ОБЛАСТЬ ТЕХНИКИ, К КОТОРОЙ ОТНОСИТСЯ ИЗОБРЕТЕНИЕ
Настоящее изобретение относится к применению катализаторов на хромовой основе с алюмоалкильными активаторами. Применение алюмоалкилов позволяет регулировать молекулярную массу, молекулярно-массовое распределение полимера и степень разветвления посредством боковых цепей при одновременном проявлении катализаторами необходимых значений производительности. Алюмоалкилы могут быть использованы в реакторе непосредственно в катализаторе или отдельно.
ПРЕДПОСЫЛКИ СОЗДАНИЯ ИЗОБРЕТЕНИЯ
Этиленовые полимеры находят массовое и широкое применение в качестве полимерных материалов для различных формованных изделий и в зависимости от метода формования и цели нуждаются в обладании разными свойствами. Так, например, полимеры, обладающие относительно низкими молекулярными массами и узкими молекулярно-массовыми распределениями, подходят для изготовления изделий, формованных по методу литья под давлением. С другой стороны, полимеры, обладающие относительно высокими молекулярными массами и широкими молекулярно-массовыми распределениями, подходят для изготовления изделий, формованных выдувным формованием или пневматическим формованием. Для многих целей необходимы полиэтилены от средней до высокой молекулярных масс. Такие полиэтилены обладают достаточной прочностью для таких целей применения, для которых требуется такая прочность (например, изготовление труб), и одновременно обладают хорошими характеристиками перерабатываемости.
Этиленовые полимеры, обладающие широкими молекулярно-массовыми распределениями, могут быть получены с использованием хромового катализатора, обычно называемого в данной области техники катализатором фирмы "Филлипс", приготовленного кальцинированием соединения хрома, проводимым на неорганическом оксидном носителе в невосстановительной атмосфере для его активации таким образом, чтобы по меньшей мере часть содержащихся атомов хрома превращалась в шестивалентные атомы хрома (Cr+6). Соответствующим материалом пропитывают диоксид кремния, псевдоожижают и нагревают в присутствии кислорода до примерно 400-860°С с переводом хрома из состояния окисления +3 в состояние окисления +6. Второй хромовый катализатор, используемый для получения полиэтилена высокой плотности, состоит из силилхромата (бис-трифенилсилилхромат), абсорбированного на обезвоженном диоксиде кремния и в дальнейшем восстановленного диэтилалюмоэтоксидом (ДЭАлЭ). Образующиеся полиэтилены, получаемые с использованием каждого из этих катализаторов, разнятся некоторыми важными свойствами. Катализаторы из оксида хрома на диоксиде кремния обладают хорошей производительностью (г ПЭ/г катализатора), также определяемой активностью (г ПЭ/г катализатора/ч), но обеспечивают получение полиэтиленов с молекулярно-массовыми распределениями, которые уже целевых. Катализаторы на основе силилхромата обеспечивают получение полиэтиленов с необходимыми молекулярно-массовыми характеристиками (более широкое молекулярно-массовое распределение с высокомолекулярным участком (участок, соответствующий высокомолекулярной фракции) на кривой молекулярно-массового распределения, указывающим на наличие популяций с двумя отчетливо различимыми молекулярными массами).
Monoi в JP 200202412 описано применение нанесенных на неорганический оксид содержащих Cr+6 твердых компонентов (А), приготовленных спеканием в невосстановительных условиях содержащих алюмодиалкильную функциональную группу алкоксидов (Б) и алюмотриалкила (В). Получаемые этиленовые полимеры обладают, как сказано, хорошим сопротивлением растрескиванию под воздействием внешних нагрузок и хорошим сопротивлением ползучести при выдувном формовании. В заявке US 2002042428 описан способ полимеризации этилена в соприсутствии водорода с использованием несущего алюмотриалкильное соединение хромового катализатора (А), где этот хромовый катализатор получают кальцинирующим активированием соединения Cr, нанесенного на неорганический оксидный носитель, в невосстановительной атмосфере с целью перевода атомов Cr в шестивалентное состояние, последующей обработкой А алюмотриалкильным соединением в инертном углеводородном растворителе и удалением растворителя в течение короткого промежутка времени.
Hasebe и др. в JP 2001294612 описаны катализаторы, содержащие нанесенные на неорганический оксид соединения Cr, кальцинированные при 300-1100°С в невосстановительной атмосфере, R3-nAlLn (R обозначает C1-С12алкил; L обозначает С1-С8алкокси, фенокси; 0<n<1) и органические соединения, являющиеся основаниями Льюиса. Эти катализаторы обеспечивают, как заявлено, образование полиолефинов с высокой молекулярной массой и узким молекулярно-массовым распределением.
Hasebe и др. в JP 2001198811 описана полимеризация олефинов с использованием катализаторов, содержащих оксиды Cr (нанесенные на огнестойкие соединения и активированные нагреванием в невосстановительных условиях) и R3-nAlLn (R обозначает C1-С6алкил; L обозначает С1-С8алкокси, фенокси; n>0,5, но <1). Этилен полимеризуют в присутствии нанесенного на SiO2 CrO3 и продукта взаимодействия в смеси МеОН-Et3Al 0,9:1 с получением полимера с показателем текучести расплава 0,18 г/10 мин при 190° под нагрузкой 2,16 кг и содержанием 1-гексеновых звеньев 1,6 мг/г полимера.
В патенте КНР 1214344, выданном на имя Da и др., речь идет о нанесенном на носитель катализаторе на хромовой основе для газофазной полимеризации этилена, приготовленном пропиткой неорганического оксидного носителя, содержащего на поверхности гидроксильную группу, водным раствором неорганического соединения хрома; сушкой на воздухе; активированием частиц в кислороде и восстановлением активированного промежуточного каталитического продукта органическим соединением алюминия. 10 г технического силикагеля смешивали с 0,05 моль/л водного раствора CrO3, сушили при 80-120°С в течение 12 ч, подвергали горячей сушке при 200°С в течение 2 ч, обжигали при 600°С в течение 4 ч и восстанавливали 25%-ным гексановым раствором диэтилэтоксиалюминия с получением порошкообразного катализатора с содержанием Cr 0,25% и соотношением Al/Cr 3.
В патенте US 5075395, выданном на имя Durand и др., речь идет о способе устранения индукционного периода в полимеризации этилена введением этилена в условиях полимеризации в псевдоожиженном слое и/или механического перемешивания в контакт с загружаемым порошком в присутствии катализатора, включающего хромоксидное соединение, связанное с гранулированным носителем и активированное термической обработкой, причем этот катализатор используют в форме форполимера. Предлагаемый Durand способ характеризуется тем, что используемый загружаемый порошок предварительно подвергают обработке введением упомянутого загружаемого порошка в контакт с алюминийорганическим соединением таким путем, что полимеризация начинается сразу же после контактирования этилена с загружаемым порошком в присутствии форполимера.
Уникальность катализа на хромовой основе в общем состоит в том, что с увеличением продолжительности реакции происходит увеличение молекулярных масс. Таким образом, увеличение продолжительности пребывания при применении катализаторов на хромоксидной основе позволяет получать более высокомолекулярные полимеры. Однако увеличение продолжительностей пребывания в реакторе приводит к снижению производительности реактора и к увеличению технологических затрат. Уменьшение продолжительностей пребывания может привести к улучшенным экономическим показателям, но при любом конкретном катализаторе на хромовой основе они приводят также к пониженным молекулярным массам полимеров. Для содействия сохранению более высоких молекулярных масс можно понизить температуру в реакторе, но это приводит к пониженному теплопереносу и более низким значениям производительности. От каталитических систем на хромовой основе требуется улучшенное регулирование характеристик получаемого полиэтилена при одновременном сохранении или повышении производительности. Необходимо сохранить требуемые молекулярные массы и показатели активности катализатора при уменьшенной продолжительности пребывания. Хотя литература, посвященная данной области техники, содержит эти и другие примеры применения в сочетании катализаторов типа катализатора фирмы "Филлипс" и алюминийорганического соединения, до сих пор не описан способ получения полиэтилена, обладающего от средней до высокой молекулярной массой, с использованием каталитической системы, обладающей хорошей производительностью, в котором можно регулировать молекулярную массу и молекулярно-массовое распределение и можно регулировать степень разветвления посредством боковых цепей. Более того, литература, посвященная данной области техники, не содержит информации об использовании добавления in-situ алюмоалкилов (непосредственно в реактор) для всестороннего решения проблем, возникающих при более высокой производительности реактора и более короткой продолжительности пребывания (молекулярная масса полимера, молекулярно-массовое распределение и производительность катализатора). Настоящее изобретение предназначено для устранения ряда недостатков полимеризации этилена с использованием катализатора на хромовой основе, ранее не представленного в литературе, посвященной данной области техники.
КРАТКОЕ ИЗЛОЖЕНИЕ СУЩНОСТИ ИЗОБРЕТЕНИЯ
Объектом настоящего изобретения являются система и способ полимеризации этилена, которые можно применять для проведения процесса с высокой объемной производительностью (более короткие продолжительности пребывания) с использованием катализаторов на хромовой основе, которые обладают хорошими значениями производительности и варьируемым регулированием молекулярной массы полимера, молекулярно-массового распределения и образования ответвлений посредством боковых цепей.
Используемая в настоящем описании ссылка на что-либо в единственном числе может служить также понятием в собирательном смысле.
Используемое в настоящем описании понятие "in situ" в ссылке на способ добавления компонента в катализатор служит для указания на добавление в катализатор в реакторе. Следовательно, когда каталитический компонент добавляют in situ, его добавляют к остальным каталитическим компонентам в реакторе, а не совмещают с другими каталитическими компонентами перед их подачей в реактор. Выражение "в реакторе" является синонимом, используемым в настоящем описании как взаимозаменяемое с выражением "in situ."
Используемое в настоящем описании понятие "в катализатор" или "на катализатор" в ссылке на способ добавления компонента в катализатор в настоящем описании служит указанием на добавление непосредственно в катализатор перед введением катализатора в реактор. Следовательно, когда компонент добавляют в катализатор ("в катализатор" или "на катализатор"), его добавляют к другим каталитическим компонентам перед подачей такого агрегата в реактор.
Используемое в настоящем описании понятие "алюмоалкил" определяют как соединение, отвечающее общей формуле R3Al, в которой R может быть любой из алкильных групп, содержащих от одного до двенадцати углеродных атомов. Группы R могут быть одинаковыми или разными.
Используемое в настоящем описании понятие "алкилалюмоалкоксид" служит для обозначения соединения, отвечающего общей формуле R2-Al-OR, в которой R может быть любой из алкильных групп, содержащих от одного до двенадцати углеродных атомов, a OR представляет собой алкокси или феноксигруппу, содержащую от одного до двенадцати углеродных атомов. Группы R могут быть одинаковыми или разными.
Используемое в настоящем описании понятие "ДЭАлЭ" означает диэтилалюмоэтоксид.
Используемое в настоящем описании понятие "ТЭАл" означает триэтилалюминий.
Используемое в настоящем описании понятие "ТЭБ" означает триэтилбор.
Используемое в настоящем описании понятие "ТИБА" означает триизобутилалюминий.
Используемое в настоящем описании понятие "ТнГАл" означает три-н-гексилалюминий.
Используемое в настоящем описании понятие "Mw" обозначает средневесовую молекулярную массу.
Используемое в настоящем описании понятие "Mn" обозначает среднечисленную молекулярную массу.
Используемое в настоящем описании понятие "Mz" обозначает z-среднюю молекулярную массу.
Используемое в настоящем описании понятие "молекулярно-массовое распределение" определяют как Mw/Mn.
В одном варианте выполнения настоящего изобретения предлагается нанесенный на носитель хромовый катализатор, включающий оксид хрома; содержащий диоксид кремния носитель, включающий диоксид кремния, выбранный из группы, включающей диоксид кремния, обладающий (а) удельным объемом пор примерно от 1,1 до 1,8 см3 /г и удельной площадью поверхности примерно от 245 до 375 м2 /г, (б) удельным объемом пор примерно от 2,4 до 3,7 см3/г и удельной площадью поверхности примерно от 410 до 620 м2/г и (в) удельным объемом пор примерно от 0,9 до 1,4 см3/г и удельной площадью поверхности примерно от 390-590 м2/г; и алюминийорганическое соединение, где нанесенный на носитель хромовый катализатор активируют при температуре от 400 до 860°С. В другом варианте алюминийорганическое соединение добавляют in situ. В еще одном варианте диоксид кремния обладает удельным объемом пор примерно от 2,4 до 3,7 см3/г и удельной площадью поверхности примерно от 410 до 620 м2/г, а алюминийорганическое соединение представляет собой алкилалюмоалкоксидное соединение. В еще одном варианте алюминийорганическое соединение представляет собой алкилалюмоалкоксидное соединение. В предпочтительном варианте алкилалюмоалкоксидное соединение представляет собой диэтилалюмоэтоксид. В еще одном варианте катализатор готовят добавлением in situ алкилалюмоалкоксидного соединения. В предпочтительном варианте алкилалюмоалкоксид, добавляемый in situ, представляет собой диэтилалюмоэтоксид. В одном варианте нанесенный на носитель катализатор активируют при температуре от 600 до 860°С. В еще одном варианте катализатор также включает тетраизопропоксид титана. В еще одном варианте каталитическое алюминийорганическое соединение представляет собой алюмоалкильное соединение. В предпочтительном варианте, в котором алюминийорганическим соединением служит алюмоалкильное соединение, это алюмоалкильное соединение представляет собой триэтилалюминий, триизобутилалюминий или три-н-гексилалюминий. В предпочтительном варианте алюмоалкильное соединение добавляют in situ. В более предпочтительном варианте катализатор готовят добавлением in situ триэтилалюминия.
В другом варианте предлагается система нанесенного на носитель хромового катализатора, включающая силилхромат, содержащий диоксид кремния носитель, обезвоженный при примерно от 400 до 860°С, включающий диоксид кремния, выбранный из группы, включающей диоксид кремния, обладающий (а) удельным объемом пор примерно от 1,1 до 1,8 см3/г и удельной площадью поверхности примерно от 245 до 375 м2/г, (б) удельным объемом пор примерно от 2,4 до 3,7 см3/г и удельной площадью поверхности примерно от 410 до 620 м2/г и (в) удельным объемом пор примерно от 0,9 до 1,4 см3/г и удельной площадью поверхности примерно от 390 до 590 м2/г; алюминийорганическое соединение; катализатор, приготовленный по способу добавления алюминийорганического соединения in situ. В еще одном варианте алюминийорганическое соединение представляет собой алкилалюмоалкоксидное соединение. В предпочтительном варианте алкилалюмоалкоксидное соединение представляет собой диэтилалюмоэтоксид. В еще одном варианте алюминийорганическое соединение представляет собой алюмоалкильное соединение. В предпочтительном варианте алюмоалкильное соединение выбирают из группы, включающей триэтилалюминий, триизобутилалюминий и три-н-гексилалюминий.
В другом варианте предлагается система нанесенного на носитель хромового катализатора, включающая силилхромат, содержащий диоксид кремния носитель, обезвоженный при примерно от 400 до 860°С, включающий диоксид кремния, выбранный из группы, включающей диоксид кремния, обладающий (а) удельным объемом пор примерно от 1,1 до 1,8 см3/г и удельной площадью поверхности примерно от 245 до 375 м2/г, (б) удельным объемом пор примерно от 2,4 до 3,7 см3/г и удельной площадью поверхности примерно от 410 до 620 м2/г и (в) удельным объемом пор примерно от 0,9 до 1,4 см3/г и удельной площадью поверхности примерно от 390 до 590 м2/г; алюминийорганическое соединение, выбранное из группы, включающей триэтилалюминий, триизобутилалюминий и три-н-гексилалюминий, причем этот катализатор готовят по способу добавления алюминийорганического соединения в катализатор.
В другом варианте предлагается система нанесенного на носитель хромового катализатора, включающая силилхромат, содержащий диоксид кремния носитель, обезвоженный при температуре от 400 до 860°С, включающий диоксид кремния, выбранный из группы, включающей диоксид кремния, обладающий удельным объемом пор примерно от 0,9 до 1,4 см3/г и удельной площадью поверхности примерно от 390 до 590 м2/г; и алюминийорганическое соединение.
В еще одном варианте предлагается система нанесенного на носитель хромового катализатора, включающая силилхромат, содержащий диоксид кремния носитель, обезвоженный при температуре от 400 до 860°С, включающий диоксид кремния, выбранный из группы, включающей диоксид кремния, обладающий (а) удельным объемом пор примерно от 1,1 до 1,8 см3/г и удельной площадью поверхности примерно от 245 до 375 м2/г, (б) удельным объемом пор примерно от 2,4 до 3,7 см3/г и удельной площадью поверхности примерно от 410 до 620 м2/г и (в) удельным объемом пор примерно от 0,9 до 1,4 см3/г и удельной площадью поверхности примерно от 390 до 590 м2/г; и триэтилбор, вводимый по способу добавления триэтилбора in situ.
В другом варианте предлагается способ получения этиленового полимера, включающий стадии контактирования этилена в полимеризационных условиях с каталитической системой, причем эта каталитическая система включает оксид хрома, алюмоалкил и содержащий диоксид кремния носитель, включающий диоксид кремния, выбранный из группы, включающей диоксид кремния, обладающий (а) удельным объемом пор примерно от 1,1 до 1,8 см3/г и удельной площадью поверхности примерно от 245 до 375 м2/г, (б) удельным объемом пор примерно от 2,4 до 3,7 см3/г и удельной площадью поверхности примерно от 410 до 620 м2/г и (в) удельным объемом пор примерно от 0,9 до 1,4 см3/г и удельной площадью поверхности примерно от 390 до 590 м2/г; и регулирования одного или нескольких из каталитической активности, степени разветвления посредством полимерных боковых цепей, Mz/Mw полимера, Mw/Mn полимера, плотности полимера и молекулярной массы получаемого этиленового полимера добавлением алкилалюмоалкоксида в количестве, достаточном для достижения конечного соотношения между эквивалентами алюминия и эквивалентами хрома от 0,1:1 до 10:1. В еще одном варианте алюмоалкил представляет собой триэтилалюминий, триизобутилалюминий или три-н-гексилалюминий. В предпочтительном варианте алкилалюмоалкоксид представляет собой диэтилалюмоэтоксид. В еще одном варианте каталитическая система далее включает тетраизопропоксид титана. В предпочтительном варианте полимеризация представляет собой газофазную полимеризацию. В предпочтительном варианте добавление диэтилалюмоэтоксида включает добавление in situ. В еще одном варианте добавление диэтилалюмоэтоксида включает добавление непосредственно в катализатор во время приготовления катализатора. В еще одном варианте значение Mw/Mn полимера превышает или равно 16, а значение упомянутого Mz/Mw полимера превышает или равно 6.
