Изобретение относится к области органической химии, а именно к способу получения производных хлорина е6 с двумя и тремя аминогруппами, и может быть использовано при синтезе противоопухолевых и противовирусных препаратов для медицины.
Известен способ получения аминохлорина, заключающийся во взаимодействии мезопирофеофорбида (а) с гексаметилендиамином с образованием мезопирофеофорбида (а) 17-(6-аминогексил)-амида (US 2004186087, МКИ А61К 031/555; C07D 487/22; А61К 031/409).
Недостатком настоящего способа введения аминогруппы на периферию хлоринового цикла является необходимость в предварительном активировании карбоксильной группы мезопирофеофорбида (а) дициклогексил-карбодиимидом.
Известен также способ получения N-замещенных аминохлоринов, заключающийся во взаимодействии пирофеофорбида (d) с N,N-диметилпропилендиамином (Synthesis and self-assembly of amphiphilic zinc chlorins possessing a 3(1)-hydroxy group. Miyatake, Tomohiro; Tamiaki, Hitoshi; Shinoda, Hiroyuki; Fujiwara, Manabu; Matsushita, Takayuki. Tetrahedron (2002), 58 (50), p.9989-10000).
Как и в предыдущем аналоге, недостатком способа введения аминогруппы на периферию хлоринового цикла является необходимость в предварительном активировании карбоксильной группы исходного соединения (в качестве активирующего агента в этом случае используется 1-(3-диметиламино-пропил)-3-этилкарбодиимид).
Для введения трех N-замещенных аминогрупп в работе (Synthesis of cationic water-soluble esters of chlorine е6. Hidetoshi Taima, Akihiro Okubo, Naoki Yoshioka, and Hidenari Inoue. Tetrahedron Letters (2005), p.4161-1464) используется реакция хлорина е6 с N,N-диметилэтаноламином.
Как и во всех описанных выше аналогах, для модификации карбоксильных групп исходного соединения проводится предварительная активация (активирующий агент, как и в предыдущем случае, 1-(3-диметиламино-пропил)-3-этилкарбодиимид).
Известно, что при синтезе лекарственных веществ необходимо, по возможности, избегать использования токсичных реагентов, каковыми являются названные в приведенных способах активирующие агенты. Кроме того, исключение использования таких дорогостоящих реактивов, как дициклогексил- и -(3-диметиламино-пропил)-3-этил-карбодиимидов, существенно удешевляет процесс.
Задачей изобретения является разработка способа синтеза дитриаминохлоринов без применения активирующих агентов.
Предлагаемый способ позволяет синтезировать хлорины, содержащие 2 и 3 аминогруппы без применения активирующих агентов с выходами, близкими и превосходящими выходы аналогичных соединений, получаемыми при использовании активирующих агентов. При использовании предлагаемого способа ди- и триаминохлорины могут быть селективно получены непосредственно из метилфеофорбида (а) без выделения промежуточных соединений.
Технический результат достигается тем, что способ синтеза аминохлоринов, включающий взаимодействие сложноэфирных групп хлорина е6 с этилендиамином и выделение целевого продукта, согласно изобретению используют чистый этилендиамин, хлорин е6 13-N-(2-аминоэтиламида)-15,17-диметиловый эфир получают in situ из метилфеофорбида (а), взаимодействие осуществляют путем перемешивания раствора метилфеофорбида (а) и 50-200-кратного мольного избытка этилендиамина в хлороформе при комнатной температуре 15-25°С в течение 1,5-2,5 часов, с последующим упариванием хлороформа на роторном испарителе при пониженном давлении 20-30 мм рт.ст. и температуре бани, не превышающей 50°С, перед выделением реакционную смесь выдерживают в темноте при комнатной температуре, при этом при выдерживании смеси в течение 20 часов в качестве целевого продукта выделяют диамид хлорина е6, содержащего две аминогруппы, при выдерживании смеси в течение 70 часов - выделяют триамид хлорина е6, содержащего, соответственно, три аминогруппы.
Выделение диаминохлорина со структурной формулой (3), представленной ниже, осуществляют путем разбавления полученной смеси хлороформом, отмывки раствора от этилендиамина водой, сушки над безводным сульфатом натрия, упаривания при пониженном давлении с последующим выделением целевого продукта колоночной хроматографией на силикагеле. В случае триаминохлорина (4) выделение целевого продукта проводят колоночной хроматографией непосредственно из реакционной смеси.
