Настоящая заявка заявляет приоритет заявки США 60/625348, находящейся в процессе одновременного рассмотрения, поданной 5 ноября 2004 г., полное раскрытие которой включено в настоящее описание в качестве ссылки.
Изобретение относится к 4-гидроксибензоморфанам, замещенным в положении 3 карбоксамидом или тиокарбоксамидом. Соединения могут быть использованы как анальгетики, антидиарейные агенты, противосудорожные средства, противозудные средства, антикокаиновые лекарства и средства, снижающие зависимость от чрезмерного употребления лекарственных средств.
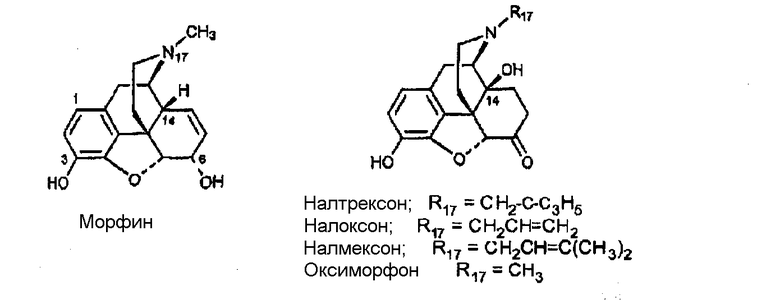
Опиаты были предметом широких исследований с момента выделения морфина в 1805 г., и идентифицированы тысячи соединений, обладающих опиатным или опиатоподобным действием. Многие взаимодействующие с опиатными рецепторами соединения, включая те, что используются для производства анальгетиков (например, морфин), и те, что используются для лечения зависимости от наркотиков (например, налтрексон и циклазоцин) используются для лечения людей. Почти все терапевтически используемые опиоиды в классах бензазоцина и морфинана содержат фенольную гидроксильную группу (ОН) в положении, которое обозначают «8» в числовой системе, используемой для 2,6-метано-3-бензазоцинах [например, циклазоцин и ЕКС (этилкетоциклазоцин)], и которая обозначена «3» в числовой системе, используемой для морфинанов (например, морфин).
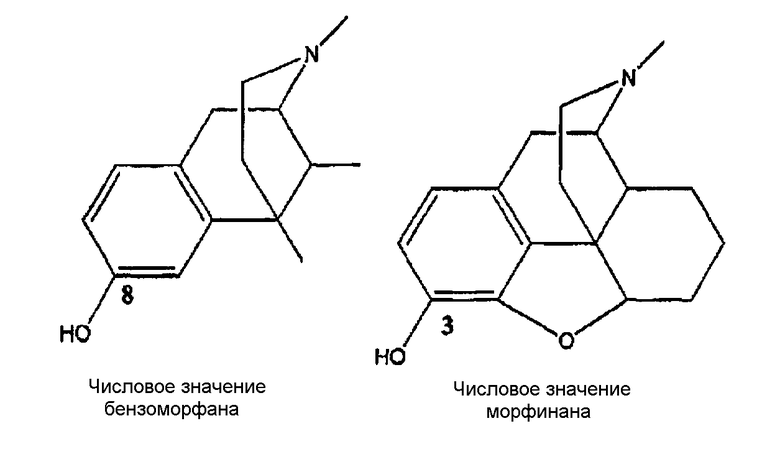
Хотя соединения настоящего изобретения не содержат фуранового кольца морфинанов, будет использована числовая система обозначения морфинана;
2,6-метано-3-бензазоцины также известны как бензоморфаны, и эта терминология будет использована попеременно в тексте настоящего описания.
До публикаций Wentland et al., BioOrg. Med. Chem. Lett. 11, 623-626 (2001) и BioOrg. Med. Chem. Lett. 11, 1717-1721 (2001)] общепринятая практика в данной области за последние семьдесят лет была основана на том, что извлечение или замещение фенольной 3-гидроксильной группы приводило к получению фармакологически неактивных соединений.
Авторами изобретения в настоящее время установлено, что когда 3-гидроксильная группа замещается рядом небольших, полярных, нейтральных остатков, таких как карбоксамидные и тиокарбоксамидные группы, соседнее 4-положение может быть замещено гидроксильной группой с получением соединений с необычайно высоким сродством к опиоидному рецептору. Поэтому соединения настоящего изобретения могут быть использованы как анальгетики, противозудные средства, антидиарейные агенты, противосудорожные средства, противокашлевые средства, аноретики и средства для снижения веса и как лечебные средства для лечения гипералгезии, зависимости от чрезмерного употребления лекарственных средств, угнетения дыхания, дискенизии, болей (включая нейропатические боли), синдрома воспаленной толстой кишки и нарушений перистальтики кишечника.
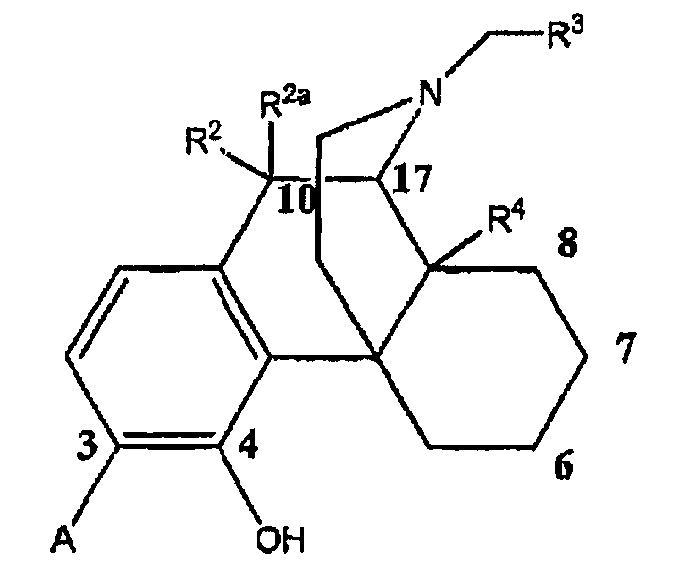
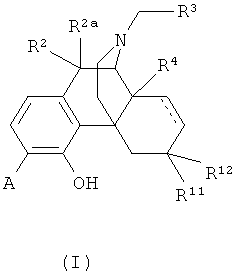
В одном аспекте изобретение относится к соединениям формулы 1:
Соединение формулы:
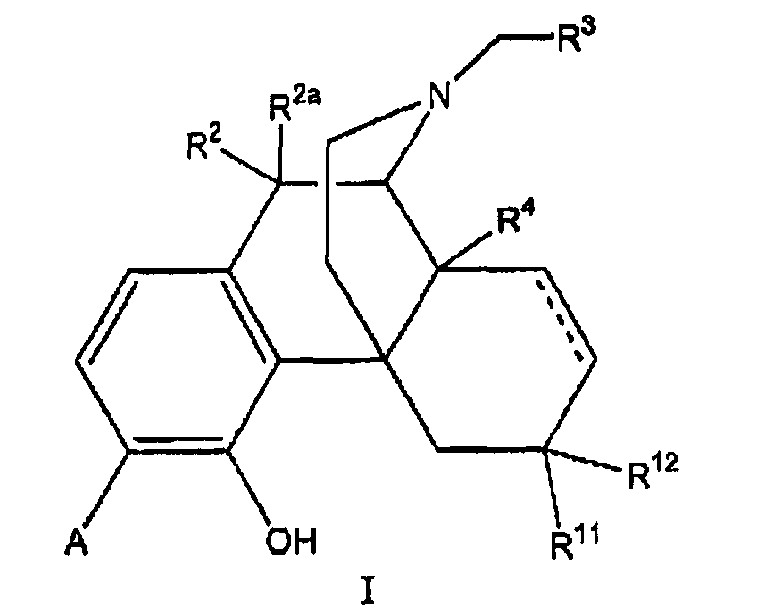
в которой
А выбран из -C(=О)NH2 и -C(=S)NH2;
R2 и R2a оба представляют собой атом водорода или, взятые вместе, R2 и R2a представляют собой =О;
R3 выбран из атома водорода, низшего алкила, алкенила, арила, гетероциклила, бензила и гидроксиалкила;
R4 выбран из атома водорода, гидрокси, амино, низшей алкокси, С1-С20алкила и С1-С20алкила, замещенного гидроксилом или карбонилом;
R11 представляет собой атом водорода;
R12 выбран из атома водорода, гидрокси, низшей алкоксигруппы и -NR13R14; или
радикалы R11 и R12 вместе образуют карбонильный или виниловый заместитель;
R13 и R14 выбраны независимо из атома водорода и C1-C7 углеводородной группы, и
пунктирными линиями обозначены необязательные двойные связи.
В другом аспекте изобретение относится к способам лечения болезней или состояний изменением отклика, обусловленного опиоидным рецептором. Способ включает осуществление контакта соединения формулы I с опиоидным рецептором. Болезни и состояния, которые поддаются лечению соединениями настоящего изобретения, включают боли, зуды, диарею, синдром раздраженной толстой кишки, нарушения перистальтики кишечника, ожирение, угнетение дыхания, конвульсии, кашель, гипералгезию и зависимость от чрезмерного употребления лекарственных средств. Зависимости от чрезмерного употребления лекарственных средств, как использовано в настоящем описании, включают алкогольную, никотиновую, опиатную и кокаиновую зависимости. Это подтверждается литературными данными, согласно которым соединения могут также быть использованы как иммуносупрессоры и противовоспалительные агенты и для снижения ишемического повреждения (и кардиозащиты), для улучшения обучаемости и памяти и для лечения недержания мочи.
Из многолетних SAR исследований известно, что гидроксил морфинанов и бензоморфанов взаимодействует по определенному положению в опиатном рецепторе. Предварительная оценка доступности данного положения для функциональных групп, отличающихся от фенольных гидроксилов, почти повсеместно привела к полной или почти полной потере опиоидного связывания. Авторы изобретения ранее сообщали (WO 02/36573), что гидроксил может быть замещен одним из нескольких биоизостеров. Хотя довольно широкий ряд первичных и вторичных карбоксамидов, а также карбоксилаты, аминометил, гидроксиметил и даже дигидроимидазолил показали связывание в желательном интервале ниже 25 нанометров, оптимальную активность наблюдали с карбоксамидо-, тиокарбоксамидо-, гидроксиамидино- или формамидогруппами. Авторы изобретения сейчас установили, что бензоморфаны, содержащие гидроксил в положении 4, и биоизостеры «А» в положении 3 обладают неожиданным уровнем опиоидной активности.
Фенольные 3-гидроксильные функциональные группы бензоморфанов и морфинанов могут быть химически превращены в амидные простым, гибким и удобным путем, описанным в WO 02/36573 и в WO 2004/007449, а тиокарбоксамиды, гидроксиамидины и формамидные соединения также легко могут быть синтезированы, как описано в данных ссылках. Предпочтительными остатками А являются -C(=O)NH2 и -C(=S)NH2.
Известно, что соединения, которые являются µ, δ и κ агонистами, проявляют анальгетическое действие; соединения, которые являются селективными µ агонистами, проявляют антидиарейное действие и могут быть использованы при лечении дискинезии; µ антагонисты и κ агонисты могут быть использованы при лечении героиновой, кокаиновой, алкогольной и никотиновой зависимости; κ агонисты являются также противозудными агентами и могут быть использованы при лечении гипералгезии. В общем, правовращающие изомеры морфинанов могут быть использованы в качестве противокашлевых агентов и противоконвульсивных средств.