В другом варианте предлагается способ получения этиленового полимера, включающий стадии контактирования этилена в полимеризационных условиях с каталитической системой, включающей силилхромат и содержащий диоксид кремния носитель, включающий диоксид кремния, выбранный из группы, включающей диоксид кремния, обладающий (а) удельным объемом пор примерно от 1,1 до 1,8 см3/г и удельной площадью поверхности примерно от 245 до 375 м2/г, (б) удельным объемом пор примерно от 2,4 до 3,7 см3/г и удельной площадью поверхности примерно от 410 до 620 м2/г и (в) удельным объемом пор примерно от 0,9 до 1,4 см3/г и удельной площадью поверхности примерно от 390 до 590 м2/г; где упомянутый содержащий диоксид кремния носитель обезвоживают при температуре примерно от 400 до 860°С; и регулирования производительности катализатора, времени индуцирования реакции и молекулярной массы получаемого этиленового полимера добавлением алюминийорганического соединения в количестве, достаточном для достижения конечного соотношения между эквивалентами алюминия и эквивалентами хрома от 0,1:1 до 10:1. В предпочтительном варианте добавление алюминийорганического соединения включает добавление диэтилалюмоэтоксида. В еще одном варианте добавление диэтилалюмоэтоксида включает добавление in situ диэтилалюмоэтоксида. В еще одном варианте добавление упомянутого диэтилалюмоэтоксида включает добавление непосредственно в катализатор во время приготовления катализатора. В предпочтительном варианте полимеризация представляет собой газофазную полимеризацию. В предпочтительном варианте силилхромат размещают на упомянутом содержащем диоксид кремния носителе в концентрации примерно от 0,15 до 1,0 мас.% хрома. В еще одном варианте добавление алюминийорганического соединения включает добавление алюмоалкильного соединения. В предпочтительном варианте алюмоалкильное соединение выбирают из группы, включающей триэтилалюминий, триизобутилалюминий и три-н-гексилалюминий.
В другом варианте предлагается способ получения этиленового полимера, включающий стадии контактирования этилена в полимеризационных условиях с каталитической системой, включающей силилхромат и содержащий диоксид кремния носитель, включающий диоксид кремния, выбранный из группы, включающей диоксид кремния, обладающий (а) удельным объемом пор примерно от 1,1 до 1,8 см3/г и удельной площадью поверхности примерно от 245 до 375 м2/г, (б) удельным объемом пор примерно от 2,4 до 3,7 см3/г и удельной площадью поверхности примерно от 410 до 620 м2/г и (в) удельным объемом пор примерно от 0,9 до 1,4 см3/г и удельной площадью поверхности примерно от 390 до 590 м2/г; где упомянутый содержащий диоксид кремния носитель обезвоживают при температуре примерно от 400 до 860°С; и регулирования активности катализатора, времени индуцирования реакции и молекулярной массы получаемого этиленового полимера добавлением сокатализатора в количестве, достаточном для достижения конечного соотношения между эквивалентами алюминия и эквивалентами хрома от 0,1:1 до 10:1. В еще одном варианте стадия контактирования включает контактирование с диэтилалюмоэтоксидом. В еще одном варианте сокатализатор выбирают из группы, включающей триэтилалюминий, триизобутилалюминий и три-н-гексилалюминий. В еще одном варианте соотношение между эквивалентами алюминия и эквивалентами хрома составляет от примерно 1:1 до примерно 3:1. В предпочтительном варианте полимеризация представляет собой газофазную полимеризацию. В еще одном варианте катализатор обрабатывают (в самом катализаторе) алюмоалкилом или алкилалюмоалкоксидом перед добавлением сокатализатора. В другом конкретном варианте алкилалюмоалкоксид представляет собой диэтилалюмоэтоксид и соотношение между эквивалентами алюминия и эквивалентами хрома находится в пределах примерно 1:1 и 10:1.
В другом варианте выполнения настоящего изобретения предлагается способ получения этиленового полимера, включающий стадии контактирования этилена в полимеризационных условиях с каталитической системой, включающей оксид хрома и содержащий диоксид кремния носитель, включающий диоксид кремния, выбранный из группы, включающей диоксид кремния, обладающий (а) удельным объемом пор примерно от 1,1 до 1,8 см3/г и удельной площадью поверхности примерно от 245 до 375 м2/г, (б) удельным объемом пор примерно от 2,4 до 3,7 см3/г и удельной площадью поверхности примерно от 410 до 620 м2/г и (в) удельным объемом пор примерно от 0,9 до 1,4 см3/г и удельной площадью поверхности примерно от 390 до 590 м2/г; и регулирования активности катализатора, Mw/Mn полимера и молекулярной массы получаемого этиленового полимера добавлением сокатализатора в количестве, достаточном для достижения конечного соотношения между эквивалентами алюминия и эквивалентами хрома от 0,1:1 до 10:1. В предпочтительном варианте сокатализатор выбирают из группы, включающей триэтилалюминий, триизобутилалюминий и три-н-гексилалюминий. В отдельном варианте соотношение между эквивалентами алюминия и эквивалентами хрома составляет от примерно 1:1 до примерно 3:1. В еще одном варианте полимеризация представляет собой газофазную полимеризацию.
Другим вариантом выполнения настоящего изобретения является способ получения этиленового полимера в реакторе, включающий контактирование этилена в полимеризационных условиях с хромовой каталитической системой; проведение полимеризации при значении объемной производительности больше 8 и проведение полимеризации при производительности катализатора больше 3000 кг полимера/кг катализатора и при реакционной температуре по меньшей мере на 2,5°С выше, чем реакционная температура, когда полимеризацию проводят с той же хромовой каталитической системой в отсутствие триэтилалюминия и получают этиленовый полимер с такими же молекулярной массой и плотностью полимера с использованием тех же значения объемной производительности, парциального давления этилена, мольного соотношения газов Н2/С2 и мольного отношения сомономера к газу С2.
Вышеизложенное в общих чертах довольно широко представляет особенности и технические преимущества настоящего изобретения, что дает возможность лучше понять приведенное ниже подробное описание изобретения. В дальнейшем описаны другие особенности и преимущества изобретения, которые составляют сущность формулы изобретения. Специалистам в данной области техники необходимо принять во внимание, что описанные концепция и конкретный вариант могут быть легко использованы в качестве основы для модификации или разработки других структур для достижения тех же целей настоящего изобретения. Специалистам в данной области техники также необходимо ясно понимать, что такие эквивалентные структуры не выходят из сущности и объема изобретения, которые представлены в прилагаемой формуле изобретения. Эти новые особенности, которые, как полагают, являются характеризующими изобретение как в организационном, так и в технологическом смысле, совместно с другими объектами и преимуществами можно лучше понять из следующего описания, если его рассматривать в связи с прилагаемыми чертежами. Однако необходимо ясно представлять себе, что каждый из этих чертежей приведен только с иллюстративными и описательными целями, а не с целью ограничения объема настоящего изобретения.
КРАТКОЕ ОПИСАНИЕ ЧЕРТЕЖЕЙ
Для более полного понимания сущности настоящего изобретения в дальнейшем приведены ссылки на следующие описания, составленные в сочетании с нижеследующими прилагаемыми чертежами.
Фиг.1. Возможная структура катализатора из оксида хрома-на-диоксиде кремния (фирмы "Филлипс").
Фиг.2. Возможная структура катализатора из силилхромата-на-диоксиде кремния.
Фиг.3. Графики молекулярной массы полиэтилена, полученного с использованием хромоксидного катализатора MS35100: (а) без ДЭАлЭ; (б) ДЭАлЭ in situ; (в) ДЭАлЭ, добавленный в катализатор.
Фиг.4. Поток этилена по времени в случае хромоксидного катализатора MS35100.
Фиг.5. Графики молекулярной массы полиэтилена, полученного с использованием хромоксидного катализатора 957HS: (а) без ДЭАлЭ; (б) ДЭАлЭ in situ; (в) ДЭАлЭ, добавленный в катализатор.
Фиг.6. Поток этилена по времени в случае хромоксидного катализатора 957HS.
Фиг.7. Графики молекулярной массы полиэтилена, полученного с использованием хромоксидного катализатора ЕР352: (а) ДЭАлЭ in-situ; (б) ДЭАлЭ, добавленный в катализатор.
Фиг.8. Поток этилена по времени в случае хромоксидного катализатора ЕР352.
Фиг.9. Графики молекулярной массы полиэтилена, полученного с использованием силилхромата на MS3050 с ДЭАлЭ, добавленным in-situ.
Фиг.10. Поток этилена по времени в случае силилхромата на диоксиде кремния MS3050.
Фиг.11. Графики молекулярной массы полиэтилена, полученного с использованием силилхромата на диоксиде кремния 955: (а) без ДЭАлЭ; (б) 5 экв. ДЭАлЭ/экв. Cr в катализатор; (в) 10 экв. ДЭАлЭ/экв. Cr в катализатор.
Фиг.12. Поток этилена по времени в случае силилхромата на диоксиде кремния 955.
Фиг.13. Активность в зависимости от числа эквивалентов сокатализатора (Al/Cr) при различных сокатализаторах для силилхроматного катализатора, содержащего 5 экв. ДЭАлЭ/экв. Cr.
Фиг.14. Показатель текучести в зависимости от числа эквивалентов сокатализатора (Al/Cr) при различных сокатализаторах для силилхроматного катализатора, содержащего 5 экв. ДЭАлЭ/экв. Cr.
Фиг.15. Активность по времени для силилхроматного катализатора, содержащего 5 экв. ДЭАлЭ/экв. Cr, без сокатализатора и в присутствии ТЭАл.
Фиг.16. Активность по времени для силилхроматного катализатора, содержащего 5 экв. ДЭАлЭ/экв. Cr, без сокатализатора и в присутствии ТИБА.
Фиг.17. Активность по времени для силилхроматного катализатора, содержащего 5 экв. ДЭАлЭ/экв. Cr, без сокатализатора и в присутствии ТнГАл.
Фиг.18. График молекулярной массы полученного полиэтилена для силилхроматного катализатора, содержащего 5 экв. ДЭАлЭ/экв. Cr, без сокатализатора.
Фиг.19. График молекулярной массы полученного полиэтилена для силилхроматного катализатора, содержащего 5 экв. ДЭАлЭ/экв. Cr, в присутствии ТИБА.
Фиг.20. График молекулярной массы полученного полиэтилена для силилхроматного катализатора, содержащего 5 экв. ДЭАлЭ/экв. Cr, в присутствии ТЭАл.
Фиг.21. График молекулярной массы полученного полиэтилена для силилхроматного катализатора, содержащего 5 экв. ДЭАлЭ/экв. Cr, в присутствии ТнГАл.
Фиг.22. Активность по времени для силилхроматного катализатора, содержащего 1,5 экв. ДЭАлЭ/экв. Cr, без сокатализатора и в присутствии ТЭАл.
Фиг.23. Активность по времени для силилхроматного катализатора, содержащего 1,5 экв. ДЭАлЭ/экв. Cr, без сокатализатора и в присутствии ТнГАл.
Фиг.24. Активность по времени для силилхроматного катализатора, содержащего 1,5 экв. ДЭАлЭ/экв. Cr, без сокатализатора и в присутствии ТИБА.
Фиг.25. График молекулярной массы полученного полиэтилена для силилхроматного катализатора, содержащего 1,5 экв. ДЭАлЭ/экв. Cr, без сокатализатора.
Фиг.26. График молекулярной массы полученного полиэтилена для силилхроматного катализатора, содержащего 1,5 экв. ДЭАлЭ/экв. Cr, в присутствии ТИБА.
Фиг.27. График молекулярной массы полученного полиэтилена для силилхроматного катализатора, содержащего 1,5 экв. ДЭАлЭ/экв. Cr, в присутствии ТЭАл.
Фиг.28. График молекулярной массы полученного полиэтилена для силилхроматного катализатора, содержащего 1,5 экв. ДЭАлЭ/экв. Cr, в присутствии ТнГАл.
Фиг.29. Активность в зависимости от числа эквивалентов сокатализатора (Al/Cr) при различных сокатализаторах для катализатора оксид хрома 957HS-ТИПТ, содержащего 5 экв. ДЭАлЭ/экв. Cr.
Фиг.30. Показатель текучести в зависимости от числа эквивалентов сокатализатора (Al/Cr) при различных сокатализаторах для катализатора оксид хрома 957Н8-ТИПТ, содержащего 1,5 экв. ДЭАлЭ/экв. Cr.
Фиг.31. График молекулярной массы полученного полиэтилена для катализатора оксид хрома 957Н8-ТИПТ без сокатализатора.
Фиг.32. График молекулярной массы полученного полиэтилена для катализатора оксид хрома 957Н8-ТИПТ в присутствии ТИБА.
Фиг.33. График молекулярной массы полученного полиэтилена для катализатора оксид хрома 957Н8-ТИПТ в присутствии ТЭАл.
Фиг.34. График молекулярной массы полученного полиэтилена для катализатора оксид хрома 957Н8-ТИПТ в присутствии ТнГАл.
Фиг.35. График молекулярной массы полученного полиэтилена для катализатора оксид хрома 957Н8-ТИПТ без сокатализатора.
Фиг.36. График молекулярной массы полученного полиэтилена для катализатора оксид хрома 957Н8-ТИПТ в присутствии ТЭБ.
ПОДРОБНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
Изобретение применимо к полимеризации олефинов проведением процесса в суспензии, растворе, шламе или газовой фазе с использованием известных оборудования и реакционных условий, а не ограничено каким-либо конкретным типом полимеризационной системы. Обычно температуры полимеризации олефинов находятся в интервале от примерно 0 до примерно 300°С под атмосферным, пониженными или повышенными давлениями. В суспензионных или растворных полимеризационных системах можно создавать пониженные или повышенные давления и температуры в интервале от примерно 40 до примерно 300°С. Эффективная жидкофазная полимеризационная система описана в US 3324095. Жидкофазные полимеризационные системы обычно включают реактор, в который вводят олефиновый мономер и каталитическую композицию и который содержит жидкую реакционную среду для растворения или суспендирования полиолефина. Жидкая реакционная среда может состоять из массы жидкого мономера или инертного жидкого углеводорода, который не реакционноспособен в создаваемых полимеризационных условиях. Хотя нет необходимости в том, чтобы такой инертный жидкий углеводород служил в качестве растворителя для каталитической композиции или полимера, получаемого проведением этого процесса, его обычно применяют в качестве растворителя для используемых в полимеризации мономеров. Среди инертных жидких углеводородов, приемлемых для этой цели, упоминают изопентан, гексан, циклогексан, гептан, бензол, толуол и т.п. Реакционный контакт между олефиновым мономером и каталитической композицией необходимо поддерживать постоянным перемешиванием или смешением. Реакционную среду, содержащую продукт полимеризации олефина и непрореагировавший олефиновый мономер, отводят из реактора непрерывно. Продукт полимеризации олефина отделяют, а непрореагировавший олефиновый мономер и жидкую реакционную среду возвращают в реактор.
Однако изобретение особенно эффективно применительно к газофазным полимеризационным системам с давлениями, превышающими атмосферное, в интервале от 1 до 1000 фунтов/кв.дюйм, предпочтительно от 50 до 400 фунтов/кв.дюйм, наиболее предпочтительно от 100 до 300 фунтов/кв.дюйм, и температурами в интервале от 30 до 130°С, предпочтительно от 65 до 110°С. Особенно эффективны газофазные полимеризационные системы с перемешиваемым или псевдоожиженным слоем. Обычный газофазный процесс с псевдоожиженным слоем в общем проводят пропусканием непрерывного потока, включающего один или несколько олефиновых мономеров, через реактор с псевдоожиженным слоем в реакционных условиях и в присутствии каталитической композиции при скорости, достаточной для того, чтобы поддерживать слой твердых частиц во взвешенном состоянии. Поток, включающий непрореагировавший мономер, непрерывно отводят из реактора, сжимают, охлаждают, необязательно частично или полностью конденсируют и возвращают в реактор. Продукт отводят из реактора и в рецикловый поток добавляют свежий мономер. При необходимости для регулирования температуры полимеризационной системы в этот газовый поток можно также добавлять любой газ, инертный в отношении каталитической композиции и реагентов. Кроме того, можно использовать вспомогательное средство для псевдоожижения, такое как углеродная сажа, диоксид кремния, глина и тальк, как это изложено в US №4994534.
Полимеризационная система может включать единственный реактор или два или большее число последовательно размещенных реакторов и процесс проводят по существу в отсутствие каталитических ядов. С целью повысить активность катализатора в качестве нейтрализующих яды агентов можно использовать металлорганические соединения. Примерами нейтрализующих яды агентов являются металлоалкилы, предпочтительно алюмоалкилы.
В таком процессе можно использовать обычные добавки при условии, что они не препятствуют работе каталитической композиции в образовании целевого полиолефина. В качестве регулятора степени полимеризации в этом процессе можно использовать водород в количествах до примерно 10 моль водорода на моль всего мономерного сырья.
Полиолефины, которые могут быть получены в соответствии с изобретением, включают, хотя ими их список не ограничен, те, которые получают из олефиновых мономеров, таких как этилен, а также линейные и разветвленные высшие альфа-олефиновые мономеры, содержащие от 3 до примерно 20 углеродных атомов. Могут быть получены гомополимеры или тройные сополимеры этилена и высших альфа-олефиновых мономеров с плотностями в интервале от примерно 0,86 до примерно 0,95. Приемлемые высшие альфа-олефиновые мономеры включают, например, пропилен, 1-бутен, 1-пентен, 1-гексен, 4-метил-1-пентен, 1-октен и 3,5,5-триметил-1-гексен. Олефиновые полимеры в соответствии с изобретением могут быть также получены на основе или содержат звенья сопряженных или несопряженных диенов, таких как линейные, разветвленные и циклические углеводородные диены, содержащие от примерно 4 до примерно 20, предпочтительно от 4 до 12, углеродных атомов. Предпочтительные диены включают 1,4-пентадиен, 1,5-гексадиен, 5-винил-2-норборнен, 1,7-октадиен, винилциклогексен, дициклопентадиен, бутадиен, изобутилен, изопрен, этилиденнорборнен и т.п. В соответствии с изобретением могут быть также полимеризованы ароматические соединения, содержащие винильные ненасыщенные группы, такие как стирол и замещенные стиролы, полярные винильные мономеры, такие как акрилонитрил, эфиры малеиновой кислоты, винилацетат, эфиры акриловой кислоты, эфиры метакриловой кислоты, винилтриалкилсиланы и т.п. Конкретные полиолефины, которые могут быть получены в соответствии с изобретением, включают, например, полиэтилен высокой плотности, полиэтилен средней плотности (включая этилен-бутеновые сополимеры и этилен-гексеновые сополимеры), гомополиэтилен, полипропилен, этилен-пропиленовые каучуки (ЭПК), этилен-пропилен-диеновые тройные сополимеры (ТЭПД), полибутадиен, полиизопрен и т.п.
