Данное изобретение относится к серии новых соединений, которые являются ингибиторами фермента дипептидилпептидазы IV, к фармацевтическим композициям, содержащим эти ингибиторы, и использованию таких композиций при лечении заболеваний человека.
Предпосылки создания изобретения
Фермент дипептидилпептидаза IV, здесь обозначаемая аббревиатурой ДП-IV (в других источниках ДАП-IV или ДПП-IV) и также известная по классификации ЕС.3.4.14.5, является серинпротеазой, которая отщепляет N-концевой дипептид от пептидов, которые начинаются с последовательности Н-Хаа-Pro (где Хаа является любой аминокислотой, хотя предпочтительно липофильной аминокислотой, a Pro является пролином). Также в качестве субстратов приемлемы пептиды, которые начинаются с последовательности H-Xaa-Ala (где Ala является аланином). ДП-IV сначала был идентифицирован как связанный с мембраной белок. Позже была идентифицирована растворимая форма.
Первоначальный интерес в отношении ДП-IV был сфокусирован на его роли в активации Т-лимфоцитов. ДП-IV идентичен Т-клеточному протеину CD26. Было сделано предположение, что ингибиторы ДП-IV были бы способны к модуляции ответной реакции Т клеток, и таким образом, могли бы разрабатываться в качестве новых иммуномодуляторов. Далее было сделано предположение, что CD26 является необходимым сорецептором для ВИЧ, и, таким образом, что ингибиторы ДП-IV могли бы быть полезны для лечения СПИДа.
Было уделено внимание роли ДП-IV вне иммунной системы. Было установлено, что ДП-IV играет ключевую роль в деградации нескольких пептидных гормонов, включая релизинг-гормон гормона роста (GHRH=РГГР) и глюкагоноподобный пептид-1 и -2 (GLP-1 и GLP-2 = ГПП-1 и ГПП-2). Так как ГПП-1, как известно, обладает потенцирующим эффектом в отношении действия инсулина при регуляции уровней глюкозы после приема пищи, ясно, что ингибиторы ДП-IV также могли бы эффективно использоваться при лечении диабета типа II и сниженной толерантности к глюкозе. По меньшей мере, два ингибитора ДП-IV в настоящее время проходят клинические испытания для изучения использования этой возможности.
Несколько групп обнаружили ингибиторы ДП-IV. В то время как некоторые примеры были найдены в результате программ случайного поиска (скрининга), большинство работ в данной области было направлено на исследования субстратных аналогов. Ингибиторы ДП-IV, которые являются аналогами субстратов, описаны, например, в US 5462928, US 5543396, WO 95/15309 (эквивалент US 5939560 и ЕР 0731789), WO 98/19998 (эквивалент US 6011155), WO 99/46272 и WO 99/61431. Наиболее сильными ингибиторами являются аминоацилпирролидинбороновые кислоты, но они являются нестабильными и имеют склонность циклизироваться, в то время как более стабильные пирролидиновые и тиазолидиновые производные имеют более низкое сродство к ферменту и, таким образом, в клинической ситуации были бы нужны большие дозы. Пирролидиннитрилы, по-видимому, представляют хороший компромисс, так как они обладают как более высоким сродством к ферменту, так и достаточно длинным полупериодом существования в растворе в виде свободного основания. Однако остается потребность в ингибиторах ДП-IV с улучшенными свойствами.
Краткое описание изобретения
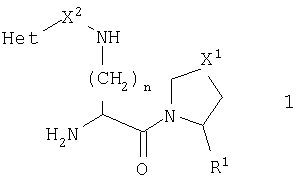
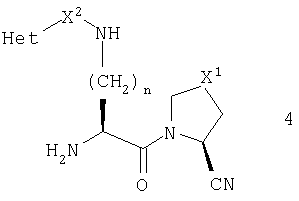
Данное изобретение относится к серии ингибиторов ДП-IV с улучшенным сродством к ферменту. Данные соединения могут использоваться для лечения ряда человеческих заболеваний, включая сниженную толерантность к глюкозе и диабет типа II. Соответственно, данное изобретение относится к использованию данных соединений для изготовления фармацевтических композиций, к самим таким композициям и к использованию таких композиций при лечении людей. Соединения данного изобретения описываются общей формулой 1
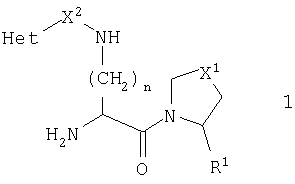
В общей формуле 1 R1 представляет или Н, или CN, Х1 представляет S, О, SO2 или CH2, X2 представляет карбонильную группу, СН2 или отсутствует, n равно 1-5 и Het является необязательно замещенным ароматическим азотосодержащим гетероциклом.
Подробное описание изобретения
В первом аспекте данное изобретение представляет серию новых соединений, которые являются ингибиторами фермента ДП-IV и пригодны для лечения некоторых заболеваний человека. Данные соединения описываются общей формулой 1
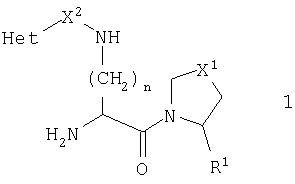
В этой общей формуле R1 представляет или атом водорода, или нитрильную группу (-C N), X1 представляет атом серы, атом кислорода, сульфонильную группу (-SO2-) или метиленовую группу (-CH2-), Х2 представляет или карбонильную группу (>С=O), метиленовую группу (-СН2-), или ковалентную связь. Переменная n может иметь любое целое значение между 1 и 5. Het представляет ароматический азотосодержащий гетероцикл, выбранный из пиридила, пиридазинила, пиримидинила, пиразинила, имидазолила, пиразолила, тиазолила, изотиазолила, оксазолила, изоксазолила и их бензоконденсированных аналогов, таких как хинолинил, изохинолинил, циннолинил, фталазинил, хиназолинил, хиноксалинил, бензимидазолил, индазолил, бензотиазолил, бензизотиазолил, бензоксазолил и бензизоксазолил. Этот гетероцикл может быть необязательно замещенным по одному атому углерода или более. Подходящими заместителями являются низшие алкильные, гидрокси, низшие алкилокси, амино, низшие алкиламино, ди(низший алкил)амино, фтор, хлор, бром, нитро, трифторметил, циано, карбокси и низшие алкилоксикарбонильные группы.
N), X1 представляет атом серы, атом кислорода, сульфонильную группу (-SO2-) или метиленовую группу (-CH2-), Х2 представляет или карбонильную группу (>С=O), метиленовую группу (-СН2-), или ковалентную связь. Переменная n может иметь любое целое значение между 1 и 5. Het представляет ароматический азотосодержащий гетероцикл, выбранный из пиридила, пиридазинила, пиримидинила, пиразинила, имидазолила, пиразолила, тиазолила, изотиазолила, оксазолила, изоксазолила и их бензоконденсированных аналогов, таких как хинолинил, изохинолинил, циннолинил, фталазинил, хиназолинил, хиноксалинил, бензимидазолил, индазолил, бензотиазолил, бензизотиазолил, бензоксазолил и бензизоксазолил. Этот гетероцикл может быть необязательно замещенным по одному атому углерода или более. Подходящими заместителями являются низшие алкильные, гидрокси, низшие алкилокси, амино, низшие алкиламино, ди(низший алкил)амино, фтор, хлор, бром, нитро, трифторметил, циано, карбокси и низшие алкилоксикарбонильные группы.
В контексте данного описания подразумевается, что термин «низший алкил» или сам по себе, или в таких сочетаниях, как низший алкокси, включает линейные, разветвленные и циклические насыщенные углеводородные группы из числа атомов углерода между одним и шестью. Примеры низших алкильных групп включают, но не ограничиваются ими, метил, этил, изопропил, трет-бутил, неопентил, циклогексил, циклопентилметил, 2-(циклопропил)этил, 3,3-диметилциклобутил и бицикло[3,1,0]гексил.
Соединения общей формулы 1 имеют, по меньшей мере, один стереогенный центр и, таким образом, могут проявлять оптическую изомерию. Все такие изомеры, включая энантиомеры, диастереомеры и эпимеры, включены в объем данного изобретения. Кроме того, данное изобретение включает такие соединения в виде отдельных изомеров и в виде смесей, включая рацематы. Некоторые соединения общей формулы 1, включая те, в которых группа Het несет гидрокси или амино заместитель, могут существовать в виде таутомеров. Такие таутомеры или отдельно, или в виде смесей также рассматриваются как находящиеся в объеме данного изобретения.
Соединения общей формулы 1 имеют, по меньшей мере, одну функциональную группу основания. Поэтому они могут образовывать соли присоединения с кислотами. Эти соли присоединения, которые образуются с фармацевтически приемлемыми кислотами, включены в объем данного изобретения. Примеры подходящих кислот включают уксусную кислоту, трифторуксусную кислоту, лимонную кислоту, фумаровую кислоту, бензойную кислоту, памовую кислоту, метансульфоновую кислоту, соляную кислоту, азотную кислоту, серную кислоту, фосфорную кислоту и т.п.
Некоторые соединения общей формулы 1 имеют кислотную группу и, таким образом, способны образовывать соли с основаниями. Примеры таких солей включают соли натрия, калия и кальция, которые образуются при реакции кислоты с соответствующим гидроксидом, оксидом, карбонатом или бикарбонатом металла. Подобным же образом соли тетраалкиламмония можно получить по реакции кислоты с гидроксидом тетраалкиламмония. Первичные, вторичные и третичные амины, такие как триэтиламин, могут образовывать соли присоединения с кислотой. Особым случаем является внутренняя соль присоединения, образованная между кислотной группой и первичной аминогруппой одной и той же молекулы, которая также называется цвиттерионом. Поскольку они фармацевтически приемлемы, все эти соли включены в объем данного изобретения.
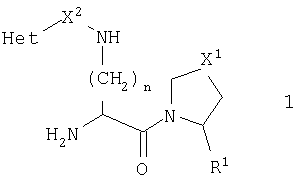
В предпочтительном осуществлении данного изобретения R1 является нитрильной группой. В данном воплощении предпочтительно, когда стереохимия нитрильной группы является такой, которая показана в общей формуле 2
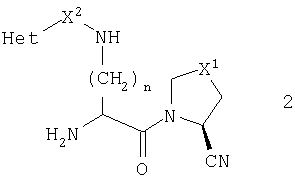
В соответствии со стандартной терминологией это представляет собой S-конфигурацию, когда Х1 является метиленом, но R-конфигурацию, когда Х1 является серой, кислородом или сульфонилом.
