Настоящее изобретение относится к химической промышленности, а именно - к способу получения гидроксиламинсульфата (ГАС), используемого в качестве основного реагента в производстве капролактама.
Известен способ получения ГАС, включающий приготовление реакционной смеси аммиака, кислорода и водяного пара, каталитическое парофазное окисление аммиака, смешение полученного нитрозного газа с водородом, стабилизацию нитрозного газа гидрированием избыточного кислорода в присутствии Ag-Mn катализатора, его двухстадийное концентрирование конденсацией водяных паров, смешение концентрированного монооксида азота с водородом и синтез ГАС в среде разбавленной серной кислоты. Конденсат второй стадии концентрирования нитрозного газа гидрируют водородом в присутствии платинового катализатора в жидкой фазе. Образующийся при этом чистый оксид азота (II) используют для синтеза ГАС, а продукт гидрирования с массовой долей азотной кислоты до 0,45 мас.% вместе с конденсатом первой ступени используют для разбавления концентрированной серной кислоты, обеспечивая тем самым безотходность технологии [заявка ВВП №0945401, МПК6 С 07 В 21/14, 1999 г.]. Преимуществом данного способа является получение концентрированного оксида азота (II) и утилизация отходов производства в виде разбавленных растворов азотной кислоты. Однако недостатком данного способа является то, что при стабилизации состава нитрозного газа гидрированием избыточного кислорода на Ag-Mn катализаторе безвозвратно теряется оксид азота (II), и образуются азот и оксид азота (I), которые снижают эффективность работы стадии синтеза ГАС.
Известен также способ получения ГАС, включающий приготовление реакционной смеси аммиака, кислорода и водяного пара, каталитическое парокислородное окисление аммиака, смешение полученного нитрозного газа с водородом, стабилизацию нитрозного газа путем гидрирования на Ag-Mn катализаторе избыточного кислорода на 70-75%, его двухстадийное концентрирование конденсацией паров воды, смешение концентрированного оксида азота (II) с водородом и синтез ГАС в среде разбавленной серной кислоты. В данном способе получения ГАС на стадии стабилизации нитрозного газа избыточный кислород гидрируют на 70-75% вместо обычных 90%. В результате этого на второй стадии концентрирования нитрозного газа образуется конденсат, содержащий в 1,5-2,5 раза больше азотной кислоты по сравнению с предыдущим способом. При его жидкофазном гидрировании образуется больше чистого оксида азота (II). Кроме того, при гидрировании избыточного кислорода на 70-75% в процессе стабилизации нитрозного газа оксид азота (II) практически не теряется и не образуются дополнительно инертные примеси, а объемная доля оксида азота (II) в концентрированном нитрозном газе на 2,3-2,5% больше, чем в предыдущем способе [Патент Украины №50681 А, МПК7 С 01 В 21/14, 2002 г.].
Общим недостатком обоих вышеописанных способов является сложность схемы в целом, предусматривающей как стадию стабилизации состава нитрозного газа гидрированием части избыточного кислорода, так и стадии жидкофазного гидрирования конденсата второй ступени концентрирования нитрозного газа. Кроме того, схема отличается жесткой взаимосвязью между стадиями окисления аммиака и синтеза ГАС, обусловленной необходимостью сжатия оксида азота (II), что усложняет управление процессом.
Наиболее близким решением поставленной технической задачи является способ получения ГАС по патенту Украины №14329, МПК7 C 01 В 21/14, в соответствии с которым процесс проводят при избыточном давлении путем смешения в реакционной зоне нитрозного газа, водорода, смеси серной кислоты и воды и катализатора "платина на электрографите". Водный раствор серной кислоты вводят одним потоком в среднюю часть реакционной зоны. Перемешивание в реакционной зоне осуществляют расположенной в центральной зоне мешалкой без дополнительной турбулизации смеси в центральной части реакционной зоны. Процесс проводят в нескольких реакционных зонах (каскаде из 5-6 реакторов смешения). Контроль за ходом процесса обычно осуществляют по остаточному содержанию NO в хвостовых газах. Увеличение остаточного содержания NO в хвостовых газах свидетельствует о частичной дезактивации катализатора за счет отравления его активных центров примесями, присутствующими в реакционной системе, прежде всего соединениями железа. Частично дезактивированный катализатор выводят из системы и регенерируют с использованием известных приемов. В среднем рабочий "пробег" катализатора до стадии его регенерации составляет 14-21 суток (2-3 недели).
Недостатком прототипа является малый межрегенерационный период работы катализатора.
