Объектом настоящего изобретения является соединение.
Одним из аспектов генной терапии является введение чужеродной нуклеиновой кислоты (такой как ДНК) в клетки, в случае, если экспрессируемый ими протеин может обладать требуемой терапевтической функцией.
Примеры терапии этого типа включают встраивание генов ТК, TSG или ILG с целью лечения рака; встраивание гена CFTR с целью лечения кистозного фиброза; встраивание генов NGF, ТН или LDL с целью лечения нейродегенеративных и сердечно-сосудистых заболеваний; встраивание антагониста гена IL-1 с целью лечения ревматоидного артрита; встраивание антигенов ВИЧ и гена ТК с целью лечения инфекций, связанных с СПИДом и вызываемых CMV (цитомегаловирусом); встраивание антигенов и цитокинов, которые могут действовать в качестве вакцин; и встраивание β-глобина с целью лечения гемоглобинопатических состояний, таких как талассемия.
Во многих современных исследованиях в области генной терапии используют аденовирусные векторы генов - такие как Ad3 или Ad5 - или другие векторы генов. Однако с их применением связаны серьезные проблемы. Это обусловливает потребность в разработке менее опасных невирусных систем для переноса генов.
Обладающая высокой эффективностью невирусная система переноса основана на использовании катионных липосом. Так, катионные липосомы, которые обычно состоят из нейтрального фосфолипида и катионного липида, использовали для переноса в клетки ДНК, мРНК, антисмысловых ологонуклеотидов, протеинов и лекарственных веществ. Большое количество катионных липосом имеются в продаже и в настоящее время синтезирован целый ряд новых катионных липидов. Эффективность этих липосом продемонстрирована как in vitro, так и in vivo.
Цитофектин, применяемый для получения катионной липосомы, представляет собой хлорид N-[1-(2,3-диолеоилокси)пропил]-N,N,N-триметиламмония, обозначенный так же, как "ДОТМА".
Одна из наиболее широко применяемых систем катионных липосом состоит из смеси нейтрального фосфолипида диолеилофосфатидинилэтаноламина (обычно обозначаемого "ДОФЭ") и катионного липида, 3β-[(N,N-диметиламиноэтан)карбамоил]холестерина (обычно обозначаемого "ДК-Хол").
Несмотря на эффективность известных катионных липосом, все еще сохраняется потребность в оптимизации эффективности переноса генов катионными липосомами, применяемыми для генной терапии человека. С учетом практически полной расшифровки генома человека применение генов для терапевтических целей, т.е. для генной терапии, должно привести к революции в медицине. В этом контексте повышается внимание исследователей к невирусному введению генов, как более безопасной альтернативы для применения на человеке, даже если оно пока остается менее эффективным, чем основанные на применении вирусов технологии.
В этой области, особенно в последнее десятилетие, начали применять сложные макромолекулярные конструкции, включая многие элементы существующих технологий (вирусные протеины или пептиды, липосомы, полимеры, стратегии направленного переноса и скрытые ранее возможности).
В находящейся в совместном рассмотрении заявке этих же авторов PCT/GB00/04767 предложена система, основанная на триплексе, состоящем из вирусного корового протеина Mu, плазмидной ДНК и катионной липосомы (LMD). Эта базисная технология позволила получить хорошие результаты in vitro и многообещающие результаты in vivo. Но так же, как и другие невирусные технологии, эта система требует дальнейшего усовершенствования для внедрения в терапию in vivo.
Это подразумевает достижение большее высокого уровня стабильности частиц в биологических жидкостях (сыворотка, слизистая легкого) наряду с сохранением способности к эффективной трансфекции.
Это требование является одним из препятствий для внедрения всех существующих технологий. Современные стабильные композиции обеспечивают невысокий уровень трансфекции, а наиболее эффективные в настоящее время агенты для трансфекции имеют очень значительные ограничения с точки зрения их применения, связанные с их нестабильностью.
После введения (в кровь в случае системного применения или в слизистую в случае местного нанесения на легкое), заряженные комплексы подвергаются воздействию соли и биологических макромолекул, что приводит к выраженной коллоидной агрегации и адсорбции биологически активных элементов (опсонины) на их поверхностях. Эти носители генов подвергаются резким изменениям, которые могут включать осаждение, связывание протеинов, что приводит к элиминации частиц макрофагами, и к нарушению поверхностей, приводящих к их деструкции.
Для разработки систем для введения лекарственных средств и генов, обладающих способностью к направленному переносу в клетку in vitro и in vivo, необходимо создание протоколов, предназначенных для получения стабильных к действию биологических жидкостей систем введения, которые обладали бы достаточной активностью для их применения в терапевтических целях. Таким образом, для разработки эффективного носителя лекарственных средств/генов должен быть создан баланс между стабильностью и активностью.
В находящейся в совместном рассмотрении заявке этих же авторов PCT/GB00/04767 предложена система, основой которой является модифицированный липид, который несет углеводный фрагмент. Было установлено, что эти модифицированные липиды являются стабильными и обладают низкой токсичностью. При создании таких систем необходимо связывание с липидом дополнительного фрагмента для получения модифицированного липида, обладающего стабильностью и низкой токсичностью. Таким образом, в данной области существует необходимость в разработке липидов, несущих группы, к которым легко присоединять дополнительные фрагменты.
Настоящее изобретение позволяет решить проблемы, существующие в данной области.
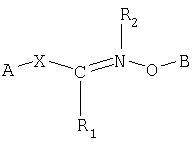
Одним из объектов настоящего изобретения является соединение формулы
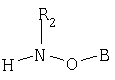
где В обозначает липид; и R2 обозначает Н или гидрокарбильную группу.
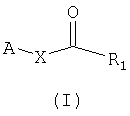
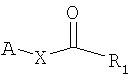
Следующим объектом настоящего изобретения является способ получения модифицированного липида формулы
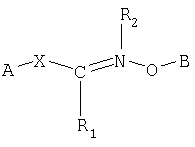
который заключается во взаимодействии (I) соединения формулы; и
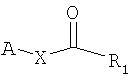
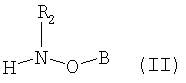
(II) соединения формулы
где В обозначает липид и А обозначает представляющий интерес фрагмент (ПИФ); где Х обозначает необязательную линкерную группу; где R1 обозначает Н или гидрокарбильную группу; и где R2 обозначает неподеленную пару или R4, где R4 обозначает приемлемый заместитель.
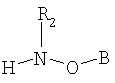
Таким образом, одним из объектов настоящего изобретения является композиция, содержащая (I) а соединение формулы
(II) соединение формулы
где В обозначает липид и А обозначает представляющий интерес фрагмент (ПИФ); где Х обозначает необязательную линкерную группу; где R1 обозначает Н или гидрокарбильную группу; и где R2 обозначает неподеленную пару или приемлемый заместитель.
Следующим объектом настоящего изобретения является соединение, композиция или соединение, полученное способом по настоящему изобретению, которые предназначены для применения в терапии.
Еще одним объектом настоящего изобретения является применение соединения, композиции или соединения, полученного способом по настоящему изобретению, для приготовления лекарственного средства, предназначенного для лечения связанного с геномом нарушения или состояния или заболевания.
Следующим объектом настоящего изобретения является липосома, содержащая соединение, композицию или соединение, полученное способом по настоящему изобретению.
Еще одним объектом настоящего изобретения является способ получения липосомы, который заключается в получении липосомы из соединения, композиции или соединения, полученного способом по настоящему изобретению.
Следующим объектом настоящего изобретения является липосома по настоящему изобретению или липосома, полученная способом по настоящему изобретению, которые предназначены для применения в терапии.
Еще одним объектом настоящего изобретения является применение липосомы по настоящему изобретению или липосомы, полученной способом по настоящему изобретению, для приготовления лекарственного средства, предназначенного для лечения связанного с геномом нарушения или состояния или заболевания.
И еще одним объектом настоящего изобретения является композиция, содержащая нуклеотидную последовательность или фармацевтически активный агент и один или несколько ингредиентов, выбранных из ряда, включающего соединение, композицию, соединение, полученное способом по настоящему изобретению, липосому по настоящему изобретению или липосому, полученную способом по настоящему изобретению.
Следующим объектом настоящего изобретения является композиция по настоящему изобретению, предназначенная для применения в терапии.
И еще одним объектом настоящего изобретения является применение композиции по настоящему изобретению для приготовления лекарственного средства, предназначенного для лечения связанного с геномом нарушения или состояния или заболевания.
Следующим объектом настоящего изобретения является фармацевтическая композиция, содержащая соединение, композицию или соединение, полученное способом по настоящему изобретению, в смеси с фармацевтическим и необязательно в смеси с фармацевтически приемлемым разбавителем, носителем или эксципиентом.
И еще одним объектом настоящего изобретения является фармацевтическая композиция, содержащая липосому по настоящему изобретению или липосому, полученную способом по настоящему изобретению, в смеси с фармацевтическим и необязательно в смеси с фармацевтически приемлемым разбавителем, носителем или эксципиентом.
Некоторые другие объекты изобретения представлены в приведенной ниже формуле изобретения.
При создании изобретения было обнаружено, что липид, содержащий аминоксигруппу, облегчает присоединение дополнительных фрагментов к липиду через аминоксигруппу. При взаимодействии с фрагментом (ПИФ), который содержит альдегидную или кетогруппу, получают соединение, в котором ПИФ и липид связаны через амидную группу. Такую связь легко можно получать путем реакции в "одной пробирке". Эта методология позволяет избежать процедуры экстенсивной очистки путем использования простого диализа избытка непрореагировавших реагентов.
Способ по настоящему изобретению нанесения покрытия после реакции в одной пробирке основан на применении селективного и обладающего высокой реакционной способностью аминоксилинкера для взаимодействия с альдегидами и кетонами с образованием -C=N- ковалентных связей (типа оснований Шиффа). Важно, что реакцию можно осуществлять в водной среде при основном или кислом значении рН. Кроме того, не происходит частичного расщепления реакционноспособной группы при обработке водной средой, как это происходит в случае активированных NHS карбоксилов и других сложных эфиров. Таким образом, стабильность реакционноспособных субстанций, например, альдегида/кетона и аминоксигруппы позволяет полностью контролировать поверхностную реакцию без потери реакционноспособных субстанций вследствие гидролиза/расщепления.
Предпочтительные варианты осуществления изобретения
Соединение по настоящему изобретению имеет формулу
где В обозначает липид; и где R2 обозначает Н или гидрокарбильную группу.
Понятие "гидрокарбильная группа" в контексте настоящего описания обозначает группу, которая содержит по меньшей мере С и Н и которая не обязательно может содержать один или несколько других приемлемых заместителей. Примерами таких заместителей могут являться галоген, алкокси-, нитро-, алкильная группа, циклическая группа и т.д. Помимо того, что заместители могут представлять собой циклическую группу, комбинация заместителей может образовывать циклическую группу. Если гидрокарбильная группа содержит более одного атома С, то не является обязательным, чтобы эти атомы углерода были связаны друг с другом. Например, по меньшей мере два из атомов углерода могут быть связаны через пригодный элемент или группу. Таким образом, гидрокарбильная группа может содержать гетероатомы. Приемлемые гетероатомы хорошо известны специалистам в данной области и включают, например, серу, азот и кислород. Примером гидрокарбильной группы (но не ограничиваясь им) является ацильная группа.
