1. Область техники, к которой относится изобретение
Изобретение относится к области органической химии, а именно к усовершенствованному способу получения 5-фенил-3-(трифторметил)-1H-пиразол-4 амина (1)
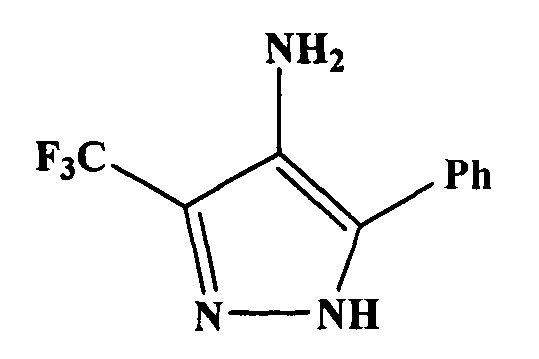
(далее - аминопиразол 1) - ключевого соединения в синтезе гибридных молекул, обладающих различными видами биологической активности (Emmadi, N.R., Bingi, С., Kotapalli, S.S., Ummanni, R., Nanubolu, J.B., Atmakur, K., Synthesis and evaluation of novel fluorinated pyrazolo-1,2,3-triazole hybrids as antimycobacterial agents, Bioorganic & Medicinal Chemistry Letters (2015), doi: http://dx.doi.Org/10.1016/j.bmcl.2015.05.044).
Изобретение может быть использовано в научно-исследовательских институтах для создания новых лекарственных препаратов и в химико-фармацевтической промышленности.
2. Уровень техники
Одно из направлений современной медицинской химии - создание «многофункциональных лекарств» путем комбинации различных фармакофоров в одной основе, которые обладают способностью взаимодействовать с несколькими «мишенями». Такие препараты имеют более предсказуемый фармакокинетический профиль, у них существенно снижен риск несовместимости с другими препаратами за счет уменьшения количества разных лекарств, прописываемых пациенту.
В настоящее время уже получены гибридные соединения, включающие фрагменты 3-трифторметилпиразола и 1,2,3-триазола, которые показали антимикобактериальную активность против М. smegmatis, активность по отношению к клеткам рака легких. Кроме того, указанные структуры можно рассматривать как потенциальные антитуберкулезные агенты с низкой цитотоксичностью (Emmadi, N.R., Bingi, С., Kotapalli, S.S., Ummanni, R., Nanubolu, J.В., Atmakur, K., Synthesis and evaluation of novel fluorinated pyrazolo-1,2,3-triazole hybrids as antimycobacterial agents, Bioorganic & Medicinal Chemistry Letters (2015), doi: http://dx.doi.org/10.1016/j.bmcl. 2015.05.044).
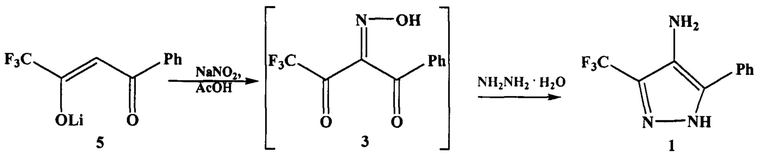
Из литературных источников известно, что аминопиразол 1 получают в три стадии с выделением и очисткой промежуточных продуктов на каждой стадии.
