Область, к которой относится изобретение
Данное изобретение относится к контролированию беспозвоночных растительноядных сельскохозяйственных вредителей, таких как членистоногие вредители, путем контактирования посевного материала (сеянцев) растения или частей посевного материала (сеянцев) с определенными антраниламидами, и к содержащим антраниламиды композициям, которые наносятся на ростки.
Предпосылки создания изобретения
Для достижения высокой урожайности чрезвычайно важен контроль за беспозвоночными вредителями, такими как членистоногие. Повреждение беспозвоночными вредителями растущих и находящихся на хранении сельскохозяйственных культур может вызывать значительное снижение продуктивности и, следовательно, приводить в результате к увеличенным затратам для потребителя. Также важен контроль за беспозвоночными вредителями в лесных хозяйствах, у тепличных культур, декоративных растений и растений в питомниках.
Растения подвергаются повреждению беспозвоночными вредителями на всех стадиях роста, начиная с семян или других сеянцев, таких как луковицы, клубни, корневища, клубнелуковицы, стебля и черенков листа, и заканчивая зрелыми растениями. Помимо расхода материалов, усилия и времени, необходимые для применения веществ для контроля за беспозвоночными вредителями, делают нежелательными повторные обработки. В идеале однократная обработка растения на стадии сеянца защитила бы растение от беспозвоночных вредителей на протяжении всей его жизни.
Известны разнообразные методики для обработки посевного материала веществами для защиты растений. Они включают в себя пропитывание сеянцев артроподицид-содержащими растворами, покрытие их пленками, дражировочными материалами и тому подобным, содержащими артроподицидные композиции и применение артроподицидных веществ для ростовой среды окружающей сеянцы. В то время как некоторые вещества могут эффективно защищать сеянцы от определенных растительноядных беспозвоночных вредителей, необходимы новые вещества, которые являются более эффективными или обладают более широким спектром активности, менее дорогостоящие, менее токсичные, более безопасные для окружающей среды или имеющие различные механизмы действия.
Особая потребность имеется в обработке регулирующей беспозвоночных вредителей, которые могут защищать растение не только на стадии сеянца, а также позже при его развитии. Для достижения этой цели требуются соединения, которые проявляют активность против беспозвоночных вредителей и могут эффективно перемещаться от локуса сеянца вверх по растущим стеблям, листьям и другим частям растения. Кроме того, данные соединения должны обладать высокой активностью против беспозвоночных вредителей для компенсации разведения, вызванного увеличивающейся массой растения. Также, данные соединения не должны быстро разрушаться и терять свою биологическую силу в среде, окружающей сосудистые ткани растения. Сочетание таких свойств встречается редко. Здесь описаны способы обработки сеянцев, эффективные для защиты от растительноядных беспозвоночных вредителей не только ростков, но также растения на более поздних стадиях роста.
Краткое изложение сущности изобретения
Данное изобретение включает в себя соединения Формулы I, их N-оксиды и их соли, приемлемые для использования в сельском хозяйстве.

где A и B представляют собой независимо О или S;
R1 представляет собой H, C1-C6 алкил, С2-С6 алкоксикарбонил или С2-С6 алкилкарбонил;
R2 представляет собой H или C1-C6 алкил;
R3 представляет собой H; C1-C6 алкил, С2-С6 алкенил, С2-С6 алкинил, или С3-С6 циклоалкил, каждый необязательно замещен одним или более заместителями, выбранными из группы, состоящей из: галогена, CN, NO2, гидрокси, C1-C4 алкила, C1-C4 алкокси, C1-C4 галогеналкокси, C1-C4 алкилтио, C1-C4 алкилсульфинила, C1-C4 алкилсульфонила, C2-C6алкоксикарбонила, C2-C6алкилкарбонила, С3-С6 триалкилсилила, фенила, фенокси, 5-членных гетероароматических колец, и 6-членных гетероароматических колец; каждый фенил, фенокси, 5-членное гетероароматическое кольцо, и 6-членное гетероароматическое кольцо необязательно замещено одним-тремя заместителями, независимо выбранными из группы, состоящей из C1-C4 алкила, С2-С4 алкенила, С2-С4 алкинила, С3-С6 циклоалкила, C1-C4 галогеналкила, С2-С4 галогеналкенила, С2-С4 галогеналкинила, С3-С6 галогенциклоалкила, галогена, CN, NO2, C1-C4 алкокси, C1-C4 галогеналкокси, C1-C4 алкилтио, C1-C4 алкилсульфинила, C1-C4 алкилсульфонила, C1-C4 алкиламино, C2-C8 диалкиламино, C3-C6 циклоалкиламино, C4-C8 (алкил)(циклоалкил)амино, С2-С4 алкилкарбонила, С2-C6 алкоксикарбонила, C2-C6 алкиламинокарбонила, C3-C8 диалкиламинокарбонила и С3-С6 триалкилсилила; C1-C4 алкокси; C1-C4 алкиламино; C2-C8 диалкиламино; C3-C6 циклоалкиламино; C2-C6 алкоксикарбонила или С2-C6 алкилкарбонила;
R4 представляет собой H, C1-C6 алкил, C2-C6 алкенил, C2-C6 алкинил, C3-C6 циклоалкил, C1-C6 галогеналкил, CN, галоген, C1-C4 алкокси, C1-C4 галогеналкокси или NO2;
R5 представляет собой H, C1-C6 алкил, C1-C6 галогеналкил, C1-C4 алкоксиалкил, C1-C4 гидроксиалкил, C(О)R10, CO2R10, C(О)NR10R11, галоген, C1-C4 алкокси, C1-C4 галогеналкокси, NR10R11, N(R11)C(O)R10, N(R11)CO2R10 или S(О)nR12;
R6 представляет собой H, C1-C6 алкил, C1-C6 галогеналкил, галоген, CN, C1-C4 алкокси или C1-C4 галогеналкокси;
R7 представляет собой C1-C6 алкил, C2-C6 алкенил, C2-C6 алкинил, C3-C6 циклоалкил, C1-C6 галогеналкил, C2-C6 галогеналкенил, C2-C6 галогеналкинил или C3-C6 галогенциклоалкил; или R7 представляет собой фенильное кольцо, бензильное кольцо, 5- или 6-членное гетероароматическое кольцо, нафтильную кольцевую систему или ароматическую 8-, 9- или 10-членную конденсированную гетеробициклическую кольцевую систему, каждое кольцо, или кольцевая система, замещено одним-тремя заместителями, независимо выбранными из R9;
R8 представляет собой H, C1-C6 алкил, C1-C6 галогеналкил, галоген, C1-C4 алкокси или C1-C4 галогеналкокси;
каждый R9 представляет собой независимо C1-C4 алкил, С2-С4 алкенил, С2-С4 алкинил, С3-С6 циклоалкил, C1-C4 галогеналкил, С2-С4 галогеналкенил, С2-С4 галогеналкинил, С3-С6 галогенциклоалкил, галоген, CN, NO2, C1-C4 алкокси, C1-C4 галогеналкокси, C1-C4 алкилтио, C1-C4 алкилсульфинил, C1-C4 алкилсульфонил, C1-C4 алкиламино, C2-C8 диалкиламино, C3-C6 циклоалкиламино, C4-C8 (алкил)(циклоалкил)амино, С2-С4 алкилкарбонил, C2-C6 алкоксикарбонил, C2-C6 алкиламинокарбонил, C3-C8 диалкиламинокарбонил или С3-С6 триалкилсилил;
R10 представляет собой H, C1-C4 алкил или C1-C4 галогеналкил;
R11 представляет собой H или C1-C4 алкил;
R12 представляет собой C1-C4 алкил или C1-C4 галогеналкил; и
n равно 0, 1 или 2.
Данное изобретение предлагает способ защиты сеянца или выросшего из него растения от беспозвоночных вредителей. Данный метод включает в себя контактирование сеянца или локуса сеянца с биологически эффективным количеством соединения Формулы I, его N-оксида или его соли, приемлемой для использования в сельском хозяйстве.
Данное изобретение также предлагает сеянец, содержащий биологически эффективное количество соединения I, его N-оксида или его соли, приемлемой для использования в сельском хозяйстве.
Данное изобретение также предлагает сеянец, контактировавший с биологически эффективным количеством соединения I, его N-оксида или его соли, приемлемой для использования в сельском хозяйстве.
Данное изобретение далее предлагает композицию для контроля за беспозвоночными вредителями для покрытия сеянца, содержащую биологически эффективное количество соединения Формулы I, его N-оксида или его соли, приемлемой для использования в сельском хозяйстве и пленкообразователь или адгезивное вещество.
Подробное описание изобретения
Указанный в настоящем изобретении и в формуле изобретения термин "посадочный материал" или "сеянец" означает семя или часть растения, способную к регенерации. Термин "часть растения, способная к регенерации" означает часть растения, иную чем семена, из которой может вырасти целое растение, или регенерировать, когда данную часть растения помещают в садовую или сельскохозяйственную ростовую среду, такую как увлажненная почва, торфяной мох, песок, вермикулит, перлит, минеральное волокно, стекловолокно, волокно скорлупы кокосового ореха, волокно древовидного папоротника и подобное, или даже полностью жидкую среду, такую как вода. Части растения, способные к регенерации, как правило включают в себя корневища, клубни, луковицы и клубнелуковицы таких геофитных видов растений, как картофель, батат, ямс, лук, георгин, тюльпан, нарцисс и т.д. Способные к регенерации части растения включают в себя части растения, которые разделяют (например, режут) для сохранения их способности вырасти в новое растение. Таким образом, части растения, способные к регенерации включают в себя жизнеспособные части корневищ, клубней, луковиц и клубнелуковиц, которые сохраняют меристемные ткани, как например глазки. Способные к регенерации части растения также могут включать в себя другие части растения, такие как разрезанные или разделенные стебли и листья, из которых могут быть выращены некоторые виды растений с использованием садоводческой или сельскохозяйственной ростовой среды. Указанный в настоящем описании и формуле изобретения, если не указано иначе, термин "семена" включает в себя как непророщенные семена, так и пророщенные семена, в которых семенная кожура (покрытие семени) по-прежнему окружает часть протастающего ростка и корня.
В вышеуказанных перечислениях, термин "алкил", используемый или самостоятельно, или в составе слов, таких как "алкилтио" или "галогеналкил", включает в себя алкил с прямой цепью или разветвленный алкил, такой как метил, этил, н-пропил, изо-пропил, или различные изомеры бутила, пентила или гексила. "Алкенил" включает в себя алкены с прямой цепью или разветвленные алкены, такие как 1-пропенил, 2-пропенил, различные изомеры бутенила, пентенила и гексенила. "Алкенил" также включает в себя полиены, такие как 1,2-пропадиенил и 2,4-гексадиенил. "Алкинил" включает в себя алкинилы с прямой цепью или разветвленные алкинилы, такие как 1-пропинил, 2-пропинил и различные изомеры бутинила, пентинила и гексинила. "Алкинил" также может включать в себя части, содержащие повторяющиеся тройные связи, такие как 2,5-гексадиинил. "Алкокси" включает в себя например метокси, этокси, н-пропилокси, изопропилокси и различные бутокси, пентокси и гексилокси изомеры. "Алкоксиалкил" означает алкокси замещение на алкил. Примеры "алкоксиалкила" включают в себя CH3OCH2, CH3OCH2CH2,CH3CH2OCH2,CH3CH2CH2CH2OCH2 и CH3CH2OCH2CH2. "Алкилтио" включает в себя части алкилтио разветвленные, или с прямой цепью, такие как метилтио, этилтио, и различные пропилтио, бутилтио, пентилтио и гексилтио изомеры. "Циклоалкил" включает в себя, например, циклопропил, циклобутил, циклопентил и циклогексил.
Термин "гетероциклическое кольцо" или "гетероциклическая кольцевая система" обозначает кольца или систему колец, в которых по меньшей мере один атом кольца не является углеродом и содержит от 1 до 4 гетероатомов независимо выбранных из группы, состоящей из азота, кислорода и серы, при условии, что каждое гетероциклическое кольцо содержит не более 4 азотов, не более 2 кислородов и не более 2 атомов серы. Гетероциклическое кольцо может быть присоединено через любой доступный углерод или азот путем замены водорода на указанный углерод или азот. Термин "ароматическая кольцевая система" обозначает полностью ненасыщенные карбоциклы и гетероциклы, в которых по меньшей мере одно кольцо полициклической кольцевой системы является ароматическим (где ароматичность указывает на то, что удовлетворяет правилу Хюккеля для кольцевой системы). Термин "гетероароматическое кольцо" означает полностью ароматические кольца, в которых по меньшей мере один атом кольца не является углеродом и содержит от 1 до 4 гетероатомов, независимо выбранных из группы, состоящей из азота, кислорода и серы, при условии, что каждое гетероциклическое кольцо содержит не более 4 азотов, не более 2 кислородов и не более 2 атомов серы (где ароматичность указывает на то, что выполняется правило Хюккеля). Гетероциклическое кольцо может быть присоединено через любой доступный углерод или азот путем замены водорода на указанный углерод или азот. Термин "ароматическая гетероциклическая кольцевая система" включает в себя полностью ароматические гетероциклы и гетероциклы, в которых по меньшей мере одно кольцо полициклической кольцевой системы является ароматическим (где термин "ароматический" означает, что выполняется правило Хюккеля). Термин "конденсированная гетеробициклическая кольцевая система" включает в себя кольцевую систему, состоящую из двух конденсированных колец, в которых по меньшей мере один атом кольца не является углеродом и может быть ароматическим или неароматическим, как определено выше.
Термин "галоген", либо самостоятельно, либо в составе слов, таких как "галогеналкил", включает в себя фтор, хлор, бром или йод. Далее, при использовании в составе слов, таких как "галогеналкил", указанный алкил может быть частично или полностью замещен атомами галогена, которые могут быть одинаковыми или различными. Примеры "галогеналкила" включают в себя F3C, ClCH2, CF3CH2 и CF3CCl2. Термины "галогеналкенил", "галогеналкинил", "галогеналкокси" и подобные определены аналогично термину "галогеналкил". Примеры "галогеналкенила" включают в себя (Cl)2C=CHCH2 и CF3CH2CH=CHCH2. Примеры "галогеналкинила" включают в себя HC≡CCHCl, CF3C≡C, CCl3C≡C и FCH2С≡CCH2. Примеры "галогеналкокси" включают в себя CF3О, CCl3CH2O, HCF2CH2CH2O и CF3CH2О.
Общее число атомов углерода в замещающей группе указано префиксом "Ci-Cj", где i и j представляют собой числа от 1 до 8. Например, C1-C4 алкилсульфонил означает метилсульфонил по бутилсульфонил; C2 алкоксиалкил означает CH3ОСН2; С3 алкоксиалкил означает, например, CH3CH(OCH3), CH3OCH2CH2 или CH3CH2OCH2; и С4 алкоксиалкил означает различные изомеры алкильной группы, замещенные алкокси группой, содержащей все черыре атома углерода, примеры включают в себя CH3CH2CH2OCH2 и CH3CH2OCH2CH2. В перечисленном выше, в случае, когда соединение Формулы I содержит герероциклическое кольцо, все заместители связаны с этим кольцом через любой доступный атом углерода или азота путем замещения водорода на указанном атоме углерода или азота.
В том случае, когда группа имеет заместитель, который может быть водородом, например R3, затем, в том случае, когда этот заместитель рассматривается как водород, это признается эквивалентом указанной незамещенной группы.
Соединения Формулы I находятся в виде одного или более стереоизомеров. Различные стереоизомеры включают в себя энантиомеры, диастереоизомеры, атропизомеры и геометрические изомеры. Специалисту в данной области будет понятно, что один стереоизомер может быть более активным и/или может проявлять полезное действие, когда обогащен относительно других стереоизомеров (стереоизомера) или когда отделен от других стереоизомеров (стереоизомера). Кроме того, специалист в данной области знает как разделить, обогатить и/или выборочно приготовить указанные стереоизомеры. Соответственно, соединения данного изобретения могут быть представлены в виде смеси стереоизомеров, отдельных стереоизомеров или в виде оптически активной формы.
Соли соединений Формулы I включают в себя кислотно-аддитивные соли (соли присоединения кислот) с неорганическими или органическими кислотами, такими как бромистоводородная, хлористоводородная, азотная, фосфорная, серная, уксусная, масляная, фумаровая, молочная, малеиновая, малоновая, щавелевая, пропионовая, салициловая, винная, 4-толуолсульфоновая или валериановая кислоты.
Способы, сеянцы и композиции данного изобретения предпочтительные из соображений стоимости, простого химического синтеза или применения и/или биологической эффективности, включают в себя следующие предпочтительные соединения:
Предпочтение 1. Соединение формулы I где A и B оба представляют собой О;
R7 представляет собой фенильное кольцо или 5- или 6-членное гетероароматическое кольцо, выбранное из группы, состоящей из

каждое кольцо необязательно замещено одним-тремя заместителями, независимо выбранными из R9;
Q представляет собой O, S, NH или NR9; и
W, X, Y и Z независимо представляют собой N, CH или CR9, при условии, что в J-3 и J-4 по меньшей мере один из W, X, Y или Z представляет собой N.
Предпочтение 2. Соединение Предпочтения 1, где
R1, R2 и R8 все представляют собой H;
R3 представляет собой C1-C4 алкил необязательно замещенный галогеном, CN, OCH3 или S(О)pCH3;
группа R4 присоединена в положении 2;
R4 представляет собой CH3, CF3, OCF3, OCHF2, CN или галоген;
R5 представляет собой H, CH3 или галоген;
R6 представляет собой CH3, CF3 или галоген;
R7 представляет собой фенил или 2-пиридинил, каждый необязательно замещен; и
p равно 0, 1 или 2.
Предпочтение 3. Соединение Предпочтения 2, где R3 представляет собой C1-C4 алкил и R6 представляет собой CF3.
Предпочтение 4. Соединение Предпочтения 2, где R3 представляет собой C1-C4 алкил и R6 представляет собой Cl или Br.
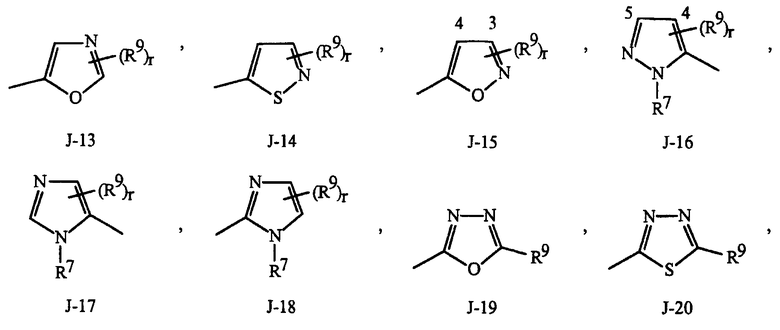
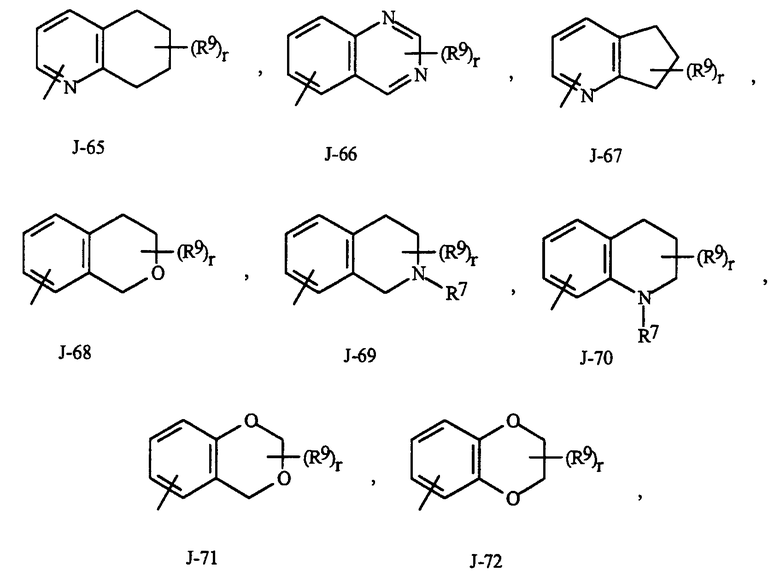
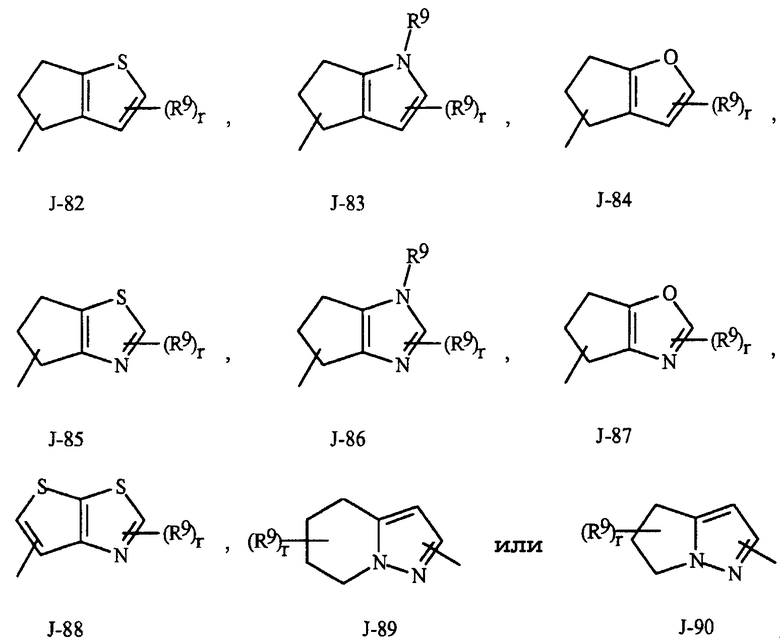
Как отмечено выше, R7 представляет собой (среди прочего) фенил, бензил, 5- или 6-членное гетероароматическое кольцо, нафтильную кольцевую систему или ароматическую 8-, 9- или 10-членную конденсированную гетеробициклическую кольцевую систему, каждое кольцо или кольцевая система необязательно замещены одним-тремя заместителями, независимо выбранными из R9. Термин "необязательно замещенные" в связи с этими R7 группами относится к группам, которые незамещены или имеют по меньшей мере один неводородный заместитель, который не гасит регулирующую активность в отношении беспозвоночных вредителей, которой обладает незамещенный аналог. Следует отметить, что с J-1 по J-4 ниже означают 5- или 6-членные гетероароматические кольца. Примером фенильного кольца, необязательно замещенного от 1 до 3. R9 является кольцо, показанное как J-5 в Представлении 1, где r представляет собой целое число от 0 до 3. Примером бензильного кольца, необязательно замещенного от 1 до 3. R9 является кольцо, показанное как J-6 в Представлении 1, где r представляет собой целое число от 0 до 3. Пример нафтильной кольцевой системы необязательно замещенной от 1 до 3. R9 показан в виде J-59 в Представлении 1, где r представляет собой целое число от 0 до 3. Примеры 5- или 6-членного гетероароматического кольца необязательно замещенного от 1 до 3. R9 включают в себя кольца с J-7 по J-58 показанные в Представлении 1, где r представляет собой целое число от 0 до 3. Следует отметить, что с J-7 по J-26 представляют собой примеры J-1, с J-27 по J-41 представляют собой примеры J-2, и с J-46 по J-58 представляют собой примеры J-3 и J-4. Атомы азота, требующие замещения до заполнения их валентности, замещены H или R9. Следует отметить, что некоторые J группы могут быть замещены только менее чем 3. R9 группами (например, J-19, J-20, J-23 по J-26 и J-37 по J-40 могут быть замещены только одним R9). Примеры ароматических 8-, 9- или 10-членных конденсированных гетеробициклических кольцевых систем, необязательно замещенных от 1 до 3. R9 включают в себя J-60 по J-90, показанные в Представлении 1, где r представляет собой целое число от 0 до 3. Хотя R9 группы показаны в структурах с J-5 по J-90, отмечено, что их присутствие не обязательно, поскольку они являются необязательными заместителями. Следует отметить, что в том случае, когда место присоединения между (R9)r и J группой показано плавающим, (R9)r может быть присоединен к любому доступному атому углерода J группы. Следует отметить, что в том случае, когда место присоединения на J группе показано плавающим, данная J группа может быть присоединена к остатку соединения Формулы I через любой доступный атом углерода J группы путем замещения атома водорода.
Представление 1





Для получения соединений Формулы I могут быть использованы один или более из следующих способов и вариантов, описанных в Схемах 1-22. Определения для A, B и R1 по R9 в соединениях Формул 2-40, приведенных ниже, даны ранее в разделе «Краткое изложение сущности изобретения», если не указано другое. Соединения Формул Ia-d, 2a-d, 3a, 4a-d, 5a-b, 17a-c, 18a и 32a-b представляют собой различные подгруппы соединений Формул I, 2, 3, 4, 5, 17, 18 и 32. В данных схемах, Het представляет собой часть молекулы, которая показана ниже:

Типичный способ получения соединения Формулы Ia описан в Схеме 1.
Схема 1
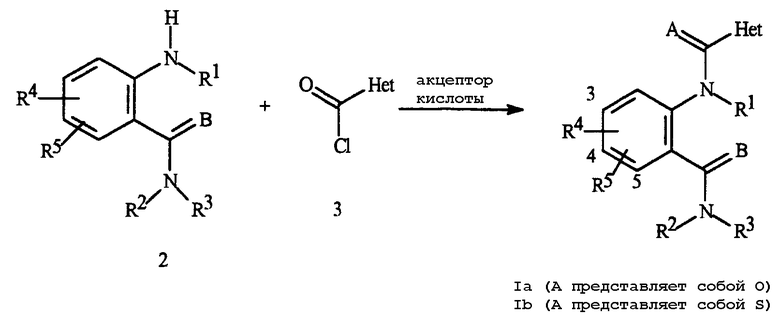
Способ Схемы 1 включает в себя взаимодействие амина Формулы 2 с хлорангидридом кислоты Формулы 3 в присутствии акцептора кислоты с получением соединения Формулы Ia. Типичные акцепторы кислоты включают в себя аминные основания, такие как триэтиламин, N,N-диизопропилэтиламин и пиридин; другие акцепторы включают в себя гидроксиды, такие как гидроксид натрия и калия и карбонаты, такие как карбонат натрия и карбонат калия. В отдельных случаях полезно использовать акцепторы кислоты на полимерном носителе, такие как полимерно-связанный N,N-диизопропилэтиламин и полимерно-связанный 4-(диметиламино)пиридин. Взаимодействие может быть проведено в подходящем инертном растворителе, таком как тетрагидрофуран, диоксан, диэтиловый эфир или дихлорметан с получением анилида Формулы Ia.
Тиоамид Формулы Ib может быть получен на последующей стадии из соответствующего амида Формулы Ia обработкой одним из различных стандартных реагентов, переносящих тиогруппу, включающих в себя фосфористый пентасульфид и реактив Лавессона (Lawesson's) (2,4-бис(4-метоксифенил)-1,3-дитиа-2,4-дифосфетан-2,4-дисульфид).
Как показано на Схеме 2, альтернативная методика получения соединений Формулы Ia включает в себя взаимодействие амина Формулы 2 с кислотой Формулы 4 в присутствии дегидратирующего агента, такого как дициклогексилкарбодиимид (DCC), 1,1'-карбонил-диимидазол, бис(2-оксо-3-оксазолидинил)фосфиновый хлорид или бензотриазол-1-илокситрис-(диметиламино)фосфоний гексафторфосфат.
Схема 2

Здесь также полезны реагенты на полимерном носителе, такие как полимерно-связанный циклогексилкарбодиимид. Данное взаимодействие может быть проведено в подходящем инертном растворителе, таком как дихлорметан или N,N-диметилформамид. Способы синтеза по схемам 1 и 2 являются только характерными примерами широкого разнообразия способов взаимодействия, используемых для получения соединений Формулы I; в литературе по синтезу много реакций такого типа взаимодействия.
Специалисту в данной области также будет понятно, что хлорангидриды кислот Формулы 3 могут быть получены из кислот Формулы 4 с помощью многочисленных хорошо известных способов. Например, хлорангидриды кислот Формулы 3 легко получают из карбоновых кислот Формулы 4 путем взаимодействия карбоновой кислоты 4 с тионилхлоридом или оксалилхлоридом в инертном растворителе, таком как толуол или дихлорметан в присутствии каталитического количества N,N-диметилформамида.
Как показано на Схеме 3, амины Формулы 2a обычно могут быть получены из соответствующих 2-нитробензамидов Формулы 5 через каталитическую гидрогенизацию нитрогруппы.
Схема 3

Типичные методики включают в себя восстановление водородом в присутствии металлического катализатора, такого как палладий на угле или оксид платины и в гидроксильных растворителях, таких как этанол и изопропанол. Амины Формулы 2a также могут быть получены путем восстановления цинком в уксусной кислоте. Эти методики хорошо описаны в химической литературе. R1 заместители, такие как С1-С6 алкил могут быть введены на этой стадии с помощью хорошо известных методик, включающих либо прямое алкилирование, либо посредством обычно предпочитаемого способа восстановительного алкилирования амина. Как показано далее на Схеме 3, обычно используемая методика представляет собой соединение амина 2а с альдегидом в присутствии восстанавливающего агента, такого как цианоборгидрид натрия с получением соединений Формулы 2b, где R1 представляет собой C1-C6 алкил.
Схема 4 показывает, что соединения Формулы Ic могут быть алкилированы или ацилированы подходящим алкилирующим или ацилирующим агентом, таким как алкил галогенид, алкилхлороформиат или ацилхлорид в присутствии основания, такого как гидрид натрия или н-бутиллитий в инертном растворителе, таком как тетрагидрофуран или N,N-диметилформамид с получением анилидов Формулы Id, где R1 иной, чем водород.
Схема 4

Промежуточные амиды Формулы 5a легко готовят из коммерчески доступных 2-нитробензойных кислот. Могут быть использованы типичные способы образования амида. Как показано на Схеме 5, эти способы включают прямое дегидративное взаимодействие кислот Формулы 6 с аминами Формулы 7 с использованием, например, DCC, и преобразование кислот в активированные формы, такие как хлорангидриды кислот или ангидриды и последующее взаимодействие с аминами с образованием амидов Формулы 5a.
Схема 5
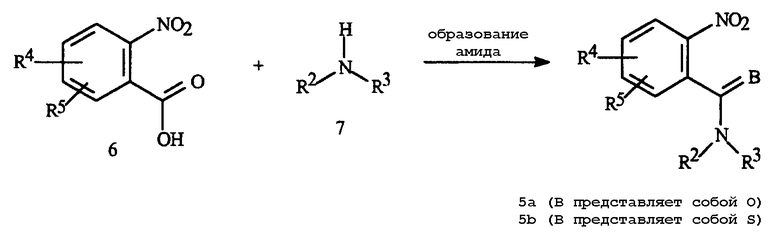
Алкильные хлороформиаты, такие как этилхлороформиат или изопропилхлороформиат, являются особенно используемыми реагентами для этого типа реакции, включающей активацию данной кислоты. В химической литературе существует большое количество способов образования амидов. Амиды Формулы 5a легко превращаются в тиоамиды Формулы 5b с использованием коммерчески доступных реагентов для переноса тиогрупп, такие как фосфорный пентасульфид и реактив Лавессона.
Промежуточные антранильные амиды Формулы 2c или 2d также могут быть получены из ангидридов изатиновой кислоты Формулы 8 или 9, соответственно, как показано на Схеме 6.
Схема 6


Типичные методики включают в себя соединение эквимолярных количеств амина 7 с ангидридом изатиновой кислоты в полярных апротонных растворителях, таких как пиридин и N,N-диметилформамид при температуре в интервале от комнатной температуры до 100°C. R1, заместители, такие как алкил и замещенный алкил, могут быть введены путем алкилирования, катализируемого основанием, ангидрида изатиновой кислоты 8 известными алкилирующими агентами R1-Lg (где Lg представляет собой нуклеофильную замещаемую уходящую группу, такую как галогенид, алкил или арилсульфонаты или алкилсульфаты) с получением алкилзамещенного промежуточного соединения 9. Ангидриды изатиновой кислоты Формулы 8 могут быть получены способами, описанными в Coppola, Synthesis 1980, 505-36.
Как показано на Схеме 7, альтернативная методика получения характерных соединений Формулы Ic включают в себя взаимодействие амина 7 с бензоксазиноном Формулы 10.
Схема 7

Реакция по Схеме 7 может быть проведена неразбавленной или в различных подходящих растворителях, включая тетрагидрофуран, диэтиловый эфир, пиридин, дихлорметан или хлороформ с оптимумом температур в интервале от комнатной температуры до температуры кипения с обратным холодильником данного растворителя. Общая реакция бензоксазинонов с аминами с образованием антраниламидов хорошо описана в химической литературе. Для просмотра химии бензоксазинонов смотрите Jakobsen et al., Biorganic and Medicinal Chemistry 2000, 8, 2095-2103 и цитируемые там ссылки. См. также Coppola, J. Heterocyclic Chemistry 1999, 36, 563-588.
Бензоксазиноны Формулы 10 могут быть получены с помощью различных методик. Две методики, используемые главным образом, подробно представлены на Схемах 8-9. На Схеме 8, бензоксазинон Формулы 10 получают напрямую через взаимодействие пиразолкарбоновой кислоты Формулы 4a с антраниловой кислотой Формулы 11.
Схема 8
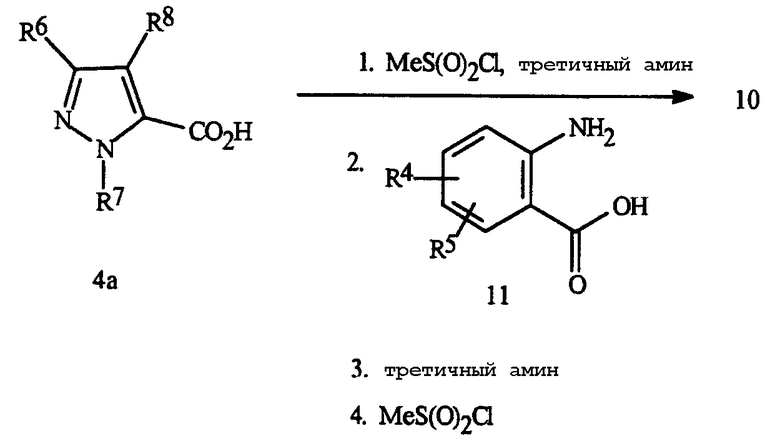
Это включает в себя последовательное добавление метансульфонилхлорида в присутствии третичного амина, такого как триэтиламин или пиридина к пиразолкарбоновой кислоте Формулы 4a, с последующим добавлением антраниловой кислоты Формулы 11, с последующим повторным добавлением третичного амина и метансульфонилхлорида. Эта методика в основном дает хорошие выходы бензоксазинона и более подробно показана в Примерах 6 и 8.
Схема 9 описывает альтернативное получение бензоксазинонов Формулы 10, включающее взаимодействие пиразольного хлорангидрида кислоты Формулы 3a с ангидридом изатиновой кислоты Формулы 8 с непосредственным получением бензоксазинона Формулы 10.
Схема 9