Способ получения ди- и триаминохлоринов включает взаимодействие сложноэфирных групп получающегося in situ из метилфеофорбида (а) (1) хлорина е6 13-N-(2-аминоэтиламида)-15,17-диметилового эфира (2) с чистым этилендиамином, в результате которого, в зависимости от времени протекания реакции, происходит образование ди- (3) или триамида хлорина е6 (4), содержащего, соответственно, 2 или 3 аминогруппы:
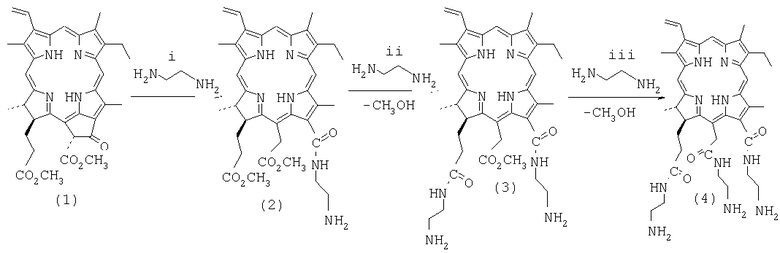
i: этилендиамин, CHCl3, 15-25°С температура, 1,5-2,5 ч; ii: этилендиамин, комнатная температура, 20 ч, без выделения исходного моноамида; iii: этилендиамин, комнатная температура, 70 ч.
Предложенный способ осуществляется следующим образом. Для получения диамида со структурной формулой (3) раствор метилфеофорбида (а) и этилендиамина в хлороформе перемешивают при температуре 15-25°С до исчезновения исходного метилфеофорбида (а) в течение 1,5-2,5 часов. Затем хлороформ упаривают на роторном испарителе при пониженном давлении (20-30 мм рт.ст.) при температуре бани, не превышающей 50°С, и оставшуюся смесь моноамида со структурной формулой (2) с этилендиамином оставляют в темноте при комнатной температуре на 20 часов. Далее процесс выделения осуществляют известным методом. Реакционную смесь разбавляют хлороформом, полученный раствор отмывают водой от этилендиамина, сушат над безводным сульфатом натрия и упаривают при пониженном давлении. Продукт реакции выделяют колоночной хроматографией на силикагеле.
Для получения триамида (4) взаимодействие этилендиамина и метилфеофорбида (а) и удаление хлороформенного проводят так же как при получении диамида (3), полученную в результате смесь моноамида (2) с этилендиамином оставляют в темноте при комнатной температуре на 70 часов. Триамид (4) выделяют из реакционной смеси колоночной хроматографией на силикагеле.
Пример 1.
Хлорин e6 13,17-N,N'-(2-аминоэтил)-диамид-15-метиловый эфир (3)
К раствору 300 мг метилфеофорбида (а) в 5 мл хлороформа прибавляют 2 мл этилендиамина. Смесь перемешивают при комнатной температуре 20°С в течение 2-х часов до исчезновения исходного метилфеофорбида (а) (контроль ТСХ, Silufol, элюэнт смесь хлороформа с метанолом 4 к 1 по объему) затем упаривают хлороформ при пониженном давлении (20 мм рт.ст.) при температуре бани, не превышающей 50°С, и оставшийся раствор моноамида (2) в этилендиамине оставляют в темноте на 20 часов. Реакционную смесь разбавляют 50 мл хлороформа и отмывают этилендиамин водой. Хлороформенный раствор сушат над безводным сульфатом натрия и хлороформ упаривают при пониженном давлении. Остаток после упаривания хроматографируют на силикагеле (Силохром С-120), элюируют сначала смесью хлороформа метанолом 10:1 (по объему) для отделения остаточного моноамида (2), затем диамид (3) элюируют смесью хлороформа с метанолом 1:1 (по объему). Контроль за разделением осуществляют при помощи ТСХ (Sorbfil, элюент:хлороформ-метанол, 4:1). Элюат, содержащий основной продукт, упаривают и остаток после упаривания переосаждают из смеси хлороформа с пентаном. Получают 135 мг соединения со структурной формулой (3), что составляет 39% от исходного метилфеофорбида (а). (Выход на каждой стадии составляет, в среднем, 62%).
