Область техники, к которой относится изобретение
Настоящее изобретение относится к производным фенилпиридинкарбонилпиперазина, полезным в качестве лечебного средства, в частности в качестве ингибиторов фосфодиэстеразы типа 4 (PDE4).
Уровень техники
Астма, которую до последнего времени рассматривали как обратимую обструкцию дыхательных путей, в настоящее время понимается как болезнь, характеризующаяся гиперчувствительностью дыхательных путей и обструкцией дыхательных путей, происходящей из-за хронического воспаления дыхательных путей с участием некоторых клеток зоны воспаления. Число больных неуклонно возрастает, и предсказывается его дальнейший рост.
Для лечения астмы в настоящее время применяют, главным образом, ингаляционные стероидные лекарственные средства как противовоспалительные средства, β-стимуляторы, такие как прокатерол, и производные ксантина, такие как аминофиллин и теофиллин, как бронходилитаторы.
Ингаляционные стероидные лекарственные средства обладают противовоспалительным действием широкого спектра и весьма полезны в качестве лекарственных средств для лечения астмы, но отмечается необходимость в указаниях по подходящему способу ингаляции и наличие больных астмой, невосприимчивых к стероидам (ASTHMA, 13-1, 69-73 (2000); Internal Medicine, 81, 485-490 (1998)).
Бронходилитаторы смягчают сокращение гладкой мускулатуры дыхательных путей за счет повышения концентрации внутриклеточного циклического аденозин-3',5'-монофосфата (сАМР) через активацию внутриклеточного продуцирующего сАМР фермента аденилатциклазы или ингибирование гидролизующего сАМР фермента фосфодиэстеразы (PDE) в гладкой мускулатуре дыхательных путей (Internal Medicine, 69, 207-214 (1992)). Известно, что повышенная концентрация внутриклеточного сАМР вызывает подавление сокращения гладкой мускулатуры дыхательных путей (Clin. Exp. Allergy, 22, 337-344 (1992); Drugs of the Future, 17, 799-807 (1992)), что эффективно для улучшения состояния при астме.
Однако известно, что производные ксантина производят системное побочное действие, такое как гипотензия, и кардиотоническое действие (J. Cyclic Nucleotide and Protein Phosphorylation Res., 10, 551-564 (1985); J. Pharmacol. Exp. Ther., 257, 741-747 (1991)), а β-стимуляторы склонны вызывать десенсибилизацию и, когда повышают дозировку, производить побочное действие, такое как дрожание пальцев и учащенное сердцебиение.
С другой стороны, хроническая обструктивная болезнь легких (COPD) является респираторным заболеванием, которое относится к аномальной воспалительной реакции и характеризуется необратимым ограничением проходимости дыхательных путей, и в настоящее время является четвертой причиной смертей в мире (Executive summary. Global Initiative for Chronic Obstructive Lung Disease (GOLD) (2000)). В настоящее время, как и в случае астмы, в качестве фармакотерапии в случае COPD используют, как правило, β-стимуляторы, антихолинергические лекарственные средства и производные ксантина, такие как аминофиллин и теофиллин, как бронходилитаторы. Кроме того, также используют ингаляционные стероидные лекарственные средства, так как внимание привлекает тот факт, что наличие хронического воспаления в дыхательных путях является частью обструктивного расстройства также и при COPD, но сообщается, что непрерывное лечение ингаляционным стероидом не улучшает длительного снижения FEV1 у больных COPD (N. Engl. J. Med., 340, 1948-53 (1999); Lancet, 353, 1819-23 (1999); BMJ, 320, 1297-303 (2000); N. Engl. J. Med., 343, 1902-9 (2000)). Таким образом, существует высокая потребность в противовоспалительном лекарственном средстве, способном улучшать состояние при COPD.
Показано, что PDE подразделяется на по меньшей мере семь семейств от PDE1 до PDE7, и каждое из них имеет разное распространение и функцию (Prog. Nucleic Acid Res. Mol. Biol., 63, 1-38 (1999)). В частности, PDE4 не действует на циклический гуанозин-3',5'-монофосфат (cGMP), но специфически из числа нуклеотидов гидролизует сАМР, и ее присутствие обнаруживают как в гладкой мускулатуре дыхательных путей, так и в инфильтрирующих клетках.
Также сообщается, что ингибиторы PDE4 показывают ингибирующее действие в отношении инфильтрации эозинофилов под действием антигенов и факторов активации тромбоцитов у морских свинок (Eur. J. Pharmacol., 255, 253-256 (1994)) и ингибируют выделение вредных белков (МВР, ЕСР) из эозинофилов (Br. J. Pharmacol., 115, 39-47 (1995)). Также сообщается, что они оказывают ингибирующее действие на сокращение гладкой мускулатуры дыхательных путей сократительными веществами (гистамином, метахолином, LTD4) (Br. J. Pharmacol., 113, 1423-1431 (1994)), ингибируют продуцирование IL-4-цитокина, о котором известно, что он играет активную роль при астме (J. Invest. Dermatol., 100, 681-684 (1993)), оказывают ингибирующее действие на ускорение проницаемости сосудов в дыхательных путях (Fundam. Clin. Pharmacol., 6, 247-249 (1992)) и оказывают ингибирующее действие в отношении гиперчувствительности дыхательных путей (Eur. J. Pharmacol., 275, 75-82 (1995)). Таким образом, ожидается, что ингибитор PDE4 будет средством для лечения астмы.
Кроме того, сообщается, что игибиторы PDE4 оказывают ингибирующее действие на инфильтрацию нейтрофилов, которые, как полагают, вовлекаются в воспаление дыхательных путей при COPD (Pulm. Pharmacol. Ther., 2001, Mar; 14(2):157-164). Кроме того, игибиторы PDE4 способны улучшать респираторную функцию у пациентов с COPD (Clin. Exp. Allergy, 1999, Jun; 29 Suppl. 2:99-109). Таким образом, также ожидается, что ингибитор PDE4 будет лекарственным средством для лечения COPD.
В качестве соединения, обладающего ингибирующей активностью в отношении PDE4, в WO 94/12461 описывается соединение
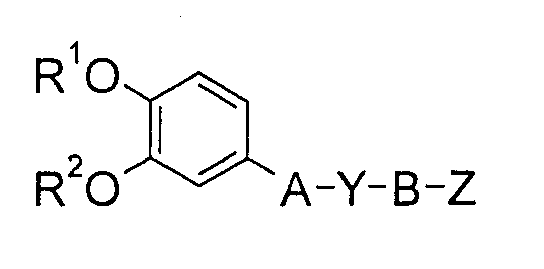
(где каждый из А, Y и В обозначает связь или подобное, Z обозначает пиридиновый цикл или подобный цикл, который может быть замещен R3, R3 обозначает CONR4R5 или подобную группу, и R4 и R5, каждый независимо, представляет (1) насыщенный или ненасыщенный пяти- или шестичленный гетероцикл, который может быть замещен одной или двумя группами, выбранными из числа С1-4-алкила, CO2R7, CONH2, CON(CH3)2, оксо, ОН, NH2 и N(CH3)2, (2) насыщенный или ненасыщенный шестичленный гетероцикл с одним гетероатомом как дополнительным атомом цикла, выбранным из числа O, S, NH, NCH3, NCOCH3 или NCH2Ph, или (3) хинолиновый цикл, который может быть замещен фтором или подобной группой). Однако в публикации в широкую формулу изобретения включена часть производных фенилпиридинкарбонилпиперазина, но конкретное соединение в ней не описывается. Даже в отношении производных фенилпиридинкарбоксамида в публикации описывается только 5-фенилпиридин-3-карбоксамид формулы
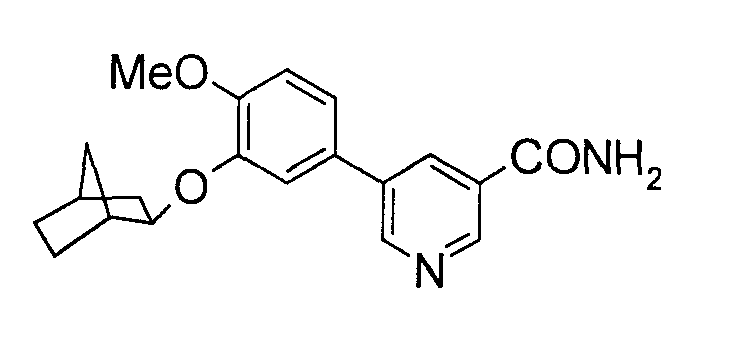
Описание изобретения
Авторы изобретения провели широкие исследования соединений с перорально доступной удовлетворительной ингибирующей активностью в отношении PDE4. В результате обнаружено, что новое производное пиридин-2-карбонилпиперазина, содержащее фенильную группу в положении 6, обладает сильной ингибирующей активностью в отношении PDE4, и, таким образом осуществили изобретение.
А именно, изобретение относится к новому производному фенилпиридинкарбонилпиперазина, представленному приведенной далее общей формулой (I), или его фармацевтически приемлемой соли и лечебному средству, содержащему указанные соединения в качестве активного ингредиента.
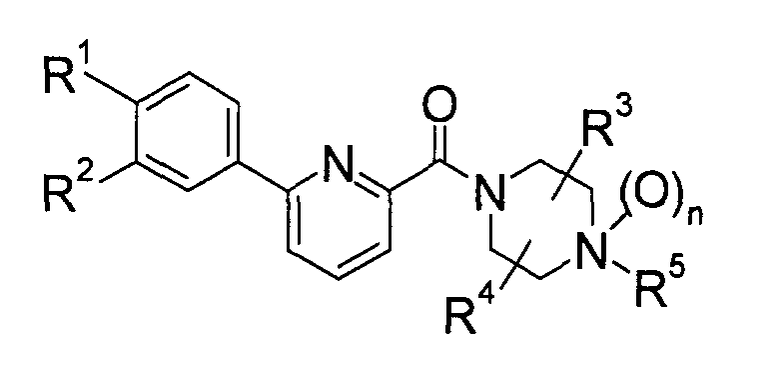
(где каждый символ имеет следующие значения:
R1 и R2: одинаковые или отличаются друг от друга и представляют собой Н, галоген, низший алкил, О-(низший алкил), О-(низший алкил, замещенный галогеном(ами)), NH2, NH-(низший алкил), N(низший алкил)2, NHCO-(низший алкил), О-(низший алкилен)-NH-(низший алкил), О-(низший алкилен)-N(низший алкил)2, О-(низший алкилен)-CO2R0, О-(низший алкилен)-(углеводородный цикл) или О-(низший алкилен)-гетероцикл, или R1 и R2 объединяются с образованием -О-(низший алкилен)-О-,
R0: Н, низший алкил или СН2-(необязательно замещенный фенил),
R3 и R4: одинаковые или отличаются друг от друга и представляют собой Н, необязательно замещенный низший алкил, галоген, CO2R0, CONH2, CON(R0)-(необязательно замещенный низший алкил), необязательно замещенный углеводородный цикл, необязательно замещенный гетероцикл, СО-(необязательно замещенный низший алкил), СО-(необязательно замещенный углеводородный цикл), СО-(необязательно замещенный гетероцикл) или CN, или R3 и R4 объединяются с образованием низшего алкилена или оксо,
R5: Н, низший алкил, CO2R0, CONH2, CON(R0)-(низший алкил), необязательно замещенный углеводородный цикл, необязательно замещенный гетероцикл, (низший алкилен)-(необязательно замещенный углеводородный цикл), (низший алкилен)-(необязательно замещенный гетероцикл), (низший алкенилен)-(необязательно замещенный углеводородный цикл), (низший алкенилен)-(необязательно замещенный гетероцикл), (низший алкилен)-R51, (низший алкилен)-CO2R0, СО-(низший алкил), СО-(необязательно замещенный углеводородный цикл), СО-(необязательно замещенный гетероцикл), СО-(низший алкилен)-(необязательно замещенный углеводородный цикл), СО-(низший алкилен)-(необязательно замещенный гетероцикл), СО-О-(низший алкилен)-(необязательно замещенный углеводородный цикл), СО-О-(низший алкилен)-(необязательно замещенный гетероцикл), CON(R0)(R56), C(R53)(R54)-R55 или (низший алкилен)-C(R53)(R54)-R55,
R51: СО-(низший алкил), СО-(необязательно замещенный углеводородный цикл), СО-(необязательно замещенный гетероцикл), СО-(низший алкилен)-(необязательно замещенный углеводородный цикл), СО-(низший алкилен)-(необязательно замещенный гетероцикл), CN, OH, О-(низший алкил), О-(необязательно замещенный углеводородный цикл), О-(необязательно замещенный гетероцикл), О-(низший алкилен)-(необязательно замещенный углеводородный цикл), О-(низший алкилен)-(необязательно замещенный гетероцикл), S-(низший алкил), S-(необязательно замещенный углеводородный цикл), S-(необязательно замещенный гетероцикл), S-(низший алкилен)-(необязательно замещенный углеводородный цикл), S-(низший алкилен)-(необязательно замещенный гетероцикл), NH(R0), N(R0)2, N(R0)-(необязательно замещенный углеводородный цикл), N(R0)-(необязательно замещенный гетероцикл), N(R0)-(низший алкилен)-(необязательно замещенный углеводородный цикл), N(R0)-(низший алкилен)-(необязательно замещенный гетероцикл), N(R0)СО-(низший алкил), N(R0)СО-(необязательно замещенный углеводородный цикл), N(R0)СО-(необязательно замещенный гетероцикл), N(R0)СО-(низший алкилен)-(необязательно замещенный углеводородный цикл), N(R0)СО-(низший алкилен)-(необязательно замещенный гетероцикл), N(R0)СО-О-(низший алкил), N(R0)СО-О-(низший алкилен)-(необязательно замещенный углеводородный цикл) или N(R0)СО-О-(низший алкилен)-(необязательно замещенный гетероцикл),
R53, R54 и R55: одинаковые или отличаются друг от друга, и представляют собой Н, низший алкил, CO2R0, CON(R0)(R56), R51 или R56,
R56: необязательно замещенный углеводородный цикл, необязательно замещенный гетероцикл, (низший алкилен)-(необязательно замещенный углеводородный цикл), (низший алкилен)-(необязательно замещенный гетероцикл), (низший алкилен)-R51 или (низший алкилен)-CO2R0,
n: 0 или 1,
при условии, что (1) когда R5 представляет собой группу, связанную с СО, или Н, n равен 0, и (2) когда каждый из R3 и R4 представляет собой Н, R5 представляет группу, иную, чем метил, ацетил или бензил;
то же самое будет верным и далее).
Изобретение также относится к лечебному средству, в частности ингибитору PDE4, содержащему производное фенилпиридинкарбонилпиперазина или его соль.
Далее изобретение описывается подробно.
Термины "алкил", "алкилен" и "алкенилен", используемые в данном описании, обозначают, каждый, линейную или разветвленную углеводородную цепь. "Низший алкил" представляет собой алкильную группу с 1-6 атомами углерода, предпочтительно алкильную группу с 1-4 атомами углерода, предпочтительнее метил или этил. "Низший алкилен" обозначает двухвалентную группу, образованную путем удаления одного атома водорода из вышеуказанного "низшего алкила", и представляет собой предпочтительно алкилен с 1-4 атомами углерода, предпочтительнее метилен, этилен или пропилен. "Низший алкенилен" обозначает группу с одной или несколькими двойными связями в любом положении в "низшем алкилене" с двумя или большим числом атомов углерода и предпочтительно представляет собой алкенилен с 2-4 атомами углерода.
"Галоген" представляет F, Cl, Br или I. "Низший алкил, замещенный галогеном(ами)" обозначает, например, низший алкил, замещенный одним или несколькими атомами галогена, и представляет собой предпочтительно С1-6-алкил, замещенный одним или несколькими атомами фтора, предпочтительнее фторметил, дифторметил, трифторметил или трифторэтил.
Термин "углеводородный цикл" обозначает одноядерный-трехъядерный углеводородный цикл с 3-14 атомами углерода и включает циклоалкил, циклоалкенил и ароматический углеводород, мостиковый циклоалкил и спироцикл. Также такие циклы могут конденсироваться друг с другом с образованием инданила, тетрагидронафтила или подобной группы.
"Циклоалкил" представляет собой предпочтительно циклоалкил с 3-8 атомами углерода, предпочтительнее циклопропил, циклопентил или циклогексил. "Циклоалкенил" представляет собой предпочтительно циклоалкенил с 5-8 атомами углерода, предпочтительнее циклогексенил. Термин "ароматический углеводород" обозначает ароматическую углеводородную группу с 6-14 атомами углерода и представляет собой предпочтительно фенил или нафтил, предпочтительнее фенил. "Мостиковый циклоалкил" представляет собой предпочтительно норборнил или адамантил.
"Гетероцикл" представляет собой насыщенный или ненасыщенный одноядерный-трехъядерный трех-восьмичленный, предпочтительно пяти-семичленный, гетероцикл, содержащий в качестве атома(ов) цикла от 1 до 4 гетероатомов, выбранных из числа атомов О, S и N, которые могут конденсироваться друг с другом или циклоалкилом или бензольным циклом с образованием двухъядерного или трехъядерного гетероцикла. Атом цикла - S или N - может быть окислен с образованием оксида или диоксида. К такому гетероциклу относятся насыщенный гетероцикл, ароматический гетероцикл и его частично насыщенный вариант, и в насыщенном гетероцикле и частично насыщенном гетероцикле любой(ые) атом(ы) углерода может(могут) быть замещен(ы) оксогруппой. Кроме того, гетероцикл может содержать мостиковую связь или может образовывать спироцикл, включающий ацетальный цикл, образованный из оксогруппы, такой как 1,3-диоксолан. Гетероцикл представляет собой предпочтительно пяти-семичленный насыщенный или ненасыщенный одноядерный гетероцикл и предпочтительнее представляет собой пирролидин, пиридин, пиперидин, морфолин, тиофен, тиазол, имидазол, тетразол, пиразин или пиперазин.