Для этой реакции подходят растворители, такие как пиридин или пиридин/ацетонитрил. Хлорангидриды кислот Формулы 3a могут быть получены из соответствующих кислот Формулы 4a различными способами синтеза, такими как хлорирование тионилхлоридом или оксалилхлоридом.
Ангидриды изатиновой кислоты Формулы 8 могут быть получены из изатинов Формулы 13, как изображено на Схеме 10.
Схема 10

Изатины Формулы 13 получают из производных анилина Формулы 12 используя способы, известные из литературы. Окисление изатина 13 перекисью водорода обычно дает хорошие выходы соответствующего ангидрида изатиновой кислоты 8 (Angew. Chem. Int. Ed. Engl. 1980, 19, 222-223). Ангидриды изатиновой кислоты также могут быть получены из антраниловых кислот 11 с помощью различных известных методик, включающих взаимодействие соединения 11 с фосгеном или эквивалентом фосгена.
Синтез характерных кислот Формулы 4 изображен на Схемах 11-16. Синтезы пиразолов Формулы 4a показаны на Схеме 11.
Схема 11

Синтез соединений Формулы 4a на Схеме 11 в качесте основной стадии включает в себя введение заместителя R7 посредством алкилирования или арилирования пиразола Формулы 14 соединениями Формулы 15 (где Lg представляет собой уходящую группу, как определено выше). Окисление метильной группы дает пиразольную карбоновую кислоту. Некоторые из более предпочтительных R6 групп включают галогеналкил.
Синтез пиразолов Формулы 4a также показан на Схеме 12.
Схема 12

Эти кислоты могут быть получены путем металлирования и карбоксилирования соединений Формулы 18, как основной этап. R7 группу вводят тем же путем, как изображено на Схеме 11, т.е. через алкилирование или арилирование соединением Формулы 15. Характерные R6 группы включают в себя, например, циано, галогеналкил и галоген.
Эту методику в частности используют для получения 1-(2-пиридинил)пиразолкарбоновых кислот Формулы 4b, как показано на Схеме 13.
Схема 13
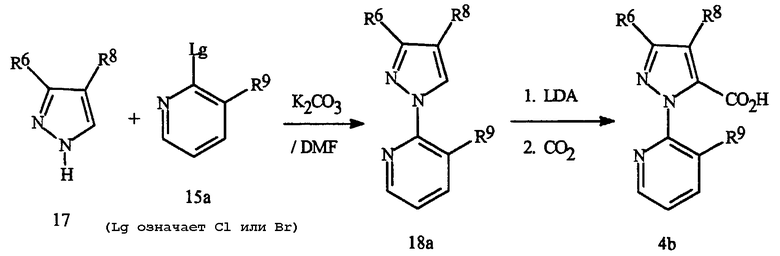
Взаимодействие пиразола Формулы 17 с 2,3-дигалопиридином Формулы 15a дает хорошие выходы 1-пиридилпиразола Формулы 18a с хорошими свойствами для требуемой региохимии. Металлирование соединения 18a диизопропиламидом лития (LDA) с последующим гашением соли лития диоксидом углерода дает 1-(2-пиридинил)пиразолкарбоновую кислоту Формулы 4b. Дополнительные подробности этих методик представлены в Примерах 1, 3, 6, 8 и 10.
Синтез пиразолов Формулы 4c описан на Схеме 14.
Схема 14

Схема 14 включает в себя взаимодействие необязательно замещенного фенилгидразина Формулы 19 с кетопируватом Формулы 20 с выходом эфиров пиразола Формулы 21. Гидролиз этих эфиров дает пиразольные кислоты Формулы 4c. Эта методика в частности используется для получения соединений, в которых R7 представляет собой необязательно замещенный фенил, а R6 представляет собой галогеналкил.
Альтернативный синтез пиразольных кислот Формулы 4c приведен на Схеме 15.
Схема 15

Способ по Схеме 15 включает в себя 3+2 циклоприсоединение соответственно замещенного иминогалогенида 22 либо к замещенным пропиолатам Формулы 23, либо акрилатам Формулы 25. Циклоприсоединение к акрилату требует дополнительного окисления промежуточного пиразолина в пиразол. Гидролиз этих эфиров дает пиразольные кислоты Формулы 4c. Предпочтительные иминогалогениды для этой реакции включают в себя трифторметил иминохлорид Формулы 26 и иминодибромид Формулы 27. Соединения, такие как 26, известны (J. Heterocycl. Chem. 1985, 22(2), 565-8). Соединения, такие как 27, могут быть получены известными способами (Tetrahedron Letters 1999, 40, 2605). Эти методики в частности используют для получения соединений, где R7 представляет собой необязательно замещенный фенил, а R6 представляет собой галогеналкил или бром.
Исходные пиразолы Формулы 17 являются известными соединениями или могут быть получены с помощью известных способов. Пиразол Формулы 17a (соединение Формулы 17, где R6 представляет собой CF3, а R8 представляет собой H) может быть получен по методикам из литературы (J. Fluorine Chem. 1991, 53(1), 61-70). Пиразолы Формулы 17c (соединения Формулы 17, где R6 представляет собой Cl или Br, а R8 представляет собой H) также могут быть получены по методикам, известным из литературы (Chem. Ber. 1966, 99(10), 3350-7). Полезный альтернативный способ получения соединения 17с представлен на Схеме 16.
Схема 16

В способе по Схеме 16 металлирование сульфамилпиразола Формулы 28 н-бутиллитием с последующим прямым галогенированием аниона либо гексахлорэтаном (для R6, являющегося Cl), либо 1,2-дибромтетрахлорэтаном (для R6, являющегося Br), дает галогенированные производные Формулы 29. Удаление сульфамильной группы трифторуксусной кислотой (TFA) при комнатной температуре протекает чисто и с хорошим выходом с получением пиразолов Формулы 17c. Специалисту в данной области будет понятно, что Формула 17c представляет собой таутомер Формулы 17b. Дополнительные подробности экспериментов по этим методикам описаны в Примерах 8 и 10.
Пиразолкарбоновые кислоты Формулы 4d, где R6 представляет собой H, C1-C6 алкил или C1-C6 галогеналкил, могут быть получены с помощью способа, представленного на Схеме 17.
Схема 17

Взаимодействие соединения Формулы 30, где R13 представляет собой C1-C4 алкил, с подходящим основанием в подходящем органическом растворителе дает циклизованный продукт Формулы 31 после нейтрализации кислотой, такой как уксусная кислота. Подходящим основанием, может быть например, но без ограничения гидрид натрия, т-бутоксид калия, димсил натрия (CH3S(О)CH2 -Na+), карбонаты щелочного металла (такого, как литий, натрий или калий), или гидроксиды, тетраалкил (такой, как метил, этил или бутил)фторидов аммония или гидроксидов, или 2-трет-бутилимино-2-диэтиламино-1,3-диметилпергидро-1,3,2-диазафосфонин. Подходящим органическим растворителем может быть, например, но без ограничения, ацетон, ацетонитрил, тетрагидрофуран, дихлорметан, диметилсульфоксид или N,N-диметилформамид. Данная реакция циклизации обычно проводится при температуре в интервале примерно от 0 до 120°C. Влияние растворителя, основания, температуры и времени добавления зависят друг от друга и выбор условий реакции важен для сведения к минимуму образования побочных продуктов. Предпочтительным основанием является тетрабутиламмоний фторид.
Дегидратация соединения Формулы 31 с получением соединения Формулы 32 с последующим превращением эфира карбоновой кислоты в карбоновую кислоту дает соединение Формулы 4d. Дегидратация осуществляется путем обработки каталитическим количеством подходящей кислоты. Такой каталитической кислотой может быть, например, но без ограничения, серная кислота. Эту реакцию проводят главным образом с использованием органического растворителя. Специалисту в данной области будет понятно, что реакции дегидратации можно проводить в широком ряду растворителей в интервале температур главным образом примерно от 0 до 200°C, более предпочтительно примерно от 0 до 100°C. Для дегидратации по способу Схемы 17, предпочтительно, чтобы растворитель содержал уксусную кислоту, а температура составляла около 65°C. Соединения эфира карбоновой кислоты могут быть превращены в соединения карбоновой кислоты множеством способов, включающих нуклеофильное расщепление в безводных условиях или гидролитическими способами, включающими использование либо кислот, либо оснований (для просмотра способов см. T. W. Greene and P. G. M. Wuts, Protective Groups in Organic Synthesis, 2nd ed., John Wiley & Sons, Inc., New York, 1991, pp. 224-269). Для способа Схемы 17 предпочтительны гидролитические способы, катализируемые основанием. Подходящие основания включают в себя гидроксиды щелочного металла (такого, как литий, натрий или калий). Например, эфир может быть растворен в смеси воды и спирта, такого как этанол. При обработке гидроксидом натрия или гидроксидом калия, данный эфир омыляется с образованием натриевой или калиевой соли карбоновой кислоты. Подкисление сильной кислотой, такой как соляная кислота или серная кислота, дает карбоновую кислоту Формулы 4d. Данная карбоновая кислота может быть выделена способами, известными специалистам в данной области, включая кристаллизацию, экстракцию и дистилляцию.
Соединения Формулы 30 могут быть получены с помощью способа, представленного на Схеме 18.
Схема 18
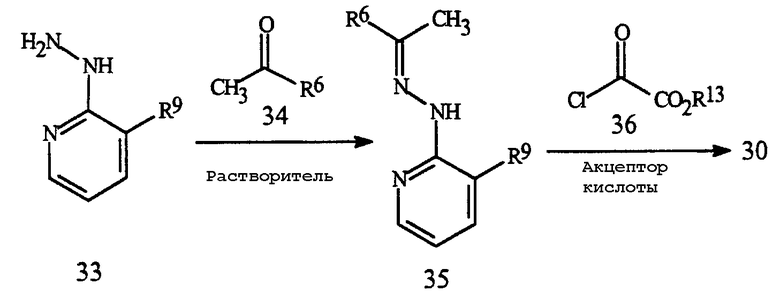
где R6 представляет собой H, C1-C6 алкил или C1-C6 галогеналкил, а R13 представляет собой С1-С4 алкил
Обработка гидразинового соединения Формулы 33 кетоном Формулы 34 в растворителе, таком как вода, метанол или уксусная кислота, дает гидразон Формулы 35. Специалисту в данной области будет понятно, что для этой реакции может требоваться катализ произвольной кислотой а также могут требоваться повышенные температуры, зависящие от молекулярного замещения структуры гидразона Формулы 35. Взаимодействие гидразона Формулы 35 с соединением Формулы 36 в подходящем органическом растворителе, таком как, например, но без ограничения, дихлорметан или тетрагидрофуран в присутствии акцептора кислоты, такого как триэтиламин, дает соединение Формулы 30. Данную реакцию обычно проводят при температуре примерно от 0 до 100°C. Дальнейшие подробности эксперимента для способа по Схеме 18 представлены в Примере 17. Гидразиновые соединения Формулы 33 могут быть получены стандартными способами, как, например, контактирование соответствующего галогенового соединения Формулы 15a с гидразином.
Пиразолкарбоновые кислоты Формулы 4d, где R6 представляет собой галоген, могут быть получены с помощью способа, представленного на Схеме 19.
Схема 19

где R13 представляет собой С1-С4 алкил.
Окисление соединения Формулы 37 необязательно в присутствии кислоты дает соединение Формулы 32, с последующим превращением группы эфира карбоновой кислоты в карбоновую кислоту, с получением соединения Формулы 4d. Окисляющим агентом может быть перекись водорода, органические пероксиды, персульфат калия, персульфат натрия, персульфат аммония, моноперсульфат калия (например, Oxone®) или перманганат калия. Для получения полного превращения следует использовать по меньшей мере один эквивалент окисляющего агента в отношении соединения Формулы 37 предпочтительно примерно от одного до двух эквивалентов. Это окисление обычно проводят в присутствии растворителя. Данным растворителем может быть простой эфир, такой как тетрагидрофуран, п-диоксан и подобное, органический сложный эфир, такой как этилацетат, диметилкарбонат и подобное, или полярный апротонный органический растворитель, такой как N,N-диметилформамид, ацетонитрил и подобные. Кислоты, подходящие для использования на данной стадии окисления, включают в себя неорганические кислоты, такие как серная кислота, фосфорная кислота и подобные и органические кислоты, такие как уксусная кислота, бензойная кислота и подобные. При использовании кислот их следует использовать более чем 0,1 эквивалента по отношению к соединению Формулы 37. Для получения полного превращения может быть использовано от одного до пяти эквивалентов кислоты. Предпочтительным окислителем является персульфат калия и данное окисление предпочтительно выполняют в присутствии серной кислоты. Данная реакция может быть выполнена перемешиванием соединения Формулы 37 в заданном растворителе, и кислоте, если используется. Затем может быть добавлен окислитель в подходящем соотношении. Температура реакции обычно варьирует уже, примерно, от 0°C до точки кипения растворителя, для получения приемлемого времени реакции, для завершения данной реакции, предпочтительно, менее 8 часов. Желаемый продукт, соединение Формулы 32, может быть выделен способами, известными специалистам в данной области, включая кристаллизацию, экстракцию и дистилляцию. Способы, подходящие для превращения сложного эфира Формулы 32 в карбоновую кислоту Формулы 4d, уже описаны для Схемы 17. Дополнительные подробности эксперимента для способа по Схеме 19 представлены в Примерах 12 и 13.
Соединения Формулы 37 могут быть получены из соответствующих соединений Формулы 38, как показано на Схеме 20.
Схема 20
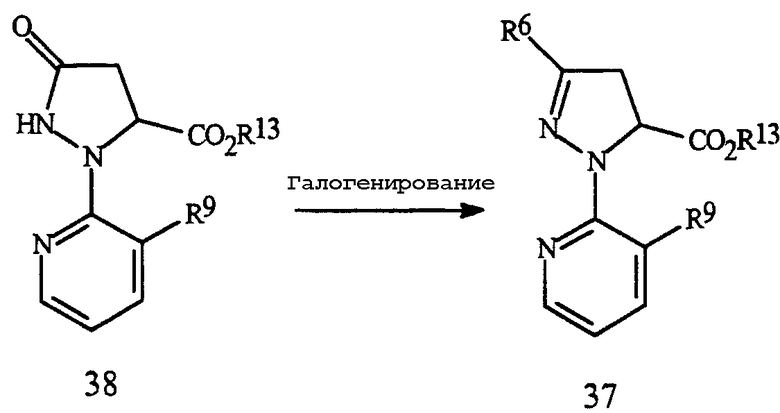
Обработка соединения Формулы 38 галогенирующим реагентом обычно в присутствии растворителя дает соответствующее галогеновое соединение Формулы 37. Галогенирующие реагенты, которые могут быть использованы, включают в себя оксигалогениды фосфора, тригалогениды фосфора, пентагалогениды фосфора, тионилхлорид, дигалотриалкилфосфораны, дигалодифенилфосфораны, оксалилхлорид и фосген. Предпочтительными являются оксигалогениды и пентагалогениды фосфора. Для получения полного превращения следует использовать по меньшей мере 0,33 эквивалентов оксигалогенида фосфора по отношению к соединению Формулы 38 (т.е. мольное отношение оксигалогенида фосфора к соединению Формулы 18 составляет по меньшей мере 0,33), предпочтительно примерно от 0,33 до 1,2 эквивалентов. Для получения полного превращения следует использовать по меньшей мере 0,20 эквивалентов пентагалогенида фосфора по отношению к соединению Формулы 38, предпочтительно примерно от 0,20 до 1,0 эквивалентов. Для этой реакции предпочтительными являются соединения Формулы 38, где R13 представляет собой C1-C4 алкил. Типичные растворители для этого галогенирования включают в себя галогенированные алканы, такие как дихлорметан, хлороформ, хлорбутан и подобные, ароматические растворители, такие как бензол, ксилен, хлорбензол и подобные, простые эфиры, такие как тетрагидрофуран, п-диоксан, диэтиловый эфир и подобные, и полярные апротонные растворители, такие как ацетонитрил, N,N-диметилформамид и подобные. Необязательно может быть добавлено органическое основание, такое как триэтиламин, пиридин, N,N-диметиланилин или подобное. Добавление катализатора, такого как N,N-диметилформамид, также по выбору. Предпочтительным является процесс, при котором растворителем является ацетонитрил, а основание отсутствует. Обычно при использовании растворителя ацетонитрила не требуется ни основания, ни катализатора. Предпочтительный процесс проводят путем перемешивания соединения Формулы 38 в ацетонитриле. Затем через подходящее время добавляют галогенирующий реагент, и данную смесь затем содержат при нужной температуре до завершения реакции. Температура реакции обычно составляет от 20°C и до точки кипения ацетонитрила, и время реакции обычно менее 2 часов. Реакционную массу затем нейтрализуют неорганическим основанием, таким как бикарбонат натрия, гидроксид натрия и подобным, или органическим основанием, таким как ацетат натрия. Желаемый продукт, соединение Формулы 37, может быть выделен способами, известными специалистам в данной области, включая кристаллизацию, экстракцию и дистилляцию.
Альтернативно, соединения Формулы 37, где R6 представляет собой галоген, могут быть получены путем обработки соответствующих соединений Формулы 37, где R6 представляет собой другой галоген (например, Cl для получения соединения Формулы 37, где R3 представляет собой Br) или сульфонатную группу, такую как п-толуолсульфонат, безолсульфонат и метансульфонат, подходящим галогенидом водорода. Посредством этого способа R6 галоген или сульфонатный заместитель исходного соединения Формулы 37 замещается, например, Br или Cl из бромида водорода или хлорида водорода, соответственно. Эту реакцию проводят в подходящем растворителе, таком как дибромметан, дихлорметан или ацетонитрил. Эта реакция может быть проведена при атмосферном давлении, или примерно при атмосферном давлении в автоклаве. В том случае, когда R6 в исходном соединении Формулы 37 представляет собой галоген, такой как Cl, эту реакцию предпочтительно проводят таким образом, чтобы галогенид водорода, образованный в результате этой реакции, удалялся путем барботирования или другими подходящими способами. Эту реакцию можно проводить примерно от 0 до 100°C, наиболее подходяще примерно при температуре окружающей среды (например, примерно от 10 до 40°C), и более предпочтительно примерно от 20 до 30°C. Добавление катализатора кислоты Льюиса (такой как трибромид алюминия для получения соединения Формулы 37, где R6 представляет собой Br) может облегчить протекание реакции. Продукт Формулы 37 выделяют обычными способами, известными специалистам в этой области, включая экстракцию, дистилляцию и кристаллизацию. Дополнительные подробности этого процесса представлены в Примере 14.
Исходные соединения Формулы 37, где R6 представляет собой Cl или Br, могут быть получены из соответствующих соединений Формулы 38, как уже было описано. Исходные соединения Формулы 37, где R6 представляет собой сульфонатную группу, могут быть получены таким же образом из соответствующих соединений Формулы 38 стандартными способами, таким как обработка сульфонилхлоридом (например, п-толуолсульфонилхлоридом) и основанием, таким как третичный амин (например, триэтиламином) в подходящем растворителе, таком как дихлорметан; дополнительные подробности этого процесса представлены в Примере 15.
Пиразолкарбоновые кислоты Формулы 4d, где R6 представляет собой C1-C4 алкокси или C1-C4 галогеналкокси также могут быть получены способом, представленным на Схеме 21.
Схема 21

где R13 представляет собой C1-C4 алкил, а X является уходящей группой.
В этом способе, вместо галогенирования, как показано на Схеме 20, соединение Формулы 38 окислено до соединения Формулы 32a. Условия реакции для этого окисления уже описаны для превращения соединения Формулы 37 в соединение Формулы 32 на Схеме 19.
Соединение Формулы 32a затем алкилируют для образования соединения Формулы 32b путем контактирования с алкилирующим агентом CF3CH2X (39) в присутствии основания. В алкилирующем агенте 39 X представляет собой уходящую группу нуклеофильной реакции, такую как галоген (например, Br, I), OS(О)2CH3 (метансульфонат), OS(О)2CF3, OS(О)2Ph-п-CH3 (п-толуолсульфонат), и подобное; хорошо подходит метансульфонат. Эту реакцию проводят в присутствии по меньшей мере одного эквивалента основания. Подходящие основания включают в себя неорганические основания, такие как карбонаты и гидроксиды щелочных металлов (таких как литий, натрий или калий) и органические основания, такие как триэтиламин, диизопропилэтиламин и 1,8-диазабицикло[5,4,0]ундек-7-ен. Эту реакцию главным образом проводят в растворителе, который может содержать спирты, такие как метанол и этанол, галогенированные алканы, такие как дихлорметан, ароматические растворители, такие как бензол, толуол и хлорбензол, простые эфиры, такие как тетрагидрофуран, и полярные апротонные растворители, такие как ацетонитрил, N,N-диметилформамид и подобное. С неорганическими основаниями предпочтительно использовать спирты и полярные апротонные растворители. Предпочтительными являются карбонат калия в качестве основания и ацетонитрил в качестве растворителя. Эту реакцию главным образом проводят примерно от 0 до 150°C, наиболее обычно от температуры окружающей среды до 100°C. Продукт Формулы 32b может быть выделен с помощью общепринятых методик, таких как экстракция. Сложный эфир Формулы 32b затем может быть превращен в карбоновую кислоту Формулы 4d с помощью способов, уже описанных для превращения соединения Формулы 32 в соединение Формулы 4d на Схеме 17. Дополнительные экспериментальные подробности для этого способа по Схеме 21 представлены в Примере 16.
Соединения Формулы 38 могут быть получены из соединений Формулы 33, как изображено на Схеме 22.
Схема 22

