ОБЛАСТЬ ИЗОБРЕТЕНИЯ
Настоящее изобретение относится к новым соединениям 3-галоген-1-арил-замещенных дигидро-1H-пиразолов и пиразолов на основе карбоновых кислот. Эти соединения являются пригодными для получения определенных соединений на основе антранил-амидов, которые представляют интерес в качестве инсектицидов (смотри, например, публикацию PCT WO 01/070671).
ПРЕДПОСЫЛКИ ИЗОБРЕТЕНИЯ
Tetrahedron Letters, 1999, 40, 2605-2606 описывает получение соединений 1-фенил-3-бромпиразол-5-карбоновых кислот, включающее получение реакционно-способного промежуточного соединения бромнитрилимина. Циклоприсоединение этого промежуточного соединения со сложным акриловым эфиром дает сложный 1-фенил-3-бром-2-пиразолин-5-карбоксилатный эфир, который затем может быть окислен до желаемого сложного 1-фенил-3-бром-2-пиразол-5-карбоксилатного эфира. Альтернативно, циклоприсоединение сложного эфира пропиолата непосредственно дает сложный 1-фенил-3-бром-2-пиразол-5-карбоксилатный эфир.
Патент США 3153654 описывает конденсацию определенного необязательно замещенного арила (например, фенил или нафтил, которые необязательно замещены низшим алкилом, низшим алкокси или галогеном) гидразинов с определенными сложными фумаровыми или малеиновыми эфирами, с получением производных 3-пиразолидинон карбоновых кислот.
Публикации непрошедших экспертизу патентов Японии 9-316055 и 9-176124 описывают получение соединений сложных пиразоловых эфиров карбоновых кислот и производных пиразолинов, соответственно, которые замещены алкилом в 1-положении.
J. Med. Chem. 2001, 44, 566-578 описывает получение 1-(3-цианофенил)-3-метил-1H-пиразол-5-карбоновой кислоты и ее использование при получении ингибиторов фактора свертывания крови Xa.
Настоящее изобретение предлагает методики, пригодные для удобного получения 3-галоген-5-карбоксилат-1-арил-замещенных дигидро-1H-пиразолов и пиразолов.
КРАТКОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
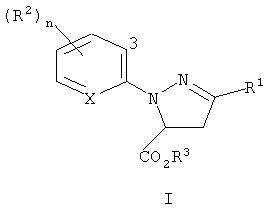
Настоящее изобретение относится к соединению формулы I
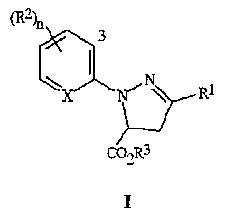
где
R1 представляет собой галоген;
каждый R2 представляет собой, независимо, С1-C4 алкил, С2-C4 алкенил, С2-C4 алкинил, C3-С6 циклоалкил, C1-C4 галогеналкил, С2-C4 галогеналкенил, С2-C4 галогеналкинил, C3-С6 галогенциклоалкил, галоген, CN, NO2, C1-C4 алкокси, C1-C4 галогеналкокси, C1-C4 алкилтио, C1-C4 алкилсульфинил, C1-C4 алкилсульфонил, C1-C4 алкиламино, C2-C8 диалкиламино, C3-С6 циклоалкиламино, C3-С6 (алкил)циклоалкиламино, С2-C4 алкилкарбонил, C2-C6 алкоксикарбонил, C2-C6 алкиламинокарбонил, C3-C8 диалкиламинокарбонил или C3-C6 триалкилсилил;
R3 представляет собой H или C1-C4 алкил;
X представляет собой N или CR4;
R4 представляет собой H или R2; и
n равно от 0 до 3, при условии, что, когда X представляет собой CH, n равно, по меньшей мере, 1.
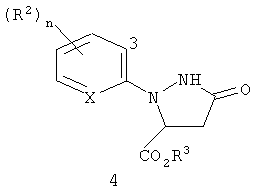
Настоящее изобретение также относится к способу получения соединения формулы I, включающему (1) обработку соединения формулы 4
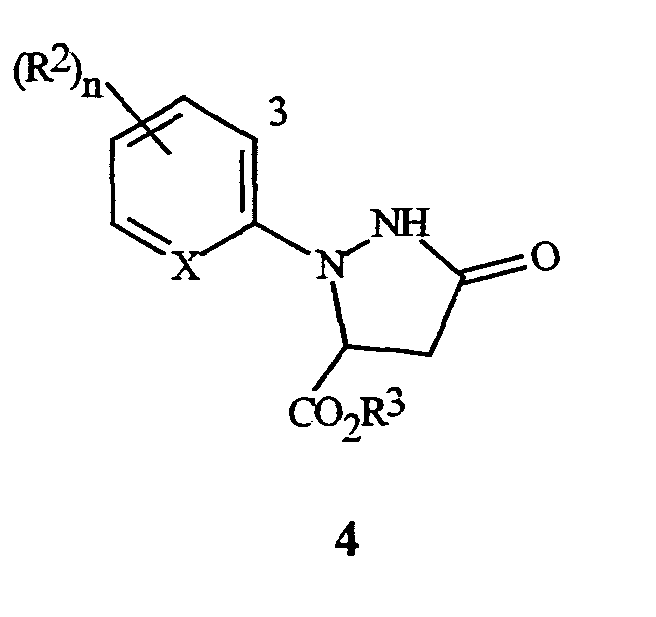
(где X, R2 и n являются такими, как описано выше для формулы I, и R3 представляет собой C1-C4 алкил) галогенирующим агентом, с получением соединения формулы I; а затем, в случае получения соединений формулы I, где R3 представляет собой H, (2) преобразование соединения, полученного в (1) в соединение, где R3 представляет собой H.
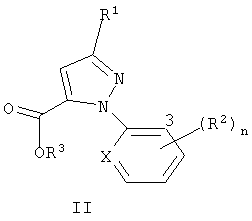
Настоящее изобретение также относится к соединению формулы II
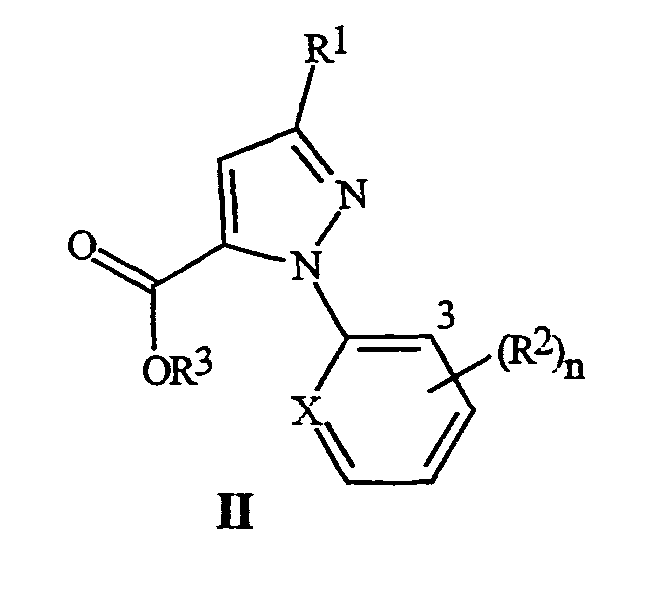
где R1 представляет собой галоген (и X, R2, R3 и n являются такими, как определено выше, для формулы I), и к способу получения соединения формулы II. Способ включает (3) обработку соединения формулы I окислителем, необязательно, в присутствии кислоты, с получением соединения формулы II; а когда соединение формулы I, где R3 представляет собой C1-C4 алкил, используется для получения соединения формулы II, где R3 представляет собой H, (4) преобразование соединения, полученного в (2), в соединение формулы II, где R3 представляет собой H.
Настоящее изобретение также предусматривает соединения формулы 4, где X представляет собой N, и их использование при получении соединений формул I и II, где X представляет собой N (и R2, R3 и n являются такими, как определено выше, для формулы I).
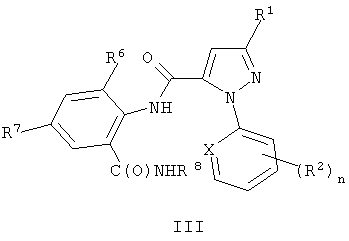
Настоящее изобретение также включает способ получения соединения формулы III
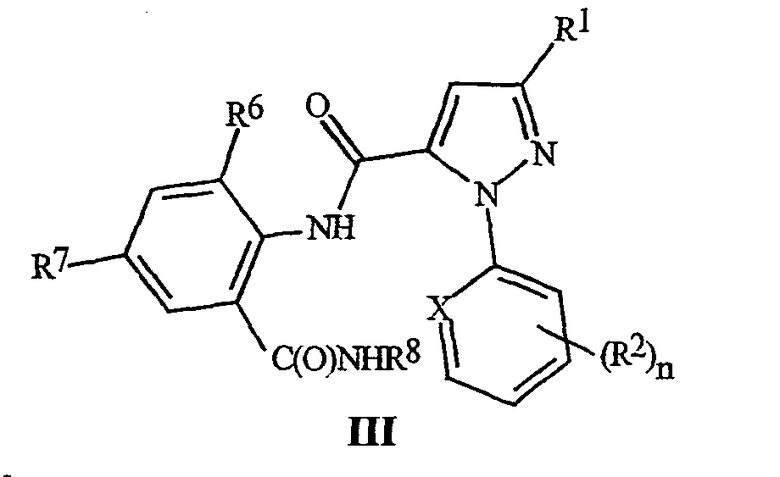
где X, R1, R2, и n являются такими, как определено выше, для формулы II; R6 представляет собой CH3, Cl или Br; R7 представляет собой F, Cl, Br, I или CF3; и R8 представляет собой C1-C4 алкил, с использованием соединения формулы II, где R6 представляет собой H. Этот способ отличается получением соединения формулы II с помощью способа, как указано выше.
ПОДРОБНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
В указанных выше перечислениях, термин "алкил", используемый либо сам по себе, либо в составных словах, таких как "алкилтио" или "галогеналкил", включает алкил с прямой или разветвленной цепью, такой как метил, этил, н-пропил, изо-пропил, или различные изомеры бутила, пентила или гексила. "Алкенил" может включать алкены с прямой или разветвленной цепью, такие как 1-пропенил, 2-пропенил, и различные изомеры бутенила, пентенила и гексенила. "Алкенил" также включает полиены, такие как 1,2-пропадиенил и 2,4-гексадиенил. "Алкинил" включает алкины с прямой или разветвленной цепью, такие как 1-пропинил, 2-пропинил, и различные изомеры бутинила, пентинила и гексинила. "Алкинил" также может включать остатки, содержащие множество тройных связей, такие как 2,5-гексадиинил. "Алкокси" включает, например, метокси, этокси, н-пропилокси, изопропилокси и различные изомеры бутокси, пентокси и гексилокси. "Алкоксиалкил" обозначает алкокси замещение группы алкила. Примеры "алкоксиалкила" включают CH3OCH2, CH3OCH2CH2, CH3CH2OCH2, CH3CH2CH2CH2OCH2 и CH3CH2OCH2CH2. "Алкилтио" включает алкилтио остатки с прямой или разветвленной цепью, такие как метилтио, этилтио, и различные изомеры пропилтио, бутилтио, пентилтио и гексилтио. "Циклоалкил" включает, например, циклопропил, циклобутил, циклопентил и циклогексил. "Циклоалкилалкил" указывает на алкильную группу, замещенную циклоалкильной группой, и включает, например, циклопропилметил, циклобутилэтил, циклопентилпропил и циклогексилметил. "Циклоалкиламино" обозначает, что атом азота амино присоединен к циклоалкильному радикалу и атому водорода и включает такие группы, как циклопропиламино, циклобутиламино, циклопентиламино и циклогексиламино. "(Алкил)циклоалкиламино" обозначает циклоалкиламино группу, где атом водорода замещен алкильным радикалом; примеры включают такие группы, как (алкил)циклопропиламино, (алкил)циклобутиламино, (алкил)циклопентиламино и (алкил)циклогексиламино. Предпочтительно, алкил в (алкил)циклоалкиламино представляет собой C1-C4 алкил, в то время как циклоалкил в циклоалкиламино и (алкил)циклоалкиламино представляет собой C3-C6 циклоалкил.
