4R,5S)-1-бутил-2-[(фенилметокси)метил]-3,4,5-трис(фенилметокси)пиперидин (4,8 г) растворяли в метаноле (100 мл) и перемешивали в течение ночи в атмосфере водорода в присутствии PdCl2 (2,5 г). ТСХ показала завершение реакции. Реакционную смесь фильтровали через целит (с последующей промывкой метанолом/водой) и концентрировали до 25 мл водного раствора. Данный раствор медленно загружали на 40 мл смолы Amberlite IR-120(plus), предварительно промытой хлористоводородной кислотой. Смолу промывали водой и затем элюировали смесью 1:7 концентрированный водный аммиак:вода (1:7) (500 мл). Продуктовые фракции концентрировали, получая (2S,3S,4R,5S)-1-бутил-2-(гидроксиметил)-3,4,5-пиперидинтриол (1,27 г, 70%) в виде бесцветного масла.
1Н ЯМР (D2О): δ 0,95(3H, т), 1,35(м, 2H), 2,61(1H, дд), 2,70(1H, м), 2,87(1H, дд), 2,95(1H, ддд), 3,76(1H, ддд), 3,78(1H, дд), 3,90(1H, дд), 3,94(1H, ддд), 4,06(1H, дд).
Пример 6. (2S,3R,4R,5S)-1-нонил-2-(гидроксиметил)-3,4,5-пиперидинтриол
a) (2S,3R,4R,5S)-1-нонил-2-[(фенилметокси)метил]-3,4,5-трис(фенилметокси)пиперидин
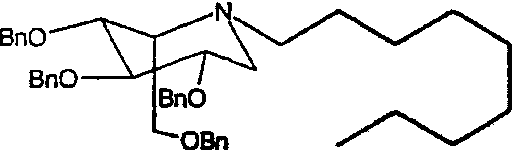
Сырой 2,3,4,6-тетра-О-бензил-1,5-ди-О-мезил-D-глюцитол (пример 1а, 1 г) растворяли в нониламине (1,2 мл) и перемешивали при 55°С в течение 5 суток. Реакционную смесь концентрировали и полученное сырое масло очищали колоночной хроматографией (градиентное элюирование 0→12% этиловый эфир/петролейный эфир), получая (2S,3R,4R,5S)-1-нонил-2-[(фенилметокси)метил]-3,4,5-трис(фенилметокси)пиперидин (660 мг, 71%).
1Н ЯМР (300 МГц, CDCl3): δ 0,88(3H, т, J=7 Гц), 1,14-1,40(12H, м), 1,40-1,55(2H, м), 2,43-2,54(2H, м), 2,60-2,71(1H, м), 2,84(1H, дд, J=12, 5 Гц); 3,30-3,36(1H, м), 3,42-3,57(2H, м), 3,64(1H, дд, 9, 5 Гц); 3,67(1H, дд, J=11, 3 Гц), 3,78(1H, дд, J=9, 6 Гц), 4,47(2H, AB кв), 4,56-4,72(4H, м), 4,78(2H, AB кв), 7,18-7,42(20H, м).
b) (2S,3R,4R,5S)-1-нонил-2-(гидроксиметил)-3,4,5-пиперидинтриол
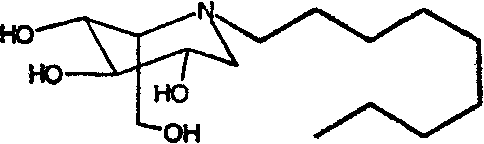
(2S,3R,4R,5S)-1-нонил-2-[(фенилметокси)метил]-3,4,5-трис(фенилметокси)пиперидин (660 мг) растворяли в МеОН (10 мл) и перемешивали в течение ночи в атмосфере водорода в присутствии PdCl2 (300 мг). Реакционную смесь фильтровали через целит (с последующей промывкой метанолом/водой) и фильтрат концентрировали. Концентрат очищали абсорбцией на смоле Dowex 50X4-200 (8 г) и элюированием смесью 1:7 водного аммиака и воды, получая (2S,3R,4R,5S)-1-нонил-2-(гидроксиметил)-3,4,5-пиперидинтриол (160 мг, 61%) в виде резиноподобного твердого вещества.
1Н ЯМР (300 МГц, CD3OD): δ 0,91(3H, м), 1,3(12H, уш.с), 1,45-1,58(2H, м), 2,53-2,69(2H, м), 2,70-2,84(2H, м), 3,00-3,07(1H, м), 3,35-3,42(1H, м), 3,49-3,58(1H, м), 3,70(1H, дд, J=9, 5 Гц), 3,78-3,89(2H, м).
МС m/z 290 (M+H)+.
Пример 7. (2S,3R,4R,5S)-1-(1-этил)пропил-2-(гидроксиметил)-3,4,5-пиперидинтриол
a) (2S,3R,4R,5S)-1-(1-этил)пропил-2-[(фенилметокси)метил]-3,4,5-трис(фенилметокси)пиперидин
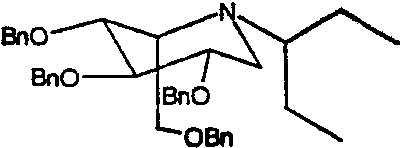
1,5-Ди-О-метансульфонил-2,3,4,6-тетра-О-бензил-D-глюцитол (4,0 г) растворяли в 1-этилпропиламине (6 мл) и перемешивали при 55°С в течение 4 суток. Реакционную смесь концентрировали и полученное коричневое масло очищали колоночной хроматографией на силикагеле (градиентное элюирование 0→15% диэтиловый эфир/петролейный эфир), получая (2S,3R,4R,5S)-1-(1-этил)пропил-2-[(фенилметокси)метил]-3,4,5-трис(фенилметокси)пиперидин (1,46 г, 33%) в виде бледно-желтого масла.