Известно, что важную роль в процессе получения ГАС играет содержание катализатора в реакторах, его активность и селективность. Поддержание массовой концентрации катализатора в пределах 30-50 г/дм3, а также необходимой его активности и селективности, обеспечивает высокую скорость реакции и стабильность ведения процесса. Для поддержания на необходимом уровне активности и селективности катализатора необходимо точное соблюдение оптимального соотношения водород оксид азота (II) в синтез-газе, регулирование степени отравления катализатора при регенерации в сочетании с периодичностью его частичной замены в реакторах. При регенерации катализатора платину частично отравляют для снижения скоростей протекания побочных реакций, прежде всего образования аммиака и сульфата аммония. Регенерированный катализатор обеспечивает селективность по целевому процессу на уровне 90-95%. Селективность процесса постепенно снижается за счет накопления в реакционной зоне соединений железа и соединений других элементов семейства железа. Эти соединения, с одной стороны, необратимо отравляют активные центры катализатора, а с другой стороны, катализируют распад целевого продукта реакции - гидроксиламина. Поэтому поиск путей вывода каталитических ядов - соединений железа и элементов его семейства представляет актуальную техническую задачу, решение которой позволит интенсифицировать процесс и увеличить межрегенерационный период работы катализатора.
Целью настоящего изобретения является интенсификация процесса и увеличение межрегенерационного периода работы катализатора.
Согласно изобретению поставленная цель достигается способом получения ГАС при избыточном давлении путем смешения в реакционном объеме синтез-газа, состоящего из оксида азота (II) и водорода, смеси серной кислоты и воды и катализатора, представляющего собой платину на электродном графите («платина на электрографите»), с последующей регенерацией катализатора. Процесс проводят в присутствии 2-30%-ного водного раствора комплексообразователя состава:
Гидроксиэтилиден-1,1-дифосфоновая кислота (I) формулы
СН3-С(ОН)=[Р(O)(ОН)2]2,
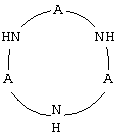
Циклический Р-комплекс (II) общей формулы

где А есть = Р(O)СН3ОН, при мольном отношении I:II, равном 1:1, при массовом отношении водного раствора комплексообразователя и железа, содержащегося в реакционной системе, равном 100-500 ppm на 1 ppm Fe.
Проведение реакции привело к существенной интенсификации процесса и значительному увеличению межрегенерационного периода работы катализатора.
Существенными отличительными признаками данного способа является проведение процесса в присутствии комплексообразователя состава:
Гидроксиэтилиден-1,1-дифосфоновая кислота (I) формулы CH3-С(ОН)=[P(O)(ОН)2]2,
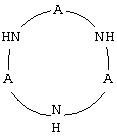
Циклический Р-комплекс (II) общей формулы
где А есть = P(O)CH3ОН, при мольном отношении I:II, равном 1:1. Используют 2-30%-ный водный раствор смеси I и II. Количество водного раствора комплексообразователя составляет 100-500 ppm на 1 ppm Fe, содержащегося в реакционной системе.
Использование этого комплексообразователя приводит к значительному снижению содержания железа и других элементов его семейства в образцах катализатора. Этот факт был подтвержден специальным анализом катализатора с применением метода масс-спектрометрии с ионизацией в индуктивно связанной плазме. Анализировали 4 образца катализатора "платина на электрографите":
Образец 1: до регенерации, каскад А.
Образец 2: после регенерации по стандартной методике, каскад А.
Образец 3: образец 1, отмытый в течение 24 часов заявляемым комплексообразователем.
Образец 4: образец 2, отмытый в течение 24 часов заявляемым комплексообразователем.
Анализ содержания элементов в образцах катализатора проводили с использованием метода масс-спектрометрии для элементного и изотопного анализа с ионизацией в индуктивно связанной плазме (ИСПМС) на приборе "VG PLASMA QUAD PQ 2-TURBO". Метод ИСПМС является эффективным аналитическим методом и позволяет определять концентрации элементов и изотопов, содержащихся в растворе, на уровне 10-9 г/мл. ИСПМС соединяет в себе достоинства высокочастотной индуктивно связанной плазмы, как источника ионов, и квадрупольного масс-анализатора, производящего разделение по массам и детектирование ионов. Образцы в виде мелкодисперсного аэрозоля вводятся в центральный канал плазменного факела. В плазме происходит их эффективное испарение, атомизация и ионизация. Степень ионизации элементов, имеющих потенциал ионизации ниже 10 эВ (а они составляют большинство в периодической таблице), приближается к 100%. Образующиеся ионы экстрагируются из плазмы через отверстие пробоотборника и, увлекаемые газодинамическим потоком, попадают в вакуумную камеру, где, после фокусировки ионно-оптической системой и разделения по массам квадрупольным масс-анализатором, детектируются вторично-электронным умножителем (ВЭУ). Импульсы ВЭУ после усиления поступают на вход многоканального анализатора (МКА) и накапливаются в течение анализа. Полученный массив данных обрабатывается микрокомпьютером.
