Настоящее изобретение относится к области биотехнологии, в частности к генетической инженерии, и может быть использовано в микробиологической промышленности при получении полусинтетических бета-лактамных антибиотиков нового поколения.
Предлагается рекомбинантная плазмида pSVH0106, содержащая фрагмент ДНК, который кодирует полную аминокислотную последовательность ацилазы глутарил-7-аминоцефалоспориновой кислоты штамма Brevundimonas diminuta BKM В-1297, и обеспечивающая высокий уровень ее экспрессии в клетках Escherichia coli, и рекомбинантный штамм E.coli BL21(DE3)/pSVH0106- продуцент Gl7АСА-ацилазы
Уровень техники
Важнейшим соединением-предшественником для получения антибиотиков из семейства цефалоспоринов является 7-аминоцефалоспориновая кислота (7-АСА) [1]. Конверсия цефалоспорина С в 7-АСА может осуществляться либо путем химического гидролиза, требующего использования крайне токсичных соединений и специальных условий инкубации при сверхнизких температурах, либо за счет энзиматического превращения. Известные процессы энзиматической трансформации могут быть разделены на 2 группы: а) одностадийные с использованием, например, цефалоспорин С-ацилазы из Pseudomonas[2] и б)двустадийные, включающие превращение цефалоспорина С в глутарил-7-АСА (Gl7ACA) с использованием фермента оксидазы D-аминоксилот (DAO; ЕС-1.4.3.3), в частности получаемой из Trigonopsis variabilis, Aspergillus, Penicillium, Neurospora, Pseudomonas, Cephalosporium, и последующий гидролиз глутарил-7-АСА с образованием 7-АСА либо под действием специфической 7-бета-(4-карбоксилбутанамино) ацилазы (Gl7АСА-ацилазы) [3], либо под действием ферментов других классов, относящихся к гамма-глутамил-траснферазам или цефалоспорин-С-деацетилазам [4].
Наибольший практический интерес для биокаталитического синтеза 7-АСА представляет фермент Cl7АСА-ацилаза [5], относящийся к обширному семейству пенициллин-амидаз (ЕС 3.5.1.11), способных превращать пенициллины и цефалоспорины в ценные промежуточные соединения, используемые для производства новых антибиотиков [6,7]. Gl7АСА-ацилазы представляют собой гетеротетрамеры, состоящие из 2-х α и 2-х β субьединиц [8] и характеризуются высокой активностью по отношению к Gl7ACA при низкой активности по отношению к цефалоспорину С[3].
К настоящему времени известны первичные структуры генов бактериальных Gl7АСА-ацилаз из различных штаммов Pseudomonas, описаны рекомбинантные плазмидные ДНК, обеспечивающие синтез этих ферментов в клетках E.coli и Bacillus subtilis, а также способы получения рекомбинантных форм ферментов с их использованием [4, 8-14].
Общими недостатками известных методов гетерологичной экспрессии Gl7ACA-ацилаз являются невысокий уровень синтеза рекомбинантного продукта [15] и во многом связанные с этим сложности процессов масштабирования ферментации штаммов [16,17], а также необходимость освобождения полученных препаратов Gl7АСА-ацилаз от примесей неспецифических бета-лактамаз [3]. В связи с этим актуальной остается задача разработки новых средств, обеспечивающих повышение выхода Gl7АСА-ацилазы при получении ее технологией рекомбинантных ДНК и упрощение методов очистки этих ферментов.
Раскрытие изобретения
Решение задачи повышения выхода активной рекомбинантной Gl7АСА-ацилазы предполагает идентификацию бактериальных штаммов, эффективно синтезирующих в естественных условиях этот фермент, выделение из них кодирующих фермент генов и создание оптимальных векторных конструкций для их эффективной и стабильной экспрессии в гетерологичной бактериальной клетке. Эти аспекты и составили цель предлагаемого изобретения.
Поставленная цель была достигнута за счет того, что
1) в коллекции микроорганизмов обнаружен штамм (Brevundimonas diminuta BKM В-1297), активно продуцирующий Gl7АСА-ацилазу (BrdGl7ACA);
2) из этого штамма получен ген Gl7АСА-ацилазы и определена последовательность ДНК, кодирующая полную аминокислотную последовательность фермента;
3) сконструирована рекомбинантная плазмидная ДНК pSVH0106, обеспечивающая синтез BrdGl7ACA в клетках кишечной палочки с высоким выходом;
4) в результате трансформации клеток экспрессирующей плазмидой pSVH0106 получен рекомбинантный штамм Escherichia coli BL21(DE3)/ pSVH0106 с высоким уровнем индуцибельного синтеза активной Gl7АСА-ацилазы.
Штамм Brevundimonas diminuta ВКМ В-1297 депонирован ранее во Всероссийской коллекции микроорганизмов и отобран нами как наиболее активный продуцент Gl7ACA-ацилазы (BrdGl7ACA).
Полная кодирующая последовательность гена BrdGl7ACA получена методом полимеразной цепной реакции (ПЦР) с использованием в качестве матрицы хромосомной ДНК, выделенной из штамма Brevundimonas diminuta ВКМ В-1297, а в качестве праймеров - синтетических олигонуклеотидов с нуклеотидными последовательностями SEQ ID №12 (праймер 7-aca-F) и SEQ ID№13 (праймер 7-aca-R), специфичными по отношению к N- и С-концевым областям кодирующей части (от положения 103 до положения 2265) изолированного фрагмента гена BrdGL7ACA с SEQ ID N06.