1Н ЯМР (CDCl3): δ 0,79-0,86(6H, м), 1,17-1,30(4H, м), 2,30-2,41(1H, м), 2,62-2,71(1H, м), 2,83(1H, дд, J=12, 5 Гц), 3,30-3,34(1H, м), 3,43-3,53(2H, м), 3,66-3,73(2H, м), 3,83(1H, дд, J=9, 6 Гц), 4,50(2H, с), 4,63-4,79(4H, м), 4,83(2H, AB кв), 7,23-7,42(20H, м).
b) (2S,3R,4R,5S)-1-(1-этил)пропил-2-(гидроксиметил)-3,4,5-пиперидинтриол
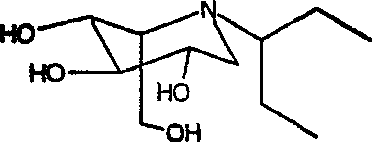
К раствору (2S,3R,4R,5S)-1-(1-этил)пропил-2-[(фенилметокси)метил]-3,4,5-трис(фенилметокси)пиперидина (1,46 г) в метаноле (15 мл) добавляли PdCl2 (750 мг). Реакционную смесь перемешивали в атмосфере водорода в течение ночи. Анализ ТСХ показал завершение реакции, и реакционную смесь фильтровали через слой целита и концентрировали. Сырой продукт очищали абсорбцией на 8,5 г смолы Dowex 50X4-200 и элюированием смесью 1:7 28% водный аммиак:вода, получая после лиофилизации (2S,3R,4R,5S)-1-(1-этил)пропил-2-(гидроксиметил)-3,4,5-пиперидинтриол (530 мг, 92%) в виде белого твердого вещества.
1Н ЯМР (D2О): δ 0,82(6H, т, J=7 Гц), 1,29-1,58(4H, м), 2,41-2,52(2H, м), 2,87(1H, дд, J=13, 5 Гц), 3,16(1H, дд, J=10, 4 Гц), 3,36-3,44(1H, м), 3,47-3,56(1H, м), 3,69-3,77(3H, м).
МС m/z 234 (M+H)+.
Пример 8. (2S,3R,4R,5S)-1-(3-метил)бутил-2-(гидроксиметил)-3,4,5-пиперидинтриол
a) (2S,3R,4R,5S)-1-(3-метил)бутил-2-[(фенилметокси)метил]-3,4,5-трис(фенилметокси)пиперидин
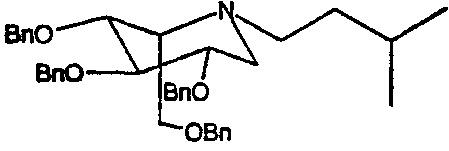
1,5-Ди-О-метансульфонил-2,3,4,6-тетра-О-бензил-D-глюцитол (4,0 г) растворяли в изоамиламине (4 мл) и перемешивали при 55°С в течение 4 суток. Реакционную смесь концентрировали и полученное коричневое масло очищали колоночной хроматографией на силикагеле (градиентное элюирование 0→20% диэтиловый эфир/петролейный эфир), получая (2S,3R,4R,5S)-1-(3-метил)бутил-2-[(фенилметокси)метил]-3,4,5-трис(фенилметокси)пиперидин (2,53 г, 67%) в виде бесцветного масла.
1Н ЯМР (CDCl3): δ 0,80(6H, д, J=6 Гц), 1,12-1,33(3H, м), 2,39-2,50(2H, м), 2,59-2,70(1H, м), 2,79(1H, дд, J=11, 4 Гц), 3,26-3,32(1H, м), 3,38-3,52(2H, м), 3,56-3,67(2H, м), 3,74(1H, дд, J=11, 6 Гц), 4,43(2H, AB кв), 4,52-4,67(4H, м), 4,75(2H, AB кв), 7,18-7,30(20H, м).
b) (2S,3R,4R,5S)-1-(3-метил)бутил-2-(гидроксиметил)-3,4,5-пиперидинтриол
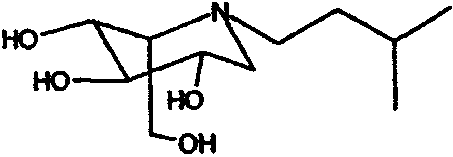
К раствору (2S,3R,4R,5S)-1-(3-метил)бутил-2-[(фенилметокси)метил]-3,4,5-трис(фенилметокси)пиперидина (2,53 г) в метаноле (30 мл) добавляли PdCl2 (1,2 г). Реакционную смесь перемешивали в атмосфере водорода в течение ночи. Анализ ТСХ показал завершение реакции, и реакционную смесь фильтровали через слой целита и концентрировали. Сырой продукт очищали абсорбцией на 12 г смолы Dowex 50X4-200 и элюированием смесью 1:7 28% водный аммиак:вода, получая после лиофилизации (2S,3R,4R,5S)-1-(3-метил)бутил-2-(гидроксиметил)-3,4,5-пиперидинтриол (960 мг, 97%) в виде резиноподобного твердого вещества.
