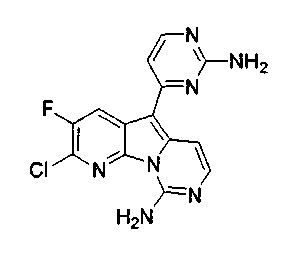
Настоящее изобретение относится к противоопухолевым соединениям и, в частности, к новым противоопухолевым аналогам вариолиновых соединений, включая вариолин B и дезоксивариолин B.
ПРЕДПОСЫЛКИ СОЗДАНИЯ ИЗОБРЕТЕНИЯ
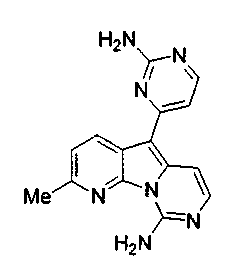
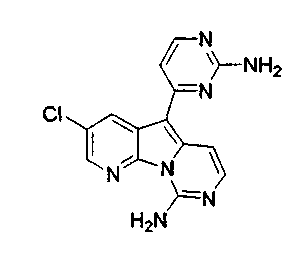
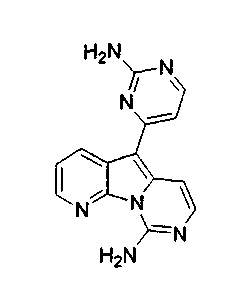
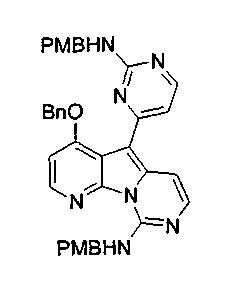
Вариолины, типичным примером которых является вариолин B (1), являются классом морских алкалоидов, выделенных из редкой и труднодоступной антарктической губки Kirkpatrickia varialosa. Все вариолины содержат конденсированный пиридо[3',2':4,5]пирроло[1,2-c]пиримидиновый остов (2) с присоединенным в положении C5 или гетероциклическим ароматическим кольцом, или сложноэфирной группой, как в вариолине B (1), вариолине D (3) и дезоксивариолине B (4).
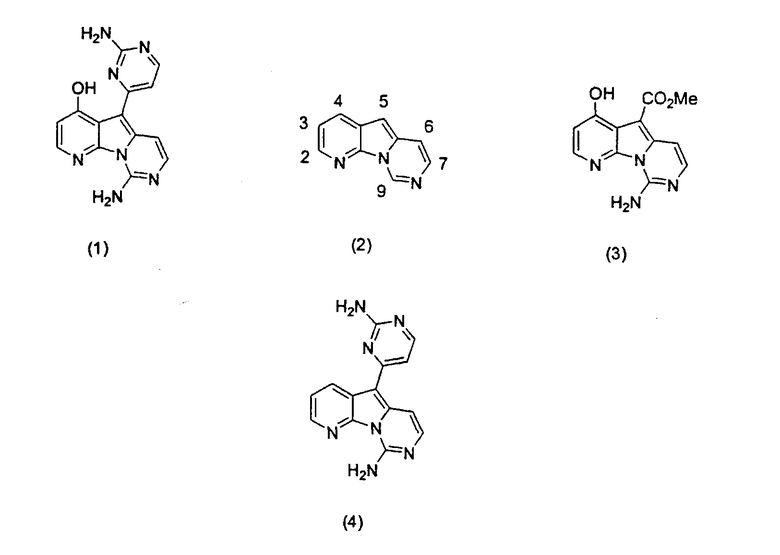
Показано, что вариолины обладают противоопухолевой активностью и другими полезными свойствами. Полное строение этих и родственных соединений представлено в работах N.B. Perry et al., Tetrahedron 1994, 50, 3987-3992 и G. Trimurtulu et al., Tetrahedron 1994, 50, 3993-4000.
Исследования синтеза 2-аминопиримидиновых алкалоидов вариолинов и меридианинов являются предметом работы Tetrahedron Lett. 2000, 41, 4777-4780. В работе M. Alvarez et al., Tetrahedron Lett. 2001, 42, 315-317 описан синтез дезоксивариолина B с применением 7-азаиндола в качестве исходного материала. Исследования полного синтеза вариолинов описаны в работе Tetrahedron Lett. 2001, 42, 311-313. Первый полный синтез вариолина B был описан в работе R.J. Anderson et al., Tetrahedron Lett. 2001, 42, 8697-8699. Позднее в работе P. Molina et al., Tetrahedron Lett. 2002, 43, 1005-1007 также был описан синтез вариолина B путем тандемной аза-Виттиг/карбодиимидной циклизации.
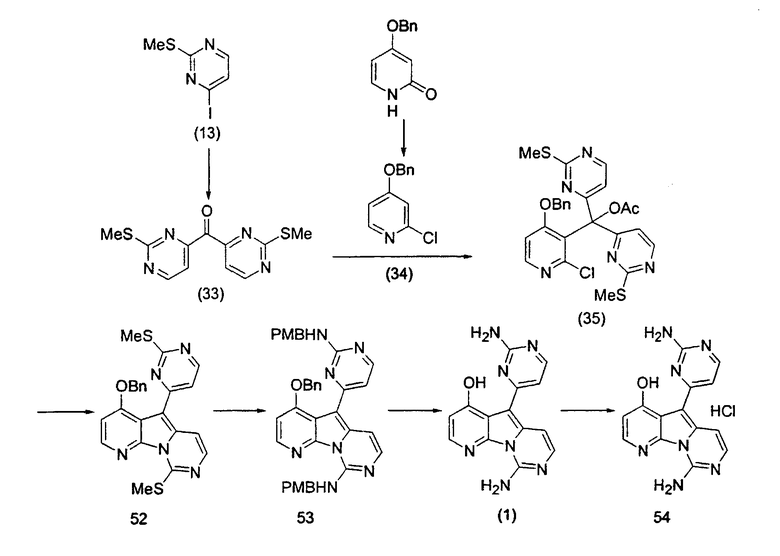
В заявке на выдачу международного патента WO 0204447, опубликованной 17 января 2002 года, вместе с получением новых производных вариолина также описан способ получения вариолина B (1) и дезоксивариолина B (4) из простых моногетероароматических исходных материалов.
Заявка на выдачу международного патента WO 0212240, опубликованная 14 февраля 2002 года, относится к производным вариолина B.
КРАТКОЕ ИЗЛОЖЕНИЕ СУЩНОСТИ ИЗОБРЕТЕНИЯ
Настоящее изобретение относится к соединениям общей формулы (5), которые содержат конденсированную пиридопирролопиримидиновую кольцевую систему вариолиновых соединений:
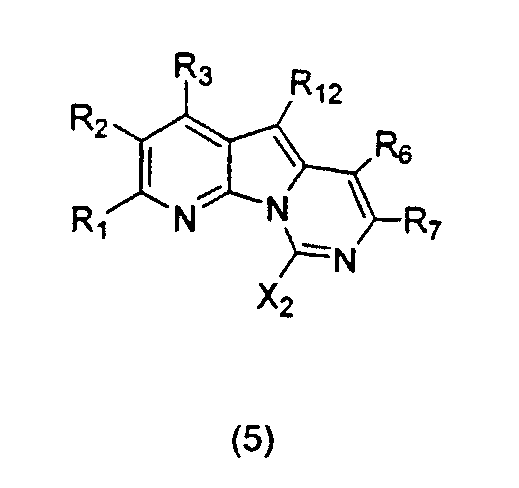
где каждый из заместителей, определенных как X2, R1, R2, R3, R6, R7 и R12, независимо выбран из группы, включающей H, OH, OR', SH, SR', SOR', SO2R', NO2, NH2, NHR', N(R')2, NHCOR', NHSO2R', CN, галоген, =O, C(=O)H, C(=O)R', CO2H, CO2R', карбоксиалкил, C1-C12-алкил, C2-C12-алкенил, C2-C12-алкинил, замещенный или незамещенный арил, замещенный или незамещенный аралкил и замещенную или незамещенную гетероароматическую группу;
где каждая из R'-групп независимо выбрана из группы, включающей H, OH, SH, NO2, NH2, CN, галоген, =O, C(=O)H, C(=O)CH3, CO2H, CO2CH3, C1-C12-алкил, C2-C12-алкенил, C2-C12-алкинил, арил, аралкил и гетероароматическую группу;
где пары групп R1 и R2, R2 и R3, R3 и R12, R12 и R6 или R6 и R7 могут быть объединены в карбоциклическую или гетероциклическую кольцевую систему.
В следующем аспекте настоящее изобретение относится к соединениям формулы (5a):
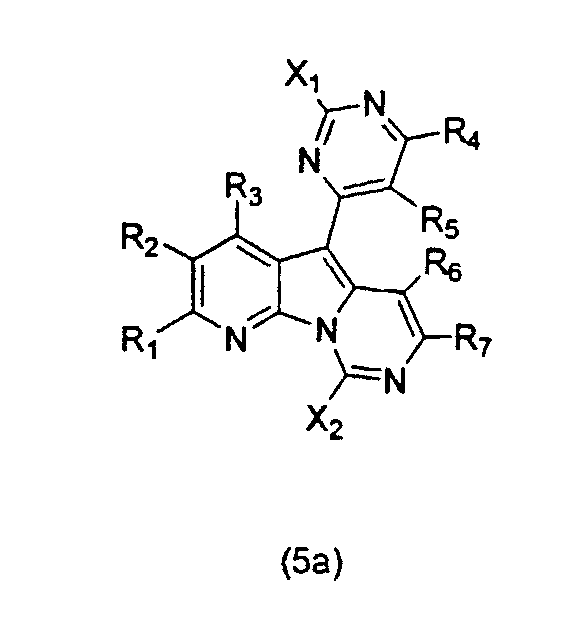
где каждый из заместителей, определенных как X1, X2, R1, R2, R3, R4, R5, R6 и R7, независимо выбран из группы, состоящей из H, OH, OR', SH, SR', SOR', SO2R', NO2, NH2, NHR', N(R')2, NHCOR', NHSO2R', CN, галогена, =O, C(=O)H, C(=O)R', CO2H, CO2R', карбоксиалкила, C1-C12-алкила, C2-C12-алкенила, C2-C12-алкинила, замещенного или незамещенного арила, замещенного или незамещенного аралкила и замещенной или незамещенной гетероароматической группы;
где каждая из R'-групп независимо выбрана из группы, состоящей из H, OH, SH, NO2, NH2, CN, галогена, =O, C(=O)H, C(=O)CH3, CO2H, CO2CH3, C1-C12-алкила, C2-C12-алкенила, C2-C12-алкинила, арила, аралкила и гетероароматической группы;
где пары групп R1 и R2, R4 и R5 или R6 и R7 могут быть объединены в карбоциклическую или гетероциклическую кольцевую систему.
Подходящие галогеновые заместители в соединениях по настоящему изобретению включают F, Cl, Br и I.
Алкильные группы, предпочтительно, содержат от 1 до приблизительно 12 атомов углерода, более предпочтительно, от 1 до приблизительно 8 атомов углерода, еще более предпочтительно, от 1 до приблизительно 6 атомов углерода и, наиболее предпочтительно, 1, 2, 3 или 4 атома углерода. Метил, этил и пропил, включая изопропил, являются особенно предпочтительными алкильными группами в соединениях по настоящему изобретению. Используемый здесь термин «алкил», если не определено иначе, относится и к циклическим и к нециклическим группам, хотя циклические группы должны содержать в составе кольца, по крайней мере, три атома углерода.
Предпочтительные алкенильные и алкинильные группы в соединениях по настоящему изобретению содержат одну или несколько ненасыщенных связей и от 2 до приблизительно 12 атомов углерода, более предпочтительно, от 2 до приблизительно 8 атомов углерода, еще более предпочтительно, от 2 до приблизительно 6 атомов углерода и, еще более предпочтительно, 2, 3 или 4 атома углерода.
Используемые здесь термины «алкенил» и «алкинил» относятся и к циклическим, и к нециклическим группам, хотя неразветвленные или разветвленные нециклические группы являются, как правило, более предпочтительными.
Предпочтительные алкоксигруппы в соединениях по настоящему изобретению включают группы, содержащие одну или несколько кислородных связей и от 1 до приблизительно 12 атомов углерода, более предпочтительно, от 1 до приблизительно 8 атомов углерода и, еще более предпочтительно, от 1 до приблизительно 6 атомов углерода и, наиболее предпочтительно, 1, 2, 3 или 4 атома углерода.
Предпочтительные алкилтиогруппы в соединениях по настоящему изобретению содержат одну или несколько тиоэфирных связей и от 1 до приблизительно 12 атомов углерода, более предпочтительно, от 1 до приблизительно 8 атомов углерода и еще более предпочтительно, от 1 до приблизительно 6 атомов углерода. Алкилтиогруппы, содержащие 1, 2, 3 или 4 атома углерода, являются особенно предпочтительными.
Предпочтительные алкилсульфинильные группы в соединениях по настоящему изобретению включают те группы, которые содержат одну или несколько сульфоксидных (SO) групп и от 1 до приблизительно 12 атомов углерода, более предпочтительно, от 1 до приблизительно 8 атомов углерода и, еще более предпочтительно, от 1 до приблизительно 6 атомов углерода. Алкилсульфинильные группы, содержащие 1, 2, 3 или 4 атома углерода, являются особенно предпочтительными.
Предпочтительные алкилсульфонильные группы в соединениях по настоящему изобретению включают те группы, которые содержат одну или несколько сульфонильных (SO2) групп и от 1 до приблизительно 12 атомов углерода, более предпочтительно, от 1 до приблизительно 8 атомов углерода и, еще более предпочтительно, от 1 до приблизительно 6 атомов углерода. Алкилсульфонильные группы, содержащие 1, 2, 3 или 4 атома углерода, являются особенно предпочтительными.
Предпочтительные аминоалкильные группы включают те группы, которые содержат первичные, вторичные и/или третичные аминогруппы и от 1 до приблизительно 12 атомов углерода, более предпочтительно, от 1 до приблизительно 8 атомов углерода, еще более предпочтительно, от 1 до приблизительно 6 атомов углерода и, еще более предпочтительно, 1, 2, 3 или 4 атома углерода. Вторичные и третичные аминогруппы являются, как правило, более предпочтительными, чем радикалы первичных аминов.
Подходящие карбоксиалкильные группы включают моно- и дикарбоксизамещенные алкильные группы. Карбоксигруппы обычно представлены в виде COOR', в особенности, где R' является водородом или алкилом, предпочтительно, водородом или метилом.
Подходящие гетероциклические группы включают гетероароматические и гетероалициклические группы. Подходящие гетероароматические группы в соединениях по настоящему изобретению содержат один, два или три гетероатома, выбранных из атомов N, O или S, и включают, например, кумаринил, включая 8-кумаринил, хинолинил, включая 8-хинолинил, пиридил, пиразинил, пиримидил, фурил, пирролил, тиенил, тиазолил, оксазолил, имидазолил, индолил, бензофуранил и бензотиазол. Подходящие гетероалициклические группы в соединениях по настоящему изобретению содержат один, два или три гетероатома, выбранных из атомов N, O или S, и включают, например, тетрагидрофуранильную, тетрагидропиранильную, пиперидинильную, морфолино- и пирролидинильную группы.
Подходящие карбоциклические арильные группы в соединениях по настоящему изобретению включают однокольцевые и многокольцевые соединения, включая многокольцевые соединения, которые содержат раздельные и/или конденсированные арильные группы. Типичные карбоциклические арильные группы содержат от 1 до 3 раздельных или конденсированных колец и от 6 до приблизительно 18 атомов углерода в составе колец. Особенно предпочтительные карбоциклические арильные группы включают фенил, включая замещенный фенил, такой как 2-замещенный фенил, 3-замещенный фенил, 4-замещенный фенил, 2,3-замещенный фенил, 2,4-замещенный фенил, 2,5-замещенный фенил, 2,6-замещенный фенил, 3,4-замещенный фенил, 3,5-замещенный фенил, 3,6-замещенный фенил, 2,3,4-замещенный, 2,3,5-замещенный, 2,3,6-замещенный, 2,4,5-замещенный, 2,4,6-замещенный и 3,4,5-замещенный фенил, включая группы, где один или несколько заместителей фенила являются группой, такой как галоген, цианогруппа, нитрогруппа, алканоил, сульфинил, сульфонил и тому подобное; нафтил, включая 1-нафтил и 2-нафтил; бифенил; фенантрил; и антрацил.
Указанные здесь ссылки на замещенные R'-группы в соединениях по настоящему изобретению относятся к конкретному радикалу, который может быть замещен в одном или нескольких доступных положениях одной или несколькими подходящими группами, например галогеном, таким как фтор, хлор, бром и йод; цианогруппой; гидроксилом; нитрогруппой; азидогруппой; алканоилом, таким как C1-6-алканоильная группа, такая как ацил, и тому подобное; карбоксамидогруппой; алкильными группами, включая те группы, которые содержат от 1 до приблизительно 12 атомов углерода, или от 1 до приблизительно 6 атомов углерода и, более предпочтительно, 1-3 атома углерода; алкенильными и алкинильными группами, включая группы, содержащие одну или несколько ненасыщенных связей и от 2 до приблизительно 12 атомов углерода, или от 2 до приблизительно 6 атомов углерода; алкоксигруппами, включая те группы, которые содержат одну или несколько кислородных связей и от 1 до приблизительно 12 атомов углерода или от 1 до приблизительно 6 атомов углерода; арилоксигруппами, такими как феноксигруппа; алкилтиогруппами, включая те радикалы, которые содержат одну или несколько тиоэфирных связей и от 1 до приблизительно 12 атомов углерода, или от 1 до приблизительно 6 атомов углерода; алкилсульфинильными группами, включая те радикалы, которые содержат одну или несколько сульфинильных связей и от 1 до приблизительно 12 атомов углерода или от 1 до приблизительно 6 атомов углерода; алкилсульфонильными группами, включая те радикалы, которые содержат одну или несколько сульфонильных связей и от 1 до приблизительно 12 атомов углерода, или от 1 до приблизительно 6 атомов углерода; аминоалкильными группами, такими как группы, содержащие один или несколько атомов азота и от 1 до приблизительно 12 атомов углерода, или от 1 до приблизительно 6 атомов углерода; карбоциклическим арилом, содержащим 6 или более атомов углерода, в частности фенилом (где, например, R является замещенным или незамещенным бифенильным радикалом); и аралкилом, таким как бензил.
Предпочтительно, из объема настоящего изобретения исключены известные соединения вариолин A, вариолин B, вариолин D, N-(3')-метилтетрагидровариолин B и дезоксивариолин B.
Соединения по настоящему изобретению могут быть синтезированы, используя метод, описанный в заявке на выдачу международного патента WO 0204447, в котором остов вариолина конструируют из простых моногетероароматических исходных материалов, основываясь на следующем ретросинтезе.
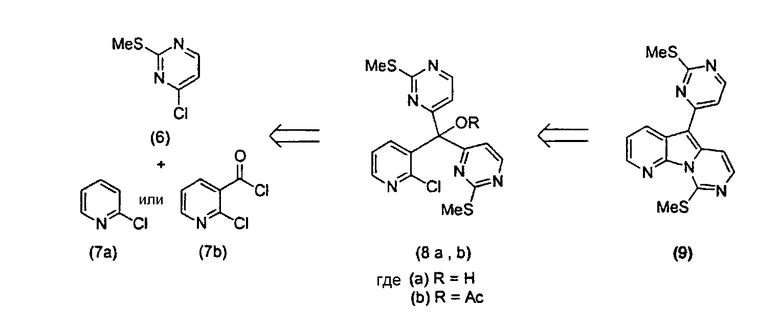
В зависимости от выбора моногетероароматических исходных материалов (6) и (7) методика может быть легко расширена для возможности получения широкого ряда аналогов вариолина, как представлено здесь в примерах.
Заявка на выдачу международного патента WO 0212240 также относится к руководству по синтезу вариолиновых соединений.
Таким образом, настоящее изобретение также относится к путям синтеза соединений по настоящему изобретению.
Противоопухолевая активность данных соединений включает лейкоз, рак легких, рак толстой кишки, рак почек, рак матки, рак предстательной железы, рак яичников, рак поджелудочной железы, эндотелиома, рак молочной железы, саркома и меланома.
Таким образом, настоящее изобретение относится к способу лечения любого страдающего от рака млекопитающего, в особенности человека, который включает введение больному терапевтически эффективного количества соединения по настоящему изобретению или его фармацевтической композиции.
Другим, особенно предпочтительным, воплощением настоящего изобретения являются как применяемые в качестве противоопухолевых средств фармацевтические композиции, которые содержат в качестве активного ингредиента соединение или соединения по настоящему изобретению, так и способы их получения.
Примеры фармацевтических композиций включают любые твердые (таблетки, пилюли, капсулы, гранулы и т.д.) или жидкие (растворы, суспензии или эмульсии) композиции с подходящим составом для перорального, местного или парентерального введения.
Введение соединений или композиций по настоящему изобретению может осуществляться любым подходящим способом, таким как препарат для внутривенного введения, препарат для перорального приема, препарат для внутрибрюшинного или внутривенного введения.
ПОДРОБНОЕ ОПИСАНИЕ ПРЕДПОЧТИТЕЛЬНЫХ ВОПЛОЩЕНИЙ
R1, предпочтительно, является водородом, алкилом или галогеном; более предпочтительно, водородом, метилом или хлором. Альтернативно, R1 и R2, предпочтительно, образуют конденсированное кольцо, более предпочтительно, конденсированное ароматическое кольцо и, наиболее предпочтительно, конденсированное бензольное кольцо. Такое кольцо может быть замещено, например, OR', NR'2 или галогеном; более предпочтительно, гидроксигруппой, алкоксигруппой, аминогруппой или галогеном; наиболее предпочтительно, гидроксигруппой, метоксигруппой, аминогруппой, фтором или хлором.
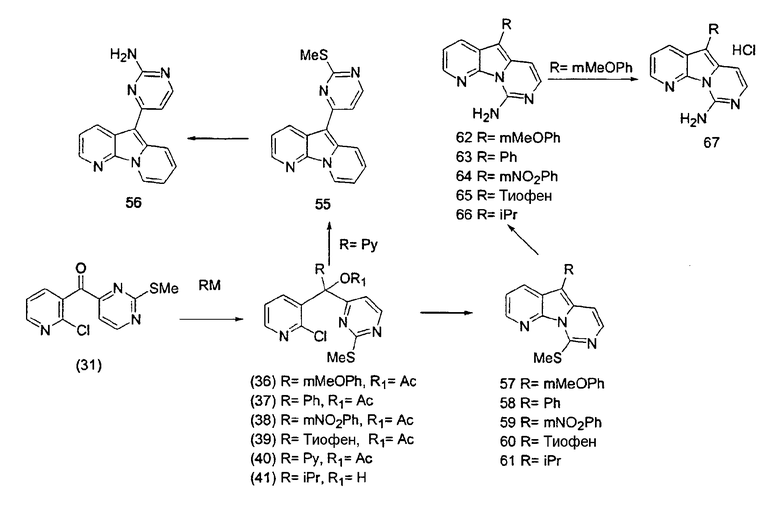
R2, предпочтительно, является водородом или галогеном; более предпочтительно, водородом, фтором или хлором. Как было упомянуто, альтернативно R1 и R2, предпочтительно, образуют конденсированное кольцо.
R3, предпочтительно, является водородом, OR', NR'2 или галогеном; более предпочтительно, водородом, гидроксигруппой, алкоксигруппой, защищенной гидроксигруппой, аминогруппой, защищенной аминогруппой или галогеном; наиболее предпочтительно, водородом, гидроксигруппой, метоксигруппой, бензилоксигруппой, аминогруппой, метоксибензиламиногруппой или хлором.
По-видимому, наибольшая активность наблюдается, когда R3 является водородом, затем гидроксигруппой, галогеном (хлором), метоксигруппой, аминогруппой.
R4, предпочтительно, является водородом.
R5, предпочтительно, является водородом.
R6, предпочтительно, является водородом.
R7, предпочтительно, является водородом.
R12, предпочтительно, является алкилом, арилом или гетероарилом; более предпочтительно, алкилом, фенилом или гетероарилом с 5 или 6 атомами в составе кольца и 1 или 2 гетероатомами, наиболее предпочтительно, изопропилом, фенилом, пиримидинилом, тиофенилом или пиридинилом. Арильные или гетероарильные группы являются незамещенными или содержат предпочтительные заместители, выбранные из OR', в особенности, алкоксигруппы, такие как метоксигруппа, или нитрогруппы, а пиримидинил может содержать определенный заместитель X1.
По-видимому, наибольшая активность наблюдается, когда R12 является 4-пиримидинилом, как в формуле (5a).
X1, предпочтительно, является водородом, алкилом, OR', NR'2, SR', SOR', SO2R', карбоксиалкилом или аралкилом; более предпочтительно, водородом, алкилом, гидроксигруппой, алкоксигруппой, арилоксигруппой, аминогруппой, защищенной аминогруппой, тиоалкилом, алкилсульфинилом, алкилсульфонилом или дикарбоксиалкилом; наиболее предпочтительно, водородом, метилом, гидроксигруппой, метоксигруппой, этоксигруппой, бензилоксигруппой, феноксигруппой, аминогруппой, метоксибензиламиногруппой, тиометилом, метилсульфинилом, метилсульфонилом или диметилкарбоксиэтилом.
По-видимому, если X1 является SR', SOR',SO2R', то наблюдается высокая избирательность в отношении рака матки. В тестах на культуре клеток Hela активность возрастала на 2-3 порядка. Конкретно, X1, предпочтительно, является S-алкилом, SO-алкилом или SO2-алкилом, где алкилом обычно является метил.
X2, предпочтительно, является NR'2 или SR'; более предпочтительно, NH2 или тиоалкилом; наиболее предпочтительно, NH2 или тиометилом.
Таким образом, предпочтительные соединения по настоящему изобретению соответствуют формуле (5), где заместители соответствуют одному или нескольким, предпочтительно всем, из указанных предпочтительных, более предпочтительных или наиболее предпочтительных определений.
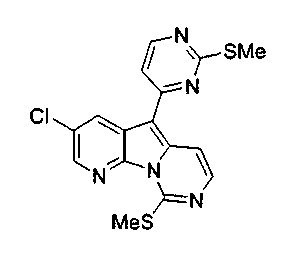
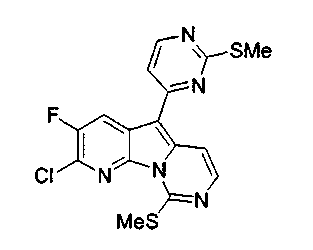
Особенно предпочтительными воплощениями настоящего изобретения являются вариолиноподобные соединения с общими формулами (10), (11) и (29).
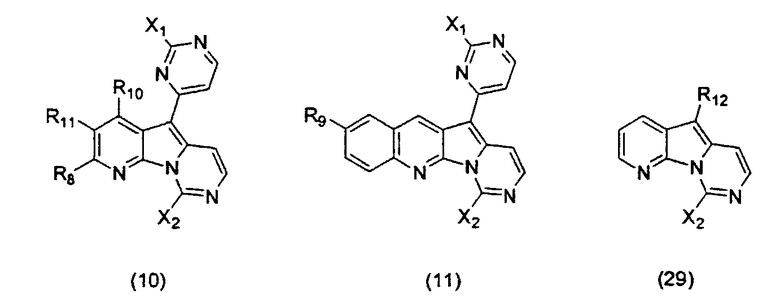
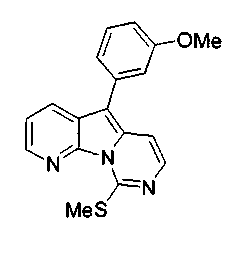
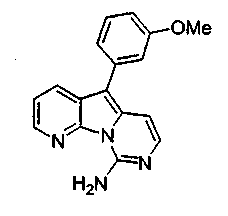
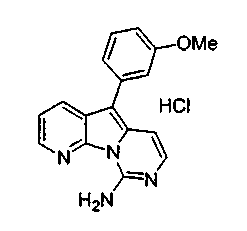
где каждый из заместителей, обозначенных как X1 и X2, независимо выбран из группы, состоящей из H, OH, OR', SH, SR', SOR', SO2R', NO2, NH2, NHR', N(R')2, NHCOR', NHSO2R', CN, галогена, =O, C(=O)H, C(=O)R', CO2H, CO2R', карбоксиалкила, C1-C12-алкила, C2-C12-алкенила, C2-C12-алкинила, замещенного или незамещенного арила, замещенного или незамещенного аралкила и замещенной или незамещенной гетероароматической группы, но наиболее предпочтительными являются NH2, SMe, SOMe или SO2Me;
где R8 выбран из группы, состоящей из H, OH, OR', SH, SR', SOR', SO2R', NO2, NH2, NHR', N(R')2, NHCOR', NHSO2R', CN, галогена, =O, C(=O)H, C(=O)R', CO2H, CO2R', C1-C12-алкила, C2-C12-алкенила, C2-C12-алкинила, замещенного или незамещенного арила, замещенного или незамещенного аралкила и замещенной или незамещенной гетероароматической группы, но наиболее предпочтительными являются H, метил или Cl;
где R9 выбран из группы, состоящей из H, OH, OR', SH, SR', SOR', SO2R', NO2, NH2, NHR', N(R')2, NHCOR', NHSO2R', CN, галогена, =O, C(=O)H, C(=O)R', CO2H, CO2R', C1-C12-алкила, C2-C12-алкенила, C2-C12-алкинила, замещенного или незамещенного арила, замещенного или незамещенного аралкила и замещенной или незамещенной гетероароматической группы, но наиболее предпочтительными являются H или OMe;
где R10 выбран из группы, состоящей из H, OH, OR', SH, SR', SOR', SO2R', NO2, NH2, NHR', N(R')2, NHCOR', NHSO2R', CN, галогена, =O, C(=O)H, C(=O)R', CO2H, CO2R', C1-C12-алкила, C2-C12-алкенила, C2-C12-алкинила, замещенного или незамещенного арила, замещенного или незамещенного аралкила и замещенной или незамещенной гетероароматической группы, но наиболее предпочтительными являются H, OH, Cl, F, NH2 или OMe;
где R11 выбран из группы, состоящей из H, OH, OR', SH, SR', SOR', SO2R', NO2, NH2, NHR', N(R')2, NHCOR', NHSO2R', CN, галогена, =O, C(=O)H, C(=O)R', CO2H, CO2R', C1-C12-алкила, C2-C12-алкенила, C2-C12-алкинила, замещенного или незамещенного арила, замещенного или незамещенного аралкила и замещенной или незамещенной гетероароматической группы, но наиболее предпочтительными являются H, Cl или F;
где R12 выбран из группы, состоящей из H, OH, OR', SH, SR', SOR', SO2R', NO2, NH2, NHR', N(R')2, NHCOR', NHSO2R', CN, галогена, =O, C(=O)H, C(=O)R', CO2H, CO2R', C1-C12-алкила, C2-C12-алкенила, C2-C12-алкинила, замещенного или незамещенного арила, замещенного или незамещенного аралкила и замещенной или незамещенной гетероароматической группы, но наиболее предпочтительными являются алкил, замещенный или незамещенный арил, или замещенной или незамещенной гетероароматической группой, и еще более предпочтительными являются алкил, замещенный или незамещенный фенил или замещенный или незамещенный тиофенил, пиридинил или пиримидинил, а предпочтительные заместители включают алкоксигруппу или нитрогруппу, в особенности метоксигруппу или нитрогруппу, вместе с разрешенными для группы X1 определениями;
где каждая из R'-групп независимо выбрана из группы, состоящей из H, OH, SH, NO2, NH2, CN, галогена, =О, C(=O)H, C(=O)CH3, CO2H, CO2CH3, C1-C12-алкила, C2-C12-алкенила, C2-C12-алкинила, арила, аралкила и гетероароматической группы.
Конкретные воплощения соединений формулы (10) включают те соединения, где один или несколько заместителей являются следующими:
значения X1 определены выше, включая предпочтительные, более предпочтительные и наиболее предпочтительные значения;
значения X2 определены выше, включая предпочтительные, более предпочтительные и наиболее предпочтительные значения;
значения R8 определены выше для R1, включая предпочтительные, более предпочтительные и наиболее предпочтительные значения;
значения R10 определены выше для R3, включая предпочтительные, более предпочтительные и наиболее предпочтительные значения;
значения R11 определены выше для R2, включая предпочтительные, более предпочтительные и наиболее предпочтительные значения.
Конкретные воплощения соединений формулы (11) включают те соединения, где один или несколько заместителей являются следующими:
значения X1 определены выше, включая предпочтительные, более предпочтительные и наиболее предпочтительные значения;
значения X2 определены выше, включая предпочтительные, более предпочтительные и наиболее предпочтительные значения;
R9 является одним из разрешенных для арильного кольца заместителей, таким как водород или алкоксигруппа, предпочтительно, водород или метоксигруппа.
Конкретные воплощения соединений формулы (29) включают те соединения, где один или несколько заместителей являются следующими:
значения X2 определены выше;
R12 не является пиримидинилом и, предпочтительно, является алкилом, арилом или гетероарилом за исключением пиримидинила; более предпочтительно, алкилом, фенилом или гетероарилом с 5 или 6 атомами в составе кольца и 1 или 2 гетероатомами; наиболее предпочтительно, изопропилом, фенилом, тиофенилом или пиридинилом. Арильные или гетероарильные группы являются незамещенными или содержат предпочтительные заместители, выбранные из OR', в особенности алкоксигруппы, такие как метоксигруппа, или нитрогруппы.
Соединения общих формул (10), (11) и (29) могут быть синтезированы с применением модификаций метода, описанного в заявках на выдачу международного патента WO 0204447 и WO 0212240.
Некоторые из предпочтительных способов получения соединений по настоящему изобретению описаны ниже в следующих схемах реакций с примерами типичных заместителей. Настоящее изобретение не ограничивается этими типичными заместителями, а способ получения следует понимать в более общем смысле без особого внимания к конкретным значениям кодовых символов.
Из этих соединений было получено множество активных противоопухолевых соединений и полагают, что с применением методик в соответствии с раскрытием сущности настоящего изобретения может быть получено еще больше соединений.
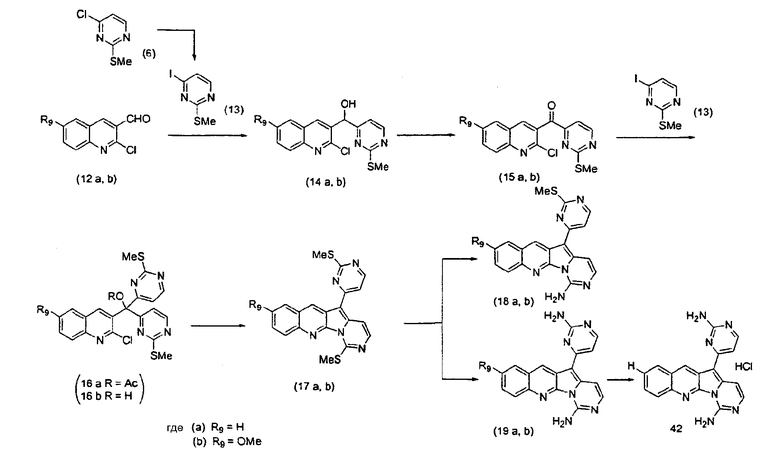
Получение соединений общей формулы (11) проиллюстрировано ниже на примере, где R9 является H или OMe.
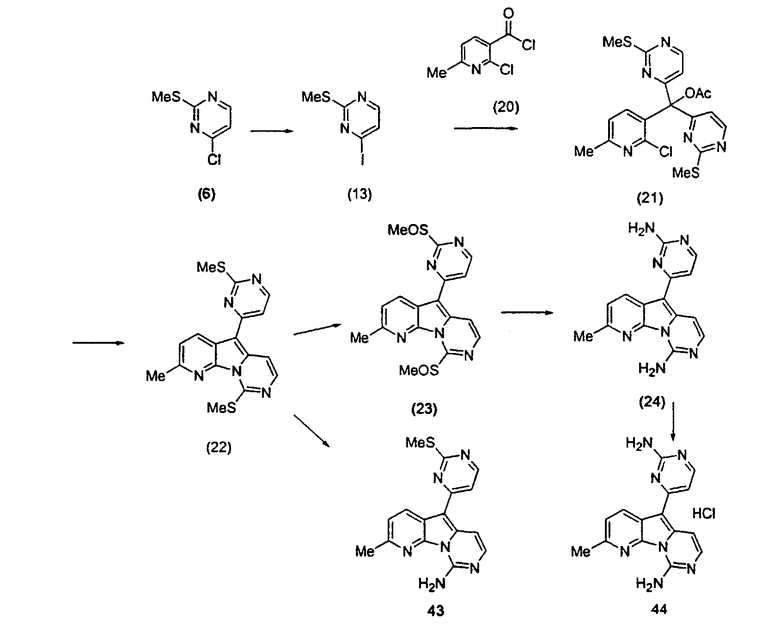
Получение соединений общей формулы (10) проиллюстрировано на примере, где R8 является метилом, а R10 и R11 являются H.
