Изобретение относится к области химии и химико-фармацевтической промышленности, а именно к способам синтеза субстанции миглустата ((2R,3R,4R,5S)-1-бутил-2-(гидроксиметил)пиперидин-3,4,5 -триола).
Наследственные болезни обмена веществ-наиболее сложная и обширная группа заболеваний, среди которых в последние годы особую актуальность приобретают лизосомные болезни накопления, такие как сфинголипидозы. Лизосомные болезни накопления относятся к редким (орфанным) заболеваниям, как правило, имеющим тяжелое течение. Все они обусловлены генетическими изменениями лизосомных ферментов, контролирующих процесс внутриклеточного расщепления макромолекул. Патогенетическими следствиями этих изменений являются внутритканевое и внутрилизосомное накопление нерасщепленных макромолекул и увеличение количества лизосом в клетках разных тканей организма. Такое накопление приводит к нарушению нормального функционирования клеток и их гибели. В случае сфинголипидозов (таких как болезни Гоше и болезни Ниманна-Пика), их развитие связано с недостаточностью фермента глюкоцереброзидазы, которая приводит к накоплению глюкоцереброзида во многих тканях, включая селезенку, печень, почки, легкие, мозг и костный мозг. Накопление этих молекул приводит к ряду клинических проявлений, включая спленомегалию, гепатомегалию, нарушения скелета, тромбоцитопению и анемию.
В настоящее время в лечении ряда сфинголипидозов достигнуты хорошие результаты, которые стали возможны благодаря разработке и внедрению в практику энзимредуцирующей терапии с использованием препарата Миглустат.
Методы базируются на способности миглустата подавлять активность энзимов первого этапа синтеза промежуточных токсичных продуктов, что приводит к положительным сдвигам, главным образом со стороны ЦНС.
Миглустат относится к классу азасахаров или иминосахаров, и представляет собой N-бутильное производное 1-дезоксинойиримицина:
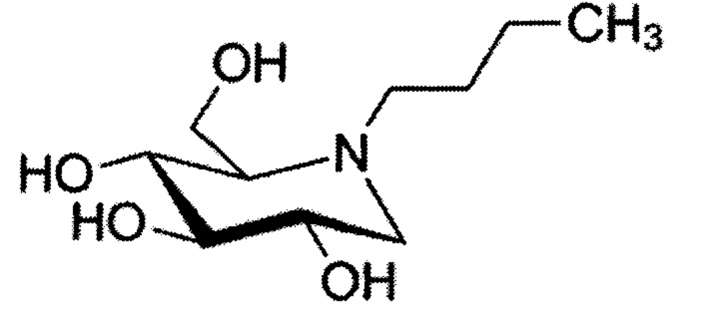
Миглустат включен в список жизненно необходимых и важнейших лекарственных препаратов (ЖНВЛП). Лечение проводится, как правило, пожизненно. Поэтому к субстанции предъявляются особые требования, касающиеся чистоты и стабильности.
Известно получение миглустата путем введения бутильной цепи в 1-дезоксинойиримицин или его производные с защищенными функциональными группами путем восстановительного аминирования с бутиральдегидом (см., например, US 4639436 и ЕР 367748).
Известен способ получения миглустата, согласно которому получают иминосахар путем восстановительного аминирования и циклизации промежуточного соединения 5-оксо-2,3,4,6-тетрабензилоксигексаналя, которое является результатом оксидирования 2,3,4,6-тетра-О-бензилглюцитола с помощью ДМСО/P2O5, снимают защиту с иминосахара путем гидрирования на Pd/C с получением целевого соединения (IT 1425538 В1).
Также в уровне техники раскрывается способ получения миглустата из 2,3,4,6-тетра-О-бензил-D-глюцитола через оксидирование и восстановительную циклоконденсацию с н-бутиламином в присутствии боран-морфолина (US 9708263 В2).
Перечисленные известные способы не позволяют получить продукт с высоким выходом.
Известен способ получения миглустата путем дебензилирования 2,3,4,6-тетра-О-бензил-D-глюцита с последующей кристаллизацией в С1-С5 алканоле при добавлении С3-С7 кетона с охлаждением смеси и извлечением твердого вещества (RU 2678085 С1). Способ обеспечивает получение определенной кристаллической формы соединения.
В качестве ближайшего аналога (прототипа) может быть указан способ по US 2014/0243369 А1. Указанный способ получения миглустата включает дебензилирование каталитическим гидрированием соединения формулы (III)- а именно N-бутил-2,3,4,6-тетра-О-бензил-1,5-дидезокси-1,5-имино-D-глюцитола. Раствор, полученный смешиванием соединения формулы (III) в метаноле в присутствии 32% HCl обрабатывают 16% Pd/С.Смесь выдерживают при интенсивном перемешивании в атмосфере водорода при 4 барах в течение примерно 4 часов, затем фильтруют через перлитовую панель и полученный раствор концентрируют при низком давлении. Полученный таким образом твердый остаток растворяют в воде, и полученный раствор кислоты пропускают через колонку Amberlite IRA 900С1. Фракции, которые дали положительный результат анализа нингидрина, объединяли и концентрировали при низком давлении, получая миглустат в виде маслянистого остатка с химической степенью чистоты более 98%.
Химическая чистота является важным потенциальным показателем активной фармацевтической субстанции ввиду потенциального влияния примесей на безопасность лекарственного препарата. Однако не менее важным является сохранение чистоты в процессе хранения и возможность воспроизводимости субстанции с заданной степенью чистоты в промышленном производстве.
Задачей настоящего изобретения является получение стабильно чистой субстанции (2R,3R,4R,5S)-1-бутил-2-(гидроксиметил)пиперидин-3,4,5-триола с высоким выходом целевого продукта.
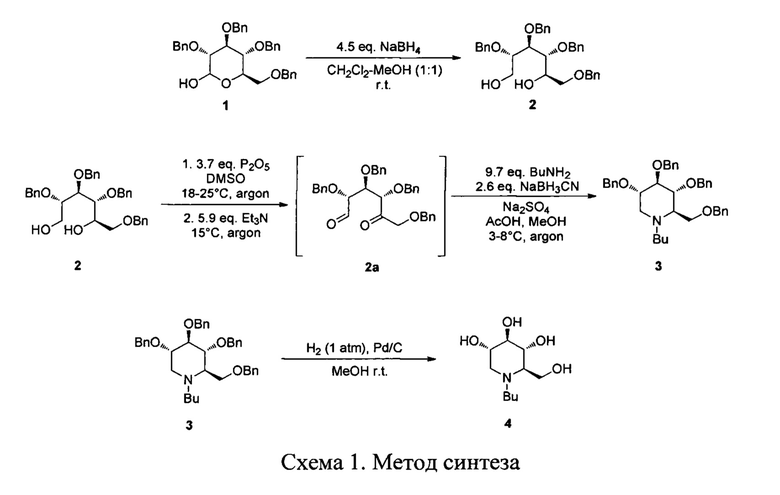
Задача решается новым усовершенствованным способом получения субстанции (2R,3R,4R,5S)-1-бутил-2-(гидроксиметил)пиперидин-3,4,5-триола, включающим следующие стадии: взаимодействие 2,3,4,6-тетра-О-бензил-Д-глюкопиранозы с борогидридом натрия в органическом растворителе с получением 2,3,4,6-Тетра-О-бензил-Д-глюцитола, который экстрагируют этилацетатом и после сушки и упаривания органического слоя удаляют следы воды с помощью азеотропной отгонки с толуолом; последовательное окисление 2,3,4,6-Тетра-О-бензил-Д-глюцитола смесью ДМСО и оксида фосфора Р2О5 с образованием 1,5-дикарбонильного производного, очистку указанного производного диэтиловым эфиром и его восстановительное аминирование с использованием цианоборгидрида натрия, очистку перекристаллизацией из метанола полученного 2,3,4,6-Тетра-O-бензил-N-бутил-1,5-дидеокси-1,5-Д-глюцитола, его взаимодействие с водородом в присутствии палладия в качестве катализатора, нанесенного на активированный уголь, при давлении водорода равном одной атмосфере с образованием целевого продукта-(2R,3R,4R,5S)-1-бутил-2-(гидроксиметил)пиперидин-3,4,5-триола с последующей очисткой силикагелем.
