Предпосылки изобретения
Данное изобретение относится к некоторым антраниламидам, их N-оксидам, сельскохозяйственно-приемлемым солям и композициям и способам их применения для борьбы с беспозвоночными вредителями, такими как членистоногие (Arthropoda), как в сельском хозяйстве, так и в несельскохозяйственных условиях.
Борьба с беспозвоночными вредителями, такими как членистоногие, является чрезвычайно важной для достижения высокой эффективности возделывания сельскохозяйственных культур. Повреждение беспозвоночными вредителями растущих и хранящихся сельскохозяйственных культур может вызывать значительное снижение продуктивности и тем самым приводить к повышению стоимости для потребителя. Важной является также борьба с беспозвоночными вредителями в лесном хозяйстве, в возделывании тепличных культур, декоративных культур, в выращивании саженцев, в хранении пищевых и волокнистых продуктов, а также вредителями скота, домашнего хозяйства и для общественного здравоохранения и охраны здоровья животных. Многие продукты являются коммерчески доступными для этих целей, но сохраняется потребность в новых соединениях, которые являются более эффективными, менее дорогостоящими, менее токсичными, более безопасными для окружающей среды или имеющими отличающиеся механизмы действия.
В NL 9202078 описаны производные N-ацилантраниловой кислоты формулы i в качестве инсектицидов
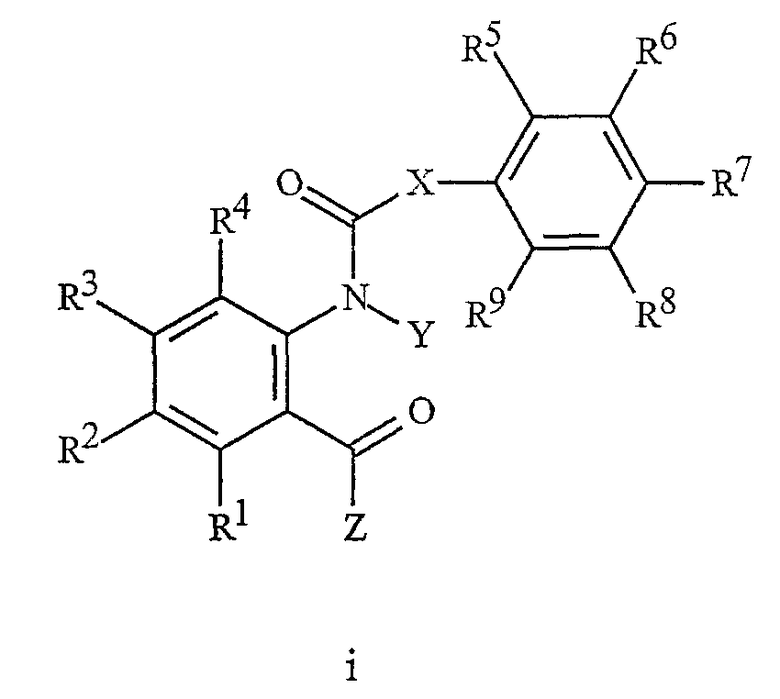
где
Х обозначает прямую связь;
Y обозначает Н или С1-С6алкил;
Z обозначает NH2, NH(С1-С3алкил) или N(С1-С3алкил)2; и
R1-R9 независимо обозначают Н, галоген, С1-С6алкил, фенил, гидрокси, С1-С6алкокси или С1-С7ацилокси.
Сущность изобретения
Данное изобретение относится к соединению формулы 1, его N-оксиду или сельскохозяйственно приемлемой соли этого соединения
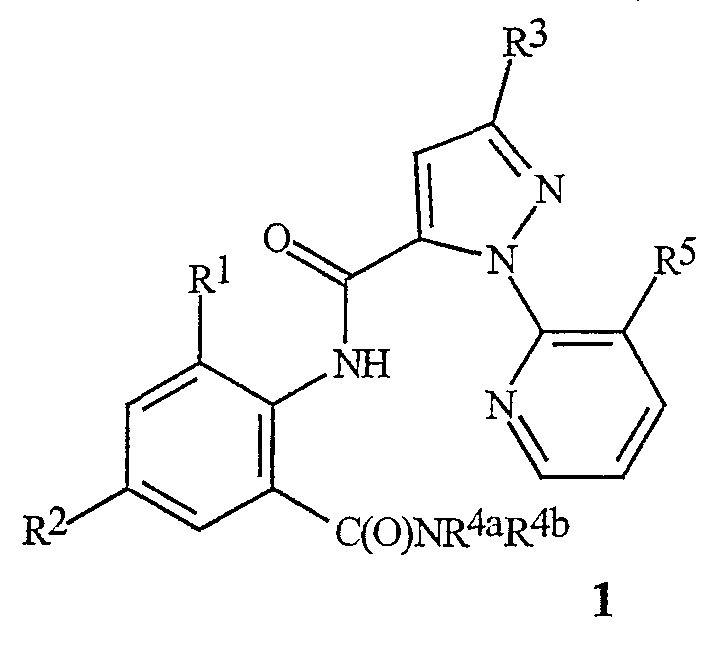
где
R1 обозначает СН3, F, Cl или Br;
R2 обозначает F, Cl, Br, I или CF3;
R3 обозначает CF3, Cl, Br или OCH2CF3;
R4a обозначает С1-С4алкил;
R4b обозначает Н или СН3 и
R5 обозначает Cl или Br.
Данное изобретение относится также к композиции для борьбы с беспозвоночными вредителями, содержащей биологически эффективное количество соединения формулы 1 и по меньшей мере один дополнительный компонент, выбранный из группы, состоящей из поверхностно-активных веществ, твердых разбавителей и жидких разбавителей. Данное изобретение относится также к композиции, содержащей биологически эффективное количество соединения формулы 1 и эффективное количество по меньшей мере одного дополнительного биологически активного компонента или агента.
Данное изобретение относится также к способу борьбы с беспозвоночными вредителями, предусматривающему контактирование беспозвоночного вредителя или окружающей его среды с биологически эффективным количеством соединения формулы 1 (например, в виде описанной здесь композиции). Данное изобретение относится также к такому способу, в котором беспозвоночного вредителя или окружающую его среду приводят в контакт с биологически эффективным количеством соединения формулы 1 или композиции, содержащей соединение формулы 1 и биологически эффективное количество по меньшей мере одного дополнительного соединения или агента для уничтожения беспозвоночных вредителей.
Данное изобретение относится также к соединению бензоксазинона формулы 2
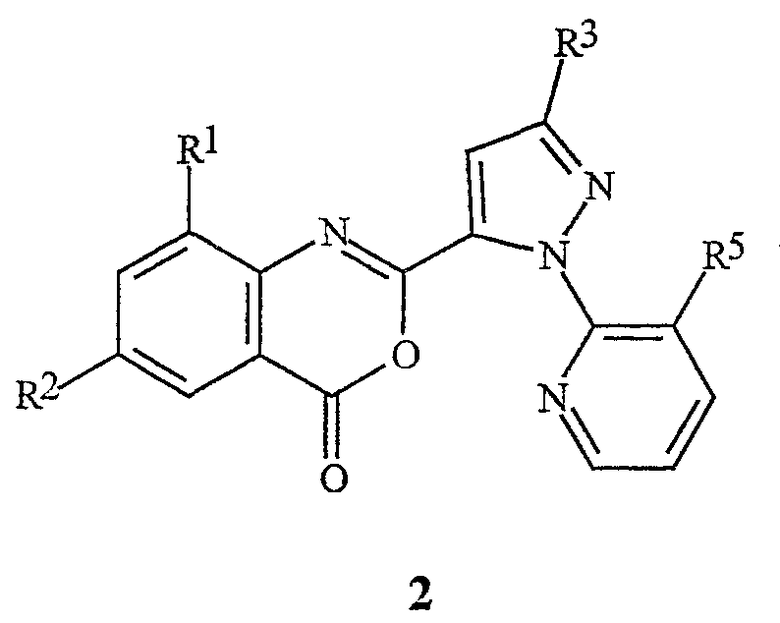
где
R1 обозначает СН3, F, Cl или Br;
R2 обозначает F, Cl, Br, I или CF3;
R3 обозначает CF3, Cl, Br или OCH2CF3;
R5 обозначает Cl или Br;
которое применимо в качестве промежуточного продукта в синтезе для получения соединения формулы 1.
Данное изобретение относится также к соединению пиразолкарбоновой кислоты формулы 4
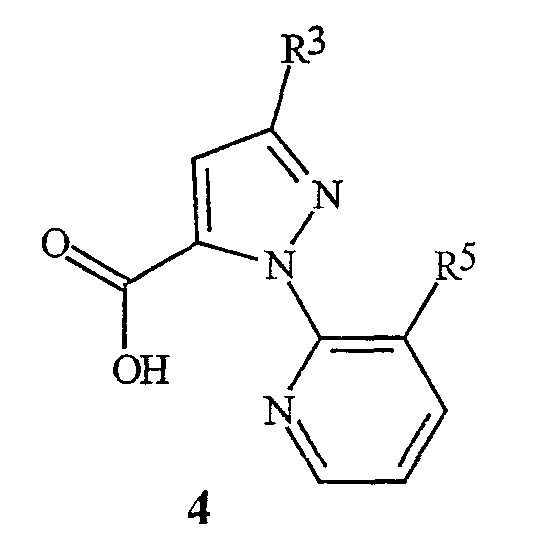
где
R3 обозначает CF3, Cl, Br или OCH2CF3; и
R5 обозначает Cl или Br;
которое применимо в качестве промежуточного продукта в синтезе для получения соединения формулы 1.
Подробное описание изобретения
В приведенных выше перечислениях термин «алкил», используемый отдельно или в составных словах, таких как «алкилтио» или «галогеналкил», обозначает имеющий прямую цепь или разветвленный алкил, такой как метил, этил, н-пропил, изопропил или различные изомеры бутила. Специалисту в данной области будет понятно, что не все содержащие азот гетероциклы могут образовывать N-оксиды, так как на азоте должна быть доступной неподеленная пара электронов для окисления до оксида; специалисту в данной области будет понятно, какие содержащие азот гетероциклы могут образовывать N-оксиды. Специалисту в данной области будет также понятно, что третичные амины могут образовывать N-оксиды. Синтетические способы для получения N-оксидов гетероциклов и третичных аминов очень хорошо известны специалисту в данной области, в том числе окисление гетероциклов и третичных аминов пероксикислотами, такими как перуксусная и м-хлорпербензойная кислота (MCPBA), пероксид водорода, алкилгидропероксиды, такие как трет-бутилгидропероксид, перборат натрия и диоксираны, такие как диметилдиоксиран. Эти способы получения N-оксидов были подробно описаны и рассмотрены в литературе, см., например: T.L. Gilchrist in Comprehensive Organic Synthesis, vol. 7, pp 748-750, S.V. Ley, Ed., Pergamon Press; M. Tisler and B. Stanovnik in Comprehensive Heterocyclic Chemistry, vol. 3, pp 18-20, A.J. Boulton and A. McKillop, Eds., Pergamon Press; M.R. Grimmett and B.R.T. Keene in Advances in Heterocyclic Chemistry, vol. 43, pp 149-161, A.R. Katritzky, Ed., Academic Press; M. Tisler and B. Stanovnik in Advances in Heterocyclic Chemistry, vol. 9, pp 285-291, A.R. Katritzky and A.J. Boulton, Eds., Academic Press; and G.W.H. Cheeseman and E.S.G. Werstiuk in Advances in Heterocyclic Cheniistly, vol. 22, pp 390-392, A.R. Katritzky and A.J. Boulton, Eds., Academic Press.
Соединения данного изобретения могут существовать в виде одного или нескольких стереоизомеров. Различные стереоизомеры включают в себя энантиомеры, диастереомеры, атропизомеры и геометрические изомеры. Специалисту в данной области будет понятно, что один стереомер может быть более активным и/или может проявить лучшее действие при обогащении им смеси относительно другого стереоизомера (других стереоизомеров) или при отделении от другого стереоизомера (других стереоизомеров). Кроме того, специалисту в данной области известно, как отделять, обогащать и/или селективно получать указанные стереоизомеры. Таким образом, данное изобретение включает в себя соединения, выбранные из соединений формулы 1, их N-оксидов и сельскохозяйственно приемлемых солей. Соединения данного изобретения могут находиться в виде смеси стереоизомеров, в виде индивидуальных стереоизомеров или в виде оптически активной формы.
Соли соединений данного изобретения включают в себя кислотно-аддитивные соли (соли присоединения) с неорганическими или органическими кислотами, такими как бромистоводородная, хлористоводородная, азотная, фосфорная, серная, уксусная, масляная, фумаровая, молочная, малеиновая, малоновая, щавелевая, пропионовая, салициловая, винная, 4-толуолсульфоновая или валериановая кислоты.
Предпочтительными соединениями по стоимости, легкости синтеза и/или биологической эффективности являются:
Предпочтительные соединения 1: соединения формулы 1, где R4а обозначает С1-С4алкил, а R4b обозначает Н; или R4а обозначает СН3 и R4b обозначает СН3.
Предпочтительные соединения 2: предпочтительные соединения 1, где R5 обозначает Cl.
Предпочтительные соединения 3: предпочтительные соединения 2, где R4b обозначает СН3, СН2СН3, СН(СН3)2 или С(СН3)3.
Предпочтительные соединения 4: предпочтительные соединения 3, где R2 обозначает Cl или Br.
Предпочтительные соединения 5: предпочтительные соединения 4, где R1 обозначает СН3.
Предпочтительные соединения 6: предпочтительные соединения 4, где R1 обозначает Cl.
Предпочтительные соединения 7: соединения формулы 1, где R1 обозначает СН3, Cl или Br; R2 обозначает F, Cl, Br, I или CF3; R3 обозначает СН3, Cl или Br; R4а обозначает С1-С4алкил; R4b обозначает Н и R5 обозначает Cl или Br.
Особенно предпочтительным является соединение формулы 1, выбранное из группы, состоящей из:
соединения формулы 1, где R1 обозначает СН3, R2 обозначает Br, R3 обозначает CF3, R4а обозначает CH(CH3)2, R4b обозначает Н и R5 обозначает Cl;
соединения формулы 1, где R1 обозначает СН3, R2 обозначает Br, R3 обозначает CF3, R4а обозначает CH3, R4b обозначает Н и R5 обозначает Cl;
соединения формулы 1, где R1 обозначает СН3, R2 обозначает Br, R3 обозначает Br, R4а обозначает CH(СН3)2, R4b обозначает Н и R5 обозначает Cl;
соединения формулы 1, где R1 обозначает СН3, R2 обозначает Br, R3 обозначает Br, R4а обозначает СН3, R4b обозначает Н и R5 обозначает Cl;
соединения формулы 1, где R1 обозначает СН3, R2 обозначает Br, R3 обозначает Cl, R4а обозначает CH(СН3)2, R4b обозначает Н и R5 обозначает Cl;
соединения формулы 1, где R1 обозначает СН3, R2 обозначает Br, R3 обозначает Cl, R4а обозначает СН3, R4b обозначает Н и R5 обозначает Cl;
соединения формулы 1, где R1 обозначает СН3, R2 обозначает Cl, R3 обозначает СН3, R4а обозначает CH(СН3)2, R4b обозначает Н и R5 обозначает Cl;
соединения формулы 1, где R1 обозначает СН3, R2 обозначает Cl, R3 обозначает СН3, R4а обозначает СН3, R4b обозначает Н и R5 обозначает Cl;
соединения формулы 1, где R1 обозначает СН3, R2 обозначает Cl, R3 обозначает Br, R4а обозначает CH(СН3)2, R4b обозначает Н и R5 обозначает Cl;
соединения формулы 1, где R1 обозначает СН3, R2 обозначает Cl, R3 обозначает Br, R4а обозначает СН3, R4b обозначает Н и R5 обозначает Cl;
соединения формулы 1, где R1 обозначает СН3, R2 обозначает Cl, R3 обозначает Cl, R4а обозначает CH(СН3)2, R4b обозначает Н и R5 обозначает Cl;
соединения формулы 1, где R1 обозначает СН3, R2 обозначает Cl, R3 обозначает Cl, R4а обозначает СН3, R4b обозначает Н и R5 обозначает Cl;
соединения формулы 1, где R1 обозначает СН3, R2 обозначает Cl, R3 обозначает OCH2CF3, R4а обозначает CH(СН3)2, R4b обозначает Н и R5 обозначает Cl;
соединения формулы 1, где R1 обозначает СН3, R2 обозначает Cl, R3 обозначает OCH2CF3, R4а обозначает СН3, R4b обозначает Н и R5 обозначает Cl;
соединения формулы 1, где R1 обозначает Cl, R2 обозначает Cl, R3 обозначает Br, R4а обозначает СН3, R4b обозначает Н и R5 обозначает Cl;
соединения формулы 1, где R1 обозначает СН3, R2 обозначает Cl, R3 обозначает OCH2CF3, R4а обозначает СН3, R4b обозначает Н и R5 обозначает Cl.
Предпочтительные композиции данного изобретения являются композициями, которые содержат приведенные выше предпочтительные соединения. Предпочтительные способы применения являются способами с использованием приведенных выше предпочтительных соединений.
Предпочтительными являются соединения формул 1, 2 и 4, в которых R1 обозначает СН3, Cl или Br; R2 обозначает F, Cl, Br, I или СН3; R3 обозначает СН3, Cl или Br; R4а обозначает С1-С4алкил; R4b обозначает Н и R5 обозначает Cl или Br.
Соединения формулы 1 могут быть получены с использованием одного или нескольких из следующих способов и вариантов, показанных на схемах 1-11. Определения R1, R2, R3, R4а, R4b и R5 в соединениях формул 1-24 ниже являются такими же, какие определены выше в разделе Сущность изобретения, если нет других указаний.
Соединения формулы 1 могут быть получены реакцией бензоксазинонов формулы 2 с С1-С4алкиламинами, как представлено в общем виде на схеме 1.
Схема 1
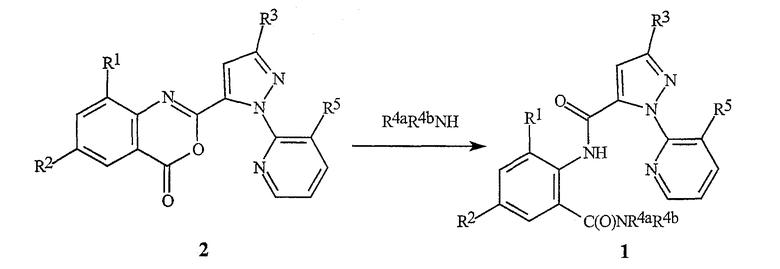
Эта реакция может протекать в неразбавленном виде или в различных подходящих растворителях, в том числе тетрагидрофуране, диэтиловом эфире, дихлорметане или хлороформе, с оптимальными температурами в диапазоне от комнатной температуры до температуры дефлегмации (кипения с обратным холодильником) растворителя. Общая реакция бензоксазинонов с аминами для получения антраниламидов хорошо описана в химической литературе. В отношении обзора см. Jakobsen et al., Bioorganic and Medicinal Chemistry 2000, 8, 2095-2103 и цитируемые в этом обзоре ссылки. См. также G.M. Coppola, J. Heterocyclic Chemistry 1999, 36, 563-588.
Бензоксазиноны формулы 2 могут быть получены различными способами. Два способа, которые являются особенно применимыми, подробно описаны на схемах 2-3. На схеме 2 бензоксазинон формулы 2 получают непосредственно связыванием пиразолкарбоновой кислоты формулы 4 с антраниловой кислотой формулы 3.
Схема 2
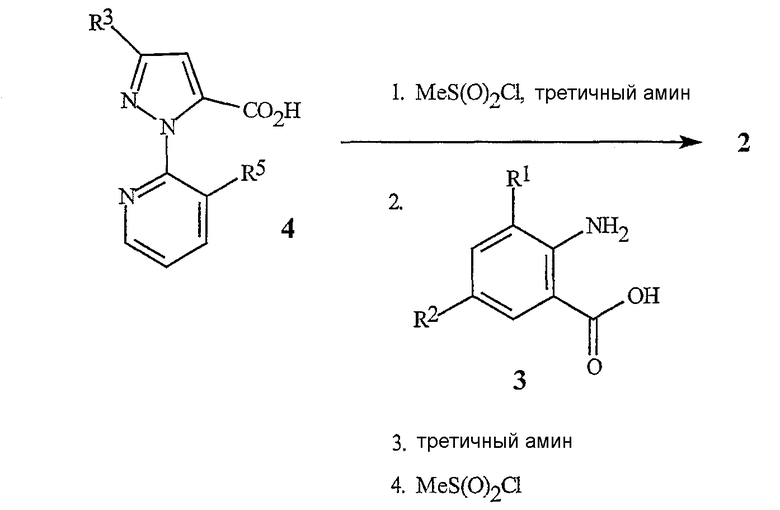
Она включает в себя последовательное добавление метансульфонилхлорида в присутствии третичного амина, такого как триэтиламин или пиридин, к пиразолкарбоновой кислоте формулы 4 с последующим добавлением антраниловой кислоты формулы 3, с последующим вторым добавлением третичного амина и метансульфонилхлорида. Этот способ обычно дает хорошие выходы бензоксазинона и иллюстрируется более подробно в примере 1.
Схема 3 отображает альтернативное получение бензоксазинонов формулы 2 с использованием связывания хлорангидрида кислоты формулы 6 с ангидридом изатиновой кислоты формулы 5 для получения непосредственно бензоксазинона формулы 2.
Схема 3
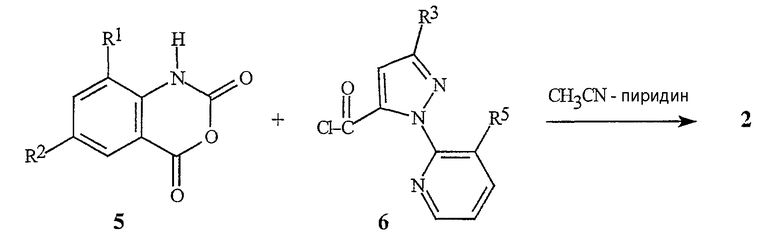
Для этой реакции применимы такие растворители, как пиридин или смесь пиридин/ацетонитрил. Хлорангидриды кислот формулы 6 могут быть получены из соответствующих кислот формулы 4 известными способами, такими как хлорирование тионилхлоридом или оксалилхлоридом.
Антраниловые кислоты формулы 3 могут быть получены различными известными способами. Многие из этих соединений являются известными. Как показано на схеме 4, антраниловые кислоты, содержащие в качестве заместителя R2 хлор, бром или иод, могут быть получены прямым галогенированием незамещенной антраниловой кислоты формулы 7 N-хлорсукцинимидом (NCS), N-бромсукцинимидом (NBS) или N-иодсукцинимидом (NIS) соответственно в растворителях, таких как N,N-диметилформамид (ДМФ), с получением соответствующей замещенной кислоты формулы 3.
Схема 4

Получение ангидридов изатиновой кислоты формулы 5 может быть выполнено из изатинов формулы 9, как представлено в общем виде на схеме 5.
Схема 5
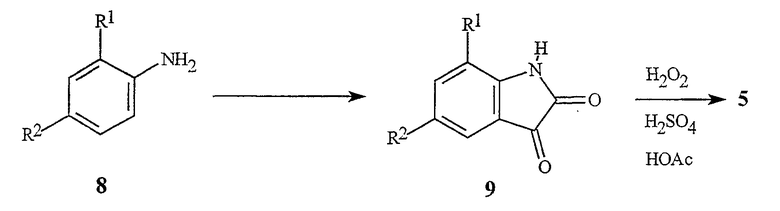
Изатины формулы 9 могут быть получены из производных анилина формулы 8 согласно описанным в литературе методикам, таким как F.D. Popp, Adv. Heterocycl. Chem. 1975, 18, 1-58 и J.F.M. Da Silva et al., Journal of the Brazilian Society 2001, 12(3), 273-324. Окисление изатина 9 пероксидом водорода обычно дает хорошие выходы соответствующего ангидрида изатиновой кислоты 5 (G. Reissenweber and D. Mangold, Angew. Chem. Int. ed. Engl. 1980, 19, 222-223). ангидриды изатиновой кислоты могут быть также получены из антраниловых кислот 3 посредством многочисленных известных методик, в том числе реакции 3 с фосгеном или эквивалентом фосгена.
Пиразолкарбоновые кислоты формулы 4 могут быть получены по способу, представленному в общем виде на схеме 6.
Схема 6
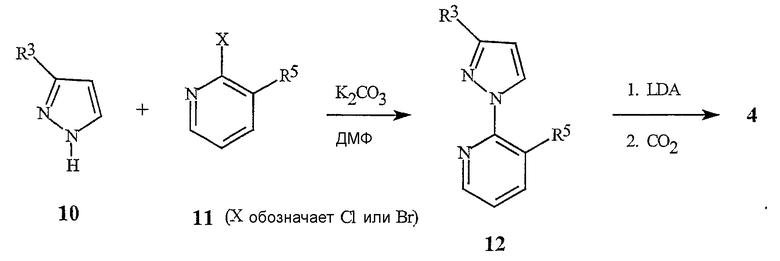 Реакция пиразола 10 с 2,3-дигалогенпиридином формулы 11 дает хорошие выходы 1-пиридилпиразола 12 с хорошей специфичностью в отношении желаемой региохимии. Металлирование 12 диизопропиламидом лития (LDA) с последующим гашением соли лития диоксидом углерода дает пиразолкарбоновую кислоту формулы 4. Дополнительные детали методик для этого способа приведены в примерах 1, 3 и 5.
Реакция пиразола 10 с 2,3-дигалогенпиридином формулы 11 дает хорошие выходы 1-пиридилпиразола 12 с хорошей специфичностью в отношении желаемой региохимии. Металлирование 12 диизопропиламидом лития (LDA) с последующим гашением соли лития диоксидом углерода дает пиразолкарбоновую кислоту формулы 4. Дополнительные детали методик для этого способа приведены в примерах 1, 3 и 5.
Исходные пиразолы 10, где R3 представляет собой СН3, Cl или Br, являются известными соединениями. Пиразол 10, в котором R3 является СН3, является коммерчески доступным. Пиразолы 10, в которых R3 является Cl или Br, могут быть получены в соответствии с описанными в литературе методиками (H. Reimlinger and A. Van Overstraeten, Chem. Ber. 1966, 99(10), 3350-7). Применимый альтернативный способ получения 10, в котором R3 представляет собой Cl или Br, изображен на схеме 7.