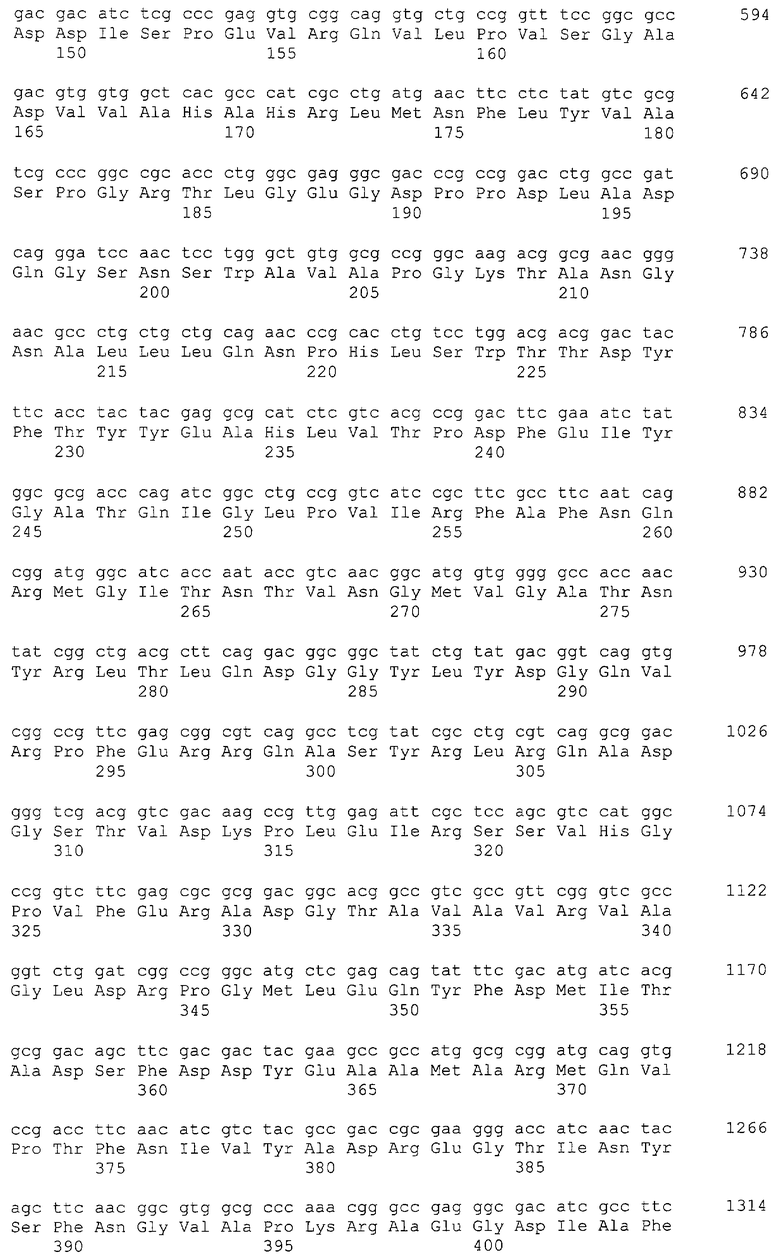
Данная последовательность кодирует белок размером 74кДа (SEQ ID N0 7), обладающий высокой степенью гомологии с другими известными предшественниками бактериальных Gl7ACA-ацилаз, и включает N-концевую сигнальную последовательность секреции и последовательность, кодирующую две субъединицы фермента, разделенные спейсерным пептидом.
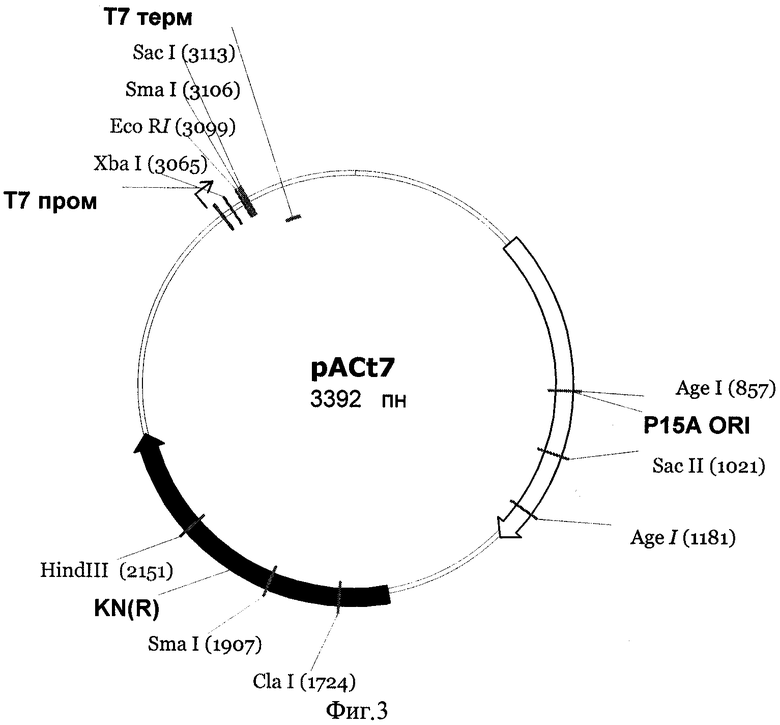
На первом этапе создания рекомбинантной плазмиды для экспрессии фрагмента ДНК, кодирующего Gl7ACA-ацилазу, был сконструирован вектор-носитель рАСТ7, физическая и генетическая карты которого представлены на фиг.3. Данный вектор включает промотор и терминатор РНК-полимеразы фага Т7, разделенные участком полилинкера, репликон p15A и kanR-ген, кодирующий аминогликозид-3-фосфотрансферазу и обеспечивающий устойчивость клеток E.coli, несущих плазмиду, к канамицину. Использование в качестве маркера гена устойчивости к канамицину, а не традиционно применяемого в подобных конструкциях гена устойчивости к ампициллину, освобождает от необходимости последующей сложной очистки целевого белка для удаления примесей бета-лактамаз.
Экспрессирующая рекомбинантная плазмида pSHV0106 получена путем встраивания кодирующей последовательности гена BrdGl7ACA в вектор рАСТ7 по сайтам рестрикции EcoRI и Sac I (фиг.4).