где R13 представляет собой С1-С4 алкил.
В этом способе гидразиновое соединение Формулы 33 приводят в контакт с соединением Формулы 40 (может быть использован эфир фумарата или малеата или их смесь) в присутствии основания и растворителя. Основанием обычно является соль алкоксида металла, такого как метоксид натрия, метоксид калия, этоксид натрия, этоксид калия, трет-бутоксид калия, трет-бутоксид лития, и подобное. Следует использовать более 0,5 эквивалентов основания по сравнению с соединением Формулы 33, предпочтительно от 0,9 до 1,3 эквивалентов. Следует использовать более 1,0 эквивалента соединения Формулы 40, предпочтительно от 1,0 до 1,3 эквивалентов. Могут быть использованы полярные протонные и полярные апротонные органические растворители, такие как спирты, ацетонитрил, тетрагидрофуран, N,N-диметилформамид, диметилсульфоксид и подобное. Предпочтительными растворителями являются спирты, такие как метанол и этанол. Особенно предпочтительно, чтобы спирт был таким же, как и составляющий фумаровый или малеиновый эфир, и алкоксидное основание. Эту реакцию обычно проводят путем перемешивания соединения Формулы 33 и основания в растворителе. Эта смесь может быть нагрета или охлаждена до нужной температуры и соединение Формулы 40 добавляют в течение определенного времени. Обычные температуры реакции составляют от 0°C до точки кипения используемого растворителя. Эти реакции можно проводить под давлением, большим чем атмосферное давление, для повышения точки кипения растворителя. Главным образом предпочтительны температуры от 30 до 90°C. Время добавления может быть настолько быстрым, насколько позволяет теплоперенос. Обычное время добавления составляет от 1 минуты до 2 часов. Оптимальная температура реакции и время добавления варьируют в зависимости от особенностей соединений Формулы 33 и Формулы 40. После добавления реакционную смесь можно сохранять в течение некоторого времени при температуре реакции. В зависимости от температуры реакции это требуемое время сохранения может быть от 0 до 2 часов. Обычно время удержания составляет от 10 до 60 минут. Реакционная масса затем может быть подкислена добавлением органической кислоты, такой как уксусная кислота и подобные, или неорганической кислоты, такой как соляная кислота, серная кислота и подобные. В зависимости от условий реакции и способов выделения, функциональная группа -CO2R13 соединения Формулы 38 может быть гидролизована до -CO2H; например, такому гидролизу может способствовать наличие воды в реакционной смеси. В случае образования карбоновой кислоты (-СО2Н), она может быть снова превращена в -CO2R13, где R13 представляет собой C1-C4 алкил, с использованием способов этерификации, хорошо известных в данной области. Желаемый продукт, соединение Формулы 38, может быть выделен с помощью способов, известных специалистам в данной области, таких как кристаллизация, экстракция или дистилляция.
Очевидно, что некоторые реагенты и условия реакции, описанные выше для получения соединений Формулы I, могут не сочетаться с некоторыми функциональными особенностями, имеющими место в промежуточных соединениях. В этих случаях получить желаемый продукт поможет включение в синтез последовательностей защиты/снятия защиты функциональных групп или взаимопревращений. Использование и выбор защищающих групп будет очевиден специалисту в области химического синтеза (см., например, Greene, T. W.; Wuts, P. G. M. Protective Groups in Organic Synthesis, 2nd ed.; Wiley: New York, 1991). Специалисту в данной области будет понятно, что в некоторых случаях, после введения заданного реагента, как показано на любой отдельной схеме, для завершения синтеза соединений Формулы I может быть необходимым выполнение дополнительных стандартных операций, не описанных в деталях. Специалисту в данной области также будет понятно, что может быть необходимо выполнить определенную последовательность стадий, показанных на вышеприведенных схемах в порядке, ином, чем следует из отдельной последовательности, представленной для получения соединений Формулы I.
Считается, что специалист в данной области, используя описание действий, может приготовить соединения Формулы I настоящего изобретения в самом полном объеме. Следующие Примеры, следовательно, предназначены только для иллюстрации, а не для ограничения раскрытия в каком бы то ни было направлении. Проценты даны по весу, исключая смеси растворителей для хроматографии или где указано особо. Части и проценты для смесей растворителей для хроматографии даны по объему, если не оговорено особо. Спектры 1Н ЯМР приведены в миллионных долях относительно тетраметилсилана; с означает синглет, д означает дублет, т означает триплет, кв означает квартет, м означает мультиплет, дд означает дублет дублетов, дт означает дублет триплетов, шир с означает широкий синглет.
ПРИМЕР 1
Получение 2-[1-этил-3-трифторметилпиразол-5-ил-карбамоил]-3-метил-N-(1-метилэтил)бензамида
Стадия A: Получение 3-Метил-N-(1-метилэтил)-2-нитробензамида
Раствор 3-метил-2-нитробензойной кислоты (2,00 г, 11,0 ммоль) и триэтиламина (1,22 г, 12,1 ммоль) в 25 мл метиленхлорида охлаждали до 10°C. Осторожно добавляли этил хлороформиат и образовывался твердый осадок. После перемешивания в течение 30 минут добавляли изопропиламин (0,94 г, 16,0 ммоль) и в результате получали гомогенный раствор. Реакционную смесь перемешивали в течение дополнительного часа, выливали в воду и экстрагировали этилацетатом. Органические экстракты промывали водой, сушили над сульфатом магния, выпаривали при пониженном давлении с получением 1,96 г желаемого промежуточного соединения в виде белого твердого вещества, плавящегося при 126-128°C.
1Н ЯМР (CDCl3)δ 1,24 (д, 6H), 2,38 (с, 3H), 4,22 (м, 1H), 5,80 (шир с, 1H), 7,4 (м, 3H).
Стадия B: Получение 2-амино-3-метил-N-(1-метилэтил)бензамида
2-нитробензамид со Стадии A (1,70 г, 7,6 ммоль) гидрогенизировали над 5% Pd/C в 40 мл этанола при 50 ф/дм2 (psi). По прекращении поглощения водорода реакционную смесь фильтровали через Celite® диатомовый слой для фильтрования и Celite® промывали простым эфиром. Фильтрат выпаривали при пониженном давлении с получением 1,41 г названного соединения в виде твердого вещества, плавящегося при 149-151°C.
1H ЯМР (CDCl3) δ 1,24 (дд, 6H), 2,16 (с, 3H), 4,25 (м, 1H), 5,54 (шир с, 2H), 5,85 (шир с, 1H), 6,59 (т, 1H), 7,13 (д, 1H), 7,17 (д, 1H).
Стадия C: Получение 1-этил-3-трифторметилпиразол-5-ил карбоновой кислоты
К смеси 3-трифторметилпиразола (5 г, 37 ммоль) и порошкообразного карбоната калия (10 г, 72 ммоль), перемешанной в 30 мл N,N-диметилформамида, добавляли по каплям йодэтан (8 г, 51 ммоль). После умеренного выделения теплоты, реакционную смесь перемешивали в течение ночи при комнатной температуре. Реакционную смесь распределяли между 100 мл диэтилового эфира и 100 мл воды. Эфирный слой отделяли, промывали водой (3X) и солевым раствором и сушили над сульфатом магния. Выпаривание растворителя в вакууме давало 4 г масла.
К 3,8 г этого масла, смешанного в 40 мл тетрагидрофурана в атмосфере азота на бане сухой лед/ацетон, добавляли по каплям 17 мл 2,5 M раствора н-бутиллития в тетрагидрофуране (43 ммоль) и данный раствор перемешивали в течение 20 минут при -78°C. В перемешанный раствор барботировали избыток газообразного диоксида углерода с умеренной скоростью в течение 10 минут. После добавления диоксида углерода реакционную смесь оставляли для медленного достижения комнатной температуры и перемешивали в течение ночи. Реакционную смесь распределяли между диэтиловым эфиром (100 мл) и 0,5 н. водным гидроксидом натрия (100 мл). Отделяли основной слой и подкисляли концентрированной соляной кислотой до рН 2-3. Эту водную смесь экстрагировали этилацетатом (100 мл) и органический экстракт промывали водой и солевым раствором и сушили над сульфатом магния. Маслянистый остаток, оставшийся после выпаривания растворителя в вакууме,растирали до сухого вещества из небольшого количества 1-хлорбутана. После фильтрования и высушивания получали образец 1-этил-3-трифторметил-пиразол-5-ил карбоновой кислоты (1,4 г) с незначительными примесями в виде плавящегося в широком температурном интервале твердого вещества.
1H ЯМР (CDCl3) δ 1,51 (т, 3H), 4,68 (кв, 2H), 7,23 (с, 1H), 9,85 (шир с, 1H).
Стадия D: Получение 2-[1-этил-3-трифторметилпиразол-5-ил карбамоил]-3-метил-N-(1-метилэтил)бензамида
К раствору 1-этил-3-трифторметил-пиразол-5-ил карбоновой кислоты (т.е. продукту со Стадии C) (0,5 г, 2,4 ммоль), перемешанной в 20 мл метиленхлорида, добавляли оксалилхлорид (1,2 мл, 14 ммоль). При добавлении 2 капель N,N-диметилформамида происходило образование пены и газообразование. Реакционную смесь в виде желтого раствора нагревали в колбе с обратным холодильником в течение 1 часа. После охлаждения удаляли растворитель в вакууме и полученный в результате остаток растворяли в 20 мл тетрагидрофурана. К перемешанному раствору добавляли 2-амино-3-метил-N-(1-метилэтил)бензамид (т.е. продукт со Стадия B) (0,7 г, 3,6 ммоль) с последующим добавлением по каплям N,N-диизопропилэтиламина (3 мл, 17 ммоль). После перемешивания при комнатной температуре в течение ночи реакционную смесь распределяли между этилацетатом (100 мл) и 1 н. водной соляной кислотой (75 мл). Отделенный органический слой промывали водой и солевым раствором и сушили над сульфатом магния. Выпаривание в вакууме дало белый твердый остаток, который при очистке с помощью флэш-хроматографии на колонке из силикагеля (2:1 гексан/этилацетат) давал 0,5 г названного соединения, соединения по настоящему изобретению, плавящегося при 223-226°C.
1Н ЯМР (DMSO-d6) δ 1,06 (д, 6H), 1,36 (т, 3H), 2,45 (с, 3H), 3,97 (м, 1H), 4,58 (кв, 2H), 7,43-7,25 (м, 3H), 7,45 (с, 1H), 8,05 (д, 1H), 10,15 (с, 1H).
ПРИМЕР 2
Получение N-[2-метил-6-[[(1-метилэтил)амино]карбонил]фенил]-1-фенил-3-(трифторметил)-1H-пиразол-5-карбоксамида
Стадия A: Получение 2-Метил-1-фенил-4-(трифторметил)-1Н-пиразола
Раствор 1,1,1-трифторпентан-2,4-диона (20,0 г, 0,130 моль) в ледяной уксусной кислоте (60 мл) охлаждали до 7°C с использованием ванны лед/вода. Фенилгидразин (14,1 г, 0,130 моль) добавляли по каплям на протяжении 60 минут. Во время добавления температура реакционной массы повышалась до 15°C. Полученный в результате оранжевый раствор сохраняли в условиях окружающей среды в течение 60 минут. Объем уксусной кислоты удаляли путем выпаривания на роторном испарителе при температуре бани 65°C. Остаток растворяли в метиленхлориде (150 мл). Раствор промывали водным бикарбонатом натрия (3 г в 50 мл воды). Пурпурно-красный органический слой отделяли, обрабатывали активированным углем (2 г) и MgSO4, затем фильтровали. Летучие компоненты удаляли на роторном испарителе. Неочищенный продукт содержал 28,0 г масла розового цвета, содержащего ˜89% желаемого продукта, и 11% 1-фенил-5-(трифторметил)-3-метилпиразола.
1Н ЯМР (DMSO-d6) δ 2,35 (с, 3H), 6,76 (с, 1H), 7,6-7,5 (м, 5H).
Стадия B: Получение 1-фенил-3-(трифторметил)-1Н-пиразол-5-карбоновой кислоты
Образец неочищенного 2-метил-1-фенил-4-(трифторметил)-1Н-пиразола (т.е. продукта со Стадии A) (˜89%, 50,0 г, 0,221 моль) смешивали с водой (400 мл) и хлоридом цетилтриметиламмония (4,00 г, 0,011 моль). Данную смесь нагревали до 95°C. Добавляли перманганат калия 10 равными порциями, с интервалами ˜8 минут. В течение этого периода реакционную массу поддерживали при 95-100°C. После добавления последней порции данную смесь (сохраняли) выдерживали в течение ˜15 минут при 95-100°C, после чего обесцвечивался пурпурный цвет перманганата. Реакционную массу фильтровали горячей (˜75°C) через 1-см слой Celite® диатомового слоя для фильтрования в 150-мл воронке со стеклофильтром. Осадок на фильтре промывали теплой (˜50°C) водой (3×100 мл). Объединенные фильтрат и смывы экстрагировали простым эфиром (2×100 мл) для удаления небольшого количества желтого водонерастворимого вещества. Водный слой продували азотом для удаления остаточного эфира. Прозрачный бесцветный щелочной раствор подкисляли добавлением концентрированной соляной кислоты по каплям до достижения рН ˜1,3 (28 г, 0,28 моль). Во время добавления первых двух третей было интенсивное выделение газа. Данный продукт собирали фильтрацией, промывали водой (3×40 мл), затем сушили в течение ночи при 55°C в вакууме. Данный продукт состоял из 11,7 г белого кристаллического порошка, который был по существу чистым исходя из 1Н ЯМР.
1Н ЯМР (CDCl3) δ 7,33 (с, 1H), 7,4-7,5 (м, 5H).
Стадия C: Получение 1-фенил-3-(трифторметил)-1Н-пиразол-5-карбонилхлорида
Образец неочищенной 1-фенил-3-(трифторметил)пиразол-5-карбоновой кислоты (т.е. продукта со Стадии B) (4,13 г, 16,1 ммоль) растворяли в метиленхлориде (45 мл). Данный раствор обрабатывали оксалилхлоридом (1,80 мл, 20,6 ммоль), затем N,N-диметилформамидом (0,010 мл, 0,13 ммоль). Газообразование началось вскоре после добавления катализатора N,N-диметилформамида. Реакционную смесь перемешивали в течение ˜20 минут в условиях окружающей среды, затем нагревали до температуры кипения с обратным холодильником в течение 35 минут. Летучие компоненты удаляли выпариванием реакционной смеси на роторном испарителе при температуре ванны 55°C. Данный продукт состоял из 4,43 г светло-желтого масла. Единственной примесью, зарегистрированной по 1Н ЯМР, был N,N-диметилформамид.
1Н ЯМР (CDCl3) δ 7,40 (м, 1H), 7,42 (с, 1H), 7,50-7,53 (м, 4H).
Стадия D: Получение N-[2-метил-6-[[(1-метилэтил)амино]карбонил]фенил]-1-фенил-3-(трифторметил)-1H-пиразол-5-карбоксамида
Образец 3-метилангидрида изатиновой кислоты (0,30 г, 1,7 ммоль), частично растворенный в пиридине (4,0 мл), обрабатывали 1-фенил-3-(трифторметилпиразол)-5-карбоксилхлоридом (т.е. продуктом со Стадии C) (0,55 г, 1,9 ммоль). Смесь нагревали до ˜95°C в течение 2 часов. Полученный в результате оранжевый раствор охлаждали до 29°C, затем обрабатывали изопропиламином (1,00 г, 16,9 ммоль). Реакционная масса экзотермически нагревалась до 39°C. Дополнительно ее нагревали до 55°C в течение 30 минут, после чего образовывался большой осадок. Реакционную массу растворяли в дихлорметане (150 мл). Данный раствор промывали водной кислотой (5 мл конц. HCl в 45 мл воды), затем водным основанием (2 г карбоната натрия в 50 мл воды). Органический слой высушивали над MgSO4, фильтровали, затем концентрировали на роторном испарителе. При уменьшении до ˜4 мл сформировались кристаллы продукта. Суспензию разводили ˜10 мл простого эфира, после чего выкристаллизовывалось больше продукта. Данный продукт выделяли фильтрацией, промывали простым эфиром (2×10 мл), затем промывали водой (2×50 мл). Влажный осадок сушили в течение 30 минут при 70°C в вакууме. Данный продукт, соединение по настоящему изобретению, состоял из 0,52 г не совсем белого порошка, плавящегося при 260-262°C.
1Н ЯМР (DMSO-d6) δ 1,07 (д, 6H), 2,21 (с, 3H), 4,02 (октет, 1H), 7,2-7,4 (м, 3H), 7,45-7,6 (м, 6H), 8,10 (д, 1H), 10,31 (с, 1H).
ПРИМЕР 3
Получение N-[2-метил-6-[[(1-метилэтил)амино]карбонил]фенил]-3-(трифторметил)-1-[3-(трифторметил)-2-пиридинил]-1Н-пиразол-5-карбоксамида
Стадия A: Получение 3-трифторметил-2-[3-(трифторметил)-1Н-пиразол-1-ил]пиридина
Смесь 2-хлор-3-трифторметилпиридина (3,62 г, 21 ммоль), 3-трифторметилпиразола (2,7 г, 20 ммоль) и карбоната калия (6,0 г, 43 ммоль) нагревали при 100°C в течение 18 ч. Охлажденную реакционную смесь добавляли к ледяной воде (100 мл). Смесь дважды экстрагировали простым эфиром (100 мл) и объединенные эфирные экстракты дважды промывали водой (100 мл). Органический слой высушивали сульфатом магния и концентрировали до масла. Хроматография на силикагеле с гексан/этилацетат от 8:1 до 4:1 в качестве элюента давала названное соединение (3,5 г) в виде масла.
1Н ЯМР (CDCl3) δ 6,75 (м, 1H), 7,5 (м, 1H), 8,2 (м, 2H), 8,7 (м, 1H).
Стадия B: Получение 3-(трифторметил)-1-[3-(трифторметил)-2-пиридинил]-1Н-пиразол-5-карбоновой кислоты
Смесь соединения, названного в ПРИМЕРЕ 3, Стадии A (3,4 г, 13 ммоль) растворяли в тетрагидрофуране (30 мл) и охлаждали до -70°C. Добавляли диизопропиламид лития (2 н. в гептан/тетрагидрофуране, (Aldrich) 9,5 мл, 19 ммоль) и полученную в результате темную смесь перемешивали в течение 10 минут. Сухой диоксид углерода барботировали через смесь в течение 15 минут. Смесь оставляли для нагревания до 23°C и обрабатывали водой (50 мл) и 1 н. гидроксидом натрия (10 мл). Данную водную смесь экстрагировали простым эфиром (100 мл), а затем этилацетатом (100 мл). Водный слой подкисляли 6 н. соляной кислотой до pH 1-2 и экстрагировали дважды дихлорметаном. Органический слой высушивали сульфатом магния и концентрировали с получением названного соединения (1,5 г).
1Н ЯМР (CDCl3) δ 7,6 (м, 1H), 7,95 (м, 1H), 8,56 (м, 1H), 8,9 (м, 1H), 14,2 (шир, 1H).
Стадия C: Получение N-[2-метил-6-[[(1-метилэтил)амино]карбонил]фенил]-3-(трифторметил)-1-[3-(трифторметил)-2-пиридинил]-1Н-пиразол-5-карбоксамида
Смесь названного соединения ПРИМЕРА 3, Стадия B (0,54 г, 1,1 ммоль), названного соединения из ПРИМЕРА 1, Стадия B (0,44 г, 2,4 ммоль) и BOP хлорида (бис(2-оксо-оксазолидинил)фосфинилхлорид, 0,54 г, 2,1 ммоль) в ацетонитриле (13 мл), обрабатывали триэтиламином (0,9 мл). Смесь встряхивали в закрытом сцинтилляционном флаконе в течение 18 ч. Реакционную смесь распределяли между этилацетатом (100 мл) и 1 н. соляной кислотой. Слой этилацетата промывали последовательно 1 н. соляной кислотой (50 мл), 1 н. гидроксидом натрия (50 мл) и насыщенным раствором хлорида натрия (50 мл). Органический слой сушили над сульфатом магния и концентрировали. Остаток подвергали флэш-хроматографии на колонке из силикагеля с гексан/этилацетат (5:1 до 3:1) в качестве элюента. Названное соединение (0,43 г), соединение по настоящему изобретению, выделяли в виде белого твердого вещества, т.пл. 227-230°C.
1Н ЯМР (CDCl3) δ 1,2 (м, 6H), 4,15 (м, 1H), 5,9 (шир д, 1H), 7,1 (м, 1H), 7,2 (м, 2H), 7,4 (с, 1H), 7,6 (м, 1H), 8,15 (м, 1H), 8,74 (м, 1H), 10,4 (шир, 1H).
ПРИМЕР 4
Получение 1-(3-хлор-2-пиридинил)-N-[2-метил-6-[[(1-метилэтил)амино]карбонил]-фенил]-3-(трифторметил)-1Н-пиразол-5-карбоксамида
Стадия A: Получение 3-хлор-2-[3-(трифторметил)-1Н-пиразол-1-ил]пиридина
К смеси 2,3-дихлорпиридина (99,0 г, 0,67 моль) и 3-(трифторметил)пиразола (83 г, 0,61 моль) в сухом N,N-диметилформамиде (300 мл) добавляли карбонат калия (166,0 г, 1,2 моль) и затем реакционную смесь нагревали до 110-125°C на протяжении 48 часов. Реакционную смесь охлаждали до 100°C и фильтровали через Celite® диатомовый слой для фильтрования для удаления твердых примесей. N,N-Диметилформамид и избыток дихлорпиридина удаляли дистилляцией при атмосферном давлении. Дистилляция данного продукта при пониженном давлении (т.к. 139-141°C, 7 мм) давала желаемое промежуточное соединение в виде светло-желтого масла (113,4 г).
1Н ЯМР (CDCl3) δ 6,78 (с, 1H), 7,36 (т, 1H), 7,93 (д, 1H), 8,15 (с, 1H), 8,45 (д, 1H).
Стадия B: Получение 1-(3-хлор-2-пиридинил)-3-(трифторметил)-1Н-пиразол-5-карбоновой кислоты
К раствору 3-хлор-2-[3-(трифторметил)-1Н-пиразол-1-ил]пиридина (т.е. продукта Стадии A) (105,0 г, 425 ммоль) в сухом тетрагидрофуране (700 мл) при -75°C добавляли через канюлю -30°C раствор диизопропиламида лития (425 ммоль) в сухом тетрагидрофуране (300 мл). Темно-красный раствор перемешивали в течение 15 минут, после этого времени барботировали диоксид углерода при -63°C до тех пор, пока раствор не становился бледно-желтым и прекращалось выделение тепла. Реакционную смесь перемешивали в течение дополнительных 20 минут и быстро охлаждали водой (20 мл). Растворитель удаляли при пониженном давлении, и реакционную смесь распределяли между простым эфиром и 0,5 н. водным раствором гидроксида натрия. Водные экстракты промывали простым эфиром (3x), фильтровали через Celite® диатомовый слой для фильтрования для удаления остаточных твердых примесей, и затем подкисляли приблизительно до рН 4, в этой точке образовывалось оранжевое масло. Водную смесь интенсивно перемешивали и добавляли дополнительное количество кислоты для понижения рН до 2,5-3. Оранжевое масло застывало в сыпучее твердое вещество, которое фильтровали, промывали последовательно водой и 1 н. соляной кислотой и сушили под вакуумом при 50°C с получением названного продукта в виде не совсем белого твердого вещества (130 г). (Продукт из другой фракции, полученный по сходной методике, плавился при 175-176°C.)
1Н ЯМР (DMSO-d6) δ 7,61 (с, 1H), 7,76 (дд, 1H), 8,31 (д, 1H), 8,60 (д, 1H).
Стадия C: Получение 8-метил-2Н-3,1-бензоксазин-2,4(1Н)диона
К раствору 2-амино-3-метилбензойной кислоты (6 г) в сухом 1,4-диоксане (50 мл) добавляли по каплям раствор трихлорметилхлороформиат (8 мл) в сухом 1,4-диоксане (25 мл) с охлаждением ледяной водой для удержания температуры реакции ниже 25°C. Во время добавления начинал образовываться белый осадок. Реакционную смесь перемешивали при комнатной температуре в течение ночи. Осевшие твердые примеси удаляли фильтрацией и промывали 1,4-диоксаном (2×20 мл) и гексаном (2×15 мл), и сушили на воздухе с выходом 6,51 г не совсем белого твердого вещества.
1Н ЯМР (DMSO-d6) δ 2,33 (с, 3H), 7,18 (т, 1H), 7,59 (д, 1H), 7,78 (д, 1H), 11,0 (шир с, 1H).
Стадия D: Получение 2-[1-(3-хлор-2-пиридинил)-3-(трифторметил)-1Н-пиразол-5-ил]-8-метил-4Н-3,1-бензоксазин-4-она
К суспензии продукта карбоновой кислоты, полученного также, как на Стадии B (146 г, 500 ммоль) в дихлорметане (приблизительно 2 л), добавляли N,N-диметилформамид (20 капель) и оксалилхлорид (67 мл, 750 ммоль) порциями приблизительно по 5 мл через, приблизительно, 2 ч. Во время добавления происходит интенсивное выделение газа. Реакционную смесь перемешивали при комнатной температуре в течение ночи. Реакционную смесь концентрировали в вакууме с получением неочищенного хлорангидрида кислоты в виде непрозрачной оранжевой смеси. Это вещество помещали в дихлорметан, фильтровали для удаления некоторых твердых примесей и затем повторно концентрировали и использовали без дальнейшей очистки. Неочищенный хлорангидрид кислоты растворяли в ацетонитриле (250 мл) и добавляли к суспензии продукт со Стадии C в ацетонитриле (400 мл). Добавляли пиридин (250 мл), смесь перемешивали в течение 15 мин при комнатной температуре, затем нагревали до температуры кипения с обратным холодильником в течение 3 ч. Полученную в результате смесь охлаждали до комнатной температуры и перемешивали в течение ночи с получением твердой массы. Добавляли дополнительное количество ацетонитрила и смесь перемешивали для образования густой кашицы. Твердые примеси собирали и промывали холодным ацетонитрилом. Твердые примеси сушили на воздухе и сушили в вакууме при 90°C в течение 5 ч с выходом 144,8 г пушистого твердого продукта.
1Н ЯМР (CDCl3) δ 1,84 (с, 3H), 7,4 (т, 1H), 7,6 (м, 3H), 8,0 (дд, 1H), 8,1 (с, 1H), 8,6 (д, 1H).
Стадия E: Получение 1-(3-хлор-2-пиридинил)-N-[2-метил-6-[[(1-метилэтил)амино]карбонил]фенил]-3-(трифторметил)-1Н-пиразол-5-карбоксамида
К суспензии продукта бензоксазинона Стадии D (124 г, 300 ммоль) в дихлорметане (500 мл) добавляли по каплям изопропиламин (76 мл, 900 ммоль) при комнатной температуре. Во время добавления температура реакционной смеси увеличивалась и суспензия разжижалась. Затем реакционную смесь нагревали до температуры кипения с обратным холодильником в течение 1,5 ч. Образовывалась новая суспензия. Реакционную смесь охлаждали до комнатной температуры и добавляли диэтиловый эфир (1,3 л) и смесь перемешивали при комнатной температуре в течение ночи. Твердые частицы собирали и промывали простым эфиром. Твердые частицы сушили на воздухе, а затем сушили в вакууме при 90°C в течение 5 ч с выходом 122 г названного соединения, соединение по настоящему изобретению в виде пушистого твердого продукта, плавящегося при 194-196°C.
1Н ЯМР (CDCl3) δ 1,23 (д, 6H), 2,21 (с, 3H), 4,2 (м, 1H), 5,9 (д, 1H), 7,2 (т, 1H), 7,3 (м, 2H), 7,31 (с, 1H), 7,4 (м, 1H), 7,8 (д, 1H), 8,5 (д, 1H), 10,4 (с, 1H).
ПРИМЕР 5
Альтернативное получение 1-(3-хлор-2-пиридинил)-N-[2-метил-6-[[(1-метилэтил)амино]-карбонил]фенил]-3-(трифторметил)-1Н-пиразол-5-карбоксамида
К раствору продукта карбоновой кислоты, полученного как в ПРИМЕРЕ 4, Стадия B (28 г, 96 ммоль) в дихлорметане (240 мл) добавляли N,N-диметилформамид (12 капель) и оксалилхлорид (15,8 г, 124 ммоль). Реакционную смесь перемешивали при комнатной температуре до прекращения выделения газа (приблизительно 1,5 ч). Реакционную смесь концентрировали в вакууме с получением неочищенного хлорангидрида кислоты в виде масла, используемого без дальнейшей очистки. Неочищенный хлорангидрид кислоты растворяли в ацетонитриле (95 мл) и добавляли к раствору бензоксазин-2,4-диона, приготовленного по способу ПРИМЕРА 4, Стадии C, в ацетонитриле (95 мл). Полученную в результате смесь перемешивали при комнатной температуре (приблизительно 30 мин). Добавляли пиридин (95 мл) и смесь нагревали примерно до 90°C (приблизительно 1 ч). Реакционную смесь охлаждали примерно до 35°C и добавляли изопропиламин (25 мл). Во время добавления реакционную смесь нагревали экзотермически, а затем удерживали примерно при 50°C (приблизительно 1 ч). Реакционную смесь затем выливали в ледяную воду и перемешивали. Полученный в результате осадок собирали фильтрацией, промывали водой и сушили в вакуумев течение ночи с получением 37,5 г названного соединения, соединения по настоящему изобретению в виде желтовато-коричневого твердого вещества.
1Н ЯМР (CDCl3) δ 1,23 (д, 6H), 2,21 (с, 3H), 4,2 (м, 1H), 5,9 (д, 1H), 7,2 (т, 1H), 7,3 (м, 2H), 7,31 (с, 1H), 7,4 (м, 1H), 7,8 (д, 1H), 8,5 (д, 1H), 10,4 (с, 1H).
ПРИМЕР 6
Получение N-[4-[хлор-2-метил-6-[[(1-метилэтил)амино]карбонил]фенил]-1-(3-хлор-2-пиридинил)-3-(трифторметил)-1Н-пиразол-5-карбоксамида.
Стадия A: Получение 2-амино-3-метил-5-хлорбензойной кислоты
К раствору 2-амино-3-метилбензойной кислоты (Aldrich, 15,0 г, 99,2 ммоль) в N,N-диметилформамиде (50 мл) добавляли N-хлорсукцинимид (13,3 г, 99,2 ммоль) и реакционную смесь нагревали до 100°C в течение 30 минут. Нагрев убирали, реакционную смесь охлаждали до комнатной температуры и оставляли в течение ночи. Реакционную смесь затем медленно выливали в ледяную воду (250 мл) для осаждения белого твердого вещества. Твердое вещество фильтровали и промывали четыре раза водой, а затем помещали в этилацетат (900 мл). Раствор этилацетата сушили над сульфатом магния, выпаривали при пониженном давлении, остаточное твердое вещество промывали простым эфиром с получением желаемого промежуточного продукта в виде белого твердого вещества (13,9 г).
1Н ЯМР (DMSO-d6) δ 2,11 (с, 3H), 7,22 (с, 1H), 7,55 (с, 1H).
Стадия В: Получение 3-хлор-2-[3-(трифторметил)-1Н-пиразол-1-ил]пиридина
К смеси 2,3-дихлорпиридина (99,0 г, 0,67 моль) и 3-трифторметил пиразола (83 г, 0,61 моль) в сухом N,N-диметилформамиде (300 мл) добавляли карбонат калия (166,0 г, 1,2 моль) и затем реакцию нагревали до 110-125°C на протяжении 48 часов. Реакцию охлаждали до 100°C, фильтровали через Celite® диатомовый слой для удаления твердых частиц. N,N-диметилформамид и избыток дихлорпиридина удаляли дистилляцией при атмосферном давлении. Дистилляция данного продукта при пониженном давлении (т.к. 139-141°C, 7 мм) давала названное соединение в виде светло-желтого масла (113,4 г). 1Н ЯМР(CDCl3) δ 6,78 (с, 1H), 7,36 (т, 1H), 7,93 (д, 1H), 8,15 (с, 1H), 8,45 (д, 1H).
Стадия C: Получение 1-(3-хлор-2-пиридинил)-3-(трифторметил)-1Н-пиразол-5-карбоновой кислоты.
К раствору пиразольного продукта со Стадии B (105,0 г, 425 ммоль) в сухом тетрагидрофуране (700 мл) при -75°C добавляли через канюлю -30°C раствор диизопропиламида лития (425 ммоль) в сухом тетрагидрофуране (300 мл). Насыщенный красный раствор перемешивали в течение 15 минут, после этого времени через раствор барботировали диоксид углерода при -63°C, до тех пор, пока раствор не становился бледно-желтым и не прекращалось выделение тепла. Реакционную смесь перемешивали в течение дополнительных 20 минут, а затем резко охлаждали водой (20 мл). Растворитель удаляли при пониженном давлении, и реакционную смесь распределяли между простым эфиром и 0,5 н. водным раствором гидроксида натрия. Водные экстракты промывали простым эфиром (3x), фильтровали через Celite® диатомовый слой для фильтрования для удаления остаточных твердых частиц и затем подкисляли приблизительно до pH 4, в этой точке образовывалось оранжевое масло. Водную смесь интенсивно перемешивали, добавляли дополнительное количество кислоты для снижения pH до 2,5-3. Оранжевое масло затвердевало в гранулярное твердое вещество, которое фильтровали, последовательно промывали водой и 1 н. соляной кислотой и сушили под вакуумом при 50°C с получением названного продукта в виде не совсем белого твердого вещества (130 г). (Продукт из другой фракции по сходной методике плавился при 175-176°C.).
1Н ЯМР (DMSO-d6) δ 7,61 (с, 1H), 7,76 (дд, 1H), 8,31 (д, 1H), 8,60 (д, 1H).
Стадия D: Получение 6-хлор-2-[1-(3-хлор-2-пиридинил)-3-(трифторметил)-1Н-пиразол-5-ил]-8-метил-4Н-3,1-бензоксазин-4-она.
К раствору метансульфонилхлорида (2,2 мл, 28,3 ммоль) в ацетонитриле (75 мл) добавляли по каплям смесь продукта карбоновой кислоты со Стадии C (7,5 г, 27,0 ммоль) и триэтиламина (3,75 мл, 27,0 ммоль) в ацетонитриле (75 мл) при 0-5°C. Затем температуру реакции поддерживали при 0°C в течение всего добавления реактивов. После перемешивания в течение 20 минут добавляли 2-амино-3-метил-5-хлорбензойную кислоту со Стадии A (5,1 г, 27,0 ммоль) и перемешивание продолжали в течении дополнительных 5 минут. Затем по каплям добавляли раствор триэтиламина (7,5 мл, 54,0 ммоль) в ацетонитриле (15 мл) и реакционную смесь перемешивали 45 минут с последующим добавлением метансульфонилхлорида (2,2 мл, 28,3 ммоль). Реакционную смесь затем нагревали до комнатной температуры и перемешивали в течение ночи. Затем добавляли приблизительно 75 мл воды для осаждения 5,8 г желтого твердого вещества. Дополнительный 1 г продукта выделяли экстракцией из фильтрата с получением в сумме 6,8 г названного соединения в виде желтого твердого вещества.
1Н ЯМР (CDCl3) δ 1,83 (с, 3H), 7,50 (с, 1H), 7,53 (м, 2H), 7,99 (м, 2H), 8,58 (д, 1H).
Стадия E: Получение N-[4-хлор-2-метил-6-[[(1-метилэтил)амино]карбонил]фенил]-1-(3-хлор-2-пиридинил)-3-(трифторметил)-1Н-пиразол-5-карбоксамида
К раствору продукта бензоксазинона со Стадии D (5,0 г, 11,3 ммоль) в тетрагидрофуране (35 мл) добавляли по каплям изопропиламин (2,9 мл, 34,0 ммоль) в тетрагидрофуране (10 мл) при комнатной температуре. Реакционную смесь нагревали до растворения всех твердых частиц и перемешивали дополнительные пять минут, в этот момент завершение реакции подтверждали с помощью тонкослойной хроматографии на силикагеле. Тетрагидрофурановый растворитель выпаривали при пониженном давлении, и оставшиеся твердые частицы очищали хроматографией на силикагеле с последующим растиранием в порошок с эфир/гексаном с получением названного соединения, соединения по настоящему изобретению, в виде твердого вещества (4,6 г), плавящегося при 195-196°C.
1Н ЯМР (CDCl3) δ 1,21 (д, 6H), 2,17 (с, 3H), 4,16 (м, 1H), 5,95 (шир д, 1H), 7,1-7,3 (м, 2H), 7,39 (с, 1H), 7,4 (м, 1H), 7,84 (д, 1H), 8,50 (д, 1H), 10,24 (шир с, 1H).
ПРИМЕР 7
Получение N-[4-хлор-2-метил-6-[(метиламино)карбонил]фенил]-1-(3-хлор-2-пиридинил)-3-(трифторметил)-1Н-пиразол-5-карбоксамида
К раствору продукта бензоксазинона ПРИМЕРА 6 Стадии D (4,50 г, 10,18 ммоль) в тетрагидрофуране (THF; 70 мл) добавляли по каплям метиламин (2,0 M раствор в THF, 15 мл, 30,0 ммоль) и реакционную смесь перемешивали при комнатной температуре в течение 5 минут. Тетрагидрофурановый растворитель выпаривали при пониженном давлении и оставшиеся твердые частицы очищали хроматографией на силикагеле с получением 4,09 г названного соединения, соединение по настоящему изобретению, в виде белого твердого вещества, плавящегося при 185-186°C.
1Н ЯМР (DMSO-d6) δ 2,17 (с, 3H), 2,65 (д, 3H), 7,35 (д, 1H), 7,46 (дд, 1H), 7,65 (дд, 1H), 7,74 (с, 1H), 8,21 (д, 1H), 8,35 (шир кв, 1H), 8,74 (д, 1H), 10,39 (с, 1H).
ПРИМЕР 8
Получение 3-хлор-N-[4-хлор-2-метил-6-[[(1-метилэтил)амино]карбонил]фенил]-1-(3-хлор-2-пиридинил)-1Н-пиразол-5-карбоксамида
Стадия A: Получение 3-хлор-N,N-диметил-1Н-пиразол-1-сульфонамида
К раствору N-диэтилсульфамоилпиразола (188,0 г, 1,07 моль) в сухом тетрагидрофуране (1500 мл) при -78°C добавляли по каплям раствор 2,5 M н-бутиллития (472 мл, 1,18 моль) в гексане, удерживая температуру ниже -65°C. По завершении добавления реакционную смесь поддерживали при -78°C в течение дополнительных 45 минут, после этого времени добавляли по каплям раствор гексахлорэтана (279 г, 1,18 моль) в тетрагидрофуране (120 мл). Реакционную смесь удерживали в течение часа при -78°C, нагревали до -20°C и затем резко охлаждали водой (1 л). Реакционную смесь экстрагировали метиленхлоридом (4x500 мл); органические экстракты сушили над сульфатом магния и концентрировали. Неочищенный продукт далее очищали хроматографией на силикагеле, используя метиленхлорид в качестве элюента, с получением названного соединения в виде желтого масла (160 г).
1Н ЯМР (CDCl3) δ 3,07 (д, 6H), 6,33 (с, 1H), 7,61 (с, 1H).
Стадия B: Получение 3-хлорпиразола
К трифторуксусной кислоте (290 мл) добавляли по каплям продукт хлорпиразола (160 г) со Стадии A, и реакционную смесь перемешивали при комнатной температуре в течение 1,5 часов и затем концентрировали при пониженном давлении. Остаток помещали в гексан, нерастворимые твердые частицы отфильтровывали и гексан концентрировали с получением неочищенного продукта в виде масла. Неочищенный продукт в дальнейшем очищали хроматографией на силикагеле используя в качестве элюента эфир/гексан (40:60) с получением названного продукта в виде желтого масла (64,44 г). 1Н ЯМР (CDCl3) δ 6,39 (с, 1H), 7,66 (с, 1H), 9,6 (шир с, 1H).
Стадия C: Получение 3-хлор-2-(3-хлор-1Н-пиразол-1-ил)пиридина
К смеси 2,3-дихлорпиридина (92,60 г, 0,629 моль) и 3-хлорпиразола (т.e. продукта со Стадии B) (64,44 г, 0,629 моль) в N,N-диметилформамиде (400 мл) добавляли карбонат калия (147,78 г, 1,06 моль), и затем реакционную смесь нагревали до 100°C в течение 36 часов. Реакционную смесь охлаждали до комнатной температуры и медленно выливали в ледяную воду. Осевшие твердые частицы фильтровали и промывали водой. Твердый осадок на фильтре помещали в этилацетат, сушили над сульфатом магния и концентрировали. Неочищенное твердое вещество хроматографировали на силикагеле с использованием в качестве элюента 20% этилацетат/гексан с получением названного продукта в виде белого твердого вещества (39,75 г).
1H ЯМР (CDCl3) δ 6,43 (с, 1H), 7,26 (м, 1H), 7,90 (д, 1H), 8,09 (с, 1H), 8,41 (д, 1H).
Стадия D: Получение 3-хлор-1-(3-хлор-2-пиридинил)-1Н-пиразол-5-карбоновой кислоты
К раствору пиразольного продукта со Стадии C (39,75 г, 186 ммоль) в сухом тетрагидрофуране (400 мл) при -78°C добавляли по каплям раствор 2,0 M диизопропиламида лития (93 мл, 186 ммоль) в тетрагидрофуране. Диоксид углерода барботировали через янтарно-желтый раствор в течение 14 минут, по истечении этого времени данный раствор становился бледным коричневато-желтым. Создавали щелочную реакцию 1 н. водным раствором гидроксида натрия и экстрагировали простым эфиром (2x500 мл). Водные экстракты подкисляли 6 н. соляной кислотой и экстрагировали этилацетатом (3×500 мл). Этилацетатные экстракты сушили над сульфатом магния и концентрировали с получением названного продукта в виде не совсем белого твердого вещества (42,96 г). (Продукт из другой фракции по сходной методике плавился при 198-199°C.)
1Н ЯМР (DMSO-d6) δ 6,99 (с, 1H), 7,45 (м, 1H), 7,93 (д, 1H), 8,51 (д, 1H).
Стадия E: Получение 6-хлор-2-[3-хлор-1-(3-хлор-2-пиридинил)-1Н-пиразол-5-ил]-8-метил-4Н-3,1-бензоксазин-4-она
К раствору метансульфонилхлорида (6,96 г, 61,06 ммоль) в ацетонитриле (150 мл) добавляли по каплям смесь продукта карбоновой кислоты со Стадии D (15,0 г, 58,16 ммоль) и триэтиламина (5,88 г, 58,16 ммоль) в ацетонитриле (150 мл) при -5°C. Реакционную смесь затем перемешивали в течение 30 минут при 0°C. Затем добавляли 2-амино-3-метил-5-хлорбензойную кислоту из ПРИМЕРА 6, Стадии A (10,79 г, 58,16 ммоль) и перемешивание продолжали в течение дополнительных 10 минут. Затем добавляли по каплям раствор триэтиламина (11,77 г, 116,5 ммоль) в ацетонитриле, сохраняя температуру ниже 10°C. Реакционную смесь перемешивали 60 минут при 0°C, а затем добавляли метансульфонилхлорид (6,96 г, 61,06 ммоль). Реакционную смесь затем нагревали до комнатной температуры и дополнительно перемешивали 2 часа. Реакционную смесь затем концентрировали и неочищенный продукт хроматографировали на силикагеле, используя в качестве элюента метиленхлорид с получением названного продукта в виде желтого твердого вещества (9,1 г).
1H ЯМР (CDCl3) δ 1,81 (с, 3H), 7,16 (с, 1H), 7,51 (м, 2H), 7,98 (д, 2H), 8,56 (д, 1H).
Стадия F: Получение 3-хлор-N-[4-хлор-2-метил]-6-[[(1-метилэтил)амино]карбонил]фенил]-1-(3-хлор-2-пиридинил)-1Н-пиразол-5-карбоксамида
К раствору продукта бензоксазинона Стадии E (6,21 г, 15,21 ммоль) в тетрагидрофуране (100 мл) добавляли изопропиламин (4,23 г, 72,74 ммоль) и реакционную смесь затем нагревали до 60°C, перемешивали в течение 1 часа, а затем охлаждали до комнатной температуры. Тетрагидрофурановый растворитель выпаривали при пониженном давлении, и оставшиеся твердые частицы очищали хроматографией на силикагеле с получением названного соединения, соединения по настоящему изобретению в виде белого твердого вещества (5,05 г), плавящегося при 173-175°C.
1Н ЯМР (CDCl3) δ 1,23 (д, 6H), 2,18 (с, 3H), 4,21 (м, 1H), 5,97 (д, 1H), 7,01 (м, 1H), 7,20 (с, 1H), 7,24 (с, 1H), 7,41 (д, 1H), 7,83 (д, 1H), 8,43 (д, 1H), 10,15 (шир с, 1H).
ПРИМЕР 9
Получение 3-хлор-N-[4-хлор-2-метил-6-[(метиламино)карбонил]фенил]-1-(3-хлор-2-пиридинил)-1Н-пиразол-5-карбоксамида
К раствору продукта бензоксазинона ПРИМЕРА 8, Стадии E (6,32 г, 15,47 ммоль) в тетрагидрофуране (50 мл) добавляли метиламин (2,0 M раствор в THF, 38 мл, 77,38 ммоль), и реакционную смесь нагревали до 60°C, перемешивали в течение 1 часа и затем охлаждали до комнатной температуры. Тетрагидрофурановый растворитель выпаривали при пониженном давлении и оставшиеся твердые частицы очищали хроматографией на силикагеле с получением названного соединения, соединения по настоящему изобретению, в виде белого твердого вещества (4,57 г), плавящегося при 225-226°C.
1Н ЯМР (CDCl3) δ 2,15 (с, 3H), 2,93 (с, 3H), 6,21 (д, 1H), 7,06 (с, 1H), 7,18 (с, 1H), 7,20 (с, 1H), 7,42 (м, 1H), 7,83 (д, 1H), 8,42 (д, 1H), 10,08 (шир с, 1H).
ПРИМЕР 10
Получение 3-Бром-N-[4-хлор-2-метил-6-[[(1-метилэтил)амино]карбонил]фенил]-1-(3-хлор-2-пиридинил)-1Н-пиразол-5-карбоксамида
Стадия A: Получение 3-бром-N,N-диметил-1Н-пиразол-1-сульфонамида
К раствору N-диметилсульфамоилпиразола (44,0 г, 0,251 моль) в сухом тетрагидрофуране (500 мл) при -78°C добавляли по каплям раствор н-бутиллития (2,5 M в гексане, 105,5 мл, 0,264 моль) поддерживая температуру ниже -60°C. Во время добавления образовывалось густое плотное вещество. По завершении добавления реакционную смесь удерживали в течение дополнительных 15 минут, по истечении этого времени добавляли по каплям раствор 1,2-дибромтетрахлорэтана (90 г, 0,276 моль) в тетрагидрофуране (150 мл), поддерживая температуру ниже -70°C. Реакционная смесь становилась ярко оранжевой; перемешивание продолжали в течение дополнительных 15 минут. Ванну -78°C убирали и реакцию резко охлаждали водой (600 мл). Реакционную смесь экстрагировали метиленхлоридом (4x), и органические экстракты сушили над сульфатом магния и концетрировали. Неочищенный продукт в дальнейшем очищали хроматографией на силикагеле, используя в качестве элюента метиленхлорид/гексан (50:50), с получением названного продукта в виде прозрачного бесцветного масла (57,04 г).
1Н ЯМР (CDCl3) δ 3,07 (д, 6H), 6,44 (м, 1H), 7,62 (м, 1H).
Стадия B: Получение 3-бромпиразола
К трифторуксусной кислоте (70 мл) медленно добавляли бромпиразольный продукт (57,04 г) со Стадии A. Реакционную смесь перемешивали при комнатной температуре в течение 30 минут и затем концентрировали при пониженном давлении. Остаток помещали в гексан, нерастворимые твердые частицы отфильтровывали и гексан выпаривали с получением неочищенного продукта в виде масла. Неочищенный продукт в дальнейшем очищали хроматографией на силикагеле, используя в качестве элюента этилацетат/дихлорметан (10:90), с получением масла. Это масло помещали в дихлорметан, нейтрализовали водным раствором бикарбоната натрия, экстрагировали метиленхлоридом (3x), сушили над сульфатом магния и концентрировали с получением названного продукта в виде белого твердого вещества (25,9 г), т.пл. 61-64°C.
1Н ЯМР (CDCl3) δ 6,37 (д, 1H), 7,59 (д, 1H), 12,4 (шир с, 1H).
Стадия C: Получение 2-(3-бром-1Н-пиразол-1-ил)-3-хлорпиридина
К смеси 2,3-дихлорпиридина (27,4 г, 185 ммоль) и 3-бромпиразола (т.e. продукта со Стадии B) (25,4 г, 176 ммоль) в сухом N,N-диметилформамиде (88 мл) добавляли карбонат калия (48,6 г, 352 ммоль), и реакционную смесь нагревали до 125°C в течение 18 часов. Реакционную смесь охлаждали до комнатной температуры и выливали в ледяную воду (800 мл). Образовывался осадок. Осевшие твердые частицы перемешивали в течение 1,5 часов, фильтровали и промывали водой (2×100 мл). Плотный осадок на фильтре помещали в метиленхлорид и промывали последовательно водой, 1 н. соляной кислотой, насыщенным водным раствором бикарбоната натрия и солевым раствором. Органические экстракты затем сушили над сульфатом магния и концентрировали с получением 39,9 г розового твердого вещества. Неочищенное твердое вещество суспендировали в гексане и интенсивно перемешивали в течение 1 часа. Твердые частицы фильтровали, промывали гексаном и сушили с получением названного продукта в виде не совсем белого порошка (30,4 г) с чистотой >94%, определенной с помощью ЯМР. Это вещество использовали без дальнейшей очистки на Стадии D.