В этой заявке термин "арил" относится к ароматическому кольцу, или к кольцевой системе, или к гетероароматическому кольцу, или кольцевой системе, при этом, каждое кольцо или кольцевая система является необязательно замещенной. Термин "ароматическая кольцевая система" обозначает полностью ненасыщенные карбоциклы и гетероциклы, в которых, по меньшей мере, одно кольцо полициклической кольцевой системы является ароматическим. Ароматический указывает на то, что каждый из атомов кольца находится по существу в одной и той же плоскости и имеет p-орбиталь, перпендикулярную плоскости кольца, и в котором (4n + 2)π электронов, где n равно 0 или положительному целому числу, являются связанными с кольцом, в соответствии с правилом Хюккеля. Термин "ароматическая карбоциклическая кольцевая система" включает полностью ароматические карбоциклы и карбоциклы, в которых, по меньшей мере, одно кольцо полициклической кольцевой системы является ароматическим (например, фенилом и нафтилом). Термин "гетероароматическое кольцо или кольцевая система" включает полностью ароматические гетероциклы и гетероциклы, в которых, по меньшей мере, одно кольцо полициклической кольцевой системы является ароматическим, и в которых, по меньшей мере, один атом кольца не является углеродом, и которые могут содержать от 1 до 4 гетероатомов, независимо выбранных из группы, состоящей из азота, кислорода и серы, при условии, что каждое гетероароматическое кольцо содержит не более чем 4 атома азота, не более чем 2 атома кислорода и не более чем 2 атома серы (где ароматический указывает на то, что выполняется правило Хюккеля). Гетероциклические кольцевые системы могут быть соединены через любой доступный атом углерода или азота, путем замещения водорода на указанном атоме углерода или азота. Более конкретно, термин "арил" относится к остатку
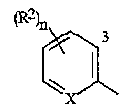
где R2 и n являются такими, как определено выше, и "3" обозначает 3-е положение для заместителей на остатке.
Термин "галоген", либо сам по себе, либо в составных словах, таких как "галогеналкил", включает фтор, хлор, бром или йод. Кроме того, когда он используется в составных словах, таких как "галогеналкил", указанный алкил может быть частично или полностью замещен атомами галогенов, которые могут быть одинаковыми или различными. Примеры "галогеналкила" включают F3C, ClCH2, CF3CH2 и CF3CC12. Термины "галогеналкенил", "галогеналкинил", "галогеналкокси", и тому подобное, определяются по аналогии с термином "галогеналкил". Примеры "галогеналкенила" включают (C1)2C = CHCH2 и CF3CH2CH = CHCH2. Примеры "галогеналкинила" включают HC ≡ CCHCl, CF3C ≡ C, CC13C ≡ C и FCH2C ≡ CCH2. Примеры "галогеналкокси" включают CF3O, CC13CH2O, HCF2CH2CH2O и CF3CH2O.
Примеры "алкилкарбонила" включают C(O)CH3, C(O)CH2CH2CH3 и C(O)CH(CH3)2. Примеры "алкоксикарбонила" включают CH3OC(=O), CH3CH2OC(=O), CH3CH2CH2OC(=O), (CH3)2CHOC(=O) и различные изомеры бутокси- или пентоксикарбонила. Термины "алкиламинокарбонил" и "диалкиламинокарбонил" включают, например, CH3NHC(=O), CH3CH2NHC(=O) и (CH3)2NC(=O).
Общее количество атомов углерода в группе-заместителе указывается с помощью приставки "Ci-Cj", где i и j представляют собой цифры от 1 до 8. Например, C1-C3 алкилсульфонил обозначает соединения от метилсульфонила до пропилсульфонила. В указанных выше перечислениях, когда соединение формулы I содержит гетероароматическое кольцо, все заместители соединяются с этим кольцом через любой доступный атом углерода или азота, путем замещения водорода на указанном атоме углерода или азота.
Когда группа содержит заместитель, который может представлять собой водород, например R4, тогда, когда этот заместитель берется как атом водорода, тогда очевидно, что это эквивалентно тому, что указанная группа является незамещенной.
Определенные соединения по настоящему изобретению могут существовать в виде одного или нескольких стереоизомеров. Различные стереоизомеры включают энантиомеры, диастереомеры, атропоизомеры и геометрические изомеры. Специалист в данной области должен помнить, что один из стереоизомеров может быть более активным и/или может проявлять полезные эффекты, при обогащении им, по отношению к другому стереоизомеру (стереоизомерам), или когда он отделен от другого стереоизомера (стереоизомеров). Кроме того, специалист в данной области знает, как выделить, увеличить содержание и/или селективно получить указанные стереоизомеры. Соответственно, соединения по настоящему изобретению могут присутствовать в виде смеси стереоизомеров, отдельных стереоизомеров, или в виде оптически активной формы.
С точки зрения стоимости, простоты синтеза и/или наибольшей полезности предпочтительными являются следующие соединения:
1. Соединения формулы I, где R1 представляет собой Cl или Br;
каждый R2 представляет собой, независимо, Cl или Br, и один R2 находится в 3-м положении; и
X представляет собой N.
2. Соединения формулы I, где
R1 представляет собой Cl или Br;
X представляет собой N; и
n равно 0.
Особенно следует отметить соединения формулы I (включая, но, не ограничиваясь предпочтительными соединениями 1), где n равно от 1 до 3.
3. Соединения формулы II, где
X представляет собой N.
4. Соединения формулы II, где
R1 представляет собой Cl или Br;
каждый R2 представляет собой независимо Cl или Br, и один R2 находится в 3-м положении; и
X представляет собой N.
5. Соединения формулы II, где
R1 представляет собой Cl или Br;
X представляет собой N; и
n равно 0.
Особенно следует отметить соединения формулы II (включая, но не ограничиваясь предпочтительными соединениями 3 и 4), где n равно от 1 до 3.
6. Соединения формулы 4 (где R3 представляет собой C1-C4 алкил), где каждый R2 представляет собой независимо Cl или Br, и один R2 находится в 3-м положении.
7. Соединения формулы 4 (где R3 представляет собой C1-C4 алкил), где
X представляет собой N; и
n равно 0.
Особенно следует отметить соединения формулы 4 (где R3 представляет собой C1-C4 алкил), включая, но, не ограничиваясь предпочтительными соединениями 6, где n равно от 1 до 3.
3-е положение определяется с помощью цифры "3", изображенной в арильном остатке, включенном выше в формулу I, формулу II и формулу 4.
Особенно предпочтительны соединения формулы II, где, когда R1 представляет собой Cl или Br, n равно 1, и R2, выбранный из Cl или Br, находится в 3-м положении; тогда X представляет собой N. Включены соединения, где n равно от 1 до 3.
Особенно предпочтительны соединения формулы II, где, когда R1 представляет собой Cl или Br, n равно 1, и R2, выбранный из Cl или Br, находится в 3-м положении; тогда X представляет собой CR4. Включены соединения, где n равно от 1 до 3.
Предпочтительные способы представляют собой такие, которые содержат предпочтительные соединения, указанные выше. Отмеченные способы являются такими, которые включают соединения, отмеченные выше. Особенно отмеченными являются способ получения соединения формулы I, где n равно от 1 до 3; и способ получения соединения формулы II, где n равно от 1 до 3.
Многостадийный способ получения соединений формулы I и формул II, предусмотренный здесь, включает (a) обработку соединения формулы 2
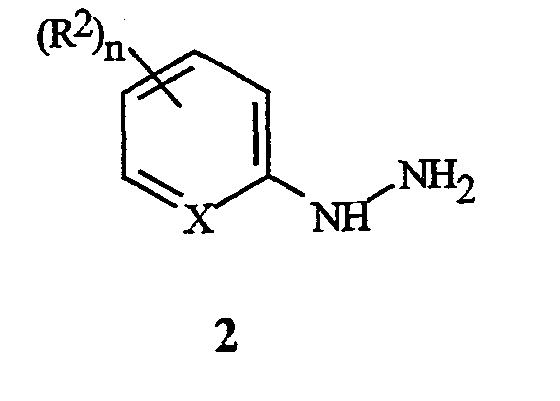
соединением формулы 3
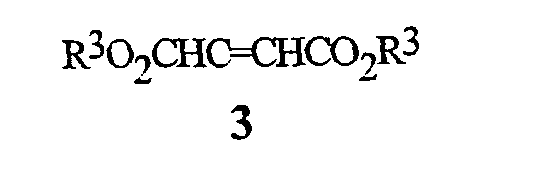
где R3 представляет собой C1-C4 алкил,
в присутствии основания, с получением соединения формулы 4
где X, R2 и n являются такими, как определено выше, и R3 представляет собой H или C1-C4 алкил.
Соединение формулы 4, где R3 представляет собой C1-C4 алкил, может затем (1) обрабатывается галогенирующим агентом, с получением соединения формулы I; а, в случае получения соединения формулы I, где R3 представляет собой H, и (2) соединения, полученные в (1), преобразуются в соединение, где R3 представляет собой H.
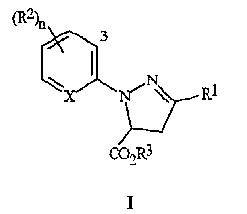
Соединение формулы I, полученное в (1) или (2), может затем (3) обрабатываться окислителем, необязательно в присутствии кислоты, с получением соединения формулы II; и когда соединения формулы I, где R3 представляет собой C1-C4 алкил, используются для получения соединений формулы II, где R3 представляет собой H, (4), осуществляют преобразование соединения, полученного в (3), в соединение формулы II, где R3 представляет собой H
Схема 1 иллюстрирует стадию (a).
Схема 1
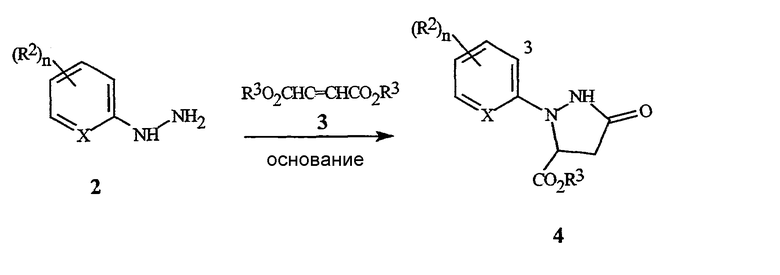
На стадии (a), соединение формулы 2 обрабатывают соединением формулы 3, где R3 представляет собой C1-C4 алкил (могут быть использованы сложные фумаратный или малеатный эфиры, или их смесь), в присутствии основания и растворителя. Основание, как правило, представляет собой соль алкоксида металла, такую как метоксид натрия, метоксид калия, этоксид натрия, этоксид калия, третбутоксид калия, третбутоксид лития, и тому подобное. Необходимо использовать более чем 0,5 эквивалента основания, по отношению к соединению формулы 2, предпочтительно, в пределах между 0,9 и 1,3 эквивалента. Необходимо использовать более чем 1,0 эквивалент соединения формулы 3, предпочтительно, в пределах между 1,0 и 1,3 эквивалента. Могут быть использованы полярные протонные и полярные апротонные органические растворители, такие как спирты, ацетонитрил, тетрагидрофуран, N,N-диметилформамид, диметилсульфоксид, и тому подобное. Предпочтительные растворители представляют собой спирты, такие как метанол и этанол. Является особенно предпочтительным, чтобы спирт был таким же, как и тот, что используется при получении сложного фумаратного или малеатного эфира и алкоксидного основания. Как правило, взаимодействие осуществляют путем смешивания соединения формулы 2 и основания в растворителе. Смесь может нагреваться или охлаждаться до желаемой температуры, и соединение формулы 3 добавляют в течение некоторого периода времени. Типичные температуры реакции находятся в пределах между 0°C и температурой кипения используемого растворителя. Взаимодействие может осуществляться при давлении, большем, чем атмосферное давление, для увеличения температуры кипения растворителя. Как правило, предпочтительными являются температуры в пределах, примерно, между 30° и 90°C. Время добавления может быть настолько быстрым, насколько позволяет изменение температуры (теплоперенос). Типичное время добавления находится в пределах между 1 минутой и 2 часами. Оптимальная температура реакции и время добавления изменяются в зависимости от характера соединений формулы 2 и формулы 3. После добавления, реакционная смесь может выдерживаться, в течение некоторого времени, при температуре реакции. В зависимости от температуры реакции, необходимое время выдерживания может находиться в пределах от 0 до 2 часов. Типичное время выдерживания составляет примерно от 10 до 60 минут. Затем реакционная масса может быть подкислена путем добавления органической кислоты, такой как уксусная кислота, и тому подобное, или неорганической кислоты, такой как хлористоводородная кислота, серная кислота, и тому подобное. В зависимости от условий реакции и средств выделения, могут быть получены соединения формулы 4, где R3 представляет собой H, или соединения формулы 4, где R3 представляет собой C1-C4 алкил. Например, соединение формулы 4, где R3 представляет собой C1-C4 алкил, может гидролизоваться in situ до соединения формулы 4, где R3 представляет собой H, когда в реакционной смеси присутствует вода. Соединения формулы 4, где R3 представляет собой H, легко могут преобразовываться в соединения формулы 4, где R3 представляет собой С1-C4 алкил, с использованием способов эстерификации, хорошо известных в данной области. Соединения формулы 4, где R3 представляет собой С1-C4 алкил, являются предпочтительными. Желаемый продукт, соединение формулы 4, может быть выделено с помощью способов, известных специалисту в данной области, таких как кристаллизация, экстракция или дистилляция.