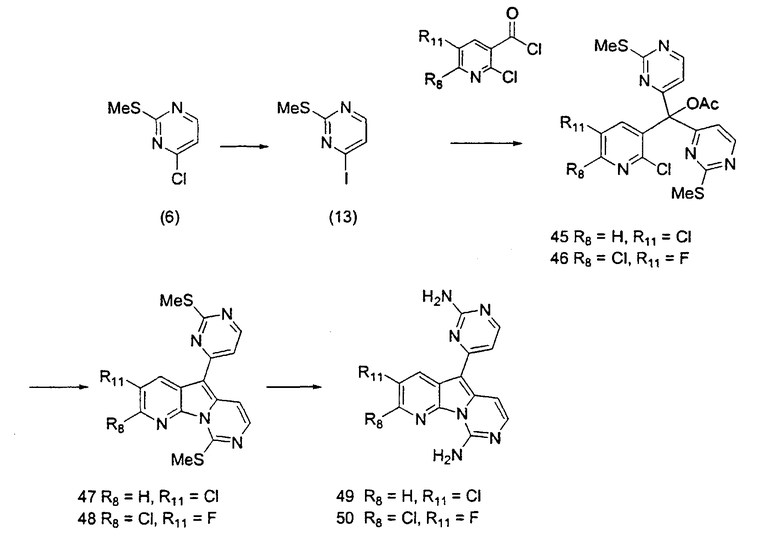
Получение соединений общей формулы (10) проиллюстрировано на примере, где R8 является H или Cl, R10 является H и R11 является F или Cl.
Получение соединений общей формулы (10) проиллюстрировано на примере, где R8, R10 и R11 являются H.
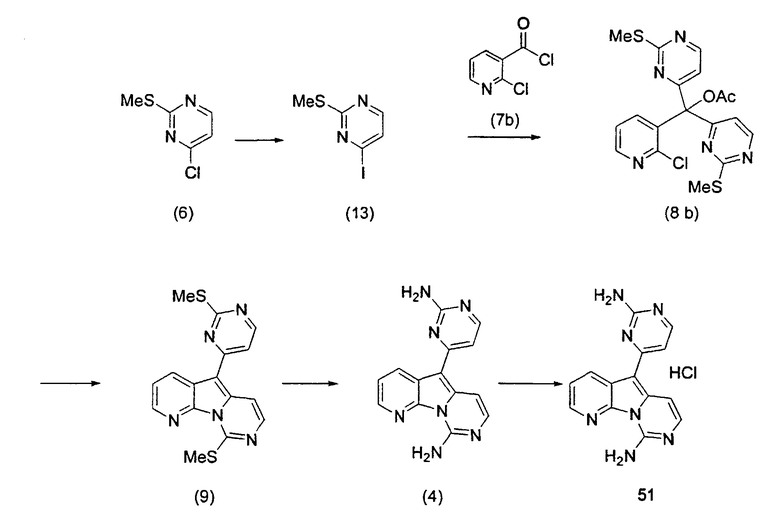
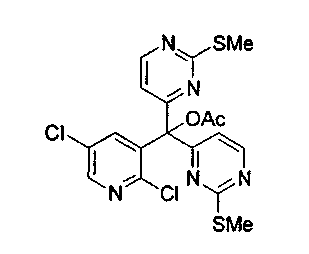
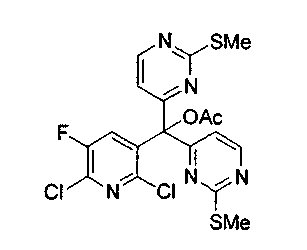
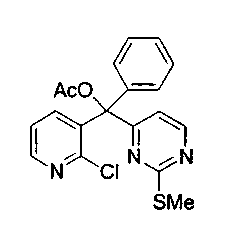
Для получения ключевого промежуточного продукта (8b, пример 17: соединение 18 в заявке на выдачу международного патента WO 0204447) с применением известного способа было необходимо осуществлять взаимодействие между 2-хлорникотиноилхлоридом (7b) и йодпиримидином (13) при -100°C. Когда авторы предприняли попытку масштабирования реакции, им пришлось увеличить времядобавлениядля удержания температуры реакционной смеси ниже -95°C. Эта большая продолжительность приводила к увеличению побочных реакций и снижала выход.
Для того чтобы избежать использования столь низкой температуры при получении соединения (8b), авторы разработали новый путь получения соединения (8b). Так, соединение (7b) преобразовывали в соответствующий амид Уайнреба (30), который вступал во взаимодействие с магниевым производным соединения (13) при -5°C с получением кетона (31). Преобразование соединения (30) в соединение (8b) осуществляли двумя способами: a) путем осуществления взаимодействия с магниевым производным соединения (13) при 0°С или b) путем осуществления взаимодействия с литиевым производным соединения (13) в условиях Барбье при -78°С. При использовании обоих способов реакцию легко масштабировали, а выходы реакции были лучше, чем в оригинальном способе.
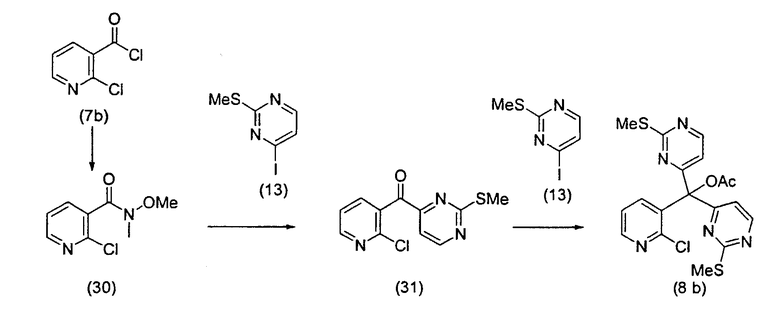
Как отражение этого открытия настоящее изобретение относится к способу получения вариолинового промежуточного продукта формулы (8z):
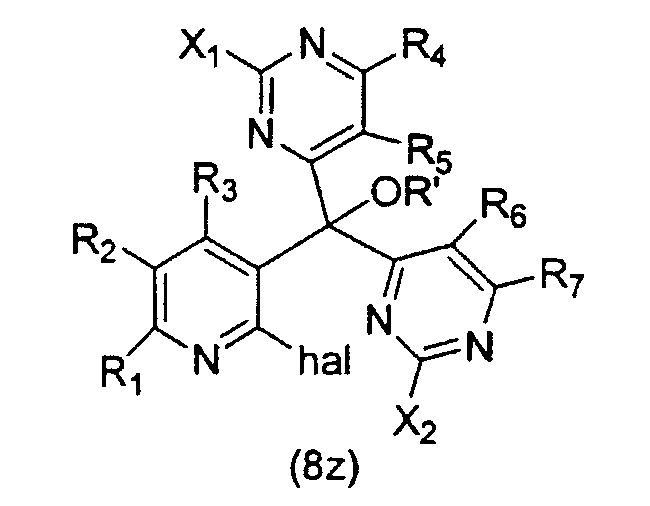
где hal является галогеном, а значения остальных заместителей определены выше; который включает осуществление взаимодействия соединения формулы (31z):
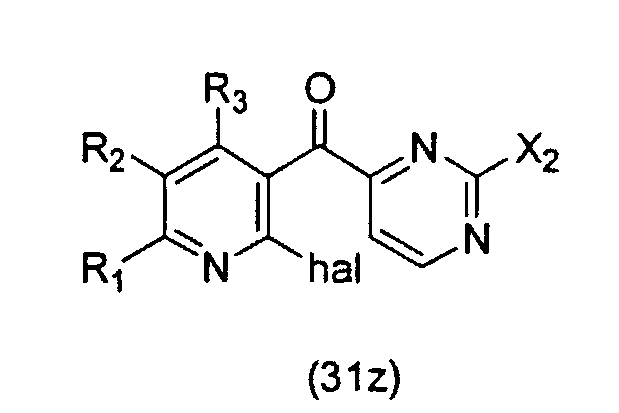
где значения заместителей определены выше, с соединением формулы (13z):
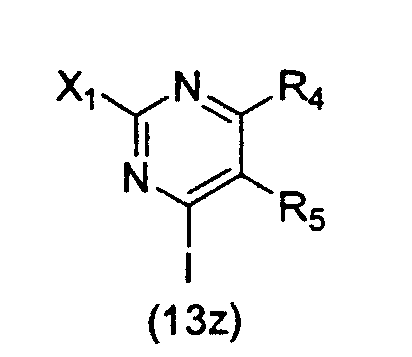
где значения заместителей определены выше.
В этой реакции заместители X1 и X2 могут быть одними и теми же или различными и, предпочтительно, оба являются -SMe; hal обычно является Cl; R' обычно является H или Ac, а остальные заместители обычно являются H или как для предпочтительных соединений по настоящему изобретению.
Соединение формулы (31z) подходящим образом получают путем осуществления взаимодействия соединения формулы (30z):
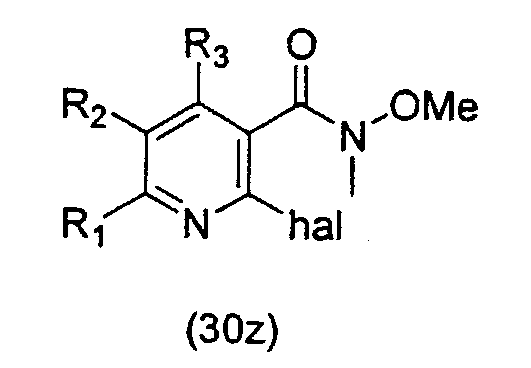
где значения заместителей определены выше, с соединением формулы (13z). Соединение формулы (13z), применяемое для осуществления взаимодействия с соединением формулы (30z), может быть одним и тем же или отличаться от соединения формулы (13z), применяемого для осуществления взаимодействия с соединением формулы (31z).
Получение соединений общей формулы (10) проиллюстрировано на примере, где R8 и R11 являются H, и R10 является OBn.
Получение соединений общей формулы (29) проиллюстрировано на примере, где R12 является замещенным или незамещенным аралкилом и замещенным или незамещенным гетероароматическим соединением.
Таким образом, существует возможность трансформировать ряд простых гетероароматических соединений во множество промежуточных продуктов и производных с потенциальной противоопухолевой терапевтической активностью.
Для соединений общей формулы (10) и (11) взаимопревращения ряда простых функциональных групп позволяют получать широкий ряд дополнительных производных с различными заместителями X1, X2 и R10, как проиллюстрировано ниже.
Примеры биологической активности соединений по настоящему изобретению включены в таблицу (см. в конце описания).
По настоящей заявке испрашивается приоритет в соответствии с заявкой на выдачу патента Великобритании. Авторы специально включают в настоящее изобретение в качестве ссылки любое раскрытие в описании указанной заявки на выдачу патента Великобритании, не содержащееся в настоящем описании.
Экспериментальные методики и физико-химические характеристики соединений являются следующими:
ОСНОВНЫЕ ЭКСПЕРИМЕНТАЛЬНЫЕ УСЛОВИЯ
Если не определено иначе, все взаимодействия осуществляли в атмосфере аргона в предварительно высушенной стеклянной посуде.
Пример 1: Соединение 13
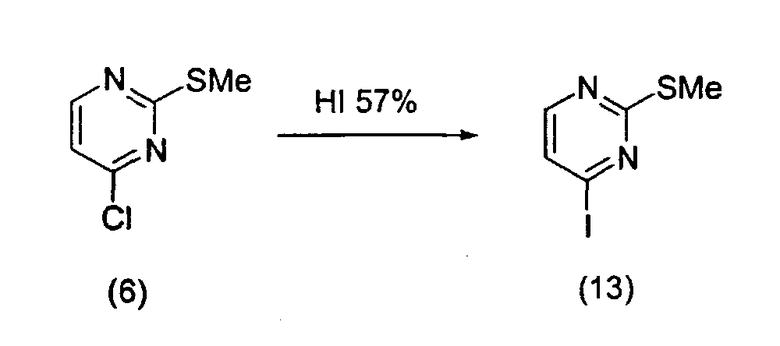
Йодпиримидин (13) получали в соответствии с экспериментальной методикой, описанной в литературе в работе A.J. Majeed et al. Tetrahedron 1989, 45, 993.
Пример 2: Соединение 14a
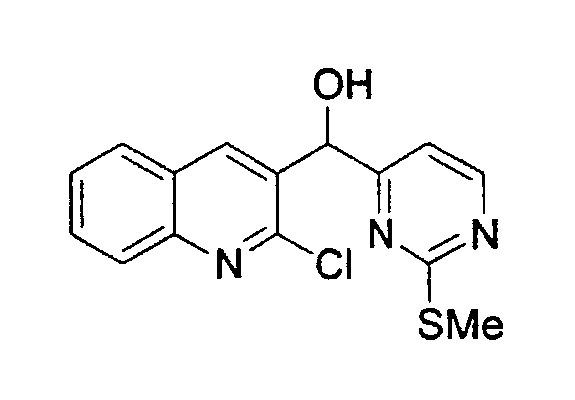
Способ A
Раствор 4-йод-2-метилтиопиримидина (соединение 13, 5,13 г, 20,3 ммоль) в тетрагидрофуране (75 мл) обрабатывали n-BuLi (8,1 мл, 20,3 ммоль, 2,5 М в гексанах) при -100°C. Реакционную смесь перемешивали при -100°C в течение 45 мин и обрабатывали раствором 2-хлор-3-хинолинкарбоксальдегида (соединение 12a, 3,0 г, 15,7 ммоль) в тетрагидрофуране (60 мл) при -100°C в течение 2,5 ч. Реакцию гасили добавлением насыщенного водного раствора хлорида аммония, нагревали до 23°C и распределяли между этилацетатом и насыщенным водным раствором хлорида аммония. Органический слой сушили над сульфатом натрия, фильтровали и упаривали. Неочищенный продукт подвергали хроматографии (элюируя гексаном:этилацетатом от 4:1 до 1:1) с получением соединения 14a в виде желтого твердого вещества (4,0 г, 81%).
1H ЯМР (CDCl3, 300 МГц) δ 8,45 (д, J=7,8 Гц, 1H), 8,25 (с, 1Н), 8,02 (д, J=8,3 Гц, 1Н), 7,81 (д, J=8,0 Гц, 1Н), 7,74 (т, J=7,3 Гц, 1Н), 7,56 (т, J=7,0 Гц, 1Н), 7,02 (д, J=7,8 Гц, 1Н), 6,24 (д, J=4,1 Гц, 1H), 4,91 (д, J=4,2 Гц, 1Н, OH), 2,58 (с, 3H).
13C ЯМР (CDCl3, 75 МГц) δ 172,4, 169,5, 157,9, 149,4, 147,0, 138,0, 133,9, 130,9, 127,9, 127,9, 127,4, 127,2, 113,5, 71,7, 14,2.
МС (ESI) m/z 318 (М+1)+.
Rf: 0,12 (гексан:этилацетат 4:1).
Способ B
Раствор 4-йод-2-метилтиопиримидина (соединение 13, 5,1 г, 20,3 ммоль) в толуоле (40 мл) обрабатывали i-PrMgCl (10 мл, 20,0 ммоль, 2 M в тетрагидрофуране) при 0°C в течение 1 ч и добавляли через канюлю к раствору 2-хлор-3-хинолинкарбоксальдегида (соединение 12a, 3,0 г, 15,7 ммоль) в толуоле (150 мл) при 0°C. Реакционную смесь перемешивали при 0°C в течение 16 ч, гасили добавлением насыщенного водного раствора хлорида аммония, нагревали до 23°C и распределяли между насыщенным водным раствором хлорида аммония и этилацетатом. Органический слой сушили над сульфатом натрия, фильтровали и упаривали. Остаток подвергали хроматографии (элюируя гексаном:этилацетатом от 4:1 до 1:1) с получением соединения 14a в виде желтого твердого вещества (3,5 г, 70%).
Пример 3: Соединение 14b
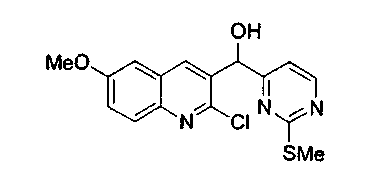
Раствор 4-йод-2-метилтиопиримидина (соединение 13, 3,4 г, 13,5 ммоль) в тетрагидрофуране (60 мл) обрабатывали n-BuLi (5,4 мл, 2,5 M в гексане, 13,5 ммоль) при -100°C и перемешивали при -100°C в течение 45 мин. Потом добавляли раствор 2-хлор-6-метокси-3-хинолинкарбоксальдегида (соединение 12b, 1,7 г, 7,9 ммоль) в тетрагидрофуране (35 мл) при -100°C и перемешивали в течение 2,5 ч. Реакцию гасили добавлением насыщенного водного раствора хлорида аммония, нагревали до 23°C и распределяли между этилацетатом и насыщенным водным раствором хлорида аммония. Органический слой сушили над сульфатом натрия, фильтровали и упаривали. Остаток подвергали хроматографии (элюируя гексаном:этилацетатом от 4:1 до 3:1) с получением соединения 14b в виде желтого твердого вещества (1,8 г, 67%).
1H ЯМР (CDCl3, 300 МГц) δ 8,42 (дд, J=5,1, 1,0 Гц, 1H), 8,11 (с, 1H), 7,86 (д, J=9,3 Гц, 1H), 7,34 (дд, J=9,3, 2,7 Гц, 1H), 7,03 (д, J=5,1 Гц, 1H), 7,00 (д, J=2,7 Гц, 1Н), 6,20 (ушир.с, 1H), 5,09 (ушир.с, 1H), 3,87 (с, 3H), 2,53 (ушир.с, 3H).
13C ЯМР (CDCl3, 75 МГц) δ 172,7, 168,4, 158,6, 157,9, 146,6, 143,5, 136,8, 133,6, 129,7, 128,6, 123,9, 113,5, 105,3, 71,2, 55,8, 14,4.
МС (ESI) m/z 370 (M+23)+.
Rf: 0,37 (гексан:этилацетат 1:1).
Пример 4: Соединение 15a
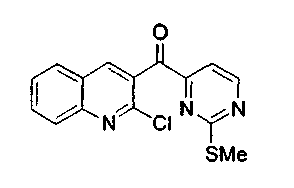
Раствор соединения 14a (4,0 г, 12,6 ммоль) и PDC (7,1 г, 18,9 ммоль) в CH2Cl2 (50 мл) перемешивали при 23°C в течение 24 ч. Реакционную смесь фильтровали через броунмиллерит, упаривали и подвергали хроматографии (элюируя гексаном:этилацетатом 4:1) с получением соединения 15a в виде белого твердого вещества (3,0 г, 75%).
1H ЯМР (CDCl3, 300 МГц) δ 8,85 (д, J=4,9 Гц, 1H), 8,42 (с, 1H), 8,11 (д, J=8,5 Гц, 1H), 7,93 (д, J=8,1 Гц, 1H), 7,88 (т, J=7,1 Гц, 1H), 7,69 (д, J=4,9 Гц, 1H), 7,66 (т, J=7,1 Гц, 1H), 2,35 (с, 3H).
13C ЯМР (CDCl3, 75 МГц) δ 192,7, 173,7, 159,7, 159,3, 148,3, 146,8, 140,6, 132,6, 131,2, 128,8, 128,6, 128,1, 126,1, 113,7, 14,3.
МС (ESI) m/z, 280 (M-35)+.
Rf: 0,23 (гексан:этилацетат 4:1).
Пример 5: Соединение 15b
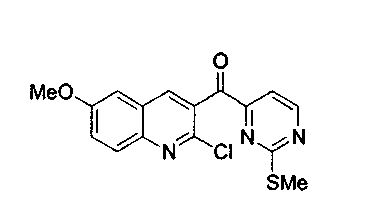
Раствор соединения 14b (1,8 г, 5,2 ммоль) в CH2Cl2 (50 мл) обрабатывали PDC (2,9 г, 7,8 ммоль) при 23°C в течение 48 ч. Реакционную смесь фильтровали через броунмиллерит, концентрировали и подвергали хроматографии (элюируя гексаном:этилацетатом 3:1) с получением соединения 15b в виде белого твердого вещества (1,5 г, 82%).
1H ЯМР (CDCl3, 300 МГц) δ 8,82 (д, J=4,9 Гц, 1H), 8,27 (с, 1H), 7,97 (д, J=9,3 Гц, 1H), 7,66 (д, J=4,9 Гц, 1H), 7,47 (дд, J=9,3, 2,9 Гц, 1Н), 7,13 (д, J=2,7 Гц, 1H), 3,93 (с, 3H), 2,34 (с, 3H).
13C ЯМР (CDCl3, 75 МГц) δ 193,0, 173,7, 159,6, 159,4, 158,9, 144,4, 144,2, 139,2, 131,3, 130,1, 127,3, 125,2, 113,7, 105,9, 55,9, 14,3.
МС (ESI) m/z, 368 (M+23)+.
Rf: 0,26 (гексан:этилацетат 3:1).
Пример 6: Соединение 16a
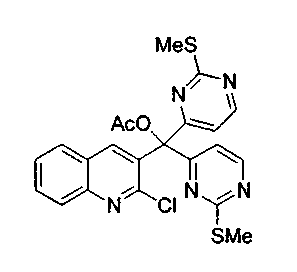
Раствор 4-йод-2-метилтиоформамида (соединение 13, 0,50 г, 2,0 ммоль) в безводном толуоле (12 мл) обрабатывали i-PrMgCl (1,0 мл, 2 M в тетрагидрофуране, 2 ммоль) при 0°C в течение 1 ч. Полученный арилмагний добавляли через канюлю к раствору соединения 15a (0,32 г, 1,0 ммоль) в безводном толуоле (30 мл) при 0°C, перемешивали в течение 25 мин, обрабатывали избытком ацетилхлорида (2,0 мл) и перемешивали в течение ночи при 23°C. Реакционную смесь распределяли между этилацетатом и насыщенным водным раствором бикарбоната натрия. Органический слой сушили над сульфатом натрия, фильтровали и упаривали. Остаток подвергали хроматографии (элюируя гексаном:этилацетатом от 4:1 до 1:1) с получением соединения 16a в виде желтого твердого вещества (150 мг, 15%).
1H ЯМР (CDCl3, 300 МГц) δ 8,53 (д, J=5,1 Гц, 2H), 8,32 (с, 1H), 8,01 (д, J=8,5 Гц, 1H), 7,81-7,77 (м, 2H), 7,58 (т, J=7,6 Гц, 1H), 7,35 (д, J=5,1 Гц, 2H), 2,36 (с, 6H), 2,30 (с, 3H).
Пример 7: Соединение 16b
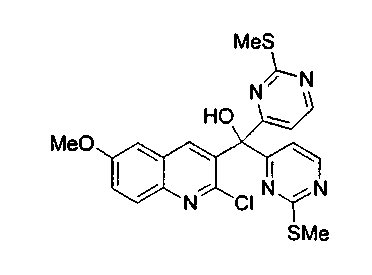
Раствор 4-йод-2-метилтиопиримидина (соединение 13, 2,1 г, 8,3 ммоль, 2,0 эквив.) в тетрагидрофуране (50 мл) обрабатывали n-BuLi (3,3 мл, 2,5 M в гексанах, 8,3 ммоль) при -100°C и перемешивали при -100°C в течение 45 мин. Потом через канюлю при -78°C медленно добавляли раствор соединения 15b (1,4 г, 4,1 ммоль) в тетрагидрофуране (20 мл), поддерживая температуру -100°C. Реакционную смесь перемешивали при -100°C в течение 3 ч. Реакцию гасили добавлением насыщенного водного раствора хлорида аммония, нагревали до 23°C и распределяли между этилацетатом и насыщенным водным раствором хлорида аммония. Органический слой сушили над сульфатом натрия, фильтровали и упаривали. Неочищенный продукт подвергали хроматографии (элюируя гексаном:этилацетатом от 4:1 до 2:1) с получением соединения 16b в виде белого твердого вещества (1,48 г, 76%).
1H ЯМР (CDCl3, 300 МГц) δ 8,57 (д, J=5,1 Гц, 2H), 7,87 (д, J=9,3 Гц, 1H), 7,47 (с, 1Н), 7,43 (д, J=5,1 Гц, 2H), 7,36 (дд, J=9,3, 2,7 Гц, 1Н), 6,92 (д, J=2,7 Гц, 1Н), 6,34 (с, 1Н), 3,87 (с, 3H), 2,47 (с, 6H).
13C ЯМР (CDCl3, 75 МГц) δ 172,1, 169,4, 158,7, 158,2, 147,5, 143,1, 138,1, 134,4, 129,7, 127,4, 124,2, 115,3, 105,7, 80,0, 55,9, 14,5.
МС (ESI) m/z: 494 (M+23)+.
Rf: 0,06 (гексан:этилацетат 4:1).
Пример 8: Соединение 17a
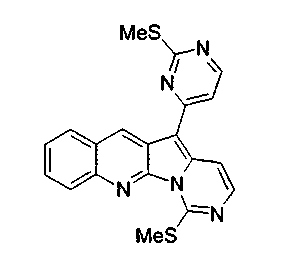
Смесь соединения 16a (680 г, 1,40 ммоль), триэтилсилана (2,70 мл, 16,9 ммоль) и трифторуксусной кислоты (0,23 мл, 2,95 ммоль) нагревали с обратным холодильником в дихлорэтане (3 мл) в герметичной пробирке при 80°C в течение 32 ч. После охлаждения красный остаток разбавляли CH2Cl2 (50 мл) и промывали насыщенным водным раствором бикарбоната натрия (50 мл) и насыщенным водным раствором NaCl (50 мл). Объединенный органический слой сушили над сульфатом натрия, фильтровали, упаривали и подвергали хроматографии (элюируя CH2Cl2:этилацетатом от 100:0 до 30:1) с получением соединения 17a в виде оранжевого твердого вещества (226 мг, 41%).
1H ЯМР (CDCl3, 300 МГц) δ 8,95 (с, 1H), 8,53 (д, J=5,4 Гц, 1H), 8,26 (д, J=8,2 Гц, 1H), 8,09-8,03 (м, 2H), 7,89 (д, J=6,6 Гц, 1H), 7,75 (т, J=7,2 Гц, 1H), 7,60 (т, J=7,9 Гц, 1H), 7,41 (д, J=5,3 Гц, 1H), 2,76 (с, 3H), 2,71 (с, 3H).
13C ЯМР (CDCl3, 75 МГц) δ 173,5, 161,0, 157,3, 156,3, 144,8, 143,1, 141,8, 141,0, 128,7, 128,4, 128,0, 126,3, 125,9, 120,0, 112,7, 108,4, 100,1, 15,1, 14,4.
МС (ESI) m/z 392 (М+1)+.
Rf: 0,83 (CH2Cl2:MeOH, 96:4).
Пример 9: Соединение 17b
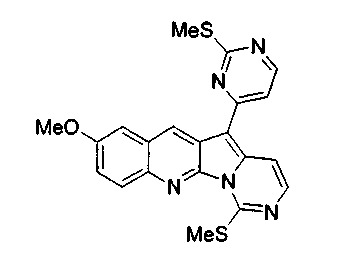
Суспензию соединения 16b (113 мг, 0,24 ммоль) и TFA (37 мкл, 0,48 ммоль) в 1,2-дихлорэтане (0,5 мл) обрабатывали Et3SiH (0,3 мл, 1,9 ммоль) в герметичной пробирке при 100°C в течение 43 ч. Реакционную смесь охлаждали и распределяли между CH2Cl2 и насыщенным водным раствором бикарбоната натрия. Органический слой сушили над сульфатом натрия, фильтровали и упаривали. Неочищенный продукт подвергали хроматографии (элюируя гексаном:этилацетатом от 4:1 до 2:1) с получением соединения 17b в виде желтого твердого вещества (34 мг, 34%).
1H ЯМР (CDCl3, 300 МГц) δ 8,87 (с, 1Н), 8,54 (д, J=5,4 Гц, 1H), 8,18 (д, J=9,0 Гц, 1H), 8,11 (д, J=6,6 Гц, 1H), 7,83 (д, J=6,6 Гц, 1H), 7,46 (д, J=5,4 Гц, 1H), 7,42 (дд, J=9,0, 2,7 Гц, 1H), 7,27 (д, J=2,7 Гц, 1H), 4,00 (с, 3H), 2,76 (с, 3H), 2,72 (с, 3H).
МС (APCI) m/z 420 (М+1)+.
Rf: 0,27 (гексан:этилацетат 4:1).
Пример 10: Соединение 18a
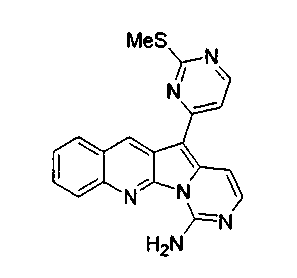
Раствор соединения 17a (15,3 мг, 0,039 ммоль) в смеси 1,4-диоксана (5 мл) и NH4OH (8 мл, 32%) нагревали в герметичной пробирке при 85°C в течение 16 ч. Реакционную смесь упаривали и подвергали хроматографии (элюируя CH2Cl2:MeOH, 98:2) с получением соединения 18a в виде желтого твердого вещества (9,3 мг, 67%).
1H ЯМР (CDCl3, 300 МГц) δ 9,02 (с, 1H), 8,49 (д, J=5,4Гц, 1H), 8,11 (д, J=8,5 Гц, 1H), 8,02 (д, J=8,3 Гц, 1H), 7,72-7,68 (м, 2H), 7,59 (т, J=8,1 Гц, 1H), 7,54 (д, J=6,8 Гц, 1H), 7,35 (д, J=5,4 Гц, 1H), 2,71 (с, 3H).
МС (ESI) m/z 359 (М+1)+.
Rf: 0,64 (CH2Cl2:MeOH, 6:1).
Пример 11: Соединение 18b
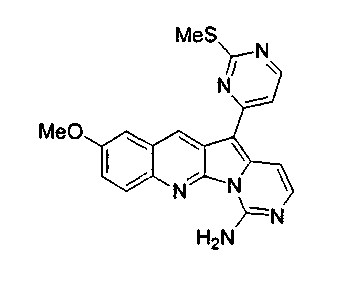
Раствор соединения 17b (21,0 мг, 0,05 ммоль) в 1,4-диоксане:NH4OH (32%), 2:3 (25 мл) нагревали при 90°C в герметичной пробирке в течение 16 ч. Реакционную смесь упаривали и подвергали хроматографии (элюируя CH2Cl2:MeOH, 98:2) с получением соединения 18b в виде желтого твердого вещества (10 мг, 52%).
1H ЯМР (CDCl3, 300 МГц) δ 8,80 (с, 1H), 8,30 (д, J=5,3 Гц, 1H), 7,91 (д, J=8,7 Гц, 1H), 7,52-7,47 (м, 2H), 7,32 (д, J=5,3 Гц, 1H), 7,27-7,25 (дд, J=8,7, 2,7 Гц, 1H), 7,16 (д, J=3,0 Гц, 1H), 3,85 (с, 3H), 2,58 (с, 3H).
МС (ESI) m/z 389 (М+1)+.
Пример 12: Соединение 19a
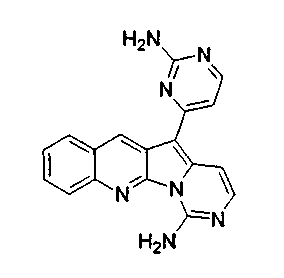
Раствор соединения 17a (26,7 мг, 0,068 ммоль) в CH2Cl2 (3 мл) по каплям обрабатывали при -30°C раствором mCPBA (12,5 мг, 0,051 ммоль, 2,4 эквив., 77%) в CH2Cl2 (1 мл) и нагревали до 0°C в течение 30 мин. Реакционную смесь обрабатывали насыщенным водным раствором Na2S2О3 и промывали насыщенным водным раствором бикарбоната натрия. Органический слой сушили над сульфатом натрия, фильтровали и упаривали. Неочищенный продукт реакции растворяли в 1,4-диоксане:NH4OH (32%) и нагревали при 80°C в герметичной пробирке в течение 16 ч. Реакционную смесь упаривали и подвергали хроматографии (элюируя CH2Cl2:CH3OH от 98:2 до 94:6) с получением соединения 19a в виде желтого твердого вещества (4,5 мг, 20%).
1H ЯМР (CDCl3, 300 МГц) δ 8,90 (с, 1H), 8,17 (д, J=5,5 Гц, 1H), 8,07 (д, J=8,6 Гц, 1H), 8,00 (д, J=7,8 Гц, 1H), 7,64 (т, J=7,2 Гц, 1H), 7,52 (т, J=7,4 Гц, 1H), 7,48 (д, J=6,7 Гц, 1H), 7,40 (д, J=6,7 Гц, 1H), 7,04 (д, J=5,5 Гц, 1H).
МС (ESI) m/z:328(М+1).
Rf: 0,79 (CH2Cl2:MeOH, 6:1).
Пример 13: Соединение 42
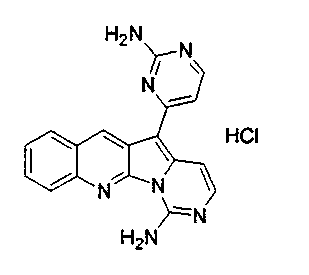
Соединение 19a (2,1 мг, 0,006 ммоль) обрабатывали HCl в 1,4-диоксане (0,5 мл, 3,8 M) при 23°C в течение 5 мин. Реакционную смесь упаривали с получением соединения 42 в виде желтого твердого вещества (2,5 мг, 100%).
1H ЯМР (CD3OD, 300 МГц) δ 9,47 (с, 1H), 8,30 (д, J=8,5 Гц, 1H), 8,23 (д, J=6,9 Гц, 1Н), 8,20 (д, J=6,6 Гц, 1Н), 7,97 (д, J=7,2 Гц, 1Н), 7,89 (т, J=6,9 Гц, 1Н), 7,79 (д, J=7,5 Гц, 1Н), 7,74 (т, J=8,4 Гц, 1Н), 7,59 (д, J=6,9Гц, 1Н).
МС (ESI) m/z, 328 (M)+.
Пример 14: Соединение 19b
Раствор соединения 17b (18,0 мг, 0,04 ммоль) в CH2Cl2 (5 мл) обрабатывали при -30°C раствором mCPBA (24,7 мг, 0,11 ммоль, 77%) в CH2Cl2 (3 мл). Реакционную смесь нагревали до 0°C в течение 30 мин и обрабатывали насыщенным водным раствором Na2S2О3. Реакционную смесь распределяли между CH2Cl2 и насыщенным водным раствором бикарбоната натрия, сушили над сульфатом натрия, фильтровали и упаривали. Остаток подвергали хроматографии (элюируя CH2Cl2:MeOH, 95:5) с получением соединения 19b в виде желтого твердого вещества (2,8 мг, 20%).
1H ЯМР (CDCl3, 300 МГц) δ 8,81 (с, 1H), 8,13 (д, J=5,6 Гц, 1H), 7,91 (д, J=9,3 Гц, 1Н), 7,45 (д, J=6,8 Гц, 1Н), 7,37 (д, J=6,8 Гц, 1Н), 7,27 (дд, J=9,3, 2,7 Гц, 1Н), 7,20 (д, J=2,7 Гц, 1Н), 6,99 (д, J=5,6 Гц, 1Н), 3,89 (с, 3H).
Пример 15: Соединение 20
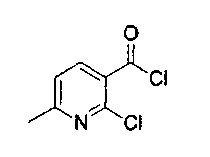
DMF (2 капли) добавляли к суспензии оксалилхлорида (5,4 мл, 10,6 ммоль) и 2-хлор-6-метилникотиновой кислоты (1,69 г, 9,8 ммоль) в CH2Cl2 (45 мл). Смесь перемешивали в течение 3 ч при 23°C и выпаривали растворитель при пониженном давлении с получением коричневого масла (1,8 г, 97%), которое использовали без дополнительной очистки.
1H ЯМР (CDCl3, 300 МГц) δ 8,35 (д, J=8,2 Гц, 1Н), 7,28 (д, J=8,0 Гц, 1Н), 2,61 (с, 3H).
Пример 16: Соединение 21
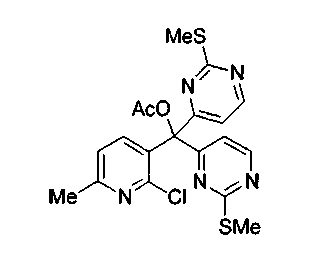
n-BuLi (11,6 мл, 2,5 M в гексане, 28,8 ммоль) добавляли по каплям к раствору 4-йод-2-метилтиопиримидина (соединение 13, 7,16 г, 28,4 ммоль) в тетрагидрофуране (60 мл) при -100°C. Черный раствор перемешивали в течение 15 мин при -100°C. Через канюлю добавляли раствор 2-хлор-6-метилникотиноилхлорида (соединение 20, 1,8 г, 9,47 ммоль) в тетрагидрофуране (10 мл) при -100°C. Ярко-красную смесь перемешивали в течение 1 ч при -95°C и осторожно добавляли ацетилхлорид (3,4 мл, 47,3 ммоль). Красную смесь перемешивали в течение 4 ч при 23°C и добавляли насыщенный водный раствор бикарбоната натрия (100 мл). Слои разделяли и экстрагировали водный слой диэтиловым эфиром (3x150 мл). Объединенные органические слои сушили над сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Красный остаток подвергали хроматографии (элюируя гексаном:этилацетатом от 1:5 до 1:1,5) с получением соединения 21 (2,3 г, 54%) в виде бледно-красного твердого вещества.
1H ЯМР (CDCl3, 300 МГц) δ 8,54 (д, J=5,2 Гц, 2H), 7,79 (д, J=8,0 Гц, 1H), 7,31 (д, J=5,3 Гц, 2 H), 7,05 (д, J=7,7Гц, 1H), 2,56 (с, 3H), 2,49 (с, 6H), 2,27 (с, 3H).
Пример 17: Соединение 22
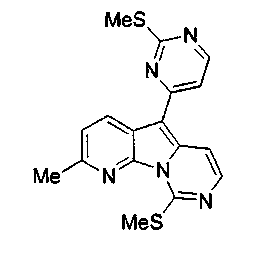
Смесь соединения 21 (2,3 г, 5,1 ммоль), Et3SiH (6,6 мл, 41,1 ммоль) и трифторуксусной кислоты (0,83 мл, 10,7 ммоль) нагревали с обратным холодильником в дихлорэтане (10 мл) в герметичной пробирке в течение 3 ч. После охлаждения красный остаток фильтровали, промывали диэтиловым эфиром и вливали в смесь CH2Cl2 (300 мл) и насыщенного водного раствора бикарбоната натрия (300 мл). Коричневую смесь перемешивали в течение 1 ч при 23°C и разделяли слои. Водный слой экстрагировали CH2Cl2 (3x200 мл), объединенные органические слои сушили над сульфатом натрия, фильтровали и концентрировали при пониженном давлении с получением соединения 22 (1,1 г, 61%) в виде желтого твердого вещества.