Одним путем к усовершенствованным каталитическим системам для получения полиэтиленов, обладающих характеристиками тех материалов, которые, как правило, получают с использованием катализаторов типа силилхромата-на-диоксиде кремния, являются катализаторы из восстановленного оксида хрома-на-диоксиде кремния. Необходимо, чтобы любая такая каталитическая система работала хорошо во время процесса с высокой объемной производительностью (т.е. процесса с получением максимального количества полимера на единицу реакторного времени и реакторного пространства) с образованием наибольшего возможного количества полиэтилена с высокой активностью катализатора при более короткой продолжительности пребывания. Катализаторы из оксида хрома обладают адекватными производительностью и активностью, но, тем не менее, полиэтилены, получаемые благодаря их применению, оказываются менее оптимальными для ряда целей применения, где требуются высокая молекулярная масса, широкое молекулярно-массовое распределение и наличие некоторой степени бимодальности молекулярно-массового распределения.
Так называемый катализатор фирмы "Филлипс", внедренный в начале 1960-х годов, был первым катализатором из оксида хрома-на-диоксиде кремния. Этот катализатор готовят пропиткой диоксида кремния материалами с Cr+3 с последующим псевдоожижением матрицы из диоксида кремния при температуре примерно от 400 до 860°С. В этих условиях Cr+3 превращают в Cr+6. Катализатор фирмы "Филлипс" в данной области техники также обычно называют "нанесенным на неорганический оксид Cr+6". Хотя катализаторы из оксида хрома-на-диоксиде кремния проявляют хорошую производительность, они обеспечивают образование полиэтиленов, обладающих относительно узким молекулярно-массовым распределением. Так называемый катализатор фирмы "Филлипс" и родственные катализаторы в настоящем описании обозначены как катализаторы "CrOx". Фиг.1 дает схематическое представление о структуре катализаторов CrOx. Катализаторы из силилхромата-на-диоксиде кремния являются катализаторами из нанесенного на неорганический оксид Cr+6 одного типа, которые обеспечивают образование полиэтиленов, не обладающих вышеупомянутыми недостатками. Катализаторы из силилхромата-на-диоксиде кремния в настоящем описании обозначают как катализаторы "СХ". Фиг.2 дает схематическое представление о структуре катализаторов СХ типа. Катализаторы СХ типа, как правило, восстанавливают алюмоалкилами, такими как ДЭАлЭ, во время стадии приготовления катализатора перед введением в реактор. Целью является сохранить или улучшить производительность катализаторов CrOx при одновременном получении полиэтилена с молекулярной массой и молекулярно-массовыми распределениями, которые в большей степени приближаются к этим же параметрам, достигаемым с использованием катализаторов СХ.
Варианты катализаторов с применением материалов с Cr+6, нанесенных на диоксид кремния, известны. В одном конкретном варианте перед активацией используют пропитанный тетраизопропоксидом титана (ТИПТ) диоксид кремния совместно с материалами с Cr+3. Этот вариант в последующем обозначен как "Ti-CrOx" (титанированный оксид хрома). Результатом использования таких модификаций являются полиэтилены с несколько более значительными молекулярно-массовыми распределениями в сравнении с теми, которых достигают без титанирования. Хотя эта система обеспечивает образование полиэтиленов, которые ближе к материалам, получаемым с использованием катализаторов типа силилхромата-на-диоксиде кремния, необходимы дальнейшие улучшения молекулярной массы и молекулярно-массового распределения, позволяющие им более плотно приблизиться к тем, которые получены с использованием силилхромата-на-диоксиде кремния.
Примеры
Эксперименты примеров с 1 по 53 проводили как реакции суспензионной полимеризации. Эксперименты примеров с 54 по 74 проводили в газофазном реакторе с псевдоожиженным слоем.
Общие процессы приготовления катализаторов
Во всех случаях, если не указано иное, все катализаторы, используемые в следующих примерах, готовили согласно следующим методам.
Общий процесс приготовления А.
Активация хромоксидного катализатора: катализаторы получали от поставщиков с уже содержавшейся на носителях хромовой пропиткой. Физические свойства катализаторов представлены в таблице 2. Активацию проводили пропусканием газа через катализатор в течение четырех часов при указанной температуре в сухом воздухе. Это обычно осуществляли в трубчатой печи. В дальнейшем катализатор до применения хранили в азотной атмосфере.
Общий процесс приготовления Б.
Процессы восстановления катализатора из оксида хрома: в типичном процессе приготовления 3 г предварительно активированного катализатора в инертной атмосфере помещали в 50-миллилитровую не содержавшую воздуха стеклянную колбу с валом мешалки. Добавляли тридцать пять миллилитров сухого дегазированного гексана и смесь нагревали до 50°С. Затем посредством шприца добавляли восстановитель (все реагенты содержались в гексане в концентрации 20-25 мас.%). Указанные числа эквивалентов всегда являлись отношениями реагента к хрому. После 30 мин начинали сушку. Ее можно было проводить под высоким вакуумом или с продувкой азотом. До применения катализатор хранили в азотной атмосфере.
Общий процесс приготовления В.
Процессы приготовления катализаторов СХ типа: перед применением все диоксиды кремния обезвоживали. Обезвоживание диоксида кремния проводили пропусканием газа через катализатор в течение четырех часов при указанной температуре в сухом воздухе или азоте. В типичном процессе приготовления 3 г предварительно обезвоженного диоксида кремния в инертной атмосфере помещали в 50-миллилитровую не содержавшую воздуха стеклянную колбу с валом мешалки. Добавляли тридцать пять миллилитров сухого дегазированного гексана и смесь нагревали до 50°С. Хроморганический источник (трифенилсилилхромат (ТФСХ)) можно было добавлять перед, одновременно или после добавления разбавителя. Смесь, как правило, перемешивали в течение 2 ч (когда это указано, перемешивание могло продолжаться в течение 10 ч). Затем посредством шприца добавляли восстановитель (все реагенты содержались в гексане в концентрации 20-25 мас.%). Указанные числа эквивалентов всегда являлись отношениями реагента к хрому. После 30 мин начинали сушку. Ее можно было проводить под высоким вакуумом или с продувкой азотом. До применения катализатор хранили в азотной атмосфере. В случаях, когда восстановитель не добавляли, сушку начинали после смешения источника хрома и диоксида кремния аналогично изложенному выше.
Описания катализаторов
В случае использования отношение восстановителя к добавляемому хрому можно обнаружить в примере; "в реактор" означает, что реагент добавляли отдельно от катализатора. "В катализатор" означает, что реагент добавляли на стадии приготовления катализатора. Указываемые значения в мас.% для хрома являлись приблизительными; фактические значения могли содержаться в интервале ±50%. Это относится как к хромоксидному, так и к силилхроматному катализаторам.
Пример 1: катализатор использовали в том виде, как он поступал от фирмы Davison Chemical, он состоял из 0,5 мас.% хрома на диоксиде кремния Davison 955 и был активирован при 825°С (общий процесс приготовления А) (см. технические условия для диоксида кремния в таблице 2).
Примеры 2-6: катализатор был таким же, как использованный в примере 1, за исключением того, что восстановители добавляли на стадии приготовления катализатора так, как в общем процессе приготовления Б. Когда использовали смесь восстановителей, мольное соотношение между каждыми из них составляло 1:1.
Пример 7: катализатор состоял из 0,5 мас.% Cr на диоксиде кремния Davison 955 (обезвоживание при 200°С), обработанном перед активацией тетраизопропоксидом титана. Добавляли достаточное количество ТИПТ, благодаря чему после активации оставалось 3,8 мас.% Ti (описание конкретных методов добавления ТИПТ см. в US 4011382).
Примеры 8-9: катализатор был таким же, как использованный в примере 7, за исключением того, что восстановитель добавляли на стадии приготовления катализатора так, как в общем процессе приготовления Б.
Примеры 10-12: MS35100 представлял собой хромоксидный катализатор, полученный от фирмы PQ, с техническими условиями, приведенными в таблице 2. Этот катализатор содержал 0,5 мас.% Cr. Катализатор активировали при 700°С (общий процесс приготовления А). В случае применения восстановитель добавляли на стадии приготовления катализатора так, как в общем процессе приготовления Б.
Примеры 13-15: катализатор был таким же, как использованный в примере 1, с добавлением ДЭАлЭ в качестве восстановителя проведением общего процесса приготовления Б.
Примеры 16-18: ЕР352 представлял собой хромоксидный катализатор, полученный из Ineos, с техническими условиями, приведенными в таблице 2. Этот катализатор содержал 0,5 мас.% Cr. Катализатор активировали при 700°С (общий процесс приготовления А). В случае применения восстановитель добавляли на стадии приготовления катализатора так, как в общем процессе приготовления Б.
Примеры 19-21: трифенилсилилхромат добавляли к носителю MS3050 (который предварительно обезвоживали при 700°С), как в общем процессе приготовления В. Добавляли достаточное количество трифенилсилилхромата, вследствие чего конечная высушенная композиция включала 0,5 мас.% Cr. В случае применения восстановитель добавляли на стадии приготовления катализатора так, как в общем процессе приготовления В.
Примеры 22-25 и 27: трифенилсилилхромат добавляли к носителю Davison 955 (который предварительно обезвоживали при 600°С), как в общем процессе приготовления В. Добавляли достаточное количество трифенилсилилхромата, вследствие чего конечная высушенная композиция включала 0,24-0,25 мас.% Cr. В случае применения восстановитель ДЭАлЭ добавляли на стадии приготовления катализатора так, как в общем процессе приготовления В.
Пример 26: трифенилсилилхромат добавляли к носителю Davison 955 (который предварительно обезвоживали при 600°С), как в общем процессе приготовления В. Добавляли достаточное количество трифенилсилилхромата, вследствие чего конечная высушенная композиция включала 0,25 мас.% Cr. Триизобутилалюминиевый восстановитель добавляли на стадии приготовления катализатора так, как в общем процессе приготовления В.
Примеры 28-34: этот катализатор готовили в промышленном масштабе. Трифенилсилилхромат добавляли к носителю Davison 955 (который предварительно обезвоживали при 600°С), как в общем процессе приготовления В. Добавляли достаточное количество трифенилсилилхромата, вследствие чего конечная высушенная композиция включала 0,24 мас.% Cr. Перед добавлением ДЭАлЭ ТФСХ давали перемешиваться с диоксидом кремния в течение 10 ч. Использовали соотношение ДЭАлЭ/Cr 5:1.
Примеры 35-38: при этом применяли такой же катализатор, как использованный в примере 28, за исключением того, что соотношение ДЭАлЭ/Cr было равным 1,5.
Примеры 39-45, 50-53: при этом применяли такой же катализатор, как использованный в примере 7. Сокатализаторы, перечисленные как добавляемые, вводили в реактор отдельно.
Примеры 46-49 и 74: при этом применяли такой же катализатор, как использованный в примере 1. Сокатализатор, указанный как добавляемый, вводили в реактор отдельно.
Примеры 54, 55, 60-68 и 72: этот катализатор готовили в промышленном масштабе (за исключением катализатора примера 55, который готовили в масштабе лабораторной пилотной установки). Трифенилсилилхромат добавляли к носителю Davison 955 (который предварительно был обезвожен при 600°С), как в общем процессе приготовления В. Добавляли достаточное количество трифенилсилилхромата, вследствие чего конечная высушенная композиция включала 0,24 мас.% Cr. Перед добавлением ДЭАлЭ ТФСХ давали перемешиваться с диоксидом кремния в течение 10 ч. Использовали соотношение ДЭАлЭ/Cr 5:1. Сокатализаторы, перечисленные как добавляемые в реактор, вводили в реактор отдельно.
Примеры 69, 70, 71, 74: этот катализатор готовили в промышленном масштабе. Бис-трифенилсилилхромат добавляли к носителю Davison 955 (который предварительно обезвоживали при 600°С), как в общем процессе приготовления В. Добавляли достаточное количество трифенилсилилхромата, вследствие чего конечная высушенная композиция включала 0,25 мас.% Cr. Перед добавлением ДЭАлЭ ТФСХ давали перемешиваться с диоксидом кремния в течение 10 ч. Использовали соотношение ДЭАлЭ/Cr 1,5:1. Сокатализаторы, перечисленные как добавляемые в реактор, вводили в реактор отдельно.
Пример 56: этот катализатор был таким же, как использованный в примере 19, но его готовили в масштабе пилотной установки. Использовали соотношение ДЭАлЭ/Cr 5:1.
Примеры 57 и 58: катализатор был таким же, как использованный в примере 13, в качестве восстановителя применяли ДЭАлЭ при соотношении ДЭАлЭ/Cr 5:1 и катализатор готовили в масштабе пилотной установки.
Пример 59: катализатор был таким же, как использованный в примере 10, в качестве восстановителя применяли ДЭАлЭ при соотношении ДЭАлЭ/Cr 5:1 и его готовили в масштабе пилотной установки.
Хотя в этих конкретных примерах указано конкретное содержание силилхромата на носителях из диоксида кремния, необходимо иметь в виду, что можно использовать хром в концентрации примерно от 0,2 до 1,0 мас.%, и это составляет часть настоящего изобретения.
Лабораторный суспензионный метод
Для проведения реакций полимеризации применяли однолитровый реактор с мешалкой. Перед каждым экспериментом реактор тщательно сушили при повышенных температурах в токе азота. В реактор при 60°С вводили 500 мл сухого дегазированного гексана. В случае применения на этом этапе добавляли гексен. Во всех случаях, если не указано иное, в каждом эксперименте использовали 10 мл 1-гексена. Затем для пассивирования всех примесей в реактор добавляли небольшое количество (0,1-0,25 г) диоксида кремния Davison 955, обезвоженного при 600°С и обработанного 0,6 ммоль/г ТЭАл. В любом эксперименте, в котором реагент добавляли в реактор отдельно от катализатора, диоксид кремния, обработанный ТЭАл, не добавляли. После перемешивания в течение 15 мин катализатор загружали с последующим введением дополнительных реагентов. Сокатализаторы добавляли непосредственно в реактор в виде разбавленных растворов, как упоминается в других местах. Реактор герметизировали и на этом этапе вводили водород. Водород использовали только в тех случаях, которые отмечены в таблицах. Подачей этилена давление в реакторе доводили до 200 фунтов/кв.дюйм. Этилену давали возможность поступать для поддержания в реакторе давления 200 фунтов/кв.дюйм. Поглощение этилена определяли с помощью электронного расходомера. Все процессы сополимеризации проводили при 85°С, процессы гомополимеризации проводили при 90°С. Процессы полимеризации проводили до тех пор, пока не получали максимум 160 г ПЭ, или завершали раньше. После сброса давления и понижения температуры реактор открывали. После того как давали испариться разбавителю, определяли массу полимера. В дальнейшем полимер охарактеризовывали проведением ряда испытаний.
Испытания
Плотность: ASTM D-1505.
Индекс расплава: (I2) ASTM D-2338, условие Е, определяли при 190°С, фиксировали в граммах на 10 мин.
Показатель текучести: (I21) ASTM D-1238, условие F, определяли под нагрузкой, которая в 10 раз превышала массу, используемую при определении вышеупомянутого индекс расплава.
СВР: соотношение вязкостей расплавов определяли как показатель текучести/индекс расплава.
ГПХ: лабораторный прибор для полимеров модели HT-GPC-220, колонки: Shodex, темп. эксперимента: 140°С, калибровочный стандарт: относимый к NIST, растворитель: 1,2,4-трихлорбензол.
ЧБО: частота бутильных ответвлений, как это определяют по 13С-ЯМР. Эта величина представляет собой число бутильных ответвлений на 1000 углеродных атомов.
При создании настоящего изобретения было установлено, что системы, в которых используют катализаторы из восстановленного оксида хрома на диоксиде кремния, проявляют целевую производительность и одновременно обеспечивают образование полиэтиленов, обладающих молекулярной массой и молекулярно-массовым распределением, аналогичными тем, которых достигают с использованием силилхромата-на-диоксиде кремния. Добавление алюмоалкильных соединений, таких как триэтилалюминий (ТЭАл), либо 1) непосредственно в катализатор перед введением в реакцию, либо 2) непосредственно в реактор (in-situ), увеличивает молекулярную массу и молекулярно-массовое распределение получаемых полиэтиленов. Обычно алкильные группы триалкилалюминия могут быть одинаковыми или разными и должны содержать от примерно 1 до примерно 12 углеродных атомов, а предпочтительно от 2 до 4 углеродных атомов. Примеры включают, хотя ими их список не ограничен, триэтилалюминий, триизопропилалюминий, триизобутилалюминий, три-н-гексилалюминий, метилдиэтилалюминий и триметилалюминий. Хотя в примерах использован почти исключительно ТЭАл, необходимо понимать, что объем изобретения им не ограничивается. Однако применение ТЭАл приводит к некоторому нерегулируемому разветвлению в полимере посредством боковых групп. Было бы лучше устранить это разветвление посредством боковых групп в тех случаях применения, в которых оно не требуется, но тем не менее его сохранить в тех случаях применения, в которых оно требуется. Это может быть достигнуто добавлением алкилалюмоалкоксидных соединений, таких как диэтилалюмоэтоксид. Применение алкилалюмоалкоксида, такого как диэтилалюмоэтоксид (ДЭАлЭ), устраняет такое разветвление посредством боковых групп.Обычно алкилалюмоалкоксид, отвечающий общей формуле R2-Al-OR, где алкильные группы могут быть одинаковыми или разными, должен содержать от примерно 1 до примерно 12 углеродных атомов, а предпочтительно от 2 до 4 углеродных атомов. Примеры включают, хотя ими их список не ограничен, диэтилалюмоэтоксид, диэтилалюмометоксид, диметилалюмоэтоксид, диизопропилалюмоэтоксид, диэтилалюмопропоксид, диизобутилалюмоэтоксид и метилэтилалюмоэтоксид. Хотя в примерах использован почти исключительно ДЭАлЭ, необходимо понимать, что объем изобретения им не ограничивается. Данные таблицы 1 иллюстрируют реакционные условия и характеристики получаемого полимера, когда ТЭАл и ДЭАлЭ используют с катализаторами CrOx (оксид хрома на диоксиде кремния). Предшествующие алюмоалкилу перечисленные числа в каждом случае указывают мольное соотношение между алюминием и хромом. Приведенный в таблице 1 катализатор CrOx получают пропиткой оксидом хрома диоксида кремния Grace 955 с последующими псевдоожижением воздухом и нагреванием до примерно 825°С. Катализатор Ti-CrOx готовят аналогичным образом за исключением того, что перед псевдоожижением и активацией к диоксиду кремния добавляют также тетраизопропоксид титана. Восстановители добавляют в виде дополнительной стадии приготовления катализатора.
Катализатор CrOx
Если обратиться к примерам в таблице 1, то пример 1 показывает, что в описанных полимеризационных условиях согласно ЯМР анализу отмечают 3,8 бутильных ответвлений на 1000 углеродных атомов. Это демонстрирует степень введения сомономера в полимер. Пример 2 показывает, что когда катализатор обрабатывают ТЭАл, количество введенного в таких же условиях гексена слегка уменьшается, тогда как показатель текучести полимера понижается. Пример 3 показывает, что значительную степень разветвления обнаруживают, когда катализатор обрабатывают ТЭАл, даже несмотря на отсутствие сомономера. В этом случае обнаруживают наличие как бутильных (2,4), так и этильных ответвлений (1,0). Когда катализатор обрабатывают ДЭАлЭ, то обнаруживают меньше боковых цепей полимера, а это указывает на то, что происходит введение меньшего количества сомономера (пример 4). Когда восстановитель катализатора представляет собой сочетание ТЭАл и ДЭАлЭ, можно видеть, что степень введения сомономера находится в пределах той, которая обнаружена в случае любого самостоятельно использованного восстановителя (пример 5). Когда при приготовлении катализатора используют это сочетание восстановителей катализатора и катализатор присутствует в реакции гомополимеризации, из данных примера 6 может видеть, что боковые цепи не обнаружены. Это показывает, что ДЭАлЭ подавляет образование ответвлений в виде боковых цепей в отсутствие сомономера. Как в присутствии, так и отсутствие гексена добавление ДЭАлЭ значительно уменьшает, а иногда устраняет образование в получаемом этиленовом полимере ответвлений в виде боковых цепей.