На стадии (1), как иллюстрируется на схеме 2, соединение формулы 4 обрабатывают с помощью галогенирующего реагента, как правило, в присутствии растворителя. Галогенирующие реагенты, которые могут быть использованы, включают оксигалогениды, тригалогениды и пентагалогениды фосфора, тионилхлорид, дигалогентриалкилфосфораны, дигалогендифенилфосфораны, оксалилхлорид и фосген. Предпочтительными являются оксигалогениды и пентагалогениды фосфора. Для получения полного преобразования, необходимо использовать, по меньшей мере, 0,33 эквивалента оксигалогенида фосфора, по отношению к соединению формулы 4, предпочтительно, в пределах между 0,33 и 1,2 эквивалента. Для получения полного преобразования, необходимо использовать, по меньшей мере, 0,20 эквивалента пентагалогенида фосфора по отношению к соединению формулы 4, предпочтительно, в пределах примерно между 0,20 и 1,0 эквивалентом. Соединения формулы 4, где R3 представляет собой C1-C4 алкил, являются предпочтительными для этой реакции.
Схема 2
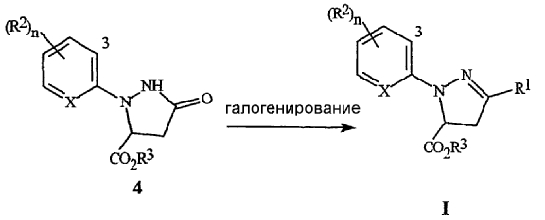
Типичные растворители для такого галогенирования включают галогенированные алканы, такие как дихлорметан, хлороформ, хлорбутан, и тому подобное, ароматические растворители, такие как бензол, ксилол, хлорбензол, и тому подобное, простые эфиры, такие как тетрагидрофуран, п-диоксан, простой диэтиловый эфир, и тому подобное, и полярные апротонные растворители, такие как ацетонитрил, N,N-диметилформамид, и тому подобное. Необязательно, может быть добавлено органическое основание, такое как триэтиламин, пиридин, N,N-диметиланилин, или что-либо подобное. Добавление катализатора, такого как N,N-диметилформамид, также является необязательным. Предпочтительным является способ, в котором растворитель представляет собой ацетонитрил, и основание отсутствует. Как правило, когда используется ацетонитрил как растворитель, не требуется ни основания, ни катализатора. Предпочтительный способ осуществляется путем смешивания соединения формулы 4 с ацетонитрилом. Затем, за удобное время, добавляется галогенирующий реагент, а затем, смесь выдерживают при желаемой температуре до завершения реакции. Температура реакции, как правило, находится в пределах между 20°C и температурой кипения ацетонитрила, а время реакции, как правило, является меньшим чем 2 часа. Затем реакционную массу нейтрализуют с помощью неорганического основания, такого как бикарбонат натрия, гидроксид натрия, и тому подобное, или органического основания, такого как ацетат натрия. Желаемый продукт, соединение формулы I, может быть выделено с помощью способов, известных специалистам в данной области, включая кристаллизацию, экстракцию и дистилляцию.
На стадии (2), соединение формулы I, где R3 представляет собой C1-C4 алкил, сложный эфир, может быть гидролизовано до соединения формулы I, где R3 представляет собой H, до карбоновой кислоты. Гидролиз может катализироваться с помощью кислот, ионов металлов и с помощью ферментов. Йодтриметилсилан отмечается как пример кислоты, которая может быть использована для катализа гидролиза (смотри Advanced Organic Chemistry, Third Ed., Jerry March, John Wiley & Sons, Inc. New York, 1985, pp. 334-338, обзор методов). Способы гидролиза, катализируемого основаниями, не рекомендуются для гидролиза соединений формулы I и могут привести к разложению. Карбоновая кислота может быть выделена с помощью способов, известных специалистам в данной области, включая кристаллизацию, экстракцию и дистилляцию.
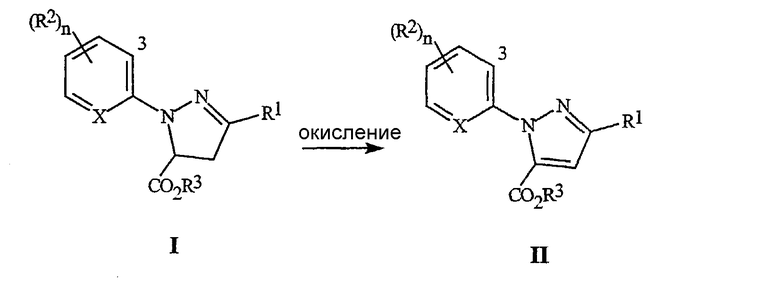
На стадии (3), как иллюстрируется на схеме 3, соединение формулы I обрабатывают окислительным агентом, необязательно, в присутствии кислоты. Соединение формулы I, где R3 представляет собой C1-C4 алкил (то есть, предпочтительный продукт стадии (1)), является предпочтительным, в качестве исходного материала для стадии (3). Окислительный агент может представлять собой перекись водорода, органические пероксиды, персульфат калия, персульфат натрия, персульфат аммония, моноперсульфат калия (например, Oxone®) или перманганат калия. Для достижения полного превращения (преобразования), необходимо использовать, по меньшей мере, один эквивалент окислительного агента, по отношению к соединению формулы I, предпочтительно, примерно от одного до двух эквивалентов. Это окисление, как правило, осуществляется в присутствии растворителя. Растворитель может представлять собой простой эфир, такой как тетрагидрофуран, п-диоксан, и тому подобное, сложный органический эфир, такой как этилацетат, диметилкарбонат, и тому подобное, или полярный апротонный органический растворитель, такой как N,N-диметилформамид, ацетонитрил, и тому подобное. Кислоты, пригодные для использования на стадии окисления, включают неорганические кислоты, такие как серная кислота, фосфорная кислота, и тому подобное, и органические кислоты, такие как уксусная кислота, бензойная кислота, и тому подобное. Кислота, когда она используется, должна быть использована в пропорции, большей чем 0,1 эквивалента, по отношению к соединению формулы I. Для достижения полного преобразования, можно использовать от одного до пяти эквивалентов кислоты. Для соединений формулы I, где X представляет собой CR2, предпочтительный окислитель представляет собой перекись водорода, и окисление предпочтительно осуществляют в отсутствие кислоты. Для соединений формулы I, где X представляет собой N, предпочтительный окислитель представляет собой персульфат калия, и окисление предпочтительно осуществляют в присутствии серной кислоты. Взаимодействие может осуществляться путем перемешивания соединения формулы I в желаемом растворителе и в кислоте, если она используется. Затем может добавляться окислитель, с удобной скоростью. Температура реакции, как правило, изменяется, самое меньшее, примерно от 0°C до температуры кипения растворителя, с целью получения разумного времени взаимодействия, для завершения реакции, предпочтительно, меньше чем за 8 часов. Желаемый продукт, соединение формулы II, где R3 представляет собой C1-C4 алкил, может быть выделено с помощью способов, известных специалистам в данной области, включая кристаллизацию, экстракцию и дистилляцию.
Схема 3
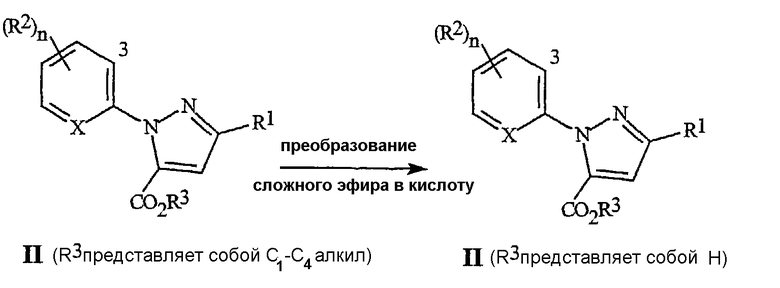
На стадии (4), как иллюстрируется на схеме 4, соединение формулы II, где R3 представляет собой C1-C4 алкил, то есть сложный эфир, может быть преобразовано в соединение формулы II, где R3 представляет собой H, то есть карбоновую кислоту. Способы преобразования сложных эфиров в карбоновые кислоты хорошо известны специалистам в данной области. Соединения формулы II (R3 представляет собой C1-C4 алкил) могут быть преобразованы в соединения формулы II (R3 представляет собой H) с помощью многочисленных способов, включая нуклеофильное расщепление в безводных условиях, или гидролитических способов, включая использование либо кислот, либо оснований (смотри T.W. Greene и P.G.M. Wuts, Protective Groups in Organic Synthesis, 2nd ed., John Wiley & Sons, Inc., New York, 1991, pp. 224-269, обзор методов). Для способа схемы 4, предпочтительными являются способы гидролиза со щелочным катализом. Пригодные для использования основания включают гидроксиды щелочных металлов (таких как литий, натрий или калий). Например, сложный эфир может быть растворен в смеси воды и спирта, такого как этанол. При обработке гидроксидом натрия или гидроксидом калия, сложный эфир омыляется, с получением натриевой или калиевой соли карбоновой кислоты. Подкисление с помощью сильной кислоты, такой как хлористоводородная кислота или серная кислота, дает карбоновую кислоту. Карбоновая кислота может быть выделена способами, известными специалистам в данной области, включая кристаллизацию, экстракцию и дистилляцию.
Схема 4
Следует отметить, что определенные соединения формулы I, где R1 представляет собой галоген, могут быть получены из других соединений формулы I, где R1 представляет собой другой галоген или представляет собой сульфонатную группу, такую как п-толуолсульфонат, бензолсульфонат и метансульфонат. Например, соединение формулы I, где R1 представляет собой Br, может быть получено путем обработки, с помощью бромистого водорода, соответствующего соединения формулы I, где R1 представляет собой Cl или п-толуолсульфонат. Взаимодействие осуществляется в соответствующем растворителе, таком как дибромметан, дихлорметан или ацетонитрил. Взаимодействие может осуществляться при атмосферном давлении или около него, или при давлении, превышающем атмосферное давление, в емкости высокого давления. Когда R1,в исходном соединении формулы I, представляет собой галоген, такой как Cl, взаимодействие предпочтительно осуществляется таким образом, что галогенид водорода, генерируемый в реакции, удаляется с помощью продувки или других соответствующих средств. Взаимодействие может осуществляться при температуре в пределах между примерно 0 и 100°C, удобнее всего, около температуры окружающей среды (например, примерно 10-40°C), а более предпочтительно, в пределах примерно между 20 и 30°C. Добавление катализатора на основе кислоты Льюиса (например, трибромида алюминия, для получения соединения формулы I, где R1 представляет собой Br) может ускорить реакцию. Продукт формулы I может быть выделен с помощью обычных способов, известных специалистам в данной области, включая экстракцию, дистилляцию и кристаллизацию.
Исходные соединения формулы I, где R1 представляет собой галоген, могут быть приготовлены так, как уже описано для схемы 2. Исходные соединения формулы I, где R1 представляет собой сульфонатную группу, могут, подобным же образом, быть получены из соответствующих соединений формулы 4 с помощью стандартных способов, таких как обработка сульфонилхлоридом (например, п-толуолсульфонилхлоридом) и основанием, таким как третичный амин (например, триэтиламин), в соответствующем растворителе, таком как дихлорметан.
Без дальнейших пояснений, предполагается, что специалист в данной области, используя предшествующее описание, может использовать настоящее изобретение в его наиболее полном объеме. По этой причине, следующие далее примеры должны рассматриваться исключительно как иллюстративные и не ограничивающие описание каким-либо образом. Исходный материал для следующих далее примеров может и не обязательно готовиться с помощью конкретного способа получения, процедура которого описывается в других примерах. Процентные содержания представляют собой проценты по массе (массовые), исключая смеси хроматографических растворителей или те места, где указано иное. Доли и проценты для смесей хроматографических растворителей указаны по объему, если не указано иного. Спектры 1H ЯМР указываются в м.д., в сторону слабого поля, от тетраметилсилана; "s" обозначает синглет, "d" обозначает дублет, "t" обозначает триплет, "q" обозначает квартет, "m" обозначает мультиплет, "dd" обозначает дублет дублетов, "dt" обозначает дублет триплетов, и "br s" обозначает уширенный синглет.