1H ЯМР (CDCl3, 300 МГц) δ 8,51 (д, J=8,4 Гц, 1H), 8,50 (д, J=5,5 Гц, 1H), 8,07 (д, J=6,5 Гц, 1H), 7,78 (д, J=6,5 Гц, 1H), 7,37 (д, J=8,4 Гц, 1H), 7,34 (д, J=5,5 Гц, 1H), 2,79 (с, 3H), 2,79 (с, 3H), 2,71 (с, 3H).
Пример 18: Соединение 24
Раствор mCPBA (45 мг, 0,18 ммоль, 77%) в предварительно высушенном над сульфатом натрия CH2Cl2 (2 мл) добавляли по каплям к охлажденному (-30°C) раствору соединения 22 (29,5 мг, 0,083 ммоль) в CH2Cl2 (4 мл). Желтый раствор перемешивали в течение 15 мин при 0°C. Добавляли насыщенный водный раствор Na2S2О3 (5 мл) и промывали насыщенным водным раствором бикарбоната натрия (5 мл). Объединенные водные слои экстрагировали CH2Cl2 (3x10 мл). Объединенные органические слои сушили над сульфатом натрия, фильтровали и концентрировали. Промежуточный продукт 23 вливали в герметичную пробирку с 1,4-диоксаном (4 мл) и добавляли 32% раствор аммиака (14 мл). Коричневую смесь нагревали в течение 14 ч при 85°C. Полученную коричневую смесь упаривали в вакууме, добавляли CH2Cl2:MeOH (10:1, 11 мл), сушили над сульфатом натрия, фильтровали и выпаривали растворитель при пониженном давлении. Желтое твердое вещество очищали по методу флэш-хроматографии, применяя в качестве элюента от CH2Cl2:MeOH (2%) до CH2Cl2:MeOH (5%), с получением соединения 24 (4 мг, 17%, в 2 этапа) в виде желтого твердого вещества.
1H ЯМР (CDCl3, 300 МГц) δ 8,50 (д, J=8,3 Гц, 1H), 8,19 (д, J=5,4 Гц, 1H), 7,46-7,73 (м, 2H), 7,29 (д, J=8,3 Гц, 1H), 6,97 (д, J=5,4 Гц, 1H), 2,68 (с, 3H).
MS (ES) m/z 292 (М+1)+.
Пример 19: Соединение 44
Соединение 24 (30 мг) суспендировали в растворе HCl в 1,4-диоксане (6 мл, 3,8н) при 0°C. Бледно-коричневую смесь перемешивали при 0°C в течение 15 мин и упаривали в вакууме. Добавляли CH2Cl2 (5 мл), перемешивали в течение 1 мин и снова упаривали. Добавляли 1,4-диоксан (5 мл), бледно-коричневое твердое вещество фильтровали и дополнительно промывали 1,4-диоксаном (3 мл) с получением 30 мг соединения 44.
1H ЯМР (CD3OD, 300 МГц) δ 8,84 (д, J=8,3 Гц, 1Н), 8,21 (д, J=6,8 Гц, 1H), 8,00 (д, J=7,3 Гц, 1H), 7,66 (д, J=7,6 Гц, 1H), 7,61 (д, J=8,3 Гц, 1H), 7,30 (д, J=6,8 Гц, 1H), 2,79 (с, 3H).
Пример 20: Соединение 43
32% раствор аммиака (4 мл) добавляли к раствору соединения 22 (11 мг, 0,031 ммоль) в 1,4-диоксане (2 мл). Коричневую смесь нагревали в течение 14 ч при 85°C в герметичной пробирке. Полученную желтую смесь упаривали в вакууме, добавляли CH2Cl2 (5 мл), сушили над сульфатом натрия, фильтровали и выпаривали растворитель при пониженном давлении. Желтое твердое вещество очищали по методу флэш-хроматографии, применяя в качестве элюента CH2Cl2:MeOH от 1 до 3%, с получением соединения 43 (3 мг, 67% BRSM) в виде желтого твердого вещества.
1H ЯМР (CDCl3, 300 МГц) δ 8,60 (д, J=8,3 Гц, 1H), 8,47 (д, J=5,4 Гц, 1H), 7,63 (д, J=6,6 Гц, 1H), 7,57 (д, J=6,6 Гц, 1H), 7,35 (д, J=8,5 Гц, 1H), 7,31 (д, J=5,4 Гц, 1H), 2,73 (с, 3H), 2,69 (с, 3H).
МС (ESI) m/z, 323 (М+1)+.
Пример 21: Соединение 45
Раствор 4-йод-2-метилтиопиримидина (соединение 13, 0,76 г, 3,0 ммоль) в безводном тетрагидрофуране (10 мл) обрабатывали n-BuLi (1,2 мл, 2,5 M в гексанах, 3,0 ммоль) при -100°C и перемешивали оранжевый раствор при -100°C в течение 20 мин. К предварительно приготовленному раствору органо-литиевого производного по каплям добавляли раствор 2,5-дихлорникотиноилхлорида (0,21 г, 1,0 ммоль) в тетрагидрофуране (5 мл) и перемешивали реакционную смесь в течение 3 ч при -100°C. Потом неочищенный продукт реакции гасили добавлением насыщенного водного раствора хлорида аммония (20 мл) и экстрагировали этилацетатом (3x20 мл). Объединенные органические слои сушили над сульфатом натрия, фильтровали и концентрировали. Остаток очищали по методу флэш-хроматографии (элюируя этилацетатом:гексаном, 50:50) с получением соответствующего карбинола (0,11 г, 26%).
Раствор этого карбинола (0,11 г, 0,26 ммоль) в тетрагидрофуране (10 мл) добавляли к суспензии NaH (16 мг, 0,4 ммоль, 60%) в тетрагидрофуране (5 мл). Темно-красную смесь перемешивали при 23°C в течение 10 мин. Затем по каплям добавляли ацетилхлорид (0,071 мл, 1,0 ммоль) и перемешивали полученную желтую взвесь в течение 5 ч при 23°C. Добавляли насыщенный водный раствор бикарбоната натрия (20 мл) и экстрагировали водный слой CH2Cl2 (3x20 мл). Объединенные органические слои сушили над сульфатом натрия, фильтровали и концентрировали с получением соединения 45 (0,11 г, 90%, общий выход 24%) в виде коричневого масла, которое использовали без дополнительной очистки.
1H ЯМР (CDCl3, 300 МГц) δ 2,30 (с, 3H), 2,40 (с, 6H), 7,28 (д, J=5,3 Гц, 2H), 7,91 (д, J=2,4 Гц, 1H), 8,34 (д, J=2,4 Гц, 1H), 8,52 (д, J=5,3 Гц, 2H).
МС (ESI) m/z 468 (M)+.
Пример 22: Соединение 46
Раствор 4-йод-2-метилтиопиримидина (соединение 13, 2,3 г, 9,0 ммоль) в безводном тетрагидрофуране (10 мл) обрабатывали n-BuLi (3,6 мл, 2,5 M в гексанах, 9,1 ммоль) при -100°C и перемешивали оранжевый раствор при -100°C в течение 20 мин. К предварительно приготовленному раствору органо-литиевого производного по каплям добавляли раствор 2,6-дихлор-5-фторникотиноилхлорида (0,69 г, 3 ммоль) в тетрагидрофуране (5 мл) и перемешивали реакционную смесь в течение 3 ч при -100°C. Потом неочищенный продукт реакции гасили добавлением насыщенного водного раствора хлорида аммония (20 мл) и экстрагировали этилацетатом (3x20 мл). Объединенные органические слои сушили над сульфатом натрия, фильтровали и концентрировали. Остаток очищали по методу флэш-хроматографии (элюируя этилацетатом:гексаном, 50:50) с получением соответствующего карбинола (0,15 г, 11%).
Раствор этого карбинола (0,15 г, 0,3 ммоль) в тетрагидрофуране (3 мл) добавляли к суспензии NaH (20 мг, 0,5 ммоль, 60%) в тетрагидрофуране (2 мл). Темно-красную смесь перемешивали при 23°C в течение 10 мин. Затем по каплям добавляли ацетилхлорид (0,10 мл, 1,3 ммоль) и перемешивали полученную желтую взвесь в течение 5 ч при 23°C. Добавляли насыщенный водный раствор бикарбоната натрия (20 мл) и экстрагировали водный слой CH2Cl2 (3x20 мл). Объединенные органические слои сушили над сульфатом натрия, фильтровали и концентрировали с получением соединения 46 (0,13 г, 83%, общий выход 9%) в виде коричневого масла, которое использовали без дополнительной очистки.
1H ЯМР (CDCl3, 300 МГц) δ 2,31 (с, 3H), 2,42 (с, 6H), 7,27 (д, J=5,3 Гц, 2H), 7,81 (д, JH-F= 8,6 Гц, 1H), 8,52 (д, J=5,3 Гц, 2H).
МС (ESI) m/z 486 (M)+.
Пример 23: Соединение 47
Раствор соединения 45 (110 мг, 0,23 ммоль) и трифторуксусной кислоты (0,04 мл, 0,52 ммоль) в 1,2-дихлорэтане (2 мл) переносили в оснащенную резиновой мембраной пробирку Юнга, содержащую триэтилсилан (0,33 мл, 2,08 ммоль). Под сильной струей аргона мембрану заменяли тефлоновой закручивающейся крышкой и нагревали герметичную пробирку при 100°C в течение 48 ч. После охлаждения реакционный сосуд открывали и разбавляли содержимое CH2Cl2 (10 мл). Раствор нейтрализовали добавлением насыщенного водного раствора бикарбоната натрия (10 мл) и разделяли слои. Водный слой несколько раз экстрагировали CH2Cl2, а объединенные органические слои сушили над сульфатом натрия, фильтровали и концентрировали. Неочищенный материал очищали по методу флэш-хроматографии (элюируя этилацетатом:гексаном, 33%) с получением двух продуктов с одинаковым значением Rf. Путем добавления диэтилового эфира (15 мл) и фильтрации получали соединение 47 в виде желтого твердого вещества (20 мг, 20%).
1H ЯМР (CDCl3, 300 МГц) δ 2,68 (с, 3H), 2,73 (с, 3H), 7,31 (д, J=5,4 Гц, 1H), 7,86 (д, J=6,5 Гц, 1H), 8,00 (д, J=6,5 Гц, 1H), 8,51 (д, J=2,1 Гц, 1Н), 8,54 (д, J=5,4 Гц, 1Н), 8,66 (д, J=2,1 Гц, 1H), МС (ESI) m/z 374 (М+1)+.
Пример 24: Соединение 48
Раствор соединения 46 (200 мг, 0,42 ммоль) и трифторуксусной кислоты (64 мкл, 0,82 ммоль) в 1,2-дихлорэтане (2 мл) переносили в оснащенную резиновой мембраной пробирку Янга, содержащую триэтилсилан (0,52 мл, 3,28 ммоль). Под сильной струей аргона мембрану заменяли тефлоновой закручивающейся крышкой и нагревали герметичную пробирку при 140°C в течение 96 ч. После охлаждения реакционный сосуд открывали и разбавляли содержимое CH2Cl2 (10 мл). Раствор нейтрализовали добавлением насыщенного водного раствора бикарбоната натрия (10 мл) и разделяли слои. Водный слой несколько раз экстрагировали CH2Cl2, а объединенные органические слои сушили над сульфатом натрия, фильтровали и концентрировали. Неочищенный материал очищали по методу флэш-хроматографии (элюируя этилацетатом:гексаном, 33%) с получением двух продуктов с одинаковым значением Rf. Путем добавления диэтилового эфира (15 мл) и фильтрации получали соединение 48 в виде желтого твердого вещества (26 мг, 16%).
1H ЯМР (CDCl3, 300 МГц) δ 2,67 (с, 3H), 2,74 (с, 3H), 7,25 (д, J=5,4 Гц, 1H), 7,85 (д, J=6,5 Гц, 1H), 7,91 (д, J=6,5 Гц, 1H), 8,47 (д, JH-F= 8,8 Гц, 1H), 8,53 (д, J=5,4 Гц, 1H).
МС (ESI) m/z, 392 (М+1)+.
Пример 25: Соединение 49
Соединение 47 (20 мг, 0,054 ммоль) растворяли в охлажденном при -30°C хлороформе (5 мл). По каплям добавляли предварительно охлажденный (-30°C) раствор мета-хлорпербензойной кислоты (19 мг, 0,11 ммоль, 77%) в CH2Cl2 (5 мл) и перемешивали при 0°C в течение 15 мин. Раствор нагревали до 23°C, нейтрализовали добавлением насыщенного водного раствора бикарбоната натрия и несколько раз экстрагировали CH2Cl2. Объединенные органические слои сушили над сульфатом натрия, фильтровали и концентрировали с получением сульфоксида в виде желтого твердого вещества, которое использовали без дополнительной очистки. Неочищенный окисленный материал нагревали с избытком пара-метоксибензиламина (1 мл) при 85°C в течение 15 ч. Реакционную смесь очищали по методу флэш-хроматографии (элюируя этилацетатом:гексаном, 50:50). Этот продукт обрабатывали трифторметансульфоновой кислотой (0,5 мл) при 23°C в течение 3 ч. Реакционную смесь охлаждали до 0°C и последовательно обрабатывали MeOH (1 мл) и водным NH4OH (1 мл, 32%).
Осадок отфильтровывали, промывали MeOH (5 мл) и диэтиловым эфиром (5 мл) и сушили с получением соединения 49 в виде желтого твердого вещества (10 мг, общий выход 60%).
1H ЯМР (CDCl3, 300 МГц) δ 6,92 (д, J=5,5 Гц, 1H), 7,34 (д, J=6,6 Гц, 1H), 7,51 (д, J=6,6 Гц, 1H), 8,17 (д, J=5,5 Гц, 1H), 8,25 (д, J=2,2 Гц, 1H), 8,64 (д, J=2,2 Гц, 1H).
МС (ESI) m/z, 312 (М+1)+.
Пример 26: Соединение 50
Соединение 48 (26 мг, 0,066 ммоль) растворяли в хлороформе (5 мл) и охлаждали при -30°C. По каплям добавляли предварительно охлажденный (-30°C) раствор мета-хлорпербензойной кислоты (23 мг, 0,132 ммоль) в CH2Cl2 (5 мл) и перемешивали при 0°C в течение 15 мин. Раствор нагревали до 23°C, нейтрализовали добавлением насыщенного водного раствора бикарбоната натрия и несколько раз экстрагировали CH2Cl2. Объединенные органические слои сушили над сульфатом натрия, фильтровали и концентрировали с получением сульфоксида в виде желтого твердого вещества, которое использовали без дополнительной очистки. Неочищенный окисленный материал нагревали с избытком пара-метоксибензиламина (1 мл) при 85°C в течение 15 ч. Реакционную смесь очищали по методу флэш-хроматографии (элюируя этилацетатом:гексаном, 50:50). Этот продукт обрабатывали трифторметансульфоновой кислотой (0,5 мл) при 23°C в течение 3 ч. Реакционную смесь охлаждали до 0°C и последовательно обрабатывали MeOH (1 мл) и водным NH4OH (1 мл, 32%).
Осадок фильтровали, промывали MeOH (5 мл) и диэтиловым эфиром (5 мл) и сушили с получением соединения 50 в виде желтого твердого вещества (3 мг, общий выход 14%).
1H ЯМР (CDCl3, 300 МГц) δ 6,86 (д, J=5,4 Гц, 1H), 7,28 (д, J=6,6 Гц, 1H), 7,48 (д, J=6,6 Гц, 1H), 8,12 (д, J=5,4 Гц, 1H), 8,50 (д, JH-F = 9,3 Гц, 1H), МС (ESI) m/z, 329 (M)+.
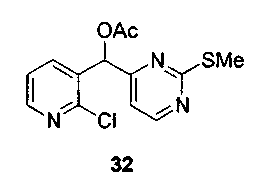
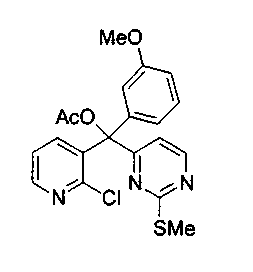
Пример 27: Соединение 8b
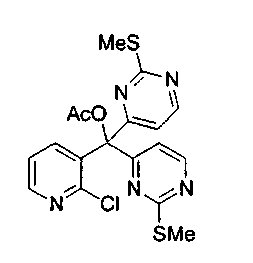
Способ A
Раствор изо-пропилмагнийбромида (5,0 мл, 10,0 ммоль, 2,0 M в тетрагидрофуране) добавляли по каплям к раствору 4-йод-2-метилтиопиримидина (соединение 13, 2,52 г, 10,0 ммоль) в толуоле (60 мл) при 0°C. Коричневый раствор перемешивали в течение 1 ч при 0°C. Этот раствор добавляли по каплям через канюлю к раствору соединения 31 (1,06 г, 4 ммоль) в толуоле (40 мл) в течение 20 мин. После завершения добавления реакционную смесь дополнительно перемешивали в течение 15 минут при 0°C и затем гасили добавлением ацетилхлорида (0,99 мл, 14,0 ммоль). Ледяную баню удаляли и перемешивали полученную коричневую взвесь в течение 3 ч при 23°C. Добавляли насыщенный водный раствор бикарбоната натрия (100 мл) и экстрагировали водный слой этилацетатом (3x50 мл). Объединенные органические слои сушили над сульфатом натрия, фильтровали и концентрировали. Неочищенный остаток очищали по методу флэш-хроматографии (элюируя этилацетатом:гексаном, 50:50) с получением соединения 8b (1,28 г, 74%) в виде пенистого бледно-оранжевого твердого вещества. Продукт выделяли вместе с небольшим количеством соединения 32, которое образуется в данной реакции в качестве побочного продукта. На следующем этапе применяли смесь обоих соединений.
Соединение 8b:
1H ЯМР (CDCl3, 300 МГц) δ 8,48 (д, J=5,1 Гц, 2H), 8,36 (дд, J=4,9, 1,9 Гц, 1Н), 7,86 (дд, J=7,8, 1,7 Гц, 1Н), 7,28 (д, J=5,1 Гц, 2H), 7,24 (дд, J=7,8, 4,6 Гц, 1Н), 2,36 (с, 6H), 2,28 (с, 3H).
МС (ESI) m/z, 456 (M+23)+, 434 (М+1)+.
1H ЯМР (CDCl3, 300 МГц) δ 8,47 (д, J=5,1 Гц, 1Н), 8,37 (дд, J=4,9, 1,9 Гц, 1Н), 7,82 (дд, J=7,8, 1,6 Гц, 1Н), 7,30 (дд, J=7,8, 4,6 Гц, 1Н), 7,10 (д, J=5,1 Гц, 1Н), 7,01 (с, 1Н), 2,36 (с, 3H), 2,28 (с, 3H).
Способ B
n-BuLi (9 мл, 22,6 ммоль, 2,5 M в гексане), предварительно охлажденный при -78°C, добавляли по каплям к раствору соединения 31 (3 г, 11,3 ммоль) и 4-йод-2-метилтиопиримидина (соединение 13, 5,71 г, 22,6 ммоль) в тетрагидрофуране (75 мл) при -78°C. Темно-коричневую смесь перемешивали в течение 15 мин при -78°C и осторожно добавляли ацетилхлорид (3,2 мл, 45,3 ммоль). Темно-зеленую взвесь перемешивали при 23°C в течение 3,5 ч, гасили добавлением насыщенного водного раствора бикарбоната натрия (150 мл) и экстрагировали диэтиловым эфиром (2x150 мл). Объединенные органические слои сушили над MgSO4, фильтровали и концентрировали. Красный остаток очищали по методу флэш-хроматографии (элюируя этилацетатом:гексаном от 1:4 до 100:0) с получением соединения 8b (3 г, 61%) в виде бледно-красного твердого вещества.
Пример 28: Соединение 9
Смесь соединения 8b (1,6 г, 3,7 ммоль), трифторуксусной кислоты (0,6 мл, 7,74 ммоль) и Et3SiH (4,7 мл, 29,5 ммоль) вливали в пробирку Янга, содержащую 1,2-дихлорэтан (8 мл). Герметичный реакционный сосуд нагревали при 90°C в течение 24 ч. После охлаждения реакционный сосуд открывали и разбавляли содержимое хлороформом (100 мл). Раствор нейтрализовали добавлением насыщенного водного раствора бикарбоната натрия (100 мл) и разделяли слои. Водный слой несколько раз экстрагировали хлороформом, а объединенные органические слои сушили над сульфатом натрия, фильтровали и концентрировали. Темно-красный остаток очищали путем добавления диэтилового эфира (100 мл) и фильтрации ярко-красного осадка с получением соединения 9 (0,85 г, 68%).
1H ЯМР (CDCl3, 300 МГц) δ 8,64 (дд, J=8,1, 1,7 Гц, 1H), 8,60 (дд, J=4,6, 1,7 Гц, 1H), 8,51 (д, J=5,4 Гц, 1H), 8,06 (д, J=6,4 Гц, 1H), 7,82 (д, J=6,6 Гц, 1H), 7,51 (дд, J=8,5, 4,6 Гц, 1Н), 7,34 (д, J=5,4 Гц, 1H), 2,73 (с, 3H), 2,68 (с, 3H).
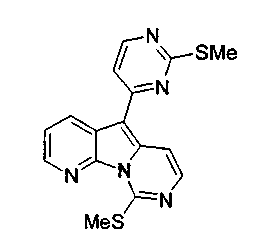
Пример 29: Дезоксивариолин (соединение 4)
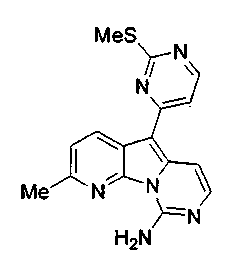
Раствор mCPBA (98 мг, 0,39 ммоль, 77%) в предварительно высушенном над сульфатом натрия CH2Cl2 (4 мл) добавляли по каплям к охлажденному (-30°C) раствору соединения 9 (61 мг, 0,18 ммоль) в CH2Cl2 (5 мл). Желтый раствор перемешивали в течение 15 мин при 0°C. Добавляли насыщенный водный раствор Na2S2О3 (5 мл) и промывали органический слой насыщенным водным раствором бикарбоната натрия (5 мл). Объединенные водные слои экстрагировали CH2Cl2 (3x10 мл). Объединенные органические слои сушили над сульфатом натрия, фильтровали и концентрировали. Соединение 28 вливали в герметичную пробирку с 1,4-диоксаном (4 мл) и добавляли 32% раствор аммиака (8 мл). Коричневую смесь нагревали в течение 14 ч при 85°C. Полученную желтую смесь упаривали в вакууме, добавляли CH2Cl2:MeOH (10:1, 11 мл), сушили над сульфатом натрия, фильтровали и выпаривали растворитель при пониженном давлении. Желтое твердое вещество очищали по методу флэш-хроматографии, применяя в качестве элюента от CH2Cl2:MeOH (2%) до CH2Cl2:MeOH (5%), с получением дезоксивариолина (соединение 4, 14 мг, 29 %, в 2 этапа) в виде желтого твердого вещества.
1H ЯМР (DMSO-d6, 300 МГц) δ 8,92 (дд, J=8,1, 1,5 Гц, 1H), 8,45 (дд, J=4,6, 1,4 Гц, 1H), 8,22 (д, J=5,5 Гц, 1H), 7,68 (д, J=6,6 Гц, 1H), 7,63 (д, J=6,6 Гц, 1H), 7,58 (дд, J=8,1, 4,6 Гц, 1H), 7,06 (д, J=5,4 Гц, 1H).
МС (ESI) m/z, 278 (М+1)+.
Пример 30: Соединение 51
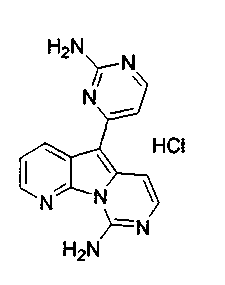
Дезоксивариолин (соединение 4, 6 мг, 0,024 ммоль) обрабатывали раствором HCl в 1,4-диоксане (2 мл, 5,3 M) и перемешивали в течение 2 ч при 23°C. Суспензию фильтровали с получением соединения 51 (3 мг) в виде желтого твердого вещества.
1H ЯМР (CD3OD, 300 МГц) δ 8,99 (дд, J=8,2, 1,2 Гц, 1Н), 8,67 (дд, J=4,9, 1,2 Гц, 1H), 8,30 (д, J=6,8 Гц, 1H), 8,05 (д, J=7,5 Гц, 1H), 7,77 (дд, J=8,3, 4,9 Гц, 1H), 7,67 (д, H, J=6,9 Гц, 1H), 7,57 (д, J=7,1 Гц, 1H).
Пример 31: Соединение 30
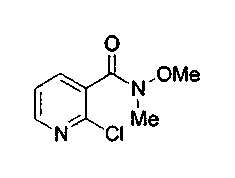
К интенсивно перемешанной суспензии N,O-диметилгидроксиамингидрохлорида (33,2 г, 0,34 моль) в тетрагидрофуране (1,2 л) добавляли триэтиламин (59 мл, 0,426 моль) и перемешивали реакционную смесь в течение 20 мин при 23°C. Потом добавляли чистый2-хлорникотиноилхлорид (соединение 7b, 50 г, 0,284 моль) и перемешивали реакционную смесь в течение ночи при 23°C. Неочищенную смесь гасили добавлением насыщенного водного раствора бикарбоната натрия и экстрагировали CH2Cl2 (3x750 мл). Объединенные органические слои сушили над сульфатом натрия, фильтровали и концентрировали с получением соединения 30 (49,4 г, 87%) в виде желтого твердого вещества, которое использовали без дополнительной очистки.
1H ЯМР (CDCl3, 300 МГц) δ 3,39 (с, 3H), 3,48 (с, 3H), 7,29 (дд, J=7,5, 4,6 Гц, 1Н), 7,68 (д, J=7,5 Гц, 1H), 8,43 (дд, J=4,6, 1,5 Гц, 1H).
МС (ESI) m/z, 201 (М+1)+.
Пример 32: Соединение 31
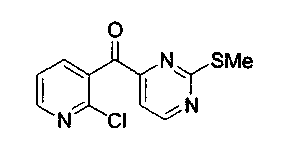
К раствору 4-йод-2-метилтиопиримидина (соединение 13, 20,3 г, 80,5 ммоль) в безводном толуоле при -4°C по каплям добавляли i-PrMgCl (40,2 мл, 80,5 ммоль, 2,0 M в тетрагидрофуране). Полученную бледно-коричневую суспензию перемешивали при -4°C в течение 45 мин. Через канюлю в течение 15 мин добавляли раствор соединения 30 (12,9 г, 64,4 ммоль) в тетрагидрофуране (50 мл) и перемешивали полученную темно-коричневую смесь при 0°C в течение 1 ч. Реакционную смесь гасили добавлением насыщенного водного раствора хлорида аммония (200 мл) и экстрагировали этилацетатом (2x200 мл). Объединенные органические слои сушили над сульфатом натрия, фильтровали и упаривали с получением коричневого твердого вещества (18 г). Остаток растирали со смесью диэтилового эфира и гексана (3:1, 40 мл) и фильтровали с получением соединения 31 в виде бледно-коричневого твердого вещества (13,6 г, 80%).
1H ЯМР (CDCl3, 300 МГц) δ 8,82 (д, J=4,9 Гц, 1H), 8,57 (дд, J=4,2, 1,9 Гц, 1H), 7,89 (дд, J=7,5, 1,8 Гц, 1H), 7,64 (д, J=5,1 Гц, 1H),7,41 (дд, J=7,6, 4,8 Гц, 1H), 2,38 (с, 3H).
13C ЯМР (CDCl3, 300 МГц) δ 198,2, 173,6, 159,6, 158,7, 151,6, 148,5, 139,4, 133,3, 122,3, 113,6, 14,2.
МС (ESI) m/z 288 (M+23)+, 266 (М+1)+.
Пример 33: Соединение 33
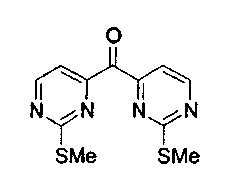
Способ A
Раствор 4-йод-2-метилтиопиримидина (соединение 13, 15,6 г, 62 ммоль) в тетрагидрофуране (200 мл) обрабатывали n-BuLi (24,8 мл, 2,5 M в гексанах, 62 ммоль) при -100°C. После добавления реакционную смесь перемешивали при -100°C в течение 30 мин и обрабатывали при -110°C раствором диэтилкарбоната (3,8 мл, 31 ммоль) в тетрагидрофуране (6 мл) в течение 30 мин. Реакционную смесь нагревали до -80°C, гасили добавлением насыщенного водного раствора хлорида аммония и распределяли между этилацетатом и насыщенным водным раствором хлорида аммония. Органический слой сушили над сульфатом натрия, фильтровали и упаривали. Остаток подвергали хроматографии (элюируя гексаном:этилацетатом от 4:1 до 3:1) с получением соединения 33 в виде желтого твердого вещества (5,3 г, 61%).
1H ЯМР (CDCl3, 300 МГц) δ 8,78 (д, J=4,9 Гц, 2H), 7,53 (д, J=4,9 Гц, 2H), 2,50 (с, 6H).
13C ЯМР (CDCl3, 75 МГц) δ 190,9, 173,5, 159,5, 159,0, 115,2, 14,4.
МС (ESI) m/z 279 (М+1)+.
Способ B
Раствор 4-йод-2-метилтиопиримидина (соединение 13, 50,4 г, 200 ммоль) в толуоле (420 мл) обрабатывали i-PrMgCl (100 мл, 2 M в тетрагидрофуране, 200 ммоль) при -10°C в течение 2 ч. Через канюлю к EtOCOOEt (72 мл, 594,4 ммоль) при -10°C добавляли пиримидинмагний и перемешивали при 0°C в течение 2,5 ч. Реакционную смесь гасили добавлением насыщенного водного раствора хлорида аммония, нагревали до 23°C и распределяли между этилацетатом и насыщенным водным раствором хлорида аммония. Органический слой сушили над сульфатом натрия, фильтровали и упаривали. Остаток растирали с CH2Cl2 и фильтровали. Фильтрат упаривали и подвергали хроматографии (элюируя гексаном:этилацетатом от 4:1 до 3:1) с получением соединения 33 в виде желтого твердого вещества (13,3 г, 48%).
Пример 34: Соединение 34
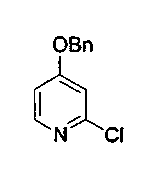
Суспензию 4-бензилокси-2-(1Н)пиридона (3,0 г, 5,0 ммоль) в свежеперегнанном POCl3 (18 мл) нагревали при 90°C в течение 15 ч. Реакционную смесь охлаждали и упаривали. Остаток вливали на лед, обрабатывали насыщенным водным раствором бикарбоната натрия и экстрагировали CH2Cl2. Органическую фазу сушили над сульфатом натрия, фильтровали и концентрировали. Полученный остаток подвергали хроматографии (элюируя CH2Cl2:MeOH, 100:1) с получением соединения 34 в виде белого твердого вещества (2,5 г, 75%).
1H ЯМР (CDCl3, 300 МГц) δ 8,19 (д, J=5,7 Гц, 1H), 7,40 (м, 5H), 6,90 (д, J=2,1 Гц, 1H), 6,80 (дд, J=5,7, 2,1 Гц, 1H), 5,10 (с, 2H).
13С ЯМР (CDCl3, 75 МГц) δ 166,6, 152,8, 150,6, 135,3, 129,0, 128,8, 127,8, 110,6, 110,5, 70,6.
МС (ESI) m/z, 219 (M)+.
Rf: 0,4 (гексан:этилацетат, 4:1).
Пример 35: Соединение 35
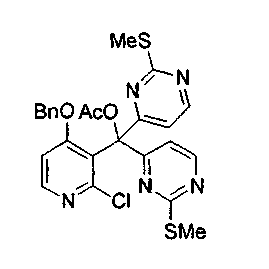
Раствор соединения 34 (1,5 г, 6,8 ммоль) в тетрагидрофуране (20 мл) обрабатывали n-BuLi (2,7 мл, 6,8 ммоль, 2,5 M в гексанах) при -100°C, оставляли нагреваться до -78°C и поддерживали эту температуру в течение 4 ч. Реакционную смесь охлаждали до -100°C и обрабатывали через канюлю раствором соединения 33 (1,9 г, 6,8 ммоль) в предварительно охлажденном при -78°C тетрагидрофуране (15 мл). Реакционную смесь перемешивали при -78°C в течение 3 ч и нагревали до -50°C в течение 30 мин, повторно охлаждали до -78°C и обрабатывали ацетилхлоридом, предварительно перегнанным из хинолина (2,0 мл, 28 ммоль), нагревали до 23°C и перемешивали в течение ночи. Реакционную смесь распределяли между этилацетатом и насыщенным водным раствором бикарбоната натрия. Органический слой сушили над сульфатом натрия, фильтровали и упаривали. Остаток подвергали хроматографии (элюируя гексаном:этилацетатом от 2:1 до 1:1) с получением соединения 35 в виде белого твердого вещества (2,9 г, 78%).
1H ЯМР (CDCl3, 300 МГц) δ 8,26 (д, J=5,6 Гц, 1H), 8,11 (д, J=5,4 Гц, 2H), 7,32-7,26 (м, 4H), 7,08 (д, J=5,4 Гц, 2H), 7,02 (дд, J=6,0, 2,4 Гц, 1H), 6,87 (д, J=5,6 Гц, 1H), 4,80 (с, 2H), 2,37 (с, 6H), 2,32 (с, 3H).
13C ЯМР (CDCl3, 75 МГц) δ 171,5, 168,3, 168,1, 165,8, 156,5, 152,5, 149,9, 133,9, 128,7, 128,7, 128,0, 122,5, 115,1, 107,8, 84,7, 71,7, 21,7, 14,2.
Пример 36: Соединение 52
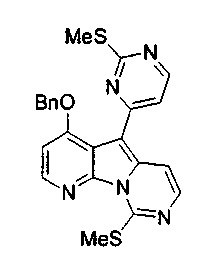
Раствор соединения 35 (740 мг, 1,37 ммоль) и TFA (221 мкл, 2,86 ммоль) в 1,2-дихлорэтане (3,0 мл) добавляли в атмосфере аргона в пробирку Янга с Et3SiH (1,8 мл, 11 ммоль). Трубку Янга герметизировали и нагревали при 80°C в течение 24 ч. Реакционную смесь растворяли в хлороформе и промывали насыщенным водным раствором бикарбоната натрия. Органический слой сушили над сульфатом натрия, фильтровали и упаривали. Остаток подвергали хроматографии (элюируя гексаном:этилацетатом, 4:1) с получением соединения 52 в виде желтого твердого вещества (420 мг, 69%).
1H ЯМР (CDCl3, 300 МГц) δ 8,47 (д, J=5,4 Гц, 1H), 8,06 (д, J=5,4 Гц, 1H), 8,00 (д, J=6,6Гц, 1H), 7,71 (д, J=6,6 Гц, 1H), 7,39 (м, 5H), 7,35 (д, J=5,4 Гц, 1H), 7,01 (д, J=5,4 Гц, 1H), 5,28 (с, 2H), 2,70 (с, 3H), 2,63 (с, 3H).
13C ЯМР (CDCl3, 75 МГц) δ 171,2, 160,9, 158,9, 155,8, 154,4, 144,7, 143,6, 138,9, 136,3, 135,1, 128,9, 128,9, 128,3, 118,3, 111,2, 108,9, 103,1, 71,2, 15,12, 14,3. Наблюдается частичное перекрывание сигнала одного ароматического углерода.
МС (APCI) m/z 446 (М+1)+.
Rf: 0,37 (гексан:этилацетат, 2:1).
Пример 37: Соединение 53
Раствор соединения 52 (1,9 г, 4,3 ммоль) в хлороформе (50 мл) обрабатывали при -30°C раствором mCPBA (2,4 г, 10,9 ммоль, 77%) в хлороформе (20 мл). После добавления реакционную смесь нагревали до 0°C, перемешивали в течение 20 мин и гасили добавлением насыщенного водного раствора Na2S2О3. Органический слой промывали насыщенным водным раствором бикарбоната натрия, сушили над сульфатом натрия, фильтровали и концентрировали. Остаток обрабатывали пара-метоксибензиламином (10 мл, 76,5 ммоль) при 90°C в течение 24 ч. Избыток пара-метоксибензиламина удаляли путем перегонки в аппарате Кугельрора (140°C, 0,5 мм рт.ст.) и подвергали хроматографии остаток (элюируя гексаном:этилацетатом от 1:1 до 0:100) с получением соединения 53 в виде желтого твердого вещества (1,5 г, 55%).
1H ЯМР (CDCl3, 300 МГц) δ 10,39 (т, J=5,0 Гц, 1H), 8,13 (д, J=5,7 Гц, 1H), 7,95 (д, J=5,2 Гц, 1H), 7,44-7,25 (м, 11H), 6,98 (д, J=5,4 Гц, 1H), 6,91-6,89 (м, 5H), 5,47 (т, J=5,0 Гц, 1H), 5,25 (с, 2H), 4,85 (д, J=5,4 Гц, 2H), 4,62 (д, J=5,7 Гц, 2H), 3,80 (с, 3H), 3,79 (с, 3H).
МС (ESI) m/z, 624 (M)+.