Как правило, гидрокарбильная группа представляет собой углеводородную группу. В контексте настоящего описания понятие "углеводород" обозначает любую алкильную группу, алкенильную группу, алкинильную группу, эти группы могут быть линейными, разветвленными или циклическими, или арильную группу. Понятие "углеводород" включает также эти же группы, но необязательно замещенные. Если углеводород имеет разветвленную структуру и несет заместитель(и), то замещение может быть либо на углеводородном скелете, либо в боковой цепи; в другом варианте замещение может быть и углеводородном скелете, и в боковой цепи.
Предпочтительно реакцию по настоящему изобретению осуществляют в водной среде.
Необязательный линкер Х
Согласно предпочтительному варианту осуществления изобретения необязательный линкер Х присутствует.
Согласно предпочтительному варианту осуществления изобретения Х обозначает гидрокарбильную группу.
Согласно предпочтительному варианту осуществления изобретения линкер Х содержит или связан с липидом через полиаминную группу.
Вероятно, наличие полиаминной группы является целесообразным, поскольку она повышает способность связывать ДНК и эффективность переноса гена полученной липосомой.
Согласно одному из предпочтительных вариантов осуществления изобретения предпочтительно полиаминная группа представляет собой не встречающийся в естественных условиях полиамин. Вероятно, полиаминная головная группа является предпочтительной, поскольку повышенная активность аминофункции увеличивает общий положительный заряд липосомы. Кроме того, как известно, полиамины обладают как способностью к высокоаффинному связыванию с ДНК, так и способностью к стабилизации ДНК. Кроме того, полиамины встречаются в клетках в естественных условиях и поэтому, вероятно, при их применении будут минимизированы проблемы, связанные с токсичностью.
Согласно другому варианту осуществления изобретения предпочтительно две или большее количество аминных групп полиаминной группы по настоящему изобретению разделены одной или несколькими группами, которые не встречаются в естественных условиях и разделяют аминные группы встречающихся в естественных условиях полиаминных соединений (т.е. предпочтительно полиаминная группа по настоящему изобретению имеет не встречающийся в естественных условиях спейсинг).
Предпочтительно полиаминная группа содержит по меньшей мере два амина полиаминной группы, которые разделены (пространственно отделены друг от друга) этиленовой (-СН2СН2-) группой.
Предпочтительно все амины полиаминной группы разделены (пространственно отделены друг от друга) этиленовой (-CH2CH2-) группой.
Типичными примерами пригодных полиаминов являются спермидин, спермин, калдопентамин, норспермидин и норспермин. Предпочтительным полиамином является спермидин или спермин, поскольку, как хорошо известно, эти полиамины взаимодействуют с одно- или двухцепочечной ДНК. Другим предпочтительным полиамином является калдопентамин.
R1
Согласно предпочтительному варианту осуществления изобретения R1 обозначает Н
C=N
Связь C=N может быть либо чувствительной к действию кислот, либо устойчивой к действию кислот.
Согласно одному из вариантов осуществления изобретения связь C=N чувствительна к действию кислот.
Согласно другому варианту осуществления изобретения связь C=N устойчива к действию кислот.
ПИФ
Представляющий интерес фрагмент (ПИФ) может представлять собой любой фрагмент, который требуется связать с липидом.
ПИФ может представлять собой углеводный фрагмент.
Согласно предпочтительному варианту осуществления изобретения углеводный фрагмент представляет собой моносахарид.
Согласно предпочтительному варианту осуществления изобретения углеводный фрагмент представляет собой фрагмент сахара.
Предпочтительно углеводный фрагмент выбирают из ряда, включающего маннозу, глюкозу (D-глюкозу), галактозу, глюкуроновую кислоту, лактозу, мальтозу, мальтотриозу, мальтотетраозу, мальтогептаозу и их смеси. Более предпочтительно углеводный фрагмент представляет собой D-глюкозу.
Согласно одному из вариантов осуществления изобретения соединение по настоящему изобретению содержит от 1 до 7 углеводных фрагментов. Предпочтительно соединение содержит один углеводный фрагмент.
Липид
Согласно предпочтительному варианту осуществления изобретения липид представляет собой или содержит холестериновую группу или глицериновый/керамидный скелет. Можно применять любые липидоподобные структуры или полиамины.
Предпочтительно холестериновая группа представляет собой холестерин.
Предпочтительно холестериновая группа связана с Х через карбамоильную связь.
Холестериновая группа может представлять холестерин или его производное. Примеры производных холестерина включают замещенные производные, в которых одна или несколько циклических СН2- или СН-групп и/или одна или несколько СН2- или СН-групп с прямой цепью соответствующим образом замещена(ы). В альтернативном или дополнительном варианте одна или несколько циклических групп и/или одна или несколько групп с прямой цепью могут(жет) быть ненасыщенными(ой).
Согласно предпочтительному варианту осуществления изобретения холестериновая группа представляет собой холестерин. Вероятно, холестерин является предпочтительным, так как он стабилизирует полученный липосомный бислой.
Предпочтительно холестериновую группу связывают с необязательной линкерной группой через карбамоильную связь. Вероятно, эта связь является предпочтительной, поскольку образовавшаяся липосома имеет низкую или минимальную цитотоксичность.
Дополнительные варианты осуществления изобретения
Предпочтительно R2 обозначает Н или гидрокарбильную группу.
Согласно предпочтительному варианту осуществления изобретения R2 обозначает гидрокарбильную группу, содержащую необязательные гетероатомы, выбранные из О, N и галогенов.
Согласно предпочтительному варианту осуществления изобретения R2 обозначает Н.
Предпочтительно способ по настоящему изобретению осуществляют в водной среде или в полностью водной среде.
Настоящее изобретение относится также к соединению, приготовленному с помощью описанного способа по настоящему изобретению, соединению, полученному с помощью описанного способа по настоящему изобретению и/или соединению, которое можно получать с помощью описанного способа по настоящему изобретению.
Предпочтительно соединение находится в смеси или связано с нуклеотидной последовательностью.
Нуклеотидная последовательность может представлять собой часть или всю систему экспрессии, которую можно применять в терапии, такой как генная терапия.
Согласно предпочтительному варианту осуществления изобретения соединение по настоящему изобретению находится в смеси с конденсированным комплексом полипептид/нуклеиновая кислота, что позволяет получать невирусный вектор для введения нуклеиновой кислоты. Конденсированный комплекс полипептид/нуклеиновая кислота предпочтительно включает комплексы, описанные в находящейся в совместном рассмотрении заявке PCT/GB00/04767. Предпочтительно полипептиды или их производные обладают способностью образовывать комплекс с нуклеиновой кислотой. Предпочтительно полипептиды или их производные обладают способностью образовывать конденсированный комплекс с нуклеиновой кислотой. Предпочтительно входящая в комплекс нуклеиновая кислота является гетерологичной относительно полипептидов или их производных.
Предпочтительно способ предусматривает использование молекулярного сита.
Предпочтительно катионную липосому получают из соединения по настоящему изобретению и нейтрального фосфолипида, такого как ДОТМА или ДОФЭ. Предпочтительно нейтральный фосфолипид представляет собой ДОФЭ.
Ниже настоящее изобретение будет описано более подробно со ссылкой на приведенные только в качестве примера чертежи, на которых показано:
на фиг.1 - схема синтеза гидроксиламинного липида 11. Реагенты: (а) СН2Cl2, Et3N, Boc2O, KT, 5 ч, 98%; (б) EtOAc, N-гидроксисукцинимид (1 экв.), ДЦК (N,N'-дициклогексилкарбодиимид) (1 экв.), 10 ч., КТ; (в) (8), EtOAC/ТГФ [95/5], Et3N (pH=8), 2 ч, КТ, 90%; (г) CH2Cl2, ТФК (15 экв.), 0°С, N2, 5 ч, 86%;
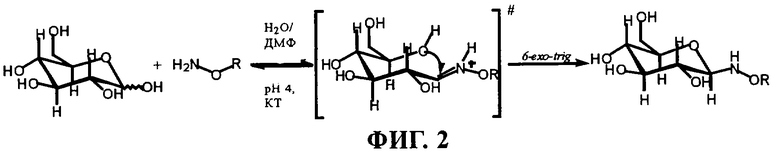
на фиг.2 - принцип хемоселективного гликозилирования O-замещенного гидроксиламина с помощью D-глюкозы (хотя показан β-аномер, должна иметь место мутаротация и также образовываться α-аномер);
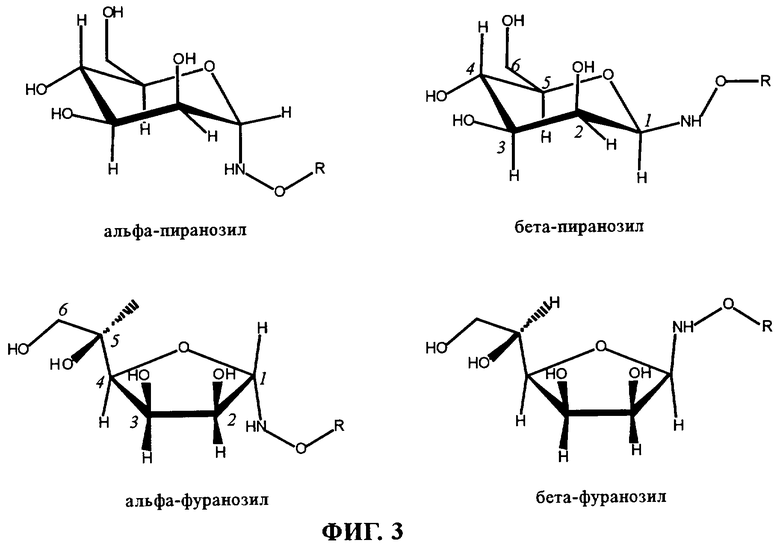
на фиг.3 - возможная структура неогликолипида, полученного из маннозы;
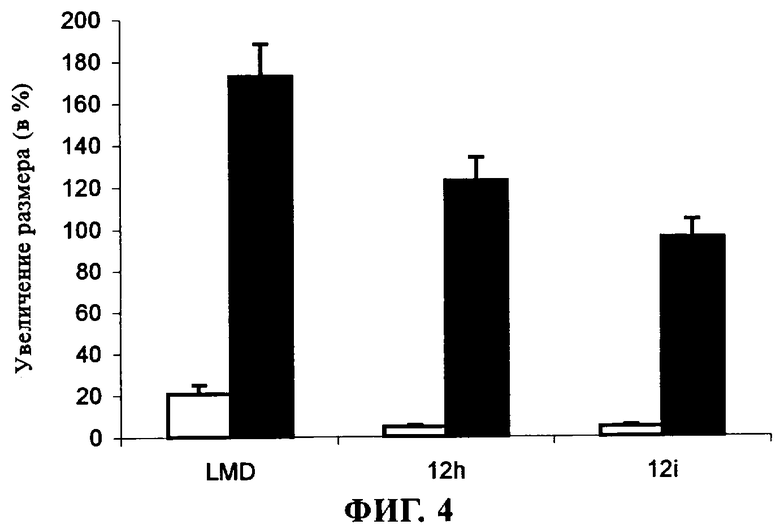
на фиг.4 - результат анализа различий в размере липоплексов, проведенного с помощью фотонно-корреляционной спектроскопии (ФКС). Размер оценивали после 30-минутной инкубации липоплексов с концентрацией [ДНК] равной 1 мкг/мл, в среде Optimem +/- 10% ФТС при 37°С. Сравнение стандартной композиции на основе LMD и LMD, модифицированного путем добавления 7,5 мол. % продукта 12h и 12i, осуществляли в среде Optimem (незакрашенные столбцы) и 10%-ной сыворотке (столбы, окрашенные в черный цвет) и выражали в виде % увеличения размера по сравнению с исходным измеренным размером 180 нм;
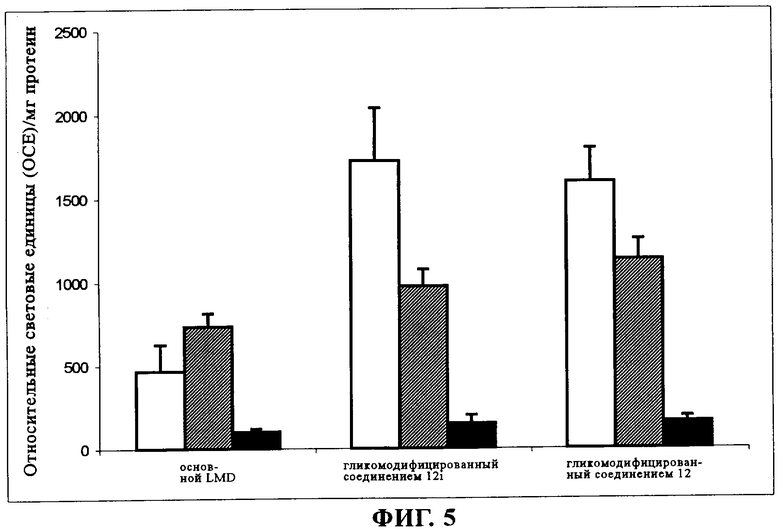
на фиг.5 - сравнение эффективности в отношении трансфекции основного LMD и LMD, гликомодифицированного с помощью 7,5 мол. % продукта 12h и 12i, клеток линии HeLa в 0% (незакрашенные столбцы), 50% (окрашенные в черный и незакрашенные столбцы) и 100%-ной сыворотке (черный цвет). Результаты выражали в виде относительных световых единиц на мг протеина (n=4);
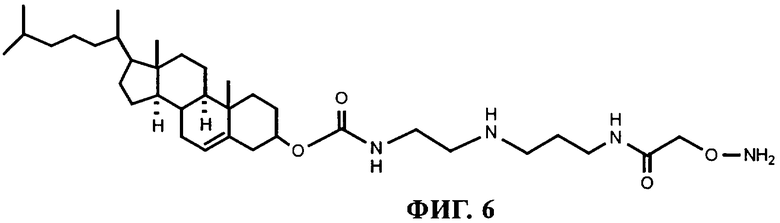
на фиг.6 - структура аминоксилипида.