ИК, см-1 (KBr): 1736 (νC=O, сложный эфир); 1608 «хлориновая полоса»; 1656 «амид I»; 1540 «амид II». ЯМР Н1 (ДМФА-D7, 400 МГц, δ (м. д.), мультиплетность, расщепления (Гц); нумерация атомов та же, что и в соединении со структурной формулой (5), представленной ниже): 9.89 (с 1Н; 10-Н); 9.87 (с 1Н; 5-Н); 9.21 (с 1Н; 20-Н); 8.88 (уш. т 1Н, 5.7 Гц; 13(1)-NH); 8.39 (д д, 1Н, 17.6 и 11.6 Гц, 3(1)-Н); 7.98 (уш. т 1Н, 5.7 Гц; 13(1)-NH); 6.49 (д д 1Н, 17.6 и 1.2 Гц; 3(2)-Н (транс)); 6.19 (д д 1Н, 11.6 и 1.2 Гц; 3(2)-Н (цис)); 15(1)-СН2: 5.75 (д, 1Н, 19.2 Гц) и 5.41 (д 1Н, 19.2 Гц); 4.70 (к 1Н, 7.6 Гц; 18-Н); 4.34 (уш. д 1Н, 9.6 Гц; 17-Н); 3.88 (к 2Н, 7.2 Гц 8(1)-СН2); 13(2)-СН2, 13(3)-СН2: 3.71-3.61 (м 1Н), 3.34-3.24 (м 3Н); 3.13-3.23 (м 4Н, 17(4)-СН2, 17(5)-СН2); 3.79 (с 3Н; 15(3)-СН3); 3.60 (с 3Н; 12(1)-СН3); 3.59 (с 3Н; 2(1)-СН3); 3.38 (с 3Н; 7(1)-СН3); 3.11 (уш т 1Н, 6.0 Гц; 13(3) и 17(5)-NH2); 2.62-2.72 (м 2Н; 17(1)-СН2); 2.35-2.22 (м 2Н; 17(1)-СН2); 1.73 (д 3Н, 7.2 Гц, 18(1)-СН3); 1.72 (т 3Н, 7.6 Гц, 8(2)-СН3); - 1.64 (уш с 1Н; I-NH); - 1.97(уш c 1H; III-NH).
Пример 2.
Хлорин e6 13,15,17-N,N',N''-(2-аминоэтил)-триамид (4)
К раствору 230 мг метилфеофорбида (а) в 4 мл хлороформа прибавляют 2 мл этилендиамина, смесь перемешивают при комнатной температуре 18°С в течение 2-х часов до исчезновения исходного метилфеофорбида (а) (контроль ТСХ, Silufol, элюэнт смесь хлороформа с метанолом 4 к 1 по объему) в течение 2-х часов. Хлороформ упаривают при пониженном давлении (30 мм рт.ст.) при температуре бани, не превышающей 50°С, и оставшийся раствор моноамида (2) в этилендиамине оставляют в темноте на 70 часов. Реакционную смесь разбавляют 1,5 мл хлороформа и хроматографируют на силикагеле (Силохром С-120), элюируют сначала смесью хлороформа метанолом 1:1 (по объему) для отделения остаточного диамида (3), затем триамид (4) элюируют смесью хлороформ-метанол-ацетон (1:1:1 по объему). Контроль за разделением осуществляют при помощи ТСХ (Sorbfil, элюент: хлороформ-метанол-NH4OH, 1:1:0.1). Элюат, содержащий основной продукт, упаривают и переосаждают из смеси хлороформа с пентаном. Получают 55 мг соединения со структурной формулой (4), что составляет 20% от исходного метилфеофорбида (а). (Выход на каждой стадии составляет, в среднем, 67%).
ИК, см-1 (KBr): 3080 (νNH-амид); 1606 «хлориновая полоса»; 1656 «амид I»; 1554 «амид II», полосы колебаний сложноэфирной группы отсутствуют.
Ввиду низкой растворимости и склонности к агрегации соединения (4) в хлороформе и ДМФА, для идентификации при помощи ЯМР спектроскопии проводят ацетилирование аминогрупп соединения (4) и получают соответствующий триацетат со структурной формулой (5).
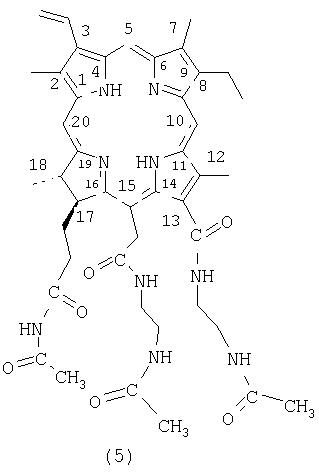
Для этого 40 мг соединения (4) растворяют в смеси 1 мл уксусного ангидрида с 1 мл пиридина. Полученный раствор оставляют в темноте на 30 минут, затем реакционную смесь разбавляют 100 мл хлороформа, пиридин и уксусный ангидрид отмывают 5% соляной кислотой (3 порции по 50 мл), хлороводород отмывают водой (до нейтральной реакции промывных вод по универсальной индикаторной бумаге). Полученный раствор сушат над безводным сульфатом натрия и хлороформ упаривают при пониженном давлении. Остаток после упаривания хроматографируют на силикагеле (Lachema-Л-100/400, элюент: хлороформ-метанол, 5:1). Получают 38 мг соединения (5).