1Н ЯМР (CDCl3) δ 6,52 (с, 1H), 7,30 (дд, 1H), 7,92 (д, 1H), 8,05 (с, 1H), 8,43 (д, 1H).
Стадия D: Получение 3-бром-1-(3-хлор-2-пиридинил)-1Н-пиразол-5-карбоновой кислоты
К раствору пиразольного продукта со Стадии C (30,4 г, 118 ммоль) в сухом тетрагидрофуране (250 мл) при -76°C добавляли по каплям раствор диизопропиламида лития (118 ммоль) в тетрагидрофуране с такой скоростью, чтобы удерживать температуру ниже -71°C. Реакционную смесь перемешивали в течение 15 минут при -76°C, а затем барботировали диоксид углерода в течение 10 минут, вызывая нагревание до -57°C. Реакционную смесь нагревали до -20°C и резко охлаждали водой. Реакционную смесь концентрировали и затем помещали в воду (1 л) и эфир (500 мл), а затем добавляли водный раствор гидроксида натрия (1 н., 20 мл). Водные экстракты промывали простым эфиром и подкисляли соляной кислотой. Осевшие твердые частицы фильтровали, промывали водой и сушили с получением названного продукта в виде желтовато-коричневого твердого вещества (27,7 г). (Продукт из другой фракции по сходной методике плавился при 200-201°C.)
1Н ЯМР (DMSO-d6) δ 7,25 (с, 1H), 7,68 (дд, 1H), 8,24 (д, 1H), 8,56 (д, 1H).
Стадия E: Получение 2-[3-бром-1-(3-хлор-2-пиридинил)-1Н-пиразол-5-ил]-6-хлор-8-метил-4Н-3,1-бензоксазин-4-она
Для превращения продукта пиразолкарбоновой кислоты из ПРИМЕРА 10, Стадии D (1,5 г, 4,96 ммоль) и 2-амино-3-метил-5-хлорбензойной кислоты (0,92 г, 4,96 ммоль) в названный продукт в виде твердого вещества была использована методика, аналогичная методике ПРИМЕРА 6, Стадии D.
1Н ЯМР (CDCl3) δ 2,01 (с, 3H), 7,29 (с, 1H), 7,42 (д, 1H), 7,95 (д, 1H), 8,04 (м, 1H), 8,25 (с, 1H), 8,26 (д, 1H).
Стадия F: Получение 3-бром-N-[4-хлор-2-метил-6-[[(1-метилэтил)амино]карбонил]фенил]-1-(3-хлор-2-пиридинил)-1Н-пиразол-5-карбоксамида
К раствору бензоксазинона со Стадии E (0,20 г, 0,44 ммоль) в тетрагидрофуране добавляли изопропиламин (0,122 мл, 1,42 ммоль), и реакционную смесь нагревали до 60°C в течение 90 минут, а потом охлаждали до комнатной температуры. Тетрагидрофурановый растворитель выпаривали при пониженном давлении, и оставшиеся твердые частицы растирали в порошок с простым эфиром, фильтровали и сушили с получением названного соединения, соединения по настоящему изобретению, в виде твердого вещества (150 мг), т.пл. 159-161°C.
1Н ЯМР (CDCl3) δ 1,22 (д, 6H), 2,19 (с, 3H), 4,21 (м, 1H), 5,99 (м, 1H), 7,05 (м, 1H), 7,22 (м, 2H), 7,39 (м, 1H), 7,82 (д, 1H), 8,41 (д, 1H).
ПРИМЕР 11
Получение 3-бром-N-[4-хлор-2-метил-6-[(метиламино)карбонил]фенил]-1-(3-хлор-2-пиридинил)-1Н-пиразол-5-карбоксамида
К раствору бензоксазинона ПРИМЕРА 10, Стадии E (0,20 г, 0,44 ммоль) в тетрагидрофуране добавляли метиламин (2,0 M раствор в THF, 0,514 мл, 1,02 ммоль), и реакционную смесь нагревали до 60°C в течение 90 минут, а затем охлаждали до комнатной температуры. Тетрагидрофурановый растворитель выпаривали при пониженном давлении, и оставшиеся твердые частицы растирали в порошок с эфиром, фильтровали и сушили с получением названного соединения, соединения по настоящему изобретению, в виде твердого вещества (40 мг), т.пл. 162-164°C.
1Н ЯМР (CDCl3) δ 2,18 (с, 3H), 2,95 (с, 3H), 6,21 (м, 1H), 7,10 (с, 1H), 7,24 (м, 2H), 7,39 (м, 1H), 7,80 (д, 1H), 8,45 (д, 1H).
Следующий ПРИМЕР 12 иллюстрирует альтернативное получение 3-хлор-1-(3-хлор-2-пиридинил)-1Н-пиразол-5-карбоновой кислоты, которая может быть использована для получения, например, 3-хлор-N-[4-хлор-2-метил-6-[[(1-метилэтил)амино]карбонил]фенил]-1-(3-хлор-2-пиридинил)-1Н-пиразол-5-карбоксамида и 3-хлор-N-[4-хлор-2-метил-6-[(метиламино)карбонил]фенил]-1-(3-хлор-2-пиридинил)-1Н-пиразол-5-карбоксамида, посредством дополнительных стадий, представленных в Примерах 8 и 9.
ПРИМЕР 12
Получение 3-хлор-1-(3-хлор-2-пиридинил)-1Н-пиразол-5-карбоновой кислоты
Стадия A: Получение этил 2-(3-хлор-2-пиридинил)-5-оксо-3-пиразолидинкарбоксилата (по-другому называемый этил 1-(3-хлор-2-пиридинил)-3-пиразолидинон-5-карбоксилат)
В 2-литровую четырехгорлую колбу, снабженную механической мешалкой, термометром, капельной воронкой, парциальным конденсатором горячего орошения (обратным холодильником) и клапаном для ввода азота, загружали абсолютный этанол (250 мл) и этанольный раствор этоксида натрия (21%, 190 мл, 0,504 моль). Смесь нагревали до температуры кипения с обратным холодильником примерно при 83°C. Затем ее обрабатывали 3-хлор-2(1Н)-пиридинон гидразоном (68,0 г, 0,474 моль). Смесь повторно нагревали до температуры кипения с обратным холодильником на протяжении 5 минут. Желтую суспензию затем обрабатывали по каплям диэтилмалеатом (88,0 мл, 0,544 моль) на протяжении 5 минут. Во время добавления заметно возрастала интенсивность кипения с обратным холодильником. К концу добавления все исходные вещества растворялись. Полученный в результате оранжево-красный раствор выдерживали с обратным холодильником в течение 10 минут. После охлаждения до 65°C реакционную смесь обрабатывали ледяной уксусной кислотой (50,0 мл, 0,873 моль). Образовывался осадок. Смесь разводили водой (650 мл), вызывая растворение осадка. Оранжевый раствор охлаждали на ледяной бане. Продукт начинал осаждаться при 28°C. Суспензию выдерживали примерно при 2°C в течение 2 часов. Данный продукт выделяли с помощью фильтрации, промывали водным этанолом (40%, 3×50 мл), и затем сушили воздухом на фильтре примерно в течение 1 часа. Названное соединение получали в виде высококристаллического светло-оранжевого порошка (70,3 г, 55% выход). Значительных примесей по данным 1Н ЯМР не наблюдалось.
1Н ЯМР (DMSO-d6) δ 1,22 (т, 3H), 2,35 (д, 1H), 2,91 (дд, 1H), 4,20 (кв, 2H), 4,84 (д, 1H), 7,20 (дд, 1H), 7,92 (д, 1H), 8,27 (д, 1H), 10,18 (с, 1H).
Стадия В: Получение этил 3-хлор-1-(3-хлор-2-пиридинил)-4,5-дигидро-1Н-пиразол-5-карбоксилата (по-другому назыаемый этил 1-(3-хлор-2-пиридинил)-3-хлор-2-пиразолин-5-карбоксилат)
В 2-литровую четырехгорлую колбу, снабженную механической мешалкой, термометром, парциальным конденсатором горячего орошения (обратным холодильником) и клапаном ввода азота, загружали ацетонитрил (1000 мл), этил 2-(3-хлор-2-пиридинил)-5-оксо-3-пиразолидинкарбоксилат (т.е. продукт Стадии A) (91,0 г, 0,337 моль) и оксихлорид фосфора (35,0 мл, 0,375 моль). При добавлении оксихлорида фосфора смесь разогревалась от 22 до 25°C и образовывался осадок. Светло-желтую суспензию нагревали до температуры кипения с обратным холодильником при 83°C на протяжении 35 минут, после чего осадок растворялся. Полученный в результате оранжевый раствор выдерживали с обратным холодильником в течение 45 минут, после чего он становился темно-зеленым. Парциальный конденсатор горячего орошения снабжали дистилляционной насадкой и удаляли 650 мл растворителя посредством дистилляции. В другую 2-литровую четырехгорлую колбу, снабженную механической мешалкой, помещали бикарбонат натрия (130 г, 1,55 моль) и воду (400 мл). Концентрированную реакционную смесь добавляли к суспензии бикарбоната натрия на протяжении 15 минут. Полученную в результате двухфазную смесь интенсивно перемешивали в течение 20 минут, в это время прекращалось выделение газа. Смесь разбавляли дихлорметаном (250 мл), а затем перемешивали в течение 50 минут. Смесь обрабатывали Celite® 545 диатомным ускорителем фильтрования (11 г), а затем фильтровали для удаления черного смолообразного вещества, которое мешало разделению фаз. Поскольку фильтрат медленно разделялся на отдельные фазы, его разбавляли дихлорметаном (200 мл) и водой (200 мл) и обрабатывали большим количеством Celite® 545 (15 г). Смесь фильтровали и фильтрат переносили в делительную воронку. Отделяли более тяжелый темно-зеленый органический слой. Рыхлый слой (50 мл) повторно фильтровали, а затем добавляли к органическому слою. Органический раствор (800 мл) обрабатывали сульфатом магния (30 г) и силикагелем (12 г), и суспензию перемешивали магнитом в течение 30 минут. Суспензию фильтровали для удаления сульфата магния и силикагеля, ставшую темного зелено-голубого цвета. Осадок на фильтре промывали дихлорметаном (100 мл). Фильтрат концентрировали на роторном испарителе. Данный продукт состоял из темно-желтого масла (92,0 г, 93% выход). Наблюдаемые посредством 1Н ЯМР только значительные примеси составили 1% исходного вещества и 0,7% ацетонитрила.
1Н ЯМР (DMSO-d6) δ 1,15 (т, 3H), 3,26 (дд, 1H), 3,58 (дд, 1H), 4,11 (кв, 2H), 5,25 (дд, 1H), 7,00 (дд, 1H), 7,84 (д, 1H), 8,12 (д, 1H).
Стадия C: Получение этил 3-хлор-1-(3-хлор-2-пиридинил)-1Н-пиразол-5-карбоксилата (по-другому называемый этил 1-(3-хлор-2-пиридинил)-3-хлорпиразол-5-карбоксилат)
В 2-литровую черырехгорлую колбу, снабженную механической мешалкой, термометром, парциальным конденсатором горячего орошения (обратным холодильником) и клапаном ввода азота, загружали этил 3-хлор-1-(3-хлор-2-пиридинил)-4,5-дигидро-1Н-пиразол-5-карбоксилат (т.е. продукт Стадии B) (95% очистки, 99,5 г, 0,328 моль), ацетонитрил (1000 мл), серную кислоту (98%, 35,0 мл, 0,661 моль). При добавлении серной кислоты смесь самонагревалась с 22 до 35°C. После перемешивания в течениеи нескольких минут смесь обрабатывали персульфатом калия (140 г, 0,518 моль). Суспензию нагревали до температуры кипения с обратным холодильником при 84°C в течение 4,5 часов. Полученную в результате оранжевую суспензию, пока еще теплую (50-65°C), фильтровали для удаления мелкого белого осадка. Осадок на фильтре промывали ацетонитрилом (50 мл). Фильтрат концентрировали примерно до 500 мл на роторном испарителе. В другую 2-литровую четырехгорлую колбу, снабженную механической мешалкой, загружали воду (1250 мл). Концентрированную реакционную массу добавляли к воде на протяжении примерно 5 минут. Продукт выделяли посредством фильтрации, промывали водным ацетонитрилом (25%, 3×125 мл), однократно промывали водой (100 мл), а затем сушили в течение ночи в вакууме при комнатной температуре. Данный продукт состоял из кристаллического оранжевого порошка (79,3 г, 82% выход). Наблюдаемые 1Н ЯМР только значительные примеси составили около 1,9% воды и 0,6% ацетонитрила.
1Н ЯМР (DMSO-d6) δ 1,09 (т, 3H), 4,16 (кв, 2H), 7,31 (с, 1H), 7,71 (дд, 1H), 8,38 (д, 1H), 8,59 (д, 1H).
Стадия D: Получение 3-хлор-1-(3-хлор-2-пиридинил)-1Н-пиразол-5-карбоновой кислоты (по-другому называемой 1-(3-хлор-2-пиридинил)-3-хлорпиразол-5-карбоновой кислотой)
В 1-литровую четырехгорлую колбу, снабженную механической мешалкой, термометром и азотным клапаном, загружали этил 3-хлор-1-(3-хлор-2-пиридинил)-1Н-пиразол-5-карбоксилат (т.е. продукт Стадии C) (97,5% очистки, 79,3 г, 0,270 моль), метанол (260 мл), воду (140 мл), гранулы гидроксида натрия (13,0 г, 0,325 моль). При добавлении гидроксида натрия смесь самонагревалась с 22 до 35°C, и исходное вещество начинало растворяться. После перемешивания в течение 45 минут в условиях окружающей среды растворились все исходные вещества. Полученный в результате темный оранжево-коричневый раствор концентрировали примерно до 250 мл на роторном испарителе. Затем концентрированную реакционную смесь разбавляли водой (400 мл). Водный раствор экстрагировали эфиром (200 мл). Затем водный слой переносили в 1-литровую колбу Erlenmeyer, снабженную магнитной мешалкой. Данный раствор обрабатывали по каплям концентрированной соляной кислотой (36,0 г, 0,355 моль) на протяжении примерно 10 минут. Продукт выделяли с помощью фильтрации, вновь суспендировали в воде (2×200 мл), однократно промывали водой (100 мл), а затем сушили на воздухе на фильтре в течение 1,5 часов. Данный продукт состоял из кристаллического светло-коричневого порошка (58,1 г, 83% выход). Единственной значительной примесью, наблюдаемой с помощью 1Н ЯМР, было 0,7% эфира.
1Н ЯМР (DMSO-d6) δ 7,20 (с, 1H), 7,68 (дд, 1H), 8,25 (д, 1H), 8,56 (д, 1H), 13,95 (шир с, 1H).
Следующий ПРИМЕР 13 иллюстрирует альтернативное получение 3-бром-1-(3-хлор-2-пиридинил)-1Н-пиразол-5-карбоновой кислоты, которая может быть использована для получения, например, 3-бром-N-[4-хлор-2-метил-6-[[(1-метилэтил)амино]карбонил]фенил]-1-(3-хлор-2-пиридинил)-1Н-пиразол-5-карбоксамида и 3-бром-N-[4-хлор-2-метил-6-[(метиламино)карбонил]фенил]-1-(3-хлор-2-пиридинил)-1Н-пиразол-5-карбоксамида, дополнительными стадиями, проиллюстрированными в Примерах 10 и 11.
ПРИМЕР 13
Получение 3-бром-1-(3-хлор-2-пиридинил)-1Н-пиразол-5-карбоновой кислоты
Стадия A1: Получение этил 3-бром-1-(3-хлор-2-пиридинил)-4,5-дигидро-1Н-пиразол-5-карбоксилата (по-другому называемый этил 1-(3-хлор-2-пиридинил)-3-бром-2-пиразолин-5-карбоксилат) с использованием оксибромида фосфора
В 1-литровую черырехгорлую колбу, снабженную механической мешалкой, термометром, парциальным конденсатором горячего орошения (обратным холодильником) и клапаном ввода азота, загружали ацетонитрил (400 мл), этил 2-(3-хлор-2-пиридинил)-5-оксо-3-пиразолидинкарбоксилат (т.е. продукт ПРИМЕРА 12, Стадии A) (50,0 г, 0,185 моль) и оксибромид фосфора (34,0 г, 0,119 моль). Оранжевую суспензию нагревали до температуры кипения с обратным холодильником при 83°C на протяжении 20 минут. Полученный в результате мутный оранжевый раствор нагревали с обратным холодильником в течение 75 минут, в это время образовывался плотный желтовато-коричневый кристаллический осадок. Парциальный конденсатор горячего орошения располагали с дистилляционной насадкой и собирали мутный бесцветный дистиллят (300 мл). В другую 1-литровую четырехгорлую колбу, снабженную механической мешалкой, загружали бикарбонат натрия (45 г, 0,54 моль) и воду (200 мл). Концентрированную реакционную смесь добавляли к суспензии бикарбоната натрия на протяжении 5 минут. Полученную в результате двухфазную смесь интенсивно перемешивали в течение 5 минут, в это время прекращалось выделение газа. Смесь разбавляли дихлорметаном (200 мл), а затем перемешивали в течение 75 минут. Смесь обрабатывали 5 г Celite® 545 диатомовым усилителем фильтрования, а затем фильтровали для удаления коричневого смолистого вещества. Фильтрат переносили в делительную воронку. Отделяли коричневый органический слой (400 мл), а затем обрабатывали сульфатом магния (15 г) и активированным углем Darco® G60 (2,0 г). Полученную в результате суспензию перемешивали магнитом в течение 15 минут, а затем фильтровали для удаления сульфата магния и угля. Зеленый фильтрат обрабатывали силикагелем (3 г) и перемешивали в течение нескольких минут. Темный зелено-голубой силикагель удаляли фильтрацией и фильтрат концентрировали на роторном испарителе. Данный продукт состоял из светлого янтарно-желтого масла (58,6 г, 95% выход), которое кристаллизовалось при стоянии. Единственной значительной примесью, наблюдаемой с помощью 1Н ЯМР, было 0,3% ацетонитрила.
1Н ЯМР (DMSO-d6) δ 1,15 (т, 3H), 3,29 (дд, 1H), 3,60 (дд, 1H), 4,11 (кв, 2H), 5,20 (дд, 1H), 6,99 (дд, 1H), 7,84 (д, 1H), 8,12 (д, 1H).
Стадия A2: Получение этил 3-бром-1-(3-хлор-2-пиридинил)-4,5-дигидро-1Н-пиразол-5-карбоксилата с использованием пентабромида фосфора
В 1-литровую черырехгорлую колбу, снабженную механической мешалкой, термометром, парциальным конденсатором горячего орошения (обратным холодильником) и клапаном ввода азота, загружали ацетонитрил (330 мл), этил 2-(3-хлор-2-пиридинил)-5-оксо-3-пиразолидинкарбоксилат (т.е. продукт ПРИМЕРА 12, Стадии A) (52,0 г, 0,193 моль), и пентабромид фосфора (41,0 г, 0,0952 моль). Оранжевую суспензию нагревали до температуры кипения с обратным холодильником при 84°C на протяжении 20 минут. Полученную в результате темно-красную смесь нагревали с обратным холодильником в течение 90 минут, в это время образовывался плотный желтовато-коричневый кристаллический осадок. Парциальный конденсатор горячего орошения располагали с дистилляционной насадкой и собирали мутный бесцветный дистиллят (220 мл). В другую 1-литровую четырехгорлую колбу, снабженную механической мешалкой, загружали бикарбонат натрия (40 г, 0,48 моль) и воду (200 мл). Концентрированную реакционную смесь добавляли к суспензии бикарбоната натрия на протяжении 5 минут. Полученную в результате двухфазную смесь интенсивно перемешивали в течение 10 минут, в это время прекратилось выделение газа. Смесь разбавляли дихлорметаном (200 мл) а затем перемешивали в течение 10 минут. Смесь обрабатывали Celite® 545 диатомовым ускорителем фильтрования (5 г), а затем фильтровали для удаления пурпурного смолообразного вещества. Осадок на фильтре промывали дихлорметаном (50 мл). Фильтрат переносили в делительную воронку. Отделяли пурпурно-красный органический слой (400 мл), а затем обрабатывали сульфатом магния (15 г) и активированным углем Darco® G60 (2,2 г). Суспензию перемешивали магнитом в течение 40 минут. Суспензию фильтровали для удаления сульфата магния и угля. Фильтрат концентрировали на роторном испарителе. Данный продукт состоял из темного янтарно-желтого масла (61,2 г, 95% выход), кристаллизующегося при стоянии. Единственной значительной примесью, наблюдаемой с помощью 1Н ЯМР, было 0,7% ацетонитрила.
1Н ЯМР (DMSO-d6) δ 1,15 (т, 3H), 3,29 (дд, 1H), 3,60 (дд, 1H), 4,11 (кв, 2H), 5,20 (дд, 1H), 6,99 (дд, 1H), 7,84 (д, 1H), 8,12 (д, 1H).
Стадия B: Получение этил 3-бром-1-(3-хлор-2-пиридинил)-1Н-пиразол-5-карбоксилата (по-другому называемого этил 1-(3-хлор-2-пиридинил)-3-бромпиразол-5-карбоксилат)
В 1-литровую черырехгорлую колбу, снабженную механической мешалкой, термометром, парциальным конденсатором горячего орошения (обратным холодильником) и клапаном ввода азота, загружали этил 3-бром-1-(3-хлор-2-пиридинил)-4,5-дигидро-1Н-пиразол-5-карбоксилат (т.е. продукт Стадий A1 и A2) (40,2 г, 0,121 моль), ацетонитрил (300 мл) и серную кислоту (98%, 13,0 мл, 0,245 моль). При добавлении серной кислоты смесь самонагревалась с 22 до 36°C. После перемешивания в течение нескольких минут, смесь обрабатывали персульфатом калия (48,0 г, 0,178 моль). Суспензию нагревали до температуры кипения с обратным холодильником при 84°C в течение 2 часов. Полученную в результате оранжевую суспензию еще теплой (50-65°C) фильтровали для удаления белого осадка. Осадок на фильтре промывали ацетонитрилом (2×50 мл). Фильтрат концентрировали примерно до 200 мл на роторном испарителе. В другую 1-литровую четырехгорлую колбу, снабженную механической мешалкой, загружали воду (400 мл). Концентрированную реакционную массу добавляли к воде на протяжении примерно 5 минут. Продукт выделяли посредством фильтрации, промывали последовательно водным ацетонитрилом (20%, 100 мл) и водой (75 мл), а затем сушили на воздухе на фильтре в течение 1 часа. Данный продукт состоял из кристаллического оранжевого порошка (36,6 г, 90% выход). Единственными значительными примесями, наблюдаемыми с помощью 1Н ЯМР, были 1% неизвестного вещества и 0,5% ацетонитрила.
1Н ЯМР (DMSO-d6) δ 1,09 (т, 3H), 4,16 (кв, 2H), 7,35 (с, 1H), 7,72 (дд, 1H), 8,39 (д, 1H), 8,59 (д, 1H).
Стадия C: Получение 3-бром-1-(3-хлор-2-пиридинил)-1Н-пиразол-5-карбоновой кислоты (по-другому называемая 1-(3-хлор-2-пиридинил)-3-бромпиразол-5-карбоновая кислота)
В 300 мл черырехгорлую колбу, снабженную механической мешалкой, термометром и азотным клапаном, загружали этил 3-бром-1-(3-хлор-2-пиридинил)-1Н-пиразол-5-карбоксилат (т.е. продукт Стадии B) (98,5% очистки, 25,0 г, 0,0756 моль), метанол (75 мл), воду (50 мл), и гранулы гидроксида натрия (3,30 г, 0,0825 моль). При добавлении гидроксида натрия смесь самонагревалась с 29 до 34°C и исходное вещество начинало растворяться. После перемешивания в течение 90 минут в условиях окружающей среды, растворились все исходные вещества. Полученный в результате темно-оранжевый раствор концентрировали примерно до 90 мл на роторном испарителе. Концентрированную реакционную смесь затем разбавляли водой (160 мл). Водный раствор экстрагировали эфиром (100 мл). Затем водный слой переносили в 500 мл колбу Erlenmeyer, снабженную магнитной мешалкой. Данный раствор обрабатывали по каплям концентрированной соляной кислотой (8,50 г, 0,0839 моль) на протяжении примерно 10 минут. Данный продукт выделяли посредством фильтрации, вновь суспендировали в воде (2×40 мл), верхний слой однократно промывали водой (25 мл), а затем сушили на воздухе на фильтре в течение 2 часов. Данный продукт состоял из кристаллического, желтовато-коричневого порошка (20,9 г, 91% выход). Единственными значительными примесями, наблюдаемыми с помощью 1Н ЯМР, были примерно 0,8% неизвестного вещества и 0,7% эфира.
1Н ЯМР (DMSO-d6) δ 7,25 (с, 1H), 13,95 (шир с, 1H), 8,56 (д, 1H), 8,25 (д, 1H), 7,68 (дд, 1H).
Следующий ПРИМЕР 14 иллюстрирует альтернативное получение этил 3-бром-1-(3-хлор-2-пиридинил)-4,5-дигидро-1Н-пиразол-5-карбоксилата, который может быть использован для приготовления, например, этил 3-бром-1-(3-хлор-2-пиридинил)-1Н-пиразол-5-карбоксилата (т.е. продукта ПРИМЕРА 13, Стадии B).
ПРИМЕР 14
Получение Этил 3-бром-1-(3-хлор-2-пиридинил)-4,5-дигидро-1Н-пиразол-5-карбоксилата из этил 3-хлор-1-(3-хлор-2-пиридинил)-4,5-дигидро-1Н-пиразол-5-карбоксилата с использованием бромида водорода.
Бромид водорода пропускали через раствор этил 3-хлор-1-(3-хлор-2-пиридинил)-4,5-дигидро-1Н-пиразол-5-карбоксилата (т.е. продукта ПРИМЕРА 12, Стадии B) (8,45 г, 29,3 ммоль) в дибромметане (85 мл). Через 90 минут прекращали поток газа и реакционную смесь промывали водным раствором бикарбоната натрия (100 мл). Органическую фазу сушили и выпаривали при пониженном давлении для получения названного продукта в виде масла (9,7 г, 99% выход), кристаллизующегося при стоянии.
1Н ЯМР (CDCl3) δ 1,19 (т, 3Н), 3,24 (1/2 АВ в образце АВХ, J=9,3, 17,3 Гц, 1Н), 3,44 (1/2 АВ в образце АВХ, J=11,7, 17,3 Гц, 1Н), 4,18 (кв, 2Н), 5,25 (Х из АВХ, 1Н, J=9,3, 11,9 Гц), 6,85 (дд, J=4,7, 7,7 Гц, 1Н), 7,65 (дд, J=1,6, 7,8 Гц), 8,07 (дд, J=1,6, 4,8 Гц, 1Н).
Следующий ПРИМЕР 15 иллюстрирует получение этил 1-(3-хлор-2-пиридинил)-4,5-дигидро-3-[[(4-метилфенил)сульфонил]окси]-1Н-пиразол-5-карбоксилата, который может быть использован для приготовления этил 3-бром-1-(3-хлор-2-пиридинил)-4,5-дигидро-1Н-пиразол-5-карбоксилата по методике, сходной с методикой, описанной в ПРИМЕРЕ 14.
ПРИМЕР 15
Получение этил 1-(3-хлор-2-пиридинил)-4,5-дигидро-3-[[(4-метилфенил)сульфонил]окси]-1Н-пиразол-5-кабоксилата
К смеси этил 2-(3-хлор-2-пиридинил)-5-оксо-3-пиразолидинкарбоксилата (т.е. продукта ПРИМЕРА 12, Стадии A) (10,0 г, 37,1 ммоль) и п-толуолсульфонилхлорида (7,07 г, 37,1 ммоль) в дихлорметане (100 мл) добавляли по каплям триэтиламин (3,75 г, 37,1 ммоль) при 0°C. В дальнейшем добавляли порции п-толуолсульфонилхлорида (0,35 г, 1,83 ммоль) и триэтиламина (0,19 г, 1,88 ммоль). Реакционную смесь затем оставляли для согревания до комнатной температуры и перемешивали в течение ночи. Затем смесь разбавляли дихлорметаном (200 мл) и промывали водой (3×70 мл). Органическую фазу сушили и выпаривали с получением названного продукта в виде масла (13,7 г, 87% выход), которое медленно образовывало кристаллы. Продукт, перекристаллизованный из этилацетат/гексанов, плавился при 99,5-100°C.
ИК (нюол) ν 1740, 1638, 1576, 1446, 1343, 1296, 1228, 1191, 1084, 1027, 948, 969, 868, 845 см-1.
1Н ЯМР (CDCl3) δ 1,19 (т, 3Н), 2,45 (с, 3Н), 3,12 (1/2 АВ в образце АВХ, J=17,39 Гц, 1Н), 3,33 (1/2 АВ в образце АВХ, J=17,5, 11,8 Гц, 1Н), 4,16 (кв, 2Н), 5,72 (Х из АВХ, 1Н, J=9, 11,8 Гц, 1Н), 6,79 (дд, J=4,6, 7,7 Гц, 1Н), 7,36 (дд, J=8,4, 2Н), 7,56 (дд, J=1,6, 7,8 Гц, 1Н), 7,95 (д, J=8,4 Гц, 2Н), 8,01 (дд, J=1,4, 4,6 Гц, 1Н).
ПРИМЕР 16
Получение N-[4-хлор-2-метил-6-[(метиламино)карбонил]фенил]-1-(3-хлор-2-пиридинил)-3-(2,2,2-трифторэтокси)-1Н-пиразол-5-карбоксамида
Стадия A: Получение этил 1-(3-хлор-2-пиридинил)-2,3-дигидро-3-оксо-1H-пиразол-5-карбоксилата
К суспензии этил 2-(3-хлор-2-пиридинил)-5-оксо-3-пиразолидинкарбоксилата (т.е. продукта ПРИМЕРА 12, Стадии A) (27 г, 100 ммоль), перемешанной в сухом ацетонитриле (200 мл), добавляли серную кислоту (20 г, 200 ммоль) за одну порцию. Реакционную смесь разжижали до образования бледно-зеленого почти прозрачного раствора, прежде чем снова сгустить до образования бледно-желтой суспензии. За одну порцию добавляли персульфат калия (33 г, 120 ммоль), а затем реакционную смесь нагревали при слабом кипении с обратным холодильником в течение 3,5 часов. После охлаждения с использованием ледяной ванны осадок белого твердого вещества удаляли посредством фильтрации и отбрасывали. Фильтрат разбавляли водой (400 мл), а затем экстрагировали три раза этиловым эфиром (всего 700 мл). Концентрирование объединенных эфирных экстрактов до уменьшенного объема (75 мл) вызывало преципитацию не совсем белого твердого вещества (3,75 г), которое собирали фильтрацией. Маточный раствор эфира в дальнейшем концентрировали с получением второго выхода не совсем белого осадка (4,2 г), который также собирали посредством фильтрации. Из водной фазы также выпадало в осадок не совсем белое твердое вещество; это твердое вещество (4,5 г) собирали фильтрацией с получением в общей сумме 12,45 г названного соединения.
1Н ЯМР (DMSO-d6) δ 1,06 (т, 3H), 4,11 (кв, 2H), 6,34 (с, 1H), 7,6 (т, 1H), 8,19 (д, 1H), 8,5 (д, 1H), 10,6 (с, 1H).
Стадия В: Получение этил 1-(3-хлор-2-пиридинил)-3-(2,2,2-трифторэтокси)-1Н-пиразол-5-карбоксилата
К суспензии этил 1-(3-хлор-2-пиридинил)-2,3-дигидро-3-оксо-1Н-пиразол-5-карбоксилата (т.е. продукта Стадии A) (0,8 г, 3 ммоль), перемешанной в сухом ацетонитриле (15 мл) при -5°C, добавляли карбонат калия (0,85 г, 6,15 ммоль). Эту суспензию перемешивали в течение 15 минут при 20°C. Перемешанную суспензию затем охлаждали до 5°C, и добавляли по каплям 2,2,2-трифторэтил трифторметансульфонат (0,8 г, 3,45 ммоль). Реакционную смесь нагревали до комнатной температуры, а затем нагревали до температуры кипения с обратным холодильником, во время которого тонкослойная хроматография показала завершение реакции. К реакционной смеси добавляли воду (25 мл), которую потом экстрагировали этиловым эфиром. Эфирный экстракт сушили над сульфатом магния и концентрировали с получением названного соединения (1,05 г) в виде бледно-желтого масла.
1Н ЯМР (CDCl3) δ 1,21 (т, 3H), 4,20 (кв, 2H), 4,63 (кв, 2H), 6,53 (с, 1H), 7,4 (т, 1H), 7,9 (д, 1H), 8,5 (д, 1H).
Стадия С: Получение 1-(3-хлор-2-пиридинил)-3-(2,2,2-трифторэтокси)-1Н-пиразол-5-карбоновой кислоты
К перемешанному раствору этил 1-(3-хлор-2-пиридинил)-3-(2,2,2-трифторэтокси)-1Н-пиразол-5-карбоксилата (т.е. продукта Стадии B) (0,92 г, 2,8 ммоль) в метаноле (15 мл) добавляли воду (5 мл), что вызывало помутнение реакционной смеси. Водный раствор гидроксида натрия (50%, 1,5 г, 19,2 ммоль) добавляли по каплям, и реакционную смесь перемешивали при комнатной температуре в течение 30 минут, во время чего реакционная смесь снова становилась прозрачной. Добавляли воду (20 мл) и реакционную смесь экстрагировали этиловым эфиром, который отбрасывали. Водную фазу подкисляли до pH 2, используя концентрированную соляную кислоту, а затем экстрагировали этилацетатом (50 мл). Этилацетатный экстракт, который промывали водой (20 мл) и солевым раствором (20 мл), сушили над сульфатом магния и концентрировали с получением названного соединения, выделенного в виде белого твердого вещества (0,8 г).
1Н ЯМР (DMSO-d6) δ 4,9 (кв, 2H), 6,75 (с, 1H), 7,6 (т, 1H), 8,2 (д, 1H), 8,55 (д, 1H), 13,7 (шир с, 1H).
Стадия D: Получение 6-хлор-8-метил-2Н-3,1-бензоксазин-2,4(1Н)-диона
К суспензии 2-амино-3-метил-5-хлорбензойной кислоты (т.е. продукта ПРИМЕРА 6, Стадии A) (97 г, 520 ммоль), перемешанной в сухом диоксане (750 мл) при комнатной температуре, добавляли по каплям трихлорметилхлорформиат (63 г, 320 ммоль). Реакционную смесь медленно экзотермически нагревали до 42°C, и твердое вещество почти полностью растворялось до того, как снова образовывалась густая суспензия. После того, как эту суспензию перемешивали при температуре окружающей среды в течение 2,5 часов, названное соединение выделяли фильтрацией, промывали этиловым эфиром, и сушили с получением продукта названного соединения, полученного в виде белого твердого вещества (98 г).
1Н ЯМР (DMSO-d6) δ 2,3 (с, 3H), 7,70 (с, 1H), 7,75 (с, 1H), 11,2 (с, 1H).
Стадия E: Получение 6-хлор-2-[1-(3-хлор-2-пиридинил)-3-(2,2,2-трифторэтокси)-1Н-пиразол-5-ил]-8-метил-4Н-3,1-бензоксазин-4-она
К суспензии 1-(3-хлор-2-пиридинил)-3-(2,2,2-трифторэтокси)-1Н-пиразол-5-карбоновой кислоты (т.е. продукта Стадии C) (7,9 г, 24 ммоль), перемешанной в дихлорметане (100 мл), добавляли N,N-диметилформамид (4 капли). Оксалилхлорид (4,45 г, 35 ммоль) добавляли по каплям на протяжении 45 минут. Полученный в результате раствор перемешивали при комнатной температуре в течение 4 часов, а затем концентрировали под вакуумом. Выделенный хлорангидрид кислоты растворяли в сухом ацетонитриле (10 мл) и добавляли к суспензии 6-хлор-8-метил-2Н-3,1-бензоксазин-2,4(1Н)-диона (т.е. продукта Стадии D) (4,9 г, 23 ммоль), перемешанной в сухом ацетонитриле (14 мл). Добавляли пиридин (10 мл) и данный раствор нагревали при температуре кипения с обратным холодильником 6 часов. После охлаждения с использованием ледяной ванны собирали осадок белого твердого вещества (9,15 г). Спектр 1Н ЯМР собранного осадка показал пики, соответствующие названному соединению и остаточному исходному веществу 6-хлор-8-метил-2Н-3,1-бензоксазин-2,4(1Н)-диону. Небольшую часть собранного осадка перекристаллизовывали из ацетонитрила с получением чистого названного продукта, плавящегося при 178-180°C.
1Н ЯМР (DMSO-d6) δ 1,72 (с, 3H), 4,96 (кв, 2H), 7,04 (с, 1H), 7,7 (т, 1H), 7,75 (с, 1H), 7,9 (с, 1H), 8,3 (д, 1H), 8,6 (д, 1H).
Стадия F: Получение N-[4-хлор-2-метил-6-[(метиламино)карбонил]фенил]-1-(3-хлор-2-пиридинил)-3-(2,2,2-трифторэтокси)-1H-пиразол-5-карбоксамида
К суспензии 6-хлор-2-[1-(3-хлор-2-пиридинил)-3-(2,2,2-трифторэтокси)-1Н-пиразол-5-ил]-8-метил-4Н-3,1-бензоксазин-4-она (т.е продукт осадка Стадии E) (3,53 г, 7,5 ммоль) в тетрагидрофуране (15 мл) добавляли по каплям метиламин (2,0 M раствор в THF, 11 мл, 22 ммоль), и результирующий раствор перемешивали при комнатной температуре в течение 45 минут. Затем тонкослойная хроматография показала, что реакция завершена. Добавляли этиловый эфир (100 мл), и реакционную смесь перемешивали в течение 2 часов пока образовывался осадок. Осадок собирали фильтрацией, а затем перекристаллизовывали из ацетонитрила с получением белого твердого вещества (0,82 г). Вторая часть белого твердого вещества (0,35 г) осаждалась из маточного раствора ацетонитрила и была собрана посредством фильтрации. Исходный маточный раствор эфир/тетрагидрофуран концентрировали до сухости и оставшиеся твердые частицы перекристаллизовывали из ацетонитрила с получением третьей порции белого твердого вещества (0,95 г). Объединяли три сбора, составляющих 2,12 г (после сушки), названного соединения, соединения по настоящему изобретению, выделенного в виде твердого белого вещества, плавящегося при 195-197°C.
1Н ЯМР (CDCl3) δ 2,18 (с, 3H), 2,92 (д, 3H), 4,66 (кв, 2H), 6,15 (кв, 1H), 6,6 (с, 1H), 7,2 (с, 1H), 7,25 (с, 1H), 7,35 (т, 1H), 7,8 (д, 1H), 8,45 (д, 1H), 10,0 (с, 1H).
Следующий ПРИМЕР 17 иллюстрирует альтернативное получение 1-(3-хлор-2-пиридинил)-3-(трифторметил)-1Н-пиразол-5-карбоновой кислоты, которая может быть использована для приготовления, например, 1-(3-хлор-2-пиридинил)-N-[2-метил-6-[[(1-метилэтил)амино]карбонил]фенил]-3-(трифторметил)-1Н-пиразол-5-карбоксамида с помощью дальнейших стадий, представленных в Примере 4.
ПРИМЕР 17
Получение 1-(3-хлор-2-пиридинил)-3-(трифторметил)-1Н-пиразол-5-карбоновой кислоты
Стадия A: Получение 3-хлор-2(1Н)-пиридинон(2,2,2-трифтор-1-метилэтилиден)гидразона
1,1,1-Трифторацетон (7,80 г, 69,6 ммоль) добавляли к 3-хлор-2(1Н)-пиридинонгидразону (по-другому называемый (3-хлорпиридин-2-ил)гидразин) (10 г, 69,7 ммоль) при 20-25°C. После завершения добавления смесь перемешивали примерно в течение 10 минут. Растворитель удаляли при пониженном давлении и смесь распределяли между этилацетатом (100 мл) и насыщенным водным раствором карбоната натрия (100 мл). Органический слой сушили и выпаривали. Хроматография на силикагеле (промытом этилацетатом) давала продукт в виде не совсем белого твердого вещества (11 г, 66% выход), т.пл. 64-64,5°C (после кристаллизации из этилацетат/гексанов).
ИК (нюол) ν 1629, 1590, 1518, 1403, 1365, 1309, 1240, 1196, 1158, 1100, 1032, 992, 800 см-1.
1Н ЯМР (CDCl3) δ 2,12 (c, 3Н), 6,91-6,86 (м, 1Н), 7,64-7,61 (м, 1Н), 8,33- 8,32 (м, 2Н).
МС m/z 237 (М+).
Стадия В: Получение этилгидроэтандиоата (3-хлор-2-пиридинил)(2,2,2-трифтор-1-метилэтилиден)гидразида (по-другому называемого этилгидроэтандиоата (3-хлор-2-пиридинил)(2,2,2-трифтор-1-метилэтилиден)гидразин)
Триэтиламин (20,81 г, 0,206 моль) добавляли к 3-хлор-2(1Н)-пиридинон(2,2,2-трифтор-1-метилэтилиден)гидразону (т.е. продукту Стадии A) (32,63 г, 0,137 моль) в дихлорметане (68 мл) при 0°C. Этилхлороксоацетат (18,75 г, 0,137 моль) в дихлорметане (69 мл) добавляли по каплям к этой смеси при 0°C. Смесь оставляли для нагревания до 25°C примерно на протяжении 2 часов. Смесь охлаждали до 0°C и добавляли по каплям дополнительные порции этилхлороксоацетата (3,75 г, 27,47 ммоль) в дихлорметане (14 мл). Примерно после дополнительного 1 часа смесь разбавляли дихлорметаном (примерно 450 мл), и смесь промывали водой (2×150 мл).
Органический слой сушили и выпаривали. Хроматография на силикагеле (промытом 1:1 этилацетат-гексаны) давала продукт в виде твердого вещества (42,06 г, 90% выход), т.пл. 73,0-73,5°C (после кристаллизации из этилацетат/гексанов).
ИК ν (нюол) 1751, 1720, 1664, 1572, 1417, 1361, 1330, 1202, 1214, 1184, 1137, 1110, 1004, 1043, 1013, 942, 807, 836 см-1.
1Н ЯМР (ДМСО-d6, 115°С) 1,19 (т, 3Н), 1,72 (бр с, 3Н), 4,25 (кв, 2Н), 7,65 (дд, J=8,3, 4,7 Гц, 1Н), 8,20 (дд, J=7,6, 1,5 Гц, 1Н), 8,55 (д, J=3,6 Гц, 1Н).
МС m/z 337 (М+).
Стадия С: Получение этил 1-(3-хлор-2-пиридинил)-4,5-дигидро-5-гидрокси-3-(трифторметил)-1Н-пиразол-5-карбоксилата
Этилгидроэтандиоата (3-хлор-2-пиридинил)(2,2,2-трифтор-1-метилэтилиден)гидразид (т.е. продукт Стадии B) (5 г, 14,8 ммоль) в диметилсульфоксиде (25 мл) добавляли к гидрату тетрабутиламмония фториду (10 г) в диметилсульфоксиде (25 мл) в течение 8 часов. По завершении добавления смесь выливали в уксусную кислоту (3,25 г) в воде (25 мл). После перемешивания при 25°C в течение ночи, смесь затем экстрагировали толуолом (4×25 мл), и объединенные толуоловые экстракты промывали водой (50 мл), сушили и выпаривали с получением твердого вещества. Хроматография на силикагеле (промытом 1:2 этилацетат-гексаны) давала продукт в виде твердого вещества (2,91 г, 50% выход, содержащего примерно 5% 3-хлор-2(1Н)-пиридинон(2,2,2-трифтор-1-метилэтилиден)гидразона), т.пл. 78-78,5°C (после перекристаллизации из этилацетат/гексанов).
ИК (нюол) ν 3403, 1726, 1618, 1582, 1407, 1320, 1293, 1260, 1217, 1187, 1150, 1122, 1100, 1067, 1013, 873, 829 см-1.
1Н ЯМР (CDCl3) δ 1,19 (с, 3Н), 3,20 (1/2 образца ABZ, J=18 Гц, 1Н), 3,42 (1/2 образца ABZ, J=18 Гц, 1Н), 4,24 (кв, 2Н), 6,94 (дд, J=7,9, 4,9 Гц, 1Н), 7,74 (дд, J=7,7, 1,5 Гц, 1Н), 8,03 (дд, J=4,7, 1,5 Гц, 1Н).
МС m/z 319 (М+).
Стадия D: Получение этил 1-(3-хлор-2-пиридинил)-3-(трифторметил)-1Н-пиразол-5-карбоксилата
Серную кислоту (концентрированную, 2 капли) добавляли к этил 1-(3-хлор-2-пиридинил)-4,5-дигидро-5-гидрокси-3-(трифторметил)-1Н-пиразол-5-карбоксилату (т.е. продукту Стадии C) (1 г, 2,96 ммоль) в уксусной кислоте (10 мл) и эту смесь нагревали до 65°C примерно в течение 1 часа. Смесь оставляли для охлаждения до 25°C и большую часть уксусной кислоты удаляли при пониженном давлении. Смесь распределяли между насыщенным водным раствором карбоната натрия (100 мл) и этилацетатом (100 мл). В дальнейшем водный слой экстрагировали этилацетатом (100 мл). Объединенные органические экстракты сушили и выпаривали с получением продукта в виде масла (0,66 г, 77% выход).
ИК (неат) ν 3147, 2986, 1734, 1577, 1547, 1466, 1420, 1367, 1277, 1236, 1135, 1031, 973, 842, 802 см-1.
1Н ЯМР (CDCl3) δ 1,23 (т, 3Н), 4,25 (кв, 2Н), 7,21 (с, 1Н), 7,48 (дд, J=8,1, 4,7 Гц, 1Н), 7,94 (дд, J=6,62 Гц, 1Н), 8,53 (дд, J=4,7, 1,5 Гц, 1Н).
МС m/z 319 (М+).
Стадия E: Получение 1-(3-хлор-2-пиридинил)-3-(трифторметил)-1Н-пиразол-5-карбоновой кислоты
Гидроксид калия (0,5 г, 85%, 2,28 ммоль) в воде (1 мл) добавляли к этил 1-(3-хлор-2-пиридинил)-3-(трифторметил)-1Н-пиразол-5-карбоксилату (т.е. продукту Стадии D) (0,66 г, 2,07 ммоль) в этаноле (3 мл). Примерно через 30 минут растворитель удаляли при пониженном давлении и смесь растворяли в воде (40 мл). Этот раствор промывали этилацетатом (20 мл). Водный слой подкисляли концентрированной соляной кислотой и экстрагировали этилацетатом (3×20 мл). Объединенные экстракты сушили и выпаривали с получением данного продукта в виде твердого вещества (0,53 г, 93% выход), т.пл. 178-179°C (после кристаллизации из гексаны-этилацетата).
ИК (нюол) ν 1711, 1586, 1565, 1550, 1440, 1425, 1292, 1247, 1219, 1170, 1135, 1087, 1059, 1031, 972, 843, 816 см-1.
1Н ЯМР (ДМСО-d6) δ 7,61 (с, 1Н), 7,77 (м, 1Н), 8,30 (д, 1Н), 8,60 (с, 1Н).
Примеры 18 и 19 иллюстрируют альтернативные условия реакции, описанные в ПРИМЕРЕ 10, Стадия E, и ПРИМЕРЕ 8, Стадия E, соответственно.
ПРИМЕР 18
Получение 2-[3-бром-1-(3-хлор-2-пиридинил)-1Н-пиразол-5-ил]-6-хлор-8-метил-4Н-3,1-бензоксазин-4-она
Метансульфонилхлорид (1,0 мл, 1,5 г, 13 ммоль) растворяли в ацетонитриле (10 мл), и эту смесь охлаждали до -5°C. Раствор 3-бром-1-(3-хлор-2-пиридинил)-1Н-пиразол-5-карбоновой кислоты (т.е. продукт пиразолкарбоновой кислоты ПРИМЕРА 10, Стадии D) (3,02 г, 10 ммоль) и пиридин (1,4 мл, 1,4 г, 17 ммоль) в ацетонитриле (10 мл), добавляли по каплям в течение 5 минут при -5 до 0°C. Во время добавления образовалась суспензия. Смесь перемешивали 5 минут при этой температуре, а затем добавляли смесь 2-амино-3-метил-5-хлорбензойной кислоты (т.е. продукта ПРИМЕРА 6 Стадии A) (1,86 г, 10 ммоль) и пиридина (2,8 мл, 2,7 г, 35 ммоль) в ацетонитриле (10 мл), снова промывая ацетонитрилом (5 мл). Смесь перемешивали 15 минут при -5 до 0°C, а затем добавляли по каплям метансульфонилхлорид (1,0 мл, 1,5 мл, 13 ммоль) в ацетонитриле (5 мл) в течение 5 минут при температуре от -5 до 0°C. Реакционную смесь перемешивали еще 15 минут при этой температуре, затем оставляли для медленного нагревания до комнатной температуры, и перемешивали 4 ч. Воду (20 мл) добавляли по каплям, и смесь перемешивали 15 минут. Затем эту смесь фильтровали и твердые частицы промывали 2:1 ацетонитрил-вода (3×3 мл), затем ацетонитрилом (2×3 мл), и сушили в атмосфере азота с получением названного продукта в виде светло-желтого порошка, 4,07 г (90,2% выход неочищенного продукта), плавящегося при 203-205°C. ВЭЖХ данного продукта с использованием Zorbax® RX-C8 хроматографической колонки (4,6 мм × 25 см, элюент 25-95% ацетонитрил/pH 3, вода), показала главный пик, соответствующий названному соединению и имеющий 95,7% всей площади пиков хроматографии.
1Н ЯМР (DMSO-d6) δ 1,72 (с, 3H) 7,52 (с, 1H), 7,72-7,78 (м, 2H), 7,88 (м, 1H), 8,37 (дд, 1H), 8,62 (дд, 1H).
ПРИМЕР 19
Получение 6-хлор-2-[3-хлор-1-(3-хлор-2-пиридинил)-1Н-пиразол-5-ил]-8-метил-4Н-3,1-бензоксазин-4-она
Метансульфонилхлорид (1,0 мл, 1,5 г, 13 ммоль) растворяли в ацетонитриле (10 мл), и смесь охлаждали до -5°C. Раствор 3-хлор-1-(3-хлор-2-пиридинил)-1Н-пиразол-5-карбоновой кислоты (т.е. продукт карбоновой кислоты ПРИМЕРА 8, Стадии D) (2,58 г, 10 ммоль) и пиридин (1,4 мл, 1,4 г, 17 ммоль) в ацетонитриле (10 мл), добавляли по каплям в течение 5 минут при -5 до 0°C. Во время добавления образовывалась суспензия. Смесь перемешивали 5 минут при этой температуре, а затем добавляли за один раз 2-амино-3-метил-5-хлорбензойную кислоту (т.е. продукт из ПРИМЕРА 6, Стадии A) (1,86 г, 10 ммоль). Затем раствор пиридина (2,8 мл, 2,7 г, 35 ммоль) в ацетонитриле (10 мл) добавляли по каплям через 5 мин при -5 до 0°C. Смесь перемешивали 15 минут при -5 до 0°C, а затем добавляли по каплям метансульфонилхлорид (1,0 мл, 1,5 мл, 13 ммоль) в ацетонитриле (5 мл) через 5 мин при -5 до 0°C. Реакционную смесь перемешивали 15 минут при этой температуре, затем оставляли для медленного нагревания до комнатной температуры и перемешивали 4 ч. Воду (15 мл) добавляли по каплям, и эту смесь перемешивали 15 минут. Затем смесь фильтровали и твердые примеси промывали 2:1 ацетонитрил-вода (3×3 мл), затем ацетонитрилом (2×3 мл), и сушили в атмосфере азота с получением названного продукта в виде бледно-желтого порошка, 3,83 г (94,0% выход неочищенного продукта), плавящегося при 199-201°C. ВЭЖХ продукта с использованием Zorbax® RX-C8 хроматографической колонки (4,6 мм × 25 см, элюент 25-95% ацетонитрил/pH 3, вода), показала главный пик, соответствующий названному соединению, и имеющий 97,8% общей площади хроматографического пика.
1Н ЯМР (DMSO-d6) δ 1,72 (с, 3H), 7,48 (с, 1H), 7,74-7,80 (м, 2H), 7,87 (м, 1H), 8,37 (дд, 1H), 8,62 (дд, 1H).
С помощью методик, описанных здесь, вместе со способами, известными в этой области, могут быть получены следующие соединения Таблиц 1-6. В Таблицах использованы следующие аббревиатуры: t означает третичный, s означает вторичный, n означает нормальный, iозначает изо, Me означает метил, Et означает этил, Pr означает пропил, i-Pr означает изопропил и Bu означает бутил.
Таблица 1

