Настоящее изобретение относится к новой фармацевтической композиции для перорального введения, содержащей основный фармацевтически активный ингредиент, имеющий рН-зависимую растворимость, который подавляет высвобождение основного фармацевтически активного ингредиента из композиции при кислом рН (предпочтительно ниже рН 3) и предпочтительно обеспечивает по существу рН-независимое регулируемое высвобождение фармацевтически активного ингредиента в широком диапазоне рН в желудочно-кишечном тракте; способу производства указанной композиции и к ее применению в медицине.
Эффективные фармацевтические композиции с регулируемым высвобождением являются подходящими фармацевтическими продуктами, поскольку они дают возможность оптимизации лекарственной терапии, уменьшения частоты дозировки и минимизации нежелательных побочных эффектов. Однако разработка таких систем с регулируемым высвобождением является непростой задачей, особенно когда фармацевтическая композиция предназначена для перорального введения и должна пройти через желудочно-кишечный тракт, который проявляет, среди других характеристик, большой разброс рН на протяжении всей длины.
Многие лекарственные средства, проявляющие основные свойства, ионизируются при низком рН и становятся значительно более растворимыми в этом диапазоне рН по сравнению с более нейтральным окружением. Это проявление рН-зависимой растворимости в желудочно-кишечном тракте может приводить к изменчивым профилям высвобождения лекарственного средства с сопутствующими проблемами биодоступности in vivo.
Описано несколько попыток преодолеть проблему рН-зависимой растворимости основных лекарств. Эти подходы включают в себя применение кишечно-растворимого полимера, который является нерастворимым при низком рН, для замедления высвобождения лекарственного средства в окружающей среде с низким рН (смотри, например, US 4968508 и A. Streubel et al. J. Controlled Release, 67, 101-110 (2000)) либо включение органической кислоты с низкой молекулярной массой для создания кислого рН микросреды внутри матрицы фармацевтической композиции, таким образом поддерживая растворимость лекарственного средства постоянной (смотри, например, К.Е.Gabr., Eur. J. Pharm. Biopharm., 38 (6), 199-202 (1992), и V.K. Thoma and Th. Zimmer, Pharm. Ind. 51 (1), 98-101 (1989)). Включение анионного полимера (например, альгината натрия), проявляющего рН-зависимую растворимость, в фармацевтическую композицию, которая также содержит нейтральный полимер, придает свойство нерастворимости и способность к гелеобразованию при низком рН, приводя к сильному диффузионному барьеру, который теоретически должен быть главным механизмом, замедляющим высвобождение лекарственного средства при низком рН [US 4792452; Р. Timmins et al. Pharmaceutical Development and Technology, 2 (1), 25-31 (1997)]. Другие способы включают в себя использование заряженных полимеров для воздействия на высвобождение лекарственного средства либо путем межионного взаимодействия с лекарственным средством [смотри С.Caramella et al. Pharm. Res. 14 (11), 531 (1997), H.Y.Park et al. Drug Delivery, 5 13-18 (1998), N.Caram-Lelham, Ph.D. thesis, Uppsala University (1996)], либо путем воздействия на способность к гелеобразованию и набухаемость этих полимеров [смотри К.М.Picker, Drug Dev. and Ind. Pharmacy, 25 (3) 339-346 (1999)]. Фармацевтические композиции, использованные здесь, обычно основаны на одном типе полимера.
В статье Baveja et al. Int J. Pharmaceutics 39, 39-45 (1987) показано, что когда полимер, не содержащий ионогенных групп (НРМС), смешан с анионным полимером (NaCMC), высвобождение замедлено. В статье Ranga Rao et al. Drug Dev. Ind Pharmacy, 14, 2299 (1988), описаны смеси метилцеллюлозы и NaCMC для получения различных профилей высвобождения. Смеси лямбда-каррагинана и активного ингредиента описаны в WO 99/21586.
Сообщали о комбинациях подходов для получения рН-независимого профиля высвобождения (смотри WO 96/26717, WO 99/29305 и WO 99/39698). Во всех этих трех публикациях описана трехкомпонентная матричная композиция, содержащая три полимера обычно с различной растворимостью в воде и набухаемостью. Состав этой композиции может варьировать, причем регулируются указанные свойства для получения регулируемых скоростей высвобождения. Два компонента включают в себя желатинирующий полимер со значительной рН-зависимой растворимостью, такой как альгинат натрия, и желатинирующий полимер с низкой или незначительной рН-зависимой растворимостью, такой как гидроксипропилметилцеллюлоза (НРМС) или полиэтиленоксид. Третий компонент включает в себя либо кишечно-растворимый покрывающий полимер, такой как сополимер метакриловой кислоты (WO 96/26717); EUDRAGIT® L или S, которые представляют собой конкретные типы полимеров метакриловой кислоты (WO 99/29305); либо водонерастворимый полимер, такой как этилцеллюлоза (WO 99/39698). Однако эти подходы, в общем, не обеспечивают специфическую доставку основных лекарственных средств и зависят от полимеров типа полимеров для кишечно-растворимого покрытия или водонерастворимых полимеров, таких как полимер метакриловой кислоты, или рН-зависимого желатинирующего полимера, такого как альгинат натрия, для замедления высвобождения лекарственного средства при низких рН окружающей среды, по меньшей мере, частично.
Согласно настоящему изобретению предложенаая фармацевтическая композиция для перорального введения, содержащая иота-каррагинан, один или более чем один нейтральный желатинирующий полимер и основный фармацевтически активный ингредиент, причем эта композиция подавляет высвобождение из нее основного фармацевтически активного ингредиента при кислом рН (предпочтительно ниже рН 3, в частности примерно рН 1).
По существу рН-независимое высвобождение означает, что скорость высвобождения значительно замедлена при рН 1 и слегка повышена или не изменяется при рН 6,8, так что количество основного фармацевтически активного ингредиента, высвобождаемого в любой момент времени, становится менее рН-зависимым.
Согласно настоящему изобретению также предложена фармацевтическая композиция для перорального введения, содержащая иота-каррагинан, один или более чем один нейтральный желатинирующий полимер и основный фармацевтически активный ингредиент.
Иота-каррагинан предпочтительно присутствует в композиции по изобретению на уровне более 15% по массе. Иота-каррагинан имеет предпочтительно естественное происхождение. Один тип фармацевтического сорта иота-каррагинана (доступен от FMC Biopolymer) имеет вязкость не менее 5 сантипуаз (сП; 5 мПа·с), предпочтительно в диапазоне 5-10 сП (5-10 мПа·с; для 1,5% раствора, нагретого до 82°С, после чего вязкость измеряют при 75°С с помощью вискозиметра Брукфилда LV, снабженного шпинделем №1, работающим со скоростью 30 об/мин). Тип технического сорта иота-каррагинана (доступен от Fluka Biochemica) предпочтительно имеет вязкость не менее 14 мПа·с, для 0,3% водного раствора, нагретого до 20°С, после чего вязкость измеряют с помощью вискозиметра с падающим шариком, типа Haake, используемого вместе с термостатом С3 Lauda и Hakke Mess-System III, и с использованием покрытых золотом шариков из нержавеющей стали плотностью 7,8 г/см3.
Нейтральный желатинирующий полимер представляет собой единственный нейтральный разрушаемый полимер, обладающий способностью к гелеобразованию и имеющий по существу рН-независимую растворимость, или смесь из более чем одного нейтрального разрушаемого полимера. Нейтральный желатинирующий полимер предпочтительно присутствует в композиции на уровне более 10%, но предпочтительно более 20% по массе. {"Разрушаемый" и "разрушение" относятся к растворению или распаду либо отдельно, либо в комбинации. Растворение можно увеличить путем перемешивания, а распад можно увеличить путем механического взаимодействия с твердым веществом}.
Подходящие нейтральные желатинирующие полимеры включают в себя полиэтиленоксид (ПЭО), производные и члены семейства ПЭО (например, полиэтиленгликоль (ПЭГ), предпочтительно существующий в природе в твердом состоянии с подходящей молекулярной массой или вязкостью). Таким образом, нейтральный желатинирующий полимер представляет собой, например, полиэтиленоксид или полиэтиленгликоль.
При использовании в качестве единственного нейтрального желатинирующего полимера ПЭО предпочтительно имеет ММ (молекулярную массу), большую или равную 4 миллионам (4М) (например, ММ от 4 до 8 миллионов), соответствующую диапазону вязкости водного раствора 1650-5500 мПа·с (либо 1650-5500 сП, измеренной для 1% водного раствора при 25°С с использованием вискозиметра Брукфилда RVF со шпинделем №2 при 2 об/мин). Другие примеры подходящих ПЭО включают в себя ПЭО с ММ примерно 5 миллионов (5М), соответствующей диапазону вязкости водного раствора 5500-7500 мПа·с, либо ПЭО с ММ примерно 8 миллионов (8М), соответствующей диапазону вязкости водного раствора 10000-15000 мПа·с. Этот диапазон охватывает величину вязкости типичного раствора (в сП), измеренную при 25°С, приведенную для этого полимера в USP (Фармакопея США) 24/NF1 (Национальный (фармацевтический) формуляр) 9, издание 2000, стр.2285-2286. Таким образом, ПЭО может иметь ММ, равную 4-8 миллионам.
Когда ПЭГ используют в качестве единственного нейтрального желатинирующего полимера, он предпочтительно имеет высокую молекулярную массу, например ММ, равную примерно 20000, соответствующую диапазону вязкости 2700-3500 мПа·с (либо 2700-3500 сП), измеренной с использованием 50% водного раствора (мас./мас.) при 20°С с использованием капиллярного вискозиметра (Ubbelohde или эквивалентного). [ссылка: European Pharmacopoeia 3rd Ed., 2000, Supplement, pp.908-909].
Другие подходящие желатинирующие полимеры включают в себя производные целлюлозы, такие как гидроксипропилметилцеллюлоза (НРМС) или гидроксиэтилцеллюлоза (НЕС), (хотя предпочтительно НРМС) с подходящими высокими вязкостями (например, "НРМС 10000 сП", "НРМС 15000 сП", "НЕС типа НН" или "НЕС типа Н"). При использовании в качестве единственного нейтрального полимера гидроксипропилметилцеллюлозные полимеры, например «НРМС 10000 сП» и «НРМС 15000 сП», имеют соответственно кажущиеся вязкости 7500-14000 мПа·с (либо 7500-14000 сП) и 11250-21000 мПа·с (либо 11250-21000 сП) при измерении при 20°С с 2% (мас./мас.) водным раствором, рассчитанные по сухому веществу, с использованием капиллярного вискозиметра (Ubbelohde или эквивалентного). Один тип гидроксиэтилцеллюлозного полимера, например "Natrosol 250 Pharma, тип НН", от Hercules Incorporated (Aqualon), проявляет обычно вязкость по Брукфилду примерно 20000 мПа·с при использовании прибора Брукфилда Synchro-Lectric Model LVF, в условиях 1% концентрации раствора, шпиндель №4, скорость шпинделя 30 об/мин, фактор 200, 25°С (см. Natrosol Physical and Chemical Properties booklet, 33.007-E6 (1993), p.21).
Когда используют смесь нейтральных желатинирующих полимеров, эта смесь может включать в себя, например, смесь или композицию двух или более чем двух ПЭО, двух или более чем двух НРМС, ПЭО и НРМС, либо ПЭО и ПЭГ. Например, ПЭО с ММ 4, 5 или 8 миллионов может быть смешан с ПЭО с ММ 1 миллион, ПЭО с ММ 400000, ПЭО с ММ 100000 или ПЭГ с ММ 6000.
Альтернативно, нейтральный желатинирующий полимер (например, ПЭО) может быть использован в комбинации с нежелатинирующим нейтральным полимером (таким как ПЭГ с низкой ММ, например ПЭГ, имеющий ММ ниже 10000). Примеры ПЭГ с низкой ММ в такой комбинации включают в себя ПЭГ с ММ 8000 (соответствующей диапазону вязкости 260-510 мПа·с) или ПЭГ с ММ 6000 (соответствующей диапазону вязкости 200-270 мПа·с).
Смесь или композиция двух или более чем двух сортов НРМС может включать в себя как сорта с более низкой вязкостью (нежелатинирующие), так и сорта с более высокой вязкостью (желатинирующие). Например, "НРМС 50 сП", "НРМС 15 сП" и "НРМС 6 сП", имеющие соответственно кажущиеся вязкости 40-60 мПа·с, 11,3-21,0 мПа·с и 4,8-7,2 мПа·с, в соответствии со способом, описанным выше, могут быть использованы в качестве композиций с "НРМС 10000 сП" или "НРМС 15000 сП".