ИК, см-1 (KBr): 3080 (νNH-амид); 1606 «хлориновая полоса»; 1656 «амид I»; 1554 «амид II», полосы колебаний сложноэфирной группы отсутствуют.
ЯМР Н1 (ДМФА-D7, 400 МГц, δ (м. д.), мультиплетность, расщепления (Гц)): 9.90 (с 1Н; 10-Н); 9.86 (с 1Н; 5-Н); 9.22 (с 1Н; 20-Н); 9.08 (уш. т 1Н, 5.2 Гц; 13(1)-NH); NH(амид) 13(3), 15(2), 15(4), 17(3) и 17(5): 8.27 (уш. т 1Н, 5.6 Гц), 8.12 (уш. т 1Н, 4.6 Гц), 7.95 (уш. т 1Н, 6.0 Гц), 7.92 (уш. т 1Н, 4.8 Гц), 7.68 (уш. т 1Н, 5.2 Гц), 8.40 (д д, 1Н, 18.0 и 11.6 Гц. 3(1)-Н); 6.49 (д д 1Н, 18.0 и 1.2 Гц; 3(2)-Н (транс)); 6.19(д д 1Н, 11.6 и 1.2 Гц; 3(2)-Н (цис)); 15(1)-СН2: 5.55 (д, 1Н, 18.0 Гц) и 5.24 (д 1Н, 18.0 Гц); 4.68 (к 1Н, 7.2 Гц; 18-Н); 4.52 (уш. д 1Н, 9.6 Гц; 17-Н); 3.89 (к 2Н, 7.2 Гц 8(1)-СН2); 3.61 (с 3Н; 12(1)-СН3); 3.59 (с 3Н; 2(1)-СН3); 3.38 (с 3Н; 7(1)-СН3); 13(1), 15(2) и 17(3) NHCH2CH2: 3.83-3.94 (м 2Н), 3.74-3.64 (м 2Н); 3.29-3.21 (м 6Н); 3.20-3.11 (м 2Н); 2.61-2.78 (м 2Н; 17(1)-СН2); 2.39-2.26 (м 2Н; 17(1)-СН2); 13(3), 15(4) и 17(5)-NHCOCH3: 2.02, 1.84 и 1.68 (все с 3Н); 1.74 (д 3Н, 6.8 Гц, 18(1)-СН3); 1.72 (т 3Н, 8.0 Гц, 8(2)-СН3); -1.64 (уш с 1Н; 1-NH); -1.99 (уш с 1Н; III-NH).
Таким образом, предлагаемый способ позволяет получить ди- и триаминохлорины не применяя активирующие агенты без выделения промежуточных соединений.
| название | год | авторы | номер документа |
|---|---|---|---|
| 13(1)-N-(4'-N'N'-ДИМЕТИЛПИПЕРАЗИНИЛ ИОДИД) АМИД, 15(2), 17(3)-ДИМЕТИЛОВЫЙ ЭФИР ХЛОРИНА Е6 | 2022 |
|
RU2794092C1 |
| Новые производные хлорина е, содержащие фрагменты галактозы | 2019 |
|
RU2706698C1 |
| ВОДОРАСТВОРИМОЕ ПРОИЗВОДНОЕ ХЛОРОФИЛЛА α, МОДИФИЦИРОВАННОЕ ФРАГМЕНТОМ МИРИСТИНОВОЙ КИСЛОТЫ | 2017 |
|
RU2680523C1 |
| ПРОИЗВОДНЫЕ 13(1)-N-{2-[N-(КЛОЗО-МОНОКАРБАДОДЕКАБОРАН-1-ИЛ)-МЕТИЛ]АМИНОЭТИЛ}АМИД-15(2),17(3)-ДИМЕТИЛОВОГО ЭФИРА ХЛОРИНА e, ПРОЯВЛЯЮЩИЕ СВОЙСТВА ФОТОСЕНСИБИЛИЗАТОРА | 2009 |
|
RU2406726C1 |
| ПРОИЗВОДНЫЕ ХЛОРОФИЛЛА α, МОДИФИЦИРОВАННЫЕ ФРАГМЕНТАМИ МИРИСТИНОВОЙ КИСЛОТЫ | 2018 |
|
RU2673888C1 |
| ФОТОСЕНСИБИЛИЗАТОР И СПОСОБ ЕГО ПОЛУЧЕНИЯ | 2007 |
|
RU2416614C2 |
| СПОСОБ ПОЛУЧЕНИЯ ХЛОРИНА e6 | 2006 |
|
RU2330037C1 |
| СПОСОБ ПОЛУЧЕНИЯ ФОТОСЕНСИБИЛИЗАТОРОВ НА ОСНОВЕ ЦИКЛОИМИДОВ ХЛОРИНА Р6 | 2016 |
|
RU2626234C1 |
| СЕНСИБИЛИЗАТОР ДЛЯ ФОТОДИНАМИЧЕСКОГО РАЗРУШЕНИЯ КЛЕТОК ЗЛОКАЧЕСТВЕННЫХ НОВООБРАЗОВАНИЙ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ С ЕГО ИСПОЛЬЗОВАНИЕМ | 2004 |
|
RU2259200C1 |
| ПРОИЗВОДНЫЕ ФТОРСОДЕРЖАЩИХ ХЛОРИНОВ, ПРОЯВЛЯЮЩИЕ ПРОТИВООПУХОЛЕВУЮ АКТИВНОСТЬ | 2019 |
|
RU2725876C1 |
