Изобретение относится к области синтеза новых аналитических реагентов комплексообразующего типа и может быть использовано в области люминесцентного анализа, в частности для клинической диагностики объектов биогенного происхождения, а также в областях науки и техники, связанных с процессами комплексообразования ионов тяжелых металлов.
Известен способ флуоресцентно-спектроскопического определения биологически активных веществ, при осуществлении которого используются маркеры на основе комплексов лантаноидов, связанных с определяемыми веществами посредством полиами-нополикарбоновых кислот типа диэтилентриаминопентауксусной кислоты (ДТПА), обладающих высокими константами связывания ионов лантаноидов (>1018 М-1) (США, патент №4808541, класс НКИ 436/501).
Недостатком этих соединений является то, что они не обладают люминесцентными свойствами. Для детектирования связанных ионов лантаноидов в этом способе приходится перекомплексовывать их в другой хелат с люминесцирующим β-дикетоном (например, нафтоилтрифторацетоном (НТА)). Метод, известный под названием DELFIA, осуществляется в среде так называемого усиливающего раствора (УР), состоящего из буфера рН≤3,5, синергического агента триоктилфосфиноксида (ТОФО) и мицеллобразователя Тритона Х-100. Недостатком НТА является то, что он обладает невысокой константой связывания иона европия (Eu3+) (˜107 М-1) и не может быть использован для прямого мечения биологических структур. Для надежного выявления малых концентраций аналита (10-13-10-8 моль/л по Eu3+) НТА в УР используется в большом избытке (1,5·10-5 моль/л). Недостатком этого комплексообразователя является то, что максимум возбуждения люминесценции комплекса НТА - Eu3+находится при ˜340 нм, что требует применения дорогостоящих источников возбуждения типа лазера или импульсных газонаполненных ламп, а также кварцевой оптики в канале возбуждения.
Известны бифункциональные соединения на основе 2,6-дизамещенных пиридинов, образующие комплексы с ионами лантаноидов, и их использование в качестве ковалентных маркеров в анализах биоспецифических взаимодействий (США, патент №6127529, класс НКИ 534/10). Эти комплексы обладают большей стабильностью по сравнению с комплексами на основе полиаминополикарбоновых кислот и могут быть использованы при повышенных температурах, в электрофоретических условиях, в присутствии дополнительных комплексообразователей и т.п. Они обладают люминесцентными свойствами и могут быть применены для анализа in situ (например, в гомогенном иммуноанализе). Вместе с тем, из-за низкой квантовой эффективности этих маркеров чувствительность определения невысока (˜10-4 моль/л аналита в реакционной среде). В основном эти соединения применяются для прямого мечения биологических объектов ионом европия с последующим перекомплексованием их в УР (см., например, Clin.Chem.,v.46, 2000, рр.658-666). Максимум возбуждения комплексов Eu3+также не превышает 330 нм.
Известны флуоресцентные комплексы ионов лантаноидов на основе производных терпиридинов, обладающих высокими константами связывания (США, патент №5457186, класс НКИ 534/15). В отличие от комплексов на основе пиридиновых производных они обладают несколько улучшенными значениями квантовой эффективности, но таким же коротковолновым возбуждением (≤340 нм). Кроме того, стоимость этих реагентов достаточно высока, так как для достижения наибольшего эффекта используются дейтерированные производные.
Известны флуоресцентные комплексы ионов лантаноидов на основе β-дикарбонильных соединений с арильными заместителями, которые обладают максимумом возбуждения в области >360 нм (РСТ, заявка №04/040534, класс МКИ C 07 D 213/46), и их применение для проведения флуоресцентного иммуноанализа с временным разрешением. Это позволяет использовать для их возбуждения дешевые источники излучения, например светодиоды. Наиболее эффективными при проведении иммуноанализа эти соединения оказались при включении в состав гидрофобных латексных частиц, где они защищены от тушащего действия воды и диссоциации их комплекса с Eu3+. Однако прямое мечение биологических молекул этими соединениями не может обеспечить высокой чувствительности определения вследствие того, что эти соединения, также как и НТА, обладают низкой дентатностью и невысокими константами связывания ионов лантаноидов.
Наиболее близким к заявляемым соединениям по технической сущности является реагент для мечения на основе комплексов ионов лантаноидов с бис-β-дикарбонильными структурами, содержащими также хромофорную (ароматическую) и электроноакцепторную (фторзамещенные алкильные группы) части (США, патент №6166251, класс НКИ 562/828). Эти комплексы обладают высокой интенсивностью флуоресценции, более чем в 10 раз превышающей интенсивность флуоресценции реагентов известных систем (LKB). Комплексы на основе бис-β-дикарбонильных структур синтезируются с высоким выходом и дешевы, позволяют проводить измерения на твердой фазе и в жидкости, так как вследствие достаточно высокой константы стабильности не требуется их перекомплексовывания. Недостатком указанных соединений является то, что максимум длины волны возбуждения комплексов также не превышает 340 нм.
Задачей является создание соединений, способных к образованию стабильных комплексов с ионами металлов с константами связывания не менее 1011 М-1.
Техническим результатом, достигаемым при использовании заявляемых соединений, являются их улучшенные люминесцентно-спектральные характеристики при образовании комплексов с ионами тяжелых металлов, что позволяет проводить чувствительное обнаружение как самих ионов, так и других аналитов, маркированных, например, комплексами этих соединений с ионами лантаноидов. В частности, некоторые из заявляемых соединений образуют стабильные длительно люминесцирующие (времена жизни люминесценции τ порядка 600-900 мкс) комплексы с ионами Eu3+ при возбуждении в области ≥360 нм.
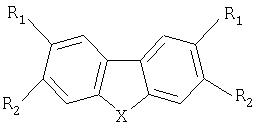
Технический результат достигается предлагаемыми соединениями, представляющими собой комплексообразующие дибензосодержащие пятичленные циклические соединения, содержащие два симметричных β-дикарбонильных заместителя с фторированными радикалами, общей формулы
 ,
,
где X - CH2, O, S, NR,
если Х - СН2, то R1=Н, R2=C(O)CH2C(O)Rf,
где Rf=(CF2)nY, n=1÷6,
при n=1 Y=Н, F, CI, Br, OCF3, ОСН3, OC3F7, OCF2CF2OCF3, С6F5, C(O)OAlk,
где Alk - углеводородный радикал,
при n=2 Y=Н, F, CI, Br, OCF3, ОС3F7, OCF2CF2OCF3, C(O)OAlk,
при n=3, 5 или 6 Y=F, C(O)OAlk,
при n=4 Y=Н, F, CI, C(O)OAlk;
если X - O, S или NR, то R=Н или Alk, R2=Н, R1=С(O)СН2С(O)Рf.
Кроме того, технический результат достигается тем, что заявляемые соединения образуют комплексы с ионами лантаноидов, в частности Eu3+.
Авторам не известны комплексообразующие дибензосодержащие пятичленные циклические соединения, содержащие два симметричных фторированных β-дикарбонильных заместителя. Кроме того, авторам не известны комплексы фторированных β-дикарбонильных соединений с ионами лантаноидов, которые при использовании, например, в условиях проведения анализа биоспецифического связывания обеспечивают указанный выше технический результат. Следовательно, заявляемое техническое решение удовлетворяет критерию "Новизна".
Известны технические решения, в которых использованы соединения, содержащие β-дикарбонильные заместители и различные ароматические хромофоры. Соединение, описанное в патенте США №6166251, класс НКИ 562/828, обеспечивает получение комплексов, обладающих достаточно высокими константами связывания Eu3+, но коротковолновым (≤340 нм) возбуждением. Соединение, описанное в заявке РСТ №04/040534, МКИ C 07 D 213/46, обеспечивает получение комплексов с Eu3+, обладающих возбуждением в области ≥360 нм, но не обеспечивает получение высокостабильных комплексов. Заявляемые соединения позволяют получать комплексы Eu3+ с высокими константами связывания (≥1011 М-1) и интенсивной длительной люминесценцией при возбуждении в области длин волн ≥360 нм. Следовательно, предлагаемое техническое решение соответствует критерию "Уровень техники".
Заявляемые соединения и их комплексы с ионами лантаноидов могут найти применение в качестве маркеров и усилителей в различных способах анализа с использованием флуориметрии с временным разрешением люминесценции. В области клинической диагностики они могут быть использованы для определения белков, гормонов, токсинов, нуклеиновых кислот и других объектов биогенного происхождения (например, микроорганизмов и вирусов), являющихся участниками биоспецифических взаимодействий. В области аналитической химии заявляемые реагенты могут быть применены для обнаружения редкоземельных элементов, их концентрирования и выделения, в том числе в хроматографии. В области техники они могут быть применены в качестве экстрагентов ионов тяжелых и редкоземельных металлов с целью их извлечения и/или очистки от их примесей сточных и контурных вод. Следовательно, заявляемое техническое решение соответствуют критерию "Промышленная применимость".