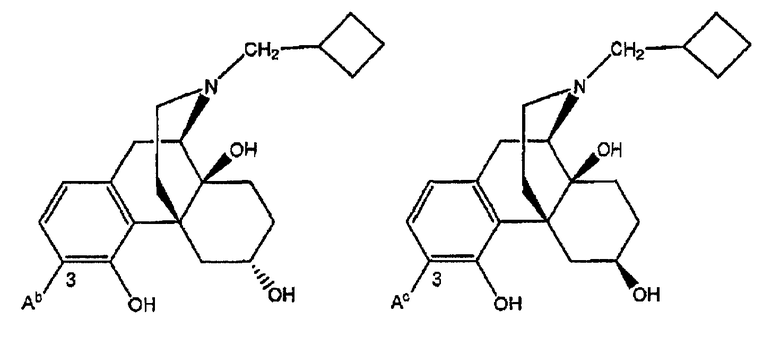
Примеры лигандов опиоидных рецепторов, обладающих известной высокой активностью, показаны на следующей диаграмме.
Замещение группы  на группу
на группу 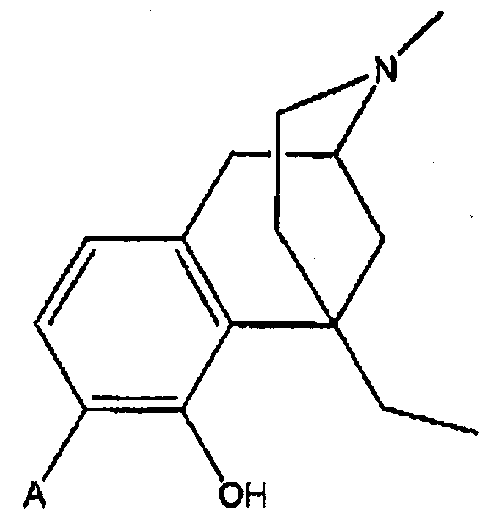 в соединениях диаграммы приводит к получению соединений, которые проявляют сильное сродство к опиоидным рецепторам.
в соединениях диаграммы приводит к получению соединений, которые проявляют сильное сродство к опиоидным рецепторам.
Диаграмма. Лиганды опиоидных рецепторов морфина и морфинанов
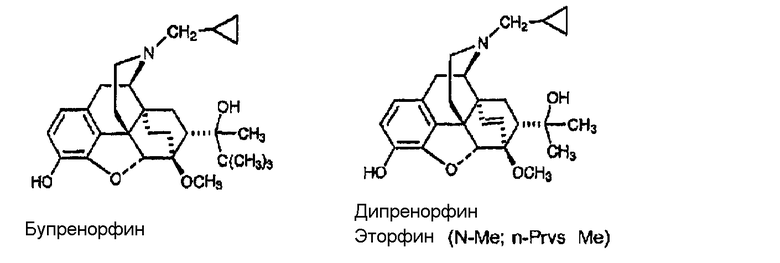
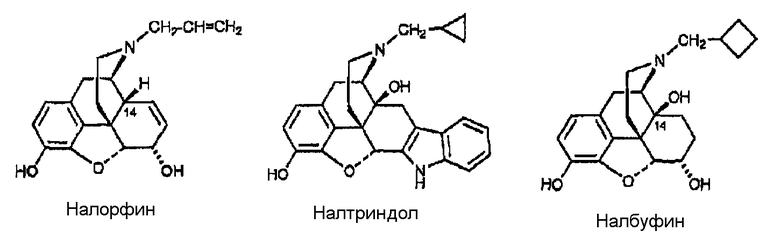
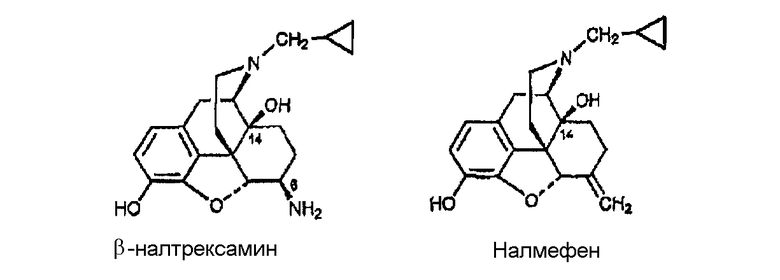
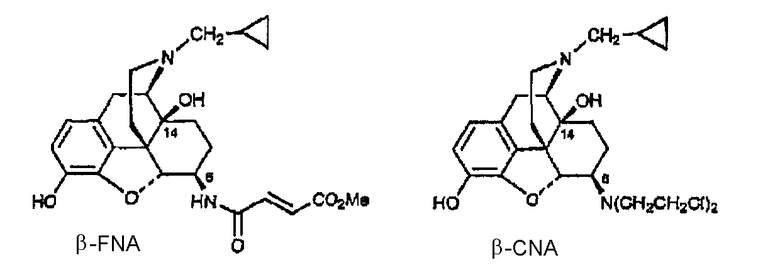
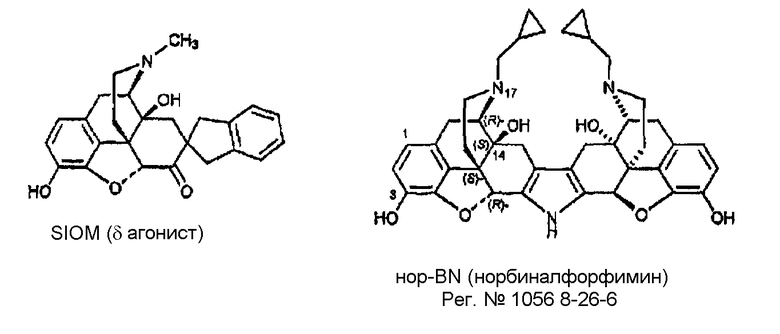
Другие опиоидные рецепторы описаны в публикации Aldrich, J.V. “Analgesics” в Burrer's Medicinal Chemistry and Drug Discovery, M.E. Wolf ed., John Wiley & Sons 1996, стр. 321-44, раскрытие которой включено в настоящее описание в качестве ссылки.
Сродство соединений согласно изобретению определяли методом, описанным в публикации Wentland et. al. [BioOrg. Med. Chem. Lett. 9, 183-187 (2000)]. Антиноцицепторую активность оценивают методом, описанным в публикации Jiang et al. [J. Pharmacol. Exp. Ther. 264, 1021-1027 (1993), page 1022] или методом, описанным в публикации Neumeyer et al. [J. Med. Chem. 46, 5162 (2003)]. Авторы изобретения изучили связывание рецепторов соединениями формулы I в серии аналогов известным соединениям, в которых ОН замещена группой А и гидроксил введен рядом с А группой. Результаты представлены в таблицах 1, 2, 3 и 4. Результаты для использованных стандартов также представлены в таблицах. Результаты данных тестов in vitro принимаются специалистами в данной области как прогноз терапевтического использования in vivo.
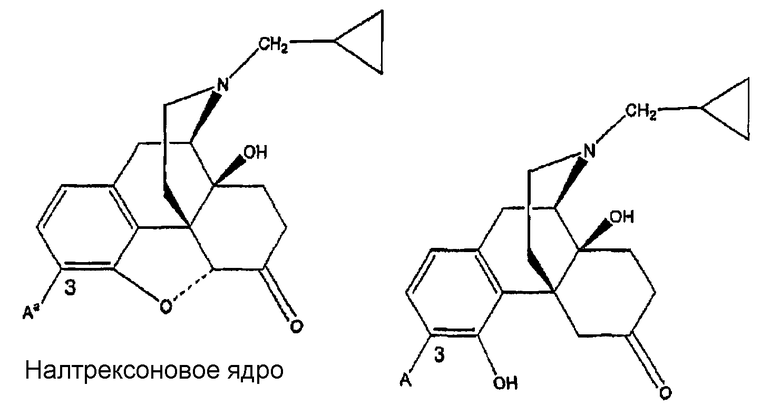
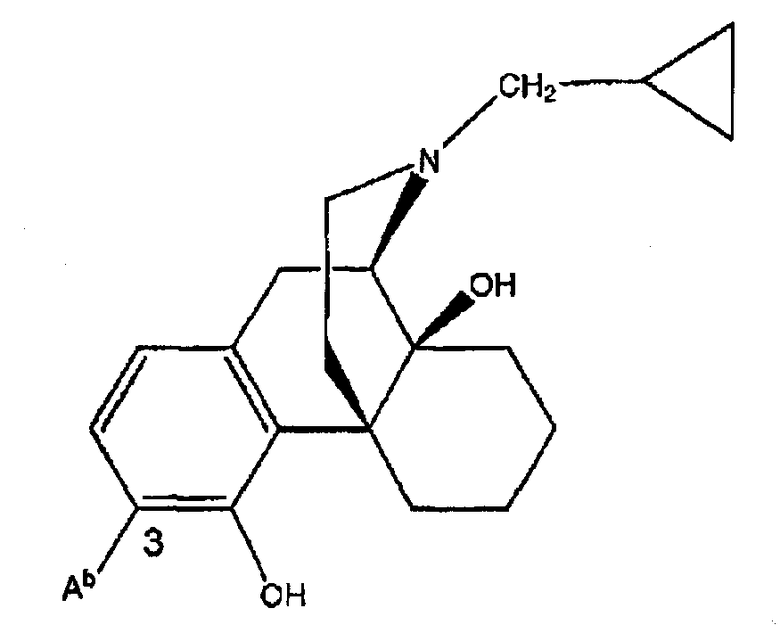
Ki(нМ±ср.кв.от.)
(налтрексон)
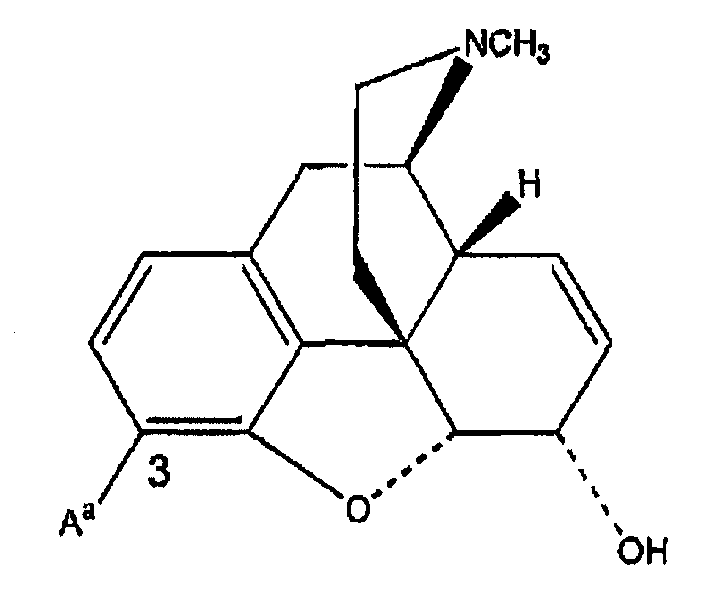
Морфиновое ядро
Ki(нМ±ср.кв.от.)