Рекомбинантный штамм-продуцент BrdGL7ACA был получен путем трансформации клеток Escherichia coli BL21(DE3) сконструированной плазмидой pSVH0106. Выбор штамма-реципиента обусловлен тем, что он несет ген РНК-полимеразы фага Т7, которая необходима для обеспечения эффективной транскрипции целевых генов, находящихся в векторной конструкции под контролем промотора фага Т7, а также тем, что данный штамм дефектен по синтезу протеаз, что существенно повышает выход синтезируемых гетерологичных белков. Синтез BrdGL7ACA в штамме BL21(DE3)/ pSVH0106 осуществляется при культивировании на обычных селективных средах с добавлением индуктора изопропил-D-тиогалактозида (ИПТГ) или лактозы.
Полученный рекомбинантный штамм Escherichia coli BL21(DE3)/ pSVH0106 характеризуется высокой физиологической стабильностью (снижение активности продуцируемого фермента после 50 генераций не превышает 20%) и высоким выходом активного продукта, который не менее чем в 1,5 раза превышает продуктивность известных штаммов.
Таким образом, настоящее изобретение включает два объекта.
Первым объектом является рекомбинантная плазмида pSVH0106, обеспечивающая синтез ацилазы глутарил-7-аминоцефалоспориновой кислоты (Gl7ACA-ацилазы) в клетках Escherichia coli, которая характеризуется тем, что содержит фрагмент ДНК с нуклеотидной последовательностью SEQ ID №6 от нуклеотида в положении 103 до нуклеотида в положении 2265, кодирующий полную аминокислотную последовательность Gl7ACA-ацилазы штамма Brevundimonas diminuta ВКМ В-1297, встроенный по сайтам рестрикции EcoRI и SacI в область полилинкера вектора рАСТ7, состоящего из фрагмента модифицированной в области полилинкера плазмиды рЕТ21d, содержащего промотор и терминатор РНК-полимеразы фага Т7, разделенные участком полилинкера, и фрагмента плазмиды рСYC117, содержащего репликон р15А и ген устойчивости к канамицину, объединенных между собой, как показано на фиг 3.
Второй объект изобретения - рекомбинантный штамм Escherichia coli BL21(DE3)/ pSVH0106-продуцент BrdGL7ACA.
Технический результат, достигаемый при осуществлении настоящего изобретения, заключается в создании новых средств (вектора экспрессии и рекомбинантного штамма), обеспечивающих повышение количества (выхода) и качества (отсутствие примесей бета-лактамазы) получаемой рекомбинантной Gl7ACA-ацилазы.
Особенности настоящего изобретения поясняются лучшими вариантами его выполнения со ссылками на фигуры.
Краткое описание фигур.
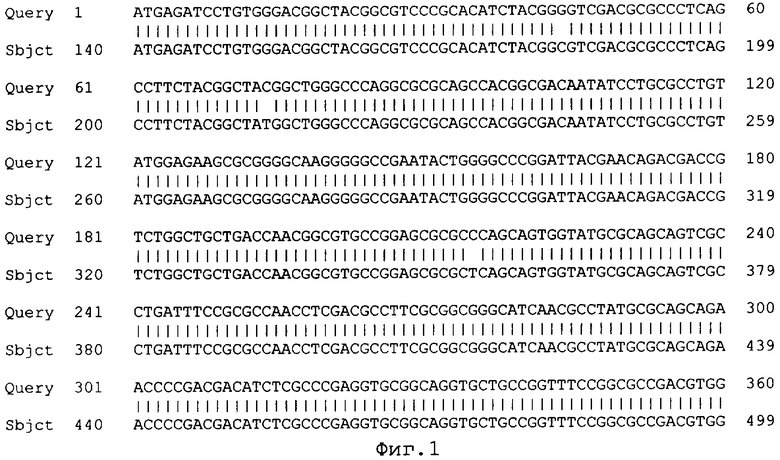
Фиг.1 Сравнение нуклеотидных последовательностей центральной кодирующей части гена Gl7ACA штамма Brevundimonas diminuta ВКМ-1267 (Query) и гомологичного участка гена глутарил ацилазы Pseudomonas SY-77 (Sbjct) с помощью программы BLAST. Степень сходства последовательностей составляет 98%.
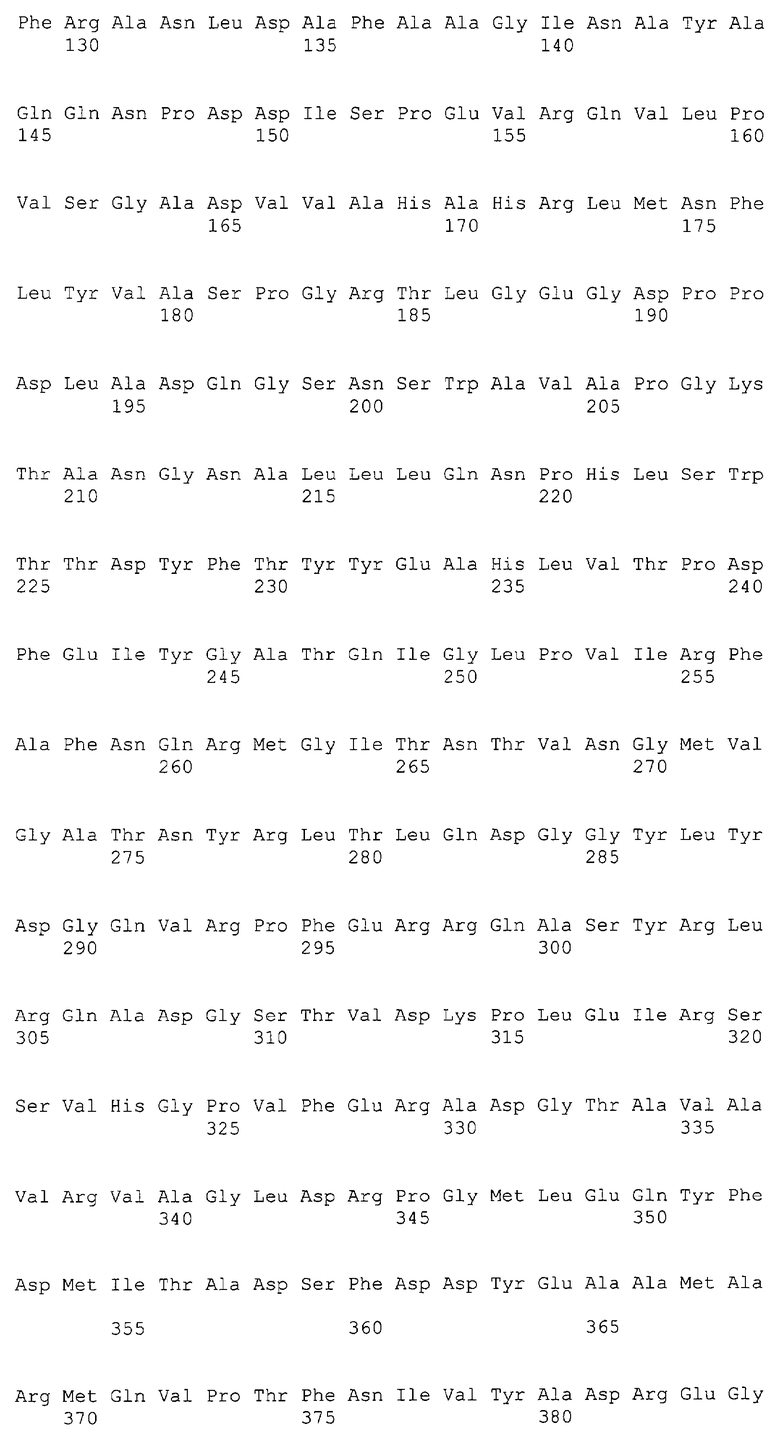
Фиг.2а и 2б Сравнение аминокислотных последовательностей известных Gl7ACA ацилаз из Pseudomonas sp.130 (AAC34685), Pseudomonas sp.SY-77 (AAN39264), Pseudomonas sp.THA1 (AAP68796) c последовательностью BrdGl7ACA. Степень сходства последовательностей составляет 98%.
Фиг.3 Физическая и генетическая карты вектора pACT7. Обозначены положения индикаторных сайтов рестрикции, участки промотора и терминатора РНК-полимеразы фага Т7 (Т7пром и Т7 терм), область начала репликации (p15Aori), ген устойчивости к канамицину (обозначен KN(R), заполнение черным).
Фиг.4 Физическая и генетическая карты плазмиды pSVH0106. Обозначено положение гена BrdGl7ACA, (заполнение серым), остальные обозначения - как на Фиг.3.
Осуществление изобретения