В общем случае заявляемые бис-β-дикарбонильные соединения получаются конденсацией Кляйзена (схема 1), хотя созданию каждого типа тетракетонов соответствуют свои оптимальные параметры: конденсирующий агент, щелочной реагент, условия реакции, стехиометрия компонентов и т.п.

В качестве конденсирующего агента можно использовать сложные эфиры и ангидриды, галогенангидриды алифатических кислот в присутствии трифторида бора, предпочтительно, однако, применять метиловые эфиры фторсодержащих карбоновых кислот.
В качестве щелочных реагентов можно применять этилат натрия, метилат натрия, металлический натрий, амид натрия, гидриды натрия и лития. Если бис-β-дикарбонильные соединения получаются в присутствии алкоголятов натрия с удовлетворительными выходами, то следует использовать именно эти реагенты, так как они менее опасны в обращении, чем металлический натрий или гидриды и менее способны к образованию побочных продуктов, чем металлический натрий или амид натрия. Предпочтительно применять в качестве реагентов метиловые эфиры фторсодержащих карбоновых кислот и метилат натрия.
В качестве растворителей при осуществлении конденсации Кляйзена используются любые инертные растворители, не меняющиеся в условиях реакции. К ним в частности принадлежат ациклические и циклические простые эфиры, ациклические и циклические углеводороды, апротонные биполярные растворители. Предпочтение отдается одному из вышеперечисленных растворителей в зависимости от конкретно взятого исходного диацетильного соединения.
При проведении реакции предпочтительным оказалось следующее соотношение компонентов: диацетильное соединение - конденсирующий агент - щелочной реагент как 1:2:2, хотя оно может изменяться в каждом конкретном случае.
Температура реакции может колебаться от -5°С до температуры кипения растворителя, однако предпочтительно от -5 до +40°С.
Реакцию можно проводить как при атмосферном, так и при повышенном или пониженном давлении, однако обычно работают при атмосферном давлении.
Заявляемые дибензосодержащие пятичленные циклические соединения, содержащие β-дикарбонильные заместители приведенной выше общей формулы, представляют собой индивидуальные химические соединения ряда бис-β-дикетонов, в которых в качестве хромофорного фрагмента выступает флуоренильный, либо дибензогетероциклический пятичленный фрагмент. В каждом из них присутствуют две симметричные β-дикарбонильные группировки (положение 2 и 7 у флуорена и положения 3 и 6 у дибензогетероциклического соединения), ковалентно связанные с фторсодержащими фрагментами, которые обеспечивают сдвиг кето-енольного равновесия в сторону енола и за счет этого облегчение образования координационной связи с ионами редкоземельных элементов. В сравнении с НТА и другими известными моно-β-дикарбонильными соединениями все предлагаемые соединения образуют комплексы повышенной стабильности, так как содержат четыре, а не две карбонильные группы в жестко фиксированных положениях двух ароматических циклов. Максимум возбуждения люминесценции комплексов предлагаемых соединений с Eu3+ варьирует, в зависимости от структуры, в интервале 340-398 нм. При этом комплексы веществ ряда флуорена имеют максимумы возбуждения ≥360 нм, а ряда карбазола ≥380 нм, что является существенным преимуществом в сравнении с известными бис-β-дикарбонильными соединениями и позволяет проводить анализ биоспецифического связывания с применением экономичных источников возбуждения люминесценции на основе светоэмиттирующих диодов или диодных лазеров.
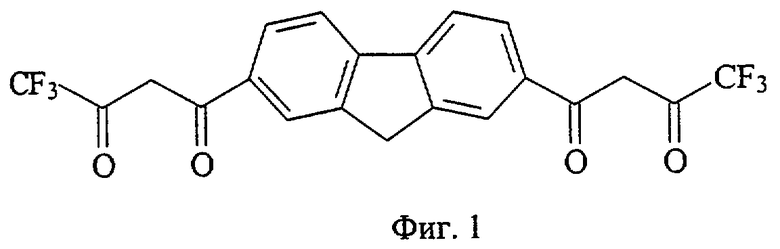
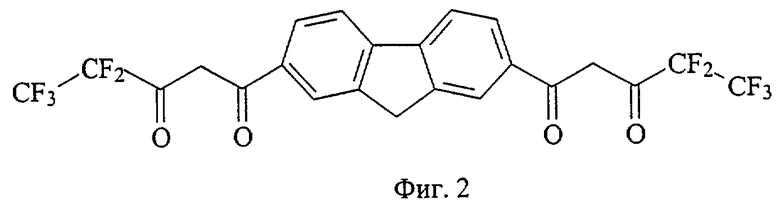
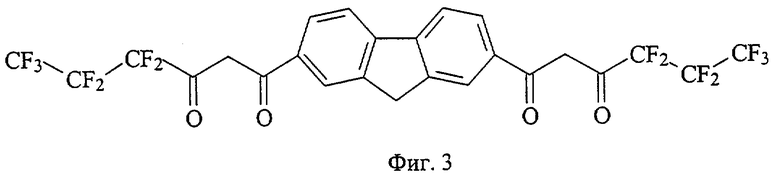
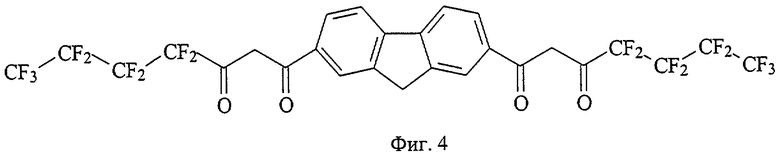
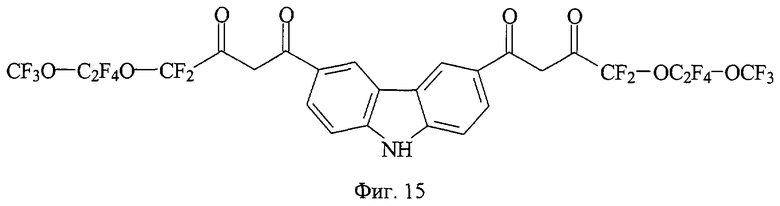
На фиг. 1 приведена структурная формула соединения 4,4,4-трифтор-1-[7-(4,4,4-трифтор-3-оксобутаноил)-9H-флуорен-2-ил]-1,3-бутандиона (а), на фиг. 2 - 4,4,5,5,5-пентафтор-1-[7-(4,4,5,5,5-пентафтор-3-оксопентаноил)-9H-флуорен-2-ил]-1,3-пентандиона (б), на фиг. 3 - 4,4,5,5,6,6,6-гептафтор-1-[7-(4,4,5,5,6,6,6-гептафтор-3-оксогексаноил)-9H-флуорен-2-ил]-1,3-гександиона (в), на фиг. 4 - 4,4,5,5,6,6,7,7,7-нонафтор-1-[6-(4,4,5,5,6,6,7,7,7-нонафтор-3-оксогептаноил)-9H-флуорен-3-ил]-1,3-гептандиона (г), на фиг. 5 - 4,4,4-трифтор-1-[6-(4,4,4-трифтор-3-оксобутаноил)-9H-карбазол-3-ил]-1,3-бутандиона (д), на фиг. 6 - 4,4,5,5,5-пентафтор-1-[6-(4,4,5,5,5-пентафтор-3-оксопентаноил)-9H-карбазол-3-ил]-1,3-пентандиона (е), на фиг. 7 - 4,4,5,5,6,6,6-гептафтор-1-[6-(4,4,5,5,6,6,6-гептафтор-3-оксобутаноил)-9H-карбазол-3-ил]-1,3-гександиона (ж), на фиг. 8 - 1-[9-этил-6-(4,4,4-трифтор-3-оксобутаноил)-9H-карбазол-3-ил]-4,4,4-трифтор-1,3-бутандиона (з), на фиг. 9 - 4,4,4-трифтор-1-[8-4,4,4-трифтор-3-оксобутаноил)дибензо[b,d] тиофен-2-ил]-1,3-бутандиона (и), на фиг. 10 - 4,4,4-трифтор-1-[6-(4,4,4-трифтор-3-оксобутаноил)дибензо[b,d]фуран-4-ил]-1,3-бутандиона (к), на фиг. 11 - 1-{6-[4,4-дифтор-3-оксо-4-(трифторметокси)бутаноил]-9H-карбазол-3-ил}-4,4-дифтор-4-(трифторметокси)-1,3-бутандиона (л), на фиг. 12 - 4,4-дифтор-1-[6-(4,4-дифтор-3-оксобутаноил)-9H-карбазол-3-ил]-1,3-бутандиона(м), на фиг. 13 - метил-10-[7-(4,4,5,5,6,6,7,7,8,8,9,9-додекафтор-10-метокси-3,10-диоксодеканоил)-9H-флуорен-2-ил]-2,2,3,3,4,4,5,5,6,6,7,7-додекафтор-8,10-диоксодеканоата (н), на фиг. 14 - метил-10-[6-(4,4,5,5,6,6,7,7,8,8,9,9-додекафтор-10-метокси-3,10-диоксодеканоил)-9H-карбазол-3-ил]-2,2,3,3,4,4,5,5,6,6,7,7-додекафтор-8,10-диоксодеканоата (о), на фиг. 15 - 1-{6-[4,4-дифтор-4-[1,1,2,2-тетрафтор-2-(трифторметокси)этоксибутаноил]-9Н-карбазол-3-ил}-4,4-дифтор-4-[1,1,2,2-тетрафтор-2- (трифторметокси)этокси]-1,3-бутандиона (п), на фиг. 16 - 2,2-дифтор-2-(2,3,4,5,6-пентафторфенил)-1-[6-2,2-дифтор-2-(2,3,4,5,6-пентафторфенил-3-оксобутаноил)-9Н-карбазол-3-ил]-1,3-бутандиона (р), на фиг. 17 - общая структурная формула заявляемых соединений, на фиг. 18 приведена зависимость интенсивности люминесценции комплексов заявляемых соединений с Eu3+ стехиометрического состава 1:1 от концентрации комплекса, на фиг. 19 приведены результаты иммуноанализа тиреотропного гормона человека по протоколу DELFIA с использованием в УР заявляемых соединений и НТА.