Термин "необязательно замещенный" означает "незамещенный" или "имеющий 1-5 заместителей, которые могут быть одинаковыми или отличаются один от другого".
Заместитель в "необязательно замещенном низшем алкиле" представляет собой предпочтительно углеводородный цикл, гетероцикл, CO2R0 или группу, описанную в определении R51.
Заместитель в "необязательно замещенном углеводородном цикле" или в "необязательно замещенном гетероцикле" представляет собой предпочтительно группу, выбранную из описанной далее группы G.
Группа G: группы, представленные (i) -Х-(С1-6-алкилен)-А, (ii) -(С1-6-алкилен)-А или (iii) -В.
Х представляет собой О, S, SO, SO2, NH, N(С1-6-алкил), SO2NH, SO2N(С1-6-алкил), NHSO2, N(С1-6-алкил)SO2, CO, СО2, О-СО, CONH, CON(С1-6-алкил), NHCO, N(С1-6-алкил)СО или NHCONH,
А представляет собой -СН, -ОН, -СО2Н, -СО2-(С1-6-алкил), -NO2, -SO3H, -NH2, -CONH2, -SO2NH2, С1-6-алкил, замещенный галогеном(ами), -NH-(С1-6-алкилен)-О-(С1-6-алкил), -N(С1-6-алкил)-((С1-6-алкилен)-О-(С1-6-алкил)), -N(-(С1-6-алкилен)-О-(С1-6-алкил))2, -(углеводородный цикл), -гетероцикл, -Х-(С1-6-алкил), -Х-(С1-6-алкил), замещенный галогеном(ами), -Х-(углеводородный цикл), -Х-гетероцикл, -Х-(С1-6-алкилен)-CN, -Х-(С1-6-алкилен)-ОН, -Х-(С1-6-алкилен)-СО2Н, -Х-(С1-6-алкилен)-СО2-(С1-6-алкил), -Х-(С1-6-алкилен)-NO2, -Х-(С1-6-алкилен)-SO3H, -Х-(С1-6-алкилен)-NH2, -Х-(С1-6-алкилен)-CONH2, -Х-(С1-6-алкилен)-SO2NH2, -Х-(С1-6-алкилен)-(углеводородный цикл) или -Х-(С1-6-алкилен)-гетероцикл,
В представляет собой -С1-6-алкил-, -галоген, С1-6-алкил, замещенный галогеном(ами), или группу, описанную в определении А.
Углеводородный цикл и гетероцикл в описанных выше А и В могут содержать от 1 до 5 заместителей, выбранных из числа С1-6-алкила, галогена, С1-6-алкила, замещенного галогеном(ами), CN, ОН, О-С1-6-алкила, NH2, -NH-С1-6-алкила, -N(С1-6-алкил)2, S-С1-6-алкила, SO-С1-6-алкила, SO2-С1-6-алкила, SO2NH2, SO2NH-С1-6-алкила, SO2N(С1-6-алкил)2, NHSO2-С1-6-алкила, СО2Н, СО2-С1-6-алкила, CONH2, CONH-С1-6-алкила, CON(С1-6-алкил)2 и NHCO-С1-6-алкила.
Заместитель в "необязательно замещенном фениле" представляет собой предпочтительно группу, представленную выше в группе G, предпочтительнее - С1-6-алкил, О-С1-6-алкил или галоген.
Предпочтительными соединениями изобретения являются следующие соединения:
соединения, где R1 представляет собой О-С1-6-алкил, предпочтительнее О-С1-4-алкил, особенно предпочтительно О-метил; соединения, где R2 представляет собой галоген, О-С1-6-алкил или О-С1-6-алкилен-(углеводородный цикл), предпочтительнее галоген, О-С1-4-алкил или О-СН2-С3-8-циклоалкил, особенно предпочтительно О-метил; соединения, где R3 и R4 представляют собой, каждый, Н, С1-6-алкил или оксо, предпочтительнее Н или метил, особенно предпочтительно Н. Особенно предпочтительны соединения, где каждый из R1 и R2 представляет собой О-метил, каждый из R3 и R4 представляет собой Н, и n равен 0. Кроме того, предпочтительными являются соединения, где R5 представляет собой необязательно замещенный углеводородный цикл или необязательно замещенный гетероцикл, предпочтительнее необязательно замещенный фенил или необязательно замещенный пиридил, фенил или пиридил с одной или двумя группами, предпочтительно одной группой, выбранными из вышеуказанной группы G.
Особенно предпочтительными соединениями изобретения являются следующие соединения:
1-[6-(3,4-диметоксифенил)пиридин-2-карбонил]-4-(4-метоксифенил)пиперазин, 1-(4-{4-[6-(3-циклопропилметокси-4-метоксифенил)пиридин-2-карбонил]пиперазин-1-ил}фенил)этанон, 1-(6-бром-2-пиридил)-4-[6-(3,4-диметоксифенил)пиридин-2-карбонил]пиперазин, 4'-{4-[6-(3,4-диметоксифенил)пиридин-2-карбонил]пиперазин-1-ил}ацетанилид, 3-диэтиламино-4'-{4-[6-(3,4-диметоксифенил)пиридин-2-карбонил]пиперазин-1-ил}пропананилид, 4-(4-{4-[6-(3,4-диметоксифенил)пиридин-2-карбонил]пиперазин-1-ил}фенил)морфолин, 1-[2-(4-{4-[6-(3,4-диметоксифенил)пиридин-2-карбонил]пиперазин-1-ил}фенокси)этил]пиперидин-4-ол, 4-{2-[(6-{4-[6-(3,4-диметоксифенил)пиридин-2-карбонил]пиперазин-1-ил}-3-пиридил)окси]этил}морфолин, транс-5-(4-{4-[6-(3,4-диметоксифенил)пиридин-2-карбонил]-2,5-диметилпиперазин-1-ил}фенил)пентановая кислота и 1-[6-(3,4-диметоксифенил)пиридин-2-карбонил]-4-{4-[(1-оксидо-4-пиридил)метокси]фенил}пиперазин.
В зависимости от вида заместителей соединения изобретения могут существовать в форме геометрических изомеров и таутомеров, и их чистые формы или смеси входят в объем изобретения.
Также соединения изобретения в некоторых случаях могут иметь асимметричные атомы углерода, и для таких атомов могут существовать формы (R) и (S) оптических изомеров. Изобретение включает все смеси и отдельные формы таких оптических изомеров.
Кроме того, к соединениям изобретения также относятся фармакологически приемлемые пролекарства. Фармакологически приемлемые пролекарства представляют собой соединения, содержащие группы, которые путем сольволиза или в условиях физиологических процессов могут превращаться в некоторые группы, такие как NH2, OH и CO2H, с образованием соединений по изобретению. Примерами групп, образующих пролекарства, являются группы, описанные в Prog. Med., 5, 2157-2161 (1985), и в "Iyakuhin no Kaihatsu (Pharmaceutical Research and Development)" (Hirokawa Publishing Co., 1990), Vol.7, Drug Design 163-198.
Соединения изобретения могут образовывать аддитивные соли кислот или, в зависимости от вида заместителей, соли с основаниями. Такие соли являются фармацевтически приемлемыми солями, и их характерными примерами являются аддитивные соли кислот, образованные с неорганическими кислотами, такими как хлористоводородная кислота, бромистоводородная кислота, иодистоводородная кислота, серная кислота, азотная кислота и фосфорная кислота, и органическими кислотами, такими как муравьиная кислота, уксусная кислота, пропионовая кислота, щавелевая кислота, малоновая кислота, янтарная кислота, фумаровая кислота, малеиновая кислота, молочная кислота, яблочная кислота, винная кислота, лимонная кислота, метансульфоновая кислота, этансульфоновая кислота, аспарагиновая кислота и глутаминовая кислота, соли с неорганическими основаниями, такие как натриевые, калиевые, магниевые, кальциевые и соли алюминия, и с органическими основаниями, такими как метиламин, этиламин, этаноламин, лизин и орнитин и соли аммония.
Кроме того, изобретение также относится к различным гидратам, сольватам и полиморфным веществам соединения (I) изобретения и его солей.
Способ получения
Соединения изобретения и их фармацевтически приемлемые соли можно получить, используя различные известные способы синтеза, с использованием особенностей, основанных на их собственной структуре или виде заместителей. В таком случае, в зависимости от вида функциональной группы, иногда эффективной с точки зрения метода получения, является защита функциональной группы подходящей защитной группой или замена такой группы на группу, которую можно легко превратить в функциональную группу, в исходном веществе или на промежуточной стадии. В качестве таких функциональных групп можно назвать, например, группы, описанные в "Protective Groups in Organic Synthesis (3rd Ed.)", edited by T.W. Greene and P.G.M. Wuts, которые можно, необязательно, использовать в условиях взаимодействия. При таком способе после введения защитной группы и последующего осуществления взаимодействия получить нужное соединение можно, удаляя защитную группу или превращая группу в нужную группу, как этого требуют обстоятельства. Кроме того, как и с вышеуказанной защитной группой, путем введения определенной группы или осуществляя взаимодействие с использованием полученного соединения изобретения в исходном веществе или на промежуточной стадии, можно получить пролекарства соединений изобретения. Взаимодействие можно осуществить с применением способа, известного специалистам в данной области техники, такого как обычная этерификация, амидирование или дегидратация.
Первый способ получения
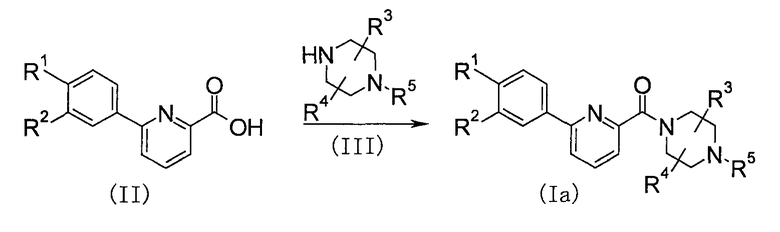
Данный способ получения представляет собой способ получения соединения (Ia) изобретения из карбоновой кислоты (II) путем амидирования.
Реакцию проводят, конденсируя соединение (II) с пиперазином (III) в присутствии агента конденсации, такого как дициклогексилкарбодиимид (DCC), диизопропилкарбодиимид (DIPC), 1-этил-3-(3-диметиламинопропил)карбодиимид (WSC) или 1,1'-карбонил-бис-1Н-имидазол (CDI), и, необязательно, еще с такой добавкой как N-гидроксисукцинимид (HONSu) или 1-гидроксибензотриазол (HOBt). Альтернативно, активный эфир соединения (II) можно сразу выделить с помощью вышеуказанной добавки и затем сконденсировать с пиперазином (III). Примерами растворителя являются ароматические углеводороды, такие как бензол, толуол и ксилол; простые эфиры, такие как диэтиловый эфир, тетрагидрофуран (ТГФ), 1,4-диоксан и диметоксиэтан; галогенированные углеводороды, такие как дихлорметан, 1,2-дихлорэтан и хлороформ; N,N-диметилформамид (ДМФА), N-метил-2-пирролидон (NMP), пиридин и подобные растворители. Указанные растворители можно использовать по одному или в виде смеси двух или большего их числа.
Второй способ получения
Соединения изобретения, в которых в группе R5 в общей формуле (I) имеются разные заместители, или соединения, где R1 или R2 представляет собой группу, иную чем алкоксигруппа, можно легко синтезировать с помощью реакций, хорошо известных специалистам в данной области техники, или их модификаций с использованием в качестве исходных веществ соединений изобретения. В частности, с использованием в качестве исходного вещества соединения, полученного вышеописанным первым способом получения, где R5 представляет собой Н, можно легко осуществить превращение R5, подвергая соединение различным взаимодействиям. Например, можно использовать взаимодействия, описанные далее.
(1) Алкилирование методом нуклеофильного замещения
Алкилирования по О-, S- или N- можно достичь взаимодействием соединения, содержащего ОН, SH или первичную-третичную аминогруппу, с алкилирующим агентом, таким как алкилгалогенид, например, алкилхлорид, или органическим сульфонатом. С другой стороны, алкилирования также можно достичь, осуществляя реакцию Мицунобу. Реакцию осуществляют в органическом растворителе, инертном по отношению к реакции, например в ароматических углеводородах, простых эфирах, спиртах (метаноле, этаноле и т.п.), ДМФА, NMP, диметилсульфоксиде (ДМСО) или подобном растворителе, в условиях от охлаждения до нагревания с использованием соединений в эквивалентных количествах или избытка одного из них. Иногда для успешного протекания реакции выгодно осуществлять реакцию в присутствии основания, такого как гидрид натрия, гидрид калия, диизопропиламид лития, гексаметилдисилазид лития, метоксид натрия, трет-бутоксид калия, гидроксид натрия, гидроксид калия, карбонат натрия или карбонат калия.
(2) Восстановительное алкилирование
Алкилирования можно достичь взаимодействием соединения, содержащего первичную или вторичную аминогруппу, с карбонилсодержащим соединением, таким как кетон или альдегид. При взаимодействии можно использовать обычный способ восстановительного алкилирования, и можно указать на способы, описанные, например, в "JIKKEN KAGAKU KOZA (4th Ed.)", edited by The Chemical Society of Japan, vol.22 (1992) (Maruzen), и подобные.
(3) Амидирование, сульфонамидирование и этерификация
С использованием карбоновой кислоты или сульфоновой кислоты можно получить соответствующее соединение, используя агент конденсации, указанный в вышеописанном первом способе получения, или способом с их реакционноспособными производными. В качестве реакционноспособных производных карбоновой кислоты или сульфоновой кислоты можно использовать галоидоангидриды, ангидриды, активные эфиры или подобные соединения. Взаимодействие можно осуществить способами, описанными, например, в "JIKKEN KAGAKU KOZA (4th Ed.)", edited by The Chemical Society of Japan, vol.22 (1992) (Maruzen), и подобными способами.
(4) Гидролиз
Соединения изобретения, содержащие карбоксильную группу, можно получить гидролизом карбоксилатов. При взаимодействии можно использовать обычный способ гидролиза, и для удаления группы, защищающей карбоксильную группу, можно применить способы, описанные, например, в указанном выше издании "Protective Groups in Organic Synthesis (3rd Ed.)".
(5) Окисление
Оксид, например, N-оксид пиридина, можно получить окислением соединения, содержащего пиридино- или аминогруппу. В качестве окислителя можно использовать неорганический окислитель, такой как пероксид водорода, Oxone (торговое наименование, Aldrich) или перборат натрия, или органический окислитель, такой как перуксусная кислота, м-хлорпербензойная кислота или диметилдиоксиран. Взаимодействие осуществляют в растворителе, инертном по отношению к реакции, выбранном из числа галогенированных углеводородов, ароматических углеводородов, простых эфиров, ДМФА, уксусной кислоты и воды, или в отсутствие растворителя в условиях от охлаждения до нагревания. При взаимодействии окислитель можно использовать в эквивалентном количестве или в избытке относительно исходного соединения. Для успешного протекания реакции иногда выгодно осуществлять взаимодействие в присутствии неорганической кислоты (предпочтительно серной кислоты, азотной кислоты, хлористоводородной кислоты или бромистоводородной кислоты), органической кислоты (предпочтительно уксусной кислоты или трифторуксусной кислоты) или неорганического основания (предпочтительно гидроксида натрия, гидроксида калия или гидрокарбоната натрия). Альтернативно сульфинил- или сульфонилсодержащее соединение можно получить путем подобного окисления с использованием сульфанилсодержащего соединения.
(6) Каталитическое восстановление
Соединение изобретения, содержащее группу ОН, можно получить, подвергая дебензилированию соединение, содержащее О-бензильную группу. Например, можно использовать обычный способ каталитического восстановления, когда взаимодействие осуществляют в атмосфере водорода в присутствии катализатора палладия-на-угле, и для удаления группы, защищающей группу ОН, также можно применить способы, описанные, например, в указанном выше издании "Protective Groups in Organic Synthesis (3rd Ed.)". Кроме того, подобным способом каталитического восстановления алкенильную группу можно превратить в алкильную группу.
Синтез исходных веществ
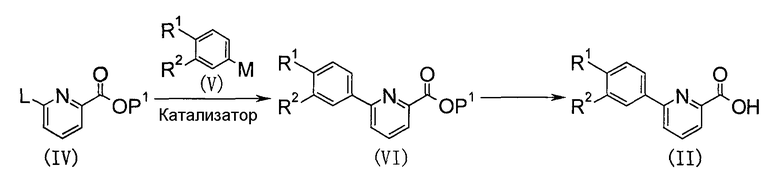
(В приведенных формулах соответственно L представляет уходящую группу, Р1 представляет группу, защищающую карбоксильную группу, и М представляет металл; те же обозначения применяются и далее).
Карбоновую кислоту (II) можно получить гидролизом соединения (VI). В качестве группы Р1, защищающей карбоксильную группу, можно использовать одну из групп, указанных в упомянутом выше издании "Protective Groups in Organic Synthesis (3rd Ed.)", которую можно удалить способом, описанным в литературе, или обычным способом, таким как гидролиз.