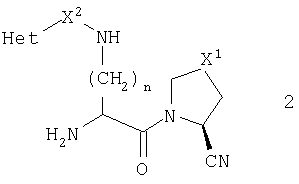
При другом предпочтительном осуществлении стереохимия у центра, соседнего с первичным амином, представляет S-конфигурацию, которая показана в общей формуле 3
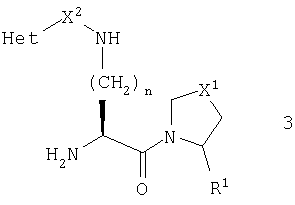
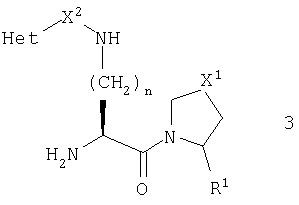
В этом воплощении более предпочтительно, чтобы R1 был нитрильной группой, и более предпочтительно, чтобы он имел абсолютную конфигурацию, изображаемую общей формулой 4
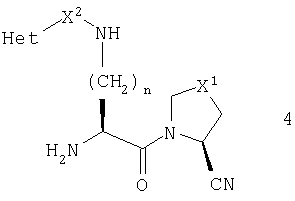
В еще одном предпочтительном воплощении данного изобретения Х1 представляет атом серы или метиленовую группу.
В другом предпочтительном осуществлении данного изобретения n равно 3 или 4.
Особенно предпочтительные соединения в данном изобретении включают:
(2S)-1-[Nω-(пиразинил-2-карбонил)-L-орнитинил]пирролидин-2-карбонитрил,
(2S)-1-[Nω-(пиразинил-2-карбонил)-L-лизинил]пирролидин-2-карбонитрил,
(2S)-1-[(2'S)-2'-амино-4'-(пиразинил-2''-карбониламино)-бутаноил]пирролидин-2-карбонитрил,
(4R)-3-[Nω-(пиразинил-2-карбонил)-L-лизинил]тиазолидин-4-карбонитрил,
1-[Nω-(пиразинил-2-карбонил)-L-орнитинил]пирролидин,
3-[Nω-(2-хлорпиридил-3-карбонил)-L-орнитинил]тиазолидин,
1-[Nω-(2-хлорпиридил-3-карбонил)-L-орнитинил]пирролидин,
(2S)-1-[Nω-(2-хлорпиридил-3-карбонил)-L-орнитинил]пирролидин-2-карбонитрил,
3-[Nω-(пиразинил-2-карбонил)-L-орнитинил]тиазолидин,
3-[Nω-(5-циано-2-пиридил)-L-лизинил]тиазолидин,
(2S)-1-[Nω-(5-циано-2-пиридил)-L-лизинил]пирролидин-2-карбонитрил,
(2S)-1-[Nω-(5-трифторметил-2-пиридил)-L-орнитинил]пирролидин-2-карбонитрил,
3-[Nω-(2-хинолинилметил)-L-лизинил]тиазолидин,
3-[Nω-(2-хинолинилметил)-L-орнитинил]тиазолидин,
3-[Nω-(2-хиноксалоил)-L-лизинил]тиазолидин,
3-[Nω-(2-хиноксалоил)-L-орнитинил]тиазолидин,
(2S)-1-[Nω-(2-хиноксалоил)-L-орнитинил]пирролидин-2-карбонитрил,
3-[Nω-(6-метилпиразинил-2-карбонил)-L-орнитинил]тиазолидин,
3-[Nω-(изохинолин-3-карбонил)-L-орнитинил]тиазолидин и
3-[Nω-(6-трифторметилникотиноил)-L-орнитинил]тиазолидин.
Во втором аспекте данное изобретение включает фармацевтическую композицию для терапевтического применения для людей. Данная композиция отличается тем, что она имеет в качестве активного вещества, по меньшей мере, одно из соединений, описанных выше. Такая композиция пригодна для лечения заболеваний человека. Композиция обычно включает один дополнительный компонент или более, выбранных из фармацевтически приемлемых вспомогательных веществ и других фармацевтически активных веществ, помимо соединений данного изобретения.
Данная композиция может быть представлена в виде твердой или жидкой лекарственной формы в зависимости от предназначенного пути введения. Примеры твердых лекарственных форм включают пилюли, таблетки, капсулы и порошки для перорального введения, суппозитории для ректального или вагинального введения, порошки для назального или внутрилегочного введения и пластыри для трансдермального введения или введения через слизистые оболочки (такие как защечные формы). Примеры жидких лекарственных форм включают растворы и суспензии для внутривенных, подкожных или внутримышечных инъекций и перорального, назального или внутрилегочного введения. Особенно предпочтительным видом являются таблетки для перорального применения. Другим предпочтительным видом, особенно для скорой помощи и интенсивной терапии, является стерильный раствор для внутривенных инъекций.
Данная композиция содержит, по меньшей мере, одно соединение, соответствующее предшествующему описанию. Композиция может содержать более одного такого соединения, но в основном предпочтительно, что она должна включать только одно соединение. Количество соединения, используемого в композиции, будет таким, что общая суточная доза активного вещества может быть введена в виде от одной до четырех удобной раздельной дозы. Например, композиция может быть таблеткой, содержащей количество соединения, равное необходимой общей суточной дозе, и указанную таблетку нужно принимать один раз в день. Альтернативно, таблетка может содержать половину (или одну треть, или одну четверть) суточной дозы, которую нужно принимать дважды (или три, или четыре раза) в день. Такая таблетка может быть также разделена рисками для облегчения деления дозы, так что, например, таблетка, содержащая полную суточную дозу, может быть разделена пополам и приниматься двумя порциями. Предпочтительно, таблетка или другой элемент дозированной формы будет содержать количество активного соединения между 0,1 мг и 1 г. Более предпочтительно, она будет содержать количество между 1 и 250 мг.
Композиция обычно включает один или более из вспомогательных веществ, выбранных из тех, о которых известно, что они фармацевтически приемлемы. Подходящие вспомогательные вещества включают, но не ограничиваются ими, наполнители, связывающие вещества, разбавители, растворители, консерванты и улучшающие вкус и запах вещества. Средства, которые модифицируют характеристики высвобождения активного вещества из композиции, такие как полимеры, которые селективно растворяются в кишечнике («энтеральные покрытия»), также рассматриваются в контексте данного изобретения как подходящие вспомогательные вещества.
Композиция может содержать в дополнение к соединению данного изобретения второе фармакологически активное вещество. Например, композиция может включать противодиабетическое средство, способствующее росту вещество, противовоспалительное вещество или противовирусное средство. Однако обычно предпочтительно, когда композиция содержит только одно активное вещество.
В третьем аспекте данное изобретение включает использование соединений и композиций, описанных выше, для лечения болезней человека. Этот аспект может в равной степени рассматриваться как включающий способ лечения таких заболеваний. Заболеваниями, поддающимися лечению, являются те, при которых подавление ДП-IV или CD26 приводит к клиническому улучшению непосредственно или опосредованно. Прямые (непосредственные) эффекты включают блокаду активации Т лимфоцитов. Непрямые эффекты включают потенцирование активности пептидного гормона предотвращением деградации этих гормонов. Примеры заболеваний включают, но не ограничиваются этим, аутоиммунные и воспалительные заболевания, такие как воспалительные заболевания кишечника и ревматоидный артрит, дефицит гормона роста, приводящий к малому росту, синдром поликистоза яичников, сниженная толерантность к глюкозе и диабет типа 2. Особенно предпочтительным является использование соединений и композиций для лечения сниженной толерантности к глюкозе и диабета типа 2, и равным образом, способ лечения этих заболеваний путем введения эффективного количества соединения или композиции, которые описаны ранее.
Точные детали лечения, включая режим дозирования, будут установлены лечащим врачом, принимающим во внимание общие данные (состояние) пациента и тяжесть заболевания. Что касается таких заболеваний, как воспалительное заболевание кишечника, которые имеют острые фазы активного заболевания, разделенные спокойными периодами, врач может выбрать относительно высокую дозу во время острой фазы и более низкую поддерживающую дозу для спокойного периода. Для хронических заболеваний, таких как диабет типа 2 и сниженная толерантность к глюкозе, дозировку, возможно, нужно будет сохранять на одном и том же уровне в течение продолжительного периода. Режим дозирования из от одной до четырех таблеток в день, причем каждая содержит между 0,1 мг и 1 г (и предпочтительно, между 1 и 250 мг) активного соединения, мог бы быть типичным в таком случае.
Соединения по данному изобретению могут быть получены известными специалистам методами. Выбранный способ будет зависеть от конкретной природы заместителей, присутствующих в запланированной молекуле. Исходный материал будет обычно производным α,ω-диаминокислоты 5
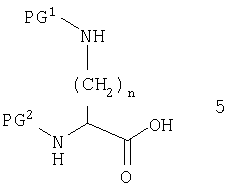
PG1 и PG2 являются «ортогональными» защитными группами - группами, которые защищают реакционноспособные аминогруппы и которые можно удалить одну в присутствии другой. Подходящие группы хорошо известны в литературе. Производные диаминокислот общей формулы 5 являются или доступными в продаже, или описаны в литературе для всех значений n в интервале от 1 до 5 и как для R-, так и S-стереоизомера.
Для некоторых стратегий синтеза предпочтительно начинать со сложного эфира вышеуказанных диаминокислот, таких как бензиловый, метиловый или трет-бутиловый сложный эфир. Сложный эфир будет выбираться так, чтобы он не гидролизовался реактивами, которые могут отщеплять PG1 или PG2.
Исходя из 5, необходимо преобразовать кислотную функциональную группу в пирролидиновое амидное производное целевой молекулы и преобразовать функциональную ω-аминогруппу в желаемое гетероарильное производное. Порядок, в котором эти две стадии выполняются, не так важен.
Схема А
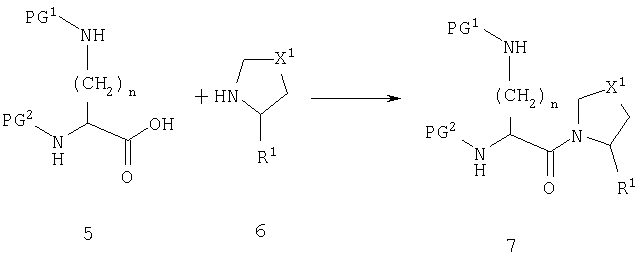
Производное диаминокислоты 5 можно вводить во взаимодействие с пирролидиновым производным 6 с получением амида 7. Условия этого превращения хорошо известны в литературе. Подходящие реагенты включают карбодиимиды, фосфорные реагенты и алкилхлорформиаты, и реакция обычно катализируется третичным амином, таким как триэтиламин и диметиламинопиридин.
Реакция, представленная на схеме А, возможна для всех комбинаций R1 и Х1. Однако для случая, когда R1 является нитрильной группой или когда Х1 является сульфонильной группой, может быть выгоднее модифицировать стратегию так, как представлено на схемах В и С.