Примеры синтеза заявляемых соединений
Пример 1
Синтез 4,4,4-трифтор-1-[7-(4,4,4-трифтор-3-оксобутаноил)-9H-флуорен-2-ил]-1,3-бутандиона (а)
39 ммоль металлического натрия растворяют в 25 мл сухого метанола, затем реакционную массу концентрируют досуха при остаточном давлении 16 Торр в течение 1 часа при 95°С. Прибавляют 80 мл сухого диэтилового эфира и в течение 10 минут 39 ммоль раствора метилового эфира трифторуксусной кислоты, синтезированного по модифицированному методу A.L.Henne and M.S.Newman, J.Amer.Chem.Soc., 1947, v.69, p.1819-1831, в 15 мл этилового эфира, затем 3,9 ммоль 2,7-диацетилфлуорена, синтезированого по модифицированному методу F.E.Ray and G.RieveshI «Organic Synthesis. An Annual Publication of Satisfactory Methods for the Preparation of Organic Chemicals», J.Wiley & Sons, 1948, v.28, p.3-14. Полученную суспензию кипятят при перемешивании в течение 31 часа, охлаждают до комнатной температуры, прибавляют 80 мл диэтилового эфира и фильтруют. Фильтрат обрабатывают раствором 11,5 г серной кислоты в 46 мл дистиллированной воды. Органический слой отделяют и промывают 4 раза по 30 мл дистиллированной водой, фильтруют, фильтрат выдерживают при остаточном давлении 20 Торр в течение 1 часа при 40°С. Сухой остаток дробно высаживают 25+80 мл гептана из 30 мл смеси диэтиловый эфир:этилацетат (1:1) и получают тетракетон а (фиг.1) - светло-желтое кристаллическое вещество с Тпл=210°С. Выход целевого продукта 0,98 ммоль (25,0%).
1Н-ЯМР: 4.11 (с, 4Н, 2СН2); 6.68 (с, 2Н, 2=СН); 7.99 (д, 2Н, 2=СН, J=10); 8.08 (д, 2Н, 2=СН, J=10); 8.21 (с, 2Н, =СН).
19F-ЯМР: 3.36 (с, 6F, 2CF3).
MB=442,31 (C21H12F6O4)
Элементный анализ: найдено С 56,95%, Н 2,77%; вычислено С 56,97%, Н 2,71%.
Пример 2
Синтез 4,4,5,5,5-пентафтор-1-[7-(4,4,5,5,5-пентафтор-3-оксопентаноил)-9H-флуорен-2-ил]-1,3-пентандиона (б)
45 ммоль металлического натрия растворяют в 30 мл сухого метанола, затем реакционную массу концентрируют досуха при остаточном давлении 18 Торр в течение 1 часа при 95°С. Прибавляют 100 мл сухого диэтилового эфира и в течение 10 минут раствор 45 ммоль метилового эфира пентафторпропионовой кислоты, синтезированного по аналогии с примером 1, в 15 мл этилового эфира, затем 4,7 ммоль диацетилфлуорена, синтезированого по аналогии с примером 1. Полученную суспензию кипятят при перемешивании в течение 43 часов, охлаждают до комнатной температуры, прибавляют 60 мл диэтилового эфира и фильтруют. Фильтрат обрабатывают раствором 15,5 г серной кислоты в 52 мл дистиллированной воды. Органический слой отделяют и промывают 4 раза по 30 мл дистиллированной водой, фильтруют, фильтрат выдерживают при остаточном давлении 15 Торр в течение 1 часа при 40°С. Сухой остаток дробно высаживают 25+80 мл гептаном из 30 мл смеси диэтиловый эфир:этилацетат (1:1) и получают тетракетон б (фиг.2) - светло-желтое кристаллическое вещество с Тпл=226-230°С. Выход целевого продукта 1,32 ммоль (28,0%).
1Н-ЯМР: 3.93 (с, 2Н, СН2); 4.10 (с, 4Н, 2СН2); 7.64 (д, 2Н, 2=СН, J=10); 7.92 (с, 2Н, =СН); 8.08 (д, 2Н, 2=СН, J=10).
19F-ЯМР: 4.91 (т, 6F, 2CF3); 45.3 (м, 4F, 2CF2).
МВ=542, 23 (C23H12F10O4)
Элементный анализ: найдено С 50,88%, Н 2,24%; вычислено С 50,90%, Н 2,21%.
Пример 3
Синтез 4,4,5,5,6,6,6-гептафтор-1-[7-(4,4,5,5,6,6,6-гептафтор-3-оксогексаноил)-9Н-флуорен-2-ил]-1,3-гександиона (в)
18 ммоль металлического натрия растворяют в 10 мл сухого метанола, затем реакционную массу концентрируют досуха при остаточном давлении 18 Торр в течение 1 часа при 95°С. Прибавляют 30 мл сухого диэтилового эфира и в течение 10 минут раствор 18 ммоль метилового эфира пентафторпропионовой кислоты, синтезированного по аналогии с примером 1, в 10 мл этилового эфира, затем 6 ммоль диацетилфлуорена, синтезированого по аналогии с примером 1. Полученную суспензию кипятят при перемешивании в течение 43 часов, охлаждают до комнатной температуры, прибавляют 60 мл диэтилового эфира и фильтруют. Фильтрат обрабатывают раствором 5,3 г серной кислоты в 22 мл дистиллированной воды. Органический слой отделяют, осадок промывают 3 раза по 5 мл диэтиловым эфиром, сушат и получают тетракетон в (фиг.3) - светло-желтое кристаллическое вещество с Тпл=166-167°С. Выход целевого продукта 1,58 ммоль (26,4%).
1Н-ЯМР: 3.97 (с, 2Н, СН2); 4.10 (с, 4Н, 2СН2); 7.71 (д, 2Н, 2=СН, J=10); 8.05 (с, 2Н, =СН); 8.11 (д, 2Н, 2=СН, J=10).
19F-ЯМР: 4.94 (т, 6F, 2CF3); 46.01 (м, 4F, 2CF2); 51.20 (ушс, 4F, 2CF2).
МВ=642, 34 (C25H12F14O4)
Элементный анализ: найдено С 46,68%, Н 1,89%; вычислено С 46,70%, Н 1,87%.
Пример 4
Синтез 4,4,5,5,6,6,7,7,7-нонафтор-1-[6-(4,4,5,5,6,6,7,7,7-нонафтор-3-оксогептаноил)-9H-флуорен-3-ил]-1,3-гептандиона (г)
26 ммоль металлического натрия растворяют в 15 мл сухого метанола, затем реакционную массу концентрируют досуха при остаточном давлении 20 Торр в течение 3 часов при 95°С. Прибавляют 30 мл сухого диэтилового эфира и в течение 10 минут раствор 26 ммоль метилового эфира перфторвалериановой кислоты, синтезированного по аналогии с примером 1, в 20 мл диэтилового эфира, затем 13 ммоль диацетилфлуорена, синтезированого по аналогии с примером 1. Полученную суспензию кипятят при перемешивании в течение 4,5 часов, охлаждают до комнатной температуры, обрабатывают раствором 5,3 г концентрированной серной кислоты в 22 мл дистиллированной воды. Органический слой отделяют, фильтруют, осадок промывают 3 раза по 5 мл диэтиловым эфиром, сушат и получают тетракетон г (фиг.4) - светло-желтое кристаллическое вещество с Тпл=141-143°С. Выход целевого продукта 3,22 ммоль (24,8%).