Исходное соединение (VI) можно получить реакцией сочетания производного пиридина (IV) и металлароматического соединения (V) в присутствии катализатора. При взаимодействии можно использовать способы, описанные в Comprehensive Organic Synthesis, Volume 3, 481, 1991, и подобные способы. В качестве уходящей группы L можно назвать галоген, трифторметансульфонилокси или подобную группу, и в качестве металла М можно назвать гидроксибор, алкилбор, алкоксибор, галогенид магния, галогенид цинка, алкилолово, алкилмедь или подобное соединение. В качестве катализатора предпочтителен палладийсодержащий комплекс, такой как тетракистрифенилфосфинпалладий, ацетат палладия или никельсодержащий комплекс, такой как дихлорбис(трифенилфосфин)никель или бис(1,5-циклооктадиен)никель. Взаимодействие осуществляют в растворителе, инертном по отношению к реакции, выбранном из числа галогенированных углеводородов, простых эфиров, ароматических углеводородов, ДМФА и воды, или в отсутствие растворителя в условиях от охлаждения до нагревания. При взаимодействии соединение (IV) и металлароматическое соединение (V) можно использовать в эквивалентных количествах, или одно из них можно использовать в избытке, и для успешного развития реакции иногда выгодно осуществлять взаимодействие в присутствии основания, такого как триэтиламин, пиридин, 4-(N,N-диметиламино)пиридин, гидроксид натрия, карбонат натрия, гидрид натрия, метоксид натрия или трет-бутоксид калия.
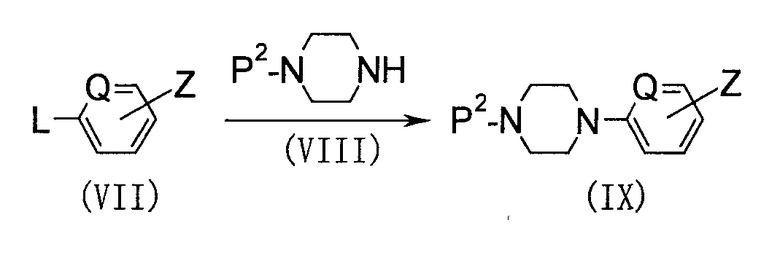
(В приведенных формулах соответственно Q представляет СН или N, Р2 представляет Н или группу, защищающую аминогруппу, и Z представляет собой группу, выбранную из группы G, или подобную группу).
Исходное соединение (IX) можно синтезировать, подвергая арильное производное (VII) реакции сочетания или самой реакции замещения пиперазином, который может содержать защитную группу. Способ получения исходного вещества (VI) можно применить к реакции сочетания. К реакции сочетания можно применить способ получения исходного соединения (VI). Условия алкилирования посредством описанного выше нуклеофильного замещения (1) можно применить к самой реакции замещения. В качестве защитной группы Р2 можно использовать защищающие аминогруппу группы, описанные в указанном выше издании "Protective Groups in Organic Synthesis (3rd Ed.)", и по окончании взаимодействия исходное соединение (IX) можно получить в свободном виде, удаляя защитную группу способом, описанным в литературе.
Продукт реакции, полученный каждым из описанных выше способов, выделяют и очищают в виде свободного соединения, соли или различных сольватов, таких как гидрат. Соль можно получить, осуществляя обычную обработку для получения соли.
Выделение и очистку осуществляют с использованием методов, обычно применяемых в химии, таких как экстракция, концентрирование, выпаривание, кристаллизация, фильтрация, перекристаллизация и различные типы хроматографии.
Различные изомеры можно выделить обычным способом, используя различие в физико-химических свойствах соответствующих изомеров. Например, оптические изомеры можно разделить общим способом оптического разделения, таким как фракционная кристаллизация или хроматография. Оптический изомер также можно получить исходя из соответствующего оптически активного исходного соединения.
Кроме того, изобретение также относится к новым промежуточным соединениям - производным карбоновых кислот, представленных общей формулой (IIa), которые полезны при получении производных фенилпиридинкарбонилпиперазина (I).
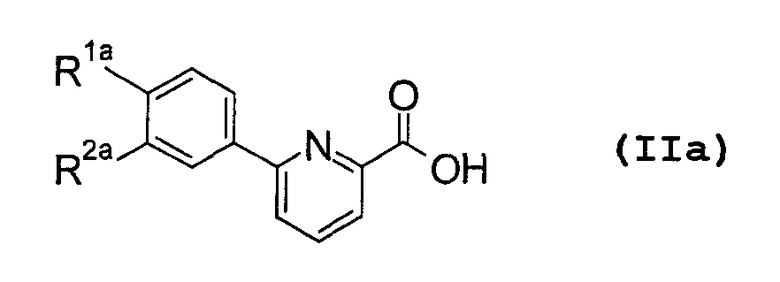
(В приведенной формуле
R1a представляет галоген, низший алкил, О-(низший алкил), О-(низший алкил, замещенный галогеном(ами)), NH2, NH-(низший алкил), N(низший алкил)2, NHCO-(низший алкил), О-(низший алкилен)-NH-(низший алкил), О-(низший алкилен)-N(низший алкил)2, О-(низший алкилен)-CO2R0, О-(низший алкилен)-(углеводородный цикл) или О-(низший алкилен)-гетероцикл,
R2a представляет Н или группу, указанную в определении R1a,
или R1a и R2a объединяются с образованием -О-(низший алкилен)-О-,
при условии, что (1) когда R2a представляет собой Н, R1a представляет группу, иную чем метил, этил, ОМе, NH2, NHMe или Cl, и (2) когда R2a представляет собой метил, R1a представляет соответственно группу, иную чем метил; то же самое будет верно и далее).
Карбоновая кислота (IIa) входит в определение карбоновой кислоты (II), описанной в качестве промежуточного соединения выше. Предпочтительными группами в случае R1a и R2a в соединении (IIa) являются те же группы, которые являются предпочтительными в случае R1 и R2 в соединении (I).
Промышленная применимость
Соединения (I) изобретения также обладают превосходной ингибирующей активностью в отношении PDE4 и, следовательно, полезны в качестве средств для предупреждения и/или лечения респираторных заболеваний (например, бронхиальной астмы (включая атопическую астму), COPD, хронического бронхита, болезней легких и респираторного дистресс-синдрома взрослых (ARDS)), которые протекают с участием PDE4. В частности, можно ожидать, что такие соединения являются средством для предупреждения и/или лечения бронхиальной астмы и COPD.
Кроме того, соединения изобретения также полезны в качестве средств для предупреждения и/или лечения других заболеваний, о которых известно, что они протекают с участием PDE4, таких, как заболевания, связанные с цитокинами (IL-1, IL-4, IL-6 и TNF (фактор некроза опухолей)) или подобными факторами (например, ревматоидный артрит, неспецифический язвенный колит, болезнь Крона, сепсис, септический шок, эндотоксиновый бактериально-токсический шок, грамотрицательный бактериальный сепсис, токсический шок, нефрит, гепатит, инфекция (бактериальная и вирусная) и недостаточность кровообращения (сердечная недостаточность, артериосклероз, инфаркт миокарда, удар) или подобные заболевания).
Пригодность соединений (I) изобретения подтверждают испытаниями, описанными далее.
Пример испытаний 1
Ингибирующая активность в отношении PDE4
1) Раствор, содержащий PDE4, очищают от желудочковой мышцы крысы следующим образом. Сердце, иссеченное из самца крысы Wistar под эфирным наркозом, промывают физиологическим раствором и затем отделяют желудочек. Отделенный таким образом желудочек мелко нарезают ножницами и суспендируют в буфере А (20 мМ бис-трис, 50 мМ ацетата натрия, 2 мМ ЭДТК, 5 мМ 2-меркаптоэтанола, 2 мМ бензамидена, 0,05 мМ фенилметилсульфонилфторида, рН 6,5), содержащем 1% протеазингибирующей смеси для экстрактов клеток млекопитающих (SIGMA). Затем клетки разрушают с использованием Polytron, подвергают ультрацентрифугированию (100000 G, 60 минут, 4°С) и получают растворимую фракцию.
2) Полученную растворимую фракцию загружают в колонку 2,6 х 10 см с Q-сефарозой, уравновешенную буфером А. Затем колонку промывают 1200 мл буфера А для удаления несвязанного белка. Белок, связанный с содержимым колонки, элюируют с использованием 750 мл буфера А, содержащего раствор ацетата натрия с линейным градиентом от 0,05 до 1,00 М, и извлекают 110 пробирок, содержащих фракции по 7 мл. Исследуют метаболизирующую активность PDE в отношении сАМР для каждой полученной фракции в присутствии или в отсутствие cGMP и кальция/калмодулина. Каждую фракцию, показывающую метаболизирующую активность в отношении сАМР, и на метаболизирующую активность которой в отношении сАМР не влияет cGMP или кальций/калмодулин, используют в качестве маточного раствора для проверки ингибирующей активности в отношении PDE4.
3) Каждое испытываемое соединение в нужной концентрации подвергают взаимодействию в течение 10 минут при 30°С в реакционной смеси, содержащей 40 мМ трис-HCl (рН 8,0), 5 мМ хлорида магния, 4 мМ 2-меркаптоэтанола, 1 мкМ сАМР, 1 мкКи/мл [3H]cAMP и маточный раствор PDE4. Реакцию останавливают, добавляя к реакционному раствору 1/2 объема 20 мг/мл суспензии гранул силиката иттрия SPA, покрытых полилизином (Amersham), содержащей 18 мМ сульфата цинка и 5 мкМ 3-изобутил-1-метилксантина (IBMX), и измеряют радиоактивность.
Концентрацию испытываемого соединения, при которой метаболическая активность PDE4 ингибируется на 50%, определяют как IC50 и вычисляют для каждого соединения. Применяя вышеописанный способ испытания и способ, описанный в WO 97/19078, подобным образом измеряют ингибирующую активность против PDЕ1, PDE2, PDE3 и PDE5.
В результате вышеописанных измерений получают, что соединения примеров 2, 10, 15, 32, 43, 45, 77, 95, 99 и 112 имеют величину IC50 для PDE4 12 нМ или менее. Кроме того, при той же концентрации они почти не обнаруживают ингибирующей активности против PDЕ1, PDE2, PDE3 и PDE5. Соответственно подтверждается, что соединения изобретения являются сильными и селективными ингибиторами PDE4.
Пример испытаний 2
Пероральная абсорбционная способность и испытание для оценки фармакокинетического профиля с использованием в качестве показателя ингибирующей активности в отношении TNF-α
1) Каждое испытываемое соединение, суспендированное в очищенной воде, содержащей 0,5% метилцеллюлозы, вводят перорально восьминедельным самцам крысы Fisher в дозе 10 мг/кг. Животным в контрольной группе таким же образом вводят растворитель (0,5% метилцеллюлозы в очищенной воде, 3 мл/кг). После перорального введения периодически из хвостовой вены каждой крысы под эфирным наркозом берут образцы крови в присутствии гепарина и обычным способом получают образцы плазмы.
2) Плазму, полученную выше (конечная концентрация 2,5%), среду RPMI1640, содержащую 10% сыворотки плода коровы, 20 мкл цельной крови самца крысы Wister и LPS (конечная концентрация 3 мкг/мл), распределяют в 96-луночном культуральном планшете таким образом, чтобы общий объем на 1 лунку составлял 200 мкл, и затем культивируют в течение ночи при 37°С с использованием инкубатора с СО2-содержащей атмосферой. По завершении культивирования планшет центрифугируют (1500 об/мин, 10 минут), супернатант извлекают и измеряют в супернатанте количество TNF-α с использованием коммерчески доступного набора для ELISA. В результате такого испытания показывают, что соединения изобретения имеют хорошую пероральную абсорбирующую способность.
На основании результатов вышеописанных испытаний на ингибирующую активность подтверждают, что соединения (I) изобретения обнаруживают селективную и сильную ингибирующую активность против PDE4, а также хорошую пероральную абсорбирующую способность, и, таким образом, очевидно, что они полезны в качестве средства для предупреждения и лечения заболеваний, к которым причастна PDE4.
Пример испытаний 3
Действие на антигениндуцированную инфильтрацию эозинофилов в дыхательных путях крысы
Содержащий ОА раствор для сенсибилизации (конечная концентрация: ОА - 1 мг/мл; Al(OH)3 - 20 мг/мл) вводят интраперитонеально четырехнедельным самкам крысы Brown Norway (Charles River Japan, Inc., Kanagawa) непрерывно в течение 3 суток в дозе 1 мл на крысу для получения антигенсенсибилизации. Первый день - день введения - обозначают как день 0. В дни 21 или 22 1% ОА/физиологический раствор переводят в аэрозоль с помощью ультразвукового распылителя (NE-U12, Omron) и сенсибилизированных крыс подвергают воздействию антигена, принуждая крыс вдыхать аэрозоль ОА в течение 20 минут для индукции инфильтрации эозинофилов в дыхательные пути. Кроме того, в качестве нормальной контрольной группы используют группу, где для воздействия животные вдыхают физиологический раствор. Испытываемое соединение суспендируют в 0,5% водном растворе МС и суспензию вводят перорально за 1 час до ингаляции и воздействия антигена. Животных не кормят со дня накануне ингаляции и воздействия антигена, а после ингаляции и воздействия антигена им предоставляют доступ к корму. Через 24 часа после ингаляции и воздействия антигена животных подвергают лапаротомии под наркозом под действием нембутала и для умервщления выпускают кровь из брюшной аорты. Затем в дыхательные пути вводят канюли (венозный катетер 6 Fr-Atom, Atom) и осуществляют бронхоальвеолярный лаваж (BAL), пять раз повторяя операцию инъецирования и извлечения 2 мл физиологического раствора, содержащего гепарин (1 единица/мл) (всего 10 мл). Затем извлеченную жидкость BAL центрифугируют при 500 х g (4°С, 10 минут), супернатант удаляют и преципитат (клеточная фракция) ресуспендируют в 500 мкл физиологического раствора, содержащего гепарин (1 единица/мл). Определяют общее содержание лейкоцитов в ресуспендированной жидкости с помощью прибора для подсчета форменных элементов крови (celltac-a, Nihon Kohden Corporation) и затем получают образец-мазок и окрашивают жидкостью, окрашивающей кровь, для дифференцировки (Dif Quick, International Reagents Corporation) и затем рассматривают под микроскопом и вычисляют относительное содержание эозинофилов из морфологической характеристики. На основании общего числа лейкоцитов и относительного содержания эозинофилов вычисляют общее число эозинофилов и посредством этого оценивают действие лекарственного средства.
Пример испытаний 4
Действие на индуцированную LPS инфильтрацию нейтрофилов в дыхательные пути
Инфильтрацию нейтрофилов в дыхательные пути вызывают введением с помощью 200-мкл зонда 10 мкг/мл раствора LPS (липополисахарид E. coli 0127:B8 Boivin, DIFCO) в физиологическом растворе в дыхательные пути шестинедельных самцов крысы Wister (Charles River Japan, Inc., Kanagawa) под наркозом, который дают интраперитонеальным введением соответствующего количества смешанного раствора кетамин/ксилазин. Кроме того, используют в качестве нормальной контрольной группы группу животных, которым в дыхательные пути вводят физиологический раствор. Испытываемое соединение суспендируют в 0,5% водном растворе МС и суспензию вводят перорально за 1 час до введения в дыхательные пути LPS. Животных не кормят со дня накануне введения в дыхательные пути LPS, а после введения LPS в дыхательные пути им предоставляют доступ к корму. Через 24 часа после введения LPS в дыхательные пути животных подвергают лапаротомии под наркозом под действием нембутала и для умерщвления выпускают кровь из брюшной аорты. Затем определяют общее содержание лейкоцитов способом, подобным описанному выше в примере испытаний 3. Затем вычисляют относительное содержание нейтрофилов из морфологической характеристики, которую наблюдают под микроскопом. На основании общего числа лейкоцитов и относительного содержания нейтрофилов вычисляют общее число нейтрофилов и посредством этого оценивают действие лекарственного средства. Фармацевтический препарат, содержащий в качестве активного ингредиента одно или два или большее число соединений изобретения или их солей, получают с использованием носителей, эксципиентов и других добавок, которые обычно используют при получении лечебных средств.
Введение может представлять собой или пероральное введение в форме, например, таблеток, пилюль, капсул, гранул, порошков или жидкостей, или парентеральное введение в форме, например, внутривенных или внутримышечных инъекций, суппозиториев, трансдермальных препаратов, трансназальных препаратов или ингаляций. Дозу определяют произвольно в зависимости от каждого случая, например, с учетом симптомов, возраста и пола каждого пациента, которого лечат, но в случае перорального введения, как правило, она составляет приблизительно от 0,001 мг/кг до 100 мг/кг в сутки для взрослого человека, и указанную дозу вводят один раз в сутки или делят на 2-4 дозы в сутки. Также когда вследствие симптомов осуществляют внутривенное введение, дозу вводят один или несколько раз в сутки, как правило, в пределах от 0,0001 мг/кг до 10 мг/кг в сутки для взрослого человека. Также в случае ингаляции дозу вводят один или несколько раз в сутки, как правило, в пределах от 0,0001 мг/кг до 1 мг/кг в сутки для взрослого человека.