Схема В
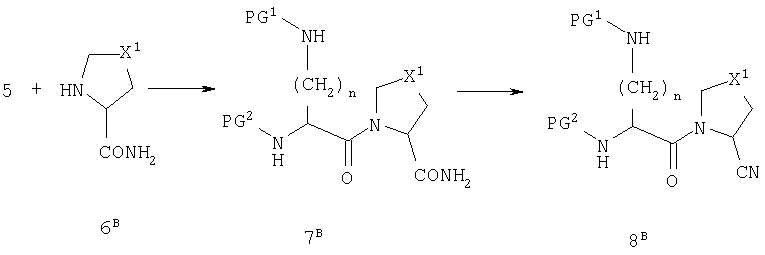
Схема С
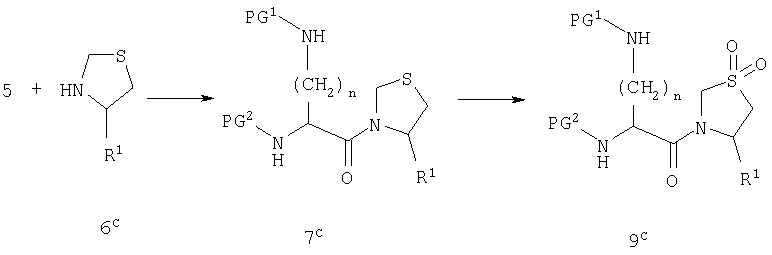
На схеме В группу R1 вводят в виде первичного амида и затем превращают в нитрил действием дегидратирующего вещества, такого как ангидрид трифторуксусной кислоты. На схеме С группу Х1 вводят в виде тиоэфира и затем превращают в сульфон действием окислителя, такого как периодат натрия.
Схема D
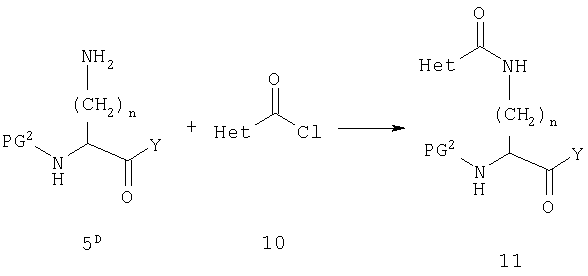
На схеме D соединение 5D представляет собой производное диаминокислоты 5 после удаления ω-защитной группы. Y может представлять ОН, но чаще представляет пирролидиновое кольцо или O-алкильную группу сложного эфира. Свободную аминогруппу вводят во взаимодействие с гетероарилкарбонилхлоридом с получением амида 11, который включает функциональность соединений данного изобретения, у которых Х2 является карбонильной группой. Гетероарилкарбонилхлориды легко получаются из соответствующих карболовых кислот, которые являются хорошо известными соединениями. Реакция схемы D обычно применима для всех вариантов группы Het при условии, что некоторые заместители у Het могут требовать защиты. Такие группы и соответствующая защита очевидны для специалистов в данной области.
Когда Х2 является ковалентной связью, возможно получить требуемую функциональную группу из амина 5D также путем прямой реакции с гетероарилхлоридом или фторидом. В некоторых случаях гетероарилхлорид или фторид не может быть легко доступен или может быть недостаточно реакционноспособен, и тогда будет необходимо использовать альтернативный путь, такой как восстановительное аминирование. Это иллюстрируется на схеме Е
Схема Е
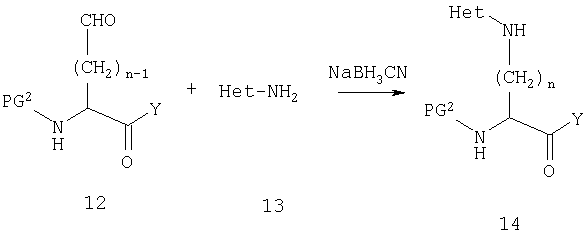
Восстановительное аминирование также является методом выбора, когда Х2 является метиленовой группой. В этом случае существуют два варианта, что иллюстрируется на схеме F
Схема F
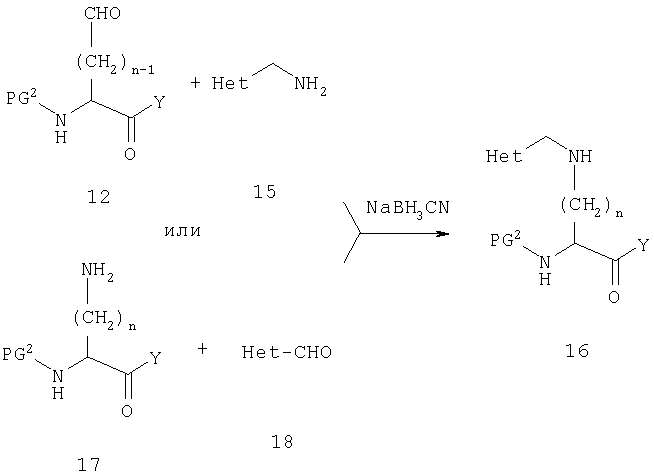
Когда все группы синтезированы, конечную защитную группу удаляют и продукт выделяют и очищают, используя стандартные методики.
Эти общие методы дополнительно иллюстрируются в следующих, не ограничивающих примерах.
ПРИМЕРЫ
Сокращения
Использованы следующие сокращения.'
ПРИМЕР 1
Трифторацетат (2S)-1-[Nω-(пиразинил-2-карбонил)-L-орнитинил]пирролидин-2-карбонитрила
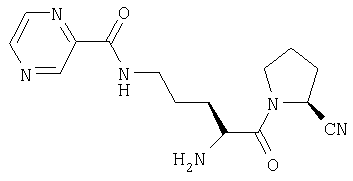
А. N-(2-нитробензолсульфенил)-L-пролин
L-пролин (25 г, 217 ммоль) растворяли в 2 М NaOH (110 мл, 220 ммоль) и диоксане (120 мл). Медленно добавляли раствор 2-нитробензолсульфенилхлорида (42 г, 222 ммоль) в диоксане (60 мл) одновременно с 2 М NaOH (110 мл, 220 ммоль). После 2 ч при комнатной температуре реакционную смесь выливали в воду (500 мл) и твердое вещество отфильтровывали. Значение рН фильтрата доводили до рН 3 с помощью 2 М HCl и раствор экстрагировали этилацетатом (3×500 мл). Объединенные органические экстракты промывали водой (4×200 мл) и насыщенным раствором соли (1×200 мл), сушили (Na2SO4) и выпаривали в вакууме с получением оранжевого твердого вещества, идентифицированного как N-(2-нитробензолсульфенил)-L-пролин (58,1 г, 217 ммоль, 100%).
В. N-(2-нитробензолсульфенил)-L-пролинсукцинимидиловый эфир
N-(2-нитробензолсульфенил)-L-пролин (57,9 г, 216 ммоль) растворяли в СН2Cl2/ДМФ (9:1, 500 мл). Добавляли N-гидроксисукцинимид (37,3 г, 324 ммоль) и водорастворимый карбодиимид (51,8 г, 260 ммоль). После 18 ч при комнатной температуре растворитель удаляли в вакууме и остаток переносили в этилацетат (1000 мл). Раствор промывали водой (4×200 мл) и насыщенным раствором соли (1×200 мл), сушили (Na2SO4) и выпаривали в вакууме с получением желтого твердого вещества, идентифицированного как N-(2-нитробензолсульфенил)-L-пролинсукцинимидиловый эфир (78,9 г, 216 ммоль, 100%).
С. N-(2-нитробензолсульфенил)-L-пролинамид
N-(2-нитробензолсульфенил)-L-пролинсукцинимидиловый эфир (78,5 г, 215 ммоль) растворяли в диоксане (500 мл). Добавляли аммиак (35%, 100 мл). После перемешивания при комнатной температуре в течение 2 ч реакционную смесь выливали в воду (700 мл). Осадок отфильтровывали, промывали водой (200 мл), сушили над Р2О5 и перекристаллизовывали из этилацетата/пет.эфира с получением желтого твердого вещества, идентифицированного как N-(2-нитробензолсульфенил)-L-пролинамид (49,6 г, 185 ммоль, 86%).
D. (2S)-N-(2-нитробензолсульфенил)пирролидин-2-карбонитрил
N-(2-нитробензолсульфенил)-L-пролинамид (49 г, 183 ммоль) растворяли в безводном ТГФ (300 мл). Раствор охлаждали до 0°С, добавляли триэтиламин (36,7 г, 367 ммоль) с последующим медленным добавлением трифторуксусного ангидрида (77 г, 367 ммоль). Значение рН доводили до 9 триэтиламином. После 30 мин реакционную смесь разбавляли этилацетатом (500 мл), промывали водой (1×200 мл) и насыщенным раствором соли, сушили (Na2SO4) и выпаривали в вакууме с получением оранжевого масла, которое очищали флэш-хроматографией (элюент: 80% пет. эфир, 20% этилацетат) с получением желтого твердого вещества, идентифицированного как (2S)-N-(2-нитробензолсульфенил)пирролидин-2-карбонитрил (38,9 г, 150 ммоль, 82%).
Е. Гидрохлорид (2S)-пирролидин-2-карбонитрила
(2S)-N-(2-нитробензолсульфенил)пирролидин-2-карбонитрил (38,5 г, 149 ммоль) растворяли в диэтиловом эфире (200 мл). Медленно добавляли 4 М HCl/диоксан (150 мл, 600 ммоль). После 2 ч при комнатной температуре реакционную смесь выливали в диэтиловый эфир (1000 мл). Твердое вещество отфильтровывали, промывали диэтиловым эфиром (500 мл) и перекристаллизовывали из метанола/диэтилового эфира с получением белого твердого вещества, идентифицированного как гидрохлорид (2S)-пирролидин-2-карбонитрила (18,9 г, 142,5 ммоль, 96%).
F. (2S)-1-[Nω-(трет-бутоксикарбонил)-Nω-(пиразинил-2-карбонил)-L-орнитинил]пирролидин-2-карбонитрил
Nω-(трет-бутоксикарбонил)-Nω-(пиразинил-2-карбонил)-L-орнитин (2,5 г, 7,4 ммоль) растворяли в CH2Cl2 (50 мл). Этот раствор охлаждали до 0°С, добавляли (2S)-пирролидин-2-карбонитрила гидрохлорид (1,2 г, 9,1 ммоль), добавляли PyBOP® (4,3 г, 8,23 ммоль) и рН доводили до 9 триэтиламином. После 18 часов при от 0°С до комнатной температуры растворитель удаляли в вакууме и остаток переносили в этилацетат (200 мл). Этот раствор промывали 0,3 М KHSO4 (2×50 мл), нас. NaHCO3 (2×50 мл), водой (2×50 мл) и насыщенным раствором соли (1×50 мл), сушили (Na2SO4) и выпаривали в вакууме с получением желтого масла. Это масло очищали флэш-хроматографией (элюент: 80% этилацетата, 20% пет. эфира) и получали бесцветное масло, идентифицированное как (2S)-1-[Nω-(трет-бутоксикарбонил)-Nω-(пиразинил-2-карбонил)-L-орнитинил]пирролидин-2-карбонитрил (2,98 г, 7,16 ммоль, 97%).