Композиция из двух или более чем двух полимеров одного и того же типа, но с разными ММ, дает лучший контроль разрушения, когда композиция по изобретению используется для изготовления таблетки. При использовании отдельно или в смеси, чем выше ММ используемого ПЭО, тем меньше этого полимера требуется для приготовления композиции по настоящему изобретению.
Конкретная композиция по изобретению зависит от молекулярной массы и молекулярно-массового распределения выбранного желатинирующего полимера, а также от качества каждого из используемых полимеров.
В соответствии с одним аспектом данного изобретения нейтральный желатинирующий полимер представляет собой ПЭО с ММ примерно 4 миллиона или более, ПЭГ с ММ примерно 20000 или более или производное целлюлозы, имеющее кажущуюся вязкость примерно 7500 сП или более (измеренную, как описано выше).
Соотношение нейтрального желатинирующего полимера (например, ПЭО, ПЭГ или НРМС, особенно ПЭО или НРМС, или их смеси друг с другом либо из двух или более чем двух ПЭО или НРМС) и иота-каррагинана предпочтительно находится в диапазоне от 20:80 до 80:20 (в частности, примерно от 40:60 до 60:40, например примерно 50:50).
Основные фармацевтически активные ингредиенты имеют одну или более чем одну основную группу, имеющую рКа предпочтительно от 1 до 12 (например, от 1 до 10 (особенно от, 1 до 7)), и возможно также имеют одну или более чем одну основную группу, имеющую рКа более 10. Таким образом, основный фармацевтически активный ингредиент может иметь одну или более чем одну величину рКа, но по меньшей мере одна составляет предпочтительно от 1 до 12 (например, от 1 до 10 (особенно от 1 до 7)). Примеры основных групп у этих основных фармацевтически активных ингредиентов, имеющие рКа от 1 до 12 (например, от 1 до 10), включают в себя гидроксиамидины, вторичные или третичные амины либо первичные и вторичные амиды.
Подходящие основные фармацевтически активные ингредиенты предпочтительно имеют растворимость в воде от низкой до средней (например, растворимость в воде вплоть до 50 мг/мл (особенно от 0,001 до 20 мг/мл) при 25°С и при рН 7,0) и являются положительно заряженными одним или более чем одним положительным зарядом (в зависимости от количества и рКа основных групп в фармацевтически активном ингредиенте) при низком рН (например, рН от 1 до 6 (особенно рН от 1 до 2)).
Подходящий основный фармацевтически активный ингредиент представляет собой, например, соединение, обладающее активностью в отношении сердечно-сосудистой системы (такое как пептидный или подобный пептиду ингибитор тромбина). Пептидные ингибиторы тромбина имеют молекулярную массу ниже 1000, имеют 1, 2, 3 или 4 пептидные связи и проявляют рН-зависимую растворимость. Они включают в себя пептидные ингибиторы тромбина (и их пролекарства), описанные в общем и более конкретно в обзорной статье Claesson в Blood Coagul. Fibrin. 5, 411 (1994), а также пептидные ингибиторы тромбина, которые описаны в патенте США №4346078, международных заявках на патент WO 97/23499, WO 97/02284, WO 97/46577, WO 98/01422, WO 93/05069, WO 93/11152, WO 95/23609, WO 95/35309, WO 96/25426, WO 94/29336, WO 93/18060 и WO 95/01168 и публикациях европейских патентов №№623596, 648780, 468231, 559046, 641779, 185390, 526877, 542525, 195212, 362002, 364344, 530167, 293881, 686642, 669317 и 601459. Пептидные ингибиторы тромбина (или их пролекарства) включают в себя, в частности, иногатран, мелагатран {НООС-CH2-RCgl-Aze-Pab-H; Глицин, N-[2-[2-[[[[4-(аминоиминометил)фенил]метил]амино]карбонил]-1-азетидинил]-1-циклогексил-2-оксоэтил]-, [2R-[2S]]-)} и Н376/95 {ксимелагатран; EtO2C-CH2-RCgl-Aze-Pab-OH; смотри пример 17 WO 97/23499; Глицин, N-[1-циклогексил-2-[2-[[[[4-[(гидроксиимино)аминометил]фенил]метил]амино]карбонил]-1-азетидинил]-2-оксоэтил]-, этиловый эфир, [S-(R*, S*)]-}.
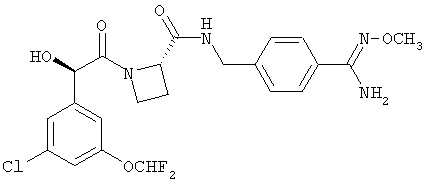
Согласно еще одному аспекту пептидные ингибиторы тромбина (или их пролекарства) включают в себя иногатран, мелагатран, Н376/95, Ph(3-Cl)(5-OCH2CH2F)-(R)CH(OH)C(O)-Aze-Pab(OMe) и Ph(3-Cl)(5-OCHF2)-(R)CH(OH)C(O)-Aze-Pab(OMe).
Согласно еще одному аспекту настоящего изобретения предложена фармацевтическая композиция, как она описана здесь, где основный фармацевтически активный ингредиент, представляет собой
Ph(3-Cl)(5-OCHF2)-(R)CH(OH)C(O)-(S)Aze-Pab(OMe) {Соединение А};
Ph(3-Cl)(5-OCHF2)-(R)(OH)C(O)-(S)Aze-Pab(2,6-диF)(OMe) (Соединение D};
Ph(3-Cl)(5-OCH2CH2F)-(R)CH(OH)C(O)-(S)Aze-Pab(OMe) {Соединение Е};
Ph(3-Cl)(5-OCHF2)-(R)CH(OH)C(O)-(S)Aze-Pab(OH) {Соединение F};
Ph(3-Cl)(5-OCHF2)-(R)CH(OH)C(O)-(S)Aze-Pab(2,6-диF)(OH) {Соединение G};
Ph(3-Cl)(5-OCH2CH2F)-(R)CH(OH)C(O)-(S)Aze-Pab(OH) {Соединение Н}.
Соединение G может быть получено способами, сходными с теми, которые описаны ниже для получения соединений F и Н.
Согласно еще одному аспекту настоящего изобретения предложена фармацевтическая композиция, где основный фармацевтически активный ингредиент представляет собой
1) 4-({3-[7-(3,3-диметил-2-оксобутил)-9-окса-3,7-диазабицикло[3.3.1]нон-3-ил]пропил}амино)бензонитрил (соединение, которое далее называют соединением В),
2) трет-бутил-2-{7-[3-(4-цианоанилино)пропил]-9-окса-3,7-диазабицикло[3.3.1]нон-3-ил}этилкарбамат,
3) трет-бутил-2-{7-[4-(4-цианофенил)бутил]-9-окса-3,7-диазабицикло[3.3.1]нон-3-ил)этилкарбамат либо
4) трет-бутил-2-{7-[(2S)-3-(4-цианофенокси)-2-гидроксипропил]-9-окса-3,7-диазабицикло[3.3.1]нон-3-ил}этилкарбамат (соединение, которое далее называют соединением С),
причем эти соединения описаны в WO 01/28992.
Согласно другому аспекту основный фармацевтически активный ингредиент представляет собой метопролол либо его соль (такую как сукцинат или тартрат).
Композиция по настоящему изобретению может включать в себя добавку, используемую в технологии лекарств, стабилизатор, пластификатор, красящее вещество, смазывающее вещество (такое как стеарилфумарат натрия), связывающее вещество, наполнитель или поверхностно-активное вещество либо другой эксципиент, обычно используемый в фармацевтической композиции.
Согласно одному конкретному аспекту композиция по настоящему изобретению включает в себя смазывающее вещество (такое как стеарилфумарат натрия).
Согласно еще одному аспекту настоящего изобретения молярное отношение иота-каррагинана к основному фармацевтически активному ингредиенту находится в диапазоне от 3:1 до 1:3.
Согласно другому аспекту фармацевтическая композиция по настоящему изобретению содержит 15-80% иота-каррагинана.
Согласно еще одному аспекту фармацевтическая композиция по настоящему изобретению содержит 15-80% одного или более чем одного нейтрального желатинирующего полимера.
Согласно другому аспекту фармацевтическая композиция по настоящему изобретению содержит 1-50% основного фармацевтически активного ингредиента.
В еще одном дополнительном аспекте фармацевтическая композиция по настоящему изобретению содержит 0-10% (в частности, 1-10%) добавки, используемой в технологии лекарств, стабилизатора, пластификатора, красящего вещества, смазывающего вещества, связывающего вещества или наполнителя либо другого эксципиента, обычно используемого в фармацевтических композициях.
Считают, что механизм подавления высвобождения основного фармацевтически активного ингредиента из композиции при кислом рН (в частности, по существу рН-независимого регулируемого высвобождения) представляет собой следующее. При низком рН ожидается, что основный фармацевтически активный ингредиент лекарства обладает относительно высокой растворимостью, поскольку он находится в сильно ионизированном состоянии, и, следовательно, ожидается, что он проявляет быстрый профиль высвобождения из любой нейтральной матрицы. Теоретически предсказывают, что при кислом рН и в присутствии иота-каррагинана существует ионное притяжение между отрицательно заряженным иота-каррагинаном и положительно заряженным лекарственным средством, что замедляет высвобождение лекарственного средства и, таким образом, вносит вклад в более постоянный профиль высвобождения. При более высоком рН, когда фармацевтическое лекарственное средство ионизировано менее сильно или не ионизировано совсем, и, следовательно, как ожидается, демонстрирует медленный профиль высвобождения из любой нейтральной матрицы, теоретически предсказывают, что межионное взаимодействие, предложенное выше, также менее значимо, и профиль высвобождения регулируется преимущественно объединенными профилями набухания, гелеобразования и разрушения нейтрального(ных) желатинирующего(щих) полимера(ов) и анионного полимера, иота-каррагинана, используемого в композиции.
Конечные набухаемость, способность к гелеобразованию и разрушению композиции по настоящему изобретению связаны с такими свойствами, как молекулярная масса и молекулярно-массовое распределение желатинирующего(щих) полимера(ов) и анионного полимера, а также связаны с рН-зависимой скоростью гидролиза анионного полимера. Таким образом, различные скорости высвобождения основного фармацевтически активного ингредиента могут быть получены путем подбора природы (например, молекулярной массы или молекулярно-массового распределения) желатинирующего полимера, количества иота-каррагинана, присутствующего в композиции и/или соотношения желатинирующего полимера и иота-каррагинана.
Композиция по настоящему изобретению может быть представлена в виде твердой лекарственной формы (такой как таблетка, капсула, гранула или порошок, диспергированный в подходящем контейнере, либо в форме композиции в виде множества частиц (таких как покрытые оболочкой гранулы, вводимые в таблетке, капсуле или саше)).
Согласно одному аспекту изобретения предложена таблетка, содержащая 20-500 мг (в частности, 40-60 мг) основного фармацевтически активного ингредиента (такого как Н376/95, или соединение А, В или С).
Когда фармацевтическая композиция по настоящему изобретению представлена в виде таблетки, таблетку предпочтительно приготавливают так, что весь основный фармацевтически активный ингредиент высвобождается в ионизированной или неионизированной форме, в зависимости от рН каждой части желудочно-кишечного тракта, в течение периода примерно 20 часов, например 18-22 часа (альтернативно в течение 20-26 часов).
Согласно еще одному аспекту предложен способ приготовления композиции по настоящему изобретению, при котором смешивают иота-каррагинан, один или более чем один нейтральный желатинирующий полимер и основный фармацевтически активный ингредиент и, возможно, прессуют указанную смесь (предпочтительно в присутствии смазывающего вещества {такого как стеарилфумарат натрия, продаваемый под товарным знаком PRUV™}) с образованием таблетки.
Композиция в форме таблетки может быть приготовлена, например, с использованием методики прямого прессования или влажного гранулирования.
Для методики прямого прессования основный фармацевтически активный ингредиент тщательно смешивают с желатинирующим полимером и иота-каррагинаном и дополнительными эксципиентами, как необходимо. Смазывающее вещество (такое как стеарилфумарат натрия) просеивают и добавляют к смеси иота-каррагинана с последующим дополнительным перемешиванием. Полученную смесь затем прессуют в таблетки.
Для методики влажного гранулирования основный фармацевтически активный ингредиент тщательно смешивают с желатинирующим полимером и иота-каррагинаном. Полученную смесь затем можно увлажнить раствором подходящего связывающего вещества (такого как поливинилпирролидон (PVP), растворенный в подходящем растворителе (таком как этанол или вода)) либо подходящим растворителем (таким как этанол или вода); полученную смесь гранулируют с использованием стандартных или модифицированных процедур гранулирования (таких как гранулирование распылением). После высушивания полученного гранулята (например, в сушильном шкафу при подходящей температуре (такой как примерно 50°С) в течение подходящего периода (такого как 20-24 часа) гранулят перемалывают (например, сухое или влажное перемалывание), смешивают со смазывающим веществом (таким как стеарилфумарат натрия, стеарат магния или тальк) и полученную композицию прессуют в таблетки. Высушенный гранулят можно использовать для наполнения капсул (таких как капсулы, изготовленные из желатина).