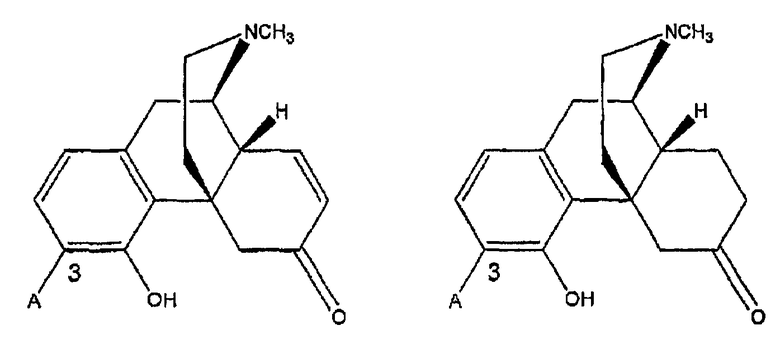
Производные оксиморфона
Ki(нМ±ср.кв.от.)
7,8-дигидро
7,8-дигидро
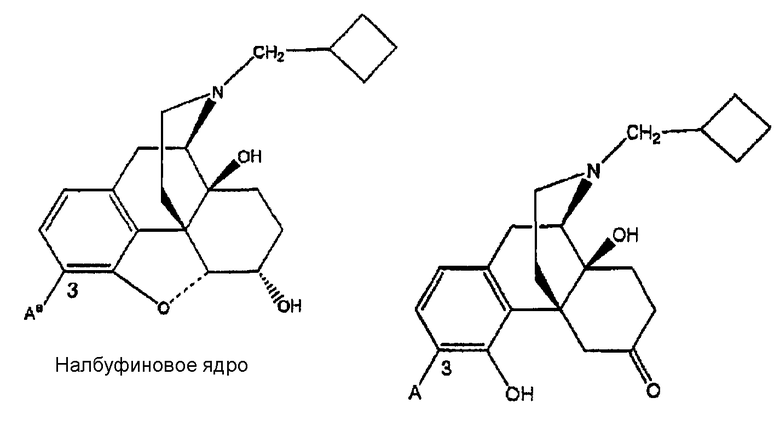
Ki(нМ±ср.кв.от.)
Определения
По всему тексту описания термины и заместители сохраняют свои определения.
Подразумевается, что алкил включают линейные, разветвленные или циклические углеводородные структуры и их комбинации. Низший алкил относится к алкильным группам, содержащим от 1 до 6 атомов углерода. Примеры низших алкильных групп включают метил, этил, пропил, изопропил, циклопропил, бутил, втор- и трет-бутил, циклопропил, циклобутил и т.п. Предпочтительными алкильными группами являются те, которые содержат С20 или ниже. Циклоалкил представляет собой подкласс алкильных групп и включает циклические углеводородные группы, содержащие от 3 до 8 атомов углерода. Примеры циклоалкильных групп включают ц-пропил, ц-бутил, ц-пентил, норборнил и т.п.
Алкокси или алкоксил относится к группам, содержащим от 1 до 8 атомов углерода линейной, разветвленной, циклической конфигурации и их комбинациям, присоединенным к основной структуре через атом кислорода. Примеры включают метокси, этокси, пропокси, изопропокси, циклопропилокси, циклогексилокси и т.п. Низшая алкоксигруппа относится к группам, содержащим от одного до четырех атомов углерода.
Арил и гетероарил означает 5- или 6-членное ароматическое или гетероароматическое кольцо, содержащее 0-3 гетероатома, выбранные из O, N или S; бициклическую 9- или 10-членную ароматическую или гетероароматическую кольцевую систему, содержащую 0-3 гетероатома, выбранные из O, N или S; или трициклическую 13- или 14-членную ароматическую или гетероароматическую кольцевую систему, содержащую 0-3 гетероатома, выбранные из O, N или S. Ароматические 6-14-членные карбоциклические кольца включают, например, бензол, нафталин, индан, тетралин и флуорены, и 5-10-членные ароматические гетероциклические кольца включают, например, имидазол, пиридин, индол, тиофен, бензопиранон, тиазол, фуран, бензимидазол, хинолин, изохинолин, хиноксалин, пиримидин, пиразин, тетразол и пиразол.
Арилалкил означает алкильный остаток, присоединенный к арильному кольцу. Примерами являются бензил, фенетил и т.п. Гетероарилалкил означает алкильный остаток, присоединенный к гетероарильному кольцу. Примеры включают, например, пиридинилметил, пиримидинилэтил и т.п.
Гетероцикл означает циклоалкильный или арильный остаток, в котором от одного до двух атомов углерода замещены гетероатомом, таким как кислород, азот или сера. Гетероарилы образуют подкласс гетероциклов. Примеры гетероциклов, которые попадают в объем притязаний изобретения, включают пирролидин, пиразол, пиррол, индол, хинолин, изохинолин, тетрагидроизохинолин, бензофуран, бензодиоксан, бензодиоксол (обычно называемый метилендиоксифенил, когда встречается в качестве заместителя), тетразол, морфолин, тиазол, пиридин, пиридазин, пиримидин, тиофен, фуран, оксазол, оксазолин, изоксазол, диоксан, тетрагидрофуран и т.п.
Замещенные алкил, арил, циклоалкил или гетероциклил относятся к алкилу, арилу, циклоалкилу или гетероциклилу, в котором до трех атомов Н в каждом остатке замещены галогеном, гидроксигруппой, низшей алкоксигруппой, карбокси, карбоалкокси, карбоксамидо, циано, карбонилом, -NO2, -NR1R2, алкилтио, сульфоксидом, сульфоном, ациламино, амидино, фенилом, бензилом, гетероарилом, фенокси, бензилокси, гетероарилокси или замещенным фенилом, бензилом, гетероарилом, фенокси, бензилокси или гетероарилокси.
Безусловно, все соединения, описанные в данном описании, содержат один или несколько центров асимметрии и, таким образом, могут способствовать возникновению энантиомеров, диастереомеров и других стереоизомерных форм, которые могут быть определены в терминах абсолютной стереохимии как (R) и (S). Подразумевается, что настоящее изобретение включает все такие возможные изомеры, а также их рацематы и оптически чистые формы. В общем, было установлено, что левовращающий изомер морфинанов и бензоморфанов является наиболее вероятным антиноцицепторным агентом, тогда как правовращающий изомер может быть использован как противокашлевый или противоспазматический агент. Оптически активные (R) и (S) изомеры могут быть получены с использованием хиральных синтонов или хиральных реагентов, или выделены с помощью обычных технологий. Когда описанные в настоящем описании соединения содержат олефиновые двойные связи или другие центры геометрической асимметрии, и если не указанно иначе, то подразумевается, что соединения включают оба E и Z геометрических изомера. Аналогично этому, подразумевается, что включены все таутомерные формы.
Как использовано в тексте настоящего описания и как будет понятно специалистам в области медицины, для которых предназначено настоящее изобретение, упоминание соединения включает фармацевтические приемлемые соли, гидраты, сольваты, клатраты и полиморфы. Термин «фармацевтическая приемлемая соль» относится к солям, полученным из фармацевтических приемлемых нетоксичных кислот или оснований, включающих неорганические кислоты и основания и органические кислоты и основания. Соли могут быть получены из фармацевтических приемлемых нетоксичных кислот, включающих неорганические и органические кислоты. Подходящие фармацевтические приемлемые аддитивные соли кислот для соединений настоящего изобретения включают соли уксусной, бензолсульфоновой (безилат), бензойной, камфорсульфоновой, лимонной, этенсульфоновой, фумаровой, глюконовой, глутаминовой, бромистоводородной, соляной, изетионовой, молочной, малеиновой, малоновой, миндальной, метансульфоновой, слизевой, азотной, памовой, пантотеновой, фосфорной, янтарной, серной, щавелевой кислот, п-толуолсульфоновой и т.п. кислот. Термин «сольват» относится к соединению - в данном случае эсзопиклону - в твердом состоянии, где молекулы подходящего растворителя вводят в форме кристаллов. Подходящим растворителем для терапевтического введения является физиологически толерантный при дозированном введении. Примерами подходящих растворителей для терапевтического введения являются этанол и вода. Когда растворителем является вода, то сольват называют гидратом. В общем, сольваты образуются при растворении соединения в соответствующем растворителе и выделении сольвата охлаждением или применением антирастворителя. Сольват обычно высушивают или получают азеотропную смесь в условиях окружающей среды.
Термин «превентивный», использованный в тексте настоящего описания, относится к введению лекарственного средства заранее, чтобы предупредить или прекратить приступ. Специалисты в области медицины (для которых предназначены пункты формулы изобретения, касающиеся способа) поймут, что термин «превентивный» не является абсолютным термином. В области медицины его понимают как профилактическое введение лекарства для существенного подавления признаков или степени тяжести состояния, и этот смысл вложен в формулировку формулы изобретения заявителя. Термин «лечение» включает профилактику, а также уменьшение интенсивности острых симптомов. Следует обратить внимание, что термин «лечение» относится либо к одному, либо к двум действиям: уменьшению интенсивности симптомов и улучшению сопутствующего состояния. При многих состояниях изобретения введение опиоида может действовать не прямо на состояние болезни, но скорее на какой-то пернициозный симптом, и облегчение данного симптома приводит к общему и желательному уменьшению интенсивности болезненного состояния.
Хотя настоящее изобретение может быть осуществлено в различных формах, но показаны предпочтительные варианты его осуществления. Однако следует понимать, что настоящее раскрытие следует рассматривать как пояснительное для принципов настоящего изобретения, а не предназначенное ограничивать изобретение до представленных вариантов изобретения. При рассмотрении изобретения можно видеть, что некоторые элементы заявленной ограничительной части не являются патентоспособными для авторов изобретения в настоящей заявке. В данном случае последующее исключение соединений из объема формулы изобретения, представленной заявителем, следует рассматривать как артефакты для рассмотрения патентоспособности, и не отражают авторской концепции или описания их изобретения; изобретение охватывает все элементы ограничительной части I, которые уже не являются достоянием общественности.
Сокращения
Следующие сокращения и термины имеют указанные значения во всем тексте описания:
Ac = ацетил
AcOH = уксусная кислота
BNB = 4-бромметил-3-нитробензойная кислота
Boc = бутилоксикарбонил
Bu = бутил
С- = цикло
DAMGO = Tyr-ala-Gly-NMePhe-NHCH2OH
DBU = диазабицикло[5.4.0]ундец-7-ен
DCM = дихлорметан = метиленхлорид = CH2Cl2
DEAD = диэтилазодикарбоксилат
DIC = диизопропилкарбодиимид
DIEA = N,N-диизопропилэтиламин
DMAP = 4-N,N-диметиламинопиридин
DMF = N,N-диметилформамид
DMSO = диметилсульфоксид
DPPF = 1,1'-бис(дифенилфосфино)ферроцен
DVB = 1,4-дивинилбензол
EEDQ = 2-этокси-1-этоксикарбонил-1,2-дигидрохинолин
Et3N = триэтиламин
EtOAc = этилацетат
Fmoc = 9-флуоренилметоксикарбонил
GC = Газовая хроматография
HATU = гексафторфосфат O-(7-азабензотриазол-1-ил)-1,1,3,3-тетраметилурония
HOBt = гидроксибензотриазол
Me = метил
Mesyl = метансульфонил
MTBE = метил-трет-бутиловый эфир
NMO = оксид N-метилморфолина
PEG = полиэтиленгликоль
Ph = фенил
PhOH = фенол
PhN(Tf)2 = N-фенилтрифторметансульфонимид
PfP = пентафторфенол
PPTS = п-толуолсульфонат пиридиния
PyBroP = гексафторфосфат бром-трис-пирролидинофосфония
Rt = комнатная температура
Sat'd = насыщенный
s- = вторичный
t- = третичный
Tf = инфлат, CF3SO2O-
TBDMS = т-бутилдиметилсилил
TFA = трифторуксусная кислота
THF = тетрагидрофуран
TMOF = триметилортоформиат
TMS = триметилсилил
Tosyl = п-толуолсульфонил
Trt = трифенилметил
В тексте описания повсеместно встречается терминология, касающаяся «введения защитных групп», «отщепления защитных групп» и «защищенных» функциональных групп. Данная терминология хорошо понятна специалистам в данной области в контексте процессов, которые включают последовательную обработку рядом реагентов. В таком контексте защитная группа относится к группе, которая использована для маскирования функциональных групп на стадии процесса, на которой в противном случае она будет взаимодействовать, но на которой реакция является нежелательной. Защитная группа предотвращает взаимодействие на этой стадии, но может быть после этого удалена с восстановлением исходной функциональности. Удаление или «удаление защитных групп» происходит после завершения реакции или реакций, в которых функциональные группы могут участвовать. Таким образом, когда определена последовательность реагентов, как это сделано в способах настоящего изобретения, специалист в данной области может легко определить те группы, которые будут подходить в качестве «защитных групп». Подходящие группы для этой цели рассмотрены в стандартном учебнике в области химии, таком как «Protective Groups in Organic Synthesis», T.W. Green [John Wiley & Sons, New York, 1991], который включен в настоящее описание в качестве ссылки.