При осуществлении изобретения помимо методов, подробно раскрытых в нижеследующих примерах, использовали хорошо известные специалистам методики, описанные в руководствах по молекулярной биологии и генетической инженерии [18, 19].
Пример 1. Идентификация штамма Brevundimonas с высоким уровнем биосинтеза Gl7ACA-ацилазы
Культуры изолятов различных штаммов лабораторной коллекции выращивали в колбах Эрленмейера объемом 100 мл в 15 мл среды, содержащей 2% гидролизат казеина, 0,5% глутамат натрия, 0,5% дрожжевой экстракт, 0,2% кукурузный экстракт, 0,1% глутаровая кислота (рН 8,2) при 28°С и постоянном перемешивании (150 об/мин) в течение 16 часов. Когда плотность культуры достигала величины 2 ОЕ при длине волны 600 нм, клетки собирали центрифугированием (6000 об/мин; 10 мин; +4°С), промывали в буфере ТЕ (50 мМ Tris pH 8,0 50 mM EDTA), суспендировали в том же буфере (0.1 г влажных клеток на 200 мкл буфера) и хранили до использования в замороженном виде.
В каждом из полученных образцов определяли глутарил-ацилазную активность, для чего использовали колориметрический метод, аналогичный методу, предложенному для определения 6-аминопенициллиновой кислоты [20]. Для этого 100 мкл 65 mM раствора глутарил-7-АСА в 0.1M фосфатном буфере (рН 7,0) добавляют к 900 мкл суспензии клеток, приготовленной на том же (0.1M фосфатном) буфере, содержащем 3 мг/мл клавулината калия, и инкубируют смесь при 37°С от 30 мин до 4 часов. Через определенные промежутки времени смесь центрифугируют, отбирают 500 мкл супернатанта и смешивают его с 3 мл смеси (2:1) 20% уксусной кислоты и 0,5 М раствора NaOH для остановки реакции. К пробе добавляют 0.5 мл p-диметиламинобензальдегида (PDAB) и после развития окраски измеряют поглощение при 415 нм. За одну единицу активности фермента принимается его количество, необходимое для превращения 1 мкмоль субстрата в указанных условиях в течение 1 мин инкубации.
Уровень определенной таким образом ацилазной активности в 64 проанализированных штаммах колебался от 0,01ед/л до примерно 4 ед/л культуры. Наиболее высокой активностью обладали штаммы Brevundiminas diminuta ВКМ B-1297 и Brevundimonas vesicularis ВКМ B-974-, полученные из Всероссийской коллекции микроорганизмов. Штамм Brevundimonas diminuta ВКМ B-1297 был отобран для дальнейшей работы.
Пример 2. Получение хромосомной ДНК из штамма Brevundimonas diminuta ВКМ B-1297
К 200 мкл суспензии клеток штамма B.diminuta ВКМ В-1297, полученной, как описано в примере 1, добавляют 10% SDS до конечной концентрации 2% и 20 мкл протеиназы К (3000 ед /мл) и инкубируют сначала 30 мин при 37°С, а затем 15 мин при 75°С. К суспензии добавляют 200 мкл промытых кислотой стеклянных бус (диаметром 0,1 мм) и подвергают интенсивному встряхиванию на вортексе в течение 5 минут. Затем к суспензии добавляют 300 мкл насыщенного буфером ТЕ раствора фенола и встряхивают на вортексе 30 сек. Смесь центрифугируют, супернатант отбирают и дважды экстрагируют водонасыщенным раствором хлороформа. Водную фазу отбирают и добавляют 5М раствор ацетата калия (рН 6,0) до конечной концентрации 1,5 М. Раствор центрифугируют и отбрасывают осадок калиевой соли SDS. К супернатанту добавляют равный объем изопропанола. Осадок нуклеиновых кислот собирают центрифугированием, растворяют в 300 мкл 0,5 М ацетата калия, повторно осаждают добавлением 3 объемов этанола и растворяют в 100 мкл деионизованной воды при нагревании в течение 30 мин при 65°. Полученную данным способом хромосомную ДНК штамма Brevundiminas diminuta ВКМ B-1297 использовали для получения гена, кодирующего Gl7ACA- ацилазу.