ПРИМЕР 1
Получение этил 5-оксо-2-фенил-3-пиразолидинкарбоксилата (альтернативное название этил 1-фенил-3-пиразолидинон-5-карбоксилат) с использованием диэтилмалеата
В 300-мл четырехгорлую колбу, оснащенную механической мешалкой, термометром, капельной воронкой для добавления реагента, обратным холодильником и вводом для азота, загружают 80 мл абсолютного этанола, 80,0 мл (0,214 моль) 21% этоксида натрия в этаноле и 20,0 мл (0,203 моль) фенилгидразина. Оранжевый раствор обрабатывают по каплям 40,0 мл (0,247 моль) диэтилмалеатом в течение примерно в 18 минут. Температура реакционной массы возрастает от 25 до 38°C в течение первых 5 минут добавления. Используют водяную баню попеременно с продолжением добавления для поддержания температуры реакции в пределах между 38-42°C. Полученный оранжево-красный раствор выдерживают в условиях окружающей среды в течение 30 минут. Затем его переносят в делительную воронку, содержащую 20,0 мл (0,349 моль) ледяной уксусной кислоты и 700 мл воды. Смесь экстрагируют 250 мл дихлорметана. Экстракт сушат над сульфатом магния, фильтруют, а затем концентрируют на роторном испарителе. Полученное черно-желтое масло (52,7 г) разбавляют 100 мл простого эфира, при этом кристаллизация продукта является достаточно быстрой для того, чтобы вызвать легкое кипение. Суспензию выдерживают в течение 2 часов в условиях окружающей среды. Затем ее охлаждают примерно до 0°C. Продукт отделяют посредством фильтрования, промывают 2 x 20 мл холодного простого эфира, а затем сушат на воздухе на фильтре в течение примерно 15 минут. Продукт состоит из 29,1 г (61%) белого порошка с высокой степенью кристалличности. По 1Н ЯМР никаких значительных примесей не обнаруживается. Фильтрат концентрируют до 20,8 г коричневого масла. Анализ масла показывает присутствие дополнительных 6,4 г (13%) желаемого продукта. Следовательно, общий выход реакции составляет 74%.
1Н ЯМР (ДМСО-d6) δ 10,25 (с, 1H), 7,32 (т, 2H), 7,15 (д, 2H), 7,00 (т, 1H), 4,61 (дд, 1H), 4,21 (кв, 2H), 2,95 (дд, 1H), 2,45 (дд, 1H), 1,25 (т, 3H).
ПРИМЕР 2
Получение этил 5-оксо-2-фенил-3-пиразолидинкарбоксилата (альтернативное название этил 1-фенил-3-пиразолидинон-5-карбоксилат) с использованием диэтилфумарата
В 500-мл четырехгорлую колбу, оснащенную механической мешалкой, термометром, капельной воронкой для добавления реагента, обратным холодильником и вводом для азота, загружают 150 мл абсолютного этанола, 15,0 г (0,212 моль) 96% этоксида натрия в этаноле и 20,0 мл (0,203 моль) фенилгидразина. Оранжевую смесь обрабатывают по каплям 40,0 мл (0,247 моль) диэтилфумарата в течение периода 75 минут. Во время добавления температура реакционной массы возрастает от 28 до 37°C максимально, и конечная температура составляет 32°C. Полученный несколько мутный оранжевый раствор выдерживают в условиях окружающей среды в течение 135 минут. Затем реакционную смесь выливают в делительную воронку, содержащую 15,0 мл (0,262 моль) ледяной уксусной кислоты и 700 мл воды. Смесь экстрагируют 150 мл дихлорметана. Экстракт сушат над сульфатом магния, фильтруют, а затем концентрируют на роторном испарителе. Полученное буро-желтое масло (41,3 г) разбавляют 100 мл простого эфира. Добавляют несколько затравочных кристаллов. Смесь выдерживают в течение 30 минут в условиях окружающей среды. Затем ее охлаждают примерно до 0°C. Продукт отделяют посредством фильтрования, промывают 2 x 10 мл холодного простого эфира, а затем сушат на воздухе на фильтре в течение примерно 15 минут. Продукт состоит из 9,5 г (20%) белого порошка с высокой степенью кристалличности. По 1Н ЯМР никаких значительных примесей не обнаруживается. Фильтрат концентрируют до 31 г коричневого масла. Анализ масла показывает присутствие дополнительных 7,8 г (16%) желаемого продукта. Следовательно, общая селективность реакции составляет 36%.
ПРИМЕР 3
Получение этил 5-оксо-2-(2-пиридинил)-3-пиразолидинкарбоксилата (альтернативное название этил 1-(2-пиридинил)-3-пиразолидинон-5-карбоксилат)
В 200-мл четырехгорлую колбу, оснащенную механической мешалкой, термометром, капельной воронкой для добавления реагента, обратным холодильником и вводом для азота, загружают 18 мл абсолютного этанола, 18,0 мл (0,0482 моль) 21% этоксида натрия в этаноле и 5,00 г (0,0458 моль) 2-гидразинопиридина. Раствор нагревают до 34°C. Затем его обрабатывают по каплям 9,0 мл (0,056 моль) диэтилмалеатом в течение периода в 20 минут. Во время добавления температура реакционной массы возрастает максимально до 48°C. Полученный оранжевый раствор выдерживают в условиях окружающей среды в течение 85 минут. Затем его выливают в делительную воронку, содержащую 4,0 мл (0,070 моль) ледяной уксусной кислоты и 300 мл воды. Смесь экстрагируют 2 x 50 мл дихлорметана. Экстракт сушат над сульфатом магния, фильтруют, а затем концентрируют на роторном испарителе. Полученное оранжевое масло (10,7 г) подвергают флэш-хроматографии на колонке с 200 г силикагеля, используя 4% метанол в хлороформе в качестве элюента (50 мл фракции). Фракции 9-12 выпаривают на роторном испарителе с получением 3,00 г оранжевого масла, которое содержит 77% желаемого продукта, 15% хлороформа и 8% диэтил 2-этоксибутандиоата. Фракции 13-17 концентрируют с получением 4,75 г оранжево-желтого масла, которое содержит 94% желаемого продукта и 6% хлороформа. Фракции 18-21 концентрируют с получением 1,51 г оливково-зеленого масла, которое содержит 80% желаемого продукта и 20% хлороформа. Общий выход желаемого продукта составляет 8,0 г (74%).
1Н ЯМР (ДМСО-d6) δ 10,68 (уш,, 1H), 8,22 (д, 1H), 7,70 (т, 1H), 6,90 (м, 2H), 5,33 (дд, 1H), 4,17 (кв, 2H), 3,05 (дд, 1H), 2,48 (дд, 1H), 1,21 (т, 3H).
ПРИМЕР 4
Получение этил 2-(2-хлорфенил)-5-оксо-3-пиразолидинкарбоксилата (альтернативное название этил 1-(2-хлорфенил)-3-пиразолидинон-5-карбоксилат)
В 250-мл четырехгорлую колбу, оснащенную механической мешалкой, термометром, капельной воронкой для добавления реагента, обратным холодильником и вводом для азота, загружают 40 мл абсолютного этанола, 40,0 мл (0,107 моль) 21% этоксида натрия в этаноле и 14,5 г (0,102 моль) (2-хлорфенил)гидразина. Пурпурный раствор нагревают до 35°C. Затем его обрабатывают по каплям 19,0 мл (0,117 моль) диэтилмалеата в течение периода примерно в 23 минуты. Водяную/ледяную баню используют попеременно с продолжением добавления для поддержания температуры реакции в пределах между 35-40°C. Реакционную смесь выдерживают при этой температуре в течение 30 минут. Затем ее добавляют в делительную воронку, содержащую 10,0 мл (0,175 моль) ледяной уксусной кислоты и 400 мл воды. Смесь экстрагируют 2 x 100 мл дихлорметана. Экстракт сушат над сульфатом магния, фильтруют, а затем концентрируют на роторном испарителе. Полученное темно-коричневое масло (31,0 г) кристаллизуется при стоянии. Материал суспендируют в 100 мл простого эфира и суспензию перемешивают в течение примерно 1 часа. Продукт отделяют путем фильтрования, промывают 50 мл простого эфира, а затем сушат в течение ночи при комнатной температуре в вакууме. Продукт состоит из 12,5 г (46%) кристаллического порошка. По 1Н ЯМР никаких значительных примесей не обнаруживается. Фильтрат концентрируют до 16,3 г коричневого масла. Анализ масла показывает присутствие дополнительных 6,7 г (25%) желаемого продукта. Следовательно, общая селективность реакции составляет 71%.
1H ЯМР (ДМСО-d6) δ 10,14 (с, 1H), 7,47 (6, 1H), 7,32 (м, 2H), 7,14 (т, 1H), 4,39 (д, 1H), 4,19 (кв, 2H), 3,07 (дд, 1H), 2,29 (д, 1H), 1,22 (т, 3H).
ПРИМЕР 5
Получение этил 2-(3-хлор-2-пиридинил)-5-оксо-3-пиразолидинкарбоксилата (альтернативное название этил 1-(3-хлор-2-пиридинил)-3-пиразолидинон-5-карбоксилат).
В 2-л четырехгорлую колбу, оснащенную механической мешалкой, термометром, капельной воронкой для добавления реагента, обратным холодильником и вводом для азота, загружают 250 мл абсолютного этанола и 190 мл (0,504 моль) 21% этоксида натрия в этаноле. Смесь нагревают с обратным холодильником примерно при 83°C. Затем обрабатывают 68,0 г (0,474 моль) 3-хлор-2(1Н)-пиридинон гидразона (альтернативное наименование 3-хлор-2-гидразинопиридин). Смесь повторно нагревают с обратным холодильником в течение периода в 5 минут. Затем желтую суспензию обрабатывают по каплям 88,0 мл (0,544 моль) диэтилмалеата в течение периода в 5 минут. Во время добавления скорость выкипания заметно увеличивается. К окончанию добавления весь исходный материал растворяется. Полученный оранжево-красный раствор выдерживают с обратным холодильником в течение 10 минут. После охлаждения до 65°C реакционную смесь обрабатывают 50,0 мл (0,873 моль) ледяной уксусной кислоты. Образуется осадок. Смесь разбавляют 650 мл воды, при этом осадок растворяется. Оранжевый раствор охлаждают на ледяной бане. Продукт начинает осаждаться при 28°C. Суспензию выдерживают примерно при 2°C в течение 2 часов. Продукт отделяют посредством фильтрования, промывают 3 x 50 мл 40% водного раствора этанола, а затем сушат на воздухе на фильтре в течение примерно 1 часа. Продукт состоит из 70,3 г (55%) светло-оранжевого порошка с высокой степенью кристалличности. По 1Н ЯМР никаких значительных примесей не обнаруживается.
1H ЯМР (ДМСО-d6) δ 10,18 (с, 1H), 8,27 (д, 1H), 7,92 (д, 1H), 7,20 (дд, 1H), 4,84 (д, 1H), 4,20 (кв, 2H), 2,91 (дд, 1H), 2,35 (д, 1H), 1,22 (т, 3H).
ПРИМЕР 6
Получение этил 3-хлор-4,5-дигидро-1-фенил-1Н-пиразол-5-карбоксилата (альтернативное название этил 1-фенил-3-хлор-2-пиразолин-5-карбоксилат)
ПРИМЕР 6A
Использование оксихлорида фосфора в ацетонитриле, в отсутствие основания
В 500-мл четырехгорлую колбу, оснащенную механической мешалкой, термометром, капельной воронкой для добавления реагента, обратным холодильником и вводом для азота, загружают 150 мл ацетонитрила, 25,0 г (0,107 моль) этил 5-оксо-2-фенил-3-пиразолидинкарбоксилата и 11,0 мл (0,118 моль) фосфор оксихлорида. Светло-желтый раствор нагревают до 78-80°C, в течение периода в 45 минут. После охлаждения до 54°C полученную сине-зеленую смесь обрабатывают по каплям раствором 25,0 г (0,298 моль) бикарбоната натрия в 250 мл воды. Во время 15-минутного добавления отделяют оранжевое масло. После перемешивания в течение примерно 5 минут pH смеси составляет примерно 1. Дополнительные 10,0 г (0,119 моль) бикарбоната натрия добавляют в виде твердого продукта в течение периода примерно в 3 минуты, что приводит к конечному значению pH, равному примерно 6. Смесь разбавляют 400 мл воды, при этом оранжевое масло кристаллизуется. Кристаллическую массу измельчают с помощью шпателя. Продукт отделяют посредством фильтрования, промывают 4 x 100 мл воды, а затем сушат на воздухе на фильтре в течение примерно 2 часов. Продукт состоит из 24,5 г (91%) рыхлого кристаллического светло-желтого порошка. По 1Н ЯМР никаких значительных примесей не обнаруживается.
1Н ЯМР (ДМСО-d6) δ 2,74 (т, 2H), 6,88 (д, 2H), 6,83 (т, 1H), 5,02 (дд, 1H), 4,14 (кв, 2H), 3,68 (дд, 1H), 3,34 (д, 1H), 1,16 (т, 3H).