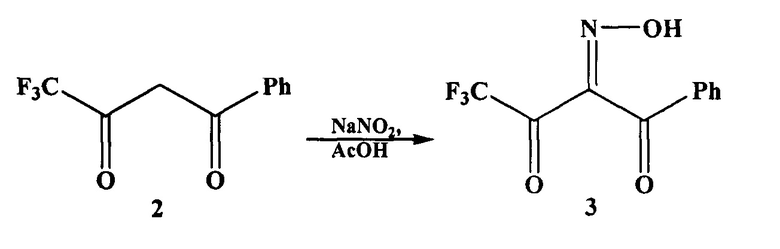
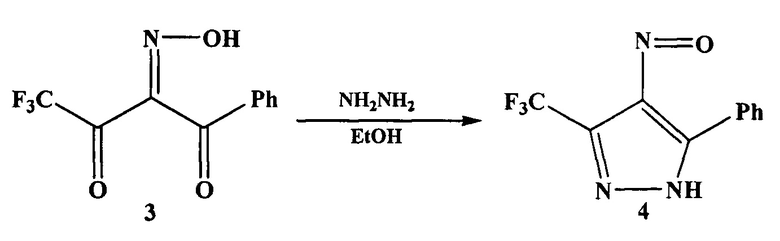
На первой стадии взаимодействием 4,4,4-трифтор-1-фенилбутандиона (дикетона 2) с нитрозирующим агентом, например нитритом натрия, в органическом растворителе, например в уксусной кислоте, получают 4,4,4-трифтор-1-фенил-1,2,3-бутантрион-2-оксим (оксим 3).

На второй стадии оксим 3 обрабатывают гидразином или гидразин-гидратом и получают 4-нитрозо-5-фенил-3-трифторметил-1H-пиразол (нитрозопиразол 4).
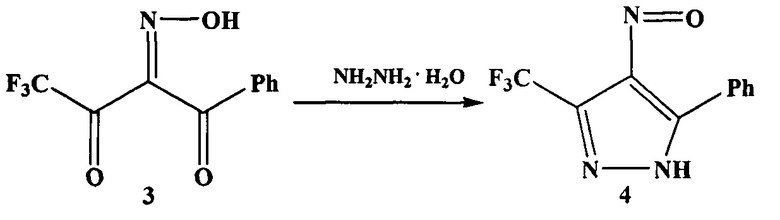
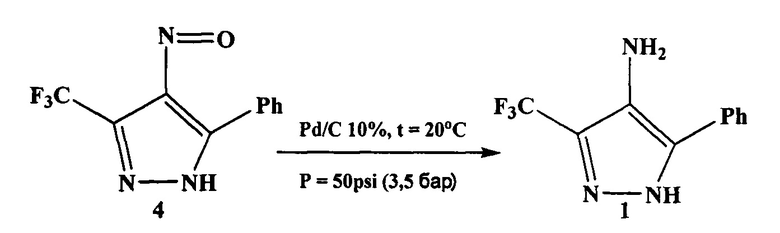
На третьей стадии нитрозопиразол 4 восстанавливают до аминопиразола 1 различными способами, например водородом с использованием палладиевого катализатора.
В патенте (Patent; Aventis Pharmaceuticals Inc.; US 2005/9859; (2005); (A1) English) аминопиразол 1 получают в три стадии. На первой стадии по методике, описанной в статье (Saloutin V.I., Burgart Y.V., Skryabina Z.E., Kuzueva O.G., J. Fluor. Chem., 1997, 84, p 107), взаимодействием дикетона 2 с нитритом натрия в уксусной кислоте получают оксим 3. По окончании реакции оксим 3 экстрагируют из реакционной массы диэтиловым эфиром (3 раза по 50 мл). Экстракт промывают насыщенным раствором гидрокарбоната натрия до pH 7, затем промывают водой и сушат при пониженном давлении 2 часа.
На второй стадии оксим 3 растворяют в этаноле и обрабатывают гидразином. После удаления этанола, переосаждения твердого остатка из диэтилового эфира гексаном и последующей перекристаллизации из хлороформа получают нитрозопиразол 4.
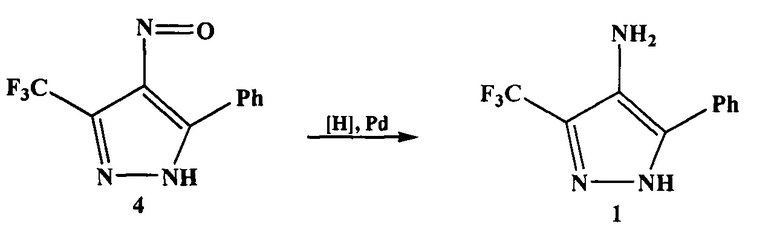
На третьей стадии нитрозопиразол 4 восстанавливают до аминопиразола 1. Для этого используют реакцию каталитического гидрирования, требующую повышенного давления. В качестве катализатора применяют дорогостоящий металлический палладий, нанесенный на углеродную подложку (Pd/C).