Пробоподготовка: образцы катализатора обрабатывали избытком смеси азотной и соляной кислот (обе - категории ОСЧ) в мольном соотношении 2:1. Растворение проводили с помощью системы микроволнового разложения MSD-2000.
Элементный анализ полученных растворов проводили на вышеуказанном приборе при следующих условиях: мощность разряда 1,3 кВт, расход транспортирующего газа (аргона) - 0,89 л/мин, расход плазмообразующего газа 12 л/мин. В качестве внутреннего стандарта в растворы вводили индий в концентрации 25 мкг/л. Калибровку прибора проводили по стандартным растворам с известным содержанием определяемых элементов.
Результаты анализа приведены в таблице 1.
Результаты анализа образцов катализатора "платина на электрографите" методом ИСПМС. Содержание элементов - в ppm. Среднее из трех параллельных испытаний.
Результаты анализа свидетельствуют об эффективном связывании заявляемым комплексообразователем соединений железа и элементов его семейства. Ниже приведены примеры осуществления процесса по прототипу и по заявляемому способу.
Пример 1. Процесс проводят в условиях прототипа. Смесь оксида азота (II) и водорода (т.н. синтез-газ) через распределительное устройство подают в каждый из реакторов каскада. Синтез ГАС проводят при интенсивном перемешивании жалюзийной мешалкой в среде разбавленной серной кислоты, которую вводят в среднюю часть реакционной зоны. Процесс синтеза ГАС ведут при избыточном давлении 0,3-0,35 МПа. Верхняя зона реактора снабжена жалюзийным пеногасителем для ликвидации образования ПГС. Отходящие (хвостовые) газы стадии синтеза ГАС направляют на сжигание. Продукты синтеза направляют в продуктовую секцию. Результаты ведения процесса приведены в табл.1.
Примеры 2-6. Процесс проводят в условиях заявляемого способа. Смесь оксида азота (II) и водорода (т.н. синтез-газ) через распределительное устройство подают в каждый из реакторов каскада. Синтез ГАС проводят при интенсивном перемешивании жалюзийной мешалкой в среде разбавленной серной кислоты, которую вводят в среднюю часть реакционной зоны. Процесс синтеза ГАС ведут при избыточном давлении 0,3-0,35 МПа. Верхняя зона реактора снабжена жалюзийным пеногасителем для ликвидации образования ПГС. Отходящие (хвостовые) газы стадии синтеза ГАС направляют на сжигание. Продукты синтеза направляют в продуктовую секцию.
Процесс синтеза ГАС проводят в присутствии комплексообразователя состава:
Гидроксиэтилиден-1,1-дифосфоновая кислота (I) формулы CH3-С(ОН)=[P(O)(ОН)2]2,
Циклический Р-комплекс (II) общей формулы
где А есть = P(O)СН3ОН, при мольном отношении I:II, равном 1:1.
Используют 2-30%-ный водный раствор смеси I и II. Количество водного раствора комплексообразователя составляет 100-500 ppm на 1 ppm Fe, содержащегося в реакционной системе.
Результаты ведения процесса приведены в табл.2.
Как видно из приведенных в табл.2 данных, организация процесса по заявленному способу позволяет более чем в 3 раза снизить содержание NO в отходящих газах, что свидетельствует о более глубоком протекании процесса синтеза ГАС по сравнению с прототипом. По сравнению с прототипом также увеличивается выход основного продукта реакции - ГАС. Значительно возрастает межрегенерационный период работы катализатора. Эти положительные результаты связаны с эффективным связыванием комплексообразователя каталитических ядов, прежде всего соединений железа и соединений других элементов его семейства. Кроме того, поскольку эти же соединения являются катализаторами распада целевого продукта - гидроксиламина, использование комплексообразователя приводит к увеличению выхода целевого продукта.