Пример 3. Выделение центрального фрагмента гена BrdGl7ACA
Предварительно было проведено сравнение аминокислотных и нуклеотидных последовательностей бактериальных Gl7ACA-ацилаз с использованием информации, доступной с помощью Интернет на сайте Национального Центра Биотехнологической Информации США. Были выявлены наиболее консервативные участки GL7ACA-ацилаз бактериального происхождения и синтезированы два синтетических олигонуклеотида brd1 (SEQID N01) и brd2 (SEQ ID N02), ограничивающие центральную кодирующую последовательность гена Gl7ACA-ацилазы:
Для осуществления ПЦР хромосомную ДНК Brevundiminas diminuta ВКМ B-1297, выделенную, как описано выше, денататурируют путем нагревания при 100°С в течение 5 минут, помещают в лед, подвергают 30 циклам в 50 мкл смеси следующего состава:
5 мкл 10-кратного Taq-SE ПЦР-буфера (Сибэнзим)
5 мкл геномной ДНК (200 нг/мкл)
5 мкл 3 мкМ праймера brd1
5 мкл 3 мкМ праймера brd2
5 мкл 2,5 мМ dNTP (смесь всех четырых видов дезоксинуклеотидтрифосфатов)
25 мкл деионизованной воды
1 мкл Taq-SE полимеразы (5 ед/мкл, Сибэнзим)
Условия проведения реакции: 94°, 5' (денатурация), 94°, 30'', 50°, 30'', 72°,1' (амплификация). После амплификации 5 мкл ПЦР смеси анализируют электрофорезом в 1% агарозном геле. При разделении в электрофорезе идентифицировали гомогенный фрагмент размером около 1 тпн. Фрагмент выделяли из геля с помощью набора Wizard PCR Preps Kit (Promega, США) (в соответствии с инструкцией производителя) и подвергали автоматическому секвенированию на приборе ABIPrizm 3100 DNA Sequencer с использованием праймеров brd1, brd2 и набора Applera "fluorescent Big dye Cycle sequencing kit".
Сравнение полученной нуклеотидной последовательности с базой данных GenBank с помощью программы BLAST показало, что она обладает высокой степенью гомологии с соответствующими кодирующими последовательностями других бактериальных Gl7ACА-ацилаз (фиг 1). На этом основании был сделан вывод, что полученный фрагмент соответствует центральной части гена Brd Gl7ACA - ацилазы штамма Brevundiminas diminuta ВКМ B-1297.
Пример 4. Получение, секвенирование и анализ фрагмента ДНК, включающего полную кодирующую последовательность гена BrdGl7ACA
Фрагмент ДНК, включающий полную кодирующую последовательность, получают с помощью ПЦР-амплификации с праймерами (BrdGl7ACA_F с SEQID N03 и BrdGl7ACA_R с SEQ ID N04), специфичными к консервативным участкам, фланкирующим полные кодирующие последовательности известных генов Gl7ACA-ацилаз псевдомонад.
1 мкг геномной ДНК подвергают 25 циклам ПЦР-амплификации с праймерами BrdGl7ACA_F / BrdGl7ACA_R и использованием набора Expand Long Template PCR system ("Roche Diagnostics GmbH", Mannheim, Германия) в соответствии с инструкцией изготовителя.
Реакционную смесь разделяют электрофорезом в 1% агарозном геле и выявляют продукт реакции - единичный фрагмент размером около 2, 25 т.п.н. Фрагмент выделяют из геля с помощью набора "Wizard PCR preps Purification System" (Promega, США) и секвенируют с использованием праймеров BrdGl7ACA_F, BrdGl7ACA_R, brd1, brd2 и brd 3 (SEQ ID N05).
Установленная таким образом нуклеотидная последовательность гена BrdGl7ACA приведена в перечне под номером SEQ ID N06. Выведенная из нее аминокислотная последовательность белка BrdGl7ACA (SEQ ID N07) имеет 98%-ное сходство с аминокислотными последовательностями других известных бактериальных GL7ACA- ацилаз (фиг 2), при этом имеющиеся отличия локализованы в сайтах, удаленных от участков активного центра фермента и областей межсубъединичных контактов [8]. Эти данные позволяют с высокой степенью вероятности заключить, что фермент BrdGl7ACA соответствует другим бактериальным Gl7ACA-ацилазам и по своим энзиматическим и физико-химическим свойствам.
Пример 5. Конструирование вектора-носителя pACT7
Векторы на основе промотора фага Т7 обеспечивают высокую экспрессию гетерологичных генов в штаммах E.coli, синтезирующих T7 полимеразу [21], и коммерчески доступны [22]. В то же время известно, что иммобилизованные препараты Gl7ACA-ацилаз, используемые для биотрансформации цефалоспориновых антибиотиков, не должны содержать примесей бета-лактамаз. Присутствие бета-лактамазы также затрудняет количественный анализ уровней продукции Gl7ACA-ацилаз в рекомбинантных штаммах-продуцентах. Поэтому для экспрессии BrdGl7ACA и других ферментов биотрансформации антибиотиков, свободных от примесей бета-лактамаз, целесообразно использовать векторы, несущие маркеры плазмидной устойчивости, отличные от AmpR. Такие векторы обладают также более высокой сегрегационной стабильностью [22], которая может быть дополнительно повышена за счет использования репликона p15A вместо репликона ColE1[23].