Раствор нитрозопиразола 4 в этаноле обрабатывают катализатором Pd/C 10% и гидрогенизируют при 50 psi (3,5 бар) в течение 7.5 часов. По окончании реакции реакционную смесь фильтруют через целит и промывают этанолом. Фильтрат концентрируют и получают аминопиразол 1 в виде желтого порошка.
Основные недостатки получения аминопиразола 1 этим способом:
1. Невозможность осуществления процесса в одном реакторе. Процесс осуществляют в три стадии, каждая их которых требует своего оборудования, реагентов и растворителей.
2. В качестве исходного соединения используют дикетон 2, который коммерчески доступен, но очень дорог, а его синтез и очистка весьма трудоемки.
4. Процесс восстановления нитрозопиразола 4 осуществляют под давлением, что требует специального оборудования.
5. В процессе восстановления нитрозопиразола 4 в качестве катализатора используют дорогостоящий металлический палладий, нанесенный на углеродную подложку (Pd/C).
3. Процесс выделения продуктов реакций на каждой стадии включает много операций, требует значительных затрат реагентов, растворителей, труда и времени.
Стадия 1. Выделение оксима 3 из реакционной массы предполагает следующие операции:
1 Экстракция сырого оксима 3 диэтиловым эфиром.
2. Промывка эфирного экстракта насыщенным раствором NaHCO3 до pH 7.
3. Промывка эфирного экстракта водой, отделение эфирного слоя.
4. Осушка эфирного экстракта при пониженном давлении в течение 2-х часов.
Стадия 2. Выделение нитрозопиразола 4 из реакционной массы предполагает следующие операции:
1. Удаление этанола
2. Переосаждение нитрозопиразола 4 из диэтилового эфира гексаном.
3. Фильтрование
4. Перекристаллизация нитрозопиразола 4 из хлороформа.
Стадия 3. Выделение аминопиразола 1 из реакционной массы предполагает следующие операции:
1. Фильтрование аминопиразола 1 через целит от катализатора.
2. Промывка целита этанолом с целью извлечения аминопиразола 1.
3. Удаление этанола.
Таким образом, только процесс выделения аминопиразола 1 и полупродуктов его синтеза (оксима 3 и нитрозопиразола 4) из реакционной массы требует осуществления 11-ти трудоемких операций.
В данном патенте авторы не приводят выход аминопиразола 1, поскольку он не является для них целевым продуктом (его используют в качестве ключевого соединения для получения более сложных структур).
Известен способ получения аминопиразола 1 (Emmadi, N.R., Bingi, С., Kotapalli, S.S., Ummanni, R., Nanubolu, J.B., Atmakur, K., Synthesis and evaluation of novel fluorinated pyrazolo-1,2,3-triazole hybrids as antimycobacterial agents, Bioorganic & Medicinal Chemistry Letters (2015), doi: http://dx.doi.org/10.1016/j.bmcl.2015.05.044) из дикетона 2 - прототип.
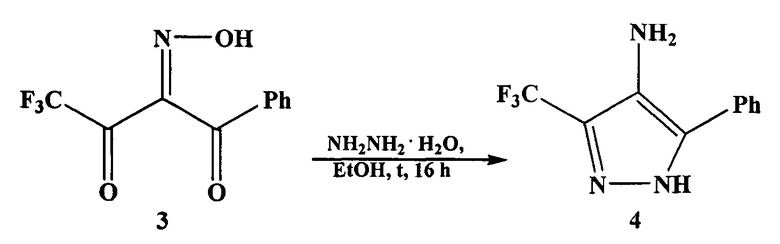
В данной работе предложен более простой способ получения аминопиразола 1. Авторы устранили стадию гидрирования (отказались от Pd/C катализатора), используя в качестве восстановителя тот же гидразин-гидрат (10-кратный избыток), который идет на получение нитрозопиразола 4. Однако, несмотря на внесенные изменения, процесс получения аминопиразола 1 включает две стадии с выделением и очисткой промежуточного оксима 3, что требует проведения ряда операций.