Твердую композицию для перорального введения согласно изобретению применяют в форме, например, таблеток, порошков или гранул. В такой твердой композиции одно или несколько активных веществ смешаны с по меньшей мере одним инертным эксципиентом, таким как лактоза, маннит, глюкоза, гидроксипропилцеллюлоза, микрокристаллическая целлюлоза, крахмал, поливинилпирролидон или алюмометасиликат магния. Как правило, композиция может содержать инертные добавки, в том числе смазывающее вещество, такое как стеарат магния, и вещество, способствующее рассыпанию, такое как карбоксиметилкрахмалнатрий, или вещество, способствующее растворению. При необходимости на таблетки или пилюли может быть нанесена пленка сахара или покрытие из вещества, растворяющегося в желудке или кишечнике.
Жидкие композиции для перорального введения включают, например, фармацевтически приемлемые эмульсии, жидкости, суспензии, сиропы и эликсиры, и содержат обычно используемые инертные растворители, такие как очищенная вода или этанол. Кроме инертных растворителей такие композиции также могут содержать вспомогательные вещества, такие как солюбилизаторы, увлажнители и суспендирующие вещества, а также подсластители, корригенты, ароматизаторы и антисептики.
Инъекции для парентерального введения включают асептические водные или неводные жидкости, суспензии и эмульсии. Примерами водного растворителя являются дистиллированная вода для инъекций и физиологический раствор. Примерами неводного растворителя являются пропиленгликоль, полиэтиленгликоль, растительное масло, такое как оливковое масло, спирт, такой как этанол, и полисорбат 80 (торговое наименование). Такая композиция также может содержать тонизирующее средство, антисептик, увлажняющее вещество, эмульгатор, диспергатор, стабилизатор и вещество, способствующее солюбилизации. Такие композиции стерилизуют, например, фильтрацией через фильтр, задерживающий бактерии, смешиванием с антимикробным препаратом или облучением. Кроме того, такие композиции можно применять, изготовляя их сначала в виде стерильных твердых композиций и растворяя их в стерильной воде или стерильном растворителе для инъекции непосредственно перед применением.
Препараты для введения через слизистую оболочку, такие как ингаляции и трансназальные препараты, применяют в форме твердых, жидких и полутвердых веществ, и их можно получить согласно способам, известным и ранее. Например, в такой препарат могут быть добавлены, необязательно, эксципиент, такой как лактоза или крахмал, а также регулятор рН, антисептик, поверхностно-активное вещество, смазывающее вещество, стабилизатор и загуститель. Для введения можно использовать устройство, подходящее для ингаляции или распыления. Например, с использованием известного устройства, такого как ингалятор с дозатором или распылитель, можно ввести одно соединение или в сочетании с фармацевтически приемлемым носителем ввести его как композицию в виде порошка или в виде раствора или суспензии. Устройство для вдыхания сухого порошка или подобное может представлять собой устройство однократного применения или устройство многократного применения, где можно использовать сухой порошок или капсулу, содержащую сухой порошок. Альтернативно, оно может представлять собой форму распылителя аэрозоля под давлением, в котором используется соответствующий пропеллент, например подходящий газ, такой как хлорфторсодержащий алкан, частично фторированный алкан или диоксид углерода.
Наилучший способ осуществления изобретения
Ниже изобретение будет описываться конкретно с помощью примеров, которые, однако, не ограничивают объем изобретения. Способы получения исходных соединений демонстрируются в ссылочных примерах.
Ссылочный пример 1
К смеси метил-6-хлорпиридин-2-карбоксилата, 3,4-диметоксифенилборной кислоты, диметоксиэтана и воды добавляют ацетат палладия, трифенилфосфин и карбонат натрия, осуществляют взаимодействие при 100°С в течение 1 часа и получают метил-6-(3,4-диметоксифенил)пиридин-2-карбоксилат. Полученное таким образом соединение подвергают взаимодействию при 60°С в течение 30 минут в смешанном растворителе ТГФ-метанол, добавляя 1 М водный раствор гидроксида натрия, и посредством этого получают 6-(3,4-диметоксифенил)пиридин-2-карбоновую кислоту.
Ссылочный пример 2
К раствору 4-бром-2-хлоранизола в ТГФ при -78°С добавляют раствор н-бутиллития в н-гексане и затем перемешивают в течение 30 минут. Затем добавляют триметилборат и смесь нагревают до комнатной температуры и затем перемешивают в течение 30 минут. С использованием вместо 3,4-диметоксифенилборной кислоты остатка, полученного после выпаривания растворителя, способом, подобным способу ссылочного примера 1, получают нужное соединение.
Ссылочный пример 3
Нужное соединение получают с использованием 1-бензилокси-4-бром-2-метоксибензола способом, подобным способу ссылочного примера 2, за исключением того, что гидролиз осуществляют в 1 М водном растворе гидроксида натрия при 100°С в течение 2,5 суток.
Ссылочный пример 4
С использованием 6-(3,4-диметоксифенил)пиридин-2-карбоновой кислоты и трет-бутоксикарбонилпиперазина получают 1-[6-(3,4-диметоксифенил)пиридин-2-карбонил]-4-(трет-бутоксикарбонил)пиперазин способом, подобным способу, описанному ниже в примере 2. Затем к полученному веществу добавляют 4 М раствор хлороводорода в этилацетате, в смеси осуществляют взаимодействие и получают нужное соединение.
Ссылочный пример 5
С использованием 1-бензилоксикарбонил-4-(трет-бутоксикарбонил)пиперазин-2-карбоновой кислоты и морфолина получают 1-бензилоксикарбонил-4-(трет-бутоксикарбонил)-2-[(морфолин-4-ил)карбонил]пиперазин способом, подобным способу, описанному ниже в примере 4. К полученному веществу в этилацетате добавляют 4 М раствор хлороводорода в этилацетате, в смеси осуществляют взаимодействие и получают 1-бензилоксикарбонил-2-[(морфолин-4-ил)карбонил]пиперазин. Соединение в течение 1 суток кипятят с обратным холодильником в толуоле в присутствии бромбензола, трис(дибензилиденацетон)дипалладия(0), 2,2'-бис(дифенилфосфино)-1,1'-бинафтила и трет-бутоксида натрия и получают 1-бензилоксикарбонил-2-морфолинокарбонил-4-фенилпиперазин. Затем полученное таким образом соединение перемешивают при комнатной температуре в течение 1,5 суток в этаноле в присутствии 10% палладия-на-угле в атмосфере водорода при нормальном давлении. После отфильтровывания нерастворимых веществ остаток, полученный после выпаривания растворителя, растворяют в этаноле, к раствору добавляют 10% палладий-на-угле и формиат аммония, все перемешивают на масляной бане при температуре 70°С в течение 2,5 суток и получают нужное соединение.
Ссылочный пример 6
К раствору 4-бром-2-этилфенола в ДМФА добавляют карбонат калия и бензилбромид, все перемешивают на масляной бане при температуре 60°С в течение 30 минут и получают бензил(4-бром-2-этилфениловый) эфир, который затем обрабатывают способом, подобным описанному в первой части ссылочного примера 2, и получают метил-6-(4-бензилокси-3-этилфенил)пиридин-2-карбоксилат. Полученное соединение перемешивают в смешанном растворителе (метанол и ТГФ) в присутствии 10% палладия-на-угле в атмосфере водорода при нормальном давлении при комнатной температуре в течение 24 часов и затем полученный таким образом продукт реакции растворяют в трифторуксусной кислоте. К раствору при охлаждении льдом добавляют пентаметилбензол и все перемешивают на масляной бане при температуре 50°С в течение 1 часа и затем при комнатной температуре в течение 4,5 суток и получают метил-6-(3-этил-4-гидроксифенил)пиридин-2-карбоксилат. Полученное соединение обрабатывают трифторметансульфоновым ангидридом в пиридине и получают метил-6-(3-этил-4-трифторметансульфонилоксифенил)пиридин-2-карбоксилат. Затем к раствору полученного выше эфира в 1,4-диоксане добавляют трибутилвинилолово, хлорид лития, тетракис(трифенилфосфин)палладий(0) и 2,6-ди-трет-бутил-4-метилфенол и все кипятят с обратным холодильником в течение 18 часов. Затем к смеси еще добавляют тетракис(трифенилфосфин)палладий(0), после чего продолжают кипячение с обратным холодильником в течение 2 суток. Затем к смеси при комнатной температуре добавляют фторид калия и все перемешивают при комнатной температуре в течение 2 суток и получают метил-6-(3-этил-4-винилфенил)пиридин-2-карбоксилат. Соединение обрабатывают в метаноле 1 М водным раствором гидроксида натрия и получают нужное соединение.
Ссылочный пример 7
К раствору метил-6-(3-этил-4-гидроксифенил)пиридин-2-карбоксилата в ДМФА добавляют карбонат калия и метилиодид, все перемешивают на масляной бане при температуре 70°С в течение 2 часов и получают метил-6-(3-этил-4-метоксифенил)пиридин-2-карбоксилат, который затем перемешивают в метаноле и 1 М водном растворе гидроксида натрия на масляной бане при температуре 60°С в течение 1 часа, и получают нужное соединение.
Ссылочный пример 8
4-Иодфенол вводят в реакцию с гидрохлоридом 2-хлордиметиламинэтана в ДМФА при нагревании в присутствии карбоната калия и получают [2-(4-иодфенокси)этил]диметиламин. Полученное соединение при нагревании в толуоле превращается в присутствии трет-бутилпиперазин-1-карбоксилата, трет-бутоксида натрия, три(2-метилфенил)фосфина и каталитического количества трис(дибензилиденацетон)палладия(0) и получают нужное соединение.
Ссылочный пример 9
2,6-Дихлорпиридин вводят в реакцию с трет-бутилпиперазин-1-карбоксилатом в N,N-диметилимидазолидиноне при нагревании в присутствии карбоната калия и получают нужное соединение.
Ссылочный пример 10
В смешанном растворителе ТГФ-метанол перемешивают метил-6-(3-бензилокси-4-метоксифенил)пиридин-2-карбоксилат в присутствии палладия-на-угле в атмосфере водорода и получают метил-6-(3-гидрокси-4-метоксифенил)пиридин-2-карбоксилат. Полученное соединение вводят в реакцию с циклопропилметилбромидом и карбонатом калия в ДМФА при нагревании и получают метил-6-(3-циклопропилметокси-4-метоксифенил)пиридин-2-карбоксилат, который затем в смешанном растворителе ТГФ-метанол при нагревании подвергают превращению, добавляя 1 М водный раствор гидроксида натрия, и получают нужное соединение.
Ссылочный пример 11
К раствору 4-бром-2-хлоранизола в толуоле добавляют 1-(трет-бутоксикарбонил)пиперазин, трис(дибензилиденацетон)палладий(0), 2,2'-бис(дифенилфосфино)-1,1'-бинафтил и трет-бутоксид натрия и затем смесь нагревают 4 часа на масляной бане при температуре 110°С. Затем обычным способом осуществляют последующую обработку и очистку и получают нужное соединение.
Ссылочный пример 12
К раствору 1-(трет-бутоксикарбонил)-4-(3-хлор-4-метоксифенил)пиперазина в хлороформе добавляют трифторуксусную кислоту и перемешивают в течение 30 минут. Затем обычным способом осуществляют последующую обработку и очистку и получают нужное соединение.Ссылочный пример 13
Раствор 6-хлорникотинонитрила и (±)-транс-2,5-диметилпиперазина в NMP перемешивают на масляной бане при температуре 120°С в течение 1 часа и получают нужное соединение.
Ссылочный пример 14
К раствору 4-фторбензальдегида и 1-(трет-бутоксикарбонил)пиперазина в NMP добавляют карбонат калия и все перемешивают при нагревании. Затем обычным способом осуществляют последующую обработку и очистку и получают нужное соединение.
Ссылочный пример 15
К расплавленному пиперазину при 150°С добавляют 2-хлорбензотиазол и затем перемешивают в течение 1 часа. Затем обычным способом осуществляют последующую обработку и очистку и получают нужное соединение.
Ссылочный пример 16
К смеси 60% гидрида натрия и ТГФ при охлаждении до 0°С добавляют по каплям этилдиэтилфосфоноацетат и затем 4-[4-(трет-бутоксикарбонил)пиперазин-1-ил]бензальдегид и затем смесь перемешивают. Затем обычным способом осуществляют последующую обработку и очистку и получают этил-3-{4-[4-(трет-бутоксикарбонил)пиперазин-1-ил]фенил}акрилат. Затем осуществляют каталитическое восстановление с использованием палладия-на-угле и получают нужное соединение.
Ссылочный пример 17
Раствор метил-6-хлорникотината и пиперазина в ДМСО перемешивают на масляной бане при температуре 120°С и получают нужное соединение.
Ссылочный пример 18
К раствору 1-(3-бензилокси-4-нитрофенил)-4-(трет-бутоксикарбонил)пиперазина в смешанном растворителе метанол-ТГФ добавляют палладий-на-угле и затем перемешивают в атмосфере водорода. К раствору в метаноле 2-амино-5-[1-(трет-бутоксикарбонил)пиперазин-4-ил]фенола, полученного обычной последующей обработкой и очисткой, добавляют метилортоформиат и п-толуолсульфоновую кислоту и затем смесь нагревают при перемешивании. Затем обычным способом осуществляют последующую обработку и очистку и получают нужное соединение.
Ссылочный пример 19
N-Бензилиминодиуксусную кислоту вводят в реакцию с CDI и 5-аминоиндолом в ТГФ и получают 4-бензил-1-(1Н-индол-5-ил)пиперазин-2,6-дион, который затем вводят в реакцию с алюмогидридом лития в ТГФ. К раствору полученного таким образом соединения в этаноле добавляют концентрированную хлористоводородную кислоту и гидроксид палладия, в смеси проводят реакцию в атмосфере водорода при 3 атм в течение 65 часов и получают нужное соединение.
Ссылочный пример 20
4-(2-Хлорпиримидин-4-ил)пиперазин-1-карбальдегид и 2-(диметиламино)этанол вводят в реакцию в ДМФА в присутствии трет-бутоксида калия. Полученное таким образом соединение в метаноле подвергают превращению при 80°С в течение 24 часов в присутствии карбоната калия и получают нужное соединение.
Ссылочный пример 21
4-[4-(трет-Бутоксикарбонил)пиперазин-1-ил]бензальдегид и бромид [3-(этоксикарбонил)пропил]трифенилфосфония вводят в реакцию в ТГФ в присутствии трет-бутоксида калия и получают этил-5-{4-[4-(трет-бутоксикарбонил)пиперазин-1-ил]фенил}-4-пентеноат, который затем подвергают каталитическому восстановлению с использованием палладия-на-угле, и получают нужное соединение.
Ссылочный пример 22
2-Бром-6-иодпиридин-3-ол вводят в реакцию с карбонатом калия и бензилбромидом и получают 3-(бензилокси)-2-бром-6-иодпиридин, который затем последовательно вводят в реакцию как в ссылочном примере 11, примере 22 и примере 4. Затем полученный продукт реакции подвергают каталитическому восстановлению с использованием палладия-на-угле и получают нужное соединение.
Ссылочный пример 23
К раствору 2-бром-6-{4-[6-(3,4-диметоксифенил)пиридин-2-карбонил]пиперазин-1-ил}пиридин-3-ола в ДМФА добавляют 60% гидрид натрия и этил-4-бромбутаноат и затем осуществляют взаимодействие в течение 1 часа при комнатной температуре. Затем обычным способом осуществляют последующую обработку и очистку и получают нужное соединение.
Ссылочный пример 24
4-(2-Хлорпиримидин-4-ил)пиперазин-1-карбальдегид и бензиловый спирт последовательно обрабатывают способами, подобными способам ссылочного примера 20 и примера 4. Затем полученный продукт реакции подвергают каталитическому восстановлению с использованием палладия-на-угле и затем обрабатывают способом, подобным способу ссылочного примера 23, и получают нужное соединение.
Ссылочный пример 25
К 4-[6-(3,4-диметоксифенил)пиридин-2-карбонил]-1-(4-гидроксифенил)пиперазину добавляют 1,2-дибромэтан, 2 М водный раствор гидроксида натрия, гидросульфат тетра-н-бутиламмония и воду и затем смесь перемешивают при 60°С. После охлаждения реакционного раствора к нему добавляют воду и хлороформ, нерастворимые вещества удаляют фильтрацией, затем полученный продукт реакции обычным способом подвергают последующей обработке и очистке и получают нужное соединение.
Ссылочный пример 26
К раствору 2,5-дибромпиридина и 2-(диметиламино)этанола в ДМФА добавляют трет-бутоксид калия, все перемешивают на масляной бане при температуре 100°С в течение 3 часов и получают N-{2-[(5-бромпиридин-2-ил)окси]этил}-N,N-диметиламин, который затем обрабатывают способами, подобными способам ссылочных примеров 11 и 12, и получают нужное соединение.
Ссылочный пример 27
2-(Бензилокси)-6-бромнафталин последовательно обрабатывают способами, подобными способам ссылочного примера 11, примера 22 и примера 4, и получают 1-[6-(бензилокси)-2-нафтил]-4-[6-(3,4-диметоксифенил)пиридин-2-карбонил]пиперазин. Соединение растворяют в трифторуксусной кислоте, к раствору при охлаждении льдом добавляют пентаметилбензол, все перемешивают при комнатной температуре в течение 2 часов и затем на масляной бане при температуре 40°С в течение 2 часов и получают нужное соединение.
Ссылочный пример 28
К раствору (±)-транс-4-(2,5-диметилпиперазин-1-ил)бензальдегида в ацетонитриле добавляют ди(трет-бутоксикарбонил)дикарбонат и 4-диметиламинопиридин и смесь перемешивают. Затем обычным способом осуществляют последующую обработку и очистку и получают нужное соединение.