Конструирование вектора pACT7, удовлетворяющего указанным условиям, проводили в несколько стадий.
А) Конструирование промежуточной плазмиды pET2ldRI.
C использованием метода "инвертированной ПЦР" и праймеров pETR1_F (SEQ ID N08) и pETR1_R (SEQ ID N09) на матрице пламиды pET21d [21] с использованием набора Expand Long Template PCR system получают уникальный ПЦР фрагмент размером 5,4 тпн, который выделяют из агарозного геля, как описано в примере 4. 100 нг полученного фрагмента гидролизуют 5 единицами рестриктазы EcoRI (Fermentas) и лигируют с помощью Т4 ДНК лигазы. Полученной лигазной смесью трансформируют компетентные клетки штамма Escherichia coli XL1-Blue recA1 endA1 gyrA96 thi-1 hsdR17 supE44 relA1 lac [F'proAB lacIqZ M15 Tn10 (Tetr)] (Stratagene, США), затем из полученных ампициллин-устойчивых трансформантов выделяют препараты плазмидной ДНК с использованием набора Wizard MiniPreps Kit (Promega, США). Полученные образцы ДНК анализируют совместным гидролизом рестриктазами EcoRI/PstI и BamHI/PstI и отбирают "положительные" клоны, содержащие EcoRI/PstI фрагменты размером 1300 и 4100 п.н. При этом за счет делеции сайта BamHI в BamHI/PstI гидролизате ДНК плазмидных клонов присутствует уникальный фрагмент размером 5400 пн, в то время как в препарате плазмидной ДНК pET21d, гидролизованной BamHI/PstI, обнаруживаются фрагменты размером 1300 и 4100 п.н. Несколько "положительных" клонов проверяют секвенированием с использованием праймеров Т7prom (SEQ ID N0 10) и T7term (SEQ ID N0 11) и отбирают клон с модифицированным таким образом участком полилинкера, не содержащий неспецифических мутаций, который обозначают как pET21dR1.
M15 Tn10 (Tetr)] (Stratagene, США), затем из полученных ампициллин-устойчивых трансформантов выделяют препараты плазмидной ДНК с использованием набора Wizard MiniPreps Kit (Promega, США). Полученные образцы ДНК анализируют совместным гидролизом рестриктазами EcoRI/PstI и BamHI/PstI и отбирают "положительные" клоны, содержащие EcoRI/PstI фрагменты размером 1300 и 4100 п.н. При этом за счет делеции сайта BamHI в BamHI/PstI гидролизате ДНК плазмидных клонов присутствует уникальный фрагмент размером 5400 пн, в то время как в препарате плазмидной ДНК pET21d, гидролизованной BamHI/PstI, обнаруживаются фрагменты размером 1300 и 4100 п.н. Несколько "положительных" клонов проверяют секвенированием с использованием праймеров Т7prom (SEQ ID N0 10) и T7term (SEQ ID N0 11) и отбирают клон с модифицированным таким образом участком полилинкера, не содержащий неспецифических мутаций, который обозначают как pET21dR1.
Б) Конструирование плазмиды pET21dRl/Tet.
Для удаления гена AmpR из плазмиды pET21dR1 и замены его геном устойчивости к тетрациклину проводили конструирование плазмиды pET21dR1/Tet. Для этого для получения "вставки" несущей ген устойчивости к тетрациклину 2 мкг плазмидной ДНК pKRP12[24] гидролизовали рестриктазой PstI и выделяли из агарозного геля фрагмент размером 1200 п.н. В качестве вектора использовали гидролизованную по сайту PstI плазмиду pET21dR1. Использовали следующие условия лигирования: 1 мкл вектора (20 нг/мкл), 1 мкл фрагмента (50 нг/мкл), 2 мкл 5-кратного лигазного буфера (Gibco-BRL), 5 мкл воды и 1 мкл Т4 ДНК лигазы (1 ед/мкл, Сибэнзим). Смесь инкубировали в течение 14 часов при 12°С, затем прогревали 15 мин при 65°С, охлаждали во льду и 5 мкл смеси использовали для трансформации компетентных клеток штамма E. coli JM109[19]. Отбирали тетрациклин-устойчивые клоны, чувствительные к ампициллину. Дополнительную селекцию нужных клонов проводили с помощью рестриктного анализа препаратов плазмидной ДНК (по образованию фрагментов размером 1200, 1300, 4300 п.н. при гидролизе EcoRI+PstI). Один из отобранных клонов обозначили как pET21dR1/Tet и использовали в дальнейшей работе.
В) Конструирование плазмиды pACYCpET.
Для получения производного вектора pET21dRI с репликоном p15A ДНК плазмиды pACYC177 [20] гидролизовали совместно рестриктазами BamHI+PstI и выделяли из агарозного геля фрагмент размером 3000 п.н., включающий область репликона р15А, ген устойчивости к канамицину и С-концевую кодирующую часть гена бета-лактамазы (устойчивости к ампициллину). Плазмиду pET21dR1 гидролизовали совместно BglII+PstI и выделяли из агарозного геля фрагмент размером 1,5 т.п.н., включающий участок полилинкера, промотор и терминатор Т7-фага и N-концевую кодирующую часть гена бета-лактамазы. Два фрагмента лигировали, полученной лигазной смесью трансформировали штамм E. Coli Xl1Blue и отбирали трансформанты, устойчивые одновременно к ампициллину и канамицину.
Выделенные из культур полученных клонов плазмидные ДНК анализировали путем рестрикции эндонуклеазой SmaI и отбирали клоны, образующие фрагменты размером 3,2 и 1,2 тпн. Один из отобранных клонов обозначили как pACYCpET.
Г) Конструирование вектора pACT7.
Для удаления гена бета-лактамазы из вектора pACYCpET препарат плазмидной ДНК гидролизуют совместно рестриктазами NaeI/FspI, выделяют из агарозного геля фрагмент размером 3,4 тпн, лигируют "сам на себя" по "тупым концам" и лигазной смесью трансформируют компетентные клетки штамма E. coli JM109. Из отобранных канамицин-устойчивых и ампициллин-чувствительных трансформантов выделяют плазмидную ДНК и идентифицируют нужный клон по данным рестриктного анализа (наличие фрагментов размером 1.2 и 2,2 тпн в SmaI-гидролизатах препаратов плазмидной ДНК).