На схеме 1 представлен общий подход к синтезу субстанции (2R,3R,4R,5S)-1-бутил-2-(гидроксиметил)пиперидин-3,4,5-триола 4 согласно заявляемому способу:
Более подробно возможность осуществления изобретения может быть продемонстрирована ниже представленными примерами.
Стадия 1
2,3,4,6-Тетра-О-бензил-Д-глюцитола (2)
Синтез проводился с использованием литературной методики. Ключевое отличие от предложенных ранее методов-экстракция продукта этилацетатом вместо хлористого метилена. Данный растворитель имеет плотность меньше, чем плотность воды, что облегчает проведение последующей промывки экстракта водными растворами. Ниже приводим оптимизированный протокол проведения реакции.
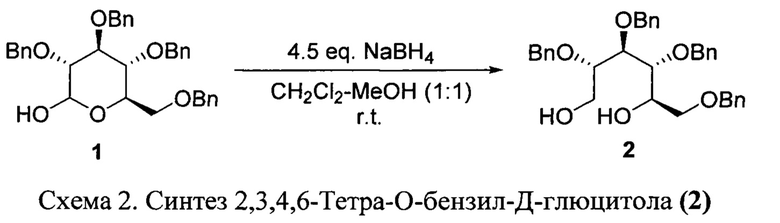
В одногорлую двухлитровую колбу, снабженную магнитной мешалкой и обратным холодильником, помещают 50 г (92.5 ммоль) исходной 2,3,4,6-тетра-О-бензил-Д-глюкопиранозы 1, добавляют 230 мл дихлорметана и 230 мл метанола. Ввиду сильного вспенивания реакционной смеси, последующее добавление 12.25 г (323.8 ммоль, 3.5 экв.) NaBH4 проводят аккуратно, порциями (3-4 порции с временным интервалом 30 мин). После полного добавления борогидрида натрия реакционную смесь перемешивают при комнатной температуре 4 часа. Затем вносят новую порцию NaBH4 в количестве 3.5 г (92.5 ммоль, 1 экв.) и перемешивают смесь еще 15 часов.
Контроль за протеканием реакции проводят с помощью ТСХ (элюент -этилацетат/толуол, объем, соотн. 1:2; Rf исх. 1-0.58, Rf продукта 2-0.35). В случае неполного протекания реакции добавляют еще одну порцию NaBH4 в количестве 3.5 г (92.5 ммоль, 1 экв.) и перемешивают смесь еще 3-4 часа.
По завершении реакции реакционную смесь разбавляют этилацетатом (800 мл) и переносят в делительную воронку. Полученную смесь последовательно промывают 3 М раствором NaOH (2 раза по 200 мл) и насыщенным раствором KCl (3 раза по 300 мл). Органический слой сушат 30 мин над 80 г безводного Na2SO4 и упаривают на роторном испарителе при пониженном давлении при температуре бани 55°С.
Так как наличие влаги в продукте 2 отрицательно влияет на ход последующей реакции окисления Олбрайта-Онодеры окончательное удаление следов воды проводят с помощью азеотропной отгонки с толуолом. Для этого глюцитол 2 растворяют в 100 мл толуола и упаривают на роторном испарителе при пониженном давлении при температуре бани 55°С. Данную процедуру необходимо повторить 4 раза. После охлаждения продукт 2 представляет собой бесцветный прозрачный сироп массой 50 г (вых. 99%).
Данный синтез проводился с различными начальными загрузками глюкопиранозы 1: 50 г, 120 г, 130 г, 140 г, 250 г. При масштабировании, кратно загрузке увеличивали массу борогидрида натрия, а также объемы используемых растворителей. При сильном вспенивании реакционной смеси можно увеличить время добавления и количество порций борогидрида натрия. Во всех случаях выход глюцитола 2 оставался количественным.
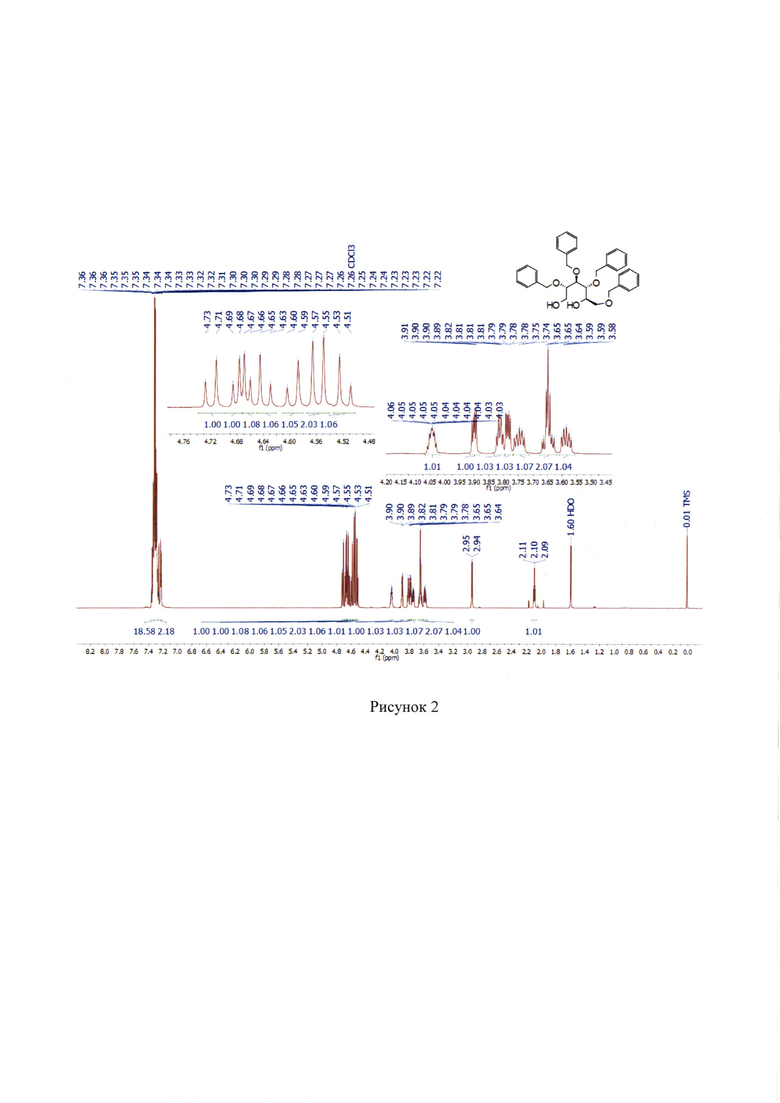
1Н NMR (700 МГц, СДСl3) δ 2.10 (т, J=6.3 Гц, 1H), 2.94 (д, J=5.2 Гц, 1H), 3.58 (ддд, J=11.5, 6.2, 4.7 Гц, 1H), 3.62-3.68 (м, 2Н), 3.75 (ддд, J=11.2, 6.3, 4.4 Гц, 1Н), 3.78 (дд, J=7.0, 3.7 Гц, 1Н), 3.80-3.83 (м, 1H), 3.90 (дд, J=6.5, 3.7 Гц, 1Н), 4.02-4.07 (м, 1H), 4.52 (д, J=11.9 Гц, 1Н), 4.54-4.57 (м, 2Н), 4.60 (д, J=11.5 Гц, 1H), 4.64 (д, J=11.6 Гц, 1H), 4.66 (д, J=6.3 Гц, 1Н), 4.68 (д, J=6.6 Гц, 1Н), 4.72 (д, J=11.3 Гц, 1Н), 7.22 -7.25 (м, 2Н), 7.26-7.37 (м, 18Н). (Рисунок 2).