Схема 7
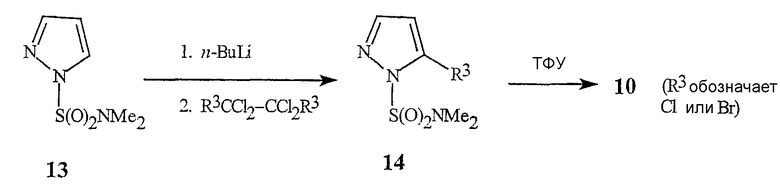
Металлирование сульфамоилпиразола 13 н-бутиллитием с последующим прямым галогенированием этого аниона либо гексахлорэтаном (если R3 является Cl), либо 1,2-дибромтетрахлорэтаном (если R3 является Br) дает галогенированные производные 14. Удаление сульфамоильной группы трифторуксусной кислотой (ТФУ) при комнатной температуре протекает без затруднений и с хорошим выходом с образованием пиразолов 10, где R3 представляет собой Cl или Br соответственно. Дополнительные экспериментальные подробности для этого способа описаны в примерах 3 и 5.
В качестве альтернативы способу, иллюстрированному на схеме 6, пиразолкарбоновые кислоты формулы 4, где R3 обозначает CF3, могут быть также получены способом, представленным в общем виде на схеме 8.
Схема 8
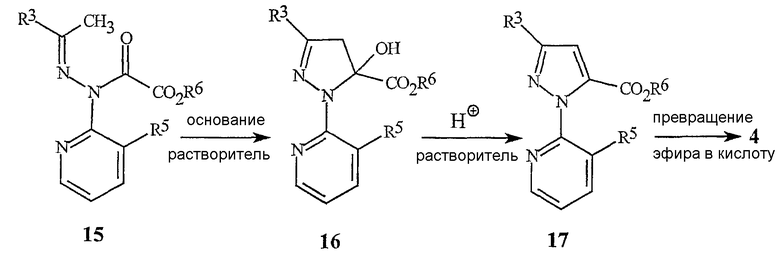
Реакция соединения формулы 15, где R6 обозначает С1-С4алкил, с подходящим основанием в подходящем органическом растворителе дает циклизированный продукт формулы 16 после нейтрализации кислотой, такой как уксусная кислота. Подходящим основанием может быть, например, но без ограничения, гидрид натрия, трет-бутоксид натрия, димсил натрия (диметилсульфоксид натрия) (CH3S(O)CH2 -Na+), карбонаты или гидроксиды щелочных металлов (таких как литий, натрий или калий), фториды или гидроксиды тетраалкил(например, метил, этил или бутил)аммония или 2-трет-бутиламино-2-диэтиламино-1,3-диметилпергидро-1,3,2-диазафосфонин. Подходящим органическим растворителем может быть, например, но без ограничения, ацетон, ацетонитрил, тетрагидрофуран, дихлорметан, диметилсульфоксид или N,N-диметилформамид. Реакцию циклизации обычно проводят в диапазоне температур от приблизительно 0 до 120°С. Влияния растворителя, основания, температуры и времени добавления являются взаимозависимыми, и выбор условий реакции является важным для минимизации образования побочных продуктов. Предпочтительным основанием является тетрабутиламмонийфторид.
Дегидратация соединения формулы 16 с получением соединения формулы 17 с последующим превращением функциональной группы эфира карбоновой кислоты в функциональную группу карбоновой кислоты дает соединение формулы 4. Дегидратацию выполняют обработкой каталитическим количеством подходящей кислоты. Этой каталитической кислотой может быть, например, но без ограничения, серная кислота. Реакцию обычно проводят с использованием органического растворителя. Как должно быть понятно специалисту в данной области, реакции дегидратации могут проводиться в широком разнообразии растворителей в диапазоне температур обычно между приблизительно 0 и 200°С, более предпочтительно между приблизительно 0 и 100°С. Для дегидратации в способе схемы 8 предпочтительными являются растворитель, содержащий уксусную кислоту, и температуры около 65°С. Соединения эфиров карбоновых кислот могут быть превращены в соединения карбоновых кислот многочисленными способами, в том числе нуклеофильным расщеплением при безводных условиях или гидролитическими способами, включающими в себя применение либо кислот, либо оснований (см. T.W. Greene and P.G.M. Wuts, Protective Groups in Organic Synthesis, 2nd ed., John Wiley & Sons, Inc., New York, 1991, pp. 224-269 в отношении обзора способов). Для способа схемы 8 предпочтительными являются катализируемые основанием гидролитические способы. Подходящие основания включают в себя гидроксиды щелочных металлов (таких как литий, натрий или калий). Например, эфир может быть растворен в смеси воды и спирта, такого как этанол. После обработки гидроксидом натрия или гидроксидом калия эфир омыляется с образованием натриевой или калиевой соли карбоновой кислоты. Подкисление сильной кислотой, такой как хлористоводородная кислота или серная кислота, дает карбоновую кислоту формулы 4. Эта карбоновая кислота может быть выделена способами, известными специалистам в данной области, в том числе кристаллизацией, экстракцией и дистилляцией.
Соединения формулы 15 могут быть получены по способу, представленному в общем виде на схеме 9.
Схема 9
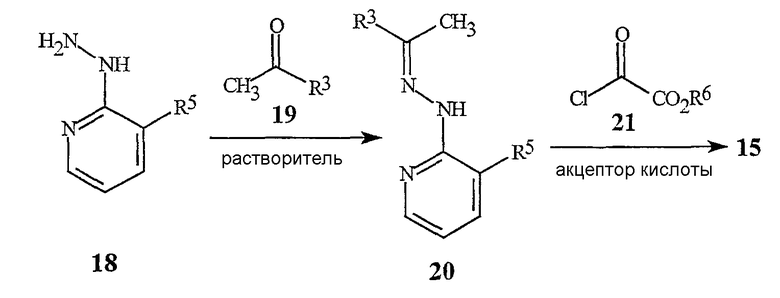
где R3 обозначает CF3 и R6 обозначает С1-С4алкил.
Обработка соединения гидразина формулы 18 кетоном формулы 19 в растворителе, таком как вода, метанол или уксусная кислота, дает гидразон формулы 20. Специалисту в данной области должно быть понятно, что эта реакция может требовать катализа выбранной кислотой и может также требовать повышенных температур в зависимости от характера молекулярного замещения гидразона формулы 20. Реакция гидразона формулы 20 с соединением формулы 21 в подходящем органическом растворителе, таком как, например, но без ограничения, дихлорметан или тетрагидрофуран, в присутствии акцептора кислоты, такого как триэтиламин, дает соединение формулы 15. Эту реакцию обычно проводят при температуре между приблизительно 0 и 100°С. Дополнительные экспериментальные подробности для способа схемы 9 иллюстрируются в примере 7. Соединения гидразина формулы 18 могут быть получены стандартными способами, такими как контактирование соответствующего галогенсодержащего соединения формулы 11 с гидразином.
В качестве альтернативы способу, иллюстрированному на схеме 6, пиразолкарбоновые кислоты формулы 4, в которых R3 представляет собой Cl или Br, могут быть также получены по способу, представленному в общем виде на схеме 10.
Схема 10
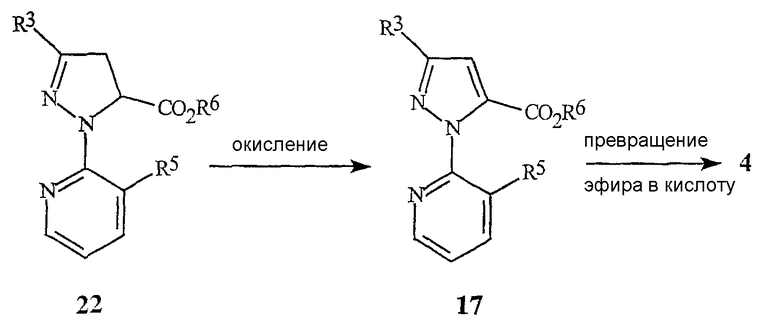
где R6 представляет С1-С4алкил.
Окисление соединения формулы 22, необязательно в присутствии кислоты, с получением соединения формулы 17 с последующим превращением функциональной группы эфира карбоновой кислоты в группу карбоновой кислоты обеспечивает соединение формулы 4. Окислительным агентом может быть пероксид водорода, органические пероксиды, персульфат калия, персульфат натрия, персульфат аммония, моноперсульфат калия (например, Oxone®) или перманганат калия. Для получения полного превращения должен использоваться по меньшей мере один эквивалент окислительного агента относительно соединения формулы 22, предпочтительно между приблизительно одним и двумя эквивалентами. Это окисление обычно проводят в присутствии растворителя. Растворителем может быть эфир, такой как тетрагидрофуран, п-диоксан и т.п., органический эфир, такой как этилацетат, диметилкарбонат и т.п., или полярный апротонный органический растворитель, такой как N,N-диметилформамид, ацетонитрил и т.п. Кислоты, пригодные для применения на стадии окисления, включают в себя неорганические кислоты, такие как серная кислота, фосфорная кислота и т.п., и органические кислоты, такие как уксусная кислота, бензойная кислота и т.п. Кислота, когда она используется, должна применяться в количестве, большем, чем 0,1 эквивалента относительно соединения формулы 22. Для получения полного превращения могут быть использованы один-пять эквивалентов кислоты. Предпочтительным окислителем является персульфат калия, и окисление предпочтительно проводят в присутствии серной кислоты. Реакция может проводиться смешиванием соединения формулы 22 в желаемом растворителе и, если используется кислота, в кислоте. Затем может быть добавлен окислитель с подходящей скоростью. Температуру реакции обычно варьируют от низкой температуры, такой как приблизительно 0°С, до точки кипения растворителя для получения приемлемой скорости реакции для завершения реакции в пределах, предпочтительно, менее 8 часов. Желаемый продукт, соединение формулы 17, может быть выделен способами, известными специалистам в данной области, в том числе кристаллизацией, экстракцией и дистилляцией. Способы, подходящие для превращения эфира формулы 17 в карбоновую кислоту формулы 4, уже описаны для схемы 8. Дополнительные экспериментальные подробности для способа схемы 10 иллюстрируются в примерах 8 и 9.
Соединения формулы 22 могут быть получены из соответствующих соединений формулы 23, как показано на схеме 11.
Схема 11
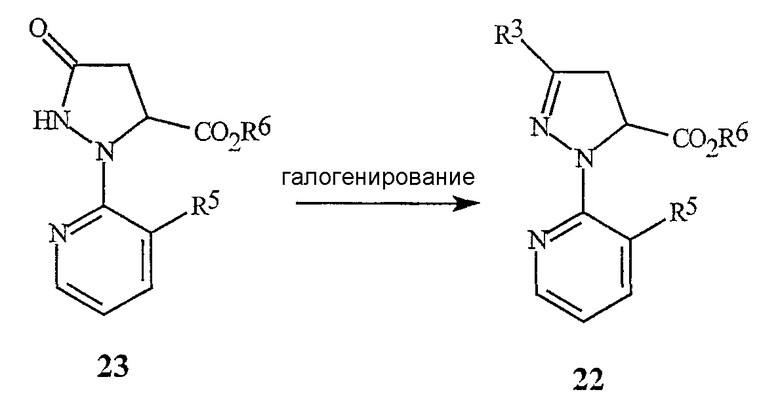
где R6 представляет С1-С4алкил.
Обработка соединения формулы 23 галогенирующим реагентом, обычно в присутствии растворителя, дает соответствующее галогенсодержащее соединение формулы 22. Галогенирующие реагенты, которые могут быть использованы, включают в себя оксигалогениды фосфора, тригалогениды фосфора, пентагалогениды фосфора, тионилхлорид, дигалогентриалкилфосфораны, дигалогендифенилфосфораны, оксалилхлорид и фосген. Предпочтительными являются оксигалогениды фосфора и пентагалогениды фосфора. Для получения полного превращения должны использоваться по меньшей мере 0,33 эквивалента оксигалогенида фосфора относительно соединения формулы 23, предпочтительно между приблизительно 0,33 и 1,2 эквивалента. Для получения полного превращения должны использоваться по меньшей мере 0,20 эквивалента пентагалогенида фосфора относительно соединения формулы 23, предпочтительно между приблизительно 0,20 и 1,0 эквивалентом. Соединения формулы 23, в которых R6 обозначает С1-С4алкил, являются предпочтительными для этой реакции. Типичные растворители для этого галогенирования включают в себя галогенированные алканы, такие как дихлорметан, хлороформ, хлорбутан и т.п., ароматические растворители, такие как бензол, ксилол, хлорбензол и т.п., эфиры, такие как тетрагидрофуран, п-диоксан, диэтиловый эфир и т.п., и полярные апротонные растворители, такие как ацетонитрил, N,N-диметилформамид и т.п. Необязательно, может быть добавлено органическое основание, такое как триэтиламин, пиридин, N,N-диметиланилин или т.п. Необязательно, может быть также добавлен катализатор, такой как N,N-диметилформамид. Предпочтительным является процесс, в котором растворителем является ацетонитрил, а основание отсутствует. Обычно ни основание, ни катализатор не требуются, когда в качестве растворителя используют ацетонитрил. Предпочтительную реакцию проводят смешиванием соединения формулы 23 в ацетонитриле. Затем добавляют галогенирующий реагент в течение удобного времени, и затем эту смесь выдерживают при желаемой температуре, пока реакция не завершится. Температура реакции обычно находится в диапазоне между 20°С и точкой кипения ацетонитрила, и время реакции обычно меньше, чем 2 часа. Затем реакционную массу нейтрализуют неорганическим основанием, таким как бикарбонат натрия, гидроксид натрия и т.п., или органическим основанием, таким как ацетат натрия. Желаемый продукт, соединение формулы 22, может быть выделен способами, известными специалистам в данной области, в том числе кристаллизацией, экстракцией и дистилляцией.
Альтернативно, соединения формулы 22, где R3 обозначает Br или Cl, могут быть получены обработкой соответствующих соединений формулы 22, где R3 обозначает отличающийся галоген (например, Cl для получения формулы 22, где R3 обозначает Br) или сульфонатную группу, такую как п-толуолсульфонат, бензолсульфонат и метансульфонат, бромидом водорода или хлоридом водорода соответственно. При помощи этого способа заместитель галоген или сульфонат R3 в исходном соединении формулы 22 заменяется Br или Cl из бромида водорода или хлорида водорода соответственно. Эту реакцию проводят в подходящем растворителе, таком как дибромметан, дихлорметан или ацетонитрил. Реакцию можно проводить примерно при атмосферном давлении или при более высоком давлении, чем атмосферное давление, в автоклаве. Когда R3 в исходном соединении формулы 22 является галогеном, таким как Cl, реакцию предпочтительно проводят таким образом, что галогенид водорода, генерируемый из этой реакции, удаляют с использованием барботера или другими подходящими средствами. Реакцию можно проводить при температуре между приблизительно 0 и 100°С, наиболее предпочтительно при температуре около температуры окружающей среды (например, при приблизительно 10-40оС) и, более предпочтительно, между приблизительно 20 и 30°С. Добавление в качестве катализатора кислоты Льюиса (такой как трибромид алюминия, для получения соединения формулы 22, где R3 является Br) может способствовать этой реакции. Продукт формулы 22 выделяют обычными способами, известными специалистам в данной области, в том числе экстракцией, дистилляцией и кристаллизацией. Дополнительные подробности для этого способа иллюстрируются в примере 10.
Исходные соединения формулы 22, где R3 обозначает Cl или Br, могут быть получены обработкой соответствующих соединений формулы 23, как уже описано. Исходные соединения формулы 22, где R3 обозначает сульфонатную группу, могут быть также получены из соответствующих соединений формулы 23 стандартными способами, такими как обработка сульфонилхлоридом (например, п-толуолсульфонилхлоридом) и основанием, таким как третичный амин (например, триэтиламин) в подходящем растворителе, таком как дихлорметан; дополнительные подробности для этого способа иллюстрируются в примере 11.
В качестве альтернативы способу, иллюстрированному на схеме 6, пиразолкарбоновые кислоты формулы 4, где R3 обозначает OCH2CF3, могут быть также получены по способу, представленному в общем виде на схеме 12.
Схема 12
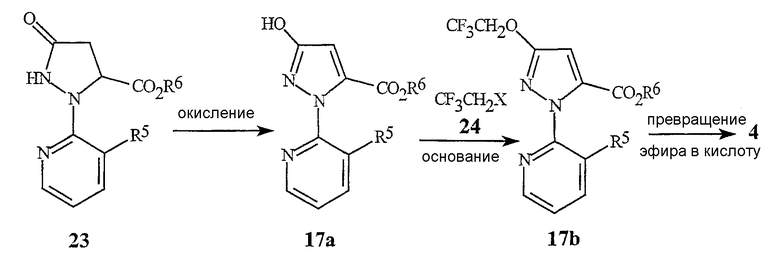
где R6 обозначает С1-С4алкил и Х обозначает уходящую (отщепляемую) группу.
В этом способе вместо галогенирования, показанного на схеме 11, соединение формулы 23 окисляют до соединения формулы 17а. Условия реакции для этого окисления уже описаны для превращения соединения формулы 22 в соединение формулы 17 на схеме 10.
Затем соединение формулы 17а алкилируют с образованием соединения формулы 17b контактированием с алкилирующим агентом СН3СН2Х (24) в присутствии основания. В алкилирующем агенте 24 Х обозначает уходящую группу нуклеофильной реакции, такую как галоген (например, Br, I), OS(O)2CH3 (метансульфонат), OS(O)2CF3, OS(O)2Ph-p-CH3 (п-толуолсульфонат) и т.п.; хорошо действует метансульфонат. Эту реакцию проводят в присутствии по меньшей мере одного эквивалента основания. Подходящие основания включают в себя неорганические основания, такие как карбонаты и гидроксиды щелочных металлов (таких как литий, натрий или калий), и органические основания, такие как триэтиламин, диизопропилэтиламин и 1,8-диазабицикло[5.4.0]ундец-7-ен. Реакцию обычно проводят в растворителе, который может содержать спирты, такие как метанол и этанол, галогенированные алканы, такие как дихлорметан, ароматические растворители, такие как бензол, толуол и хлорбензол, эфиры, такие как тетрагидрофуран, и полярные апротонные растворители, такие как ацетонитрил, N,N-диметилформамид и т.п. Спирты и полярные апротонные растворители являются предпочтительными для применения с неорганическими основаниями. Предпочтительными являются карбонат калия в качестве основания и ацетонитрил в качестве растворителя. Реакцию обычно проводят при температуре между приблизительно 0 и 150°С, наиболее часто между температурой окружающей среды и 100°С. Продукт формулы 17b может быть выделен общепринятыми способами, такими как экстракция. Затем эфир формулы 17b превращают в карбоновую кислоту формулы 4 способами, уже описанными для превращения соединения формулы 17 в соединение формулы 4 на схеме 8. Дополнительные подробности для способа схемы 12 иллюстрируются в примере 12.
Соединения формулы 23 могут быть получены из соединений формулы 18, как представлено в общем виде на схеме 13.
Схема 13
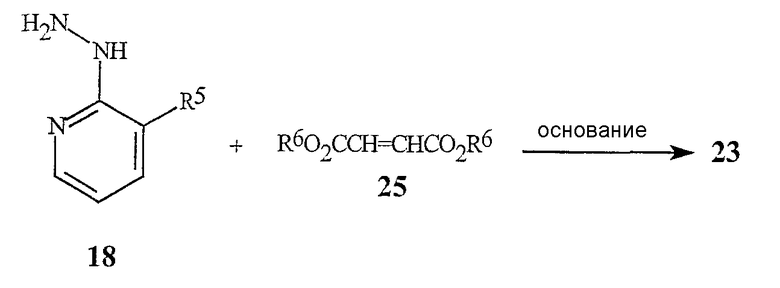
где R6 обозначает С1-С4алкил.
В этом способе соединение гидразина формулы 18 контактируют с соединением формулы 25 (фумаратным эфиром или малеатным эфиром или может быть использована их смесь) в присутствии основания и растворителя. Основанием является обычно соль алкоксида металла, такая как метоксид натрия, метоксид калия, этоксид натрия, этоксид калия, трет-бутоксид калия, трет-бутоксид лития и т.п. Следует использовать больше, чем 0,5 эквивалента основания относительно соединения формулы 18, предпочтительно между 0,9 и 1,3 эквивалента. Следует использовать больше, чем 1,0 эквивалент соединения формулы 25, предпочтительно между 1,0 и 1,3 эквивалента. Могут быть использованы полярные протонные и полярные апротонные органические растворители, такие как спирты, ацетонитрил, тетрагидрофуран, N,N-диметилформамид, диметилсульфоксид и т.п. Предпочтительными растворителями являются спирты, такие как метанол и этанол. Особенно предпочтительно, чтобы спирт был тем же самым спиртом, которым разбавляют фумаратный или малеатный эфир и алкоксидное основание. Реакцию обычно проводят смешиванием соединения формулы 18 и основания в растворителе. Эта смесь может быть нагрета или охлаждена до желаемой температуры, и соединение формулы 25 добавляют в течение некоторого периода времени. Обычно температуры реакции находятся в диапазоне между 0°С и точкой кипения используемого растворителя. Реакцию можно проводить при давлении, более высоком, чем атмосферное давление, для увеличения точки кипения растворителя. Обычно предпочтительными являются температуры между приблизительно 30 и 90°С. Время добавления может быть настолько быстрым, насколько это позволяет теплопередача. Типичные периоды добавления находятся между 1 минутой и 2 часами. Оптимальные температура реакции и время добавления варьируются в зависимости от конкретных соединений формулы 18 и формулы 25. После добавления реакционная смесь может быть выдержана в течение некоторого времени при температуре реакции. В зависимости от температуры реакции требуемое время выдерживания может быть от 0 до 2 часов. Обычные периоды выдерживания равны 10-60 минутам. Затем масса реакции может быть подкислена добавлением органической кислоты, такой как уксусная кислота и т.п., или неорганической кислоты, такой как хлористоводородная кислота, серная кислота и т.п. В зависимости от условий реакции и способов выделения группа -СО2R6 на соединении формулы 23 может гидролизоваться до -СО2Н; например, такому гидролизу может способствовать присутствие воды в реакционной смеси. Если образуется карбоновая кислота (-СО2Н), она может быть превращена обратно в -СО2R6, где R6 обозначает С1-С4алкил, с использованием способов этерификации, хорошо известных в данной области. Желаемый продукт, соединение формулы 23, может быть выделен способами, известными специалистам в данной области, такими как кристаллизация, экстракция или дистилляция.
Понятно, что некоторые реагенты и условия реакции, описанные выше, для получения соединений формулы 1, могут не быть совместимыми с некоторыми функциональными группами в промежуточных продуктах. В этих случаях включение последовательностей реакций введения/снятия защиты или взаимопревращений функциональной группы в этот синтез будет способствовать получению желаемых продуктов. Применение и выбор защитных групп будут очевидными специалисту в области химического синтеза (см., например, T.W. Greene and P.G.M. Wuts, Protective Groups in Organic Synthesis, 2nd ed., John Wiley: New York, 1991). Специалисту в данной области должно быть понятно, что, в некоторых случаях, после введения конкретного реагента, как это изображено на любой отдельной схеме, может быть необходимым выполнение дополнительных рутинных синтетических стадий, не описанных подробно, для завершения синтеза соединений формулы 1. Специалисту в данной области будет также понятно, что может быть необходимым выполнение комбинации стадий, иллюстрированных в приведенных выше схемах, в другом порядке, чем порядок, подразумеваемый конкретной последовательностью реакций, представленных для получения соединений формулы 1.
Авторы данного изобретения считают, что специалист в данной области, используя предшествующее описание, сможет использовать данное изобретение в его наибольшей степени. Таким образом, следующие далее примеры должны рассматриваться лишь как иллюстративные, но не как ограничивающие каким бы то ни было образом это описание. Стадии в следующих примерах иллюстрируют методику для каждой стадии в общем синтетическом превращении, и исходный материал для каждой стадии может не быть обязательно полученным в конкретном препаративном опыте, методика которого описана в других примерах или стадиях. Приводятся проценты по весу (мас.%), за исключением процентов для хроматографических смесей растворителей или особо оговоренных случаев. Части и проценты для хроматографических смесей растворителей приводятся по объему, если нет других указаний. 1Н-ЯМР-спектры даются в м.д. в направлении слабого поля от тетраметилсилана; "с" означает синглет, "д" означает дублет, "т" означает триплет, "к" означает квартет, "м" означает мультиплет, "дд" означает дублет дублетов, "дт" означает дублет триплетов и "шир.с" означает широкий синглет.
ПРИМЕР 1
Получение N-[4-хлор-2-метил-6-[[(1-метилэтил)амино]карбонил]фенил]-1-(3-хлор-2-пиридинил)-3-(трифторметил)-1Н-пиразол-5-карбоксамид
Стадия А: Получение 2-амино-3-метил-5-хлорбензойной кислоты
К раствору 2-амино-3-метилбензойной кислоты (Aldrich, 15,0 г, 99,2 ммоль) в N,N-диметилформамиде (50 мл) добавляли N-хлорсукцинимид (13,3 г, 99,2 ммоль) и реакционную смесь нагревали до 100°С в течение 30 минут. Нагревание прекращали, реакционную смесь охлаждали до комнатной температуры и оставляли на ночь. Затем реакционную смесь медленно выливали в смесь воды со льдом (250 мл) для осаждения белого твердого вещества. Твердое вещество отфильтровывали и промывали четыре раза водой и затем растворяли в этилацетате (900 мл). Этилацетатный раствор сушили над сульфатом магния, упаривали при пониженном давлении и оставшееся твердое вещество промывали эфиром с получением желаемого промежуточного продукта в виде белого твердого вещества (13,9 г).
1Н ЯМР (ДМСО-d6) δ 2,11 (с, 3H), 7,22 (с, 1H), 7,55 (с, 1H).
Стадия В: Получение 3-хлор-2-[3-(трифторметил)-1Н-пиразол-1-ил]пиридина
К смеси 2,3-дихлорпиридина (99,0 г, 0,67 моль) и 3-(трифторметил)пиразола (83 г, 0,61 моль) в сухом N,N-диметилформамиде (300 мл) добавляли карбонат калия (166,0 г, 1,2 моль) и затем реакционную смесь нагревали до 110-125°С в течение 48 часов. Реакционную смесь охлаждали до 100°С и фильтровали через диатомовый фильтрующий слой Celite® для удаления твердых веществ. N,N-диметилформамид и избыток дихлорпиридина удаляли перегонкой при атмосферном давлении. Перегонка продукта при пониженном давлении (т.кип. 139-141°С, 7 мм) давала желаемый промежуточный продукт в виде прозрачного желтого масла (113,4 г).
1Н ЯМР (CDCl3) δ 6,78 (с, 1H), 7,36 (т, 1H), 7,93 (д, 1H), 8,15 (с, 1Н), 8,45 (д, 1Н).