Если производить сопоставления, используя производительность (г полиэтилена/г катализатора) или активность (г полиэтилена/г катализатора/ч), то присутствие гексена становится благотворным, повышая производительность и активность. Тенденции в области молекулярной массы получаемых полимеров могут быть выявлены из обзора результатов определения показателя текучести (ПТ). Сопоставление значений ПТ у полимера, полученного с катализатором CrOx в отсутствие ТЭАл, с результатами, полученными в присутствии ТЭАл, демонстрирует увеличение молекулярной массы, на что указывает уменьшение показателя текучести. Таким образом, целесообразное применение ТЭАл и ДЭАлЭ во время приготовления катализатора создает возможность модифицировать молекулярную массу и молекулярно-массовое распределение и одновременно регулировать степень разветвления посредством боковых цепей с помощью этих катализаторов на хромоксидной основе. Эта технология обычно эффективна при получении полимеров более высокой плотности.
Если суммировать, то добавление ДЭАлЭ уменьшает степень разветвления и увеличивает молекулярную массу в случаях полимеров, полученных с CrOx. Когда сомономер не содержится, добавление ТЭАл увеличивает молекулярную массу получаемого полимера и увеличивает образование ответвлений посредством боковых цепей.
Катализатор Ti-CrOx
Катализатор Ti-CrOx является таким же, как CrOx, за исключением того, что перед активацией диоксид кремния совместно пропитывают тетраизопропоксидом титана и оксидом хрома (примеры 7-9 в таблице 1). В случае катализатора CrOx наблюдается такая же тенденция в области молекулярной массы, как и отмечаемая в случае катализатора Ti-CrOx в присутствии ТЭАл в сравнении со случаями отсутствия восстановителя.
Влияние добавления ДЭАлЭ
Было установлено также, что производительность катализаторов на хромовой основе может быть увеличена добавлением активатора, такого как ДЭАлЭ, непосредственно в реактор или в качестве части стадии приготовления катализатора. В соответствии с вышеприведенным обсуждением регулирование молекулярной массы и молекулярно-массового распределения полимера является другой особенностью изобретения.
Катализаторы на хромоксидной основе обладают высокой активностью при умеренных индукционных периодах. Эти катализаторы обеспечивают получение полимеров с промежуточным молекулярно-массовым распределением. Добавление реагентов, таких как ДЭАлЭ, в полимеризационный реактор с этими катализаторами устраняет индукционный период и повышает активность (повышение производительности). Присутствие ДЭАлЭ также модифицирует молекулярно-массовое распределение. В случае катализаторов типа силилхромата-на-диоксиде кремния (СХ) в отсутствие восстановителей вследствие длительных индукционных периодов производительность оказывается особенно плохой. Было установлено, что добавление ДЭАлЭ in-situ эффективно устраняет индукционные периоды в каталитических системах типа силилхромата-на-диоксиде кремния.
В таблице 2 представлено несколько иллюстративных технических носителей из диоксида кремния с их физическими свойствами. Было изучено влияние присутствия ДЭАлЭ и применяемого метода восстановления (прямое добавление к катализатору перед полимеризацией в сравнении с прямым добавлением (in-situ) в реактор). Эти носители из диоксида кремния являются иллюстрирующими примерами, а не исчерпывающим списком типов диоксида кремния, которые можно использовать при выполнении настоящего изобретения. При этом могут быть также использованы другие носители из диоксида кремния, которые обычно применяют и известны специалистам в данной области техники. В таблице 2 представлены приблизительные удельный объем пор, удельная площадь поверхности, средний диаметр пор, средний размер пор и количество в процентах титана для носителей из диоксида кремния, использованных в этом исследовании. Обозначение приведено в том виде, в котором его использует поставщик для идентификации носителя. Число без скобок является обозначением носителя, поставляемого в качестве диоксида кремния как такового. Число в скобках является обозначением носителя, когда его поставляют с хромовой солью, которой носитель уже пропитан. Хотя от поставщиков получали именно эти кремнеземы, полагают, что любой диоксид кремния, соответствующий приведенным ниже техническим требованиям, выполнил бы свои функции аналогичным образом. Объем настоящего изобретения не ограничен каким-либо конкретным техническим носителем из диоксида кремния, поскольку его можно выполнять с использованием любых кремнеземов, обладающих удельным объемом пор от примерно 1,1 до примерно 1,8 см3/г и удельной площадью поверхности примерно от 245 до 375 м2/г или удельным объемом пор от примерно 2,4 до примерно 3,7 см3/г и удельной площадью поверхности примерно от 410 до 620 м2/г, или удельным объемом пор от примерно 0,9 до примерно 1,4 см3/г и удельной площадью поверхности примерно от 390 до 590 м2/г.
Технические носители из диоксида кремния и физические свойства
диаметр пор (П)
Изучали эксплуатационные свойства MS 35100 как катализатора CrOx (оксид хрома на диоксиде кремния) 1) в отсутствие ДЭАлЭ, 2) когда ДЭАлЭ добавляли непосредственно к катализатору и 3) когда его вводили в реактор in situ. Реакции проводили в 500 мл гексановой суспензии с 10 мл добавленного 1-гексена; реакцию осуществляли при 85°С и под общим давлением 200 фунтов/кв.дюйм. Фиг.3 иллюстрирует молекулярно-массовое распределение получаемого полимера в отсутствие и присутствии ДЭАлЭ. В отсутствие ДЭАлЭ (фиг.3(а)) полученный полимер обладал молекулярно-массовым распределением 16,9. Когда ДЭАлЭ добавляли in-situ (фиг.3(б)), наблюдали расширение молекулярно-массового распределения, причем при молекулярно-массовом распределении 23,8 наличие плечевого участка становилось очевидным. Аналогичных, но менее заметных результатов добивались, когда ДЭАлЭ добавляли в катализатор перед полимеризацией (фиг.3(в)), причем высокомолекулярный участок был слегка менее видимым. Когда ДЭАлЭ добавляли непосредственно к катализатору, фиксировали значение молекулярно-массового распределения полимера 32,4. Аналогичную тенденцию значения Mz/Mw наблюдали, когда добавляли ДЭАлЭ. Значение Mz/Mw служило указанием на высокомолекулярный участок: по мере увеличения Mz/Mw целевой высокомолекулярный участок становился более заметным. Данные Mz/Mw получали в результате анализа полимера эксклюзивной хроматографией. В отсутствие ДЭАлЭ (фиг.3(а)) фиксировали значение Mz/Mw 5,7. Когда ДЭАлЭ добавляли in-situ и к катализатору (фиг.3(6) и 3(в)), получали соответственно значения Mz/Mw примерно от 7,7 и 9,6.
Как свидетельствуют данные таблицы 3, повышений плотности полимера и активности катализатора достигали как при прямом добавлении к катализаторам (к катализатору), так и при добавлении in-situ (в реактор). Что касается введения сомономера, то, о чем свидетельствует параметр разветвления (ЧБО), в сравнении с отсутствием ДЭАлЭ он указывает на уменьшение степени введения сомономера в случаях как добавления ДЭАлЭ in-situ, так и добавления ДЭАлЭ к катализатору. При этом происходило небольшое уменьшение молекулярной массы, о чем свидетельствует увеличение показателя текучести при применении ДЭАлЭ. Как продемонстрировано на фиг.4, когда ДЭАлЭ добавляли либо in-situ, либо непосредственно к катализатору перед полимеризацией, индукционные периоды фактически устранялись. Устранение индукционных периодов в случаях добавления ДЭАлЭ in-situ или к катализатору контрастирует с длительными индукционными периодами, которые наблюдали при той же каталитической системе в отсутствие ДЭАлЭ. И, наконец, в случаях катализатора CrOx при добавлении ДЭАлЭ in-situ он ведет себя не так, как при добавлении ДЭАлЭ к катализатору перед полимеризацией.
Аналогичные эксперименты проводили с хромоксидными катализаторами 957HS. Реакции осуществляли в 500 мл гексановой суспензии с 10 мл добавленного 1-гексена; реакцию проводили при 85°С и под общим давлением 200 фунтов/кв.дюйм. Фиг.5 иллюстрирует молекулярно-массовое распределение получаемого полимера в отсутствие и присутствии ДЭАлЭ. В отсутствие ДЭАлЭ (фиг.5(а)) полученный полимер проявлял молекулярно-массовое распределение 9,7, а молекулярная масса была намного меньше 500000. Когда ДЭАлЭ добавляли in-situ (фиг.5(б)), увеличение молекулярно-массового распределения полимера наблюдали до значения примерно 12,0. Значения Mz/Mw демонстрировали, что при добавлении ДЭАлЭ появлялся высокомолекулярный участок, причем значение Mz/Mw составляло примерно 4,5 в отсутствие ДЭАлЭ и примерно 8,6 и примерно 8,3 соответственно в случаях ДЭАлЭ, добавленного in-situ, и ДЭАлЭ, добавленного к катализатору. Повышения плотности и пониженная степень образования ответвлений посредством боковых цепей возникали как при прямом добавлении к катализаторам, так и при добавлении in-situ (в реактор), что видно из данных в таблице 4. Умеренное уменьшение молекулярной массы было продемонстрировано увеличением показателя текучести. Аналогично тому эффекту, который наблюдали в случае MS35100 как катализатора CrOx, добавление ДЭАлЭ к 957HS как катализатору CrOx либо путем введения in-situ, либо прямым добавлением к катализатору приводило к фактическому устранению индукционного периода и вследствие этого к повышению активности катализатора (фиг.6). В заключение, добавление ДЭАлЭ in-situ к этой каталитической системе с CrOx приводило к более высокой активности, более низкой молекулярной массе, сопоставимому молекулярно-массовому распределению и сопоставимому введению сомономера с тем, что происходило в случае, когда ДЭАлЭ добавляли непосредственно к катализатору перед полимеризацией. Как добавление in-situ, так и прямое добавление в полимер приводило к по существу нулевому индукционному периоду в сравнении с ограниченными индукционными периодами, наблюдаемыми в отсутствие ДЭАлЭ.
Исследовали также эксплуатационные свойства ЕР352 как катализатора CrOx 1) в отсутствие ДЭАлЭ, 2) когда ДЭАлЭ добавляли непосредственно к катализатору и 3) когда его вводили в реактор in situ. Реакции осуществляли в 500 мл гексановой суспензии с 10 мл добавленного 1-гексена; реакцию проводили при 85°С и под общим давлением 200 фунтов/кв.дюйм. Фиг.7 иллюстрирует молекулярно-массовое распределение получаемого полимера в присутствии ДЭАлЭ. Когда ДЭАлЭ добавляли in-situ (фиг.7(а)), наблюдали более широкое молекулярно-массовое распределение в сравнении со случаем с ДЭАлЭ, добавленным непосредственно к катализатору (фиг.7(б)), при наличии высокомолекулярного участка в обоих случаях аналогично тому, что отмечали в случае ЕР352 как катализатора CrOx без ДЭАлЭ. Увеличения плотности полимера и более низкая степень образования ответвлений посредством боковых цепей возникали как при прямом добавлении к катализаторам (в катализатор), так и при добавлении in-situ (в реактор), что видно из данных в таблице 5. Однако добавление ДЭАлЭ in-situ к ЕР352 как катализатору CrOx приводило к небольшому изменению активности относительно того, что отмечали в отсутствие ДЭАлЭ. Это находится в полном противоречии со случаем добавления ДЭАлЭ непосредственно к катализатору перед полимеризацией, когда наблюдали существенное повышение активности катализатора. Фиг.8 демонстрирует улучшение индукционного периода в присутствии ДЭАлЭ, причем это улучшение происходило как когда дЭАлЭ добавляли in-situ, так и когда его добавляли к катализатору. В заключение, добавление ДЭАлЭ in-situ к этой каталитической системе с CrOx приводило к более высокой активности, более широкому молекулярно-массовому распределению и сопоставимому введению сомономера с тем, что отмечали, когда ДЭАлЭ добавляли непосредственно к катализатору перед полимеризацией. Индукционный период улучшался при любом методе добавления ДЭАлЭ, если сравнивать с отсутствием ДЭАлЭ.
Аналогичные данные для СХ катализатора на MS3050 проиллюстрированы на фиг.9 и 10 и в таблице 6. Как можно видеть из фиг.10, добавление ДЭАлЭ влияло на абсолютное улучшение индукционного периода: фактическое устранение индукционного периода для СХ катализатора. Это также очевидно из значительного повышения активности, как это продемонстрировано данными в таблице 6. Длительные индукционные периоды являются главными недостатками катализаторов силилхромат-на-диоксиде кремния, добавление in-situ ДЭАлЭ или других алюмоалкильных соединений значительно повышало активность благодаря устранению индукционного периода. Молекулярная масса получаемого полимера уменьшалась, о чем свидетельствовало значительное увеличение показателя текучести. Хотя молекулярная масса получаемого полимера уменьшалась, катализатор характеризовался улучшенной применимостью в двухкатализаторной системе с использованием дополнительного катализатора для получения высокомолекулярного полимера.
Изучали также СХ катализатор на диоксиде кремния Grace 955. Когда добавляли ДЭАлЭ, вновь наблюдали заметное улучшение индукционного периода. Это имеет важное значение, поскольку, когда используют катализаторы типа силилхромата-на-диоксиде кремния, длительный индукционный период является основным недостатком. Как продемонстрировано на фиг.11, характеристики молекулярной массы и молекулярно-массового распределения при добавлении ДЭАлЭ в этот СХ катализатор изменялись незначительно. Из оценки данных в таблице 7 можно видеть, что, когда ДЭАлЭ добавляли in-situ, этого не происходило. Во всех случаях добавление ДЭАлЭ фактически устраняло индукционный период (фиг.12). Добавление in-situ значительно повышало активность и уменьшало молекулярную массу полимера. Применение ТИБА с катализаторами СХ типа создает каталитическую систему, которая обладает высокой производительностью и обеспечивает образование полимера с более высокой молекулярной массой, чем достигаемая, когда ДЭАлЭ используют в качестве восстановителя. Это имеет особенно важное значение для сохранения молекулярной массы полимера при более кратковременных продолжительностях пребывания. Аналогичного действия ожидают, по-видимому, и от других алюмоалкильных соединений, таких как триэтилалюминий и три-н-гексилалюминий.
В заключение, применение ДЭАлЭ или ТИБА с силилхроматными катализаторами приводило к достижению характеристик молекулярной массы полимера (молекулярная масса, молекулярно-массовое распределение, высокомолекулярные участки и т.д.), аналогичным характеристикам, достигнутым без применения ДЭАлЭ или ТИБА, но при более лучших значениях производительности, чем в отсутствие этих соединений алюминия. Таким образом, при использовании ДЭАлЭ или ТИБА сохраняются позитивные свойства молекулярной массы полученных с помощью силилхромата полимеров с сопутствующим повышением активности. Применение ТЭАл и ДЭАлЭ с катализаторами CrOx приводило к образованию полимеров, более схожих с теми, которые получены с использованием CX катализаторов, при одновременном сохранении целевых показателей активности, присущих CrOx, при получении полимеров. Постоянное варьирование ТЭАл и ДЭАлЭ в каталитических системах как с CrOx, так и с СХ позволяет создать механизм для целевого изменения характеристик получаемого таким образом полиэтилена при одновременном сохранении хороших показателей активности. Таким путем для получения ряда полиэтиленов разных сортов может быть оптимизирована объемная производительность (масса полимера на единицу реакторного объема на единицу времени).
Влияние сокатализатора на эксплуатационные свойства
Влияние сокатализатора на эксплуатационные свойства СХ катализатора (обработанного 5 экв. ДЭАлЭ/Cr) было изучено с использованием следующих сокатализаторов: ТЭАл, ТИБА (триизобутилалюминий) и ТнГАл (три-н-гексилалюминий). Хотя примеры ограничены конкретными сокатализаторами, необходимо уяснить себе, что возможно использование других алюмоалкильных соединений, которые при этом составляют часть изобретения. В таблице 8 и на фиг.13-21 представлены показатель текучести, активность, плотность, индукционный период и различные связанные с молекулярной массой данные для полимеров, полученных, когда варьировали сокатализатор. Базовой каталитической системой, результаты изучения которой отражают данные таблицы 8 и фиг.13-21, является СХ катализатор с 5 экв. ДЭАлЭ на эквивалент Cr (обозначен в настоящем описании как SC-500). Тенденция в области показателя текучести в таблице 8 указывает на увеличение молекулярной массы при добавлении сокатализатора. Таблица 8 также показывает, что активность катализатора при добавлении сокатализатора возрастает. Необходимо отметить, что в качестве сокатализатора для СХ катализаторов может быть также использован ТЭБ (триэтилбор). Следует уяснить себе, что по определению сокатализатор всегда добавляют "в реактор".
Фиг.13 и 14 демонстрируют общее повышение активности катализатора и молекулярной массы с максимальным эффектом при примерно от 1 до 2 экв. Al на эквивалент Cr. Не основываясь на какой-либо теории, полагают, что в более высоких концентрациях сокатализатор начинает в более высокой степени отравлять катализатор. Фиг.15-17 иллюстрируют влияние сокатализатора на индукционный период. Во всех случаях можно видеть, что, когда содержится сокатализатор, пики активности являются более высокими и в значительной мере остаются более высокими. Благодаря присутствию сокатализатора для системы с SC-500 индукционные периоды по существу устраняются.
Фиг.18-21 демонстрируют влияние присутствия сокатализатора на молекулярно-массовое распределение получаемого полимера. Несмотря на то что по предыдущим наблюдениям благодаря сокатализатору молекулярная масса возрастала, молекулярно-массовое распределение оставалось в значительной мере неизменным. Более того, интенсивность высокомолекулярного участка, на что указывало значение Mz/Mw, также оставалась неизменной относительно этого параметра у полиэтилена, полученного с использованием SC-500 в отсутствие сокатализатора. В заключение, сокатализатор повышал активность катализатора и молекулярную массу полимера в случае катализатора SC-500, но молекулярно-массовое распределение полимера оказывалось в значительной мере неизменным. Эти особенности необходимы для проведения процесса с кратковременной продолжительностью пребывания.