G. Трифторацетат (2S)-1-[Nω-(пиразинил-2-карбонил)-L-орнитинил]пирролидин-2-карбонитрила
(2S)-1-[Nω-(трет-бутоксикарбонил)-Nω-(пиразинил-2-карбонил)-L-орнитинил]пирролидин-2-карбонитрил (2,8 г, 6,7 ммоль) растворяли в трифторуксусной кислоте (5 мл). После 1 ч при комнатной температуре растворитель удаляли в вакууме. Остаток очищали препаративной ЖХВД (Vydac C18, 5-50% 0,1% ТФУ/ацетонитрил в 0,1% ТФУ/воде в течение 40 мин при 3 мл/мин). Фракции, содержащие продукт, лиофилизировали с получением бесцветного масла, идентифицированного как трифторацетат (2S)-1-[Nω-(пиразинил-2-карбонил)-L-орнитинил]пирролидин-2-карбонитрила (1,5 г, 3,48 ммоль, 52%).
[М+Н]+=317,3
ПРИМЕР 2
Трифторацетат (2S)-1-[Nω-(пиразинил-2-карбонил)-L-лизинил]пирролидин-2-карбонитрила
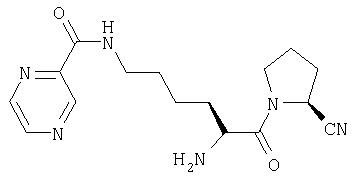
А. (Nα-(трет-бутилоксикарбонил)-Nω-(9-флуоренилметилоксикарбонил)-L-лизинил)-L-пролинамид
Nω-(трет-бутилоксикарбонил)-Nω-(9-флуоренилметилоксикарбонил)-L-лизин (5 г, 10,7 ммоль) растворяли в СН2Cl2 (100 мл). Данный раствор охлаждали до 0°С, добавляли L-пролинамид (1,78 г, 11,7 ммоль) и PyBOP® (6,7 г, 12,8 ммоль) и рН доводили до 9 триэтиламином. После 18 ч при от 0°С до комнатной температуры растворитель удаляли в вакууме и остаток переносили в этилацетат (200 мл). Раствор промывали 0,3 М KHSO4 (2×50 мл), нас. NaHCO3 (2×50 мл), водой (2×50 мл) и насыщенным раствором соли (1×50 мл), сушили (Na2SO4) и выпаривали в вакууме. Остаток очищали флэш-хроматографией (элюент: 2% метанола, 98% хлороформа) с получением бесцветного масла, идентифицированного как (Nα-(трет-бутилоксикарбонил)-Nω-(9-флуоренилметилоксикарбонил)-L-лизинил)-L-пролинамид (4,05 г, 7,2 ммоль, 67%).
В. (2S)-1-(Nα-(трет-бутилоксикарбонил)-Nω-(9-флуоренилметилоксикарбонил)-L-лизинил)пирролидин-2-карбонитрил
(Nα-(трет-бутилоксикарбонил)-Nω-(9-флуоренилметилоксикарбонил)-L-лизинил)-L-пролинамид (3,95 г, 7,02 ммоль) растворяли в сухом ТГФ (100 мл). Раствор охлаждали до 0°С, добавляли триэтиламин (1,4 г, 14 ммоль) с последующим медленным добавлением трифторуксусного ангидрида (2,97 г, 14,1 ммоль). Значение рН доводили до 9 триэтиламином. Через 30 мин реакционную смесь разбавляли этилацетатом (100 мл), промывали водой (1×50 мл) и насыщенным раствором соли (1×50 мл), сушили (Na2SO4) и выпаривали в вакууме с получением оранжевого масла. Остаток очищали флэш-хроматографией (элюент: 60% пет. эфир, 40% этилацетат) с получением бесцветного масла, идентифицированного как (2S)-1-(Nα-(трет-бутилоксикарбонил)-Nω-(9-флуоренилметилоксикарбонил)-L-лизинил)пирролидин-2-карбонитрил (3,3 г, 6,11 ммоль, 87%).
С. (2S)-1-(Nα-(трет-бутилоксикарбонил)-L-лизинил)пирролидин-2-карбонитрил
(2S)-1-(Nα-(трет-бутилоксикарбонил)-Nω-(9-флуоренилметилоксикарбонил)-L-лизинил)пирролидин-2-карбонитрил (3,1 г, 5,7 ммоль) растворяли в ТГФ (80 мл). Добавляли диэтиламин (20 мл). После 2 ч при комнатной температуре растворитель удаляли в вакууме. Остаток очищали флэш-хроматографией (элюент: 90% хлороформ, 7% метанол, 3% триэтиламин) с получением бесцветного масла, идентифицированного как (2S)-1-(Nα-(трет-бутилоксикарбонил)-L-лизинил)пирролидин-2-карбонитрил (1,63 г, 5,03 ммоль, 89%).
D. (2S)-1-(Nα-(трет-бутилоксикарбонил)-Nω-(пиразинил-2-карбонил)-L-лизинил)пирролидин-2-карбонитрил
(2S)-1-(Nα-(трет-бутилоксикарбонил)-L-лизинил)пирролидин-2-карбонитрил (100 мг, 0,31 ммоль) растворяли в СН2Cl2/ДМФ (9:1, 20 мл). К этому раствору при 0°С добавляли гидрат 1-гидроксибензотриазола (84 мг, 0,62 ммоль), водорастворимый карбодиимид (76 мг, 0,38 ммоль), 2-пиразинкарбоновую кислоту (43 мг, 0,35 ммоль) и триэтиламин (65 мг, 0,65 ммоль). После 18 ч при от 0°С до комнатной температуры растворитель удаляли в вакууме и остаток переносили в этилацетат (70 мл). Этот раствор промывали 0,3 М KHSO4 (2×20 мл), нас. NaHCO3 (2×20 мл), водой (2×20 мл) и насыщенным раствором соли (1×20 мл), сушили (Na2SO4) и выпаривали в вакууме с получением желтого масла. Остаток очищали флэш-хроматографией (элюент: 2% метанола, 98% хлороформа) с получением бесцветного масла, идентифицированного как (2S)-1-(Nα-(трет-бутилоксикарбонил)-Nω-(пиразинил-2-карбонил)-L-лизинил)пирролидин-2-карбонитрил (124 мг, 0,29 ммоль, 93%).
Е. Трифторацетат (2S)-1-[Nω-(пиразинил-2-карбонил)-L-лизинил]пирролидин-2-карбонитрила
(2S)-1-(Nα-(трет-бутилоксикарбонил)-Nω-(пиразинил-2-карбонил)-L-лизинил)пирролидин-2-карбонитрил (110 мг, 0,26 ммоль) растворяли в трифторуксусной кислоте (5 мл). После 1 ч при комнатной температуре растворитель удаляли в вакууме. Остаток очищали препаративной ЖХВД (Vydac С18, 5-50% 0,1% ТФУ/ацетонитрил в 0,1% ТФУ/воде в течение 40 мин при 3 мл/мин). Фракции, содержащие продукт, лиофилизировали с получением бесцветного масла, идентифицированного как трифторацетат (2S)-1-[Nω-(пиразинил-2-карбонил)-L-лизинил]пирролидин-2-карбонитрила (66 мг).
[М+Н]+=331,1
ПРИМЕР 3
Трифторацетат (4R)-3-[Nω-(пиразинил-2-карбонил)-L-лизинил]тиазолидин-4-карбонитрила
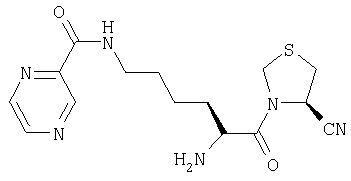
А. (4R)-3-(трет-бутилоксикарбонил)тиазолидин-4-карбоксамид
(4R)-3-(трет-бутилоксикарбонил)тиазолидин-4-карбоновую кислоту (12,5 г, 54,1 ммоль) растворяли в СН2Cl2/ДМФ (9:1, 150 мл). К этому раствору при 0°С добавляли гидрат 1-гидроксибензотриазола (14,6 г, 108 ммоль) и водорастворимый карбодиимид (13,0 г, 65 ммоль). После 1 ч при 0°С добавляли аммиак (35%, 50 мл). После 18 ч при от 0°С до комнатной температуры растворитель удаляли в вакууме и остаток переносили в этилацетат (500 мл). Раствор промывали 0,3 М KHSO4 (2×100 мл), нас. NaHCO3 (2×100 мл), водой (2×100 мл) и насыщенным раствором соли (1×100 мл), сушили (Na2SO4) и выпаривали в вакууме с получением желтого масла. Остаток очищали флэш-хроматографией (элюент: 2% метанола, 98% хлороформа) с получением бесцветного масла, идентифицированного как (4R)-3-(трет-бутилоксикарбонил)тиазолидин-4-карбоксамид (8,9 г, 38,4 ммоль, 71%).
В. Гидрохлорид (4R)-тиазолидин-4-карбоксамида
(4S)-3-(трет-бутилоксикарбонил)тиазолидин-4-карбоксамид (8,6 г, 37,1 ммоль) растворяли в 4 М HCl/диоксане (50 мл). После 1 ч при комнатной температуре растворитель выпаривали в вакууме с получением белого твердого вещества, идентифицированного как гидрохлорид (4R)-тиазолидин-4-карбоксамида (6,2 г, 36,8 ммоль, 99%).
С. (4R)-3-[Nα-(трет-бутилоксикарбонил)-Nω-(9-флуоренилметилоксикарбонил)-L-лизинил]тиазолидин-4-карбоксамид
Nα-(трет-бутилоксикарбонил)-Nω-(9-флуоренилметилоксикарбонил)-L-лизин (5 г, 10,7 ммоль) растворяли в CH2Cl2 (100 мл). Этот раствор охлаждали до 0°С, добавляли гидрохлорид (4R)-тиазолидин-4-карбоксамида (1,78 г, 11,7 ммоль), PyBOP® (6,7 г, 12,8 ммоль) и рН доводили до 9 триэтиламином. После 18 ч при от 0° до комнатной температуры растворитель удаляли в вакууме и остаток переносили в этилацетат (200 мл). Раствор промывали 0,3 М KHSO4 (2×50 мл), нас. NaHCO3 (2×50 мл), водой (2×50 мл) и насыщенным раствором соли (1×50 мл), сушили (Na2SO4) и выпаривали в вакууме с получением желтого масла. Остаток очищали флэш-хроматографией (элюент: 2% метанол, 98% хлороформ) с получением бесцветного масла, идентифицированного как (4R)-3-[Nα-(трет-бутилоксикарбонил)-Nω-(9-флуоренилметилоксикарбонил)-L-лизинил]тиазолидин-4-карбоксамид (2,81 г, 4,8 ммоль, 44%).