Таблица 2










Таблица 3

Таблица 4

Таблица 5

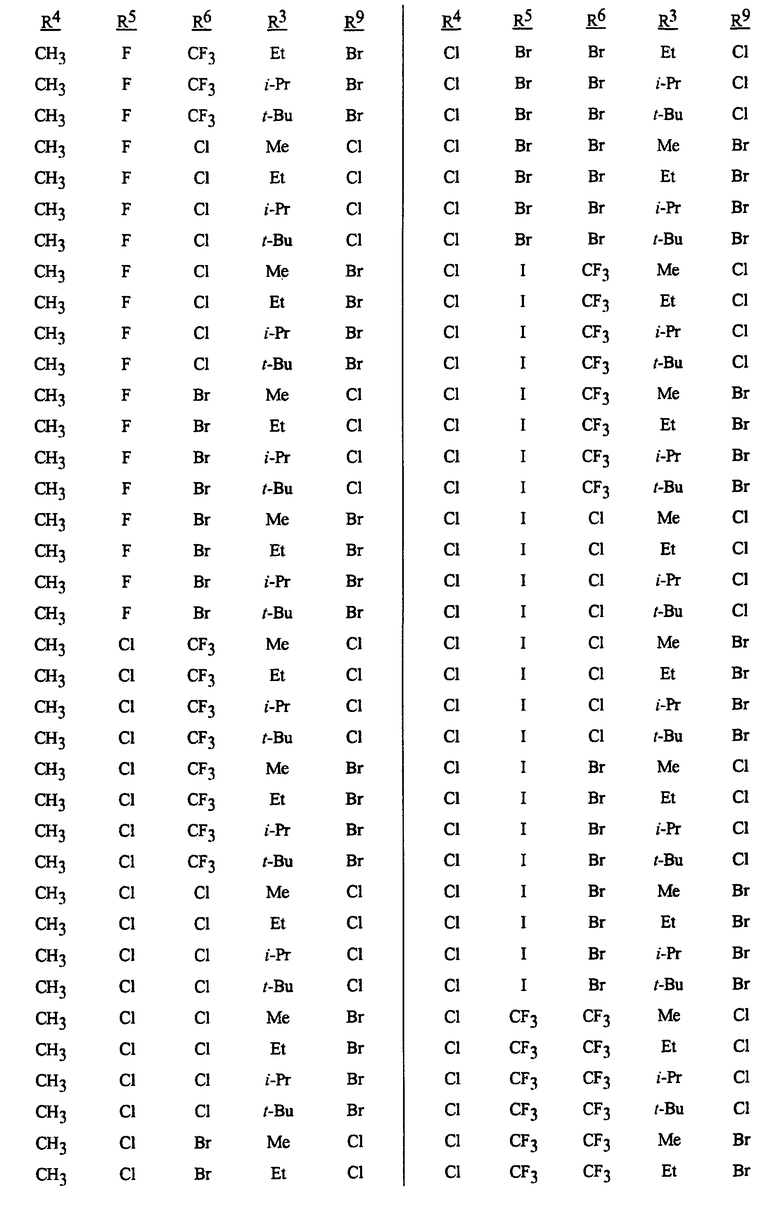




Таблица 6


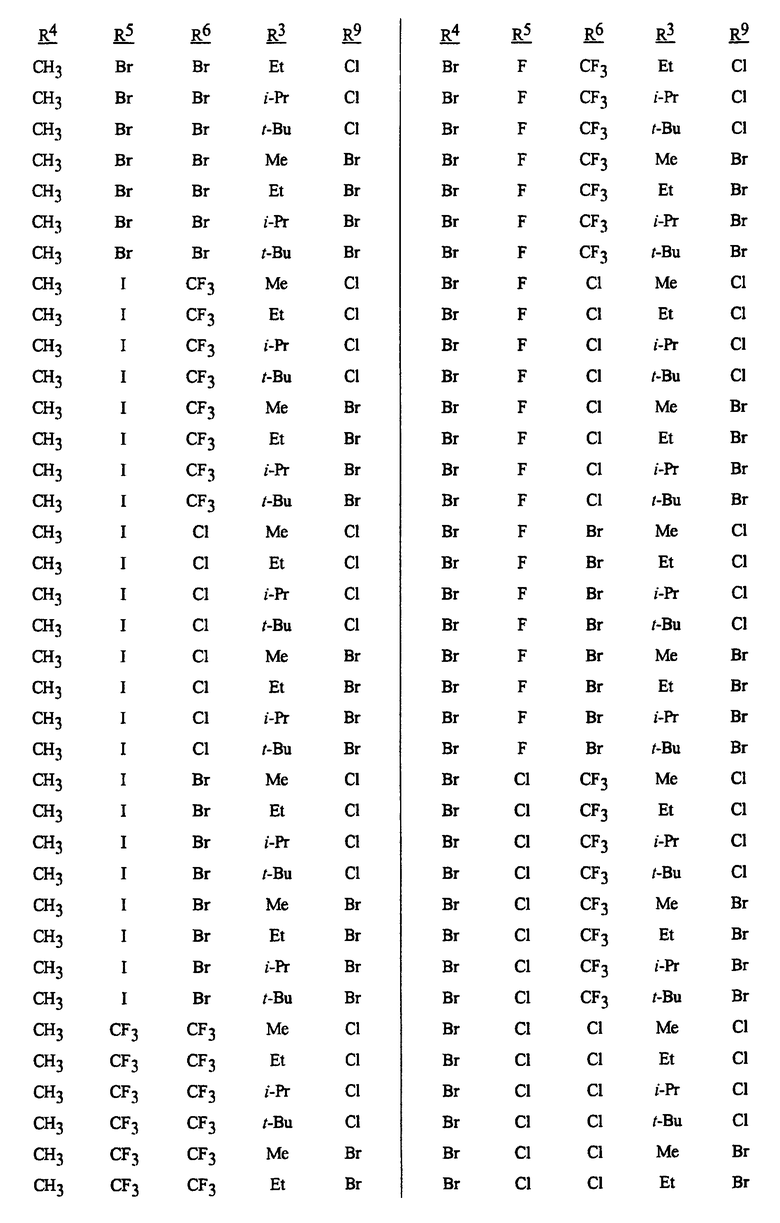
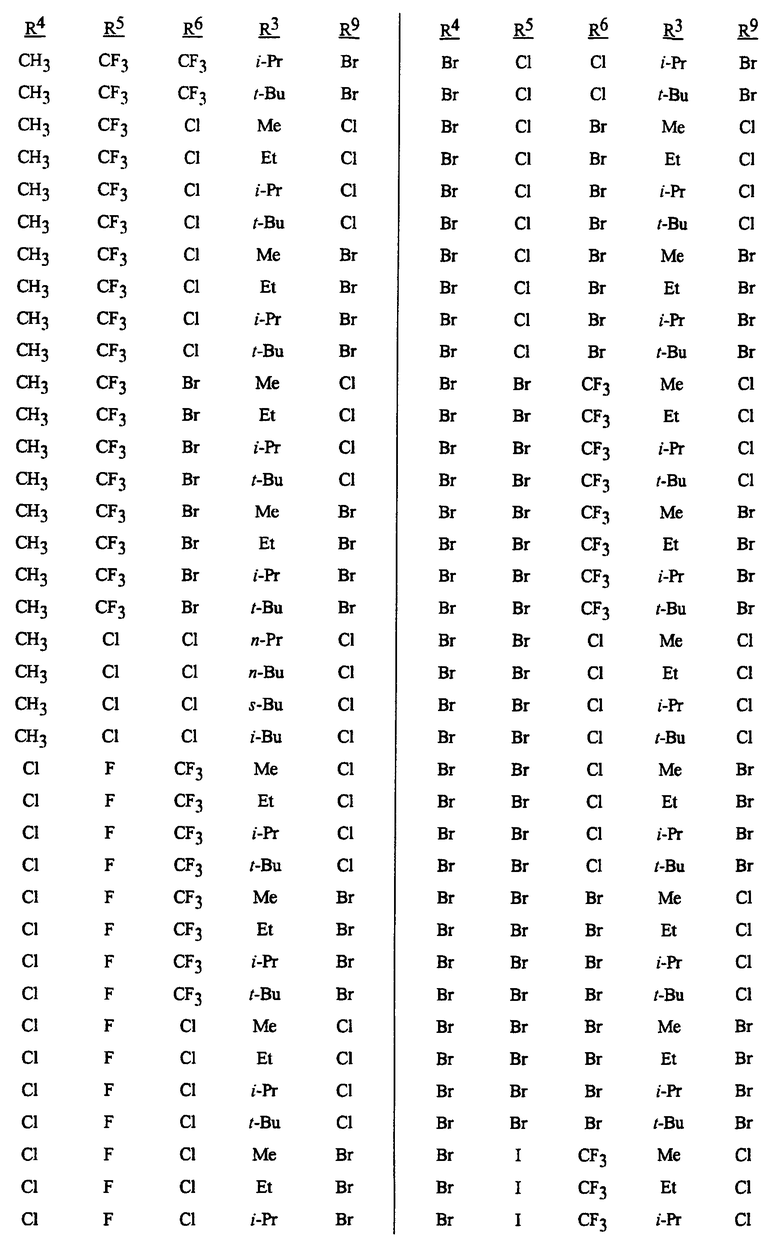