Тот же эффект отмечался при использовании СХ катализатора, включавшего 1,5 экв. ДЭАлЭ/эквивалент Cr (в настоящем описании обозначен как SC-150). В таблице 9 и на фиг.22-28 представлены индукционный период, активность и различные связанные с молекулярной массой данные у полимеров, полученных, когда варьировали сокатализатор. Ранее отмеченные тенденции для SC-500 очевидны и в случае SC-150. При добавлении сокатализаторов в эти каталитические системы индукционные периоды (см. фиг.22-24) фактически устранялись. Фиг.25-28 продемонстрировали, что на молекулярно-массовое распределение сокатализатор в существенной степени не влиял. Интенсивность высокомолекулярного участка, на что указывает значение Mz/Mw, также оставалась неизменной, если сравнивать с полиэтиленом, полученным с помощью SC-150 в отсутствие сокатализатора. Если суммировать сказанное, то сокатализатор повышает активность катализатора в случае катализатора SC-150, но молекулярно-массовое распределение полимера остается в существенной степени неизменным. Следовательно, целесообразный выбор сокатализатора позволяет модифицировать молекулярную массу и повысить активность катализатора.
Добавление сокатализатора также оказывает благоприятное влияние на катализаторы CrOx. В таблице 10 и на фиг.29-34 представлены данные, демонстрирующие влияние сокатализатора на эксплуатационные свойства Ti-CrOx (на диоксиде кремния Grace 955). Таблица 10 показывает, что показатель текучести при добавлении ТЭАл уменьшается и, следовательно, благодаря применению 5 экв. сокатализатора в случае катализатора Ti-CrOx молекулярная масса полимера возрастает. Активность Ti-CrOx реагирует на использование сокатализатора таким же образом, как и в случаях вышеописанных катализаторов SC-500 и SC-150.
Повышение активности заметно, особенно при содержании от 1 до 2 экв. Al на эквивалент Cr. Как видно из фиг.31-34, когда содержится сокатализатор, молекулярно-массовое распределение расширяется, а резко выраженный высокомолекулярный участок не появляется. Расширение молекулярно-массового распределения полимера обычно улучшает физические свойства без увеличения набухания полимера.
Более того, при создании изобретения было установлено, что для выполнения настоящего изобретения могут быть также использованы различные сокатализаторы, не основанные на алюминии. Так, например, на предмет его влияния на эксплуатационные свойства катализатора был изучен ТЭБ (триэтилбор). Таблица 11 демонстрирует влияние сокатализатора ТЭБ на эксплуатационные свойства CrOx (оксид хрома на диоксиде кремния Grace 955) и каталитические системы с Ti-CrOx.
Фиг.35-36 иллюстрируют связанные с молекулярной массой данные для полиэтилена, полученного с помощью катализатора Ti-CrOx самостоятельно (фиг.35), а также Ti-CrOx с сокатализатором ТЭБ (фиг.36). Молекулярная масса полимера возрастает, на что указывает уменьшение показателя текучести при применении ТЭБ в сравнении со случаем без сокатализатора для систем как с CrOx, так и с Ti-CrOx в отсутствие водорода. Применение ТЭБ на активность катализатора в обеих каталитических системах существенного влияния не оказывало, однако ТЭБ расширял молекулярно-массовое распределение. Более того, расширение молекулярно-массового распределения, на которое оказывало влияние применение ТЭБ, происходило в сопровождении роста только участка умеренной молекулярной массы (фиг.36), как и в случае, когда в качестве сокатализатора использовали ДЭАлЭ.
Выполнение настоящего изобретения позволяет манипулировать молекулярной массой, молекулярно-массовым распределением, активностью катализатора, а также другими свойствами получаемого полиэтилена благодаря целесообразному применению сокатализатора в общем, а конкретно алюмоалкильных сокатализаторов. Алюмоалкильные соединения, которые специально обсуждались в настоящем описании, рассматривались только в качестве неограничивающего примера; как часть настоящего изобретения могут быть также использованы другие алюмоалкилы. Подобным же образом при выполнении настоящего изобретения также могут быть использованы алкилалюмоалкоксиды, отличные от ДЭАлЭ. К ним относятся, хотя ими их список не ограничен, диэтилалюмоэтоксид, диметилалюмоэтоксид, дипропилалюмоэтоксид, диэтилалюмопропоксид и метилэтилалюмоэтоксид. Благодаря целесообразному применению сокатализатора существует возможность модифицировать эти свойства и подогнать получаемый полимер к конкретным целям применения. Важное значение имеет то, что по изобретению предлагается получение высокомолекулярных полиэтиленов с помощью катализаторов на хромовой основе с высокими показателями активности, что создает возможность проводить процесс при более кратковременных продолжительностях пребывания в реакторе. Это создает предпосылки для улучшений объемной производительности при получении полиэтилена с использованием катализаторов на хромовой основе при одновременном поддержании высоких реакционных температур.
Примеры газофазного процесса в псевдоожиженном слое
Ниже приведены примеры газофазного процесса в псевдоожиженном слое по настоящему изобретению. Для непрерывного получения этилен-гексенового сополимера высокой плотности применяли газофазный полимеризационный реактор с псевдоожиженным слоем технологической конструкции UNIPOL™, обладавший номинальным диаметром 14 дюймов. В этих случаях вентилятор рециклового газа размещали в технологической линии перед теплообменником для рециклового газа в рециркуляционном контуре для газа, но размещение обоих можно был поменять на обратное с целью понизить температуру газа, когда он входил в теплообменник. Рециркуляционный патрубок обладал диаметром примерно 2 дюйма, а скорость потока в нем варьировали шаровым клапаном на рециркуляционной линии для поддержания на целевом уровне расхода газа на единицу сечения потока в псевдоожиженном слое. Мономеры и газообразные компоненты вводили в технологической линии до холодильника и перед вентилятором, по месту рабочего колеса вентилятора или после вентилятора. Сухой катализатор непрерывно вводили отдельными небольшими аликвотами по патрубку в 1/8 дюйма непосредственно в псевдоожиженный слой на высоте от примерно 0,1 до 2 м над распределительной плитой, а наиболее предпочтительно в интервале от примерно 0,2 до 1,2 м с использованием потока азотного газообразного носителя на участке, находившемся на расстоянии от примерно 15 до 50% диаметра реактора. Для поддержания целевых приблизительных средних уровня или массы псевдоожиженного слоя полимерный продукт периодически отводили из реактора посредством разгрузочной выделительной емкости аликвотами примерно от 0,2 до 5 кг. В некоторых экспериментах для варьирования молекулярной массы и молекулярно-массового распределения полимера предусматривали наличие и использовали разбавленный поток кислорода в азоте (200 об. част./млн). Его вводили в рецикловый газ перед теплообменником, когда в реакционной системе не содержался свободный алюмоалкил, но когда содержались свободные ТЭАл и ДЭАлЭ, то с целью избежать возможности взаимодействия некоторого количества кислорода с алюмоалкилом в рециркуляционной линии или теплообменнике перед поступлением в псевдоожиженный слой точку его добавления переключали на псевдоожиженный слой. Это являлось мерой предосторожности и не препятствовало его добавлению в рециркуляционную линию или перед теплообменником.
В отдельные промежутки времени проводили различные серии экспериментов, и каждая серия включала сравнительный эксперимент. С течением времени между экспериментальными сериями в потоке исходных материалов и в реакторах варьировали окружающие примеси, которые вызывали небольшие сдвиги реакционной температуры и производительности катализатора. Сравнительные эксперименты включали катализатор, приготовленный на промышленном технологическом оборудовании, а также катализаторы, приготовленные в лабораториях. Приготовленные в лабораториях катализаторы требовали более низкой реакционной температуры, а их применение являлось сравнительным случаем для экспериментальных катализаторов, также приготовленных в лаборатории.
Примеры с 54 по 59 в таблице 12 демонстрируют результаты применения различных носителей и источников хрома. Во всех примерах реактор работал хорошо, без образования отложений или комков. Примеры 54 и 55 демонстрируют результаты для сравнительного катализатора (силилхромат, полученный на диоксиде кремния 955, обезвоженном при температуре 600°С, и восстановленный 5 экв. ДЭАлЭ). Экспериментальные катализаторы сравнивали с катализатором примера 55. СХ катализатор, приготовленный на носителе MS3050 (пример 56), проявлял значительно более высокую производительность катализатора и обеспечивал получение полимера с широким молекулярно-массовым распределением с высокомолекулярным участком. Катализатор, использованный в примерах 57 и 58, готовили на основе CrOx на диоксиде кремния 955, активировали при 825°С и затем восстанавливали 5 экв. ДЭАлЭ. В обоих случаях достигали более высоких производительностей катализатора и для получения полимера требовались более высокие реакционные температуры. Это показывает, что такие катализаторы по существу обеспечивали получение полимера более высокой молекулярной массы и обычно эффективны в процессе с кратковременной продолжительностью пребывания. В примере 58 также использовали повторно вводимый в реактор кислород, который при данной температуре понижал молекулярную массу полимера и увеличивал значения соотношения вязкостей расплавов полимера (СВР) (что указывало на более широкое молекулярно-массовое распределение полимера). Пример 59 демонстрирует результаты для катализатора PQ CrOx (CrOx на MS3050), активированного при 700°С с последующим восстановлением 5 экв. ДЭАлЭ. При этом вновь достигали более высокой производительности катализатора, и для получения полимера требовались более высокие реакционные температуры.
В заключение, эти результаты газофазных экспериментов подтверждают наблюдения, сделанные в предыдущих примерах. С применением альтернативных носителей для приготовления силилхроматных катализаторов могут быть достигнуты более высокие производительности катализаторов и получены более высокомолекулярные полимеры. Аналогичных улучшений можно также добиться с использованием восстановленных катализаторов CrOx. Во всех случаях получали полимеры с широким молекулярно-массовым распределением и целевым высокомолекулярным участком.
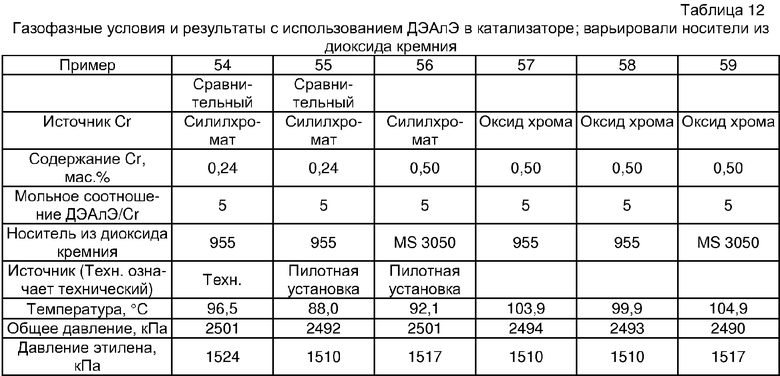

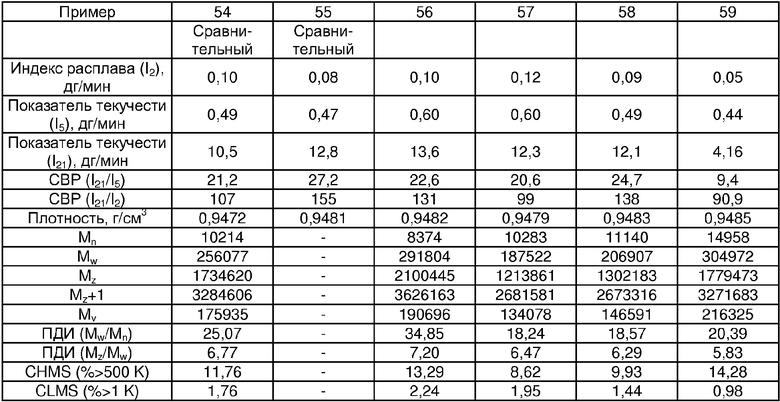
СРЧ - средний размер частиц
Эксперименты примеров с 60 по 64 в таблице 13 проводили в реакторе аналогично тем, которые представлены в таблице 12. Пример 60 является сравнительным примером. Примеры с 61 по 64 демонстрируют влияние добавления ТЭАл к стандартному силилхроматному катализатору (силилхромат, полученный на диоксиде кремния 955, обезвоженном при температуре 600°С, и восстановленный 5 экв. ДЭАлЭ). В таблице 13 результаты демонстрируют оптимум в отношении количества ТЭАл, добавленного в газофазную полимеризацию в псевдоожиженном слое, относительно силилхроматного катализатора в пересчете на производительность, характеристик частиц смолы, повышенной реакционной температуры и СВР. Для указанных катализатора и реакционных условий этот оптимум находился в интервале приблизительно от 0,5 до 3 ТЭАл/Cr, а более предпочтительно в интервале от 1 до 2 ТЭАл/Cr. В этой серии экспериментов катализатор был одинаковым. Значения производительности представляли, основываясь на скорости добавления катализатора и материальном балансе производительности по смоле. Хром, остающийся в смоле, аналогичен тенденциям в производительности. Мольное соотношение при добавлении ТЭАл/Cr было основано на скорости подачи ТЭАл и содержании Cr в смоле, как это определяли по рентгенографическому методу. ТЭАл добавляли в слой с использованием трубки 1/8 дюйма, смонтированной подобно трубке для инжекции катализатора, но без продувки азотом. ТЭАл вводили в виде разбавленного раствора в очищенном изопентане, и контейнер, в котором его готовили, перед наполнением предварительно обработали ТЭАл с целью уменьшить возможность попадания реакционноспособных примесей, таких как вода в контейнере, которые расходовали бы небольшое количество содержавшегося ТЭАл. Во время добавления ТЭАл реактор работал хорошо, без образования отложений, чешуек иди комков. Определение статического электричества в слое с помощью обладавшего высоким сопротивлением и большой емкостью электронного зонда демонстрировало пониженные уровни, когда содержался ТЭАл - сохранялась статическая нейтральность, но в более тонком слое. Настеночные поверхностные термопары, размещенные на различных расстояниях выше плиты в псевдоожиженном слое и в части, находившейся выше слоя, давали превосходные данные в случае отсутствия ТЭАл, и эти данные казались даже улучшенными в присутствии ТЭАл, с малыми колебаниями и сдвигом на примерно от 1 до 2°С в направлении ближе (от низа) к средней температуре сердцевины слоя.
В заключение, добавление сокатализатора (ТЭАл) приводило к более высокой активности катализатора и для получения полимера с такой же молекулярной массой позволяло проводить процесс в реакторе при более высоких температурах. Во всех этих примерах молекулярно-массовое распределение полимера оставалось неизменным.

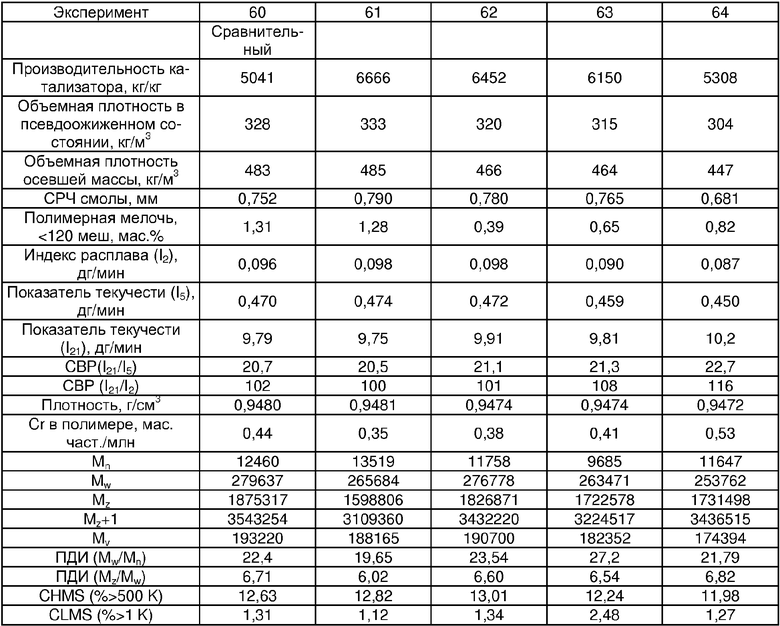
Эксперименты примеров 65-73 (данные сведены в таблицу 14) и примера 74 (обсуждаются в приведенном ниже тексте) проводили в газофазных полимеризационных реакторах, аналогичных реакторам предыдущих экспериментов. В экспериментах примеров с 65 по 71 проверяли влияние добавления сокатализатора ТЭАл в предпочтительном интервале при высокой и низкой объемной производительности и с катализаторами, приготовленными при двух содержаниях ДЭАлЭ/Cr катализатора (5 экв. ДЭАлЭ/Cr и 1,5 экв. ДЭАлЭ/Cr). При каждой изученной ОПР ТЭАл повышал производительность катализатора примерно на 35%, а также повышал реакционную температуру при каждой объемной производительности на примерно от 3 до 5°С. ТЭАл позволял проводить процесс при более высоких значениях объемной производительности с производительностью катализатора, сопоставимой или превышающей производительность катализатора при более низкой объемной производительности без ТЭАл. Когда процесс проводили при более высокой объемной производительности в присутствии ТЭАл, размер частиц смолы увеличивался, а содержание мелочи уменьшалось в сравнении с достигаемыми без него. С повышением объемной производительности СВР увеличивалось. В присутствии ТЭАл эксплуатационные свойства катализаторов с низким и высоким содержанием ДЭАлЭ были аналогичными, но в его отсутствие - разными. Как можно видеть, при проведении процесса без присутствия сокатализатора (прим. 67 и 70) производительность катализатора и требуемая температура в реакторе при высокой объемной производительности (малые продолжительности пребывания) оказывались неадекватными. Эти результаты газофазных процессов подтверждают результаты предыдущих примеров, демонстрирующих применение сокатализатора в сочетании с силилхроматными катализаторами.
Пример 72 демонстрирует применение повторно вводимого кислорода с добавлением сокатализатора. При добавлении кислорода в реактор показатель текучести полимера увеличивался. Кислород можно добавлять для регулирования молекулярной массы и молекулярно-массового распределения полимера.
В примере 73 вместо ТЭАл в реактор добавляли ДЭАлЭ с использованием катализатора с более высоким содержанием оксида хрома (0,5 мас.% Cr на диоксиде кремния 955, активирован при 825°С), что приводило к повышенной производительности катализатора и повышенной реакционной температуре в сравнении со стандартными условиями процесса с силилхроматом совместно или без ТЭАл.
Пример 74: добавление ТЭАл в проводимую реакцию полимеризации с использованием низкого соотношения ДЭАлЭ/Cr силилхроматный катализатор (ДЭАлЭ/Cr 1,5:1) в псевдоожиженном слое дважды приводило к образованию отложений и агломератов полимера, которые блокировали разгрузочное отверстие для смолы, вынуждая останавливать реактор.
В экспериментах с 65 по 72 реактор работал хорошо. ТЭАл успешно вводили в свободную от ТЭАл систему с использованием силилхроматного катализатора с соотношением ДЭАлЭ/Cr 5:1. Эксперименты с ТЭАл при соотношении ДЭАлЭ/Cr катализатор 1,5:1 успешно проводили с переходом от соотношения 5:1 до 1,5:1 катализатора в случаях, когда ТЭАл уже содержался в реакторе с псевдоожиженным слоем. В предпочтительном варианте инициировали добавление катализатора, в особенности при более низких соотношениях ДЭАлЭ/Cr катализаторы в слой, который уже содержал достаточное количество ТЭАл.