1Н-ЯМР: 3.96 (с, 2Н, СН2); 4.11 (с, 4Н, 2СН2); 7.82 (д, 2Н, 2=СН, J=10); 8.13 (с, 2Н, =СН); 8.21 (д, 2Н, 2=СН, J-10).
19F-ЯМР: 5.04 (т, 6F, 2CF3); 48.7 (м, 4F, 2CF2); 50.7 (м, 4F, 2CF2); 54.6 (ушс, 4F, 2CF2).
МВ=742, 35 (C27H12F18O4)
Элементный анализ: найдено С 43,61%, Н 1,59%; вычислено С 43,65%, Н 1,62%.
Пример 5
Синтез 4,4,4-трифтор-1-[6-(4,4,4-трифтор-3-оксобутаноил)-9H-карбазол-3-ил]-1,3-бутандиона (д)
39 ммоль металлического натрия растворяют в 25 мл сухого метанола, затем реакционную массу концентрируют досуха при остаточном давлении 16 Торр в течение 1 часа при 95°С. Прибавляют 80 мл сухого диэтилового эфира и в течение 10 минут раствор 39 ммоль метилового эфира трифторуксусной кислоты, синтезированного по аналогии с примером 1, в 15 мл этилового эфира, затем 3,9 ммоль диацетилкарбазола, синтезированого по модифицированному методу Plant and Rogers, J.Chem.Soc., 1935, v.65, p.741-752. Полученную суспензию кипятят при перемешивании в течение 31 часа, охлаждают до комнатной температуры, прибавляют 80 мл диэтилового эфира и фильтруют. Фильтрат обрабатывают раствором 11,5 г серной кислоты в 46 мл дистиллированной воды. Органический слой отделяют и промывают 4 раза по 30 мл дистиллированной водой, фильтруют, фильтрат выдерживают при остаточном давлении 20 Торр в течение 1 часа при 40°С. Сухой остаток дробно высаживают 25+80 мл гептана из 30 мл смеси диэтиловый эфир:этилацетат (1:1) и получают тетракетон д (фиг.5) - желтовато-зеленое кристаллическое вещество с Тпл=260°С (с разложением). Выход целевого продукта 1,12 ммоль (28,7%).
1Н-ЯМР: 7.1 (с, 2Н, =СН); 7.6 (д, 2Н,=СН, J=11.4); 8.2 (д, 2Н, 2=СН, J=11.4); 9.2 (с, 2Н, 2=СН); 12.4 (ушс, 1Н, NH).
19F-ЯМР: 3.3 (с, 6F, 2CF3).
МВ=443,30 (C20H11F6NO4)
Элементный анализ: найдено С 54,18%, Н 2,50%, N 3,18%; вычислено С 54,14%, Н 2,48%, N 3,16%.
Пример 6
Синтез 4,4,5,5,5-пентафтор-1-[6-(4,4,5,5,5-пентафтор-3-оксопентаноил)-9H-карбазол-3-ил]-1,3-пентандиона (е)
78 ммоль металлического натрия растворяют в 50 мл сухого метанола, затем реакционную массу концентрируют досуха при остаточном давлении 19 Торр в течение 1,5 часа при 95°С. Прибавляют 120 мл сухого диэтилового эфира и в течение 10 минут раствор 78 ммоль метилового эфира пентафторпропионовой кислоты, синтезированного по аналогии с примером 1, в 30 мл этилового эфира, затем 7,9 ммоль диацетилкарбазола, синтезированного по аналогии с примером 5. Полученную суспензию кипятят при перемешивании в течение 42 часов, охлаждают до комнатной температуры, прибавляют 120 мл диэтилового эфира и фильтруют. Фильтрат обрабатывают раствором 19,5 г серной кислоты в 78 мл дистиллированной воды. Органический слой отделяют и промывают 4 раза по 30 мл дистиллированной водой, фильтруют, фильтрат выдерживают при остаточном давлении 25 Торр в течение 2 часов при 45°С. Сухой остаток дробно высаживают 25+80 мл гептана из 50 мл смеси диэтиловый эфир:этилацетат (1:1) и получают тетракетон е (фиг.6) - желтовато-зеленое кристаллическое вещество с Tпл=248°C. Выход целевого продукта 2,32 ммоль (29,4%).
1Н-ЯМР: 7.14 (с, 2Н, =СН); 7.7 (д, 2Н, =СН, J=11.4); 8.3 (д, 2Н, 2=СН, J=11.4); 9.24 (с, 2Н, 2=СН); 12.3 (ушс, 1Н, NH).
19F-ЯМР: 4.78 (т, 6F, 2CF3); 45.1 (м, 4F, 2CF2).
МВ=543,31 (C22H11F10NO4)
Элементный анализ: найдено С 48,56%, Н 2,23%, N 2,60; вычислено С 48,59%, Н 2,21%, N 2,58.
Пример 7
Синтез 4,4,5,5,6,6,6-гептафтор-1-[6-(4,4,5,5,6,6,6-гептафтор-3-оксобутаноил)-9H-карбазол-3-ил]-1,3-гександиона (ж)
39 ммоль металлического натрия растворяют в 25 мл сухого метанола, затем реакционную массу концентрируют досуха при остаточном давлении 16 Торр в течение 1 часа при 95°С. Прибавляют 80 мл сухого диэтилового эфира и в течение 10 минут раствор 39 ммоль метилового эфира перфтормасляной кислоты, синтезированного по аналогии с примером 1, в 15 мл этилового эфира, затем 3,9 ммоль диацетилкарбазола, синтезированного по аналогии с примером 5. Полученную суспензию кипятят при перемешивании в течение 45 часов, охлаждают до комнатной температуры, прибавляют 90 мл диэтилового эфира и фильтруют. Фильтрат обрабатывают раствором 11,5 г серной кислоты в 46 мл дистиллированной воды. Органический слой отделяют и промывают 4 раза по 30 мл дистиллированной водой, фильтруют, фильтрат выдерживают при остаточном давлении 20 Торр в течение 1 часа при 40°С. Сухой остаток дробно высаживают 25+80 мл гептана из 30 мл смеси диэтиловый эфир:этилацетат (1:1) и получают тетракетон ж - зеленовато-желтое кристаллическое вещество с Тпл=238°С. Выход целевого продукта 1,22 ммоль (31,2%).
1Н-ЯМР: 7.19 (с, 2Н, =СН); 7.81 (д, 2Н,=СН, J=11.4); 8.44 (д, 2Н, 2=СН, J=11.4); 9.27 (с, 2Н, 2=СН); 12.4 (ушс, 1Н, NH).
19F-ЯМР: 4.81 (т, 6F, 2CF3); 45.3 (м, 4F, 2CF2); 49.8 (м, 4F, 2CF2).
MB=643,33 (C24H11F14NO4)
Элементный анализ: найдено С 44,80%, Н 1,73%, N 2,20%; вычислено С 44,77%, H 7,71%, N 2,18%.
Пример 8
Синтез 1-[9-этил-6-(4,4,4-трифтор-3-оксобутаноил)-9H-карбазол-3-ил]-4,4,4-трифтор-1,3-бутандиона (з)
Синтез 1 -(6-ацетил-9-этил-9H-карбазол-3-ил)-1-этанона
К суспензии 0,09 моль хлорида алюминия и 40 мл дихлорметана прибавляют (0,05 моль) хлористого ацетила, охлаждают до 0,5°С. Прибавляют раствор 0,02 моль 9-этил-9H-карбазола, синтезированого по модифицированному методу Meitsener, J. Am. Chem. Soc., 1935, v.57, p.2327-2334, в 25 мл дихлорметана, поддерживая температуру смеси +2 - +4°С. Перемешивание продолжают 23 часа. Реакционную массу переносят в смесь 50 мл концентрированной соляной кислоты и 200 г колотого льда, греют до таяния льда и сепарируют. Водный слой промывают 3 раза по 20 мл дихлорметаном. Органические слои объединяют и промывают 8 раз по 50 мл водой до нейтральной реакции, сушат и упаривают. Твердый остаток кристаллизуют из 108 мл этанола с применением активированного угля и получают диацетил-N-этилкарбазол.