Стадия С: Получение 1-(3-хлор-2-пиридинил)-3-(трифторметил)-1Н-пиразол-5-карбоновой кислоты
К раствору 3-хлор-2-[3-(трифторметил)-1Н-пиразол-1-ил]пиридина (т.е. продукта пиразола из стадии В) (105,0 г, 425 ммоль) в сухом тетрагидрофуране (700 мл) при -75°С добавляли через канюлю раствор (-30°С) диизопропиламида лития (425 ммоль) в сухом тетрагидрофуране (300 мл). Темно-красный раствор перемешивали в течение 15 минут, после чего диоксид углерода барботировали через раствор при -63°С, пока раствор не становился бледно-желтым и не прекращалось выделение тепла. Реакционную смесь перемешивали в течение дополнительных 20 минут и затем гасили водой (20 мл). Растворитель удаляли при пониженном давлении и реакционную смесь распределяли между эфиром и 0,5 н. раствором гидроксида натрия. Водные экстракты промывали эфиром (3х), фильтровали через диатомовый фильтрующий слой Celite® для удаления оставшихся твердых веществ и затем подкисляли до рН приблизительно 4, причем при этом рН образовывалось оранжевое масло. Водную смесь перемешивали энергично и добавляли дополнительное количество кислоты для снижения рН до 2,5-3. Оранжевое масло застывало в гранулированное твердое вещество, которое фильтровали, промывали последовательно водой и 1 н. хлористоводородной кислотой и сушили в вакууме при 50°С с получением указанного в заголовке продукта в виде твердого вещества не совсем белого цвета (130 г). (Продукт из другого опыта по той же методике плавился при 175-176°С).
1Н ЯМР (ДМСО-d6) δ 7,61 (с, 1H), 7,76 (дд, 1H), 8,31 (д, 1H), 8,60(д, 1Н).
Стадия D: Получение 6-хлор-2-[1-(3-хлор-2-пиридинил)-3-(трифторметил)-1Н-пиразол-5-ил]-8-метил-4Н-3,1-бензоксазин-4-она
К раствору метансульфонилхлорида (2,2 мл, 28,3 ммоль) в ацетонитриле (75 мл) добавляли по каплям смесь 1-(3-хлор-2-пиридинил)-3-(трифторметил)-1Н-пиразол-5-карбоновой кислоты (т.е. продукта карбоновой кислоты стадии С) (7,5 г, 27,0 ммоль) и триэтиламин (3,75 мл, 27,0 ммоль) в ацетонитриле (75 мл) при 0-5°С. Затем температуру реакции поддерживали при 0°С на протяжении последовательного добавления реагентов. После перемешивания в течение 20 минут добавляли 2-амино-3-метил-5-хлорбензойную кислоту (т.е. продукт из стадии А) (5,1 г, 27,0 ммоль) и перемешивание продолжали в течение дополнительных 5 минут. Затем добавляли по каплям раствор триэтиламина (7,5 мл, 54,0 ммоль) в ацетонитриле (15 мл) и реакционную смесь перемешивали в течение 45 минут с последующим добавлением метансульфонилхлорида (2,2 мл, 28,3 ммоль). Затем реакционную смесь нагревали до комнатной температуры и перемешивали в течение ночи. Затем к осадку 5,8 г желтого твердого вещества добавляли приблизительно 75 мл воды. Дополнительное количество 1 г продукта выделяли экстракцией из фильтрата с получением всего 6,8 г указанного в заголовке соединения в виде желтого твердого вещества.
1H ЯМР (CDCl3) δ 1,83 (с, 3H), 7,50 (с, 1H), 7,53 (м, 2H), 7,99 (м, 2H), 8,58 (д, 1H).
Стадия Е: Получение N-[4-хлор-2-метил-6-[[(1-метилэтил)амино]карбонил]фенил]-1-(3-хлор-2-пиридинил)-3-(трифторметил)-1Н-пиразол-5-карбоксиамида
К раствору 6-хлор-2-[1-(3-хлор-2-пиридинил)-3-(трифторметил)-1Н-пиразол-5-ил]-8-метил-4Н-3,1-бензоксазин-4-она (т.е. продукта бензоксазинона стадии D) (5,0 г, 11,3 ммоль) в тетрагидрофуране (35 мл) добавляли по каплям изопропиламин (2,9 мл, 34,0 ммоль) в тетрагидрофуране (10 мл) при комнатной температуре. Затем реакционную смесь нагревали до растворения всех твердых веществ и перемешивали дополнительно в течение пяти минут, причем в этой временной точке тонкослойная хроматография подтвердила завершение реакции. Растворитель тетрагидрофуран выпаривали при пониженном давлении и оставшееся твердое вещество очищали хроматографией на силикагеле с последующим растиранием со смесью эфир/гексан и получением указанного в заголовке соединения, соединения данного изобретения, в виде твердого вещества (4,6 г), плавящегося при 195-196°С.
1H ЯМР (CDCl3) δ 1,21 (д, 6H), 2,17 (с, 3H), 4,16 (м, 1H), 5,95 (уш.д, 1H), 7,1-7,3 (м, 2H), 7,39 (с, 1H), 7,4 (м, 1H), 7,84 (д, 1H), 8,50 (д, 1H), 10,24 (уш.с, 1H).
ПРИМЕР 2
Получение N-[4-хлор-2-метил-6-[(метиламино)карбонил]фенил]-1-(3-хлор-2-пиридинил)-3-(трифторметил)-1Н-пиразол-5-карбоксамида
К раствору 6-хлор-2-[1-(3-хлор-2-пиридинил)-3-(трифторметил)-1Н-пиразол-5-ил]-8-метил-4Н-3,1-бензоксазин-4-она (т.е. продукта бензоксазинона примера 1, стадии D) (4,50 г, 10,18 ммоль) в тетрагидрофуране (ТГФ; 70 мл) добавляли метиламин (2,0 М раствор в ТГФ, 15 мл, 30,0 ммоль) по каплям и реакционную смесь перемешивали при комнатной температуре в течение 5 минут. Растворитель тетрагидрофуран выпаривали при пониженном давлении и оставшееся твердое вещество очищали хроматографией на силикагеле с получением 4,09 г указанного в заголовке соединения, соединения данного изобретения, в виде белого твердого вещества, плавящегося при 185-186°С.
1Н ЯМР (ДМСО-d6) δ 2,17 (с, 3H), 2,65 (д, 3H), 7,35 (д, 1H), 7,46 (дд, 1H), 7,65 (дд, 1H), 7,74 (с, 1H), 8,21 (д, 1H), 8,35 (уш.кв, 1H), 8,74 (д, 1H), 10,39 (с, 1H).
ПРИМЕР 3
Получение 3-хлор-N-[4-хлор-2-метил-6-[[(1-метилэтил)амино]карбонил]фенил]-1-(3-хлор-2-пиридинил)-1Н-пиразол-5-карбоксамида
Стадия А: Получение 3-хлор-N,N-диметил-1Н-пиразол-1-сульфонамида
К раствору N-диметилсульфамоилпиразола (188,0 г, 1,07 моль) в сухом тетрагидрофуране (1500 мл) при -78°С добавляли по каплям раствор 2,5 М н-бутиллития (472 мл, 1,18 моль) в гексане при поддержании температуры ниже -65°С. После завершения добавления реакционную смесь поддерживали при -78°С в течение дополнительных 45 минут, после чего добавляли по каплям раствор гексахлорэтана (279 г, 1,18 моль) в тетрагидрофуране (120 мл). Реакционную смесь поддерживали при -78°С в течение часа, нагревали до -20°С и затем гасили водой (1 л). Реакционную смесь экстрагировали метиленхлоридом (4х500 мл); органические экстракты сушили над сульфатом магния и концентрировали. Неочищенный продукт дополнительно очищали хроматографией на силикагеле с использованием метиленхлорида в качестве элюента и получением указанного в заголовке соединения в виде желтого масла (160 г).
1H ЯМР (CDCl3) δ 3,07 (д, 6H), 6,33 (с, 1H), 7,61 (с, 1H).
Стадия В: Получение 3-хлорпиразола
К трифторуксусной кислоте (290 мл) добавляли по каплям 3-хлор-N,N-диметил-1Н-пиразол-1-сульфонамид (т.е. продукт хлорпиразола стадии А) (160 г) и реакционную смесь перемешивали при комнатной температуре в течение 1,5 часа и затем концентрировали при пониженном давлении. Остаток помещали в гексан, нерастворимые твердые вещества отфильтровывали и гексан концентрировали с получением неочищенного продукта в виде масла. Неочищенный продукт дополнительно очищали хроматографией на силикагеле с использованием смеси эфир/гексан (40:60) в качестве элюента и получением указанного в заголовке продукта в виде желтого масла (64,44 г).
1H ЯМР (CDCl3) δ 6,39 (с, 1H), 7,66 (с, 1H), 9,6 (уш.с, 1H).
Стадия С: Получение 3-хлор-2-(3-хлор-1Н-пиразол-1-ил)пиридина
К смеси 2,3-дихлорпиридина (92,60 г, 0,629 моль) и 3-хлорпиразола (т.е. продукта стадии В) (64,44 г, 0,629 моль) в N,N-диметилформамида (400 мл) добавляли карбонат калия (147,78 г, 1,06 моль) и затем реакционную смесь нагревали до 100°С в течение 36 часов. Реакционную смесь охлаждали до комнатной температуры и медленно выливали в ледяную воду. Осадившиеся твердые вещества отфильтровывали и промывали водой. Твердый отфильтрованный осадок растворяли в этилацетате, сушили над сульфатом магния и концентрировали. Неочищенное твердое вещество хроматографировали на силикагеле с использованием смеси 20% этилацетат/гексан в качестве элюента и получением указанного в заголовке продукта в виде белого твердого вещества (39,75 г).
1H ЯМР (CDCl3) δ 6,43 (с, 1H), 7,26 (м, 1H), 7,90 (д, 1H), 8,09 (с, 1H), 8,41 (д, 1H).
Стадия D: Получение 3-хлор-1-(3-хлор-2-пиридинил)-1Н-пиразол-5-карбоновой кислоты
К раствору 3-хлор-2-(3-хлор-1Н-пиразол-1-ил)пиридина (т.е. продукта пиразола стадии С) (39,75 г, 186 ммоль) в сухом тетрагидрофуране (400 мл) при -78°С добавляли по каплям раствор 2,0 М диизопропиламида лития (93 мл, 186 ммоль) в тетрагидрофуране. Через раствор янтарного цвета барботировали диоксид углерода в течение 14 минут, после чего этот раствор становился бледно-коричневато-желтым. Реакционную смесь подщелачивали 1 н. раствором гидроксида натрия и экстрагировали эфиром (2 х 500 мл). Водные экстракты подкисляли 6 н. хлористоводородной кислотой и экстрагировали этилацетатом (3 х 500 мл). Этилацетатные экстракты сушили над сульфатом магния и концентрировали с получением указанного в заголовке соединения в виде твердого вещества не совсем белого цвета (42,96 г). (Продукт из другого опыта по той же методике плавился при 198-199°С).
1Н ЯМР (ДМСО-d6) δ 6,99 (с, 1H), 7,45 (м, 1H), 7,93 (д, 1H), 8,51 (д, 1H).
Стадия Е: Получение 6-хлор-2-[3-хлор-1-(3-хлор-2-пиридинил)-1Н-пиразол-5-ил]-8-метил-4Н-3,1-бензоксазин-4-она
К раствору метансульфонилхлорида (6,96 г, 61,06 ммоль) в ацетонитриле (150 мл) добавляли по каплям смесь 3-хлор-1-(3-хлор-2-пиридинил)-1Н-пиразол-5-карбоновой кислоты (т.е. продукта карбоновой кислоты стадии D) (15,0 г, 58,16 ммоль) и триэтиламин (5,88 г, 58,16 ммоль) в ацетонитриле (150 мл) при -5°С. Затем реакционную смесь перемешивали в течение 30 минут при 0°С. Затем добавляли 2-амино-3-метил-5-хлорбензойной кислоты (т.е. продукта из примера 1, стадии А) (10,79 г, 58,16 ммоль) и перемешивание продолжали в течение дополнительных 10 минут. Затем добавляли по каплям раствор триэтиламина (11,77 г, 116,5 ммоль) в ацетонитриле при поддержании температуры ниже 10°С. Реакционную смесь перемешивали в течение 60 минут при 0°С и затем добавляли метансульфонилхлорид (6,96 г, 61,06 ммоль). Затем реакционную смесь нагревали до комнатной температуры и перемешивали в течение дополнительных 2 часов. Затем реакционную смесь концентрировали и неочищенный продукт хроматографировали на силикагеле с использованием метиленхлорида в качестве элюента с получением указанного в заголовке соединения в виде желтого твердого вещества (9,1 г).
1H ЯМР (CDCl3) δ 1,81 (с, 3H), 7,16 (с, 1H), 7,51 (м, 2H), 7,98 (д, 2H), 8,56 (д, 1H).
Стадия F: Получение 3-хлор-N-[4-хлор-2-метил-6-[[(1-метилэтил)амино]карбонил]фенил]-1-(3-хлор-2-пиридинил)-1Н-пиразол-5-карбоксамида
К раствору 6-хлор-2-[3-хлор-1-(3-хлор-2-пиридинил)-1Н-пиразол-5-ил]-8-метил-4Н-3,1-бензоксазин-4-она (например, продукта бензоксазинона стадии Е) (6,21 г, 15,21 ммоль) в тетрагидрофуране (100 мл) добавляли изопропиламин (4,23 г, 72,74 ммоль) и затем реакционную смесь нагревали до 60°С, перемешивали в течение 1 часа и затем охлаждали до комнатной температуры. Растворитель тетрагидрофуран выпаривали при пониженном давлении и оставщееся твердое вещество очищали хроматографией на силикагеле с получением указанного в заголовке соединения, соединения данного изобретения, в виде белого твердого вещества (5,05 г), плавящегося при 173-175°С.
1H ЯМР (CDCl3) δ 1,23 (д, 6H), 2,18 (с, 3H), 4,21 (м, 1H), 5,97 (д, 1H), 7,01 (м, 1H) 7,20 (с, 1H), 7,24 (с, 1H), 7,41 (д, 1H), 7,83 (д, 1H), 8,43 (д, 1H), 10,15 (уш.с, 1H).
ПРИМЕР 4
Получение 3-хлор-N-[4-хлор-2-метил-6-[(метиламино)карбонил]фенил]-1-(3-хлор-2-пиридинил)-1Н-пиразол-5-карбоксамида
К раствору 6-хлор-2-[3-хлор-1-(3-хлор-2-пиридинил)-1Н-пиразол-5-ил]-8-метил-4Н-3,1-бензоксазин-4-она (т.е. продукта бензоксазинона примера 3, стадии Е) (6,32 г, 15,47 ммоль) в тетрагидрофуране (50 мл) добавляли метиламин (2,0 М раствор в ТГФ, 38 мл, 77,38 ммоль) и реакционную смесь нагревали до 60°С, перемешивали в течение 1 часа и затем охлаждали до комнатной температуры. Растворитель тетрагидрофуран выпаривали при пониженном давлении и оставшееся твердое вещество очищали хроматографией на силикагеле с получением указанного в заголовке соединения, соединения данного изобретения, в виде белого твердого вещества (4,57 г), плавящегося при 225-226°С.
1H ЯМР (CDCl3) δ 2,15 (с, 3H), 2,93 (с, 3H), 6,21 (д, 1H), 7,06 (с, 1H), 7,18 (с, 1H), 7,20 (с, 1H), 7,42 (м, 1H), 7,83 (д, 1H), 8,42 (д, 1H), 10,08 (уш.с, 1H).
ПРИМЕР 5
Получение 3-бром-N-[4-хлор-2-метил-6-[[(1-метилэтил)амино]карбонил]фенил]-1-(3-хлор-2-пиридинил)-1Н-пиразол-5-карбоксамида
Стадия А: Получение 3-бром-N,N-диметил-1Н-пиразол-1-сульфонамида
К раствору N-диметилсульфамоилпиразола (44,0 г, 0,251 моль) в сухом тетрагидрофуране (500 мл) при -78°С добавляли по каплям раствор н-бутиллития (2,5 М в гексане, 105,5 мл, 0,264 моль) при поддержании температуры ниже -60°С. Густое твердое вещество образовывалось во время добавления. После завершения добавления реакционную смесь выдерживали в течение дополнительных 15 минут, после чего добавляли по каплям раствор 1,2-дибромтетрахлорэтана (90 г, 0,276 моль) в тетрагидрофуране (150 мл) при поддержании температуры ниже -70°С. Реакционная смесь приобретала оранжевый цвет и становилась прозрачной; перемешивание продолжали в течение дополнительных 15 минут. Баню -78°С удаляли и реакцию гасили водой (600 мл). Реакционную смесь экстрагировали метиленхлоридом (4х) и органические экстракты сушили над сульфатом магния и концентрировали. Неочищенный продукт дополнительно очищали хроматографией на силикагеле с использованием смеси метиленхлорид-гексан (50:50) в качестве элюента и получением указанного в заголовке соединения в виде прозрачного бесцветного масла (57,04 г).
1H ЯМР (CDCl3) δ 3,07 (д, 6H), 6,44 (м, 1H), 7,62 (м, 1H).
Стадия В: Получение 3-бромпиразола
К трифторуксусной кислоте (70 мл) медленно добавляли 3-бром-N,N-диметил-1Н-пиразол-1-сульфонамид (т.е. продукт бромпиразола стадии А) (57,04 г). Реакционную смесь перемешивали при комнатной температуре в течение 30 минут и затем концентрировали при пониженном давлении. Остаток помещали в гексан, нерастворимые твердые вещества отфильтровывали и гексан выпаривали с получением неочищенного продукта в виде масла. Неочищенный продукт дополнительно очищали хроматографией на силикагеле с использованием смеси этилацетат/дихлорметан (10:90) в качестве элюента и получением масла. Это масло помещали в дихлорметан, нейтрализовали водным раствором бикарбоната натрия, экстрагировали метиленхлоридом (3х), сушили над сульфатом магния и концентрировали с получением указанного в заголовке соединения в виде белого твердого вещества (25,9 г), т.пл. 61-64°С.
1H ЯМР (CDCl3) δ 6,37 (д, 1H), 7,59 (д, 1H), 12,4 (уш.с, 1H).
Стадия С: Получение 2-(3-бром-1Н-пиразол-1-ил)-3-хлорпиридина
К смеси 2,3-дихлорпиридина (27,4 г, 185 ммоль) и 3-бромпиразола (т.е. продукта стадии В) (25,4 г, 176 ммоль) в сухом N,N-диметилформамиде (88 мл) добавляли карбонат калия (48,6 г, 352 ммоль) и реакционную смесь нагревали до 125°С в течение 18 часов. Реакционную смесь охлаждали до комнатной температуры и выливали в ледяную воду (800 мл). Образовывался осадок. Осажденные твердые вещества перемешивали в течение 1,5 часов, фильтровали и промывали водой (2 х 100 мл). Твердый фильтровальный осадок помещали в метиленхлорид и промывали последовательно водой, 1 н. хлористоводородной кислотой, насыщенным водным раствором бикарбоната натрия и солевым раствором. Затем органические экстракты сушили над сульфатом магния и концентрировали с получением 39,9 г розового твердого вещества. Неочищенное твердое вещество суспендировали в гексане и перемешивали энергично в течение 1 часа. Твердые вещества фильтровали, промывали гексаном и сушили с получением указанного в заголовке соединения в виде порошка не совсем белого цвета (30,4 г), который имел чистоту >94% согласно ЯМР. Этот материал использовали без дополнительной очистки на стадии D.
1H ЯМР (CDCl3) δ 6,52 (с, 1H), 7,30 (дд, 1H), 7,92 (д, 1H), 8,05 (с, 1H), 8,43 (д, 1H).
Стадия D: Получение 3-бром-1-(3-хлор-2-пиридинил)-1Н-пиразол-5-карбоновой кислоты
К раствору 2-(3-бром-1Н-пиразол-1-ил)-3-хлорпиридина (т.е. продукта пиразола стадии С) (30,4 г, 118 ммоль) в сухом тетрагидрофуране (250 мл) при -76°С добавляли по каплям раствор диизопропиламида лития (118 ммоль) в тетрагидрофуране при такой скорости, чтобы поддерживалась температура ниже -71°С. Реакционную смесь перемешивали в течение 15 минут при -76°С и затем диоксид углерода барботировали через нее в течение 10 минут, что вызывало нагревание до -57°С. Реакционную смесь нагревали до -20°С и реакцию гасили водой. Реакционную смесь концентрировали и затем помещали в воду (1 л) и эфир (500 мл) и затем добавляли водный раствор гидроксида натрия (1 н. 20 мл). Водные экстракты промывали эфиром и подкисляли хлористоводородной кислотой. Осадившиеся твердые вещества фильтровали, промывали водой и сушили с получением указанного в заголовке соединения в виде рыжевато-коричневого твердого вещества (27,7 г). (Продукт из другого опыта по той же методике плавился при 200-201°С).
1Н ЯМР (ДМСО-d6) δ 7,25 (с, 1H), 7,68 (дд, 1H), 8,24 (д, 1H), 8,56 (д, 1H).
Стадия Е: Получение 2-[3-бром-1-(3-хлор-2-пиридинил)-1Н-пиразол-5-ил]-6-хлор-8-метил-4Н-3,1-бензоксазин-4-она
Методику, аналогичную методике примера 1, стадии Е, использовали для превращения 3-бром-1-(3-хлор-2-пиридинил)-1Н-пиразол-5-карбоновой кислоты (т.е. продукта пиразолкарбоновой кислоты примера 5, стадии D) (1,5 г, 4,96 ммоль) и 2-амино-3-метил-5-хлорбензойной кислоты (т.е. продукта примера 1, стадии А) (0,92 г, 4,96 ммоль) в указанное в заголовке соединение в виде твердого вещества (1,21 г).
1H ЯМР (CDCl3) δ 2,01,(с, 3H), 7,29 (с, 1H), 7,42 (д, 1H), 7,95 (д, 1H), 8,04 (м, 1H), 8,25 (с, 1H), 8,26 (д, 1H).
Стадия F: Получение 3-бром-N-[4-хлор-2-метил-6-[[(1-метилэтил)амино]карбонил]фенил]-1-(3-хлор-2-пиридинил)-1Н-пиразол-5-карбоксамида
К раствору 2-[3-бром-1-(3-хлор-2-пиридинил)-1Н-пиразол-5-ил]-6-хлор-8-метил-4Н-3,1-бензоксазин-4-она (т.е. продукта бензоксазинона стадии Е) (0,20 г, 0,44 ммоль) в тетрагидрофуране добавляли изопропиламин (0,122 мл, 1,42 ммоль) и реакционную смесь нагревали до 60°С в течение 90 минут и затем охлаждали до комнатной температуры. Растворитель тетрагидрофуран выпаривали при пониженном давлении, и оставшееся твердое вещество растирали с эфиром, фильтровали и сушили с получением указанного в заголовке соединения, соединения данного изобретения, в виде твердого вещества (150 мг), т.пл. 159-161°С.
1H ЯМР (CDCl3) δ 1,22 (д, 6H), 2,19 (с, 3H), 4,21 (м, 1H), 5,99 (м, 1H), 7,05 (м, 1H), 7,22 (м, 2H), 7,39 (м, 1H), 7,82 (д, 1H), 8,41 (д, 1H).
ПРИМЕР 6
Получение 3-бром-N-[4-хлор-2-метил-6-[(метиламино)карбонил]фенил]-1-(3-хлор-2-пиридинил)-1Н-пиразол-5-карбоксамида
К раствору 2-[3-бром-1-(3-хлор-2-пиридинил)-1Н-пиразол-5-ил]-6-хлор-8-метил-4Н-3,1-бензоксазин-4-она (т.е. продукту бензоксазинона примера 5, стадии Е) (0,20 г, 0,44 ммоль) в тетрагидрофуране добавляли метиламин (2,0 М раствор в ТГФ, 0,514 мл, 1,02 ммоль) и реакционную смесь нагревали до 60°С в течение 90 минут и затем охлаждали до комнатной температуры. Растворитель тетрагидрофуран выпаривали при пониженном давлении и оставшееся вещество растирали с эфиром, фильтровали и сушили с получением указанного в заголовке соединения, соединения данного изобретения, в виде твердого вещества (40 мг), т.пл. 162-164°С.
1H ЯМР (CDCl3) δ 2,18 (с, 3H), 2,95 (с, 3H), 6,21 (м, 1H), 7,10 (с, 1H), 7,24 (м, 2H), 7,39 (м, 1H), 7,80 (д, 1H), 8,45 (д, 1H).
Следующий пример 7 иллюстрирует альтернативное получение 1-(3-хлор-2-пиридинил)-3-(трифторметил)-1Н-пиразол-5-карбоновой кислоты, которая может быть использована для получения, например, N-[4-хлор-2-метил-6-[[(1-метилэтил)амино]карбонил]фенил]-1-(3-хлор-2-пиридинил)-3-(трифторметил)-1Н-пиразол-5-карбоксамида и N-[4-хлор-2-метил-6-[(метиламино)карбонил]фенил]-1-(3-хлор-2-пиридинил)-3-(трифторметил)-1Н-пиразол-5-карбоксамида с использованием дополнительных стадий, иллюстрированных в примерах 1 и 2.
ПРИМЕР 7
Получение 1-(3-хлор-2-пиридинил)-3-(трифторметил)-1Н-пиразол-5-карбоновой кислоты
Стадия А: Получение (2,2,2-трифтор-1-метилэтилиден)гидразона 3-хлор-2(1Н)-пиридинона
1,1,1-трифторацетон (7,80 г, 69,6 ммоль) добавляли к гидразону 3-хлор-2(1Н)пиридинона (альтернативно называемого (3-хлорпиридин-2-ил)гидразином) (10 г, 69,7 ммоль) при 20-25°С. После завершения добавления смесь перемешивали в течение приблизительно 10 минут. Растворитель удаляли при пониженном давлении и смесь распределяли между этилацетатом (100 мл) и насыщенным водным раствором бикарбоната натрия (100 мл). Органический слой сушили и упаривали. Хроматография на силикагеле (с элюцией этилацетатом) давала продукт в виде твердого вещества не совсем белого цвета (11 г, выход 66%), т.пл. 64-64,5°С (после кристаллизации из смеси этилацетат/гексаны).
ИК (нуйол) ν 1629, 1590, 1518, 1403, 1365, 1309, 1240, 1196, 1158, 1100, 1032, 992, 800 см-1.
1H ЯМР (CDCl3) δ 2,12 (с, 3H), 6,91-6,86 (м, 1H), 7,64-7,61 (м, 1H), 8,33-8,32 (м, 2H).
МС m/z 237 (М+).