На первой стадии взаимодействием дикетона 2 с нитритом натрия в ледяной уксусной кислоте после отгонки уксусной кислоты при пониженном давлении, обработки остатка водным раствором NaHCO3, экстракции сырого продукта этилацетатом, осушки полученного раствора над безводным Na2SO4, фильтрования от Na2SO4 и отгонки этилацетета при пониженном давлении получают оксим 3 (Выход оксима 3 в работе не приведен).

На второй стадии раствор оксима 3 в этаноле обрабатывают гидразин-гидратом. Реакционную массу нагревают при перемешивании 16 часов. По окончании реакции (контроль методом ТСХ) этанол концентрируют, остаток растворяют в этилацетате и промывают 3Н раствором соляной кислоты. Органический слой отделяют, водную фракцию нейтрализуют 10 Н раствором NaOH и экстрагируют этилацетатом. Органический экстракт сушат над Na2SO4, фильтруют от Na2SO4 и концентрируют. Получают светло желтый осадок аминопиразола 1. Выход 75%. Т.пл. 123-125°C.
Методика синтеза (Emmadi, N.R., Bingi, С., Kotapalli, S.S., Ummanni, R., Nanubolu, J.B., Atmakur, K., Synthesis and evaluation of novel fluorinated pyrazolo-1,2,3-triazole hybrids as antimycobacterial agents, Bioorganic & Medicinal Chemistry Letters (2015), doi: http://dx.doi.org/10.1016/i.bmcl. 2015.05.044) - прототип:
Стадия 1. К раствору 5 г (23.1 ммоль) 4,4,4-трифтор-1-фенилбутадиона в 8 мл. ледяной уксусной кислоты при 0°C добавляют 1.91 г (27.7 ммоль) нитрита натрия в 8 мл. воды. Реакционную массу перемешивают при 0°C 1 час. По окончании реакции (ТСХ-контроль) уксусную кислоту отгоняют при пониженном давлении. Сырой продукт встряхивают с водным раствором NaHCO3, (5 мл.), экстрагируют этилацетатом и выдерживают над безводным Na2SO4 и отфильтровывают раствор. После отгонки этилацетата при пониженном давлении получают 4,4,4-трифтор-1-фенил-1,2,3-бутантрион-2-оксим (оксим 3).
Стадия 2. К раствору 4,4,4-трифтор-1-фенил-1,2,3-бутантрион-2-оксима в 50 мл этанола добавляют по каплям 11.7 г (230.00 ммоль) гидразин-гидрата, нагревают при перемешивании 16 часов. По окончании реакции (ТСХ-контроль) реакционную массу концентрируют, растворяют в этилацетате и промывают 3Н раствором соляной кислоты. Органический слой отделяют, водную фракцию нейтрализуют 10Н раствором NaOH и далее экстрагируют этилацетатом. Органический экстракт высушивают над Na2SO4 и концентрируют. Получают 4 г (75%) 5-фенил-3-(трифторметил)-1H-пиразол-4 амина (аминопиразола 1) в виде светло-желтого осадка. Тпл. 123-125°C.
Основные недостатки получения аминопиразола 1 данным способом:
1. Невозможность осуществления процесса в одном реакторе. Процесс осуществляют в две стадии, каждая их которых требует своего оборудования, реагентов и растворителей.
2. В качестве исходного соединения используют дикетон 2, который коммерчески доступен, но очень дорог, а его синтез и очистка весьма трудоемки.
3. Процесс выделения продуктов реакций на каждой стадии включает много операций, требует значительных затрат реагентов, растворителей, труда и времени.
Стадия 1. Выделение оксима 3 из реакционной массы предполагает следующие операции:
1. Отгонка уксусной кислоты при пониженном давлении.