Ссылочный пример 29
Раствор фтор-4-нитробензола и (±)-транс-2,5-диметилпиперазина в NMP перемешивают на масляной бане при температуре 120°С в течение 3 часов и получают (±)-транс-2,5-диметил-1-(4-нитрофенил)пиперазин, который затем обрабатывают способом, подобным способу примера 4, и получают нужное соединение.
Ссылочный пример 30
К раствору 6-хлорхинолин-1-оксида в уксусном ангидриде добавляют метил-3-оксобутират и затем перемешивают в течение 30 минут на масляной бане при температуре 40°С. Полученное таким образом соединение добавляют к 10% хлористоводородной кислоте и в результате реакции в смеси при комнатной температуре получают метил-(6-хлорхинолин-2-ил)ацетат. Затем соединение последовательно обрабатывают способами, подобными способам ссылочного примера 11, примера 22 и примера 4, и получают нужное соединение.
Способами, подобными описанным в приведенных выше ссылочных примерах или в примерах, следующих далее, получают соответственно соединения ссылочных примеров 31-69, представленные в таблицах 1-5. Строение и физико-химические свойства соединений ссылочных примеров 1-69 приведены в таблицах 1-5.
Пример 1
К раствору 740 мг 2-оксо-3-фенилпиперазина в ТГФ (20 мл) добавляют 638 мг алюмогидрида лития и затем смесь 3 часа кипятят с обратным холодильником. Реакционный раствор охлаждают на льду и добавляют декагидрат сульфата натрия до тех пор, пока гель в реакционном растворе не исчезнет. После перемешивания на протяжении всего этого времени нерастворимое вещество удаляют фильтрацией. Неочищенный 2-фенилпиперазин, полученный после выпаривания растворителя, добавляют к раствору 500 мг 6-(3,4-диметоксифенил)пиридин-2-карбоновой кислоты в ТГФ (20 мл) и затем к смеси добавляют 556 мг гидрохлорида WSC и 260 мг HOBt и затем смесь 2 суток перемешивают при комнатной температуре. К реакционному раствору добавляют этилацетат и смесь промывают водой и рассолом. После сушки над безводным сульфатом магния растворитель выпаривают. Полученный остаток очищают колоночной хроматографией на силикагеле (хлороформ-метанол) и получают бесцветные бесформенные кристаллы (670 мг). Соединение растворяют в этаноле и к раствору добавляют 192 мг фумаровой кислоты для образования его соли - фумарата, которую затем перекристаллизовывают из этанола-этилацетата, и получают 607 мг полуфумарата 2-(3,4-диметоксифенил)-6-(3-фенилпиперазин-1-карбонил)пиридина в виде бесцветного кристаллического вещества.
Пример 2
К раствору 500 мг 6-(3,4-диметоксифенил)пиридин-2-карбоновой кислоты в ТГФ (20 мл) при охлаждении льдом добавляют 0,18 мл оксалилхлорида и одну каплю ДМФА. После перемешивания в течение 30 минут реакционный раствор при охлаждении льдом по каплям добавляют к раствору 370 мг 4-(4-метоксифенил)пиперазина в пиридине (10 мл). Смесь нагревают до комнатной температуры и затем перемешивают в течение 30 минут. К реакционному раствору добавляют воду и затем смесь экстрагируют этилацетатом. Органический слой промывают рассолом и сушат над безводным сульфатом магния и затем выпаривают растворитель. Остаток очищают колоночной хроматографией на силикагеле (хлороформ-метанол), затем осуществляют перекристаллизацию из ацтетата-ацетонитрила и получают 370 мг 1-[6-(3,4-диметоксифенил)пиридин-2-карбонил]-4-(4-метоксифенил)пиперазина в виде бесцветного кристаллического вещества.
Пример 3
В 15 мл 4 М раствора хлороводорода в этилацетате проводят реакцию с 0,62 г трет-бутил-4-[4-(2-диметиламиноэтокси)фенил]пиперазин-1-карбоксилата. К раствору в ДМФА (15 мл) 0,86 г неочищенного продукта реакции, полученного после выпаривания растворителя, добавляют 0,34 г гидрохлорида WSC, 0,24 г HOBt и 0,41 г 6-(3,4-диметоксифенил)пиридин-2-карбоновой кислоты и затем реакцию проводят в течение 65 часов при комнатной температуре. Затем к смеси добавляют 0,34 г гидрохлорида WSC, 0,24 г HOBt и 0,50 мл триэтиламина и затем смесь перемешивают в течение 8,5 часов при комнатной температуре. К реакционному раствору добавляют воду и затем смесь экстрагируют этилацетатом. Органический слой промывают водой и рассолом и сушат над безводным сульфатом магния и затем выпаривают растворитель. Остаток очищают колоночной хроматографией на силикагеле (этилацетат) и затем полученное таким образом соединение вводят в реакцию с 106 мг щавелевой кислоты для образования соли. Осуществляют перекристаллизацию (этанол) и получают 253 мг диоксалата 1-[6-(3,4-диметоксифенил)пиридин-2-карбонил]-4-[4-(2-диметиламиноэтокси)фенил]пиперазина в виде бледно-желтого кристаллического вещества.
Пример 4
К раствору 500 мг 6-(3,4-диметоксифенил)пиридин-2-карбоновой кислоты и 500 мг 1-(5-хлортиазол-2-ил)пиперазина в ТГФ (20 мл) при комнатной температуре добавляют 400 мг гидрохлорида WSC, 320 мг HOBt и 0,3 мл триэтиламина. После перемешивания в течение 4 часов к смеси добавляют воду и затем смесь экстрагируют этилацетатом. Органический слой промывают водой и рассолом и сушат над безводным сульфатом магния. Остаток после выпаривания растворителя очищают колоночной хроматографией на силикагеле (хлороформ-метанол), затем осуществляют перекристаллизацию из диизопропилового эфира и ацетонитрила и получают 560 мг 1-(5-хлортиазол-2-ил)-4-[6-(3,4-диметоксифенил)пиридин-2-карбонил]пиперазина в виде бесцветного кристаллического вещества.
Пример 5
К раствору 4-[N-{4-[6-(3,4-диметоксифенил)пиридин-2-карбонил]пиперазин-1-ил}фенил)амино]бутаноата в ТГФ добавляют 36% водный раствор формалина, уксусную кислоту и триацетоксиборогидрид натрия и затем смесь перемешивают. Затем обычным способом осуществляют последующую обработку и очистку и получают этил-[N-(4-{4-[6-(3,4-диметоксифенил)пиридин-2-карбонил]пиперазин-1-ил}фенил)-N-метиламино]бутаноат.
Пример 6
К раствору 1,01 г этил-3-(4-{4-[6-(3,4-диметоксифенил)пиридин-2-карбонил]пиперазин-1-ил}фенил)пропаноата в смешанном растворителе (ТГФ (5 мл) и метанола (5 мл)) добавляют 5 мл 1 М водного раствора гидроксида натрия и затем смесь перемешивают 1 час при комнатной температуре. К реакционному раствору добавляют 5 мл 1 М хлористоводородной кислоты и затем смесь экстрагируют этилацетатом. Органический слой промывают рассолом и сушат над безводным сульфатом магния. Полученное после выпаривания растворителя неочищенное кристаллическое вещество перекристаллизовывают из этанола и получают 673 мг 3-(4-{4-[6-(3,4-диметоксифенил)пиридин-2-карбонил]пиперазин-1-ил}фенил]пропановой кислоты в виде бесцветного кристаллического вещества.
Пример 7
При комнатной температуре в течение 7 часов перемешивают 0,71 г 2-хлор-6-(4-трет-бутоксикарбонилпиперазин-1-ил)пиразина в 15 мл 4 М раствора хлороводорода в этилацетате. Выпаривают растворитель и получают неочищенный продукт реакции гидрохлорид 2-хлор-6-(пиперазин-1-ил)пиразина. Полученный неочищенный продукт и 0,62 г 6-(3,4-диметоксифенил)пиридин-2-карбоновой кислоты обрабатывают способом, подобным способу примера 4, и получают 594 мг 2-хлор-6-{4-[6-(3,4-диметоксифенил)пиридин-2-карбонил]пиперазин-1-ил}пиразина в виде бледно-желтого кристаллического вещества.
Пример 8
К раствору 353 мг 1-[6-(3,4-диметоксифенил)пиридин-2-карбонил]-4-(пиридин-4-ил)пиперазина в дихлорметане (10 мл) добавляют 195 мг м-хлорпербензойной кислоты и затем смесь перемешивают 1 час при 5°С. К реакционному раствору добавляют водный раствор тиосульфата натрия и затем смесь экстрагируют хлороформом. Органический слой промывают водой и рассолом и затем сушат над безводным сульфатом магния, после чего выпаривают растворитель. Остаток очищают колоночной хроматографией на силикагеле (хлороформ-метанол), затем осуществляют перекристаллизацию (этанол-этилацетат) и получают 294 мг 1,5-гидрата 1-[6-(3,4-диметоксифенил)пиридин-2-карбонил]-4-(1-оксидопиридин-4-ил)пиперазина в виде бледно-желтого кристаллического вещества.
Пример 9
К раствору 2,5 г 1-[6-(3,4-диметоксифенил)пиридин-2-карбонил]-4-(4-нитрофенил)пиперазина в смешанном растворителе (этанол (70 мл) и вода (25 мл)) добавляют 0,15 г хлорида аммония и 3,1 г восстановленного железа и затем смесь 2 часа кипятят с обратным холодильником. Реакционный раствор фильтруют через целит и фильтрат концентрируют при пониженном давлении. К полученному таким образом остатку добавляют водный раствор гидрокарбоната натрия и затем смесь экстрагируют хлороформом. Органический слой промывают рассолом и затем сушат над безводным сульфатом магния, после чего выпаривают растворитель. Остаток очищают колоночной хроматографией на силикагеле (хлороформ-метанол), затем осуществляют кристаллизацию из ацетонитрила-этилацетата и получают 2,1 г 1-[6-(3,4-диметоксифенил)пиридин-2-карбонил]-4-(4-аминофенил)пиперазина в виде бледно-розового кристаллического вещества.
Пример 10
К раствору 1,50 г 4-{4-[6-(3,4-диметоксифенил)пиридин-2-карбонил]пиперазин-1-ил}фенола в ДМФА (10 мл) добавляют 1,00 г 4-хлорметилпиридин-N-оксида и 3,00 г карбоната цезия и затем смесь 30 минут перемешивают при комнатной температуре. После нагревания до 60°С смесь перемешивают еще в течение 30 минут. Затем к смеси добавляют 1,00 г 4-хлорметилпиридин-N-оксида и 1,50 г карбоната цезия и все перемешивают при 60°С в течение 1 часа. После охлаждения до комнатной температуры к смеси добавляют воду и затем смесь экстрагируют этилацетатом. Органический слой промывают водой и рассолом и затем сушат над безводным сульфатом магния, после чего выпаривают растворитель. Остаток очищают колоночной хроматографией на силикагеле (хлороформ-метанол), затем осуществляют перекристаллизацию из этанола и получают 440 мг 1-[6-(3,4-диметоксифенил)пиридин-2-карбонил]-4-[4-(1-оксидо-4-пиридилметокси)фенил]пиперазина в виде бледно-желтого кристаллического вещества.
Пример 11
К раствору 327 мг моногидрохлорида 1-[6-(3,4-диметоксифенил)пиридин-2-карбонил]пиперазина в этаноле (6 мл) добавляют 0,28 мл триэтиламина и 148 мг 2,4-дихлорпиримидина и затем смесь 2 часа перемешивают на масляной бане при температуре 90°С. После выпаривания растворителя к остатку добавляют воду и затем смесь экстрагируют хлороформом. Органический слой промывают водой и затем сушат над безводным сульфатом магния, после чего выпаривают растворитель. Остаток очищают колоночной хроматографией на силикагеле (гексан-этилацетат), затем осуществляют перекристаллизацию из ацетонитрила-диизопропилового эфира и получают 70 мг моногидрата 2-хлор-4-{4-[6-(3,4-диметоксифенил)пиридин-2-карбонил]пиперазин-1-ил}пиримидина в виде бесцветного кристаллического вещества.
Пример 12
К раствору 171 мг 4-{4-[6-(3,4-диметоксифенил)пиридин-2-карбонил]пиперазин-1-ил}бензойной кислоты в ТГФ (5 мл) добавляют 63 мг CDI и затем смесь перемешивают при 60°С. Затем к смеси в два приема добавляют 52 мг CDI и все перемешивают при 60°С в целом в течение 24 часов. После охлаждения реакционного раствора до комнатной температуры к нему добавляют 0,25 мл водного раствора аммиака и затем смесь перемешивают 6 часов при комнатной температуре. Затем к смеси добавляют 0,5 мл водного раствора аммиака и все перемешивают при комнатной температуре. Сырое кристаллическое вещество, выпавшее в результате в осадок, отфильтровывают и перекристаллизовывают из метанола-ТГФ и получают 68 мг 4-{4-[6-(3,4-диметоксифенил)пиридин-2-карбонил]пиперазин-1-ил}бензамида в виде бесцветного кристаллического вещества.
Пример 13
К раствору бензил-4-(4-{4-[6-(3,4-диметоксифенил)пиридин-2-карбонил]пиперазин-1-ил}фенилкарбамоил)пиперидин-1-карбоксилата в смешанном растворителе (этанол (8 мл) и ТГФ (8 мл)) в атмосфере аргона добавляют 18 мг 10% палладия-на-угле. После перемешивания в течение 2 часов при комнатной температуре в атмосфере водорода при нормальном давлении смесь фильтруют через целит и фильтрат концентрируют при пониженном давлении. Остаток очищают колоночной хроматографией на силикагеле (хлороформ-метанол-водный аммиак), затем осуществляют перекристаллизацию из ацетонитрила и получают 70 мг 4'-{4-[6-(3,4-диметоксифенил)пиридин-2-карбонил]пиперазин-1-ил}пиперидин-4-карбоксианилида в виде бесцветного кристаллического вещества.
Пример 14
К раствору 1,20 г 1-(бензофуран-5-ил)-4-(трет-бутоксикарбонил)пиперазина в хлороформе (5 мл) при 0°С добавляют 5 мл трифторуксусной кислоты, все нагревают до комнатной температуры и затем перемешивают 1 час. После нейтрализации 1 М водным раствором гидроксида натрия осуществляют экстракцию хлороформом. Органический слой промывают рассолом. После сушки над безводным сульфатом магния выпаривают растворитель. С использованием порции в 500 мг из 910 мг полученного таким образом 1-(бензофуран-5-ил)пиперазина получают 420 мг 1-(бензофуран-5-ил)-4-[6-(3,4-диметоксифенил)пиридин-2-карбонил]пиперазина в виде бесцветного кристаллического вещества.
Пример 15
К раствору 355 мг 1-(4-аминофенил)-4-[6-(3,4-диметоксифенил)пиридин-2-карбонил]пиперазина в ДМФА (3 мл) добавляют 130 мг 1-хлор-2-(2-хлорэтокси)этана, 77 мг иодида натрия и 249 мг карбоната калия и затем смесь перемешивают в течение ночи при 100°С. После охлаждения до комнатной температуры реакционный раствор концентрируют при пониженном давлении, затем к остатку добавляют воду и затем смесь экстрагируют хлороформом. Органический слой промывают рассолом и затем сушат над безводным сульфатом магния, после чего выпаривают растворитель. Остаток очищают колоночной хроматографией на силикагеле (хлороформ-метанол), затем осуществляют перекристаллизацию из этанола-диэтилового эфира и получают 210 мг 4-(4-{4-[6-(3,4-диметоксифенил)пиридин-2-карбонил]пиперазин-1-ил}фенил)морфолина в виде желтого кристаллического вещества.
Пример 16
К раствору 211 мг 1-(4-аминофенил)-4-[6-(3,4-диметоксифенил)пиридин-2-карбонил]пиперазина в ТГФ (2,5 мл) добавляют 63,5 мг метансульфонилхлорида и 76,8 мкл триэтиламина и затем смесь перемешивают в течение ночи при комнатной температуре. Затем к смеси в два приема добавляют 79 мг метансульфонилхлорида и 103 мкл триэтиламина и все перемешивают при комнатной температуре в течение 3 часов. К реакционному раствору добавляют воду и затем смесь экстрагируют этилацетатом. Органический слой промывают рассолом и затем сушат над безводным сульфатом магния, после чего выпаривают растворитель. Остаток очищают колоночной хроматографией на силикагеле (хлороформ-метанол), затем осуществляют кристаллизацию из этилацетата-диизопропилового эфира и получают 175 мг 4'-{4-[6-(3,4-диметоксифенил)пиридин-2-карбонил]пиперазин-1-ил}метансульфонанилида в виде светло-фиолетового кристаллического вещества.
Пример 17
К 233 мг этил-[(4-{4-[6-(3,4-диметоксифенил)пиридин-2-карбонил]пиперазин-1-ил}бензоил)амино]ацетата добавляют 0,8 мл концентрированной хлористоводородной кислоты и затем смесь перемешивают в течение ночи при комнатной температуре. После концентрирования реакционного раствора при пониженном давлении осуществляют кристаллизацию из 2-пропанола-диизопропилового эфира и отфильтровывают гидрохлорид [(4-{4-[6-(3,4-диметоксифенил)пиридин-2-карбонил]пиперазин-1-ил}бензоил)амино]уксусной кислоты. Фильтрат концентрируют при пониженном давлении, остаток кристаллизуют из гексана и получают 88 мг гидрата [(4-{4-[6-(3,4-диметоксифенил)пиридин-2-карбонил]пиперазин-1-ил}бензоил)амино]уксусной кислоты в виде светло-коричневого кристаллического вещества.