Изобретение относится к области органической химии, а именно к способу получения производных хлорина е6 с двумя и тремя аминогруппами, и может быть использовано при синтезе противоопухолевых и противовирусных препаратов для медицины. Предлагаемый способ позволяет синтезировать хлорины, содержащие 2 и 3 аминогруппы без применения активирующих агентов с выходами, близкими и превосходящими выходы аналогичных соединений, получаемыми при использовании активирующих агентов. При использовании предлагаемого способа ди- и триаминохлорины могут быть селективно получены непосредственно из метилфеофорбида (а) без выделения промежуточных соединений. Технический результат достигается тем, что способ синтеза аминохлоринов, включающий взаимодействие сложноэфирных групп хлорина е6 с этилендиамином и выделение целевого продукта, согласно изобретению используют чистый этилендиамин, хлорин ее 13-N-(2-аминоэтиламида)-15,17-диметиловый эфир получают in situ из метилфеофорбида (а), взаимодействие осуществляют путем перемешивания раствора метилфеофорбида (а) и 50-200-кратного мольного избытка этилендиамина в хлороформе при комнатной температуре 15-25°С в течение 1,5-2,5 часов, с последующим упариванием хлороформа на роторном испарителе при пониженном давлении 20-30 мм рт.ст. и температуре бани, не превышающей 50°С, перед выделением реакционную смесь выдерживают в темноте при комнатной температуре, при этом при выдерживании смеси в течение 20 часов в качестве целевого продукта выделяют диамид хлорина е6, содержащего две амино-группы, при выдерживании смеси в течение 70 часов - выделяют триамид хлорина е6, содержащего, соответственно, три аминогруппы.
Способ синтеза аминохлоринов, включающий взаимодействие сложноэфирных групп хлорина e6 с этилендиамином и вьщеление целевого продукта, согласно изобретения, используют чистый этилендиамин, хлорин е6 13-N-(2-аминоэтиламид)-15,17-диметиловый эфир получают in situ из метилфеофорбида (а), взаимодействие осуществляют путем перемешивания раствора метилфеофорбида (а) и 50-200-кратного мольного избытка этилендиамина в хлороформе при комнатной температуре 15-25°С в течение 1,5-2,5 ч, с последующим упариванием хлороформа на роторном испарителе при пониженном давлении 20-30 мм рт.ст. и температуре бани, не превышающей 50°С, перед выделением реакционную смесь выдерживают в темноте при комнатной температуре, при этом при выдерживании смеси в течение 20 ч в качестве целевого продукта выделяют диамид хлорина е6, содержащий две аминогруппы, при выдерживании смеси в течение 70 ч выделяют триамид хлорина e6, содержащий соответственно три аминогруппы.
| Belykh D.V | |||
| et all "Preparation of chlorophyll derivatives" | |||
| ЩИТОВОЙ ДЛЯ ВОДОЕМОВ ЗАТВОР | 1922 |
|
SU2000A1 |
| Способ приготовления мыла | 1923 |
|
SU2004A1 |
| СПОСОБ ПОЛУЧЕНИЯ ВОДОРАСТВОРИМЫХ ХЛОРИНОВ | 1998 |
|
RU2144538C1 |
| Способ изготовления фанеры-переклейки | 1921 |
|
SU1993A1 |
| ФОТОСЕНСИБИЛИЗАТОР И СПОСОБ ЕГО ПОЛУЧЕНИЯ | 2004 |
|
RU2276976C2 |