Согласно другому аспекту настоящего изобретения предложен способ приготовления композиции, как описано выше.
Соединения, активные в отношении тромбина и их пролекарства могут быть использованы для лечения и/или профилактики тромбоза и гиперкоагуляции в крови и/или тканях животных, включая человека. Известно, что гиперкоагуляция может приводить к тромбоэмболическим заболеваниям. Состояния, связанные с гиперкоагуляцией и тромбоэмболическими заболеваниями, которые могут быть упомянуты, включают в себя наследственную или приобретенную устойчивость к активированному протеину С, такую как мутация фактора V (лейденовская мутация фактора V), и наследственные или приобретенные недостаточности антитромбина III, протеина С, протеина S, гепаринового кофактора II. Другие состояния, про которые известно, что они связаны с гиперкоагуляцией и тромбоэмболическими заболеваниями, включают в себя циркуляцию антифосфолипидных антител (волчаночный антикоагулянт), гомоцистеинемию, тромбоцитопению, индуцированную гепарином, и дефекты фибринолиза, а также синдромы коагуляции (например, синдром диссеминированной внутрисосудистой коагуляции (DIC)) и сосудистое повреждение в целом (например, вследствие оперативного вмешательства).
Согласно еще одному аспекту настоящего изобретения предложена композиция, как она описана выше, для применения в терапии (как лечебной, так и профилактической), например, в качестве лекарства (такого как лекарство для сердечно-сосудистых расстройств, например тромбоэмболии).
Композиция по изобретению полезна в производстве лекарства для применения в терапии.
Согласно еще одному аспекту настоящего изобретения предложен способ лечения сердечно-сосудистого расстройства (например, тромбоэмболии) у теплокровного животного, страдающего от указанного расстройства или имеющего риск заболеть им, при котором животному, нуждающемуся в таком лечении, вводят терапевтически эффективное количество композиции по изобретению.
Некоторые пептидные ингибиторы тромбина либо их пролекарства могут быть получены с помощью мотодик, описанных ниже.
Общие методики
TLC осуществляли на силикагеле. Анализ с помощью хиральной HPLC осуществляли с использованием колонки Chiralcel OD 46 мм × 250 мм с предохранительной колонкой 5 см. Температуру колонки поддерживали при 35°С. Использовали скорость потока 1,0 мл/мин. Использовали ультрафиолетовый детектор Gilson 115 при 228 нм. Подвижная фаза состояла из гексанов, этанола и трифторуксусной кислоты, и соответствующие соотношения приведены для каждого соединения. Обычно продукт растворяли в минимальном количестве этанола и эту смесь разбавляли подвижной фазой.
В получениях ниже LC-MS/MS осуществляли, используя прибор HP-1100, снабженный инжектором CTC-PAL, и колонку 5 Tm, 4×100 мм ThermoQuest, Hypersil BDS-C18. Использовали детектор API-3000 (Sciex) MS. Скорость потока составляла 1,2 мл/мин и подвижная фаза (градиент) состояла из 10-90% ацетонитрила с 90-10% 4 мМ водн. ацетата аммония, причем оба содержали 0,2% муравьиную кислоту. В других случаях масс-спектры низкого разрешения (LRMS) записывали с ипользованием спектрометра Micromass ZQ в ESI поз./нег. режиме ионного переключения (массовый диапазон m/z 100-800), а масс-спектры высокого разрешения (HRMS) записывали с использованием спектрометра Micromass LCT в режиме ES отрицательной ионизации (массовый диапазон m/z 100-1000) с использованием лейцин-энкефалина (C28H37N5O7) в качестве внутреннего массового стандарта.
1Н-ЯМР-спектры записывали с использованием тетраметилсилана в качестве внутреннего стандарта.
Получение Ph(3-Cl)(5-OCHF2)-(R)CH(OH)C(O)-(S)Aze-Pab(OMe) {Соединение A}
(1) 3-Хлор-5-метоксибензальдегид
3,5-Дихлоранизол (74,0 г, 419 ммоль) в THF (200 мл) по каплям добавляли к металлическому магнию (14,2 г, 585 ммоль, предварительно промытому 0,5 н. HCl) в THF (100 мл) при 25°С. После добавления по каплям добавляли 1,2-дибромэтан (3,9 г, 20,8 ммоль). Полученную темно-коричневую смесь нагревали с обратным холодильником в течение 3 часов. Смесь охлаждали до 0°С и одной порцией добавляли N,N-диметилформамид (60 мл). Смесь разделяли с помощью диэтилового эфира (3×400 мл) и 6 н. HCl (500 мл). Объединенные органические экстракты промывали рассолом (300 мл), сушили (Na2SO4), фильтровали и концентрировали в вакууме с получением масла. Посредством флэш-хроматографии (2х) на силикагеле с элюцией смесью Нех:EtOAc (4:1) получили соединение, указанное в подзаголовке (38,9 г, 54%), в виде желтого масла.
1H-ЯМР (300 МГц, CDCl3): δ 9.90 (s, 1H), 7.53 (s, 1Н), 7.38 (s, 1H), 7.15 (s, 1H),3.87(s, 3H).
(2) 3-Хлор-5-гидроксибензальдегид
Раствор 3-хлор-5-метоксибензальдегида (22,8 г, 134 ммоль; см. стадию (1) выше) в CH2Cl2 (250 мл) охлаждали до 0°С. Трибромид бора (15,8 мл, 167 ммоль) по каплям добавляли в течение 15 минут. После перемешивания реакционной смеси в течение 2 часов медленно добавляли Н2О (50 мл). Раствор затем экстрагировали Et2O (2×100 мл). Органические слои объединяли, сушили (Na2SO4), фильтровали и концентрировали в вакууме. Посредством флэш-хроматографии на силикагеле с элюцией смесью Нех:EtOAc (4:1) получили соединение, указанное в подзаголовке (5,2 г, 25%).
1H-ЯМР (300 МГц, CDCl3): δ 9.85 (s, 1H), 7.35 (s, 1Н), 7.20 (s, 1H), 7.10 (s, 1H),3.68 (s, 1H).
(3) 3-Хлор-5-дифторметоксибензальдегид
Раствор 3-хлор-5-гидроксибензальдегида (7,5 г, 48 ммоль; см. стадию (2) выше) в 2-пропаноле (250 мл) и 30% КОН (100 мл) нагревали до температуры дефлегмации. При перемешивании в реакционную смесь барботировали CHClF2 в течение 2 часов. Реакционную смесь охлаждали, подкисляли 1 н. HCl и экстрагировали EtOAc (2×100 мл). Органические экстракты промывали рассолом (100 мл), сушили (Na2SO4), фильтровали и концентрировали в вакууме. Посредством флэш-хроматографии на силикагеле с элюцией смесью Нех:EtOAc (4:1) получили соединение, указанное в подзаголовке (4,6 г, 46%).
1H-ЯМР (300 МГц, CDCl3): δ 9.95 (s, 1H), 7.72 (s, 1H), 7,52 (s, 1H), 7.40 (s, 1 Н), 6.60 (t, JH-F=71,1 Гц, 1H).
(4) Ph(3-Cl)(5-OCHF2)-(R,S)CH(OTMS)CN
Раствор 3-хлор-5-дифторметоксибензальдегида (4,6 г, 22,3 ммоль; см. стадию (3) выше) в СН2Cl2 (200 мл) охлаждали до 0°С. Добавляли Znl2 (1,8 г, 5,6 ммоль) и триметилсилилцианид (2,8 г, 27,9 ммоль) и реакционную смесь оставляли нагреваться до комнатной температуры и перемешивали в течение 15 часов. Смесь частично концентрировали в вакууме, получая соединение, указанное в подзаголовке, в виде жидкости, которую использовали непосредственно на стадии (5) ниже без дополнительной очистки или идентификации.
(5)Ph(3-Cl)(5-OCHF2)-(R,S)CH(OH)C(NH)OEt
Ph(3-Cl)(5-OCHF2)-(R,S)CH(OTMS)CN (6,82 г, предположительно 22,3 ммоль; см. стадию (4) выше) по каплям добавляли к смеси HCl/EtOH (500 мл). Реакционную смесь перемешивали 15 часов, затем частично концентрировали в вакууме, получая соединение, указанное в подзаголовке, в виде жидкости, которую использовали на стадии (6) без дополнительной очистки или идентификации.
(6) Ph(3-Cl)(5-OCHF2)-(R,S)CH(OH)C(O)OEt
Ph(3-Cl)(5-OCHF2)-(R,S)CH(OH)C(NH)OEt (6,24 г, предположительно 22,3 ммоль; см. стадию (5) выше) растворяли в THF (250 мл), добавляли 0,5 М H2SO4 (400 мл) и реакционную смесь перемешивали при 40°С в течение 65 ч, охлаждали и затем частично концентрировали в вакууме для удаления большей части THF. Реакционную смесь затем экстрагировали Et2O (3×100 мл), сушили (Na2SO4), фильтровали и концентрировали в вакууме с получением соединения, указанного в подзаголовке, в виде твердого вещества, которое использовали на стадии (7) без дополнительной очистки или идентификации.
(7) Ph(3-Cl)(5-OCHF2)-(R,S)CH(OH)C(O)OH
Раствор Ph(3-Cl)(5-OCHF2)-(R,S)CH(OH)C(O)OEt (6,25 г, предположительно 22,3 ммоль; см. стадию (6) выше) в 2-пропаноле (175 мл) и 20% КОН (350 мл) перемешивали при комнатной температуре 15 часов. Реакционную смесь затем частично концентрировали в вакууме для удаления большей части 2-пропанола. Оставшуюся смесь подкисляли 1 М H2SO4, экстрагировали Et2O (3×100 мл), сушили (Na2SO4) и концентрировали в вакууме с получением твердого вещества. Посредством флэш-хроматографии на силикагеле с элюцией смесью CHCl3 : МеОН : концентрированный NH4OH (6:3:1) получали аммониевую соль соединения, указанного в подзаголовке. Аммониевую соль затем растворяли в смеси EtOAc (75 мл) и Н2O (75 мл) и подкисляли 2 н. HCl. Органический слой отделяли и промывали рассолом (50 мл), сушили (Na2SO4) и концентрировали в вакууме с получением соединения, указанного в подзаголовке (3,2 г, 57% от стадий (4)-(7)).
1H-ЯМР (300 МГц, CD3OD) δ 7.38 (s, 1Н), 7.22 (s, 1H), 7.15 (s, 1H), 6.89 (t, JH-F=71,1 Гц, 1H), 5.16 (s, 1H).
(8) Ph(3-Cl)(5-OCHF2)-(R)СН(OH)С(O)OH (а) и Ph(3-Cl)(5-OCHF2)-(S)СН(OAc)С(O)OH (б)
Смесь Ph(3-Cl)(5-OCHF2)-(R,S)СН(OH)С(O)OH (3,2 г, 12,7 ммоль; см. стадию (7) выше) и липазы PS "Amano" (˜2,0 г) в винилацетате (125 мл) и МТВЕ (125 мл) нагревали с обратным холодильником в течение 48 часов. Реакционную смесь охлаждали, фильтровали через Celite® и осадок на фильтре промывали EtOAc. Фильтрат концентрировали в вакууме и подвергали флэш-хроматографии на силикагеле, элюируя смесью CHCl3 : МеОН : концентрированный NH4OH (6:3:1), с получением аммониевых солей соединений (а) и (б), указанных в подзаголовке. Соединение (а) в виде соли растворяли в H2O, подкисляли 2 н. HCl и экстрагировали EtOAc. Органический слой промывали рассолом, сушили (Na2SO4), фильтровали и концентрировали в вакууме с получением соединения (а), указанного в подзаголовке (1,2 г, 37%).
Для соединения (а), указанного в подзаголовке
1H-ЯМР (300 МГц, CD3OD): δ 7.38 (s, 1Н), 7.22 (s, 1H), 7.15 (s, 1H), 6.89 (t, JH-F=71,1 Гц, 1Н), 5.17 (s, 1H).