D. (4R)-3-[Nα-(трет-бутилоксикарбонил)-Nω-(9-флуоренилметилоксикарбонил)-L-лизинил]тиазолидин-4-карбонитрил
(4R)-3-[Nα-(трет-бутилоксикарбонил)-Nω-(9-флуоренилметилоксикарбонил)-L-лизинил]тиазолидин-4-карбоксамид (2,7 г, 4,7 ммоль) растворяли в безводном ТГФ (100 мл). Раствор охлаждали до 0°С, добавляли триэтиламин (1,0 г, 10 ммоль) с последующим медленным добавлением трифторуксусного ангидрида (2,0 г, 9,5 ммоль). Значение рН доводили до 9 триэтиламином. Через 30 мин реакционную смесь разбавляли этилацетатом (100 мл), промывали водой (1×50 мл) и насыщенным раствором соли (1×50 мл), сушили (Na2SO4) и выпаривали в вакууме. Остаток очищали флэш-хроматографией (элюент: 60% пет. эфира, 40% этилацетата) с получением бесцветного масла, идентифицированного как (4R)-3-[Nα-(трет-бутилоксикарбонил)-Nω-(9-флуоренилметилоксикарбонил)-L-лизинил]тиазолидин-4-карбонитрил (2,14 г, 3,81 ммоль, 82%).
Е. (4R)-3-[Nα-(трет-бутилоксикарбонил)-L-лизинил]тиазолидин-4-карбонитрил
(4R)-3-[Nα-(трет-бутилоксикарбонил)-Nω-(9-флуоренилметилоксикарбонил)-L-лизинил]тиазолидин-4-карбонитрил (1,9 г, 3,4 ммоль) растворяли в ТГФ (40 мл). Добавляли диэтиламин (10 мл). После 2 ч при комнатной температуре растворитель удаляли в вакууме. Остаток очищали флэш-хроматографией (элюент: 90% хлороформа, 7% метанола, 3% триэтиламина) с получением бесцветного масла, идентифицированного как (4R)-3-[Nα-(трет-бутилоксикарбонил)-L-лизинил]тиазолидин-4-карбонитрил (863 мг, 2,5 ммоль, 75%).
F. (4R)-3-[Nα-(трет-бутилоксикарбонил)-Nω-(пиразинил-2-карбонил)-L-лизинил]тиазолидин-4-карбонитрил
(4R)-3-[Nα-(трет-бутилоксикарбонил)-L-лизинил]тиазолидин-4-карбонитрил (100 мг, 0,29 ммоль) растворяли в СН2Cl2 (20 мл). К этому раствору при 0°С добавляли 2-пиразинкарбоновую кислоту (43 мг, 0,35 ммоль), PyBOP® (170 мг, 0,33 ммоль) и рН доводили до 9 триэтиламином. После 18 ч при от 0°С до комнатной температуры растворитель удаляли в вакууме и остаток переносили в этилацетат (70 мл). Раствор промывали 0,3 М KHSO4 (2×20 мл), нас. NaHCO3 (2×20 мл), водой (2×20 мл) и насыщенным раствором соли (1×20 мл), сушили (Na2SO4) и выпаривали в вакууме. Остаток очищали флэш-хроматографией (элюент: 2% метанола, 98% хлороформа) с получением бесцветного масла, идентифицированного как (4R)-3-[Nα-(трет-бутилоксикарбонил)-Nω-(пиразинил-2-карбонил)-L-лизинил]тиазолидин-4-карбонитрил (112 мг, 0,25 ммоль, 86%).
G. Трифторацетат (4R)-3-[Nω-(пиразинил-2-карбонил)-L-лизинил]тиазолидин-4-карбонитрила
(4R)-3-[Nα-(трет-бутилоксикарбонил)-Nω-(пиразинил-2-карбонил)-L-лизинил]тиазолидин-4-карбонитрил (110 мг, 0,26 ммоль) растворяли в трифторуксусной кислоте (5 мл). После 1 ч при комнатной температуре растворитель удаляли в вакууме. Остаток очищали препаративной ЖХВД (Vydac C18, 5-50% 0,1% ТФУ/ацетонитрил в 0,1% ТФУ/воде в течение 40 мин при 3 мл/мин). Фракции, содержащие продукт, лиофилизировали с получением бесцветного масла, идентифицированного как трифторацетат (4R)-3-[Nω-(пиразинил-2-карбонил)-L-лизинил]тиазолидин-4-карбонитрила (57 мг).
[М+Н]+=349,1
ПРИМЕР 4
Трифторацетат 1-[Nω-(пиразинил-2-карбонил)-L-орнитинил]пирролидина
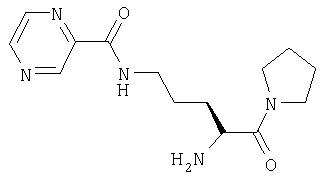
А. 1-[Nω-(бензилоксикарбонил)-Nα-(трет-бутилоксикарбонил)-L-орнитинил]пирролидин
Nω-(бензилоксикарбонил)-Nα-(трет-бутилоксикарбонил)-L-орнитин (5,49 г, 15 ммоль) растворяли в СН2Cl2/ДМФ (9:1, 100 мл). К этому раствору при 0°С добавляли гидрат 1-гидроксибензотриазола (3,37 г, 22 ммоль), водорастворимый карбодиимид (3,46 г, 18 ммоль), пирролидин (1,28 г, 18 ммоль) и триэтиламин (200 мг, 20 ммоль). После 18 ч при от 0°С до комнатной температуры растворитель удаляли в вакууме и остаток переносили в этилацетат (200 мл). Раствор промывали 0,3 М KHSO4 (2×50 мл), нас. NaHCO3 (2×50 мл), водой (2×50 мл) и насыщенным раствором соли (1×50 мл), сушили (Na2SO4) и выпаривали в вакууме. Остаток очищали флэш-хроматографией (элюент: 90% этилацетата, 10% пет. эфира) с получением бесцветного масла, идентифицированного как 1-[Nω-(бензилоксикарбонил)-Nα-(трет-бутилоксикарбонил)-L-орнитинил]пирролидин (5,15 г, 12,3 ммоль, 82%).
В. 1-[Nα-(трет-бутилоксикарбонил)-L-орнитинил]пирролидин
1-[Nω-(бензилоксикарбонил)-Nα-(трет-бутилоксикарбонил)-L-орнитинил]пирролидин (2,15 г, 5,13 ммоль) растворяли в метаноле (80 мл). Этот раствор гидрировали над 10% Pd/C (400 мг). Через 2 ч катализатор отфильтровывали и промывали метанолом (50 мл). Объединенные фильтраты выпаривали в вакууме с получением не совсем белого твердого вещества, идентифицированного как 1-[Nα-(трет-бутилоксикарбонил)-L-орнитинил]пирролидин (1,35 г, 4,74 ммоль, 94%).
С. 1-[Nα-(трет-бутилоксикарбонил)-Nω-(пиразинил-2-карбонил)-L-орнитинил]пирролидин
1-[Nα-(трет-бутилоксикарбонил)-L-орнитинил]пирролидин (100 мг, 0,35 ммоль) растворяли в CH2Cl2 (20 мл). К этому раствору при 0°С добавляли PyBOP® (195 мг, 0,4 ммоль), 2-пиразинкарбоновую кислоту (50 мг, 0,4 ммоль) и триэтиламин (100 мг, 1,0 ммоль). После 18 ч при от 0°С до комнатной температуры растворитель удаляли в вакууме и остаток переносили в этилацетат (70 мл). Раствор промывали 0,3 М KHSO4 (2×20 мл), нас. NaHCO3 (2×20 мл), водой (2×20 мл) и насыщенным раствором соли (1×20 мл), сушили (Na2SO4) и выпаривали в вакууме. Остаток очищали флэш-хроматографией (элюент: 3% метанола, 97% хлороформа) с получением липкого белого твердого вещества, идентифицированного как 1-[Nα-(трет-бутилоксикарбонил)-Nω-(пиразинил-2-карбонил)-L-орнитинил]пирролидин (90 мг, 0,25 ммоль, 66%).
D. Трифторацетат 1-[Nω-(пиразинил-2-карбонил)-L-орнитинил]пирролидина
1-[Nα-(трет-бутилоксикарбонил)-Nω-(пиразинил-2-карбонил)-L-орнитинил]пирролидин (90 мг, 0,23 ммоль) растворяли в 4 М HCl/диоксане (15 мл). После 45 мин при комнатной температуре растворитель удаляли в вакууме. Остаток очищали препаративной ЖХВД (Vydac C18, 5-50% 0,1% ТФУ/ацетонитрил в 0,1% ТФУ/воде в течение 40 мин при 3 мл/мин). Фракции, содержащие продукт, лиофилизировали с получением бесцветного масла, идентифицированного как трифторацетат 1-[Nω-(пиразинил-2-карбонил)-L-орнитинил]пирролидина (51 мг).
[М+Н]+=292,1
ПРИМЕР 5
Трифторацетат 3-[Nω-(пиразинил-2-карбонил)-L-орнитинил]тиазолидина
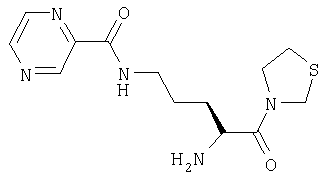
А. 3-[Nα-(трет-бутилоксикарбонил)-Nω-(9-флуоренилметилоксикарбонил)-L-орнитинил]тиазолидин
Nα-(трет-бутилоксикарбонил)-Nω-(9-флуоренилметилоксикарбонил)-L-орнитин (2,73 г, 6 ммоль) растворяли в СН2Cl2/ДМФ (9:1, 100 мл). К этому раствору при 0°С добавляли гидрат 1-гидроксибензотриазола (1,53 г, 10 ммоль), водорастворимый карбодиимид (1,34 г, 7 ммоль), тиазолидин (1,28 г, 18 ммоль) и триэтиламин (80 мг, 20 ммоль). После 18 ч при от 0°С до комнатной температуры растворитель удаляли в вакууме и остаток переносили в этилацетат (100 мл). Раствор промывали 0,3 М KHSO4 (2×25 мл), нас. NaHCO3 (2×25 мл), водой (2×25 мл) и насыщенным раствором соли (1×25 мл), сушили (Na2SO4) и выпаривали в вакууме. Остаток очищали флэш-хроматографией (элюент: 75% этилацетата, 25% пет. эфира) с получением белого твердого вещества, идентифицированного как 3-[Nα-(трет-бутилоксикарбонил)-Nω-(9-флуоренилметилоксикарбонил)-L-орнитин]тиазолидин (2,55 г, 4,85 ммоль, 81%).
В. 3-[Nα-(трет-бутилоксикарбонил)-L-орнитинил]тиазолидин
3-[Nα-(трет-бутилоксикарбонил)-Nω-(9-флуоренилметилоксикарбонил)-L-орнитинил]тиазолидин (1,15 г, 2,13 ммоль) растворяли в ацетонитриле (20 мл). Добавляли диэтиламин (5 мл). После 90 мин при комнатной температуре растворитель удаляли в вакууме и остаток очищали флэш-хроматографией (элюент: 90% хлороформ, 7% метанола, 3% триэтиламина) с получением желтого масла, идентифицированного как 3-[Nα-(трет-бутилоксикарбонил)-L-орнитинил]тиазолидин (530 мг, 1,67 ммоль, 78%).