ПРИМЕР 6B
Использование оксихлорида фосфора в хлороформе в отсутствие основания
В 100-мл двугорлую колбу, оснащенную магнитной мешалкой, термометром, обратным холодильником и вводом для азота, загружают 50 мл хлороформа, 5,00 г (0,0213 моль) этил 5-оксо-2-фенил-3-пиразолидинкарбоксилата, 2,10 мл (0,0225 моль) оксихлорида фосфора и 2 капли N,N-диметилформамида. Красно-оранжевый раствор нагревают с обратным холодильником при 64°C в течение периода в 60 минут. Полученную смесь, желто-коричневую жидкость и темно-зеленый смолистый твердый продукт, выдерживают при кипении с обратным холодильником в течение 140 минут. Затем разбавляют 100 мл дихлорметана и переносят в делительную воронку. Раствор промывают дважды 50 мл 6% водного раствора бикарбоната натрия. Органический слой сушат над сульфатом магния, фильтруют, затем концентрируют на роторном испарителе. Сырой продукт состоит из 1,50 г оранжевого масла, который кристаллизуется при стоянии. Анализ сырого продукта с помощью 1H ЯМР показывает, что он составляет примерно 65% желаемого продукта и 35% исходного материала. Выход желаемого продукта, следовательно, составляет примерно 18%.
ПРИМЕР 6C
Использование оксихлорида фосфора в хлороформе в присутствии триэтиламина
В 100-мл двухгорлую колбу, оснащенную магнитной мешалкой, термометром, обратным холодильником и вводом для азота, загружают 20 мл хлороформа, 2,00 г (0,00854 моль) этил 5-оксо-2-фенил-3-пиразолидинкарбоксилата, 1,30 мл (0,00933 моль) триэтиламина, 2 капли N,N-диметилформамида и 0,0850 мл (0,00912 моль) оксихлорида фосфора. Когда добавляют оксихлорид фосфора, имеет место мгновенная и бурная реакция. Смесь нагревают с обратным холодильником при 64°C, в течение 25 минут. Полученный желтый раствор разбавляют 50 мл воды, а затем обрабатывают 3,0 г (0,036 моль) твердого бикарбоната натрия. Двухфазную смесь перемешивают в течение 50 минут в условиях окружающей среды. Затем переносят в делительную воронку и разбавляют 100 мл дихлорметана. Органический слой отделяют, а затем промывают, в свою очередь, 50 мл 5,5% водного раствора хлористоводородной кислоты и 50 мл 3,8% водного раствора карбоната натрия. Промытый органический слой сушат над сульфатом магния, фильтруют, а затем концентрируют на роторном испарителе. Сырой продукт состоит из 1,90 г желтого масла, которое кристаллизуется при стоянии. Анализ сырого продукта с помощью 1H ЯМР показывает, что он составляет примерно 94% желаемого продукта, 2% исходного материала и 2% неопределенной примеси. Выход желаемого продукта, следовательно, составляет примерно 83%.
ПРИМЕР 7
Получение этил 3-хлор-4,5-дигидро-1-(2-пиридинил)-1Н-пиразол-5-карбоксилата (альтернативное название этил 1-(2-пиридинил)-3-хлор-2-пиразолин-5-карбоксилат)
В 250-мл четырехгорлую колбу, оснащенную механической мешалкой, термометром, обратным холодильником и вводом для азота, загружают 50 мл ацетонитрила, 4,70 г (0,0188 моль) 5-оксо-2-(2-пиридинил)-3-пиразолидинкарбоксилата и 2,00 мл (0,0215 моль) оксихлорида фосфора. Смесь нагревается сама по себе от 22 до 33°C. После выдерживания в течение 60 минут в условиях окружающей среды отбирают образец. Анализ с помощью 1H ЯМР показывает 70% преобразование исходного материала в желаемый продукт. Смесь нагревают с обратным холодильником при 85°C в течение 80 минут. Нагревательный кожух удаляют. Полученный желто-оранжевый раствор разбавляют 50 мл воды. Затем обрабатывают по каплям 3,9 г (0,049 моль) 50% водного раствора каустической соды, с получением pH, равного примерно 7,5. После перемешивания в течение 20 минут pH смеси падает примерно до 3. Добавляют дополнительные 3,0 г (0,038 моль) 50% водного раствора каустической соды, при этом pH увеличивается примерно до 9,0. Для доведения pH примерно до 7,5 добавляют малое количество концентрированной хлористоводородной кислоты. Нейтрализованную смесь переносят в делительную воронку, содержащую 300 мл воды и 100 мл дихлорметана. Органический слой отделяют, сушат над сульфатом магния, фильтруют, а затем концентрируют на роторном испарителе. Продукт состоит из 4,10 г (84%) бледно-желтого масла, которое кристаллизуется при стоянии. Единственные заметные примеси, обнаруживаемые с помощью 1Н ЯМР, представляют собой 1,0% исходного материала и 0,6% ацетонитрила.
1Н ЯМР (ДМСО-d6) δ 8,18 (д, 1H), 8,63 (т, 1H), 8,13 (д, 1H), 7,80 (т, 1H), 5,08 (дд, 1H), 4,11 (м, 2H), 3,65 (дд, 1H), 3,27 (дд, 1H), 1,14 (т, 3H).
ПРИМЕР 8
Получение этил 3-хлор-1-(3-хлор-2-пиридинил)-4,5-дигидро-1Н-пиразол-5-карбоксилата (альтернативное название этил 1-(3-хлор-2-пиридинил)-3-хлор-2-пиразолин-5-карбоксилат)
В 2-л четырехгорлую колбу, оснащенную механической мешалкой, термометром, обратным холодильником и вводом для азота, загружают 1000 мл ацетонитрила, 91,0 г (0,337 моль) этил 2-(3-хлор-2-пиридинил)-5-оксо-3-пиразолидинкарбоксилата и 35,0 мл (0,375 моль) оксихлорида фосфора. При добавлении оксихлорида фосфора смесь нагревается сама по себе от 22 до 25°C и образуется осадок. Светло-желтую суспензию нагревают с обратным холодильником при 83°C в течение периода в 35 минут, при этом осадок растворяется. Полученный оранжевый раствор выдерживают при кипении с обратным холодильником в течение 45 минут, при этом она становится черно-зеленой. Обратный холодильник заменяют насадкой для дистилляции и удаляют 650 мл растворителя отгонкой. Во вторую 2-х литровую четырехгорлую колбу, оснащенную механической мешалкой, загружают 130 г (1,55 моль) бикарбоната натрия и 400 мл воды. Концентрированную реакционную смесь добавляют к суспензии бикарбоната натрия в течение периода в 15 минут. Полученную двухфазную смесь энергично перемешивают в течение 20 минут, за это время выделение газа прекращается. Смесь разбавляют 250 мл дихлорметана, а затем перемешивают в течение 50 минут. Смесь обрабатывают 11 г диатомовой земли Celite 545®, а затем фильтруют, для удаления черного смолистого вещества, которое мешает разделению фаз. Поскольку фильтрат медленно разделяется на различные фазы, его разбавляют 200 мл дихлорметана и 200 мл воды и обрабатывают дополнительными 15 г Celite 545®. Смесь фильтруют и фильтрат переносят в делительную воронку. Более тяжелый темно-зеленый органический слой отделяется. 50 мл крупнозернистого слоя повторно фильтруют, а затем добавляют к органическому слою. Органический раствор (800 мл) обрабатывают 30 г сульфата магния и 12 г силикагеля и суспензию перемешивают с помощью магнитной мешалки в течение 30 минут. Суспензию фильтруют для удаления сульфата магния и силикагеля, при этом она становится сине-зеленой. Осадок на фильтре промывают 100 мл дихлорметана. Фильтрат концентрируют на роторном испарителе. Продукт состоит из 92,0 г (93%) темно-янтарного масла. Единственные заметные примеси, обнаруживаемые с помощью 1H ЯМР, представляют собой 1% исходного материала и 0,7% ацетонитрила.
1Н ЯМР (ДМСО-d6) δ 8,12 (д, 1H), 7,84 (д, 1H), 7,00 (дд, 1H), 5,25 (дд, 1H), 4,11 (кв, 2H), 3,58 (дд, 1H), 3,26 (дд, 1H), 1,15 (т, 3Н).
ПРИМЕР 9
Получение этил 3-бром-1-(3-хлор-2-пиридинил)-4,5-дигидро-1Н-пиразол-5-карбоксилата (альтернативное название этил 1-(3-хлор-2-пиридинил)-3-бром-2-пиразолин-5-карбоксилат)
ПРИМЕР 9A
Использование оксибромида фосфора
В 1-л четырехгорлую колбу, оснащенную механической мешалкой, термометром, обратным холодильником и вводом для азота, загружают 400 мл ацетонитрила, 50,0 г (0,185 моль) этил 2-(3-хлор-2-пиридинил)-5-оксо-3-пиразолидинкарбоксилата и 34,0 г (0,119 моль) оксибромида фосфора. Оранжевую суспензию нагревают с обратным холодильником при 83°C в течение периода в 20 минут. Полученный мутный оранжевый раствор выдерживают при кипении с обратным холодильником в течение 75 минут, за это время образуется плотный желто-коричневый кристаллический осадок. Обратный холодильник заменяют насадкой для отгонки и собирают 300 мл непрозрачного бесцветного дистиллята. Во вторую 1-л четырехгорлую колбу, оснащенную механической мешалкой, загружают 45 г (0,54 моль) бикарбоната натрия и 200 мл воды. Концентрированную реакционную смесь добавляют к суспензии бикарбоната натрия в течение периода в 5 минут. Полученную двухфазную смесь энергично перемешивают в течение 5 минут, за это время выделение газа прекращается. Смесь разбавляют 200 мл дихлорметана, а затем перемешивают в течение 75 минут. Смесь обрабатывают 5 г Celite 545®, а затем фильтруют для удаления коричневого смолистого вещества. Фильтрат переносят в делительную воронку. Коричневый органический слой (400 мл) отделяют, а затем обрабатывают 15 г сульфата магния и 2,0 г активированного угля Darco G60. Полученную суспензию перемешивают с помощью магнитной мешалки в течение 15 минут, а затем фильтруют для удаления сульфата магния и угля. Зеленый фильтрат обрабатывают 3 г силикагеля и загущают в течение нескольких минут. Сине-зеленый силикагель удаляют с помощью фильтрования и фильтрат концентрируют на роторном испарителе. Продукт состоит из 58,6 г (95%) светло-янтарного масла, которое кристаллизуется при стоянии. Единственная заметная примесь, наблюдаемая с помощью 1H ЯМР, представляет собой 0,3% ацетонитрила.
1H ЯМР (ДМСО-d6) δ 8,12 (д, 1H), 7,84 (д, 1H), 6,99 (дд, 1H), 5,20 (дд, 1H), 4,11 (кв, 2H), 3,60 (дд, 1H), 3,29 (дд, 1H), 1,15 (т, 3H).
ПРИМЕР 9B
Использование пентабромида фосфора
В 1-л четырехгорлую колбу, оснащенную механической мешалкой, термометром, обратным холодильником и вводом для азота, загружают 330 мл ацетонитрила, 52.0 г (0.193 моль) этил 2-(3-хлор-2-пиридинил)-5-оксо-3-пиразолидинкарбоксилата и 41,0 г (0.0952 моль) пентабромида фосфора. Оранжевую суспензию нагревают с обратным холодильником при 84°C в течение периода в 20 минут. Полученную кирпично-красную смесь выдерживают при кипении с обратным холодильником в течение 90 минут, за это время образуется плотный желто-коричневый кристаллический осадок. Обратный холодильник заменяют насадкой для отгонки и собирают 220 мл непрозрачного бесцветного дистиллята. Во вторую 1-л четырехгорлую колбу, оснащенную механической мешалкой, загружают 40 г (0,48 моль) бикарбоната натрия и 200 мл воды. Концентрированную реакционную смесь добавляют к суспензии бикарбоната натрия в течение периода в 5 минут. Полученную двухфазную смесь энергично перемешивают в течение 10 минут, за это время выделение газа прекращается. Смесь разбавляют 200 мл дихлорметана, а затем перемешивают в течение 10 минут. Смесь обрабатывают 5 г Celite 545®, а затем фильтруют для удаления пурпурного смолистого вещества. Осадок на фильтре промывают 50 мл дихлорметана. Фильтрат переносят в делительную воронку. Пурпурно-красный органический слой (400 мл) отделяют, затем обрабатывают 15 г сульфата магния и 2,2 г активированного угля Darco G60. Суспензию перемешивают с помощью магнитной мешалки в течение 40 минут. Суспензию фильтруют для удаления сульфата магния и угля. Фильтрат концентрируют на роторном испарителе. Продукт состоит из 61,2 г (95%) темно-янтарного масла, которое кристаллизуется при стоянии. Единственная заметная примесь, наблюдаемая с помощью 1Н ЯМР, представляет собой 0,7% ацетонитрила.