2. Обработка сырого оксима 3 водным раствором NaHCO3.
3. Экстракция сырого оксима 3 этилацетатом.
4. Осушка полученного раствора над безводным Na2SO4.
5. Фильтрование от Na2SO4.
6. Отгонка этилацетата при пониженном давлении.
Стадия 2. Выделение аминопиразола 1 из реакционной массы предполагает следующие операции:
1. Промывка раствора аминопиразола 1 в этилацетате 3Н раствором соляной кислоты.
2. Отделение органического слоя.
3. Нейтрализация водной фракции 10 раствором NaOH.
4. Экстракция аминопиразола 1 из водной фракции этилацетатом.
5. Выдерживание с целью осушки органического экстракта над Na2SO4.
6. Фильтрование от Na2SO4.
7. Удаление растворителя (этилацетата).
Таким образом, процесс выделения аминопиразола 1 и полупродукта его синтеза (оксима 3) из реакционной массы требует осуществления 13-ти трудоемких операций. Выход целевого аминопиразола 1 составляет 75%.
Задачей данного изобретения является разработка простого и удобного способа получения аминопиразола 1, который позволил бы повысить выход конечного продукта, сократить количество операций и осуществлять процесс в одном реакторе.
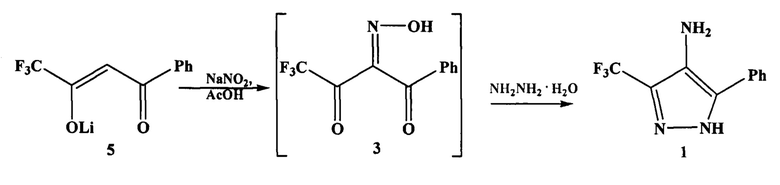
Поставленная задача решается предложенным способом получения аминопиразола 1 взаимодействием 4,4,4-трифтор-1-фенилбутадионата лития (далее - дикетоната 5) вместо соответствующего дикетона 2 с нитритом натрия в ледяной уксусной кислоте и последующей обработкой реакционной массы (без выделения оксима 3) 8-кратным избытком гидразин-гидрата.
3. Сущность изобретения
Сущность изобретения заключается в том, что в предлагаемом способе получения аминопиразола 1 вместо дикетона 2 используют более доступный 4,4,4-трифтор-1-фенилбутадионат лития дикетонат 5, а в качестве растворителя используют уксусную кислоту, что позволяет проводить процесс в одном реакторе, без выделения промежуточного оксима 3.
На первой стадии после обработки дикетоната 5 нитритом натрия, не выделяя оксим 3, в реакционную массу добавляют 8-кратный избыток гидразин гидрата и выдерживают ее при комнатной температуре до полного восстановления нитрозопроизводного 4 до аминопиразола 1(контроль состава реакционной массы методом ТСХ и ГХ-МС).
Использование уксусной кислоты в качестве органического растворителя позволяет проводить процесс в одном реакторе, без выделения промежуточного оксима 3, без применения повышенного или пониженного давления, при температуре от 10 до 20°C, что приводит к повышению селективности процесса (согласно данным ГХ-МС побочные продукты практически отсутствуют) и обеспечивает получение целевого аминопиразола 1 с выходом 93%. Процесс выделения целевого продукта предельно прост: по окончании реакции выпадает осадок, который отфильтровывают, промывают на фильтре водой и сушат на воздухе. Это позволяет сократить практически все операции, которые используют в способе-прототипе.
Способ осуществляют следующим образом. В плоскодонную колбу, снабженную мешалкой, баней со льдом, обратным холодильником и капельной воронкой помещают 4,4,4-трифтор-1-фенилбутадионат лития, ледяную уксусную кислоту и при перемешивании охлаждают до 10-16°C. К полученному раствору при интенсивном перемешивании по каплям добавляют водный раствор нитрита натрия. Реакционную массу перемешивают 1 час, охлаждают на ледяной бане до 10-16°C, после чего при интенсивном перемешивании по каплям добавляют 8-кратный избыток гидразин-гидрата, перемешивают 2-3 часа. Далее реакционную массу выдерживают при комнатной температуре 20 часов. Выпавший осадок отфильтровывают, промывают на фильтре водой и сушат на воздухе.