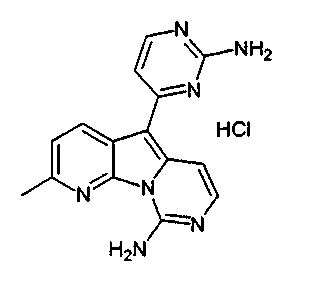
Пример 38: Вариолин B (соединение 1)
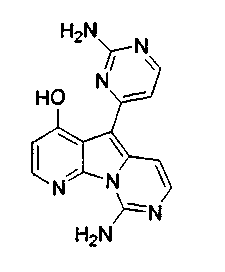
Раствор соединения 53 (204 мг, 0,33 ммоль) в TfOH (0,5 мл) перемешивали при 23°C в течение 4 ч. Реакционную смесь охлаждали до 0°C и по каплям обрабатывали MeOH (3 мл) и раствором NH4OH (4 мл, 32%). Реакционную смесь фильтровали и промывали H2О, MeOH и диэтиловым эфиром. Полученное твердое вещество подвергали хроматографии (элюируя CH2Cl2:MeOH от 95:5 до 85:15) с получением вариолина B (соединение 1) в виде желтого твердого вещества (70 мг, 72%).
1H ЯМР (CDCl3, 300 МГц) δ 8,12 (д, J=5,9 Гц, 1H), 8,07 (д, J=5,4 Гц, 1H), 7,48 (д, J=6,8 Гц, 1H), 7,03-6,99 (м, 2H), 6,76 (д, J=5,4 Гц, 1H).
Пример 39: Соединение 54
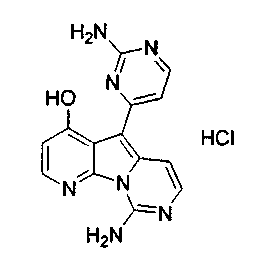
Вариолин B (19 мг, 0,065 ммоль) обрабатывали безводным HCl в 1,4-диоксане (3,0 мл, 3,8 M) в течение 5 ч при 23°C. Реакционную смесь упаривали и промывали диэтиловым эфиром с получением соединения 54 в виде оранжевого твердого вещества.
1H ЯМР (CD3OD, 300 МГц) δ 8,37 (д, J=5,6 Гц, 1Н), 8,26 (д, J=6,8 Гц, 1Н), 7,78 (д, J= 7,6 Гц, 1Н), 7,68 (д, J=7,6 Гц, 1Н), 7,57 (д, J=6,8 Гц, 1Н), 7,07 (д, J=5,6 Гц, 1Н).
МС (ESI) m/z 294 (М+1)+.
Пример 40: Соединение 36
Раствор 3-метоксифенилмагнийбромида (3,2 мл, 3,2 ммоль, 1 M в тетрагидрофуране) добавляли по каплям к раствору соединения 31 (0,7 г, 2,65 ммоль) в тетрагидрофуране (10 мл) при 0°C. Коричневую смесь перемешивали при 0°C в течение 2 ч. Добавляли ацетилхлорид (0,75 мл, 10,6 ммоль) и перемешивали смесь в течение 2 ч при 23°C. Добавляли насыщенный водный раствор хлорида аммония (50 мл) и экстрагировали водный слой диэтиловым эфиром (3x40 мл). Объединенные органические слои сушили над MgSO4, фильтровали и упаривали. Оранжевый остаток очищали по методу флэш-хроматографии (элюируя этилацетатом:гексаном от 1:3 до 1:1,5) с получением соединения 36 (0,6 г, 59%) в виде бесцветного масла.
1H ЯМР (300 МГц, CDCl3) δ 8,48 (д, J=5,2 Гц, 1Н), 8,37 (дд, J=4,6, 1,7 Гц, 1Н), 7,99 (дд, J=7,8, 1,6 Гц, 1Н), 7,28-7,20 (м, 3H), 6,95-6,91 (м, 2 H), 6,85-6,81 (м, 1Н), 3,77 (с, 3H), 2,33 (с, 3H), 2,31 (с, 3H).
Пример 41: Соединение 57
Смесь соединения 36 (0,38 г, 0,91 ммоль), трифторуксусной кислоты (0,15 мл, 1,9 ммоль) и триэтилсилана (1,17 мл, 7,3 ммоль) вливали в пробирку Янга, содержащую 1,2-дихлорэтан (4 мл). Герметичный реакционный сосуд нагревали при 90°C в течение 22 ч. После охлаждения реакционный сосуд открывали и разбавляли содержимое хлороформом (50 мл). Раствор нейтрализовали добавлением насыщенного водного раствора бикарбоната натрия (60 мл) и разделяли слои. Водный слой несколько раз экстрагировали хлороформом, объединенные органические слои сушили над сульфатом натрия, фильтровали и концентрировали. Темно-красный остаток очищали по методу флэш-хроматографии (элюируя этилацетатом:гексаном от 1:4 до 1:3) с получением соединения 57 (20 мг, 54%) в виде желтого твердого вещества.
1H ЯМР (300 МГц, CDCl3) δ 8,59 (дд, J=4,6, 1,7 Гц, 1H), 8,29 (дд, J=8,2, 1,5 Гц, 1Н), 7,57 (д, J=6,6 Гц, 1Н), 7,48-7,41 (м, 2H), 7,33 (д, J=6,6 Гц, 1Н), 7,24-7,16 (м, 2H), 6,94-6,90 (м, 1Н), 3,90 (с, 3H), 2,74 (с, 3H).
13C ЯМР (CDCl3, 75 МГц) δ 160,4, 141,7, 136,8, 134,9, 132,8, 130,3, 127,0, 126,5, 121,6, 120,2, 114,9, 112,1, 107,2, 55,6, 14,8.
МС (ESI) m/z 344 (M+23)+, 322 (М+1)+.
Пример 42: Соединение 62
Водный NH4OH (15 мл, 32%) добавляли к раствору соединения 57 (150 мг, 0,46 ммоль) в 1,4-диоксане (10 мл). Желтую смесь перемешивали в течение 30 ч при 90°C в герметичной пробирке. Полученную желтую смесь упаривали при пониженном давлении и очищали полученный желтый остаток по методу флэш-хроматографии, применяя в качестве элюента от CH2Cl2:MeOH (2%) до CH2Cl2:MeOH (3%), с получением соединения 62 (64 мг, 65% BRSM) в виде желтого твердого вещества.
1H ЯМР (300 МГц, CDCl3) δ 8,38 (дд, J=4,6, 1,5 Гц, 1H), 8,27 (дд, J=8,1, 1,5 Гц, 1H), 7,47-7,38 (м, 3 H), 7,27-7,19 (м, 2 H), 7,00 (д, J=6,8 Гц, 1H), 6,90 (дд, J=8,3, 2,5 Гц, 1H), 3,90 (с, 3H).
13C ЯМР (75 МГц, CDCl3) δ 160,1, 149,3, 142,6, 139,7, 139,6, 135,1, 133,6, 130,0, 126,5, 122,3, 121,1, 119,7, 114,3, 111,5, 103,8, 100,8, 55,3.
МС (ESI) m/z, 291 (М+1)+.
Пример 43: Соединение 67
Соединение 62 (10 мг, 0,03 ммоль) обрабатывали раствором HCl в 1,4-диоксане (3 мл, 3,5 M) и перемешивали в течение 2 ч при 23°C. Бесцветный раствор упаривали, промывали диэтиловым эфиром и фильтровали с получением соединения 67 (11 мг) в виде желтого твердого вещества.
1H ЯМР (300 МГц, CD3OD) δ 8,65 (дд, J=4,9, 1,5 Гц, 1Н), 8,41 (дд, J=8,1, 1,3 Гц, 1H), 7,69 (дд, J=8,1, 4,6 Гц, 1H), 7,52 (т, J=7,8 Гц, 1H), 7,24-7,18 (м, 3H), 7,09 (д, J=8,1 Гц, 1H), 7,06 (дд, J=8,3, 2,4 Гц, 1Н), 3,90 (с, 3H).
Пример 44: Соединение 37
Раствор PhLi (0,58 мл, 1,04 ммоль, 1,8 M в циклогексане:эфире) добавляли по каплям к раствору соединения 31 (190 мг, 0,73 ммоль) в тетрагидрофуране (6 мл) при -78°C. Темно-красную смесь перемешивали при -78°C в течение 3 ч. Добавляли насыщенный водный раствор хлорида аммония (25 мл) и экстрагировали водный слой диэтиловым эфиром (3x40 мл). Объединенные органические слои сушили над MgSO4, фильтровали и упаривали. Оранжевый остаток очищали по методу флэш-хроматографии (элюируя этилацетатом:гексаном от 25% до 35%) с получением спирта (0,11 г, 44%) в виде бледно-желтого масла. Раствор спирта (94 мг, 0,27 ммоль) в тетрагидрофуране (2 мл) добавляли к 60% суспензии NaH (22 мг, 0,54 ммоль) в тетрагидрофуране (1 мл). Темно-красную смесь перемешивали при 23°C в течение 10 мин. По каплям добавляли ацетилхлорид (0,6 мл, 8,5 ммоль) и перемешивали полученную желтую взвесь в течение 5 ч при 23°C. Добавляли насыщенный водный раствор бикарбоната натрия (20 мл) и экстрагировали водный слой CH2Cl2 (3x25 мл). Объединенные органические слои сушили над сульфатом натрия, фильтровали и упаривали. Коричневый остаток очищали по методу флэш-хроматографии (элюируя этилацетатом:гексаном от 25 до 35%) с получением соединения 37 (52 мг, 50%, 22% в 2 этапа) в виде бесцветного масла.
1H ЯМР (300 МГц, CDCl3) δ 8,49 (д, J=5,4 Гц, 1H), 8,36 (дд, J=4,6, 1,5 Гц, 1Н), 7,97 (дд, J=7,9, 1,5 Гц, 1Н), 7,35-7,10 (м, 7H), 2,31 (ушир.с, 6H).
МС (ESI) m/z, 408 (M+23)+, 386 (М+1)+.
Пример 45: Соединение 58
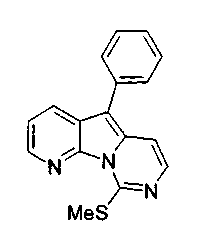
Смесь соединения 37 (51 мг, 0,13 ммоль), трифторуксусной кислоты (22 мкл, 0,28 ммоль) и триэтилсилана (0,17 мл, 1,07 ммоль) вливали в пробирку Янга, содержащую 1,2-дихлорэтан (0,5 мл). Герметичный реакционный сосуд нагревали при 85°C в течение 22 ч. После охлаждения реакционный сосуд открывали и разбавляли содержимое хлороформом (25 мл). Раствор нейтрализовали добавлением насыщенного водного раствора бикарбоната натрия (30 мл) и разделяли слои. Водный слой несколько раз экстрагировали хлороформом, объединенные органические слои сушили над сульфатом натрия, фильтровали и концентрировали. Темно-красный остаток очищали по методу флэш-хроматографии (элюируя этилацетатом:гексаном от 1:4 от 1:3) с получением соединения 58 (0,19 г, 66%) в виде желтого твердого вещества.
1H ЯМР (300 МГц, CDCl3) δ 8,62 (дд, J=4,6, 1,5 Гц, 1H), 8,29 (дд, J=8,2, 1,6 Гц, 1Н), 7,67-7,63 (м, 2H), 7,58 (д, J=6,5 Гц, 1Н), 7,57-7,53 (м, 2H), 7,45 (дд, J=8,1, 4,6 Гц, 1Н), 7,42-7,37 (м, 1Н), 7,33 (д, J=6,6 Гц, 1Н), 2,76 (с, 3H).
13C ЯМР (75 МГц, CDCl3) δ 154,3, 141,8, 136,5, 133,3, 129,0, 128,9, 126,6, 121,4, 119,9, 106,9, 14,5.
МС (ESI) m/z, 292 (М+1)+.
Пример 46: Соединение 63
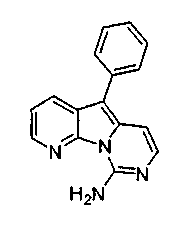
Водный NH4OH (5 мл, 32%) добавляли к раствору соединения 58 (19 мг, 0,065 ммоль) в 1,4-диоксане (2 мл). Желтую смесь перемешивали в течение 24 ч при 90°C в герметичной пробирке. Полученную желтую смесь упаривали при пониженном давлении и очищали желтый остаток по методу флэш-хроматографии, применяя в качестве элюента от CH2Cl2:MeOH (1%) до CH2Cl2:MeOH (2%), с получением соединения 63 (8 мг, 98% BRSM) в виде желтого твердого вещества.
1H ЯМР (300 МГц, CDCl3) δ 8,35 (дд, J=4,3, 1,4 Гц, 1Н), 8,23 (дд, J=7,9, 1,3 Гц, 1Н), 7,62-7,58 (м, 2H), 7,54-7,37 (м, 2H), 7,39 (дд, J=8,1, 4,4 Гц, 1Н), 7,38-7,31 (м, 2H), 6,92 (д, J=6,6 Гц, 1Н).
МС (ESI) m/z: 261 (М+1)+.
Пример 47: Соединение 38
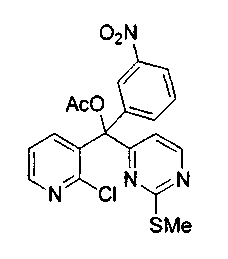
Раствор n-BuLi (0,83 мл, 2,075 ммоль, 2,5 M в гексане) добавляли по каплям к раствору 1-йод-2-нитробензола (0,51 г, 2,0 ммоль) в тетрагидрофуране (8 мл) при -78°C. Черную смесь перемешивали при -78°C в течение 10 мин и через канюлю добавляли раствор соединения 31 (0,45 г, 1,7 ммоль) в тетрагидрофуране (8 мл). Темную смесь медленно нагревали до -40°C в течение 2 ч. Добавляли ацетилхлорид (0,36 мл, 5,0 ммоль) и перемешивали смесь в течение 2 ч при 23°C. Добавляли насыщенный водный раствор бикарбоната натрия (50 мл) и экстрагировали водный слой диэтиловым эфиром (2x50 мл). Объединенные органические экстракты сушили над MgSO4, фильтровали и упаривали. Коричневый остаток очищали по методу флэш-хроматографии (элюируя этилацетатом:гексаном от 1:4 до 1:1) с получением соединения 38 (0,14 г, 19%) в виде бледно-коричневого масла.
1H ЯМР (300 МГц, CDCl3) δ 8,52 (д, J=5,9 Гц, 1H), 8,41 (дд, J=4,6, 1,7 Гц, 1H), 8,34 (т, J=1,9 Гц, 1H), 8,17-8,14 (м, 1H), 8,03 (дд, J=7,8, 1,7 Гц, 1H), 7,78-7,72 (м, 1H), 7,50 (т, J=8,1 Гц, 1H), 7,31 (дд, J=8,1, 4,6 Гц, 1H), 7,19 (д, J=5,2 Гц, 1H), 2,37 (с, 3H), 2,35 (с, 3H).
МС (ESI) m/z 453 (M+23)+, 431 (М+1)+.
Пример 48: Соединение 59
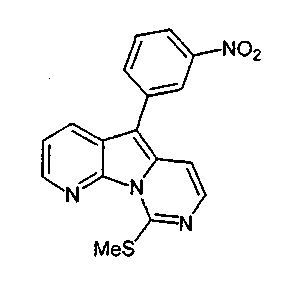
Смесь соединения 38 (140 мг, 0,32 ммоль), трифторуксусной кислоты (53 мкл, 0,68 ммоль) и триэтилсилана (0,42 мл, 2,6 ммоль) вливали в пробирку Янга, содержащую 1,2-дихлорэтан (2 мл). Реакционный сосуд герметизировали и нагревали при 90°C в течение 22 ч. После охлаждения реакционный сосуд открывали и разбавляли содержимое хлороформом (100 мл). Раствор нейтрализовали добавлением насыщенного водного раствора бикарбоната натрия (50 мл) и разделяли слои. Водный слой несколько раз экстрагировали хлороформом, объединенные органические слои сушили над сульфатом натрия, фильтровали и концентрировали. Темно-красный остаток очищали по методу флэш-хроматографии (элюируя этилацетатом:гексаном от 1:4 до 1:3) с получением соединения 59 (72 мг, 23%) в виде оранжевого твердого вещества.
1H ЯМР (300 МГц, CDCl3) δ 8,65 (дд, J=4,6, 1,5 Гц, 1Н), 8,50 (ушир.т, J=2,0 Гц, 1H), 8,27 (дд, J=8,2, 1,5 Гц, 1H), 8,21 (ддд, J=8,3, 2,4, 1,0 Гц, 1H), 7,97 (дт, J=7,8, 1,7 Гц, 1H), 7,73-7,67 (м, 2H), 7,50 (дд, J=8,3, 4,6 Гц, 1H), 7,31 (д, J=6,8 Гц, 1H), 2,76 (с, 3H).
MS(ESI) m/z:337(М+1)+.
Пример 49: Соединение 64
Водный NH4OH (6 мл, 32%) добавляли к раствору соединения 59 (48 мг, 0,14 ммоль) в 1,4-диоксане (5 мл). Желтую смесь нагревали в течение 48 ч при 105°C в герметичной пробирке. Полученную коричневую смесь упаривали при пониженном давлении и очищали желтый остаток по методу флэш-хроматографии (элюируя CH2Cl2:MeOH от 1 до 3%) с получением соединения 64 (5 мг, 12%) в виде желтого твердого вещества.
1H ЯМР (300 МГц, CDCl3) δ 8,50 (ушир.т, J=1,8 Гц, 1H), 8,39 (дд, J=4,6, 1,5 Гц, 1H), 8,22 (дд, J=8,1, 1,5 Гц, 1H), 8,19-8,12 (м, 1H), 7,92 (дт, J=7,6, 1,5 Гц, 1H), 7,69-7,61 (м, 1H), 7,48-7,41 (м, 2H), 6,91 (д, J=6,8 Гц, 1H).
МС (ESI) m/z: 306 (М+1)+.
Пример 50: Соединение 39
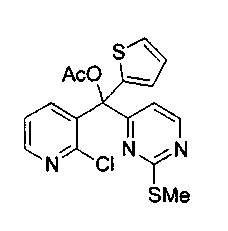
Раствор n-BuLi (1,4 мл, 3,50 ммоль, 2,5 M в гексане) добавляли по каплям к раствору 2-йодтиофена (0,37 мл, 3,40 ммоль) в тетрагидрофуране (12 мл) при -78°C. Реакционную смесь перемешивали при -78°C в течение 15 мин и через канюлю добавляли раствор соединения 31 (0,75 г, 2,83 ммоль) в тетрагидрофуране. Коричневую смесь перемешивали при -78°C в течение 2 ч. Добавляли ацетилхлорид (0,6 мл, 17,0 ммоль) и перемешивали смесь в течение 2 ч при 23°C. Добавляли насыщенный водный раствор бикарбоната натрия (50 мл) и экстрагировали водный слой CH2Cl2 (2x50 мл). Объединенные органические слои сушили над сульфатом натрия, фильтровали и концентрировали. Коричневый остаток очищали по методу флэш-хроматографии (элюируя этилацетатом:гексаном от 1:3 до 1:2) с получением соединения 39 (0,61 г) в виде бледно-коричневого твердого вещества. Обнаружено, что соединение 39 является слегка нестабильным, и его сразу использовали на следующем этапе.
МС (ESI) m/z: 392 (М+1)+.
Пример 51: Соединение 60
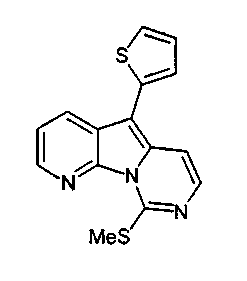
Смесь соединения 39 (610 мг, 1,56 ммоль), трифторуксусной кислоты (0,25 мл, 3,27 ммоль) и триэтилсилана (2 мл, 12,4 ммоль) вливали в пробирку Янга, содержащую 1,2-дихлорэтан (2,5 мл). Реакционный сосуд герметизировали и нагревали при 90°C в течение 16 ч. После охлаждения реакционный сосуд открывали и разбавляли содержимое CH2Cl2 (250 мл). Раствор нейтрализовали добавлением насыщенного водного раствора бикарбоната натрия (200 мл) и разделяли слои. Водный слой несколько раз экстрагировали CH2Cl2, объединенные органические слои сушили над сульфатом натрия, фильтровали и концентрировали. Темно-красный остаток очищали по методу флэш-хроматографии (элюируя этилацетатом:гексаном от 1:4 до 1:2) с получением соединения 60 (72 мг, 23%) в виде желтого твердого вещества.
1H ЯМР (300 МГц, CDCl3) δ 8,61 (дд, J=4,6, 1,7 Гц, 1H), 8,40 (дд, J=8,3, 1,7 Гц, 1Н), 7,63 (д, J=6,7 Гц, 1Н), 7,51-7,45 (м, 2H), 7,39 (д, J=5,4 Гц, 1Н), 7,31-7,29 (м, 1Н), 7,22 (дд, J=5,1, 3,6 Гц, 1Н).
МС (ESI) m/z, 298 (М+1)+.
Пример 52: Соединение 65
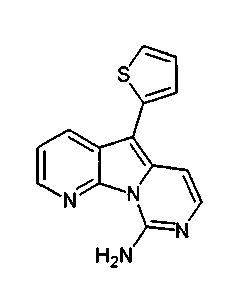
Водный NH4OH (2,5 мл, 32%) добавляли к раствору соединения 60 (13,5 мг, 0,045 ммоль) в 1,4-диоксане (1,5 мл). Желтую смесь нагревали в течение 12 ч при 90°C в герметичной пробирке. Полученную желтую смесь упаривали при пониженном давлении и очищали желтый остаток по методу флэш-хроматографии (элюируя CH2Cl2:MeOH от 2 до 3%) с получением соединения 65 (8 мг, 67%) в виде желтого твердого вещества.
1H ЯМР (300 МГц, CDCl3) δ 8,39-8,32 (м, 2H), 7,42 (дд, J=7,8, 4,8 Гц, 1Н), 7,38 (д, J=6,6 Гц, 1Н), 7,34 (д, J=5,4 Гц, 1Н), 7,29-7,26 (м, 1Н), 7,19 (дд, J=5,1, 3,7 Гц, 1Н), 7,08 (д, J=6,6 Гц, 1Н).
МС (ESI) m/z 267 (М+1)+.
Пример 53: Соединение 40
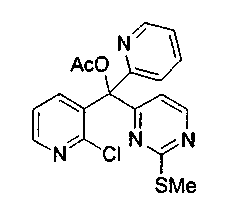
Раствор n-BuLi (0,94 мл, 2,36 ммоль, 2,5 M в гексане) добавляли по каплям к раствору 2-бромпиридина (0,21 мл, 2,27 ммоль) в тетрагидрофуране (10 мл) при -78°C. Коричнево-красную смесь перемешивали при -78°C в течение 15 мин и через канюлю добавляли раствор соединения 31 (0,5 г, 1,89 ммоль) в тетрагидрофуране. Коричневую смесь перемешивали при -78°C в течение 1 ч. Добавляли ацетилхлорид (0,4 мл, 5,67 ммоль) и перемешивали смесь в течение 2 ч при 23°C. Добавляли насыщенный водный раствор бикарбоната натрия (50 мл) и экстрагировали водный слой CH2Cl2 (2x50 мл). Объединенные органические слои сушили над сульфатом натрия, фильтровали и концентрировали. Оранжевый остаток очищали по методу флэш-хроматографии (элюируя этилацетатом:гексаном от 1:3 до 1:1) с получением соединения 40 (0,25 г, 35%) в виде бесцветного масла.
1H ЯМР (300 МГц, CDCl3) δ 8,49 (дт, J=4,6, 1,5 Гц, 1H), 8,44 (д, J=5,3, 1H), 8,35 (дд, J=4,9, 2,0 Гц, 1H), 7,91 (дд, J=8,1, 2,0 Гц, 1H), 7,71-7,61 (м, 2H), 7,30 (д, J=5,4 Гц, 1Н), 7,26-7,15 (м, 3H), 2,40 (с, 3H), 2,29 (с, 3H).
МС (ESI) m/z: 387(M+1)+.
Пример 54: Соединение 55
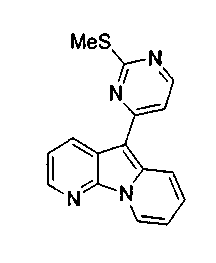
Смесь соединения 40 (250 мг, 0,65 ммоль), трифторуксусной кислоты (0,1 мл, 1,36 ммоль) и триэтилсилана (0,83 мл, 5,18 ммоль) вливали в пробирку Янга, содержащую 1,2-дихлорэтан (2 мл). Реакционный сосуд герметизировали и нагревали при 90°C в течение 24 ч. После охлаждения реакционный сосуд открывали и разбавляли содержимое CH2Cl2 (150 мл). Раствор нейтрализовали добавлением насыщенного водного раствора бикарбоната натрия (100 мл) и разделяли слои. Водный слой несколько раз экстрагировали CH2Cl2, объединенные органические слои сушили над сульфатом натрия, фильтровали и концентрировали. Темно-красный остаток очищали по методу флэш-хроматографии (элюируя этилацетатом:гексаном от 1:3 до 1:1) с получением соединения 55 (34 мг, 19%) в виде желтого твердого вещества.
1H ЯМР (300 МГц, CDCl3) δ 8,94 (дт, J=7,1, 1,2 Гц, 1H), 8,64-8,58 (м, 2H), 8,48 (дд, J=4,6, 1,4 Гц, 1Н), 8,46 (д, J=5,4 Гц, 1Н), 7,49 (дд, J=8,3, 4,6 Гц, 1Н), 7,40 (м, 2H), 6,85 (тд, J=7,1, 1,3 Гц, 1Н), 2,70 (с, 3H).
МС (ESI) m/z 293 (М+1)+.
Пример 55: Соединение 56
Раствор mCPBA (34 мг, 0,14 ммоль, 70%) в предварительно высушенном над сульфатом натрия CH2Cl2 (2 мл) по каплям добавляли к раствору соединения 55 (34 мг, 0,12 ммоль) в CH2Cl2 (3 мл) при 0°C. Желтый раствор перемешивали в течение 30 мин при 0°C. Добавляли насыщенный водный раствор Na2S2О3 (5 мл) и промывали органический слой насыщенным водным раствором бикарбоната натрия (10 мл). Объединенные водные слои экстрагировали CH2Cl2 (3x10 мл). Объединенные органические слои сушили над сульфатом натрия, фильтровали и концентрировали. Желтый остаток вливали в герметичную пробирку с 1,4-диоксаном (4 мл) и добавляли раствор NH4OH (8 мл, 32%). Коричневую смесь нагревали в течение 14 ч при 85°C. Полученную желтую смесь упаривали в вакууме. Желтое твердое вещество очищали по методу флэш-хроматографии (элюируя CH2Cl2:MeOH от 1 до 3%) с получением соединения 56 (6 мл, 20% в 2 этапа) в виде желтого твердого вещества.
1H ЯМР (300 МГц, CDCl3) δ 8,94 (д, J=6,8 Гц, 1Н), 8,64 (дд, J=8,0, 1,4 Гц, 1Н), 8,54 (д, J=9,3 Гц, 1Н), 8,46 (дд, J=4,4, 1,5 Гц, 1Н), 8,21 (д, J=5,6 Гц, 1Н), 7,48 (дд, J=8,3, 4,6 Гц, 1Н), 7,36 (ддд, J=9,3, 6,6, 1,3 Гц, 1Н), 7,09 (д, J=5,6 Гц, 1Н), 6,85 (тд, J=6,8, 1,1 Гц, 1Н).
МС (ESI) m/z, 262 (М+1)+.
Пример 56: Соединение 41
К раствору соединения 31 (0,40 г, 1,5 ммоль) в тетрагидрофуране (5 мл) при 0°C по каплям добавляли i-PrMgCl (0,75 мл, 1,5 ммоль, 2,0 M в тетрагидрофуране) и перемешивали смесь при 0°C в течение 3 ч. Добавляли насыщенный водный раствор хлорида аммония (25 мл) и экстрагировали водный слой этилацетатом (3x25 мл). Объединенные органические слои сушили над сульфатом натрия, фильтровали и концентрировали. Остаток очищали по методу флэш-хроматографии (элюируя этилацетатом:гексаном, 33%) с получением соединения 41 (0,28 г, 61%) в виде прозрачного масла.
1H ЯМР (CDCl3, 300 МГц) δ 0,84 (д, J=6,6 Гц, 3H), 0,98 (д, J=6,6 Гц, 3H), 2,53 (с, 3H), 3,01 (септет, J=6,6 Гц, 1Н), 5,17 (с, 1Н), 7,18 (д, J=5,5 Гц, 1Н), 7,30 (дд, J=7,8, 4,6 Гц, 1Н), 8,24 (дд, J=7,8, 1,7 Гц, 1Н), 8,30 (дд, J=4,6, 1,7 Гц, 1Н), 8,47 (д, J=5,5Гц, 1Н).
МС (ESI) m/z, 258 (М+1)+.
Пример 57: Соединение 61
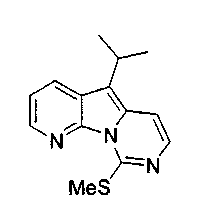
Раствор соединения 41 (0,28 г, 0,91 ммоль) и трифторуксусной кислоты (0,15 мл, 2,0 ммоль) в 1,2-дихлорэтане (3 мл) переносили в оснащенную резиновой мембраной пробирку Янга, содержащую триэтилсилан (1,28 мл, 8 ммоль). Под сильной струей аргона мембрану заменяли тефлоновой закручивающейся крышкой, герметизировали и нагревали реакционный сосуд при 100°C в течение 48 ч. После охлаждения реакционный сосуд открывали и разбавляли содержимое CH2Cl2 (20 мл). Раствор нейтрализовали добавлением насыщенного водного раствора бикарбоната натрия (20 мл) и разделяли слои. Водный слой несколько раз экстрагировали CH2Cl2, объединенные органические слои сушили над сульфатом натрия, фильтровали и концентрировали. Неочищенный материал очищали по методу флэш-хроматографии (элюируя этилацетатом:гексаном, 5%) с получением соединения 61 в виде желтого масла (176 мг, 75%).
1H ЯМР (CDCl3, 300 МГц) δ 1,47 (д, J=7,1 Гц, 6H), 2,69 (с, 3H), 3,44 (септет, J=7,1 Гц, 1Н), 7,06 (д, J=6,6 Гц, 1Н), 7,35 (дд, J=8,1, 4,6 Гц, 1Н), 7,44 (д, J=6,6 Гц, 1Н), 8,16 (дд, J=8,1, 1,5 Гц, 1Н), 8,52 (дд, J=4,6, 1,5 Гц, 1Н).
МС (ESI) m/z, 258, (М+1)+.
Пример 58: Соединение 66
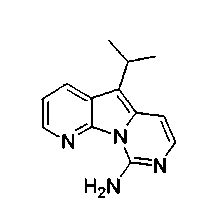
Раствор соединения 61 (146 мг, 0,56 ммоль) в 1,4-диоксане (15 мл) обрабатывали водным NH4OH (30 мл, 32%) в герметичной пробирке при 90°C в течение 24 ч. Реакционную смесь упаривали и подвергали хроматографии (элюируя этилацетатом:гексаном, 33%) с получением соединения 66 (50 мг, 39%) в виде желтого твердого вещества.
1H ЯМР (CDCl3, 300 МГц) δ 1,46 (д, J=7,1 Гц, 6H), 3,41 (септет, J=7,1 Гц, 1Н), 6,70 (д, J=6,8 Гц, 1Н), 7,27 (д, J=6,8 Гц, 1Н), 7,31 (дд, J=8,1, 4,6 Гц, 1Н), 8,11 (д, J=8,1 Гц, 1Н), 8,29 (д, J=4,6, 1,5 Гц, 1Н).
МС (ESI) m/z, 227, (М+1)+.
Пример 59: Соединение 25
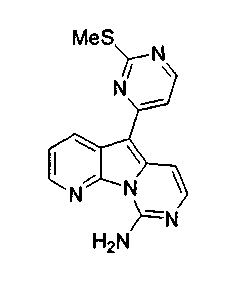
Раствор NH4OH (3 мл, 32%) добавляли к раствору соединения 9 (12 мг, 0,035 ммоль) в 1,4-диоксане (2 мл). Коричневую смесь нагревали в течение 14 ч при 85°C в герметичной пробирке. Полученную желтую смесь упаривали в вакууме, добавляли CH2Cl2 (5 мл), сушили над сульфатом натрия, фильтровали и выпаривали растворитель при пониженном давлении. Желтое твердое вещество очищали по методу флэш-хроматографии (элюируя CH2Cl2:MeOH от 2% до 3%) с получением соединения 25 (8 мл, 73%) в виде желтого твердого вещества.
1H ЯМР (300 МГц, CDCl3) δ 8,72 (дд, J=8,1, 1,5 Гц, 1H), 8,48 (д, J=5,4 Гц, 1H), 8,39 (дд, J=4,8, 1,6 Гц, 1H), 7,66 (д, J=6,8 Гц, 1H), 7,56 (д, J=6,7 Гц, 1H), 7,48 (дд, J=8,1, 4,6 Гц, 1H), 7,32 (д, J=5,3 Гц, 1H), 2,67 (с, 3H).
МС (ESI) m/z, 309 (М+1)+.
Пример 60: Соединение 68
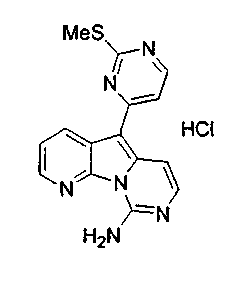
Соединение 25 (0,6 г, 1,95 ммоль) обрабатывали раствором HCl в 1,4-диоксане (45 мл, 5,0н) и перемешивали в течение 20 ч при 23°C. Коричневую суспензию упаривали, промывали диэтиловым эфиром (30 мл), диоксаном (30 мл) и фильтровали с получением соединения 68 (0,65 г) в виде оранжевого твердого вещества.
1H ЯМР (300 МГц, CD3OD) δ 8,89 (дд, J=8,3, 1,3 Гц, 1H), 8,69 (д, J=5,4 Гц, 1H), 8,68 (м, 1H), 7,89 (д, J=7,5 Гц, 1H), 7,85 (д, J=5,9 Гц, 1Н), 7,78 (дд, J=7,8, 4,9 Гц, 1H), 7,59 (д, J=7,9 Гц, 1H), 2,81 (с, 3H).
Пример 61: Соединения 69 и 26
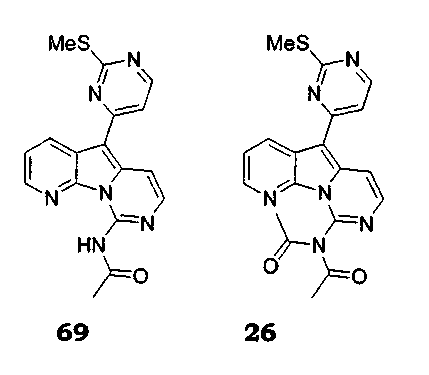
Ацетилхлорид (1,5 мл, 0,019 ммоль) добавляли к раствору соединения 25 (6 мг, 0,019 ммоль) и Et3N (3 мл, 0,029 ммоль) в тетрагидрофуране (2 мл). Желтый раствор перемешивали при 23°C в течение ночи и упаривали при пониженном давлении. Желтый остаток растворяли в CH2Cl2 (5 мл) и промывали насыщенным водным раствором бикарбоната натрия (4 мл). Органический слой сушили над сульфатом натрия, фильтровали и упаривали при пониженном давлении. Желтый остаток очищали по методу флэш-хроматографии (элюируя CH2Cl2:MeOH от 0,5 до 1%) с получением соединения 69 (2 мг, 39% BRSM) и соединения 26 (2,5 мг, 47% BRSM) в виде желтых масел.
Соединение 69:
1H ЯМР (300 МГц, CDCl3) δ 8,77 (дд, J=8,2, 1,6 Гц, 1H), 8,54 (д, J=5,3 Гц, 1H), 8,52 (дд, J=4,9, 1,5 Гц, 1H), 7,93 (д, J=6,6 Гц, 1H), 7,83 (д, J=6,7 Гц, 1H), 7,58 (дд, J=8,2, 4,6 Гц, 1H), 7,37 (д, J=5,4 Гц, 1H), 2,69 (с, 3H), 2,67 (с, 3H).
Соединение 26:
1H ЯМР (300 МГц, CDCl3) δ 8,65 (дд, J=8,3, 1,7 Гц, 1H), 8,58 (д, J=5,4 Гц, 1H), 8,54 (дд, J=6,1, 1,7 Гц, 1H), 8,39 (д, J=6,6Гц, 1H), 7,92 (д, J=6,7 Гц, 1H), 7,56 (дд, J=8,2, 4,6 Гц, 1H), 7,38 (д, J=5,6 Гц, 1Н), 2,69 (с, 3H), 2,42 (с, 6H).
МС (ESI) m/z 415 (M+23)+.
Пример 62: Соединение 28
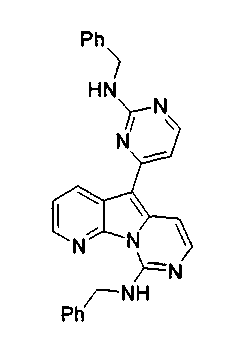
Смесь бензиламина (0,5 мл, 4,5 ммоль) и соединения 27 (160 мг, 0,45 ммоль) в тетрагидрофуране (2,5 мл) нагревали с обратным холодильником в течение 14 ч. Растворитель выпаривали и очищали желтый остаток по методу флэш-хроматографии (элюируя CH2Cl2:MeOH от 99:1 до 97:3) с получением соединения 28 (90 мг, 44%) в виде желтого масла.