Добавление ТЭАл и ДЭАлЭ в реакторы осуществляли с предварительно рассчитанной скоростью, а затем, когда эксперимент завершали, рассчитывали соотношение Al/Cr. При предопределенном соотношении Al/Cr, основываясь на скорости добавления катализатора, можно было бы регулировать или точно определять приблизительную постоянную скорость подачи ТЭАл или ДЭАлЭ. Скорость их подачи можно было бы также соразмерить с производительностью по смоле для поддержания их концентрации на некотором указанном уровне, предпочтительно на таком, который позволяет добиться целевых результатов с использованием минимального количества реагента.
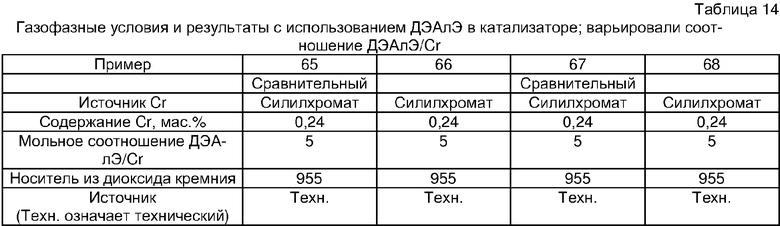
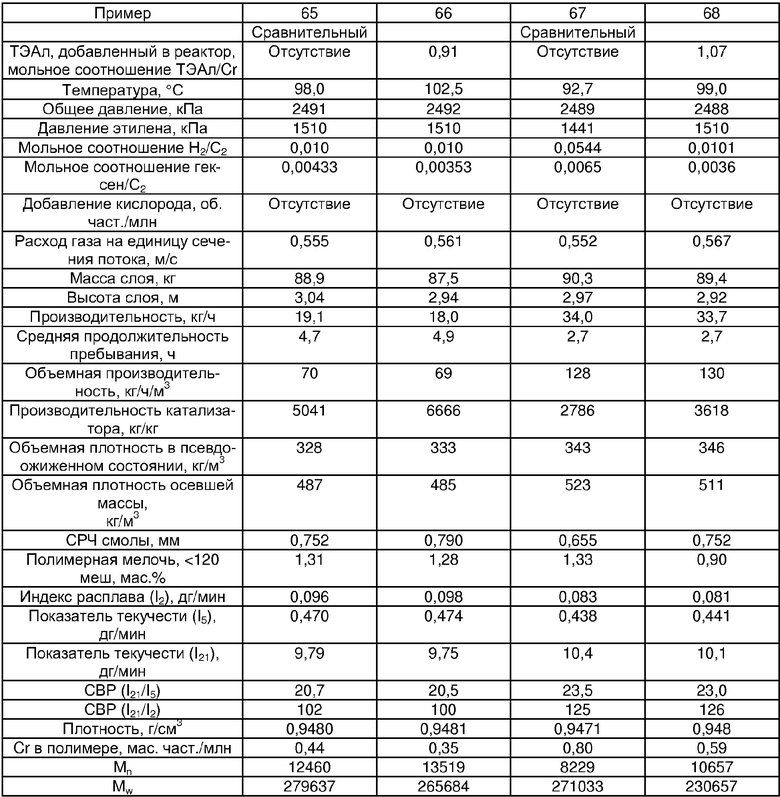
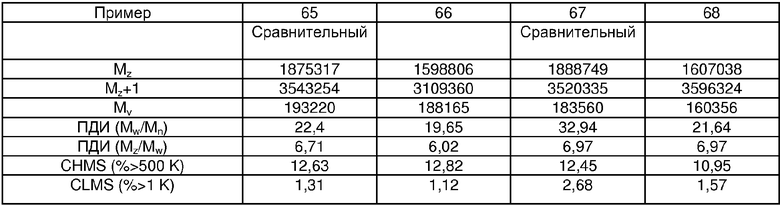
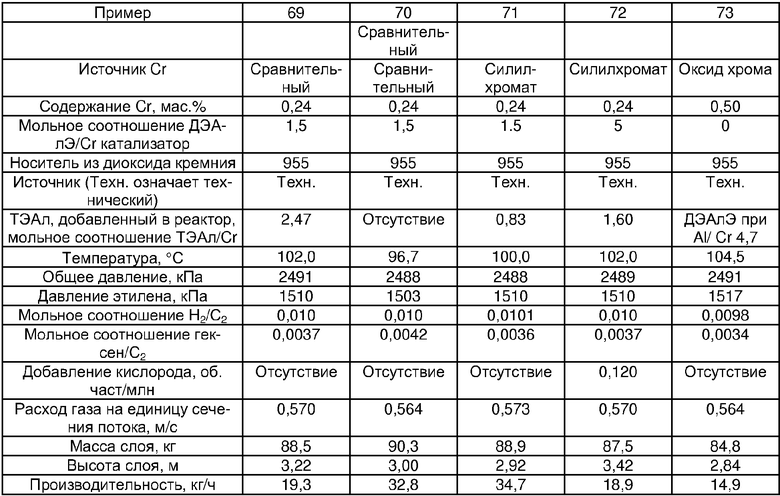
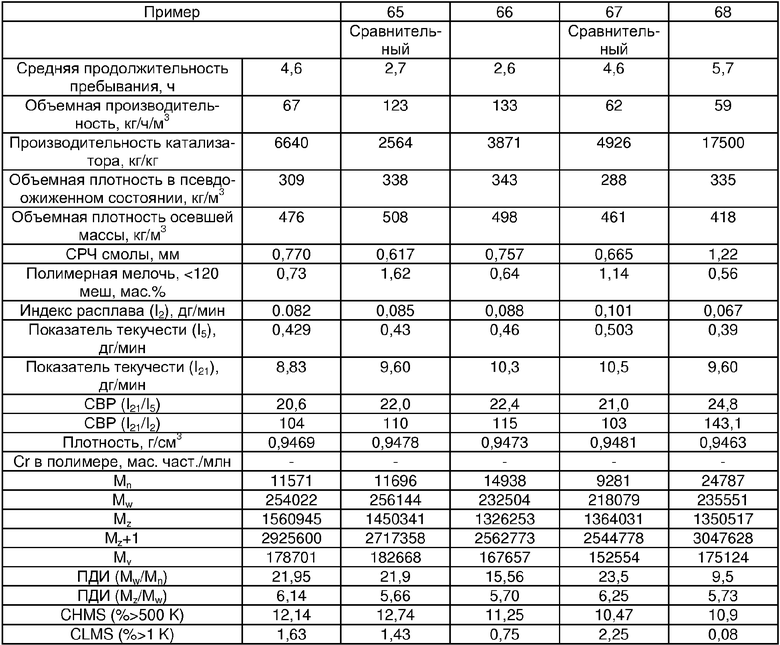
Хотя настоящее изобретение и его преимущества описаны подробно, необходимо уяснить себе, что при этом можно осуществлять различные изменения, замены и другие варианты, не выходя из сущности и объема изобретения, которые определены прилагаемой формулой изобретения. Более того, отсутствует какое-либо намерение ограничить объем данной заявки конкретными вариантами, представленными в описании. Для обычного специалиста в данной области техники из описания настоящего изобретения вполне очевидно, что в соответствии с настоящим изобретением можно использовать способы, устройства, процессы приготовления, обсуждаемые композиции, средства, методы или стадии, которые уже существуют или будут разработаны или созданы в дальнейшем и которые выполняют по существу те же функции или обеспечивают достижение по существу того же результата, что и соответствующие варианты, представленные в настоящем описании. Таким образом, подразумевается, что прилагаемая формула изобретения в своем объеме охватывает такие способы, устройства, процессы приготовления, обсуждаемые композиции, средства, методы или стадии.