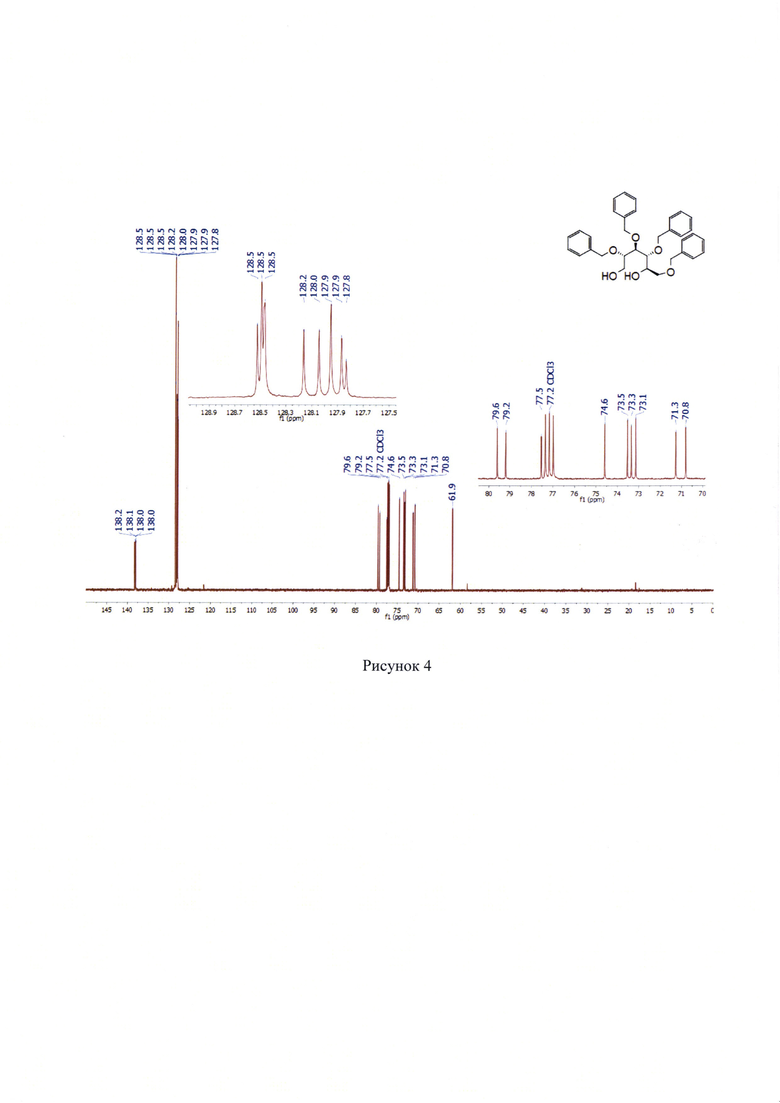
13С NMR (176 МГц, СДСl3) δ 61.9, 70.8, 71.3, 73.1, 73.3, 73.5, 74.6, 77.5, 79.2, 79.6, 127.8, 127.9, 127.9, 128.0, 128.2, 128.4-128.6, 138.0, 138.0, 138.1, 138.2. (Рисунок 3).
Спектры совпадают с описанными ранее в Qing R. L., Seung I. K., Sook J. P., Hye R. Y., A R. В., In S. K., Young H. J. Total synthesis of (+)-valienamine and (-)-l-epi-valienamine via a highly diastereoselective allylic amination of cyclic polybenzyl ether using chlorosulfonyl isocyanate // Tetrahedron. 2013. Vol. 69(48). P. 10384-10390.
Масс-спектр высокого разрешения. Найдено, m/z: 543.2825 [М+Н]+. С34Н39О6. Вычислено, m/z: 543.2741. (Рисунок 4).
Стадия 2-1.
Синтез 2,3,4,6-Тетра-O-бензил-N-бутил-1,5-дидеокси-1,5-Д-глюцитола (3).
Метод предполагает последовательное окисление глюцитола 2 с образованием 1,5-дикарбонильного производного 2а и его восстановительное аминирование (Схема 3).
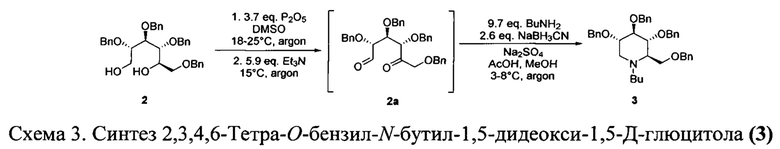
Новый подход к данной стадии не требует использования криогенного оборудования. Предложено использование реакции Олбрайта-Онодеры, то есть окисления диола смесью ДМСО и оксида фосфора, которое можно проводить при более высокой температуре (от 0 градусов до комнатной температуры).
Образующееся в результате реакции дикарбонильное соединение 2а нестабильно, в связи с чем его выделение в индивидуальном виде невозможно, однако оно может быть использовано на следующей стадии в виде раствора. Данная процедура была описана ранее в патенте-US 2014/0243369 А1, однако нами была предложена модификация описанного метода: полученный после окисления раствор дикарбонильного соединения разбавляют диэтиловым эфиром и промывают водным раствором карбоната калия и хлоридом калия. Такая процедура позволяет удалить остатки диметилсульфоксида, фосфорной кислоты и ее солей с триэтиламином, что позволяет избежать побочных процессов на стадии восстановительного аминирования.
Проведение стадии восстановительного аминирования проводилось по протоколу близкому к описанному в патенте US 2014/0243369 А1 с использованием цианоборгидрида натрия. Возможность использования боран-морфолинового комплекса не подтвердилась.
В трехгорлую литровую колбу, снабженную магнитной мешалкой, ртутным термометром и краном, соединенным с аргоновой линией, помещают раствор исходного глюцитола 2 50.7 г (93.4 ммоль) в диметилсульфоксиде (270 мл). Колбу помещают в водяную баню с холодной водой (10-15°С) и продувают аргоном. При достижении температуры реакционной смеси +18°С ток аргона останавливают и начинают добавление оксида фосфора. 49.4 г (348 ммоль) оксида фосфора предварительно взвешивают примерно равными порциями в 4 пробирках с завинчивающейся крышкой. Данные порции оксид фосфора добавляют к охлажденному раствору 2 так, чтобы температура реакционной смеси не поднималась выше +25°С, продувая аргоном колбу каждый раз после добавления. После полного добавления Р2О5 смесь перемешивают с охлаждением удерживая температуру в интервале 18÷25°С в течении 1 часа. Реакционную смесь охлаждают до +18°С и добавляют по каплям 76 мл (547 ммоль) триэтиламина с такой скоростью, чтобы температура реакционной смеси не поднималась выше +25°С. После полного добавления триэтиламина смесь нагревают до+25°С и перемешивают 1 час. Далее содержимое колбы переносят в делительную воронку, добавляют 500 мл диэтилового эфира и 400 мл холодной воды со льдом и перемешивают. Водный слой дополнительно экстрагируют 1 раз 100 мл диэтилового эфира. Органические растворы объединяют и промывают последовательно насыщенным раствором K2CO3 (1 раз по 300 мл) и насыщенным раствором KCl (3 раза по 200 мл). Полученный раствор сушат 50 мин над 80 г безводного Na2SO4. Далее этот раствор будет использован в следующей стадии без дополнительной очистки. ТСХ этого раствора (элюент-ЕтОАс/PhMe, объем, соотн. 1:2) указывает на отсутствие исходных реагентов.