1Н ЯМР (D2О): δ 0,83(6H, дд, J=7, 1 Гц), 1,26-1,42(2H, м), 1,43-1,55(1H, м), 2,46(1H, дд, J=12, 10 Гц), 2,57(1H, ддд, J=12, 10, 6 Гц), 2,68(1H, ддд, J=12, 10, 6 Гц), 2,81(1H, дд, J=12, 5 Гц), 3,08(1H, дд, J=10, 5 Гц), 3,36(1H, т, J=9 Гц), 3,54(1H, ддд, J=10, 9, 5 Гц), 3,68(1H, дд, J=10, 6 Гц), 3,75-3,87(2H, м).
МС m/z 234 (M+H)+.
Пример 9. (2S,3R,4R,5S)-1-(2-фенил)этил-2-(гидроксиметил)-3,4,5-пиперидинтриол
a) (2S,3R,4R,5S)-1-(2-фенил)этил-2-[(фенилметокси)метил]-3,4,5-трис(фенилметокси)пиперидин
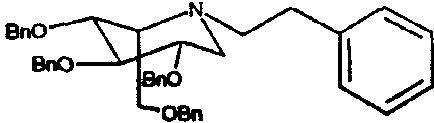
1,5-Ди-О-метансульфонил-2,3,4,6-тетра-О-бензил-D-глюцитол (5,0 г) растворяли в фенетиламине (10 мл) и перемешивали при 55°С в течение 3 суток. Реакционную смесь концентрировали и полученное коричневое масло очищали колоночной хроматографией на силикагеле (градиентное элюирование 0→30% диэтиловый эфир/петролейный эфир), получая (2S,3R,4R,5S)-1-(2-фенил)этил-2-[(фенилметокси)метил]-3,4,5-трис(фенилметокси)пиперидин (3,2 г, 71%) в виде бесцветного масла.
1Н ЯМР (CDCl3): δ 0,86-0,98(2H, м), 1,16-1,28(2H, м), 2,56-2,7(1H, м), 2,70-2,88(1H, м), 2,91(1H, дд, J=11, 4 Гц), 2,98-3,06(1H, м), 3,41-3,46(1H, м), 3,46-3,60(2H, м); 3,66(1H, дд, J=9, 6 Гц); 3,75(1H, дд, J=10, 3 Гц), 3,87(1H, дд, J=10, 6 Гц), 4,52(2H, AB кв), 4,60-4,74(4H, м), 4,83(2H, AB кв), 7,10-7,38(25H, м).
b) (2S,3R,4R,5S)-1-(2-фенил)этил-2-(гидроксиметил)-3,4,5-пиперидинтриол
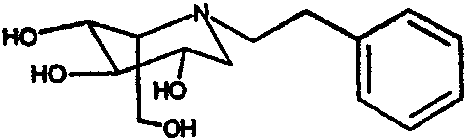
К раствору (2S,3R,4R,5S)-1-(2-фенил)этил)-2-[(фенилметокси)метил]-3,4,5-трис(фенилметокси)пиперидина (4,0 г) в метаноле (30 мл) добавляли PdCl2 (1,4 г). Реакционную смесь перемешивали в атмосфере водорода в течение ночи. Анализ ТСХ показал завершение реакции, и реакционную смесь фильтровали через слой целита и концентрировали. Сырой продукт очищали абсорбцией на 20 г смолы Dowex 50X4-200 и элюированием смесью 1:7 28% водный аммиак:вода. Продуктовые фракции лиофилизировали и затем очищали колоночной хроматографией на силикагеле (градиентное элюирование 0-20% МеОН/дихлорметан), получая (2S,3R,4R,5S)-1-(2-фенил)этил-2-(гидроксиметил)-3,4,5-пиперидинтриол (970 мг, 81%) в виде резиноподобного твердого вещества.
1Н ЯМР (D2О): δ 2,50(1H, дд, J=12, 10Гц), 2,68-2,97(5H, м), 3,3(1H, дд, J=9, 5Гц), 3,36(1H, т, J=9Гц), 3,51-3,61(1H, м), 3,66-3,73(2H, м), 3,74-3,83(1H, м), 7,18-7,37(5H, м).
МС m/z 268 (M+H)+.
Пример 10. (2S,3R,4R,5S)-1-(3-фенил)пропил-2-(гидроксиметил)-3,4,5-пиперидинтриол
a) (2S,3R,4R,5S)-1-(3-фенил)пропил-2-[(фенилметокси)метил]-3,4,5-трис(фенилметокси)пиперидин
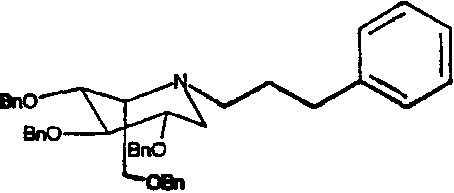
1,5-Ди-О-метансульфонил-2,3,4,6-тетра-О-бензил-D-глюцитол (5,0 г) растворяли в 3-фенилпропиламине (5 мл) и перемешивали при 55°С в течение 3 суток. Реакционную смесь концентрировали и полученное коричневое масло очищали колоночной хроматографией на силикагеле (градиентное элюирование 0→25% диэтиловый эфир/петролейный эфир), получая (2S,3R,4R,5S)-1-(3-фенил)пропил-2-[(фенилметокси)метил]-3,4,5-трис(фенилметокси)пиперидин (4,25 г, 100%) в виде бледно-желтого масла.