Ниже настоящее изобретение более подробно проиллюстрировано на примерах.
Примеры
Экспериментальный раздел
Синтез неогликолипидов
Общие сведения: 1Н-ЯМР спектр определяли при температуре окружающей среды с помощью спектрометров Brucker DrX400, DrX300 или Jeol GX-270Q с использованием в качестве внутреннего стандарта неизотопично меченного растворителя (например, CHCl3, δН=7,26). 13С-ЯМР-спектры определяли с помощью таких же спектрометров при 100, 75 и 68,5 МГц соответственно также с использованием остаточного меченного без использования изотопов растворителя (например, CHCl3, δC=77,2) в качестве внутреннего стандарта. Инфракрасный спектр определяли с использованием устройства типа Jasco FT/Ir 620 с помощью пластин NaCl, а масс-спектры (электроспрей положительных ионов) оценивали с помощью устройств типа VG-7070B или JEOL SX-10. Хроматография включала экспресс-хроматографию на колонках, которую осуществляли на Merck-Kieselgel 60 (230-400 меш) с использованием общепринятого растворителя. Тонкослойную хроматографию (ТСХ) осуществляли на пластинах на алюминиевой основе, предварительно покрытых Merck-Kieselgel 60 F254, и осуществляли обнаружение с помощью ультрафиолета, йода, кислого молибдата(IV) аммония, кислого этанольного раствора ванилина или при необходимости других агентов. Чистоту неогликолипидов оценивали с помощью аналитической жидкостной хроматографии высокого давления (ЖХВД) на системе фирмы Hitachi с использованием колонки с заделанным концом типа Purospher RP-18 (5 мкм). Элюирование осуществляли с изократической скоростью потока 1 мл/мин с помощью CH3CN/H2O (60:40), и фракции выявляли при длине волны 205 нм перед сбором и анализом массы. Безводный CH2Cl2 перед применением дистиллировали в присутствии фосфорного ангидрида. Все другие безводные растворители и химические агенты получали от фирмы Sigma-Aldrich Company LTD (Пул, графство Дорсет, Великобритания).
Сокращения: Boc: трет-бутоксикарбонил; br: широкий; Хол: холестерин; ДМФ: N,N-диметилформамид; ДМСО: диметилсульфоксид; ТФК: трифторуксусная кислота; ТГФ: тетрагидрофуран.
2-(Холестерилоксикарбонил)аминоэтанол (2): Раствор хлорформиата холестерила (99,89 г, 0,218 моля) в CH2Cl2 (600 мл) добавляли к перемешиваемому раствору 2-аминоэтанола (29,5 мл, 0,489 моля, 2,2 экв.) в СН2Cl2 (450 мл) при 0°С в течение 2 ч. Реакционной смеси давали нагреться до комнатной температуры и перемешивание продолжали в течение еще 14 ч. Реакционную смесь промывали насыщенным раствором NaHCO3 (2×200 мл), водой (2×200 мл), сушили (MgSO4) и растворители удаляли при пониженном давлении. Полученный твердый продукт перекристаллизовывали (CH2Cl2/MeOH), получая соединение 2 в виде твердого вещества белого цвета.
Выход: 99,67 г (97%); tпл: 180°C; Rf=0,26 (ацетон/простой эфир 1:9); ИК (CH2Cl2): νmax=3353, 2942, 2870, 1693, 1674, 1562, 1467, 1382, 1264 см-1; 1Н-ЯМР (270 МГц, CDCl3): δ=5,35 (d, J=6,5 Гц, 1Н, Н6'), 5,25-5,29 (m, 1H, NH), 4,42-4,57 (1H, m, Н3'), 3,70-3,62 (m, 2Н, H1), 3,25-3,35 (m, 2H, Н2), 3,12 (s, 1H, ОН), 2,28-2,38 (m, 2H, Н4'), 1,77-2,03 (m, 5H, Н2', Н7', Н8'), 1,59-0,96 (m, 21Н, H1', H9', Н11', H12', H14'-H17', H22'-H25'), 1 (3Н, s, Н-19'), 0,9 (d, J=6,5 Гц, 3Н, Н21'), 0,87 (d, J=6,5 Гц, 6Н, Н26' и Н27') и 0,67 (s, 3Н, H18'); МС (FAB+): m/z=496 [M+Na]+, 474 [М+Н]+, 369 [Хол]+, 255, 175, 145, 105, 95, 81, 43.
2-[(Холестерилоксикарбонил)амино]этилметансульфонат (3): К раствору соединения 2 (25 г, 52,3 ммоля) и триэтиламина (22 мл, 0,16 моля, 3 экв.) в CH2Cl2 (500 мл) при 0°С добавляли по каплям раствор метансульфонилхлорида (10,5 мл, 0,13 моля, 2,5 экв.). Реакционной смеси давали нагреться до комнатной температуры и перемешивали в течение 1 ч 30 мин. После того как анализ с помощью ТСХ показал, что реакция подходит к завершению, для прекращения реакции добавляли лед. Реакционную смесь добавляли к насыщенному водному раствору NH4Cl (600 мл) и экстрагировали простым эфиром (3×300 мл). Объединенные органические слои промывали водой (2×300 мл), соляным раствором (250 мл) и сушили (Na2SO4). Растворитель удаляли при пониженном давлении, получая твердое вещество белого цвета, которое очищали хроматографией (простой эфир), получая соединение 3. Выход: 28,3 г (98%); ИК (CH2Cl2): νmax=3453, 3342, 1716, 1531, 1377, 1137 и 798 см-1; 1H-ЯМР (270 МГц, CDCl3): δ=5,34 (d, J=6,5 Гц, 1H, Н6'), 5-5,1 (m, 1H, NH). 4,41-4,53 (1H, m, Н3'), 4,29-4,25 (t, J=5 Гц, 2H, H1), 3,47-3,52 (m, 2H, H2), 3,01 (s, 3Н, Н3), 2,24-2,36 (m, 2H, Н4'), 1,74-2 (m, 5H, Н2', Н7', Н8'), 0,9-1,6 (m, 21H, Н1', Н9', H11', H12', H14'-H17', H22'-H25'), 0,98 (3Н, s, Н-19'), 0,84 (d, J=6,5 Гц, 3Н, Н21'),0,83 (d, J=6,5 Гц, 6H, H26' и H27') и 0,65 (s, 3H, H18'); MC (FAB+): m/z=1104 [2M+H]+, 574 [M+Na]+, 552 [М+Н]+, 369 [Хол]+, 255, 175, 145, 95, 81.
4-аза-N6(холестерилоксикарбониламино)гексанол (4): К перемешиваемому раствору соединения 3 (28,3 г, 51 ммоль), растворенному в минимальном количестве ТГФ, добавляли аминопропанол (160 мл, 2 моля, 39 экв.). После того как ТСХ показала, что реакция завершена (12 ч), добавляли CHCl3 (350 мл) и К2СО3 (20 г) и раствор интенсивно перемешивали в течение 30 мин. Затем суспензию фильтровали через подушку из целита (Celite®), тщательно промывали CHCl3. Затем промывали насыщенным раствором бикарбоната натрия и сушили (Na2СО3). Растворитель удаляли, получая соединение 4 в виде твердого вещества белого цвета. Выход: 26,1 г (96%); ИК (СН2Cl2): νmax=3350-3210, 2937, 2850, 1531, 1460, 1380, 1220, 1120, 1040 см-1; 1H-ЯМР (270 МГц, CDCl3): δ=5,33-5,35 (m, 1H, H6'), 4,92-4,96 (m, 1H, NH), 4,42-4,51 (1H, m, H3'), 3,7-3,83 (m, 2H, H5), 3,23-3,29 (m, 2H, H1), 2,73-2,57 (m, 6H, H2, H3, H4), 2,2-2,36 (m, 2H, H4'), 1,7-2 (m, 5H, H2', H7', H8'), 0,85-1,58 (m, 21H, H1', H9', Н11', H12', H14'-H17', H22'-H25'), 0,98 (3H, s, H-19'), 0,84 (d, J=6,5 Гц, 3Н, Н21'), 0,8 (d, J=6,5 Гц, 6H, Н26' и Н27') и 0,61 (s, 3H, Н18'); МС (FAB+): m/z=543 [M+Na]+, 530 [М+Н]+, 485 [М-СО2]+, 369 [Хол]+, 144 [М-ОХол]+, 69,55.