Стадия В: Получение (3-хлор-2-пиридинил)(2,2,2-трифтор-1-метилэтилиден)гидразида этилгидроэтандиоата (альтернативно называемого (3-хлор-2-пиридинил)(2,2,2-трифтор-1-метилэтилиден)гидразином этилгидроэтандиоата)
Триэтиламин (20,81 г, 0,206 моль) добавляли к (2,2,2-трифтор-1-метилэтилиден)гидразону 3-хлор-2(1Н)-пиридинона (т.е. продукту стадии А) (32,63 г, 0,137 моль) в дихлорметане (68 мл) при 0°С. К этой смеси добавляли по каплям этилхлороксоацетат (18,75 г, 0,137 моль) в дихлорметане (69 мл) при 0°С. Смеси давали нагреваться до 25°С на протяжении приблизительно 2 часов. Смесь охлаждали до 0°С и добавляли по каплям дополнительную порцию этилхлороксоацетата (3,75 г, 27,47 ммоль) в дихлорметане (14 мл). Спустя приблизительно еще один час, смесь разбавляли дихлорметаном (приблизительно 450 мл) и смесь промывали водой (2 х 150 мл). Органический слой сушили и упаривали. Хроматография на силикагеле (с элюцией смесью 1:1 этилацетат-гексаны) давала продукт в виде твердого вещества (42,06 г, выход 90%), т.пл. 73,0-73,5°С (после кристаллизации из смеси этилацетат/гексаны).
ИК (нуйол) ν 1751, 1720, 1664, 1572, 1417, 1361, 1330, 1202, 1214, 1184, 1137, 1110, 1004, 1043, 1013, 942, 807, 836 см-1.
1Н ЯМР (ДМСО-d6 115°C) 1,19 (т, 3H), 1,72 (уш.с, 3H), 4,25 (кв, 2H), 7,65 (дд, J=8,3, 4,7 Гц, 1H), 8,20 (дд, J=7,6, 1,5 Гц, 1H), 8,55 (д, J=3,6 Гц, 1H).
МС m/z 337 (М+).
Стадия С: Получение этил 1-(3-хлор-2-пиридинил)-4,5-дигидро-5-гидрокси-3-(трифторметил)-1Н-пиразол-5-карбоксилата
(3-хлор-2-пиридинил)(2,2,2-трифтор-1-метилэтилиден)гидразид этилгидроэтандиоата (т.е. продукт стадии В) (5 г, 14,8 ммоль) в диметилсульфоксиде (25 мл) добавляли к гидрату тетрабутиламмонийфторида (10 г) в диметилсульфоксиде (25 мл) в течение 8 часов. После завершения добавления смесь выливали в уксусную кислоту (3,25 г) в воде (25 мл). Затем после перемешивания при 25°С в течение ночи смесь экстрагировали толуолом (4 х 25 мл) и объединенные экстракты в толуоле промывали водой (50 мл), сушили и упаривали с получением твердого вещества. Хроматография на силикагеле (с элюцией смесью 1:2 этилацетат-гексаны) давала продукт в виде твердого вещества (2,91 г, выход 50%, содержащий приблизительно 5% (2,2,2-трифтор-1-метилэтилиден)гидразона 3-хлор-2(1Н)-пиридинона), т.пл. 78-78,5°С (после перекристаллизации из смеси этилацетат/гексаны).
ИК (нуйол) ν 3403, 1726, 1618, 1582, 1407, 1320, 1293, 1260, 1217, 1187, 1150, 1122, 1100, 1067, 1013, 873, 829 см-1.
1H ЯМР (CDCl3) δ 1,19 (с, 3H), 3,20 (1/2 ABZ pattern, J=18 Гц, 1H), 3,42 (1/2 ABZ pattern, J=18 Гц, 1H), 4,24 (кв, 2H), 6,94 (дд, J=7,9, 4,9 Гц, 1H), 7,74 (дд, J= 7,7, 1,5 Гц, 1H), 8,03 (дд, 7=4,7, 1,5 Гц, 1H).
МС m/z 319(М+).
Стадия D: Получение этил 1-(3-хлор-2-пиридинил)-3-(трифторметил)-1Н-пиразол-5-карбоксилата
Серную кислоту (концентрированную, 2 капли) добавляли к этил 1-(3-хлор-2-пиридинил)-4,5-дигидро-5-гидрокси-3-(трифторметил)-1Н-пиразол-5-карбоксилату (т.е. продукту стадии С) (1 г, 2,96 ммоль) в уксусной кислоте (10 мл) и смесь нагревали до 65°С в течение приблизительно 1 часа. Смеси давали охладиться до 25°С и большую часть уксусной кислоты удаляли при пониженном давлении. Смесь распределяли между насыщенным водным раствором карбоната натрия (100 мл) и этилацетатом (100 мл). Водный слой дополнительно экстрагировали этилацетатом (100 мл). Объединенные органические экстракты сушили и упаривали с получением продукта в виде масла (0,66 г, выход 77%).
ИК (чистый) ν 3147, 2986, 1734, 1577, 1547, 1466, 1420, 1367, 1277, 1236, 1135, 1082, 1031, 973, 842, 802 см-1.
1H ЯМР (CDCl3) δ 1,23 (т, 3H), 4,25 (кв, 2H), 7,21 (с, 1H), 7,48 (дд, J=8,1, 4,7 Гц, 1H), 7,94 (дд, J=6,6, 2 Гц, 1H), 8,53 (дд, J=4,7, 1,5 Гц, 1H).
МС m/z 319(М+).
Стадия Е: Получение 1-(3-хлор-2-пиридинил)-3-(трифторметил)-1Н-пиразол-5-карбоновой кислоты
Гидроксид калия (0,5 г, 85%, 2,28 ммоль) в воде (1 мл) добавляли к этил 1-(3-хлор-2-пиридинил)-3-(трифторметил)-1Н-пиразол-5-карбоксилату (т.е. продукту стадии D) (0,66 г, 2,07 ммоль) в этаноле (3 мл). Спустя приблизительно 30 минут растворитель удаляли при пониженном давлении и эту смесь растворяли в воде (40 мл). Раствор промывали этилацетатом (20 мл). Водный слой подкисляли концентрированной хлористоводородной кислотой и экстрагировали этилацетатом (3 х 20 мл). Объединенные экстракты сушили и упаривали с получением продукта в виде твердого вещества (0,53 г, выход 93%), т.пл. 178-179°С (после кристаллизации из смеси гексаны-этилацетат).
ИК (нуйол) ν 1711, 1586, 1565, 1550, 1440, 1425, 1292, 1247, 1219, 1170, 1135, 1087, 1059, 1031, 972, 843, 816 см-1.
1Н ЯМР (ДМСО-d6) δ 7,61 (с, 1H), 7,77 (м, 1H), 8,30 (д, 1H), 8,60 (с, 1H).
Следующий пример 8 иллюстрирует альтернативное получение 3-хлор-1-(3-хлор-2-пиридинил)-1Н-пиразол-5-карбоновой кислоты, которая может быть использована для получения, например, 3-хлор-N-[4-хлор-2-метил-6-[[(1-метилэтил)амино]карбонил]фенил]-1-(3-хлор-2-пиридинил)-1Н-пиразол-5-карбоксамида и 3-хлор-N-[4-хлор-2-метил-6-[(метиламино)карбонил]фенил]-1-(3-хлор-2-пиридинил)-1Н-пиразол-5-карбоксамида, с использованием дополнительных стадий, иллюстрированных в примерах 3 и 4.
ПРИМЕР 8
Получение 3-хлор-1-(3-хлор-2-пиридинил)-1Н-пиразол-5-карбоновой кислоты
Стадия А: Получение этил 2-(3-хлор-2-пиридинил)-5-оксо-3-пиразолидинкарбоксилата (альтернативно называемого этил 1-(3-хлор-2-пиридинил)-3-пиразолидинон-5-карбоксилатом)
Четырехгорлую колбу на 2 л, снабженную механической мешалкой, термометром, капельной воронкой, конденсатором для дефлегмации и входным отверстием для азота, загружали абсолютным этанолом (250 мл) и этанольным раствором этоксида натрия (21%, 190 мл, 0,504 моль). Эту смесь нагревали с обратным холодильником для дефлегмации при приблизительно 83°С. Затем ее обрабатывали гидразоном 3-хлор-2(1Н)-пиридинона (68,0 г, 0,474 моль). Смесь повторно нагревали с обратным холодильником для дефлегмации (кипении с обратным холодильником) в течение 5 минут. Затем желтую суспензию обрабатывали по каплям диэтилмалеатом (88,0 мл, 0,544 моль) в течение 5 минут. Скорость дефлегмации увеличивалась заметно во время этого добавления. В конце добавления весь исходный материал растворялся. Полученный оранжево-красный раствор поддерживали при дефлегмации (кипении с обратным холодильником) в течение 10 минут. После охлаждения до 65°С реакционную смесь обрабатывали ледяной уксусной кислотой (50,0 мл, 0,873 моль). Образовывался осадок. Смесь разбавляли водой (650 мл), что вызывало растворение осадка. Оранжевый раствор охлаждали на бане со льдом. Продукт начинал осаждаться при 28°С. Суспензию выдерживали при приблизительно 2°С в течение 2 часов. Продукт выделяли фильтрованием, промывали водным этанолом (40%, 3 х 50 мл) и затем сушили на воздухе на фильтре в течение приблизительно 1 часа. Указанное в заголовке соединение получали в виде высоко кристаллического, светло-оранжевого порошка (70,3 г, выход 55%). Не наблюдали значимых примесей согласно 1Н-ЯМР.
1Н ЯМР (ДМСО-d6) δ 1,22 (т, 3H), 2,35 (д, 1H), 2,91 (дд, 1H), 4,20 (кв, 2H), 4,84 (д, 1H), 7,20 (дд, 1H), 7,92 (д, 1H), 8,27 (д, 1H), 10,18 (с, 1H).
Стадия В: Получение этил 3-хлор-1-(3-хлор-2-пиридинил)-4,5-дигидро-1Н-пиразол-5-карбоксилата (альтернативно называемого этил 1-(3-хлор-2-пиридинил)-3-хлор-3-пиразолин-5-карбоксилатом)
В четырехгорлую колбу на 2 л, снабженную механической мешалкой, термометром, конденсатором для дефлегмации и входным отверстием для азота, загружали ацетонитрил (1000 мл), этил 2-(3-хлор-2-пиридинил)-5-оксо-3-пиразолидинкарбоксилат (т.е. продукт стадии А) (91,0 г, 0,377 моль) и оксихлорид фосфора (35,0 мл, 0,375 моль). После добавления оксихлорида фосфора смесь самонагревалась с 22 до 25°С и образовывался осадок. Светло-желтую суспензию нагревали с обратным холодильником для дефлегмации при 83°С в течение 35 минут, после чего осадок растворялся. Полученный оранжевый раствор выдерживали при дефлегмации в течение 45 минут, после чего он становился черно-серым. Конденсатор, применяемый для дефлегмации, заменяли дистилляционной насадкой и 650 мл растворителя удаляли дистилляцией. Вторую четырехгорлую колбу на 2 л, снабженную механической мешалкой, загружали бикарбонатом натрия (130 г, 1,55 моль) и водой (400 мл). Концентрированную реакционную смесь добавляли к суспензии бикарбоната натрия в течение 15 минут. Полученную двухфазную смесь перемешивали энергично в течение 20 минут, после чего выделение газа прекращалось. Эту смесь разбавляли дихлорметаном (250 мл) и затем перемешивали в течение 50 минут. Смесь обрабатывали фильтровальным веществом из диатомовой земли Celite® 545 (11 г) и затем фильтровали для удаления черного, смолистого вещества, которое подавляло разделение фаз. Поскольку этот фильтрат медленно разделялся на заметные различающиеся фазы, его разбавляли дихлорметаном (200 мл) и водой (200 мл) и обрабатывали дополнительным количеством Celite® 545 (15 г). Смесь фильтровали и фильтрат переносили в делительную воронку. Более тяжелый темно-зеленый органический слой отделяли. Слоистую фазу (неровный слой) (50 мл) повторно фильтровали и затем добавляли к органическому слою. Этот органический раствор (800 мл) обрабатывали сульфатом магния (30 г) и силикагелем (12 г) и суспензию перемешивали магнитной мешалкой в течение 30 минут. Суспензию фильтровали для удаления сульфата магния и силикагеля, которые становились темно-сине-зелеными. Фильтровальный осадок промывали дихлорметаном (100 мл). Фильтрат концентрировали на роторном испарителе. Продукт состоял из темно-янтарного масла (92,0 г, выход 93%). Единственными поддающимися определению примесями, наблюдаемыми с использованием 1Н-ЯМР, были 1% исходный материал и 0,7% ацетонитрил.
1Н ЯМР (ДМСО-d6) δ 1,15 (т, 3H), 3,26 (дд, 1H), 3,58 (дд, 1H), 4,11 (кв, 2H), 5,25 (дд, 1H), 7,00 (дд, 1H), 7,84 (д, 1H), 8,12 (д, 1H).
Стадия С: Получение этил 3-хлор-1-(3-хлор-2-пиридинил)-1Н-пиразол-5-карбоксилата (альтернативно называемого этил 1-(3-хлор-2-пиридинил)-3-хлорпиразол-5-карбоксилатом)
Четырехгорлую колбу на 2 л, снабженную механической мешалкой, термометром, конденсатором для дефлегмации и входным отверстием для азота, загружали этил 3-хлор-1-(3-хлор-2-пиридинил)-4,5-дигидро-1Н-пиразол-5-карбоксилатом (т.е. продуктом стадии В) (с чистотой 95%, 99,5 г, 0,328 моль), ацетонитрилом (1000 мл) и серной кислотой (98%, 35,0 мл, 0,661 моль). Эта смесь самонагревалась с 22 до 35°С при добавлении серной кислоты. После перемешивания в течение нескольких минут эту смесь обрабатывали персульфатом калия (140 г, 0,518 моль). Суспензию нагревали с обратным холодильником для дефлегмации при 84°С в течение 4,5 часов. Полученную оранжевую суспензию, пока она была еще теплой (50-65°С), фильтровали для удаления мелкого белого осадка. Фильтровальный осадок промывали ацетонитрилом (50 мл). Фильтрат концентрировали до приблизительно 500 мл на роторном испарителе. Вторую четырехгорлую колбу на 2 л, снабженную механической мешалкой, загружали водой (1250 мл). Концентрированную реакционную массу добавляли к воде в течение приблизительно 5 минут. Продукт выделяли фильтрованием, промывали водным ацетонитрилом (25%, 3 х 125 мл), промывали один раз водой (100 мл) и затем сушили в течение ночи в вакууме при комнатной температуре. Продукт состоял из кристаллического оранжевого порошка (79,3 г, выход 82%). Единственными поддающимися определению примесями, наблюдаемыми с использованием 1Н-ЯМР, были 1,9% воды и 0,6% ацетонитрила.
1Н ЯМР (ДМСО-d6) δ 1,09 (т, 3H), 4,16 (кв, 2H), 7,31 (с, 1H), 7,71 (дд, 1H), 8,38 (д, 1H), 8,59 (д, 1H).
Стадия D: Получение 3-хлор-1-(3-хлор-2-пиридинил)-1Н-пиразол-5-карбоновой кислоты (альтернативно называемой 1-(3-хлор-2-пиридинил)-3-хлорпиразол-5-карбоновой кислотой)
Четырехгорлую колбу на 1 л, снабженную механической мешалкой, термометром и входным отверстием для азота, загружали этил 3-хлор-1-(3-хлор-2-пиридинил)-1Н-пиразол-5-карбоксилатом (т.е. продуктом стадии С) (с чистотой 97,5%, 79,3 г, 0,270 моль), метанолом (260 мл), водой (140 мл) и гранулами гидроксида натрия (13,0 г, 0,325 моль). После добавления гидроксида натрия эта смесь самонагревалась с 22 до 35°С и исходный материал начинал растворяться. После перемешивания в течение 45 минут в условиях окружающей среды весь исходный материал растворялся. Полученный темно-оранжево-коричневый раствор концентрировали до приблизительно 250 мл на роторном испарителе. Затем концентрированную реакционную смесь разбавляли водой (400 мл). Водный раствор экстрагировали эфиром (200 мл). Затем водный слой переносили в колбу Эрленмейера на 1 л с магнитной мешалкой. Раствор обрабатывали по каплям концентрированной хлористоводородной кислотой (36,0 г, 0,355 моль) в течение приблизительно 10 минут. Продукт выделяли фильтрованием, повторно суспендировали в воде (2 х 200 мл), промывали на поверхности один раз водой (100 мл) и затем сушили на воздухе на фильтре в течение 1,5 часов. Продукт состоял из кристаллического светло-коричневого порошка (58,1 г, выход 83%). Единственной поддающейся определению примесью, наблюдаемой с использованием 1Н-ЯМР, было 0,7% эфира.
1Н ЯМР (ДМСО-d6) δ 7,20 (с, 1H), 7,68 (дд, 1H), 8,25 (д, 1H), 8,56 (д, 1H), 13,95 (уш.с, 1H).
Следующий пример 9 иллюстрирует альтернативное получение 3-бром-1-(3-хлор-2-пиридинил)-1Н-пиразол-5-карбоновой кислоты, которая может быть использована для получения, например, 3-бром-N-[4-хлор-2-метил-6-[[(1-метилэтил)амино]карбонил]фенил]-1-(3-хлор-2-пиридинил)-1Н-пиразол-5-карбоксамида и 3-бром-N-[4-хлор-2-метил-6-[(метиламино)карбонил]фенил]-1-(3-хлор-2-пиридинил)-1Н-пиразол-5-карбоксамида, с использованием дополнительных стадий, иллюстрированных в примерах 5 и 6.
ПРИМЕР 9
Получение 3-бром-1-(3-хлор-2-пиридинил)-1Н-пиразол-5-карбоновой кислоты
Стадия А1: Получение этил 3-бром-1-(3-хлор-2-пиридинил)-4,5-дигидро-1Н-пиразол-5-карбоксилата (альтернативно называемого этил 1-(3-хлор-2-пиридинил)-3-бром-2-пиразолин-5-карбоксилатом) с использованием оксибромида фосфора
Четырехгорлую колбу на 1 л, снабженную механической мешалкой, термометром, конденсатором для дефлегмации и входным отверстием для азота, загружали ацетонитрилом (400 мл), этил 2-(3-хлор-2-пиридинил)-5-оксо-3-пиразолидинкарбоксилатом (т.е. продуктом примера 8, стадии А) (50,0 г, 0,185 моль) и оксибромидом фосфора (34,0 г, 0,119 моль). Оранжевую суспензию нагревали с обратным холодильником для дефлегмации при 83°С в течение 20 минут. Полученный мутный оранжевый раствор поддерживали при дефлегмации в течение 75 минут, причем в это время образовывался плотный рыжевато-коричневый кристаллический осадок. Конденсатор для дефлегмации заменяли дистилляционной насадкой и собирали мутный бесцветный дистиллят (300 мл). Вторую четырехгорлую колбу на 1 л, снабженную механической мешалкой, загружали бикарбонатом натрия (45 г, 0,54 моль) и водой (200 мл). Концентрированную реакционную смесь добавляли к суспензии в бикарбонате натрия в течение 5 минут. Полученную двухфазную смесь энергично перемешивали в течение 5 минут, после чего выделение газа прекращалось. Смесь разбавляли дихлорметаном (200 мл) и затем перемешивали в течение 75 минут. Смесь обрабатывали фильтровальным веществом из диатомовой земли Celite® 545 (5 г) и затем фильтровали для удаления коричневого смолистого вещества. Фильтрат переносили в делительную воронку. Коричневый органический слой (400 мл) отделяли и затем обрабатывали сульфатом магния (15 г) и активированным углем Darco® G60 (2,0 г). Полученную суспензию перемешивали магнитной мешалкой в течение 15 минут и затем фильтровали для удаления сульфата магния и угля. Зеленый фильтрат обрабатывали силикагелем (3 г) и перемешивали в течение нескольких минут. Темный сине-зеленый силикагель удаляли фильтрованием и фильтрат концентрировали на роторном испарителе. Продукт состоял из масла светло-янтарного цвета (58,6 г, выход 95%), которое кристаллизовалось при стоянии. Единственной поддающейся определению при помощи 1Н-ЯМР примесью было 0,3% ацетонитрила.
1Н ЯМР (ДМСО-d6) δ 1,15 (т, 3H), 3,29 (дд, 1H), 3,60 (дд, 1H), 4,11 (кв, 2H), 5,20 (дд, 1H), 6,99 (дд, 1H), 7,84 (д, 1H), 8,12 (д, 1H).
Стадия А2: Получение этил 3-бром-1-(3-хлор-2-пиридинил)-4,5-дигидро-1Н-пиразол-5-карбоксилата с использованием пентабромида фосфора
Четырехгорлую колбу на 1 л, снабженную механической мешалкой, термометром, конденсатором для дефлегмации и входным отверстием для азота, загружали ацетонитрилом (330 мл), этил 2-(3-хлор-2-пиридинил)-5-оксо-3-пиразолидинкарбоксилатом (т.е. продуктом примера 8, стадии А) (52,0 г, 0,193 моль) и пентабромидом фосфора (41,0 г, 0,0952 моль). Оранжевую суспензию нагревали с обратным холодильником для дефлегмации при 84°С в течение 20 минут. Полученную кирпично-красную смесь поддерживали при дефлегмации в течение 90 минут, причем в это время образовывался плотный рыжевато-коричневый кристаллический осадок. Конденсатор для дефлегмации заменяли дистилляционной насадкой и собирали мутный бесцветный дистиллят (220 мл). Вторую четырехгорлую колбу на 1 л, снабженную механической мешалкой, загружали бикарбонатом натрия (40 г, 0,48 моль) и водой (200 мл). Концентрированную реакционную смесь добавляли к суспензии в бикарбонате натрия в течение 5 минут. Полученную двухфазную смесь энергично перемешивали в течение 10 минут, после чего выделение газа прекращалось. Смесь разбавляли дихлорметаном (200 мл) и затем перемешивали в течение 10 минут. Смесь обрабатывали фильтровальным веществом из диатомовой земли Celite® 545 (5 г) и затем фильтровали для удаления пурпурного смолистого вещества. Фильтровальный осадок промывали дихлорметаном (50 мл). Фильтрат переносили в делительную воронку. Пурпурно-красный органический слой (400 мл) отделяли и затем обрабатывали сульфатом магния (15 г) и активированным углем Darco® G60 (2,2 г). Эту суспензию перемешивали магнитной мешалкой в течение 40 минут. Суспензию фильтровали для удаления сульфата магния и угля. Фильтрат концентрировали на роторном испарителе. Продукт состоял из масла темно-янтарного цвета (61,2 г, выход 95%), которое кристаллизовалось при стоянии. Единственной поддающейся определению при помощи 1Н-ЯМР примесью было 0,7% ацетонитрила.
1Н ЯМР (ДМСО-d6) δ 1,15 (т, 3H), 3,29 (дд, 1H), 3,60 (дд, 1H), 4,11 (кв, 2H), 5,20 (дд, 1H), 6,99 (дд, 1H), 7,84 (д, 1H), 8,12 (д, 1H).
Стадия В: Получение этил 3-бром-1-(3-хлор-2-пиридинил)-1Н-пиразол-5-карбоксилата (альтернативно называемого этил 1-(3-хлор-2-пиридинил)-3-бромпиразол-5-карбоксилатом)
Четырехгорлую колбу на 1 л, снабженную механической мешалкой, термометром, конденсатором для дефлегмации и входным отверстием для азота, загружали этил 3-бром-1-(3-хлор-2-пиридинил)-4,5-дигидро-1Н-пиразол-5-карбоксилатом (т.е. продуктом стадий А1 и А2) (40,2 г, 0,121 моль), ацетонитрилом (300 мл) и серной кислотой (98%, 13,0 мл, 0,245 моль). Смесь самонагревалась с 22 до 36°С после добавления серной кислоты. После перемешивания в течение нескольких минут смесь обрабатывали персульфатом калия (48,0 г, 0,178 моль). Суспензию нагревали с обратным холодильником для дефлегмации при 84°С в течение 2 часов. Полученную оранжевую суспензию, пока она была еще теплой (50-65°С), фильтровали для удаления белого осадка. Фильтровальный осадок промывали ацетонитрилом (2 х 50 мл). Фильтрат концентрировали до приблизительно 200 мл на роторном испарителе. Вторую четырехгорлую колбу на 1 л, снабженную механической мешалкой, загружали водой (400 мл). Концентрированную реакционную массу добавляли к воде в течение приблизительно 5 минут. Продукт выделяли фильтрованием, промывали последовательно водным ацетонитрилом (20%, 100 мл) и водой (75 мл) и затем сушили на воздухе на фильтре в течение 1 часа. Продукт состоял из кристаллического оранжевого порошка (36,7 г, выход 90%). Единственными поддающимися определению примесями, наблюдаемыми с использованием 1Н-ЯМР, были 1% неизвестного вещества и 0,5% ацетонитрила.
1Н ЯМР (ДМСО-d6) δ 1,09 (т, 3H), 4,16 (кв, 2H), 7,35 (с, 1H), 7,72 (дд, 1H), 8,39 (д, 1H), 8,59 (д, 1H).
Стадия С: Получение 3-бром-1-(3-хлор-2-пиридинил)-1Н-пиразол-5-карбоновой кислоты (альтернативно называемой 1-(3-хлор-2-пиридинил)-3-бромпиразол-5-карбоновой кислотой)
Четырехгорлую колбу на 300 мл, снабженную механической мешалкой, термометром и входным отверстием для азота, загружали этил 3-бром-1-(3-хлор-2-пиридинил)-1Н-пиразол-5-карбоксилатом (т.е. продуктом стадии В) (с чистотой 98,5%, 25,0 г, 0,0756 моль), метанолом (75 мл), водой (50 мл) и гранулами гидроксида натрия (3,30 г, 0,0825 моль). После добавления гидроксида натрия эта смесь самонагревалась с 29 до 34°С и исходный материал начинал растворяться. После перемешивания в течение 90 минут в условиях окружающей среды весь исходный материал растворялся. Полученный темно-оранжевый раствор концентрировали до приблизительно 90 мл на роторном испарителе. Затем концентрированную реакционную смесь разбавляли водой (160 мл). Водный раствор экстрагировали эфиром (100 мл). Затем водный слой переносили в колбу Эрленмейера на 500 мл, снабженную магнитной мешалкой. Раствор обрабатывали по каплям концентрированной хлористоводородной кислотой (8,50 г, 0,0839 моль) в течение приблизительно 10 минут. Продукт выделяли фильтрованием, повторно суспендировали в воде (2 х 200 мл), промывали на поверхности один раз водой (25 мл) и затем сушили на воздухе на фильтре в течение 2 часов. Продукт состоял из кристаллического рыжевато-коричневого порошка (20,9 г, выход 91%). Единственными поддающимися определению примесями, наблюдаемыми с использованием 1Н-ЯМР, были 0,8% неизвестного вещества и 0,7% эфира.