1Н ЯМР (CDCl3): δ 1,72-1,84(2H, м), 2,54-2,64(4H, м), 2,70-2,80(1H, м), 2,87(1H, дд, J=11, 5 Гц), 3,34-3,39(1H, м), 3,46-3,62(2H, м), 3,65-3,74(2H, м), 3,84(1H, дд, J=10, 6 Гц), 4,52(2H, AB кв), 4,62-4,75(4H, м), 4,84(2H, AB кв), 7,12-7,39(25H, м).
b) (2S,3R,4R,5S)-1-(3-фенил)пропил-2-(гидроксиметил)-3,4,5-пиперидинтриол
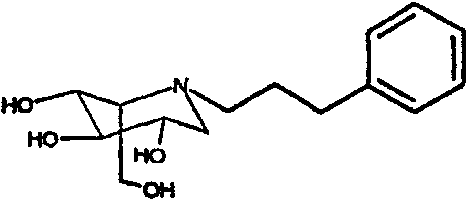
К раствору (2S,3R,4R,5S)-1-(3-фенил)пропил-2-[(фенилметокси)метил]-3,4,5-трис(фенилметокси)пиперидина (4,2 г) в метаноле (40 мл) добавляли PdCl2 (1,6 г). Реакционную смесь перемешивали в атмосфере водорода в течение ночи. Анализ ТСХ показал завершение реакции, и реакционную смесь фильтровали через слой целита и концентрировали. Сырой продукт абсорбировали на 20 г смолы Dowex 50X4-200 и элюированием смесью 1:7 28% водный аммиак:вода. Продуктовые фракции лиофилизировали и затем очищали колоночной хроматографией на силикагеле (градиентное элюирование 0-20% МеОН/дихлорметан), получая (2S,3R,4R,5S)-1-(3-фенил)пропил-2-(гидроксиметил)-3,4,5-пиперидинтриол (1,36 г, 71%) в виде прозрачной смолы.
1Н ЯМР (D2О): δ 1,69-1,82(2H, м), 2,44(1H, дд, J=12, 10 Гц), 2,51-2,72(4H, м), 2,78(1H, дд, J=13, 5 Гц), 3,05(1H, дд, J=11, 5 Гц), 3,34(1H, т, J=9 Гц), 3,52(1H, ддд, J=10, 9, 5Гц), 3,66(1Н, дд, J=10, 5 Гц), 3,71-3,81(2H, м), 7,17-7,35(5Н, м).
МС m/z 282 (M+H)+.
Пример 11. (2S,3R,4R,5S)-1-(1-этил)гексил-2-(гидроксиметил)-3,4,5-пиперидинтриол
a) (2S,3R,4R,5S)-1-(2-этил)гексил-2-[(фенилметокси)метил]-3,4,5-трис(фенилметокси)пиперидин
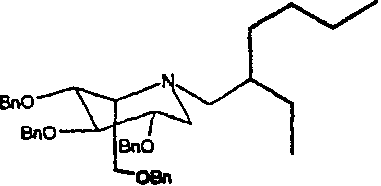
1,5-Ди-О-метансульфонил-2,3,4,6-тетра-О-бензил-D-глюцитол (5,0 г) растворяли в 2-этилгексиламине (5 мл) и перемешивали при 55°С в течение 4 суток. Реакционную смесь концентрировали и полученное коричневое масло очищали колоночной хроматографией на силикагеле (градиентное элюирование 0-17,5% диэтиловый эфир/петролейный эфир), получая (2S,3R,4R,5S)-1-(2-этил)гексил-2-[(фенилметокси)метил]-3,4,5-трис(фенилметокси)пиперидин (2,6 г, 57%) в виде бесцветного масла.
1Н ЯМР (CDCl3): δ 0,75-0,93(6H, м), 1,17-1,38(9H, м), 2,16(1H, дд, J=13, 6 Гц), 2,25-2,36(2H, м), 2,52-2,60(1H, м), 3,02-3,09(1H, м), 3,24-3,36(2H, м), 3,40-3,51(2H, м), 3,60(1H, дд, J=10, 6 Гц), 4,53(2H, AB кв), 4,62-4,76(4H, м), 4,85(2H, AB кв), 7,18-7,31(20H, м).
b) (2S,3R,4R,5S)-1-(2-этил)гексил-2-(гидроксиметил)-3,4,5-пиперидинтриол
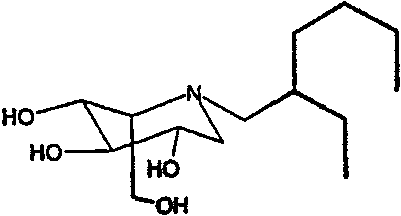
К раствору (2S,3R,4R,5S)-1-(2-этил)гексил-2-[(фенилметокси)метил]-3,4,5-трис(фенилметокси)пиперидина (2,6 г) в метаноле (20 мл) добавляли PdCl2 (900 мг). Реакционную смесь перемешивали в атмосфере водорода в течение ночи. Анализ ТСХ показал завершение реакции, и реакционную смесь фильтровали через слой целита и концентрировали. Сырой продукт очищали абсорбцией на 13 г смолы Dowex 50X4-200 и элюированием смесью 1:7 28% водный аммиак:вода. Затем продуктовые фракции очищали колоночной хроматографией на силикагеле (градиентное элюирование 0-10% МеОН/дихлорметан), получая после лиофилизации (2S,3R,4R,5S)-1-(1-этил)гексил-2-(гидроксиметил)-3,4,5-пиперидинтриол (320 мг, 28%) в виде резиноподобного твердого вещества.