Пример 18
К раствору 1,51 г 2,5-дихлорпиразина в NMP (7,5 мл) добавляют 2,00 г 1-(трет-бутоксикарбонил)пиперазина и 2,00 г карбоната калия и затем смесь перемешивают 1 час при нагревании при 100°С. Смесь охлаждают до комнатной температуры, добавляют воду и затем экстрагируют этилацетатом. Органический слой промывают водой и рассолом и затем сушат над безводным сульфатом магния, после чего выпаривают растворитель. Остаток очищают колоночной хроматографией на силикагеле (хлороформ-метанол) и получают 2,73 г 2-хлор-5-(4-трет-бутоксикарбонилпиперазин-1-ил)пиразина. С использованием полученного соединения способом, подобным способу примера 14, получают 2-хлор-5-{4-[6-(3,4-диметоксифенил)пиридин-2-карбонил]пиперазин-1-ил}пиразин в виде бесцветного кристаллического вещества.
Пример 19
К раствору 460 мг моногидрата 2-хлор-4-{4-[6-(3,4-диметоксифенил)пиридин-2-карбонил]пиперазин-1-ил}пиримидина в метаноле (20 мл) добавляют 150 мг 10% палладия-на-угле и затем смесь перемешивают в течение 23 часов при комнатной температуре в атмосфере водорода при нормальном давлении. Нерастворимое вещество удаляют фильтрацией и остаток, полученный после выпаривания растворителя, очищают колоночной хроматографией на силикагеле (хлороформ-метанол) и затем осуществляют перекристаллизацию из ацетонитрила-диизопропилового эфира и получают 83 мг 4-{4-[6-(3,4-диметоксифенил)пиридин-2-карбонил]пиперазин-1-ил}пиримидина в виде бесцветного кристаллического вещества.
Пример 20
К 297 мг 4-[6-(3,4-диметоксифенил)пиридин-2-карбонил]-1-(4-гидроксифенил)пиперазина добавляют 623 мг [1,3]диоксолан-2-она и 147 мг карбоната калия и затем смесь 1,5 часа перемешивают при 100°С. После охлаждения смеси до комнатной температуры к реакционному раствору добавляют воду и затем 1 М хлористоводородную кислоту, которую затем нейтрализуют насыщенным водным раствором гидрокарбоната натрия, и затем смесь экстрагируют хлороформом. Органический слой промывают рассолом и затем сушат над безводным сульфатом магния, после чего выпаривают растворитель. Остаток очищают колоночной хроматографией на силикагеле (хлороформ-метанол), затем осуществляют перекристаллизацию из этилацетата и получают 41 мг 2-(4-{4-[6-(3,4-диметоксифенил)пиридин-2-карбонил]пиперазин-1-ил}фенокси)этанола в виде бледно-желтого кристаллического вещества.
Пример 21
К раствору 213 мг 6-{4-[6-(3,4-диметоксифенил)пиридин-2-карбонил]пиперазин-1-ил}пиридин-3-ола в ДМФА (5 мл) при охлаждении льдом добавляют 81 мг гидрохлорида (2-хлорэтил)диметиламина и 43 мг 60% гидрида натрия. После перемешивания в течение 1 часа на масляной бане при температуре 70°С к смеси добавляют воду и затем экстрагируют этилацетатом. Органический слой промывают рассолом и затем сушат над безводным сульфатом магния, после чего выпаривают растворитель. Остаток очищают колоночной хроматографией на силикагеле (хлороформ-хлороформ-метанол) и полученный таким образом продукт реакции (110 мг) растворяют в этаноле и превращают в его оксалат, добавляя 40 мг щавелевой кислоты. Затем соль перекристаллизовывают из этанола и получают 81 мг оксалата 1-[6-(3,4-диметоксифенил)пиридин-2-карбонил]-4-[5-(2-диметиламиноэтокси)-2-пиридил]пиперазина в виде бесцветного кристаллического вещества.
Пример 22
К раствору трет-бутил-4-[2-(4-{4-[6-(3,4-диметоксифенил)пиридин-2-карбонил]пиперазин-1-ил}фенокси)этил]пиперазин-1-карбоксилата в хлороформе (3 мл) добавляют 0,427 мл 4 М раствора хлороводорода в этилацетате и затем смесь 2 суток перемешивают при комнатной температуре. Затем к смеси добавляют 2 мл хлороформа и 1 мл 4 М раствора хлороводорода в этилацетате и все перемешивают при комнатной температуре в течение ночи. К реакционной смеси добавляют этанол, неочищенное кристаллическое вещество отфильтровывают и перекристаллизовывают из метанола и получают 114 мг гидрата тетрагидрохлорида 1-[6-(3,4-диметоксифенил)пиридин-2-карбонил]-4-[4-(2-пиперазин-1-илэтокси)фенил]пиперазина в виде бледно-желтого кристаллического вещества.
Пример 23