С. 3-[Nα-(трет-бутилоксикарбонил)-Nω-(пиразинил-2-карбонил)-L-орнитинил]тиазолидин
3-[Nα-(трет-бутилоксикарбонил)-L-орнитинил]тиазолидин (80 мг, 0,27 ммоль) растворяли в CH2Cl2 (20 мл). К этому раствору при 0°С добавляли PyBOP® (146 мг, 0,3 ммоль), 2-пиразинкарбоновую кислоту (37 мг, 0,3 ммоль) и триэтиламин (90 мг, 0,9 ммоль). После 18 ч при от 0°С до комнатной температуры растворитель удаляли в вакууме и остаток переносили в этилацетат (70 мл). Раствор промывали 0,3 М KHSO4 (2×20 мл), нас. NaHCO3 (2×20 мл), водой (2×20 мл) и насыщенным раствором соли (1×20 мл), сушили (Na2SO4) и выпаривали в вакууме. Остаток очищали флэш-хроматографией (элюент: 3% метанол, 97% хлороформ) с получением липкого белого твердого вещества, идентифицированного как 3-[Nα-(трет-бутилоксикарбонил)-Nω-(пиразинил-2-карбонил)-L-орнитинил]тиазолидин (45 мг, 0,11 ммоль, 41%).
D. Трифторацетат 3-[Nω-(пиразинил-2-карбонил)-L-орнитинил]тиазолидина
3-[Nα-(трет-бутилоксикарбонил)-Nω-(пиразинил-2-карбонил)-L-орнитинил]тиазолидин (45 мг, 0,11 ммоль) растворяли в 4 М HCl/диоксане (10 мл). После 45 мин при комнатной температуре растворитель удаляли в вакууме. Остаток очищали препаративной ЖХВД (Vydac С18, 5-50% 0,1% ТФУ/ацетонитрил в 0,1% ТФУ/воде в течение 40 мин при 3 мл/мин). Фракции, содержащие продукт, лиофилизировали с получением бесцветного масла, идентифицированного как трифторацетат 3-[Nω-(пиразинил-2-карбонил)-L-орнитинил]тиазолидина (14 мг).
[М+Н]+=310,0
ПРИМЕР 6
Трифторацетат (2S)-1-[Nω-(2-хлорпиридил-3-карбонил)-L-орнитинил]пирролидин-2-карбонитрила
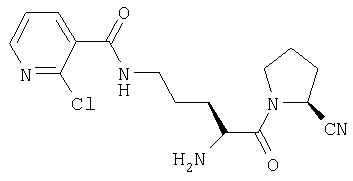
A. (2S)-1-[Nα-(трет-бутилоксикарбонил)-L-орнитинил]пирролидин-2-карбонитрил
(2S)-1-[Nα-(трет-бутилоксикарбонил)-L-орнитинил]пирролидин-2-карбонитрил получали методом, описанным для производного лизина в примере 2.
В. (2S)-1-(Nα-(трет-бутилоксикарбонил)-Nω-(2-хлорпиридил-3-карбонил)-L-орнитинил)пирролидин-2-карбонитрил
(2S)-1-[Nα-(трет-бутилоксикарбонил)-L-орнитинил]пирролидин-2-карбонитрил (80 мг, 0,26 ммоль) растворяли в CH2Cl2 (20 мл). К этому раствору добавляли 2-хлорпиридин-3-карбонилхлорид (55 мг, 0,32 ммоль) и рН доводили до 9 триэтиламином. После 18 часов при комнатной температуре растворитель удаляли в вакууме и остаток переносили в этилацетат (70 мл). Раствор промывали 0,3 М KHSO4 (2×20 мл), нас. NaHCO3 (2×20 мл), водой (2×20 мл) и насыщенным раствором соли (1×20 мл), сушили (Na2SO4) и выпаривали в вакууме. Остаток очищали флэш-хроматографией (элюент: 95% этилацетат, 5% пет. эфир) с получением бесцветного масла, идентифицированного как (2S)-1-(Nα-(трет-бутилоксикарбонил)-Nω-(2-хлорпиридил-3-карбонил)-L-орнитинил)пирролидин-2-карбонитрил (60 мг, 0,14 ммоль, 53%).
С. Трифторацетат (2S)-1-[Nω-(2-хлорпиридил-3-карбонил)-L-орнитинил]пирролидин-2-карбонитрила
(2S)-1-[Nα-(трет-бутилоксикарбонил)-Nω-(2-хлорпиридил-3-карбонил)-L-орнитинил]пирролидин-2-карбонитрил (60 мг, 0,14 ммоль) растворяли в трифторуксусной кислоте (5 мл). После 1 ч при комнатной температуре растворитель удаляли в вакууме. Остаток очищали препаративной ЖХВД (Vydac C18, 5-50% 0,1% ТФУ/ацетонитрил в 0,1% ТФУ/воде в течение 40 мин при 3 мл/мин). Фракции, содержащие продукт, лиофилизировали с получением белого твердого вещества, идентифицированного как трифторацетат (2S)-1-[Nω-(2-хлорпиридил-3-карбонил)-L-орнитинил]пирролидин-2-карбонитрила (52 мг).
[М+Н]+=350,1
ПРИМЕР 7
Гидрохлорид 1-[Nω-(2-хлорпиридил-3-карбонил)-L-орнитинил]пирролидина
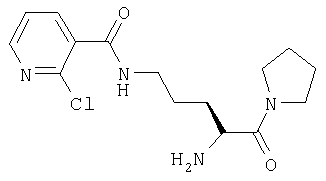
А. 1-(Nα-(трет-бутилоксикарбонил)-Nω-(2-хлорпиридил-3-карбонил)-L-орнитинил]пирролидин
1-(Nα-(трет-бутилоксикарбонил)-L-орнитинил]пирролидин (20 мг, 0,069 ммоль) растворяли в CH2Cl2 (5 мл). К этому раствору добавляли 2-хлорпиридин-3-карбонилхлорид (14 мг, 0,076 ммоль) и рН доводили до 9 триэтиламином. После 1 ч при комнатной температуре растворитель удаляли в вакууме и остаток переносили в этилацетат (70 мл). Раствор промывали 0,3 М KHSO4 (2×20 мл), нас. NaHCO3 (2×20 мл), водой (2×20 мл) и насыщенным раствором соли (1×20 мл), сушили (Na2SO4) и выпаривали в вакууме. Остаток очищали флэш-хроматографией (элюент: 10% метанола, 90% дихлорметана) с получением бесцветного масла, идентифицированного как 1-(Nα-(трет-бутилоксикарбонил)-Nω-(2-хлорпиридил-3-карбонил)-L-орнитинил]пирролидин (19 мг, 0,045 ммоль, 63%).
В. Гидрохлорид 1-[Nω-(2-хлорпиридил-3-карбонил)-L-орнитинил]пирролидина
1-(Nα-(трет-бутилоксикарбонил)-Nω-(2-хлорпиридил-3-карбонил)-L-орнитинил]пирролидин (19 мг, 0,045 ммоль) растворяли в 4 М HCl/диоксане (10 мл). После 45 мин при комнатной температуре растворитель удаляли в вакууме с получением белого твердого вещества, идентифицированного как гидрохлорид 1-[Nω-(2-хлорпиридил-3-карбонил)-L-орнитинил]пирролидина (15 мг).
[М+Н]+=325,1
ПРИМЕР 8
Гидрохлорид 3-[Nω-(2-хлорпиридил-3-карбонил)-L-орнитинил]тиазолидина
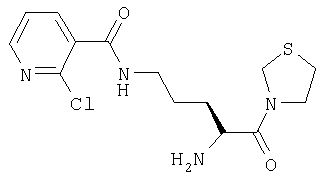
А. 3-(Nα-(трет-бутилоксикарбонил)-Nω-(2-хлорпиридил-3-карбонил)-L-орнитинил)тиазолидин
3-(Nα-(трет-бутилоксикарбонил)-L-орнитинил)тиазолидин (136 мг, 0,45 ммоль) растворяли в СН2Cl2 (10 мл). К этому раствору добавляли 2-хлорпиридин-3-карбонилхлорид (88 мг, 0,5 ммоль) и рН доводили до 9 триэтиламином. После 1 ч при комнатной температуре растворитель удаляли в вакууме и остаток переносили в этилацетат (70 мл). Раствор промывали 0,3 M KHSO4 (2×20 мл), нас. NaHCO3 (2×20 мл), водой (2×20 мл) и насыщенным раствором соли (1×20 мл), сушили (Na2SO4) и выпаривали в вакууме. Остаток очищали флэш-хроматографией (элюент: 1,5% метанола, 98,5% дихлорметана) с получением бесцветного масла, идентифицированного как 3-(Nα-(трет-бутилоксикарбонил)-Nω-(2-хлорпиридил-3-карбонил)-L-орнитинил)тиазолидин (30 мг, 0,068 ммоль, 15%).
В. Гидрохлорид 3-[Nω-(2-хлорпиридил-3-карбонил)-L-орнитинил]тиазолидина
3-(Nα-(трет-бутилоксикарбонил)-Nω-(2-хлорпиридил-3-карбонил)-L-орнитинил)тиазолидин (30 мг, 0,068 ммоль) растворяли в 4 М HCl/диоксане (10 мл). После 45 мин при комнатной температуре растворитель удаляли в вакууме с получением белого твердого вещества, идентифицированного как гидрохлорид 3-[Nω-(2-хлорпиридил-3-карбонил)-L-орнитинил]тиазолидина (25 мг).
[М+Н]+=342,1
ПРИМЕР 9
Гидрохлорид 3-[Nω-(5-циано-2-пиридил)-L-лизинил]тиазолидина
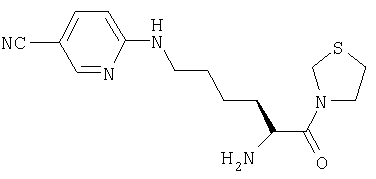
А. 3-(Nα-(трет-бутилоксикарбонил)лизинил)тиазолидин
3-(Nα-(трет-бутилоксикарбонил)лизинил)тиазолидин получали в две стадии способом, описанным в отношении соответствующего производного орнитина в примере 5.
В. 3-(Nα-(трет-бутилоксикарбонил)-Nω-(5-циано-2-пиридил)-L-лизинил)тиазолидин
3-(Nα-(трет-бутилоксикарбонил)лизинил)тиазолидин (52 мг, 0,165 ммоль) растворяли в ДМФ (10 мл). Добавляли 6-хлорникотинонитрил (22,8 мг, 0,165 ммоль) и карбонат калия (45,8 мг, 0,3 ммоль). Реакционную смесь перемешивали при 70°С в течение 18 часов и растворитель удаляли в вакууме. Остаток очищали флэш-хроматографией (элюент: 97% хлороформа, 3% метанола) с получением бесцветного масла, идентифицированного как 3-(Nα-(трет-бутилоксикарбонил)-Nω-(5-циано-2-пиридил)-L-лизинил)тиазолидин (30 мг, 0,067 ммоль, 43%).