(9) Ph(3-Cl)(5-OCHF2)-(R)СН(OH)С(O)-Aze-Pab(Teoc)
К раствору Ph(3-Cl)(5-OCHF2)-(R)СН(OH)С(O)OH (1,1 г, 4,4 ммоль; см. стадию (8) выше) и H-Aze-Pab(Teoc) (смотри международную патентную заявку WO 00/42059, 2,6 г, 5,7 ммоль) в DMF (50 мл) при 0°С добавляли РуВОР (2,8 г, 5,3 ммоль) и коллидин (1,3 г, 10,6 ммоль). Реакционную смесь перемешивали при 0°С в течение 2 часов, а затем при комнатной температуре в течение дополнительных 15 часов. Реакционную смесь концентрировали в вакууме и подвергали флэш-хроматографии на силикагеле (3х), элюируя сначала смесью CHCl3:EtOH (9:1), затем смесью EtOAc:EtOH (20:1) и, наконец, элюируя смесью СН2Cl2 : СН3ОН (95:5) с получением соединения, указанного в подзаголовке (1,0 г, 37%) в виде белого твердого вещества.
1H-ЯМР (300 МГц, CD3OD, смесь ротамеров): δ 7.79-7.85 (d, J=8,7 Гц, 2Н), 7.15-7.48 (m, 5H), 6.89 и 6.91 (t, JH-F=71,1 Гц, 1H), 5.12 и 5.20 (s, 1H), 4.75-4.85 (m, 1H), 3.97-4.55 (m, 6H), 2.10-2.75 (m, 2H), 1.05-1.15 (m, 2H), 0.09 (s, 9H).
MS(m/z) 611 (M+1)+.
(10) Ph(3-Cl)(5-OCHF2)-(R)СН(OH)С(O)-Aze-Pab(Ome, Teoc)
Ph(3-Cl)(5-OCHF2)-(R)СН(OH)С(O)-Aze-Pab(Teoc) (0,40 г, 0,65 ммоль; см. стадию (9) выше) растворяли в 20 мл ацетонитрила и добавляли 0,50 г (6,0 ммоль) гидрохлорида О-метилгидроксиламина. Смесь нагревали при 70°С в течение 2 часов. Растворитель выпаривали и остаток распределяли между водой и этилацетатом. Водную фазу экстрагировали еще дважды этилацетатом и объединенную органическую фазу промывали водой, рассолом, сушили (Na2SO4), фильтровали и упаривали. Выход 0,41 г (91%).
1H-ЯМР (400 МГц; CDCl3): δ 7.83 (bt, 1H), 7.57 (bs, 1H), 7.47 (d, 2H), 7.30 (d, 2H), 7.20 (m, 1H), 7.14 (m, 1H), 7.01 (m, 1H), 6.53 (t, 1H), 4.89 (s, 1H), 4.87 (m, 1H), 4.47 (m, 2H), 4.4-4.2 (b, 1H), 4.17-4.1 (m, 3Н), 3.95 (s, 3Н), 3.67 (m, 1H), 2.68 (m, 1H), 2.42 (m, 1H), 0.97 (m, 2H), 0.01 (s, 9H).
(11) Ph(3-Cl)(5-OCHF2)-(R)СН(OH)С(O)-(S)Aze-Pab(OMe)
Ph(3-Cl)(5-OCHF2)-(R)СН(OH)С(O)-Aze-Pab(OMe, Teoc) (0,40 г, 0,62 ммоль; см. стадию (10) выше) растворяли в 5 мл TFA и оставляли взаимодействовать в течение 30 минут. TFA выпаривали и остаток распределяли между этилацетатом и NaHCO3 (водн.). Водную фазу экстрагировали еще дважды этилацетатом и объединенную органическую фазу промывали водой, рассолом, сушили (Na2SO4), фильтровали и упаривали. Продукт сушили сублимацией из воды/ацетонитрила. Необходимости в очистке не было. Выход 0,28 г (85%).
1H-ЯМР (600 МГц; CDCl3): δ 7.89 (bt, 1H), 7.57 (d, 2H), 7.28 (d, 2H), 7.18 (m, 1H), 7.13 (m, 1H), 6.99 (m, 1H), 6.51 (t, 1H), 4.88 (s, 1H), 4.87 (m, 1H), 4.80 (bs, 2H), 4.48 (dd, 1H), 4.43 (dd, 1H), 4.10 (m, 1H), 3.89 (s, 3Н), 3.68 (m, 1H), 2.68 (m, 1H), 2.40 (m, 1H).
13С-ЯМР (125 МГц; CDCl3) (углероды карбонила и/или амидина, ротамеры): δ 172.9, 170.8, 152.7, 152,6.
HRMS, вычислено для С22Н23CIF2N4O5 (M-Н)- 495,1242, найдено 495,1247.
Получение соединения D (Ph(3-Cl)(5-OCHF2)-(R)СН(OH)С(O)-(S)Aze-Pab(2,6-диF)(OMe))
(1) 2,6-Дифтор-4-[(метилсульфенил)(метилтио)метил]бензонитрил
(Метилсульфинил)(метилтио)метан (7,26 г, 0,0584 моль) растворяли в 100 мл сухого THF в атмосфере аргона и охлаждали до -78°С. Бутиллитий в гексане (16 мл 1,6 М, 0,0256 моль) по каплям добавляли при перемешивании. Смесь перемешивали в течение 15 минут. Тем временем раствор 3,4,5-трифторбензонитрила (4,0 г, 0,025 ммоль) в 100 мл сухого THF охлаждали до -78°С в атмосфере аргона и упомянутый выше раствор добавляли через канюлю к последнему раствору в течение периода 35 минут. Через 30 минут охлаждающую баню удаляли, и когда реакционная смесь достигала комнатной температуры, ее выливали в 400 мл воды. THF выпаривали и оставшийся водный слой экстрагировали трижды диэтиловым эфиром. Объединенную эфирную фазу промывали водой, сушили (Na2SO4) и упаривали. Выход 2,0 г (30%).
1H-ЯМР (500 МГц; CDCl3): δ 7.4-7.25 (m, 2Н), 5.01 (s, 1H, диастереомер), 4.91 (s, 1H, диастереомер), 2.88 (s, 3Н, диастереомер), 2.52 (s, 3Н, диастереомер), 2.49 (s, 3Н, диастереомер), 2.34 (s, 3Н, диастереомер), 1.72 (широкий, 1H).
(2) 2,6-Дифтор-4-формилбензонитрил
2,6-Дифтор-4-[(метилсульфинил)(метилтио)метил]бензонитрил (2,17 г, 8,32 ммоль; см. стадию (1) выше) растворяли в 90 мл THF и добавляли 3,5 мл концентрированной серной кислоты. Смесь оставляли при комнатной температуре в течение 3 суток и затем выливали в 450 мл воды. Затем трижды проводили экстракцию EtOAc и объединенную эфирную фазу промывали дважды водным бикарбонатом натрия и рассолом, сушили (Na2SO4) и упаривали. Выход 1,36 г (98%). Положение формильной группы определяли с помощью 13С-ЯМР. Сигнал от фторированных углеродов при 162,7 млн-1 проявлял ожидаемую картину сочетания с двумя константами сочетания в порядке 260 и 6,3 Гц соответственно соответствующую ипсо- и мета-сочетаниям от атомов фтора.
1H-ЯМР (400 МГц; CDCl3): δ 10.35 (s, 1H), 7.33 (m, 2Н).
(3) 2,6-Дифтор-4-гидроксиметилбензонитрил
2,6-Дифтор-4-формилбензонитрил (1,36 г, 8,13 ммоль; см. стадию (2) выше) растворяли в 25 мл метанола и охлаждали на ледяной бане. Борогидрид натрия (0,307 г, 8,12 ммоль) добавляли порциями при перемешивании и реакционную смесь оставляли на 65 минут. Растворитель выпаривали и остаток распределяли между диэтиловым эфиром и водным бикарбонатом натрия. Эфирный слой промывали дополнительным количеством водного бикарбоната натрия и рассолом, сушили (Na2SO4) и упаривали. Неочищенный продукт вскоре кристаллизовался и мог быть использован без дополнительной очистки. Выход 1,24 г (90%).
1H-ЯМР (400 МГц; CDCl3): δ 7.24 (m, 2Н), 4.81 (s, 2Н), 2.10 (широкий, 1Н).
(4) 4-Циано-2,6-дифторбензилметансульфонат
К охлажденному на льду раствору 2,6-дифтор-4-гидроксиметилбензонитрила (1,24 г, 7,32 ммоль; см. стадию (3) выше) и метансульфонилхлорида (0,93 г, 8,1 ммоль) в 60 мл метиленхлорида при перемешивании добавляли триэтиламин (0,81 г, 8,1 ммоль). Через 3 часа после нахождения при 0°С смесь промывали дважды 1 М HCl и один раз водой, сушили (Na2SO4) и упаривали. Продукт мог быть использован без дополнительной очистки. Выход 1,61 г (89%).
1H-ЯМР (300 МГц; CDCl3): δ 7.29 (m, 2Н), 5.33 (s, 2H), 3.07 (s, 3Н).
(5) 4-Азидометил-2,6-дифторбензонитрил
Смесь 4-циано-2,6-дифторбензилметансульфоната (1,61 г, 6,51 ммоль; см. стадию (4 выше) и азида натрия (0,72 г, 0,0111 моль) в 10 мл воды и 20 мл DMF перемешивали при комнатной температуре в течение ночи. Полученную смесь затем выливали в 200 мл воды и экстрагировали три раза диэтиловым эфиром. Объединенную эфирную фазу промывали пять раз водой, сушили (Na2SO4) и упаривали. Небольшой образец упаривали для целей ЯМР и продукт кристаллизовался. Остальное осторожно упаривали, но не до полной сухости. Выход (теоретически 1,26 г) предположительно был почти количественным, исходя из ЯМР и аналитической HPLC.
1H-ЯМР (400 МГц; CDCl3): δ 7.29 (m, 2H), 4.46 (s, 2H).
(6) 4-Аминометил-2,6-дифторбензонитрил
Эту реакцию осуществляли в соответствии с методикой, описанной в J. Chem. Res. (M) (1992) 3128. К суспензии 520 мг 10% Pd/C (50% влажность) в 20 мл воды добавляли раствор борогидрида натрия (0,834 г, 0,0221 моль) в 20 мл воды. В результате наблюдалось выделение небольшого количества газа. 4-Азидометил-2,6-дифторбензонитрил (1,26 г, 6,49 ммоль; см. стадию (5) выше) растворяли в 50 мл THF и в течение 15 минут добавляли к водной смеси на ледяной бане. Смесь перемешивали в течение 4 часов, после чего добавляли 20 мл 2 М HCl и смесь фильтровали через Celite. Celite промывали дополнительным количеством воды и объединенную водную фазу промывали EtOAc, а затем подщелачивали 2 М NaOH. Затем три раза проводили экстракцию метиленхлоридом и объединенную органическую фазу промывали водой, сушили (Na2SO4) и упаривали. Выход 0,87 г (80%).
1H-ЯМР (400 МГц; CDCl3): δ 7.20 (m, 2H), 3.96 (s, 2Н), 1.51 (широкий, 2Н).
(7) 2,6-Дифтор-4-трет-бутоксикарбониламинометилбензонитрил
Раствор 4-аминометил-2,6-дифторбензонитрила (0,876 г, 5,21 ммоль; см. стадию (6) выше) растворяли в 50 мл THF и добавляли ди-трет-бутилдикарбонат (1,14 г, 5,22 ммоль) в 10 мл THF. Смесь перемешивали в течение 3,5 часов. THF выпаривали и остаток распределяли между водой и EtOAc. Органический слой три раза промывали 0,5 М HCl и водой, сушили (Na2SO4) и упаривали. Продукт мог быть использован без дополнительной очистки. Выход 1,38 г (99%).
1H-ЯМР (300 МГц; CDCl3): δ 7.21 (m, 2H), 4.95 (широкий, 1Н), 4.43 (широкий, 2H), 1.52 (s, 9H).
(8) Вос-Pab(2,6-диF)(OH)
Смесь 2,6-дифтор-4-трет-бутоксикарбониламинометилбензонитрила (1,38 г, 5,16 ммоль; см. стадию (7) выше), гидрохлорида гидроксиламина (1,08 г, 0,0155 моль) и триэтиламина (1,57 г, 0,0155 моль) в 20 мл этанола перемешивали при комнатной температуре в течение 36 часов. Растворитель выпаривали и остаток распределяли между водой и метиленхлоридом. Органический слой промывали водой, сушили (Na2SO4) и упаривали. Продукт мог быть использован без дополнительной очистки. Выход 1,43 г (92%).
1H-ЯМР (500 МГц; CD3OD): δ 7.14 (m, 2H), 4.97 (широкий, 1Н), 4.84 (широкий, 2H), 4.40 (широкий, 2H), 1.43 (s, 9H).