106 ммоль металлического натрия растворяют в 25 мл сухого метанола, затем реакционную массу концентрируют досуха при остаточном давлении 25 Торр в течение 3 часов при 95°С. Прибавляют 60 мл сухого диэтилового эфира и в течение 10 минут раствор 106 ммоль метилового эфира трифторуксусной кислоты, синтезированного по аналогии с примером 1, в 15 мл этилового эфира, затем прибавляют 22 ммоль диацетил-N-этилкарбазола. Полученную суспензию кипятят при перемешивании в течение 18 часов, охлаждают до комнатной температуры, обрабатывают раствором 16,2 г концентрированной серной кислоты в 85 мл дистиллированной воды. Органический слой отделяют и промывают дистиллированной водой 3 раза по 25 мл, фильтруют, выдерживают при остаточном давлении 15 Торр в течение 2 часов при 85°С. Сухой остаток перекристалли-зовывают из этилацетата (45 мл) и получают тетракетон з (фиг.8) - зеленовато-желтое кристаллическое вещество с Тпл=203-206°С. Выход целевого продукта 6,14 ммоль (27,9%).
1Н-ЯМР: 1.58 (т, 3Н, СН3); 4.21 (м, 2Н, СН2); 7.1 (с, 2Н,=СН); 7.62 (д, 2Н,=СН, J=11.4); 8.23 (д, 2Н, 2=СН, J=11.4); 9.16 (с, 2Н, 2=СН).
19F-ЯМР: 3.32 (с, 6F, 2CF3).
МВ=471, 35 (C22H15F6NO4)
Элементный анализ: найдено С 55,97%, Н 3,20%, N 2,99%; вычислено С 56,01%, H 3,18%, N 2,97%.
Пример 9
Синтез 4,4,4-трифтор-1-[8-(4,4,4-трифтор-3-оксобутаноил)дибензо[b,d]тиофен-2-ил]-1,3-бутандиона (и)
53 ммоль металлического натрия растворяют в 15 мл сухого метанола, затем реакционную массу концентрируют досуха при остаточном давлении 35 Торр в течение 3 часов при 95°С. Прибавляют 80 мл сухого диэтилового эфира и в течение 10 минут раствор 53 ммоль метилового эфира трифторуксусной кислоты, синтезированного по аналогии с примером 1, в 15 мл этилового эфира, затем прибавляют 11 ммоль 1-(8-ацетилдибензо[b,d]тиофен-2-ил)-1-этанона, синтезированого по модифицированному методу W.LAlbrecht and D.H.Gustafson, J. Org. Chem., 1972, v.21, p.3355-3362. Полученную суспензию кипятят при перемешивании в течение 16 часов, охлаждают до комнатной температуры, обрабатывают раствором 7,8 г концентрированной серной кислоты в 45 мл дистиллированной воды. Органический слой отделяют и промывают 3 раза по 15 мл дистиллированной водой, фильтруют, выдерживают при остаточном давлении 15 Торр в течение 2 часов при 85°С. Сухой остаток перекристаллизовывают из этилацетата (25 мл) и получают тетракетон и (фиг.9) - 1 зеленовато-желтое кристаллическое вещество с Тпл=203-206°С. Выход целевого продукта 3,4 ммоль (30,9%).
1Н-ЯМР: 6.6 (с, 2Н, 2=СН); 7.74 (д, 2Н,=СН, J=10.3); 7.71 (с, 2Н, 2=СН); 8.34 (д, 2H, 2=CH, J=10.3).
19F-ЯМР: 3.34 (с, 6F, 2CF3).
МВ=460,35 (C20H10F6O4S)
Элементный анализ: найдено С 52,11%, Н 2,19%; вычислено С 52,14%, Н 2,17%.
Пример 10
Синтез 4,4,4-трифтор-1-[6-(4,4,4-трифтор-3-оксобутаноил)дибензо[b,d(]фуран-4-ил]-1,3-бутандиона (к)
106 ммоль металлического натрия растворяют в 40 мл сухого метанола, затем реакционную массу концентрируют досуха при остаточном давлении 25 Торр в течение 3 часов при 95°С. Прибавляют 150 мл сухого диэтилового эфира и в течение 20 минут раствор 106 ммоль метилового эфира трифторуксусной кислоты, синтезированного по аналогии с примером 1, в 40 мл этилового эфира, затем прибавляют 23 ммоль 1-(8-ацетилдибензо[b,d]фуран-2-ил)-1-этанона, синтезированного по модифицированному методу W.M.Whaley and C.White, J. Org. Chem., 1953, v.18, p.309-322. Полученную суспензию кипятят при перемешивании в течение 18 часов, охлаждают до комнатной температуры, обрабатывают раствором 13,8 г концентрированной серной кислоты в 65 мл дистиллированной воды. Органический слой отделяют и промывают 4 раза по 15 мл дистиллированной водой, фильтруют, выдерживают при остаточном давлении 13 Торр в течение 2 часов при 95°С. Сухой остаток перекристаллизовывают из 45 мл этилацетата и получают тетракетон к (фиг.10) - желтое кристаллическое вещество с Tпл=234°C. Выход целевого продукта 7,18 ммоль (31,2%).
1Н-ЯМР: 6.7 (с, 2Н, =СН); 7.7 (д, 2Н,=СН, J=10.3); 7.7 (с, 2Н, 2=СН); 8.2 (д, 2Н, 2=CH, J=10.3).
19F-ЯМР: 3.3 (с, 6F, 2CF3).
МВ=444,28 (С20Н10F6O5)
Элементный анализ: найдено С 54,06%, Н 2,28%; вычислено С 54,02%, Н 2,25%.
Пример 11
Синтез 1 - {6-[4,4-дифтор-3-оксо-4-(трифторметокси)бутаноил]-9H-карбазол-3-ил}-4,4-дифтор-4-(трифторметокси)-1,3-бутандиона (л)
39 ммоль металлического натрия растворяют в 25 мл сухого метанола, затем реакционную массу концентрируют досуха при остаточном давлении 16 Торр в течение 1 часа при 95°С. Прибавляют 80 мл сухого диэтилового эфира и в течение 10 минут раствор 39 ммоль метилового эфира трифторметоксиперфторуксусной кислоты, в 15 мл этилового эфира, затем 3,9 ммоль диацетилкарбазола, синтезированого по модифицированному методу Plant and Rogers, J.Chem.Soc., 1935, v.65, p.741-752. Полученную суспензию кипятят при перемешивании в течение 31 часа, охлаждают до комнатной температуры, прибавляют 80 мл диэтилового эфира и фильтруют. Фильтрат обрабатывают раствором 11,5 г серной кислоты в 46 мл дистиллированной воды. Органический слой отделяют и промывают 4 раза по 30 мл дистиллированной водой, фильтруют, фильтрат выдерживают при остаточном давлении 20 Торр в течение 1 часа при 40°С. Сухой остаток дробно высаживают 25+80 мл гептана из 30 мл смеси диэтиловый эфир:этилацетат (1:1), перекристаллизовывают из смеси гептан:этилацетат (2:3) и получают тетракетон л (фиг.11) - желтовато-зеленое кристаллическое вещество с Tпл=170-172°C. Выход целевого продукта 1,12 ммоль (28,7%).
1Н-ЯМР: 7.1 (ушс, 2Н, =СН); 7.75 (д, 2Н, =СН, J=11.4); 8.3 (д, 2Н, 2=СН, J=11.4); 9.2 (с, 2Н, 2=СН); 11.3 (ушс, 1Н, NH).
19F-ЯМР: -20.8 (м, 6F, CF3); 4.9 (м, 4F, CF2).
Элементный анализ: найдено С 45,91%, Н 1,91%; вычислено С 45,92%, Н 1,93%.
Пример 12
Синтез 4,4-дифтор-1-[6-(4,4-дифтор-3-оксобутаноил)-9H-карбазол-3-ил]-1,3-бутандиона (м)
39 ммоль металлического натрия растворяют в 25 мл сухого метанола, затем реакционную массу концентрируют досуха при остаточном давлении 20 Торр в течение 1 часа при 95°С. Прибавляют 60 мл сухого диэтилового эфира и в течение 10 минут раствор 39 ммоль метилового эфира 2,2,3,3,4,4,5,5-октафторпентановой кислоты в 20 мл этилового эфира, затем 3,9 ммоль диацетилдибензокарбазола, синтезированого по модифицированному методу Plant and Rogers, J.Chem.Soc., 1935, v.65, p.741-752. Полученную суспензию кипятят при перемешивании в течение 18 часов, охлаждают до комнатной температуры, прибавляют 50 мл диэтилового эфира и фильтруют. Фильтрат обрабатывают раствором 10,5 г серной кислоты в 41 мл дистиллированной воды. Органический слой отделяют и промывают 4 раза по 30 мл дистиллированной водой, фильтруют, фильтрат выдерживают при остаточном давлении 16 Торр в течение 1 часа при 40°С. Сухой остаток дробно высаживают 25+80 мл гептана из 30 мл смеси диэтиловый эфир:этилацетат (1:1), перекристаллизовывают из смеси гептан:этилацетат (2:3) и получают тетракетон м (фиг.12) - желтовато-зеленое кристаллическое вещество с Тпл=195-198°С. Выход целевого продукта 0,95 ммоль (24,4%).