| название | год | авторы | номер документа |
|---|---|---|---|
| СМЕШАННЫЕ СОВМЕСТИМЫЕ КАТАЛИЗАТОРЫ ЦИГЛЕРА-НАТТЫ/ХРОМОВЫЕ ДЛЯ ПОЛУЧЕНИЯ УЛУЧШЕННЫХ ПОЛИМЕРНЫХ ПРОДУКТОВ | 2013 |
|
RU2662936C2 |
| ПОЛИОЛЕФИНОВАЯ ПЛЕНКА С УЛУЧШЕННОЙ УДАРНОЙ ПРОЧНОСТЬЮ | 2017 |
|
RU2739839C2 |
| СПОСОБЫ ПОЛУЧЕНИЯ ХРОМОВЫХ КАТАЛИЗАТОРОВ НА ПОДЛОЖКЕ С ПОВЫШЕННОЙ АКТИВНОСТЬЮ ПОЛИМЕРИЗАЦИИ | 2019 |
|
RU2827812C2 |
| МОДИФИКАТОР ИНДЕКСА ТЕКУЧЕСТИ ПОЛИМЕРА | 2017 |
|
RU2736067C2 |
| СПОСОБЫ РЕГУЛИРОВАНИЯ СВОЙСТВ ПОЛИМЕРА | 2014 |
|
RU2670954C9 |
| СПОСОБЫ РЕГУЛИРОВАНИЯ СВОЙСТВ ПОЛИМЕРА | 2014 |
|
RU2815015C2 |
| ПОЛИЭТИЛЕНЫ С РАСШИРЕННЫМ МОЛЕКУЛЯРНО-МАССОВЫМ РАСПРЕДЕЛЕНИЕМ | 2014 |
|
RU2672730C2 |
| ПОЛИМЕРНЫЕ ФОРМОВОЧНЫЕ КОМПОЗИЦИИ | 2005 |
|
RU2397185C2 |
| ПОЛУЧЕНИЕ ДИОКСИДОВ КРЕМНИЯ С КРУПНЫМИ ПОРАМИ И ИХ ПРИМЕНЕНИЕ В ХРОМОВЫХ КАТАЛИЗАТОРАХ ДЛЯ ПОЛИМЕРИЗАЦИИ ОЛЕФИНОВ | 2020 |
|
RU2790065C1 |
| ХРОМСОДЕРЖАЩИЙ КАТАЛИЗАТОР НА ОСНОВЕ ДИОКСИДА КРЕМНИЯ, СПОСОБ ЕГО ПОЛУЧЕНИЯ И СПОСОБ ПОЛУЧЕНИЯ ПОЛИЭТИЛЕНА С ИСПОЛЬЗОВАНИЕМ ДАННОГО КАТАЛИЗАТОРА | 2000 |
|
RU2262512C2 |
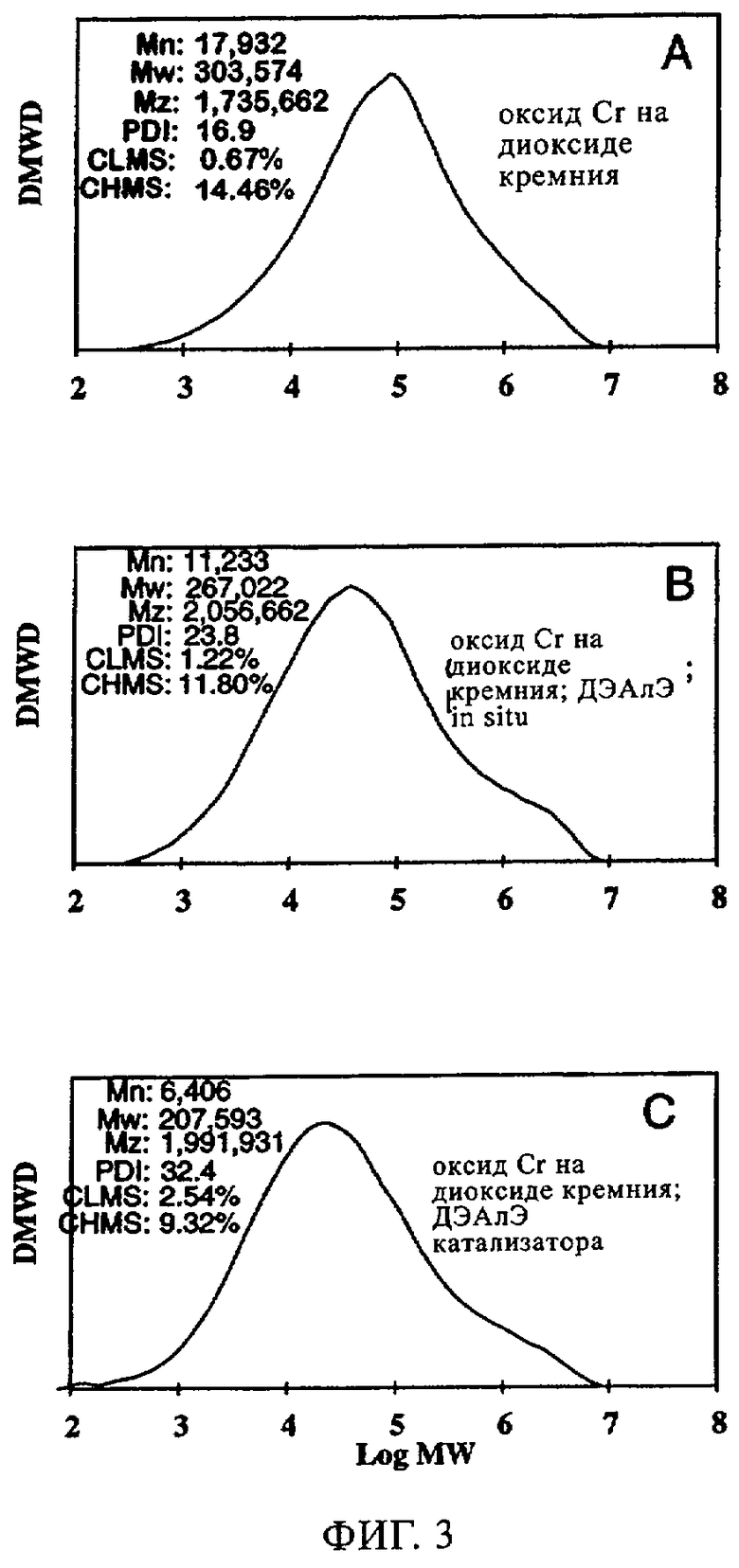
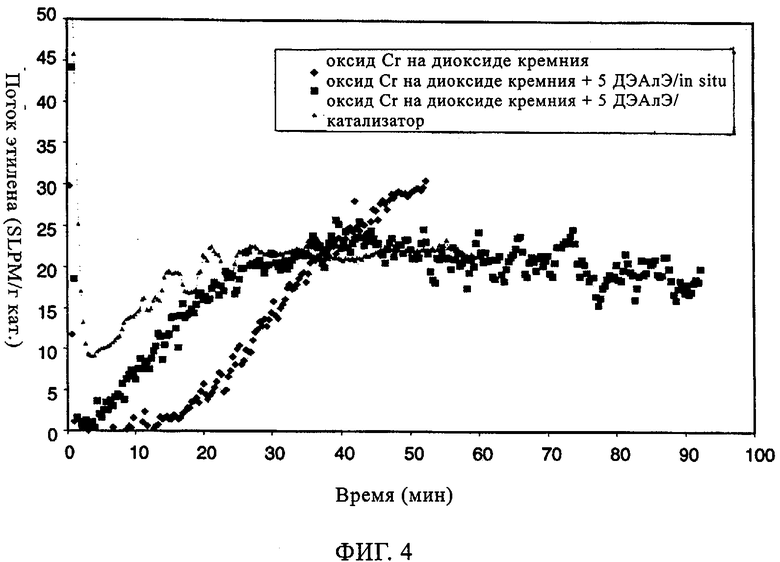
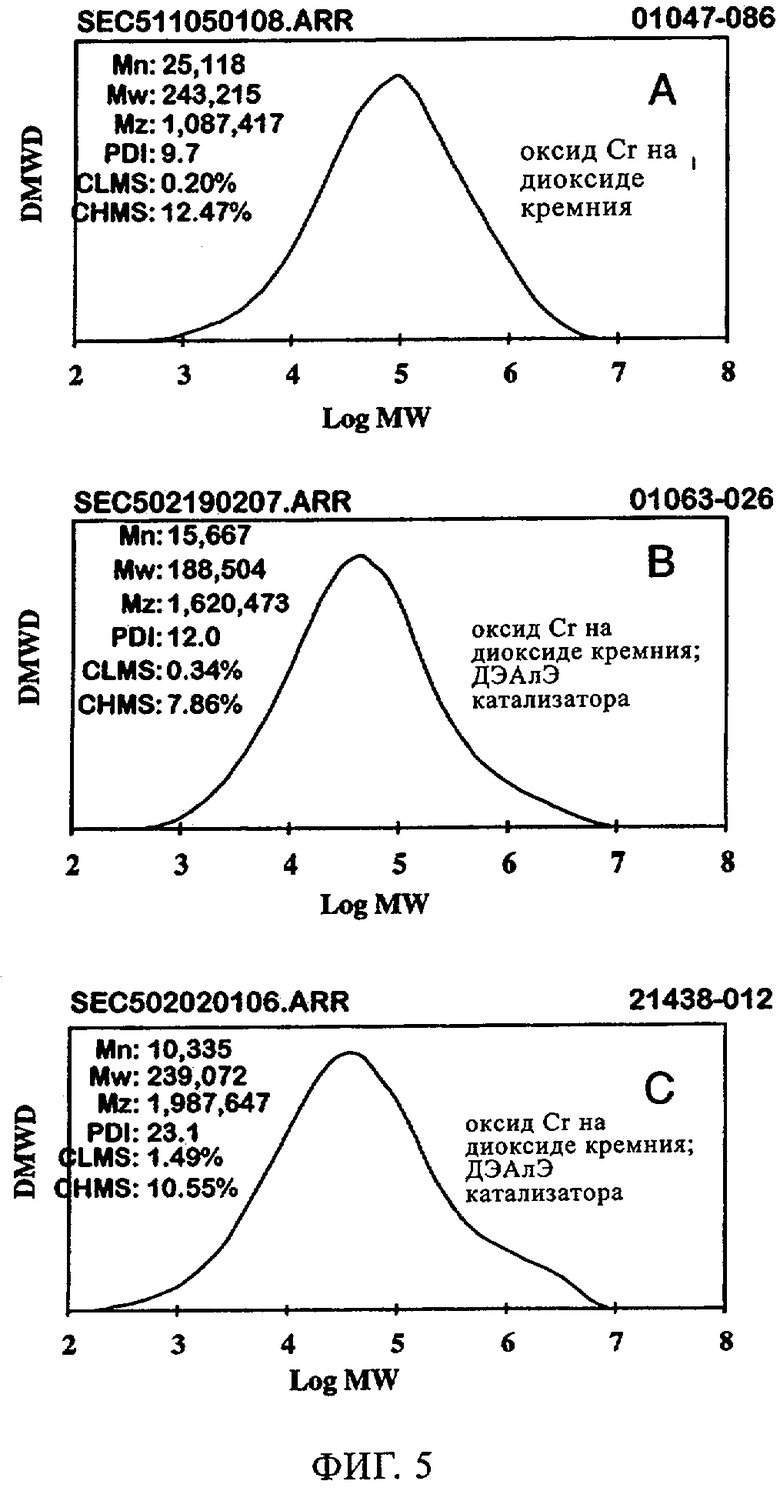
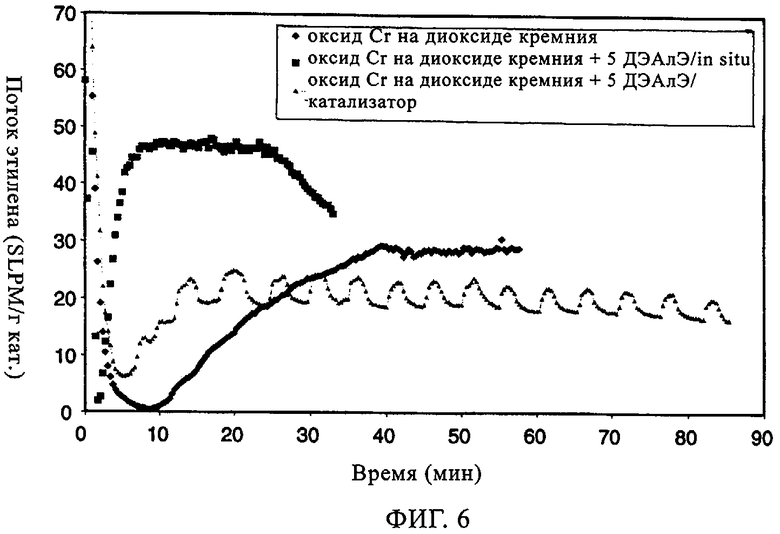
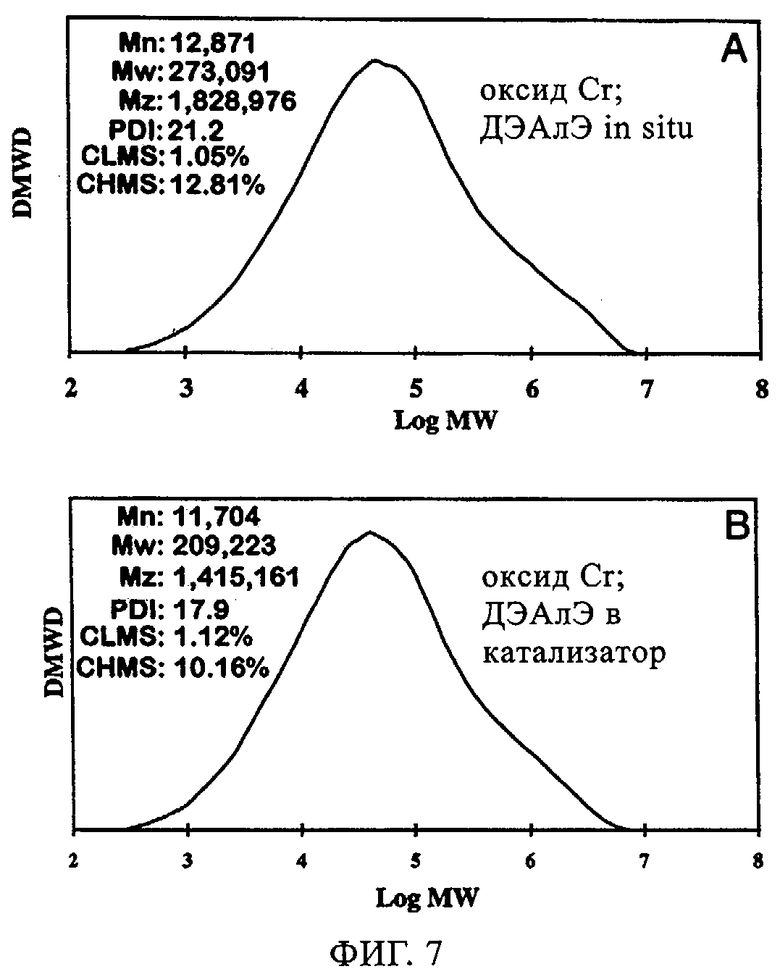
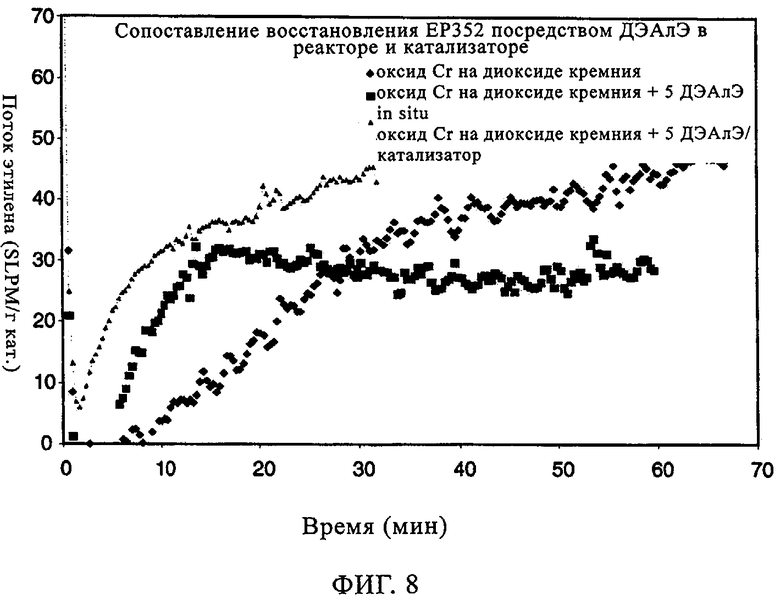
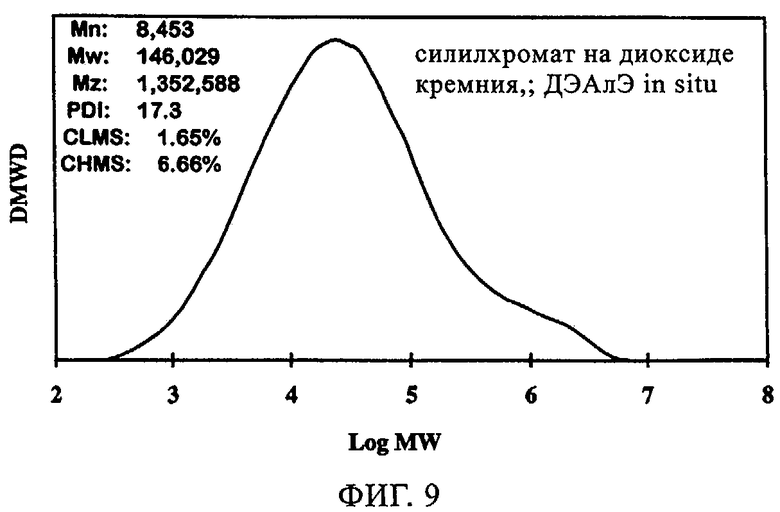
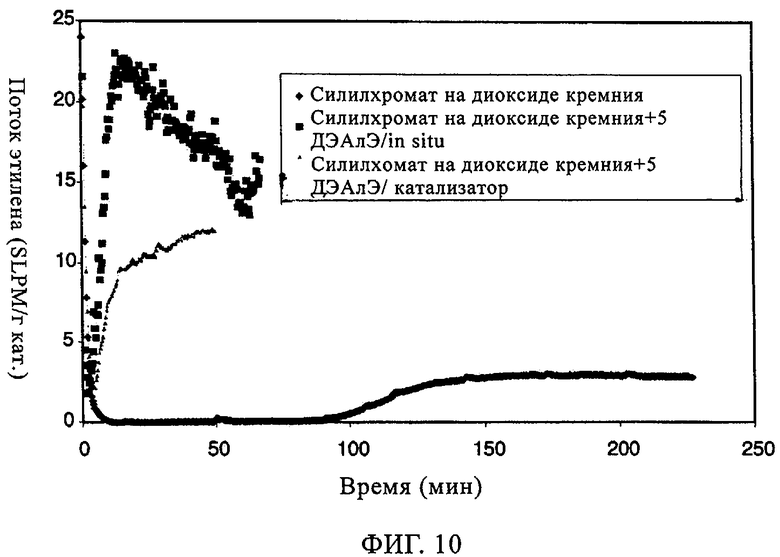
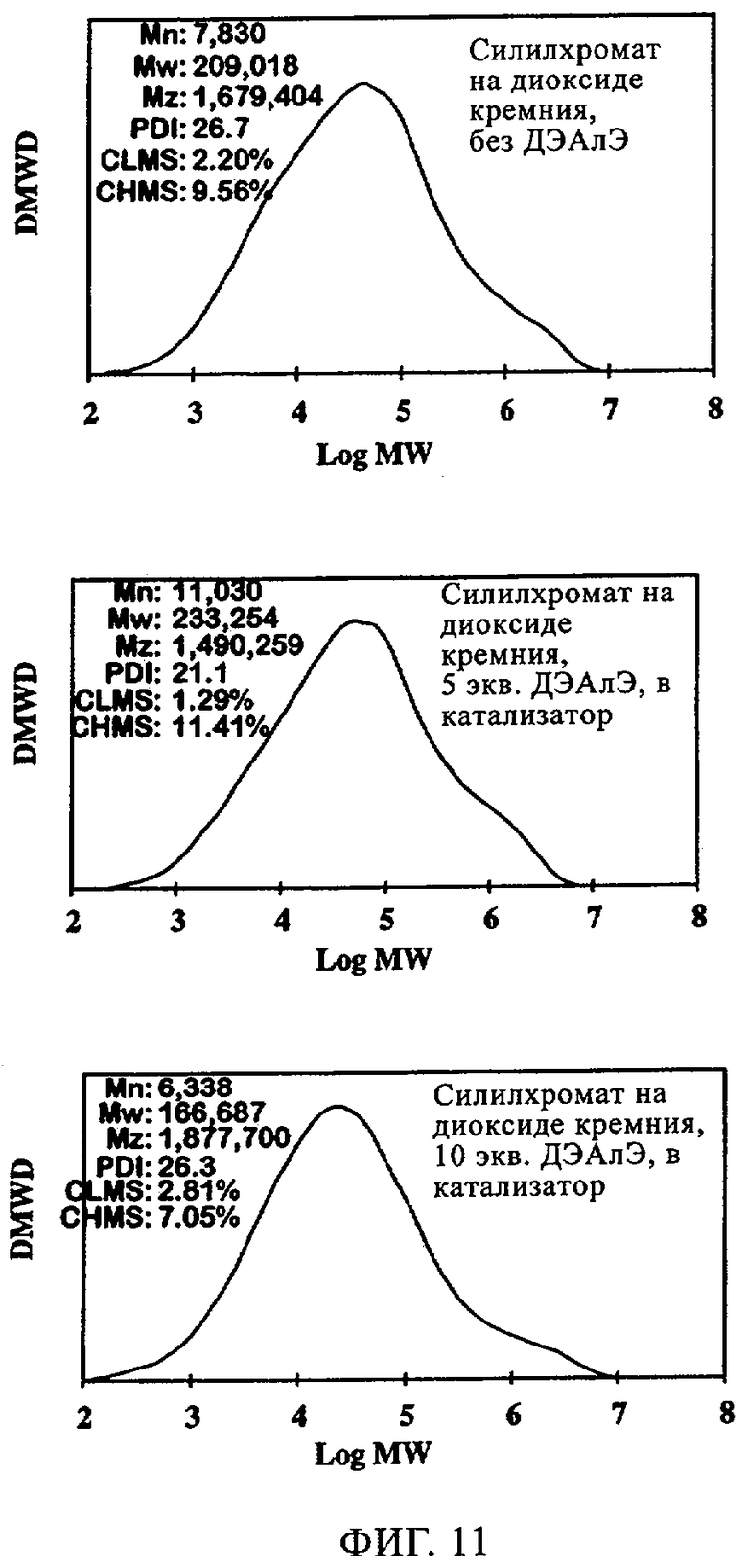
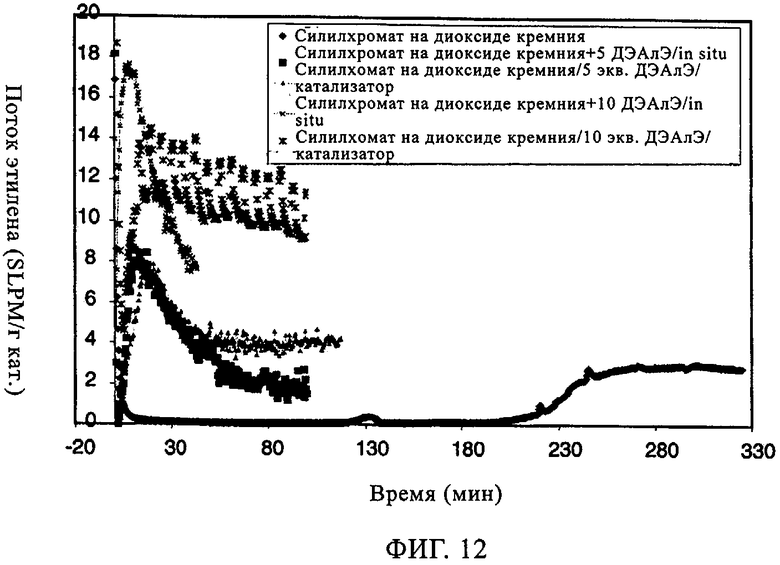
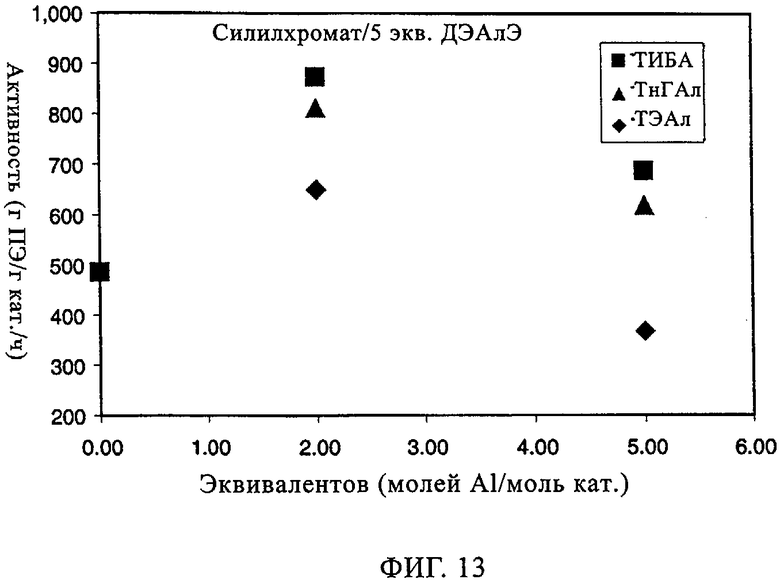
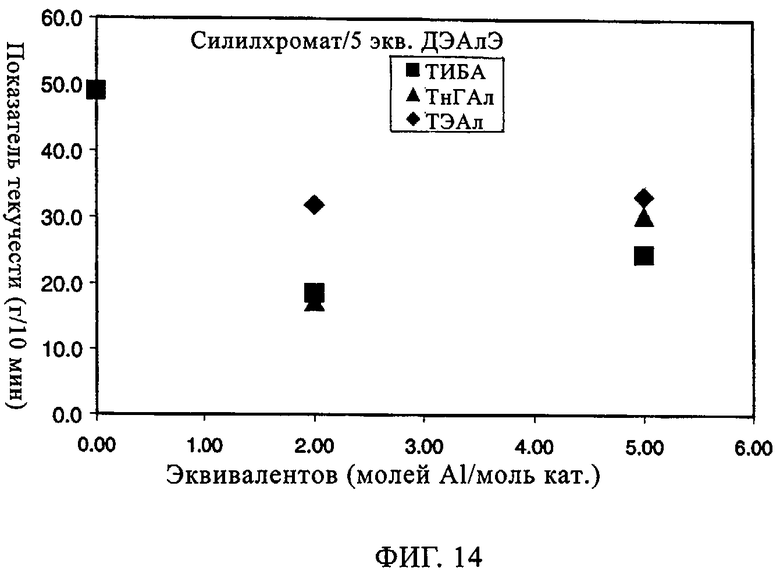

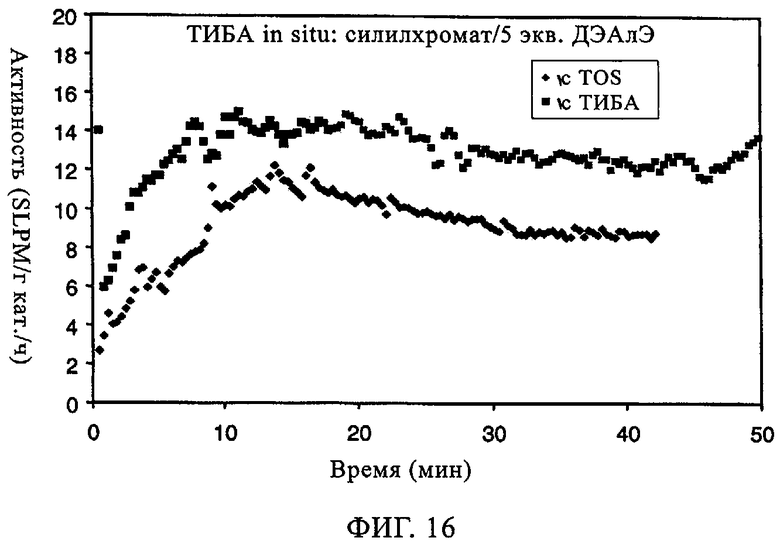
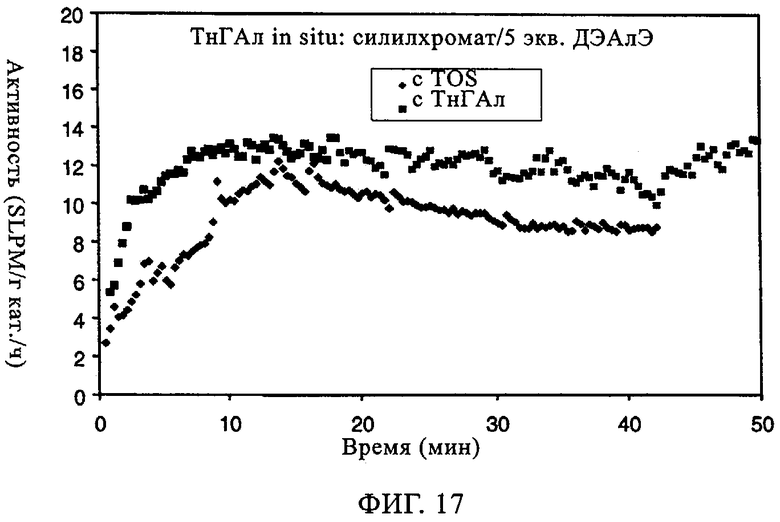
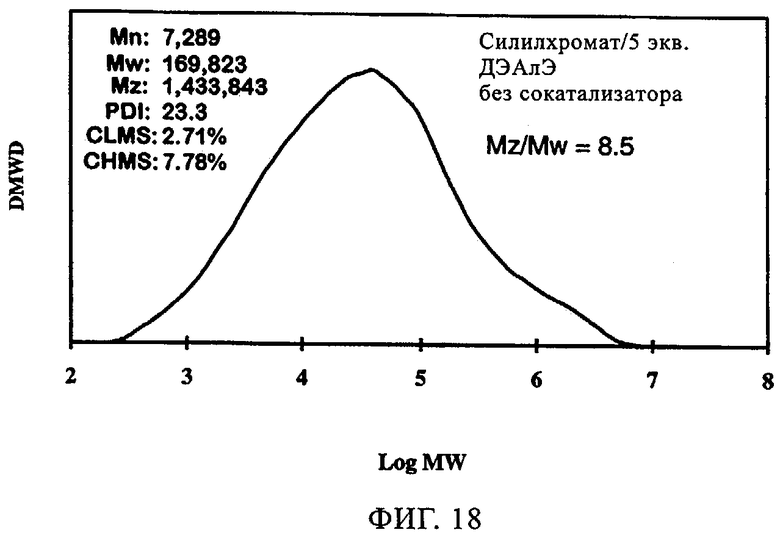

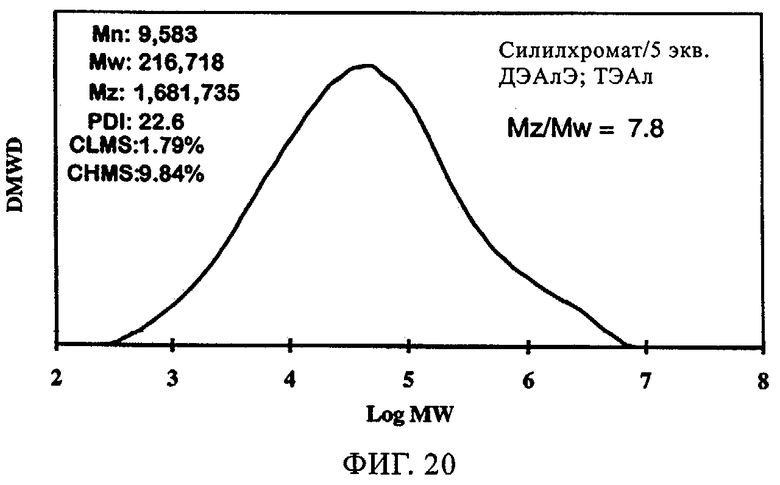
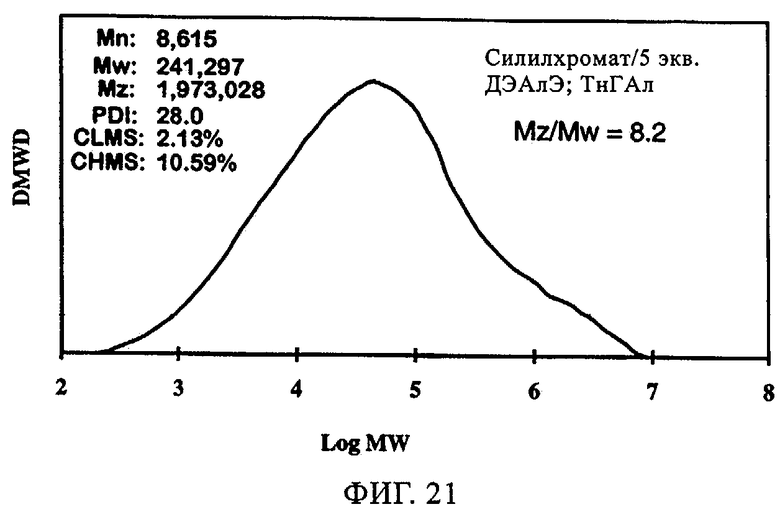
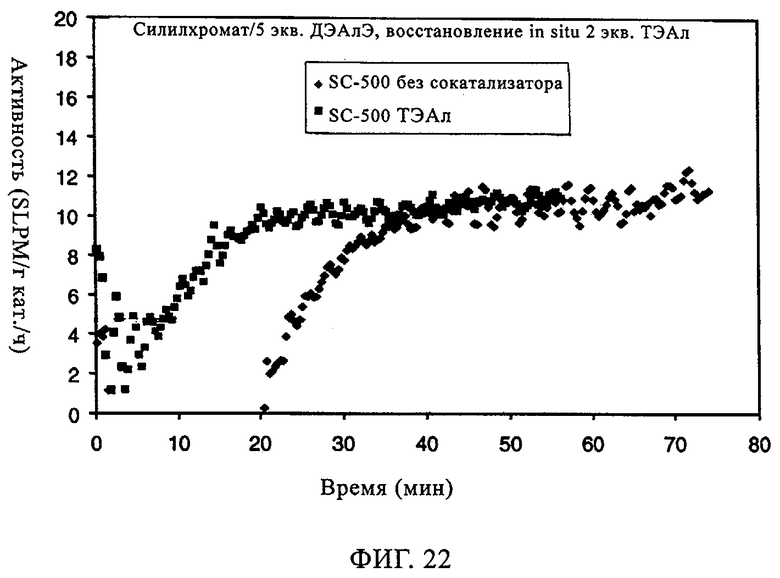
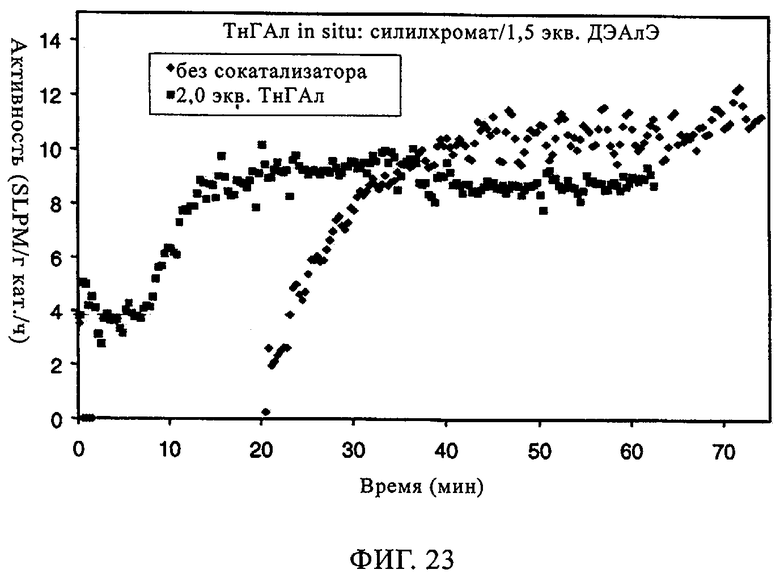

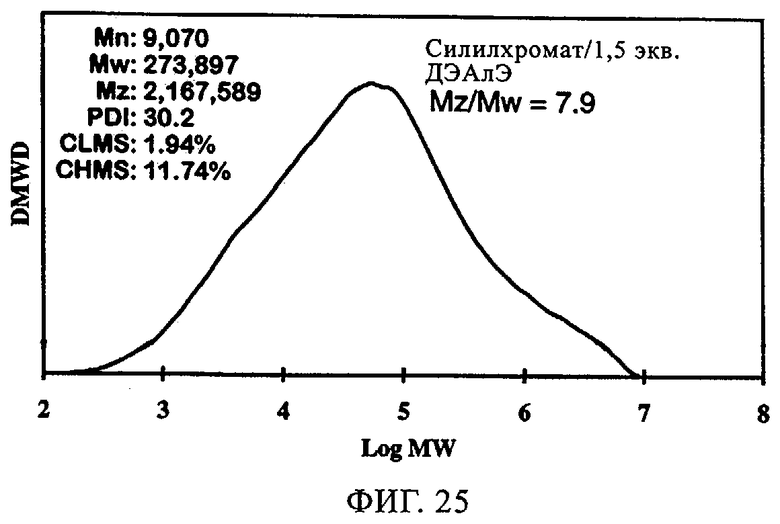