(9) Вос-Pab(2,6-диF)×НОАс
Эту реакцию осуществляли в соответствии с методикой, описанной Judkins et al. Synth. Comm. (1998) 4351. Вос-Pab(2,6-диF)(ОН) (1,32 г, 4,37 ммоль; см. стадию (8) выше), уксусный ангидрид (0,477 г, 4,68 ммоль) и 442 мг 10% Pd/C (50% влажность) в 100 мл уксусной кислоты гидрировали при давлении 5 атм (506,6 кПа) в течение 3,5 часов. Смесь фильтровали через Celite, промывали этанолом и упаривали. Остаток сушили сублимацией из ацетонитрила, воды и нескольких капель этанола. Продукт, указанный в подзаголовке, мог быть использован без дополнительной очистки. Выход 1,49 г (99%).
1H-ЯМР (400 МГц; CD3OD): δ 7.45 (m, 2H), 4.34 (s, 2H), 1.90 (s, 3Н), 1.40 (s, 9H).
(10) Вос-Pab(2,6-диF)(Теос)
К раствору Вос-Pab(2,6-диF)×НОАс (1,56 г, 5,49 ммоль; см. стадию (9) выше) в 100 мл THF и 1 мл воды добавляли 2-(триметилсилил)этил-пара-нитрофенилкарбонат (1,67 г, 5,89 ммоль). В течение 5 минут по каплям добавляли раствор карбоната калия (1,57 г, 0,0114 моль) в 20 мл воды. Смесь перемешивали в течение ночи. THF выпаривали и остаток распределяли между водой и метиленхлоридом. Водный слой экстрагировали метиленхлоридом и объединенную органическую фазу промывали дважды водным бикарбонатом натрия, сушили (Na2SO4) и упаривали. Посредством флэш-хроматографии на силикагеле с использованием смеси гептан:EtOAc=2:1 получили 1,71 г (73%) чистого соединения.
1H-ЯМР (400 МГц; CDCl3): δ 7.43 (m, 2H), 4.97 (широкий, 1Н), 4.41 (широкий, 2H), 4.24 (m, 2H), 1.41 (s, 9H), 1.11 (m, 2H), 0.06 (s, 9H).
(11) Boc-Aze-Pab(2,6-диF)(Teoc)
Вос-Pab(2,6-диF)(Теос) (1,009 г, 2,35 ммоль; см. стадию (10) выше) растворяли в 50 мл EtOAc, насыщенного HCl (газ). Смесь оставляли на 10 минут, упаривали и растворяли в 18 мл DMF, а затем охлаждали на ледяной бане. Добавляли Boc-Aze-OH (0,450 г, 2,24 ммоль), РуВОР (1,24 г, 2,35 ммоль) и, наконец, диизопропилэтиламин (1,158 г, 8,96 ммоль). Реакционную смесь перемешивали в течение 2 часов и затем выливали в 350 мл воды и экстрагировали три раза EtOAc. Объединенную органическую фазу промывали рассолом, сушили (Na2SO4) и упаривали. Посредством флэш-хроматографии на силикагеле с использованием смеси гептан : EtOAc (1:3) получили 1,097 г (96%) желаемого соединения.
1H-ЯМР (500 МГц; CDCl3): δ 7.46 (m, 2Н), 4.65-4.5 (m, 3H), 4.23 (m, 2H), 3.87 (m, 1H), 3.74 (m, 1Н), 2.45-2.3 (m, 2H), 1.40 (s, 9H), 1.10 (m, 2H), 0.05 (s, 9H).
(12) Ph(3-Cl)(5-OCHF2)-(R)CH(OH)C(O)-Aze-Pab(2,6-диF)(Teoc)
Вос-Aze-Pab(2,6-диF)(Теос) (0,256 г, 0,500 ммоль; см. стадию (11) выше) растворяли в 20 мл EtOAc, насыщенного HCl (газ). Смесь оставляли на 10 минут, упаривали и растворяли в 5 мл DMF. Добавляли Ph(3-Cl)(5-OCHF2)-(R)CH(OH)C(O)OH (0,120 г, 0,475 ммоль; см. получение А (8) выше), РуВОР (0,263 г, 0,498 ммоль) и, наконец, диизопропилэтиламин (0,245 г, 1,89 ммоль). Реакционную смесь перемешивали в течение 2 часов и затем выливали в 350 мл воды и экстрагировали три раза EtOAc. Объединенную органическую фазу промывали рассолом, сушили (Na2SO4) и упаривали. Посредством флэш-хроматографии на силикагеле с использованием EtOAc получили 0,184 г (60%) желаемого соединения, указанного в подзаголовке.
1H-ЯМР (400 МГц; CD3OD, смесь ротамеров): δ 7.55-7.45 (m, 2H), 7.32 (m, 1H, основной ротамер), 7.27 (m, 1H, второстепенный ротамер), 7.2-7.1 (m, 2H), 6.90 (t, 1H, основной ротамер), 6.86 (t, 1H, второстепенный ротамер), 5.15 (s, 1H, основной ротамер), 5.12 (m, 1H, второстепенный ротамер), 5.06 (s, 1H, второстепенный ротамер), 4.72 (m, 1H, основной ротамер), 4.6-4.45 (m, 2H), 4.30 (m, 1H, основной ротамер), 4.24 (m, 2H), 4.13 (m, 1H, основной ротамер), 4.04 (m, 1H, второстепенный ротамер), 3.95 (m, 1H, второстепенный ротамер), 2.62 (m, 1H, второстепенный ротамер), 2.48 (m, 1H, основной ротамер), 2.22 (m, 1H, основной ротамер), 2.10 (m, 1H, второстепенный ротамер), 1.07 (m, 2H), 0.07 (m, 9H).
(13) Ph(3-Cl)(5-OCHF2)-(R)CH(OH)C(O)-Aze-Pab(2,6-диF)(OMe, Teoc)
Смесь Ph(3-Cl)(5-OCHF2)-(R)CH(OH)C(O)-Aze-Pab(2,6-диF)(Teoc) (64 мг, 0,099 ммоль; см. стадию (12) выше) и гидрохлорида О-метилгидроксиламина (50 мг, 0,60 ммоль) в 4 мл ацетонитрила нагревали при 70°С в течение 3 часов. Растворитель выпаривали и остаток распределяли между водой и EtOAc. Водный слой экстрагировали дважды EtOAc и объединенную органическую фазу промывали водой, сушили (Na2SO4) и упаривали. Продукт мог быть использован без дополнительной очистки. Выход 58 мг (87%).
1H-ЯМР (400 МГц; CDCl3): δ 7.90 (bt, 1Н), 7.46 (m, 1H), 7.25-6.95 (m, 5H), 6.51, t, 1H), 4.88 (s, 1H), 4.83 (m, 1H), 4.6-4.5 (m, 2H), 4.4-3.9 (m, 4H), 3.95 (s, 3H), 3.63 (m, 1H), 2.67 (m, 1H), 2.38 (m, 1H), 1.87 (широкий, 1H), 0.98 (m, 2H), 0.01, s, 9Н).
(14) Ph(3-Cl)(5-OCHF2)-(R)CH(OH)C(O)-Aze-Pab(2,6-диF)(OMe)
Ph(3-Cl)(5-OCHF2)-(R)CH(OH)C(O)-Aze-Pab(2,6-диF)(ОМе, Теос) (58 мг, 0,086 ммоль; см. стадию (13) выше) растворяли в 3 мл TFA, охлаждали на ледяной бане и оставляли взаимодействовать в течение 2 часов. TFA выпаривали и остаток растворяли в EtOAc. Органический слой промывали дважды водным карбонатом натрия и водой, сушили (Na2SO4) и упаривали. Остаток сушили сублимацией из воды и ацетонитрила с получением 42 мг (92%) соединения, указанного в заголовке.
1H-ЯМР (300 МГц; CDCl3): δ 7.95 (bt, 1H), 7.2-7.1 (m, 4H), 6.99 (m, 1H), 6.52 (t, 1H), 4.88 (s, 1H), 4.85-4.75 (m, 3H), 4.6-4.45 (m, 2H), 4.29 (широкий, 1H), 4.09 (m, 1H), 3.89 (s, 3H), 3.69 (m, 1H), 2.64 (m, 1H), 2.38 (m, 1H), 1.85 (широкий, 1H).
13С-ЯМР (100 МГц; CDCl3) (углероды карбонила и/или амидина): δ 172.1, 169.8,151.9
APCI-MS: (M+1)=533/535 m/z.
Получение соединения Е (Ph(3-Cl)(5-OCH2CH2F)-(R)CH(OH)C(O)-(S)Aze-Pab(OMe))
(1) (2-Монофторэтил)метансульфонат
К перемешиваемому с помощью магнитной мешалки раствору 2-фторэтанола (5,0 г, 78,0 ммоль) в CH2Cl2 (90 мл) в атмосфере азота при 0°С добавляли триэтиламин (23,7 г, 234 ммоль) и метансульфонилхлорид (10,7 г, 93,7 ммоль). Смесь перемешивали при 0°С в течение 1,5 часов, разбавляли CH2Cl2 (100 мл) и промывали 2 н. HCl (100 мл). Водный слой экстрагировали CH2Cl2 (50 мл) и объединенные органические экстракты промывали рассолом (75 мл), сушили (Na2SO4), фильтровали и концентрировали в вакууме с получением соединения, указанного в подзаголовке (9,7 г, 88%), в виде желтого масла, которое использовали без дополнительной очистки.
1H-ЯМР (300 МГц; CDCl3): δ 4.76 (t, J=4 Гц, 1Н), 4.64 (t, J=4 Гц, 1Н), 4.52 (t, J=4 Гц, 1 Н), 4.43 (t, J=4 Гц, 1 Н), 3.09 (s, 3H).
(2) 3-Хлор-5-монофторэтоксибензальдегид
К раствору 3-хлор-5-гидроксибензальдегида (8,2 г, 52,5 ммоль; см. получение А (2) выше) и карбоната калия (9,4 г, 68,2 ммоль) в DMF (10 мл) в атмосфере азота при комнатной температуре по каплям добавляли раствор (2-монофторэтил)метансульфоната (9,7 г, 68,2 ммоль; см. стадию (1) выше) в DMF (120 мл). Смесь нагревали до 100°С в течение 5 часов и затем перемешивали в течение ночи при комнатной температуре. Реакционную смесь охлаждали до 0°С, выливали в охлажденную на льду 2 н. HCl и экстрагировали EtOAc. Объединенные органические экстракты промывали рассолом, сушили (Na2SO4), фильтровали и концентрировали в вакууме. Коричневое масло подвергали хроматографии на силикагеле, элюируя смесью Нех : EtOAc (4:1), с получением соединения, указанного в подзаголовке (7,6 г, 71%), в виде желтого масла.
1H-ЯМР (300 МГц; CDCl3): δ 9.92 (s, 1Н), 7.48 (s, 1Н), 7.32 (s, 1Н), 7.21 (s, 1Н), 4.87 (t, J=4 Гц, 1Н), 4.71 (t, J=3 Гц, 1Н), 4.33 (t, J=3 Гц, 1Н), 4.24 (t, J=3 Гц,1Н).
(3) Ph(3-Cl)(5-OCH2CH2F)-(R,S)CH(OTMS)CN
К раствору 3-хлор-5-монофторэтоксибензальдегида (7,6 г, 37,5 ммоль; см. стадию (2) выше) и иодида цинка (3,0 г, 9,38 ммоль) в CH2Cl2 (310 мл) по каплям в атмосфере азота при 0°С добавляли триметилсилилцианид (7,4 г, 75,0 ммоль). Смесь перемешивали при 0°С в течение 3 часов и при комнатной температуре в течение ночи. Реакционную смесь разбавляли Н2О (300 мл), органический слой отделяли, сушили (Na2SO4), фильтровали и концентрировали в вакууме с получением соединения, указанного в подзаголовке (10,6 г, 94%), в виде коричневого масла, которое использовали без дополнительной очистки или идентификации.
(4) Ph(3-Cl)(5-OCH2CH2F)-(R,S)CH(ОН)C(O)OH
К Ph(3-Cl)(5-OCH2CH2F)-(R,S)CH(OTMS)CN (10,6 г, 5,8 ммоль; см. стадию (3) выше) добавляли концентрированную соляную кислоту (100 мл) и раствор перемешивали при 100°С в течение 3 часов. После охлаждения до комнатной температуры реакционную смесь далее охлаждали до 0°С, медленно подщелачивали 3 н. NaOH (˜300 мл) и промывали Et2O (3×200 мл). Водный слой подкисляли 2 н. HCl (80 мл) и экстрагировали EtOAc (3×300 мл). Объединенные EtOAc экстракты сушили (Na2SO4), фильтровали и концентрировали в вакууме с получением соединения, указанного в подзаголовке (8,6 г, 98%), в виде бледно-желтого твердого вещества, которое использовали без дополнительной очистки.