К раствору 1,42 г (±)-транс-1-[6-(3,4-диметоксифенил)пиридин-2-карбонил]-2,5-диметил-4-(4-нитрофенил)пиперазина в смешанном растворителе (этанол (37 мл) и воде (13 мл)) добавляют 0,16 г хлорида аммония и 1,66 г восстановленного железа и затем смесь 0,5 часа кипятят с обратным холодильником. Реакционный раствор фильтруют через целит и фильтрат концентрируют при пониженном давлении. К полученному таким образом остатку добавляют водный насыщенный раствор гидрокарбоната натрия и затем смесь экстрагируют хлороформом. Органический слой промывают рассолом и затем сушат над безводным сульфатом магния, после чего выпаривают растворитель. Остаток очищают колоночной хроматографией на силикагеле (хлороформ-метанол) и полученное соединение обрабатывают 4 М раствором хлороводорода в этилацетате для образования его соли. Затем выпаривают растворитель, остаток промывают этилацетатом и получают 582 мг гидрата гидрохлорида (±)-транс-4-{4-[6-(3,4-диметоксифенил)пиридин-2-карбонил]-2,5-диметилпиперазин-1-ил}анилина в виде бледно-желтого кристаллического вещества.
Способами, подобными способам, описанным выше в примерах, получают соответственно соединения примеров 24-115, представленные в приведенных далее таблицах 6-8. Строение и физико-химические свойства соединений примеров 1-115 приводятся в таблицах 6-8.
Примеры 116-147
К раствору 13 мг (0,05 ммоль) 6-(3,4-диметоксифенил)пиридин-2-карбоновой кислоты в ДМФА (0,7 мл) добавляют раствор в ДМФА (0,06 мл) соответствующего амина (0,06 ммоль) и 25 мг диизопропилэтиламина. Затем к реакционному раствору добавляют раствор 23 мг гексафторфосфата 2-(1Н-бензотриазол-1-ил)-1,1,3,3-тетраметилурония в ДМФА (0, 3 мл) и затем смесь перемешивают 24 часа при комнатной температуре. Добавляют PS-изоцианат (1,55 ммоль/г, 100 мг; Argonaut) и перемешивают при комнатной температуре в течение 14 часов. Реакционный раствор фильтруют, к фильтрату добавляют 3 мл хлороформа и 3 мл 1 М водного раствора гидроксида натрия и затем смесь перемешивают. Хлороформный слой сушат над безводным сульфатом натрия, затем выпаривают растворитель и получают соответственно соединения примеров 116-147, указанные в приведенной далее таблице 9. Строение и физико-химические свойства отдельных соединений приводятся в таблице 9.
Кроме того, в таблицах 10-13 показано строение других соединений изобретения. Их можно легко синтезировать с использованием вышеописанных способов получения, способов, описанных в примерах, и способов, очевидных для специалистов в данной области техники, или модификаций таких способов.
В приведенных далее таблицах используются следующие аббревиатуры: С.пр. - ссылочный пример, №, Пр. - пример, №, № - номер соединения, Данные - физико-химические свойства (F - FAB-МС (M+H)+, FN - FAB-МС (M-H)-, EI - EI-MC (M+), Т.пл. - температура плавления (°С), ЯМР1 - δ (м.д.) характеристических пиков 1Н-ЯМР в CDCl3, ЯМР2 - δ (м.д.) характеристических пиков 1Н-ЯМР в ДМСО-d6, Соль - соль и содержащийся растворитель (Ox - оксалат, Fum - фумарат, пустая колонка - свободное соединение, число перед компонентом (обозначает число молей присоединенной кислоты), например, 2 HCl обозначает дигидрохлорид), Сп.с. - способ получения (каждое число указывает номер примера, где описывается подобный способ получения, или номер ссылочного примера), Ме - метил, Et - этил, cPr - циклопропил, tBu - трет-бутил, Ph - фенил, Bn - бензил, Ас - ацетил, Pip - пиперидин-1-ил, Pip4 - пиперидин-4-ил, Mor - морфолин-4-ил, Pipr - пиперазин-1-ил, и Pirr - пирролидин-1-ил. Кроме того, число перед каждым заместителем показывает положение замещения, например, 2-Cl означает 2-хлор, 3,4-диМе означает 3,4-диметил, 2,3,4-триМе означает 2,3,4-триметил, 4-Ме-Pipr означает 4-метилпиперазин-1-ил, и 3,4-(ОСН2О) означает 3,4-метилендиоксигруппу соответственно.
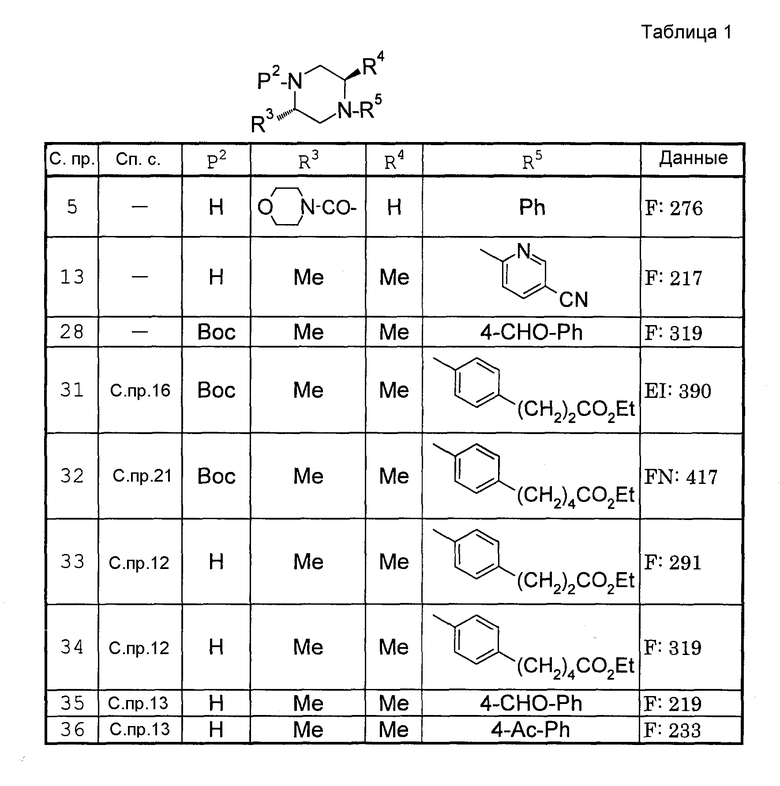
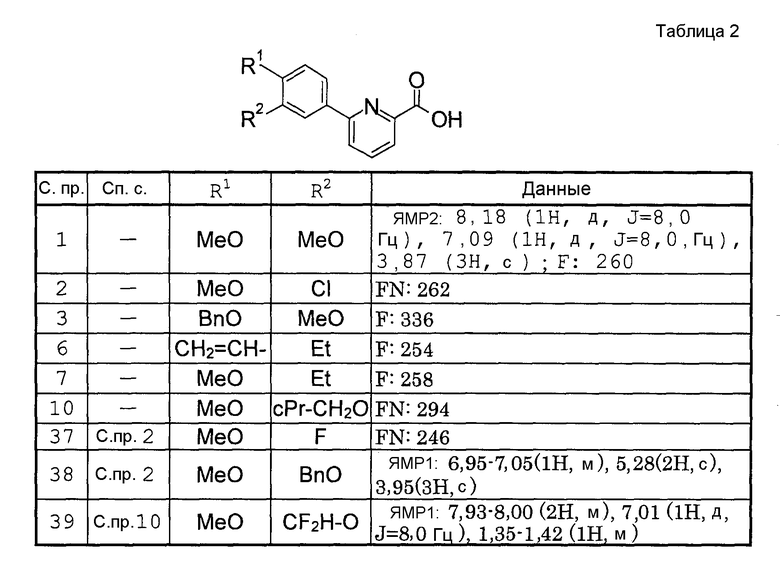
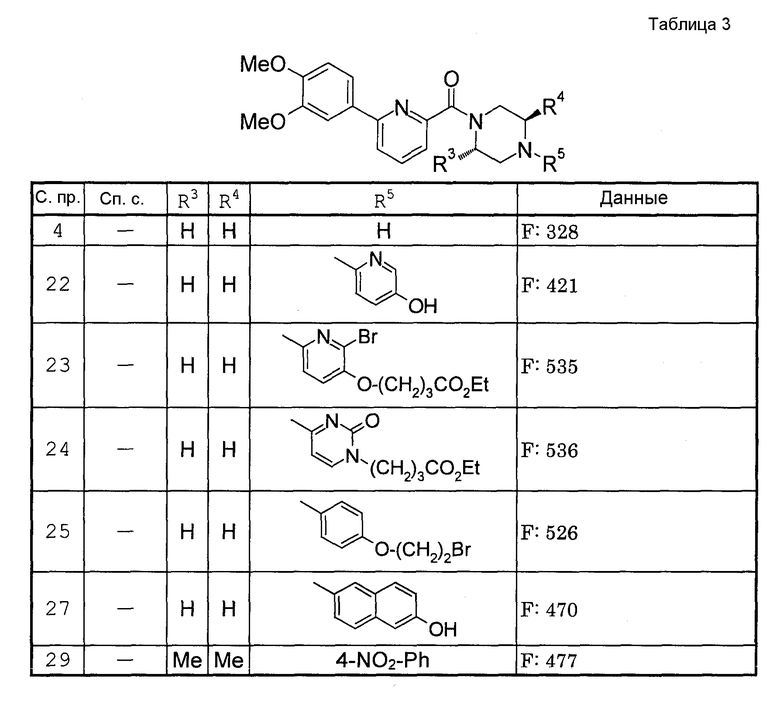
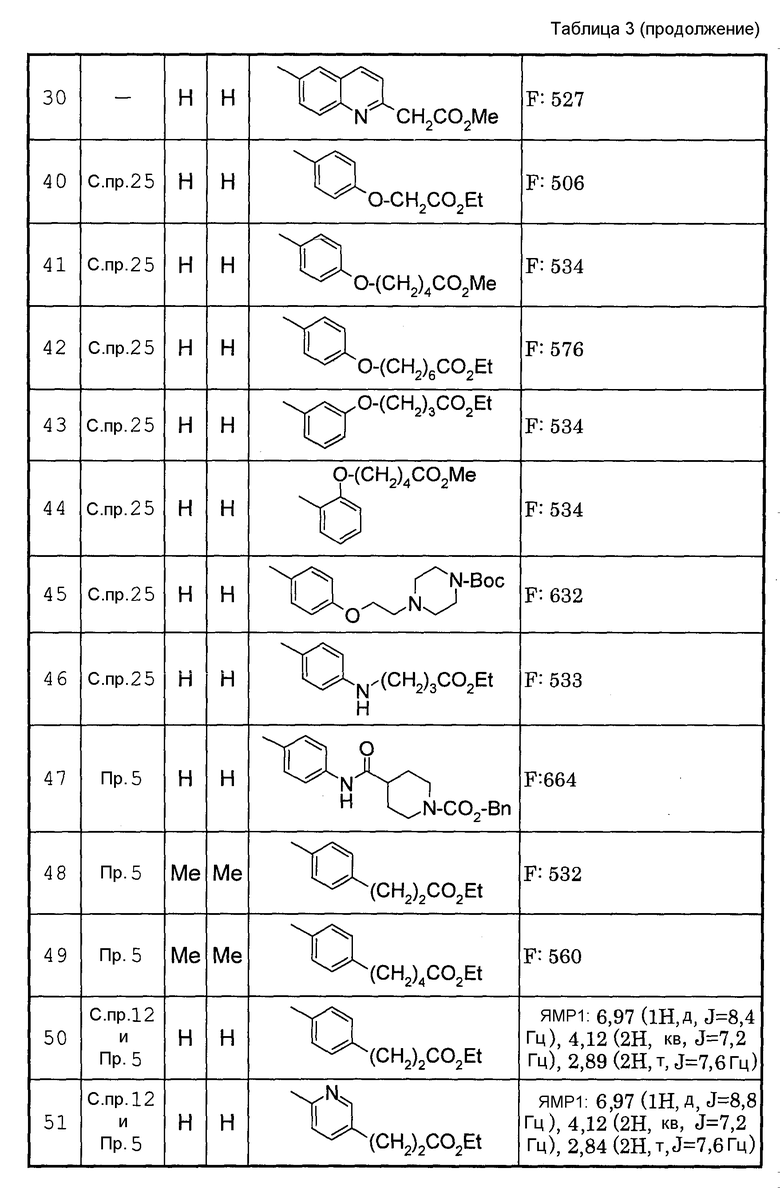
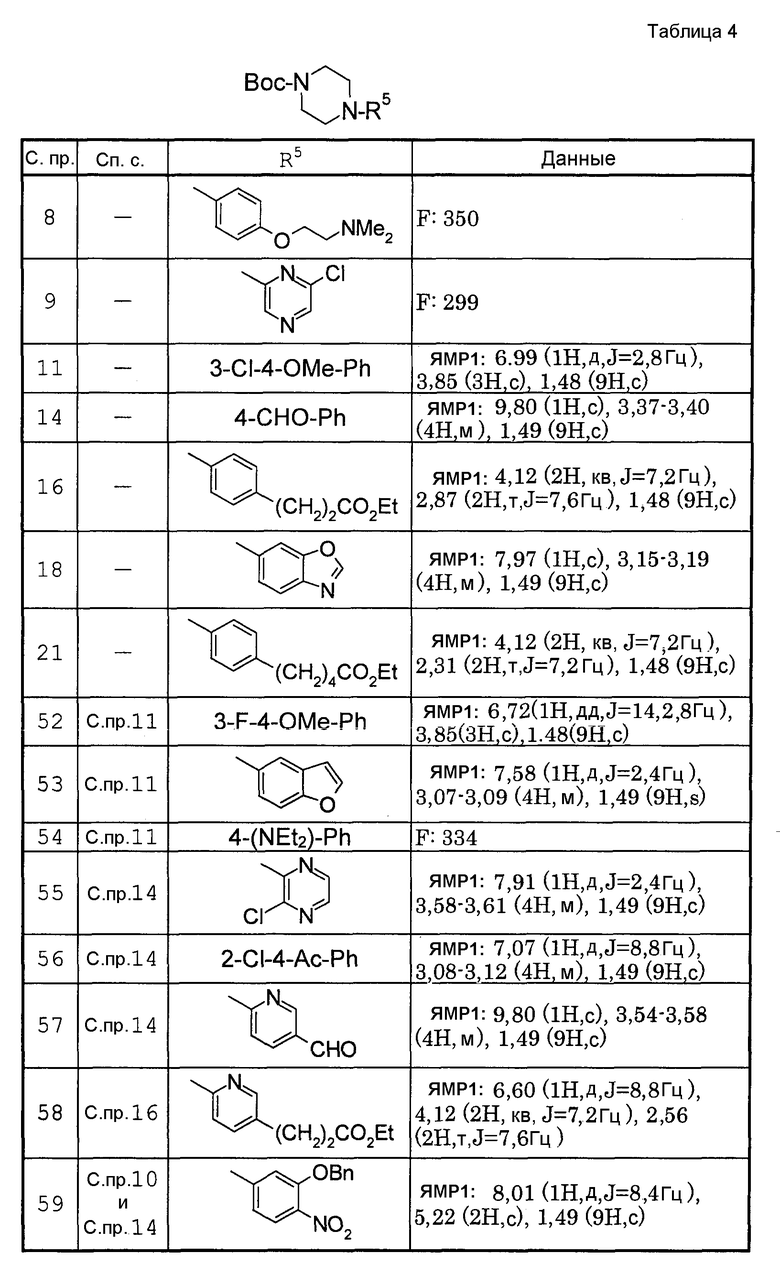
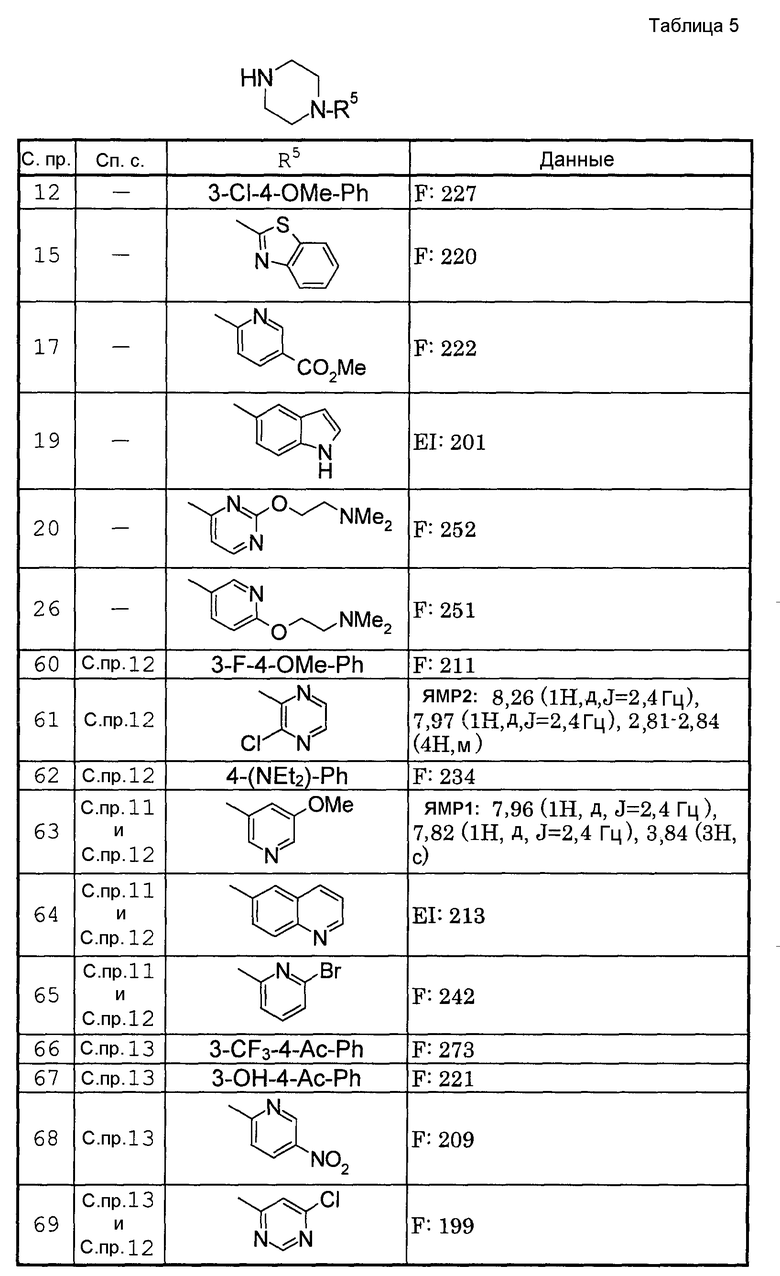
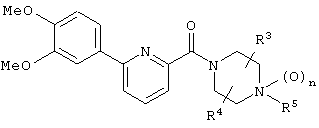
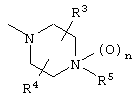
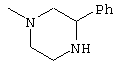
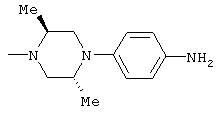
Н2О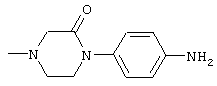
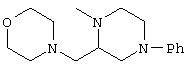
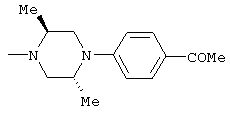
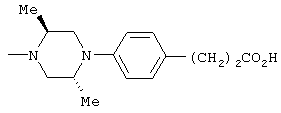
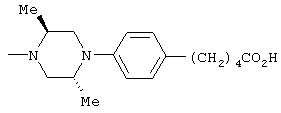
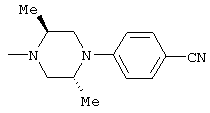
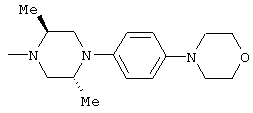
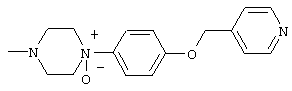
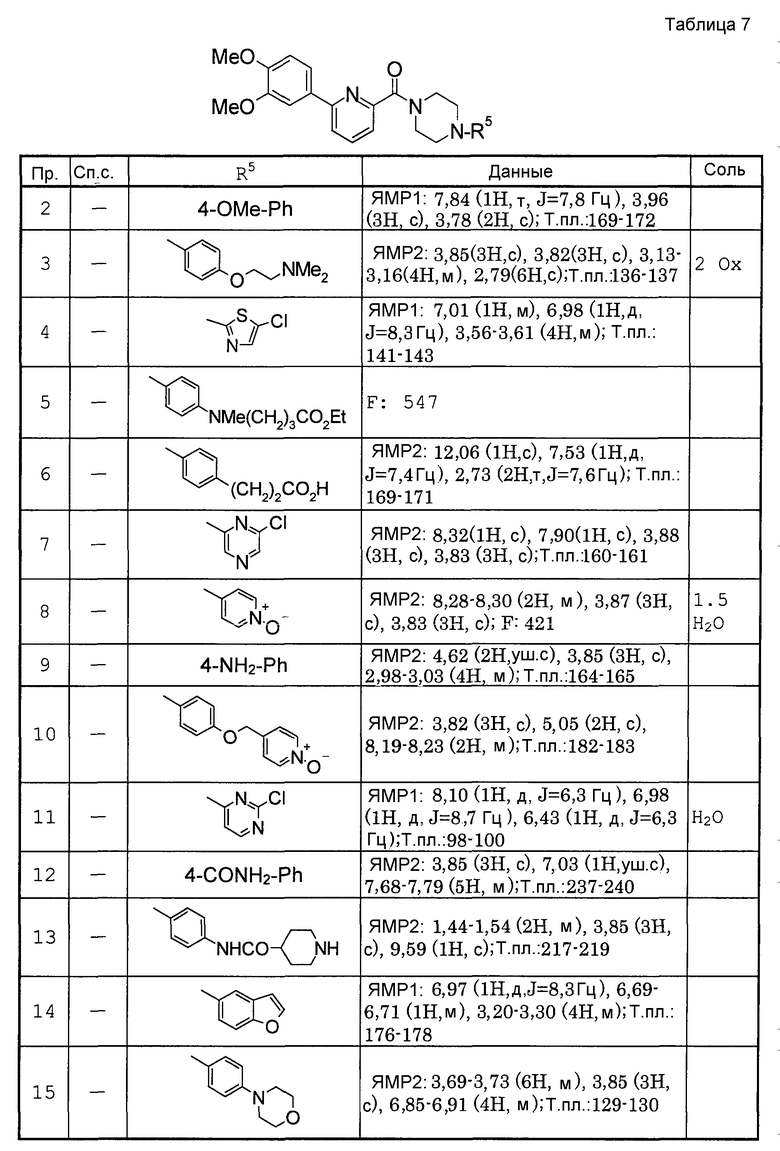

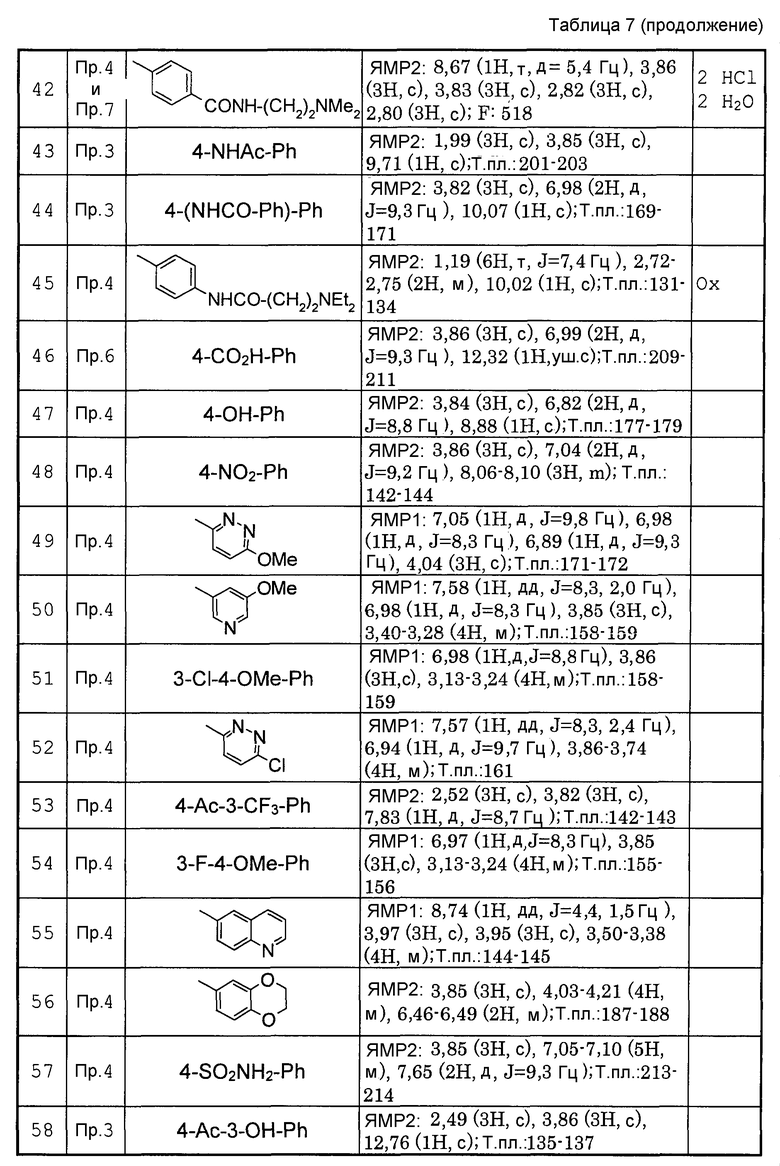
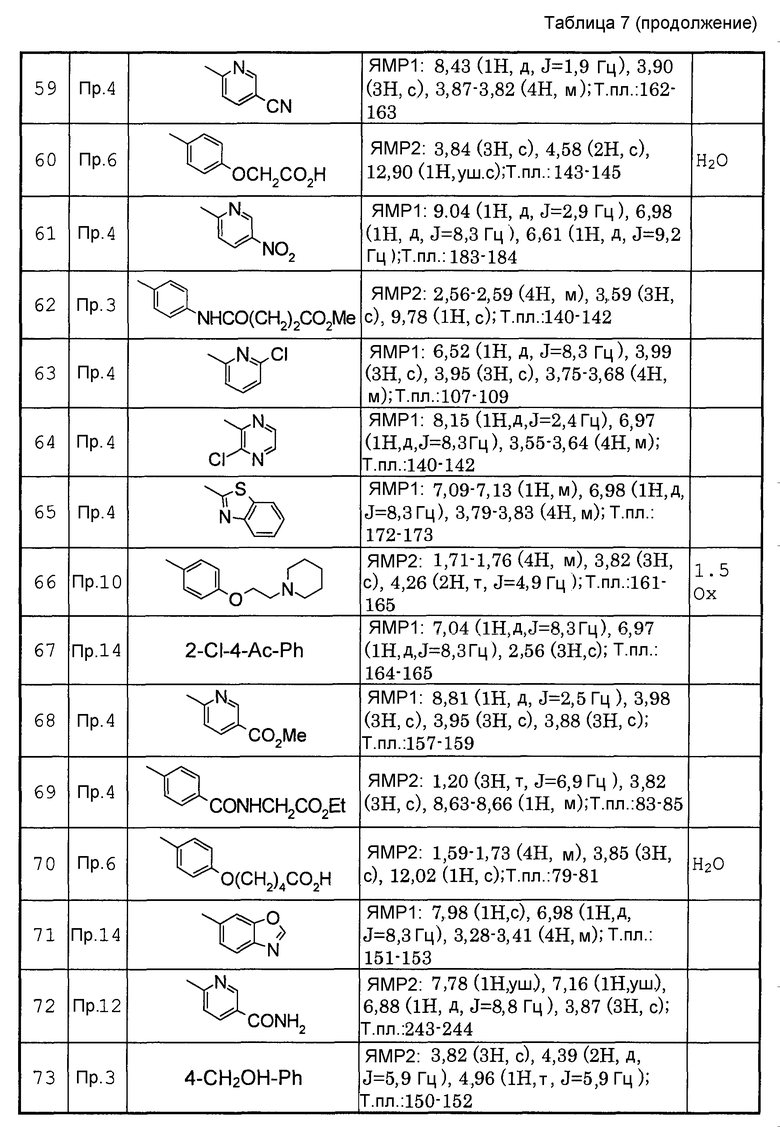
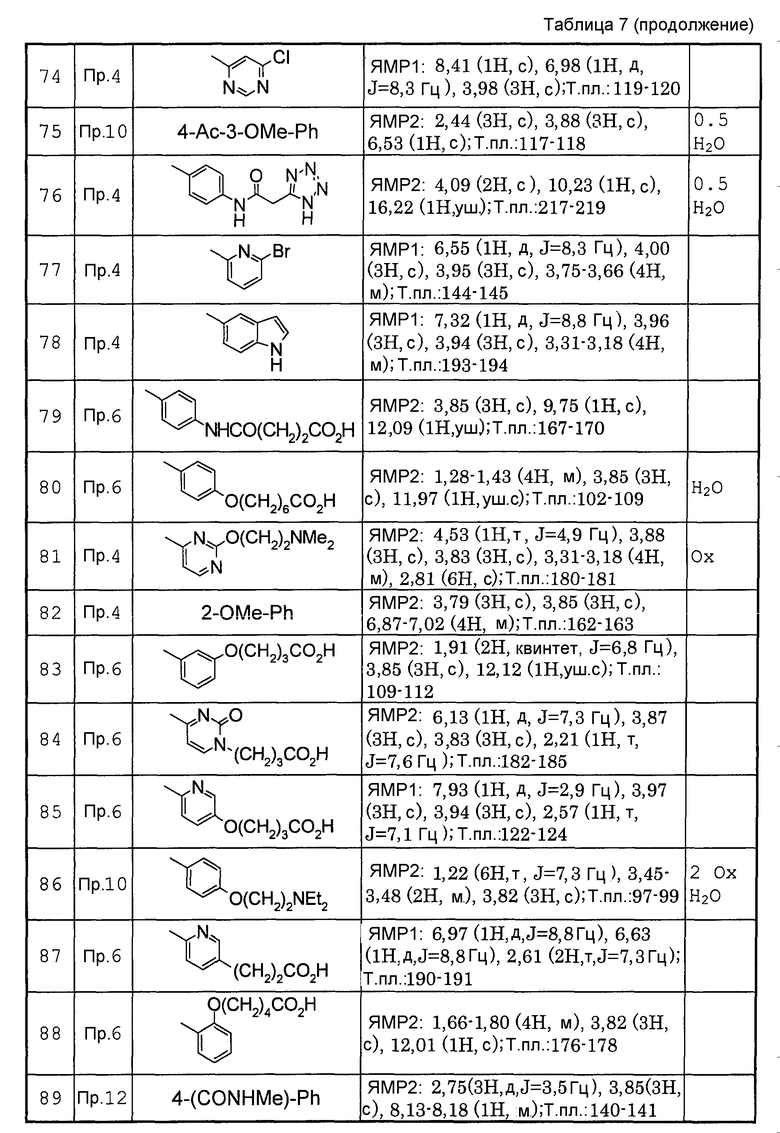
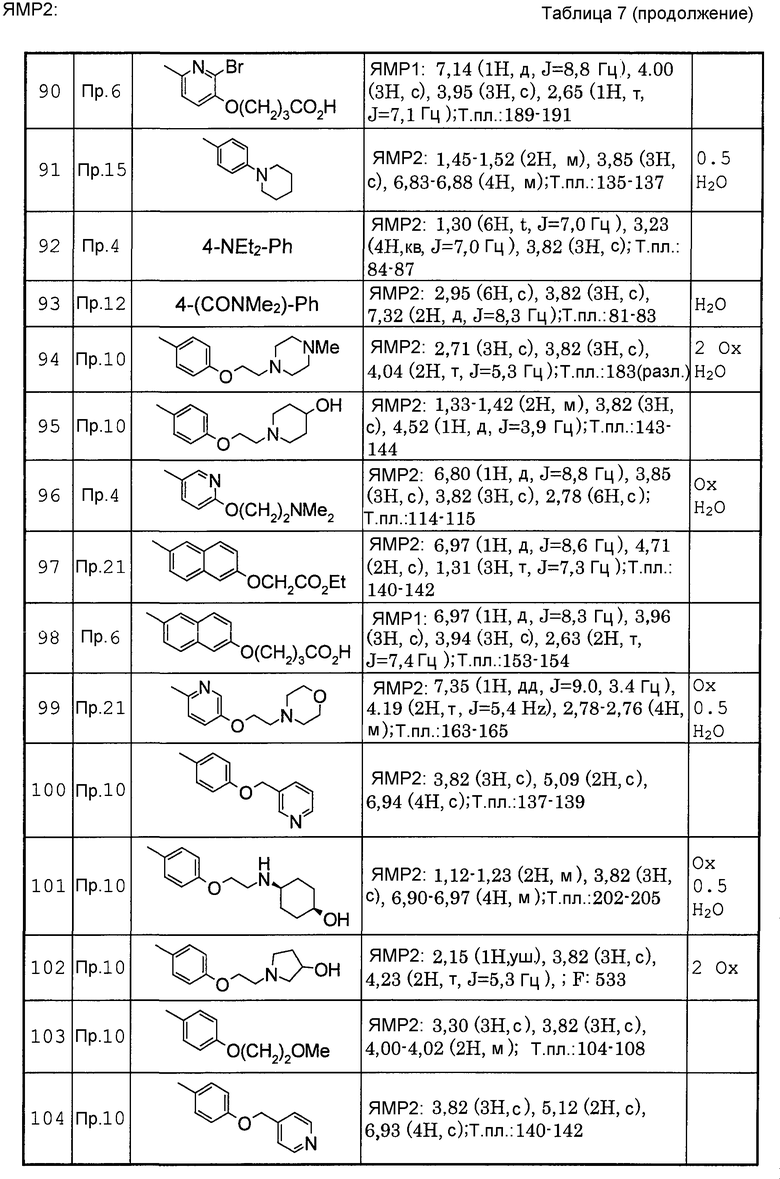
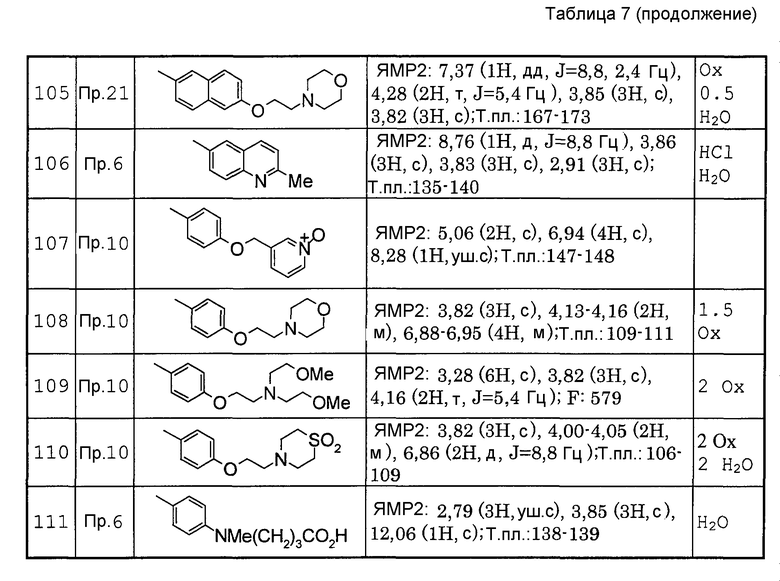
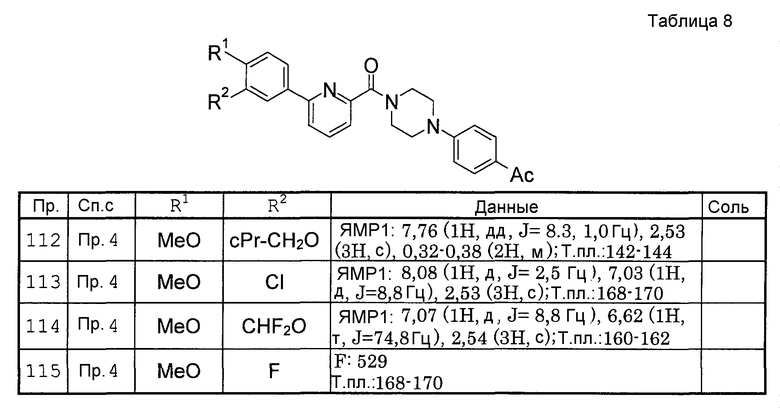
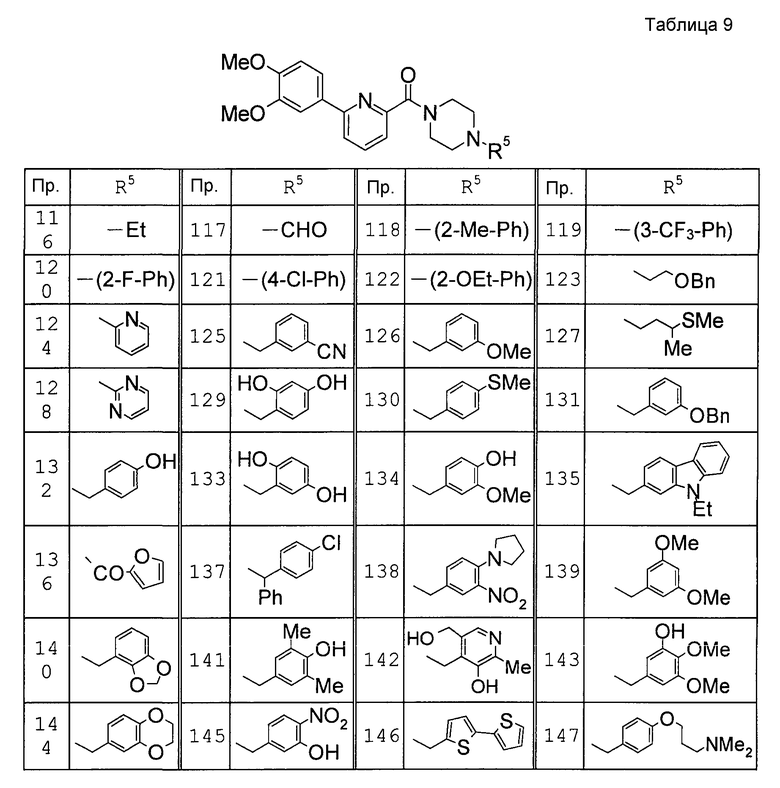
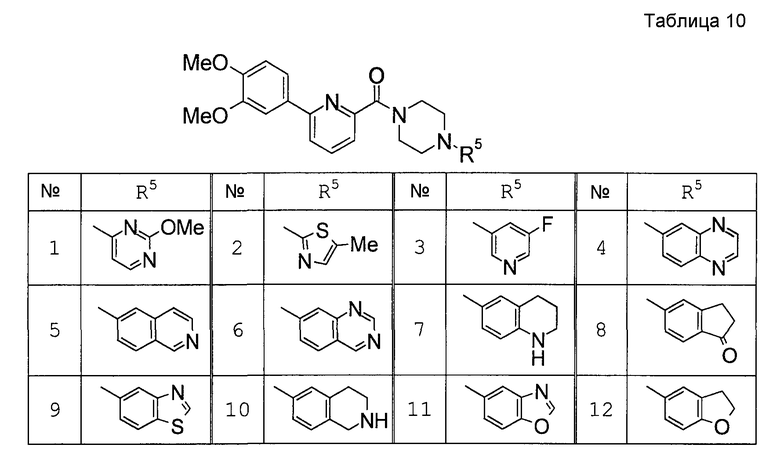

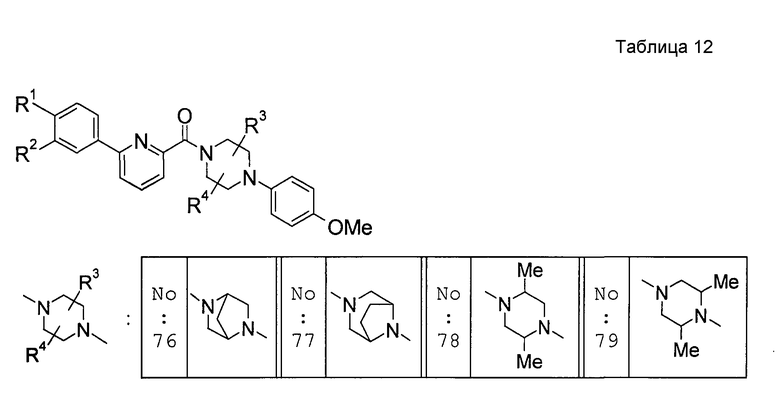
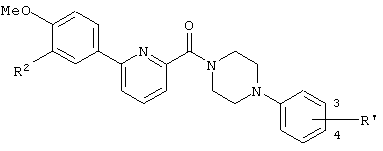