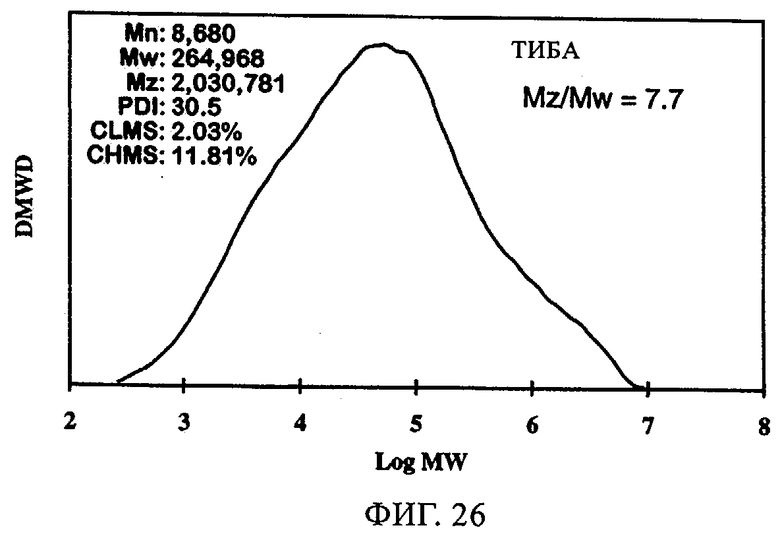

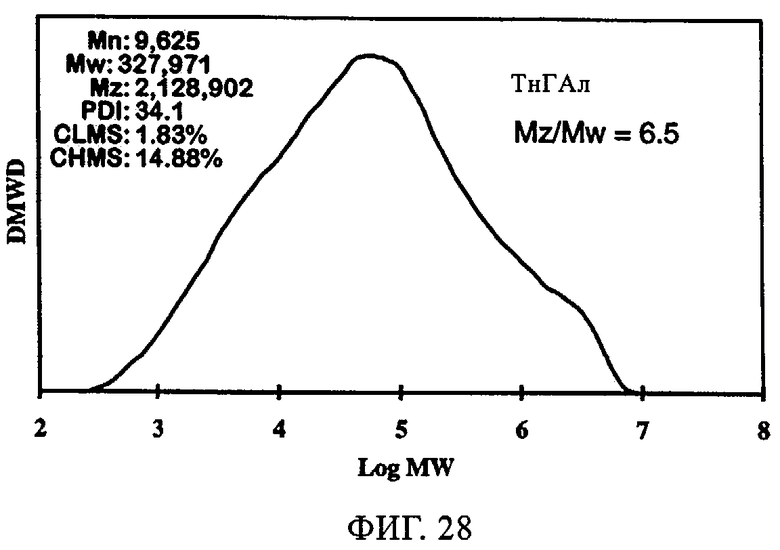
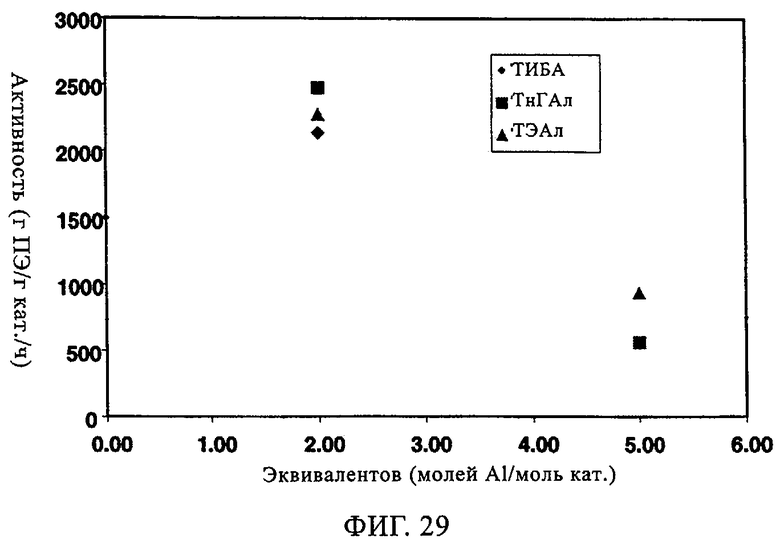
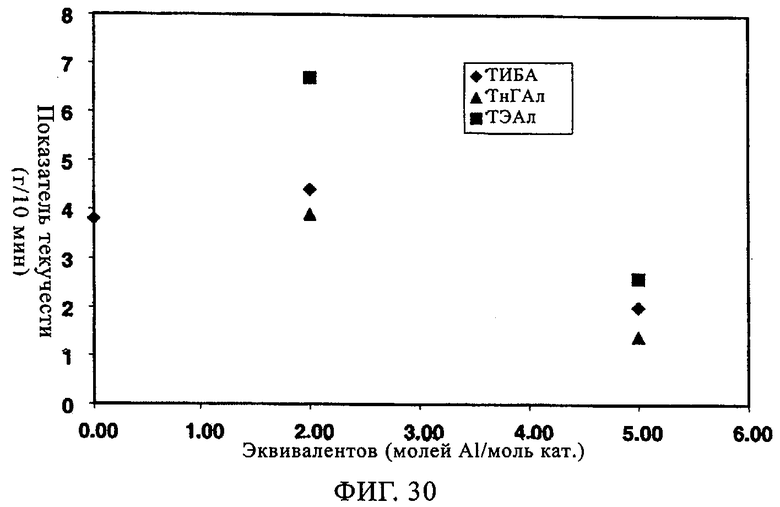
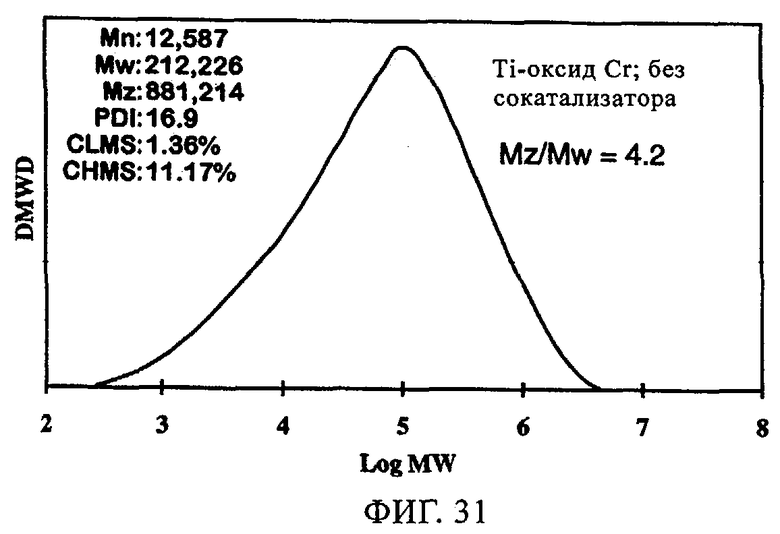



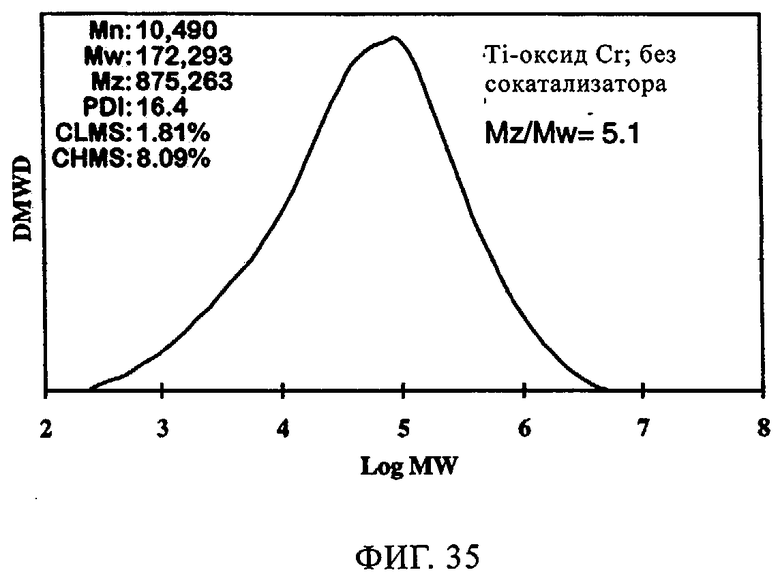

Изобретение относится к применению катализаторов на хромовой основе с алюмоалкильными активаторами. Описан нанесенный на носитель хромовый катализатор, включающий: оксид хрома, содержащий диоксид кремния носитель, включающий диоксид кремния, выбранный из группы, включающей диоксид кремния, и алюминийорганическое соединение, где упомянутое алюминийорганическое соединение добавляют в полимеризационный реактор in situ. Технический результат - улучшение эксплуатационных свойств катализаторов на хромовой основе, регулирование степени разветвления посредством боковых групп при одновременном проявлении необходимых значений производительности. 3 н. и 11 з.п.ф-лы, 14 табл., 36 ил.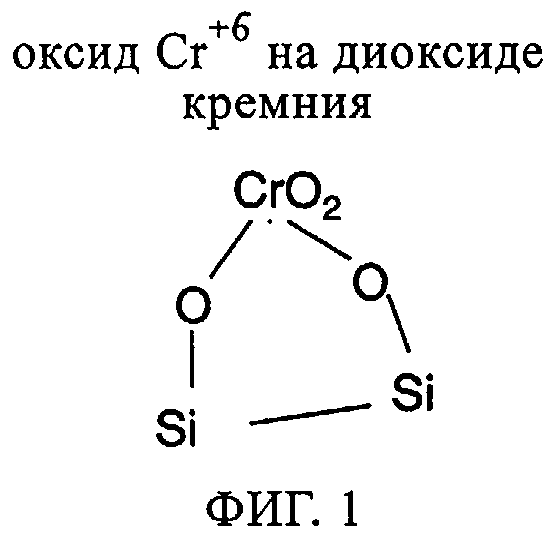
(а) удельным объемом пор от примерно 1,1 до примерно 1,8 см3/г и удельной площадью поверхности от примерно 245 до примерно 375 м2/г, (б) удельным объемом пор от примерно 2,4 до примерно 3,7 см3/г и удельной площадью поверхности от примерно 410 до примерно 620 м2/г и (в) удельным объемом пор от примерно 0,9 до примерно 1,4 см3/г и удельной площадью поверхности от примерно 390 до примерно 590 м2/г;
и алюминийорганическое соединение, где упомянутое алюминийорганическое соединение добавляют в полимеризационный реактор in situ,
где упомянутый нанесенный на носитель хромовый катализатор активируют при температуре от примерно 400 до примерно 860°С.
| US 5198400 А, 30.03.1993 | |||
| US 6326443 В1, 04.12.2001 | |||
| СПОСОБ ПОЛУЧЕНИЯ ПОЛИЭТИЛЕНА НИЗКОГО ДАВЛЕНИЯ | 2001 |
|
RU2177954C1 |
| US 5910299 A, 08.06.1999 | |||
| ГИДРАВЛИЧЕСКАЯ МНОГОКАНАЛЬНАЯ ВЫЗОВА И УПРАВЛЕНИЯСИСТЕМА | 0 |
|
SU194428A1 |