4-аза-(Boc)-N6(холестерилоксикарбониламино)гексанол (5): К раствору соединения 4 (26,1 г, 49 ммолей), добавляли Et3N (8,3 мл, 1,1 экв.) и Вос2O (10,7 г, 1 экв.) в CH2Cl2 (200 мл) и образовавшийся раствор анализировали с помощью ТСХ. После завершения реакции реакционную смесь сливали на NH4Cl (100 мл), промывали водой и сушили (Na2SO4). Растворитель удаляли в вакууме, получая соединение 5 в виде твердого вещества белого цвета. Растворитель удаляли при пониженном давлении, получая твердое вещество белого цвета, которое очищали хроматографией (СН2Cl2/МеОН/NH3 92:7:1), получая соединение 3. Выход: (27,9 г, 90%); ИК (CH2Cl2): νmax=3352, 3054, 2937, 1675, 1530, 1455, 1380, 1220, 1120; 1Н-ЯМР (270 МГц, CDCl3): δ=5,33-5,35 (m, 1H, Н6'), 4,86 (m, 1H, NH), 4,42-4,5 (1H, m, H3'), 3,62-3,7 (m, 2H, H5), 3,27-3,38 (m, 6H, H1, H2, H3), 2,18-2,33 (m, 2H, H4'), 1,73-2 (m, 5H, H2', H7', H8'), 1,45 (s, 9H, Boc), 1-1,65 (m, 23H, H4, H1', H9', Н11', H12', H14'-H17', H22'-H25'), 0,97 (3H, s, H-19'), 0,93 (d, J=6,5 Гц, 3Н, Н21'), 0,8 (d, J=6,5 Гц, 6H, Н26' и Н27') и 0,65 (s, 3Н, Н18'); МС (FAB+): m/z=654 [M+Na]+, 543 [М-Вос]+, 369 [Хол]+, 145, 121, 95, 69, 57.
4-аза-(Boc)-N6(холестерилоксикарбониламино)гексилметансульфонат (6): Этот эксперимент осуществляли аналогично получению 2-[(холестерилоксикарбонил)амино]этилметансульфоната 3 из расчета 44 ммолей с получением соединения 6. Выход: (28 г, 90%); ИК (СН2Cl2): νmax=3305, 2980, 2900, 2865, 1675, 1530, 1455, 1350, 1150; 1H-ЯМР (270 МГц, CDCl3): δ=5,33-5,35 (m, 1H, Н6'), 4,86 (m, 1H, NH), 4,35-4,55 (m, 1H, H3'), 4,22 (t, 2H, J=6,5 Гц, H5), 3,2-3,4 (m, 6Н, H1, Н2, H3), 3,01(s, 3Н, Н6), 2,15-2,33 (m, 2H, Н4'), 1,73-2 (m, 5H, Н2', Н7', Н8'), 1,44 (s, 9Н, Boc), 1-1,67 (m, 23Н, Н4, Н1', Н9', Н11', Н12', Н14'-Н17', Н22'-Н25'), 0,97 (3Н, s, Н-19'), 0,94 (d, J=6,5 Гц, 3Н, Н21'), 0,8 (d, J=6,5 Гц, 6Н, Н26' и Н27') и 0,65 (s, 3Н, Н18'); МС (FAB+): m/z=722 [M+Na]+, 609 [М-Вос]+, 369[Хол]+, 145, 121, 95, 69, 55.
4-аза-(Boc)-N6(холестерилоксикарбониламино)гексанамин (7): К раствору, содержащему соединение 6 (25 г, 35 ммолей), азид натрия (11,49 г, 175,7 ммоля, 5 экв.) и йодид натрия (5 г, 35 ммолей, 1 экв.), в атмосфере азота добавляли при перемешивании безводный ДМФ (200 мл). После присоединения конденсатора обратного холодильника и выдерживания при 80°С в течение 2 ч реакция завершалась. Реакционной смеси давали охладиться до комнатной температуры, ДМФ удаляли при пониженном давлении и остаток растворяли в EtOAc. Смесь промывали водой (2×100 мл), соляным раствором (100 мл) и сушили (Na2SO4), получая после очистки хроматографией (гексан/простой эфир 1:1) соединение 7 в виде твердого вещества белого цвета. Выход: (22 г, 95%); 1Н-ЯМР (270 МГц, CDCl2): δ=5,34-5,36 (m, 1H, Н6'), 4,35-4,55 (m, 1H, Н3'), 4,25 (t, 2H, J=6,5 Гц, H5), 3,2-3,5 (m, 6H, H1, H2, H3), 2,25-2,33 (m, 2H, Н4'), 1,7-2,05 (m, 5H, H2', H7', Н8'), 1,45 (s, 9H, Boc), 1-1,72 (m, 23H, H4, H1', H9', Н11', Н12', Н14-Н17', Н22'-Н25'), 0,98 (3Н, s, Н-19'), 0,94 (d, J=6,5 Гц, 3Н, Н21'), 0,83 (d, J=6,5 Гц, 6H, Н26' и Н27') и 0,64 (s, 3Н, H18'); МС (FAB+): m/z=568 [M+Na-Вос]+, 556 [M-Boc]+, 369 [Хол]+, 145, 121, 95, 69, 57.
4-аза-(Boc)-N6(холестерилоксикарбониламино)гексиламин (8): В круглодонную колбу, содержащую соединение 7 (22,75 г, 34,6 моля) в ТГФ (230 мл), добавляли триметилфосфин в ТГФ (1М, 40 мл, 1,15 экв.) и прохождение реакции оценивали с помощью ТСХ. После завершения реакции реакционную смесь перемешивали с водой (3 мл) и водным аммиаком (3 мл) в течение 1 ч и растворитель удаляли при пониженном давлении. После хроматографии (CH2Cl2/MeOH/NH3 от 92:7:1 до 75:22:3) получали соединение 8 в виде кристаллов белого цвета. Выход: (19,1 г, 88%); ИК (CH2Cl2): νmax=3689, 3456, 3155, 2948, 2907, 2869, 2253, 1793, 1709, 1512, 1468, 1381, 1168; 1Н-ЯМР (270 МГц, CDCl3): δ=5,32-5,35 (m, 1H, Н6'), 4,35-4,51 (m, 1H, H3'), 3,45-3,05 (m, 8H, H1, H2, H3, H5), 2,18-2,4 (m, 2H, H4'), 1,8-2,1 (m, 5H, H2', H7', H8'), 1,46 (s, 9H, Boc), 1,01-1,72 (m, 23H, H4, H1', H9', H11', Н12', H14'-H17', H22'-H25'), 0,97 (3H, s, H-19'), 0,85 (d, J=6,5 Гц, 3H, Н21'), 0,82 (d, J=6,5 Гц, 6Н, Н26' и Н27') и 0,64 (s, 3H, Н18'); МС (FAB+): m/z=630 [М+Н]+, 530 [M-Boc]+, 369 [Хол]+, 145, 121, 95, 69, 57.
(Вос)аминооксиуксусная кислота (9): Полугидрохлорид O-(карбоксиметил)гидроксиламина (1,16 г, 5,3 ммоля) растворяли в CH2Cl2 (40 мл) и значение рН доводили до 9, добавляя триэтиламин (3 мл). Затем добавляли ди-трет-бутилдикарбонат (2,36 г, 10,6 ммоля, 2,0 экв.) и смесь перемешивали при комнатной температуре до тех пор, пока ТСХ не показывала завершение реакции. Значение рН понижали до 3, добавляя разбавленную HCl. Реакционную смесь распределяли между насыщенным водным раствором NH4Cl (20 мл) и CH2Cl2 (30 мл). Водную фазу экстрагировали CH2Cl2 (3×100 мл). Объединенные органические экстракты промывали Н2О (2×100 мл) и сушили (Na2SO4). Растворитель удаляли в вакууме, получая соединение 9 в виде твердого вещества белого цвета. Выход: (1,86 г, 97%); ИК (CH2Cl2): νmax=3373, 2983, 2574, 2461, 1724, 1413, 1369, 1235; 1H-ЯМР (270 МГц, CDCl3): δ=4,48 (s, 2H, CH2), 1,48 (s, 9H, Boc); МС (FAB+): m/z=214 [М+Na]+, 192 [М+Н]+, 135, 123, 109, 69.
(Вос)аминооксипроизводное (10): N-гидроксисукцинимид (0,36 г, 3,13 ммоля, 1 экв.), соединение 9 (0,6 г, 3,13 ммоля, 1 экв.) и N,N'-дициклогексилкабодиимид (0,68 г, 3,13 ммоля, 1 экв.) растворяли в EtOAc (90 мл) и гетерогенной смеси давали перемешиваться при комнатной температуре в течение ночи. Смесь фильтровали через подушку из целита (Celite®) для удаления дициклогексилмочевины, которая образовывалась в виде белого осадка (промывали 60 мл EtOAc), и добавляли к раствору соединения 8 (1,97 г, 3,13 ммоля, 1 экв.) в ТГФ (10 мл). Значение рН этой гетерогенной реакционной смеси поддерживали на уровне 8, добавляя триэтаноламин (6 мл). Образовавшейся смеси давали перемешиваться при комнатной температуре в течение ночи. После завершения реакции смесь фильтровали и растворитель удаляли при пониженном давлении, получая после очистки экспресс-хроматографией (СН2Cl2/МеОН/NH3 92:7:1) соединение 10 в виде твердого вещества белого цвета. Выход: (2,3 г, 90%); 1H-ЯМР (270 МГц, CDCl3): δ=5,33-5,35 (m, 1Н, Н6'), 4,4-4,52 (m, 1H, H3'), 4,3 (s, 2H, H90, 3,2-3,42 (m, 8H, H1, H2, H4, H6), 2,23-2,35 (m, 2H, H4'), 1,7-2,1 (m, 7H, H2', H7', H8', H5), 1,44-1,46 (m, 18H, 2 Boc), 1-1,73 (m, 21H, H1', H9', H11', H12', H14'-H17', H22'-H25'), 0,98 (3H, s, H-19'), 0,85 (d, J=6,5 Гц, 3H, H21'), 0,83 (d, J=6,5 Гц, 6Н, Н26' и Н27') и 0,65 (s, 3H, H18'); МС (FAB+): m/z=803 [М+Н]+ 703 [M-Boc]+, 647, 603 [M-2Boc]+, 369, 279, 255, 235, 204, 145, 95, 69.
Гидроксиламин (11): К раствору соединения 10 (1,1 г, 1,36 ммоля, 1 экв.) в СН2Cl2 (10 мл) добавляли ТФК (2 мл, 20,4 ммоля, 15 экв.) при 0°С. Раствору давали перемешиваться при комнатной температуре в течение 5 ч. После завершения реакции добавляли толуол до получения азеотропа ТГФ из реакционной смеси. Растворители удаляли в вакууме, получая после очистки хроматографией (СН2Cl2/МеОН/NH3 от 92:7:1 до 75:22:3) соединении 11 в виде твердого вещества белого цвета (709 мг, выход: 86%); ИК (CHCl3): νmax=3306, 2948, 2850, 2246, 1698, 1647, 1541, 1467, 1253, 1133; 1Н-ЯМР (270 МГц, CDCl3): δ=5,26-5,4 (m, 1H, Н6'), 4,4-4,52 (m, 1H, H3'), 4,12 (s, 2H, H9), 3,34-3,41 (m, 2H, H2), 3,15-3,3 (m, 2H, H4), 2,6-2,74 (m, 4H, H1&H6), 2,14-2,39 (m, 2H, H4'), 1,62-2,1 (m, 7H, Н2', H7', Н8', H5), 1,02-1,6 (m, 21H, H1', H9', Н11', Н12', Н14'-Н17', Н22'-Н25'), 0,96 (3H, s, H-19'), 0,86 (d, J=6,5 Гц, 3H, Н21'), 0,83 (d, J=6,5 Гц, 6Н, Н26' и Н27') и 0,66 (s, 3H, H18'); МС (FAB+): m/z=603 [М+Н]+, 369 [Хол]+, 160, 137, 109, 95, 81, 69, 55.