1Н ЯМР (CDCl3): δ 0,68-0,80(6H, м), 1,08-1,32(9H, м), 2,30-2,46(3H, м), 2,60(1H, дд, J=13, 5 Гц), 2,90(1H, дд, J=12, 6 Гц), 3,30-3,38(1H, м), 3,40-3,49(1H, м), 3,55(1H, дд, J=13, 9Гц), 3,66(1H, дд, J=9, 5Гц), 3,74(1H, дд, J=11, 7Гц).
МС m/z 276 (M+H)+.
Пример 12. (2S,3R,4R,5S)-1-(2-этил)бутил-2-(гидроксиметил)-3,4,5-пиперидинтриол
a) (2S,3R,4R,5S)-1-(2-этил)бутил-2-[(фенилметокси)метил]-3,4,5-трис(фенилметокси)пиперидин
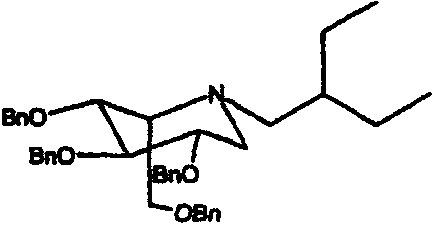
1,5-Ди-О-метансульфонил-2,3,4,6-тетра-О-бензил-D-глюцитол (3,0 г) растворяли в 2-этилбутиламине (2,5 мл) и перемешивали при 55°С в течение 4 суток. Реакционную смесь концентрировали и полученное коричневое масло очищали колоночной хроматографией на силикагеле (градиентное элюирование от 0 до 12%, диэтиловый эфир/петролейный эфир), получая (2S,3R,4R,5S)-1-(2-этил)бутил-2-[(фенилметокси)метил]-3,4,5-трис(фенилметокси)пиперидин (1,93 г, 74%) в виде бесцветного масла (Rf: 0,25, 20% этилацетат/петролейный эфир), которое непосредственно использовали на следующей стадии.
b) (2S,3R,4R,5S)-1-(2-этил)бутил-2-(гидроксиметил)-3,4,5-пиперидинтриол
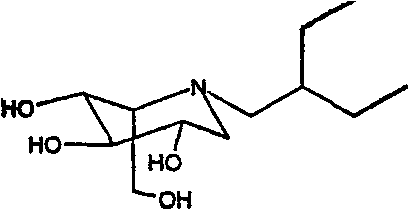
К раствору (2S,3R,4R,5S)-1-(2-этил)бутил-2-[(фенилметокси)метил]-3,4,5-трис(фенилметокси)пиперидина (1,93 г) в метаноле (20 мл) добавляли PdCl2 (800 мг). Реакционную смесь перемешивали в атмосфере водорода в течение ночи. Анализ ТСХ показал завершение реакции, и реакционную смесь фильтровали через слой целита и концентрировали. Сырой продукт очищали абсорбцией на 10 г смолы Dowex 50X4-200 и элюированием смесью 1:7 28% водный аммиак:вода, получая после лиофилизации (2S,3R,4R,5S)-1-(2-этил)бутил-2-(гидроксиметил)-3,4,5-пиперидинтриол (735 мг, 94%) в виде белого твердого вещества.
1Н ЯМР (CDCl3): δ 0,78(6H, т, J=7 Гц), 1,14-1,30(5H, м), 2,32-2,48(3H, м), 2,63(1H, дд, J= 13, 5 Гц), 2,92(1H, дд, J=13, 6 Гц), 3,37(1H, т, J=9 Гц), 3,47(1H, ддд, J=10, 9, 4 Гц), 3,57(1H, дд, J=11, 7 Гц), 3,68(1H, дд, J=9, 6 Гц), 3,77(1H, дд, J=11, 4 Гц).
МС m/z 248 (M+H)+.
Пример 13. (2S,3R,4R,5S)-1-[(2R)-(2-метил-2-фенил)этил]-2-(гидроксиметил)-3,4,5-пиперидинтриол
a) (2S,3R,4R,5S)-1-[(2R)-(2-метил-2-фенил)этил]-2-[(фенилметокси)метил]-3,4,5-трис(фенилметокси)пиперидин
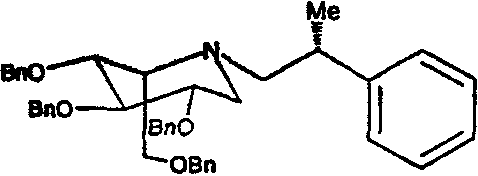
1,5-Ди-О-метансульфонил-2,3,4,6-тетра-О-бензил-D-глюцитол (2,5 г) растворяли в ДМФ (3 мл). Добавляли диизопропилэтиламин (1,5 мл) и R(+)-β-метилфенетиламин (1 г), и реакционную смесь перемешивали при 55°С в течение 5 суток. Реакционную смесь концентрировали и полученное коричневое масло очищали колоночной хроматографией на силикагеле (градиентное элюирование от 0 до 25% диэтиловый эфир/петролейный эфир), получая (2S,3R,4R,5S)-1-[(2R)-(2-метил-2-фенил)этил]-2-[(фенилметокси)метил]3,4,5-трис(фенилметокси)пиперидин (740 мг, 32%) в виде бледно-желтого масла.