Состав и структуру полученного продукта устанавливают при помощи данных хромато-масс-спектрометрии (ГХ-МС), элементного анализа, ИК и ЯМР (1Н, 19F)-спектроскопиии.
4. Сведения, подтверждающие сущность изобретения
4.1. Синтез 5-фенил-3-(трифторметил)-1H-пиразол-4 амина 1.
Способ получения включает одну химическую стадию: взаимодействие 4,4,4-трифтор-1-фенилбутадионата лития с нитритом натрия в ледяной уксусной кислоте в мольном соотношении 1:1.15 с последующей обработкой реакционной массы 8-кратным избытком гидразин-гидрата без выделения оксима 3.
Аналитический образец аминопиразола 1 получают перекристаллизацией из хлороформа. Аминопиразол 1 имеет следующие физико-химические характеристики: белое вещество, Т.пл.=123-125°C, растворимо в этаноле, ацетоне, хлороформе, диэтиловом эфире, этилацетате; не растворимо в воде. Согласно данным ГХ/МС вещество не содержит примесей: хроматограмма содержит один пик с τуд=19,78.
Найдено, %: С 52,89; Н 3,63; N 18,36; F 24,82. Брутто-формула: C10H8F3N3. Вычислено, %: 52,87; Н 3,55; N 18,49; F 25,09. ИК спектр (dra), v/см-1: 3378 ср, 3236 с, 3158 с, 3059 с, (N-Hвал.); 1608 сл., 1591 ср. (C=N, N-Ндеф.); 1313 с, 1163 с, 1141 с, 1105 с. (C-F). Спектр ЯМР 1Н (500 МГц, DMSO d6), δ (м.д.): 4.25 с (2Н, NH2), [7.31-7.38 м (1Н), 7.45-7.52 м (2Н), 7.64-7.69 м (2Н) Ph], 13.34 уш. с. (1H, NH). Спектр ЯМР 19F (470.5 МГц, DMSO d6), δ (м.д.): 103.08 с (CF3).
Масс-спектр, m/z (Iотн, %): 227 (58) [М]+; 208 (9) [М-F]+; 178 (9), 132 (10) [М-CF3CN]+; 104 (100) [Ph-C=NH]+; 103 (9) [Ph-CN]+; 77 (50) [Ph]+; 69 (6%) [CF3]+; 51 (19); 28 (13).
Способ получения аминопиразола 1 иллюстрируется следующим примером.
Пример 1. Получение 5-фенил-3-(трифторметил)-1H-пиразол-4 амина 1.
К раствору 1 г (4.5 ммоль) 4,4,4-трифтор-1-фенилбутадионата лития в 3 мл ледяной уксусной кислоты при перемешивании добавляют 0.36 г (5.2 ммоль) нитрита натрия в 3 мл воды. Реакционную массу перемешивают 1 час, после чего добавляют 1.8 г (36.0 ммоль) гидразин-гидрата и перемешивают 3 часа. Далее реакционную массу выдерживают при комнатной температуре 20 часов. Выпавший осадок фильтруют, промывают водой на фильтре, сушат на воздухе. Получают 0.95 г 5-фенил-3-(трифторметил)-1H-пиразол-4 амина 1 в виде желтого порошка. Выход 93%. Т. пл.=123-125°C.
Данное техническое решение позволяет получить аминопиразол 1 в одном реакторе (one-pot), повысить выход целевого продукта с 75 до 93% и существенно сократить количество операций при выделении целевого продукта (с 13-ти в способе-прототипе до 3-х).
Следует отметить, что технический образец аминопиразола 1, полученный предложенным способом, согласно данным ГХ-МС содержит 96% основного вещества (τуд=19.80) и может быть использован в дальнейших синтезах без дополнительной очистки.