Составы/Применение
Было обнаружено, что соединения Формулы I не только обладают отличным действием, контролирующим растительноядных беспозвоночных вредителей, но и также имеют подходящую картину остаточного содержания и передвижения веществ по растению обеспечивает защиту растения, развивающегося из растительного посадочного материала (сеянца), такого как семя, луковица, корневище, клубень, клубнелуковица, или стебля, или черенка листа. (В контексте данного описания "контроль за беспозвоночными вредителями" означает подавление развития беспозвоночного вредителя (в том числе смертность), вызывающего значительное снижение поедания и других повреждений и поражений, вызываемых вредителями; родственные выражения определены аналогично.) Таким образом, данное изобретение предлагает способ защиты растительных сеянцев от растительноядных беспозвоночных вредителей путем контактирования сеянца или локуса сеянца с биологически эффективным количеством соединения Формулы I. Способ по данному изобретению, используя достаточное количество соединения Формулы I, также был описан для защиты не только самого сеянца, а также нового ростка, развивающегося из сеянца.
Как описано здесь, "обработка" сеянца или локуса сеянца означает нанесение соединения Формулы I, или композиции, содержащей данное соединение, на сеянец или локус сеянца, таким образом, что соединение Формулы I приводят в контакт с сеянцем; родственные понятия, такие как "пропитка", определены аналогично. Таким образом, когда сеянец приведен в контакт с биологически эффективным количеством соединения Формулы I, данное соединение защищает его от повреждения растительноядными беспозвоночными вредителями. Соединение Формулы I защищает не только наружную поверхность сеянца, но оно будет абсорбироваться сеянцем, образуя сеянец, содержащий соединение Формулы I. Если сеянец приводили в контакт с достаточным количеством соединения Формулы I, адсорбируется достаточное количество для образования биологически эффективной концентрации соединения Формулы I внутри сеянца, и, следовательно, сеянец заключает в себе биологически эффективное количество соединения Формулы I. Если достаточное количество соединения Формулы I применяют для повышения концентрации соединения Формулы I в сеянце до концентрации выше, чем минимальная для биологической эффективности, в таком случае передвижение веществ по растениям может перемещать биологически эффективную концентрацию соединения Формулы I к развивающимся росткам и корням, также для их защиты.
Как указано в этом описании, термин "беспозвоночные вредители" включают в себя членистоногих, брюхоногих моллюсков и круглых червей, как вредителей, имеющих экономическое значение. Термин "растительноядные беспозвоночные вредители" относится к беспозвоночным вредителям, вызывающих повреждения растений путем их поедания, как например поедание листвы, стебля, листа, плода или ткани семени, или путем высасывания васкулярных соков растения. Термин "членистоногие" включает в себя насекомых, клещей, многоножек, двупарноногих, мокриц и симфланов. Термин "брюхоногие моллюски" включает в себя улиток, слизней и других Стебельчатоглазых. Термин "круглые черви" включает в себя растительноядных нематод (Тип или Класс Нематода). Экономически значимые растительноядные беспозвоночные вредители включают в себя: личинки отряда Чешуекрылых, как например, "походные черви", совки, капустные совки, и гелиотины в семействе Совки (например, листопадный "походный червь" (Spodoptera fugiperda J. E. Smith), свекловичный "походный червь" (Spodoptera exigua Hübner), черный озимый червь (Agrotis ipsilon Hufnagel), совка ни (Trichoplusia ni Hübner), гусеница листовертки-почкоеда табачного (Heliothis virescens Fabricius)); сверлильщики, чехликовые моли, гусеницы, строящие паутинное гнездо, мермитиды и вредители, склелетирующие листья, из семейства Огневки (например, мотылек кукурузный (Ostrinia nubilalis Hübner), гусеница, пожирающая апельсин (Amyelois transitella Walker), совка огневая (Crambus caliginosellus Clemens), луговые мотыльки (Herpetogramma licarsisalis Walker)); листовертки, листвертка-почкоед, плодожорка, и гусеница - вредитель плодов в семействе Листовертки (например, плодожорка яблонная (Cydia pomonella L. (L. означает Linnaeus)), листовертка виноградная (Endopiza viteana Clemens), листовертка восточная персиковая (Grapholita molesta Busck)); и многие другие экономически значимые чешуекрылые (например, моль капустная (Plutella xylostella L.), розовый коробочный червь хлопчатника (Pectinophora gossypiella Saunders), непарный шелкопряд (Lymantria dispar L.)); поедающие листву личинки и взрослые особи отряда Жесткокрылых, в том числе долгоносики из семейств Ложнослоники, Зерновки, и Долгоносики (например, долгоносик хлопковый (Anthonomus grandis Boheman), долгоносик рисовый водяной (Lissorhoptrus oryzophilus Kuschel), долгоносик рисовый (Sitophilus oryzae L.)); мелкие листоеды, огуречный жук-блошка, корневые черви, листоеды, колорадские жуки и блошки-минеры семействе Листоеды (например, Колорадский картофельный жук (Leptinotarsa decemlineata Say), блошка длинноусая западная (Diabrotica virgifera virgifera LeConte)); хрущи и другие жуки из семейства Пластинчатоусые (например, хрущик японский (Popillia japonica Newman) и хрущ европейский (Rhizotrogus majalis Razoumowsky)); проволочники из семейства Щелкуны и короеды из семейства Короеды; взрослые особи и личинки отряда Уховертки, в том числе уховертки из семейства Уховертки (например, уховертка обыкновенная (Forficula auricularia L.), черная уховертка (Chelisoches morio Fabricius)); взрослые особи и нимфы отрядов Полужесткокрылые и Равнокрылые, как например, клопы-слепняки из семейства Слепняки, цикады из семейства Певчие цикады, цикадки (например, Empoasca spp.) из семейства Цикадки, дельфациды из семейств Фульгориды и Дельфацины, горбатки из семейства Горбатки, листоблошки из семейства Псиллиды, белокрылки из семейства Белокрылки, тли из семейства Афидиды, Филлоксера из семейства Филлоксеры, мучнистые червецы из семейства Войлочники, щитовки из семейств Подушечницы, Щитовки и Гигантские червецы, клопы-кружевницы из семейства Кружевницы, клопы-щитники из семейства Щитники, клопы-белокрылки (например, Blissus spp.) и другие наземники из семейства Наземники, пенницы из семейства Церкопиды, клопы тыквенные из семейства Краевики, и красноклопы и красноклопы хлопковые из семейства Красноклопы; взрослые особи и личинки отряда Acari (клещи), как например, клещик паутинный и красные клещи в семействе Паукообразные клещи (например, паутинный клещ (Panonychus ulmi Koch), клещик паутинный двупятнистый (Tetranychus urticae Koch), клещ McDaniel (Tetranychus mcdanieli McGregor)), клещи в семействе Плоскотелки (например, цитрусовый клещик (Brevipalpus lewisi McGregor)), галловые клещи и почковые клещи в семействе Галлообразующие и другие клещи, поедающие листву; взрослые и неполовозрелые особи отряда Прямокрылые, в том числе кузнечиковые, настоящая саранча и сверчки (например, кобылки (например, Melanoplus sanguinipes Fabricius, M. differentialis Thomas), саранча американская (например, Schistocerca americana Drury), пустынная саранча (Schistocerca gregaria Forskal), мигрирующая саранча (Locusta migratoria L.), медведки (Gryllotalpa spp.)); взрослые и неполовозрелые особи отряда Двукрылые, в том числе минирующие мушки, галлицы, дрозофиллы (Tephritidae), злаковые мушки (например, Oscinella frit L.), почвенные личинки и другие Длинноусые; взрослые и неполовозрелые особи отряда Бахромчатокрылые, в том числе трипс табачный (Thrips tabaci Lindeman) и другие трипы, поедающие листву; и многоножки в отряде Скутигери; и представители Типа или Класса Нематода, в том числе такие важные сельскохозяйственные вредители как нематоды корневых наростов рода Meloidogyne, нематоды вредители рода Pratylenchus, stubby root nematodes рода Trichodorus, и т.д.
Специалистам в данной области будет понятно, что не все соединения в равной степени эффективны против всех вредителей. Соединения по данному изобретению проявляют особую активность в отношении вредителей в отряде Чешуекрылые (например, Alabama argillacea Hübner (червь хлопкового листа), Archips argyrospila Walker (листовертка), A. rosana L. (Листвертка розанная (золотистая) и другие виды Archips (русский эквивалент отсутствует), Огневка азиатская стеблевая (сверлильщик рисового стебля), Cnaphalocrosis medinalis Guenee (rice leaf roller), Crambus caliginosellus Clemens (огневка гусеница кукурузных коней), Crambus teterrellus Zincken (огневка гусеница-пырея), Cydia pomonella L. (плодожорка яблонная), Earias insulana Boisduval (spiny bollworm), Earias vittella Fabricius (совка (spotted bollworm)), Helicoverpa armigera Hübner (американский коробочный червь), Helicoverpa zea Boddie (совка хлопковая), Heliothis virescens Fabricius (совка), Herpetogramma licarsisalis Walker (луговой мотылек), Lobesia botrana Denis & Schiffermüller (листовертка виноградная), Pectinophora gossypiella Saunders (розовый коробочный червь хлопчатника), Phyllocnistis citrella Stainton (citrus leafminer (русский эквивалент отсутствует)), Pieris brassicae L. (большая белая бабочка), Pieris rapae L. (малая белая бабочка), Plutella xylostella L. (моль капустная), Spodoptera exigua Hübner (свекловичный "походный червь"), Spodoptera litura Fabricius (табачный озимый червь, cluster caterpillar), Spodoptera frugiperda J. E. Smith (листопадный "походный червь"), Trichoplusia ni Hübner (совка ни) и Tuta absoluta Meyrick (tomato leafminer (русский эквивалент отсутствует). Соединения данного изобретения также имеют коммерчески значимое действие в отношении представителей отряда Равнокрылые, в том числе: Acyrthisiphon pisum Harris (тля гороховая), Aphis craccivora Koch (тля черная люцерновая). Aphis fabae Scopoli (тля черная бобовая), Aphis gossypii Glover (тля хлопковая), Aphis pomi De Geer (тля яблонная), Aphis spiraecola Patch (тля таволговая), Aulacorthum solani Kaltenbach (тля вьюнковая), Chaetosiphon fragaefolii Cockerell (тля земляничная), Diuraphis noxia Kurdjumov/Mordvilko (тля русская пшеничная), Dysaphis plantaginea Paaserini (тля розовая), Eriosoma lanigerum Hausmann (тля коровяная яблонная), Hyalopterus pruni Geoffroy (тля мучнистая сливовая), Lipaphis erysimi Kaltenbach (тля ложнокапустная), Metopolophium dirrhodum Walker (тля злаковая), Macrosipum euphorbiae Thomas (тля картофельная листовая), Myzus persicae Sulzer (тля персиковая), Nasonovia ribisnigri Mosley (тля салатная), Pemphigus spp. (корневая тля и галлообразующая тля), Rhopalosiphum maidis Fitch (тля кукурузная листовая), Rhopalosiphum padi L. (тля черемуховая обыкновенная), Schizaphis graminum Rondani (тля злаковая обыкновенная), Sitobion avenae Fabricius (тля злаковая английская), Therioaphis maculata Buckton (spotted alfalfa aphid (русский эквивалент отсутствует)), Toxoptera aurantii Boyer de Fonscolombe (тля чайная), и Toxoptera citricida Kirkaldy (тля цитрусовая); Adelges spp. (херлис); Phylloxera devastatrix Pergande (филлоксера гикори); Bemisia tabaci Gennadius (белокрылка табачная, белокрылка бататная), Bemisia argentifolii Bellows & Perring (белокрылка магнолии крупнолистной), Dialeurodes citri Ashmead (белокрылка цитрусовая) и Trialeurodes vaporariorum Westwood (белокрылка тепличная); Empoasca fabae Harris (цикадка картофельная), Laodelphax striatellus Fallen (цикада коричневая мелкая), Macrolestes quadrilineatus Forbes (цикадка астровая), Nephotettix cinticeps Uhler (green leafhopper (русский эквивалент отсутствует)), Nephotettix nigropictus Stal (цикадка рисовая), Nilaparvata lugens Stal (brown planthopper (русский эквивалент отсутствует)), Peregrinus maidis Ashmead (цикада кукурузная), Sogatella furcifera Horvath (white-backed planthopper (русский эквивалент отсутствует)), Sogatodes orizicola Muir (rice delphacid (русский эквивалент отсутствует), Typhlocyba pomaria McAtee (листовертка белая яблонная), Erythroneoura spp. (листовертка виноградная); Magicidada septendecim L. (periodical cicada (русский эквивалент отсутствует)); Icerya purchasi Maskell (червец австралийский желобчатый), Quadraspidiotus perniciosus Comstock (щитовка калифорнийская); Planococcus citri Risso (мучнистый червец цитрусовый); Pseudococcus spp. (мучнистые червецы других систем); Cacopsylla pyricola Foerster (медянка грушевая), Trioza diospyri Ashmead (листоблошка хурмовая). Эти соединения также обладают активностью в отношении представителей из отряда Полужесткокрылые, в том числе: Acrosternum hilare Say (green stink bug (русский эквивалент отсутствует)), Anasa tristis De Geer (клоп тыквенный), Blissus leucopterus leucopterus Say (клоп белокрылый), Corythuca gossypii Fabricius (клоп хлопковый), Cyrtopeltis modesta Distant (клоп томатовый), Dysdercus suturellus Herrich-Schäffer (красноклоп хлопковый), Euchistus servus Say (brown stink bug (русский эквивалент отсутствует)), Euchistus variolarius Palisot de Beauvois (one-spotted stink bug (русский эквивалент отсутствует)), Graptosthetus spp. (комплекс наземников), Leptoglossus corculus Say (leaf-footed pine seed bug), Lygus lineolaris Palisot de Beauvois (клопик луговой), Nezara viridula L. (клоп хлопково-огородный), Oebalus pugnax Fabricius (rice stink bug (русский эквивалент отсутствует)), Oncopeltus fasciatus Dallas (large milkweed bug (русский эквивалент отсутствует)), Pseudatomoscelis seriatus Reuter (cotton fleahopper). Другие отряды насекомых, контролируемые соединениями по данному изобретению, включают в себя Бахромчатокрылых (например, Frankliniella occidentalis Pergande (трипс пшеничный западный), Scirthothrips citri Moulton (трипс цитрусовый), Sericothrips variabilis Beach (трипс соевый), и Thrips tabaci Lindeman (трипс табачный); и отряд Жесткокрылые (например, Leptinotarsa decemlineata Say (жук колорадский картофельный), Epilachna varivestis Mulsant (жук бобовый мексиканский) и проволочники родов Agriotes (Щелкун посевной), Athous или Limonius).
Способ по настоящему изобретению применим фактически для всех видов растений. Семена, которые можно обрабатывать, включают в себя, например, семена пшеницы (Triticum aestivum L.), пшеницы твердой (Triticum durum Desf.), ячменя (Hordeum vulgare L.) овса (Avena sativa L.), ржи посевной (Secale cereale L.), маиса (Zea mays L.), сорго (Sorghum vulgare Pers.), риса (Oryza sativa L.), дикого риса (Zizania aquatica L.), хлопка (Gossypium barbadense L. и G. hirsutum L.), льна (Linum usitatissimum L.), подсолнечника (Helianthus annuus L.), сои (Glycine max Merr.), фасоли обыкновенной (Phaseolus vulgaris L.), фасоли лима (Phaseolus limensis Macf.), кормовых бобов (Vicia faba L.), садового гороха (Pisum sativum L.), арахиса (Arachis hypogaea L.), люцерны (Medicago sativa L.), свеклы (Beta vulgaris L.), садового салата (Lactuca sativa L.), рапса (Brassica rapa L. и В. napus L.), культур капусты, таких как кочанная капуста, цветная капуста и брокколи (Brassica oleracea L.), репы (Brassica rapa L.), горчицы сарептской (Brassica juncea Coss.), горчицы черной (Brassica nigra Koch), томатов (Lycopersicon esculentum Mill.), картофеля (Solanum tuberosum L.), перца (Capsicum frutescens L.), баклажанов (Solanum melongena L.), табака (Nicotiana tabacum), огурцов (Cucumis sativus L.), дыни мускусная (Cucumis melo L.), арбуза (Citrullus vulgaris Schrad.), тыквы (Curcurbita pepo L., C. moschata Duchesne. и C. maxima Duchesne.), моркови (Daucus carota L.), цинии (Zinnia elegans Jacq.), космеи (например, Cosmos bipinnatus Cav.), хризантемы (Chrysanthemum spp.), мелколепестника (Scabiosa atropurpurea L.), львиного зева (Antirrhinum majus L.), герберы (Gerbera jamesonii Bolus), качима метельчатого (Gypsophila paniculata L., G. repens L. и G. elegans Bieb.), гвоздичника (например, Limonium sinuatum Mill., L. sinense Kuntze.), лиатриса (например, Liatris spicata Willd., L. pycnostachya Michx., L. scariosa Willd.), лизиантуса (например, Eustoma grandiflorum (Raf.) Shinn), тысячелистника (например, Achillea filipendulina Lam., A. millefolium L.), бархатцев (например, Tagetes patula L., T. erecta L.), фиалки трехцветной (например, Viola cornuta L., V. tricolor L.), бальзамина (например, Impatiens balsamina L.) петунии (Petunia spp.), герани (Geranium spp.) и колеуса (например, Solenostemon scutellarioides (L.) Codd). Не только семена, но также корневища, клубни, луковицы или клубнелуковицы, в том числе их жизнеспособные срезы, могут быть обработны в соответствии с изобретением, например, от картофеля (Solanum tuberosum L.), батата (Ipomoea batatas L.), ямса (Dioscorea cayenensis Lam. и D. rotundata Poir.), садового лука (например, Allium cepa L.), тюльпана (Tulipa spp.), гладиолуса (Gladiolus spp.), лилии (Lilium spp.), нарцисса (Narcissus spp.), георгина (например, Dahlia pinnata Cav.), ириса (Iris germanica L. и другие виды), крокуса (Crocus spp.), ветреницы (Anemone spp.), гиацинта (Hyacinth spp.), гадючего лука кистистого (Muscari spp.), фрезии (например, Freesia refracta Klatt, F. armstrongii W. Wats), декоративного лука (Allium spp.), кислицы обыкновенной (Oxalis spp.), пролески двулистной (Scilla peruviana L. и другие виды), цикламена (Cyclamen persicum Mill. и другие виды), хионодокса (Chionodoxa luciliae Boiss. и другие виды), морского лука (Puschkinia scilloides Adams), цантедексии (Zantedeschia aethiopica Spreng., Z. elliottiana Engler и другие виды), глоксинии (Sinnigia speciosa Benth. & Hook.) и клубневой бегонии (Begonia tuberhybrida Voss.). В соответствии с данным изобретением могут быть обработаны части стебля, в том числе таких растений, как сахарный тростник (Saccharum officinarum L.), гвоздика (Dianthus caryophyllus L.), florists chrysanthemum (Chrysanthemum mortifolium Ramat.), бегония (Begonia spp.), герань (Geranium spp.), колеуса (например, Solenostemon scutellarioides (L.) Codd) и пуансеттия (Euphorbia pulcherrima Willd.). В соответствии с данным изобретением могут быть обработаны срезы листьев, в том числе листьев бегонии (Begonia spp.), африканской фиалки (например, Saintpaulia ionantha Wendl.) и очитка (Sedum spp.). Вышеперечисленные злаковые, овощные, декоративные (включая цветы) и фруктовые культуры являются иллюстративными, и не должны рассматриваться как ограничивающие ни в каких случаях. Предпочтительными вариантами осуществления данного изобретения в отношении спектра контроля беспозвоночных вредителей и экономического значения являются обработка семян хлопка, маиса, соевых бобов и риса, и обработка клубней и луковиц картофеля, батата, садового лука, тюльпанов, желтого нарцисса, крокуса и гиацинта.
Локусы сеянцев могут быть обработаны соединением Формулы I с помощью многих различных способов. Все, что необходимо, это нанести на сеянец, или достаточно близко к сеянцу, биологически эффективное количество соединения Формулы I, так, чтобы это соединение могло абсорбироваться этим сеянцем. Соединение Формулы I можно нанести такими методами, как орошение ростовой среды, содержащей сеянец, раствором или дисперсией соединения Формулы I, смешиванием соединения Формулы I с ростовой средой и выращивание сеянца в обработанной ростовой среде (например, обработка рассадных горшков), или разнообразные виды обработки сеянцев, посредством которых соединение Формулы I наносят на сеянец перед тем, как его выращивают в ростовой среде.
В этих способах соединение Формулы I главным образом используют в виде состава или композиции с носителем, приемлемым для сельского хозяйства, включающим по меньшей мере один из жидких разбавителей, сухих разбавителей или поверхностно-активных веществ. Для этого изобретения подходит большое количество составов, наиболее подходящие типы составов зависят от способа применения. Как хорошо известно специалистам в данной области, назначением состава является обеспечение безопасности и удобные способы транспортировки, измерения и распределения химиката, защищающего сельскохозяйственные культуры, а также оптимизация его биологической эффективности.
В зависимости от способа применения используемые составы включают в себя жидкости, такие как растворы (в том числе эмульгирующиеся концентраты), суспензии, эмульсии (в том числе микроэмульсии и/или суспоэмульсии) и подобные, которые необязательно могут быть сгущены в гели. Используемые составы дополнительно включают в себя твердые вещества, такие как дусты, порошки, гранулы, пеллеты, таблетки, пленки и подобное, которые могут быть вододиспергируемыми ("смачиваемые") или водорастворимыми. Активный компонент может быть (микро)инкапсулирован и в дальнейшем сформирован в суспензию или твердый состав; альтернативно может быть инкапсулирован весь состав активного компонента (или "покрыт"). Инкапсулирование может контролировать или задерживать высвобождение активного компонента. Распыляемые составы могут быть распространены в подходящей среде и использованы в объемах распыления примерно от одного до нескольких сотен литров на гектар. Высококонцентрирующие композиции в первую очередь используют как промежуточные составы для приготавливаемых составов.
Составы обычно содержат эффективные количества активного компонента, разбавитель и поверхностно-активное вещество в пределах следующих приблизительных интервалов, которые дополняют до 100 процентов по массе.
Типичные твердые разбавители описаны у Watkins et al., Handbook of Insecticide Dust Diluents and Carriers, 2nd Ed., Dorland Books, Caldwell, New Jersey. Типичные жидкие разбавители описаны у Marsden, Solvents Guide, 2nd Ed., Interscience, New York, 1950. McCutcheon's Emulsifiers and Detergents and McCutcheon's Functional Materials (North America and International Editions, 2001), The Manufactuing Confection Publ.Co., Glen Rock, New Jersey, а также Sisely and Wood, Encyclopedia of Surface Active Agents, Chemical Publ. Co., Inc., New York, 1964, перечисляют поверхностно-активные вещества и рекомендуемое их использование. Все составы могут содержать незначительные количества добавок для уменьшения пенообразования, комкования, окисления, роста микроорганизмов и подобного, или загустителей для повышения вязкости.
Поверхностно-активные вещества включают в себя, например, этоксилированные спирты, этоксилированные алкилфенолы, этоксилированные эфиры сорбита и жирных кислот, этоксилированные амины, этоксилированные жирные кислоты, сложные эфиры и масла, диалкилсульфосукцинаты, алкилсульфаты, алкиларилсульфонаты, кремнийорганические вещества, N,N-диалкилаураты, гликолевые сложные эфиры, фосфатные сложные эфиры, лигнинсульфонаты, нафталинсульфонаты, конденсаты формальдегида, поликарбоксилаты и блоксополимеры, в том числе полиоксиэтилен/полиоксипропиленовые блоксополимеры. Твердые разбавители включают в себя, например, глины, такие как бентонит, монтмориллонит, аттапульгит и каолин, крахмал, сахар, кремнезем, тальк, инфузорную землю, мочевину, карбонат кальция, карбонат натрия и бикарбонат, и сульфат натрия. Жидкие разбавители включают в себя, например, воду, N,N-диметилформамид, диметилсульфоксид, N-алкилпирролидон, этиленгликоль, полипропиленгликоль, пропиленкарбонат, двухосновные сложные эфиры, парафины, алкилбензолы, алкилнафталины, оливковое, касторовое масло, масло льняного семени, тунговое масло, кунжутное масло, кукурузное, арахисовое, масло хлопкового семени, соевое, масло рапсового семени, и кокосовое масло, эфиры жирных кислот, кетоны, такие как циклогексанон, 2-гептанон, изофорон и 4-гидрокси-4-метил-2-пентанон, и спирты, такие как метанол, циклогексанол, деканол, бензиловый и тетрагидрофурфуриловый спирт.
Растворы, в том числе эмульгирующиеся концентраты, могут быть приготовлены простым смешиванием компонентов. Дусты и порошки могут быть приготовлены путем смешивания, и, обычно, измельчения, как в молотковой мельнице, так и в гидравлической мельнице. Суспензии обычно готовят путем влажного помола; см., например, U.S. 3060084. Гранулы и пеллеты могут быть приготовлены путем напыления активного вещества на подготовленные гранулярные носители или с помощью методики аггломерации. См. Browning, "Agglomeration", Chemical Engineering, December 4, 1967, pp 147-48, Perry's Chemical Engineer's Handbook, 4th Ed., McGraw-Hill, New York, 1963, страницы 8-57 и следующие, и публикацию международной заявки WO 91/13546. Пеллеты могут быть приготовлены как описано в US 4172714. Вододиспергируемые и водорастворимые гранулы могут быть приготовлены как представлено в US 4144050, US 3920442 и DE 3246493. Таблетки могут быть приготовлены, как представлено в US 5180587, US 5232701 и US 5208030. Пленки могут быть приготовлены как указано в GB 2095558 и US 3299566.
Для дополнительной информации, касающейся области составов, смотрите T. S. Woods, "The Formulator's Toolbox - Product Forms for Modern Agriculture" в Pesticide Chemistry and Bioscience, The Food-Environment Challenge, T. Brooks and T. R. Roberts, Eds., Proceedings of the 9th International Congress on Pesticide Chemistry, The Royal Society of Chemistry, Cambridge, 1999, pp. 120-133. См. также US 3235361, Кол. 6, строка 16 по Кол. 7, строка 19 и Примеры 10-41; US 3309192, Кол. 5, строка 43 по Кол. 7, строка 62 и Примеры 8, 12, 15, 39, 41, 52, 53, 58, 132, 138-140, 162-164, 166, 167 и 169-182; US 2891855, Кол. 3, строка 66 по Кол. 5, строка 17 и Примеры 1-4; Klingman, Weed Control as a Science, John Wiley and Sons, Inc., New York, 1961, pp 81-96; и Hance et al, Weed Control Handbook, 8th Ed., Blackwell Scientific Publications, Oxford, 1989.
Сеянец (посадочный материал) или растение, выросшее из него, может быть защищен от беспозвоночных вредителей в соответствии с настоящим изобретением, способом, включающим контактирование сеянца или локуса сеянца с композицией, содержащей биологически эффективное количество соединения Формулы I, его N-оксида или его соли, приемлемой для сельского хозяйства. Данное изобретение включает в себя и сеянец, контактировавший с композицией, содержащей биологически эффективное количество соединения Формулы I, его N-оксида или его соли, приемлемой для сельского хозяйства, и эффективное количество по меньшей мере одного другого биологически активного соединения или агента. Композиции, используемые для обработки сеянцев (или растения, выросшего из него) согласно данному изобретению, также могут содержать (кроме компонента Формулы I) эффективное количество одного или более других биологически активных соединений или агентов. Подходящие дополнительные соединения или агенты включают в себя инсектициды, фунгициды, нематоциды, бактерициды, акарициды, регуляторы роста, такие как стимуляторы роста корней, хемостерилизаторы, полихимикаты, репелленты, аттрактанты, феромоны, кормовые стимуляторы, другие биологически активные соединения или энтомопатогенные бактерии, вирусы или грибы для образования многокомпонентных пестицидов, дающих еще более широкий спектр сельскохозяйственного использования. Примерами таких биологически эффективных соединений или агентов, с которыми могут быть изготовлены составы соединения по данному изобретению, являются: инсектициды, такие как абамектин, ацефат, ацетамиприд, амидофлюмет (S-1955), авермектин, азадиракхтин, азинфос-метил, бифентрин, бинфеназат, бупрофезин, карбофуран, хлорфенапир, хлорфлуазурон, хлорпирифос, хлорпирифос-метил, хромафенозид, клотианидин, цифлютрин, бета-цифлютрин, цигалотрин, лямбда-цигалотрин, циперметрин, циромазин, дельтаметрин, диафентиурон, диазинон, дифлюбензурон, диметоан, диофенолан, эмамектин, эндосульфан, эсфенвалерат, этиопрол, фенотикарб, феноксикарб, фенпропатрин, фенпроксимат, фенвалерат, фипронил, флоникамид, флуцитринат, тауфлувалинат, флуфенерим (UR-50701), флуфеноксурон, фонофос, галофенозид, гексафлумурон, имидаклоприд, индоксакарб, изофенфос, луфенурон, малатион, метальдегид, метамидофос, метидатион, метомил, метопрен, метоксихлор, монокротофос, метоксифенозид, нитиазин, новалурон, новифлумурон (XDE-007), оксамил, паратион, паратионметил, перметрин, форат, фосалон, фосмет, фосфамидон, пиримикарб, профенофос, пиметрозин, пиридалил, пирипроксифен, ротенон, спиносад, спиромезифин (BSN 2060), сульпрофос, тебуфенозид, тефлюбензурон, тефлютрин, тербуфос, тетрахлорвинфос, тиаклоприд, тиаметоксам, тиодикарб, тиосультап-натрий, тралометрин, трихлорфон и трифлюмурон; фунгициды, такие как ацибензолар, азоксистробин, беномил, бластицидин-S, Бордосская смесь (сульфат трехосновной меди), бромуконазол, карпропамид, каптафол, каптан, карбендазим, хлоронеб, хлороталонил, оксихлорид меди, соли меди, цифлюфенамид, цимоксанил, ципроконазол, ципродинил, (S)-3,5-дихлор-N-(3-хлор-1-этил-1-метил-2-оксопропил)-4-метилбензамид (RH 7281), диклоцимет (S-2900), дикломезин, диклоран, дифеноконазол, (S)-3,5-дигидро-5-метил-2-(метилтио)-5-фенил-3-(фениламино)-4Н-имидазол-4-он (RP 407213), диметоморф, димоксистробин, диниконазол, диниконазол-M, додин, эдифенфос, эпоксиконазол, фамоксадон, фенамидон, фенаримол, фенбуконазол, фенкарамид (SZX0722), фенпиклонил, фенпропидин, фенпропиморф, фентинацетат, фентингидроксид, флуазинам, флудиоксонил, флуметовер (RPA 403397), флуморф/флуморлин (SYP-L190), флуоксастробин (HEC 5725), флухинконазол, флузилазол, флутоланил, флутриафол, фолпет, фосетилалюминий, фуралаксил, фураметапир (S-82658), гексаконазол, ипконазол, ипробенфос, ипродион, изопротиолан, касугамицин, крезоксим-метил, манкозеб, манеб, мефеноксам, мепронил, металаксил, метконазол, метоминостробин/феноминостробин (SSF-126), метрафенон (AC 375839), миклобутанил, нео-азозин (метанарсонат трехвалентного железа), никобифен (BAS 510), орисастробин, оксадиксил, пенконазол, пенцикурон, пробеназол, прохлораз, пропамокарб, пропиконазол, прохиназид (DPX-KQ926), протиоконазол (JAU 6476), пирифенокс, пираклостробин, пириметанил, прохилон, хиноксифен, спироксамин, сера, тебуконазол, тетраконазол, тиабендазол, тифлузамид, тиофанат-метил, тирам, тиадинил, триадимефон, триадименол, трициклазол, трифлоксистробин, тритиконазол, валидамицин и винклозолин; нематоциды, такие как алдикарб, оксамил и фенамифос; бактерициды, такие как стрептомицин; акарициды, такие как амитраз, хинометионат, хлоробензилат, цигексатин, дикофол, диенохлор, этоксазол, феназаквин, фенбутатиноксид, фенпропатрин, фенпироксимат, гекситиазокс, пропаргит, пиридабен и тебуфенпирад; и биологические агенты, такие как Bacillus thuringiensis, в том числе ssp. aizawai и kurstaki, эндотоксин дельта Bacillus thuringiensis, бакуловирусы и энтомопатогенные бактерии, вирусы и грибы.
Общей ссылкой для этих сельскохозяйственных средств защиты растений является The Pesticide Manual, 12th Edition, C. D. S. Tomlin, Ed., British Crop Protection Council, Farnham, Surrey, U.K., 2000.
Предпочтительные инсектициды и акарициды для смешивания с соединениями Формулы I включают в себя пиретроиды, такие как циперметрин, цигалотрин, цифлутрин и бета-цифлутрин, эсфенвалерат, фенвалерат и тралометрин; карбаматы, такие как фенотикарб, метомил, оксамил и тиодикарб; неоникотиноиды, такие как клотианидин, имидаклоприд и тиаклоприд; блокаторы нейрональных натриевых каналов, такие как индоксакарб, инсектицидные макроциклические лактоны, такие как спиносад, абамектин, абамецин, авермектин и эмамектин; антагонисты γ-аминомасляной кислоты (ГАМК), такие как эндосульфан, этипрол и фипронил; инсектицидные мочевины, такие как флуфеноксурон и трифлумурон; имитаторы юношеских гормонов, такие как диофенолан и пирипроксифен; пиметрозин; и амитраз. Предпочтительные биологические агенты для смешивания с соединениями по данному изобретению включают в себя Bacillus thuringiensis и Bacillus thuringiensis эндотоксин дельта, а также и природные и генетически модифицированные вирусные инсектициды, в том числе представители семейства Baculoviridae, а также энтомопатогенные грибы.
Предпочтительными регуляторами роста растения для смешивания с соединениями Формулы I в композициях для обаботки срезов стеблей являются 1Н-индол-3-уксусная кислота, 1Н-индол-3-бутановая кислота и 1-нафталинуксусная кислота и их соли, приемлемые для сельского хозяйства, сложные эфиры и амидные производные, такие как 1-нафталинацетамид. Предпочтительные фунгициды для смешивания с соединениями Формулы I включают в себя фунгициды, используемые для обработки семян, такие как тирам, манеб, манкозеб и каптан.
В следующих примерах все проценты даны по массе и все составы приготовлены обычными способами. Номера соединений относятся к соединениям из таблицы-указателя А.
ПРИМЕР A
ПРИМЕР B
ПРИМЕР C
ПРИМЕР D
Для орошения ростовой среды данные составы должны доставлять соединения Формулы I, главным образом после разведения водой, в растворе или в виде частиц, достаточно мелких для того, чтобы они остались диспергированными в жидкости. Вододиспергируемые или растворимые порошки, гранулы, таблетки, эмульгирующиеся концентраты, концентраты водных суспензий и подобное являются составами, подходящими для водного орошения ростовой среды. Орошения наиболее подходят для обработки ростовой среды относительно высокой пористости, такой как легкие почвы или искусственная ростовая среда, содержащая пористые материалы, такие как торфяной мох, перлит, вермикулит и подобное. Жидкость для орошения, содержащая соединение Формулы I, также может быть добавлена к жидкой ростовой среде (т.е. гидропонике), что делает соединение Формулы I частью жидкой ростовой среды. Специалисту в данной области будет понятно, что количество соединения Формулы I, необходимое в жидкости для орошения для эффективного контроля за беспозвоночными вредителями (т.е. биологически эффективное количество) будет изменяться в зависимости от типа сеянца соединения Формулы I, продолжительности и степени требуемой защиты беспозвоночного вредителя, за которым осуществляется контроль и факторов окружающей среды. Концентрация соединения Формулы I в жидкости для орошения в основном составляет от 0,01 ч/млн (ч/млн) до 10,000 ч/млн, более обычно примерно от 1 ч/млн до 100 ч/млн. Специалист в данной области легко сможет определить биологически эффективную концентрацию, необходимую для требуемого уровня контроля за беспозвоночными вредителями.
Для обработки ростовой среды соединение Формулы I может быть использовано путем смешивания его в виде сухого порошка или гранулированного состава с ростовой средой. Поскольку этот способ применения не требует предварительного диспергирования или растворения в воде, сухой порошок или гранулированные составы не должны быть растворимыми или диспергируемыми в высокой степени. В то время, как в горшке для рассады может быть обработана вся ростовая среда, на сельскохозяйственных полях по экономическим и экологическим причинам обычно обрабатывают только почву вблизи сеянца. Для сведения к минимуму трудностей и издержек, связанных с его применением, состав соединения Формулы I наиболее эффективно применять одновременно с выращиванием сеянца (например, высеванием). Для применения внутри борозд, состав Формулы I (наиболее удобен гранулированный состав) наносят вслед "ботинок сеяльщика". Для T-полосного применения составы Формулы I наносят в полосу через ряд позади ботинка сеяльщика и позади или обычно перед заделывающим каточком. Специалисту в данной области будет понятно, что количество соединения Формулы I, необходимое в локусе ростовой среды для эффективного контроля за беспозвоночными вредителями (т.е. биологически эффективное количество), будет изменяться в зависимости от типа сеянца, соединения Формулы I, требуемой продолжительности и степени защиты, беспозвоночного вредителя, за которым осуществляют контроль и факторов окружающей среды. Концентрация соединения Формулы I в локусе ростовой среды обычно составляет от 0,0001 ч/млн до 100 ч/млн, более обычно примерно от 0,01 ч/млн до 10 ч/млн. Специалист в данной области легко определит биологически эффективное количество, необходимое для желаемого уровня контроля за растительноядными беспозвоночными вредителями.
Сеянец может быть обработан непосредственно путем замачивания его в растворе или дисперсии соединения Формулы I. Хотя этот способ применения используют для всех типов сеянцев, для обеспечения защитного контроля за беспозвоночными вредителями, для развивающегося растения обработка крупных семян (например, имеющих средний диаметр по меньшей мере 3 мм) является более эффективной, чем обработка мелких семян. Обработка сеянцев, таких как клубни, луковицы, клубнелуковицы, корневища и стеблей и черенков листьев, в дополнение к сеянцу также может обеспечить эффективную обработку развивающегося растения. Составы, используемые для орошения ростовой среды, главным образом также используются для пропитывающих обработок. Пропитывающая среда содержит нефитотоксическую жидкость, главным образом на водной основе, хотя она может содержать нефитотоксические количества других растворителей, таких как метанол, этанол, изопропанол, этиленгликоль, пропиленгликоль, пропиленкарбонат, бензиловый спирт, двухосновные сложные эфиры, ацетон, метилацетат, этилацетат, циклогексанон, диметилсульфоксид и N-метилпирролидон, которые могут быть использованы для усиления растворимости соединения Формулы I и проникновения в сеянец. Поверхностно-активные вещества могут облегчать намокание сеянца и проникновение соединения Формулы I. Специалисту в данной области будет понятно, что количество соединения Формулы I, необходимое для пропитывающей среды для эффективного контроля за беспозвоночными вредителями (т.е. биологически эффективное количество), будет изменяться в зависимости от типа сеянца, соединения Формулы I, требуемой продолжительности и степени защиты, беспозвоночного вредителя, за которым осуществляют контроль, и факторов окружающей среды. Концентрация соединения Формулы I в пропитывающей среде обычно составляет от 0,01 ч/млн до 10000 ч/млн, более обычно примерно от 1 ч/млн до 100 ч/млн. Специалист в данной области легко определит биологически эффективную концентрацию, необходимую для желаемого уровня контроля за растительноядными беспозвоночными вредителями. Время замачивания может изменяться от 1 минут до 1 дня или дольше. Фактически сеянец может оставаться в обрабатывающей жидкости во время прорастания или проращивания (например, проращивание семян риса перед непосредственным высевом). Поскольку ростки и корни выходят из семенной кожуры (покрова семян), росток и корень напрямую контактируют с раствором, содержащим соединение Формулы I. Для обработки прорастающих семян, культуры с крупными семенами, такие как рис, время обработки составляет примерно от 8 до 48 часов, например, типичным является примерно 24 часа. Более короткое время обработки наибольшей степени употребляется для обработки мелких семян.
Сеянцы также могут быть покрыты композицией, содержащей биологически эффективное количество соединения Формулы I. Материалы для покрытия по данному изобретению способны осуществлять медленное высвобождение соединения Формулы I путем диффузии в сеянец и окружающую среду. Материалы для покрытия включают в себя сухие дусты или порошки, адгезирующиеся на сеянце под действием клейких веществ, таких как метилцеллюлоза или аравийская камедь. Материалы для покрытия также могут быть приготовлены из суспензионных концентратов, вододиспергируемых порошков или эмульсий, суспендированных в воде, распыленных на сеянец во вращающемся барабане, а затем высушенных. Соединения Формулы I, растворенные в растворителе, могут быть распылены на обрабатываемый сеянец, с последующим выпариванием растворителя. Такие композиции предпочтительно включают в себя компоненты, способствующие адгезии материалов покрытия на сеянце. Эти композиции также могут содержать поверхностно-активные вещества, способствующие смачиванию сеянца. Используемые растворители не должны быть фитотоксичными для сеянца; главным образом используют воду, но могут быть использованы другие летучие растворители низкой фитотоксичности, такие как метанол, этанол, метилацетат, этилацетат, ацетон и т.д., по отдельности или в комбинации. Летучими являются растворители, у которых точка кипения в нормальных условиях примерно менее 100°C. Высушивание должно проводиться способом, не повреждающим росток или не вызывающим преждевременное прорастание или рост.
Загустители материалов для покрытия могут варьировать от адгезивной дусты до тонких пленок и пеллетных слоев примерно толщиной от 0,5 до 5 мм. Материалы для покрытия сеянцев по данному изобретению могут содержать более чем один адгезивный слой, только один из которых должен содержать соединение Формулы I. Для мелких семян наиболее подходящими являются, главным образом пеллеты, поскольку их способность поставлять биологически эффективное количество соединения Формулы I не ограничена поверхностью семени, и дражирование мелких семян также облегчает перенос семян и процедуру посадки. Из-за большого размера и площади поверхности крупные семена и луковицы, клубни, клубнелуковицы и корневища и их жизнеспособные срезы обычно не пеллетируют, но вместо этого покрывают порошками или тонкими пленками.
Сеянцы, контактирующие с соединениями Формулы I, в соответствии с данным изобретением включают в себя семена. Подходящие семена включают в себя семена пшеницы, пшеницы твердой, ячменя, овса, ржи посевной, маиса, сорго, риса, дикого риса, хлопка, льна, подсолнечника, сои, фасоли обыкновенной, фасоли лима, кормовых бобов, садового гороха, арахиса, люцерны, свеклы, садового салата, рапса, культур капусты, репы, горчицы сарептской, горчицы черной, томатов, картофеля, перца, баклажанов, табака, огурцов, дыни мускусной, арбуза, тыквы, моркови, цинии, космеи, хризантемы, мелколепестника, львиного зева, герберы, качима метельчатого, гвоздичника, лиатриса, лизиантуса, тысячелистника, бархатцев, фиалки трехцветной, бальзамина, петунии, герани и колеуса. Известными являются семена хлопка, маиса, соевых бобов и риса. Сеянцы, контактирующие с соединениями Формулы I, в соответствии с данным изобретением также включают в себя корневища, клубни, луковицы или клубнелуковицы, или их жизнеспособные срезы. Подходящие корневища, клубни, луковицы или клубнелуковицы, или их жизнеспособные срезы включают в себя таковые от картофеля, батата, ямса, садового лука, тюльпана, гладиолуса, лилии, нарцисса, георгина, ириса, крокуса, ветреницы, гиацинта, гадючего лука кистистого, фрезии, декоративного лука, кислицы обыкновенной, пролески двулистной, цикламена, хионодокса, морского лука, цантедексии, глоксинии и клубневой бегонии. Характерными являются корневища, клубни, луковицы и клубнелуковицы или их жизнеспособные части картофеля, батата, садового лука, тюльпана, желтого нарцисса, крокуса и гиацинта. Сеянцы, контактировавшие с соединениями Формулы I, в соответствии с данным изобретением также включают в себя стебли и черенки листьев.