1Н ЯМР (CDCl3): δ 1,21-1,27(3H, м), 2,52-2,59(1H, м), 2,70-2,95(4H, м), 3,35-3,40(1H, м), 3,44-3,52(2H, м), 3,64(1H, дд, J=12, 9 Гц), 3,74(1H, дд, J=11, 3 Гц), 3,86(1H, дд, J=9, 6Гц), 4,47-4,69(6H, м), 4,83(2H, AB кв), 7,17-7,37(25H, м).
b) (2S,3R,4R,5S)-1-[(2R)-(2-метил-2-фенил)этил]-2-(гидроксиметил)-3,4,5-пиперидинтриол
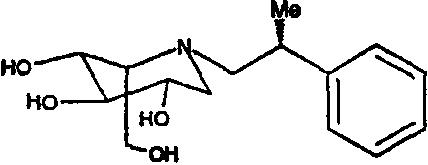
К раствору (2S,3R,4R,5S)-1-[(2R)-(2-метил-2-фенил)этил]-2-[(фенилметокси)метил]-3,4,5-трис(фенилметокси)пиперидина (740 мг) в метаноле (15 мл) добавляли PdCl2 (300 мг). Реакционную смесь перемешивали в атмосфере водорода в течение ночи. Анализ ТСХ показал завершение реакции, и реакционную смесь фильтровали через слой целита и концентрировали. Сырой продукт очищали абсорбцией на 10 г смолы Dowex 50X4-200 и элюированием смесью 1:7 28% водный аммиак:вода, получая после лиофилизации (2S,3R,4R,5S)-1-[(2R)-(2-метил-2-фенил)этил]-2-(гидроксиметил)-3,4,5-пиперидинтриол (300 мг, 92%) в виде смолоподобного твердого вещества.
1Н ЯМР (CDCl3): δ 1,18(3H, д, J=5 Гц), 2,42(1H, дд, J=12, 9 Гц), 2,56-2,87(5H, м), 3,29(1H, т, J=9 Гц), 3,39-3,66(4H, м), 7,07-7,23(5H, м).
МС m/z 282 (M+H)+.
Пример 14. (2S,3R,4R,5S)-1-[(2S)-(2-метил-2-фенил)этил]-2-(гидроксиметил)-3,4,5-пиперидинтриол
a) (2S,3R,4R,5S)-1-[(2S)-(2-метил-2-фенил)этил]-2-[(фенилметокси)метил]-3,4,5-трис(фенилметокси)пиперидин
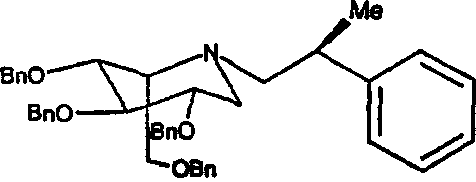
1,5-Ди-О-метансульфонил-2,3,4,6-тетра-О-бензил-D-глюцитол (2,5 г) растворяли в ДМФ (3 мл). Добавляли диизопропилэтиламин (1,5 мл) и S(-)-β-метилфенетиламин (1 г), и реакционную смесь перемешивали при 55°С в течение 5 суток. Реакционную смесь распределяли между водным NaOH (1M, 30 мл) и этилацетатом (50 мл). Органическую фазу промывали насыщенным водным NaHCO3, сушили над Na2SO4 и концентрировали. Полученное сырое масло очищали колоночной хроматографией на силикагеле (градиентное элюирование от 0 до 17% диэтиловый эфир/петролейный эфир), получая (2S,3R,4R,5S)-1-[(2S)-(2-метил-2-фенил)этил]-2-[(фенилметокси)метил]-3,4,5-трис(фенилметокси)пиперидин (700 мг, 31%) в виде бледно-желтого масла.
1Н ЯМР (CDCl3): δ 1,17-1,21(3H, м), 2,55-2,64(1H, м), 2,79(1H, дд, J=12, 7 Гц), 2,87(1H, дд, J=13, 6 Гц), 2,98(1H, дд, J=13, 7 Гц), 3,20-3,26(1H, м), 3,40-3,54(3H, м), 3,69(1H, дд, J=10, 2Гц), 3,84(1H, дд, J=13, 7 Гц), 4,46-4,70(6Н, м), 4,8(2H, AB кв), 7,09-7,38(25H, м).
b) (2S,3R,4R,5S)-1-[(2S)-(2-метил-2-фенил)этил]-2-(гидроксиметил)-3,4,5-пиперидинтриол
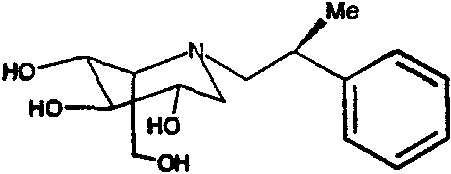
К раствору (2S,3R,4R,5S)-1-[(2S)-(2-метил-2-фенил)этил]-2-[(фенилметокси)метил]-3,4,5-трис(фенилметокси)пиперидина (700 мг) в метаноле (15 мл) добавляли PdCl2 (300 мг). Реакционную смесь перемешивали в атмосфере водорода в течение ночи. Анализ ТСХ показал завершение реакции, и реакционную смесь фильтровали через слой целита и концентрировали. Сырой продукт очищали абсорбцией на 10 г смолы Dowex 50X4-200 и элюированием смесью 1:7 28% водный аммиак:вода, получая после лиофилизации (2S,3R,4R,5S)-1-[(2S)-(2-метил-2-фенил)этил]-2-(гидроксиметил)-3,4,5-пиперидинтриол (250 мг, 81%) в виде смолоподобного твердого вещества.
1Н ЯМР (CDCl3): δ 1,17(3H, д, J=5 Гц), 2,45(1H, дд, J=13, 10 Гц), 2,59(1H, дд, J=13, 4 Гц), 2,64-2,86(3H, м), 2,95(1H, дд, J=14, 6 Гц), 3,34(1H, т, J=8 Гц), 3,42-3,55(2H, м), 3,64 (1H, дд, J=8,5 Гц), 3,74(1H, дд, J=11, 6 Гц), 7,08-7,23(5H, м).