Rf=0,28 (90:8:2 CHCl3 : МеОН : концентрированный NH4OH).
1H-ЯМР (300 МГц; CD3OD): δ 7.09 (s, 1H), 7.02 (s, 1H), 6.93 (s, 1H), 5.11 (s, 1H), 4.77-4.81 (m, 1H), 4.62-4.65 (m, 1H), 4.25-4.28 (m, 1H). 4.15-4.18 (m, 1H).
(5) Ph(3-Cl)(5-OCH2CH2F)-(S)CH(OAc)C(O)OH (а) и Ph(3-Cl)(5-OCH2CH2F)-(R)CH(OH)C(O)OH (б)
Раствор Ph(3-Cl)(5-OCH2CH2F)-(R,S)CH(OH)C(O)OH (8,6 г, 34,5 ммоль; см. стадию (4) выше) и липазы PS "Amano" (4,0 г) в винилацетате (250 мл) и МТВЕ (250 мл) нагревали при 70°С в атмосфере азота в течение 3 суток. Реакционную смесь охлаждали до комнатной температуры и фермент удаляли путем фильтрования через Celite®. Осадок на фильтре промывали EtOAc и фильтрат концентрировали в вакууме. Посредством хроматографии на силикагеле с элюцией смесью CHCl3 : МеОН : Et3N (90:8:2) получили соль соединения (а), указанного в подзаголовке, с триэтиламином в виде желтого масла. В дополнение получили соль соединения (б), указанного в подзаголовке, с триэтиламином (4,0 г). Соль соединения (б), указанного в подзаголовке, растворяли в Н2O (250 мл), подкисляли 2 н. HCl и экстрагировали EtOAc (3×200 мл). Объединенные органические экстракты сушили (Na2SO4), фильтровали и концентрировали в вакууме с получением соединения (b), указанного в подзаголовке (2,8 г, 32%), в виде желтого масла.
Данные для соединения (b), указанного в подзаголовке
Rf=0,28 (90:8:2 CHCl3 : МеОН : концентрированный NH4OH)
1H-ЯМР (300 МГц; CD3OD): δ 7.09 (s, 1H), 7.02 (s, 1H), 6.93 (s, 1H), 5.11 (s, 1H), 4.77-4.81 (m, 1H), 4.62-4.65 (m, 1H). 4.25-4.28 (m, 1H), 4.15-4.18 (m, 1H).
(6) Ph(3-Cl)(5-OCH2CH2F)-(R)CH(OH)C(O)-(S)Aze-Pab(OMe)
К раствору Ph(3-Cl)(5-OCH2CH2F)-(R)CH(OH)C(O)OH (818 мг, 3,29 ммоль; см. стадию (5) выше) в DMF (30 мл) в атмосфере азота при 0°С добавляли HAze-Pab(OMe)·2HCl (1,43 г, 4,27 ммоль, см. международную патентную заявку WO 00/42059), РуВОР (1,89 г, 3,68 ммоль) и DIPEA (1,06 г, 8,23 ммоль). Реакционную смесь перемешивали при 0°С в течение 2 часов, а затем при комнатной температуре в течение ночи. Смесь концентрировали в вакууме и остаток два раза подвергали хроматографии на силикагеле, элюируя сначала смесью CHCl3 : EtOH (15:1), а затем EtOAc : EtOH (20:1), с получением соединения, указанного в заголовке (880 мг, 54%).
Rf=0,60 (10:1 CHCl3:EtOH)
1H-ЯМР (300 МГц; CD3OD, сложная смесь ротамеров): δ 7.58-7.60 (d, J=8 Гц, 2Н), 7.34 (d, J=7 Гц, 2Н), 7.05-7.08 (m, 2H), 6.95-6.99 (m, 1H), 5.08-5.13 (m, 1H), 4.77-4.82 (m, 1H), 4.60-4.68 (m, 1H), 3.99-4.51 (m, 7H), 3.82 (s, 3H), 2.10-2.75 (m, 2H).
13C-ЯМР (150 МГц; CD3OD) (углероды карбонила и/или амидина): δ 173.3, 170.8, 152.5.
APCI-MS: (M+1)=493 m/z.
Получение соединения F (Ph(3-Cl)(5-OCHF2)-(R)CH(OH)C(O)-Aze-Pab(OH))
(1) Ph(3-Cl)(5-OCHF2)-(R)СН(OH)С(O)-Aze-Pab(OH,Теос)
Ph(3-Cl)(5-OCHF2)-(R)CH(OH)C(O)-Aze-Pab(Teoc) (0,148 г, 0,24 ммоль; см. получение D, стадию (9) выше) растворяли в 9 мл ацетонитрила и добавляли 0,101 г (1,45 ммоль) гидрохлорида гидроксиламина. Смесь нагревали при 70°С в течение 2,5 часов, фильтровали через Celite® и упаривали. Неочищенный продукт (0,145 г; 75% чистота) использовали непосредственно на следующей стадии без дополнительной очистки.
(2) Ph(3-Cl)(5-OCHF2)-(R)CH(OH)C(O)-Aze-Pab(OH)
Ph(3-Cl)(5-OCHF2)-(R)CH(OH)C(O)-Aze-Pab(OH, Teoc) (0,145 г, 0,23 ммоль; см. стадию (1) выше) растворяли в 0,5 мл CH2Cl2 и 9 мл TFA. Реакции позволяли протекать в течение 60 минут. TFA выпаривали и остаток очищали, используя препаративную HPLC. Интересующие фракции объединяли и сушили сублимацией (2х), получая 72 мг (выход за две стадии 62%) соединения, указанного в заголовке.
MS (m/z) 482 (N-1)-; 484 (М+1)+.
1H-ЯМР (400 МГц; CD3OD): δ 7.58 (d, 2H), 7.33 (m, 3Н), 7.15 (m, 2H), 6.89 (t, 1Н основной ротамер), 6.86 (t, 1H второстепенный ротамер), 5.18 (s, 1H основной ротамер; m, 1H второстепенный ротамер), 5.12 (s, 1H второстепенный ротамер), 4.77 (m, 1H основной ротамер), 4.42 (m, 2H), 4.34 (m, 1H основной ротамер), 4.14 (m, 1H основной ротамер), 4.06 (m, 1H второстепенный ротамер), 3.95 (m, 1H второстепенный ротамер), 2.66 (m, 1H второстепенный ротамер), 2.50 (m, 1H основной ротамер), 2.27 (m, 1H основной ротамер), 2.14 (m, 1H второстепенный ротамер).
13С-ЯМР (100 МГц; CD3OD) (углероды карбонила и/или амидина, ротамеры): δ 172.4, 172.3, 172.0, 171.4 152.3, 152.1
Получение соединения Н (Ph(3-Cl)(5-OCH2CHF2)-(R)CH(OH)C(O)-Aze-Pab(OH))
(1) Ph(3-Cl)(5-OCH2CHF2)-(R)CH(OH)C(O)-Aze-Pab(Z)
Boc-Aze-Pab(Z) (см. международную патентную заявку WO 97/02284; 92 мг, 0,197 ммоль) растворяли в 10 мл EtOAc, насыщенного HCl (газ), и оставляли взаимодействовать в течение 10 минут. Растворитель выпаривали и остаток смешивали с Ph(3-Cl)(5-OCH2CHF2)-(R)CH(OH)C(O)OH (50 мг, 0,188 ммоль), РуВОР (109 мг, 0,209 ммоль) и, наконец, диизопропилэтиламином (96 мг, 0,75 ммоль) в 2 мл DMF. Смесь перемешивали в течение 2 часов, а затем выливали в 50 мл воды и экстрагировали три раза EtOAc. Объединенную органическую фазу промывали водой, сушили (Na2SO4) и упаривали. Неочищенный продукт подвергали флэш-хроматографии на силикагеле с использованием смеси EtOAc : МеОН (9:1). Выход 100 мг (87%).
1H-ЯМР (300 МГц; CD3OD, смесь ротамеров): δ 7.85-7.75 (m, 2H), 7.45-7.25 (m, 7H), 7.11 (m, 1H, основной ротамер), 7.08 (m, 1H, второстепенный ротамер), 7.05-6.9 (m, 2H), 6.13 (bt, 1H), 5.25-5.05 (m, 3Н), 4.77 (m, 1H, частично скрытый сигналом CD3ОН), 4.5-3.9 (m, 7H), 2.64 (m, 1H, второстепенный ротамер), 2.47 (m, 1H, основной ротамер), 2.25 (m, 1H, основной ротамер), 2.13 (m, 1H, второстепенный ротамер).
(2) Ph(3-Cl)(5-OCH2CHF2)-(R)CH(OH)C(O)-Aze-Pab(OH)
Гидрохлорид гидроксиламина (65 мг, 0,94 ммоль) и триэтиламин (0,319 г, 3,16 ммоль) смешивали в 8 мл THF и диспергировали с помощью ультразвука в течение 1 часа при 40°С. Ph(3-Cl)(5-OCH2CHF2)-(R)CH(OH)C(O)-Aze-Pab(Z) (96 мг, 0,156 ммоль; см. стадию (1) выше) добавляли с дополнительными 8 мл THF. Смесь перемешивали при 40°С в течение 4,5 суток. Растворитель выпаривали и неочищенный продукт очищали с помощью препаративной RPLC с использование смеси СН3CN : 0,1 М NH4OAc (40:60). Выход 30 мг (38%). Чистота 99%.
1H-ЯМР (300 МГц; CD3OD, смесь ротамеров): δ 7.6-7.55 (m, 2H), 7.35-7.3 (m, 2H), 7.12 (m, 1H, основной ротамер), 7.09 (m, 1H, второстепенный ротамер), 7.05-6.9 (m, 2H), 6.15 (триплет мультиплетов, 1H), 5,15 (m, 1H, второстепенный ротамер), 5.13 (s, 1H, основной ротамер), 5.08 (s, 1H, второстепенный ротамер), 4.77 (m, 1H, основной ротамер), 4.5-4.2 (m, 5H), 4.08 (m, 1H, основной ротамер), 3.97 (m, 1H, второстепенный ротамер), 2.66 (m, 1H, второстепенный ротамер), 2.50 (m, 1H основной ротамер), 2.27 (m, 1H, основной ротамер), 2.14 (m, 1H, второстепенный ротамер).
13С-ЯМР (100 МГц; CD3OD) (углероды карбонила и/или амидина, смесь ротамеров): δ 172.8, 172.2, 171.4, 159.1, 158.9, 154.2.
APCI-MS: (M+1)=497/499 m/z.
Сокращения
Ac = ацетил
APCI = химическая ионизация при атмосферном давлении (в отношении MS)
API = ионизация при атмосферном давлении (в отношении MS)
водн. = водный
Aze (и (S)-Aze) = (S)-азетидин-2-карбоксилат (если не указано иначе)
Boc = трет-бутилоксикарбонил
br = широкий (в отношении ЯМР)
Cl=химическая ионизация (в отношении MS)
d = день (дни)
d = дублет (в отношении ЯМР)
DCC = дициклогексилкарбодиимид
dd = дублет дублетов (в отношении ЯМР)
DIBAL-H = гидрид диизобутилалюминия
DIPEA = диизопропилэтиламин
DMAP = 4-(N,N-диметиламино)пиридин
DMF = N,N-диметилформамид
DMSO = диметилсульфоксид
DSC = дифференциальная сканирующая калориметрия
DVT = тромбоз глубоких вен
EDC = гидрохлорид 1-(3-диметиламинопропил)-3-этилкарбодиимида
экв. = эквиваленты
ES = электрораспыление
ESI = интерфейс электрораспыления
Et = этил
эфир = диэтиловый эфир
EtOAc = этилацетат
EtOH = этанол
Et2O = диэтиловый эфир
HATU = гексафторфосфат O-(азабензотриазол-1-ил)-N,N,N',N'-тетраметилурония
HBTU = [гексафторфосфат N,N,N',N'-тетраметил-O-(бензотриазол-1-ил)урония]
HCl = соляная кислота, газ хлороводорода или соль гидрохлорид (в зависимости от контекста)
Hex = гексаны
НОАс = уксусная кислота
HPLC = высокоэффективная жидкостная хроматография
LC = жидкостная хроматография
m = мультиплет (в отношении ЯМР)
Me = метил
МеОН = метанол
MS = масс-спектроскопия
МТВЕ = метил-трет-бутиловый эфир
ЯМР = ядерный магнитный резонанс
ОАс = ацетат
Pab = пара-амидинобензиламино
Н-Pab = пара-амидинобензиламин
Pd/C = палладий на углероде
Ph = фенил
РуВОР = гексафторфосфат (бензотриазол-1-илокси)трипирролидинофосфония
q = квартет (в отношении ЯМР)
QF = фторид тетрабутиламмония
rt/RT = комнатная температура
s = синглет (в отношении ЯМР)
t = триплет (в отношении ЯМР)
TBTU = [тетрафторборат N,N,N',N'-тетраметил-O-(бензотриазол-1-ил)урония]
TEA = триэтиламин
Теос = 2-(триметилсилил)этоксикарбонил
TEMPO = свободный радикал 2,2,6,6-тетраметил-1-пиперидинилокси
TFA = трифторуксусная кислота
TGA = термогравиметрический анализ
THF = тетрагидрофуран
TLC = тонкослойная хроматография
УФ = ультрафиолет
Префиксы н-, втор-, изо- и трет- имеют свои обычные значения: нормальный, вторичный, изо- и третичный.