Маннозильное производное (12а): Смешивали раствор D-маннозы (266 мг, 4,8 ммоля) в водном ацетатном буфере (ацетат натрия/уксусная кислота, 0.1М, рН 4, 7 мл) и раствор соединения 11 (290 мг, 0,48 ммоля, 10 экв.) в ДМФ (7 мл) и перемешивали в течение 3 дней при комнатной температуре. Растворитель удаляли в вакууме путем сушки вымораживанием и хроматографировали (CH2Cl2/MeOH/NH3 75:22:3), получая продукт 21 в виде твердого вещества белого цвета (233 мг, выход: 65%). Чистоту дополнительно подтверждали с помощью ЖХВД. Конечный продукт содержал β-пиранозную форму (82%) и α-пиранозную форму (18%), которые не выделяли, но характеризовали в смеси. МС (FAB+): m/z=765 [М+Н]+, 787 [M+Na]+, 397, 369 [Хол]+, 322, 240, 121, 109, 95, 81, 69, 57. β-пиранозная форма: 1H-ЯМР (400 МГц, CD3OD/CDCl3 [75/25]): δ=7,64-7,62 (d, 3J1a-2a=7 Гц, 1H, H1a), 5,35-5,36 (m, 1H, H6'), 4,45-4,5 (s, 2H, H9), 4,35-4,5 (m, 1H, H3'), 4,19-4,24 (dd, 1H, H2a, 3J1a-2a=7,4 Гц, 3J2а-3а=7,7 Гц), 3,81-3,9 (m, 1H, Н3а), 3,73-3,8 (m, 2H, H4a, Н6аха), 3,63-3,71 (m, 2H, H5a, Heq6a), 3,34-3,42 (m, 2H, H2), 3,27-3,30 (m, 2H, Н4), 3-3,08 (m, 2H, H1), 2,9-2,98 (m, 2H, Н6), 2,25-2,35 (m, 2H, H4'), 1,78-2,07 (m, 7Н, H2', Н7', Н8', Н5), 1,03-1,65 (m, 21H, H1', H9', H11', H12', H14'-H17', H22'-H25'), 1,01 (3Н, s, H-19'), 0,91 (d, J=6,5 Гц, 3Н, Н21'), 0,85 (d, J=6,5 Гц, 6Н, Н26' и Н27') и 0,69 (s, 3Н, Н18'); 13С-ЯМР (400 МГц, CDCl3/CD3OD [25/75]): 12,33 (С18'), 19,20 (С21'), 19,74 (С19'), 21,91 (С11'), 22,91 (С27'), 23,17 (С26'), 24,67 (С23'), 25,07 (С15'), 27,37 (С5), 28,85 (С25'), 28,96 (С2'), 29,07 (С12'), 32,76 (С7'), 32,87 (С8'), 36,38 (С2), 36,78 (С20'), 37,09 (С1) 37,76 (С22'), 37,95 (С1'), 38,4 (С4), 39,36 (С4'), 40,41 (С24'), 40,76 (С16'), 46,16 (С6), 51,19 (С9'), 57,19 (С17'), 57,75 (С14'), 64,62 (С6а), 70,19 (С2а), 70,58 (С4а), 72,12 (С3а), 72,37 (С5а), 73,11 (С9), 75,91 (C3'), 123,39 (С6'), 140,72 (С5'), 155,02 (C1a), 158,69 (NHCOOХол), 173,1 (С8); α-пиранозная форма: идентичные данные за исключением 1Н -ЯМР (400 МГц, CD3OD/CDCl3 [75/25]): δ=6,90-6,88 (d, 3J1a-2a=7 Гц, 1H, H1a), 5-5,05 (dd, 1H, H2a, 3J1a-2a=7,3 Гц, 3J2а-3а=7,6 Гц); 13С-ЯМР (400 МГц, CDCl3/CD3OD [25/75]): 65,33 (С2а), 155,79 (C1a), 1H-ЯМР (400, CD3OD/CDCl3 [75/25]): (m, 1H, H3') отсутствует, находится под пиком растворителя, подтверждено 1Н-ЯМР (300 МГц, ДМСО): δ=4,67-4,82 (m, 1H, H3'). 13С-ЯМР (400 МГц, CDCl3/CD3OD [25/75]): C1 отсутствует, находится под пиком МеОН, подтверждено 1Н/13С, корреляция при 400 МГц примерно 49. Данные протонного резонанса подтверждали с помощью 1Н-DQF-COSY градиентного типа и TOCSY; 1H/13C-корреляцию и DEPT 135 использовали для однозначного определения резонансов углерода. α-Пиранозная форма давала 1J13C1a-H1a=177 Гц, а β-пиранозная форма давала 1J13C1a-H1a=167 Гц. Конформацию подтверждали с помощью 1Н-фазочувствительного NOESY.
Глюкозильное производное (12b): Это соединение получали с использованием раствора D-глюкозы (150 мг, 0,82 ммоля) и соединения 11 (100 мг, 0,16 ммоля) аналогично методу получения соединения 12а, осуществляя перемешивание в течение 1 дня и очистку с помощью хроматографии (СН2Cl2/МеОН/NH3 75:22:3), получали продукт 12b в виде твердого вещества белого цвета (103 мг, выход: 82%). Чистоту дополнительно подтверждали с помощью ЖХВД. Конечный продукт содержал аномер α-пиранозы (11%) и аномер β-пиранозы (89%), которые не выделяли, а характеризовали в смеси. (FAB+): m/z=765 [М+Н]+, 787 [М+Na]+, 391, 369 [Хол]+, 309, 290, 171, 152, 135, 123, 109, 95, 81, 69; β-пиранозная форма: (300 МГц, CDCl2/CD3OD [90/10]): δ=7,53-7,56 (d, J=5,6 Гц, 1Н, H1a), 5,26-5,36 (m, 1H, Н6'), 4,2-4,45 (m, 3H, Н9, Н3'), 4,05-4,15 (m, 1H, Н2а), 3,45-3,85 (m, 5H, Н6а, Н3а, Н5а, Н4а), 2,9-3,4 (m, Н2, Н4, МеОН), 2,9-3,15 (m, 4Н, H1, Н6), 2,15-2,3 (m, 2Н, Н4'), 1,65-2 (m, 5H, Н2', Н7', Н8'), 0,95-1,55 (m, 23Н, Н5, H1', H9', Н11', Н12', Н14'-Н17', Н22'-Н25'), 0,93 (3H, s, Н-19'), 0,84 (d, J=6,5 Гц, 3H, Н21'), 0,78 (d, J=6,5 Гц, 6Н, Н26' и Н27') и 0,62 (s, 3H, H18'); α-пиранозная форма: идентичные данные за исключением, 1Н-ЯМР (300 МГц, CDCl2/CD3OD [90/10]): δ=7,22-7,24 (d, J=6,61 Гц, 1H, H1a), 4,95-5,07 (m, 1H, H2a); 1H-ЯМР (300 МГц, CD3OD): (m, 1H, Н3') отсутствует, находится под пиком растворителя, подтверждено 1Н-ЯМР (300 МГц, ДМСО): δ=4,7-4,86 (m, 1H, Н3').
Галактозильное производное (12с): Соединение получали с использованием раствора D-галактозы (50 мг, 0,27 ммоля) и соединения 11 (40 мг, 0,066 ммоля) аналогично методу получения соединения 12а, осуществляя перемешивание в течение 1 дня и очистку с помощью хроматографии (СН2Cl2/МеОН/NH3 75:22:3), получали продукт 12с в виде твердого вещества белого цвета (35 мг, выход: 70%). Чистоту дополнительно подтверждали с помощью ЖХВД. Конечный продукт содержал аномер α-пиранозы (15%) и аномер β-пиранозы (85%), которые не выделяли, а характеризовали в смеси. МС (FAB+): m/z=765 [М+Н]+, 588, 391, 369 [Хол]+, 322, 290, 165, 152, 135, 121, 109, 95, 81, 69; β-пиранозная форма 1Н-ЯМР (270 МГц, ДМСО): δ=7,78-7,82 (m, 1H, NHCO C8), 7,55-7,58 (d, J=7,2 Гц, 1H, H1a), 6,95-7,1 (m, 1H, NHCOOХол), 5,25-5,37 (m, 1H, Н6'), 4,2-4,43 (m, 3H, H9, Н3'), 3,2-3,9 (m, H2a, Н6а, Н3а, Н5а, Н4а, ОН), 2,9-3,18 (m, 4Н, Н2, Н4), 2,4-2,65 (m, 4Н, H1, Н6), 2,15-2,3 (m, 2Н, Н4'), 1,67-2 (m, 5H, Н2', Н7', Н8'), 0,92-1,6 (m, 23Н, Н5, Н1', H9', Н11', Н12', Н14-Н17', Н22'-Н25'), 0,96 (3H, s, Н-19'), 0,89 (d, J=6,5 Гц, 3H, Н21'), 0,84 (d, J=6,5 Гц, 6Н, Н26' и Н27') и 0,65 (s, 3H, H18'), α-пиранозная форма: идентичные данные за исключением, 1H-ЯМР (270 МГц, ДМСО): 6,86-6,88 (d, J=6 Гц, 1Н, H1a).
Глюкуроновое производное (12d): Соединение получали с использованием раствора моногидрата натриевой соли D-глюкуроновой кислоты (30 мг, 0,128 ммоля, 1,5 экв.) и соединения 11 (50 мг, 0,08 ммоля) аналогично методу получения соединения 12а, осуществляя перемешивание в течение 1 дня, очистку с помощью хроматографии (СН2Cl2/МеОН/NH3 75:22:3), получали натриевую соль 12d в виде твердого вещества белого цвета (41 мг, выход: 60%). Чистоту дополнительно подтверждали с помощью ЖХВД. Конечный продукт содержал аномер α-пиранозы (85%) и аномер β-пиранозы (15%), которые не выделяли, а характеризовали в смеси. МС (FAB+): m/z=779 [М+Н]+, 733, 588, 411, 369 [Хол]+, 336, 290, 240, 214, 159, 145, 135, 121, 109, 95, 81, 69, 55. β-пиранозная форма: 1Н-ЯМР (300 МГц, CDCl2/CD3OD [75/25]): δ=7,51-7,53 (d, J=5,9 Гц, 1H, H1a), 5,25-5,33 (m, 1H, H6'), 4,2-4,45 (m, 3H, H9, H3'), 3,8-4,1 (m, 3H, H2a, H3a, H4a), 3,6-3,75 (m, 1H, H5a), 3,2-3,55 (m, H2, H4, MeOH). 2,7-3,15 (m, 4H, H1, H6), 2,18-2,32 (m, 2H, H4'), 1,62-2 (m, 5H, H2', H7', H8'), 0,9-1,6 (m, 23H, H5, H1', H9', H11', H12', H14'-H17', H22'-H25'), 0,93 (3H, s, H-19'), 0,83 (d, J=6,5 Гц, 3H, Н21'), 0,77 (d, J=6,5 Гц, 6Н, Н26' и Н27') и 0,6 (s, 3H, Н18')); α-пиранозная форма: идентичные данные за исключением, 1Н-ЯМР (300 МГц, CD3OD): δ=7,22-7,24 (d, J=6,3 Гц, 1H, H1a), 5-5,1 (m, 1H, H2a).