1H ЯМР (300 МГц, CDCl3) δ 10,44 (ушир.с, 1H), 8,51 (д, J=6,0 Гц, 1Н), 8,29 (д, J=6,2 Гц, 1Н), 8,26 (д, J=1,1 Гц, 1Н), 7,60 (д, J=6,6 Гц, 1Н), 7,48-7,28 (м, 12H), 6,99 (д, J=5,5 Гц, 1Н), 5,81 (ушир.с, 1Н), 4,97 (д, J=5,7 Гц, 2H), 4,76 (д, J=5,8 Гц, 2H).
Пример 63: Соединения 72 и 73
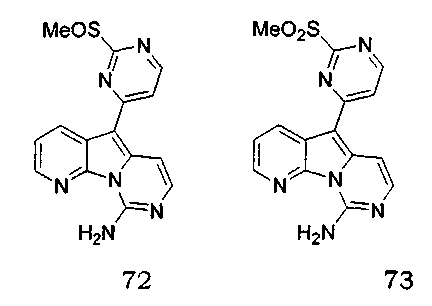
Раствор mCPBA (70 мг, 0,30 ммоль, 70%) в предварительно высушенном над сульфатом натрия CH2Cl2 (3 мл) добавляли по каплям к раствору соединения 25 (39 мг, 0,13 ммоль) в CH2Cl2 (7 мл). Желтый раствор перемешивали в течение 2 ч при 23°C. Добавляли насыщенный водный раствор Na2S2О3 (5 мл) и промывали органический слой насыщенным водным раствором бикарбоната натрия (5 мл). Объединенные водные слои экстрагировали CH2Cl2 (3x20 мл). Объединенные органические слои сушили над сульфатом натрия, фильтровали и концентрировали. Желтый остаток очищали по методу флэш-хроматографии (элюируя CH2Cl2:MeOH от 2 до 5%) с получением соединения 72 (15 мг, 35%) в виде желтого масла и соединения 73 (25 мг, 58%) в виде желтого твердого вещества.
Соединение 72:
1H ЯМР (300 МГц, CDCl3) δ 8,86 (дд, J=8,1, 1,5 Гц, 1H), 8,74 (д, J=5,6 Гц, 1H), 8,43 (дд, J=4,6, 1,3 Гц, 1H), 7,77 (д, J=6,8 Гц, 1H), 7,72 (д, J=6,6 Гц, 1H), 7,67 (д, J=5,8 Гц, 1H), 7,54 (дд, J=8,0, 4,6 Гц, 1H), 3,02 (с, 3H).
МС (ESI) m/z: 325 (М+1)+.
Соединение 73:
1H ЯМР (300 МГц, CDCl3) δ 8,80 (дд, J=8,1, 1,4 Гц, 1H), 8,71 (д, J=5,6 Гц, 1H), 8,41 (дд, J=4,8, 1,4 Гц, 1H), 7,76 (д, J=5,6 Гц, 1H), 7,74 (д, J=6,4 Гц, 1H), 7,64 (д, J=6,4 Гц, 1H), 7,52 (дд, J=8,2, 4,8 Гц, 1H), 3,40 (с, 3H).
Пример 64: Соединение 70
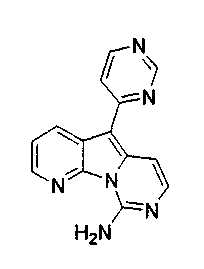
Суспензию соединения 25 (29 мг, 0,09 ммоль) в 1,2-дихлорэтане (0,5 мл) обрабатывали Pd/C (3 мг, 10%), TFA (14 мкл, 0,18 ммоль) и Et3SiH (110 мкл, 0,7 ммоль) в герметичной пробирке при 100°C в течение 72 ч. Реакционную смесь растворяли в CH2Cl2:MeOH (20:1) и промывали насыщенным водным раствором бикарбоната натрия. Органический слой сушили над сульфатом натрия, фильтровали и упаривали. Остаток подвергали хроматографии (элюируя CH2Cl2:MeOH от 50:1 до 95:5) для восстановления соединения 25 (6,5 мг) и получения соединения 70 в виде желтого твердого вещества (7,5 мг, 39% BRSM).
1H ЯМР (CDC13:CD3OD 9:1, 300 МГц) δ 9,02 (с, 1Н), 8,60 (дд, J=8,3, 1,5 Гц, 1H), 8,50 (д, J=5,6 Гц, 1H), 8,29 (дд, J=4,9, 1,5 Гц, 1H), 7,59 (д, J=5,6 Гц, 1Н), 7,49-7,43 (м, 2H), 7,38 (дд, J=8,3, 4,9 Гц, 1H).
МС (ESI) m/z, 263 (М+1)+.
Пример 65: Соединение 71
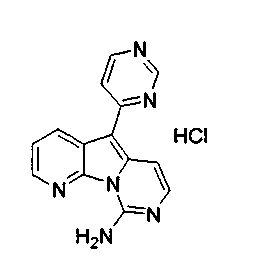
Соединение 70 (6,5 мг, 0,025 ммоль) обрабатывали HC1 в 1,4-диоксане (1,0 мл, 3,8 M) в течение 15 мин. Реакционную смесь упаривали, промывали диэтиловым эфиром, растворяли в MeOH и упаривали с получением соединения 71 в виде оранжевого твердого вещества (7,0 мг, 94%).
1H ЯМР (CD3OD, 300 МГ) δ 9,35 (с, 1H), 8,97 (дд, J=8,1, 1,5 Гц, 1H), 8,88 (д, J=6,1 Гц, 1H), 8,68 (дд, J=4,9, 1,5 Гц, 1H), 8,27 (дд, J=6,3, 1,2 Гц, 1H), 8,04 (д, J=7,6 Гц, 1H), 7,78 (дд, J=8,3, 5,1 Гц, 1H), 7,57 (д, J=7,3 Гц, 1H).
МС (ESI) m/z, 263 (M)+.
Пример 66: Соединение 74
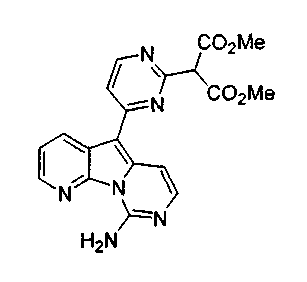
Диметилмалонат (0,013 мл, 0,11 ммоль) добавляли по каплям к суспензии NaH (4,4 мг, 0,1 ммоль, 60%) в тетрагидрофуране (2,5 мл) при 23°C. Через 10 мин одной порцией добавляли соединение 73 (4 мг, 0,011 ммоль) и перемешивали желтую взвесь при 23°C в течение ночи. Реакционную смесь подвергали ТСХ и обнаруживали только один исходный материал. Смесь нагревали с обратным холодильником в течение 3 ч, охлаждали и гасили добавлением насыщенного водного раствора хлорида аммония. Водный слой экстрагировали CH2Cl2 (3x10 мл), объединенные органические слои сушили над сульфатом натрия, фильтровали и упаривали. Желтый остаток очищали по методу флэш-хроматографии (элюируя CH2Cl2:MeOH от 1 до 3%) с получением соединение 74 (1,5 мг, 36%) в виде желтого твердого вещества.
1H ЯМР (300 МГц, CDCl3) δ 8,72 (дд, J=8,2, 1,5 Гц, 1H), 8,69 (д, J=5,3 Гц, 1H), 8,39 (дд, J=4,6, 1,5 Гц, 1H), 7,69 (д, J=5,9 Гц, 1H), 7,59 (д, J=6,6 Гц, 1H), 7,58 (д, J=6,6 Гц, 1H), 7,50 (дд, J=8,2, 4,7 Гц, 1H), 4,23 (с, 1H), 3,85 (с, 6H).
МС (ESI) m/z 415 (M+23)+, 393 (М+1)+.
Пример 67: Соединение 75
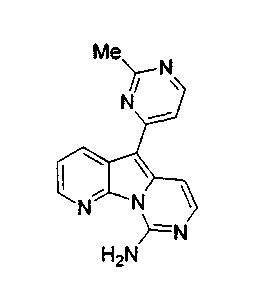
Раствор метилмагнийбромида (0,037 мл, 3 M в тетрагидрофуране) добавляли по каплям к раствору соединения 73 (25 мг, 0,073 ммоль) в тетрагидрофуране (5 мл) при 0°C. Коричневую смесь перемешивали в течение 2 ч при 0°C и в течение 1 ч при 23°C. Добавляли насыщенный водный раствор хлорида аммония (25 мл) и экстрагировали водный слой CH2Cl2 (3x20 мл). Объединенные органические слои сушили над сульфатом натрия, фильтровали и концентрировали. Оранжевый остаток очищали по методу флэш-хроматографии (элюируя CH2Cl2:MeOH от 1 до 3%) с получением соединения 75 (14 мг, 69%) в виде желтого твердого вещества.
1H ЯМР (300 МГц, CDCl3) δ 8,75 (дд, J=8,1, 1,5 Гц, 1H), 8,60 (д, J=5,6 Гц, 1H), 8,39 (дд, J=4,6, 1,5 Гц, 1H), 7,67 (д, J=6,6 Гц, 1H), 7,60 (д, J=6,6 Гц, 1H), 7,49 (дд, J=8,1, 5,0 Гц, 1H), (д, J=5,6 Гц, 1H), 2,81 (с, 3H).
МС (ESI) m/z 277 (М+1)+.
Пример 68: Соединение 76
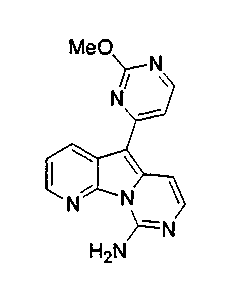
Раствор соединения 72 (5,8 мг, 0,017 ммоль) в MeOH (2 мл) добавляли к раствору NaMeO в MeOH (2 мл) при 0°C. Желтый раствор перемешивали при 23°C в течение 4 ч, гасили добавлением насыщенного водного раствора хлорида аммония и экстрагировали этилацетатом (3x10 мл). Объединенные органические слои сушили над сульфатом натрия, фильтровали и упаривали при пониженном давлении. Желтый остаток очищали по методу флэш-хроматографии (элюируя CH2Cl2:MeOH от 1 до 3%) с получением соединения 76 (2,6 мг, 53%) в виде желтого твердого вещества.
1H ЯМР (300 МГц, CDCl3) δ 8,78 (дд, J=8,1, 1,5 Гц, 1H), 8,51 (д, J=5,4 Гц, 1H), 8,41 (дд, J=4,6, 1,5 Гц, 1H), 7,69 (д, J=6,6 Гц, 1H), 7,63 (д, J=6,6 Гц, 1H), 7,50 (дд, J=8,1, 4,6 Гц, 1H), 7,34 (д, J=5,4 Гц, 1H), 4,14 (с, 3H).
МС (ESI) m/z, 293 (М+1)+.
Пример 69: Соединение 81
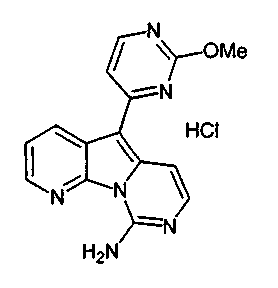
Соединение 76 (30 мг) суспендировали в 5,3 н. растворе HCl в 1,4-диоксане (7 мл). Бледно-коричневую смесь перемешивали при 23°C в течение 2 ч и упаривали в вакууме. Добавляли CH2Cl2 (5 мл), перемешивали в течение 1 мин и снова упаривали. Добавляли 1,4-диоксан (5 мл), бледно-коричневое твердое вещество фильтровали и промывали 1,4-диоксаном (3 мл) с получением соединения 81 (30 мг).
1H ЯМР (300 МГц, CD3OD) δ 8,92 (дд, J=8,0, 1,1 Гц, 1H), 8,78-8,70 (м, 2H), 8,01 (д, J=7,8 Гц, 1H), 7,98 (д, J=6,3 Гц, 1Н), 7,82 (дд, J=8,1, 4,6 Гц, 1H), 7,67 (д, J=5,4 Гц, 1H), 4,38 (с, 3H).
Пример 70: Соединение 77
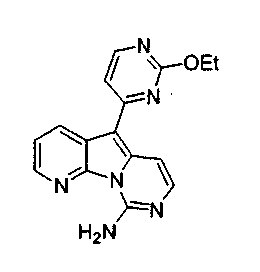
Раствор соединения 72 (150 мг, 0,46 ммоль) в EtOH (2 мл) добавляли при 0°C к раствору EtONa в EtOH (3 мл), свежеприготовленному путем добавления избытка Na к EtOH. Желтый раствор перемешивали при 23°C в течение 5 ч, гасили добавлением насыщенного водного раствора хлорида аммония и экстрагировали хлороформом (3x10 мл). Объединенные органические слои сушили над сульфатом натрия, фильтровали и упаривали при пониженном давлении. Желтый остаток очищали по методу флэш-хроматографии (элюируя CH2Cl2:MeOH от 1 до 3%) с получением соединения 77 (96 мг, 68%) в виде желтого твердого вещества.
1H ЯМР (300 МГц, CDCl3) δ 8,74 (дд, J=8,2, 1,5 Гц, 1H), 8,47 (д, J=5,3 Гц, 1Н), 8,39 (дд,J=4,6, 1,5 Гц, 1Н), 7,67 (д, J=6,6 Гц, 1Н), 7,59 (д, J=6,6 Гц, 1Н), 7,48 (дд, J=8,2, 4,7 Гц, 1Н), 7,29 (д, J=5,4 Гц, 1Н), 4,55 (c, J=7,1 Гц, 2H), 1,52 (т, J=7,1 Гц, 3H).
МС (ESI) m/z, 329 (M+23)+, 307 (М+1)+.
Пример 71: Соединение 82
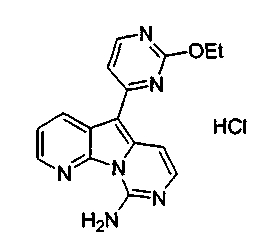
Соединение 77 (35 мг, 0,11 ммоль) обрабатывали раствором HCl в 1,4-диоксане (4 мл, 5,3 M) и перемешивали в течение 3 ч при 23°C. Суспензию упаривали, промывали диэтиловым эфиром и фильтровали с получением соединения 82 (32 мг) в виде желтого твердого вещества.
1H ЯМР (300 МГц, CD3OD) δ 8,91 (дд, J=8,2, 1,2 Гц, 1Н), 8,77-8,63 (м, 2H), 7,98 (д, J=7,7 Гц, 1Н), 7,98 (д, J=7,0 Гц, 1Н), 7,81 (дд, J=7,9, 4,8 Гц, 1Н), 7,64 (д, J=7,9 Гц, 1Н), 4,69 (c, J=7,0 Гц, 2H), 1,54 (т, J=7,0 Гц, 3H).
Пример 72: Соединение 78
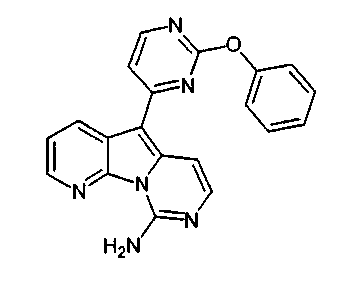
PhOH (116 мг, 1,2 ммоль) добавляли к суспензии NaH (50 мг, 1,2 ммоль, 60%) в тетрагидрофуране (3 мл) при 0°C. При 0°C добавляли суспензию соединения 72 (50 мг, 0,15 ммоль) в тетрагидрофуране (5 мл), перемешивали желтый раствор при 23°C в течение 24 ч, гасили добавлением насыщенного водного раствора NaCl (50 мл) и экстрагировали хлороформом (3x50 мл). Объединенные органические слои сушили над сульфатом натрия, фильтровали и упаривали при пониженном давлении. Желтый остаток очищали по методу флэш-хроматографии (элюируя CH2Cl2:MeOH от 0,5 до 1,5%) с получением соединения 78 (20 мг, 53% BRSM) в виде желтого твердого вещества.
1H ЯМР (300 МГц, CDCl3) δ 8,54 (д, J=5,4 Гц, 1Н), 8,36 (дд, J=8,1, 1,3 Гц, 1Н), 8,32 (дд, J=4,6, 1,2 Гц, 1Н), 7,54-7,46 (м, 3H), 7,39 (д, J=5,4 Гц, 1Н), 7,40-7,30 (м, 3H), 7,29-7,25 (м, 2H).
МС (ESI) m/z, 355 (М+1)+.
Пример 73: Соединение 79
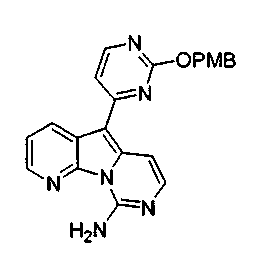
Пара-метоксибензиловый спирт (0,085 мл, 0,67 ммоль) добавляли к суспензии NaH (27 мг, 0,67 ммоль, 60%) в тетрагидрофуране (1 мл) при 0°C. При 0°C добавляли суспензию соединения 72 (23 мг, 0,06 ммоль) в тетрагидрофуране (2 мл), перемешивали желтый раствор при 23°C в течение 5 ч, гасили добавлением насыщенного водного раствора хлорида аммония (10 мл) и экстрагировали хлороформом (3x10 мл). Объединенные органические слои сушили над сульфатом натрия, фильтровали и упаривали при пониженном давлении. Желтый остаток очищали по методу флэш-хроматографии, применяя в качестве элюента CH2Cl2:MeOH (2%). Желтый остаток промывали диэтиловым эфиром с получением соединения 79 (12 мг, 46%) в виде желтого твердого вещества.
1H ЯМР (300 МГц, CDCl3) δ 8,72 (дд, J=8,0, 1,6 Гц, 1H), 8,56 (д, J=5,8 Гц, 1Н), 8,39 (дд, J=4,4, 1,6 Гц, 1Н), 7,61-7,45 (м, 5H), 7,32 (д, J=5,4 Гц, 1Н), 6,92 (д, J=7,2 Гц, 2H), 5,42 (ушир.с, 2H), 3,82 (с, 3H).
МС (ESI) m/z, 399 (М+1)+.
Пример 74: Соединение 80
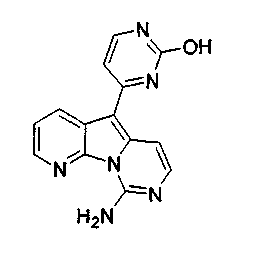
Соединение 79 (10 мг, 0,025 ммоль) растворяли в чистой трифторметансульфоновой кислоте (0,5 мл) и перемешивали при 23°C в течение 3 ч. Реакционный сосуд охлаждали до 0°C и по каплям добавляли MeOH (1 мл). После добавления водного NH4OH (1 мл, 32%) получали ярко-желтый осадок, который фильтровали и промывали эфиром с получением соединения 80 (6 мг, 78%).
1H ЯМР (300 МГц, DMSO-d6) δ 8,85 (дд, J=8,5, 1,4 Гц), 8,45 (дд, J=4,6, 1,5 Гц), 7,85 (д, J=6,8 Гц, 1Н), 7,76 (д, J=6,6 Гц, 1H), 7,66 (д, J=6,6 Гц, 1Н), 7,60 (дд, J=8,1, 4,6 Гц), 6,88 (д, J=6,8 Гц, 1Н).
13C ЯМР (75 МГц, DMSO-d6) δ 156,3, 153,1, 149,8, 145,7, 145,1, 143,1, 140,3, 140,1, 129,1, 121,6, 121,0, 101,3, 101,2, 99,0.
МС (ESI) m/z: 279 (М+1)+.
Пример 75: Соединение 84
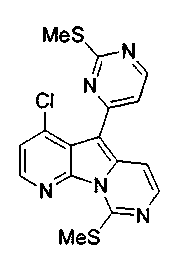
Раствор соединения 52 (250 мг, 0,56 ммоль) в POCl3 (2 мл) перемешивали при 100°C в герметичной пробирке в течение 48 ч. Реакционную смесь вливали на лед, обрабатывали насыщенным водным раствором бикарбоната натрия до pH 7 и экстрагировали хлороформом. Органический слой сушили над сульфатом натрия, фильтровали и упаривали. Остаток подвергали хроматографии (элюируя гексаном:этилацетатом, 4:1) с получением соединения 84 в виде желтого твердого вещества (154 мг, 74%).
1H ЯМР (CDCl3, 300 МГц) δ 8,55 (д, J=5,1 Гц, 1Н), 8,46 (д, J=5,1 Гц, 1Н), 7,75 (д, J=6,6 Гц, 1Н), 7,62 (д, J=6,6 Гц, 1Н), 7,49 (д, J=5,1 Гц, 1Н), 7,24 (д, J=5,1 Гц, 1Н), 2,72 (с, 3H), 2,64 (с, 3H).
13C ЯМР (CDCl3, 75 МГц) δ 172,2, 160,1, 156,2, 155,0, 141,5, 139,7, 137,7, 135,1, 122,1, 119,0, 107,4, 102,8, 15,2, 14,4. Наблюдается частичное перекрывание сигнала двух четвертичных атомов.
МС (ESI) m/z, 374 (М+1)+.
Пример 76: Соединение 85
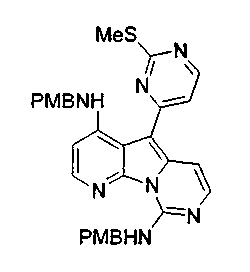
Раствор соединения 84 (20 мг, 0,053 ммоль) в пара-метоксибензиламине (0,2 мл) перемешивали при 100°C в течение 16 ч. Реакционную смесь подвергали хроматографии (элюируя гексаном:этилацетатом от 4:1 до 1:1) с получением соединения 85 в виде желтого твердого вещества (29 мг, 97%).
1H ЯМР (CDCl3, 300 МГц) δ 11,02 (т, J=4,8 Гц, 1H), 9,28 (т, J=5,7 Гц, 1H), 8,42 (д, J=5,5 Гц, 1H), 7,86 (д, J=5,7 Гц, 1H), 7,56 (д, J=6,7 Гц, 1H), 7,38 (д, J=8,7 Гц, 2H), 7,31 (д, J=6,6 Гц, 1H), 6,94 (д, J=6,9 Гц, 1H), 6,91-6,85 (м, 4H), 6,35 (д, J=5,7 Гц, 1H), 4,82 (д, J=5,4 Гц, 2H), 4,52 (д, J=5,7 Гц, 2H), 3,80 (с, 3H), 3,78 (с, 3H), 2,50 (с, 3H).
МС (ESI) m/z, 564 (М+1)+.
Пример 77: Соединение 86
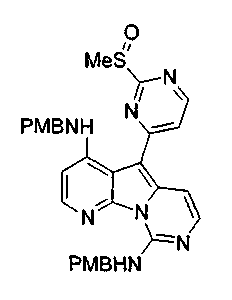
Раствор соединения 85 (40 мг, 0,071 ммоль) в CH2Cl2 (5 мл) обрабатывали при -30°C раствором mCPBA (20 мг, 0,09 ммоль, 1,25 эквив., 77%) в CH2Cl2 (1 мл), нагревали до 0°C в течение 30 мин и гасили добавлением насыщенного водного раствора Na2S2О3. Реакционную смесь четырежды промывали насыщенным водным раствором бикарбоната натрия. Органический слой сушили над сульфатом натрия, фильтровали и упаривали. Остаток подвергали хроматографии (элюируя CH2Cl2:MeOH, 100:3) с получением соединения 86 в виде желтого твердого вещества (26 мг, 63%).
1H ЯМР (CDCl3, 300 МГц) δ 11,26 (т, J=5,7 Гц, 1Н), 10,18 (т, J=5,5 Гц, 1H), 8,58 (д, J=5,5 Гц, 1H), 7,83 (д, J=5,7 Гц, 1H), 7,67 (д, J=5,9 Гц, 2H), 7,38 (д, J=8,9 Гц, 2H), 7,29 (д, J=8,7 Гц, 2H), 7,02 (д, J=6,7 Гц, 1H), 6,89 (д, J=8,9 Гц, 2H), 6,82 (д, J=8,7 Гц, 2H), 6,35 (д, J=5,9 Гц, 1H), 4,83 (д, J=5,5 Гц, 2H), 4,70-4,63 (м, 2H), 3,80 (с, 3H), 3,76 (с, 3H), 2,90 (с, 3H).
МС (ESI) m/z 602 (M+23)+, 580 (М+1)+.
Пример 78: Соединение 89
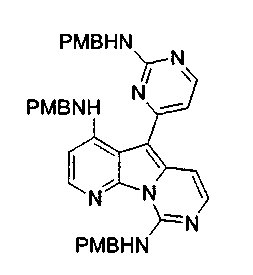
Раствор соединения 84 (14 мг, 0,037 ммоль) в безводном CH2Cl2 (3 мл) обрабатывали при -30°C раствором mCPBA (18 мг, 0,08 ммоль, 2,2 эквив., 77%) в CH2Cl2 (2 мл). Желтый раствор нагревали до 0°C в течение 30 мин, обрабатывали насыщенным водным раствором Na2S2О3 и трижды промывали насыщенным водным раствором бикарбоната натрия. Органический слой сушили над сульфатом натрия, фильтровали и упаривали с получением неочищенного бис-сульфоксида, который обрабатывали 4-метоксибензиламином (0,1 мл, 0,76 ммоль) при 100°C в течение 15 ч. Реакционную смесь подвергали хроматографии (элюируя CH2Cl2:MeOH, 98:2) с получением соединения 89 в виде желтого масла (11 мг, 46%).
1H ЯМР (CDCl3, 300 МГц) δ 10,99 (т, J=5,5 Гц, 1H), 9,38 (ушир.с, 1H), 8,22 (д, J=5,4 Гц, 1Н), 7,96 (д, J=5,7 Гц, 1Н), 7,51 (д, J=6,7 Гц, 1Н), 7,40 (д, J=8,7 Гц, 2H), 7,33 (д, J=8,4 Гц, 2H), 7,08 (д, J=8,7 Гц, 2H), 6,97-6,80 (м, 8H), 6,44 (д, J=5,7 Гц, 1Н), 4,84 (д, J=5,5 Гц, 2H), 4,35 (д, J=4,5 Гц, 2H), 4,29 (д, J=5,4 Гц, 2H), 3,81 (с, 3H), 3,78 (с, 3H), 3,70 (с, 3H). Один NH сигнал не наблюдается.
МС (ESI) m/z: 653 (М+1)+.
Пример 79: Соединение 90
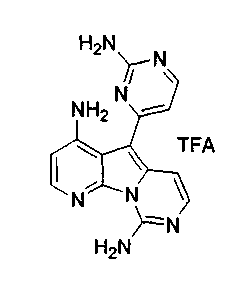
Соединение 89 (10 мг, 0,015 ммоль) обрабатывали трифторметансульфоновой кислотой (0,2 мл) при 23°C в течение 2 ч. Реакционную смесь охлаждали до 0°C и обрабатывали MeOH (1 мл) и водным NH4OH (1 мл, 32%). Реакционную смесь упаривали, растворяли в MeOH:H2О (1:4) и наносили на предварительно обработанный MeOH:H2О (1:1) силикагель колонки для хроматографии с обращенной фазой. Колонку промывали MeOH:H2О (1:4) для удаления солей и элюировали соединение 90 MeOH:H2О (4:1) (1,5 мл, 34%).
Соединение 90 является нерастворимым в MeOH и для ЯМР-анализа получали его трифторацетатную соль.
1H ЯМР (CD3OD плюс одна капля TFA, 300 МГц) δ 8,28 (д, J=6,8 Гц, 1H), 8,10 (д, J=5,7 Гц, 1H), 7,39-7,29 (м, 3H), 6,80 (д, J=5,6 Гц, 1H).
МС (ESI) m/z, 293 (M+1)+.
Пример 80: Соединение 91
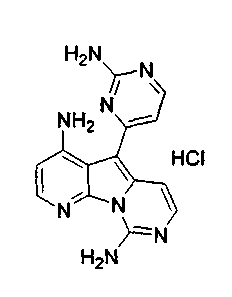
Соединение 90 (19 мг, 0,065 ммоль) обрабатывали безводным HCl в 1,4-диоксане (3 мл, 3,8 M) при 23°C в течение 1 ч. Реакционную смесь упаривали и промывали этиловым эфиром с получением соединения 91 в виде оранжевого твердого вещества (20 мг).
1H ЯМР (CD3OD, 300 МГц) δ 8,31 (д, J=6,8 Гц, 1H), 8,11 (д, J=5,6 Гц, 1Н), 7,41-7,32 (м, 3H), 6,82 (д, J=5,6 Гц, 1Н).
МС (ESI) m/z, 293 (M)+.
Пример 81: Соединение 92
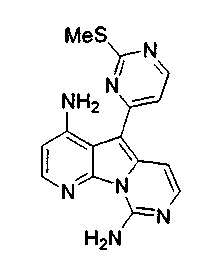
Раствор соединения 85 (29 мг, 0,051 ммоль) в трифторметансульфоновой кислоте (0,2 мл) перемешивали при 23°C в течение 2 ч. Реакционную смесь охлаждали при 0°C, обрабатывали MeOH (1 мл) и водным NH4OH (32%) (1 мл), фильтровали и промывали H2О, MeOH и диэтиловым эфиром с получением соединения 92 в виде желтого твердого вещества (10 мг, 61%).
1H ЯМР (CDCl3, 300 МГц) δ 8,63 (д, J=5,6 Гц, 1H), 8,07 (д, J=5,9 Гц, 1H), 7,48 (д, J=5,4 Гц, 1H), 7,18-7,17 (м, 2H), 6,72 (д, J=5,9 Гц, 1H), 2,63 (с, 3H).
МС (ESI) m/z 324 (М+1)+.
Пример 82: Соединение 93
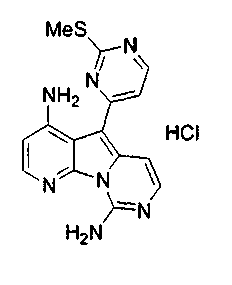
Соединение 92 обрабатывали безводным HCl в 1,4-диоксане (1 мл, 3,8 M) в течение 15 мин при 23°C. Реакционную смесь упаривали и промывали диэтиловым эфиром с получением соединения 93 в виде оранжевого твердого вещества (10 мг).
1H ЯМР (CD3OD, 300 МГц) δ 8,66 (д, J=5,1 H, 1H), 8,09 (д, J=5,9 Гц, 1H), 7,50 (д, J=5,1 Гц, 1H), 7,21 (с, 2H), 6,74 (д, J=5,6 Гц, 1H), 2,64 (с, 3H).
МС (ESI) m/z 324 (M)+.
Пример 83: Соединение 87
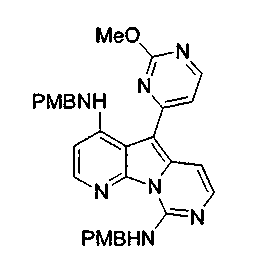
Раствор соединения 86 обрабатывали раствором MeONa в MeOH, приготовленным путем добавления Na (30 мг, 1,3 ммоль) к MeOH (4 мл), при 23°C в течение 16 ч. Реакционную смесь упаривали, растворяли в CH2Cl2 и промывали насыщенным водным раствором хлорида аммония. Органический слой сушили над сульфатом натрия, фильтровали и упаривали. Остаток подвергали хроматографии (элюируя CH2Cl2:MeOH, 100:3) с получением соединения 87 в виде желтого твердого вещества (24 мг, 97%).
1H ЯМР (CDCl3, 300 МГц) δ 11,06 (т, J=5,4 Гц, 1H), 9,44 (т, J=5,1 Гц, 1H), 8,40 (д, J=5,4 Гц, 1Н), 7,92 (д, J=5,7 Гц, 1H), 7,57 (д, J=6,6 Гц, 1H), 7,39 (д, J=9,0 Гц, 2H), 7,32 (д, J=8,7 Гц, 2H), 7,31 (д, J=5,4 Гц, 1H), 6,96 (д, J=6,9 Гц, 1H), 6,90 (д, J=8,7 Гц, 2H), 6,87 (д, J=8,7 Гц, 2H), 6,40 (д, J=6,0 Гц, 1H), 4,84 (д, J=5,4 Гц, 2H), 4,47 (д, J=5,4 Гц, 2H), 3,83 (с, 3H), 3,81 (с, 3H), 3,80 (с, 3H).
МС (ESI) m/z, 548(M+1)+.
Пример 84: Соединение 88
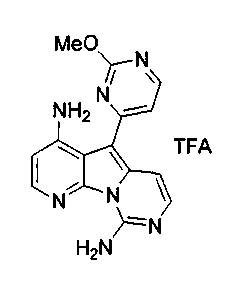
Соединение 87 (23 мг, 0,04 ммоль) обрабатывали TfOH (0,2 мл) при 23°C в течение 2,5 ч. Реакционную смесь охлаждали до 0°C и обрабатывали MeOH (1 мл) и водным NH4OH (2 мл, 32%). Полученное желтое твердое вещество фильтровали через тефлоновый фильтр и промывали H2О (5 мл), EtOH (5 мл) и диэтиловым эфиром (5 мл) с получением соединения 88 (15 мг) в виде оранжевого твердого вещества. Это соединение было нерастворимо в хлороформе и было охарактеризовано как его трифторацетатная соль.
1H ЯМР (CDC13:CD3OD 9:1 плюс 2 капли дейтерированной ТФУ, 300 МГц) δ 8,56 (д, J=5,4 Гц, 1H), 8,02 (д, J=5,6 Гц, 1H), 7,31 (д, J=5,4 Гц, 1H), 7,12 (д, J=7,8 Гц, 1H), 6,97 (д, J=7,8 Гц, 1H), 6,62 (д, J=5,6 Гц, 1H), 4,07 (с, 3H).
МС (ESI) m/z 308 (M+1)+.
Пример 85: Соединение 94
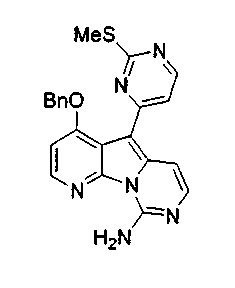
Раствор соединения 52 (100 мг, 0,22 ммоль) в 1,4-диоксане (10 мл) обрабатывали 32% водным NH4OH (20 мл) в герметичной пробирке при 90°C в течение 24 ч. Реакционную смесь упаривали и подвергали хроматографии (элюируя CH2Cl2:MeOH, 97:3) с получением соединения 94 в виде желтого твердого вещества (83 мг, 89%).
1H ЯМР (CDCl3, 300 МГц) δ 8,22 (д, J=5,5 Гц, 1H), 7,95 (д, J=6,8 Гц, 1H), 7,52 (д, J=6,8 Гц, 1H), 7,40 (д, J=6,5 Гц, 1H), 7,38-7,26 (м, 6H), 6,94 (д, J=5,5 Гц, 1H), 5,25 (с, 2H), 2,56 (с, 3H).
МС (ESI) m/z 415 (М+1)+.
Пример 86: Соединение 95
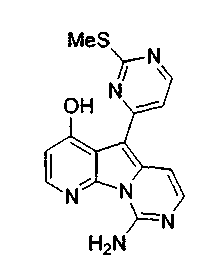
Соединение 94 (62 мг, 0,15 ммоль) обрабатывали TfOH (0,3 мл) при 23°C в течение 1 ч. Реакционную смесь обрабатывали MeOH (1 мл), водным NH4OH (2 мл, 32%), фильтровали и промывали H2О (5 мл), MeOH (5 мл) и диэтиловым эфиром (4 мл). Полученное твердое вещество подвергали хроматографии (элюируя CH2Cl2:MeOH, 95:5) с получением соединения 95 в виде желтого твердого вещества (45 мг, 92%).
1H ЯМР (CDC13:CD3OD 9:1, 300 МГц) δ 8,38 (д, J=5,7 Гц, 1H), 8,09 (д, J=5,5 Гц, 1H), 7,53 (д, J=6,7 Гц, 1H), 7,34 (д, J=5,7 Гц, 1H), 7,00 (д, J=6,7 Гц, 1H), 6,82 (д, J=5,5 Гц, 1H), 2,58 (с, 3H).
МС (ESI) m/z: 325 (М+1)+.
Пример 87: Соединение 97
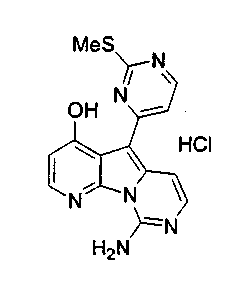
Соединение 95 (45 мг, 0,14 ммоль) обрабатывали раствором HCl в 1,4-диоксане (3 мл, 3,8 M) в течение 15 мин при 23°C. Реакционную смесь упаривали и дважды промывали диэтиловым эфиром с получением соединения 97 в виде желтого твердого вещества (36 мг, 71%).
1H ЯМР (CDC13:CD3OD 95:5, 300 МГц) δ 8,71 (д, J=5,9 Гц, 1H), 8,37 (д, J=4,9 Гц, 1H), 7,86 (д, J=6,1 Гц, 1H), 7,59 (д, J=7,3 Гц, 1H), 7,44 (д, J=7,3 Гц, 1Н), 7,04 (д, J=5,9 Гц, 1H), 2,72 (с, 3H).
МС (ESI) m/z: 325 (M)+.
Пример 88: Соединение 98
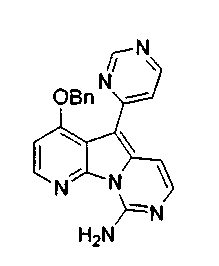
Смесь соединения 94 (65 мг, 0,157 ммоль), Pd/C (6 мг, 10%) и TFA (28 мкл, 0,36 ммоль) в 1,2-дихлорэтане (1 мл) в пробирке Янга обрабатывали Et3SiH (220мкл, 1,4 ммоль) при 100°C в течение 72 ч. Реакционную смесь обрабатывали насыщенным водным раствором бикарбоната натрия и экстрагировали CH2Cl2. Органический слой сушили над сульфатом натрия, фильтровали и упаривали. Остаток подвергали хроматографии (элюируя CH2Cl2:MeOH, 98:2) с получением соединения 98 в виде бледно-желтого твердого вещества (13 мг, 23%).