Следующие примеры поясняют синтез различных соединений настоящего изобретения, имеющих формулу I, многие из которых находятся в таблицах. Остальные соединения, перечисленные в таблицах, были получены аналогичным образом. Кроме того, изобретение не ограничено соединениями, полученными в примерах или указанными в таблицах, и аналогичные операции могут быть использованы для получения дополнительных соединений, имеющих формулу I.
Если не указанно иначе, реактанты и реагенты, использованные в примерах, представляют собой легко доступные материалы. Такие материалы могут быть традиционно получены в соответствии с обычными методами получения или получены из коммерческих источников. Данные 1Н ЯМР обозначены как с (синглет), д (дуплет), т (триплет), к (квартет), м (мультиплет) и уш. (уширенный).
Пример 1
Синтез 3-карбоксиамидо-4-гидроксиналтрексона, производного 3
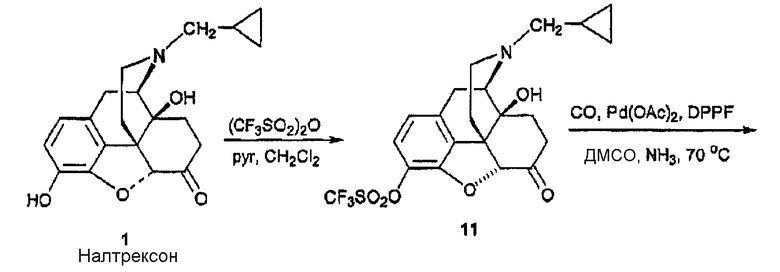
(А) Синтез 3-карбоксиамидоналтрексона 2
Трифлат 11 налтрексона получали в соответствии с методом Wentland et al. (Bioorg. Med. Chem. Lett. 9 183-187 (2000), и карбоксамид 2 получали методом, описанным Wentland et. al. [(Bioorg. Med. Chem. Lett. b, 623-626 (2001); и Bioorg. Med. Chem. Lett. 11, 1717-1721 (2001)], включающим катализированное Pd карбонилирование трифлата 11 в присутствии аммиака и Pd(O) лиганда, DPPF ([1,1'-бис(дифенилфосфино)ферроцен]) и ДМСО.
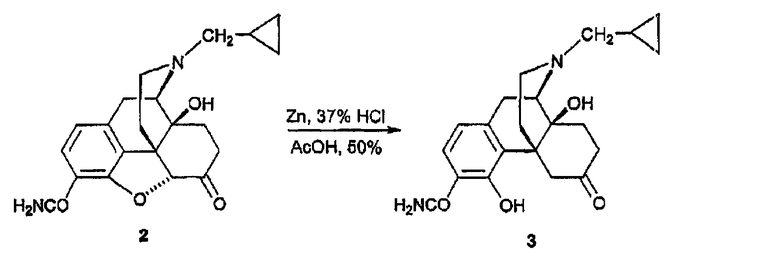
(В) Синтез 3-карбоксиамидо-4-гидроксиналтрексона, производного 3
Порошок цинка (26 мг, 0,40 ммоль) порциями добавляли к раствору 2 (50 мг, 0,14 ммоль) в HCl (37%, 0,2 мл) и AcOH (2 мл) в колбе с обратным холодильником. После кипячения в колбе с обратным холодильником еще в течение 15 минут реакционную смесь охлаждали добавлением смеси лед/вода (10 мл) и подщелачивали (рН=9) с помощью NH3/H2O, раствор экстрагировали BtOAc (3×10 мл). Органические экстракты промывали насыщенным раствором соли, сушили и концентрировали. Остаток очищали колоночной хроматографией (SiO2, CH2Cl2, CH3OH:NH3/H2O = 15:1:0,01) и получали соединение 3 в виде вспененного вещества (25 мг, 50%). 1Н ЯМР (CDCl3) δ 13,28 (c, 1H, 4-OH), 7,15 (д, 1H, J=8,1, H-2), 6,47 (д, 1H, J=8,4, H-1), 6,10 (уш., 1H, N-Н), 4,35 (уш., 1H, N-H), 4,04 (дд, 1H, J=1,8, 13,5, Н-5), 3,1l (д, 1H, J=6), 2,99 (д, 1H, J=5,7), 2,94 (c, 1H), 2,86 (д, 1H, J=6), 2,84-2,75 (м, 2H), 2,65-2,61 (м, 2Н), 2,17-2,05 (м, 1H), 1,89-1,84 (м, 2H), 0,85 (м, 1H), 0,56-0,50 (м, 2H), 0,13-0,09 (м, 2H), [α]D 25=-98,4° (c=0,6, CH2Cl2). MC m/z (ESI) 371 (MH-).
Пример 2
Синтез 3-метокси-4-гидроксиналтрексона, производного 4
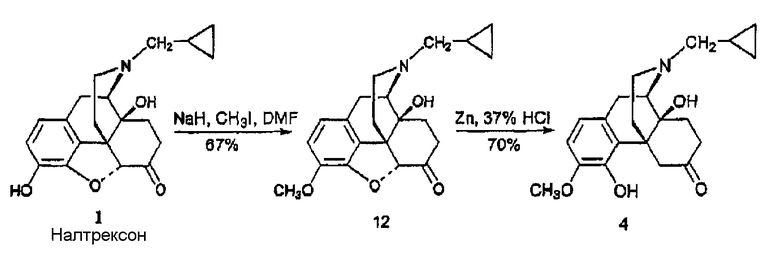
(А) Синтез 3-метоксиналтрексона, производного 12
Используя методику Nan et al., J. Heterocyclic Chem. 34, 1195-1203 (1997), 95%-ый гидрид натрия (22 мг, 0,87 ммоль) добавляли к раствору налтрексона 1 (200 мг, 0,58 ммоль) в сухом DMF (1 мл) при комнатной температуре. После перемешивания в течение 15 мин раствор охлаждали до 5°С на ледяной бане и добавляли метилиодид (40 мкл, 99 мг, 0,70 ммоль). После перемешивания еще в течение 15 мин реакционный раствор концентрировали в вакууме. Остаток очищали флэш-хроматографией (SiO2, CH2Cl2:NH3/H2O=100:1) и получали производное 12 в виде вспененного вещества (131 мг, 67%). 1Н ЯМР (CDCl3) δ 6,69 (д, 1H, J=8,0, H-2), 6,61 (д, 1H, J=8,0, H-1), 4,67 (c, 1H, H-5), 3,89 (с, 3H, 3-OCH3), 3,18 (м, 1H), 3,06 (м, 2Н), 2,99 (c, 1H), 2,87 (c, 1H), 2,70 (м, 1H), 2,59 (м, 1H), 2,40 (м, 2H), 2,41 (м, 2Н), 2,31 (м, 2H), 2,12 (м, 2Н), 1,89 (м, 2H), 1,59 (м, 1Н), 0,87 (м, 1H), 0,55 (м, 2Н), 0,15 (м, 2H), [α]D 25=-181,7° (c-0,12, CH2Cl2). MC m/z (ESI) 356 (MH-).
(В) Синтез 3-метокси-4-гидроксиналтрексона, производного 4
Для этого синтеза использовали модифицированную методику Coop et al., J. Med. Chem. 42, 1673-1679 (1999). Порошок цинка (114 мг, 1,72 ммоль) порциями добавляли к раствору производного 12 (122 мг, 0,34 ммоль) в HCl (37%, 0,2 мл) и AcOH (2 мл) в колбе с обратным холодильником. После кипячения в колбе с обратным холодильником еще в течение 15 минут реакционную смесь охлаждали добавлением смеси лед/вода (20 мл) и подщелачивали (рН=9) с помощью NH3/H2O, раствор экстрагировали EtOAc (3×10 мл). Органические экстракты промывали насыщенным раствором соли, сушили и концентрировали. Остаток очищали колоночной хроматографией (SiO2, CH2Cl2: CH3OH:NH3/H2O = 20:1:0,01) и получали соединение 4 в виде вспененного вещества (85 мг, 70%). 1Н ЯМР (CDCl3) δ 6,67 (д, 1H, J=8,0, H-2), 6,56 (д, 1H, J=8,0, H-1), 6,12 (c, 1H, 4-OH), 3,94 (д, 1H, J=13,0), 3,82 (c, 3H, 3-OCH3), 3,10 (м, 1Н), 2,97 (м, 1Н), 2,80 (м, 2Н), 2,61 (м, 1Н), 2,36 (м, 2Н), 2,15 (м, 1Н), 2,05 (м, 2Н), 1,82 (м, 1H), 0,54 (м, 2Н), 0,12 (м, 2Н), [a]D 25=-96,2° (c=0,5, CH2Cl2). MC m/z (ESI) 888 (MH-).