Примеры 1-5 и 8-14 иллюстрируют изобретение. Примеры 6 и 7 представлены только для сравнительных целей и не являются частью настоящего изобретения. В примерах и графических материалах соотношения, приведенные в скобках, относятся к массовым % соотношениям нейтрального желатинирующего полимера к иота-каррагинану и не учитывают основный фармацевтически активный ингредиент или любой другой компонент, который мог бы присутствовать. Описание сопровождается графическими материалами.
ФИГ.1: высвобождение Н376/95 из смесей с варьирующим соотношением композиции иота-каррагинана и ПЭО, 4 М. Таблетки анализировали в течение 2 часов при рН 1, а в течение оставшегося времени при рН 6,8.
ФИГ.2: высвобождение Н376/95 из смесей с соотношением композиции (20:80) ПЭО с различной молекулярной массой и иота-каррагинана. Таблетки анализировали в течение 2 часов при рН 1, а в течение оставшегося времени при рН 6,8.
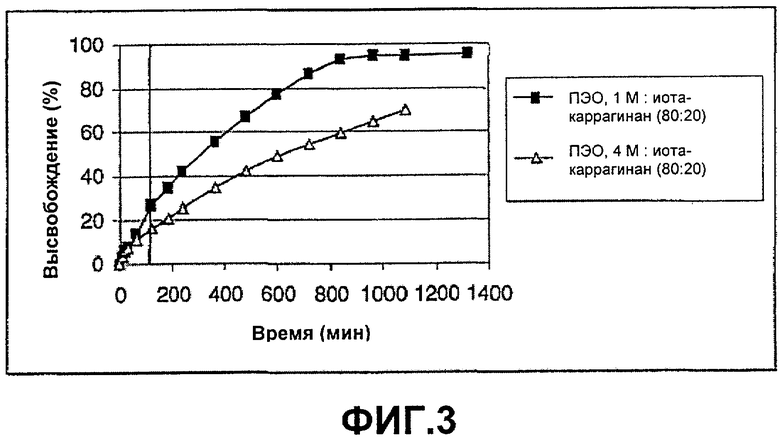
ФИГ.3: высвобождение Н376/95 из смесей с соотношением композиции (80:20) ПЭО с различной молекулярной массой и иота-каррагинана. Таблетки анализировали в течение 2 часов при рН 1, а в течение оставшегося времени при рН 6,8.
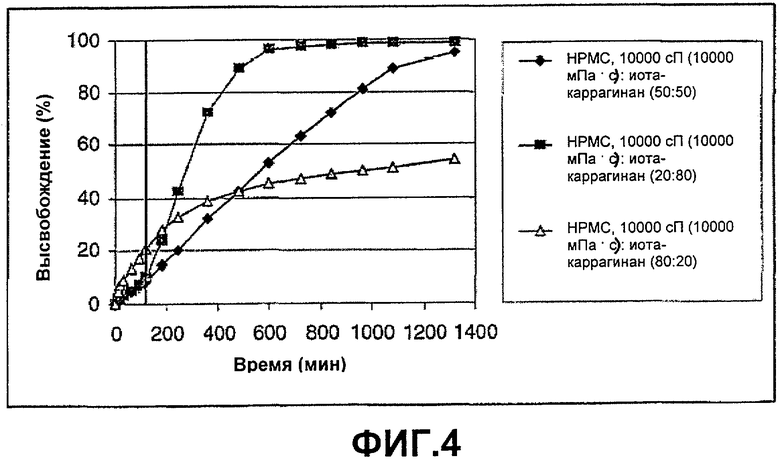
ФИГ.4: высвобождение Н376/95 из смесей с варьирующим соотношением композиции иота-каррагинана и НРМС, 10000 сП (10000 мПа·с). Таблетки анализировали в течение 2 часов при рН 1, а в течение оставшегося времени при рН 6,8.
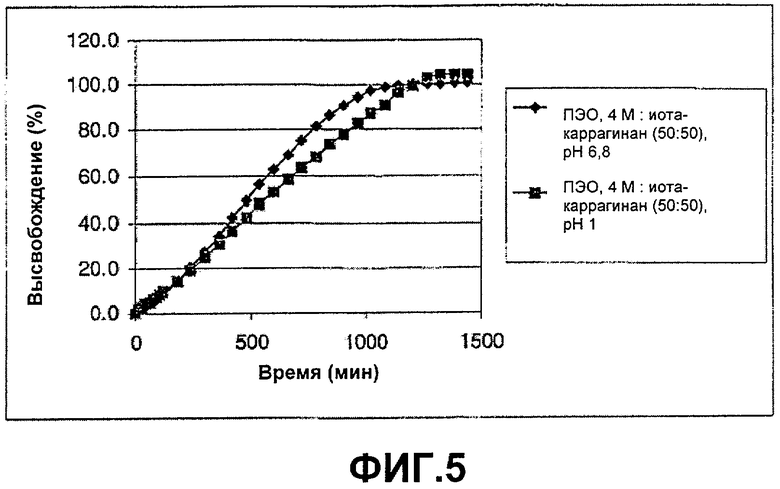
ФИГ.5: высвобождение Н376/95 из смесей с соотношением композиции (50:50) ПЭО, 4 М, и иота-каррагинана. Таблетки анализировали в течение 24 часов в различных искусственных средах.
ФИГ.6: высвобождение Н376/95 из нейтрального желатинирующего полимера ПЭО, 4 М, анализировали в различных искусственных средах.
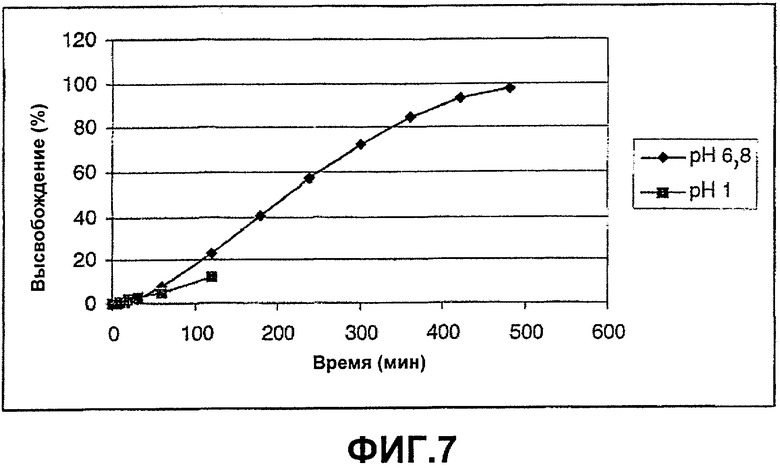
ФИГ.7: высвобождение Н376/95 из анионного полимера иота-каррагинана анализировали в различных искусственных средах.
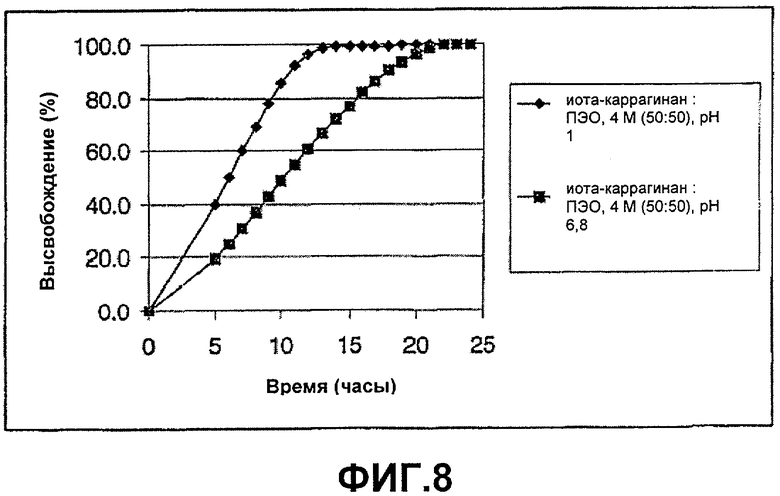
ФИГ.8: высвобождение соединения А из смесей с соотношением композиции (50:50) иота-каррагинана и ПЭО, 4 М, при рН 1 и 6,8. Таблетки анализировали в течение 24 часов в различных искусственных средах.
ФИГ.9: высвобождение соединения А из смесей с соотношением композиции (50:50) иота-каррагинана и НРМС, 10000 сП (10000 мПа·с), при рН 1 и 6,8. Таблетки анализировали в течение 24 часов в различных искусственных средах.
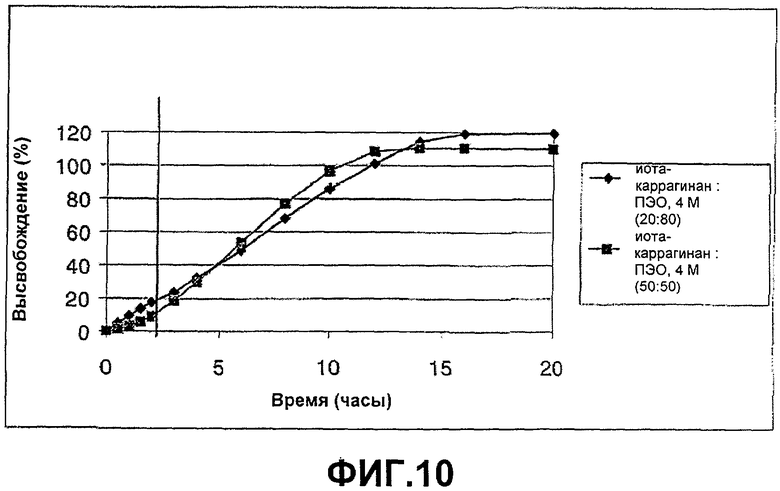
ФИГ.10: высвобождение соединения В из иота-каррагинана, смешанного с нейтральным полимером ПЭО, 4 М, в соотношении (50:50) и (80:20). Таблетки анализировали в течение 2 часов при рН 1, а оставшееся время при рН 6,8.
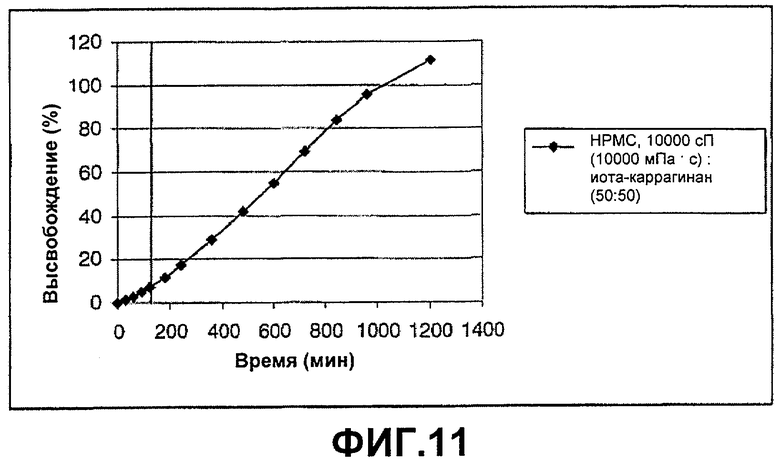
ФИГ.11: высвобождение соединения В из иота-каррагинана, смешанного с нейтральным полимером НРМС, 10000 сП (10000 мПа·с), в соотношении (50:50). Таблетки анализировали в течение 2 часов при рН 1, а оставшееся время при рН 6,8.
Пример 1
Этот пример демонстрирует высвобождение Н376/95 из смесей с варьирующим соотношением композиции ПЭО, 4 М, и иота-каррагинана. Таблетки анализировали в течение 2 часов при рН 1, а в течение оставшегося времени при рН 6,8.