1Н-ЯМР: 5,5 (т, 2Н, =CHF2, J=45,0), 6.8 (2Н, =СН); 7.75 (д, 2Н, -СН, J=11.4); 8.4 (д, 2Н, 2=СН, J=11.4); 9.1 (с, 2Н, 2=СН); 11.6 (ушс, 1Н, NH).
19F-ЯМР: 38.5 (д, 4F, 2CF2H, J=45.0).
Элементный анализ: найдено С 57,83%, Н 3,39%; вычислено С 58,92%, Н 3,19%.
Пример 13
Синтез метил-10-[7-(4,4,5,5,6,6,7,7,8,8,9,9-додекафтор-10-метокси-3,10-диоксодеканоил)-9H-флуорен-2-ил]-2,2,3,3,4,4,5,5,6,6,7,7-додекафтор-8,10-диоксодеканоата (н)
39 ммоль металлического натрия растворяют в 25 мл сухого метанола, затем реакционную массу концентрируют досуха при остаточном давлении 16 Торр в течение 1 часа при 95°С. Прибавляют 80 мл сухого диэтилового эфира и в течение 10 минут раствор 97,5 ммоль диметилового эфира 2,2,3,3,4,4,5,5,6,6,7,7-додекафтороктандиовой кислоты, в 50 мл этилового эфира, затем 3,9 ммоль диацетилдибензокарбазола, синтезированого по модифицированному методу Plant and Rogers, J.Chem.Soc., 1935, v.65, p.741-752. Полученную суспензию кипятят при перемешивании в течение 30 часов, охлаждают до комнатной температуры, прибавляют 50 мл диэтилового эфира и фильтруют.
Фильтрат обрабатывают раствором 14 г серной кислоты в 35 мл дистиллированной воды. Органический слой отделяют и промывают 6 раз по 20 мл дистиллированной водой, фильтруют, фильтрат выдерживают при остаточном давлении 15 Торр в течение 1 часа при 40°С. Сухой остаток дробно высаживают 20+60 мл гептана из 60 мл смеси диэтиловый эфир:этилацетат (1:1), перекристаллизовывают из смеси гептан:этилацетат (2:3) и получают тетракетон н (фиг.13) - желтовато-зеленое кристаллическое вещество с Тпл=230°С (разл). Выход целевого продукта 0,61 ммоль (15,2%).
1Н-ЯМР: 4.0 (с, 3Н, СН3); 4.1 (с, 4Н, 2СН2); 6.7 (с, 2Н, =СН); 7.7 (д, 2Н, 2=СН, J=10); 8.1 (д, 2Н, 2=СН, J=10); 8.2 (с, 2Н, 2=СН).
19F-ЯМР: -40.3 (т, 2F, CF2, J=6.5); -41.8 (т, 2F, CF2, J=6.5); -42.9 (м, 2F, CF2); -43.6 (м, 2F, CF2); -43.8 (м, 2F, CF2); -44.5 (м, 2F, CF2).
Элементный анализ: найдено С 40,31%, Н 1,43%; вычислено С 41,08%, Н 1,76%.
Пример 14
Синтез метил-10-[6-(4,4,5,5,6,6,7,7,8,8,9,9-додекафтор-10-метокси-3,10-диоксодеканоил)-9H-карбазол-3-ил]-2,2,3,3,4,4,5,5,6,6,7,7-додекафтор-8,10-диоксодеканоата (о)
39 ммоль металлического натрия растворяют в 25 мл сухого метанола, затем реакционную массу концентрируют досуха при остаточном давлении 16 Торр в течение 1 часа при 95°С. Прибавляют 80 мл сухого диэтилового эфира и в течение 10 минут раствор 97,5 ммоль метилового эфира диметилового эфира 2,2,3,3,4,4,5,5,6,6,7,7-додекафтороктандиовой кислоты, в 50 мл этилового эфира, затем 3,9 ммоль диацетилдибензокарбазола, синтезированого по модифицированному методу Plant and Rogers, J.Chem.Soc., 1935, v.65, p.741-752. Полученную суспензию кипятят при перемешивании в течение 70 часов в запаянной ампуле под воздействием ультразвука (35 кГц), охлаждают до комнатной температуры, прибавляют 50 мл диэтилового эфира и фильтруют. Фильтрат обрабатывают раствором 14 г серной кислоты в 35 мл дистиллированной воды. Органический слой отделяют и промывают 6 раз по 20 мл дистиллированной водой, фильтруют, фильтрат выдерживают при остаточном давлении 17 Торр в течение 1 часа при 40°С. Сухой остаток дробно высаживают 30+70 мл гептана из 60 мл смеси диэтиловый эфир:этилацетат (1:1), перекристаллизовывают из смеси гептан:этилацетат (2:3) и получают тетракетон н (фиг.14) - желтовато-зеленое кристаллическое вещество с Тпл=310°С (разл). Выход целевого продукта 0,27 ммоль (7,2%).
1Н-ЯМР: 4.0 (с, 6Н, ОСН3); 7.2 (с, 2Н,=СН); 7.85 (д, 2Н, =СН, J=10.3); 8.4 (д, 2Н, =СН, J=10.3); 9.3 (с, 2Н, -СН); 11.5 (ушс, 1Н, NH).
19F-ЯМР: -40.6 (т, 2F, CF2, J=6.5); -41.9 (т, 2F, CF2, J=6.5); -43.3 (м, 2F, CF2); -43.4 (м, 2F, CF2); -43.8 (м, 2F, CF2); -44.7 (м, 2F, CF2).
Элементный анализ: найдено С 40,51%, Н 1,45%; вычислено С 39,87%, Н 1,66%.
Пример 15
Синтез 1-{-6-[4,4-дифтор-4-[1,1,2,2-тетрафтор-2-(трифторметокси)этоксибутаноил]-9Н-карбазол-3-ил}-4,4-дифтор-4-[1,1,2,2-тетрафтор-2-(трифторметокси)этокси]]-1,3-бутандиона (п)
39 ммоль металлического натрия растворяют в 35 мл сухого метанола, затем реакционную массу концентрируют досуха при остаточном давлении 18 Торр в течение 1 часа при 95°С. Прибавляют 60 мл сухого диэтилового эфира и в течение 10 минут раствор 39 ммоль метилового эфира 2,2-дифтор-2-[-1,1,2,2-тетрафтор-2-(трифторметокси)этокси] уксусной кислоты, в 20 мл этилового эфира, затем 3,9 ммоль диацетилдибензокарбазола, синтезированого по модифицированному методу Plant and Rogers, J.Chem.Soc., 1935, v.65, p.741-752. Полученную суспензию кипятят при перемешивании в течение 18 часов, охлаждают до комнатной температуры, прибавляют 50 мл диэтилового эфира и фильтруют. Фильтрат обрабатывают раствором 10,5 г серной кислоты в 41 мл дистиллированной воды. Органический слой отделяют и промывают 4 раза по 30 мл дистиллированной водой, фильтруют, фильтрат выдерживают при остаточном давлении 16 Торр в течение 1 часа при 40°С. Сухой остаток дробно высаживают 25+80 мл гептана из 30 мл смеси диэтиловый эфир:этилацетат (1:1), перекристаллизовывают из смеси гептан:этилацетат (2:3) и получают тетракетон п (фиг.15) - желтовато-зеленое кристаллическое вещество с Тпл=278-291°С. Выход целевого продукта 0,55 ммоль (14,1%).
1Н-ЯМР: 7.1 (ушс, 2Н, =СН); 7.75 (д, 2Н,=СН, J=11.4); 8.3 (д, 2Н, 2=СН, J=11.4); 9.2 (с, 2Н, 2=СН); 11.3 (ушс, 1Н, NH).
19F-ЯМР: -19.8 (м, 6F, CF3); 2.4 (м, 8F, CF2), 4.9 (м, 4F, CF2).
Элементный анализ: найдено С 36,91%, Н 1,56%; вычислено С 38,65%, Н 1,36%.