Пример 3
Синтез 3-метокси-4-гидрокси-6-оксоморфина, производного 7
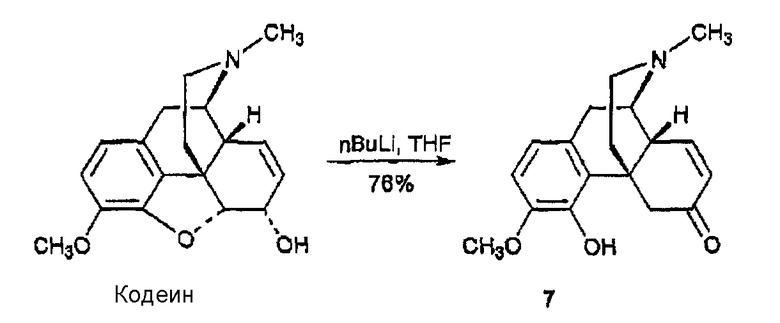
Используя методику Coop et al. (J. Med. Chem. 42, 1673-1679 (1999) и Heterocycles 50, 39-42 (1999), н-бутиллитий (1,52 М в гексане, 1,6 мл, 2,50 ммоль) добавляли к раствору кодеина (150 мг, 0,501 ммоль) в THF при -78°С. После перемешивания при -78°С в течение 1 ч слегка желтый раствор нагревали до комнатной температуры, а затем перемешивали в течение 20 мин. Реакцию останавливали водой (10 мл). Смесь три раза экстрагировали CHCl3. Объединенные органические фазы промывали насыщенным раствором соли, сушили над сульфатом натрия, фильтровали и концентрировали, в результате чего получали твердый остаток, который очищали флэш-хроматографией (CH2Cl2:MeOH:NH4OH 15:1:0,1) и получали дигидросоединение 7 в виде белого вспененного вещества (114 мг, 0,38 ммоль, 76%); 1Н ЯМР (500 МГц, CDCl3) δ 6,68 (дд, 1H, J=10,0, 2,0 Гц), 6,64 (д, 1Н, J=8,0 Гц), 6,55 (д, 1Н, J=8,5 Гц), 6,00 (уш., 1Н), 5,89 (дд, 1Н, J=10,0, 3,0 Гц), 4,26 (д, 1Н, 7=15,5 Гц), 3,81 (c, 3Н), 3,22 (м, 1Н), 3,02 (д, 1Н, J=18,5 Гц), 2,89 (c, 1Н), 2,65 (1Н, 1Н), 2,54 (м, 1Н), 2,43 (c, 3Н), 2,38 (д, 1Н, J=15,0 Гц), 2,07 (м, 1Н), 1,90 (м, 2Н); 13C ЯМР (125 МГц, CDCl3) δ 199,38, 149,53, 144,91, 144,58, 130,75, 130,18, 122,86, 118,10, 108,71, 55,93, 55,80, 48,88, 47,02, 46,95, 42,52, 40,47, 36,19, 24,32; MC (ESI) m/z 300 (M+H)+; Элементный анализ: Рассчитано для C18H21NO3·0,5H2O: C 70,11; H 7,19; N 4,54. Найдено: С 69,94; Н 6,87; N 4,38.
Пример 4
Синтез 3-метокси-4-гидрокси-6-оксо-7,8-дигидроморфина, производного 8
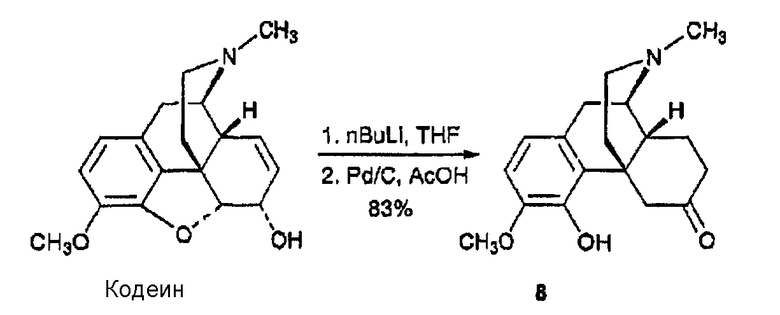
н-Бутиллитий (1,52М в гексане, 1,6 мл, 2,50 ммоль) добавляли к раствору кодеина (150 мг, 0,501 ммоль) в THF при -78°С. После перемешивания при -78°С в течение 1 ч светло-желтый раствор нагревали до комнатной температуры, а затем перемешивали в течение 20 мин. Реакцию останавливали добавлением воды (10 мл). Смесь три раза экстрагировали CHCl3. Объединенные органические фазы промывали насыщенным раствором соли, сушили над сульфатом натрия, фильтровали и концентрировали, в результате чего получали твердый остаток, который растворяли в AcOH (10 мл) и перемешивали с 10% Pd/C (54 мг) в атмосфере водорода (30 фунт/кв. дюйм) в течение 20 ч. Реакционную смесь фильтровали и концентрировали, в результате чего получали не совсем белый остаток, который очищали флэш-хроматографией (CH2Cl2:MeOH:NH4OH 14:1:0,1), в результате чего получали соединение 8 в виде белого твердого вещества (125 мг, 0,415 ммоль, 83%); 1Н ЯМР (500 МГц, CDCl3) δ 6,67 (д, 1H, J=8,0 Гц), 6,60 (д, 1H, J=8,0 Гц), 6,09 (c, 1Н), 4,23 (дд, 1Н, J=13,5, 2,5 Гц), 3,83 (c, 3Н), 2,98 (д, 1Н, J=18,5 Гц), 2,66 (м, 1Н), 2,44 (м, 2Н), 2,42 (c, 3Н), 2,24 (м, 3Н), 2,06 (м, 1Н), 1,86 (м, 3Н), 1,69 (м, 2Н); MC (ESI) m/z 302 (М+Н)+; Элементный анализ: Рассчитано для C18H23NO3·0,5H2O: C 69,65; H 7,79; N 4,51. Найдено: С 70,04; Н 7,68; N 4,39.
Пример 5
Синтез 3-карбоксиамидо-4-гидроксигидрокодона, производного 17
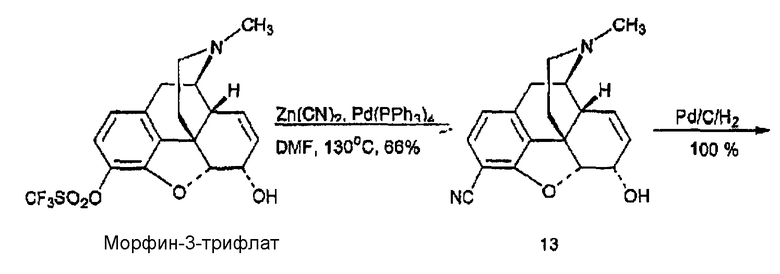
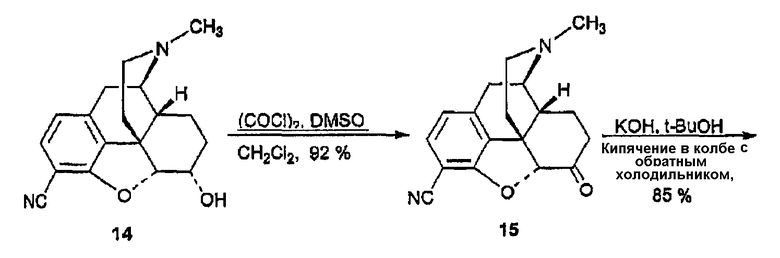

(А) Синтез морфин-3-карбонитрила, производного 13
Морфин-3-трифлат получали в соответствии с методикой, описанной Wentland et al. (J. Med. Chem. 3, 3558-3565 (2000)), а затем загружали (420 мг, 1,007 ммоль) в сухую колбу вместе с цианидом цинка (354 мг, 3,022 ммоль) и тетракис(трифенилфосфин)палладием (0) (116 мг, 0,101 ммоль) в атмосфере азота. Затем колбу снабжали холодильником, закрывали и вакуумировали, после чего заполняли аргоном за 5 циклов. Сухой DMF (2,0 мл) вводили шприцем и образующуюся смесь перемешивали в течение 20 ч при 120°С. Затем реакционную смесь охлаждали до 25°С, разбавляли EtOAc (30 мл), однократно промывали насыщенным раствором бикарбоната, дважды водой и один раз насыщенным раствором соли. Органическую фазу сушили над сульфатом натрия, фильтровали и концентрировали, в результате чего получали твердый остаток, который очищали флэш-хроматографией (CH2Cl2:MeOH:NH4OH 30:1:0,1), в результате чего получали 13 в виде белого твердого вещества (195 мг, 0,663 ммоль, 66%): 1Н ЯМР (500 МГц, CDCl3) δ 1,20 (д, 1H, J=8,1 Гц), 6,68 (д, 1H, J=8,1 Гц), 5,71 (м, 1H), 5,30 (м, 1H), 5,02 (м, 1H), 4,24 (уш.с, 1H), 3,38 (м, 1H), 3,12 (д, 1H, J=19,8 Гц), 2,68 (м, 3H), 2,44 (c, 3H), 2,33 (м, 2Н), 2,10 (м, 1H), 1,85 (м, 1H); MC (ESI) m/z 295 (M+H)-; Элементный анализ: Рассчитано для C18H18N2O2·0,125H2O: C 72,89; H 6,20; N 9,44. Найдено: С 72,74; Н 6,14; N 9,28.
(B) Синтез 7,8-дигидроморфин-3-карбонитрила, производного 14
Раствор соединения 13 (81 мг, 0,28 ммоль) и 10% Pd/C в 5 мл MeOH гидрировали под давлением 40 фунт/кв.дюйм в течение 4 ч при комнатной температуре. Реакционную смесь фильтровали через целит, и растворитель удаляли, в результате чего получали соединение 14 в виде вспененного вещества (81 мг; 100%). 1Н ЯМР (CDCl3) δ 7,20 (д, 1H, J=8,1 Гц), 6,69 (д, 1H, J=8,1 Гц), 4,7 (c, 1H), 3,12-3,09 (м, 1H), 3,0 (д, 1H, J=19,5 Гц), 2,55 (м, 1H), 2,44 (м, 1H), 2,4 (м, 1H), 2,35 (c, 3H), 2,25 (м, 2H), 2,1 (дд, 1H, J=4,2, 12,0), 1,94-1,84 (м, 2H), 1,55 (м, 1H), 1,4 (м, 1H). [α]D 25=-50,6° (c=0,64, CH2Cl2). MC m/z (ESI) 297 (MH-).
(С) Синтез гидрокодон-3-карбонитрила, производного 15
Оксалилхлорид (41,9 мкл, 0,47 ммоль) растворяли в 1 мл безводного CH2Cl2 в потоке аргона при -78°С. Затем добавляли сухой DMSO (66,9 мкл, 0,95 ммоль). Реакционную смесь перемешивали в течение 5 мин и шприцем добавляли раствор 14 (70 мг, 0,24 ммоль) в 1 мл сухого CH2Cl2. Смесь перемешивали в течение 20 мин при -78°С и к реакционной смеси добавляли 164 мкл Et3N и нагревали до комнатной температуры. Смесь распределяли между водой (10 мл) и CH2Cl2 (10 мл×3). Объединенный органический растворитель сушили (MgSO4), затем концентрировали в вакууме. Образующееся соединение очищали флэш-хроматографией (силикагель, CH2Cl2:CH3OH:NH3/H2O = 20:1:0,01), в результате чего получали 63,7 мг (92%) соединение 15 в виде вспененного вещества. 1Н ЯМР (CDCl3) δ 7,28 (д, 1H, J=8,1 Гц), 6,84 (д 1H, J=8,1 Гц), 4,83 (c, 1H), 3,24 (т, 1H, J=2,4 Гц), 3,1 (д, 1H, J=19,5 Гц), 2,66 (м, 1H), 2,61 (дт, 2Н, J=2,4, 5,7 Гц), 2,46 (м, 1H), 2,44 (c, 3H), 2,33 (м, 1H), 2,1 (м, 1H), 1,92-1,87 (м, 1Н), 1,75 (м, 1H), 1,18 (м, 1Н) [α]D 25=-64,4° (c=0,87, CH2Cl2). MC m/z (ESI) 295 (MH-).