1Н ЯМР (ДМСО-d6) δ 8,12 (д, 1H), 7,84 (д, 1H), 6,99 (дд, 1H), 5,20 (дд, 1H), 4,11 (кв, 2H), 3,60 (дд, 1H), 3,29 (дд, 1H), 1,15 (т, 3H).
ПРИМЕР 10
Получение этил 3-хлор-1-фенил-1Н-пиразол-5-карбоксилата (альтернативное название этил 1-фенил-3-хлорпиразол-5-карбоксилат)
ПРИМЕР 10A
Использование перекиси водорода
В 100-мл двухгорлую колбу, оснащенную магнитной мешалкой, термометром, обратным холодильником и вводом для азота, загружают 1,50 г (0,00594 моль) этил 3-хлор-4,5-дигидро-1-фенил-1Н-пиразол-5-карбоксилата и 15 мл ацетонитрила. Смесь нагревают до 80°C. Затем ее обрабатывают 0,700 мл (0,00685 моль) 30% водного раствора перекиси водорода. Смесь выдерживают при 78-80°C в течение 5 часов. Затем реакционную массу добавляют к 70 мл воды. Осадившийся продукт отделяют посредством фильтрования, а затем промывают 15 мл воды. Влажную лепешку осадка растворяют в 100 мл дихлорметана. Раствор сушат над сульфатом магния, фильтруют, а затем концентрируют на роторном испарителе. Продукт состоит из 1,24 г (примерно 79%) оранжевого масла, которое кристаллизуется при стоянии. Как показывает 1H ЯМР, полученный материал содержит примерно 95% чистого продукта.
1H ЯМР (ДМСО-d6) δ 7,50 (с, 5H), 7,20 (с, 1H), 7,92 (д, 1H), 4,18 (кв, 2H), 1,14 (т, 3H).
ПРИМЕР 10B
Использование двуокиси марганца
В 100-мл двухгорлую колбу, оснащенную магнитной мешалкой, термометром, обратным холодильником и вводом для азота, загружают 3,00 г (0,0119 моль) этил 3-хлор-4,5-дигидро-1-фенил-1Н-пиразол-5-карбоксилата, 25 мл хлороформа и 2,50 г (0,0245 моль) активированной двуокиси марганца. Смесь нагревают при кипении с обратным холодильником при 62°C в течение периода в 1 час. Анализ образца реакционной массы с помощью 1H ЯМР показывает примерно 6% преобразование исходного материала, в основном, в желаемый этил 1-фенил-3-хлорпиразол-5-карбоксилат. Смесь выдерживают при кипении с обратным холодильником в течение дополнительных 5 часов. Анализ второго образца показывает примерно 9% преобразование.
ПРИМЕР 10C
Использование гипохлорита натрия
В 100-мл двухгорлую колбу, оснащенную магнитной мешалкой, термометром, обратным холодильником и вводом для азота, загружают 1,00 г (0,00396 моль) этил 3-хлор-4,5-дигидро-1-фенил-1Н-пиразол-5-карбоксилата, 10 мл ацетонитрила, 0,55 г (0,0040 моль) натрия дигидрофосфата моногидрата и 5,65 г (0,00398 моль) 5,25% водного раствора гипохлорита натрия. Оранжевый раствор выдерживают в условиях окружающей среды в течение 85 минут. Анализ образца реакционной массы c помощью 1Н ЯМР показывает примерно 71% преобразование исходного материала в два основных продукта. Раствор нагревают до 60°C в течение 60 минут. Анализ второго образца не показывает никакого роста преобразования относительно первого образца. Реакционную смесь обрабатывают дополнительными 3,00 г (0,00211 моль) 5,25% водного раствора гипохлорита натрия. После выдерживания в течение 60 минут при 60°C реакционную массу добавляют к 100 мл воды. Смесь экстрагируют 100 мл дихлорметана. Экстракт отделяют, сушат над сульфатом магния, фильтруют, а затем концентрируют на роторном испарителе. Сырой продукт состоит из 0,92 г красно-оранжевого масла. 1Н ЯМР показывает, что сырой продукт состоит, в основном, из этил 3-хлор-1-(4-хлорфенил)-4,5-дигидро-1Н-пиразол-5-карбоксилата (альтернативное название этил 1-(4-хлорфенил)-3-хлор-2-пиразолин-5-карбоксилат) и этил 3-хлор-1-(2-хлорфенил)-4,5-дигидро-1Н-пиразол-5-карбоксилата (альтернативное название этил 1-(2-хлорфенил)-3-хлор-2-пиразолин-5-карбоксилат), в соотношении 2:1. Изомер может быть выделен с помощью хроматографии на силикагеле, с использованием 10% этилацетата в гексане, в качестве элюента. 1Н ЯМР для этил 3-хлор-1-(4-хлорфенил)-4,5-дигидро-1H-пиразол-5-карбоксилата (ДМСО-d6) δ 7,28 (д, 2H), 6,89 (д, 2H), 5,08 (дд, 1H), 4,14 (кв, 2H), 3,71 (дд, 1H), 3,37 (дд, 1H), 1,16 (т, 3H). 1H ЯМР для этил 3-хлор-1-(2-хлорфенил)-4,5-дигидро-1Н-пиразол-5-карбоксилата (ДМСО-d6) δ 7,41 (д, 1H), 7,30 (м, 2H), 7,14 (м, 1H), 5,22 (дд, 1H), 3,90 (кв, 2H), 3,68 (дд, 1H), 3,38 (дд, 1H), 0,91 (т, 3H).
ПРИМЕР 11
Получение этил 3-хлор-1-(3-хлор-2-пиридинил)-1Н-пиразол-5-карбоксилата (альтернативное название этил 1-(3-хлор-2-пиридинил)-3-хлорпиразол-5-карбоксилат)
В 2-л четырехгорлую колбу, оснащенную механической мешалкой, термометром, обратным холодильником и вводом для азота, загружают 99,5 г (0,328 моль) этил 3-хлор-1-(3-хлор-2-пиридинил)-4,5-дигидро-1Н-пиразол-5-карбоксилата, 1000 мл ацетонитрила с чистотой 95% и 35,0 мл (0,661 моль) 98% серной кислоты. Смесь нагревается сама по себе от 22 до 35°C при добавлении серной кислоты. После перемешивания в течение нескольких минут смесь обрабатывают 140 г (0,518 моль) персульфата калия. Суспензию нагревают с обратным холодильником при 84°C в течение 4,5 часов. Полученную оранжевую суспензию фильтруют в то время, пока она еще теплая (50-65°C), для удаления мелкодисперсного белого осадка. Осадок на фильтре промывают 50 мл ацетонитрила. Фильтрат концентрируют на роторном испарителе примерно до 500 мл. Во вторую 2-л четырехгорлую колбу, оснащенную механической мешалкой, загружают 1250 мл воды. К воде добавляют концентрированную реакционную массу в течение периода примерно в 5 минут. Продукт отделяют посредством фильтрования, промывают 3 x 125 мл 25% водного раствора ацетонитрила, промывают один раз 100 мл воды, а затем сушат в течение ночи в вакууме при комнатной температуре. Продукт состоит из 79,3 г (82%) кристаллического оранжевого порошка. Единственные заметные примеси, обнаруживаемые с помощью 1Н ЯМР, представляют собой примерно 1,9% воды и 0,6% ацетонитрила.
1Н ЯМР (ДМСО-d6) δ 8,59 (д, 1H), 8,38 (д, 1H), 7,71 (дд, 1H), 7,31 (с, 1H), 4,16 (кв, 2H), 1,09 (т, 3H).
ПРИМЕР 12
Получение этил 3-бром-1-(3-хлор-2-пиридинил)-1H-пиразол-5-карбоксилата (альтернативное название этил 1-(3-хлор-2-пиридинил)-3-бромпиразол-5-карбоксилат)
В 1-л четырехгорлую колбу, оснащенную механической мешалкой, термометром, обратным холодильником и вводом для азота, загружают 40,2 г (0,121 моль) этил 3-бром-1-(3-хлор-2-пиридинил)-4,5-дигидро-1Н-пиразол-5-карбоксилата, 300 мл ацетонитрила и 13,0 мл (0,245 моль) 98% серной кислоты. Смесь нагревается сама по себе от 22 до 36°C, при добавлении серной кислоты. После перемешивания в течение нескольких минут смесь обрабатывают 48,0 г (0,178 моль) персульфата калия. Суспензию нагревают при кипении с обратным холодильником при 84°C в течение 2 часов. Полученную оранжевую суспензию фильтруют в то время, пока она еще теплая (50-65°C), для удаления белого осадка. Осадок на фильтре промывают 2 x 50 мл ацетонитрила. Фильтрат концентрируют на роторном испарителе примерно до 200 мл. Во вторую 1-л четырехгорлую колбу, оснащенную механической мешалкой, загружают 400 мл воды. К воде добавляют концентрированную реакционную массу в течение периода примерно в 5 минут. Продукт отделяют посредством фильтрования, промывают 100 мл 20% водного раствора ацетонитрила, промывают 75 мл воды, а затем сушат на воздухе на фильтре в течение 1 часа. Продукт состоит из 36,6 г (90%) кристаллического оранжевого порошка. Единственные заметные примеси, обнаруживаемые с помощью 1H ЯМР, представляют собой примерно 1% неизвестного продукта и 0,5% ацетонитрила.
1H ЯМР (ДМСО-d6) δ 8,59 (д, 1H), 8,39 (д, 1H), 7,72 (дд, 1H), 7,35 (с, 1H), 4,16 (кв, 2H), 1,09 (т,3H).
ПРИМЕР 13
Получение 3-хлор-1-(3-хлор-2-пиридинил)-1Н-пиразол-5-карбоновой кислоты (альтернативное название 1-(3-хлор-2-пиридинил)-3-хлорпиразол-5-карбоновая кислота)
В 1-л четырехгорлую колбу, оснащенную механической мешалкой, термометром и вводом для азота, загружают 79,3 г (0,270 моль) 97,5% этил 3-хлор-1-(3-хлор-2-пиридинил)-1H-пиразол-5-карбоксилата, 260 мл метанола, 140 мл воды и 13,0 г (0,325 моль) гранул гидроксида натрия. Смесь нагревается сама по себе от 22 до 35°C, и исходный материал начинает растворяться при добавлении гидроксида натрия. После перемешивания в течение 45 минут в условиях окружающей среды весь исходный материал растворяется. Полученный оранжево-коричневый раствор концентрируют на роторном испарителе примерно до 250 мл. Затем концентрированную реакционную смесь разбавляют 400 мл воды. Водный раствор экстрагируют 200 мл простого эфира. Водный слой переносят в 1-л колбу Эрленмейера, оснащенную магнитной мешалкой. Затем раствор обрабатывают по каплям 36,0 г (0,355 моль) концентрированной хлористоводородный кислоты в течение периода примерно в 10 минут. Продукт отделяют посредством фильтрования, повторно суспендируют 2 x 200 мл воды, верхний слой промывают один раз 100 мл воды, а затем сушат на воздухе на фильтре в течение 1,5 часов. Продукт состоит из 58,1 г (83%) кристаллического светло-коричневого порошка. Примерно 0,7% простого эфира представляет собой единственную заметную примесь, обнаруживаемую с помощью 1Н ЯМР.
1Н ЯМР (ДМСО-d6) δ 13,95 (уш.с, 1H), 8,56 (д, 1H), 8,25 (д, 1H), 7,68 (дд, 1H), 7,20 (с, 1H).
ПРИМЕР 14
Получение 3-бром-1-(3-хлор-2-пиридинил)-1Н-пиразол-5-карбоновой кислоты (альтернативное название 1-(3-хлор-2-пиридинил)-3-бромпиразол-5-карбоновая кислота)
В 300-мл четырехгорлую колбу, оснащенную механической мешалкой, термометром и вводом для азота, загружают 25,0 г (0,0756 моль) этил 3-бром-1-(3-хлор-2-пиридинил)-1Н-пиразол-5-карбоксилата с чистотой 98,5%, 75 мл метанола, 50 мл воды и 3,30 г (0,0825 моль) гранул гидроксида натрия. Смесь нагревается сама по себе от 29 до 34°C, и исходный материал начинает растворяться при добавлении гидроксида натрия. После перемешивания в течение 90 минут в условиях окружающей среды весь исходный материал растворяется. Полученный темно-оранжевый раствор концентрируют примерно до 90 мл на роторном испарителе. Затем концентрированную реакционную смесь разбавляют 160 мл воды. Водный раствор экстрагируют 100 мл простого эфира. Водный слой переносят в 500-мл колбу Эрленмейера, оснащенную магнитной мешалкой. Затем раствор обрабатывают по каплям 8,50 г (0,0839 моль) концентрированной хлористоводородной кислотой в течение периода примерно в 10 минут. Продукт отделяют посредством фильтрования, повторно суспендируют 2 x 40 мл воды, верхний слой промывают один раз 25 мл воды, а затем сушат на воздухе на фильтре в течение 2 часов. Продукт состоит из 20,9 г (91%) кристаллического желто-коричневого порошка. Единственные заметные примеси, обнаруживаемые с помощью 1Н ЯМР, представляют собой примерно 0,8% неизвестного продукта и 0,7% простого эфира.1Н ЯМР (ДМСО-d6) δ 13,95 (уш.с, 1H), 8,56 (д, 1H), 8,25 (д, 1H), 7,68 (дд, 1H), 7,25 (с, 1H).