Результаты проведения синтеза ГАС
Гидроксиэтилиден-1,1-дифосфоновая кислота (I) формулы CH3 -С(ОН)=[P(O)(ОН)2]2,
Циклический Р-комплекс (II) общей формулы
где А есть = P(O)CH3ОН, при мольном отношении I:II, равном 1:1, при массовом отношении водного раствора комплексообразователя и железа, содержащегося в реакционной системе, равном 100-500 ppm на 1 ppm Fe.
| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБ ПОЛУЧЕНИЯ ГИДРОКСИЛАМИНСУЛЬФАТА | 2018 |
|
RU2690931C1 |
| СПОСОБ ПОЛУЧЕНИЯ ГИДРОКСИЛАМИНСУЛЬФАТА | 2005 |
|
RU2287482C1 |
| СПОСОБ УПРАВЛЕНИЯ ПРОЦЕССОМ ПОЛУЧЕНИЯ ГИДРОКСИЛАМИНСУЛЬФАТА | 2006 |
|
RU2305657C1 |
| СПОСОБ ПОЛУЧЕНИЯ ГИДРОКСИЛАМИНСУЛЬФАТА | 2019 |
|
RU2717515C1 |
| СПОСОБ ПОЛУЧЕНИЯ ГИДРОКСИЛАМИНСУЛЬФАТА | 2003 |
|
RU2257339C1 |
| СПОСОБ ПОЛУЧЕНИЯ ГИДРОКСИЛАМИНСУЛЬФАТА | 2003 |
|
RU2257340C1 |
| СПОСОБ ПОЛУЧЕНИЯ ГИДРОКСИЛАМИНСУЛЬФАТА | 2002 |
|
RU2241662C2 |
| СПОСОБ ПОЛУЧЕНИЯ ГИДРОКСИЛАМИНСУЛЬФАТА | 2003 |
|
RU2259940C1 |
| СПОСОБ ПОЛУЧЕНИЯ ГИДРОКСИЛАМИНСУЛЬФАТА И УСТАНОВКА ДЛЯ ЕГО ОСУЩЕСТВЛЕНИЯ | 1991 |
|
RU2045471C1 |
| СПОСОБ УПРАВЛЕНИЯ ПРОЦЕССОМ СИНТЕЗА ГИДРОКСИЛАМИНСУЛЬФАТА | 2019 |
|
RU2702575C1 |
Изобретение относится к способу получения гидроксиламинсульфата, применяемого в синтезе капролактама. Способ заключается в получении гидроксиламинсульфата (ГАС) при избыточном давлении путем смешения в реакционном объеме синтез-газа, состоящего из оксида азота (II) и водорода, смеси серной кислоты и воды и катализатора "платина на электрографите", т.е. платина, нанесенная на электродный графит. Процесс проводят в присутствии 2-30%-ного водного раствора комплексообразователя состава: Гидроксиэтилиден-1,1-дифосфоновая кислота (I) формулы СН3-С(ОН)=[Р(O)(ОН)2]2,
Циклический Р-комплекс (II) общей формулы
где А есть = Р(O)СН3ОН, при мольном отношении I:II, равном 1:1, при массовом отношении водного раствора комплексообразователя и железа, содержащегося в реакционной системе, равном 100-500 ppm на 1 ppm Fe. Способ позволяет более чем в 3 раза снизить содержание NO в отходящих газах и увеличить выход основного продукта реакции - ГАС, а также возрастает межрегенерационный период работы катализатора. 2 табл.
Способ получения гидроксиламинсульфата (ГАС) при избыточном давлении путем смешения в реакционном объеме синтез-газа, состоящего из оксида азота (II) и водорода, смеси серной кислоты и воды и катализатора "платина на электрографите", отличающийся тем, что процесс проводят в присутствии 2-30%-ного водного раствора комплексообразователя состава:
Гидроксиэтилиден-1,1-дифосфоновая кислота (I) формулы
СН3-С(ОН)=[Р(O)(ОН)2]2,
Циклический Р-комплекс (II) общей формулы
где А есть = Р(O)СН3ОН, при мольном соотношении I:II, равном 1:1, при массовом отношении водного раствора комплексообразователя и железа, содержащегося в реакционной системе, равном 100-500 ppm комплексообразователя на 1 ppm железа.
| Способ получения гидроксиламинсульфата | 1988 |
|
SU1599302A1 |
| Способ получения гидроксиламинсульфата | 1983 |
|
SU1214584A1 |
| Способ получения гидроксиламинсульфата | 1984 |
|
SU1237629A1 |
| Способ получения гидроксиламинсульфата в барботажном колонном реакторе | 1988 |
|
SU1627508A1 |
| Цифровое индикаторное устройство | 1975 |
|
SU535514A1 |
| Устройство для испытания изделий на ударные нагрузки | 1984 |
|
SU1179120A2 |