В двухлитровой одногорлой колбе, снабженной магнитной мешалкой, готовят раствор 90 мл бутиламина (910 ммоль) в 300 мл метанола. Колбу помещают в баню с водно-ледовой смесью и охлаждают до температуры 3÷9°С. Затем к смеси добавляют 57 мл уксусной кислоты, 50 г сульфата натрия и охлаждают до температуры 3÷9°С. К полученной смеси добавляют полученный ранее раствор 2а из первой стадии через капельную воронку в течение 25 мин, постоянно охлаждая колбу. После полного добавления раствора 2а с помощью индикаторной полоски необходимо убедиться, что рН реакционной смеси находится в интервале 5.0-5.5. При необходимости к смеси добавляют еще некоторое количество уксусной кислоты, так чтобы рН оказался в указанном диапазоне. Далее смесь перемешивают еще 30 мин при этой же температуре, а затем добавляют 15.1 г цианоборогидрида натрия и перемешивают еще 4 часа при той же температуре (3÷9°С). Затем реакционную смесь нагревают до комнатной температуры (вынимают из бани) и перемешивают еще 15 часов.
Реакционную смесь разбавляют 500 мл этилацетата и переносят в делительную воронку. Органический слой последовательно промывают насыщенным раствором NaHCO3 (2 раза по 300 мл), водой (1 раз по 200 мл) и насыщенным раствором хлорида калия (3 раза по 200 мл). Органический слой высушивают 30 мин над 80 г безводного Na2SO4 и упаривают на роторном испарителе при пониженном давлении при температуре бани не выше+45°С.
Остаток после упаривания представляет собой желтое вязкое масло массой 50.6 г, которое при стоянии и охлаждении до комнатной температуры кристаллизуется. Спектр ЯМР 1Н указывает на содержание целевого N-бутилиминоглюцитола 3 не менее 60 мол. % в смеси.
Синтез N-бутилиминоглюцитола 3 проводили несколько раз, варьируя начальные загрузки глюцитола 2: 50 г, 67 г, 90-93 г. Количество других реагентов и объемы растворителей увеличивали кратно загрузке. Масса остатков после упаривания экстрактов составляет 46-59 г для пятидесятиграммовой загрузки, 66 г для шестидесятисемиграммовой загрузки, 92-98 г для девяностограммовых загрузок. Содержание продукта 3 в этих остатках составляет 61-75 мол. % (по данным ПМР).
См. Рисунок 3.
Стадия 2-2.
2,3,4,6-Тетра-O-бензил-N-бутил-1,5-дидеокси-1,5-Д-глюцитол (3). Очистка
Очистка полученного соединения 3 также проводилась по процедуре близкой у описанной в патенте (US 2014/0243369 А1) однако изопропиловый спирт был заменен на метанол. Метанол имеет меньшую вязкость, что облегчает работу со смесью и позволяет ее охладить. Ниже приводим оптимизированный протокол проведения перекристаллизации, который позволяет получить N-бутилиминоглюцитол 3 с чистотой не менее 95% (по спектрам ЯМР примеси не видны).
Неочищенный продукт прошлой стадии общей массой 156.6 г разбавляли 150 мл метанола и тщательно перемешивали. Смесь осторожно нагревали до полного растворения кристаллов (40-50 градусов). Затем смесь охлаждали до комнатной температуры, добавляли затравку, закрывали пробкой, охлаждали до+4°С и выдерживали 15 часов.
Содержимое колбы растирали шпателем, переносили на фильтр и отжимали досуха с использованием колбы Бюхнера и вакуума, обеспечиваемого водоструйным насосом. Осадок на фильтре тщательно (измельчая слипшиеся куски вещества шпателем до однородной массы) суспендировали в 150 мл холодного (охлажден до -20°С) метанола и снова отжимали досуха. Процедуру повторяли еще один раз. Полученный продукт 3 сушили на воздухе до постоянной массы (1 день). Продукт представляет собой белый рассыпчатый порошок. Масса 72.2 г, выход продукта (в пересчете на диол 2) составляет 45%. В аналогичном опыте с увеличенной начальной загрузкой грязного продукта 3 (348.4 г) и двумя последовательными промывками осадка холодным метанолом (200 мл каждая) на фильтре, было получено 140.6 г чистого продукта 3 (общий выход-39%).
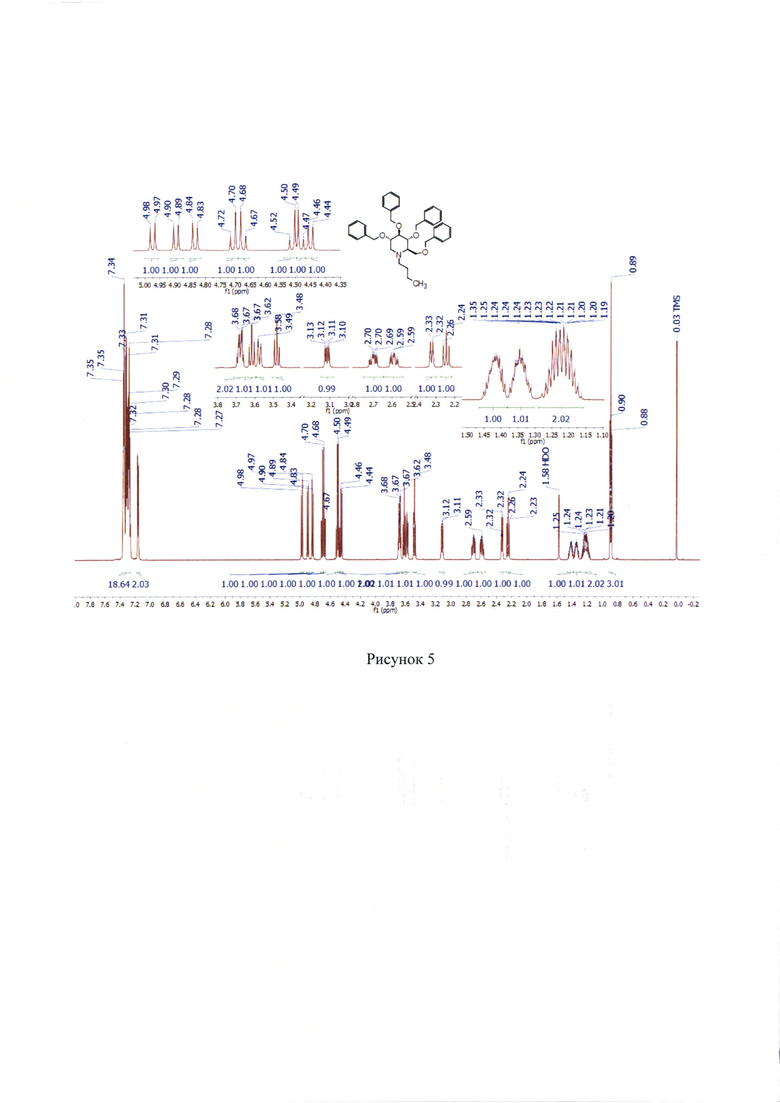
lH NMR (700 МГц, СДСl3) δ 0.89 (т, J=7.3 Гц, 3Н), 1.15-1.29 (м, 2Н), 1.30-1.38 (м, 1H), 1.38-1.46 (м, 1H), 2.24 (т, J=10.8 Гц, 1Н), 2.32 (дт, J=9.6, 2.4 Гц, 1Н), 2.59 (ддд, J=13.5, 10.9, 4.7 Гц, 1Н), 2.70 (ддд, J=13.4, 11.0, 5.3 Гц, 1Н), 3.11 (дд, J=11.2, 4.9 Гц, 1Н), 3.48 (т, J=9.1 Гц, 1H), 3.57 (дд, J=10.5, 2.4 Гц, 1H), 3.62 (т, J=9.3 Гц, 1Н), 3.65-3.71 (м, 2Н), 4.45 (д, J=10.8 Гц, 1Н), 4.48 (д, 12.2 Гц, 1H), 4.51 (д, J=12.3 Гц, 1H), 4.67 (д, J=11.6 Гц, 1H), 4.71 (д, J=11.6 Гц, 1Н), 4.83 (д, J=11.1 Гц, 1Н), 4.90 (д, J=10.8 Гц, 1H), 4.97 (д, J=11.1 Гц, 1H), 7.14-7.18 (м, 2Н), 7.26-7.37 (м, 18Н). (Рисунок 5).