ПРИМЕР 15
Получение этил 3-бром-1-(3-хлор-2-пиридинил)-4,5-дигидро- 1H-пиразол-5-карбоксилата из этил 3-хлор-1-(3-хлор-2-пиридинил)-4,5-дигидро-1H-пиразол-5-карбоксилата с использованием бромистого водорода
Бромистый водород пропускают через раствор этил 3-хлор-1-(3-хлор-2-пиридинил)-4,5-дигидро-1Н-пиразол-5-карбоксилата (8,45 г, 29,3 ммоль) в дибромметане (85 мл). Через 90 минут поток газа прекращают и реакционную смесь промывают водным раствором бикарбоната натрия (100 мл). Органическую фазу сушат и выпаривают при пониженном давлении с получением указанного в заголовке продукта в виде масла (9,7 г, выход 99%), которое кристаллизуется при стоянии.
1H ЯМР (CDC13) δ 8,07 (дд, J =1,6, 4,8Гц, 1H), 7,65 (дд, J =1,6, 7,8Гц, 1H), 6,85 (дд, J =4,7, 7,7Гц, 1H), 5,25 (X из ABX, 1H, J =9,3, 11,9Гц), 4,18 (кв, 2H), 3,44 (1/2 AB в серии ABX, J =11,7, 17,3Гц, 1H), 3,24 (1/2 AB в серии ABX, J =9,3, 17,3Гц, 1H), 1,19 (т, 3H).
Следующий далее пример 16 иллюстрирует получение этил 1-(3-хлор-2-пиридинил)-4,5-дигидро-3-[[(4-метилфенил)-сульфонил]окси]-1Н-пиразол-5-карбоксилата, который может быть использован для получения этил 3-бром-1-(3-хлор-2-пиридинил)-4,5-дигидро-1Н-пиразол-5-карбоксилата, с помощью методик, подобных тем, что описаны в примере 15.
ПРИМЕР 16
Получение этил 1-(3-хлор-2-пиридинил)-4,5-дигидро-3-[[4-метилфенил)сульфонил]окси]-1Н-пиразол-5-карбоксилата
Триэтиламин (3,75 г, 37,1 ммоль) добавляют по каплям в смесь этил 2-(3-хлор-2-пиридинил)-5-оксо-3-пиразолидинкарбоксилата (10,0 г, 37,1 ммоль) и п-толуолсульфонил хлорида (7,07 г, 37,1 ммоль) в дихлорметане (100 мл), при 0°C. Добавляют дополнительные порции п-толуолсульфонил хлорида (0,35 г, 1,83 ммоль) и триэтиламина (0,19 г, 1,88 ммоль). Затем реакционной смеси дают возможность нагреться до комнатной температуры и перемешивают ее в течение ночи. Затем смесь разбавляют дихлорметаном (200 мл) и промывают водой (3 x 70 мл). Органическую фазу сушат и выпаривают с получением указанного в заголовке продукта в виде масла (13,7 г, выход 87%), которое медленно образует кристаллы. Продукт, перекристаллизованный из этил ацетата/гексана, плавится при 99,5-100°C.
ИК (нуджол): 1740, 1638, 1576, 1446, 1343, 1296, 1228, 1191, 1178, 1084, 1027,948, 969, 868, 845 см-1.
1H ЯМР (CDC13) δ 8,01 (дд, J =1,4, 4,6Гц, 1H), 7,95 (д, J =8,4Гц, 2H), 7,56 (дд, J =1,6, 7,8Гц, 1H), 7,36 (д, J =8,4Гц, 2H), 6,79 (дд, J =4,6, 7,7Гц, 1H), 5,72 (X из ABX, J =9, 11,8Гц, 1H), 4,16 (кв, 2H), 3,33 (1/2 AB в серии ABX, J =17,5, 11,8 Гц, 1H), 3,12 (1/2 AB в серии ABX, J =17,3, 9Гц, 1H), 2,45 (с, 3H), 1,19 (т, 3H).
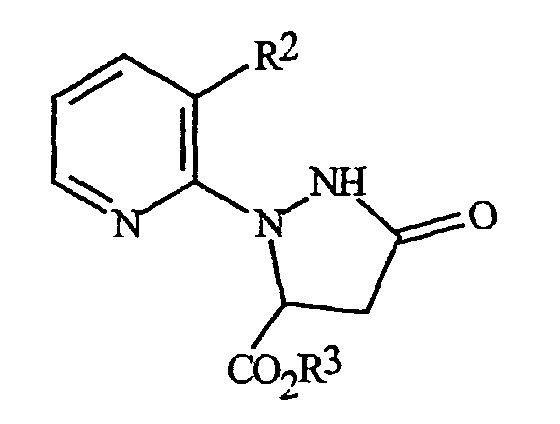
С помощью процедур, описанных здесь, вместе со способами, известными в данной области, могут быть получены следующие далее соединения из таблиц 1-3. В таблицах используются следующие сокращения: t представляет собой третичный, s представляет собой вторичный, n представляет собой нормальный, i представляет собой изо, Me представляет собой метил, Et представляет собой этил, Pr представляет собой пропил, i-Pr представляет собой изопропил и t-Bu представляет собой третичный бутил.
Таблица 1
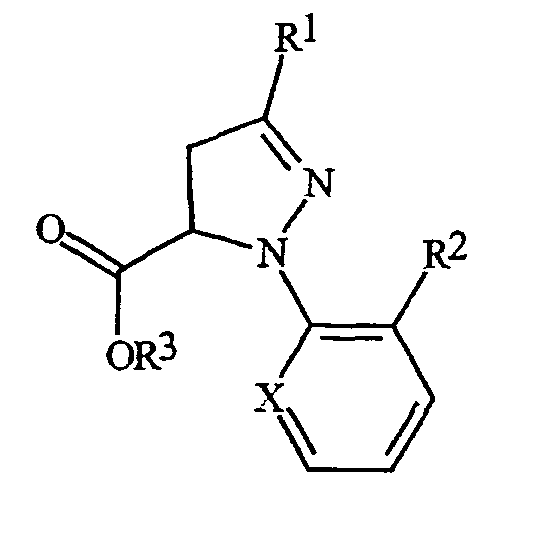
Таблица 2
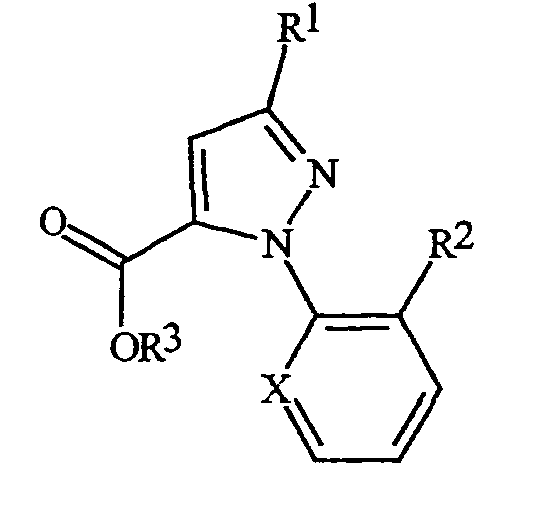
Таблица 3
Применимость
Соединения формул I, II и 4 являются пригодными для использования, в качестве синтетических промежуточных соединений, для получения соединения формулы III
где X, R1, R2 и n являются такими, как определено выше; R6 представляет собой CH3, Cl или Br; R7 представляет собой F, Cl, Br, I или CF3; и R8 представляет собой C1-C4 алкил.
Соединения формулы III являются пригодными для использования в качестве инсектицидов.
Соединения формулы III могут быть получены из соединений формулы II (и, в свою очередь, из соединений формулы 4 и I) с помощью способов, изображенных на схемах 5-7.
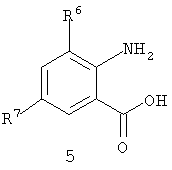
Соединение пиразолкарбоновой кислоты формулы IIa (соединение формулы II, где R3 представляет собой H) с антраниловой кислотой формулы 5 дает бензоксазинон формулы 6. На схеме 5 бензоксазинон формулы 6 готовят непосредственно путем последовательного добавления метансульфонил хлорида в присутствии третичного амина, такого как триэтиламин или пиридин, к пиразолкарбоновой кислоте формулы IIa, с последующим добавлением антраниловой кислоты формулы 5, с последующим вторым добавлением третичного амина и метансульфонил хлорида. Эта процедура, как правило, дает хороший выход бензоксазинона.
Схема 5
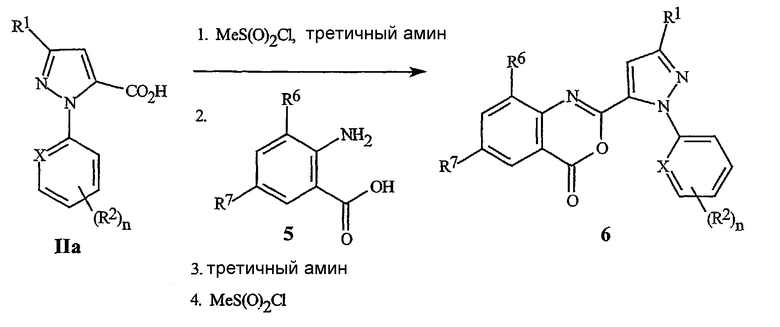
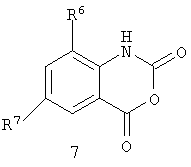
Схема 6 изображает альтернативное получение для бензоксазинонов формулы 6, включающее соединение хлорангидрида пиразоловой кислоты формулы 8 с ангидридом изотоновой кислоты формулы 7, с непосредственным получением бензоксазинона формулы 6.
Схема 6
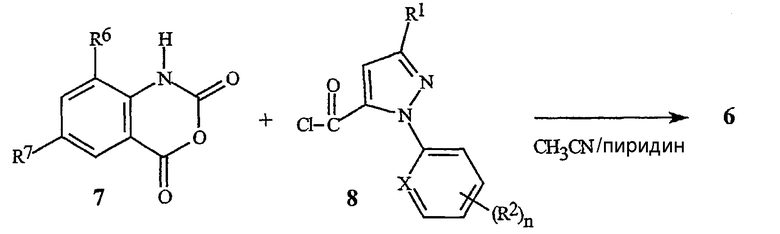
Такие растворители, как пиридин или пиридин/ацетонитрил, являются пригодными для использования в этой реакции. Хлорангидриды формулы 8 являются доступными из соответствующих кислот формулы IIa с помощью известных процедур, таких как хлорирование с помощью тионилхлорида или оксалилхлорида.
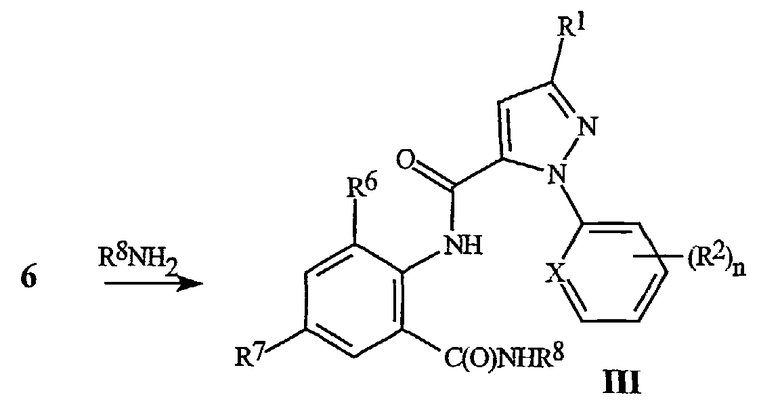
Соединения формулы III могут быть получены путем взаимодействия бензоксазинонов формулы 6 с C1-C4 алкиламинами, как изображено на схеме 7. Взаимодействие может осуществляться само по себе или в разнообразных соответствующих растворителях, включающих тетрагидрофуран, простой диэтиловый эфир, дихлорметан или хлороформ, при оптимальных температурах, находящихся в пределах от комнатной температуры до температуры дефлегмации растворителя. Общее взаимодействие бензоксазинонов с аминами, с получением антраниламидов, является хорошо известным в химической литературе. Обзор химии бензоксазинонов смотри в Jakobsen et al., Biorganic and Medicinal Chemistry 2000, 8, 2095-2103 и в ссылках, цитируемых там. Смотри также Coppola, J. Heterocyclic Chemistry 1999, 36, 563-588.