1H ЯМР (CDCl3, 300 МГц) δ 9,14 (с, 1H), 8,27 (д, J=5,6 Гц, 1H), 8,22 (д, J=5,6 Гц, 1H), 7,67-7,63 (м, 2H), 7,54 (д, J=6,6 Гц, 1H), 7,38 (с, 5H), 6,97 (д, J=5,6 Гц, 1H), 5,28 (с, 2H).
МС (ESI) m/z 369 (М+1)+.
Пример 89: Соединение 100
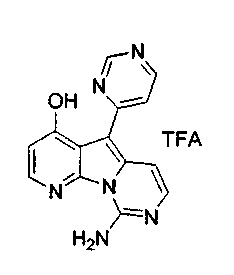
Соединение 98 (11 мг, 0,030 ммоль) обрабатывали TfOH (0,3 мл) при 23°C в течение 1 ч. Реакционную смесь охлаждали до 0°C и обрабатывали MeOH (2 мл) и 32% водным NH4OH (2 мл). Полученное оранжевое твердое вещество отфильтровывали, промывали H2О, EtOH и диэтиловым эфиром и обрабатывали TFA с получением соединения 100 (9,0 мг, 76%).
1H ЯМР (CD3OD, 300 МГц) δ 9,30 (с, 1H), 8,88 (д, J=6,3 Гц, 1H), 8,40 (д, J=6,1 Гц, 1H), 8,34 (д, J=5,6 Гц, 1H), 7,72 (д, J=7,8 Гц, 1H), 7,49 (д, J=7,8 Гц, 1H), 7,04 (д, J=5,6 Гц, 1H).
МС (ESI) m/z, 279 (M)+.
Пример 90: Соединение 101
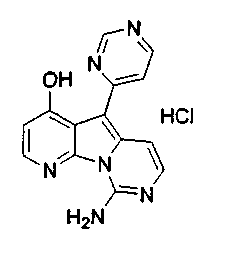
Соединение 100 (35 мг, 0,13 ммоль) обрабатывали HCl в 1,4-диоксане (4 мл, 3,8 M) при 0°C в течение 10 мин. Реакционную смесь упаривали, растворяли в MeOH и упаривали с получением соединения 101 в виде желтого твердого вещества (36 мг, 88%).
1H ЯМР (300 МГц, CD3OD) δ 9,35 (с, 1H), 8,92 (д, J=6,4 Гц, 1H), 8,44 (д, J=6,3 Гц, 1H), 8,38 (д, J=5,9 Гц, 1H), 7,79 (д, J=7,8 Гц, 1H), 7,54 (д, J=7,8 Гц, 1H), 7,08 (д, J=5,6 Гц, 1H).
МС (ESI) m/z, 279 (M)+.
Пример 91: Соединение 99
Раствор соединения 94 (83 мг, 0,20 ммоль) в CH2Cl2 (10 мл) обрабатывали при 0°C раствором mCPBA (112 мг, 0,5 ммоль, 2,5 эквив., 77%) и нагревали до 23°C в течение 3 ч. Реакционную смесь гасили добавлением насыщенного водного раствора Na2S2О3 и трижды промывали насыщенным водным раствором бикарбоната натрия. Органический слой сушили над сульфатом натрия, фильтровали и упаривали. Остаток подвергали хроматографии (элюируя CH2Cl2:MeOH, 95:5) с получением соединения 99 в виде желтого твердого вещества (25 мг, 28%).
1H ЯМР (CDC13:CD3OD 95:5, 300 МГц) δ 8,21 (д, J=5,6 Гц, 1H), 8,03 (д, J=5,4 Гц, 1H), 7,81 (д, J=5,6 Гц, 1H), 7,74 (д, J=6,6 Гц, 1H), 7,48 (д, J=6,8 Гц, 1H), 7,31 (с, 5H), 6,98 (д, J=5,6 Гц, 1H), 5,21 (с, 2H), 3,24 (с, 3H).
МС (ESI) m/z 447 (M+1)+.
Пример 92: Соединение 102
Суспензию соединения 99 (10 мг, 0,022 ммоль) обрабатывали при 0°C раствором MeONa в MeOH, предварительно приготовленным путем добавления Na (15 мг, 0,66 ммоль) к 1 мл MeOH. Реакционную смесь перемешивали при 23°C в течение 5 ч. Затем смесь распределяли между CH2Cl2 и насыщенным водным раствором хлорида аммония. Органический слой сушили над сульфатом натрия, фильтровали и упаривали с получением соединения 102 в виде желтого твердого вещества (5,6 мг, 64%).
1H ЯМР (CDC13:CD3OD 95:5, 300 МГц) δ 8,26 (д, J=5,6 Гц, 1H), 8,06 (д, J=5,4 Гц, 1H), 7,69 (д, J=6,6 Гц, 1H), 7,53 (д, J=6,6 Гц, 1H), 7,42-7,34 (м, 6 H), 6,97 (д, J=5,6 Гц, 1H), 5,29 (с, 2H), 4,06 (с, 3H).
МС (ESI) m/z 399 (М+1)+.
Пример 93: Соединение 103
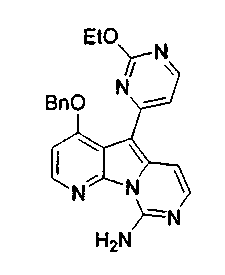
Суспензию соединения 99 (10 мг, 0,022 ммоль) обрабатывали при 0°C раствором EtONa в EtOH, предварительно приготовленным путем добавления Na (15 мл, 0,66 ммоль) к EtOH (1 мл), в течение 6 ч. Реакционную смесь распределяли между хлороформом и насыщенным водным раствором хлорида аммония. Органический слой сушили над сульфатом натрия, фильтровали и упаривали. Остаток подвергали хроматографии (элюируя CH2Cl2:MeOH, 95:5) с получением соединения 103 в виде желтого твердого вещества (8 мг, 88%).
1H ЯМР (CDC13:CD3OD 95:5, 300 МГц) δ 8,19 (д, J=5,6 Гц, 1H), 7,90 (д, J=5,4 Гц, 1H), 7,48 (д, J=6,6 Гц, 1H), 7,34 (д, J=6,6 Гц, 1H), 7,27-7,24 (м, 6H), 6,92 (д, J=6,8 Гц, 1H), 5,20 (с, 2H), 4,36 (q, J=6,8 Гц, 2H), 1,35 (т, J=6,8 Гц, 3H).
МС (ESI) m/z:413(М+1)+.
Пример 94: Соединение 104
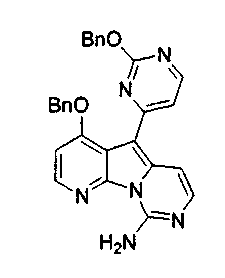
Суспензию NaH (18 мг, 0,44 ммоль, 60%) в тетрагидрофуране (2 мл) обрабатывали бензиловым спиртом (45 мкл, 0,44 ммоль). По каплям добавляли раствор соединения 99 (10 мг, 0,022 ммоль) в безводном тетрагидрофуране (3 мл) и перемешивали при 23°C в течение 6 ч. Реакционную смесь распределяли между хлороформом и насыщенным водным раствором хлорида аммония. Органический слой сушили над сульфатом натрия, фильтровали и упаривали. Остаток подвергали хроматографии (элюируя от CH2Cl2 до CH2Cl2:MeOH 95:5) с получением соединения 104 в виде желтого твердого вещества (9,0 мг, 86%).
1H ЯМР (CDC13:CD3OD 95:5, 300 МГц) δ 8,18 (д, J=5,6 Гц, 1H), 7,93 (д, J=5,4 Гц, 1H), 7,36 (д, J=7,8 Гц, 1H), 7,36-7,19 (м, 12H), 6,92 (д, J=5,9 Гц, 1H), 5,40 (с, 2H), 5,20 (с, 2H).
МС (ESI) m/z: 475(М+1)+.
Пример 95: Соединение 105
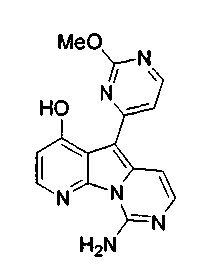
Раствор соединения 102 (4,0 мг, 0,01 ммоль) в трифторметансульфоновой кислоте (0,2 мл) перемешивали при 23°C в течение 1 ч. Реакционную смесь охлаждали до 0°C и обрабатывали по каплям MeOH (1 мл) и водным NH4OH (32%) (1 мл). Реакционную смесь фильтровали, промывали H2О, MeOH и диэтиловым эфиром и подвергали хроматографии (элюируя CH2Cl2:MeOH, 95:5) с получением соединения 105 в виде желтого твердого вещества (1,8 мг, 58%).
1H ЯМР (CDC13:CD3OD 95:5, 300 МГц) δ 8,26 (д, J=5,6 Гц, 1H), 7,99 (д, J=5,4 Гц, 1H), 7,43 (д, J=6,8 Гц, 1H), 7,25 (д, J=5,6 Гц, 1H), 6,94 (д, J=6,8 Гц, 1H), 6,70 (д, J=5,4 Гц, 1H), 3,91 (с, 3H), МС (ESI) m/z 309 (М+1)+.
Пример 96: Соединение 106
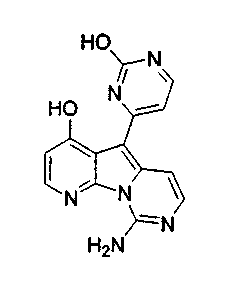
Раствор соединения 104 (7 мг, 0,017 ммоль) в трифторметансульфоновой кислоте (0,2 мл) перемешивали при 23°C в течение 1,5 ч. Реакционную смесь обрабатывали при 0°C MeOH (1 мл) и водным NH4OH (32%) (1 мл), фильтровали и промывали H2О, EtOH и диэтиловым эфиром с получением соединения 106 в виде оранжевого твердого вещества (6,6 мг, 95%).
1H ЯМР (CDC13:CD3OD 95:5, 300 МГц) δ 8,35 (д, J=5,9 Гц, 1H), 8,20 (д, J=6,6 Гц, 1H), 7,60 (д, J=7,6 Гц, 1Н), 7,43 (д, J=7,6 Гц, 1H), 7,18 (д, J=6,8 Гц, 1H), 7,03 (д, J=5,4 Гц, 1H).
МС (ESI) m/z 295(M+1)+.
Пример 97: Соединение 96
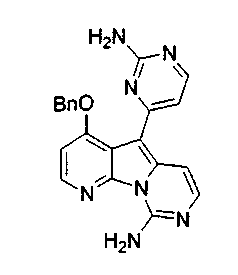
Раствор соединения 52 (24 мг, 0,05 ммоль) в CH2Cl2 (3 мл) обрабатывали при -30°C раствором mCPBA (30 мг, 0,14 ммоль, 77%) в CH2Cl2. Реакционную смесь нагревали до 0°C и перемешивали в течение 30 мин. Реакционную смесь промывали насыщенным водным раствором Na2S2О3 и насыщенным водным раствором бикарбоната натрия. Органический слой сушили над сульфатом натрия, фильтровали и упаривали. Неочищенный продукт растворяли в 1,4-диоксане (6 мл), обрабатывали водным NH4OH (32%) (10 мл) в герметичной пробирке и перемешивали при 100°C в течение 16 ч. Реакционную смесь упаривали и подвергали хроматографии (элюируя CH2Cl2:MeOH от 98:2 до 95:5) с получением соединения 96 в виде желтого твердого вещества (6 мг, 29%).
1H ЯМР (CDC13:CD3OD 9:1, 300 МГц) δ 8,18 (д, J=5,4 Гц, 1H), 7,78 (д, J=5,4 Гц, 1Н), 7,30-7,25 (м, 7H), 6,90 (д, J=5,4 Гц, 2H), 5,19 (с, 2H).
МС (APCI) m/z 384 (М+1)+.
Rf: 0,64 (CH2Cl2:MeOH, 6:1).
Пример 98: Соединение 107
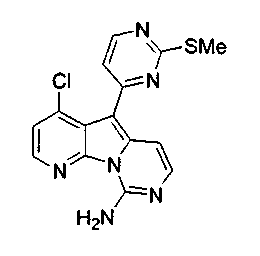
Раствор соединения 84 (50 мг, 0,135 ммоль) в смеси 1,4-диоксана:H2О (50:50, 8 мл) перемешивали в герметичной пробирке при 100°C в течение 16 ч. Реакционную смесь упаривали и подвергали хроматографии (элюируя CH2Cl2:MeOH, 98:2) с получением соединения 107 в виде желтого твердого вещества (40 мг, 86%).
1H ЯМР (CDCl3, 300 МГц) δ 8,54 (д, J=5,4 Гц, 1H), 8,27 (д, J=5,4 Гц, 1H), 7,59 (д, J=6,6 Гц, 1H), 7,46 (д, J=5,1 Гц, 1H), 7,26 (д, J=6,3 Гц, 1H), 7,23 (д, J=5,4 Гц, 1H), 2,64 (с, 3H).
13C ЯМР (CDCl3, 75 МГц) δ 172,0, 160,3, 156,1, 149,5, 144,0, 143,1, 139,9, 139,0, 135,0, 122,0, 119,9, 118,6, 101,4, 101,1, 14,3.
МС (ESI) m/z, 343 (М+1)+.
Пример 99: Соединение 108
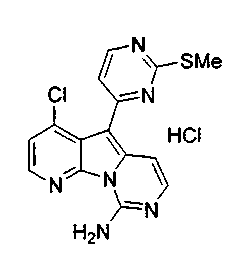
Соединение 107 (25 мл, 0,073 ммоль) обрабатывали безводным HCl (0,5 мл, 3,8 н. в 1,4-диоксане) в течение 10 мин при 23°C. Реакционную смесь упаривали, растворяли в MeOH и упаривали с получением соединения 108 в виде желтого твердого вещества (28 мг, 100%).
1H ЯМР (CD3OD, 300 МГц) δ 8,75 (д, J=5,6 Гц, 1H), 8,59 (д, J=5,4 Гц, 1H), 7,79 (д, J=5,1 Гц, 1H), 7,65 (д, J=5,6 Гц, 1H), 7,46 (д, J=7,8 Гц, 1H), 7,33 (д, J=7,8 Гц, 1H), 2,71 (с, 3H).
МС (ESI) m/z, 343 (M)+.
Пример 100: Соединение 109
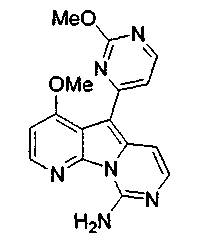
Соединение 107 (10 мл, 0,029 ммоль) обрабатывали MeONa в MeOH, свежеприготовленным путем добавления Na (30 мг, 1,3 ммоль) к MeOH (4 мл), при 80°C в течение 22 ч. Реакционную смесь упаривали и распределяли между CH2Cl2 и насыщенным водным раствором хлорида аммония. Органический слой сушили над сульфатом натрия, фильтровали и упаривали. Остаток подвергали хроматографии (элюируя CH2Cl2:MeOH, 95:5) с получением соединения 109 (15 мг, 53%) в виде желтого твердого вещества.
1H ЯМР (CDCl3, 300 МГц) δ 8,45 (д, J=5,4 Гц, 1H), 8,28 (д, J=5,4 Гц, 1H), 7,7 (д, J=6,6 Гц, 1Н), 7,54 (д, J=6,6 Гц, 1Н), 7,4 (д, J=5,4 Гц, 1Н), 6,91 (д, J=5,7 Гц, 1Н), 4,10 (с, 3H), 4,04 (с, 3H).
МС (ESI) m/z, 323 (М+1)+.
Пример 101: Соединения 110 и 111
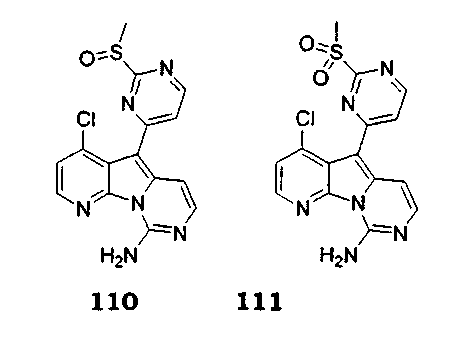
Раствор соединения 107 (50 мг, 0,146 ммоль) в CH2Cl2 (10 мл) обрабатывали при -30°C раствором mCPBA (82 мг, 77%, 0,37 ммоль) в CH2Cl2 (1 мл). Реакционную смесь перемешивали при 23°C в течение 2 ч, обрабатывали насыщенным водным раствором Na2S2О3 и распределяли между насыщенным водным раствором бикарбоната натрия и CH2Cl2. Органический слой сушили над сульфатом натрия, фильтровали и упаривали. Остаток подвергали хроматографии (элюируя CH2Cl2:MeOH, 95:5) с получением соединения 110 (23 мг, 44%) и соединения 111 (29 мг, 53%) в виде желтых твердых веществ.
Соединение 110:
1H ЯМР (CDCl3, 300 МГц) δ 8,84 (д, J=5,1 Гц, 1H), 8,30 (д, J=5,4 Гц, 1H), 7,68 (д, J=6,6 Гц, 1H), 7,63 (д, J=5,4 Гц, 1H), 7,50 (д, J=5,1 Гц, 1H), 7,46 (д, J=6,6 Гц, 1H), 3,04 (с, 3H).
МС (ESI) m/z, 381 (M+23)+, 359 (М+1)+.
Соединение 111:
1H ЯМР (CDCl3, 300 МГц) δ 8,84 (д, J=5,4 Гц, 1H), 8,32 (д, J=5,1 Гц, 1H), 7,75 (д, J=5,4 Гц, 1Н), 7,72 (д, J=6,6 Гц, 1Н), 7,53 (д, J=6,6 Гц, 1Н), 7,52 (д, J=5,4 Гц, 1Н), 3,43 (с, 3H).
МС (ESI) m/z, 397 (M+23)+, 375 (М+1)+.
Пример 102: Соединение 112
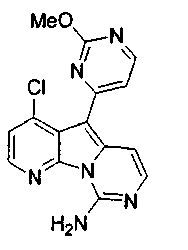
Соединение 111 (29 мг, 0,077 ммоль) обрабатывали раствором MeONa в MeOH, свежеприготовленным путем добавления Na (30 мг, 1,3 ммоль) к MeOH (4 мл), в течение 6 ч при 23°C. Реакционную смесь упаривали и распределяли между CH2Cl2 и насыщенным водным раствором хлорида аммония. Органический слой сушили над сульфатом натрия, фильтровали и упаривали. Остаток подвергали хроматографии (элюируя CH2Cl2:MeOH, 95:5) с получением соединения 112 (17 мг, 68%) в виде желтого твердого вещества.
1H ЯМР (CDCl3, 300 МГц) δ 8,54 (д, J=5,1 Гц, 1H), 8,27 (д, J=5,1 Гц, 1H), 7,58 (д, J=6,9 Гц, 1H), 7,47 (д, J=5,1 Гц, 1H), 7,29 (д, J=6,6 Гц, 1H), 7,21 (д, J=5,1 Гц, 1H), 4,10 (с, 3H).
МС (ESI) m/z, 327 (М+1)+.
Пример 103: Соединение 113
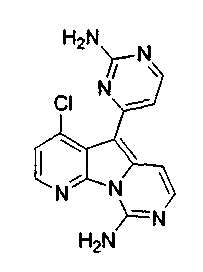
Раствор соединения 84 (50 мг, 0,14 ммоль) в CH2Cl2 (10 мл) обрабатывали при -30°C раствором mCPBA (76 мг, 0,34 ммоль, 77%) в CH2Cl2 (2 мл). Реакционную смесь нагревали до 0°C и перемешивали в течение 30 мин. Реакционную смесь обрабатывали насыщенным водным раствором Na2S2О3 и насыщенным водным раствором бикарбоната натрия. Органический слой сушили над сульфатом натрия, фильтровали и упаривали. Неочищенный продукт реакции растворяли в 1,4-диоксане (4 мл) и обрабатывали 32%-ным водным NH4OH (4 мл). Реакционную смесь вливали в герметичную пробирку и перемешивали при 85°C в течение 16 ч. Реакционную смесь упаривали и подвергали хроматографии (элюируя CH2Cl2:MeOH, 95:5) с получением соединения 113 в виде желтого твердого вещества (17 мг, 40%).
1H ЯМР (CDCl3:CD3OD 9:1, 300 МГц) δ 8,17 (д, J=5,4 Гц, 2H), 7,36-7,34 (м, 2H), 6,98 (д, J=6,8 Гц, 1H), 6,78 (д, J=5,1 Гц, 1H).
13C ЯМР (CDC13:CD3OD 9:1, 75 МГц) δ 162,5, 161,6, 156,9, 149,6, 143,9, 141,8, 140,1, 138,1, 135,0, 121,9, 119,8, 114,0, 101,7, 100,7.
МС (ESI) m/z 312 (М+1)+.
Пример 104: Соединение 83
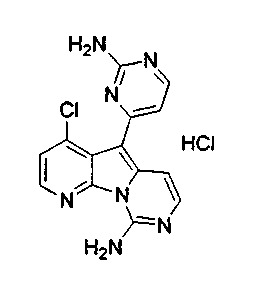
Соединение 113 (12 мг, 0,038 ммоль) обрабатывали безводным HCl в 1,4-диоксане (0,5 мл, 3,8 н.) в течение 10 мин при 23°C. Реакционную смесь упаривали, растворяли в MeOH (1 мл) и упаривали с получением соединения 83 в виде желтого твердого вещества (13,9 мг, 95%).
1H ЯМР (CD3OD, 300 МГц) δ 8,61 (д, J=5,1 Гц, 1H), 8,37 (д, J=6,6 Гц, 1H), 7,82 (д, J=5,1 Гц, 1H), 7,55 (с, 2H), 7,36 (д, J=6,6 Гц, 1H).
МС (ESI) m/z 312 (M)+.
Пример 105: Соединение 115
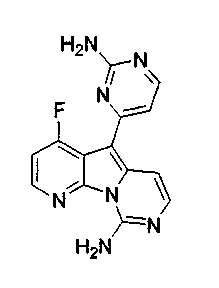
Смесь соединения 113 (5,0 мг, 0,016 ммоль), 18-краун-6 (10 мг, 0,038 ммоль) и безводного KF (60 мг, 1,2 ммоль) в DMSO (0,5 мл) нагревали в герметичной пробирке в течение 16 ч при 140°C. Реакционную смесь упаривали и подвергали хроматографии (элюируя CH2Cl2:MeOH, 95:5) с получением соединения 115 в виде желтого твердого вещества (3,5 мг, 74%).
1H ЯМР (CDCl3:CD3OD 9:1, 300 МГц) δ 8,26 (дд, J=7,1, 5,4 Гц, 1H), 8,13 (д, J=5,4 Гц, 1H), 7,48 (д, J=6,6 Гц, 1H), 7,41 (д, J=6,6 Гц, 1H), 7,13 (д, J=11,0, 5,4 Гц, 1H), 6,94 (дд, J=5,4, 3,2 Гц, 1H).
МС (ESI) m/z, 296 (M+1)+.
Пример 106: Соединение 114
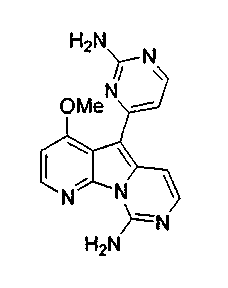
Соединение 113 (10 мг, 0,032 ммоль) обрабатывали при 90°C в течение 40 ч раствором MeONa, приготовленным путем добавления Na (30 мг, 1,3 ммоль) к MeOH, в MeOH:тетрагидрофуране (2:1, 6 мл). Реакционную смесь упаривали, растворяли в CH2Cl2 и промывали насыщенным водным раствором хлорида аммония. Органический слой сушили над сульфатом натрия, фильтровали и упаривали. Остаток подвергали хроматографии (элюируя CH2Cl2:MeOH, 95:5) с получением соединения 114 в виде желтого твердого вещества (4,0 мг, 41%).
1H ЯМР (CDCl3:CD3OD 9:1, 300 МГц) δ 8,21 (д, J=5,4 Гц, 1H), 8,12 (д, J=5,6 Гц, 1H), 7,32 (с, 2H), 6,96 (д, J=5,9 Гц, 1H), 6,84 (д, J=5,6 Гц, 1H), 3,95 (с, 3H).
МС (ESI) m/z, 308 (М+1)+.
Пример 107: Соединение 116
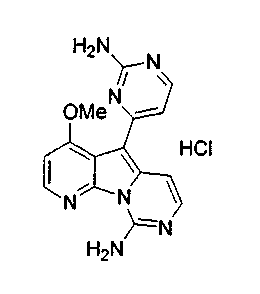
Соединение 114 (1-7 мг, 0,006 ммоль) при 23°C обрабатывали HCl в 1,4-диоксане (0,5 мл, 5н) в течение 20 мин. Реакционную смесь упаривали, суспендировали в диэтиловом эфире и упаривали с получением соединения 116 в виде желтого твердого вещества (1,2 мг, 57%).
1H ЯМР (300 МГц, CD3OD) δ 8,57 (д, J=5,8 Гц, 1H), 8,26 (д, J=6,8 Гц, 1H), 7,94-7,90 (м, 2H), 7,68 (д, J=6,8 Гц, 1H), 7,49 (д, J=7,8 Гц, 1H), 7,36 (д, J=5,8 Гц, 1H), 4,16 (с, 3H).
БИОАНАЛИЗ ДЛЯ СКРИНИНГА ПРОТИВООПУХОЛЕВОЙ АКТИВНОСТИ
Заключительным действием данных методов анализа является прекращение роста культуры опухолевых клеток in vitro путем продолжительного воздействия тестируемого образца на клетки.
КЛЕТОЧНЫЕ ЛИНИИ
1. Оценка ингибирования роста клеток путем подсчета клеток
В данной разновидности анализа применяли 24-луночные планшеты с диаметром лунки 16 мм (Bergeron, 1984; Schroeder, 1981). Применяли следующие линии опухолевых клеток: P-388 (ATCC CCL 46), суспензия культуры лимфоидных опухолевых клеток от мышей линии DBA/2; A-549 (ATCC CCL 185), монослойная культура клеток карциномы легких человека; HT-29 (ATCC HTB-38), монослойная культура клеток карциномы толстого кишечника человека; MEL-28 (ATCC HTB-72), монослойная культура клеток меланомы человека и DU-145 (ATCC HTB-81), монослойная культура клеток карциномы предстательной железы человека.
Клетки поддерживали в логарифмической фазе роста в минимальной поддерживающей среде Игла с добавлением сбалансированных солей Эрла, заменимых аминокислот, 2,0 мМ L-глутамина, не содержащей бикарбоната натрия (EMEM/neaa), с добавлением 10% фетальной бычьей сыворотки (FCS), 10-2 M бикарбоната натрия, 0,1 ед./л пенициллина G и 0,1 г/л стрептомицинсульфата. Для проведения экспериментов клетки получали из субконфлюэнтных культур с применением трипсина и ресуспендировали в свежей среде перед добавлением в лунки.
Клетки P-388 высевали в лунки диаметром 16 мм в концентрации 1x104 клеток на лунку в 1 мл аликвоты EMEM с 5% FCS, содержащие различные концентрации тестируемого образца. Для того чтобы убедиться в том, что клетки находятся в экспоненциальной фазе роста контроля роста, в качестве контроля высевали отдельную серию культур. Все определения выполняли в двукратном повторе. После инкубации клеток в течение трех суток при 37°C в атмосфере с 5% CO2 и 98% влажностью, определяли приблизительное значение IC50 путем сравнения роста в лунках, содержащих лекарство, с ростом в контрольных лунках.
Клетки A-549, HT-29, MEL-28 и DU-145 высевали в лунки диаметром 16 мм в концентрации 1x104 клеток на лунку в 1 мл аликвоты EMEM с 5% FCS, содержащие различные концентрации тестируемого образца. Для того чтобы убедиться в том, что клетки находятся в экспоненциальной фазе роста контроля роста, в качестве контроля высевали отдельную серию культур. Все определения выполняли в двукратном повторе. После инкубации в течение трех суток при 37°C в атмосфере, содержащей 5% CO2, и с 98% влажностью, клетки окрашивали 0,1% кристаллическим фиолетовым. Приблизительное значение IC50 определяли путем сравнения роста в лунках, содержащих лекарство, с ростом в контрольных лунках.
Для количественной оценки активности после инкубации клетки обрабатывали трипсином и посчитывали в Coulter Counter ZM. Общее количество (клеток на лунку) представляли в виде среднего значения из двух повторов. Рассчитывали значение процента роста относительно роста культур в отсутствие лекарства (%G). Результаты данных методов анализа использовали для построения кривых дозовых зависимостей, из которых определяли более точные значения IC50 (концентрацию образца, которая ингибирует рост клеток на 50%).
Полученные результаты могут быть использованы для предсказания применимости конкретного лекарства в качестве потенциального противоопухолевого средства. Для данной методики для дальнейших исследований отбирали соединения со значениями IC50, не превышающими 1 мкг/мл. Значения IC50 могут позволить предсказать обладание лекарством не только цитостатических качеств, но также и потенциала в отношении торможения развития опухоли.
2. Колориметрический метод анализа ингибирования роста клеток
Для количественной оценки роста и жизнеспособности клеток был адаптирован колориметрический метод анализа с применением реакции сульфородамина B (SRB) [следуя методике, описанной Philip Skehan, et al. (1990), New colorimetric cytotoxicity assay for anticancer drug screening, J. Natl. Cancer Inst., 82: 1107-1112].
В этом способе анализа применяли 96-луночные микропланшеты с диаметром лунок 9 мм (Faircloth, 1988; Mosmann, 1983). Большинство клеточных линий, происходящих из различных типов опухолевых клеток человека, были получены от American Type Culture Collection (ATCC).
Клетки поддерживали в среде RPMI 1640 с 10% FBS с добавлением 0,1 г/л пенициллина и 0,1 г/л стрептомицинсульфата и затем инкубировали при 37°C в атмосфере с 5% CO2 и 98% влажностью. Для проведения экспериментов клетки получали из субконфлюэнтных культур с применением трипсина и ресуспендировали в свежей среде перед добавлением в лунки.
Клетки высевали в 96-луночные микропланшеты в концентрации 5x103 клеток на плашку в 195 мкл аликвоты среды и оставляли на 18 часов прикрепляться к поверхности микропланшета в свободной от лекарства среде. Потом в аликвоты добавляли 5 мкл образцов в диапазоне концентраций от 10 до 10-8 мкг/мл, растворенных в DMSO/EtOH/PBS (0,5/0,5/99). После 48 часов воздействия измеряли противоопухолевый эффект по SRB-методике: клетки фиксировали добавлением 50 мкл холодной 50% (мас./об.) трихлоруксусной кислоты (TCA) и инкубировали в течение 60 минут при 4°C. Плашки промывали деионизированной водой и сушили. В каждую лунку микропланшета добавляли 100 мкл раствора SRB (0,4 мас.%/об. раствор в 1% уксусной кислоте) и инкубировали в течение 10 минут при комнатной температуре. Несвязавшийся SRB удаляли путем промывания 1% уксусной кислотой. Микропланшеты сушили на воздухе и солюбилизировали связавшийся краситель трис-буфером. Оптические плотности считывали на автоматическом спектрофотометрическом планшетном ридере при длине волны 490 нм.
Из результатов экспериментов в трех повторах рассчитывали значения среднего ± SD. Рассчитывали некоторые параметры клеточного ответа: GI = ингибирование роста, TGI = общее ингибирование роста (цитостатический эффект) и LC = гибель клеток (цитотоксический эффект).
Полученные результаты могут быть использованы для предсказания применимости конкретного лекарства в качестве потенциального противоопухолевого средства. Для данной методики для дальнейших исследований отбирали соединения со значениями GI50 не превышающими 10 мкг/мл. Значения GI50 могут позволить предсказать обладание лекарством не только цитостатических качеств, но также и потенциала в отношении торможения развития опухоли.