Таблетки изготавливали путем прямого прессования. Активный ингредиент, ПЭО 4 М и иота-каррагинан тщательно смешивали и через 0,7-миллиметровое сито добавляли смазывающее вещество - стеарилфумарат натрия. Осуществляли дополнительное окончательное перемешивание и смесь прессовали с помощью 9-миллиметровое пуансона в однопуансонном таблеточном прессе. Таблетки анализировали в ванне для растворения, UPS II (50 об/мин, 37°С, искусственная среда), содержащей 0,1 М HCl, рН 1, в течение двух часов. После этого таблетки перемещали в ванну для растворения с 0,1 М фосфатным буфером, рН 6,8, и далее анализировали. Результаты анализа представлены на фиг.1. Данная композиция (50:50) показывает по существу рН-независимый профиль высвобождения в диапазоне рН между 1 и 6,8. Дополнительно можно сделать вывод, что при смешивании различных соотношений анионного полимера, иота-каррагинана и нейтрального желатинирующего полимера ПЭО 4 М скорость высвобождения в средах с различным рН может изменяться.
Пример 2
Этот пример демонстрирует высвобождение Н376/95 из смесей с соотношением композиции (20:80) ПЭО с различной молекулярной массой и иота-каррагинана. Таблетки анализировали в течение 2 часов при рН 1, а в течение оставшегося времени при рН 6,8.
Таблетки изготавливали и анализировали в соответствии с примером 1. Результаты анализа представлены на фиг.2 и показывают, что использование более высокой молекулярной массы нейтрального желатинирующего полимера приводит к более медленной скорости высвобождения при нейтральном рН. Скорость высвобождения в области низкого рН не затрагивается, поскольку в композицию включено достаточное количество анионного полимера.
Пример 3
Этот пример демонстрирует высвобождение Н376/95 из смесей с соотношением композиции (80:20) ПЭО с различной молекулярной массой и иота-каррагинана. Таблетки анализировали в течение 2 часов при рН 1, а оставшееся время при рН 6,8.
Таблетки приготавливали и анализировали в соответствии с примером 1. На фиг.3 показано, как использование желатинирующего полимера с более высокой молекулярной массой может уменьшить скорость высвобождения при нейтральном рН. В то же время эффект замедления высвобождения при рН 1 менее четкий, поскольку используют меньшее количество иота-каррагинана по сравнению с примерами, показанными на фиг.2.
Пример 4
Этот пример демонстрирует высвобождение Н376/95 из смесей с варьирующим соотношением композиции иота-каррагинана и НРМС, 10000 сП (10000 мПа·с). Таблетки анализировали в течение 2 часов при рН 1, а в течение оставшегося времени при рН 6,8.
Таблетки изготавливали и анализировали в соответствии с примером 1. Результаты анализа в различных средах растворения представлены на фиг.4. Данная композиция (50:50) вновь показала по существу рН-независимый профиль высвобождения в диапазоне рН между 1 и 6,8. Можно сделать вывод, что скорость высвобождения вновь может быть изменена путем смешивания различных соотношений различных других нейтральных желатинирующих полимеров, в данном случае НРМС, 10000 сП (10000 мПа·с), с анионным полимером, иота-каррагинаном.
Пример 5
Этот пример демонстрирует высвобождение Н376/95 из смеси с соотношением композиции (50:50) ПЭО 4 М и иота-каррагинана. Таблетки анализировали в течение 24 часов в различных средах.
Таблетки изготавливали путем прямого прессования в соответствии с примером 1. Анализы осуществляли в ваннах для растворения (прибор 2 USP (Фармакопея США) с использованием таблеток, помещенных в корзинку1 по ходу струи пара), причем три таблетки анализировали в течение 24 часов в каждой среде, 0,1 М HCl и 0,1 М фосфатном буфере, рН 6,8, с использованием 5% этанола (EtOH), добавленного для улучшения растворимости лекарственного средства. Результаты, представленные на фиг.5, ясно показывают, что таблетка с рН-независимым профилем высвобождения может быть приготовлена при использовании композиции с равными частями ПЭО, 4 М, и иота-каррагинана. [1 Изготовленная по заказу четырехугольная корзинка из проволочной сетки, припаянная одной из ее верхних узких сторон к концу стального стержня. Стержень введен через крышку сосуда для растворения и зафиксирован посредством двух тефлоновых гаек, на расстоянии 3,2 см от центра сосуда. Нижний край дна корзинки отрегулирован так, чтобы быть зафиксированным на 1 см выше лопасти. Корзинку с тестируемой таблеткой, находящейся на дне корзинки, направляют по ходу струи пара].
Пример 6
Этот пример демонстрирует высвобождение Н376/95 из нейтрального желатинирующего полимера ПЭО, 4 М, при отсутствии иота-каррагинана; анализ проводили в различных искусственных средах.
Таблетки изготавливали путем прямого прессования в соответствии с примером 1. Анализы осуществляли отдельно в различных ваннах для растворения. Таблетки в сосудах, содержащих 0,1 М HCl, анализировали в течение 2 часов. При использовании 0,1 М фосфатного буфера, рН 6,8, в качестве среды растворения таблетки анализировали в течение 20 часов. Результаты, представленные на фиг.6, показывают, что скорость высвобождения при рН 1 значительно выше, чем высвобождение при рН 6,8, указывая на то, что использования одного нейтрального полимера недостаточно для получения рН-независимого профиля высвобождения для основного лекарственного средства, обладающего рН-зависимой растворимостью.
Пример 7
Этот пример демонстрирует высвобождение Н376/95 из анионного полимера иота-каррагинана при отсутствии нейтрального желатинирующего полимера; анализ проводили в различных искусственных средах.
Таблетки изготавливали путем прямого прессования в соответствии с примером 1. Анализы осуществляли отдельно в различных ваннах для растворения аналогично примеру 6. На фиг.7 показано, что скорость высвобождения при рН 1 замедлена по сравнению с высвобождением при рН 6,8. Этот эффект не показан при использовании любого другого гомополимера, который авторы изобретения протестировали в качестве полимера матрицы.
Пример 8
Этот пример демонстрирует высвобождение соединения А из смеси с соотношением композиции (50:50) ПЭО, 4 М, и иота-каррагинана. Таблетки анализировали в течение 24 часов в различных средах.
Активный ингредиент вручную смешивали с полимерами и смазывающим веществом. Смесь непосредственно прессовали в таблетки.
Пример 9
Этот пример демонстрирует высвобождение соединения А из смеси с соотношением композиции (50:50) НРМС, 10000 сП (10000 мПа·с), и иота-каррагинана. Таблетки анализировали в течение 24 часов в различных средах.
Активный ингредиент вручную смешивали с полимерами и смазывающим веществом. Смесь непосредственно прессовали в таблетки.
Оценка кумулятивного высвобождения соединения А из таблеток по примерам 8 и 9
Две отдельные таблетки тестировали на высвобождение лекарственного средства в 900 мл среды с использованием прибора для растворения 2 USP (лопасть + корзинка1) при 50 об/мин и 37°С. Используемые среды растворения представляли собой 0,1 М соляную кислоту (рН 1) и 0,1 М натрий-фосфатный буфер (рН 6,8) с 5% этанолом, добавленным для улучшения растворимости лекарственного средства. Было подтверждено, что добавление этанола незначительно влияет на скорость высвобождения этих композиций. Поточный количественный анализ осуществляли с использованием системы волоконной оптики С Technologies с 235 нм в качестве аналитической длины волны, когда 0,1 М HCl использовали в качестве среды растворения, и с 250 нм в качестве аналитической длины волны, когда модифицированный фосфатный буфер рН 6,8 использовали в качестве среды растворения. 350 нм использовали в качестве эталонной длины волны с обеими средами. [1 Изготовленная по заказу четырехугольная корзинка из проволочной сетки, припаянная одной из ее верхних узких сторон к концу стального стержня. Стержень введен через крышку сосуда для растворения и зафиксирован посредством двух тефлоновых гаек, на расстоянии 3,2 см от центра сосуда. Нижний край дна корзинки отрегулирован так, чтобы быть зафиксированным на 1 см выше лопасти. Корзинку с тестируемой таблеткой, находящейся на дне корзинки, направляют по ходу струи пара].
Пример 10
Этот пример демонстрирует высвобождение соединения В из смеси с соотношением композиции (50:50) ПЭО, 4 М, и иота-каррагинана.
Таблетки приготавливали в соответствии с примером 9. Данные по высвобождению показаны на фиг.10.
Пример 11
Этот пример демонстрирует высвобождение соединения В из смеси с соотношением композиции (80:20) ПЭО, 4 М, и иота-каррагинана.
Таблетки приготавливали в соответствии с примером 9. Данные по высвобождению показаны на фиг.10.
Пример 12
Этот пример демонстрирует высвобождение соединения В из смеси с соотношением композиции (50:50) НРМС, 10000 сП (10000 мПа·с), и иота-каррагинана.
Таблетки приготавливали в соответствии с примером 9. Данные по высвобождению показаны на фиг.11.
Пример 13
Прямое прессование соединения С с НРМС, 10000 сП (10000 мПа·с)
Активное вещество и эксципиенты смешивали в чане (beeting vat). В смесь добавляли смазывающее вещество - стеарилфумарат натрия - и прессовали в таблетки с использованием эксцентриковой машины.
Прямое прессование соединения С с НРМС, 10000 сП (10000 мПа·с), и иота-каррагинаном, соотношение 50:50
Активное вещество и эксципиенты смешивали в чане (beeting vat). В смесь добавляли смазывающее вещество - стеарилфумарат натрия - и прессовали в таблетки с использованием эксцентриковой машины.
Скорости высвобождения определяли следующим образом. Три отдельные таблетки тестировали на высвобождение лекарственного средства в 900 мл среды с использованием прибора для растворения 2 USP (лопасть + корзинка1) при 50 об/мин и 37°С. Используемые среды растворения представляли собой 0,1 М соляную кислоту (рН 1) и 0,1 М натрий-фосфатный буфер (рН 6,8). Поточный количественный анализ осуществляли с использованием системы волоконной оптики С Technologies с 220 нм в качестве аналитической длины волны, когда 0,1 М HCl использовали в качестве среды растворения, и с 260 нм в качестве аналитической длины волны, когда фосфатный буфер рН 6,8 использовали в качестве среды растворения. 350 нм использовали в качестве эталонной длины волны с обеими средами. В течение первых двух часов анализа величину высвобождения измеряли каждые 15 минут, а затем каждый час в течение оставшегося времени анализа. [1 Изготовленная по заказу четырехугольная корзинка из проволочной сетки, припаянная одной из ее верхних узких сторон к концу стального стержня. Стержень введен через крышку сосуда для растворения и зафиксирован посредством двух тефлоновых гаек на расстоянии 3,2 см от центра сосуда. Нижний край дна корзинки отрегулирован так, чтобы быть зафиксированным на 1 см выше лопасти. Корзинку с тестируемой таблеткой, находящейся на дне корзинки, направляют по ходу струи пара].
Пример 14
Эту композицию можно приготовить, как описано в примере 13.




Фармацевтическая композиция для перорального введения содержит иота-каррагинан, один или более чем один нейтральный желатинирующий полимер и основный фармацевтически активный ингредиент. Нейтральный желатинирущий полимер представляет собой полиэтиленоксид, полиэтиленликоль, гидроксипропилметилцеллюлозу или смесь гидроксипропилметилцеллюлозы и полиэтиленоксида. Основный фармацевтически активный ингредиент предпочтительно представляет собой метопролол. Композиция по изобретению подавляет высвобождение основного фармацевтически активного ингредиента при кислом рН и создает независимый от рН профиль высвобождения. 2 н. и 8 з.п. ф-лы, 11 ил.
4-({3-[7-(3,3-диметил-2-оксобутил)-9-окса-3,7-диазабицикло[3.3.1]нон-3-ил]пропил}амино)бензонитрил,
трет-бутил-2-{7-[3-(4-цианоанилино)пропил]-9-окса-3,7-диазабицикло-[3.3.1]нон-3-ил} этилкарбамат,
трет-бутил-2-{7-[4-(4-цианофенил)бутил]-9-окса-3,7-диазабицикло-[3.3.1]нон-3-ил)этилкарбамат либо
трет-бутил-2-{7-[(2S)-3-(4-цианофенокси)-2-гидроксипропил]-9-окса-3,7-диазабицикло[3.3.1]нон-3-ил}этилкарбамат.
| Bonferoni M.C | |||
| et al: «On the employment of carrageenan in matrix system | |||
| II-Carrageenan and hydroxypropylmethylcellulose mixtures», Journal of Controlled Release, Vol.30, 1994, p.175-182 | |||
| US 4792452 A, 20.12.1988 | |||
| Проникающая жидкость для цветной дефектоскопии | 1975 |
|
SU539059A1 |
| WO 9626717 A1, 06.09.1996 | |||
| WO 9929305 А1, 17.06.1996 | |||
| WO 9939698 A1, 12.08.1999 | |||
| WO 9921586 А2, 06.05.1999 | |||
| Кирпичеделательная машина | 1929 |
|
SU13710A1 |