Пример 6. Конструирование плазмид для экспрессии BrdGL7ACA в E.coli.
Сначала методом ПЦР получают фрагмент ДНК штамма Brevundiminas diminuta ВКМ B-1297, содержащий полную кодирующую последовательность BrdGl7ACA (без прилежащих не кодирующих последовательностей) и фланкированный уникальными сайтами рестрикции.
Для этого 1 мкг геномной ДНК штамма ВКМ B-1297 подвергают 25 циклам ПЦР-амплификации с праймерами 7-aca_F/7aca_R (SEQ ID N012,13) с использованием набора Expand Long Template PCR system ("Roche Diagnostics GmbH", Mannheim, Германия) в соответствии с инструкцией изготовителя. Использованные праймеры ограничивают фрагмент гена BrdGL7ACA, включающий инициаторный кодон трансляции, расположенного в положении (+103) последовательности SEQID N06, и терминаторный кодон TGA, расположенный в положении+2265 той же последовательности и несут сайты для рестриктаз EcoRI и SacI, отсутствующие в кодирующей последовательности гена BrdGl7АСА.
Реакционную смесь разделяют электрофорезом в 1% агарозном геле и выявляют продукт реакции - единичный фрагмент размером около 2,16 т.п.н. Выделенный из агарозного геля фрагмент гидролизуют рестриктазами EcoRI/SacI и клонируют в EcoRI/SacI вектор pET21dR1/Tet (см. пример 5 "Б"). Отбирают тетрациклин-устойчивые трансформанты штамма E.coli JM109 по критерию образования фрагментов размером 2,2 и 6,5 тпн в EcoRI/SacI гидролизатах полученных препаратов плазмидной ДНК.
С целью выявления клона, несущего вставку с интактным геном BrdGL7ACA без возможных мутаций, привнесенных за счет ПЦР-амплификации, проверяют способность отобранных плазмид направлять синтез функционально-активной BrdGL7ACA при их переносе в штамм E.coli BL21(DE3) [22].
Для этого индивидуальные трансформанты штамма E.coli BL21(DE3), несущие полученные плазмиды, выращивают на LB-среде, содержащей 10 мкг/мл тетрациклина в объеме 5 мл до ОП600˜ 1,0. После этого в культуры вносят ИПТГ до конечной концентрации 1 мкМ для индукции промотора фага Т7 и продолжают культивирование в течение 18 часов при температуре 25-30°С. Клетки собирают центрифугированием и проводят определение активности Gl7ACA-ацилазы, как описано в примере 1. Отобранные "положительные клоны" секвенируют с использованием праймеров T7prom, T7term, brd1, brd2, brd3 и идентифицируют клон, несущий вставку с интактным геном BrdGl7ACA, кодирующая последовательность которого идентична последовательности фрагмента ДНК от положения+103 до положения+2265 определенной кодирующей последовательности гена BrdGl7ACA, приведенной в перечне последовательностей под номером SEQ ID N07. Плазмиду, выделенную из отобранного клона, обозначают pSVH18.
Для получения экспрессирующей BrdGl7ACA плазмиды c репликоном p15A EcoR1/Sac1 фрагмент с геном BrdGl7ACA из плазмиды pSVH18 клонируют в вектор pACТ7 с образованием плазмиды pSVH0106.
Пример 7 Получение рекомбинантных штаммов E. coli с использованием плазмид pSVH18 и pSVH0106.
Для получения штаммов-продуцентов рекомбинатной BrdGl7ACA с использованием стандартных методов [18] проводят трансформацию реципиентного штамма E.coli BL21(DE3) [22] полученными рекомбинантными плазмидными ДНК. На чашках с LB средой, содержащей 10 мг/л тетрациклина, отбирают индивидуальные тетрациклин-устойчивые трансформанты, полученные с использованием плазмидной ДНК pSVH18. Первичные трансформанты культивируют в жидкой среде LB, содержащей 10 мг/л тетрациклина, проводят индукцию синтеза BrdGl7ACA с использованием ИПТГ, как описано в примере 6. Один из отобранных клонов обозначают как E.coli BL21(DE3)/pSVH18, выращивают его в 5 мл жидкой среды LB, содержащей 10 мг/л тетрациклина до ОП600˜ 5,0, добавляют к культуре равный объем стерильного 30% глицерина, разливают по 500 мкл в стерильные 2 мл пробирки, замораживают и хранят при - 70°С.
Аналогичным образом получают первичные трансформанты штамма E.coli BL21(DE3) плазмидой pSVH0106. Индивидуальные канамицин-устойчивые трансформанты выращивают в жидкой среде LB с добавлением 30 мг/л канамицина, проверяют на способность к синтезу функционально активной BrdGl7ACA, отбирают отдельный клон E.coli BL21(DE3)/ pSVH0106 и хранят при - 70°С.
Пример 8 Определение уровней продукции рекомбинантной BrdGl7ACA и физиологической стабильности продуцирующей способности рекомбинантных штаммов.
Для сравнительного количественного определения уровня экспрессии BrdGl7ACA в полученных рекомбинантных штаммах E.coli BL21(DE3)/ pSVH18 и E.coli BL21(DE3)/ pSVH0106 клетки выращивают в колбах Эрленмейера объемом 300 мл в 50 мл LB среды с добавлением соответствующего антибиотика при температуре 25°С до А600 1,0 ОЕ. Далее вносят индуктор - ИПТГ до концентрации 0,2mM и проводят инкубацию в течение 22 часов. Периодически отбирают аликвоты суспензии клеток и используют их для определения роста культуры и активности фермента, как описано в примере 1.