Пример 16
Синтез 2,2-дифтор-2-(2,3,4,5,6-пентафторфенил)-1-[6-2,2-дифтор-2-(2,3,4,5,6-пентафторфенил-3-оксобутаноил)-9H-карбазол-3-ил]-1,3-бутандиона (р)
39 ммоль металлического натрия растворяют в 25 мл сухого метанола, затем реакционную массу концентрируют досуха при остаточном давлении 15 Торр в течение 1 часа при 95°С. Прибавляют 40 мл сухого диэтилового эфира и в течение 10 минут раствор 39 ммоль метилового эфира 2,3,4,5,6-пентафторбензойной кислоты в 20 мл этилового эфира, затем 3,9 ммоль диацетилдибензокарбазола, синтезированого по модифицированному методу Plant and Rogers, J.Chem.Soc., 1935, v.65, p.741-752. Полученную суспензию кипятят при перемешивании в течение 10 часов, охлаждают до комнатной температуры, прибавляют 40 мл диэтилового эфира и фильтруют. Фильтрат обрабатывают раствором 11,5 г серной кислоты в 40 мл дистиллированной воды. Органический слой отделяют и промывают 4 раза по 30 мл дистиллированной водой, фильтруют, фильтрат выдерживают при остаточном давлении 19 Торр в течение 1 часа при 40°С. Сухой остаток дробно высаживают 35+60 мл гептана из 30 мл смеси диэтиловый эфир:этилацетат (1:1), перекристаллизовывают из смеси гептан:этилацетат (2:3) и получают тетракетон р (фиг.16) - желтовато-зеленое кристаллическое вещество с Тпл=122-126°С. Выход целевого продукта 0,85 ммоль (21,8%).
1H-ЯМР: 7.1 (ушс, 2Н,=СН); 7.75 (д, 2Н, =СН, J=11.4); 8.3 (д, 2Н, 2=СН, J=11.4); 9.2 (с, 2Н, 2=СН); 11.3 (ушс, 1Н, NH).
19F-ЯМР: 84.3 (м, 10F, CF).
Элементный анализ: найдено С 51,94%, Н 1,78%; вычислено С 52,56%, Н 1,73%.
Пример 17
Люминесцентно-спектральные свойства комплексов заявляемых соединений с Eu3+
Регистрацию спектрально-люминесцентных характеристик комплексов осуществляли в диапазоне длин волн 230-800 нм на флуориметре с временным разрешением люминесценции - модель Perkin-Elmer LS-5B, США. Соответствующие результаты приведены в таблице, где λвозб - длина волны возбуждения люминесценции комплекса при регистрации эмиссии на 615 нм, I - интенсивность люминесценции в максимуме возбуждения и регистрации в относительных единицах (время задержки регистрации эмиссии люминесценции td - 0,1 мс, время регистрации эмиссии люминесценции tg - 1 мс), τ - время жизни люминесценции.
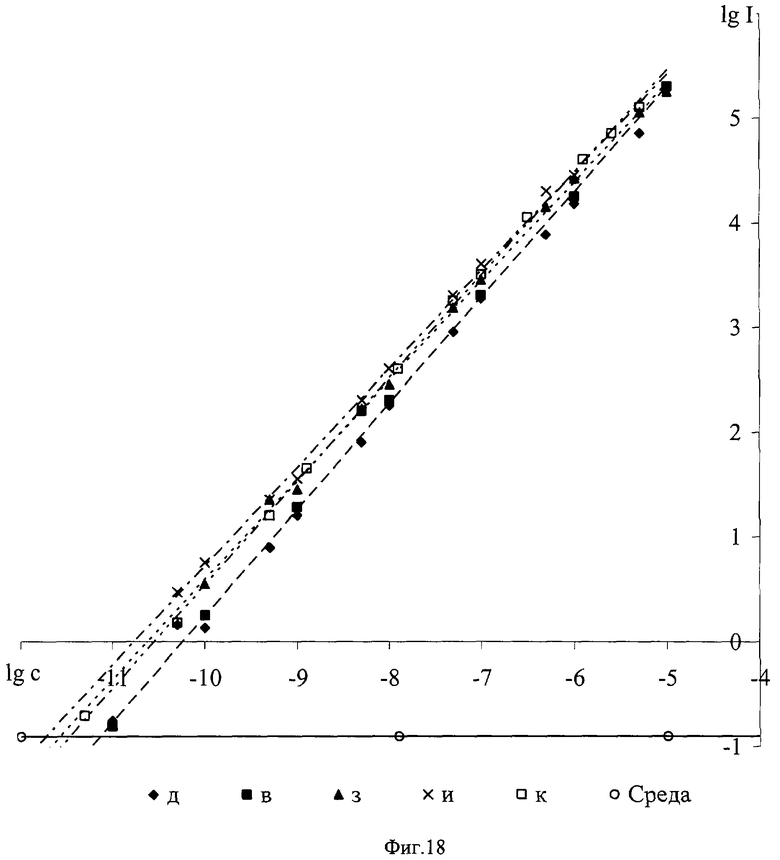
Все характеристики получены в водных растворах, содержащих 5·10-5 моль/л ТО-ФО и 0,1% Тритона Х-100. Для нейтральной среды (0,05 моль/л Трис-буфер, рН 7,2) приведены результаты, полученные от комплексов лиганд - Eu3+ стехиометрического состава 1:1 (концентрации лиганд:ион=10-8:10-8 моль/л), для кислой (среда УР рН 3,2) - состава n:1, где n≥2 (концентрации лиганд:ион=1,5.10-5:10-8 моль/л).
Из данных, приведенных в таблице, следует, что заявляемые соединения образуют интенсивно и длительно люминесцирующие комплексы с Eu3+ как в нейтральных средах, так и в кислой среде. Последнее предполагает возможность их использования в качестве перекомплексообразователей в способе иммуноанализа на основе DELFIA. При этом соединения а, в, д, ж, з на основе производных флуоренила и карбазола имеют максимумы возбуждения люминесценции, расположенные в области свыше 360 нм.
Все заявляемые соединения в нейтральной среде 0,05 М Трис-буфера, рН 7,2, в присутствии 5·10-5 М ТОФО и 0,1% Тритон Х-100 образуют стабильные 1:1 комплексы с Eu3+, не диссоциирующие вплоть до концентраций 10-11 - 10-12 моль/л, что следует из данных, приведенных на фиг.18, где на примере некоторых из заявляемых соединений в логарифмическом масштабе представлена зависимость интенсивности люминесценции комплексов от их концентрации (моль/л). Отсутствие излома на этих зависимостях свидетельствует о том, что концентрация комплекса остается равной концентрации лиганда и Eu3+ даже при самых малых их значениях (регистрируемых спектрофлуориметром LS-5В).
Примеры использования заявляемых соединений
Пример 18
Синтез латексных частиц, нагруженных комплексами заявляемых соединений с Eu3+
Синтез частиц, нагруженных комплексами заявляемых соединений с Eu3+, осуществляют двумя способами. Первый заключается во введении хелата в частицы в процессе их образования, второй - во введении хелата в готовые частицы.
Латексы представляют собой взвешенную в водной фазе мелкодисперсную суспензию твердых частиц меламино-формальдегидной смолы, изготовляемую на основе опытно-промышленного регламента, разработанного в ГосНИИ биологического приборостроения.
Способ 1. В стакан на 400 мл наливают 180 мл дистиллированной воды и 20 мл 36% раствора формальдегида в воде, после чего помешают в кипящую водяную баню и закрывают крышкой, содержащей механическую мешалку и термометр. Включают мешалку и по достижении температуры 70°С добавляют 2 г меламина. Продолжают следить за температурой и по достижении 89°С добавляют в реакционную среду 2 мл раствора соединения ж в концентрации 3,3 мг/мл тетрагидрофурана, содержащего также 0,6 мг/мл EuCl3·6Н2O. Реакционную массу выдерживают при температуре 90°С в течение 15 мин, после чего стакан вынимают из горячей бани и помещают в ледяную баню. Образовавшуюся суспензию частиц (⊘ср=1 мкм), содержащую комплекс ж-Eu3+, отмывают дистиллированной водой путем центрифугирования при 3000 об/мин в течение 5 мин и последующего ресуспендирования до тех пор, пока надосадочная жидкость не станет прозрачной и не будет давать сигнала, превышающего фоновый при возбуждении 385 нм и регистрации люминесценции на 615 нм.
Способ 2. К 1 мл водной латексной суспензии, содержащей 3·1010 частиц (⊘ср=0,5 мкм), добавляют 1 мл тетрагидрофурана и при постоянном перемешивании магнитной мешалкой выдерживают в течение 30 мин при комнатной температуре. Затем к суспензии добавляют 0,17 мл раствора соединения ж в концентрации 3,3 мг/мл тетрагидрофурана, содержащего также 0,6 мг/мл EuCl3·6Н2O. Перемешивание продолжают в течение 4 час при комнатной температуре, после чего латексы с сорбированным на поверхности частиц комплексом ж-Eu3+ подвергают отмывке дистиллированной водой по способу, аналогичному вышеописанному.