(D) Синтез 3-карбоксиамидогидрокодона, производного 16
Раствор 15 (72 мг, 0,25 ммоль) и KOH в t-BuOH (10 мл) кипятили в колбе с обратным холодильником при перемешивании в течение 2 ч. После охлаждения реакционную смесь фильтровали через целит, и фильтрат концентрировали. Остаток очищали на флэш-колонке (силикагель, CH2Cl2:CH3OH:NH3/H2O = 20:1:0,01), в результате чего получали 64,9 мг (85%) соединения 16 в виде вспененного вещества. 1Н ЯМР (CDCl3) δ 7,77 (д, 1H, J=8,1 Гц), 7,46 (c, 1H), 6,82 (д, 1H, J=8,1 Гц), 5,89 (c, 1H), 4,80 (c, 1H), 3,2 (дд, 1Н, J=2,7, 6,0 Гц), 3,1 (д, 1H, J=19,5 Гц), 2,66 (м, 1H,), 2,62 (м, 2H), 2,46 (м, 1H), 2,44 (c, 3H), 2,33 (д, 1Н, J=5,4 Гц), 2,1 (м, 1H), 1,92-1,87 (м, 1Н), 1,75 (м, 1Н), 1,18 (м, 1Н). [α]D 25=-96,6° (c=0,23, CH2Cl2). MC m/z (ESI) 313 (MH-).
(E) Синтез 3-карбоксиамидо-4-гидроксигидрокодона, производного 17
Смесь 16 (46 мг, 0,15 ммоль), NH4Cl (78,9 мг, 0,88 ммоль), порошка цинка (57,3 мг, 0,88 ммоль) и EtOH (95%, 15 мл) кипятили в колбе с обратным холодильником в течение 4 ч. После охлаждения смесь фильтровали и твердый остаток промывали NH3/H2O (2 мл). Объединенные фильтраты и промывные фракции концентрировали и экстрагировали CH2Cl2 (10 мл×3). Органические экстракты сушили (MgSO4) и концентрировали. Остаток очищали колоночной хроматографией (SiO2, CH2Cl2:CH3OH:NH3/H2O = 10:1:0,01), в результате чего получали 29 мг (63%) соединения 17 в виде вспененного вещества. 1H ЯМР (CDCl3) δ 13,1 (c, 1H), 7,12 (дд, 1H, J=1,2, 8,1 Гц), 7,46 (c, 1H), 6,54 (д, 1Н, J=8,1 Гц), 6,02 (уш., 2H), 4,35 (д, 1H, J=13,5 Гц), 2,99 (м, 2Н), 2,92 (м, 1H), 2,7 (дд, 1H, J=4,7, 13,9 Гц), 2,46 (м, 2Н), 2,4 (c, 3H), 2,24 (м, 2Н), 1,98 (м, 1H), 1,87 (м, 1H), 1,6 (м, 1H). [a]D 25=-25,9° (c=0,7, CHCl3). MC m/z (ESI) 315 (MH-).
Пример 6
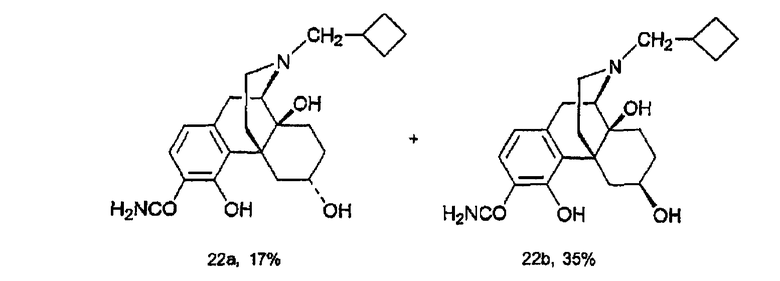
Синтез 3-карбоксамидо-4-гидрокси-6α-гидроксиналбуфина, производного 22а, и 3-карбоксамидо-4-гидрокси-6β-гидроксиналбуфина, производного 22b
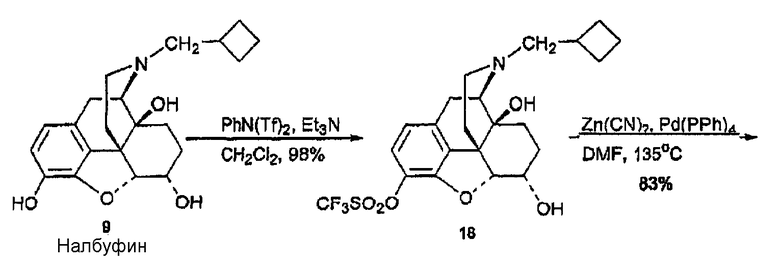
(А) Синтез налбуфин-3-трифлата 18
К дисперсии дигидрохлорида налбуфина (714 мг, 1,812 ммоль) в CH2Cl2 (30 мл) добавляли триэтиламин (600 мкл, 4,53 ммоль) при 0°С, после чего PhN(Tf)2 (654 мг, 1,812 ммоль) за один прием. Смеси давали возможность нагреться до комнатной температуры и перемешивали в течение ночи. Растворитель удаляли при пониженном давлении, и остаток распределяли между 6 н раствором NH4OH (50 мл) и CH2Cl2 (3Ч50 мл). CH2Cl2 экстракты объединяли, и объем их снижали до 50 мл при пониженном давлении. Органическую фазу промывали насыщенным водным раствором Na2CO3 (3×50 мл), затем сушили (Na2SO4) и концентрировали, в результате чего получали соединение 18 (886 мг, 1,812 ммоль, 100%). 1Н ЯМР (500 МГц, CDCl3) δ 6,95 (д, 1H, J=8,5 Гц), 6,69 (д, 1H, J=8,5 Гц), 4,97 (уш., 1H), 4,75 (д, 1H, J=5,0 Гц), 4,19 (м, 1H), 3,12 (д, 1H, J=19,0 Гц), 2,85 (д, 1H, J=6,0 Гц), 2,66 (дд, 1H, J=19,0, 6,0 Гц), 2,52-2,44 (м, 4Н), 2,25 (тд, 1H, J=12,5, 5,0 Гц), 2,17 (тд, 1H, J=12,5, 3,0 Гц), 2,07 (м, 1H), 1,98-1,81 (м, 3H), 1,73-1,44 (м, 5H), 1,26 (м, 1Н); 13C ЯМР (125 МГц, CDCl3) δ 149,5, 134,4, 134,3, 130,2, 121,8, 119,6, 92,9, 69,8, 66,6, 62,7, 60,8, 47,0, 43,4, 33,8, 32,8, 27,6, 27,1, 26,9, 23,8, 23,7, 18,9; MC (ESI) m/z 490 (М+Н)+ .
(В) Синтез налбуфин-3-карбонитрила, производного 19
В трехгорлую колбу, снабженную холодильником, вводили соединение 18 (886 мг, 1,812 ммоль), Zn(CN)2 (6,38 мг, 5,436 ммоль) и Pd(PPH3)4 (419 мг, 0,362 ммоль) в атмосфере азота. Колбу закрывали и извлекали из эксикатора. Через пробку вводили безводный DMF (6 мл). Смесь нагревали при 135°С в течение 24 часов. DMF удаляли при пониженном давлении, и остаток распределяли между насыщенным водным раствором NaHCO3 (100 мл) и этилацетатом (3×100 мл). Органические экстракты объединяли, сушили (Na2SO4) и концентрировали, в результате чего получали сырой продукт, который очищали флэш-хроматографией [(гексан/этилацетат/гидроксид аммония (1:1:0,01)] и получали соединение 19 в виде белого вспененного материала (549 мг, 1,50 ммоль, 83%). 1Н ЯМР (500 МГц, CDCl3) δ 7,25 (д, 1H, J=8,0 Гц), 6,73 (д, 1Н, J=8,0 Гц), 4,77 (д, 1Н, J=5,0 Гц), 4,23 (м, 1Н), 3,15 (д, 1Н, J=19,5 Гц), 2,86 (д, 1Н, J=6,0 Гц), 2,69 (дд, 1Н, J=19,5, 6,0 Гц), 2,49 (м, 4Н), 2,26 (тд, J=13,0, 5,0 Гц), 2,15 (тд, 1Н, J=11,5, 3,0 Гц), 2,06 (м, 3Н), 1,90 (м, 1Н), 1,84 (м, 2Н), 1,65 (м, 3Н), 1,47 (м, 1Н), 1,41 (м, 1Н), 1,18 (м, 1Н); 13C ЯМР (125 МГц, CDCl3) δ 161,3, 139,8, 131,7, 131,3, 119,1, 115,8, 92,5, 90,4, 69,5, 66,4, 62,3, 60,6, 46,1, 43,0, 33,5, 32,8, 27,7, 26,9, 26,7, 24,2, 23,4, 18,7; MC (ESI) m/z 367 (M+H)+.
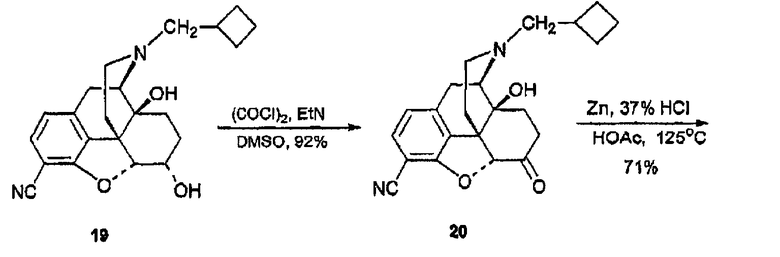
(С) Синтез 6-оксоналбуфин-3-карбонитрила, производного 20
Оксалилхлорид (143 мкл, 1,64 ммоль) в CH2Cl2 (5 мл) охлаждали до -78°С в атмосфере азота и добавляли безводный DMSO (232 мкл, 3,27 ммоль) с помощью шприца. Через 2 минуты добавляли соединение 19 (335 мг, 0,915 ммоль) в сухом CH2Cl2 (5 мл) и перемешивание продолжали в течение 15 минут. Добавляли сухой триэтиламин (570 мкл, 4,097 ммоль) и перемешивание продолжали в течение 5 минут. После нагревания до комнатной температуры реакционную смесь распределяли между насыщенным водным раствором NaHCO3 (50 мл) и CH2Cl2 (3×50 мл). Объединенный органический слой промывали насыщенным раствором соли (100 мл), сушили (Na2SO4) и концентрировали, в результате чего получали сырой продукт, который очищали флэш-хроматографией [CH2Cl2:MeOH (25:1)] и получали соединение 20 (308 мг, 0,846 ммоль, 92%). 1H ЯМР (500 МГц, CDCl3) δ 7,28 (д, 1H, J=8,0 Гц), 6,80 (д, 1H, J=8,0 Гц), 5,13 (уш., 1H), 4,81 (c, 1H), 3,19 (д, 1H, J=19,5 Гц), 3,03 (тд, 1H, J=14,5, 6,0 Гц), 2,97 (д, 1H, J=6,0 Гц), 2,67 (дд, 1H, J=19,5, 6,0 Гц), 2,60-2,48 (м, 4Н), 2,44 (тд, J=12,5, 5,5 Гц), 2,32 (м, 1H), 2,16-2,02 (м, 6Н), 1,70 (м, 2Н), 1,53 (м, 2H); 13C ЯМР (125 МГц, CDCl3) δ 206,2, 159,2, 138,8, 132,0, 129,4, 119,5, 115,0, 92,7, 91,2, 69,8, 62,2, 60,3, 50,0, 43,2, 35,9, 33,5, 31,2, 30,6, 26,9, 26,7, 24,0, 18,7; MC (ESI) m/z 365 (М+Н)+.