1Н ЯМР (ДМСО-d6) δ 7,25 (с, 1H), 13,95 (уш.с, 1H), 8,56 (д, 1H), 8,25 (д, 1H), 7,68 (дд, 1H).
Следующий пример 10 иллюстрирует альтернативное получение этил 3-бром-1-(3-хлор-2-пиридинил)-4,5-дигидро-1Н-пиразол-5-карбоксилата, который может быть использован для получения, например, этил 3-бром-1-(3-хлор-2-пиридинил)-1Н-пиразол-5-карбоксилата (т.е. продукта примера 9, стадии В).
ПРИМЕР 10
Получение этил 3-бром-1-(3-хлор-2-пиридинил)-4,5-дигидро-1Н-пиразол-5-карбоксилата из этил 3-хлор-1-(3-хлор-2-пиридинил)-4,5-дигидро-1Н-пиразол-5-карбоксилата с использованием бромида водорода
Бромид водорода пропускали через раствор этил 3-хлор-1-(3-хлор-2-пиридинил)-4,5-дигидро-1Н-пиразол-5-карбоксилата (т.е. продукта примера 8, стадии В) (8,45 г, 29,3 ммоль) в дибромметане (85 мл). Спустя 90 минут ток газа останавливали и реакционную смесь промывали водным раствором бикарбоната натрия (100 мл). Органическую фазу сушили и упаривали при пониженном давлении с получением указанного в заголовке продукта в виде масла (9,7 г, выход 99%), которое кристаллизовалось при стоянии.
1H ЯМР (CDCl3) δ 1,19 (т, 3H), 3,24 (1/2 AB в ABX pattern, J=9,3, 17,3 Гц, 1H), 3,44 (1/2 AB в ABX pattern, J=11,7, 17,3 Гц, 1H), 4,18 (кв, 2H), 5,25 (X ABX, 1H, J=9,3, 11,9 Гц), 6,85 (дд, J=4,7, 7,7 Гц, 1H), 7,65 (дд, J=1,6, 7,8 Гц, 1H), 8,07 (дд, J=1,6, 4,8 Гц, 1H).
Следующий пример 11 иллюстрирует получение этил 1-(3-хлор-2-пиридинил)-4,5-дигидро-3-[[(4-метилфенил)сульфонил]окси]-1Н-пиразол-5-карбоксилата, который может быть использован для получения этил 3-бром-1-(3-хлор-2-пиридинил)-4,5-дигидро-1Н-пиразол-5-карбоксилата при помощи методик, сходных с методиками, описанными в примере 10.
ПРИМЕР 11
Получение этил 1-(3-хлор-2-пиридинил)-4,5-дигидро-3-[[(4-метилфенил)сульфонил]окси]-1Н-пиразол-5-карбоксилата
Триэтиламин (3,75 г, 37,1 ммоль) добавляли по каплям к смеси этил 2-(3-хлор-2-пиридинил)-5-оксо-3-пиразолидинкарбоксилата (т.е. продукта примера 8, стадии А) (10,0 г, 37,1 ммоль) и п-толуолсульфонилхлорида (7,07 г, 37,1 ммоль) в дихлорметане (100 мл) при 0°С. Добавляли дополнительные порции п-толуолсульфонилхлорида (0,35 г, 1,83 ммоль) и триэтиламина (0,19 г, 1,88 ммоль). Затем реакционной смеси давали нагреться до комнатной температуры и перемешивали в течение ночи. Затем эту смесь разбавляли дихлорметаном (200 мл) и промывали водой (3 х 70 мл). Органическую фазу сушили и упаривали с оставлением указанного в заголовке продукта в виде масла (13,7 г, выход 87%), которое медленно образовывало кристаллы. Продукт перекристаллизовывали из смеси этилацетат/гексаны, плавится при 99,5-100°С.
ИК (нуйол) ν: 1740, 1638, 1576, 1446, 1343, 1296, 1228, 1191, 1178, 1084, 1027, 948, 969, 868, 845 см-1.
1H ЯМР (CDCl3) δ 1,19 (т, 3H), 2,45 (с, 3H), 3,12 (1/2 AB в ABX pattern, J=17,3, 9 Гц, 1H), 3,33 (1/2 AB в ABX pattern, J=17,5, 11,8 Гц, 1H), 4,16 (кв, 2H), 5,72 (X ABX, J=9, 11,8 Гц, 1H), 6,79 (дд, J=4,6, 7,7 Гц, 1H), 7,36 (д, J=8,4 Гц, 2H), 7,56 (дд, J=1,6, 7,8 Гц, 1H), 7,95 (д, J=8,4 Гц, 2H), 8,01 (дд, J=1,4, 4,6 Гц, 1H).
ПРИМЕР 12
Получение N-[4-хлор-2-метил-6-[(метиламино)карбонил]фенил]-1-(3-хлор-2-пиридинил)-3-(2,2,2-трифторэтокси)-1Н-пиразол-5-карбоксамида
Стадия А: Получение этил 1-(3-хлор-2-пиридинил)-2,3-дигидро-3-оксо-1Н-пиразол-5-карбоксилата
К суспензии этил 2-(3-хлор-2-пиридинил)-5-оксо-3-пиразолидинкарбоксилата (т.е. продукта примера 8, стадии А) (27 г, 100 ммоль), перемешиваемой в сухом ацетонитриле (200 мл), добавляли серную кислоту (20 г, 200 мл) в виде одной порции. Реакционная смесь разжижалась с образованием бледно-зеленого почти прозрачного раствора перед загущением опять с образованием бледно-желтой суспензии. Добавляли персульфат калия (33 г, 120 ммоль) в виде одной порции и затем реакционную смесь нагревали с обратным холодильником для спокойной дефлегмации в течение 3,5 часов. После охлаждения при помощи бани со льдом осадок белого твердого вещества удаляли фильтрованием и выбрасывали. Фильтрат разбавляли водой (400 мл) и затем экстрагировали три раза этиловым эфиром (700 мл всего). Концентрирование объединенных эфирных экстрактов до уменьшенного объема (75 мл) вызывало осаждение твердого вещества не совсем белого цвета (3,75 г), которое собирали фильтрованием. Эфирный маточный раствор дополнительно концентрировали с получением второй партии осадка не совсем белого цвета (4,2 г), который также собирали фильтрованием. Твердое вещество не совсем белого цвета осаждалось также из водной фазы; это твердое вещество (4,5 г) собирали фильтрованием с получением в целом 12,45 г указанного в заголовке соединения.
1Н ЯМР (ДМСО-d6) δ 1,06 (т, 3H), 4,11 (кв, 2H), 6,34 (с, 1H), 7,6 (т, 1H), 8,19 (д, 1H), 8,5 (д, 1H), 10,6 (с, 1H).
Стадия В: Получение этил 1-(3-хлор-2-пиридинил)-3-(2,2,2-трифторэтокси)-1Н-пиразол-5-карбоксилата
К суспензии этил 1-(3-хлор-2-пиридинил)-2,3-дигидро-3-оксо-1Н-пиразол-5-карбоксилата (т.е. продукта стадии А) (0,8 г, 3 ммоль), перемешиваемой в сухом ацетонитриле (15 мл) при -5°С, добавляли карбонат калия (0,85 г, 6,15 ммоль). Суспензию перемешивали в течение 15 минут при 20°С. Затем перемешиваемую суспензию охлаждали до 5оС и добавляли по каплям 2,2,2-трифторэтилтрифторметансульфонат (0,8 г, 3,45 ммоль). Реакционную смесь нагревали до комнатной температуры и затем нагревали с обратным холодильником для дефлегмации, после чего тонкослойная хроматография показала завершение реакции. К реакционной смеси добавляли воду (25 мл) и затем смесь экстрагировали этиловым эфиром. Эфирный экстракт сушили над сульфатом магния и концентрировали с получением указанного в заголовке соединения-продукта (1,05 г) в виде бледно-желтого масла.
1H ЯМР (CDCl3) δ 1,21 (т, 3H), 4,20 (кв, 2H), 4,63 (кв, 2H), 6,53 (с, 1H), 7,4 (т, 1H), 7,9 (д, 1H), 8,5 (д, 1H).
Стадия С: Получение 1-(3-хлор-2-пиридинил)-3-(2,2,2-трифторэтокси)-1Н-пиразол-5-карбоновой кислоты
К перемешиваемому раствору этил 1-(3-хлор-2-пиридинил)-3-(2,2,2-трифторэтокси)-1Н-пиразол-5-карбоксилата (т.е. продукта стадии В) (0,92 г, 2,8 ммоль) в метаноле (15 мл) добавляли воду (5 мл), что вызывало помутнение реакционной смеси. Добавляли по каплям водный раствор гидроксида натрия (50%, 1,5 г, 19,2 ммоль) и реакционную смесь перемешивали при комнатной температуре в течение 30 минут, причем во время этого периода времени реакционная смесь становилась опять прозрачной. Добавляли воду (20 мл) и реакционную смесь экстрагировали этиловым эфиром, который выбрасывали. Водную фазу подкисляли до рН 2 при помощи концентрированной хлористоводородной кислоты и затем экстрагировали этилацетатом (50 мл). Этилацетатный экстракт, который промывали водой (20 мл) и солевым раствором (20 мл), сушили над сульфатом магния и концентрировали с получением указанного в заголовке соединения, выделенного в виде белого твердого вещества (0,8 г).
1Н ЯМР (ДМСО-d6) δ 4,9 (кв, 2H), 6,75 (с, 1H), 7,6 (т, 1H), 8,2 (д, 1H), 8,55 (д, 1H), 13,7 (уш.с, 1H).
Стадия D: Получение 6-хлор-8-метил-2Н-3,1-бензоксазин-2,4(1Н)-диона
К суспензии 2-амино-3-метил-5-хлорбензойной кислоты (т.е. продукта примера 1, стадии А) (97 г, 520 ммоль), перемешиваемой в сухом диоксане (750 мл) при комнатной температуре, добавляли по каплям трихлорметилхлорформиат (63 г, 320 ммоль). Реакционная смесь экзотермически нагревалась медленно до 42°С, и твердое вещество почти полностью растворялось перед тем, как снова образовать густую суспензию. После перемешивания этой суспензии при температуре окружающей среды в течение 2,5 часов указанное в заголовке соединение выделяли фильтрованием, промывали этиловым эфиром и сушили с получением указанного в заголовке соединения-продукта, полученного в виде белого твердого вещества (98 г).
1Н ЯМР (ДМСО-d6) δ 2,3 (с, 3H), 7,70 (с, 1H), 7,75 (с, 1H), 11,2 (с, 1H).
Стадия Е: Получение 6-хлор-2-[1-(3-хлор-2-пиридинил)-3-(2,2,2-трифторэтокси)-1Н-пиразол-5-ил]-8-метил-4Н-3,1-бензоксазин-4-она
К суспензии 1-(3-хлор-2-пиридинил)-3-(2,2,2-трифторэтокси)-1Н-пиразол-5-карбоновой кислоты (т.е. продукта стадии С) (7,9 г, 24 ммоль), перемешиваемой в дихлорметане (100 мл), добавляли N,N-диметилформамид (4 капли). Добавляли по каплям оксалилхлорид (4,45 г, 35 ммоль) в течение 45 минут. Полученный раствор перемешивали при комнатной температуре в течение 4 часов и затем концентрировали в вакууме. Выделенный хлорангидрид кислоты растворяли в сухом ацетонитриле (10 мл) и добавляли к суспензии 6-хлор-8-метил-2Н-3,1-бензоксазин-2,4(1Н)-диона (т.е. продукта стадии D) (4,9 г, 23 ммоль), перемешиваемой в сухом ацетонитриле (14 мл). Добавляли пиридин (10 мл) и этот раствор нагревали с обратным холодильником для дефлегмации в течение 6 часов. После охлаждения на бане со льдом собирали осадок белого твердого вещества (9,15 г). 1Н-ЯМР-спектр собранного осадка показал пики, согласующиеся с присутствием указанного в заголовке соединения и остаточного исходного материала 6-хлор-8-метил-2Н-3,1-бензоксазин-2,4(1Н)-диона. Небольшую порцию собранного осадка перекристаллизовывали из ацетонитрила с получением чистого указанного в заголовке продукта, плавящегося при 178-180°С.
1Н ЯМР (ДМСО-d6) δ 1,72 (с, 3H), 4,96 (кв, 2H), 7,04 (с, 1H), 7,7 (т, 1H), 7,75 (с, 1H), 7,9 (с, 1H), 8,3 (д, 1H), 8,6 (д, 1H).
Стадия F: Получение N-[4-хлор-2-метил-6-[(метиламино)карбонил]фенил]-1-(3-хлор-2-пиридинил)-3-(2,2,2-трифторэтокси)-1Н-пиразол-5-карбоксамида
К суспензии 6-хлор-2-[1-(3-хлор-2-пиридинил)-3-(2,2,2-трифторэтокси)-1Н-пиразол-5-ил]-8-метил-4Н-3,1-бензоксазин-4-она (т.е. продукта в виде осадка стадии Е) (3,53 г, 7,5 ммоль) в тетрагидрофуране (15 мл) добавляли по каплям метиламин (2,0 М раствор в ТГФ, 11 мл, 22 ммоль), и полученный раствор перемешивали при комнатной температуре в течение 45 минут. После этого тонкослойная хроматография показала завершение реакции. Добавляли этиловый эфир (100 мл) и реакционную смесь перемешивали в течение 2 часов при образовании осадка. Осадок собирали фильтрованием и затем перекристаллизовывали из ацетонитрила с получением белого твердого вещества (0,82 г). Вторую партию белого твердого вещества (0,35 г) осаждали из маточного раствора в ацетонитриле и собирали фильтрованием. Исходный маточный раствор эфир/тетрагидрофуран концентрировали досуха и оставшееся твердое вещество перекристаллизовывали из ацетонитрила с получением третьей партии белого твердого вещества (0,95 г). Эти три партии объединяли с получением в целом 2,12 г (после высушивания) указанного в заголовке соединения, выделенного в виде белого твердого вещества, плавящегося при 207-208°С.
1H ЯМР (CDCl3) δ 2,18 (с, 3H), 2,92 (д, 3H), 4,66 (кв, 2H), 6,15 (кв, 1H), 6,6 (с, 1H), 7,2 (с, 1H), 7,25 (с, 1H), 7,35 (т, 1H), 7,8 (д, 1H), 8,45 (д, 1H), 10,0 (с, 1H).
Примеры 13 и 14 иллюстрируют альтернативы условиям реакций, описанным в примере 5, стадии Е, и в примере 3, стадии Е, соответственно.
ПРИМЕР 13
Получение 2-[3-бром-1-(3-хлор-2-пиридинил)-1Н-пиразол-5-ил]-6-хлор-8-метил-4Н-3,1-бензоксазин-4-она
Метансульфонилхлорид (1,0 мл, 1,5 г, 13 ммоль) растворяли в ацетонитриле (10 мл) и эту смесь охлаждали до -5°С. Добавляли по каплям раствор 3-бром-1-(3-хлор-2-пиридинил)-1Н-пиразол-5-карбоновой кислоты (т.е. продукта пиразолкарбоновой кислоты примера 5, стадии D) (3,02 г, 10 ммоль) и пиридина (1,4 мл, 1,4 г, 17 ммоль) в ацетонитриле (10 мл) в течение 5 минут при -5°С - 0°С. Во время этого добавления образовывалась суспензия. Смесь перемешивали в течение 5 минут при этой температуре и затем добавляли смесь 2-амино-3-метил-5-хлорбензойной кислоты (т.е. продукта примера 1, стадии А) (1,86 г, 10 ммоль) и пиридина (2,8 мл, 2,7 г, 35 ммоль) в ацетонитриле (10 мл), с промывкой дополнительным количеством ацетонитрила (5 мл). Эту смесь перемешивали в течение 15 минут при -5°С - 0°С и затем добавляли по каплям метансульфонилхлорид (1,0 мл, 1,5 мл, 13 ммоль) в ацетонитриле (5 мл) в течение 5 минут при температуре -5°С - 0°С. Реакционную смесь перемешивали еще в течение 15 минут при этой температуре, затем смеси давали нагреваться медленно до комнатной температуры и перемешивали в течение 4 часов. Добавляли по каплям воду (20 мл) и смесь перемешивали в течение 15 минут. Затем эту смесь фильтровали, и твердые вещества промывали смесью 2:1 ацетонитрил-вода (3 х 3 мл), затем ацетонитрилом (2 х 3 мл) и сушили в атмосфере азота с получением указанного в заголовке продукта в виде светло-желтого порошка, 4,07 г (выход неочищенного продукта 90,2%), плавящегося при 203-205°С. ВЭЖХ этого продукта с использованием хроматографической колонки Zorbax® RX-C8 (4,6 мм х 25 см, элюент 25-95% ацетонитрил/вода рН 3) показала основной пик, соответствующий указанному в заголовке соединению и имеющий 95,7% общей площади пиков хроматограммы.
1Н ЯМР (ДМСО-d6) δ 1,72 (с, 3H), 7,52 (с, 1H), 7,72-7,78 (м, 2H), 7,88 (м, 1H), 8,37 (дд, 1H), 8,62 (дд, 1H).
ПРИМЕР 14
Получение 6-хлор-2-[3-хлор-1-(3-хлор-2-пиридинил)-1Н-пиразол-5-ил]-8-метил-4Н-3,1-бензоксазин-4-она
Метансульфонилхлорид (1,0 мл, 1,5 г, 13 ммоль) растворяли в ацетонитриле (10 мл) и эту смесь охлаждали до -5°С. Добавляли по каплям раствор 3-хлор-1-(3-хлор-2-пиридинил)-1Н-пиразол-5-карбоновой кислоты (т.е. продукта карбоновой кислоты примера 3, стадии D) (2,58 г, 10 ммоль) и пиридина (1,4 мл, 1,4 г, 17 ммоль) в ацетонитриле (10 мл) в течение 5 минут при -5°С - 0°С. Во время этого добавления образовывалась суспензия. Смесь перемешивали в течение 5 минут при этой температуре и затем добавляли 2-амино-3-метил-5-хлорбензойную кислоту (т.е. продукт из примера 1, стадии А) (1,86 г, 10 ммоль), все количество сразу. Затем добавляли по каплям раствор пиридина (2,8 мл, 2,7 г, 35 ммоль) в ацетонитриле (10 мл) в течение 5 минут при -5°С - 0°С. Эту смесь перемешивали в течение 15 минут при -5°С - 0°С и затем добавляли по каплям метансульфонилхлорид (1,0 мл, 1,5 мл, 13 ммоль) в ацетонитриле (5 мл) в течение 5 минут при -5°С - 0°С. Реакционную смесь перемешивали в течение 15 минут при этой температуре, затем давали ей нагреваться медленно до комнатной температуры и перемешивали в течение 4 часов. Добавляли по каплям воду (15 мл) и эту смесь перемешивали в течение 15 минут. Затем смесь фильтровали, и твердые вещества промывали смесью 2:1 ацетонитрил-вода (3 х 3 мл), затем ацетонитрилом (2 х 3 мл) и сушили в атмосфере азота с получением указанного в заголовке продукта в виде бледно-желтого порошка, 3,83 г (выход неочищенного продукта 94,0%), плавящегося при 199-201°С. ВЭЖХ этого продукта с использованием хроматографической колонки Zorbax® RX-C8 (4,6 мм × 25 см, элюент 25-95% ацетонитрил/вода рН 3) показала основной пик, соответствующий указанному в заголовке соединению и имеющий 97,8% общей площади пиков хроматограммы.
1H ЯМР (ДМСО-d6) δ 1,72 (с, 3Н), 7,48 (с, 1Н), 7,74-7,80 (м, 2Н), 7,87 (м, 1Н), 8,37 (дд, 1Н), 8,62 (дд, 1Н).
ПРИМЕР 15
Получение 3-бром-1-(3-хлор-1-оксидо-2-пиридинил)-N-[2,4-дихлор-6-[[(1-метилэтил)амино]карбонил]фенил]-1H-пиразол-5-карбоксамида
3-бром-1-(3-хлор-2-пиридинил)-N-[2,4-дихлор-6-[[(1-метилэтил)амино]карбонил]фенил]-1H-пиразол-5-карбоксамид (соединение 25, полученное по методике, аналогичной методике из примера 5, 487 мг, 0,916 ммоль) добавляли к смеси метанола (1 мл) и метиленхлорида (9 мл) с последующим добавлением 30%-ного водного раствора Н2O2 (1 мл) и метилтриоксорения (VII) (0,100 г, 0,401 ммоль). Реакционную смесь интенсивно перемешивали в течение 88 часов, а по истечении данного времени для реакционной смеси проводили разделение между этилацетатом и насыщенным водным раствором NaHCO3. Объединенные органические экстракты высушивали (MgSO4), концентрировали и очищали по методу колоночной хроматографии с получением указанного в заголовке продукта - соединения настоящего изобретения - в виде твердой фазы (54.8 мг), температура плавления 224-226°С.
1H ЯМР (ДМСО) δ 1,02 (дд, 6Н), 3,88 (дд, 1Н), 7,44, (с, 1 Н) 7,50-7,57 (м, 2Н), 7,66 (д, 1Н), 8,17 (д, NH), 7,83 (с, 1Н), 10,51 (с, NH) 8,36 (д, 1Н).
При помощи описанных здесь методик и с использованием способов, известных в данной области, могут быть получены следующие соединения таблицы 1. В таблицах, которые следуют далее, использовали следующие аббревиатуры: t означает третичный, s означает вторичный, n означает нормальный, i означает изо. Me означает метил, Et означает этил, Pr означает пропил, i-Pr означает изопропил и Bu означает бутил.
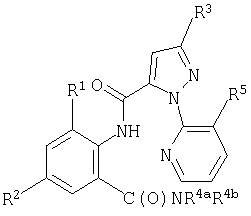
Как показано на схеме 1 и дополнительно иллюстрировано в примерах 1-10, бензоксазины формулы 2, такие как перечисленные в таблице 2, применимы для получения соединений формулы 1, в том числе соединений, перечисленных в таблице 1.
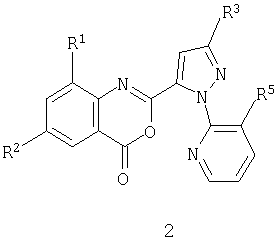
Как показано на схеме 2 и дополнительно иллюстрировано в примерах 1-10, пиразолкарбоновые кислоты формулы 4, такие как перечисленные в таблице 3, применимы в получении соединений формулы 1, в том числе соединений, перечисленных в таблице 1.
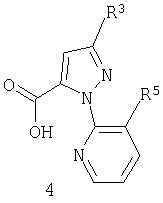
Приготовление композиций/применимость
Соединения данного изобретения обычно будут использоваться в виде готовой формы или композиции с сельскохозяйственно приемлемым носителем, содержащим по меньшей мере один жидкий разбавитель, твердый разбавитель или поверхностно-активное вещество. Ингредиенты готовой формы или композиции выбирают таким образом, чтобы они были совместимы с физическими свойствами активного ингредиента, способом введения и факторами окружающей среды, такими как тип почвы, влажность и температура. Применимые готовые формы включают в себя жидкости, такие как растворы (в том числе эмульгируемые концентраты), суспензии, эмульсии (в том числе микроэмульсии и/или суспензии-эмульсии) или т.п., которые могут быть необязательно загущены с образованием гелей. Кроме того, применимые готовые формы включают в себя твердые вещества, такие как дусты, порошки, гранулы, шарики, таблетки, пленки и т.п., которые могут быть вододиспергируемыми ("смачиваемыми") или водорастворимыми. Активный ингредиент может быть (микро)инкапсулированным и дополнительно приготовленным в виде суспензии или твердой готовой формы; альтернативно, вся готовая форма активного ингредиента может быть инкапсулирована (или "заключена в защитное покрытие"). Инкапсулирование может регулировать или задерживать высвобождение активного ингредиента. Пригодные для разбрызгивания композиции могут быть распределены в подходящих средах и использованы в объемах аэрозолей от приблизительно одного до нескольких сотен литров на гектар. Композиции с высокой концентрацией прежде всего используются в качестве промежуточных продуктов для дальнейшего приготовления готовых форм.
Готовые формы обычно содержат эффективные количества активного ингредиента, разбавитель и поверхностно-активное вещество в следующих примерных диапазонах, которые добавляются до 100 процентов по весу.
Типичные твердые разбавители описаны в Watkins, et al., Handbook of Insecticide Dust Diluents and Carriers, 2nd Ed., Dorland Books, Caldwell, New Jersey. Типичные жидкие разбавители описаны в Marsden, Solvents Guide, 2nd Ed., Interscience, New York, 1950. McCutcheon's Detergents and Emulsifiers Annual, Allured Publ. Corp., Ridgewood, New Jersey, а также Sisely and Wood, Encyclopedia of Surface Active Agents, Chemical Publ. Co., Inc., New York, 1964, перечни поверхностно-активных веществ и рекомендуемые применения. Все готовые формы могут содержать минорные количества добавок для уменьшения пенообразования, слеживания, коррозии, микробиологического роста и т.п., или загустители для увеличения вязкости.
Поверхностно-активные вещества включают в себя, например, полиэтоксилированные спирты, полиэтоксилированные алкилфенолы, полиэтоксилированные эфиры жирных кислот сорбитана, диалкилсульфосукцинаты, алкилсульфаты, алкилбензолсульфонаты, органосиликоны, N,N-диалкилтаураты, лигнинсульфонаты, конденсаты нафталинсульфоната-формальдегида, поликарбоксилаты и блок-сополимеры полиоксиэтилена и полиоксипропилена. Твердые разбавители включают в себя, например, глины, такие как бентонит, монтмориллонит, аттапульгит и каолин, крахмал, сахар, диоксид кремния, тальк, диатомовую землю, мочевину, карбонат кальция, карбонат и бикарбонат натрия и сульфат натрия. Жидкие разбавители включают в себя, например, воду, N,N-диметилформамид, диметилсульфоксид, N-алкилпирролидон, этиленгликоль, полипропиленгликоль, полипропиленкарбонат, эфиры двухосновных кислот, парафины, алкилбензолы, алкилнафталины, оливковое, касторовое, льняное, тунговое, кунжутное, кукурузное, арахисовое, хлопковое, соевое, расповое и кокосовое масла, эфиры жирных кислот, кетоны, такие как циклогексанон, 2-гептанон, изофорон и 4-гидрокси-4-метил-2-пентанон, и спирты, такие как метанол, циклогексанол, деканол, бензиловый и тетрагидрофурфуриловый спирт.