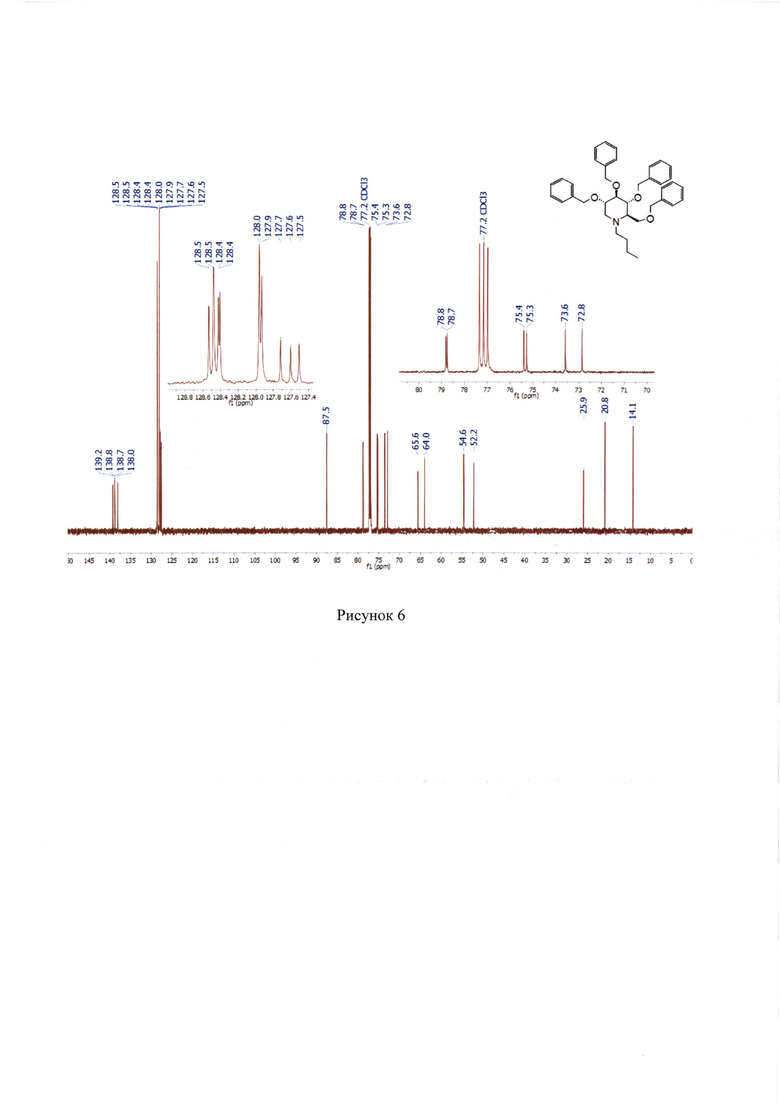
13С NMR (176 МГц, СДСl3) δ 14.1, 20.8, 25.9, 52.2, 54.6, 64.0, 65.6, 72.8, 73.6, 75.3, 75.4, 78.7, 78.8, 87.5, 127.5, 127.6, 127.7, 127.9-128.0, 128.3-128.6, 138.0, 138.7, 138.8, 139.2. (Рисунок 6).
Спектры ЯМР совпадают с описанными ранее в литературе (Zhen-Xing Z., Baolin W., Bin W., Tie-Hai L., Peng-Fei Z., Li-Na G., Wen-jun W., Wei Z., Peng G. W. Facile апд STereo-соптгоИед synThesis of 2^eoxynojirimycin, MiglusTaT and MigliTol // ТетгаЬедгоп Leirers. 2011. Vol. 52 (29). P. 3802-3804).
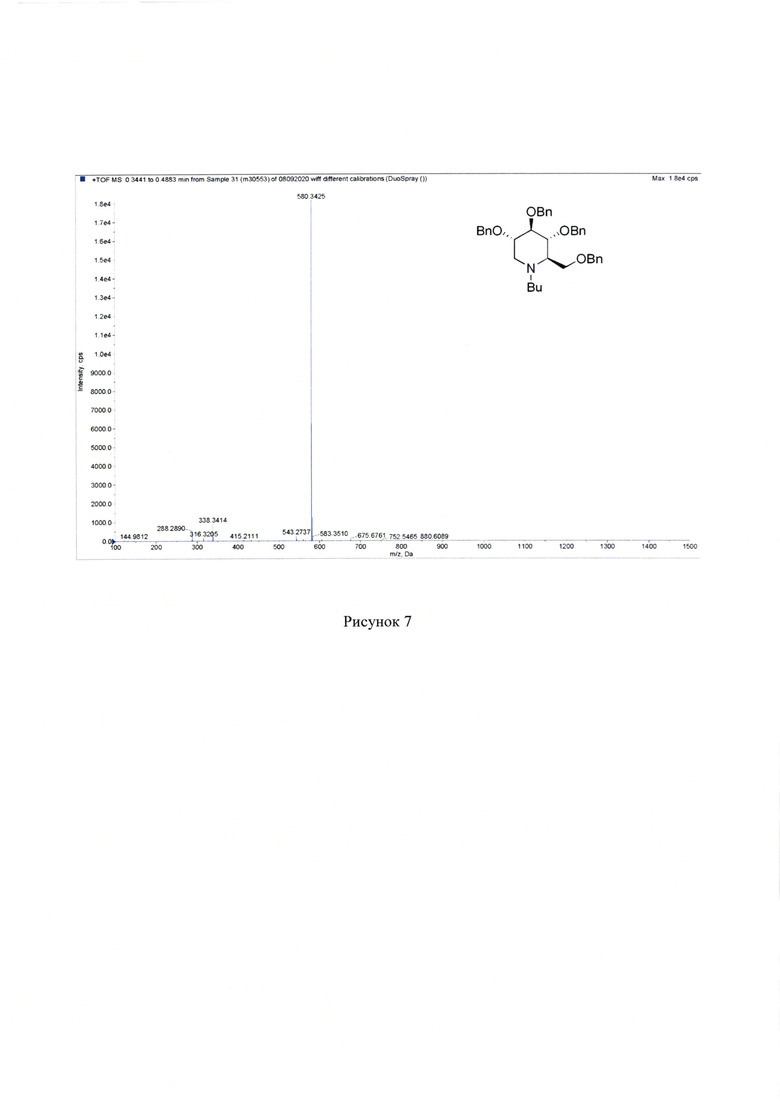
Масс-спектр высокого разрешения. Найдено, m/z: 580.3425 [М+Н]+. C38H46NO4. Вычислено, m/z: 580.3421. (Рисунок 7).
Стадия 3-1.
(2R,3R,4R,5S)-1-бутил-2-(гидроксиметил)пиперидин-3,4,5-триол (4). Синтез
Синтез данного соединения проводился по классической методике с использованием водорода и каталитических количеств палладия, нанесенного на активированный уголь. Аналогичная методика описана в патенте (US 2014/0243369 А1), однако нам удалось показать, что данный синтез может быть приведен при более низком давлении водорода-всего при одной атмосфере.
В двухлитровую одногорлую колбу помещают 7.5 грамм палладия на активированном угле (10% палладия по массе), добавляют дистиллированную воду (50 мл) и водную соляную кислоту (30 мл, около 0.3 моль). Смесь перемешивают до полного смачивания палладия на угле и прибавляют к ней 250 мл метанола, затем присыпают исходное безилированное производное 3 и приливают еще 250 мл метанола. В горло колбы устанавливают кран и подвергают смесь вакуумированию действием водоструйного насоса до того момента, как визуально будет наблюдаться закипание метанола. Затем кран перекрывают, подключают к нему сосуд с водородом (резиновая камера) и открывают. Смесь перемешивают в атмосфере водорода при комнатной температуре в течении 4-10 дней. Контроль за протеканием реакции ведут по ТСХ (элюент-хлористый метилен/метанол/водный аммиак, объем, соотн. 85:15:1) или отбором проб на ЯМР (0,5 мл смеси упаривают на роторном испарителе, растворяют в дейтерированном ДМСО и регистрируют спектр). Последний метод предпочтительнее, так как позволяет оценить наличие следовых количеств полупродуктов.