(D) Синтез 3-карбоксамидо-4-гидрокси-6-оксоналбуфина, производного 21
В колбу, содержащую соединение 20 (252 мг, 0,692 ммоль) добавляли Zn-пыль (900 мг, 13,85 ммоль), ледяную уксусную кислоту (5 мл) и концентрированную HCl (0,69 мл, 83 ммоль). После кипячения в колбе с обратным холодильником при 125°С в течение 3 часов реакционную смесь охлаждали до 0°С и концентрировали, добавляли раствор NH4OH для доведения величины рН до 10. Суспензионную смесь экстрагировали CH2Cl2 (3×100 мл). Органические экстракты объединяли, сушили (Na2SO4) и концентрировали, в результате чего получали 253 мг сырого продукта. Флэш-хроматография дала соединение 21 (187 мг, 0,487 ммоль, 71%). 1H ЯМР (500 МГц, CDCl3) δ 13,14 (c, 1H), 7,13 (д, 1H, J=8,0 Гц), 6,56 (д, 1H, J=8,0 Гц), 6,30-5,40 (уш., 2H), 4,65 (c, 1H), 4,04 (дд, 1H, J=11,0, 2,0 Гц), 3,02 (м, 1H), 2,94 (д, 1H, J=13,0 Гц), 2,89 (м, 1H), 2,86 (м, 1H), 2,50 (м, 1H), 2,45 (м, 1Н), 2,16-1,71 (м, 9Н), 1,68 (м, 3H); 13C ЯМР (125 МГц, CDCl3) δ 212,5, 173,3, 162,0, 144,3, 127,2, 124,9, 117,5, 111,0, 68,9, 60,4, 59,9, 45,6, 44,7, 43,9, 37,7, 33,8, 32,7, 32,1, 27,0, 26,8; 26,7, 18,7; ИК (пленка) νmax 3354, 2928, 1709, 1653, 1617, 1429 см-1; MC (ESI) m/z 385 (М+Н)+.
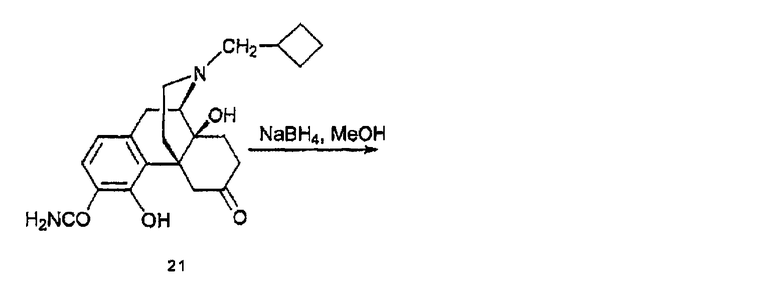
(E) Синтез 3-карбоксамидо-4-гидрокси-6α-гидроксиналбуфина, производного 22а, и 3-карбоксамидо-4-гидрокси-6β-гидроксиналбуфина, производного 22b
Соединение 21 (1,15 мг, 0,3 ммоль) растворяли в MeOH (2 мл) и охлаждали до 0°С. Добавляли NaBH4 (46 мг, 1,2 ммоль) за один прием. Реакционную смесь перемешивали при 0°С в течение двух часов и останавливали добавлением насыщенного водного раствора NH4Cl. MeOH удаляли при пониженном давлении и концентрировали. Добавляли раствор NH4OH для доведения величины рН до 10. Водную фазу экстрагировали CHCl3 (4×50 мл) и органические экстракты объединяли, сушили (NaSO4) и концентрировали, в результате чего получали 97 мг сырого продукта. Флэш-хроматография [CHCl3/MeOH/NH4OH (10:1:0,1)] давала изомеры 22а (31,8 мг, 0,082 ммоль, 17%) и 22b (40,7 мг, 0,105 ммоль, 35%). 22а: 1Н ЯМР (500 МГц, CDCl3) δ 13,43 (c, 1H), 7,12 (д, 1H, J=8,0 Гц), 6,62 (д, 1H, J=8,0 Гц), 6,30-5,30 (уш., 2H), 4,60 (c, 1H), 4,18 (c, 1H), 3,47 (м, 1H), 3,01 (д, 1H, J=19,0 Гц), 2,95 (тд, 1H, J=19,0, 6,0 Гц), 2,66 (д, 1H, J=5,5 Гц), 2,47-2,37 (м, 4H), 2,10-1,85 (м, 10H), 1,66-1,47 (м, 4H), 1,27 (м, 1H); 13C ЯМР (125 МГц, CDCl3) δ 173,6, 161,9, 144,3, 131,4, 123,9, 118,4, 110,5, 69,5, 67,8, 60,8, 60,4, 44,4, 39,5, 35,2, 33,7, 33,1, 27,7, 27,00, 26,96, 26,93, 26,7, 18,7; ИК (пленка) νmax 3445 (уш.), 2929, 1653, 1425 см-1; MC (ESI) m/z 387 (М+H)+; 22b: 1Н ЯМР (500 МГц, CDCl3) δ 13,10 (c, 1H), 7,15 (д, 1H, J=8,0 Гц), 6,60 (д, 1H, J=8,0 Гц), 6,30-5,30 (уш., 2H), 4,46 (c, 1H), 3,53 (м, 1H), 3,38 (м, 1H), 3,00 (д, 1H, J=19,5 Гц), 2,84 (тд, 1H, J=19,5, 6,5 Гц), 2,71 (д, 1H, J=6,0 Гц), 2,46-2,38 (м, 4H), 2,07-1,49 (м, 14H), 1,34 (д, 1H, J=5,0 Гц); 13C ЯМР (125 МГц, CDCl3) δ 173,6, 161,0, 143,9, 127,5, 124,5, 117,2, 110,3, 68,5, 66,7, 59,7, 59,6, 43,6, 41,4, 37,3, 33,1, 31,6, 29,8, 29,7, 26,2, 25,9 (2C), 17,8; ИК (пленка) νmax 3410 (уш.), 2929, 1653, 1617, 1425 см-1; MC (ESI) m/z 387 (м+H)+.
Пример 7
Синтез 3-карбоксамид-4-гидроксиналтрексона, производного 24
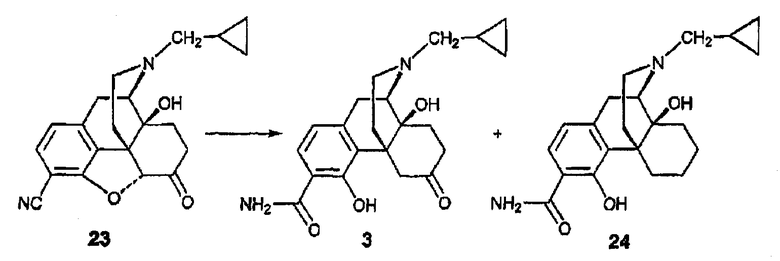
В колбу объемом 50 мл, содержащую нитрил 23 (полученный при использовании методики Kubota et al., Tetrahedron Letters 39(19), 2907-2910 (1998) (452 мг, 1,29 ммоль), загружали цинковую пыль калибром 325 меш (1679 мг, 25,83 ммоль), а затем добавляли 8 мл ледяной уксусной кислоты и 1,29 мл 12М HCl. Устанавливали обратный холодильник и реакционную смесь затем кипятили при 125°С в течение 3-х ч. На дне колбы образовывалось некоторое количество цинковых шариков. Реакционную смесь охлаждали до 0°С и концентрированный раствор NH4OH по каплям добавляли для доведения величины рН примерно до 10. Наблюдали образование белой суспензии. Смесь экстрагировали метиленхлоридом (100 мл×3). Органические фазы сушили над сульфатом натрия и концентрировали, в результате чего получали светло-желтый вспененный материал (484 мг), который очищали с использованием флэш-хроматографии (25:1:0,1 CH2Cl2:MeOH:NH4OH), в результате чего получали соединение 3 в виде белого вспененного вещества (264 мг, 0,713 ммоль, 55%) и соединение 24 в виде белого твердого вещества (100 мг, 0,281 ммоль, 22%); Т.п. 268-270°С. 1Н ЯМР (500 МГц, CDCl3) δ 12,99 (c, 1H), 7,15 (д, 1H, J=8,0 Гц), 6,60 (д, 1H, J=8,0 Гц), 6,60-5,40 (уш.c, 2H), 4,52 (уш.с, 1H), 3,11 (м, 1H), 3,00-2,80 (м, 1H), 2,60 (м, 1H), 2,31 (м, 2Н), 2,10-1,70 (м, 4Н), 1,60-1,35 (м, 5Н), 1,18 (м, 1H), 0,83 (м, 1H), 0,50 (м, 2Н), 0,10 (м, 2Н); MC (ESI) m/z 300 (M+H)+. Элементный анализ: Рассчитано для C21H28N2O3·0,375H2O: С 69,44; Н 7,98; N 7,71. Найдено: С 69,46; Н 8,11; N 7,42. [ОС]25 D=-85,0° (c=0,40; CHCl3).
Пример 8
Синтез 3-тиокарбоксиамидо-4-гидроксиналтрексона, производного 26
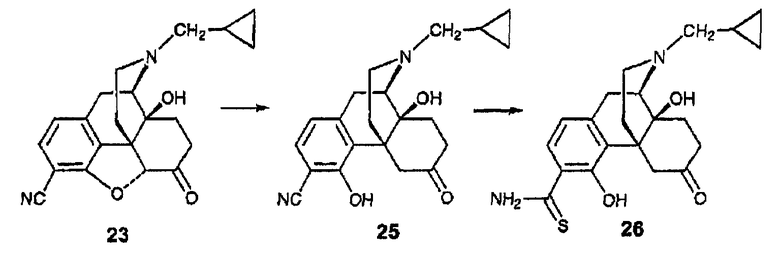
(А) Синтез 3-карбонитрил-4-гидроксиналтрексона, производного 25
В колбу объемом 50 мл, содержащую нитрил 23 (101 мг, 0,28 ммоль), загружали цинковую пыль калибром 325 меш (126 мг, 1,94 ммоль), а затем добавляли гидрохлорид аммония (148 мг, 2,77 ммоль), а затем 4 мл EtOH:H2O (20:1). Устанавливали обратный холодильник и реакционную смесь затем кипятили при 95°С в течение 3 часов. Реакционную смесь охлаждали до комнатной температуры и фильтровали через слой целлита. Целлит промывали MeOH. Фильтрат концентрировали, а затем распределяли между CH2Cl2 (40 мл×3) и 40 мл OfNH4OH в воде (рН 8-9). Органические фазы объединяли, сушили над сульфатом натрия и концентрировали, в результате чего получали твердое вещество (106 мг), которое очищали с использованием флэш-хроматографии (25:1:0,1 CH2Cl2:MeOH:NH4OH) и получали соединение 25 в виде белого твердого вещества (63 мг, 0,17 ммоль, 62%). 1Н ЯМР (500 МГц, CDCl3) δ 7,25 (д, 1H, J=9,3 Гц), 7,40 (д, 1H, J=7,8 Гц), 5,12 (уш.с, 1H), 3,81 (д, 1H, J=12,6 Гц), 3,40-2,60 (м, 6Н), 2,41 (c, 2Н), 2,30-1,75 (м, 5H), 1,60 (м, 1H), 0,88 (м, 1H), 0,56 (м, 2Н), 0,14 (м, 2H); MC (ESI) m/z 300 (M+H)+. [α]25 D=-64,3 (c=0,56°; EtOH).