С. Гидрохлорид 3-[Nω-(5-циано-2-пиридил)-L-лизинил]тиазолидина
3-(Nα-(трет-бутилоксикарбонил)-Nω-(5-циано-2-пиридил)-L-лизинил)тиазолидин (30 мг, 0,067 ммоль) растворяли в 4 М HCl/диоксане (20 мл). После 1 ч при комнатной температуре растворитель удаляли в вакууме с получением белого твердого вещества, идентифицированного как гидрохлорид 3-[Nω-(5-циано-2-пиридил)-L-лизинил]тиазолидина (24 мг, 0,067 ммоль, 100%).
[М+Н]+=348,2
ПРИМЕР 10
Трифторацетат (2S)-1-[Nω-(5-циано-2-пиридил)-L-лизинил]пирролидин-2-карбонитрила
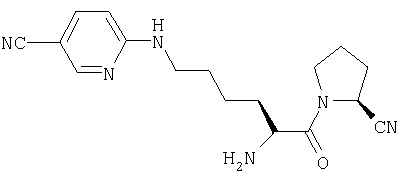
A. (2S)-1-[Nα-(трет-бутилоксикарбонил)-Nω-(5-циано-2-пиридил)-L-лизинил]пирролидин-2-карбонитрил
(2S)-1-(Nα-(трет-бутилоксикарбонил)лизинил)пирролидин-2-карбонитрил (150 мг, 0,46 ммоль) растворяли в ДМФ (10 мл). Добавляли 6-хлорникотинонитрил (70 мг, 0,51 ммоль) и карбонат калия (130 мг, 0,94 ммоль). Реакционную смесь перемешивали при 70°С в течение 18 часов и растворитель удаляли в вакууме. Остаток очищали флэш-хроматографией (элюент: 97% хлороформа, 3% метанола) с получением бесцветного масла, идентифицированного как (2S)-1-[Nα-(трет-бутилоксикарбонил)-Nω-(5-циано-2-пиридил)-L-лизинил]пирролидин-2-карбонитрил (71 мг, 0,17 ммоль, 37%).
В. Трифторацетат (2S)-1-[Nω-(5-циано-2-пиридил)-L-лизинил]пирролидин-2-карбонитрила
(2S)-1-[Nα-(трет-бутилоксикарбонил)-Nω-(5-циано-2-пиридил)-L-лизинил]пирролидин-2-карбонитрил (71 мг, 0,17 ммоль) растворяли в 4 М HCl/диоксане (20 мл). После 1 ч при комнатной температуре растворитель удаляли в вакууме с получением белого твердого вещества, идентифицированного как гидрохлорид (2S)-1-[Nω-(5-циано-2-пиридил)-L-лизинил]пирролидин-2-карбонитрила (62 мг, 0,17 ммоль, 100%).
[М+Н]+=327,1
ПРИМЕР 11
Трифторацетат (2S)-1-[Nω-(5-трифторметил-2-пиридил)-L-орнитинил]пирролидин-2-карбонитрила
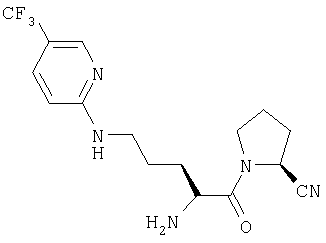
А. (2S)-1-[Nα-(трет-бутилоксикарбонил)-Nω-(5-трифторметил-2-пиридил)-L-орнитинил]пирролидин-2-карбонитрил
(2S)-1-[Nα-(трет-бутилоксикарбонил)орнитинил]пирролидин-2-карбонитрил (140 мг, 0,45 ммоль) растворяли в ДМФ (10 мл). Добавляли 2-хлор-5-(трифторметил)пиридин (90 мг, 0,49 ммоль) и карбонат калия (130 мг, 0,92 ммоль). Реакционную смесь перемешивали при 70°С в течение 18 часов и растворитель удаляли в вакууме. Остаток очищали флэш-хроматографией (элюент: 97% хлороформа, 3% метанола) с получением бесцветного масла, идентифицированного как (2S)-1-[Nα-(трет-бутилоксикарбонил)-Nω-(5-трифторметил-2-пиридил)-L-орнитинил]пирролидин-2-карбонитрил (58 мг, 0,13 ммоль, 28%).
В. Трифторацетат (2S)-1-[Nω-(5-трифторметил-2-пиридил)-L-орнитинил]пирролидин-2-карбонитрила
(2S)-1-[Nα-(трет-бутилоксикарбонил)-Nω-(5-трифторметил-2-пиридил)-L-орнитинил]пирролидин-2-карбонитрил (58 мг, 13 ммоль) растворяли в 4 М HCl/диоксане (20 мл). После 1 ч при комнатной температуре растворитель удаляли в вакууме с получением твердого белого вещества, идентифицированного как гидрохлорид (2S)-1-[Nω-(5-трифторметил-2-пиридил)-L-орнитинил]пирролидин-2-карбонитрила (51 мг, 0,13 ммоль, 100%).
[М+Н]+=356,2
ПРИМЕР 12
Гидрохлорид 3-[Nω-(2-хинолинилметил)-L-лизинил]тиазолидина
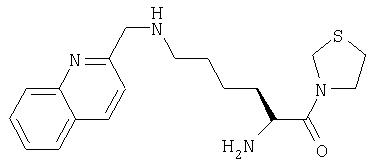
А. 3-[Nα-(трет-бутилоксикарбонил)-Nω-(2-хинолинилметил)-L-лизинил]тиазолидин
3-[Nα-(трет-бутилоксикарбонил)лизинил]тиазолидин (100 мг, 0,32 ммоль) растворяли в метаноле (10 мл). Добавляли 2-хинолинкарбоксальдегид (61 мг, 0,39 ммоль). Через 1 час добавляли ацетоксиборогидрид натрия (138 мг, 0,65 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 18 часов и растворитель удаляли в вакууме. Остаток очищали флэш-хроматографией (элюент: 93% хлороформа, 7% метанола) с получением бесцветного масла, идентифицированного как 3-[Nα-(трет-бутилоксикарбонил)-Nω-(2-хинолинилметил)-L-лизинил]тиазолидин (38 мг, 0,083 ммоль, 26%).
В. Гидрохлорид 3-[Nω-(2-хинолинилметил)-L-лизинил]тиазолидина
3-[Nα-(трет-бутилоксикарбонил)-Nω-(2-хинолинилметил)-L-лизинил]тиазолидин (38 мг, 0,083 ммоль) растворяли в 4 М HCl/диоксане (20 мл). После 1 ч при комнатной температуре растворитель удаляли в вакууме с получением твердого белого вещества, идентифицированного как гидрохлорид 3-[Nω-(2-хинолинилметил)-L-лизинил]тиазолидина (31 мг, 0,078 ммоль, 94%).
[М+Н]+=358,2
ПРИМЕР 13
Гидрохлорид 3-[Nω-(2-хинолинилметил)-L-орнитинил]тиазолидина
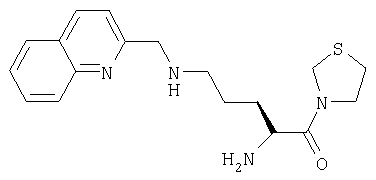
А. 3-[Nα-(трет-бутилоксикарбонил)-Nω-(2-хинолинилметил)-L-орнитинил]тиазолидин
3-[Nα-(трет-бутилоксикарбонил)орнитинил]тиазолидин (98 мг, 0,33 ммоль) растворяли в метаноле (10 мл). Добавляли 2-хинолинкарбоксальдегид (52 мг, 0,33 ммоль). Через 1 час добавляли ацетоксиборогидрид натрия (119 мг, 0,56 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 18 часов и растворитель удаляли в вакууме. Остаток очищали флэш-хроматографией (элюент: 93% хлороформа, 7% метанола) с получением бесцветного масла, идентифицированного как 3-[Nα-(трет-бутилоксикарбонил)-Nω-(2-хинолинилметил)-L-орнитинил]тиазолидин (45 мг, 0,10 ммоль, 36%).
В. Гидрохлорид 3-[Nω-(2-хинолинилметил)-L-орнитинил]тиазолидина
3-[Nα-(трет-бутилоксикарбонил)-Nω-(2-хинолинилметил)-L-орнитинил]тиазолидин (45 мг, 0,1 ммоль) растворяли в 4 М HCl/диоксане (20 мл). После 1 ч при комнатной температуре растворитель удаляли в вакууме с получением твердого белого вещества, идентифицированного как гидрохлорид 3-[Nω-(2-хинолинилметил)-L-орнитинил]тиазолидина (38 мг, 0,098 ммоль, 98%).
[М+Н]+=345,2
ПРИМЕР 14
Гидрохлорид 3-[Nω-(2-хиноксалоил)-L-лизинил]тиазолидина
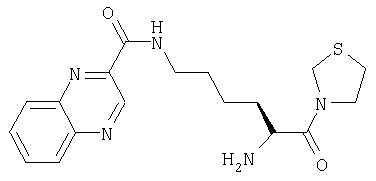
А. 3-[Nα-(трет-бутилоксикарбонил)-Nω-(2-хиноксалоил)-L-лизинил]тиазолидин
3-[Nα-(трет-бутилоксикарбонил)лизинил]тиазолидин (128 мг, 0,4 ммоль) растворяли в СН2Cl2 (10 мл). Добавляли 2-хиноксалоилхлорид (85 мг, 0,44 ммоль) и карбонат калия (45,8 мг, 0,3 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 18 часов и растворитель удаляли в вакууме. Остаток очищали флэш-хроматографией (элюент: 99,5% хлороформа, 0,5% метанола) с получением бесцветного масла, идентифицированного как 3-[Nα-(трет-бутилоксикарбонил)-Nω-(2-хиноксалоил)-L-лизинил]тиазолидин (140 мг, 0,296 ммоль, 74%).
В. Гидрохлорид 3-[Nω-(2-хиноксалоил)-L-лизинил]тиазолидина
3-[Nα-(трет-бутилоксикарбонил)-Nω-(2-хиноксалоил)-L-лизинил]тиазолидин (140 мг, 0,296 ммоль) растворяли в 4 М HCl/диоксане (20 мл). После 1 ч при комнатной температуре растворитель удаляли в вакууме с получением твердого белого вещества, идентифицированного как гидрохлорид 3-[Nω-(2-хиноксалоил)-L-лизинил]тиазолидина (128 мг, 0,296 ммоль, 100%).
[М+Н]+=374,2
Примеры, представленные в следующих таблицах, были получены способами, аналогичными представленным выше.