По окончании реакции к смеси прибавляют раствор карбоната калия (55 грамм, 0.4 моль) в дистиллированной воде (150 мл) и перемешивают 30 минут. Затем смесь фильтруют, а осадок на фильтре промывают метанолом (2 раза по 100 мл). Полученный раствор упаривают на роторном испарителе при пониженном давлении при температуре бани не выше +40°С. В ходе этой процедуры важно обеспечить такое давление, чтобы при данной температуре происходило удаление из смеси не только метанола, но и воды.
Полученную смесь растворяют в 200 мл метанола, перемешивают 20 минут при комнатной температуре для полного формирования осадка неорганических солей, а затем фильтруют. Осадок на фильтре промывают метанолом (2 раза по 75 мл). Полученный раствор упаривают на роторном испарителе при пониженном давлении при температуре бани не выше +40°С.
Данную процедуру повторяют еще два раза, таким образом, из смеси удается удалить большую часть влаги и неорганических солей. К сожалению, полного удаления достичь не удается, и полученная смесь требует проведения еще одной стадии очистки. После упаривания на роторном испарителе смесь дополнительно сушат на вакууме мембранного или масляного насоса (вакуум ниже 1 мм рт.с.т), после чего она приобретает вид «твердой пены» с массой около 25 грамм (более 100% от теоретического выхода).
Стадия 3-2.
(2R,3R,4R,5S)-1-бутил-2-(гидроксиметил)пиперидин-3,4,5-триол (4). Очистка
Для очистки данного соединения нами был предложен альтернативный подход с использованием силикагеля.
На фильтр Шотта диаметром 90 мм помещают 175 грамм силикагеля, смачивают его смесью хлористого метилена, метанола и триэтиламина (соотношение 90 к 10 к 1) и промывают дополнительно 400 мл данной смеси.
Неочищенный продукт 4 растворяют в смеси хлористого метилена и метанола (9 к 1, 100 мл) и переносят на смоченный силикагель с помощью еще 50 мл данной смеси. С помощью воронки Бюхнера и вакуума водоструйного насоса силикагель на фильтре промывают смесью хлористого метилена, метанола и триэтиламина (соотношение 80 к 20 к 1) детектируя наличие продукта в выходящем растворе с помощью ТСХ (элюент - хлористый метилен/метанол/водный аммиак, объем, соотн. 85:15:1). Как правило, продукт не наблюдается в первых 400 мл раствора, а затем полностью смывается в последующих 1500-2000 мл.
Полученный раствор упаривают на роторном испарителе при пониженном давлении при температуре бани не выше +40°С. На последних моментах упаривания возможна кристаллизация продукта. Вне зависимости от того, кристаллизуется продукт или нет, к полученной смеси прибавляют 100 мл ацетона и интенсивно перемешивают. Если кристаллизации не наблюдает, то к смеси можно добавить затравку продукта. После начала кристаллизации смесь выдерживают при комнатной температуре 30 минут и фильтруют. Осадок на фильтре промывают ацетоном (3 раза по 50 мл) и сушат под вакуумом до постоянной массы.
Получают 18-20 грамм белого кристаллического продукта (выход около 90-92%).
Чистота по ЯМР выше 98%. Примесей не детектируется вовсе.
Анализ чистоты субстанции проводился различными методами.
1. Анализ методом ЯМР
Спектры ЯМР:
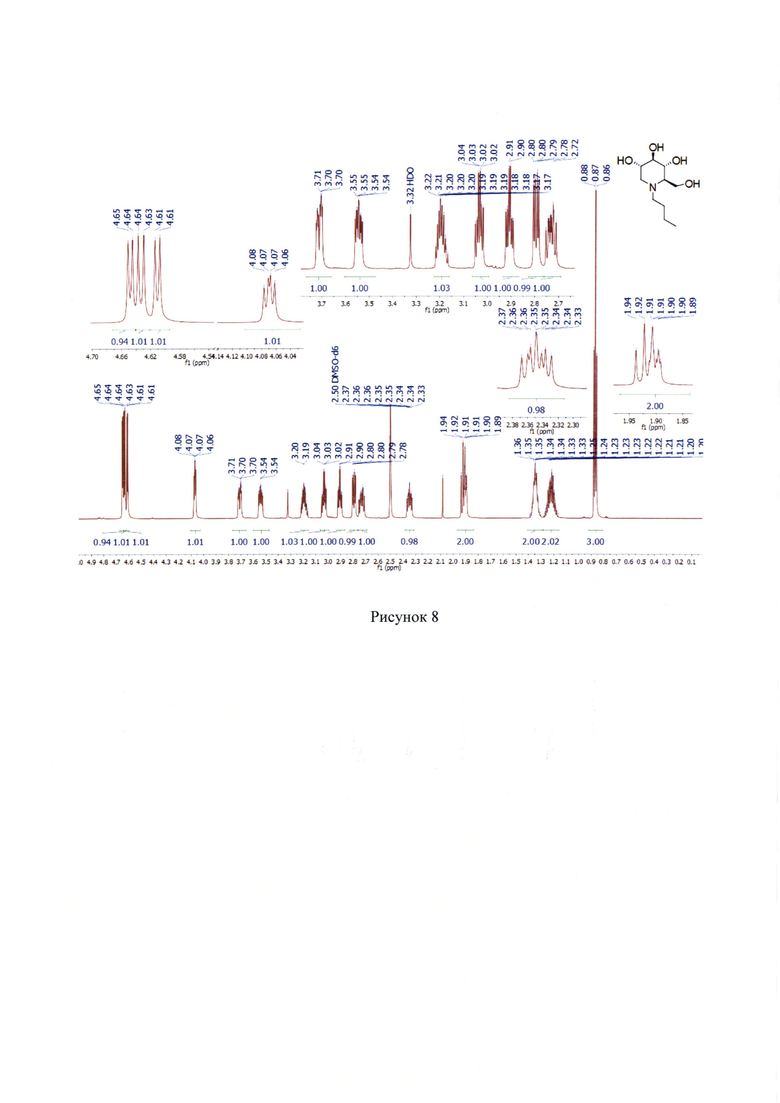
1Н NMR (700 МГц, ДМСО-d6): δ 4.64 (д, J=4.2 Гц, 1 Н), 4.63 (д, J=5.3 Гц, 1H), 4.61 (д, J=4.8 Гц, 1Н), 4.07 (дд, J=6.3, 2.4 Гц, 1H), 3.72 (ддд, J=11.6, 4.0, 2.5 Гц, 1Н), 3.55 (ддд, J=11.6, 7.6, 3.6 Гц, 1H), 3.22-3.17 (м, 1Н), 3.04 (дт, J=9.1, 5.2 Гц, 1Н), 2.90 (дт, J=9.1, 4.2 Гц, 1H), 2.00 (дд, J=10.9, 5.0 Гц, 1Н), 2.75-2.71 (м, 1Н), 2.37-2.33 (м, 1Н), 1.94-1.88 (м, 2Н), 1.39-1.31 (м, 2Н), 1.28-1.18 (м, 2Н), 0.87 (т, J=7.4 Гц, 3Н). (Рисунок 8).