Растворы, включающие в себя эмульгируемые концентраты, могут быть приготовлены простым смешиванием ингредиентов. Дусты и порошки могут быть приготовлены смешиванием и обычно помолом, например, в молотковой мельнице или струйной мельнице. Суспензии обычно готовят мокрым помолом (в бегунковой мельнице мокрого помола); см., например, патент США 3060084. Гранулы и шарики могут быть приготовлены распылением активного материала на предварительно сформованные гранулированные носители или методом аггломерации. См. Browning, "Agglomeration", Chemical Engineering, December 4, 1967, pp.147-48, Perry's Chemical Engineer's Handbook, 4th Ed., McGraw-Hill, New York, 1963, pages 8-57 и следующие страницы, и РСТ Публикацию WO 91/13546. Шарики могут быть приготовлены, как описано в патенте США 4172714. Вододиспергируемые и водорастворимые гранулы могут быть приготовлены, как описано в патентах США с номерами 4144050, 3920442 и патенте Германии 3246493. Таблетки могут быть приготовлены, как описано в патентах США с номерами 5180587, 5232701 и 5208030. Пленки могут быть приготовлены, как описано в патенте Великобритании 2095558 и патенте США 3299566.
В отношении дополнительной информации, касающейся области приготовления, см. T.S.Woods, "The Formulator's Toolbox - Product Forms for Modern Agriculture" in Pesticide Chemistry and Bioscience, The Food-Environment Challenge, T.Brooks and T.R.Roberts, Eds., Proceedings of the 9th International Congress on Pesticide Chemistry, The Royal Society of Chemistry, Cambridge, 1999, pp.120-133. См. также U.S. 3235361, Столбец 6, строка 16 по Столбец 7, строка 1-9 и примеры 10-41; U.S. 3309192, Столбец 5, строка 43 по Столбец 7, строка 62 и примеры 8, 12, 15, 39, 41, 52, 53, 58, 132, 138-140, 162-164, 166, 167 и 169-182; U.S. 2891855, Столбец 3, строка 66 по Столбец 5, строка 17 и примеры 1-4; Klingman, Weed Control as a Science, John Wiley and Sons, Inc., New York, 1961, pp 81-96; and Hance et al., Weed Control Handbook, 8th Ed., Blackwell Scientific Publications, Oxford, 1989.
В следующих примерах все проценты являются процентами по весу (мас.%) и все готовые формы готовят общепринятыми способами. Номера соединений относятся к соединениям в Таблице Индексов А.
Соединения данного изобретения отличаются благоприятным характером метаболического и/или почвенного остаточного распределения и обнаруживают активность подавления спектра сельскохозяйственных и несельскохозяйственных беспозвоночных вредителей. (В данном контексте это выражение "подавление беспозвоночных вредителей" означает ингибирование развития беспозвоночных вредителей (в том числе их умерщвление), которое вызывает значимое уменьшение поедания или другого повреждения или ущерба, вызываемого этими вредителями; родственные выражения определяются аналогично). Термин "беспозвоночный вредитель", используемый в данном изобретении, включает в себя членистоногих, брюхоногих моллюсков и нематод (круглых червей) экономического значения в качестве вредителей. Термин "членистоногие" (Arthropoda) включает в себя насекомых, клещей, пауков, скорпионов, губоногих (многоножек), двупарноногих, мокриц и симфилл. Термин "брюхоногие моллюски" (Gastropoda) включает в себя улиток, слизней и других Stylommatophora. Термин "нематода" (круглые черви) включает в себя гельминтов, таких как: круглые черви, гельминты, паразитирующие в сердце, и растительноядные нематоды (Nematoda), трематоды (Trematoda), скребни (Acanthocephala) и ленточные черви (Cestoda). Специалистам в данной области должно быть понятно, что не все соединения являются равным образом эффективными против всех вредителей. Соединения данного изобретения проявляют активность против экономически важных сельскохозяйственных и несельскохозяйственных вредителей. Термин "сельскохозяйственные" относится к продукции полевых культур, например, для пищевых продуктов и волокон, и включает в себя выращивание зерновых культур (например, пшеницы, овса, ячменя, ржи, риса, кукурузы), сои, овощных культур (например, салата латука, капусты, томатов, фасоли), картофеля, сладкого картофеля (батата), винограда, хлопка, и получению плодов плодовых деревьев (например, мясистых (семечковых) плодов, косточковых плодов (костянок) и плодов цитрусовых или померанцевых культур). Термин "несельскохозяйственные" относится к другим садоводческим растениям (например, лесным, тепличным растениям, выращиваемым в питомниках растениям или декоративным растениям, не растущим в поле), общественному здоровью (человека) и здоровью животных, структурам для одомашнивания животных и коммерческим структурам, домашнему хозяйству и к применениям для хранящихся продуктов или вредителям. Ввиду широкого спектра подавления беспозвоночных вредителей и экономической важности защита (от ущерба или повреждения, вызываемых беспозвоночными вредителями) сельскохозяйственных культур хлопчатника, кукурузы, сои, риса, овощных культур, картофеля, сладкого картофеля (батата), винограда и плодов плодовых деревьев посредством уничтожения беспозвоночных вредителей является предпочтительными вариантами данного изобретения. Сельскохозяйственные или несельскохозяйственные вредители включают в себя личинки (гусеницы) отряда Lepidoptera (чешуекрылые (бабочки)), такие как "походные (ратные) черви", т.е. гусеницы некоторых насекомых, образующие крупные, подвижные скопления), насекомые, подгрызающие растения, пяденицы и совки в семействе Noctuidae (например, совка травяная (Spodoptera fugiperda J.E. Smith), совка малая (Spodoptera exigua Hübner), совка ипсилон (Agrotis ipsilon Hufnagel), моль капустная (совка ни) (Trichoplusia ni Hübner), совка табачная (Heliothis virescens Fabricius)); сверлильщики (точильщики), чехлоноски, гусеницы, строящие паутинное гнездо, конусообразные черви, мерметиды (Mermithidae) и вредители, скелетирующие листья (оставляющие только жилки), из семейства Pyralidae (например, мотылек кукурузный (Ostrinia nubilalis Hübner), гусеница, поражающая апельсин Навель (пупочный апельсин) (Amyelois transitella Walker), совка огневая (Crambus caliginosellus Clemens), луговой мотылек (Herpetogramma licarsisalis Walker)); листовертки, гусеницы листовертки-почкоеда и гусеницы - вредители семян и гусеницы - вредители плодов (плодожорки) в семействе Tortricidae (например, плодожорка яблонная (Cydia pomonella Linnaeus), листовертка виноградная (Endopiza viteana Clemens), листовертка восточная персиковая (Grapholita molesta Busck)); и многие другие экономически важные чешуекрылые (например, моль капустная (Plutella xylostella Linnaeus), розовый коробочный червь хлопчатника (Pectinophora gossypiella Saunders), шелкопряд непарный (непарник) (Lymantria dispar Linnaeus)); нимфы и взрослые особи отряда Blattodea (Тараканы), в том числе тараканы из семейств Blattellidae и Blattidae (например, таракан черный (Blatta orientalis Linnaeus), таракан азиатский (Blatella asahinai Mizukubo), таракан рыжий (прусак) (Blattella germanica Linnaeus), таракан коричнево-полосатый (Supella longipalpa Fabricius), таракан американский (Periplaneta americana Linnaeus), таракан коричневый (Periplaneta brunnea Burmeister), таракан мадейрский (Leucophaea madera Fabricius)); питающиеся листвой гусеницы и взрослые особи отряда Coleoptera (жесткокрылые), в том числе долгоносики (слоники) из семейств Anthribidae, Bruchidae и Curculionidae (например, долгоносик хлопковый (Anthonomus grandis Boheman), долгоносик рисовый водяной (Lissorhoptrus oryzophilus Kuschel), долгоносик амбарный (слоник зерновой) (Sitophilus granarius Linnaeus), долгоносик рисовый (Sitophillus oryzae Linnaeus)); земляные (огородные) мошки, жуки-блошки огуречные, листоеды-корнееды, листоеды, жуки картофельные и минирующие мушки (мушки-минеры) в семействе Chrysomelidae (например, колорадский жук (Leptinotarsa decemlineata Say), блошка кукурузная (Diabrotica virgifera virgifera LeConte)); хрущи и другие жуки из семейства Scaribaeidae (например, хрущик японский (Popillia Japonica Newman) и хрущик европейский (Rhizotrogus majalis Razoumowsky)); кожееды из семейства Dermestidae; жуки-щелкуны из семейства Elateridae; жуки-короеды из семейства Scolytidae и хрущаки мучные большие из семейства Tenebrionidae. Кроме того, сельскохозяйственные и несельскохозяйственные вредители включают в себя: взрослых особей и личинок (гусениц) отряда Dermaptera (уховертки), в том числе уховерток из семейства Forficulidae (например, уховертку обыкновенную (Forficula auricularia Linnaeus), уховертку черную (Chelisoches morio Fabricius)); взрослых особей и нимф отрядов Hemiptera (полужесткокрылые (клопы)) и Homoptera (равнокрылые), таких как клопы-слепняки из семейства Miridae, цикады из семейства Cicadidae, цикадки (например, Empoasca spp.) их семейства Cicadellidae, кобылки и дельфациды из семейств Fulgoroidae и Delphacidae, горбатки из семейства Membracidae, листоблошки из семейства Psyllidae, белокрылки из семейства Aleyrodidae, тля из семейства Aphididae, филлоксера из семейства Phylloxeridae, мучнистые червецы (войлочники) из семейства Pseudococcidae, щитовки (червецы) из семейств Coccidae, Diaspididae и Margarodidae, клопы-кружевницы из семейства Tingidae, клопы-щитники из семейства Pentatomidae, клопы белокрылые (например, Blissus spp.) и другие вредители семян из семейства Lygaeidae, пенницы из семейства Cercopidae, клопы тыквенные из семейства Coreidae и красноклопы и красноклопы хлопковые из семейства Pyrrhocoridae. В перечень вредителей включены также взрослые особи и личинки отряда Acari (акаровые клещи), такие как клещи паутинные и клещи красные в семействе Tetranychidae (например, клещ красный европейский (Panonychus ulmi Koch), клещ двупятнистый паутинный (Tetranychus urticae Koch), клещ МакДаниеля (Tetranychus mcdanieli McGregor)), плоскотелки в семействе Tenuipalpidae (например, плоскотелка цитрусовая (Brevipalpus lewisi McGregor)), клещи галлообразующие и почковые в семействе Eriophydae и другие питающиеся листвой клещи и клещи, влияющие на здоровье человека и животных, т.е. клещи домашней пыли в семействе Epidermoptidae, железницы (угрицы) в семействе Demodicidae, клещи зерновые в семействе Glycyphagidae, клещи иксодовые в семействе Ixodidae (например, клещ олений (Ixodes scapularis Say), клещ паралитический австралийский (Ixodes holocyclus Neumann), клещ собачий американский (Dermacentor variabilis Say), клещ амблиома (Amblyomma americanum Linnaeus) и клещи конские и зудни чесоточные в семействах Psoroptidae, Pyemotidae и Sarcotidae; взрослые и не развитые полностью особи отряда Orthoptera (прямокрылые), в том числе кузнечиковые, саранчовые и сверчки (например, кобылки мексиканские (например, Melanoplus sanguinipes Fabricius, M. differentialis Thomas), саранча американская (например, Schistocerca americana Drury), саранча пустынная (Schistocerca gregaria Forskal), саранча перелетная (Locusta migratoria Linnaeus), сверчок домовой (Acheta domesticus Linnaeus), медведки обыкновенные (рак земляной) (Gryllotalpa spp.)); взрослые особи и не развитые полностью особи отряда Diptera (двукрылые), в том числе минирующие мушки, галлицы, плодовые мушки (Tephritidae), шведские мушки (например, Oscinella frit Linnaeus), почвенные личинки мух, мухи комнатные (например, Musca domestica Linnaeus), мухи малые комнатные (например, Fannia canicularis Linnaeus, F. femoralis Stein), жигалки обыкновенные (жигалки осенние) (например, Stomoxis calcitrans Linnaeus), мухи осенние, жигалки коровьи малые, мухи мясные синие (например, Chrysomya spp., Phormia spp.), и другие настоящие мухи-вредители, оводы лошадиные (например, Tabanus spp.), оводы носоглоточные лошадиные (например, Gastrophilus spp., Oestrus spp.), личинки бычьего полосатого овода (например, Hypoderma spp.), мухи оленьи (например, Chrysops spp.), кровососки (например, Melophagus ovinus Linnaeus) и другие Brachycera (короткоусые), москиты (комары) (например, Aedes spp., Anopheles spp., Culex spp.), мошки черные (например, Prosimulium spp., Simulium spp.), кусающие мокрецы, мошки, комарики, и другие Nematocera; взрослые и не развитые полностью особи отряда Thysanoptera (трипсы), в том числе трипсы луковые (табачные) (Thrips tabaci Lindeman) и другие питающиеся листвой трипсы; насекомые-вредители отряда Hymenoptera (перепончатокрылые), в том числе муравьи (например, муравей-древоточец красный (Camponotus ferruginous Fabricius), муравей-древоточец черный (Camponotus pennsylvanicus De Geer), фараонов муравей (Monomorium pharaonis Linnaeus), васмания (Wasmannia auropunctata Roger), муравей Рихтера (Solenopsis geminata Fabricius), муравей лесной рыжий (Solenopsis invicta Buren), муравей аргентинский (Iridomyrmex humilis Mayr), паратрехина (Paratrechina longicornis Latreille), муравей дерновый (Tetramorium caespitum Linnaeus), лазий американский (Lasius alienus Forster), муравей пахучий домашний (Tapinoma sessile Say)), пчелы (в том числе пчелы-плотники), шершни, золотистые каранксы и осы; насекомые-вредители отряда Isoptera (термиты), в том числе термит желтоногий средиземноморский (Reticulitermes flavipes Kollar), термит средиземноморский западный (Reticulitermes hesperus Banks), термит средиземноморский тайванский (Coptotermes formosanus Shiraki), термит древоядный западно-индийский (Incisitermes immigrans Snyder) и другие термиты экономического значения; насекомые-вредители отряда Thysanura (щетинохвостки), такие как чешуйница обыкновенная (Lepisma saccharina Linnaeus) и чешуйница домашняя (Thermobia domestica Packard); насекомые-вредители отряда Mallophaga (пухоеды) и, в том числе, вошь головная (Pediculus humanus capitis De Geer), вошь платяная (Pediculus humanus humanus Linnaeus), пухоед (Menacanthus stramineus Nitszsch), вошь собачья (Trichodectes canis De Geer), пухоед куриный перстробрюхий (Goniocotes gallinae De Geer), власоед овечий (Bovicola ovis Schrank), вошь крупного рогатого скота коротконосая кровососущая (Haematopinus eurysternus Nitzsch), вошь крупного рогатого скота длинноносая кровососущая (Linognathus vituli Linnaeus) и другие кровососущие вши и пухоеды-паразиты, которые атакуют людей и животных; насекомые-вредители отряда Siphonoptera (блохи), в том числе блоха крысиная (Xenopsylla cheopis Rotschild), блоха кошачья (Ctenocephalides felis Bouche), блоха собачья (Ctenocephalides canis Curtis), блоха куриная (Ceratophyllus gallinae Schrank), блоха присасывающаяся (Echidnophaga gallinacea Westwood), блоха человеческая (Pulex irritans Linnaeus) и другие блохи, беспокоящие млекопитающих и птиц. Дополнительные охватываемые членистоногие вредители включают в себя: пауков отряда Агапеае (пауки), таких как паук-отшельник коричневый (Loxosceles reclusa Gertsch & Mulaik) и паук-ткач черный ("черная вдова") (Latrodectus mactans Fabricius), и губоногих в отряде Scutigeromorpha (Скутигеры), таких как мухоловка обыкновенная (Scutigera coleoptrata Linnaeus). Соединения данного изобретения являются также активными в отношении членов классов Nematoda (Нематоды, Круглые черви), Cestoda (цестоды, ленточные черви), Trematoda (трематоды) и Acanthocephala (скребни), включающие в себя экономически важных членов отрядов Strongylida, Ascaridida, Oxyurida, Rhabditida, Spirurida и Enoplida, таких как, но не только, экономически важные сельскохозяйственные вредители (т.е. образующие корневые наросты, галловые нематоды в роде Meloidogyne, повреждающие нематоды в роде Pratylenchus, вызывающие повреждения корней нематоды в роде Trichodoris, и т.д.) и влияющие на здоровье животных и человека вредители (т.е. все экономически важные трематоды, ленточные черви и круглые черви, такие как Strongylus vulgaris в лошадях, Toxocara canis в собаках, Haemonchus contortus в овцах, Dirofilaria immitis Leidy в собаках, Haemonchus contortus в овцах, Dirofilaria immitis Leidy в собаках, Anoplocephala perfoliata в лошадях, Fasciola hepatica Linnaeus в жвачных, и т.д.)
Соединения данного изобретения обнаруживают особенно высокую активность против вредителей отряда Lepidoptera (чешуекрылые) (например, Alabama agrillacea Hübner (гусеницы совки хлопковой американской), Archips argyrospila Walker (листовертки плодовых деревьев), A. rosana Linnaeus (гусеницы горностаевой моли) и других видов Archips, Chilo suppressalis Walker (сверлильщика рисового стеблевого), Cnaphalocrosis medinalis Guenee (листовертки рисовой), Crambus caliginosellus Clemens (бабочки-огневки корней кукурузы), Crambus teterrellus Zincken (огневки мятличной), Cydia pomonella Linnaeus (плодожорки яблоневой), Earias insulana Boisduval (шиповатого червя), Earias vitella Fabricius (совки пятнистой), Helicoverpa armigera Hübner (совки американской), Helicoverpa zea Boddie (совки хлопковой), Heliothis virescens Fabricius (совки табачной), Herpetogramma licarsisalis Walker (лугового мотылька), Lobesia botrana Denis & Schiffermüller (листовертки гроздевой (виноградной), Pectinophora gossypiella Saunders (розового коробочного червя хлопчатника), Phyllocnistis citrella Stainton (сокоедки цитрусовой), Pieris brassicae Linnaeus (белянки капустной), Pieris rapae Linnaeus (белянки репной (репницы)), Plutella xylostella Linnaeus (моли капустной), Spodoptera exigua Hübner (походный червь свекольный), Spodoptera litura Fabricius (совки табачной, коконопряда, гусеницы которого строят общие паутинные гнезда), Spodoptera frugiperda J.E. Smith (совки травяной), Trichoplusia ni Hübner (совки капустной, совки ни) и Tuta absoluta Meyrick (мушки минирующей томатной)). Соединения данного изобретения проявляют также коммерчески значимую активность на членах из отряда Homoptera (равнокрылые), включающих в себя: Acyrthisiphon pisum Harris (тлю гороховую), Aphis craccivora Koch (тлю люцерновую), Aphis fabae Scopoli (тлю бобовую), Aphis gossypii Glover (тлю хлопковую, тлю бахчевую), Aphis pomi De Geer (тлю яблонную), Aphis spiraecola Patch (тлю таволговую), Aulacorthum solani Kaltenbach (тлю вьюнковую), Chaetosiphon fragaefolii Cockerell (тлю земляничную), Diuraphus noxia Kurdjumov/MordviIko (тлю пшеничную русскую), Dysaphis plantaginea Paaserini (тлю яблоневую розовую), Eriosoma lanigerum Hausmann (тлю яблонную кровяную), Hyalopterus pruni Geoffroy (тлю сливовую опыленную), Lipaphis erysimi Kaltenbach (тлю ложнокапустную), Metopolophium dirrhodum Walker (тлю злаковую), Macrosipum euphorbiae Thomas (тлю картофельную листовую), Myzus persicae Sulzer (тлю персиковую-картофельную, тлю персиковую зеленую), Nasanovia ribisnigri Mosley (тлю салатную). Pemphigus spp. (корневую тлю и галловую тлю), Rhopalosiphum maidis Fitch (тлю кукурузную листовую), Rhopalosiphum padi Linnaeus (тлю черемуховую-овсовую), Schizaphis graminum Rondani (тлю злаковую обыкновенную), Sitobion avenae Fabricius (тлю листовую), Therioaphis maculata Buckton (тлю люцерновую пятнистую), Toxoptera aurantii Boyer de Fonscolombe (тлю цитрусовую черную) и Toxoptera citricida Kirkaldy (тлю цитрусовую коричневую); Adelges spp. (хермес, адельгес); Phylloxera devastatrix Pergande (филлоксеру гикори), Bemisia tabaci Gennadius (белокрылку табачную, белокрылку батата (сладкого картофеля)), Bemisia argentifolii Bellows & Perring (белокрылку магнолиевую), Dialeurodes citri Ashmead (белокрылку цитрусовую) и Trialeurodes vaporariorum Westwood (белокрьшку тепличную); Empoasca fabae Harris (цикадку картофельную), Laodelphax striatellus Fallen (цикадку малую коричневую), Macrolestes quadrilineatus Forbes (цикадку астровую), Nephotettix cinticeps Uhler (цикадку зеленую), Nephotettix nigropictus Stal (цикадку рисовую), Nilaparvata lugens Stal (цикадку коричневую), Peregrinus maidis Ashmead (цикадку кукурузную), Sogatella furcifera Horvath (цикадку белоспинную), Sogatodes orizicola Muir (дельфацида рисового), Typhlocyba pomaria McAtee (цикадку яблонную), Erythroneoura spp. (цикадки виноградные); Magicidada septendecim Linnaeus (цикадку 17-летнюю); Icerya purchasi Maskell (червеца австралийского чешуйчатого), Quadraspidiotus perniciosus Comstock (щитовку калифорнийскую), Planococcus citri Risso (червеца цитрусового); Pseudococcus spp. (комплекс других червецов); Cacopsylla pyricola Foerster (медяницу грушевую), Trioza diospyri Ashmead (листоблошку хурмовую). Эти соединения проявляют также активность на членах из отряда Hemiptera (полужесткокрылые (клопы)), включающих в себя: Acrosternum hilare Say (щитника зеленого), Anasa tristis De Geer (клопа-ромбовника печального), Blissus leucopterus leucopterus Say (клопа белокрылого), Corythuca gossypii Fabricius (клопа хлопкового), Cyrtopeltis modesta Distant (клопа томатного), Dysdercus suturellus Herrich-Schäffer (красноклопа хлопкового), Euchistus servus Say (клопа-щитника коричневого), Euchistus variolarius Palisot de Beauvois (клопа-щитника с одним пятном), Graptosthetus spp. (комплекс клопов-вредителей семян), Leptoglossus corculus Say (клопа-краевика семян сосны), Lygus lineolaris Palisot de Beauvois (клопика лугового), Nezara viridula Linnaeus (клопа хлопково-огородного), Oebalus pugnax Fabricius (клопа рисового), Oncopeltus fasciatus Dallas (клопа молочайного), Pseudatomoscelis seriatus Reuter (клопа прыгающего хлопкового). Другие отряды насекомых, подавляемые соединениями данного изобретения, включают в себя отряд Thysanoptera (трипсы) (например, Frankliniella occidentalis Pergande (трипса западного), Scirthothrips citri Moulton (трипса цитрусового), Sericothrips variabilis Beach (трипса соевого) и Thrips tabaci Lindeman (трипса лукового, трипса табачного); и отряд Coleoptera (жесткокрылые) (например, Leptinotarsa decemlineata Say (колорадского жука), Epilachna varivestis Mulsant (жука фасолевого мексиканского) и личинки щелкунов родов Agriotes, Athous или Limonius).
Соединения данного изобретения могут быть также смешаны с одним или несколькими другими биологически активными соединениями или агентами, в том числе инсектицидами, фунгицидами, нематоцидами, бактерицидами, акарицидами, регуляторами роста, такими как стимуляторы укоренения, химиостерилизаторы, полухимикалии, репелленты, аттрактанты, феромоны, стимуляторы питания, другие биологически активные соединения или энтомопатогенные бактерии, вирус или грибы, для образования многокомпонентного пестицида, обеспечивающего еще более широкий спектр сельскохозяйственной применимости. Таким образом, данное изобретение относится также к композиции, содержащей биологически эффективное количество соединения формулы 1 и эффективное количество по меньшей мере одного дополнительного биологически активного соединения или агента, и эта композиция может содержать дополнительно по меньшей мере одно поверхностно-активное вещество, по меньшей мере один твердый разбавитель или жидкий разбавитель. Примерами таких биологически активных соединений или агентов, с которыми могут быть приготовлены соединения данного изобретения, являются: инсектициды, такие как абамектин, ацефат, ацетамиприд, амидофлумет (S-1955), авермектин, азадирахтин, азинофос-метил, бифентрин, бинфеназат, бупрофезин, карбофуран, хлорфенапир, хлорфлуазурон, хлорпирифос, хлорпирифос-метил, хромафенозид, клотианидин, цифлутрин, бета-цифлутрин, цигалотрин, лямбда-цигалотрин, циперметрин, циромазин, дельтаметрин, диафентиурон, диазинон, дифлубензурон, диметоат, диофенолан, эмамектин, эндосульфан, эсфенвалерат, этипрол, фенотикарб, феноксикарб, фенпропатрин, фенвалерат, фипронил, флоникамид, флуцитринат, тау-флувалинат, флуфенерим (UR-50701), флуфеноксурон, фонофос, галофенозид, гексафлумурон, имидаклоприд, индоксакарб, изофенфос, луфенурон, малатион, метальдегид, метамидофос, метидатион, метомил, метопрен, метоксихлор, монокротофос, метоксифенозид, нитиазин, новалурон, новифлумурон (XDE-007), оксамил, паратион, паратион-метил, перметрин, форат, фосалон, фосмет, фосфамидон, пиримикарб, профенофос, пиметрозин, пиридалил, пирипроксифен, ротенон, спиносад, спиромезифин (BSN 2060), сульпрофос, тебуфенозид, тефлубензурон, тефлутрин, тербуфос, тетрахлорвинфос, тиаклоприд, тиаметоксам, тиодикарб, тиосультап-натрий, тралометрин, трихлорфон и трифлумурон; фунгициды, такие как ацибензолар, азоксистробин, беномил, бластицидин-3, бордосская жидкость (трехосновный сульфат меди), бромуконазол, карпропамид, каптафол, каптан, карбендазим, хлоронеб, хлороталонил, оксихлорид меди, соли меди, цифлуфенамид, цимоксанил, ципроконазол, ципродинил, (S)-3,5-дихлор-N-(3-хлор-1-этил 1-метил-2-оксопропил)-4-метилбензамид (RH 7281), диклоцимет (S-2900), дикломезин, диклоран, дифеноконазол, (S)-3,5-дигидро-5-метил-2-(метилтио)-5-фенил-3-(фениламино)-4Н-имидазол-4-он (RP 407213), диметоморф, димоксистробин, диниконазол, диниконазол-М, додин, эдифенфос, эпоксиконазол, фамоксадон, фенамидон, фенаримол, фенбуконазол, фенкарамид (SZX0722), фенпиклонил, фенпропидин, фенпропиморф, фентинацетат, фентингидроксид, флуазинам, флудиоксонил, флуметовер (RPA 403397), флуморф/флуморлин (SYP-L190), флуоксастробин (НЕС 5725), флухинконазол, флузилазол, флутоланил, флутриафол, фольпет, фозетил-алюминий, фуралаксил, фураметапир (S-82658), гексаконазол, ипконазол, ипробенфос, ипродион, изопротиолан, казугамицин, крезоксим-метил, манкозеб, манеб, мефеноксам, мепронил, металаксил, метконазол, метоминостробин/феноминостробин (SSF-126), метрафенон (АС375839), миклобутанил, нео-азоцин (метанарсонат железа (III)), никобифен (BAS 510), оризастробин, оксадиксил, пенконазол, пенцикурон, пробеназол, прохлораз, пропамокарб, пропиконазол, прохиназид (DPX-KQ926), протиоконазол (JAU 6476), пирифенокс, пираклостробин, пириметанил, пирохилон, хиноксифен, спироксамин, сера, тебуконазол, тетраконазол, тиабендазол, тифлузамид, тиофанат-метил, тирам, тиадинил, триадимефон, триадименол, трициклазол, трифлоксистробин, тритиконазол, валидамицин и винклозолин; нематоциды, такие как алдикарб, оксамил и фенамифос; бактерициды, такие как стерптомицин; акарициды, такие как амитраз, хинометионат, хлорбензилат, цигексатин, дикофол, диенохлор, этоксазол, феназахин, оксид фенбутатина, фенпропатрин, фенпироксимат, гекситиазокс, пропаргит, пиридабен и тебуфенпирад; и биологические агенты, такие как Bacillus thuringiensis, в том числе подвиды aizawai и kurstaki, дельта-эндотоксин Bacillus thuringiensis, бакуловирус, и энтомопатогенные бактерии, вирус и грибы. Соединения данного изобретения и их композиции могут наноситься на растения, генетически трансформированные для экспрессии протеинов, токсичных в отношении беспозвоночных вредителей (таких как токсин Bacillus thuringiensis). Действие экзогенно нанесенных соединений данного изобретения, подавляющих беспозвоночных вредителей, может быть синергическим с экспрессируемыми белками токсинов.