СХЕМА 7
Пример 17
Получение 6,8-дихлор-2-[1-(3-хлор-2-пиридинил)-3-бром-1H-пиразол-5-ил]-4Н-3,1-бензоксазин-4-она
Стадия А: Получение 6,8-дихлор-2Н-3,1-бензоксазин-2,4(1Н)-диона
К суспензии 2-амино-3,5-дихлорбензойной кислоты (104 г, 500 ммоль), перемешиваемой в сухом диоксане (750 мл) при комнатной температуре, добавляли по каплям трихлорметилхлорформиат (70 г, 350 ммоль). Реакционная смесь экзотермически нагревалась медленно до 30°С, и твердое вещество почти полностью растворялось перед тем, как снова образовать густую суспензию. После перемешивания этой суспензии при температуре окружающей среды в течение 2,5 часов указанное в заголовке соединение выделяли фильтрованием, промывали этиловым эфиром и сушили с получением указанного в заголовке соединения-продукта, полученного в виде белого твердого вещества (82 г).
1H ЯМР (ДМСО-d6) δ 7,88(d, 1Н), 8,07 (d, 1Н), 11,6 (с, 1Н).
Стадия В: Получение 6,8-дихлор-2-[1-(3-хлор-2-пиридинил)-3-бром-1H-пиразол-5-ил]-4Н-3,1-бензоксазин-4-она
К суспензии 1-(3-хлор-2-пиридинил)-3-бром-1Н-пиразол-5-карбоновой кислоты (т.е. продукта стадии С) (118 г, 390 ммоль), перемешиваемой в дихлорметане (1 л), добавляли N,N-диметилформамид (8 капли). Добавляли по каплям оксалилхлорид (63,8 г, 500 ммоль) в течение 1.5 час. Полученный раствор перемешивали при комнатной температуре в течение ночи и затем концентрировали в вакууме. Выделенный хлорангидрид кислоты растворяли в сухом ацетонитриле (200 мл) и добавляли к суспензии 6,8-дихлор-2Н-3,1-бензоксазин-2,4(1Н)-дион (т.е. продукта стадии А) (82 г, 350 ммоль), перемешиваемой в сухом ацетонитриле (200 мл). Добавляли пиридин (175 мл) и этот раствор нагревали с обратным холодильником для дефлегмации в течение 4 часов. После охлаждения на бане со льдом собирали осадок белого твердого вещества (152 г).
1H ЯМР (CDCl3) δ 7,27 (d, 1Н), 7,38 (кв, 1H), 7,72 (с, 1Н), 7,9 (d, 1Н), 8,05 (с, 1Н), 8,55 (d, 1Н).
Пример 18
Получение 2-[3-бром-1-(3-хлор-2-пиридинил)-1Н-пиразол-5-ил]-6-хлор-8-метил-4Н-3,1-бензоксазин-4-она
К раствору К раствору метансульфонилхлорида (0,40 мл, 5,2 ммоль) в ацетонитриле добавляли по каплям смесь 3-бром-1-(3-хлор-2-пиридинил)-1Н-пиразол-5-карбоновой кислоты (1,5 г, 4,96 ммоль) и триэтиламин (0,64 мл, 4,96 ммоль) в ацетонитриле при 0-5°С. Затем температуру реакции поддерживали при 0°С на протяжении последовательного добавления реагентов. После перемешивания в течение 20 минут добавляли 2-амино-3-метил-5-хлорбензойную кислоту (0,92 г, 4,96 ммоль) и перемешивание продолжали в течение дополнительных 5 минут. Затем добавляли по каплям раствор триэтиламина (10,0 ммоль) в ацетонитриле и реакционную смесь перемешивали в течение 45 минут с последующим добавлением метансульфонилхлорида (0,40 мл, 5,2 ммоль). Затем реакционную смесь нагревали до комнатной температуры и перемешивали в течение ночи.
Получали указанное в заголовке соединение в виде твердого вещества (1,21 г).
1H ЯМР (CDCl3) δ 2,01, (с, 3Н), 7,29 (с, 1Н), 7,42 (д, 1Н), 7,95 (д, 1Н), 8,04 (м, 1Н), 8,25 (с, 1Н), 8,26 (д, 1Н).
Пример 19
Получение 3-бром-N-[4-хлор-2-метил-6-[[(1-метилэтил)амино]-карбонил]фенил]-1-(3-хлор-2-пиридинил)-1Н-пиразол-5-карбоксамида
К раствору 2-[3-бром-1-(3-хлор-2-пиридинил)-1Н-пиразол-5-ил]-6-хлор-8-метил-4Н-3,1-бензоксазин-4-она (т.е. продукта бензоксазинона, полученного выше) (0,20 г, 0,44 ммоль) в тетрагидрофуране добавляли изопропиламин (0,122 мл, 1,42 ммоль) и реакционную смесь нагревали до 60°С в течение 90 минут и затем охлаждали до комнатной температуры. Растворитель тетрагидрофуран выпаривали при пониженном давлении и оставшееся твердое вещество растирали с эфиром, фильтровали и сушили с получением указанного в заголовке соединения, соединения данного изобретения, в виде твердого вещества (150 мг), т.пл. 159-161°С.
1H ЯМР (CDCl3) δ 1,22 (д, 6Н), 2,19 (с, 3Н), 4,21 (м, 1Н), 5,99 (м, 1Н), 7,05 (м, 1Н), 7,22 (м, 2Н), 7,39 (м, 1Н), 7,82 (д, 1Н), 8,41 (д, 1Н).
| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБ БОРЬБЫ С КОНКРЕТНЫМИ НАСЕКОМЫМИ-ВРЕДИТЕЛЯМИ ПУТЕМ НАНЕСЕНИЯ АНТРАНИЛАМИДНЫХ СОЕДИНЕНИЙ | 2002 |
|
RU2262231C1 |
| ЦИАНОАНТРАНИЛАМИДНЫЕ ИНСЕКТИЦИДЫ | 2004 |
|
RU2343151C2 |
| ОРТОЗАМЕЩЕННЫЕ АРИЛАМИДЫ, СПОСОБ БОРЬБЫ С НАСЕКОМЫМИ, КОМПОЗИЦИЯ ДЛЯ БОРЬБЫ С НАСЕКОМЫМИ, ПРОМЕЖУТОЧНОЕ СОЕДИНЕНИЕ | 2002 |
|
RU2283839C2 |
| АНТРАНИЛАМИДНОЕ СОЕДИНЕНИЕ, КОМПОЗИЦИЯ ДЛЯ БОРЬБЫ С НАСЕКОМЫМИ, КОМПОЗИЦИЯ ДЛЯ БОРЬБЫ С БЕСПОЗВОНОЧНЫМИ ВРЕДИТЕЛЯМИ, СПОСОБЫ БОРЬБЫ С НАСЕКОМЫМИ, ПРОМЕЖУТОЧНЫЕ СОЕДИНЕНИЯ | 2002 |
|
RU2283840C2 |
| СПОСОБ ЗАЩИТЫ СЕЯНЦЕВ ИЛИ РАСТЕНИЙ, ВЫРОСШИХ ИЗ НИХ, ОТ НАСЕКОМЫХ, КОМПОЗИЦИЯ ДЛЯ КОНТРОЛЯ ЗА НАСЕКОМЫМИ | 2002 |
|
RU2292138C2 |
| СПОСОБ ПОЛУЧЕНИЯ N-ФЕНИЛПИРАЗОЛ-1-КАРБОКСАМИДОВ | 2005 |
|
RU2397165C2 |
| СПОСОБ ПОЛУЧЕНИЯ 3-ГАЛОГЕН-4,5-ДИГИДРО-1Н-ПИРАЗОЛОВ | 2003 |
|
RU2326877C2 |
| ПИРАЗОЛОПИРИМИДИНОВЫЕ СОЕДИНЕНИЯ-ИНГИБИТОРЫ JAK И СПОСОБЫ | 2009 |
|
RU2539568C2 |
| ИНСЕКТИЦИДНЫЕ АНТРАНИЛАМИДЫ | 2001 |
|
RU2278852C2 |
| СОЕДИНЕНИЕ, КОМПОЗИЦИЯ ДЛЯ БОРЬБЫ С НАСЕКОМЫМИ, СПОСОБЫ БОРЬБЫ С НАСЕКОМЫМИ | 2002 |
|
RU2298007C2 |
Изобретение относится к новым соединениям формулы I и формулы II
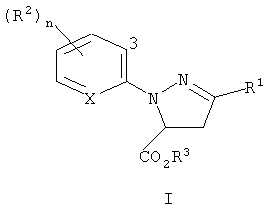
где R1 представляет собой галоген; R2 представляет галоген; R3 представляет C1-C4 алкил; Х представляет N или СН; и n равно 0-3, при условии, что когда Х представляет собой СН, тогда n равно, по меньшей мере, 1.
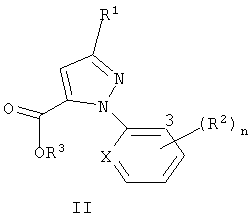
где R1 представляет галоген; R2 представляет галоген; R3 представляет собой Н или C1-C4 алкил; Х представляет собой N или СН; и n равно от 0 до 3, при условии, что когда Х представляет собой СН, тогда n равно, по меньшей мере, 1. Изобретение также относится к способу получения соединения формулы I, способу получения соединения формулы II, способ получения соединения формулы III. Также описываются промежуточные соединения формулы 4. Технический результат - получение новых биологически активных соединений, которые представляют интерес в качестве инсектицидов, а также способ их получения. 6 н. и 18 з.п. ф-лы, 3 табл.
где R1 представляет собой галоген;
R2 представляет галоген;
R3 представляет C1-C4 алкил;
Х представляет N или СН; и
n равно 0-3, при условии, что когда Х представляет собой СН, тогда n равно, по меньшей мере, 1.
где R3 представляет собой C1-C4 алкил; галогенирующим агентом.
где R1 представляет галоген;
R2 представляет галоген;
R3 представляет собой Н или C1-C4 алкил;
Х представляет собой N или СН; и
n равно от 0 до 3, при условии, что когда Х представляет собой СН, тогда n равно, по меньшей мере, 1.
R1 представляет собой Cl или Br;
каждый R2 представляет собой, независимо, Cl или Br, и один R2 находится в 3-м положении; Х представляет собой N.
R2 представляет Cl или Br, и R2 находится в 3-м положении и R3 представляет Н.
где каждый R2 представляет галоген;
Х представляет N;
R3 представляет C1-C4 алкил; и
n равно от 0 до 3.
окислителем, необязательно, в присутствии кислоты, с получением соединения формулы II; и когда соединение формулы I, где R3 представляет собой C1-C4 алкил, используют для получения соединения формулы II, где R3 представляет собой Н,
(4) преобразование соединения, полученного в (3), в соединение формулы II, где R3 представляет собой Н.
где R3 представляет собой C1-C4 алкил;
галогенирующим агентом с получением соединения формулы I.
где R1 представляет галоген;
каждый R2 представляет галоген;
Х представляет N или СН;
R6 представляет СН3, Cl или Br;
R7 представляет F, Cl, Br, I или CF3;
R8 представляет C1-C4 алкил и
n равно 0, 1, 2 или 3; при условии, что когда Х представляет собой СН, тогда n равно, по меньшей мере, 1;
с использованием в качестве промежуточного соединения формулы II
где R3 представляет собой Н;
отличающийся тем, что способ включает следующие стадии:
(а) получение соединения формулы II в соответствии с методом, описанным в п.16;
(в) получение соединения формулы 6
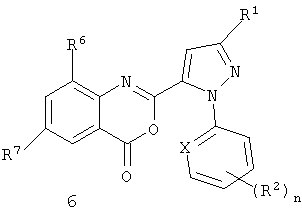
или (i) путем сочетания соединения II с соединением формулы 5
или (ii) хлорированием соединения формулы II, получая соединение формулы 8, и сочетание соединения формулы 8 с соединением формулы 7
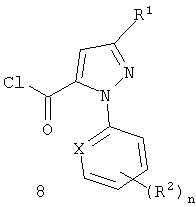
(в) взаимодействие соединения формулы 6 с соединением формулы R8NH2.
| NL 9202078 А, 16.06.1994 | |||
| FOTI F | |||
| ET AL ELSEVIER SCIENCE PUBLISHERS, vol | |||
| Приспособление с иглой для прочистки кухонь типа "Примус" | 1923 |
|
SU40A1 |
| RU 97102760/04 C2, 20.08.2000. | |||