Благодаря простоте аппаратурного оформления, данное техническое решение можно масштабировать для получения более крупных партий аминопиразола 1 (вплоть до промышленных).
Исходное соединение, дикетонат 5, доступнее соответствующего дикетона 2, поскольку является интермедиатом синтеза последнего. Дикетонат 5 получают из доступных реагентов: конденсацией этилового эфира трифторуксусной кислоты с ацетофеноном под действием гидрида лития и используют без очистки в виде технического продукта. Кроме того, дикетонат 5 устойчивее при хранении и удобнее для использования в химических процессах чем соответствующий дикетон 2. Характеристики дикетоната 5 приведены в работе (Н.С. Болтачева, В.И. Филякова, Е.Ф. Хмара, О.В. Корякова, В.Н. Чарушин «Синтез и строение фторалкилсодержащих 1,3-дикетонатов лития» Рос. хим. ж. (Ж. Рос. хим. об-ва им. Д.И. Менделеева), 2009, т. LIII, №1, с. 54-63).
| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБ ЛЕЧЕНИЯ ВОСПАЛЕНИЯ ИЛИ СВЯЗАННОГО С ВОСПАЛЕНИЕМ ЗАБОЛЕВАНИЯ У СОБАК | 1996 |
|
RU2253456C2 |
| 4-Амино-3-метоксиметил-5-фенил-1Н-пиразол | 2015 |
|
RU2642060C2 |
| ЛЮМИНЕСЦИРУЮЩИЕ КОМЛЕКСНЫЕ СОЕДИНЕНИЯ РЕДКОЗЕМЕЛЬНЫХ ЭЛЕМЕНТОВ С ПИРАЗОЛСОДЕРЖАЩИМИ ФТОРИРОВАННЫМИ 1,3-ДИКЕТОНАМИ И СПОСОБ ИХ ПОЛУЧЕНИЯ | 2011 |
|
RU2485163C1 |
| ПИРАЗОЛЬНЫЕ СОЕДИНЕНИЯ, ЗАМЕЩЕННЫЕ ГЕТЕРОАРИЛОМ, И ИХ ПРИМЕНЕНИЕ В ФАРМАЦЕВТИКЕ | 2019 |
|
RU2805312C2 |
| ПРОИЗВОДНЫЕ 1-(4-ИЗОКСАЗОЛ-5-ИЛ)-1Н-ПИРАЗОЛ-1-ИЛ)-2-МЕТИЛПРОПАН-2-ОЛА И РОДСТВЕННЫЕ СОЕДИНЕНИЯ В КАЧЕСТВЕ ИНГИБИРОРОВ ИЛ-17 И ИФН-ГАММА ДЛЯ ЛЕЧЕНИЯ АУТОИММУННЫХ ЗАБОЛЕВАНИЙ И ХРОНИЧЕСКОГО ВОСПАЛЕНИЯ | 2018 |
|
RU2785342C2 |
| ПИРАЗОЛИЛЗАМЕЩЕННЫЙ БЕНЗОЛСУЛЬФОНАМИД ИЛИ ЕГО ФАРМАЦЕВТИЧЕСКИ ПРИЕМЛЕМАЯ СОЛЬ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ, СПОСОБ ЛЕЧЕНИЯ ОТ ВОСПАЛЕНИЯ ИЛИ СВЯЗАННОГО С ВОСПАЛЕНИЕМ ЗАБОЛЕВАНИЯ | 1994 |
|
RU2139281C1 |
| 3-МЕТИЛ-4-НИТРОЗОПИРАЗОЛЫ И СПОСОБ ИХ ПОЛУЧЕНИЯ | 2010 |
|
RU2440343C1 |
| ИНГИБИТОРЫ ФЕРМЕНТА ДИАЦИЛГЛИЦЕРИН О-АЦИЛТРАНСФЕРАЗА ТИПА 1 | 2007 |
|
RU2474576C2 |
| ПРОИЗВОДНЫЕ ПИРАЗОЛА, СПОСОБЫ ИХ ИСПОЛЬЗОВАНИЯ, ГЕРБИЦИДНАЯ КОМПОЗИЦИЯ И СПОСОБ БОРЬБЫ | 1991 |
|
RU2137761C1 |
| СИНТЕЗ ДИАРИЛПИРАЗОЛОВ | 2003 |
|
RU2319695C2 |
Предлагается способ получения 5-фенил-3-(трифторметил)-1H-пиразол-4 амина, который имеет формулу 1, приведенную ниже. Это соединение является ключевым в синтезе гибридных молекул, обладающих различными видами биологической активности. Сущность способа заключается в том, что 4,4,4-трифтор-1-фенилбутадионат лития обрабатывают нитритом натрия в ледяной уксусной кислоте при охлаждении, затем избытком гидразин-гидрата. При этом в качестве органического растворителя используют уксусную кислоту. Способ позволяет проводить процесс в одном реакторе, без выделения промежуточных продуктов, без применения повышенного или пониженного давления, при температуре от 10 до 20°C, что приводит к повышению селективности процесса, сокращению количества операций при выделении целевого соединения и обеспечивает получение целевого соединения с выходом 93%. 1 пр.