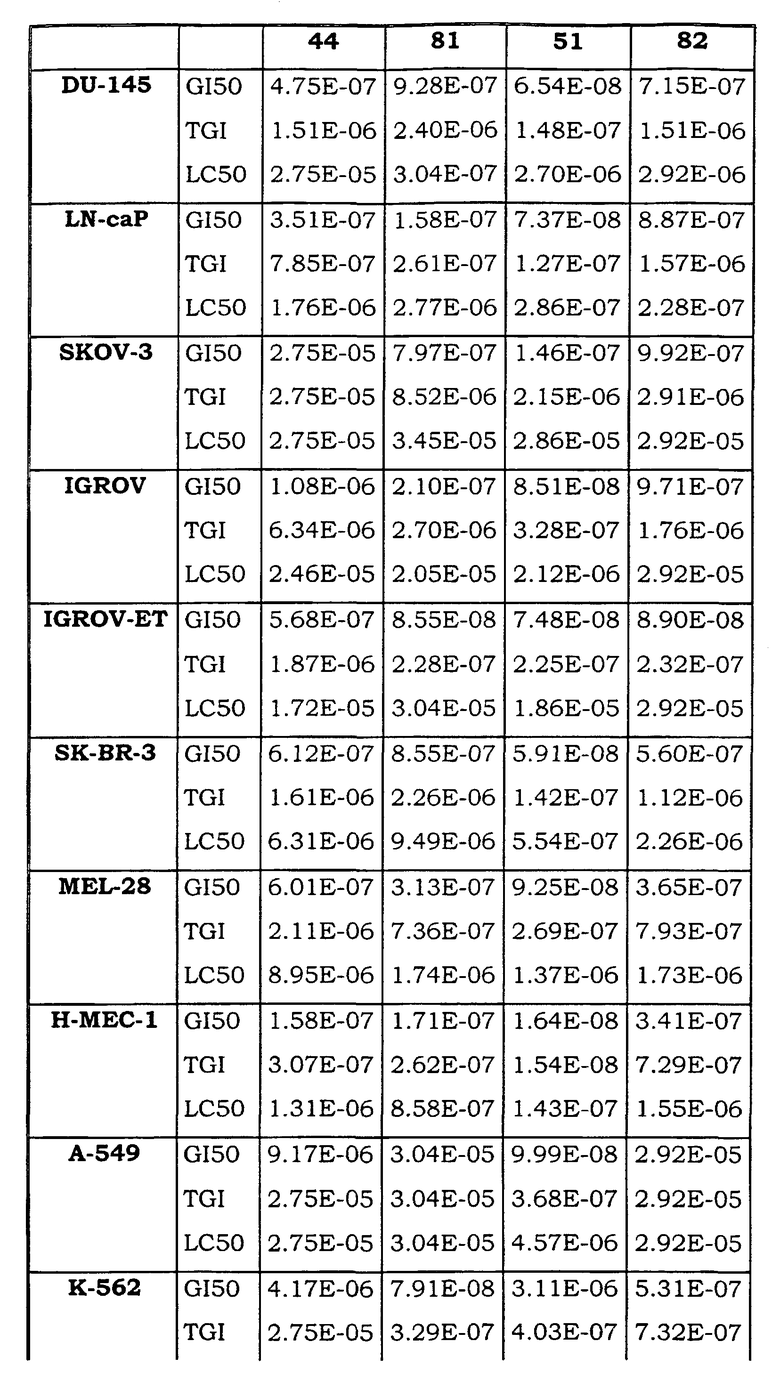
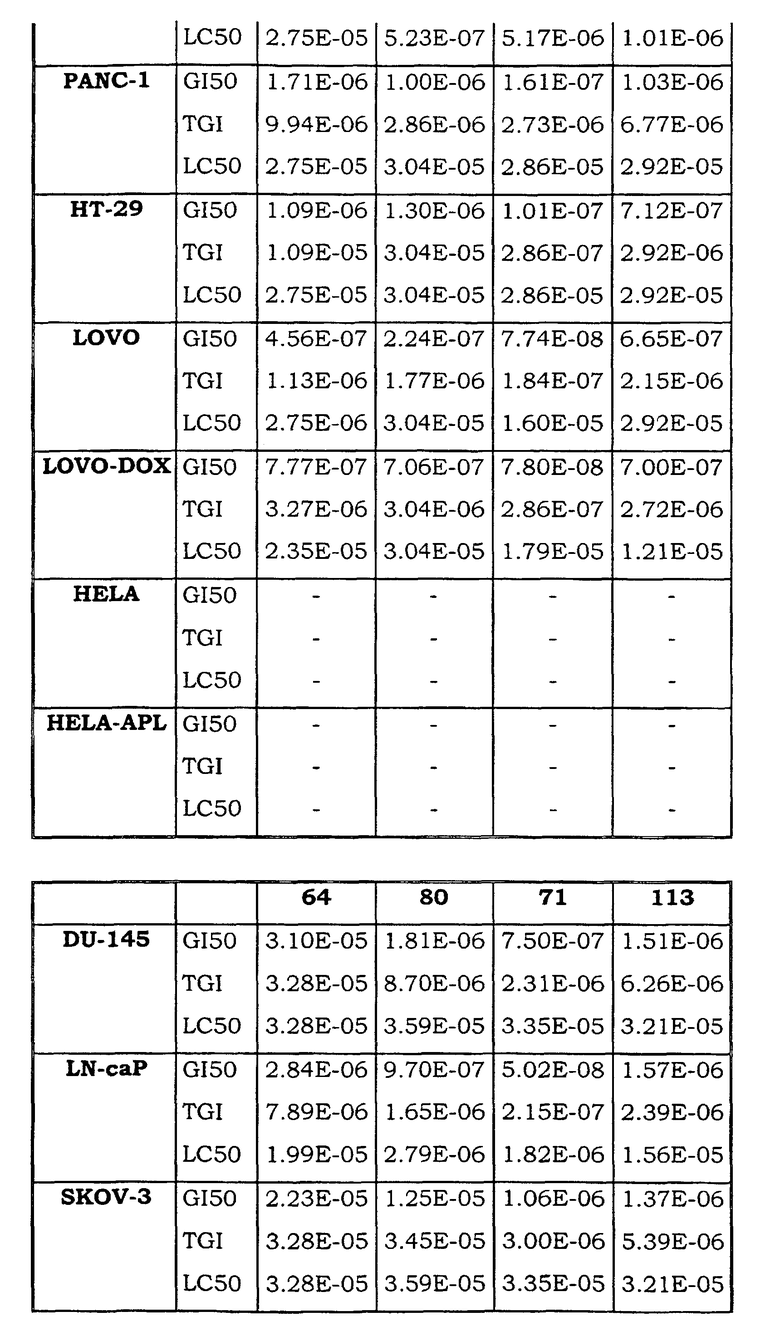
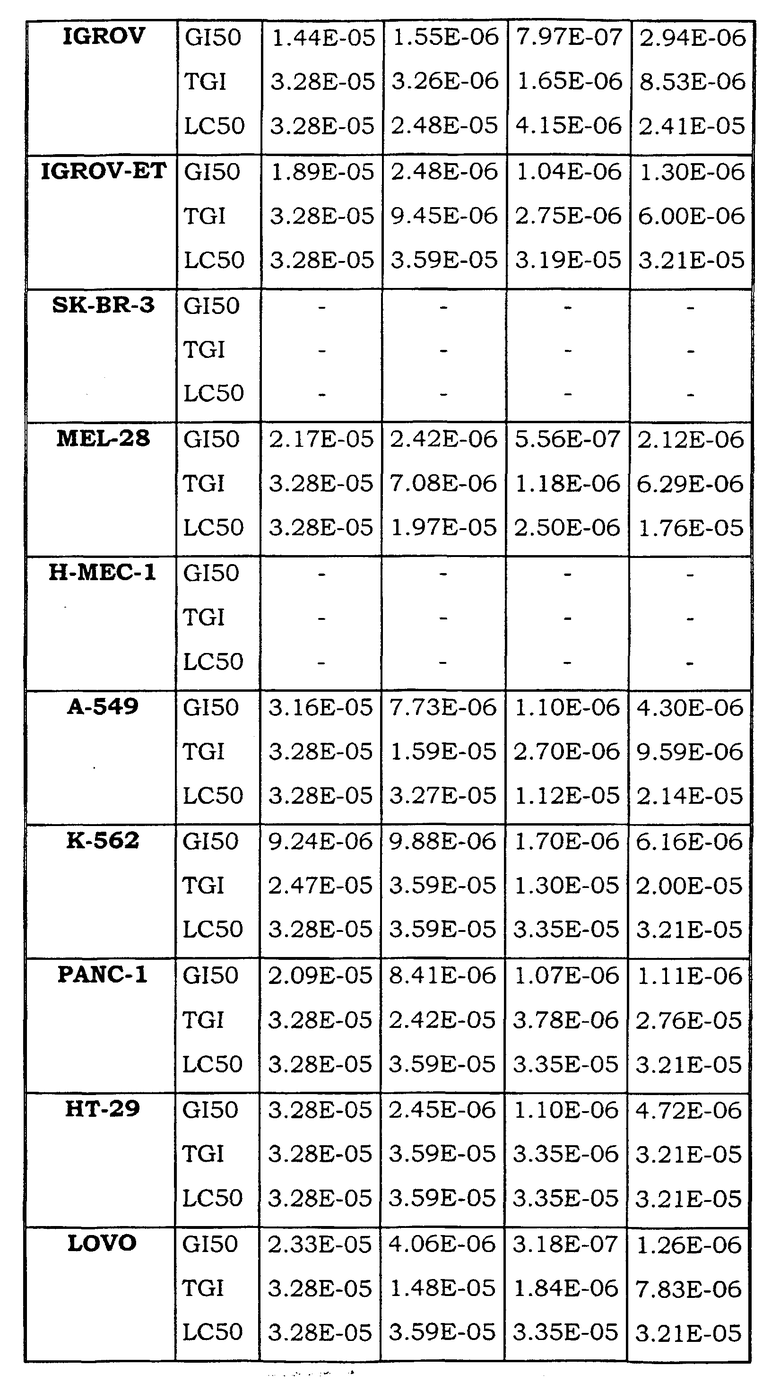
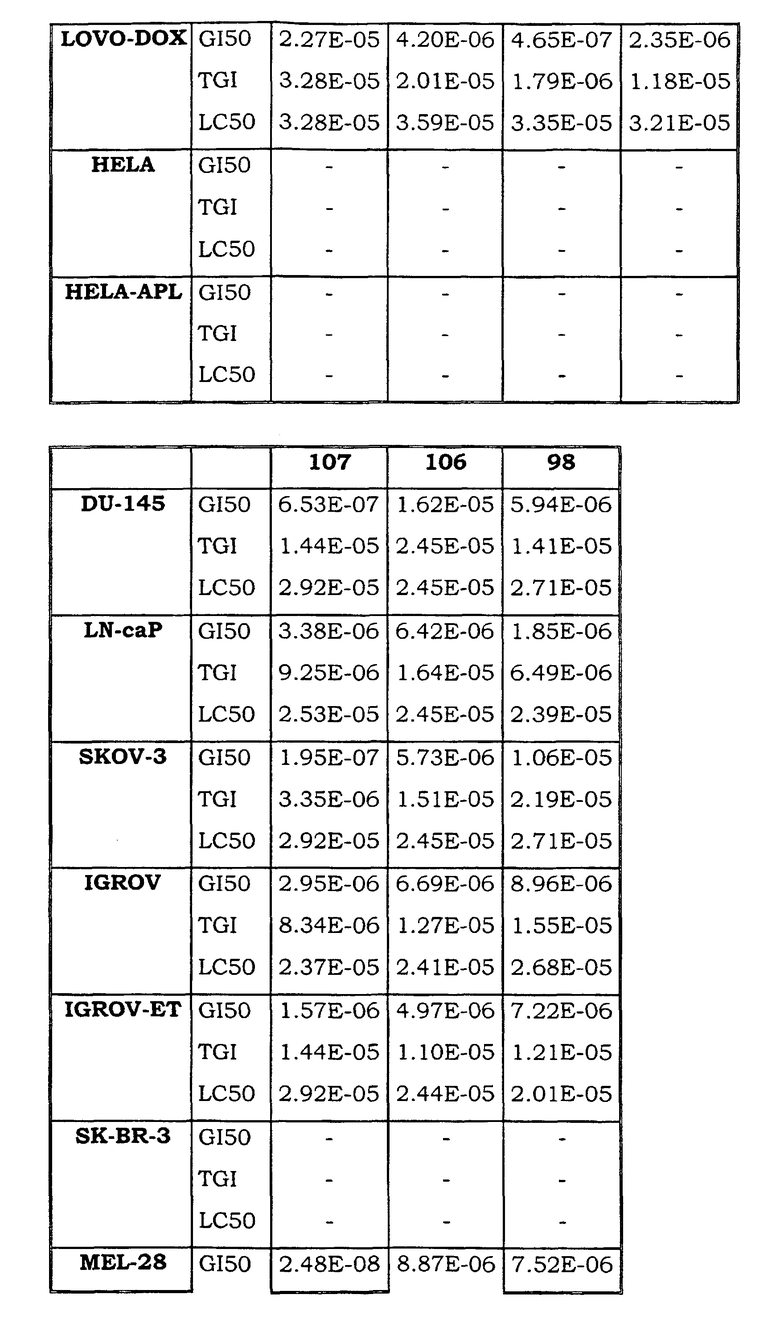
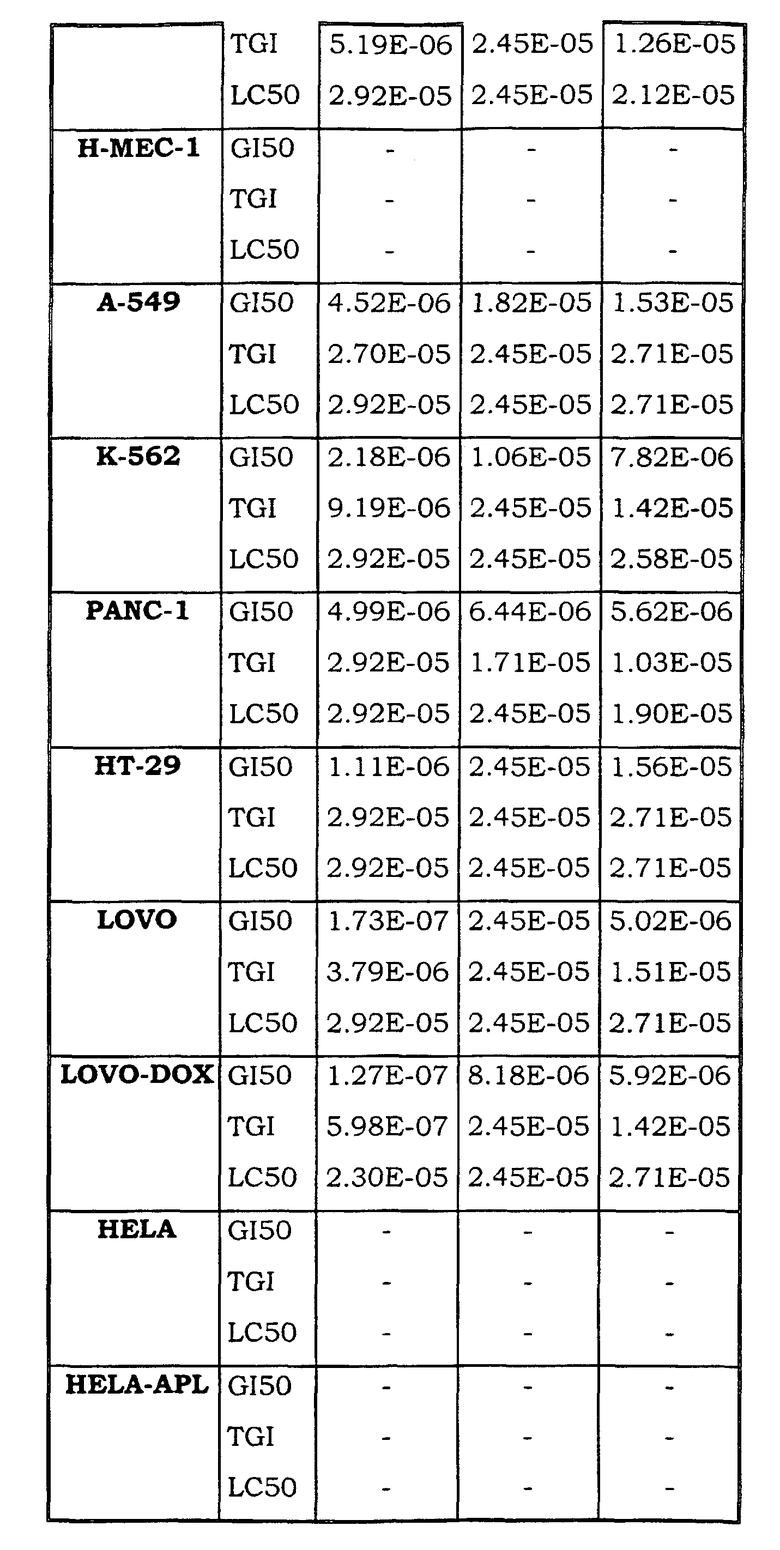
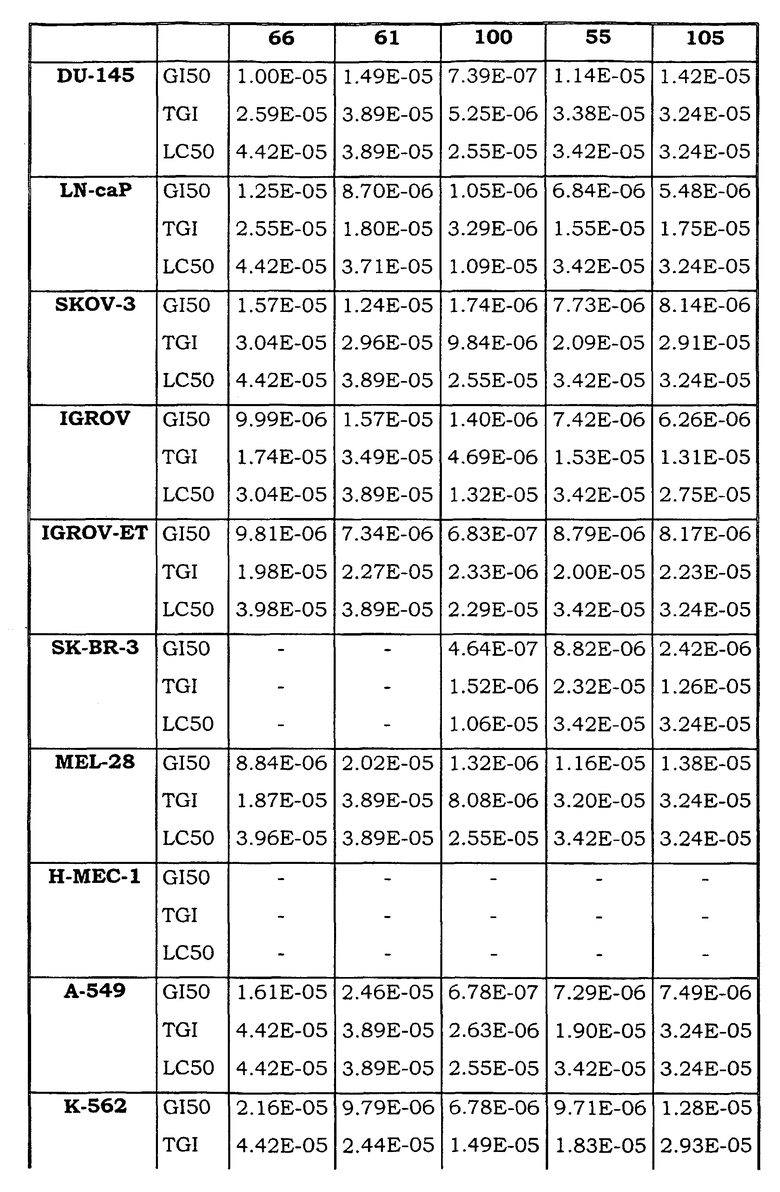
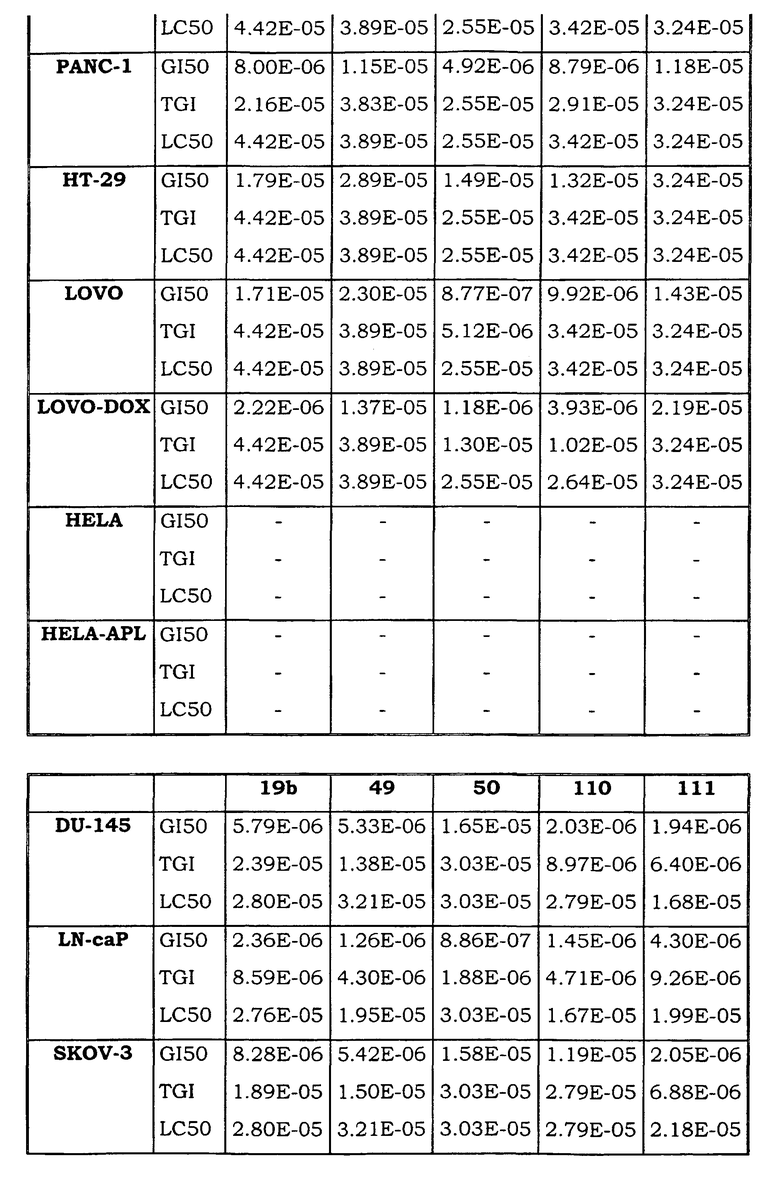
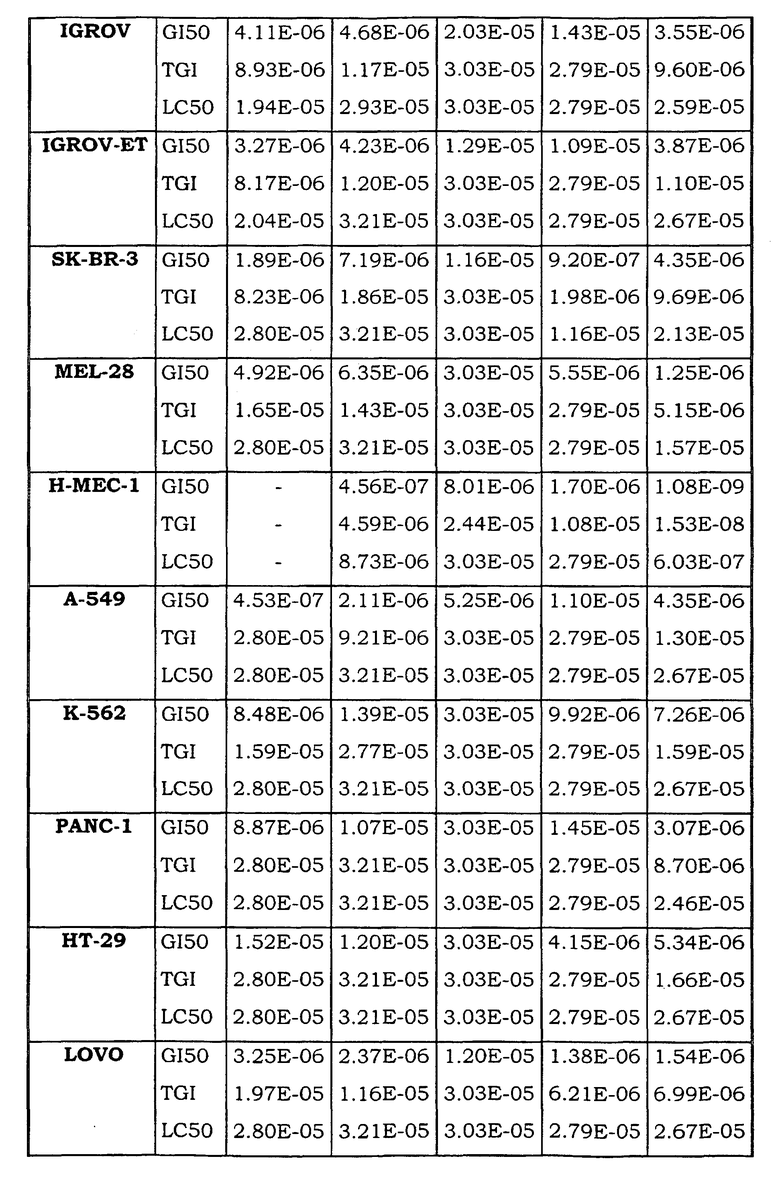
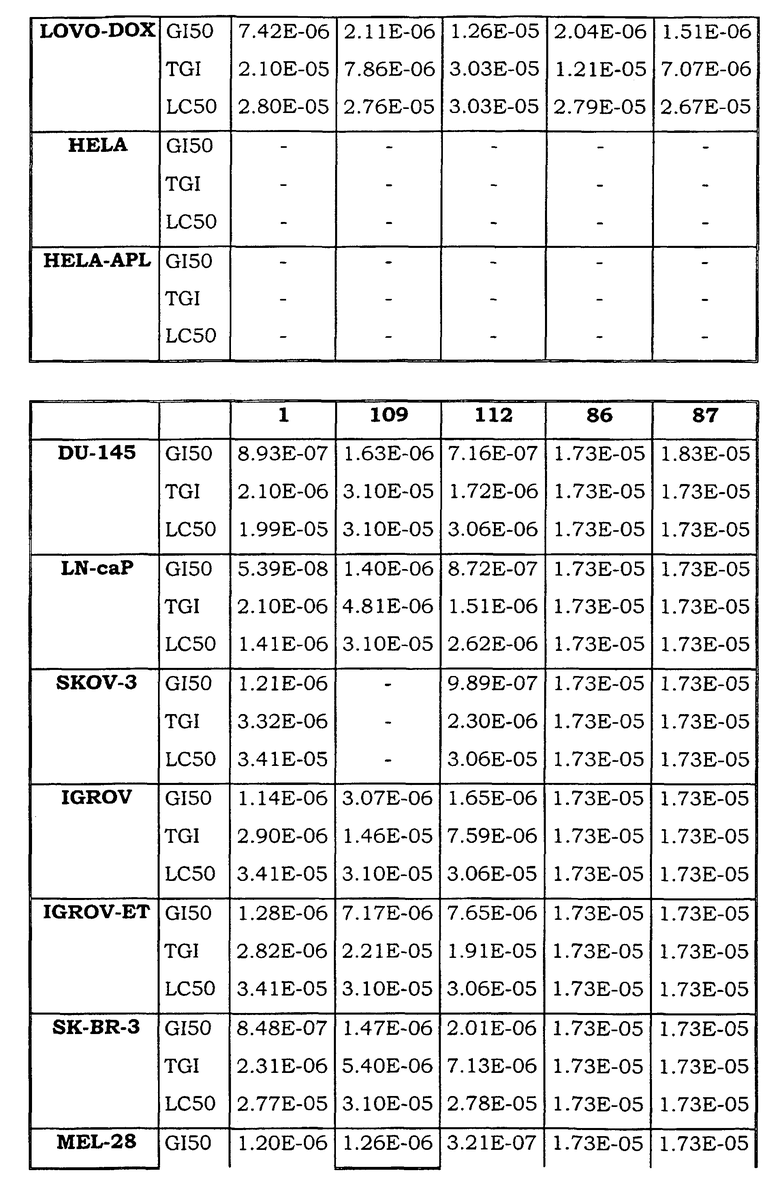
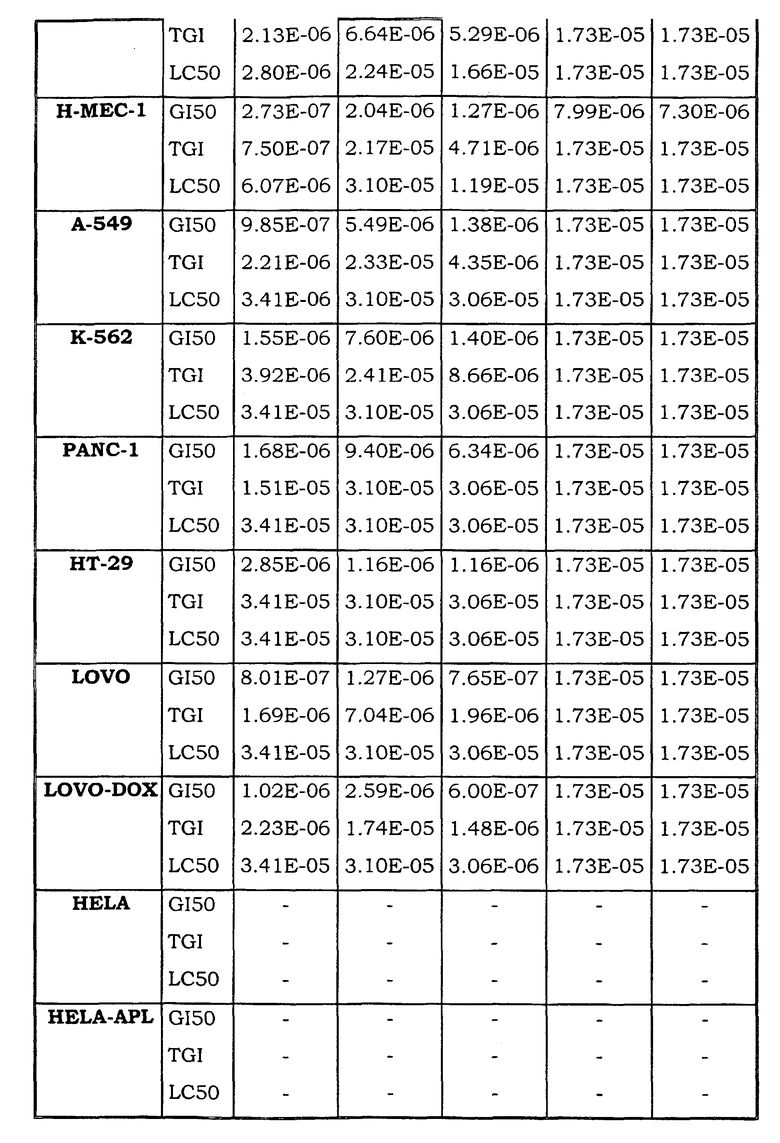
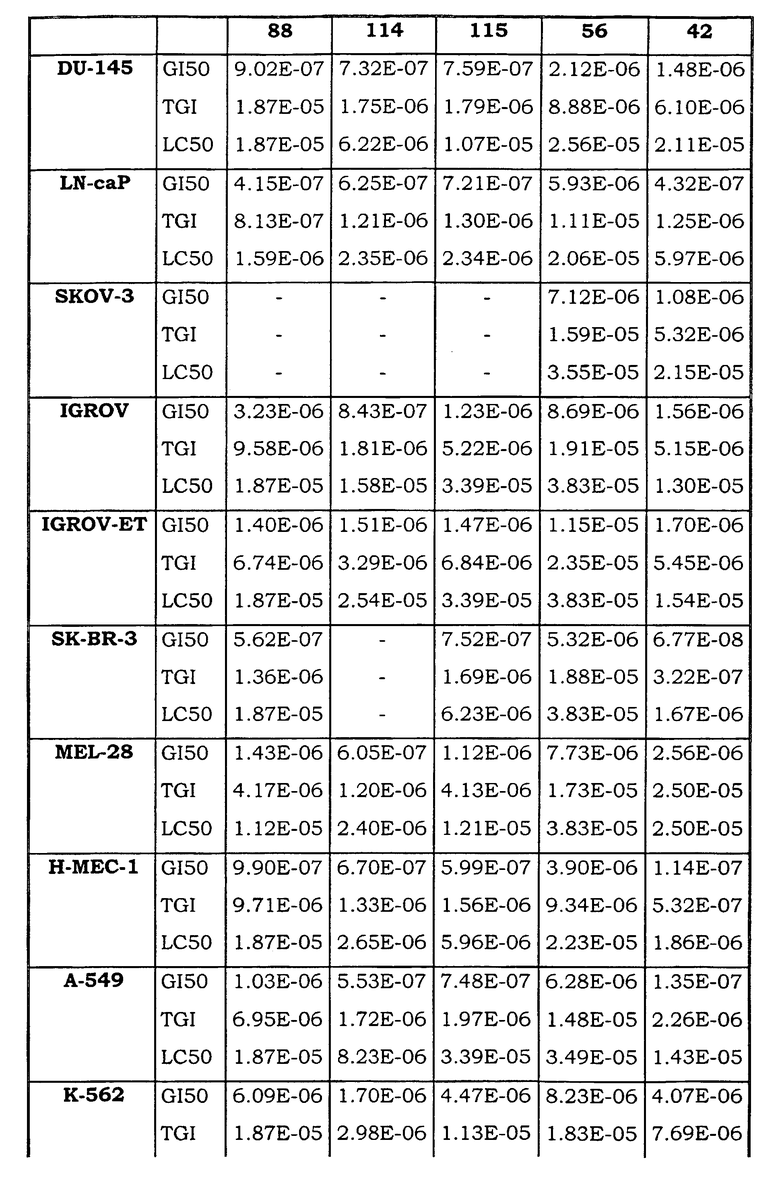
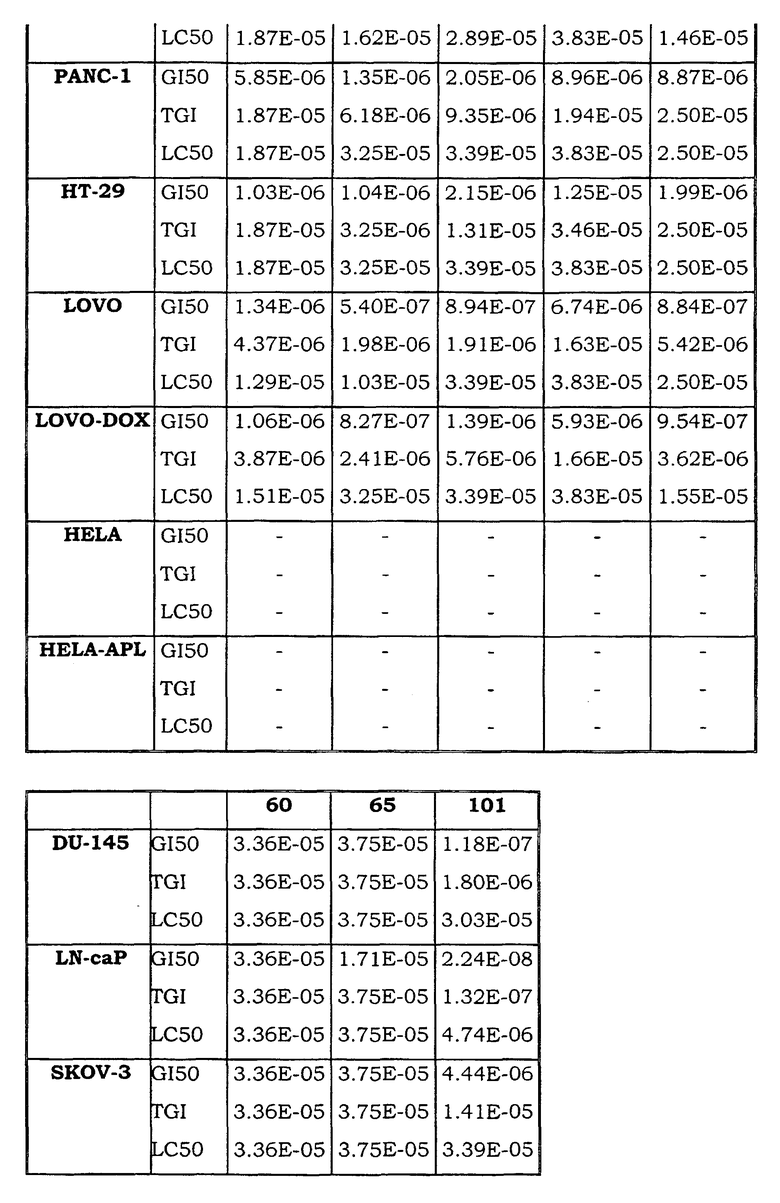
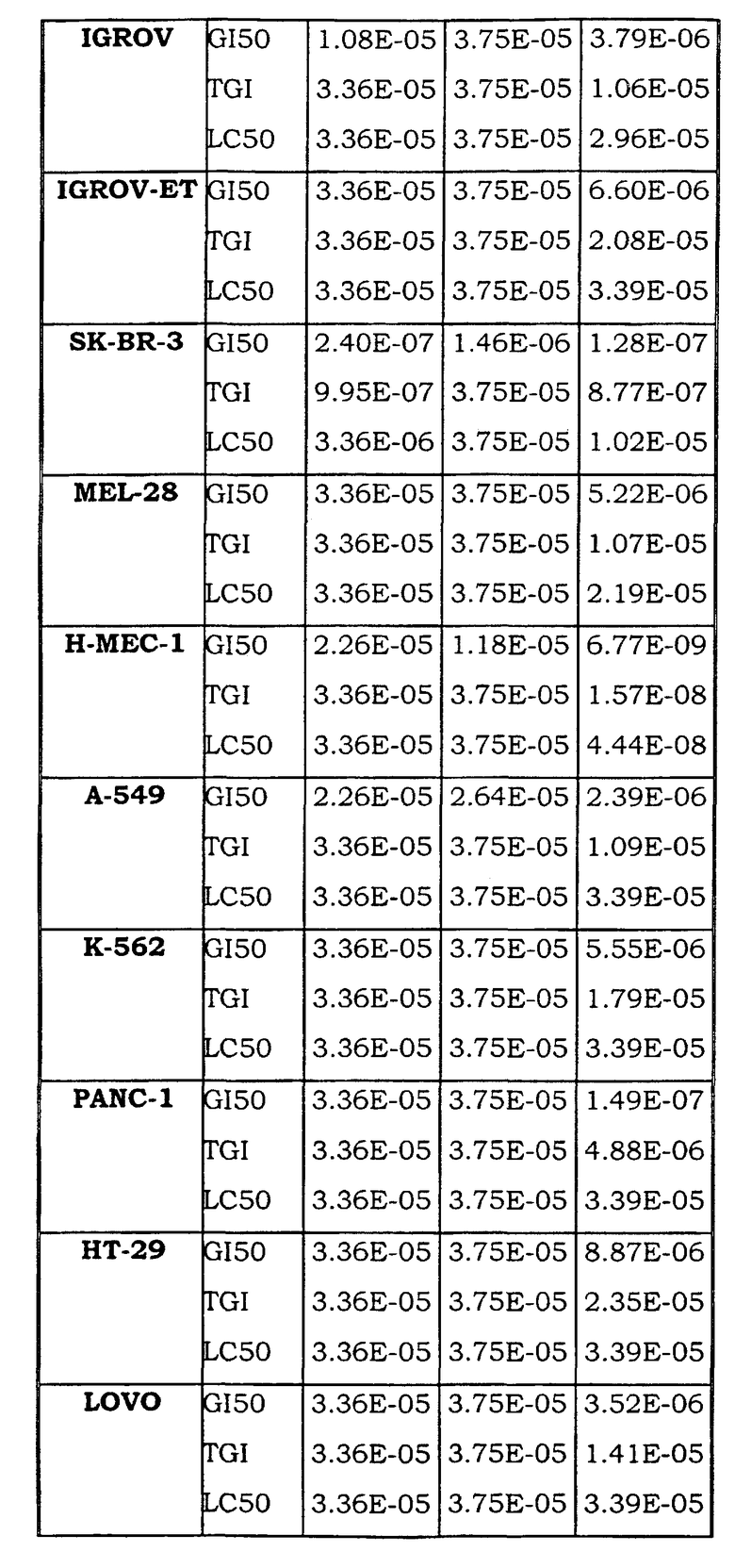
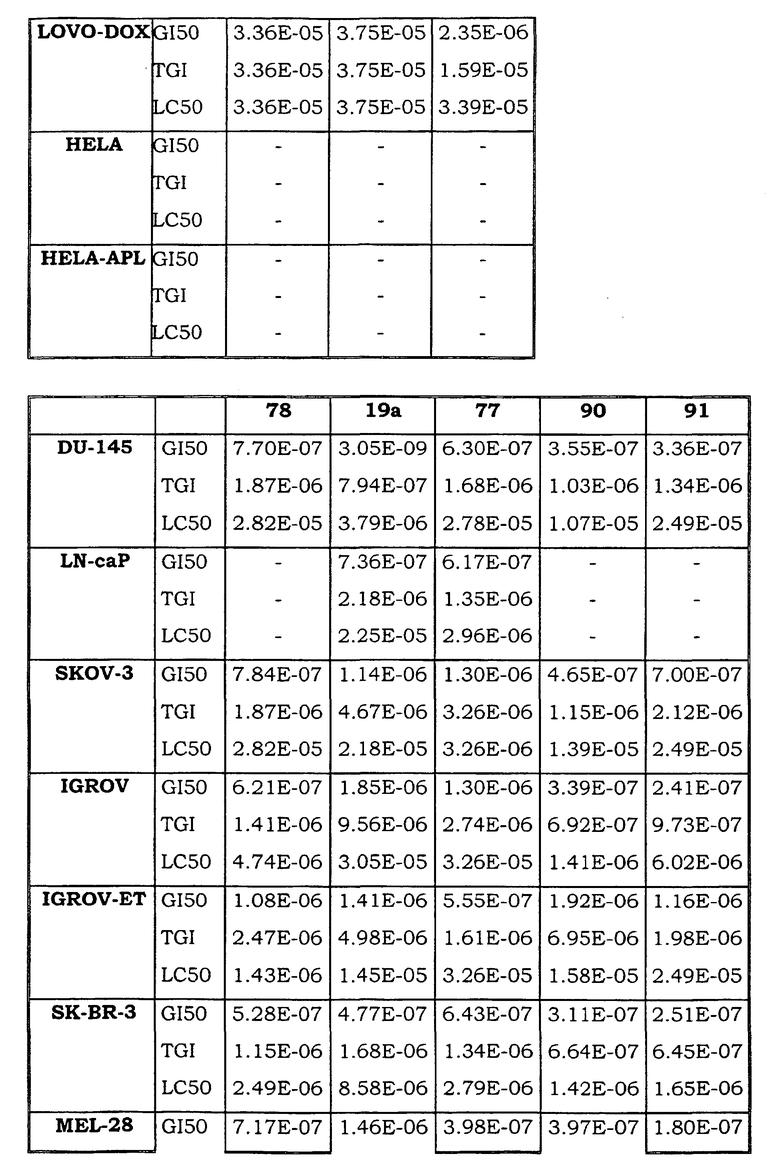
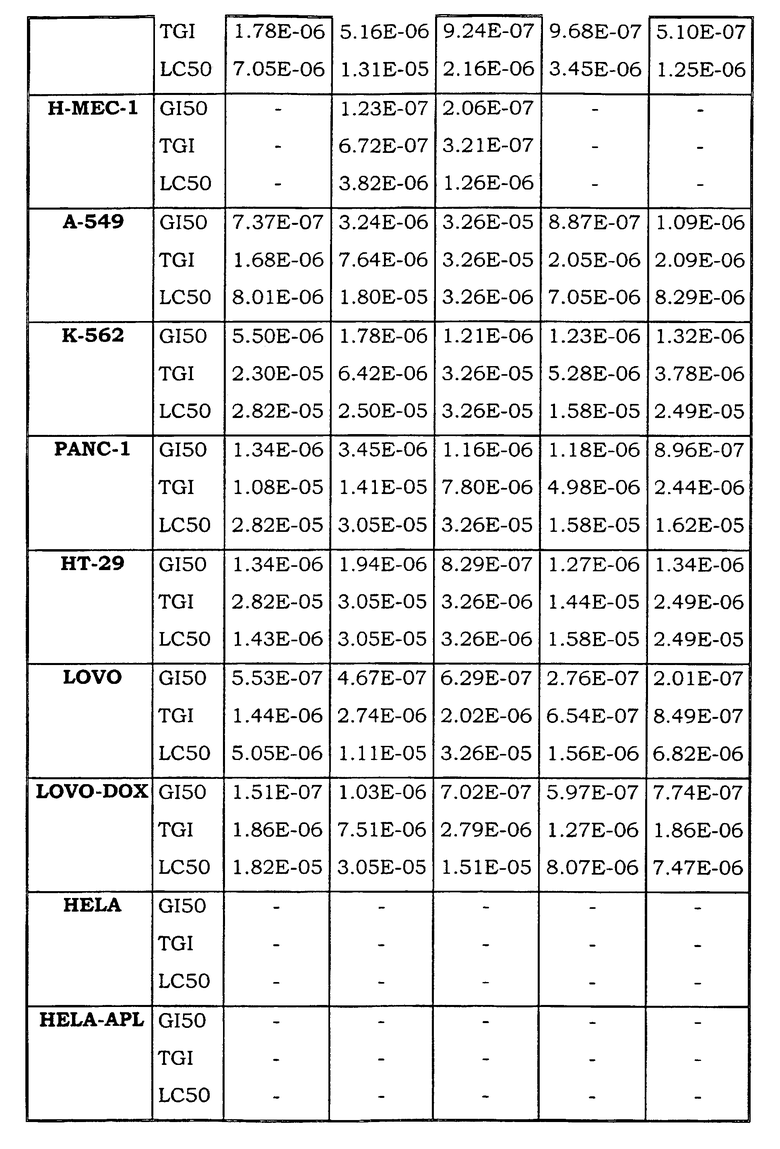
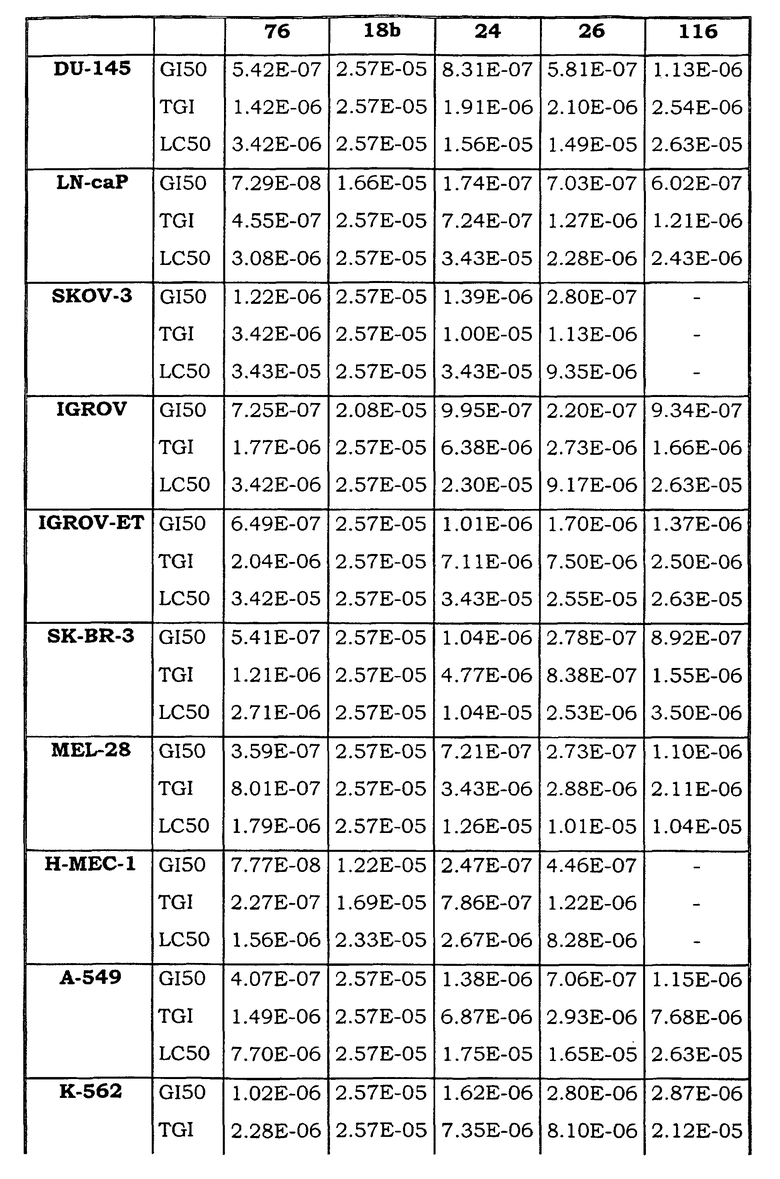
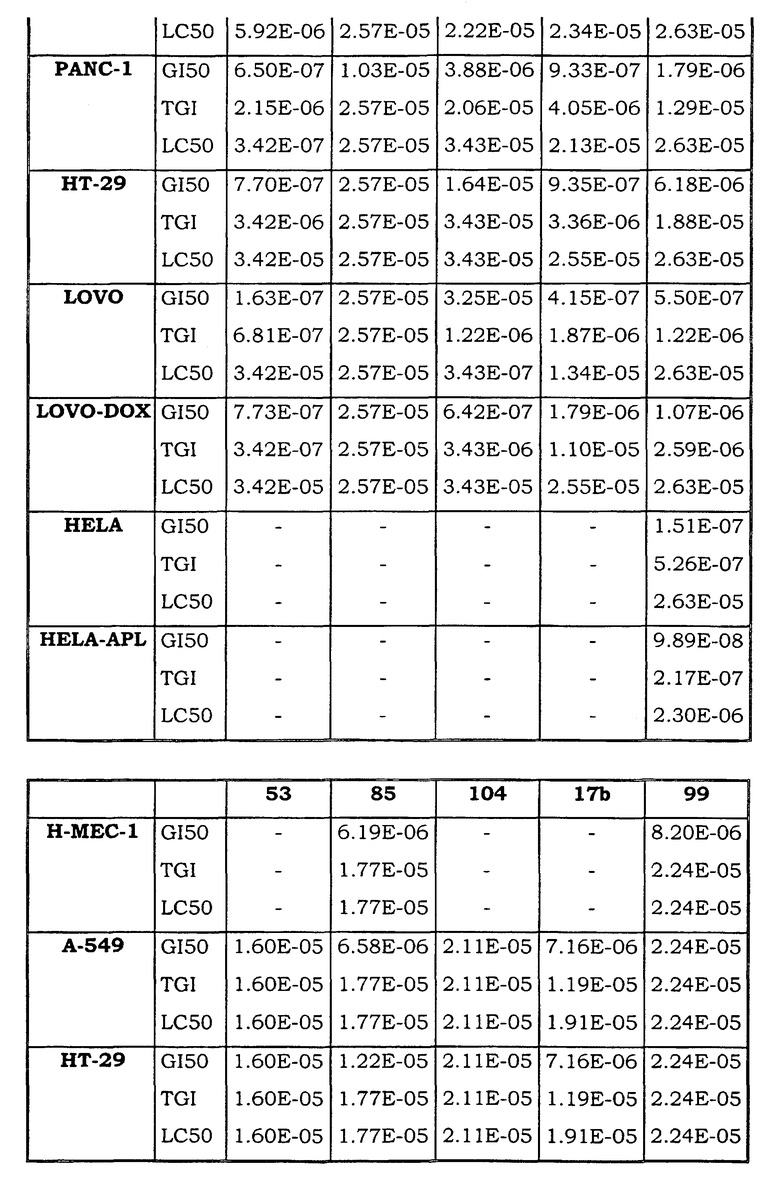
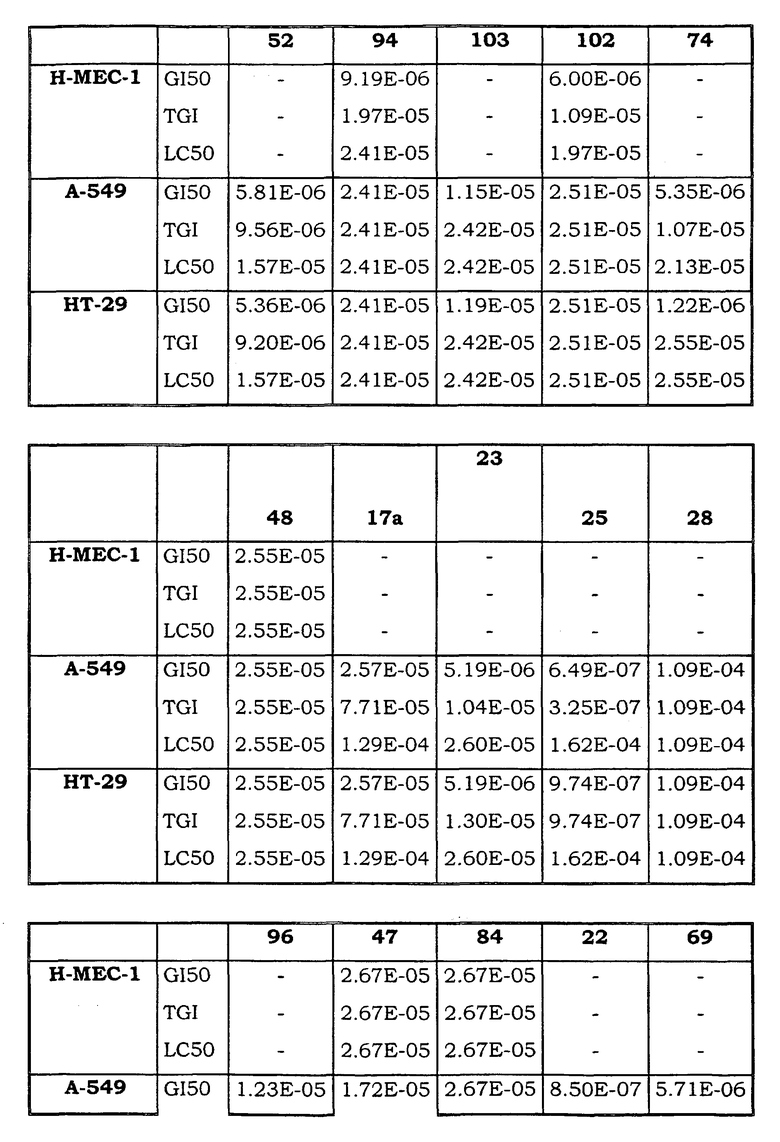
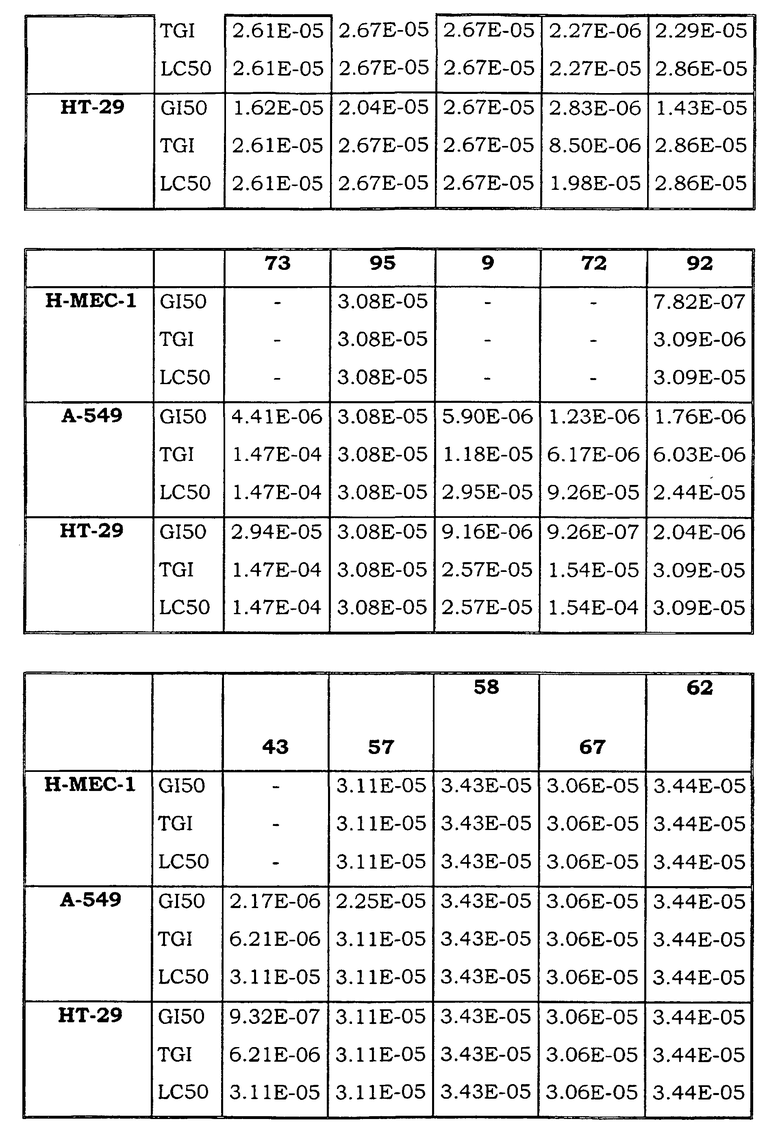
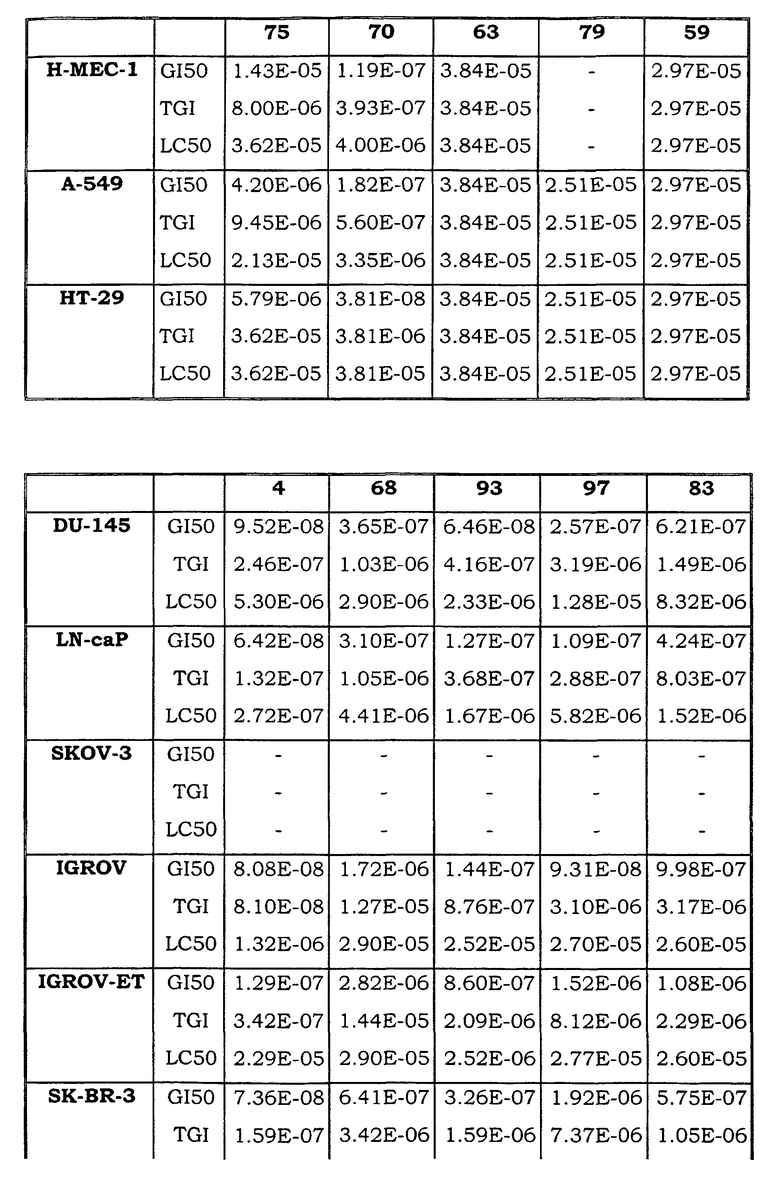
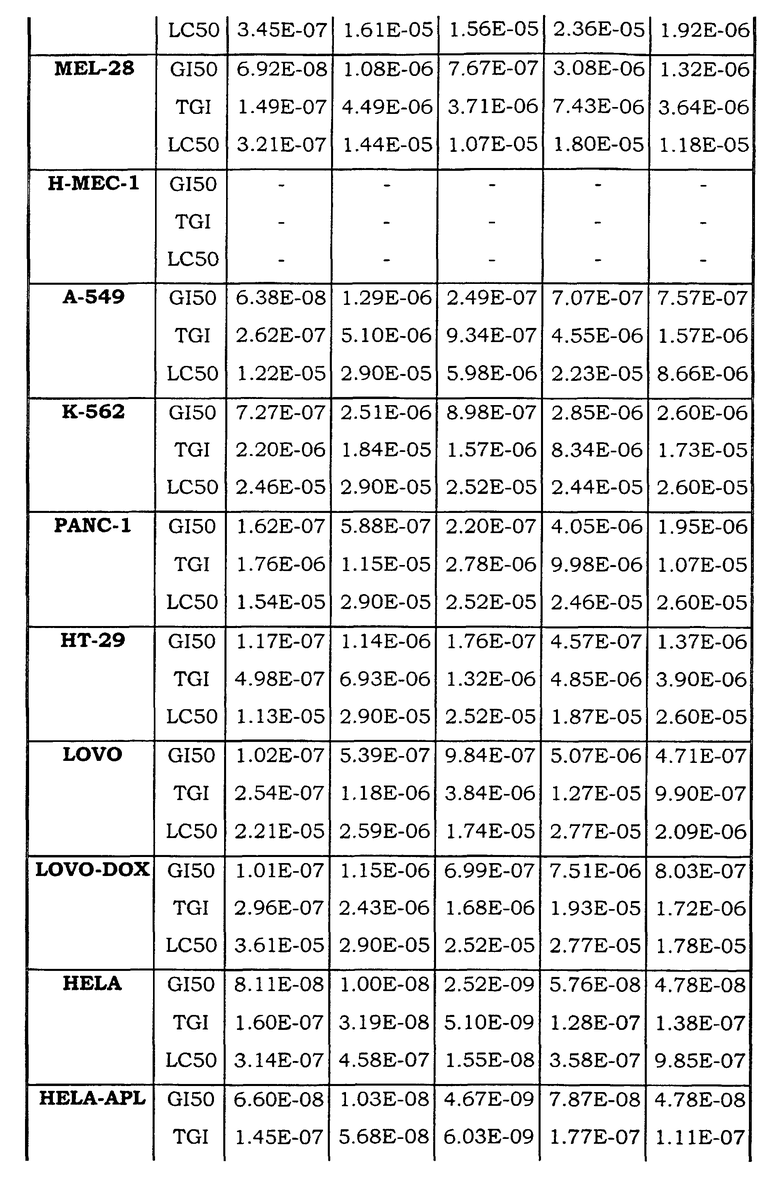

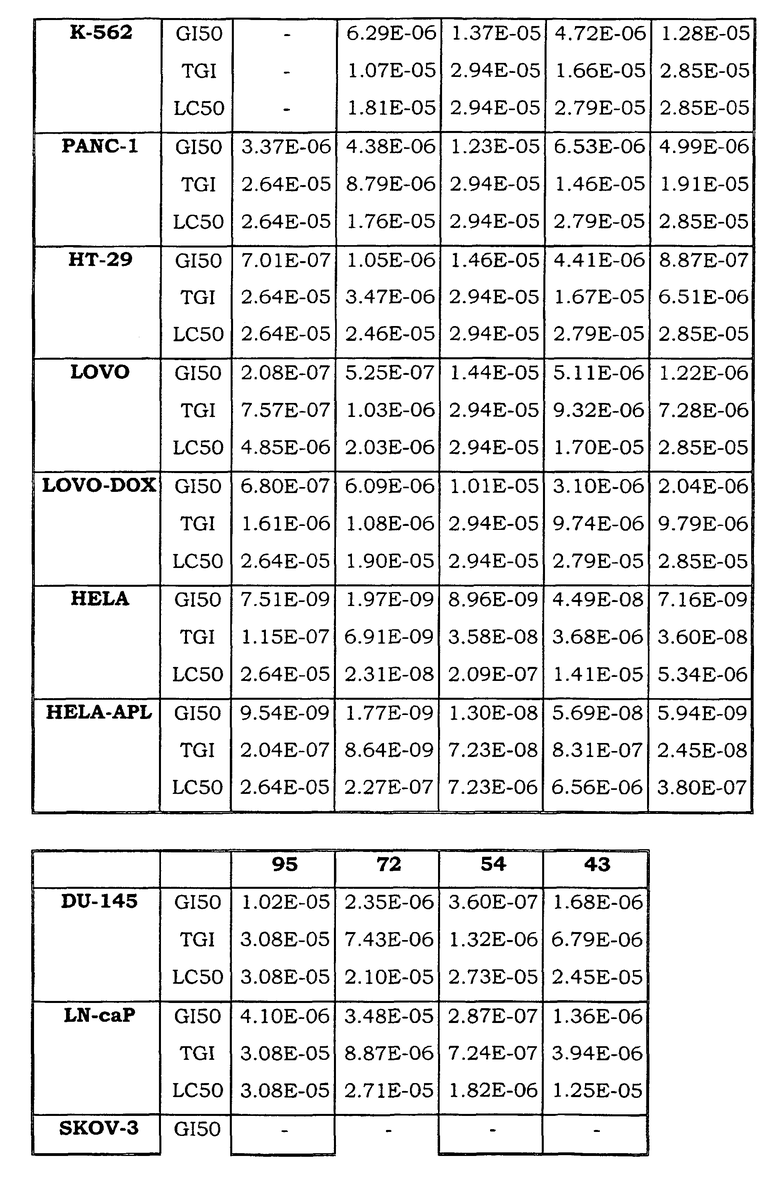
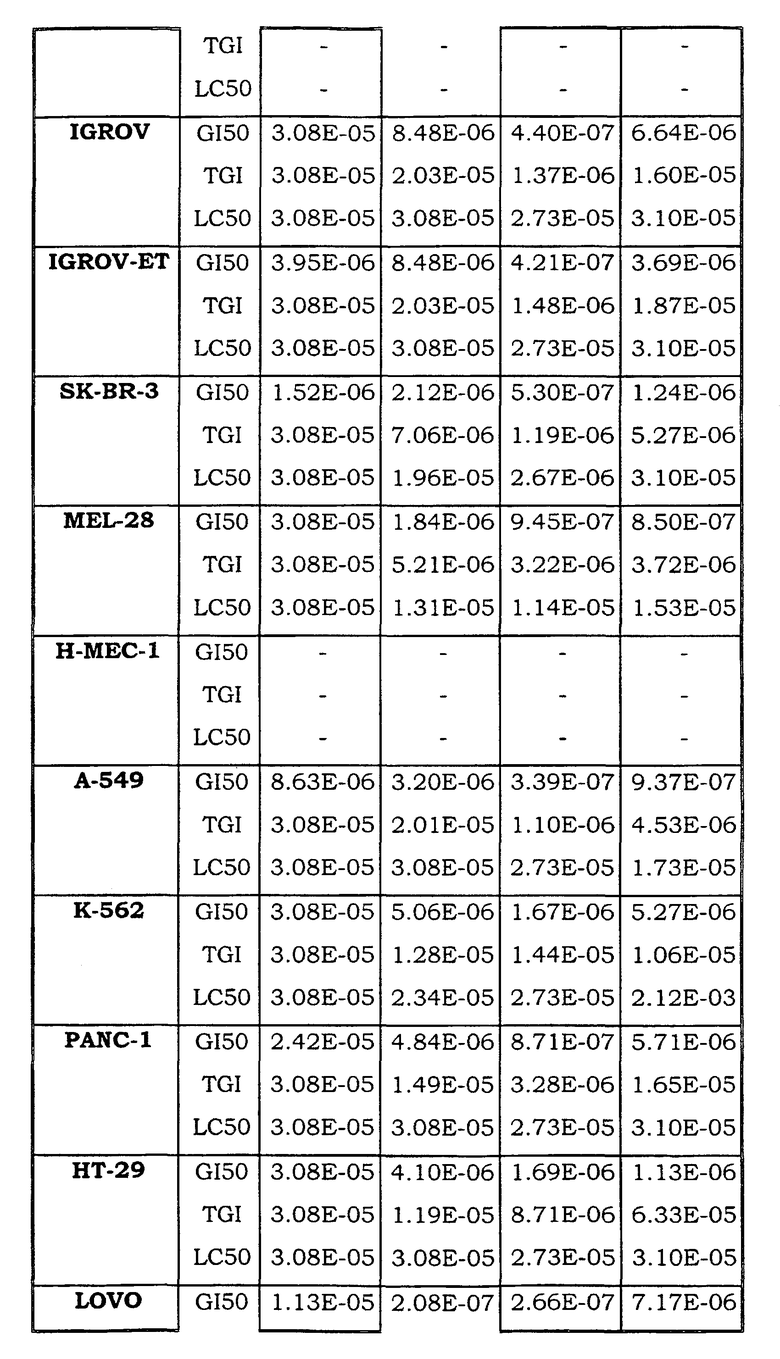
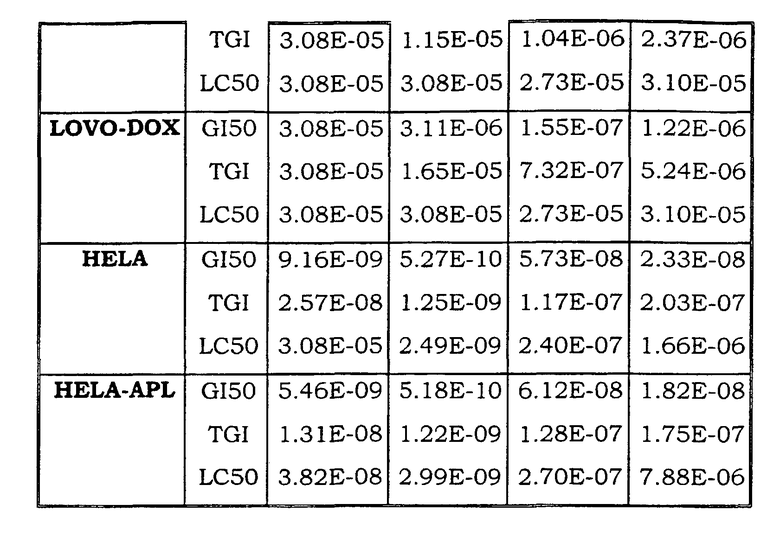
ССЫЛКИ
Bergeron, R.J.; Cavanaugh, P.F. Jr.; Kline, S.J.; Hughes, R.G. Jr.; Elliott, G.T. and Porter C.W. (1984). Antineoplastic and antiherpetic activity of spermidine catecholamide irons chelators. Biochemical and Biophisical Research Communications, 121 (3): 848-854.
Faircloth, G.T.; Stewart, D. and Clement, J.J. (1988). A simple screening procedure for the quantitative measurement of cytotoxicity assay. Journal of Tissue and Culture Methods, 11 (4): 201-205.
Monks, A.; Scudiero, D.; Skehan, Ph.; Shoemaker, R.; Paull, K.; Vistica, D.; Hose, C.; Langley, J.; Cronise, P.; Vaigro-Wolf, A.; Gray-Goodrich, M.; Campbell, H.; Mayo, J.; Boyd, M. (1991). Feasibility of a high-flux anticancer drug screen using a diverse panel of cultured human tumor cell lines. Articles, 83 (11): 757-766.
Mosmann, T. (1983). Rapid colorimetric assay for cellular growth and survival: application to proliferation and cytoto-xicity assays. Journal of Immunological Methods, 65: 55-63.
Skehan, P.A. et al. (1990). New colorimetric cytotoxicity assay for anticancer drug screening. J. Natl. Cancer Inst., 82: 1107-1112.
Schroeder, A.C.; Hughes, R.C. Jr and Bloch, A. (1981). Synthesis and Biological Effects of Acyclic Pyrimidine Nucleoside Analoges. J. Med. Chem., 24: 1078-1083.
| название | год | авторы | номер документа |
|---|---|---|---|
| 2-ПИРИДИЛЗАМЕЩЕННЫЕ ИМИДАЗОЛЫ В КАЧЕСТВЕ ИНГИБИТОРОВ ALK5 И/ИЛИ ALK4 | 2012 |
|
RU2612958C2 |
| ПРОТИВООПУХОЛЕВЫЕ АНАЛОГИ ЛАМЕЛЛАРИНА | 2003 |
|
RU2328500C2 |
| ОБЩИЙ СИНТЕЗ МИРИАПОРОНОВ | 2003 |
|
RU2328492C2 |
| 4-ГИДРОКСИБЕНЗОМОРФАНЫ | 2005 |
|
RU2415131C2 |
| 4-ГИДРОКСИБЕНЗОМОРФАНЫ | 2005 |
|
RU2480455C2 |
| ИНГИБИТОРНЫЕ СОЕДИНЕНИЯ | 2013 |
|
RU2673079C2 |
| АНТИПРОЛИФЕРАТИВНЫЕ СОЕДИНЕНИЯ И ИХ ПРИМЕНЕНИЯ | 2019 |
|
RU2811969C2 |
| СПОСОБ ПОЛУЧЕНИЯ 6-АРИЛ-4-АМИНОПИКОЛИНАТОВ | 2010 |
|
RU2525918C2 |
| ГЕТЕРОЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ, МОДЕЛИРУЮЩИЕ АКТИВНОСТЬ ХЕМОКИНОВОГО РЕЦЕПТОРА, ИХ ПРИМЕНЕНИЕ И СОДЕРЖАЩАЯ ИХ ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 2001 |
|
RU2297413C2 |
| ПЕСТИЦИДНЫЕ КОМПОЗИЦИИ И ОТНОСЯЩИЕСЯ К НИМ СПОСОБЫ | 2013 |