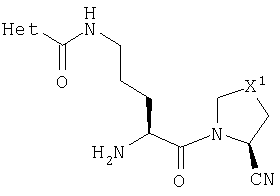
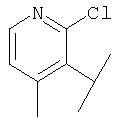
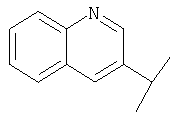
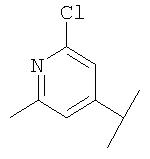
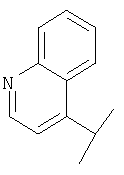
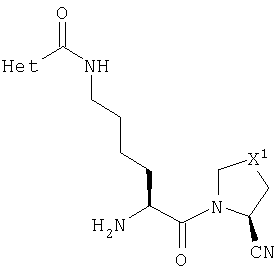
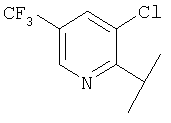
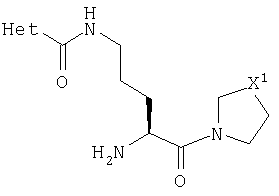
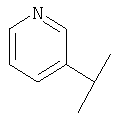
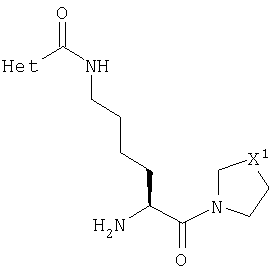
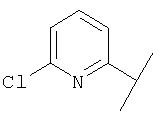
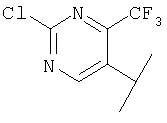
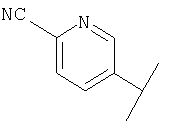
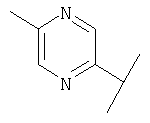
В Таблице 6 приведены значения молекулярных масс соединений, синтезированных в соответствии с изобретением, определенные масс-спектрометрически.
ПРИМЕР 121
Определение активности in vitro
Соединения испытывали как ингибиторы ДП-IV методами, описанными в WO 95/15309. Все соединения, описанные в предшествующих примерах, были конкурентными ингибиторами ДП-IV со значениями Ki менее 300 нМ.
ПРИМЕР 122
Определение активности in vivo
Антидиабетическое действие избранных соединений было продемонстрировано на крысах Цукера, страдающих ожирением, с использованием стандартного теста толерантности к глюкозе при пероральном введении. Контрольным крысам давали раствор глюкозы путем перорального вливания через зонд и определяли уровни глюкозы в плазме. У этих крыс выявлена значительная гипергликемия. Соединения по данному изобретению растворяли в растворе глюкозы в различных концентрациях так, чтобы крысам можно было бы дать различные дозы соединения вместе с глюкозной нагрузкой. Гипергликемическое отклонение снижалось зависящим от дозы образом у животных, получивших дозу между 0,1 и 100 мг/кг ингибитора ДП-IV.
ПРИМЕР 123
Фармацевтический препарат
Таблетки, содержащие 100 мг соединения из примера 1 в качестве активного вещества, изготавливали из следующего:
Данные материалы перемешивают и затем прессуют с получением 2000 таблеток по 250 мг, причем каждая содержит 100 мг соединения из примера 1.
Вышеизложенное демонстрирует, что соединения по данному изобретению являются ингибиторами ДП-IV in vitro и эффективными антигипергликемическими средствами in vivo. Соответственно, можно ожидать, что они будут пригодны в качестве терапевтических средств для лечения сниженной толерантности к глюкозе, диабета типа II и других заболеваний, где подавление этого фермента приводит к улучшению по лежащей в основе патологии или симптомам.
Данное изобретение далее определяется следующей формулой изобретения.
| название | год | авторы | номер документа |
|---|---|---|---|
| ИНГИБИТОРЫ ДИПЕПТИДИЛПЕПТИДАЗЫ IV | 2001 |
|
RU2283303C2 |
| НОВЫЕ АНТИДИАБЕТИЧЕСКИЕ АГЕНТЫ | 2000 |
|
RU2265012C2 |
| ЗАМЕЩЕННЫЕ ГЕТЕРОЦИКЛИЧЕСКИЕ СУЛЬФОНАМИДНЫЕ СОЕДИНЕНИЯ, ПОЛЕЗНЫЕ В КАЧЕСТВЕ МОДУЛЯТОРОВ TRPA 1 | 2014 |
|
RU2675792C2 |
| ПЕПТИД, ОБЛАДАЮЩИЙ БИОЦИДНОЙ АКТИВНОСТЬЮ | 2000 |
|
RU2183643C1 |
| Спироконденсированные пирролидиновые производные в качестве ингибиторов деубиквитилирующих ферментов (DUB) | 2017 |
|
RU2730552C2 |
| ПРОИЗВОДНЫЕ ПИРРОЛИДИНА ДЛЯ ПРИМЕНЕНИЯ В КАЧЕСТВЕ ИНГИБИТОРОВ КАТЕПСИНА | 2011 |
|
RU2548684C2 |
| ТРИЦИКЛИЧЕСКИЕ АЗОТСОДЕРЖАЩИЕ ПРОИЗВОДНЫЕ ИМИДАЗО[4,5-с]ПИРИДИНА, ОБЛАДАЮЩИЕ ИНГИБИРУЮЩЕЙ АКТИВНОСТЬЮ В ОТНОШЕНИИ РЕЦЕПТОРА ГИСТАМИНА 4 (hH4R) | 2012 |
|
RU2628074C2 |
| ЗАМЕЩЕННЫЕ ПИРИДОПИРАЗИНЫ КАК НОВЫЕ ИНГИБИТОРЫ Syk | 2012 |
|
RU2569635C9 |
| АНАЛОГ ЭНКЕФАЛИНА, МОДУЛИРУЮЩИЙ ФУНКЦИИ МОЗГА, ОСЛАБЛЯЮЩИЙ СУДОРОЖНУЮ РЕАКЦИЮ, УСКОРЯЮЩИЙ КОНСОЛИДАЦИЮ И ВОСПРОИЗВЕДЕНИЕ ИНФОРМАЦИИ В СТРЕССОВЫХ УСЛОВИЯХ | 1991 |
|
RU1818826C |
| ЗАМЕЩЕННЫЕ БЕНЗИМИДАЗОЛЫ И ЛЕКАРСТВЕННОЕ СРЕДСТВО НА ИХ ОСНОВЕ | 2000 |
|
RU2261248C2 |
Настоящее изобретение относится к серии новых соединений, которые являются ингибиторами фермента дипептидилпептидазы IV. Предложено соединение, выбранное из производных формулы 1, его таутомеров и стереомеров и фармацевтически приемлемых солей указанных производных, таутомеров и изомеров
где R1 представляет Н или CN, X1 представляет S, О, SO2 или СН2, Х2 представляет СО, СН2 или ковалентную связь, Het представляет азотосодержащий гетероцикл и n равно 1-5. Предложены фармацевтическая композиция, обладающая свойствами ингибитора ДП-IV, а также способ лечения, по меньшей мере, одного из: диабета типа 2, сниженной толерантности к глюкозе, дефицита гормона роста, синдрома поликистоза яичников и аутоиммунных и воспалительных заболеваний. Достигнутый технический результат заключается в получении серии ингибиторов ДП-IV с улучшенным сродством к ферменту, к изготовлению фармацевтических композиций и использовании их для лечения ряда человеческих заболеваний, включая сниженную толерантность к глюкозе и диабет типа II. 3 н. и 11 з.п. ф-лы, 6 табл.
где R1 представляет атом водорода или нитрильную группу;
Х1 выбран из атома серы и метиленовой группы;
Х2 представляет карбонильную группу, метиленовую группу или ковалентную связь;
Het представляет ароматический азотосодержащий гетероцикл, выбранный из пиридила, пиридазинила, пиразинила, тиазолила, оксазолила и его бензоконденсированных аналогов, все из которых могут необязательно быть замещенными по одному или более из атомов углерода, и где заместители выбраны из C1-С6-алкильной, гидрокси, C1-С6-алкилокси, хлора, брома, трифторметильной, циано, карбокси и C1-С6-алкилоксикарбонильной групп, и
n равно 1-5.
(2S)-1-[Nω-(пиразинил-2-карбонил)-L-орнитинил]пирролидин-2-карбонитрила,
(2S)-1-[Nω-(пиразинил-2-карбонил)-L-лизинил]пирролидин-2-карбонитрила,
(2S)-1-[(2'S)-2'-амино-4'-(пиразинил-2''-карбониламино)бутаноил]пирролидин-2-карбонитрила,
(4R)-3-[Nω-(пиразинил-2-карбонил)-L-лизинил]тиазолидин-4-карбонитрила,
1-[Nω-(пиразинил-2-карбонил)-L-орнитинил]пирролидина,
3-[Nω-(2-хлорпиридил-3-карбонил)-L-орнитинил]тиазолидина,
1-[Nω-(2-хлорпиридил-3-карбонил)-L-орнитинил]пирролидина,
(2S)-1-[Nω-(2-хлорпиридил-3-карбонил)-L-орнитинил]пирролидин-2-карбонитрила,
3-[Nω-(пиразинил-2-карбонил)-L-орнитинил]тиазолидина,
3-[Nω-(5-циано-2-пиридил)-L-лизинил]тиазолидина,
(2S)-1-[Nω-(5-циано-2-пиридил)-L-лизинил]пирролидин-2-карбонитрила,
(2S)-1-[Nω-(5-трифторметил-2-пиридил)-L-орнитинил]пирролидин-2-карбонитрила,
3-[Nω-(2-хинолинилметил)-L-лизинил]тиазолидина,
3-[Nω-(2-хинолинилметил)-L-орнитинил]тиазолидина,
3-[Nω-(2-хиноксалоил)-L-лизинил]тиазолидина,
3-[Nω-(2-хиноксалоил)-L-орнитинил]тиазолидина,
(2S)-1-[Nω-(2-хиноксалоил)-L-орнитинил]пирролидин-2-карбонитрила,
3-[Nω-(6-метилпиразинил-2-карбонил)-L-орнитинил]тиазолидина,
3-[Nω-(изохинолин-3-карбонил)-L-орнитинил]тиазолидина и
3-[Nω-(6-трифторметилникотиноил)-L-орнитинил]тиазолидина.
| СЛИТОЙ БЕЛОК И СПОСОБ ВЫДЕЛЕНИЯ СЛИТОГО БЕЛКА | 1993 |
|
RU2114119C1 |
| ПРОИЗВОДНЫЕ ПЕПТИДОВ - АНАЛОГИ GRF ИЛИ ИХ НЕТОКСИЧНЫЕ СОЛИ | 1990 |
|
RU2096416C1 |
| WO 9818763 A1, 07.05.1998 | |||
| WO 9819998 A2, 14.05.1998 | |||
| US 6011155 А, 04.01.2000. | |||