МС m/z 282,3 (M+H)+.
Пример 15. (2S,3R,4R,5S)- 1-[(4-метоксифенил)метил]-2-[(фенилметокси)метил]-3,4,5-трис(фенилметокси)пиперидин (защищенное промежуточное соединение)
1,5-Ди-О-метансульфонил-2,3,4,6-тетра-О-бензил-D-глюцитол (25 г) растворяли в 4-метоксибензиламине (50 мл) и перемешивали при 55°С в течение 4 суток. Реакционную смесь концентрировали и полученное коричневое масло очищали колоночной хроматографией на силикагеле (градиентное элюирование от 0 до 23% диэтиловый эфир/петролейный эфир), получая (2S,3R,4R,5S)-1-[(4-метоксифенил)метил]-2-[(фенилметокси)метил]-3,4,5-трис(фенилметокси)пиперидин (17,1 г, 75%) в виде бледно-желтого масла.
1Н ЯМР (CDCl3): δ 2,50-2,60(1H, м), 2,83(1H, дд, J=13,4 Гц), 3,39-3,44(1H, м), 3,51-3,61(2H, м), 3,64-3,80(3H, м), 3,84(3H, с), 3,70-3,77(2H, м), 4,54(2H, с), 4,58-4,69(4H, м), 4,85(2H, AB кв), 6,87(2H, д, J=7 Гц), 7,18(2H, д, J=7 Гц), 7,26-7,40(20H, м).
Биологические данные
Соединения по изобретению испытывали (таблица 1) для определения их концентрации IC50 в отношении галактозидазы и глюкозилцерамидсинтазы. В первом случае испытания проводили согласно методам, описанным в Jacob and Scudder, Methods in Enzymology (1994), 230, 280. В случае глюкозилцерамидсинтазы испытания проводили согласно методу, описанному в Platt et al., J.Biol.Chem. (1994), 269,27108.
(IC50, мкМ)
(IC50, мкМ)
(Ki, мкМ)
(IC50, мкМ)
(IC50, мкМ)
Таблица 2 показывает данные по ферментам человека. Испытания на ингибирование ГЦС были проведены, в основном, так, как описано в Platt et al., J. Biol. Chem. (1994), 269, 27108, причем источником фермента являлась рекомбинантная ГЦС человека, экспрессированная в клетках насекомого. Тесты на глюкозидазу проводили, как описано (Biochemical Genetics, A Laboratory Manual, Oxford University Press), за исключением того, что вместо субстратов, соединенных с метилумбеллифероном использовали субстраты, соединенные с п-нитрофенилом.
(IC50, мкМ)
(IC50, мкМ)
(IC50, мкМ)
(Ki, мкМ)
Таким образом, соединения по изобретению проявляют меньшую ингибирующую активность против и глюкозидаз и галактозидаз (благодаря чему уменьшают побочные эффекты), чем такие соединения как NB-DNJ и NB-DGJ, оставаясь в то же время активными против глюкозилцерамидсинтаз.
| название | год | авторы | номер документа |
|---|---|---|---|
| ПРОИЗВОДНЫЕ МАННОЗЫ ДЛЯ ЛЕЧЕНИЯ БАКТЕРИАЛЬНЫХ ИНФЕКЦИЙ | 2013 |
|
RU2667060C2 |
| БЕТА-ЛАКТАМЫ, СПОСОБ ПОЛУЧЕНИЯ УКАЗАННЫХ СОЕДИНЕНИЙ И СЫВОРОТОЧНЫЕ ГИПОХОЛЕСТЕРИНЕМИЧЕСКИЕ СРЕДСТВА, СОДЕРЖАЩИЕ ТАКИЕ СОЕДИНЕНИЯ | 2002 |
|
RU2301799C2 |
| АЗУЛЕНОВОЕ ПРОИЗВОДНОЕ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ЕГО ОСНОВЕ И СПОСОБ ЛЕЧЕНИЯ ДИАБЕТА | 2003 |
|
RU2295522C2 |
| НОВЫЕ КОМПОЗИЦИИ ДЛЯ ПРЕДУПРЕЖДЕНИЯ И/ИЛИ ЛЕЧЕНИЯ ЛИЗОСОМНЫХ БОЛЕЗНЕЙ НАКОПЛЕНИЯ | 2010 |
|
RU2608520C2 |
| СПОСОБ ПОЛУЧЕНИЯ ПРОИЗВОДНОГО ДИФЕНИЛМЕТАНА | 2017 |
|
RU2797392C2 |
| КОМПОЗИЦИЯ, СОДЕРЖАЩАЯ ИНГИБИТОРЫ КОТРАНСПОРТЕРОВ НАТРИЯ-ГЛЮКОЗЫ 1 И 2, И СПОСОБЫ ИХ ПРИМЕНЕНИЯ | 2012 |
|