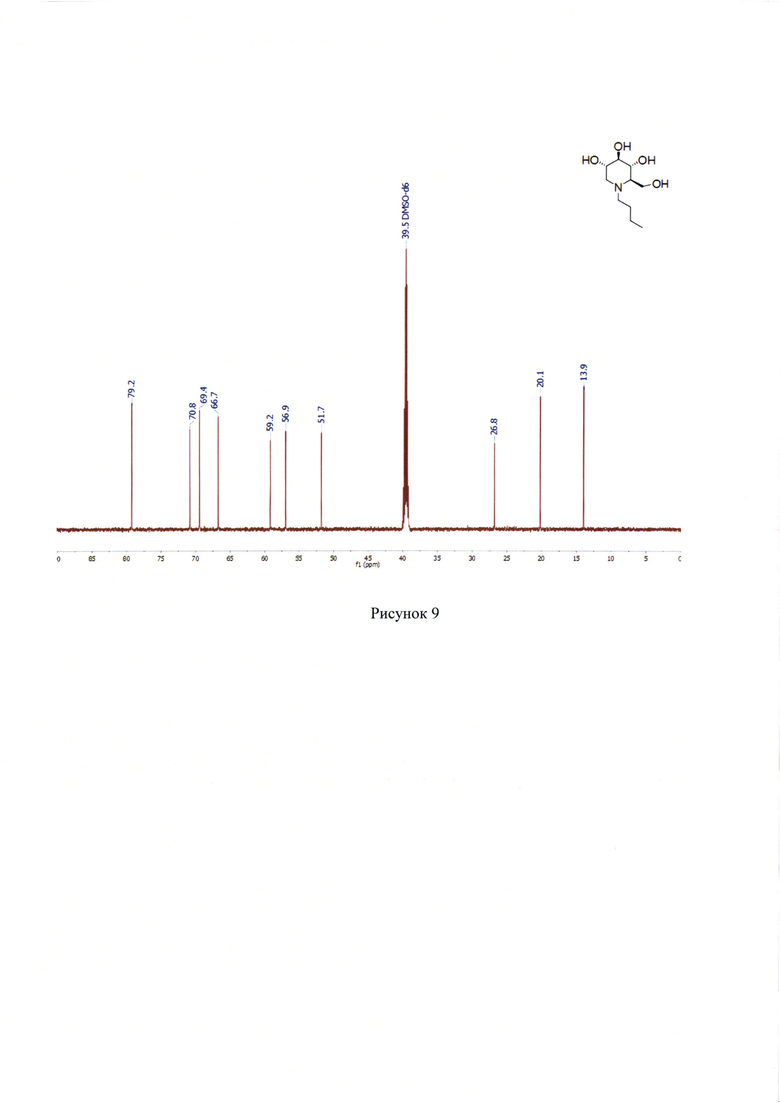
13С ЯМР (176 МГц, ДМСО-d6): δ 79.2, 70.8, 69.4, 66.7, 59.1, 56.9, 51.7, 26.8, 20.1, 13.9. (Рисунок 9).
Масс-спектры:
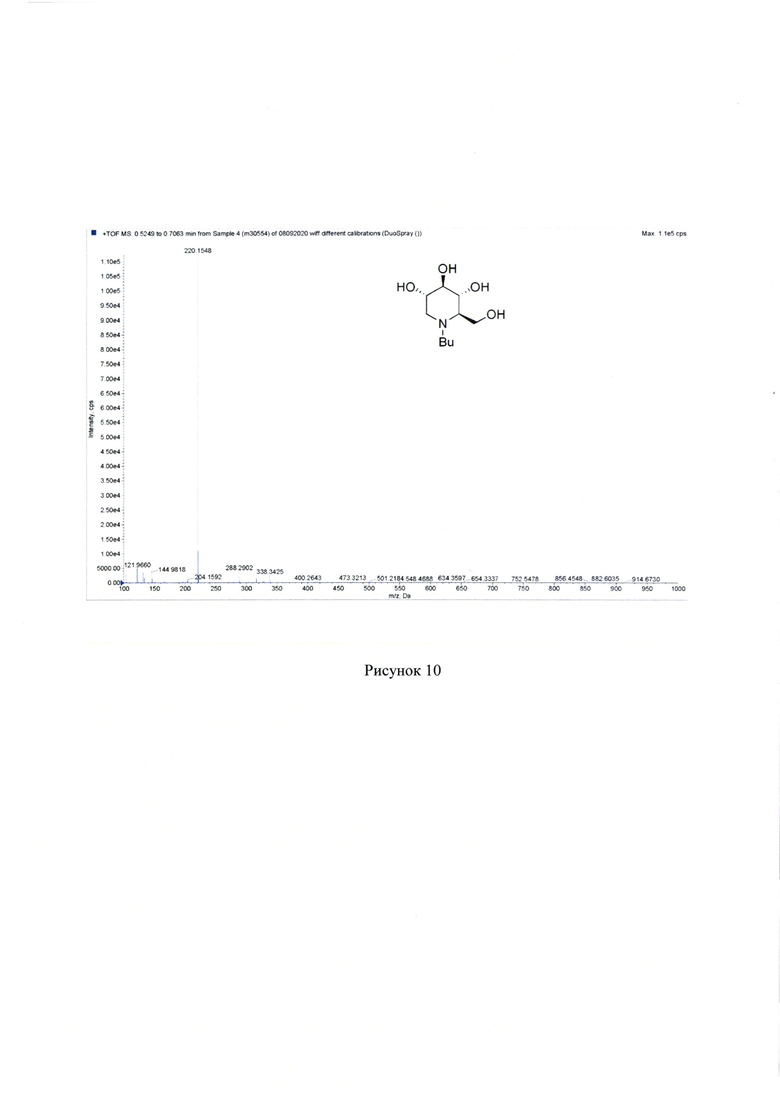
Найдено, m/z: 220.1548 [М+Н]+. C10H22NO4+. Вычислено, m/z: 220.1549. см. Рисунок 10.
2. Анализ методом ВЭЖХ
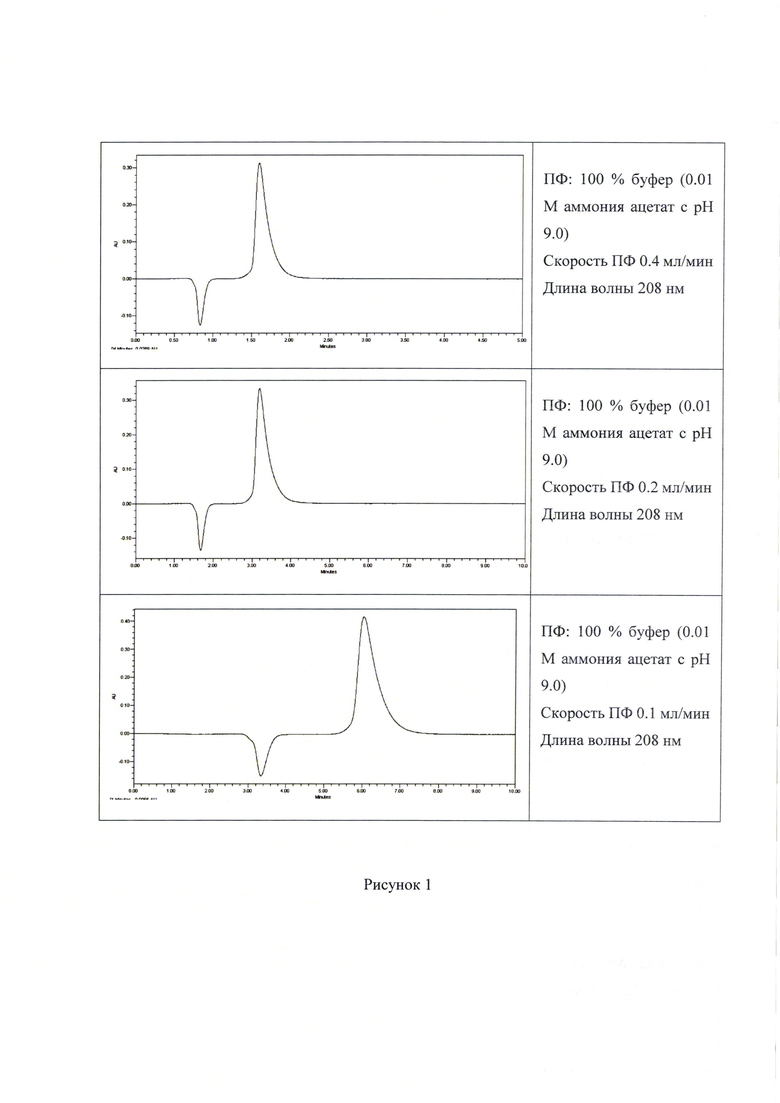
Анализ соединения 4 был проведен при использовании колонки «Waters» XBridge Amide, 30*4.6 мм, 3.5 мкм с использованием в качестве подвижной фазы раствора ацетата аммония с рН=9.0. Детектирование подобных веществ, как правило проводится с помощью рефрактометрического детектора, однако мы показали, что также может быть использовано и оптическое детектирование на длине волны 208 нм.