Общей ссылкой в отношении этих сельскохозяйственных защитных агентов является The Pesticide Manual, 12th Edition, C.D.S. Tomlin, Ed., British Crop Protection Council, Farnham, Surrey, U.K., 2000.
Предпочтительные инсектициды и акарициды для смешивания с соединениями этого изобретения включают в себя пиретроиды, такие как циперметрин, цигалотрин, цифлутрин, бета-цифлутрин, эсфенвалерат, фенвалерат и тралометрин; карбаматы, такие как фенотикарб, метомил, оксамил и тиодикарб; неоникотиноиды, такие как клотианидин, имидаклоприд и тиаклоприд; блокаторы нейронных натриевых каналов, такие как индоксакарб; инсектицидные макроциклические лактоны, такие как спиносад, абамектин, авермектин и эмамектин; антагонисты γ-аминомасляной кислоты (GABA), такие как эндосульфан, этипрол и фипронил; инсектицидные мочевины, такие как флуфеноксурон и тирфлумурон; миметики ювенильных гормонов, такие как диофенолан и пирипроксифен; пиметрозин; и амитраз. Предпочтительные биологические агенты для смешивания с соединениями данного изобретения включают в себя Bacillus thuringiensis и дельта-эндотоксин Bacillus thuringiensis, а также природно встречающиеся и генетически модифицированные вирусные инсектициды, в том числе члены семейства Baculoviridae, а также насекомоядные грибы.
Наиболее предпочтительные смеси включают в себя смесь соединения данного изобретения с цигалотрином; смесь соединения данного изобретения с бета-цифлутрином; смесь соединения данного изобретения с эсфенвалератом; смесь соединения данного изобретения с метомилом; смесь соединения данного изобретения с имидаклопридом; смесь соединения данного изобретения с тиаклопридом; смесь соединения данного изобретения с индоксакарбом; смесь соединения данного изобретения с абамектином; смесь соединения данного изобретения с эндосульфаном; смесь соединения данного изобретения с этипролом; смесь соединения данного изобретения с фипронилом; смесь соединения данного изобретения с флуфеноксуроном; смесь соединения данного изобретения с пирипроксифеном; смесь соединения данного изобретения с пиметрозином; смесь соединения данного изобретения с амитразом; смесь соединения данного изобретения с Bacillus thuringiensis и смесь соединения данного изобретения с дельта-эндотоксином Bacillus thuringiensis.
В некоторых случаях для преодоления резистентности вредителей особенно предпочтительными будут комбинации с другими соединениями для борьбы с беспозвоночными вредителями. Таким образом, композиция согласно изобретению может дополнительно содержать биологически эффективное количество по меньшей мере одного дополнительного соединения или агента для борьбы с беспозвоночными вредителями, имеющего сходный спектр уничтожения вредителей, но отличающийся механизм действия. Контактирование растения, генетически модифицированного для экспрессии защищающего растение соединения (например, белка), или локуса этого растения с биологически эффективным количеством соединения данного изобретения может также обеспечивать более широкий спектр защиты растений и быть полезным для преодоления резистентности вредителей.
Количественные соотношения при использования таких соединений либо агентов вместе с соединением, описываемым формулой 1, определяются нормами применения последних, хорошо известными специалистам из общедоступных источников информации, в частности вышеупомянутого справочного источника (1) "The Pesticide Manual", а также в других общих справочных руководствах (подробные выходные данные цитируемых справочных руководств приведены ниже после таблицы 4).
Приведенная ниже Таблица 4 включает данные по нормам применения известных биологически активных соединений либо агентов в соответствии с указанными общедоступными источниками информации и рассчитанные массовые соотношения для использования таких соединений либо агентов вместе с соединением, описываемым формулой 1. В таблице 4 представлены наименование известного биологически активного соединения либо агента (столбец 1), класс, к которому он относится по типу своего пестицидного действия (столбец 2), норму внесения в граммах на гектар (г/га) для известного биологически активного соединения либо агента (столбец 3), ссылку на известный справочный источник, из которого получена норма внесения (столбец 4) и математически полученное соотношение по массе между соединением, описываемым формулой 1, и известным биологически активным соединением либо агентом (столбец 5).
Например, вторая строка в таблице 4 показывает, что абамектин - макроциклический лактон - характеризуется нормой внесения 5-28 г/га. Данная информация имеется в справочном источнике 1 - "The Pesticide Manual". Массовое соотношение между соединением, описываемым формулой 1, и абамектином приводится в виде диапазона в пределах от 400:1 до 1:2,8. Первое соотношение 400:1 получают в результате деления наибольшей нормы для соединения, описываемого формулой 1 (2000 г/га), на наименьшую норму для абамектина (5 г/га). Подобно этому деление наибольшей нормы для абамектина (28 г/га) на наименьшую норму для соединения, описываемого формулой 1, (10 г/га) дает в результате соотношение 1:2,8. Массовые соотношения, перечисленные в таблице 4 для других известных биологически активных соединений либо агентов, рассчитаны аналогичным образом.
(1) The Pesticide Manual, 12th Edition, C.D.S. Tomlin, Ed., British Crop Protection Council, Farnham, Surrey, UK., 2000.
(2) Global Insecticide Directory, 2nd Edition, R.Bryant, M.G.Bite and W.L.Hopkins, Ed., Agranova, Orpington, Kent, UK., 1999.
(3) Insect and Disease Control Guide, Vol. 1, R.Т.Meister Ed., Meister Publishing Company, Willoughby, OH, USA, 1999.
ПРИМЕР Е
В таблице Е перечисляются конкретные композиции, содержащие соединение, описываемое формулой 1, вместе с другими биологически активными компонентами, предназначенные для подавления развития беспозвоночных сельскохозяйственных вредителей в соответствии со способами настоящего изобретения. Номера соединений, перечисленные в первом столбце таблицы Е, соответствуют номерам соединений, приведенным в ТАБЛИЦЕ ИНДЕКСОВ А.
Во втором столбце таблицы Е перечисляются конкретные дополнительные биологически активные компоненты (например, "абамектин" в первой строке). В третьем столбце таблицы Е приводится класс, к которому относится конкретное соединение. В четвертом столбце таблицы Е приводится типичный диапазон массовых соотношений для количеств, в которых используют дополнительный компонент в отношении к соединению, описываемому формулой 1, (например, "От 50:1 до 1:10" абамектина по отношению к соединению 1 при расчете на массу). Таким образом, первая строка таблицы Е, например, конкретно описывает комбинацию соединения, описываемого формулой 1, - соединения 1 - и абамектина, и в ней указывается, что абамектин является представителем макроциклических лактонов, и сообщается, что абамектин и соединение 1 обычно используют при массовом соотношении в диапазоне от 50:1 до 1:10. Информацию в остальных строках в таблице Е следует толковать подобным же образом.
Таблица Е
Композиция, содержащая соединение, описываемое формулой 1, и, по меньшей мере, одно дополнительное биологически активное соединение либо агент.
Смеси, содержащие комбинации активных компонентов, перечисленных в таблице Е, с массовыми соотношениями 1:1, составляли при использовании раствора, содержащего 10% ацетона, 90% воды и 300 ч./млн неионного поверхностно-активного вещества Х-77® Spreader Lo-Foam Formula, содержащего алкиларилполиоксиэтилен, свободные жирные кислоты, гликоли и изопропанол, (от компании Loveland Industries, Inc.), и испытывали в соответствии с протоколом, описанным для ИСПЫТАНИЯ А, в котором концентрация каждого активного компонента составляла 50 ч./млн Каждая из смесей обеспечивала достижение превосходных уровней защиты растений (10% или менее повреждений вследствие поедания).
Беспозвоночные вредители уничтожаются при сельскохозяйственных и несельскохозяйственных применениях посредством нанесения одного или нескольких соединений данного изобретения в эффективном количестве на среду этих вредителей, в том числе на сельскохозяйственный и/или несельскохозяйственный очаг заражения вредителями, на подлежащую защите зону или непосредственно на подлежащих уничтожению вредителей. Таким образом, данное изобретение дополнительно относится к способу борьбы с беспозвоночными вредителями, предусматривающему контактирование беспозвоночных вредителей или окружающей их среды с биологически эффективным количеством одного или нескольких соединений данного изобретения или с композицией, содержащей по меньшей мере одно такое соединение, или с композицией, содержащей по меньшей мере одно такое соединение и эффективное количество по меньшей мере одного дополнительного биологически активного соединения или агента. Примеры подходящих композиций, содержащих соединение данного изобретения и эффективное количество по меньшей мере одного дополнительного биологически активного соединения или агента, включают в себя гранулированные композиции, в которых дополнительное биологически активное соединение или агент присутствует на той же самой грануле, что и соединение данного изобретения, или на гранулах, присутствующих отдельно от гранул соединения данного изобретения.
Предпочтительным способом контакта является опрыскивание. Альтернативно, гранулированная композиция, содержащая соединение данного изобретения, может наноситься на листву растения или на почву. Соединения данного изобретения эффективно доставляются также через поглощение растением посредством контактирования этого растения с композицией, содержащей соединение данного изобретения, применяемой в виде пропитывания почвы жидкой готовой формой, внесения гранулированной готовой формы в почву, обработки ящика для саженцов или погружения рассады. Соединения являются также эффективными при местном нанесении композиции, содержащей соединение данного изобретения, на очаг заражения вредителями. Другие способы контактирования включают в себя нанесение соединения или композиции данного изобретения с использованием растворов для контактного опрыскивания прямого действия или растворов с последействием, воздушных растворов для опрыскивания, гелей, дражирования семян (нанесения на семена), микроинкапсулирования, системного поглощения, приманок, клипс, болюсов, туманообразователей, фумигаитов, аэрозолей, дустов и многих других средств. Соединениями данного изобретения могут быть также импрегнированы материалы для изготовления устройств для борьбы с беспозвоночными вредителями (например, сеток для насекомых).
Соединения данного изобретения могут быть включены в приманки, которые поедаются беспозвоночными, или могут находиться в таких устройствах, как ловушки и т.п. Гранулы или приманки, содержащие 0,01-5% активного ингредиента, 0,05-10% удерживающего влагу агента (агентов) и 40-99% овощной муки, являются эффективными в уничтожении почвенных насекомых при очень низких нормах применения, в частности, при дозах активного ингредиента, которые являются летальными при проглатывании, а не при прямом контакте.
Соединения данного изобретения могут применяться в чистом виде, но наиболее часто применяют готовую форму, содержащую одно или несколько соединений с подходящими носителями, разбавителями и поверхностно-активными веществами и, возможно, в комбинации с кормом, в зависимости от предполагаемого конечного использования. Предпочтительный способ применения включает в себя разбрызгивание водной дисперсии или раствора в рафинированном масле этих соединений. Комбинации с разбрызгиваемыми маслами, концентрации разбразгиваемых масел, повышающие липкость агенты для разбрасывателей, адъюванты, другие растворители и синергисты, такие как пиперонилбутоксид, часто повышают эффективность соединений.
Норма нанесения, требующаяся для эффективного подавления (т.е. "биологически эффективное количество"), будет зависеть от таких факторов, как вид беспозвоночного, подлежащего уничтожению, жизненный цикл вредителя, стадия жизни, его размер, местоположение, время года, культура- или животное-хозяин, "кормовое" поведение, поведение при спаривании, влажность, температура окружающей среды, и т.п. При нормальных обстоятельствах нормы применения приблизительно 0,01-2 кг активного ингредиента на гектар являются достаточными для уничтожения вредителей в сельскохозяйственных экологических системах, но может быть достаточным такое небольшое количество, как 0,0001 кг/гектар, или может потребоваться такое большое количество, как 8 кг/гектар. Для несельскохозяйственных применений эффективные нормы применения будут находиться в диапазоне от приблизительно 0,1 до 50 мг/квадратный метр, но может быть достаточным такое небольшое количество, как 0,1 мг/квадратный метр, или может потребоваться такое большое количество, как 150 мг/квадратный метр. Специалист в данной области может легко определить биологически эффективное количество, необходимое для желаемого уровня подавления беспозвоночного вредителя.
Следующие Тесты в разделе Биологические примеры данного изобретения демонстрируют эффективность подавления соединениями данного изобретения на конкретных вредителях. "Эффективность подавления" представляет ингибирование развития членистоногих (в том числе их смертность), которое вызывает значимо уменьшенное поедание. Однако защита от вредителей, осуществляемая этими соединениями, не ограничивается этими видами. См. Таблицу Индексов А в отношении описаний соединений. В этой таблице индексов используются следующие аббревиатуры: t означает третичный, n означает нормальный, i означает изо, s означает вторичный. Me означает метил, Et означает этил, Pr означает пропил и Bu означает бутил; соответственно, i-Pr означает изопропил, sBu означает вторичный бутил и т.д. Аббревиатура "Ех" означает "Пример" и сопровождается номером, указывающим, в каком примере получено это соединение.
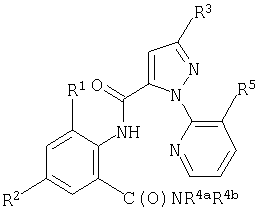
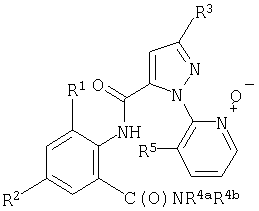
БИОЛОГИЧЕСКИЕ ПРИМЕРЫ ИЗОБРЕТЕНИЯ
ИСПЫТАНИЕ А
Для оценки уничтожения моли капустной (Plutella xylostella) устройство для испытания состояло из небольшого открытого контейнера, внутри которого находились 12-14-дневные растения редиса. Оно было предварительно заражено 10-15 новорожденными гусеницами на кусочке рациона насекомых с использованием пробоотборника с внутренней полостью (стержнем) для забора "пробки" из слоя затвердевшего рациона насекомых, на котором растут многочисленные гусеницы, и переноса этого образца-"пробки", содержащей гусениц и рацион, в устройство для испытания.
Испытуемые соединения (тест-соединения) готовили с использованием раствора, содержащего 10% ацетона, 90% воды и 300 ч./млн смеси неионогенного поверхностно-активного вещества Х-77® Spreader Lo-Foam, содержащей алкиларилполиоксиэтилен, свободные жирные кислоты, гликоли и изопропанол (Loveland Industries, Inc.), если нет других указаний. Приготовленные соединения наносили в 1 мл жидкости через наконечник для мелкокапельного распыления SUJ2 с изготовленным на заказ корпусом 1/8 JJ (Spraying Systems Co.), помещаемым на расстоянии 1,27 см (0,5 дюйма) над верхней частью каждого устройства для испытания. Все экспериментальные соединения в этом скрининге распыляли при норме 50 ч/млн, и испытывали в трех повторностях. После опрыскивания приготовленным тест-соединением каждому устройству для испытания давали высохнуть в течение 1 часа и затем на него помещали сверху черную сетчатую крышку. Эти тест-устройства выдерживали в течение 6 дней в камере для выращивания при 25°С и 70% относительной влажности. Затем визуально оценивали повреждение, вызываемое поеданием растения.
Из испытанных соединений следующие соединения обеспечивали превосходные уровни защиты растений (10% или меньшее повреждение в результате поедания): 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, 20, 21, 22, 23, 24, 25, 26, 27, 28, 29, 30, 31, 32, 33, 34, 35, 36, 37, 38, 39, 40, 41, 42, 43, 44, 45, 46, 47, 48, 49, 50, 51, 52, 53, 54, 55, 56, 57, 58, 59, 60, 61, 62, 63, 64, 65, 66, 67, 68, 69, 70, 71, 72, 73, 74, 75, 76, 77, 78, 79, 80, 81, 82, 83, 84, 85, 86, 87, 88, 89, 90, 91, 92, 93, 94, 95, 96, 97, 98, 99, 100, 101, 102, 103, 104, 105, 106, 107, 108, 109, 110, 111, 112, 113, 114, 115, 116, 117, 119, 120, 121, 122, 123, 124, 125, 126, 127, 128, 129, 130, 131, 132, 133, 134, 135, 136, 137.
ИСПЫТАНИЕ В
Для оценки уничтожения совки травяной (Spodoptera frugiperda) устройство для испытания состояло из небольшого открытого контейнера с 4-5-дневными растениями кукурузы внутри него, предварительно зараженного 10-15 однодневными гусеницами на кусочке рациона насекомых с использованием пробоотборника с внутренним стержнем, как описано в испытании А.
Испытуемые соединения (тест-соединения) готовили и распыляли при норме 50 ч./млн, как описано для теста А. Опрыскивания проводили трижды (в трех повторностях). После опрыскивания устройства для испытаний выдерживали в камере для выращивания и затем визуально оценивали, как описано для теста А.
Из испытанных соединений следующие соединения обеспечивали превосходные уровни защиты растений (10% или меньшее повреждение в результате поедания): 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, 20, 21, 22, 23, 24, 25, 26, 27, 28, 29, 30, 31, 32, 33, 34, 35, 36, 37, 38, 39, 40, 41, 42, 43, 44, 45, 46, 47, 48, 49, 50, 51, 52, 53, 54, 55, 56, 57, 58, 59, 60, 61, 62, 63, 64, 65, 66, 67, 68, 69, 70, 71, 72, 73, 74, 75, 76, 77, 78, 79, 80, 81, 82, 83, 84, 85, 86, 87, 88, 89, 90, 92, 93, 94, 95, 96, 97, 98, 99, 100, 101, 102, 103, 104, 105, 106, 107, 108, 109, 110, 111, 112, 113, 114, 115, 116, 117, 119, 120, 121, 122, 123, 124, 125, 126, 127, 128, 129, 130, 131, 132, 133, 134, 135, 136, 137.
ИСПЫТАНИЕ С
Для оценки уничтожения совки табачной (Heliothis virescens) устройство для испытания состояло из небольшого открытого контейнера с 6-7-дневными растениями хлопчатника внутри него, предварительно зараженного 8 двухдневными гусеницами на кусочке рациона насекомых с использованием пробоотборника с внутренним стержнем, как описано для испытания А.
Испытуемые соединения (тест-соединения) готовили и распыляли при норме 50 ч./млн, как описано для теста А. Опрыскивания проводили трижды (в трех повторностях). После опрыскивания устройства для испытаний выдерживали в камере для выращивания и затем визуально оценивали, как описано для теста А.
Из испытанных соединений следующие соединения обеспечивали превосходные уровни защиты растений (10% или меньшее повреждение в результате поедания): 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, 20, 21, 22, 23, 24, 25, 26, 27, 28, 29, 30, 31, 32, 33, 34, 35, 36, 37, 38, 39, 40, 41, 42, 43, 44, 45, 46, 47, 48, 49, 50, 51, 52, 53, 54, 55, 56, 57, 58, 59, 60, 61, 62, 63, 64, 65, 66, 67, 68, 69, 70, 71, 72, 73, 74, 75, 76, 77, 78, 79, 80, 81, 82, 83, 84, 85, 86, 87, 88, 89, 90, 92, 93, 94, 95, 96, 97, 98, 99, 100, 101, 102, 103, 104, 105, 106, 107, 108, 109, 110, 111, 112, 113, 114, 115, 116, 117, 119, 120, 121, 122, 123, 124, 125, 126, 127, 128, 129.
ИСПЫТАНИЕ D
Для оценки уничтожения совки малой (Spodoptera exigua) устройство для испытания состояло из небольшого открытого контейнера с 4-5-дневными растениями кукурузы внутри него, предварительно зараженного 10-15 однодневными гусеницами на кусочке рациона насекомых с использованием пробоотборника с внутренним стержнем, как описано для испытания А.
Испытуемые соединения (тест-соединения) готовили и распыляли при норме 50 ч./млн, как описано для теста А. Опрыскивания проводили трижды (в трех повторностях). После опрыскивания устройства для испытаний выдерживали в камере для выращивания и затем визуально оценивали, как описано для теста А.
Из испытанных соединений следующие соединения обеспечивали превосходные уровни защиты растений (10% или меньшее повреждение в результате поедания): 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, 20, 21, 22, 23, 24, 25, 26, 27, 28, 29, 30, 31, 32, 33, 34, 35, 36, 37, 39, 40, 41, 42, 43, 44, 45, 46, 47, 48, 49, 50, 51, 52, 53, 54, 55, 56, 57, 58, 59, 60, 61, 62, 63, 64, 65, 66, 67, 68, 69, 70, 71, 72, 73, 74, 75, 76, 77, 78, 79, 80, 81, 82, 83, 84, 85, 86, 87, 88, 89, 90, 91, 92, 93, 94, 95, 96, 97, 98, 100 101, 102, 103, 104, 105, 106, 107, 108, 109, 110, 111, 112, 113, 114, 115, 116, 117, 119, 120, 121, 122, 123, 124, 125, 126, 127, 128, 129.
ИСПЫТАНИЕ Е
Для оценки уничтожения тли персиковой зеленой (Myzus persicae) посредством контактирования и/или системным образом устройство для испытания состояло из небольшого открытого контейнера с 12-15-дневными растениями редьки внутри него, предварительно зараженного помещением на лист испытуемого растения 30-40 насекомых на кусочке листа, вырезанном из культивируемого растения (способ отрезанного листа). Гусеницы перемещались на испытуемые растения по мере обезвоживания этого кусочка листа. После предварительного заражения почву устройства для испытания покрывали слоем песка.
Испытуемые соединения (тест-соединения) готовили с использованием раствора, содержащего 10% ацетона, 90% воды и 300 ч./млн смеси неионогенного поверхностно-активного вещества Х-77® Spreader Lo-Foam, содержащей алкиларилполиоксиэтилен, свободные жирные кислоты, гликоли и изопропанол (Loveland Industries, Inc.), если нет других указаний. Приготовленные соединения наносили в 1 мл жидкости через наконечник для мелкокапельного распыления SUJ2 с изготовленным на заказ корпусом 1/8 JJ (Spraying Systems Co.), помещаемым на расстоянии 1,27 см (0,5 дюйма) над верхней частью каждого устройства для испытания. Все экспериментальные соединения в этом скрининге распыляли при 250 ч/млн, и испытывали в трех повторностях. После опрыскивания приготовленным тест-соединением каждому устройству для испытания давали высохнуть в течение 1 часа и затем на него помещали сверху черную сетчатую крышку. Эти тест-устройства выдерживали в течение 6 дней в камере для выращивания при 19-21°С и 50-70% относительной влажности. Затем каждое устройство для испытания визуально оценивали на смертность насекомых.
Из испытанных соединений следующие соединения приводили по меньшей мере к 80% смертности: 1, 2, 3,4, 5, 6, 7, 8, 10, 11, 12, 13, 14, 16, 20, 21, 22, 23, 25, 26, 27, 28, 29, 31, 32, 33, 36, 38, 39, 41, 42, 43, 45, 46, 47, 49, 51, 52, 54, 55, 56, 58, 59, 60, 61, 62, 63, 64, 65, 66, 67, 69, 72, 74, 77, 78, 79, 80, 81, 82, 83, 84, 85, 86, 87, 88, 90, 91, 93, 98, 99, 100, 101, 102, 103, 104, 105, 106, 108, 109, 111, 112, 113, 114, 115, 116, 126, 127, 128, 131, 135.
ИСПЫТАНИЕ F
Для оценки уничтожения тли хлопковой (тли бахчевой) (Aphis gossypii) посредством контактирования и/или системным образом устройство для испытания состояло из небольшого открытого контейнера с 6-7-дневными растениями хлопчатника внутри него, предварительно зараженного 30-40 насекомыми на кусочке листа в соответствии со способом отрезанного листа, описанным для испытания Е, и почву устройства для испытания покрывали слоем песка.
Испытуемые соединения (тест-соединения) готовили и распыляли при норме 250 ч./млн, как описано для теста А. Опрыскивания испытывали в трех повторностях. После опрыскивания устройства для испытаний выдерживали в камере для выращивания и затем визуально оценивали, как описано для теста Е.
Из испытанных соединений следующие соединения приводили по меньшей мере к 80% смертности: 1, 2, 4, 5, 7, 8, 10, 11, 12, 13, 14, 16, 20, 21, 22, 23, 25, 26, 27, 28, 29, 31, 32, 33, 34, 36, 38, 39, 42, 43, 45, 46, 47, 49, 51, 52, 53, 54, 55, 56, 58, 59, 62, 63, 65, 66, 67, 77, 78, 79, 80, 81, 82, 87, 88, 90, 91, 93, 94, 96, 98, 99, 100, 101, 102, 103, 104, 105, 106, 107, 108, 109, 111, 112, 113, 114, 115, 116, 117, 119, 120, 121, 122, 123, 124, 125, 126, 127, 128, 131, 133, 135, 136.