В одном варианте осуществления сеянец, контактировавший с соединением Формулы I, представляет собой сеянец, покрытый композицией, содержащей соединение Формулы I, его N-оксид или их соль, приемлемую для сельского хозяйства и пленкообразователь или адгезивный агент. Композиции по данному изобретению, содержащие биологически эффективное количество соединения Формулы I, его N-оксид или его соль, приемлемую для сельского хозяйства и пленкообразователь или адгезивный агент, дополнительно могут содержать эффективное количество по меньшей мере одного дополнительного биологически активного соединения или агента. Известными являются композиции, содержащие (в дополнение к компоненту Формулы I и пленкообразователю и адгезивному агенту) антроподицидные вещества группы, состоящей из пиретроидов, карбаматов, неоникотиноидов, блокаторов нейрональных натриевых каналов, инсектицидных макроциклических лактонов, антагонистов γ-аминомасляной кислоты (ГАМК), инсектицидной мочевины и имитаторов юношеских гормонов. Также характерными являются композиции, содержащие (в дополнение к компоненту Формулы I и пленкообразователю или адгезивному агенту) по меньшей мере одно дополнительное биологически активное соединение или агент, выбранные из группы, содержащей абамектин, ацефат, ацетамиприд, амидофлюмет (S-1955), авермектин, азадиракхтин, азинфос-метил, бифентрин, бинфеназат, бупрофезин, карбофуран, хлорфенапир, хлорфлуазурон, хлорпирифос, хлорпирифос-метил, хромафенозид, клотианидин, цифлютрин, бета-цифлютрин, цигалотрин, лямбда-цигалотрин, циперметрин, циромазин, дельтаметрин, диафентиурон, диазинон, дифлюбензурон, диметоан, диофенолан, эмамектин, эндосульфан, эсфенвалерат, этиопрол, фенотикарб, феноксикарб, фенпропатрин, фенпроксимат, фенвалерат, фипронил, флоникамид, флуцитринат, тауфлувалинат, флуфенерим (UR-50701), флуфеноксурон, фонофос, галофенозид, гексафлумурон, имидаклоприд, индоксакарб, изофенфос, луфенурон, малатион, метальдегид, метамидофос, метидатион, метомил, метопрен, метоксихлор, монокротофос, метоксифенозид, нитиазин, новалурон, новифлумурон (XDE-007), оксамил, паратион, паратион-метил, перметрин, форат, фосалон, фосмет, фосфамидон, пиримикарб, профенофос, пиметрозин, пиридалил, пирипроксифен, ротенон, спиносад, спиромезифин (BSN 2060), сульпрофос, тебуфенозид, тефлюбензурон, тефлютрин, тербуфос, тетрахлорвинфос, тиаклоприд, тиаметоксам, тиодикарб, тиосультап-натрий, тралометрин, трихлорфон и трифлюмурон, алдикарб, оксамил фенамифос, амитраз, хинометионат, хлоробензилат, цигексатин, дикофол, диенохлор, этоксазол, феназаквин, фенбутатиноксид, фенпропатрин, фенпироксимат, гекситиазокс, пропаргит, пиридабен и тебуфенпирад; и биологические агенты, такие как Bacillus thuringiensis, в том числе ssp. aizawai и kurstaki, эндотоксин дельта Bacillus thuringiensis, бакуловирусы и энтомопатогенные бактерии, вирусы и грибы. Также характерными являются композиции, содержащие (в дополнение к компоненту формулы I и пленкообразователю или адгезивному агенту) по меньшей мере одно дополнительное биологически активное соединение или агент, выбранный из фунгицидов группы, содержащей ацибензолар, азоксистробин, беномил, бластицидин-S, Бордосскую смесь (сульфат трехосновной меди), бромуконазол, карпропамид, каптафол, каптан, карбендазим, хлоронеб, хлороталонил, оксихлорид меди, соли меди, цифлюфенамид, цимоксанил, ципроконазол, ципродинил, (S)-3,5-дихлор-N-(3-хлор-1-этил-1-метил-2-оксопропил)-4-метилбензамид (RH 7281), диклоцимет (S-2900), дикломезин, диклоран, дифеноконазол, (5)-3,5-дигидро-5-метил-2-(метилтио)-5-фенил-3-(фениламино)-4Н-имидазол-4-он (RP 407213), диметоморф, димоксистробин, диниконазол, диниконазол-M, додин, эдифенфос, эпоксиконазол, фамоксадон, фенамидон, фенаримол, фенбуконазол, фенкарамид (SZX0722), фенпиклонил, фенпропидин, фенпропиморф, фентинацетат, фентингидроксид, флуазинам, флудиоксонил, флуметовер (RPA 403397), флуморф/флуморлин (SYP-L190), флуоксастробин (HEC 5725), флухинконазол, флузилазол, флутоланил, флутриафол, фолпет, фосетил-алюминий, фуралаксил, фураметапир (S-82658), гексаконазол, ипконазол, ипробенфос, ипродион, изопротиолан, касугамицин, крезоксим-метил, манкозеб, манеб, мефеноксам, мепронил, металаксил, метконазол, метоминостробин/феноминостробин (SSF-126), метрафенон (AC 375839), миклобутанил, нео-азозин (метанарсонат трехвалентного железа), никобифен (BAS 510), орисастробин, оксадиксил, пенконазол, пенцикурон, пробеназол, прохлораз, пропамокарб, пропиконазол, прохиназид (DPX-KQ926), протиоконазол (JAU 6476), пирифенокс, пираклостробин, пириметанил, прохилон, хиноксифен, спироксамин, сера, тебуконазол, тетраконазол, тиабендазол, тифлузамид, тиофанат-метил, тирам, тиадинил, триадимефон, триадименол, трициклазол, трифлоксистробин, тритиконазол, валидамицин и винклозолин (особенно композиции, где по меньшей мере одно биологически активное соединение или агент выбрано из фунгицидов группы, состоящей из тирама, манеба, макозеба и каптана).
В основном материал по настоящему изобретению, покрывающий сеянец, содержит соединение Формулы I, пленкообразователь или адгезивное вещество. Материал для покрытия может дополнительно содержать вспомогательные вещества, такие как диспергирующие вещества, поверхностно-активные вещества, носители и необязательно противопенные вещества и красители. Специалисту в данной области будет понятно, что количество соединения Формулы I, необходимое для материала для покрытия для эффективного контроля за беспозвоночными вредителями (т.е. биологически эффективное количество), будет изменяться в зависимости от типа сеянца, соединения Формулы I, требуемой продолжительности и степени защиты, беспозвоночного вредителя, за которым осуществляют контроль, и факторов окружающей среды. Материал для покрытия не должен задерживать прорастание или проращивание сеянца и должен быть постоянно эффективным для снижения повреждения растения во время повреждающей растения фазы жизненного цикла беспозвоночного вредителя, против которого проводят обработку. Материал для покрытия, содержащий достаточное количество соединения Формулы I, может обеспечить контролирующую защиту от беспозвоночного вредителя примерно до 120 дней или даже дольше. В основном количество соединения Формулы I находится в интервале примерно от 0,001 до 50% массы сеянца, для семян чаще в интервале примерно от 0,01 до 50% массы семени, и наиболее обычно для крупных семян в интервале примерно от 0,1 до 10% массы семени. Однако могут быть использованы большие количества, вплоть до 100% или более, в частности для дражирования мелкого семени для продления защитного контроля беспозвоночного вредителя. Для сеянцев, таких как луковицы, клубни, клубнелуковицы и корневища и их жизнеспособных срезов, и стеблей и черенков листьев, в основном, количество соединения Формулы I находится в интервале примерно от 0,001 до 5% массы сеянца, с использованием более высоких процентов для более мелких сеянцев. Специалист в данной области может легко определить биологически эффективное количество, необходимое для желаемого уровня контроля за растительноядными беспозвоночными вредителями.
Пленкообразователь или адгезивный агент, входящий в состав материала для покрытия сеянца, предпочтительно состоит из адгезивного полимера, который может быть природным или синтетическим и не обладает фитотоксическим действием на покрываемый сеянец. Пленкообразователь или адгезивный агент может быть выбран из поливинилацетатов, сополимеров поливинилацетата, гидролизованных поливинилацетатов, сополимеров поливинилпирролидонвинилацетата, поливинильных спиртов, сополимеров поливинильных спиртов, поливинилметилового эфира, сополимера поливинилметиловый эфир-малеиновый ангидрид, восков, латексных полимеров, целлюлоз, в том числе этилцеллюлозы и метилцеллюлозы, гидроксиметилцеллюлозы, гидроксипропилцеллюлозы, гидроксиметилпропилцеллюлозы, поливинилпирролидонов, альгинатов, декстринов, мальтодекстринов, полисахаридов, жиров, масел, белков, карайской камеди, гуаровой камеди, трагаканта, полисахаридных камедей, клейких веществ, аравийской камеди, шеллака, полимеров и сополимеров винилиденхлорида, полимеров и сополимеров, основанных на белках соевых бобов, лигносульфонатов, акриловых сополимеров, крахмалов, поливинилакрилатов, зеинов, желатина, карбоксиметилцеллюлозы, хитозана, полиэтиленоксида, полимеров и сополимеров акрилимида, полигидроксиэтилакрилата, мономеров метилакрилимида, альгината, этилцеллюлозы, полихлоропрена и сиропов или их смесей. Предпочтительные пленкообразователи и адгезивные агенты включают в себя полимеры и сополимеры винилацетата, сополимер поливинилпирролидонвинилацетат и водорастворимые воски. Особенно предпочтительными являются сополимеры поливинилпирролидонвинилацетат и водорастворимые воски. Определенные выше полимеры включают в себя полимеры, известные в этой области и для примера некоторые из них определены как Agrimer® VA 6 и Licowax® KST. Количество пленкообразователя или адгезивного агента в данном составе главным образом находится в интервале примерно от 0,001 до 100% массы сеянца. Для крупных семян количество пленкообразователя или адгезивного агента обычно находится в интервале примерно от 0,05 до 5% массы семени; для мелких семян количество обычно находится в интервале примерно от 1 до 100%, но может быть больше 100% массы семени при дражировании. Для других сеянцев количество пленкообразователя или адгезивного агента обычно находится в интервале от 0,001 до 2% массы сеянца.
Материалы, известные как вспомогательные вещества, также могут быть использованы в материалах для покрытия по данному изобретению для обработки сеянцев для контроля за беспозвоночными вредителями и хорошо известны специалистам в данной области. Вспомогательные вещества способствуют получению или способу обработки сеянцев и включают в себя, но не ограничиваются ими, диспергирующие вещества, поверхностно-активные вещества, носители, противопенные вещества и красители. Используемые диспергирующие вещества могут включать анионные поверхностно-активные вещества, водорастворимые в высокой степени, подобные Borresperse™ CA, Morwet® D425 и подобные. Используемые поверхностно-активные вещества могут включать неионные поверхностно-активные вещества с высокой степенью водорастворимости, как Pluronic® F108, Brij® 78 и подобные. Используемые носители могут включать жидкости, подобные воде и маслам, являющиеся водорастворимыми, такие как спирты. Используемые носители также могут включать в себя наполнители, как древесная мука, глина, активированный уголь, инфузорная земля, мелко измельченные неорганические частицы, карбонат кальция и подобное. Глины и неорганические частицы, которые могут быть использованы, включают в себя бентонит кальция, каолин, китайскую глину, тальк, перлит, слюду, вермикулит, кремнеземы, порошок кварца, мортмориллонит и их смеси. Противопенные вещества могут включать в себя вододиспергируемые жидкости, содержащие полиорганические силоксаны, как Rhodorsil® 416. Красители могут включать вододиспергируемые жидкие красящие композиции, как Pro-lzed® Colorant Red. Специалисту в данной области будет понятно, что это не исчерпывающее перечисление вспомогательных веществ для составов, и что могут быть использованы другие общеизвестные материалы, в зависимости от покрываемого сеянца, соединения Формулы I, используемого в покровном материале. Подходящие примеры вспомогательных веществ для составов включают в себя перечисленные здесь и перечисленные в McCutcheon's 2001, Volume 2: Functional Materials, published by MC Publishing Company. Количество вспомогательных веществ, используемых для состава, может быть различным, но в основном масса компонентов будет находиться в интервале примерно от 0,001 до 10000% массы сеянца, проценты выше 100% главным образом используют для дражирования мелких семян. Для непеллетируемых семян количество вспомогательных веществ в составе в основном составляет примерно от 0,01 до 45% массы семени и обычно примерно от 0,1 до 15% массы семени. Для сеянцев, отличных от семян, количество вспомогательных веществ в композиции в основном примерно составляет от 0,001 до 10% массы сеянца.
Для выполнения покрытия семян по настоящему изобретению могут быть использованы обычные методы нанесения покровных материалов. Дусты или порошки могут быть нанесены путем галтовки сеянца составом, содержащим соединение Формулы I и адгезивного агента для адгезии дусты или порошка к сеянцу и не ссыпания во время упаковки или транспортировки. Дусты или порошки также могут быть нанесены путем добавления дуста или порошка прямо к барабану для дражирования сеянцев с последующим распылением жидкого носителя на семя и высушиванием. Дусты и порошки, содержащие соединение Формулы I, также могут быть нанесены путем обработки (например, погружения) по меньшей мере части сеянца растворителем, таким как вода, необязательно содержащим адгезивный агент, и погружения обработанной части в дуст или порошок. Этот способ может быть в частности использован для покрытия срезов стеблей. Сеянцы также могут быть погружены в композиции, содержащие составы Формулы I из смоченных порошков, растворов, суспоэмульсий, эмульгирующихся концентратов и эмульсий в воде, а затем высушены или непосредственно посажены в ростовую среду. Сеянцам, таким как луковицы, клубни, клубнелуковицы и корневища, нужен только один покрывающий слой для обеспечения биологически эффективного количества соединения Формулы I.
Сеянцы также могут быть покрыты путем распыления концентрата суспензии прямо в аппарате для дражирования сеянцев, а затем сушки сеянца. Альтернативно, на сеянцы могут быть распылены другие типы составов, как смоченные порошки, растворы, суспоэмульсии, эмульгирующиеся концентраты и эмульсии в воде. Этот способ в частности используют для нанесения пленочных материалов для покрытия на семена. Специалистам в данной области доступны различные механизмы и способы для покрытия. Подходящие способы включают в себя способы, перечисленные у P. Kosters et al., Seed Treatment: Progress and Prospects, 1994 BCPC Monograph No. 57 и в ссылках, перечисленных там. Три хорошо известные методики включают в себя использование покрытия в барабане, методику кипящего слоя и фонтанирующих слоев. Сеянцы, такие как семена, перед покрытием могут быть предварительно отсортированы по величине. После покрытия сеянцы сушат, а затем необязательно загрунтовывают путем переноса в сортировочную машину. Эти машины известны в уровне техники, например, типичная машина используется при калибровании семян кукурузы (маиса) в сельскохозяйственном производстве.
Для покрытия семян, семя и материал для покрытия перемешивают в любом обычном аппарате для покрытия семян. Скорость вращения и нанесения зависит от семени. Для крупных продолговатых семян, таких как семена хлопка, подходящий аппарат для покрытия семян содержит вращающийся сосуд с поднимающимися лопастями, вращающийся с достаточными оборотами в минуту для поддержания вращения семян, облегчающим равномерное покрытие. Для составов покрытия семян, применяемых в виде жидкости, покрытие семян должно происходить на протяжении достаточного времени, чтобы дать семенам просохнуть, сводя к минимуму слипание семян. Использование нагнетаемого воздуха или нагретого нагнетаемого воздуха может увеличить скорость нанесения. Специалисту в данной области будет понятно, что этот способ может быть однократным или непрерывным процессом. Как следует из названия, непрерывный процесс позволяет семенам непрерывно двигаться через поток вещества. Новые семена входят в сосуде в стационарный поток и смещают покрытые семена к выходу из сосуда.
Процесс покрытия семян по настоящему изобретению не ограничивается тонкопленочным покрытием и также может включать в себя дражирование семян. Процесс дражирования обычно увеличивает массу семени в 2-100 раз и может быть использован также для улучшения формы семени для использования в механических сеялках. Композиции для дражирования главным образом содержат твердый разбавитель, который обычно является нерастворимым сыпучим материалом, таким как глина, известняк, порошкообразный кремнезем и т.п. для обеспечения объема в добавление к связующему веществу, такому как искусственный полимер (например, поливиниловый спирт, гидролизованные поливинилацетаты, сополимер поливинилметиловый эфир-малеиновый ангидрид, и поливинилпирролидинон) или природный полимер (например, альгинаты, карайская камедь, гуаровая камедь, трагакант, полисахаридная камедь, адгезивное вещество). После нанесения достаточного количества слоев, покрытие сушат и пеллеты сортируют. Способ получения пеллет (драже) описан у Agrow, The семя Treatment Market, Chapter 3, PJB Publications Ltd., 1994.
Для дальнейшего описания компонентов композиции и способов, подходящих для покрытия сеянцев соединением Формулы I, см. патенты США 4443637, 5494709, 5527760, 5834006, 5849320, 5876739, 6156699, 6199318, 6202346 и 6230438 и публикацию европейского патента EP-1078563 A1.
Следующие примеры E-H иллюстрируют способ покрытия семян. Номера соединений относятся к соединениям Таблицы-указателя A.
ПРИМЕР E
Приготовление серии семян хлопка, покрытых композицией, содержащей Соединение 208
Стадия 1: Приготовление текучей суспензии, содержащей Соединение 208
Готовили текучую суспензию, содержащую компоненты, перечисленные в Таблице 7.
Agrimer® VA 6 является водорастворимым в высокой степени, пленкообразующее адгезивное вещество, имеющее точку размягчения 106°C, содержит сополимер поливинилпирролидинонвинилацетат и продаваемый International Specialty Products (ISP). Licowax® KST представляет собой водорастворимое в высокой степени пленкообразующее адгезивное вещество, имеющее точку каплеобразования 59°C, содержащее эфир полиэтиленгликоля кислоты горного воска, продаваемый Clariant. Borresperse™ CA представляет собой водорастворимый в высокой степени анионный дисперсант, имеющий точку размягчения 132°C, содержащий обессахаренный лигносульфонат кальция, продаваемый Borregaard LignoTech. Pluronic® F-108 представляет собой водорастворимый в высокой степени, неионный дисперсант, имеющий точку плавления 57°C, содержащий полиоксипропилен-полиоксиэтилен блоксополимер, продаваемый BASF. Brij® 78 представляет собой водорастворимый в высокой степени, неионный дисперсант, имеющий точку текучести 38°C, содержащий стеариловый спирт (POE 20), продаваемый Uniqema. Rhodorsil® 416 представляет собой вододиспергируемый жидкий противопенный агент, содержащий полиорганосилоксаны и эмульгирующийся агент, продаваемый Rhodia. Pro-lzed® Colorant Red представляет собой вододиспергируемую красящую композицию, содержащую красный пигмент, глину каолин, и неионное поверхностно-активное вещество и продается Gustafson.
Суспензионный носитель (253,20 г) готовили, первоначально растворяя Brij® 78 (6,00 г) в теплой воде (210,48 г), с последующим интенсивным перемешиванием в Agrimer® VA 6 (15,00 г), Licowax® KST (15,00 г), Borresperse™ CA (3,00 г), Pluronic® F-108 (3,00 г), Brij® 78 (6,00 г), Rhodorsil® 416 (0,6 г) и Pro-lzed® Colorant Red (0,12 г). В лабораторный стакан добавляли Соединение 208 (15,6 г), затем добавляли часть тщательно перемешанного суспензионного носителя (84,4 г), и Соединение 208 складывали в суспензионный носитель с помощью шпателя.
Эту смесь затем дополнительно гомогенизировали с использованием высокоскоростного роторного статорного диспергатора Политрон (продаваемого Brinkman Instruments Inc., Cantiague Rd., Westbury, NY 11590 U.S.A.) с 10 мм генераторным преобразователем, который дробил агрегаты Соединения 208.
Полученную в результате суспензию затем переносили в бегунковую мельницу, загруженную до 80% вместимости 0,5 мм одноразмерной керамической средой высокой плотности, и охлаждали пропуская охлажденный водный 33% раствор этиленгликоля через охлаждающую рубашку мельничной камеры. Суспензию рециркулировали через мельничную камеру в течение 13 минут с вращением лопастей мешалки 4300 об/мин. Конец циркуляторной трубы затем перемещали из питательной воронки мельницы в колбу для сбора для получения окончательной розовой высокопористой текучей суспензии (89,5 г).
Диаметры микронизированных (измельченных) частиц в данной суспензии анализировали с использованием прибора лазерной дифракции. Используя среднее значение двух измерений, среднее арифметическое значение диаметра частиц составило 2,03 мкм, 90% были меньше 5,21 мкм в диаметре, 10% частиц были менее 0,30 мкм в диаметре, и средний диаметр частиц составил 1,0 мкм.
Стадия 2: Покрытие семян хлопка композицией, содержащей Соединение 208
Семена хлопка (Stoneville 4793 RR, 122,5 г) добавляли в бак из нержавеющей стали (12 см в диаметре, 11 см глубиной), содержащий две противоположно направленных поднимающихся лопасти для подъема семени при повороте бака. Бак ориентировали под углом 40-45° к горизонту и механически вращали при 640 об/мин, вызывая хорошее перемешивание и переворачивание внутри бака.
Приготовленную на Стадии 1 текучую суспензию распыляли непосредственно на семена в дражировочном барабане, обеспечивая давление воздуха 10-11 ф/дм2 (psi) (69-76 кПа) с получением мелких капель. Посредством измерения массы резервуара можно определить количество текучей суспензии, распыленной на семена. При переворачивании семян ручной пульверизатор направляли внутрь бака для непосредственного распыления на центр переворачивающегося слоя семени. Распыление продолжали до тех пор, пока поверхности семян не становились липкими, вызывая склеивание семян вместе. Пульверизатор затем выключали и покрытие семян быстро сушили продувкой воздухом при низком давлении при комнатной температуре из насадки, установленной для прямого потока воздуха внутрь бака. Усиливающийся звук переворачивающихся семян обеспечивает аудиосигнал того, что покрытые семена достаточно высушены. Высушивающий поток воздуха затем закрывали и продолжали распыление с использованием ручного пульверизатора. Циклы распыления и высушивания продолжали до нанесения на семена желаемого количества текучей суспензии. Затем завершали просушиванием покрытия семян, подвергая их слабому потоку окружающего воздуха, в течение 60 часов.
Массу нанесенного Соединения 208 на каждые десять семян из каждой партии определяли путем вымачивания каждого семени в шаровой мельнице, а затем добавления экстрагирующего растворителя ацетонитрила. Экстракты центрифугировали и аликвоты супернатанта (супернатантной жидкости) разводили 10000:1, а затем анализировали посредством LC/MS. Результаты анализа перечислены в Таблице 8.
* на основании 10 повторов
ПРИМЕР F
Получение партии семян кукурузы, покрытых композицией, содержащей соединения 208, 484, 486, 502, 509 или 515
Стадия 1: Получение 6 Текучих суспензий, содержащих Соединения 208, 484, 486, 502, 509 или 515
Готовили шесть текучих суспензий, содержащих один из шести активных компонентов вышеуказанных соединений, по рецепту, как показано в Таблице 9.
Все компоненты, иные, чем активные компоненты соединений, описаны в примере ПРИМЕР E.
Текучую суспензию каждого соединения готовили способом, описанном в примере E, Стадия 1. Диаметры (т.е. Диа. в Таблице 10) частиц в суспензии анализировали способом, также описанном в ПРИМЕРЕ E, Стадия 1. Распределение диаметра частиц, полученное после влажного измельчения, показано в Таблице 10.
* среднее двух измерений
"<" означает менее чем
Стадия 2: Покрытие семян кукурузы отдельными композициями, содержащими соединения 208, 484, 486, 502, 509 или 515
Семена кукурузы (маиса) (Pioneer 3146 Lot # C92FA (Parent), 65g) добавляли в бак из нержавеющей стали (8 см в диаметре, 8,3 см глубина), содержащий две противоположно направленных поднимающих лопасти для подъема семени при повороте бака. Бак ориентировали под углом 40-45° к горизонту и механически вращали при 110 об/мин, что давало хорошее перемешивание и переворачивание внутри бака.
Каждую из 6 текучих суспензий, приготовленных на Стадии 1, распыляли непосредственно на дражировочный слой кукурузного семени, следуя общей методике, описанной в ПРИМЕРЕ E, Стадия 2. Затем просушивание семян завершали, оставляя семена сохнуть в течение ночи в химическом вытяжном шкафу. Достигались номинальные 3% по весу покрытия каждого микронизированного соединения на кукурузное семя, как показано в Таблице 11.
* на основании 10 повторов
ПРИМЕР G
Получение партий семян, покрытых композициями, содержащими соединения 208, 276 или 483
Стадия 1: Получение 3 текучих суспензий, содержащих соединения 208, 276 или 483
Готовили три текучие суспензии, каждая содержащая одно из трех вышеупомянутых соединений, по рецепту, показанному в Таблице 9 ПРИМЕРА F. Текучую суспензию каждого соединения готовили способом, описанным в ПРИМЕРЕ E, Стадия 1. Диаметры (т.е. Диа. в Таблице 10) частиц в суспензии анализировали способом, также описанным в ПРИМЕРЕ E, Стадия 1. Достигаемое распределение диаметров частиц после влажного измельчения показано в Таблице 12.
* среднее двух измерений
"<" означает менее чем
Стадия 2: Покрытие семян хлопка отдельными композициями, содержащими Соединения 208, 276 или 483
Семена хлопка (Stoneville 4793 RR, 33g) добавляли в бак из нержавеющей стали (6,5 см в диаметре, 7,5 см глубина), содержащий две противоположно направленных поднимающих лопасти для подъема семени при повороте бака. Бак ориентировали под углом 40-45° к горизонту и механически вращали при 100 об/мин, что обеспечивало хорошее перемешивание и переворачивание внутри бака.
Три текучие суспензии, приготовленные на Стадии 1, распыляли на партию переворачивающихся семян хлопка, следуя общей методике, описанной в ПРИМЕРЕ E, Стадия 2. Затем просушивание семян завершали, оставляя семена сохнуть в течение ночи в химическом вытяжном шкафу. Достигались номинальные 3% по весу покрытия каждого микронизированного соединения на хлопковое семя, как показано в Таблице 13.
ПРИМЕР H
Приготовление партии кукурузных семян, покрытых композицией, содержащей соединение 502
Стадия 1: Получение текучей суспензии, содержащей 15 мас.%/мас. Соединения 502
Готовили 15%-ную текучую суспензию соединения 502, содержащую те же компоненты, кроме соединений, перечисленных в Таблице 9, ПРИМЕР F. Текучую суспензию соединения 502 готовили по способу, описанному в ПРИМЕРЕ E, Стадия 1. Диаметры (т.е. Диа. в Таблице 10) частиц в суспензии анализировали методом, также описанным в ПРИМЕРЕ E, Стадия 1. Полученное в результате распределение диаметров частиц после влажного измельчения показано в Таблице 14.
Размеры частиц текучей суспензии
"<" означает менее чем
Стадия 2: Покрытие кукурузного семени композицией, содержащей Соединение 502
Семена кукурузы (маиса) (Pioneer 34M94 Hybrid Field Corn, 575 г) добавляли в бак из нержавеющей стали (17 см в диаметре, 16 см глубина), содержащий две противоположно направленных поднимающих лопасти для подъема семени при повороте бака. Бак ориентировали под углом 40-45° к горизонту и механически вращали при 200 об/мин, получая хорошее перемешивание и переворачивание внутри бака.
15 мас.%/мас. текучую суспензию, приготовленную на Стадии 1, распыляли непосредственно на отдельные партии переворачивающихся семян, следуя общей методике, описанной ПРИМЕРЕ E, Стадия 2. Затем просушивание семян завершали, оставляя семена сохнуть в течение ночи в химическом вытяжном шкафу. Достигались номинальные 0,15, 0,29, 0,58, 1,09, 1,75% по весу покрытия каждого микронизированного соединения 502 на кукурузное семя, как показано в Таблице 15. Средний мас.% Соединения 502 на покрытом семени измеряли с помощью LC/MS, следуя способу со Стадии 2 ПРИМЕРА E.
* на основании 10 повторов
ПРИМЕР I
Получение семян хлопчатника, семян кукурузы, пеллетизированных семян свеклы и семян сои, покрытых композицией, включающей соединение 855.
Стадия 1: Приготовление текучей суспензии, содержащей 15% (мас./мас.) соединения 855
Готовили текучую суспензию, содержащую ингредиенты, представленные в таблице 16.
Количества ингредиентов в текучей суспензии
Agrimer® VA представляет собой пленкообразующее адгезивное вещество с высокой степенью растворимости в воде и температурой размягчения 106°С, включающее сополимер поливинилпирролидона и винилацетата и продаваемое International Specialty Products (ISP). Licowax®KST представляет собой пленкообразующее адгезивное вещество с высокой степенью растворимости в воде и температурой каплеобразования 59°С, включающее кислоту буроугольного воска и сложный полиэтиленгликолевый эфир и продаваемое Clariant. Borresperse™ CA представляет собой анионогенный диспесант с высокой степенью растворимости в воде и температурой размягчения 132°С, включающий обессахаренный лигносульфонат кальция и продаваемый Borregaard Ligno Tech. Pluronic® F-108 представляет собой неионогенный дисперсант с высокой степенью растворимости в воде и температурой плавления 57°С, включающий блоксополимер полиоксипропилена и полиоксиэтилена и продаваемый BASF. Rhodorsil® 416 представляет собой вододиспергируемый жидкий противопенный агент, включающий полиорганосилоксаны и эмульгатор и продаваемый Rhodia. Color Coat Green представляет собой вододиспергируемый жидкий краситель, продаваемый Becker Underwood.
Суспензионный носитель (42 г) получали смешением с помощью магнитной мешалки всех инертных ингредиентов: Agrimer® VA (2,5 г), Licowax®KST (2,5 г), Borresperse™ CA (0,75 г), Pluronic® F-108 (0,75 н), Emery 912 Glycerine (1,23 г), Rhodorsil® 416 (0,10 г) и Color Coat Green (0,18 г) - с водой (34,5 г) до растворения. Соединение 855 (3,5 г) загружали в емкость объемом 60 мл с широким входным отверстием. Сверху добавляли 28 г измельчающей среды из натриево-кальциево-силикатного стекла с диаметром частиц 0,5-0,75 мм. Вставляли 25 мм абразивный импеллер. И наконец, к суспензии добавляли тщательно смешанный суспензионный носитель (19,833 г) с получением пульпы. Полученную пульпу перемешивали с высокой скоростью мешалкой с верхним приводом, охлаждая на ледяной бане. Зеленую суспензию с высокой степенью текучести отделяли от среды измельчения. Всю методику дважды повторяли с получением 43,34 г суспензии тонкого помола.
Диаметры измельченных частиц данной суспензии измеряли с использованием прибора лазерной дифракции. Среднее арифметическое значение диаметра частиц составляло 1,94 мкм, 90% частиц имели диаметр менее 2,28 мкм, 10% частиц имели диаметр менее 0,28 мкм, и медианное значение диаметра частиц равнялось 0,73 мкм.
Стадия 2: Покрытие семян кукурузы композицией, включающей соединение 855
Примерно 67 г семян помещали в бак из нержавеющей стали со шпенделем (внутренний диаметр 8,5 см, глубина 8 см). Бак ориентировали под углом 40-45°С к горизонтали и вращали механически со скоростью 640 об/мин, что обеспечивало хорошее перемешивание и переворачивание семян внутри бака. Текучую суспензию, полученную на стадии 1, распыляли непосредственно на переворачивающийся слой семян, используя двухжидкостной распылитель. Регулируемое давление воздуха 69-86 КПа обеспечивало получение тонкодисперсных капель суспензии. При переворачивании семян ручной распылитель помещали внутри бака для прямого опрыскивания в центр переворачивающегося слоя семян. Опрыскивание продолжали до тех пор, пока поверхность семян не становилась липкой, что приводило к слипанию семян. После этого распылитель выключали и семена с нанесенным на них покрытием быстро сушили обдувом воздухом низкого давления с температурой окружающей среды. Сушку воздухом выключали и продолжали опрыскивание с помощью ручного распылителя. Цикл распыления и сушки повторяли до тех пор, пока нужное количество текучей суспензии не было нанесено на семена. Взвешиванием полного бака до и после опрыскивания можно определить количество текучей суспензии, нанесенной на семена. Сушку семян с нанесенным на них покрытием завершали дополнительным обдувом воздухом в направлении, перпендикулярном направлению вращения семян. Покрытие наносили на две партии семян кукурузы, получая номинальные активные дозы 1 мг а.и. на семя и 3 мг а.и. на семя.
Характеристики семян кукурузы с покрытием композиции соединения 855
Стадия 3: Покрытие семян хлопчатника композицией, включающей соединение 855
Примерно 32,6 г семян хлопчатника помещали в небольшой бак из нержавеющей стали со шпенделем (внутренний диаметр 6,5 см, глубина 7,5 см). Бак ориентировали под углом 40-45°С к горизонтали и механически вращали со скоростью 70 об/мин, что обеспечивало хорошее смешение и переворачивающее действие внутри бака. Текучую суспензию, полученную на стадии 1, распыляли непосредственно на переворачивающийся слой семян, используя двухжидкостной распылитель. Покрытие наносили на две партии семян хлопчатника в соответствии со способом, описанным в стадии 2 примера 1, получая номинальные активные дозы 1 мг а.и. на семя и 5 мг а.и. на семя.
Характеристики семян хлопчатника с покрытием композиции, включающей соединение 855
Стадия 4: Покрытие пеллетизированных семян сахарной свеклы композицией, включающей соединение 855
Примерно 25,0 г зеленых пеллетизированных семян сахарной свеклы помещали в небольшой бак из нержавеющей стали со шпенделем (внутренний диаметр 6,5 см, глубина 7,5 см). Бак ориентировали под углом 40-45°С к горизонтали и механически вращали со скоростью 70 об/мин, что обеспечивало хорошее смешение и переворачивающее действие внутри бака. Текучую суспензию, полученную на стадии 1, распыляли непосредственно на переворачивающийся слой семян, используя двухжидкостной распылитель. Покрытие наносили на одну партию пеллетизированных семян сахарной свеклы в соответствии со способом, описанным в примере I (стадия 2), получая номинальную активную дозу 1,25 мг а.и. на семя.
Характеристики пеллетизированных семян сахарной свеклы с покрытием, содержащим соединение 855
Стадия 5: Покрытие семян сои композицией, включающей соединение 855
Примерно 75,65 г семян сои загружали в небольшой бак из нержавеющей стали со шпенделем (внутренний диаметр 8,5 см, глубина 8,0 см). Бак ориентировали под углом 40-45°С к горизонтали и механически вращали со скоростью 70 об/мин, что вызывало хорошее смешение и переворачивание семян внутри бака. Текучую суспензию, полученную на стадии 1, распыляли непосредственно на переворачивающийся слой семян, используя двухжидкостной распылитель. Покрытие наносят на две партии семян сои в соответствии со способом, описанным в примере I (стадия 2), получая номинальные активные дозы 0,625 мг а.и. на семя и 1,25 мг на семя.
Характеристики семян сои с покрытием, содержащим соединение 855
ПРИМЕР J
Получение партий семян кукурузы и семян хлопчатника с покрытием композиции, включающей соединение 854
Стадия 1: Получение текучей суспензии, содержащей соединение 854
Приготавливали текучую суспензию, содержащую ингредиенты, представленные в таблице 21.
Все ингредиенты, отличные от активного ингредиента, описаны в примере 1.
Текучую суспензию, включающую соединение 854, получали способом, описанным в Примере I (стадия 1).
Диаметры измельченных частиц данной суспензии определяли с использованием прибора лазерной дифракции. Среднее арифметическое значение диаметра частиц составляло 0,85 мкм, 90% частиц имели диаметр менее 1,81 мкм, 10% частиц имели диаметр менее 0,25 мкм, и медианное значение диаметра частиц равнялось 0,73 мкм.
Стадия 2: Покрытие семян хлопчатника композицией, включающей соединение 854
Примерно 32,65 г семян хлопчатника помещали в бак из нержавеющей стали со шпенделем (внутренний диаметр 6,5 см, глубина 7,5 см). Бак ориентировали под углом 40-45°С к горизонтали и механически вращали со скоростью 70 об/мин, что вызывало хорошее смешение и переворачивание семян внутри бака. Текучую суспензию, полученную на стадии 1, распыляли непосредственно на переворачивающийся слой семян, используя двухжидкостной распылитель. Покрытие наносили на три партии семян в соответствии с общей методикой, описанной в примере 1 (стадия 2), получая номинальные активные дозы 0,5 мг, 1,0 мг и 2,0 мг а.и. на семя.
Характеристики семян кукурузы с покрытием, включающим соединение 854
ПРИМЕР К Получение цельных клубней картофеля с покрытием из композиции, содержащей соединение 855
Стадия 1: Приготовление текучей суспензии, включающей соединение 855
Приготавливали текучую суспензию номинальной концентрации 250 г/л, содержащую ингредиенты, представленные в таблице 23.
Количества ингредиентов в текучей суспензии, содержащей соединение 855
Все ингредиенты, кроме соединения, представляющего собой активный ингредиент, описаны в примере I. Суспензионный носитель (1095,53 г) для соединения 855 получали перемешиванием с помощью магнитной мешалки всех инертных твердых ингредиентов с водой в лабораторном стакане объемом 1 л; Agrimer® VA 6 (64,27 г), Licowax®KST (64,27 г), Borresperse™ CA (17,14 г), Pluronic® F-108 (17,14 г) смешивали с водой (858,14 г) до полного растворения или диспергирования. На следующий день добавляли жидкие ингредиенты: пропиленгликоль (32,14 г), Emery 912 Glycerine (32,14 г), Legend MK (1,0 г), Rhodorsil® 416 (4,28 г) и Color Coat Green (5,0 г) - и смешивали их с помощью магнитной мешалки в течение 1 часа до завершения получения суспензионного носителя.
Соединение 855 (322,80 г) загружали в лабораторный стакан объемом 2 л. Добавляли тщательно смешанный суспензионный носитель (1095,53 г) с получением суспензии конечного состава. Суспензию перемешивали мешалкой с верхним приводом с умеренной скоростью. Затем суспензию подвергали мокрому помолу в горизонтальной бисерной мельнице Eiger 50 Mini VSE. В камеру мельницы на 80% ее объема загружали измельчающую среду из натриево-кальциево-силикатного стекла с диаметрами частиц 0,5-0,75 мм. Суспензию подвергали мокрому помолу рециркуляционным способом со скоростью вращения мешалки 4000 об/мин в течение 140 минут. Данная методика мокрого помола приводила к получению 1403 г суспензии зеленого цвета с высокой степенью текучести.
Диаметры измельченных частиц данной суспензии определяли с использованием прибора лазерной дифракции. Среднее арифметическое значение диаметра частиц составляло 0, 96 мкм, 90% частиц имели диаметр менее 2,16 мкм, 10% частиц имели диаметр менее 0,27 мкм, и медианное значение диаметра частиц равнялось 0,60 мкм.
Стадия 2: Нанесение покрытия на клубни картофеля
Приготовленную композицию, включающую соединение 855, наносили на цельные клубни картофеля (red Pontiac, размер В). Препарат наносили из капельной воронки, расположенной в середине потока воздуха, нагнетаемого под давлением 55 кПа, что приводило к дроблению капли и получению тонкодисперсного тумана. Туман распыляли на один клубень картофеля, расположенный на 4-6 см ниже кончика пипетки. В процессе нанесения клубень картофеля осторожно вращали, так что химическая обработка покрывала третью часть поверхности картофеля. Нанесение покрытия с нормами расхода 2 и 8 мкл на каждый клубень приводило к получению покрытия номинальной дозы 0,5 и 2 мг а.и./картофель.
ПРИМЕР L
Получение цельных клубней картофеля с покрытием из композиции, включающей соединение 502
Стадия 1: Получение текучей суспензии, включающей соединение 502
Приготавливали текучую суспензию номинальной концентрации 250 г/л, содержащую ингредиенты, представленные в таблице 24.
Количества ингредиентов в текучей суспензии, содержащей соединение 502
Все ингредиенты, кроме соединения, представляющего собой активный ингредиент, описаны в примере I.
Суспензионный носитель (1741,03 г) для соединения 502 получали смешением с помощью магнитной мешалки всех инертных ингредиентов в твердой и жидкой форме в лабораторном стакане объемом 3 л в течение ночи. Agrimer® VA 6 (99,88 г), Licowax®KST (99,88 г), Borresperse™ CA (20,43 г), Pluronic® F-108 (20,43 г), Brij 78 (38,59 г), пропиленгликоль (51,08 г), Emery 912 Glycerine (51,08 г). Legend MK (1,59 г), Rhodorsil® 416 (6,81 г) и Colorfast Purple (6,75 г) перемешивали с помощью магнитной мешалки в течение ночи до полного растворения /диспергирования с водой (1344,52 г).
Соединение 502 (322,80 г) загружали ложкой непосредственно на суспензионный носитель и давали возможность массе самопроизвольно смачиваться внутри стакана объемом 3 л. В процессе смачивания суспензию непрерывно перемешивали с умеренной скоростью мешалкой с верхним приводом. Затем суспензию подвергали мокрому помолу в горизонтальной бисерной мельнице Eiger 50 Mini VSE. В камеру мельницы на 80% ее объема загружали измельчающую среду из натриево-кальциево-силикатного стекла с размерами частиц 0,5-0,75 мм. Суспензию подвергали мокрому помолу рециркуляционным способом при скорости вращения мешалки 4000 об/мин в течение 189 минут. Теплоту фрикционного нагрева удаляли с использованием циркулирующей в рубашке корпуса мельницы смеси 50:50 этиленгликолевый антифриз:вода. Данная методика мокрого помола приводила к получению 2222,7 г суспензии зеленого цвета с высокой степенью текучести.
Диаметры измельченных частиц данной суспензии определяли с использованием прибора лазерной дифракции. Среднее арифметическое значение диаметра частиц составляло 0,86 мкм, 90% частиц имели диаметр менее 1,92 мкм, 10% частиц имели диаметр менее 0,26 мкм, и медианное значение диаметра частиц равнялось 0,55 мкм.
Стадия 2: Покрытие клубней картофеля текучей суспензией, включающей соединение 502
Приготовленные композиции, включающие соединение 502, наносили на цельные клубни картофеля (red Pontiac, размер В). Препарат наносили из капельной воронки, расположенной в середине потока воздуха, нагнетаемого под давлением 55 кПа, что приводило к дроблению капли и получению тонкодисперсного тумана. Туман распыляли на один клубень картофеля, расположенный на 4-6 см ниже кончика пипетки. В процессе нанесения клубень картофеля осторожно вращали, так что химическая обработка покрывала третью часть площади поверхности картофеля. Нанесение покрытия с нормами расхода 2 и 8 мкл на каждый клубень приводило к получению покрытия номинальной дозы 0,5 и 2 мг а.и./картофель.
ПРИМЕР М Приготовление текучей суспензии, включающей соединение 502 и имидаклоприд
Приготавливали текучую суспензию, содержащую ингредиенты, представленные в таблице 25.
Количества ингредиентов, включенных в текучую суспензию
Все ингредиенты, кроме соединения, представляющего собой активный ингредиент, описаны в стадии 1 примера Е.
Текучую суспензию, включающую соединение 502 и имидаклоприд, приготавливали в соответствии со способом, описанным в примере Е (стадия 1).
ПРИМЕР N
Приготовление текучей суспензии, включающей соединение 855 и каптан
Приготавливали текучую суспензию, содержащую ингредиенты, представленные в таблице 26.
Количества ингредиентов, включенных в текучую суспензию
Все ингредиенты, кроме соединения, представляющего собой активный ингредиент, описаны в примере Е (стадия 1).
Текучую суспензию, включающую соединение 855 и каптан, получали способом, описанным в примере Е (стадия 1).
Следующие испытания в Биологических примерах данного изобретения демонстрируют эффективность способов и композиций данного изобретения для защиты растений от отдельных членистоногих вредителей. Контролирующая защита от вредителей, предоставляемая данными соединениями, не ограничена, однако, этими видами. Для описания соединений см. Таблицу-указатель А. В таблице индексов использованы следующие аббревиатуры: t означает третичный, n означает нормальный, i означает изо, s означает вторичный, с означает цикл, Me означает метил, Et означает этил, Pr означает пропил и Bu означает бутил; соответственно i-Pr означает изопропил, s-Bu означает вторичный бутил, и т.д. Аббревиатура "Ex." стоит для "Пример", за которым следует указание, в каком ПРИМЕРЕ получают данное соединение.
Таблица индексов А