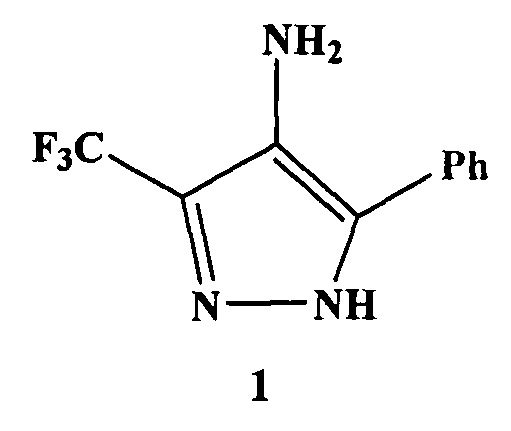
Способ получения 5-фенил-3-(трифторметил)-1H-пиразол-4 амина формулы 1
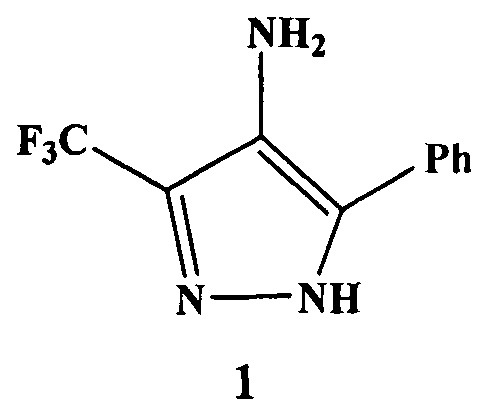
взаимодействием 1,3-дикарбонильного соединения с нитритом натрия в ледяной уксусной кислоте при охлаждении с последующей обработкой избытком гидразин-гидрата в органическом растворителе, отличающийся тем, что в качестве дикарбонильного соединения используют 4,4,4-трифтор-1-фенилбутадионат лития, который обрабатывают нитритом натрия, затем избытком гидразин-гидрата, а в качестве органического растворителя используют уксусную кислоту.
| EMMADI, NARENDER REDDY ET AL, Synthesis and evaluation of novel fluorinated pyrazolo-1,2,3-triazole hybrids as antimycobacterial agents, Bioorganic and Medicinal Chemistry Letters, 2015, 25(15), 2918-2922 | |||
| Способ обработки целлюлозных материалов, с целью тонкого измельчения или переведения в коллоидальный раствор | 1923 |
|
SU2005A1 |
| V.I | |||
| SALOUTIN ET AL, Synthesis of fluoroalkyl-containing 2-oxyimino-1,3-dicarbonyl compounds and their reaction with hydrazine hydrate, Journal of Fluorine Chemistry, 1997, 84, 107-111 | |||
| ПРОИЗВОДНЫЕ ПИРАЗОЛА И СПОСОБ ИХ ПОЛУЧЕНИЯ | 2003 |
|
RU2315758C2 |