Условия хроматографирования:
- температура колонки 30°С;
- объем вводимой пробы 10 мкл (термостат/автосамплер для пробы при 20°С);
- детектирование от 190 до 800 нм, включая длины волн 208 нм и 254 нм;
- состав ПФ: 100% буфер (0.01 М аммония ацетат с рН 9,0);
- скорость ПФ от 0.2 мл/мин до 1.0 мл/мин.
- ПФ: 100% буфер (0,01 М аммония ацетат с рН 9.0
Полученные хроматограммы представлены на Рисунке 1. Хроматограммы на колонке Waters» XBridge Amide, 30*4.6 мм, 3.5 мкм. Отрицательный сигнал соответствует выходу мертвого объема и связан с использованным для пробы растворителем-очищенная вода.
| название | год | авторы | номер документа |
|---|---|---|---|
| ПРОИЗВОДНЫЕ ПИПЕРИДИНА, СПОСОБ ИХ ПОЛУЧЕНИЯ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ИХ ОСНОВЕ | 2002 |
|
RU2279425C2 |
| Миглустат N-бутил-1,5-дидезокси-1,5-имино-D-глюцит | 2019 |
|
RU2743694C1 |
| НОВОЕ ПРОИЗВОДНОЕ ГЛЮЦИТОЛА, ЕГО ПРОЛЕКАРСТВО И ЕГО СОЛЬ И СОДЕРЖАЩИЙ ИХ ТЕРАПЕВТИЧЕСКИЙ АГЕНТ ДЛЯ ЛЕЧЕНИЯ ДИАБЕТА | 2005 |
|
RU2386631C2 |
| ПРОИЗВОДНЫЕ МАННОЗЫ ДЛЯ ЛЕЧЕНИЯ БАКТЕРИАЛЬНЫХ ИНФЕКЦИЙ | 2013 |
|
RU2667060C2 |
| БЕТА-ЛАКТАМЫ, СПОСОБ ПОЛУЧЕНИЯ УКАЗАННЫХ СОЕДИНЕНИЙ И СЫВОРОТОЧНЫЕ ГИПОХОЛЕСТЕРИНЕМИЧЕСКИЕ СРЕДСТВА, СОДЕРЖАЩИЕ ТАКИЕ СОЕДИНЕНИЯ | 2002 |
|
RU2301799C2 |
| КОМПОЗИЦИЯ, СОДЕРЖАЩАЯ ИНГИБИТОРЫ КОТРАНСПОРТЕРОВ НАТРИЯ-ГЛЮКОЗЫ 1 И 2, И СПОСОБЫ ИХ ПРИМЕНЕНИЯ | 2012 |
|
RU2669921C2 |
| С-ГЛИКОЗИДНЫЕ ПРОИЗВОДНЫЕ, СОДЕРЖАЩИЕ КОНДЕНСИРОВАННОЕ ФЕНИЛЬНОЕ КОЛЬЦО, ИЛИ ИХ ФАРМАЦЕВТИЧЕСКИ ПРИЕМЛЕМЫЕ СОЛИ, СПОСОБ ПОЛУЧЕНИЯ ТАКОВЫХ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ, СОДЕРЖАЩАЯ ТАКОВЫЕ | 2017 |
|
RU2739024C2 |
| НОВОЕ ЦИКЛОГЕКСАНОВОЕ ПРОИЗВОДНОЕ, ЕГО ПРОЛЕКАРСТВО И ЕГО СОЛЬ И СОДЕРЖАЩЕЕ ИХ ТЕРАПЕВТИЧЕСКОЕ СРЕДСТВО ОТ ДИАБЕТА | 2005 |
|
RU2394015C2 |
| СПОСОБ ПОЛУЧЕНИЯ ПРОИЗВОДНОГО ДИФЕНИЛМЕТАНА | 2017 |
|
RU2797392C2 |
| СПОСОБ ПОЛУЧЕНИЯ ПРОИЗВОДНОГО ДИФЕНИЛМЕТАНА | 2023 |
|
RU2814846C1 |

Изобретение относится к области химии и химико-фармацевтической промышленности, а именно к способу получения субстанции миглустата ((2R,3R,4R,5S)-1-бутил-2-(гидроксиметил)пиперидин-3,4,5-триола), который включает взаимодействие 2,3,4,6-тетра-О-бензил-Д-глюкопиранозы с борогидридом натрия в смеси дихлорметана и метанола, с получением 2,3,4,6-Тетра-О-бензил-Д-глюцитола, который экстрагируют этилацетатом и после сушки и упаривания органического слоя очищают от следов воды с помощью азеотропной отгонки с толуолом; последовательное окисление 2,3,4,6-Тетра-О-бензил-Д-глюцитола смесью диметилсульфоксида (ДМСО) и оксида фосфора Р2О5 с образованием 1,5-дикарбонильного производного, очистку указанного производного диэтиловым эфиром и его восстановительное аминирование с использованием цианоборгидрида натрия; очистку перекристаллизацией из метанола, полученного реакцией аминирования 2,3,4,6-Тетра-O-бензил-N-бутил-1,5-дидеокси-1,5-Д-глюцитола; его взаимодействие с водородом в присутствии палладия в качестве катализатора, нанесенного на активированный уголь, при давлении водорода, равном одной атмосфере, с образованием (2R,3R,4R,5S)-1-бутил-2-(гидроксиметил)пиперидин-3,4,5-триола с последующей очисткой целевого продукта силикагелем. Технический результат: высокий выход стабильной субстанции миглустата. 10 ил., 1 пр.
Способ получения субстанции (2R,3R,4R,5S)-1-бутил-2-(гидроксиметил)пиперидин-3,4,5-триола, характеризующийся тем, что он включает взаимодействие 2,3,4,6-тетра-О-бензил-Д-глюкопиранозы с борогидридом натрия в органическом растворителе, представляющем собой смесь дихлорметана и метанола, с получением 2,3,4,6-Тетра-О-бензил-Д-глюцитола, который экстрагируют этилацетатом и после сушки и упаривания органического слоя очищают от следов воды с помощью азеотропной отгонки с толуолом; последовательное окисление 2,3,4,6-Тетра-О-бензил-Д-глюцитола смесью диметилсульфоксида (ДМСО) и оксида фосфора Р2О5 с образованием 1,5-дикарбонильного производного, очистку указанного производного диэтиловым эфиром и его восстановительное аминирование с использованием цианоборгидрида натрия; очистку перекристаллизацией из метанола, полученного реакцией аминирования 2,3,4,6-Тетра-O-бензил-N-бутил-1,5-дидеокси-1,5-Д-глюцитола; его взаимодействие с водородом в присутствии палладия в качестве катализатора, нанесенного на активированный уголь, при давлении водорода, равном одной атмосфере, с образованием (2R,3R,4R,5S)-1-бутил-2-(гидроксиметил)пиперидин-3,4,5-триола с последующей очисткой целевого продукта силикагелем.
| US 2014243369 A1, 28.08.2014 | |||
| EP 3081555 B1, 19.10.2016 | |||
| ИМИНОСАХАР В КРИСТАЛЛИЧЕСКОЙ ФОРМЕ | 2014 |
|
RU2678085C2 |
| Способ получения производных 3,4,5-триоксипиридина или их солей | 1978 |
|
SU917697A3 |