ИСПЫТАНИЕ G
Для оценки уничтожения белокрылки магнолиевой (Bemisia tabaci) устройство для испытания состояло из 14-21-дневных растений хлопчатника, выращиваемых в среде Redi-earth® (Scotts Co.), с по меньшей мере двумя настоящими листьями, зараженными нимфами 2-ой и 3-ей возрастных стадий на нижней стороне этих листьев.
Испытуемые соединения (тест-соединения) готовили в ацетоне (в количестве не более 2 мл) и затем разбавляли водой до 25-30 мл. Приготовленные соединения наносили с использованием распыливающего наконечника с плоским соплом для распыла (Spraying Systems 122440) при расходе 10 фунтов на квадратный дюйм (69 кПа). Растения опрыскивали до стекания с использованием опрыскивателя на поворотном круге (заявка на патент ЕР-1110617-А1). Все экспериментальные соединения в этом скрининге распыляли при 250 ч./млн и испытывали в трех повторностях. После опрыскивания тест-соединением устройства для испытания выдерживали в течение 6 дней в камере для выращивания при 50-60% относительной влажности и при температуре 28°С в дневное время и 24°С в ночное время суток. Затем листья удаляли и подсчитывали мертвых и живых нимф для расчета процента смертности.
Из испытанных соединений следующие соединения приводили по меньшей мере к 80% смертности: 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 16, 19, 20, 21, 22, 23, 24, 25, 26, 27, 29, 31, 32, 33, 34, 39, 42, 43, 45, 46, 47, 49, 51, 52, 53, 54, 55, 56, 59, 77, 78, 79, 80, 81, 82, 87, 90, 93.
ИСПЫТАНИЕ Н
Для оценки почвенного системного подавления совки табачной (Heliothis virescens) растения хлопчатника выращивали в сассафрасовой почве в сосудах с диаметром 15 см в алюминиевых поддонах. Когда растения достигали стадии трех подцветников (чашелистиков) цветка хлопчатника (образования почек на растении), эти растения обрабатывали испытуемыми соединениями.
Испытуемые соединения готовили в 0,25 мл ацетона и затем разбавляли водой для получения растворов 1, 5, 10 и 50 ч/млн. 10 мл растворов для обработки добавляли в сосуды еженедельно в течение четырех недель, с четырьмя повторностями для каждой нормы обработки. Спустя один день после второй, третьей и четвертой обработок 35-50 личинок Heliothis virescens первой возрастной стадии стряхивали кистью на каждое растение и помещали на верхушечную зону, три подцветника (чашелистика) цветка хлопчатника или семенные коробочки. Спустя пять дней после последнего заражения личинками эти растения оценивали на повреждение. Из испытанных соединений следующие соединения обеспечивали превосходные уровни защиты растений при норме 10 ч./млн (10% или меньшее повреждение в результате поедания): 16.
Из испытанных соединений следующие соединения обеспечивали также превосходную защиту трех подцветников и семенных коробочек при норме 10 ч./млн, без повреждения в результате поедания или с минимальным повреждением чашелистиков: 16.
ИСПЫТАНИЕ I
Испытание I проводили по альтернативному протоколу для оценки почвенного системного подавления совки табачной (Heliothis virescens). Растения хлопчатника выращивали в сассафрасовой почве в сосудах с диаметром 15 см в условиях оранжереи. Когда растения достигали стадии трех подцветников цветка хлопчатника (стадии образования почек на растении), поверхность почвы обрабатывали испытуемыми соединениями.
Испытуемые соединения (тест-соединения) готовили в 0,25 мл ацетона и затем разбавляли водой. Десять мл раствора для обработки, содержащие 3 мг соединения, добавляли на поверхность почвы каждого сосуда. Растения поливали на следующий день и каждый день после этого по мере необходимости. На 1, 2 и 4 дни после обработки листья срезали для оценки. Из каждого растения выбирали две группы листьев: верхние листья при приблизительно втором узле от верхушечного и с площадью, большей, чем 25 см2, и более низкие листья при приблизительно третьем узле от нижней части и с площадью, большей, чем 25 см2. Срезанные листья нарезали на секции 3 см × 2 см и помещали в тест-лотки, изготовленные из высокопрочного стирола, состоящие из шестнадцати смежных лунок, каждая шириной 6 см, длиной 4 см и глубиной 3 см, с прозрачной пластиковой крышкой, отлитой таким образом, что она блокирует вход в каждую лунку посредством трения. В нижнюю часть каждой лунки помещали отверждаемый агар для поддержания влажности для растительного материала. Одну совку табачную второй возрастной стадии помещали в каждую лунку с растительным материалом; лунки герметизировали и выдерживали при 25°С, обеспечивая 16 часов освещения в сутки. Для листьев, срезанных на 1, 2 и 4 день, смертность наблюдали 4 дня спустя после обработки одной совки табачной второй возрастной стадии.
Из испытанных соединений следующие соединения обеспечивали превосходные уровни смертности (большие, чем 70% смертность) на верхних листьях, срезанных 4 дня спустя после обработки при испытуемой норме: 2, 27, 33.
ИСПЫТАНИЕ J
Для оценки почвенного системного подавления совки травяной (Spodoptera frugiperda) растения кукурузы (маиса) (Pioneer #3394) выращивали в небольших сосудах в течение 5 дней, пока они не достигали высоты по меньшей мере 4 см и первый лист не развертывался.
Испытуемые соединения (тест-соединения) растворяли в 0,25 мл ацетона и разбавляли водой с получением растворов 1, 10, 50 и 200 ч./млн 1 мл испытуемого раствора наносили пипеткой на поверхность почвы в каждом сосуде с использованием восьми растений для каждого соединения/каждой нормы. Сосуды закрывали и выдерживали при 25°С, обеспечивая 16 часов освещения в сутки. Растения поливали на следующий день и каждый день после этого по мере необходимости. Спустя 6 дней растительную массу выше первого листа срезали и нарезали на сегменты длиной 3 см. Каждое устройство для испытания было высокопрочным стироловым лотком (Поставщик: Clearpack Company, 11610 Copengagen Court, Franklin Park, IL 60131), состоящим из шестнадцати смежных лунок, каждая шириной 6 см, длиной 4 см и глубиной 3 см, с прозрачной пластиковой крышкой, отлитой таким образом, что она блокирует вход в лунку посредством трения. В нижнюю часть каждой лунки помещали отверждаемый агар (2-4 мл) для поддержания влажности во время испытания. Каждый сегмент 3 см растения кукурузы помещали в лоток таким образом, что этот растительный материал содержался в двух лунках. Одну совку травяную (Spodoptera frugiperda) второй возрастной стадии помещали в каждую лунку, лоток закрывали и затем эти устройства для испытания выдерживали при 25°С с 16 часами освещения в сутки. Смертность наблюдали после четырех дней.
Концентрации LC90 (концентрации тест-соединений, обеспечивающие 90% убивания личинок (гусениц)) рассчитывали на основе пробит-анализа (логарифмической линейной регрессии) с использованием общей линеаризованной модели (GLIM) пакета SAS статистического компьютерного анализа Института SAS (Cary, NC, U.S.A.). Из испытанных соединений следующие соединения обеспечивали превосходные уровни смертности с величинами LC90 10 ч./млн, или менее: 1, 2, 4, 9, 11, 12, 14, 16, 20, 22, 24, 31, 32, 33, 34.
ИСПЫТАНИЕ К
Для оценки подавления колорадского жука (Leptinotarsa decemlineate) пробы 5 мг испытуемого соединения (тест-соединения) растворяли в 1 мл ацетона. Затем этот раствор разбавляли до 100 мл общего объема с использованием водного раствора 500 ч/млн, поверхностно-активного вещества Ortho X-77™. Готовили серийные разведения для получения 50 мл концентрации 10 ч/млн.
Разведенные растворы тест-соединений разбрызгивали до стекания на трехнедельные растения картофеля или томатов. Эти растения помещали на вращающийся опрыскиватель на поворотном круге (10 об/мин). Тест-соединения наносили с использованием распыливающего наконечника с плоским соплом распыла (Spraying Systems 122440) при норме 10 фунтах на квадратный дюйм (69 кПа). После высыхания каждого обработанного растения срезали листья этого обработанного растения. Листья нарезали на куски, которые помещали по одному в ячейки 5,5 см × 3,5 см шестнадцатиячеечного пластикового лотка. Каждая ячейка содержала квадрат 2,5 см увлажненной хроматографической бумаги для предотвращения высыхания. Одну личинку второй возрастной стадии помещали в каждую ячейку. Через три дня после заражения регистрировали общее число мертвых колорадских жуков.
Из испытанных соединений следующие соединения приводили к по меньшей мере 90% смертности при норме 10 ч/млн: 2, 4, 27, 33, 34, 41, 61, 85.
ИСПЫТАНИЕ L
Для оценки уничтожения долгоносика хлопкового (Anthonomus g. grandis) пробы испытуемых соединений растворяли в 1 мл ацетона. Затем этот раствор разбавляли до 100 мл общего объема с использованием водного раствора 500 ч/млн, поверхностно-активного вещества Ortho X-77™. Готовили серийные разведения для получения 50 мл концентрации 50 ч./млн.
Разведенные растворы тест-соединений разбрызгивали до стекания на трехнедельные растения хлопчатника. Эти растения помещали на вращающийся опрыскиватель на поворотном круге (10 об/мин). Тест-соединения наносили с использованием распиливающего наконечника с плоским соплом распыла (Spraying Systems 122440) при норме 10 фунтов на квадратный дюйм (69 кПа). Опрысканные и высушенные растения помещали в пластиковый цилиндр. Двадцать долгоносиков помещали в каждый цилиндр, содержащий целое растение хлопчатника. Через три дня после заражения оценивали повреждение в результате поедания.
Из испытанных соединений следующие соединения обеспечивали превосходные уровни защиты растений при 50 ч./млн (10% или меньшее повреждение в результате поедания): 20, 27.
ИСПЫТАНИЕ М
Для оценки уничтожения трипсов (Frankliniella sp.) пробы испытуемых соединений растворяли в 1 мл ацетона. Затем этот раствор разбавляли до 100 мл общего объема с использованием водного раствора 500 ч./млн поверхностно-активного вещества Ortho Х-77™. Готовили серийные разведения для получения 50 мл концентрации 10 ч/млн.
Разведенные растворы тест-соединений разбрызгивали до стекания на трехнедельные растения хлопчатника или сои, зараженные трипсами. Эти растения помещали на вращающийся опрыскиватель на поворотном круге (10 об/мин). Тест-соединения наносили с использованием распыливающего наконечника с плоским соплом распыла (Spraying Systems 122440) при норме 10 фунтов на квадратный дюйм (69 кПа). Опрысканные и высушенные растения заключали в пластиковый цилиндр. Через три дня после нанесения оценивали общее количество мертвых трипсов.
Из испытанных соединений следующие соединения приводили к по меньшей мере 90% смертности при норме 10 ч./млн: 32.
ИСПЫТАНИЕ N
Для оценки подавления развития нематоды корневых наростов (Meloidogyne incognita) при использовании средств контактного и/или системного действия применяли испытательное устройство, состоящее из небольшого открытого контейнера с помещенной внутрь рассадой помидоров возрастом 7-9 дней. Композиции испытываемых соединений получали и распыляли при содержании 250 ч./млн, повторяя операцию три раза, как это описывается для испытания А. После распыления испытательные устройства оставляли в покое для высыхания в течение 1 часа, а после этого пипетированием вносили в почву приблизительно 250 личинок молодой стадии 2 (J2) и затем поверх накладывали черную экранирующую крышку. Испытательные устройства выдерживали в течение 6 дней при 25°С и 65-70% относительной влажности. После этого для каждого испытательного устройство проводили визуальную оценку повреждения корней.
Среди испытанных соединений превосходные уровни защиты растений (70% или более уменьшения корневого галлообразования) продемонстрировало следующее: 20.
ИСПЫТАНИЕ Р
Для оценки борьбы с цикадкой картофельной (Empoasca fabae Harris) посредством контакта и/или системными способами, каждая испытываемая единица состояла из небольшого открытого контейнера, содержащего внутри 5-6-дневное бобовое растение Longio (с появившимися первыми листьями). Поверх почвы добавляли белый песок и один из первых листьев отрезали до нанесения. Испытываемые соединения формулировали, как описано выше, и распыляли в трех повторах, как описано для Испытания А. После распыления испытываемым единицам давали высохнуть в течение 1 часа, а затем их заражали 5 картофельными цикадками (взрослые особи 18-21 дня). Поверх каждого контейнера помещали черную загораживающую крышку. Испытываемые единицы выдерживали в течение 6 дней в камере для роста при 19-21°С и 50-70% относительной влажности. Каждую испытываемую единицу затем визуально оценивали на смертность насекомых.
ИСПЫТАНИЕ Q
Для оценки борьбы с цикадами кукурузными (Peregrinus maidis) посредством контакта и/или системными способами, каждая испытываемая единица состояла из небольшого открытого цилиндрического контейнера, содержащего внутри 3-4-дневное растение кукурузы (маис). Поверх почвы добавляли белый песок, и один из первых листьев отрезали до нанесения. Испытываемые соединения формулировали, как описано выше, и распыляли в трех повторах, как описано для Испытания А. После распыления испытываемым единицам давали высохнуть в течение 1 часа, а затем их заражали 10-20 цикадами кукурузными (нимфы 18-21 дня), разбрызгивая их на песок при помощи шейкера. Поверх каждого контейнера помещали черную загораживающую крышку. Испытываемые единицы выдерживали в течение 6 дней в камере для роста при 19-21°С и 50-70% относительной влажности. Каждую испытываемую единицу затем визуально оценивали на смертность насекомых.
ИСПЫТАНИЕ R
Для оценки подавления цикадки картофельной (Empoasca fabae Harris) с использованием средств контактного и/или системного действия каждое испытательное устройство состояло из небольшого открытого контейнера с помещенной внутрь рассадой бобов Лонджо возрастом от 5 до 6 дней (с появившимися первичными листьями). Поверх почвы наносили чистый кварцевый песок, а один из первичных листьев перед нанесением средства отрезали. Композиции для испытаний составляли так, как описывается выше, и распыляли, повторяя операцию 3 раза, так, как это описывается для испытания А. После распыления испытательные устройства оставляли в покое для высыхания в течение 1 часа перед тем, как провести их заражение, используя 5 цикадок картофельных (взрослые особи возрастом от 18 до 21 дня). Поверх каждого контейнера размещали черную экранирующую крышку. Испытательные устройства выдерживали в течение 6 дней в ростовой камере при 19-21°С и 50-70% относительной влажности. После этого для каждого испытательного устройство проводили визуальную оценку смертности насекомых.
Далее представлены данные, иллюстрирующие дополнительные эффекты, наблюдаемые при введении известного активного соединения или агента в композицию, содержащую соединение формулы 1 (таблицы G, Н и J1-J6).
Активные ингредиенты, перечисленные в Таблице G, испытывали в соответствии с протоколом, описанным для ИСПЫТАНИЯ А, ИСПЫТАНИЯ Е, ИСПЫТАНИЯ F и ИСПЫТАНИЯ G, как описано выше; протоколы ИСПЫТАНИЯ Р и ИСПЫТАНИЯ Q описаны выше. Соединения готовили в виде композиций, используя раствор, содержащий 10% ацетона, 90% воды и 300 ч./млн неионогенного поверхностно-активного вещества Х-77® Spreader Lo-Foam Formula, содержащего алкиларилполиоксиэтилен, свободные жирные кислоты, гликоли и изопропанол (Loveland Industries, Inc.), с получением концентрации испытываемых растворов 1, 10, 100 и 250 ч./млн. Затем композиции испытываемых растворов разбрызгивали и оценивали в соответствии с протоколами.
Номера соединений (отличные от названий соединений), перечисленные в первой графе таблицы G, указывают номер соединения, указанный в Таблице Индексов А. Инсектицидная активность указана в Таблице G по категориям А, В, С, D и Е. "А" означает, что наименьшая концентрация активного ингредиента, необходимая для подавления конкретного вредителя более чем на 90%, составляет 1 ч./млн или меньше; "В" означает 10 ч./млн или меньше; "С" означает 100 ч./млн или меньше; "D" означает 250 ч./млн или меньше; и "Е" означает более чем 250 ч./млн. Другими словами, соединение категории "А" является, по меньшей мере, в 100 раз более активным, чем соединение категории "С".
Данные, представленные в таблицах от J до J6 ниже, демонстрируют, что обработка вредителя с использованием смеси либо композиции известного биологически активного соединения либо агента и нового соединения, описываемого формулой 1 (т.е., соединения 20), очевидным образом приводит к повышенному % смертности вредителя в сопоставлении с результатами обработки с использованием каждого ингредиента, взятого отдельно. Кроме того, данные смеси либо композиции также иллюстрируют массовые соотношения между соединением, описываемым формулой 1, и дополнительным биологически активным соединением либо агентом в диапазоне от 250: 1 до 1: 500.
| название | год | авторы | номер документа |
|---|---|---|---|
| ОРТОЗАМЕЩЕННЫЕ АРИЛАМИДЫ, СПОСОБ БОРЬБЫ С НАСЕКОМЫМИ, КОМПОЗИЦИЯ ДЛЯ БОРЬБЫ С НАСЕКОМЫМИ, ПРОМЕЖУТОЧНОЕ СОЕДИНЕНИЕ | 2002 |
|
RU2283839C2 |
| СОЕДИНЕНИЕ, КОМПОЗИЦИЯ ДЛЯ БОРЬБЫ С НАСЕКОМЫМИ, СПОСОБЫ БОРЬБЫ С НАСЕКОМЫМИ | 2002 |
|
RU2298007C2 |
| СПОСОБ БОРЬБЫ С КОНКРЕТНЫМИ НАСЕКОМЫМИ-ВРЕДИТЕЛЯМИ ПУТЕМ НАНЕСЕНИЯ АНТРАНИЛАМИДНЫХ СОЕДИНЕНИЙ | 2002 |
|
RU2262231C1 |
| ХИНАЗОЛИН(ДИ)ОНЫ, КОМПОЗИЦИЯ ДЛЯ БОРЬБЫ С НАСЕКОМЫМИ-ВРЕДИТЕЛЯМИ, СПОСОБ БОРЬБЫ С НАСЕКОМЫМИ-ВРЕДИТЕЛЯМИ, ПРОМЕЖУТОЧНОЕ СОЕДИНЕНИЕ | 2003 |
|
RU2315765C2 |
| ЦИАНОАНТРАНИЛАМИДНЫЕ ИНСЕКТИЦИДЫ | 2004 |
|
RU2343151C2 |
| СПОСОБ ПОЛУЧЕНИЯ N-ФЕНИЛПИРАЗОЛ-1-КАРБОКСАМИДОВ | 2005 |
|
RU2397165C2 |
| ДИАМИДЫ АНТРАНИЛОВОЙ КИСЛОТЫ, СПОСОБ БОРЬБЫ С НАСЕКОМЫМИ, КОМПОЗИЦИЯ ДЛЯ БОРЬБЫ С НАСЕКОМЫМИ | 2002 |
|
RU2299198C2 |
| ЗАМЕЩЕННЫЕ ДИГИДРО 3-ГАЛОГЕН-1H-ПИРАЗОЛ-5-КАРБОКСИЛАТЫ, ИХ ПОЛУЧЕНИЕ И ИСПОЛЬЗОВАНИЕ | 2002 |
|
RU2317983C2 |
| ИЗОКСАЗОЛИНЫ ДЛЯ БОРЬБЫ С БЕСПОЗВОНОЧНЫМИ ВРЕДИТЕЛЯМИ | 2006 |
|
RU2433123C2 |
| ПЕСТИЦИДНАЯ КОМПОЗИЦИЯ, СПОСОБ КОНТРОЛЯ ВРЕДИТЕЛЕЙ, СПОСОБ КОНТРОЛЯ ЭНДОПАРАЗИТОВ, ЭКТОПАРАЗИТОВ ИЛИ ОБОИХ И СПОСОБ УСИЛЕНИЯ ЖИЗНЕСТОЙКОСТИ РАСТЕНИЙ | 2011 |
|
RU2576316C2 |
Описывается антраниламидное соединение, выбранное из соединения формулы 1 или их N-оксидов
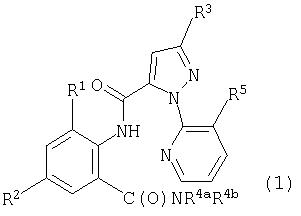
где R1 - СН3, F, Cl, Br, R2 - F, Cl, Br, I, CF3, R3 - CF3, Cl, Br, ОСН2CF3, R4a - C1-С4алкил; R4b-Н, СН3 и R5-Cl, Br, или его приемлемая для использования в сельском хозяйстве соль; композиция для борьбы с насекомыми, содержащая биологически эффективное количество соединения формулы (1) и, по меньшей мере, один дополнительный компонент, выбранный из группы, состоящей из поверхностно-активных веществ, твердых и жидких разбавителей; композиция для борьбы с беспозвоночными вредителями, содержащая биологически эффективное количество соединения формулы (1) и эффективное количество, по меньшей мере, одного дополнительного биологически активного соединения или агента. Описываются способы борьбы с насекомыми, а также промежуточные соединения: производные бензоксазинона и пиразолкарбоновой кислоты. Технический результат - используемые соединения проявляют инсектицидное действие, что позволяет применять их в сельском хозяйстве. 7 н. и 13 з.п. ф-лы, 16 табл.
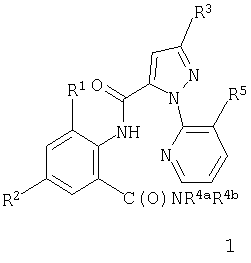
где R1 означает СН3, F, Cl или Br;
R2 означает F, Cl, Br, I или CF3;
R3 означает CF3, Cl, Br или OCH2CF3;
R4a означает С1-С4алкил;
R4b означает Н или СН3; и
R5 означает Cl или Br,
или его приемлемая для использования в сельском хозяйстве соль.
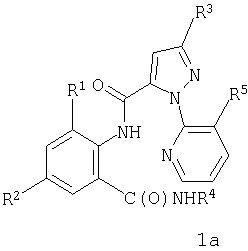
где R1 означает СН3, Cl или Br;
R2 означает F, Cl, Br, I или CF3;
R3 означает CF3, Cl или Br;
R4 означает С1-С4алкил; и
R5 означает Cl или Br,
или его приемлемая для использования в сельском хозяйстве соль.
соединения формулы 1, где R1 означает СН3, R2 означает Br, R3 означает CF3, R4a означает СН(СН3)2, R4b означает Н и R5 означает Cl;
соединения формулы 1, где R1 означает СН3, R2 означает Br, R3 означает CF3, R4a означает СН3, R4b означает Н и R5 означает Cl;
соединения формулы 1, где R1 означает СН3, R2 означает Br, R3 означает Br, R4a означает СН(СН3)2, R4b означает Н и R5 означает Cl;
соединения формулы 1, где R1 означает СН3, R2 означает Br, R3 означает Br, R4a означает СН3, R4b означает Н и R5 означает Cl;
соединения формулы 1, где R1 означает СН3, R2 означает Br, R3 означает Cl, R4a означает СН(СН3)2, R4b означает Н и R5 означает Cl;
соединения формулы 1, где R1 означает СН3, R2 означает Br, R3 означает Cl, R4a означает СН3, R4b означает Н и R5 означает Cl;
соединения формулы 1, где R1 означает СН3, R2 означает Cl, R3 означает CF3, R4a означает СН(СН3)2, R4b означает Н и R5 означает Cl;
соединения формулы 1, где R1 означает СН3, R2 означает Cl, R3 означает CF3, R4a означает СН3, R4b означает Н и R5 означает Cl;
соединения формулы 1, где R1 означает СН3, R2 означает Cl, R3 означает Br, R4a означает СН(СН3)2, R4b означает Н и R5 означает Cl;
соединения формулы 1, где R1 означает СН3, R2 означает Cl, R3 означает Br, R4a означает СН3, R4b означает Н и R5 означает Cl;
соединения формулы 1, где R1 означает СН3, R2 означает Cl, R3 означает Cl, R4a означает СН(СН3)2, R4b означает Н и R5 означает Cl;
соединения формулы 1, где R1 означает СН3, R2 означает Cl, R3 означает Cl, R4a означает СН3, R4b означает Н и R5 означает Cl;
соединения формулы 1, где R1 означает СН3, R2 означает Cl, R3 означает OCH2CF3, R4a означает СН(СН3)2, R4b означает Н и R5 означает Cl;
соединения формулы 1, где R1 означает СН3, R2 означает Cl, R3 означает ОСН2CF3, R4a означает СН3, R4b означает Н и R5 означает Cl;
соединения формулы 1, где R1 означает Cl, R2 означает Cl, R3 означает Br, R4a означает СН3, R4b означает Н и R5 означает Cl;
соединения формулы 1, где R1 означает СН3, R2 означает Cl, R3 означает ОСН2CF3, R4a означает СН3, R4b означает Н и R5 означает Cl.
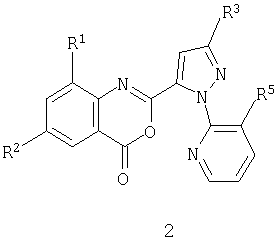
где R1 означает СН3, F, Cl или Br;
R2 означает F, Cl, Br, I или CF3;
R3 означает CF3, Cl, Br или OCH2CF3; и
R5 означает Cl или Br.
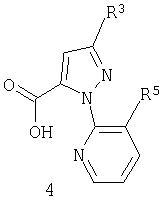
где R3 означает CF3, Cl, Br или ОСН2CF3; и
R5 означает Cl или Br.
Приоритет по пунктам и признакам:
| NL 9202078 А, 16.06.1994 | |||
| СПОСОБ ПОЛУЧЕНИЯ СМОЛ | 0 |
|
SU170671A1 |
| ПРОИЗВОДНЫЕ 1-(2-ПИРИДИЛ) ПИРАЗОЛА ИЛИ ИХ КИСЛОТНО-АДДИТИВНЫЕ СОЛИ, СПОСОБ ПОЛУЧЕНИЯ ПРОИЗВОДНЫХ 1-(2-ПИРИДИЛ) ПИРАЗОЛА ИЛИ ИХ КИСЛОТНО-АДДИТИВНЫХ СОЛЕЙ (ВАРИАНТЫ), ИНСЕКТОАКАРИЦИДОНЕМАТОЦИДНАЯ КОМПОЗИЦИЯ И СПОСОБ БОРЬБЫ С НАСЕКОМЫМИ, КЛЕЩАМИ И НЕМАТОДАМИ | 1992 |
|
RU2088580C1 |