RU2669921C2 |
| ПРОИЗВОДНЫЕ БЕНЗИЛФЕНИЛЦИКЛОГЕКСАНА И СПОСОБЫ ИХ ПРИМЕНЕНИЯ | 2008 |
|
RU2505521C2 |
| ЗАМЕЩЕННЫЕ БЕНЗИМИДАЗОЛЬНЫЕ СОЕДИНЕНИЯ, СПОСОБЫ ПОЛУЧЕНИЯ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ИХ ОСНОВЕ, ОБЛАДАЮЩАЯ АНТИВИРУСНЫМ ДЕЙСТВИЕМ | 1993 |
|
RU2141952C1 |
| СПОСОБ ПОЛУЧЕНИЯ ПРОИЗВОДНОГО ДИФЕНИЛМЕТАНА | 2023 |
|
RU2814846C1 |
| Способ получения (2R,3R,4R,5S)-1-бутил-2-(гидроксиметил)пиперидин-3,4,5-триола | 2020 |
|
RU2762605C1 |
Изобретение относится к новым производным пиперидина общей формулы (I)
где R представляет C1-16 алкил или C1-10 алкилфенил, в котором фенил необязательно замещен OR1, где R1 представляет C1-6 алкил, или их фармацевтически приемлемым солям, которые обладают ингибиторной активностью в отношении глюкозилцерамидсинтазы, и могут быть использованы в медицине для лечения заболеваний, обусловленных накоплением гликолипида. 9 н. и 6 з.п. ф-лы, 2 ил., 2 табл.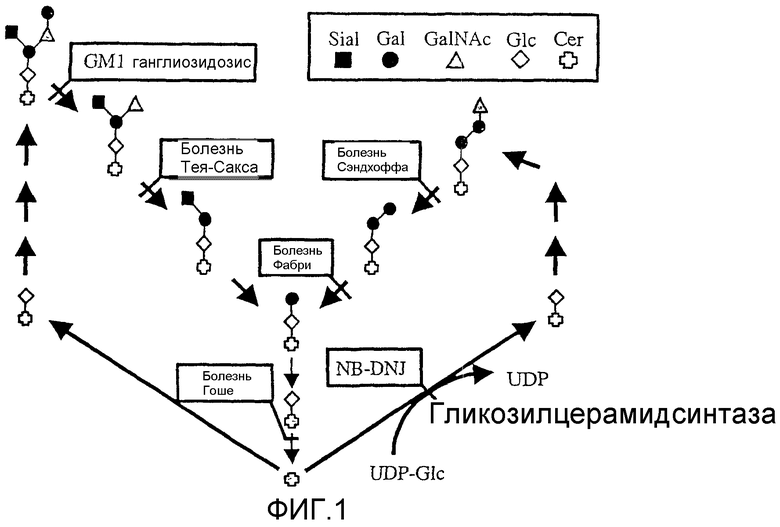
где
R представляет С1-16 алкил с прямой или разветвленной цепью, или C1-10 алкилфенил, в котором фенил необязательно замещен OR1, где R1 представляет C1-6 алкил с прямой или разветвленной цепью;
при условии, что соединение не представляет собой:
a) (2S,3R,4R,5S)-1-бутил-2-(гидроксиметил)-3,4,5-пиперидинтриол;
b) (2S,3R,4R,5S)-1-фенилметил-2-(гидроксиметил)-3,4,5-пиперидинтриол;
c) (2S,3S,4R,5S)-1-нонил-2-(гидроксиметил)-3,4,5-пиперидинтриол;
d) (2S,3R,4R,5S)-1-додецил-2-(гидроксиметил)-3,4,5-пиперидинтриол;
e) (2S,3R,4R,5S)-1-(1-фенил)этил-2-(гидроксиметил)-3,4,5-пиперидинтриол.
(2S,3R,4R,5S)-1-пропил-2-(гидроксиметил)-3,4,5-пиперидинтриол;
(2S,3R,4R,5S)-1-пентил-2-(гидроксиметил)-3,4,5-пиперидинтриол;
(2S,3R,4R,5S)-1-гептил-2-(гидроксиметил)-3,4,5-пиперидинтриол;
(2S,3S,4R,5S)-1-бутил-2-(гидроксиметил)-3,4,5-пиперидинтриол;
(2S,3R,4R,5S)-1-нонил-2-(гидроксиметил)-3,4,5-пиперидинтриол;
(2S,3R,4R,5S)-1-(1-этил)пропил-2-(гидроксиметил)-3,4,5-пиперидинтриол;
(2S,3R,4R,5S)-1-(3-метил)бутил-2-(гидроксиметил)-3,4,5-пиперидинтриол;
(2S,3R,4R,5S)-1-(2-фенил)этил-2-(гидроксиметил)-3,4,5-пиперидинтриол;
(2S,3R,4R,5S)-1-(3-фенил)пропил-2-(гидроксиметил)-3,4,5-пиперидинтриол;
(2S,3R,4R,5S)-1-(1-этил)гексил-2-(гидроксиметил)-3,4,5-пиперидинтриол;
(2S,3R,4R,5S)-1-(2-этил)бутил-2-(гидроксиметил)-3,4,5-пиперидинтриол;
(2S,3R,4R,5S)-1-[(2R)-(2-метил-2-фенил)этил]-2-(гидроксиметил)-3,4,5-пиперидинтриол;
(2S,3R,4R,5S)-1-[(2S)-(2-метил-2-фенил)этил]-2-(гидроксиметил)-3,4,5-пиперидинтриол;
или их фармацевтически приемлемые соли.
в которой R является таким, как определено в п.1, и P является бензильной группой.
в которой R является таким, как определенно в п.1, и P является бензильной группой, при условии, что соединение не является:
i) (2S,3R,4R,5S)-1-фенилметил-2-[(фенилметокси)метил)]-3,4,5-трис(фенилметокси)пиперидином.
| Способ получения производных 3,4,5-триоксипиридина или их солей | 1978 |
|
SU917697A3 |
| Экономайзер | 0 |
|
SU94A1 |
| Дорожная спиртовая кухня | 1918 |
|
SU98A1 |