R2=Cl
Пример композиции 1 (дополнительный): Фармацевтическая композиция для перорального введения.
Соединение в соответствии с изобретением (20 г) в качестве активного ингредиента смешивают с 51 г лактозы и 21,8 г кукурузного крахмала. С использованием аппарата для гранулирования, работающего в режиме псевдоожижения, полученную смесь подвергают гранулированию с 20 г 10% раствора гидроксипропилцеллюлозы, а затем высушивают. Полученный сухой продукт смешивают с низкозамещенной гидроксипропилцеллюлозой, а затем - с 0,2 г стеарата магния с получением порошка для таблетирования. Полученную смесь таблетируют в роторной машине для таблетирования (производитель - Kikusui Seisakusyo) с пуансоном диаметром 7 мм, получая таблетки массой 100 мг.
Пример композиции 2 (дополнительный): Фармацевтическая композиция для парентерального введения.
Соединение в соответствии с изобретением (1 г) в качестве активного ингредиента смешивают с 700 мл соды для инъекций. Затем добавляют 9 г хлорида натрия, получая раствор. Устанавливают требуемое значение рН раствора, добавляя растворы HCl/NaOH, после чего объем раствора доводят до 1000 мл. Затем раствор асептически фильтруют через фильтр с диаметром пор 0,2 мкм и заполняют им в апирогенных условиях ампулы из борсиликатного стекла. Ампулы стерилизуют общепринятым способом в паровом автоклаве с получением препарата для инъекций.
| название | год | авторы | номер документа |
|---|---|---|---|
| ГЕТЕРОЦИКЛИЧЕСКИЕ ДИГИДРОПИРИМИДИНОВЫЕ СОЕДИНЕНИЯ | 2000 |
|
RU2296766C2 |
| N-ГЕТЕРОЦИКЛИЧЕСКИЕ ПРОИЗВОДНЫЕ И ИХ ПРИМЕНЕНИЕ В КАЧЕСТВЕ ИНГИБИТОРОВ СИНТАЗЫ ОКСИДА АЗОТА | 1998 |
|
RU2241708C2 |
| НОВЫЕ ПРОИЗВОДНЫЕ ПИРРОЛИДИН-3,4-ДИКАРБОКСАМИДА | 2005 |
|
RU2379288C2 |
| ПРОИЗВОДНЫЕ ГЕТЕРОАРИЛАЛКИЛПИПЕРАЗИНА И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ИХ ОСНОВЕ | 2001 |
|
RU2243970C1 |
| ИСПОЛЬЗОВАНИЕ ИНГИБИТОРОВ PDE7 ДЛЯ ЛЕЧЕНИЯ НАРУШЕНИЙ ДВИЖЕНИЙ | 2010 |
|
RU2600869C2 |
| ПРОИЗВОДНЫЕ ТИОФЕНА | 2010 |
|
RU2536865C2 |
| ИНГИБИТОРЫ ТИРОЗИНКИНАЗЫ БРУТОНА | 2010 |
|
RU2542585C2 |
| НОВЫЕ ФЕНИЛПИРАЗИНОНЫ В КАЧЕСТВЕ ИНГИБИТОРОВ КИНАЗЫ | 2009 |
|
RU2507202C2 |
| НОВЫЕ ПИРИДИНОНЫ И ПИРИДАЗИНОНЫ | 2009 |
|
RU2505538C2 |
| ПРОИЗВОДНЫЕ 4-АМИНО-5-ЦИАНОПИРИМИДИНА | 2005 |
|
RU2386628C2 |
Настоящее изобретение относится к производному пиридина общей формулы I
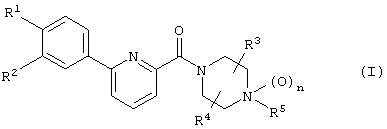
где каждый символ имеет следующие значения: R1 и R2 - водород, галоген, низший алкил, низший алкокси, R3 и R4 - водород, низший алкил, галоген, R5 - водород, низший алкил, n=0 или 1, или его фармацевтически приемлемые соли. Описаны также фармацевтическая композиция на основе соединений I и промежуточные продукты, используемые в синтезе. Соединения обладают ингибирующим действием в отношении фосфодиэстеразы типа 4. 3 з. и 6 з.п. ф-лы, 13 табл.
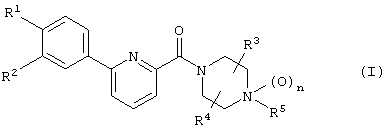
где каждый символ имеет следующие значения:
R1 и R2: одинаковые или отличаются друг от друга и представляют собой Н, галоген, низший алкил, O-(низший алкил), O-(низший алкил, замещенный галогеном(ами)), O-(низший алкилен)-(углеводородный цикл), выбранный из группы, включающей циклоалкил,
R3 и R4: одинаковые или отличаются друг от друга и представляют собой Н, низший алкил, низший алкил, замещенный гетероциклом(ами), выбранным(и) из группы, включающей морфолин, необязательно замещенный углеводородный цикл, выбранный из группы, включающей фенил, или R3 и R4 объединяются с образованием оксо,
R5: H, необязательно замещенный углеводородный цикл, выбранный из группы, включающей фенил и нафтил, необязательно замещенный гетероцикл, выбранный из группы, включающей пиридин, пиразин, пиримидин, пиридазин, хинолин, тиазол, бензоксазол, бензофуран, бензодиоксан, индол, бензотиазол и пиперидин.
n: 0 или 1,
или его фармацевтически приемлемая соль.
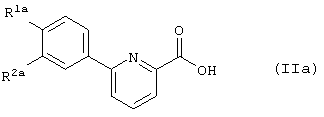
где
R1a представляет галоген, низший алкил, O-(низший алкил), O-(низший алкил, замещенный галогеном(ами)),
R2a представляет Н или группу, указанную в определении R13, или R1a и R2a объединяются с образованием -O-(низший алкилен)-O-,
при условии, что (1) когда R2a представляет собой Н, R1a представляет группу, иную чем метил, этил, ОМе, NH2, NHMe или Cl, и (2) когда R2a представляет собой метил, R1a представляет соответственно группу иную чем метил.
| Экономайзер | 0 |
|
SU94A1 |
| Приспособление в пере для письма с целью увеличения на нем запаса чернил и уменьшения скорости их высыхания | 1917 |
|
SU96A1 |