R1, R5, и R8 представляют собой Н, исключая где указано; В означает О, исключая где указано. "CN" связан через углерод, не азот; например "CN-Ph" означает цианофенил, не изоцианофенил.





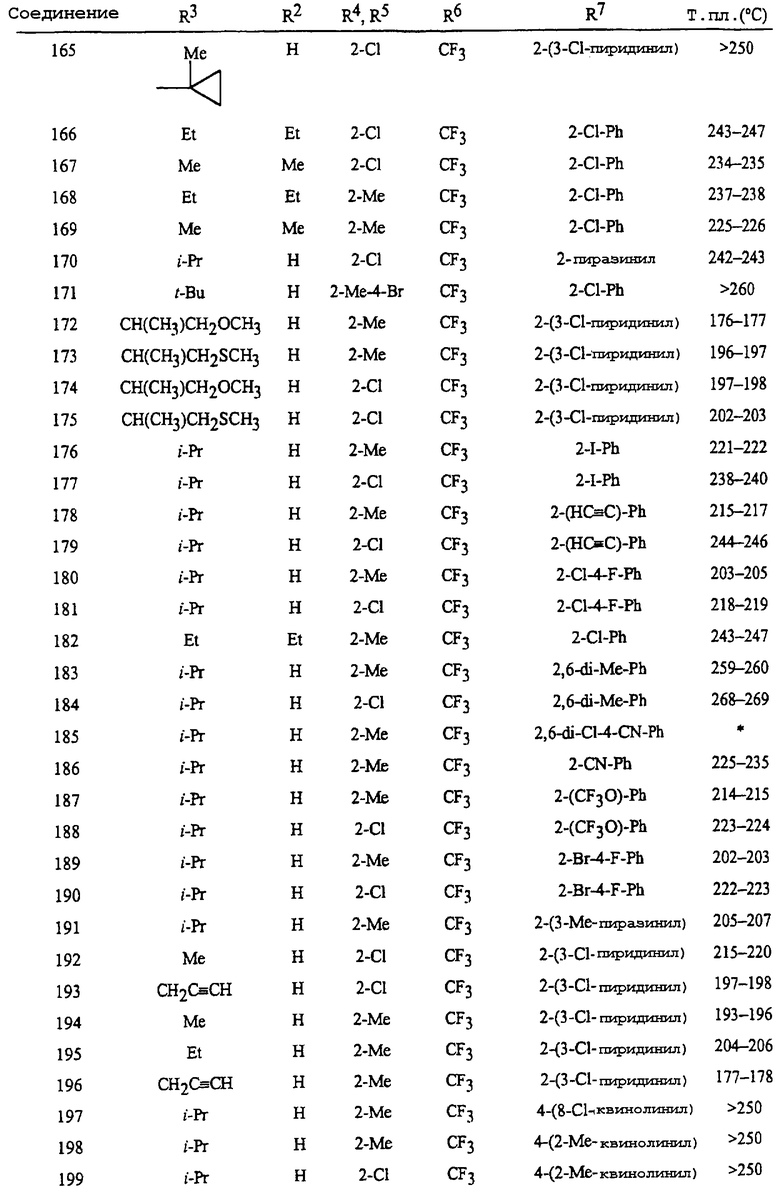








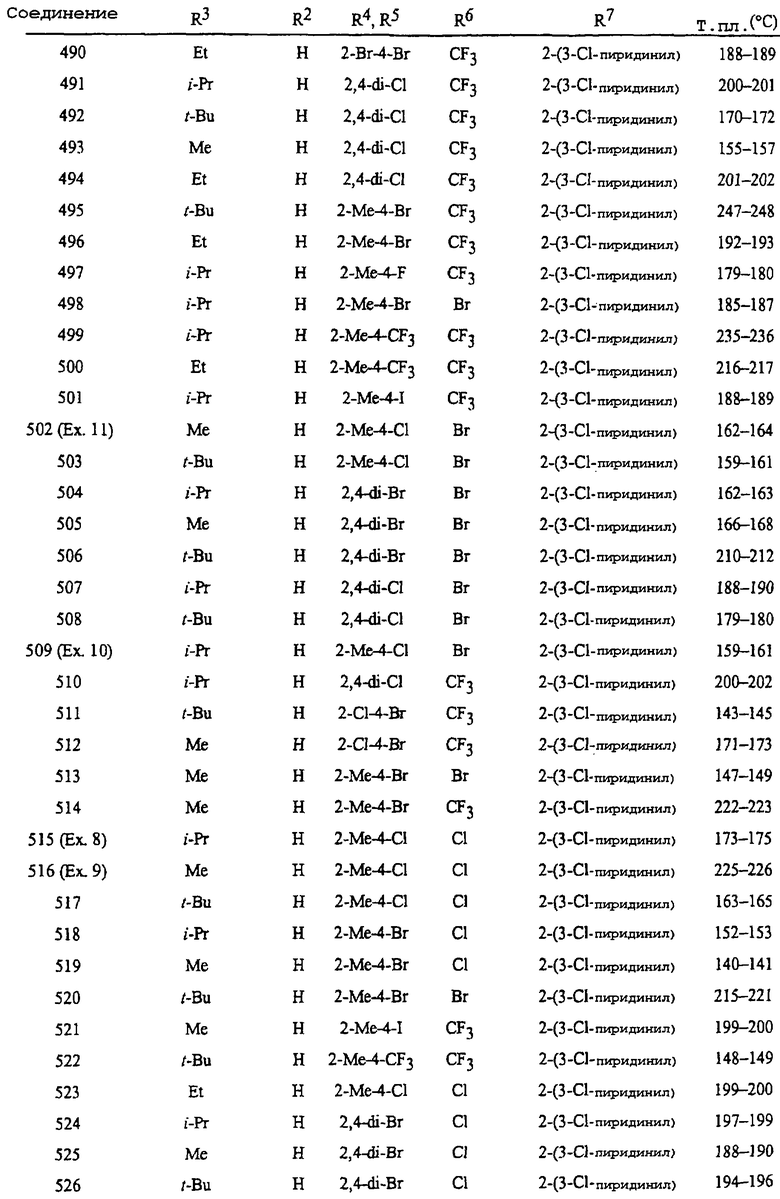


















БИОЛОГИЧЕСКИЕ ПРИМЕРЫ ПО ДАННОМУ ИЗОБРЕТЕНИЮ
ТЕСТ А
Семена хлопка, покрытые композицией Соединения 208 из серий концентраций номинала 1%, номинала 2% и номинала 3%, приготовленных как описано в ПРИМЕРЕ Е, и необработанные семена для сравнения, сажали в горшки с использованием стерильной почвы Sassafras, и выращивали в ростовой камере с 16 часовым световым днем при 28°С и 8 часовой фазой темноты при 24°С и 50% относительной влажности. После 31 дня два растения, каждое имеющее настоящие листья, отбирали из каждой партии семян и удаляли семядоли. Взрослых особей Bemisia argentifolii (белокрылка silverleaf) добавляли для отложения яиц на растениях и пластиковые цилиндры, покрытые тонкой бумагой помещали в горшки. Спустя три дня, взрослых особей удаляли и осматривали листья для установления факта отложения яиц. Спустя пятнадцать дней (примерно шесть дней после выведения яиц), инфицированные листья удаляли с растения и 49-дневные результаты получали путем подсчета мертвых и живых нимф на нижней стороне листьев. Взрослых особей Bemisia argentifolii повторно вводили для повторного отложения яиц на верхние листья растений, и пластиковые цилиндры с тонкой бумагой помещали внутрь горшков, как и ранее. Три дня спустя, взрослых особей удаляли и листья проверяли на наличие кладки яиц. Пятнадцать дней спустя (примерно через шесть дней после выведения яиц), листья удаляли с растения и 66-дневные результаты получали путем подсчета мертвых и живых нимф на нижней стороне листьев. Результаты двух значений от времени суммированы в Таблице А.
Контроль белокрылки Silverleaf путем покрытия семян хлопка композициями соединения 208
Это испытание демонстрирует, что покрытия семян по данному изобретению могут защищать растения хлопка от равнокрылых вредителей Bemisia argentifolii в течение более 9 недель после посева.
ТЕСТ В
Семена хлопка, покрытые композицией Соединения 208 из серий концентраций номинала 1%, номинала 2% и номинала 3%, приготовленных, как описано в ПРИМЕРЕ Е, и необработанные семена для сравнения, сажали в 10 см горшки с использованием стерильной почвы Sassafras, и выращивали в ростовой камере с 16-часовым световым днем и 8-часовой фазой темноты при 25°С и 50% относительной влажности. С некоторых растений собирали листья через 14 дней после посадки, разрезали на 3-4 части и помещали по одной части на лунку покрытых 16-луночных полупрозрачных пластиковых лотков в ростовой камере. Личинки Heliothis virescens (табачная листовертка-почкоед) второй возрастной стадии добавляли к частям листа (1 личинка/лунку, 6-10 личинок на обработку/тип листа), и смертность насекомых определяли через 48 часов и 96 часов после инвазии. Собирали листья с другого растения через 64 дня после посадки, разрезали на 3-4 части, и помещали по одной части на лунку покрытых 16-луночных полупрозрачных пластиковых лотков в ростовой камере. Личинки Heliothis virescens (табачная листовертка-почкоед) во второй возрастной стадии добавляли к частям листьев (1 личинка/лунку, 6-16 личинок на обработку/расположение листа), и смертность насекомых определяли через 72 часа и 96 часов после инвазии. Результаты суммированы в Таблицах В1 и В2.
Контроль табачной листовертки через 14 дней после посадки с помощью покрытия семян хлопка композициями соединения 208
Контроль табачной листовертки через 64 дня после посадки с помощью покрытия семян хлопка композициями соединения 208
Этот тест демонстрирует, что покрытия семян в соответствии с настоящим изобретением может защищать растения хлопка от чешуекрылых вредителей Heliothis virescens на протяжении более 9 недель после посадки.
ТЕСТ С
Семена хлопка, обработанные соединением 208, приготовлены как в ПРИМЕРЕ Е (номинальная 3% серия) и соединениями 276, 486 и 502, приготовленными в ПРИМЕРЕ G, и необработанные семена для сравнения, сажали в горшки с использованием либо стерильной почвы Sassafras, либо почвы Drummer. Растения выращивали в теплице и отбирали на испытание, когда они начали давать почки (squares). Листья со второго узла и конечные листья более 15 см2 отбирали для испытания (у растения было приблизительно 5 листьев). Обрезанный лист с каждого растения разрезали на 4 части и каждую часть помещали в лунку с одной личинкой Heliothis virescens (листовертка-почкоед табачная) во второй возрастной стадии. Смертность личинок записывали через 96 часов после взятия образцов.
Смертность личинок от поедания листьев из обработанных семян, выращенных на почвах двух типов
ТЕСТ D
Семена кукурузы, обработанные соединениями 208, 484, 486, 502, 509 и 515, приготовленные в ПРИМЕРЕ F, сажали в горшки с почвой Sassafras. Растения выращивали до высоты кольца листьев (9-й лист) в теплице и заражали 25 особями листопадных "походных червей" (личинки первой возрастной стадии) внизу кольца листьев. Через шесть дней после инвазии регистрировали повреждение растения, связанное с поеданием. Повреждение растения оценивали 0-100% (0 означает отсутствие поедания).
Процент повреждения растения от поедания личинками на растениях кукурузы с различными обработками семян
ТЕСТ Е
Семена кукурузы, обработанные соединением 502, как приготовлено в ПРИМЕРЕ Н, в пяти дозах (в процентах) (номинально 1,75%, 1,09%, 0,58%, 0,29% и 0,15%) высевали на сельскохозяйственные поля вблизи Newark, DE и Donna, TX. Когда растения давали 5-й лист по меньшей мере длиной 10 см, его срезали. Для каждого процента брали один срезанный лист по меньшей мере с 16 растениями, и помещали в лунку с одной личинкой листопадного "походного червя" второй возрастной стадии. Смертность личинок регистрировали через 72 часа после инвазии.
Растения кукурузы на участке Donna измеряли для определения роста растения. Личинок заворачивали в цилиндр и регистрировали высоту от земли до самого дальнего кончика листа в цилиндре.
Смертность личинок от поедания 5-го листа кукурузы, при обработках семян соединением 502
Высота растения кукурузы при обработках семян соединением 502 в Donna, ТХ
Как видно из Таблицы Е2, обработка соединением 502 в этом тесте способствует росту растения.
ТЕСТ F
Партии семян кукурузы с покрытием из композиции соединения 855 номиналом 1 мг а.и. и 3 мг а.и., полученным в соответствии с методикой стадии 2 примера I, и для сравнения необработанные семена высаживали в небольшие террариумы со стерильной почвой Matapeake и выращивали в течение 14 дней. Через 14 дней одну партию растений испытывали в отношении Pereprinus maidis (цикады кукурузной), а другую в отношении Spodoptera frugiperda (листопадные «походные черви»).
Для испытания в отношении Р.maidis вокруг растения устанавливали цилиндр, и на растение помещали 15-20 нимф третьей возрастной стадии. Сверху цилиндр закрывали экраном для предотвращения выхода личинок, но с возможностью доступа воздуха. Растения выращивали в ростовой камере с 16-часовым световым периодом и 8-часовой фазой темноты при 20°С и относительной влажности 50%. Через шесть дней подсчитывали мертвых и живых личинок для определения процента смертности.
У второй группы растений кукурузы срезали второй лист, разрезали его на четыре равных части и каждую полученную часть листа помещали в отдельную лунку покрытых 16-луночных полупрозрачных пластиковых лотков в ростовой камере. На каждую часть листа помещали личинку S.frugiperda второй возрастной стадии (1 личинка на лунку), и через 48 часов после инфицирования определяли смертность насекомых.
Контроль цикады кукурузной и листопадных «походных червей» через 14 дней после посадки кукурузы с покрытием из композиций, включающих соединение 855
ТЕСТ G
Семена хлопчатника с покрытием из композиции соединения 855 номиналом 1 мг а.и. и 5 а.и., полученные в соответствии с методикой примера I (стадия 3), и для сравнения необработанные семена высаживали в горшки диаметром 10 см со стерильной почвой Matapeake и выращивали с 16-часовым световым периодом и 8- часовым периодом темной фазы при 25°С и относительной влажности 50%. Растения испытывали в отношении Aphis fossypii (тля хлопковая), Bemisia argentifolii (белокрылка silver), Heliothis virescens (листовертка табачная) и Frankliniella occidentalis (трипс пшеничный западный), когда первый истинный лист достигал в длину примерно 2 см.
Все растения имели две удаленные семядоли. Лист растений, испытываемый в отношении A.gossypii, заражали примерно 30 нимфами, которые переносили на растение с предварительно зараженного срезанного листа. Каждое растение окружали пластиковым цилиндром и закрывали тонкой бумагой. Через шесть дней подсчитывали количество живых и мертвых личинок для определения процента смертности. Другую группу растений заражали взрослыми особями В.argentifolii для кладки яиц на растения, горшки ограждали пластиковыми цилиндрами, закрытыми тонкой бумагой, для выдерживания взрослых особей в течение 24 часов при 28°С с 16 часовым световым периодом и 8 часовым периодом темной фазы и относительной влажности 50%. Взрослых насекомых удаляли и листья обследовали для проверки наличия кладки яиц. Спустя тринадцать дней после заражения лист удаляли и подсчитывали количество погибших и живых личинок на нижней стороне листа для определения процента смертности. Третью группу растений использовали для испытания в отношении H.virescens. Первый настоящий лист удаляли, разрезали пополам и помещали в лунку (один кусочек листа на лунку) 16-луночного полупразрачного пластикового лотка с личинкой Н.virescens второй возрастной стадии (1 личинка на лунку). Лотки выдерживали в ростовой камере с 16 часовым световым периодом и 8 часовым периодом темной фазы при 25°С и относительной влажности 50%. Смертность определяли через 96 часов после заражения.
Контроль тли хлопковой, белокрылки silver и листовертки табачной при использовании семян хлопчатника с покрытием из композиции, включающей соединение 855
ТЕСТ Н
Семена хлопчатника с покрытием композиции соединения 855 номиналом 1 мг а.и. и 5 мг а.и., полученные в соответствии с методикой примера I (стадия 3), и необработанные семена для сравнения высаживали в горшки диаметром 10 см со стерильной почвой Matapeake и выращивали с 16-часовым световым периодом и 8-часовым периодом темной фазы при 25°С и относительной влажности 50%. Растения испытывали, когда первый настоящий лист достигал длины примерно 2 см.
У растений хлопчатника, испытываемых в отношении F.occidentalis, удаляли семядоли и в горшках вокруг растений устанавливали пластиковые цилиндры. Каждое растение заражали 50 трипсами и цилиндры закрывали тонкой бумагой для удерживания насекомых внутри цилиндра. Через семь дней после внесения трипсов определяли повреждение растения для определения степени защиты растения в процентах. Растения срезали и пропитывали 70% этиловым спиртом для подсчитывания удаленных трипсов и определения эффективности соединения.
Контроль трипса пшеничного западного при использовании семян хлопчатника с покрытием композиции, включающей соединение 855
ТЕСТ I
Семена сахарной свеклы с покрытием композиции соединения 855 номиналом 1,25 мг а.и., полученные в соответствии с методикой примера I (стадия 4), и для сравнения необработанные семена высаживали в горшки диаметром 10 см со стерильной почвой Matapeake и выращивали в теплице. После формирования 5-го настоящего листа растения испытывали в отношении Aphis fabae (тля свекловичная) или Spodoptera exiqua (свекловичные «походные черви»).
Для испытания в отношении А.fabae листья заражали примерно 30 нимфами, которые переносили на растения с предварительно зараженного срезанного листа. Каждое растение окружали пластиковым цилиндром и закрывали тонкой бумагой. Через шесть дней подсчитывали количество живых и мертвых нимф для определения процента смертности.
Вторую группу растений заражали шестью личинками S.exigua третьей возрастной стадии. Каждое растение окружали пластиковым цилиндром. Через шесть дней определяли степень поражения растения по шкале 0-100 (0 = отсутствие повреждения; 100 = весь материал листа съеден). Все растения после заражения выдерживали в ростовой камере с 16-часовым световым периодом и 8-часовым периодом темной фазы при 25°С и относительной влажности 50%.
Контроль тли свекловичной или свекловичного «походного червя» при применении семян сахарной свеклы с покрытием композиции, включающей соединение 855
ТЕСТ J
Семена сои с покрытием из композиции соединения 855 номиналом 0,625 мг а.и. и 1,25 мг а.и., полученные в соответствии с методикой примера I (стадия 4), и необработанные семена для сравнения высаживали в горшки диаметром 10 см. Растения заражали Aphis alycines (тля соевая) на стадии развития третьего трилистника и выращивали в теплице. Примерно 30 личинок тли сои переносили на растение с предварительного зараженного срезанного листа. Каждое растение окружали пластиковым цилиндром и закрывали тонкой бумагой. Зараженные растения переносили в среду ростовой камеры с 16-часовым световым днем и 8-часовой темной фазой при 25°С и относительной влажности 50%. Через шесть дней подсчитывали количество живых и мертвых особей тли для определения эффективности, выраженной в процентах.
Контроль тли соевой семенами сои с покрытием композиции, включающей соединение 855
ТЕСТ К
Семена хлопка с покрытием композициями соединения 854 номиналом 0,5 мг а.и., 1 мг а.и. и 2 мг а.и., полученные как описано в примере J (стадия 2), и для сравнения необработанные семена вываживали в 10 см горшки с использованием стерильной почвы Matapeake и выращивали в условиях с 16-часовым световым днем и 8-часовой фазой темноты при 25°С и относительной влажности 50%. Растения испытывали на стадии развития второго или третьего настоящего листа. Их испытывали в отношении Aphis gossypii (тля хлопковая), Bemisia argentifolii (белокрылка silverleaf) и Frankliniella occodentalis (трипс пшеничный западный).
Растения, испытываемые в отношении A.gossypii, имели второй настоящий лист длиной примерно 2 см. Две семядоли и первый настоящий лист у растений были удалены. Оставшийся лист заражали примерно 30 нимфами A.gossypii, которые переносили с предварительно зараженного срезанного листа. Каждое растение окружали пластиковым цилиндром и закрывали тонкой бумагой. Через шесть дней подсчитывали количество живых и мертвых нимф для определения процента смертности.
При испытании в отношении В.argentifolii или F.occidentalis растения использовали на стадии развития настоящего третьего листа до длины примерно 2 см. Все нижние листья удаляли (семядоли и другие настоящие листья) и растения подразделяли на две группы. Первую случайную выборку растений помещали в садок со взрослыми особями В.Argentifolii на 24 часа для кладки яиц в условиях с 16-часовым световым днем и 8-часовой фазой темноты при 28°С и относительной влажности 50%. Взрослых особей насекомых удаляли и растения обследовали на наличие, по меньшей мере, 50 отложенных яиц на каждом листе. Через тринадцать дней после инвазии растений подсчитывали количество погибших и живых нимф на нижних сторонах листьев для вычисления процента смертности. У второй группы растений, испытываемых в отношении F.occodentali, вокруг растений устанавливали пластиковые цилиндры. Каждое растение заражали 25 личинками трипса и цилиндр закрывали тонкой бумагой для удерживания насекомых. Через семь дней после внесения трипсов растения срезали и пропитывали 70% этиловым спиртом для подсчета удаленных трипсов и вычисления эффективности соединения.
Контроль тли хлопчатника, белокрылки silverleaf и трипса пшеничного западного применением семян хлопчатника с покрытием композициями соединения 854
ТЕСТ L
Обработанные клубни картофеля высаживали ручным способом в полевых условиях в день обработки клубней. Оценку Leptinotarsa decemloneata (колорадский жук) проводили через 33 дня после посадки (ДПП). Посчитывали общее количество личинок на растение и определяли процент контроля. Через сорок четыре для после посадки записывали среднее количество растений, поврежденных Ostrinia nubilalis (мотылек кукурузный), и определяли процент контроля.
Контроль колорадского жука и мотылька кукурузного применением покрытия клубней картофеля композициям и соединения 855 или соединения 502
Описывается способ защиты сеянцев или растений, выросших из них, от насекомых путем контактирования посевного материала с биологически эффективным количеством соединения формулы I или его солью, приемлемой для сельского хозяйства,

где А и В представляют собой О; R1 представляет собой Н; R2 представляет собой Н; R3 представляет собой C1-С6 алкил; R4 представляет собой C1-С6 алкил или CN; R5 представляет собой Н или галоген; R6 представляет собой C1-С6 галогеналкил или галоген; R7 представляет собой пиридинил, замещенный от одного до трех заместителями, независимо выбранных из R9; R8 предоставляет собой Н; каждый R9 представляет собой независимо галоген. Описывается также композиция для контроля за насекомыми, покрывающая сеянец, содержащая в качестве активного агента биологически эффективное количество соединения формулы I по п.1 или его соли, пленкообразователь или адгезивный агент. Технический результат - способы обработки сеянцев эффективны для защиты от насекомых не только ростков, но и растений на более поздних стадиях роста. 2 н. и 14 з.п. ф-лы, 42 табл.

где А и В представляют собой О;
R1 представляет собой Н;
R2 представляет собой Н;
R3 представляет собой C1-С6 алкил;
R4 представляет собой C1-С6 алкил или CN;
R5 представляет собой Н, C1-С6 алкил или галоген;
R6 представляет собой C1-С6 галогеналкил или галоген;
R7 представляет собой пиридинил, замещенный R9;
R8 представляет собой Н;
каждый R9 представляет собой независимо галоген.
| СПОСОБ СВАРКИ ДЕТАЛЕЙ КОСВЕННОЙ ДУГОЙ | 0 |
|
SU289879A1 |
| ПРОИЗВОДНЫЕ 1-(2-ПИРИДИЛ) ПИРАЗОЛА ИЛИ ИХ КИСЛОТНО-АДДИТИВНЫЕ СОЛИ, СПОСОБ ПОЛУЧЕНИЯ ПРОИЗВОДНЫХ 1-(2-ПИРИДИЛ) ПИРАЗОЛА ИЛИ ИХ КИСЛОТНО-АДДИТИВНЫХ СОЛЕЙ (ВАРИАНТЫ), ИНСЕКТОАКАРИЦИДОНЕМАТОЦИДНАЯ КОМПОЗИЦИЯ И СПОСОБ БОРЬБЫ С НАСЕКОМЫМИ, КЛЕЩАМИ И НЕМАТОДАМИ | 1992 |
|
RU2088580C1 |