β-D-лактозильное производное (12е): Раствор β-D-лактозы, содержащий 25-30% α-лактозы (1,13 г, 3,3 ммоля), и соединение 11 (200 мг, 0,33 ммоля) в 14 мл ДМФ/водный ацетатный буфер, перемешивали в течение 4 дней при комнатной температуре. Растворитель удаляли в вакууме путем сушки вымораживанием и хроматографировали (СН2Cl2/МеОН/NH3 75:22:3), получая продукт 12е в виде твердого вещества белого цвета (145 мг, выход: 47%). Чистоту дополнительно подтверждали с помощью ЖХВД. Конечный продукт содержал аномер α-пиранозы (15%) и аномер β-пиранозы (85%), которые не выделяли, а характеризовали в смеси. МС (FAB+): m/z=927 [М+Н]+, 588, 482, 369 [Хол]+, 290, 243, 216, 178, 152, 135, 121, 109, 95, 81, 69, 55; β-пиранозная форма: 1H-ЯМР (400 МГц, CDCl2/CD3OD [20/80]): δн=7,69-7,71 (d, 3J1a-2a=5,8-22 Гц, 1Н, H1a β-лактозы). 7,66-7,68 (d, 3J1a-2a=6,2 Гц, 1Н, H1a α-лактозы), 5,35-5,37 (m, 1H, H6'), 4,37-4,6 (m, 4H, H9, H3', H2a), 4,2-4,37 (m, 1H, H1b), 3,65-4,05 (m, 7 H, H3a, H4a, H5a, H4b, H5b, H6b), 3,25-3,6 (m, 8H, H2, H4, H6a, H2b, H3b, MeOH), 3-3,2 (m, 4H, H1, H6), 2,25-2,42 (m, 2H, H4'), 1,8-2,15 (m, 5H, H2', H7', H8'), 1-1,65 (m, 23H, H5, H1', H9', H11', H12', H14'-H17', H22'-H25'), 1,01 (3H, s, H-19'), 0,91 (d, J=6,5 Гц, 3H, Н21'), 0,85 (d, J=6,5 Гц, 6Н, Н26' и Н27') и 0,69 (s, 3H, H18'); 13С-ЯМР (400 МГц, CDCl2/CD3OD [20/80]): 13С-ЯМР (400 МГц, CDCl3/CD3OD [20/80]): 12,32 (С18'), 19,2 (С21'), 19,76 (С19'), 21,94 (С11'), 22,91 (С27'), 23,17 (С26'), 24,7 (С23'), 25,1 (С15'), 27,22 (С5), 28,89 (С25'), 29 (С2'), 29,1 (С12'), 32,8 (С7'), 32,92 (С8'), 36,29 (С22'), 36,81 (С10'), 37,12 (С1'), 37,99 (С6), 38,11 (С1), 39,48 (С2), 40,45 (С24'), 40,80 (С16'), 46,13 (С4'), 51,23 (С9'), 57,22 (С17'), 57,80 (С14'), 62,41 (С6а), 63,4 (С6а), 70,02 (C5b), 70,63 (С2а), 72,8 (С3а), 73 (C3'), 73,18 (С9), 74,75 (C2b), 76,8 (С3а), 81 (C4b), 92,39 (C1b), 105,2 (C3'), 123,42 (С6'), 140,72 (С5'), 154,8 (C1a), 156,2 (NHCOOХол), 173,17 (С8). α-пиранозная форма: идентичные данные за исключением, 1Н-ЯМР (400 МГц, CD3OD/CDCl3 [80/20]): δH=7,04-7,05 (d, 3J1a-2a=5,6 Гц, 1H, H1a), 5,05-5,07 (m, 1H, H2a), 4,09-4,11 (m, 1H, Н3а); 1H-ЯМР (270 МГц, CD3OD): (m, 1H, H3') отсутствует, вероятно находится под пиком растворителя, подтверждено 1Н-ЯМР (300 МГц, ДМСО): δ=4,7-4,85 (m, 1H, H3'). Данные протонного резонанса подтверждали с помощью 1Н-DQF-COSY градиентного типа и TOCSY; 1Н/13С-корреляцию и DEPT 135 использовали для однозначного определения резонансов углерода. Конформацию подтверждали с помощью 1Н-фазочувствительного NOESY.
Мальтозильное производное (12f): Соединение получали с использованием раствора моногидрата D-мальтозы (30 мг, 1,8 ммоля, 5 экв.) и соединения 11 (100 мг, 0,16 ммоля) аналогично методу получения соединения 12е, осуществляя перемешивание в течение 1 дня и очистку с помощью хроматографии (CH2Cl2/MeOH/NH3 75:22:3), получали продукт 12f в виде твердого вещества белого цвета (100 мг, выход: 65%). Чистоту дополнительно подтверждали с помощью ЖХВД. Конечный продукт содержал аномер α-пиранозы (87%) и аномер β-пиранозы (13%), которые не выделяли, а характеризовали в смеси. МС (FAB+): m/z=927 [M+H]+, 765, 588, 559, 484, 369[Хол]+, 322, 290, 213, 167, 161, 143, 135, 121, 109, 95, 81, 69, 55. β-пиранозная форма: 1H-ЯМР (300 МГц, CDCl3/CD3OD [80/20]): S=7,55-7,57 (d, 3J1а-2а=5,3 Гц, 1H, H1a), 5,3 (s, 1H, H6'), 4,85-5,02 (m, 1H, H3'), 4,09-4,22 (m, 1H, H1b), 3,57-4 (m, 7 H, H3a, H4a, H5a, H4b, H5b, H6b), 3,2-3,6 (m, 8H, H2, H4, H6a, H2b, H3b, MeOH), 2,8-3,1 (m, 4H, H1, H6), 2,1-2,36 (m, 2H, H4'), 1,6-2,05 (m, 5H, H2', H7', H8'), 1-1,6 (m, 23H, H5, H1', H9', Н11', Н12', H14'-H17', H22'-H25'), 0,93 (3H, s, H-19'), 0,83 (d, J=6,5 Гц, 3Н, Н21'), 0,78 (d, J=6,5 Гц, 6Н, Н26' и Н27') и 0,6 (s, 3Н, H18'); α-пиранозная форма: идентичные данные за исключением, 1H-ЯМР (300 МГц, CD3OD/CDCl3 [80/20]): δ=6,92-6,94 (d, J=4,62 Гц, 1H, H1a), 5,02-5,15 (m, 1H, H2a), 4,04-4,08 (m, 1H, H3a)
Мальтотриозильное производное (12g): Соединение получали с использованием раствора мальтотриозы (246,4 мг, 0,46 ммоля, 7 экв.) и соединения 11 (40 мг, 0,066 ммоля) аналогично методу получения соединения 12е, осуществляя перемешивание в течение 5 дней и очистку с помощью хроматографии (СН2Cl2/МеОН/NH3 75:22:3), получали продукт 12f в виде твердого вещества белого цвета (61 мг, выход: 85%). Чистоту дополнительно подтверждали с помощью ЖХВД. Конечный продукт содержал аномер α-пиранозы (15%) и аномер β-пиранозы (85%), которые не выделяли, а характеризовали в смеси. МС (FAB+): m/z=1111 [M+Na]+, 1089 [М+Н]+, 588, 423, 391, 369 [Хол]+, 240, 171, 159, 145, 121, 105. 95, 81, 69; β-пиранозня форма: 1Н-ЯМР (300 МГц, CDCl3/МеОН [20/80]): δ=7,56-7,58 (d, J=6 Гц, 1Н, H1a), 5,2-5,27 (m, 1H, H6'), 4,9-4,95 (m, 1H, H3'), 4,2-4,45 (m, 4H, H9, H3', H2a), 4,05-4,2 (m, 2H, H1b, H1e), 2,95-4 (m, 21H, H2, H4, H6a, H3a, H5a, H4a, H2b-6b, H2c-6c, MeOH), 2,85-2,95 (m, 4H, H1, H6), 2,2-2,3 (m, 2H, H4'), 1,8-2,1 (m, 5H, H2', H7', H8'), 0,98-1,6 (m, 23H, H5, H1', H9', Н11', Н12', H14'-H17', H22'-H25'), 0,94 (3Н, s, H-19'), 0,84 (d, J=6,5 Гц, 3H, H21'), 0,78 (d, J=6,5 Гц, 6H, H26' и H27') и 0,61 (s, 3Н, H18'); α-пиранозная форма: идентичные данные за исключением, 1Н-ЯМР (300 МГц, CDCl3/МеОН[20/80]): δ=6,85 (d, J=5,6 Гц, 1H, H1a).
Мальтотетраозильное производное (12h): Соединение получали с использованием раствора D-мальтотетраозы (200 мг, 0,3030 ммоля) и соединения 11 (80 мг, 0,133 ммоля), осуществляя перемешивание в течение 5 дней и очистку с помощью хроматографии (СН2Cl2/МеОН/NH3 75:22:3), получали продукт 12h в виде твердого вещества белого цвета (67,5 мг, выход: 41%). Чистоту дополнительно подтверждали с помощью ЖХВД. Конечный продукт содержал аномер α-пиранозы (15%) и аномер β-пиранозы (85%), которые не выделяли, а характеризовали в смеси. МС (FAB+): m/z=1273 [М+Na]+, 1251 [М+Н]+ 588, 369 [Хол]+ 159, 145, 121, 109, 95, 81, 69; HR-MC (FAB+) C59H102N4O24Na: [M+Na]+, рассчитано 1273, 6782, обнаружено 1273, 6821. β-пиранозная форма: 1Н-ЯМР (300 МГц, CDCl3/МеОН[20/80]): δ=7,56-7,58 (d, 1Н, Н1а), 5,15-5,25 (m, 1Н, Н6'), 4,95-5,1 (m, 1H, Н3'), 4,38-4,5 (m, 4H, Н9, Н3', Н2а), 4,04-4,22 (m, 3Н, Н1b, Н1с, H1d), 3,1-3,95 (m, 27Н, Н2, Н4, Н6а, Н3а, Н5а, Н4а, H2b-6b, H2c-6c, H2d-6d, МеОН), 2,85-3,1 (m, 4H, H1, Н6), 2,2-2,33 (m, 2Н, Н4'), 1,75-2,1 (m, 5Н, Н2', Н7', Н8'), 1-1,6 (m, 23Н, Н5, Н1', Н9', Н11', Н12', Н14'-Н17', Н22'-Н25'), 0,92 (3Н, s, Н-19'), 0,82 (d, J=6,5 Гц, 3Н, Н21'), 0,78 (d, J=6,5 Гц, 6Н, Н26' и Н27') и 0,68 (s, 3Н, H18'); α-пиранозная форма: идентичные данные за исключением, 1Н-ЯМР (300 МГц, CDCl3/МеОН[20/80]): δ=7 (d, 1H, H1a).