Пример 19
Покрытие латексных частиц стрептавидином
Аликвоту латексной суспензии, меченной комплексом Eu по способу 2 примера 12, разводят 0,05 М Трис-буфером, рН 7,2, в 10 раз и добавляют раствор стрептавидина 10 мг/мл того же буфера из расчета 10 мкл/1 мл суспензии 109 частиц/мл. Адсорбцию продолжают в течение 18 час при 4°С и постоянном перемешивании, после чего суспензию трижды отмывают от несвязавшегося стрептавидина центрифугированием (3000 об/мин, 10 мин) и ресуспендированием в 0,05 М Трис-буфере, рН 7,2. Хранят суспензию покрытых стрептавидином латексных частиц в том же буфере с добавлением 0,1% бычьего сывороточного альбумина и 0,15% натрия азида.
Пример 20
Проведение иммуноанализа тиреотропного гормона человека (ТТГ) по протоколу DELFIA (вариант «ТТГ-неоскрин» ЗАО «Иммуноскрин») с использованием в усиливающем растворе заявляемых соединений
В сенсибилизированные моноклональными антителами против α-субъединицы ТТГ лунки стрипа в двух повторах вносят калибровочные пробы, представляющие собой высушенные на фильтровальной бумаге образцы крови с известным содержанием ТТГ. Затем в них добавляют по 200 мкл моноклональных антител против β-субъединицы ТТГ, меченных ДТПА-Eu. Инкубируют сначала при комнатной температуре (10 мин), затем - при температуре +2 - +8°С в течение 18 час и снова - при комнатной температуре на встряхивателе в течение 1 часа. По окончании инкубации фильтровальные диски удаляют и производят 6-кратную отмывку стрипа промывным раствором. Вносят в каждую лунку по 200 мкл усиливающего раствора, содержащего в качестве перекомплексообразователя НТА, либо заявляемое соединение а, либо д, либо ж, либо к. Инкубируют образцы в течение 20 мин при комнатной температуре, после чего производят измерение интенсивностей люминесценции на флуориметре с временным разрешением Arcus 1234, предназначенном для регистрации люминесценции НТА. Из результатов анализа, приведенных на фиг.19, следует, что заявляемые соединения на основе дибензофуранильного (к) и карбазольного (д, ж) хромофоров образуют более интенсивно люминесцирующие комплексы с Ей3+ в условиях данного анализа, что позволяет более чувствительно по сравнению с НТА обнаруживать ТТГ.
| название | год | авторы | номер документа |
|---|---|---|---|
| N-СПЕЙСЕРСОДЕРЖАЩИЕ 3,6-БИС-β-ДИКАРБОНИЛЗАМЕЩЕННЫЕ КАРБАЗОЛЫ С ФТОРИРОВАННЫМИ ЗАМЕСТИТЕЛЯМИ, ДЛЯ ИСПОЛЬЗОВАНИЯ В КАЧЕСТВЕ МАРКЕРОВ И КОМПЛЕКСОНОВ | 2019 |
|
RU2709194C1 |
| КОМПЛЕКСООБРАЗУЮЩИЕ БЕНЗОСОДЕРЖАЩИЕ ГЕТЕРОЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ, СОДЕРЖАЩИЕ β-ДИКАРБОНИЛЬНЫЙ ЗАМЕСТИТЕЛЬ С ФТОРИРОВАННЫМИ РАДИКАЛАМИ | 2007 |
|
RU2373200C2 |
| СПОСОБ ПОЛУЧЕНИЯ АДАМАНТИЛСОДЕРЖАЩИХ β-ДИКЕТОНОВ И КЕТОЭФИРОВ | 2000 |
|
RU2187493C1 |
| СПОСОБ ПОЛУЧЕНИЯ 1-(2-МЕТИЛ-4-ФЕНОКСИФЕНИЛ)-БУТАН-1,3-ДИОНА | 2012 |
|
RU2478606C1 |
| СПОСОБ ПОЛУЧЕНИЯ 1-(3-ФЕНОКСИФЕНИЛ)БУТАН-1,3-ДИОНА | 2013 |
|
RU2529029C1 |
| СПОСОБ ПОЛУЧЕНИЯ 3-АЛКИЛИРОВАННОГО ИНДОЛА И (2-НИТРОФЕНИЛ)-АЦЕТАЛЬДЕГИД | 1991 |
|
RU2054417C1 |
| СПОСОБ ПОЛУЧЕНИЯ 5-ФЕНИЛ-3-(ТРИФТОРМЕТИЛ)-1Н-ПИРАЗОЛ-4-АМИНА | 2017 |
|
RU2642924C1 |
| 1,3-ДИКАРБОНИЛЬНЫЕ ПРОИЗВОДНЫЕ АДАМАНТАНОВ И СПОСОБ ИХ ПОЛУЧЕНИЯ | 2010 |
|
RU2458911C2 |
| ЗАМЕЩЕННЫЕ 3-ФЕНИЛПРОПИОНОВЫЕ КИСЛОТЫ И ИХ ПРИМЕНЕНИЕ | 2010 |
|
RU2553263C2 |
| РЕАГЕНТ ДЛЯ ВВЕДЕНИЯ ПЕРФТОР-ТРЕТ-БУТИЛЬНОЙ ГРУППЫ, СПОСОБ ЕГО ПОЛУЧЕНИЯ И СПОСОБ ПРИМЕНЕНИЯ ДЛЯ ПОЛУЧЕНИЯ ПЕРФТОР-ТРЕТ-БУТИЛ-ЗАМЕЩЕННЫХ СОЕДИНЕНИЙ | 2015 |
|
RU2602240C1 |













Изобретение относится к области синтеза новых аналитических реагентов комплексообразующего типа и может быть использовано в области люминесцентно-спектрального анализа, в частности для клинической диагностики объектов биогенного происхождения, а также в области техники для применения в качестве экстрагентов ионов тяжелых и редкоземельных металлов с целью их извлечения и/или очистки от их примесей сточных и контурных вод. Полученные соединения представляют собой комплексообразующие дибензосодержащие пятичленные циклические соединения, содержащие два симметричных β-дикарбонильных заместителя, ковалентно связанных с фторсодержащими радикалами, общей формулы
где Х - СН2, О, S, NR, если X - CH2, то R1=H, R2=С(O)CH2С(O)Rf, где Rf=(CF2)nY, n=1-6, при n=1 Y=H, F, CI, Br, OCF3, ОСН3, ОС3F7, OCF2CF2OCF3, С6F5, С(O)OAlk, где Alk - углеводородный радикал; при n=2 Y=H, F, CI, Br, OCF3, ОС3F7, OCF2CF2OCF3, C(O)OAlk; при n=3, 5 или 6 Y=F, C(O)OAlk; при n=4 Y=H, F, CI, C(O)OAlk, если X - O, S или NR, то R=H или Alk, R2=H, R1=C(O)CH2C(O)Rf. 19 ил., 1 табл.
Дибензосодержащие пятичленные циклические соединения, содержащие два симметричных β-дикарбонильных заместителя с фторированными радикалами для использования в качестве комплексообразующих соединений, общей формулы 1
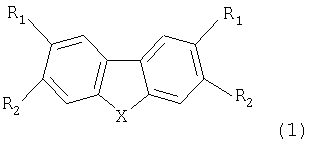
где Х - СН2, О, S, NR,
если Х - СН2, то R1 - H, R2 - C(O)CH2C(O)Rf,
где Rf -(CF2)nY, n=1÷6,
при n=1 Y -H, F, CI, Br, OCF3, ОСН3, ОС3F7, OCF2CF2OCF3, С6F5, C(O)OAlk,
где Alk - углеводородный радикал;
при n=2 Y - H, F, CI, Br, OCF3, ОС3F7, OCF2CF2OCF3, C(O)OAlk, при n=3, 5 или 6 Y - F, C(O)OAlk, при n=4 Y - H, F, CI, C(O)OAlk;
если X - O, S или NR, то R - H или Alk, R2 - H, R1 -C(O)CH2C(O)Rf.
| US 6127529 А, 03.10.2000 | |||
| US 4808541 A, 28.02.1989 | |||
| US 6166251 A, 26.12.2000 | |||
| US 5457186 A, 10.10.1995 | |||
| RU 94011565 A, 27.10.1996 | |||
| Способ получения 2,3,7,8-тетраметилдибензотиофена | 1976 |
|
SU579274A1 |