(B) Синтез 3-тиокарбоксиамидо-4-гидроксиналтрексона, производного 26
Смесь нитрила 25 (49 мг, 0,139 ммоль) и О,О-диэтилдитиофосфорной кислоты (475 мкл, 2,78 ммоль) в воде (2 мл) и этаноле (4 мл) нагревали при 80°С в течение 22 ч. Реакционную смесь охлаждали до комнатной температуры и распределяли между насыщенным раствором NaHCO3 (20 мл) и CH2Cl2 (20 мл×3). Органические фазы сушили над сульфатом натрия и концентрировали с получением соединения 26 в виде желтого твердого вещества (56 мг), которое очищали с использованием флэш-хроматографии (40:1:0,1 EtOH:MeOH:NH4OH), в результате чего получали желтое вспененное вещество (36 мг, 0,093 ммоль, 67%) 1H ЯМР (500 МГц, CDCl3) δ 12,24 (c, 1H), 7,20-7,06 (м, 3H), 6,59 (д, 1H, J=8,5 Гц), 4,72 (уш.c, 1H), 4,02 (д, 1H, J=14,0 Гц), 3,14 (м, 1H), 2,94 (м, 2H), 2,94-2,70 (м, 2H), 2,65 (м, 1H), 2,20-1,70 (м, 6H), 0,87 (м, 1H), 0,55 (м, 2H), 0,12 (м, 2Н); MC (ESI) m/z 300 (М+H)-; Элементный анализ: рассчитано для C21H26N2O3S·0,25H2O: C 64,51; H 6,83; N 7,16. Найдено: С 64,50; Н 6,61; N 6,94. [α]25D=+85,0° (c=0,20, CHCl3).
Каждый патент, патентная заявка и ссылка, упомянутая в тексте настоящего описания, включены в него в качестве ссылки во всей полноте.
Хотя типичные варианты осуществления представлены в пояснительных целях, предшествующие описание изобретения и примеры не должны рассматриваться как ограничивающие объем притязаний изобретения. Соответственно, различные модификации, изменения и альтернативные варианты могут встретиться специалистам в данной области без отклонения от существа и объема притязаний настоящего изобретения.
| название | год | авторы | номер документа |
|---|---|---|---|
| 4-ГИДРОКСИБЕНЗОМОРФАНЫ | 2005 |
|
RU2480455C2 |
| ТРИЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ БЕНЗОПИРАНА В КАЧЕСТВЕ ПРОТИВОАРИТМИЧЕСКИХ АГЕНТОВ | 2005 |
|
RU2380370C2 |
| ЗАМЕЩЕННЫЕ ХРОМАНЫ | 2015 |
|
RU2718060C2 |
| ОБЩИЙ СИНТЕЗ МИРИАПОРОНОВ | 2003 |
|
RU2328492C2 |
| НОВОЕ ПРОИЗВОДНОЕ КУМАРИНА, ОБЛАДАЮЩЕЕ ПРОТИВООПУХОЛЕВОЙ АКТИВНОСТЬЮ | 2007 |
|
RU2428420C2 |
| ПРОИЗВОДНЫЕ ВАРИОЛИНА И ИХ ПРИМЕНЕНИЕ В КАЧЕСТВЕ ПРОТИВООПУХОЛЕВЫХ СРЕДСТВ | 2002 |
|
RU2302419C2 |
| ПРОИЗВОДНЫЕ ЭКТЕИНАСЦИДИНОВ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ИХ ОСНОВЕ И ПРИМЕНЕНИЕ | 2003 |
|
RU2306316C9 |
| 2-АМИНОБЕНЗОКСАЗОЛКАРБОКСАМИДЫ В КАЧЕСТВЕ МОДУЛЯТОРОВ 5-НТ3 | 2007 |
|
RU2448105C2 |
| ПРОИЗВОДНЫЕ АМИНА И ИХ ПРИМЕНЕНИЕ ДЛЯ ЛЕЧЕНИЯ ОФТАЛЬМОЛОГИЧЕСКИХ ЗАБОЛЕВАНИЙ И РАССТРОЙСТВ | 2008 |
|
RU2536040C2 |
| НОВЫЕ СОЕДИНЕНИЯ, ИХ ИЗОМЕР ИЛИ ИХ ФАРМАЦЕВТИЧЕСКИ ПРИЕМЛЕМЫЕ СОЛИ В КАЧЕСТВЕ АНТАГОНИСТА ВАНИЛОИДНОГО РЕЦЕПТОРА И СОДЕРЖАЩАЯ ИХ ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 2007 |
|
RU2448108C2 |
Изобретение относится к соединениям формулы (I), в которой А выбран из -C(=O)NH2 и -C(=S)NH2; R2 и R2a оба представляют собой атом водорода; R3 выбран из атома водорода и С3-С4циклоалкила; R4 выбран из атома водорода и гидрокси; R11 представляет собой атом водорода; R12 выбран из атома водорода и гидрокси; или радикалы R11 и R12 вместе образуют карбонильный заместитель; и двойные связи, обозначенные пунктирными линиями, отсутствуют. Соединения формулы (I) применяют для получения лекарственного средства для лечения болезни или состояния, опосредованного опиоидным рецептором. 2 н. и 7 з.п. ф-лы, 4 табл.
1. Соединение формулы (I):
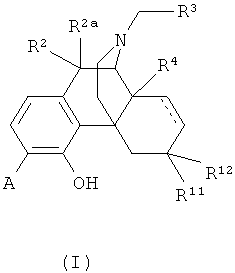
в которой А выбран из -C(=O)NH2 и -C(=S)NH2;
R2 и R2a оба представляют собой атом водорода;
R3 выбран из атома водорода и С3-С4циклоалкила;
R4 выбран из атома водорода и гидрокси;
R11 представляет собой атом водорода;
R12 выбран из атома водорода и гидрокси; или
радикалы R11 и R12 вместе образуют карбонильный заместитель;
и двойные связи, обозначенные пунктирными линиями, отсутствуют.
2. Соединение по п.1, в котором
R2 и R2a представляют собой атом водорода;
R3 выбран из атома водорода, циклопропила и циклобутила;
R4 выбран из атома водорода и гидрокси;
R11 представляет собой атом водорода;
R12 выбран из атома водорода и гидрокси; или
взятые вместе R11 и R12 образуют карбонил.
3. Соединение по п.1 формулы
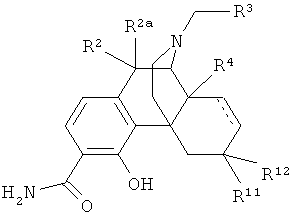
в которой R2 и R2a оба представляют собой атом водорода;
R3 выбран из атома водорода и С3-С4циклоалкила;
R4 выбран из атома водорода и гидрокси;
R11 представляет собой атом водорода;
R12 выбран из атома водорода и гидрокси; или
радикалы R11 и R12 вместе образуют карбонильный заместитель;
и двойные связи, обозначенные пунктирными линиями, отсутствуют.
4. Соединение по п.3, в котором
R2 и R2a представляют собой атом водорода;
R3 выбран из атома водорода, циклопропила и циклобутила;
R4 выбран из атома водорода и гидрокси;
R11 представляет собой атом водорода;
R12 выбран из атома водорода и гидрокси; или
взятые вместе R11 и R12 образуют карбонил.
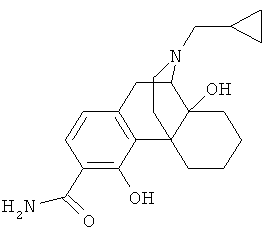
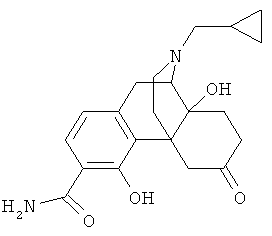
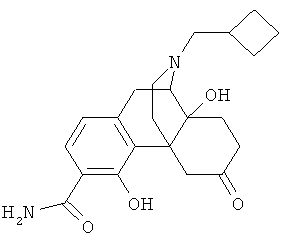
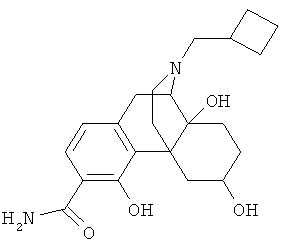
5. Соединение по п.4, выбранное из группы
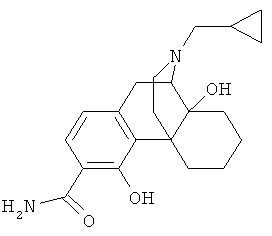

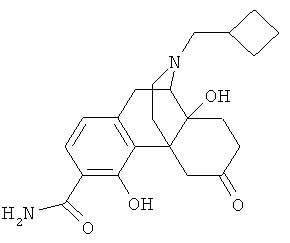
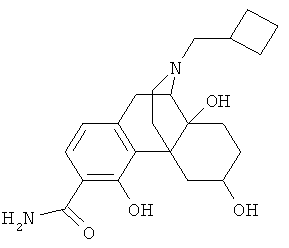
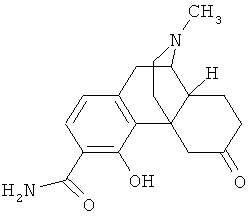
6. Соединение по п.1 формулы
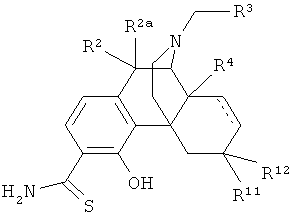
в которой R2 и R2a оба представляют собой атом водорода;
R3 выбран из атома водорода и С3-С4циклоалкила;
R4 выбран из атома водорода и гидрокси;
R11 представляет собой атом водорода;
R12 выбран из атома водорода и гидрокси; или
радикалы R11 и R12 вместе образуют карбонильный заместитель;
и двойные связи, обозначенные пунктирными линиями, отсутствуют.
7. Соединение по п.6,
в котором R2 и R2a представляют собой атом водорода;
R3 выбран из атома водорода, циклопропила и циклобутила;
R4 выбран из атома водорода и гидрокси;
R11 представляет собой атом водорода;
R12 выбран из атома водорода и гидрокси; или
взятые вместе R11 и R12 образуют карбонил.
8. Применение соединения формулы (I) для получения лекарственного средства для лечения болезни или состояния, опосредованного опиоидным рецептором,
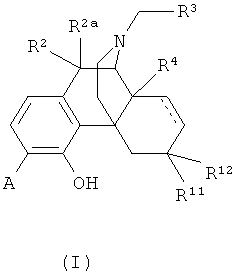
в которой А выбран из -C(=O)NH2 и -C(=S)NH2;
R2 и R2a оба представляют собой атом водорода;
R3 выбран из атома водорода и С3-С4циклоалкила;
R4 выбран из атома водорода и гидрокси;
R11 представляет собой атом водорода;
R12 выбран из атома водорода и гидрокси; или
радикалы R11 и R12 вместе образуют карбонильный заместитель;
и двойные связи, обозначенные пунктирными линиями, отсутствуют.
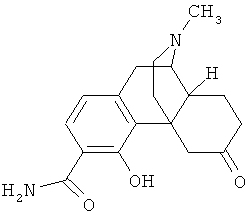
9. Применение по п.8, в котором указанное соединение формулы (I) выбрано из группы
| Способ и приспособление для нагревания хлебопекарных камер | 1923 |
|
SU2003A1 |
| MARK P.WENTLAND et al | |||
| Топка с несколькими решетками для твердого топлива | 1918 |
|
SU8A1 |
| MARK P.WENTLAND et al | |||
| Переносная печь для варки пищи и отопления в окопах, походных помещениях и т.п. | 1921 |
|
SU3A1 |