Мальтогептаозильное производное (12i): Соединение получали с использованием раствора D-мальтогептозы (100 мг, 0,08673 ммоля) и соединения 11 (30 мг, 0,0497 ммоля), осуществляя перемешивание в течение 7 дней и очистку с помощью хроматографии (СН2Cl2/МеОН/NH3 75:22:3), получали продукт 12i в виде твердого вещества белого цвета (46 мг, выход: 53%). Чистоту дополнительно подтверждали с помощью ЖХВД. Конечный продукт содержал аномер α-пиранозы (15%) и аномер β-пиранозы (85%), которые не выделяли, а характеризовали в смеси. МС (FAB+): m/z=1759 [M+Na]+, 1737 [М+Н]+, 369 [Хол]+, 145, 121, 109, 95, 81. β-пиранозная форма:
1H-ЯМР (300 МГц, CDCl3/МеОН [20/80]): δ=7,53-7,58 (d, 1H, H1a), 5,35-5,37 (m, 1H, Н6'), 4,97-5,12 (m, 1H, Н3'), 4,45-4,6 (m, 4H, Н9, Н3', Н2а), 4-4,5 (m, 6H, Н1b, H1c-g), 3,1-3,9 (m, 45Н, Н2, Н4, Н6а, Н3а, Н5а, Н4а, H2b-6b, H2c-6c, H2d-6d, Н2е-6е, H2f-6f, H2g-6g, МеОН), 2,7-3 (m, 4H, H1, H6), 2,15-2,35 (m, 2Н, Н4'), 1,7-2,1 (m, 5Н, Н2', Н7', Н8'), 1-1,6 (m, 23Н, Н5, H1', H9', Н11', Н12', Н14'-Н17', H22'-H25'), 0,94 (3H, s, H-19') 0,84 (d, J=6,5 Гц, 3H, H21'), 0,77 (d, J=6,5 Гц, 6H, Н26' и Н27') и 0,63 (s, 3Н, H18'); α-пиранозная форма: идентичные данные за исключением, 1H-ЯМР (300 МГц, CDCl3/МеОН [20/80]): δ=6,9 (d, 1H, H1a).
Биологическая и биофизическая оценка:
Общие методы: Диолеоилфосфатидинилэтаноламин (ДОФЭ) получали от фирмы Avanti Lipid (Алабастер, штат Алабама, США). Плазмиду pCMVβ получали согласно методу фирмы Bayou Biolabs (Харахан, штат Луизиана, США). ДК-Хол синтезировали в лаборатории заявителей. Пептид Mu синтезировали согласно методу М.Keller с помощью стандартного метода твердофазного синтеза пептидов Merrifield, основного на применении Fmoc, с использованием смолы Wang. Все другие химические агенты имели чистоту, соответствующую чистоте реагентов.
Получение липосом: ДК-Хол (7,5 мг, 15 мкмолей) и ДОФЭ (7,5 мг, 10 мкмолей) объединяли в дихлорметане. Раствор переносили в круглодонную колбу (как правило, объемом 50 мл) и органический растворитель удаляли при пониженном давлении (роторный испаритель), получая тонкую липидную пленку, которую сушили 2-3 ч в вакууме. Затем в круглодонную колбу добавляли 4мМ HEPES-буфер, рН 7,2 (3 мл) для гидратации тонкой липидной пленки. После непродолжительной обработки ультразвуком (2-3 мин) в атмосфере аргона образовавшуюся суспензию катионных липосом (концентрация липида 5 мг/мл) экструдировали с помощью экструдера (фирма Northern lipid). Сначала экструдировали трижды через два пакета поликарбонатных фильтров (0,2 мкм), а затем десять раз через два пакета поликарбонатных фильтров (0,1 мкм) с получением небольших однослойных катионных липосом (средний диаметр 105 нм согласно данным ФКС-анализа). Концентрации липидов (примерно 4-4,8 мг/мл) определяли методом Стюарда (Stewart).
Получение комплексов липосома:Mu:ДНК (LMD) и липосома:ДНК (LD):
Первоначально получали частицы mu:ДНК (MD) путем смешения следующих ингредиентов. Маточные растворы плазмидной ДНК (как правило, 1,2 мг/мл) добавляли к интенсивно перемешиваемому разбавленному раствору пептида mu (1 мг/мл) в 4мМ HEPES-буфере, рН 7,2. Конечное соотношение mu:ДНК составляло 0,6:1 (мас./мас.), если не указано иное, конечная концентрация плазмидной ДНК составляла 0,27 мг/мл. Содержащие MD растворы затем медленно добавляли при перемешивании к суспензиям экструдированных катионных липосом (как правило, примерно 4 мг/мл), полученных согласно описанному выше методу, что позволяло получать небольшие частицы LMD с небольшим разбросом размеров (120±30 нм) по данным ФКС. Конечное соотношение липид:mu:ДНК составляло 12:0,6:1 (мас./мас./мас). Затем добавляли раствор сахарозы (100%, мас./об.) в 4 мМ HEPES-буфере, рН 7,2, получая суспензии частиц LMD в 4 мМ HEPES-буфере, рН 7,2, содержащем 10% (мас./об.) сахарозы с требуемой концентрацией ДНК (конечная концентрация ДНК, как правило, составляла 0,14 мг/мл), и хранили при -80°С. Предназначенные для экспериментов комплексы липосома:ДНК (LD) (липоплексы) получали с соотношением липид:ДНК 12:1 (мас./мас.) согласно такому же протоколу, но без добавления пептида Mu.
Оценки размеров частиц: Размеры липоплексов оценивали после 30-минутной экспозиции при 37°С в биологических средах с помощью фотонно-корреляционной спектроскопии (тип N4 плюс, фирма Coulter). Выбранная конкретная концентрация ДНК была сопоставима с концентрацией в условиях in vitro (1 мкг/мл ДНК). Применяли следующие параметры: 20°С, 0,089 сП, показатель преломления 1,33, угол 90°, 632,8 нм. Для оценки среднего размера частиц в среде Optimem использовали унимодальный анализ. Для отделения субпопуляции небольших частиц сыворотки, имеющих размер менее 50 нм, и для оценки рассчитанного размера липоплексов в среде Optimem + 10% ФТС использовали программу для определения разброса размеров частиц на основе алгоритма CONTIN.
Трансфекция клеток линии HeLa: Клетки высевали в 24-луночные культуральные планшеты в среду DMEM, дополненную 10% ФТС, и выращивали примерно до 70% конфлюэнтности в течение 24 ч при 37°С в присутствии 5% CO2. Клетки промывали ЗФР перед внесением сред для трансфекции в каждую лунку (0,5 мл раствора, содержащего 0, 50 или 100% фетальной телячьей сыворотки в среде Dubelco OptiMem). 5 мкл LMD с концентрацией 100 мкг/мл ДНК (nis βgal) трансфектировали клетки линии HeLa в течение 30 мин. Затем клетки трижды промывали ЗФР и инкубировали в течение еще 48 ч в среде DMEM, дополненной 10% ФТС, перед процессингом для экспрессии (β-Gal с использованием стандартного хемилюминесцентного набора для анализа репортерного гена (фирма Roche Diagnostics, GmbH, каталожный No. 1758241).
Результаты и их обсуждение:
Синтез неогликолипидов: Каждый представитель исследуемого семейства неогликолипидов состоит из несущего холестерин липида и молекулы олигосахарида, связанных друг с другом с помощью линкера. Весь процесс синтеза разделяли на 2 части; сначала осуществляли синтез липида, содержащего линкер, а затем осуществляли хемоселективное сочетание этого липида с выбранными молекулами сахара. Решающую роль в этой стратегии имеет образование гидроксиламина (фиг.1).
Этот синтез защищенного с помощью Boc липида (8) был разработан впервые на основе аналогичной методологии с использованием легко доступных аминоспиртов в качестве исходных продуктов с помощью стратегии применения комплементарной блокирующей группы аминогруппы. Эта опубликованная ранее методология при использовании небольшой модификации позволяет получать предназначенный для переноса генов липид, основой которого является полиамид.
Как отмечалось выше, гликозилирование гидроксиламинопроизводных позволяет рационально удовлетворить требования к системе синтеза. Имеющийся в продаже гидрохлорид O-(карбоксиметил)гидроксиламина сначала защищали с помощью Boc, а затем подвергали взаимодействию с N-гидроксисукцинимидом и N,N'-дициклогексилкарбодиимидом (ДЦК) с получением соответствующего активированного сложного эфира. Это соединение немедленно обрабатывали in situ липидом (8) в ТГФ при рН 8, получая защищенный гидроксиламин. После очень простого удаления защитной группы с помощью трифторуксусной кислоты синтез гидроксиламинолипида (11) был завершен.
На этой стадии была исследована эффективной примененного хемоселективного сочетания путем взаимодействия липида (11) с целым рядом имеющихся в продаже незащищенных олигосахаридов. Эту реакцию осуществляли в мягких условиях с использованием в качестве растворителя системы ДМФ и водный буфер на основе уксусной кислоты с рН 4(1:1), что облегчает растворимость как сахара, так и липида. Как показано на фиг.2, реактанты находились в динамическом равновесии с открытым имеющим решающее значение протонированным промежуточным продуктом. Для сдвига равновесия в сторону образования продукта добавляли избыток сахара. Было обнаружено, что из-за амфифильной природы неогликолипидного продукта, выделение в процессе обработки было затруднено из-за образования мицелл и пены. Проблемы, связанные с растворимостью, затрудняли также выделение, очистку и процесс аналитического контроля. Продолжительность реакции и выходы варьировали в зависимости от примененного углевода, (таблица 1).
Выходы, продолжительность реакций и диастереоселективность продукта гликозилирования соединения 11.
Конформация неогликолида: Конформации углеводов можно оценивать с помощью ЯМР в растворе. Наиболее ценные данные о конформации аномерного центра (С1а), вероятно, можно получить на основе 3J13C1a-H1a. Абсолютное значение этой константы сочетания зависит от ориентации связи углерод-водород относительно неподеленных пар кольцевого кислорода, величины отрицательного электрического заряда заместителя на С1 и природы электроотрицательных заместителей, присоединенных к остальной части молекулы. Различия 3J13C1-H1 между α- и β-аномерами пираноз можно использовать для определения конфигурации аномеров. Первоначально было установлено, что 1J(C1-H1eq)>1J(C1-H1ax), причем различие составляет 10 Гц. 1J(C1-H1eq) составляет, как правило, примерно 170 Гц, a 1J(C1-H1ax) примерно 160 Гц. Более высокие значения были получены, когда 0-1 заменяли несущим более высокий отрицательный электрический заряд элементом, таким как хлор или фтор, но различие в 10 Гц, как правило, было обнаружено и в этих случаях. Были определены константы сочетания углерод-водород фуранозидов, и в этом случае 1J(C1-H1eq)>1J(C1-H1ax), но различие существенно ниже (1-3 Гц).
Характеристики описаны ниже на примере маннозы, но эту же процедуру анализа использовали и для других сахаридов, если в этом случае можно было применять ЯМР-анализ. Можно предположить наличие 4 различных кольцевых структур (фиг.3). Следует ожидать, что пиранозные формы должны быть более предпочтительными, чем фуранозные формы с точки зрения стерического строения. Поэтому из двух изученных с помощью ЯМР соединений основным, вероятно, является пираноза. Второе изученное соединение не может быть связано с мутаротационным равновесием, поскольку фазочувствительный анализ NOESY не показал наличие кросс-пика между двумя сигналами С1а (что доказывает присутствие другой молекулы). Таким образом, это соединение не относится к фуранозной форме, поскольку не было обнаружено сдвига 13С5а, а у 13С1а не была удалена защита, что было продемонстрировано для аналогичных замещенных эквивалентов фуранозы.
Было установлено, что 1J13C1a-H1a=167 Гц для основного соединения и 1J13C1a-H1a=177 Гц для минорного (второго) соединения. Абсолютное значение этого 1J13C1-H1 превышает на 10 Гц соответствующее значение, ожидаемое для классической конформации 4C1, однако это объясняется высоким отрицательным зарядом O-замещенной гидроксиламинной группы, что приводит к слабой деформации основной структуры. Для пиранозных колец было установлено, что [1J(C1-H1eq)-1J(C1-H1ax)]≈10 Гц, отсюда можно легко сделать вывод о том, что основное соединение представляет собой β-аномер (H1ах), а минорное соединение представляет собой α-аномер (H1eq).