|
RU2623233C2 |
Настоящее изобретение относится к производным вариолина формулы (5а), где R1 выбран из группы, состоящей из галогена и С1-С12алкила; R2 выбран из водорода и галогена; R3, R4, R4, R5; R6 и R7 представляют собой водород; X1 является аминогруппой, незамещенной или замещенной C1-С12алкоксигруппой бензиламиногруппой, С1-С12алкилсульфинильной группой или С1-С12тиоалкилом; Х2 является аминогруппой, C1-С12тиоалкилом, С1-С12алкилсульфинильной группой или незамещенной или замещенной С1-С12алкоксигруппой бензиламиногруппой; или пары групп R1 и R2 могут быть объединены в конденсированную бензольную кольцевую систему, которая может быть замещена С1-С12алкоксигруппой. Вышеописанные соединения обладают противоопухолевым действием. Кроме того, объектами изобретения являются фармацевтическая композиция на основе этих соединений, применение этих соединений для получения лекарственного средства для примения при лечении рака и способ получения промежуточного вариолинового соединения. 4 н. и 12 з.п. ф-лы, 2 табл.
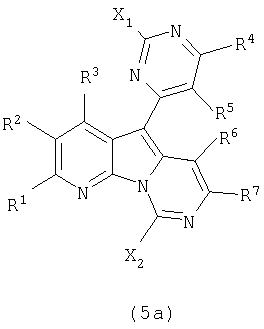
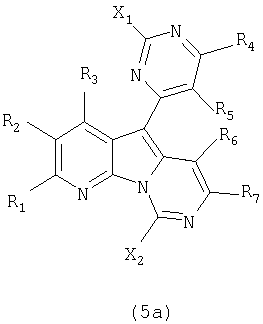
где R1 выбран из группы, состоящей из галогена и С1-С12алкила;
R2 выбран из водорода и галогена;
R3, R4, R5, R6 и R7 представляют собой водород;
X1 является аминогруппой, незамещенной или замещенной C1-С12алкоксигруппой бензиламиногруппой, С1-С12алкилсульфинильной группой или С1-С12тиоалкилом;
Х2 является аминогруппой, С1-С12тиоалкилом, С1-С12алкилсульфинильной группой, или незамещенной или замещенной С1-С12алкоксигруппой бензиламиногруппой;
или пары групп R1 и R2 могут быть объединены в конденсированную бензольную кольцевую систему, которая может быть замещена C1-С12алкоксигруппой.
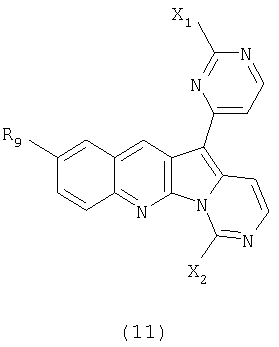
причем X1 и Х2 имеют значения, определенные в предыдущих пунктах, R9 выбирают из водорода или С1-С12алкокси.
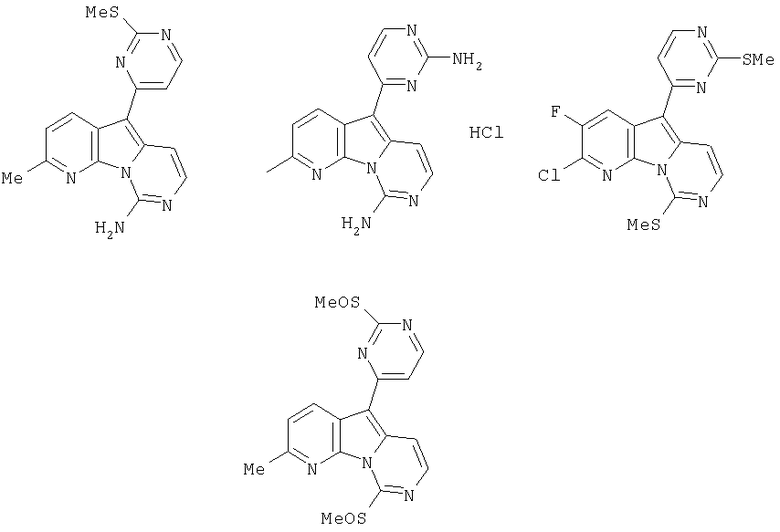
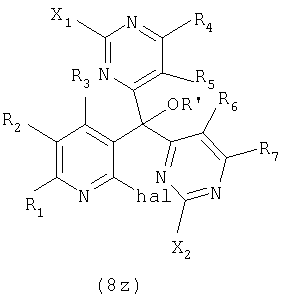
где hal является галогеном, и значения X1, Х2, R1, R2, R3, R4, R5, R6 и R7 определены в предыдущих пунктах, и R' представляет собой Н или Ас, заключающийся во взаимодействии соединения формулы (31z)
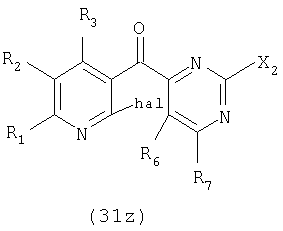
где значения Х2, R1, R2, R3, R6, R7 и hal определены в этом пункте, с соединением формулы (13z)
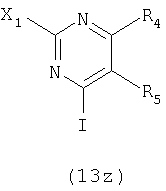
где значения X1, R4 и R5 определены в этом пункте.
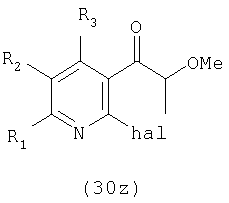
где значения hal, R1, R2 и R3 определены в пункте 15, с соединением формулы (13у)
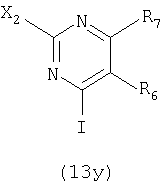
где значения Х2, R6 и R7 определены в пункте 15.
| MERCEDES ALVARES et al | |||
| Synthesis of deoxyvariolin B | |||
| Tetrahedron Letters | |||
| Перекатываемый затвор для водоемов | 1922 |
|
SU2001A1 |
| REGAN J | |||
| ANDERSON et al | |||
| Studies toward the total synthesis of the variolins: rapid entry to the core structure | |||
| Tetrahedron Letters | |||
| Перекатываемый затвор для водоемов | 1922 |
|
SU2001A1 |
| PILAR M | |||
| FRESNEDA et al | |||
| Аппарат для очищения воды при помощи химических реактивов | 1917 |
|
SU2A1 |