Как видно из представленных данных (Таблица 1), активность фермента достигает максимума через 20 часов инкубации; при этом в штамме E.coli BL21(DE3)/ pSVH0106 она существенно выше (максимальное значение составляет 1200 мЕ/мл культуры), чем в штамме E.coli BL21(DE3)/ pSVH18.
Динамика накопления BrdGl7ACA при культивировании рекомбинантных штаммов E.coli BL21(DE3)/pSVH18 и E.coli BL21(DE3)/pSVH0106
5
10
20
200
450
800
150
400
1200
1,5
2,1
4,8
1,7
2,5
5,6
Проверку физиологической стабильности продуцирующей активности рекомбинантных штаммов в условиях продолжительного культивирования осуществляют следующим образом.
1. Проводят первый цикл культивирования на среде LB так, как это описано выше, и в конце культивирования определяют активность ацилазы (1-й цикл культивирования, соответствует примерно 30 генерациям).
2. 1 мл культуры, полученной после первого цикла, используют в качестве посевного материала, который вносят в 50 мл свежей среды LB и повторяют весь цикл еше раз (2-й цикл культивирования, примерно 5 генераций).
3. Повторяют процедуру еще 4 раза - до 50 генераций.
Как следует из приведенных в Таблице 2 данных, штамм BL21(DE3)/ pSVH0106 обладает более высокой физиологической стабильностью по сравнению со штаммом BL21(DE3)/ pSVH18.
Физиологическая стабильность продукции BrdGl7ACA в клетках рекомбинантных штаммов E.coli BL21(DE3)/pSVH18 и E.coli BL21(DE3)/pSVH0106
Пример 9. Характеристика рекомбинантного штамма E.coli BL21(DE3)/pSVH0106
С учетом того, что удельная активность ацилаз, гомологичных BrdGL7ACA, в среднем составляет около 10000 мЕ/мг белка, выход рекомбинатной BrdGL7ACA в штамме BL21(DE3)/pSVH0106 равен примерно 100 мг белка на литр культуры продуцента. Сопоставление полученных данных с известными ранее литературными сведениями о характеристиках рекомбинантных штаммов E.coli, продуцирующих бактериальные Gl7ACA_ацилазы, позволяют утверждать, что уровень продукции рекомбинантной Gl7ACA_ацилазы в штамме BL21(DE3)/ pSVH0106 не менее чем в 1,5 раза превышает продуктивность известных штаммов [9,12].
Морфологические признаки
Клетки имеют продолговатую палочковидную форму, при делении не почкуются.
Культуральные признаки
Клетки хорошо растут на обычно используемых питательных средах. Время генерации около 30 мин в жидкой LB-среде. На 2-2,5% питательном агаре "Difco" образуются круглые, гладкие, желтоватые колонии с ровными краями. При выращивании на жидких LB- и YT-средах образуется интенсивная ровная мутность.
Физиолого-биохимические признаки
Оптимальная температура культивирования - от 25 до 30°C, оптимум рН - 7,6. Источником азота служат органические соединения (в виде триптона, дрожжевого экстракта).
Уровень синтеза BrdGL7ACA (по данным определения активности в образцах биомассы штамма-продуцента) составляет около 100 мг/л при титре культуры 1×109 кл/мл.












Настоящее изобретение относится к области биотехнологии, в частности к генетической инженерии, и может быть использовано в микробиологической промышленности при получении полусинтетических бета-лактамных антибиотиков нового поколения. Сконструирована рекомбинантная плазмида pSVH0106, содержащая природную последовательность гена ацилазы глутарил-7-аминоцефалоспориновой кислоты штамма Brevundimonas diminuta BKM В-1297, которая кодирует полноразмерный предшественник фермента. В результате трансформации штамма E.coli предложенной рекомбинантной плазмидой и селекции трансформированных клонов получен новый рекомбинантный штамм E.coli ВL21(DЕ3)/рSVН0106-продуцент Gl7ACA-ацилазы, обеспечивающий высокий выход активного фермента. Благодаря эффективной и стабильной экспрессии рекомбинантной Gl7АСА-ацилазы в предложенной системе применение настоящего изобретения позволяет масштабировать процесс получения данного фермента, широко используемого в производстве антибиотиков. 2 с.п. ф-лы, 4 ил., 2 табл.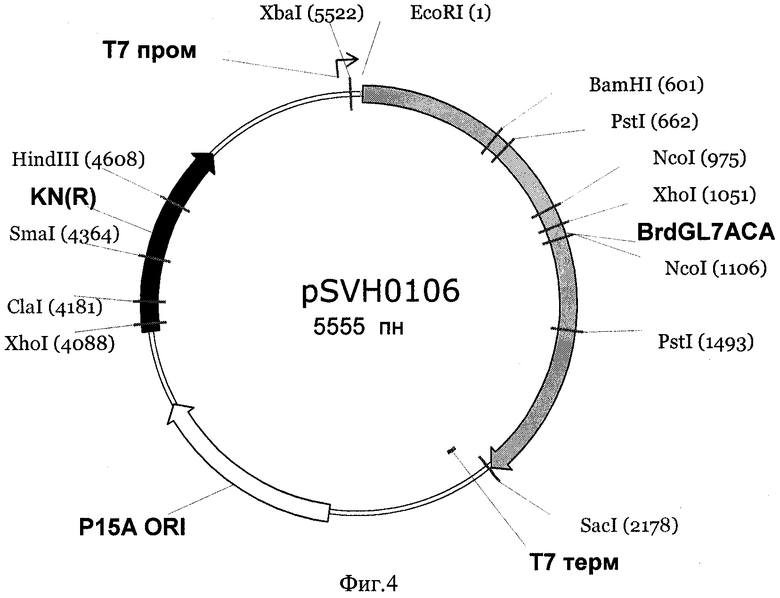
| Способ определения концентрации нефтепродуктов в воде | 1972 |
|
SU469919A1 |
| ISHIYE М., NIWA M., Biochim | |||
| Biophys | |||
| Acta, 1132, 233-239,1992 | |||
| Захватно-срезающее устройство валочно-трелевочной машины | 1974 |
|
SU496993A1 |