Результаты 1Н-фазочувствительного анализа NOESY подтвердили этот вывод. Для основного соединения наблюдался ядерный эффект Оверхаузера между H1а и Н2а в сумме с Н3а. Принимая во внимание подробно описанную выше структуру, это соединение не может представлять собой α-пиранозильный аномер, поскольку экваториальный H1 не может взаимодействовать в пространстве с Н3, в то время как в β-аномере вполне могут происходить такие взаимодействия. Для H1а второго соединения ядерный эффект Оверхаузера не был обнаружен из-за недостаточной чувствительности метода. Таким образом, данные, полученные на основе 1J13C1-H1 и NOESY, позволяют сделать вывод о том, что были получены две формы (α/β, 20/80) маннозы пиранозы.
Очень близкое соотношение аномеров (β/α), обнаруженное для неогликолипидов, не является неожиданным (таблица 1), все сахара, кроме манозы, имеют экваториальный гидроксил в положении С2. Соотношения, полученные для этого последнего соединения (маннозы) являются неожиданными, поскольку по имеющимся в литературе данным β-аномер, как правило, стерически является менее предпочтительным по сравнению с α-аномером. Возможным объяснением этого факта является то, что реакция может регулироваться определенными вторичными взаимодействиями (водородная связь) между сахаром и гидроксиламинным линкером, что стабилизирует β-аномер (это согласуется с данными о том, что ЯМР-сигнал β-аномера всегда является менее защищенным по сравнению с α-аномером). Маловероятно, чтобы эта смесь аномеров синтезированных гликолипидов оказывала большое влияние на изучаемые виды биологической активности липосомных конструкций, поэтому не были предприняты попытки тщательного разделения этих диастереоизомеров с помощью препаративной жидкостной хроматографии высокого давления.
Биологическая применимость: Гликомодификация LMD основана на природной способности мицелярной суспензии включаться в липидные мембраны Сначала LMD получали с помощью стандартного протокола, а затем суспензию синтезированных мицелл неогликолипидов в Hepes-буфере, 4 мМ, рН 7 добавляли в LMD и инкубировали в течение 30 мин при комнатной температура и хранили до применения при -80°С. Различные процентные концентрации всех полученных неогликолипидов тестировали в отношении их стабилизирующего действия, но только у соединений с более длинной цепью (мальтотетраоза 12h и мальтогептоза 12i) обнаружена выраженная активность в концентрации ниже 10% (данные не приведены).
Стабилизирующее действие модифицированного неогликолипидом LMD, было продемонстрировано путем включения соединения 12h или 12i в концентрации 7,5 мол.%. Как известно, липидные слои композиций, основой которых являются липосомы, агрегируют после обработки солью или сывороткой. Это явление можно оценивать путем измерения увеличения среднего размера частиц после фиксированного промежутка времени; любая стабилизация частиц LMD должна проявляться в снижении этого параметра. Он был выбран для оценки размера липоплексов с помощью фотонно-корреляционной спектроскопии (ФКС) после 30-минутной экспозиции при 37°С в среде OptiMem или OptiMem + 10% ФТС, что имитирует стандартные условия in vitro. С помощью ФКС оказалось невозможно анализировать воздействие более высоких концентраций сыворотки, эти условия оказались слишком экстремальными для того, чтобы позволять осуществлять значимые измерения. На фиг.4 показано увеличение размера этих липоплексов (в %).
Полученные результаты свидетельствуют о четко выраженной стабилизацией частиц при использовании LMD и стандартной композиции липосом. Введение неогликолипидов в концентрации 7,5% обеспечивает преимущество при использовании среды OptiMem и 10% сыворотки. Включение соединения 12i обусловливает наиболее высокую эффективность. Этот результат свидетельствует о необходимости использования длинных цепей углеводов для создания эффективных молекулярных щеток на поверхности их катионных липидных слоев.
Даже если продемонстрирована определенная степень стабилизации, как правило, это приводит к снижению аффинности положительно заряженного LMD с отрицательно заряженной клеточной мембраной, вызывая снижение способности конструкции к трансфекции. Однако в этом случае результаты опытов по трансфекции in vitro свидетельствуют о повышении эффективности трансфекции, связанной с модификацией неогликолипидами, как без сыворотки, так и в присутствии 50% сыворотки (фиг.5). Этот результат связан с небольшим диапазоном защитного действия вследствие того, что эти неогликолипиды препятствуют взаимодействиям, основанным на силах Ван дер Ваальса с небольшим радиусом действия, между липидными бислоями, имеющими одинаковые полярности, но не влияют на взаимодействия, имеющие больший радиус действия, между мембранами, несущими разные заряды. Агрегация, индуцированная сывороткой, первично базируется на взаимодействиях LMD с отрицательно заряженными протеинами, благоприятное действие неогликолипидов по изобретению снижалось также при увеличении процентного содержания сыворотки (отсутствие выраженного преимущества в 100% сыворотке).
Все упомянутые в описании публикации включены в него в качестве ссылки. Как должно быть очевидно специалистам в данной области, могут быть сделаны различные модификации и вариации описанных методов и системы по изобретению без отклонения от сущности и объема изобретения. Хотя изобретение описано со ссылкой на конкретные предпочтительные варианты осуществления изобретения, очевидно, что заявленный объем изобретения не ограничен в большой степени этими конкретными вариантами его осуществления. Так, под объем приведенной ниже формулы изобретения подпадают различные модификации осуществления изобретение, которые очевидны специалистам в биологии, химии или родственных областях.
| название | год | авторы | номер документа |
|---|---|---|---|
| ИНГИБИТОРЫ АРГИНАЗЫ И ИХ ТЕРАПЕВТИЧЕСКИЕ ПРИМЕНЕНИЯ | 2011 |
|
RU2586219C2 |
| ДИГИДРОНАФТИРИДИНЫ И РОДСТВЕННЫЕ СОЕДИНЕНИЯ, ПОДХОДЯЩИЕ В КАЧЕСТВЕ ИНГИБИТОРОВ КИНАЗ ДЛЯ ЛЕЧЕНИЯ ПРОЛИФЕРАТИВНЫХ ЗАБОЛЕВАНИЙ | 2018 |
|
RU2804468C2 |
| Новые 3-индол замещенные производные, фармацевтические композиции и способы применения | 2016 |
|
RU2672252C1 |
| ИНГИБИТОРЫ ФУРИНА | 2019 |
|
RU2799824C2 |
| ПРОИЗВОДНЫЕ АЗОТСОДЕРЖАЩИХ ГЕТЕРОЦИКЛИЧЕСКИХ СОЕДИНЕНИЙ, СПОСОБЫ ИХ ПОЛУЧЕНИЯ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ИХ ОСНОВЕ И СПОСОБЫ ЛЕЧЕНИЯ ВОСПАЛИТЕЛЬНЫХ ЗАБОЛЕВАНИЙ И ЗАБОЛЕВАНИЙ ДЫХАТЕЛЬНЫХ ПУТЕЙ | 2001 |
|
RU2265011C2 |
| ПИРАЗОЛБЕНЗОДИАЗЕПИНЫ В КАЧЕСТВЕ ИНГИБИТОРОВ CDK2, ПРОМЕЖУТОЧНЫЕ СОЕДИНЕНИЯ ДЛЯ ИХ ПОЛУЧЕНИЯ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ, ИХ СОДЕРЖАЩАЯ | 2000 |
|
RU2249593C2 |
| Новые модулирующие регуляцию дыхания соединения и способы их получения и применения | 2016 |
|
RU2734506C2 |
| ГЕКСАЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ, СОДЕРЖАЩАЯ ИХ И ИХ ПРИМЕНЕНИЕ В КАЧЕСТВЕ ПРОТИВООПУХОЛЕВЫХ СРЕДСТВ, СПОСОБ ПОЛУЧЕНИЯ ГЕКСАЦИКЛИЧЕСКИХ СОЕДИНЕНИЙ | 2002 |
|
RU2299212C2 |
| ПРОИЗВОДНЫЕ 1,3,4-ОКСАДИАЗОЛА В КАЧЕСТВЕ ИНГИБИТОРА ГИСТОНДЕАЦЕТИЛАЗЫ 6 И СОДЕРЖАЩАЯ ИХ ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 2021 |
|
RU2822180C1 |
| ЗАМЕЩЕННЫЕ СОЕДИНЕНИЯ ПИРИДИН АЗОЛОПИРИМИДИН-5-(6Н)-ОНА | 2013 |
|
RU2653054C2 |
Описан способ получения соединения формулы
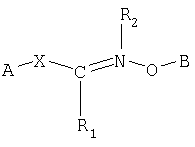
где В - липид, предпочтительно холестерин, А - представляющий интерес фрагмент (ПИФ), Х - линкер, действием соединения I
На соединение (II) в смеси или связанные с нуклеотидной последовательностью или фармацевтически активным агентом, R1-H и R2 - неподеленная пара или Н. Полученные соединения предназначены для лечения генетического нарушения или заболевания. 2 н. и 21 з.п. ф-лы, 1 табл., 6 ил.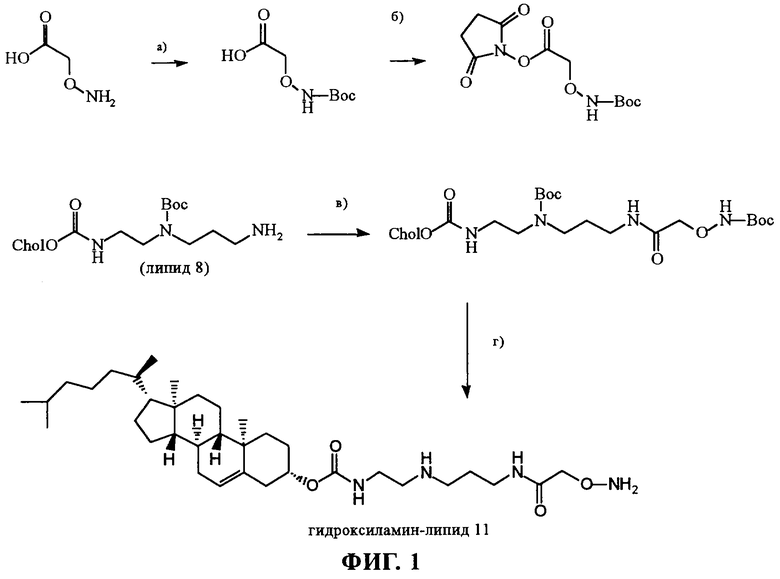
который заключается во взаимодействии (I) соединения формулы
(II) и соединения формулы
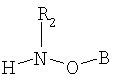
в смеси или связанными с нуклеотидной последовательностью или фармацевтически активным агентом,
где В обозначает липид;
А обозначает представляющий интерес фрагмент (ПИФ);
Х обозначает необязательную линкерную группу;
R1 обозначает Н;
R2 обозначает неподеленную пару или водород.
(II) соединение формулы
(III) нуклеотидную последовательность или фармацевтически активный агент,
где В обозначает липид;
А обозначает представляющий интерес фрагмент (ПИФ);
Х обозначает необязательную линкерную группу;
R1 обозначает Н;
R2 обозначает неподеленную пару или водород.
| US 5283185, A, 01.02.1994 | |||
| US 5510510, A, 23.04.1996 | |||
| Способ получения на волокне оливково-зеленой окраски путем образования никелевого лака азокрасителя | 1920 |
|
SU57A1 |

