Изобретение относится к фармакологии, а именно к получению биологически активных соединений, конкретно к улучшенному способу получения хлорина е6, который может найти применение в качестве исходного полупродукта для получения многочисленных фотосенсибилизаторов для фотодинамической терапии рака и других новообразований, флуоресцентного диагностического средства, а также фармакологических препаратов с широким спектром действия.
Известны несколько способов получения в основном водорастворимых солей хлорина е6, в которых непосредственно хлорин е6 образуется в результате последовательных химических процедур и используется в качестве промежуточного полупродукта без специального выделения и характеристики его физико-химических и спектральных свойств, что значительно затрудняет получение стандартизированных лекарственных препаратов на его основе.
Так, например, известен способ получения тринатриевых и трикалиевых солей хлорина е6, который заключается в обработке биомассы цианобактерий рода Spirulina водно-спиртовым раствором щелочи с последующим выделением из экстракта многочисленных хлориновых производных, их совместным щелочным гидролизом в вакууме, нейтрализацией, выделением выпавшего осадка, содержащего наряду с многочисленными хлориновыми производными также и хлорин е6, из которого при добавлении NaOH или КОН получают водорастворимый продукт (1). В данном способе состав и взаимное содержание водорастворимых хлоринов не определено, что затрудняет получение из него лекарственного препарата для медицинского использования. Основным и принципиальным недостатком данного способа получения водорастворимых солей хлорина е6 является тот факт, что из-за использования биомассы спирулины в качестве исходного сырья образующийся продукт не может иметь постоянный состав.
Известен способ получения водорастворимых хлоринов (2), согласно которому хлорины, содержащие три карбоксилатных остатка (оксикарбонилэтильную, оксикарбонилметильную и ядерную карбоксильную группы), образуют водорастворимые соли с линейным или циклическим первичным или вторичным амином или же с основной аминокислотой. В качестве примера был использован хлорин, выделяемый путем специальной переработки спирулины с выходом около 50%. К недостаткам этого метода следует отнести тот факт, что для получения водных растворов хлоринов был использован продукт, содержащий смесь различных хлоринов, в котором содержание хлорина е6 варьировалось в широких пределах и составляло 80-90%. Относительно невысокое содержание хлорина е6 обусловлено принципиальными трудностями при выделении этого вещества по приведенной технологии из биомассы спирулины.
Другим известным способом получения водорастворимых солей хлорина е6 является гидролиз соответствующего триметилового эфира в тетрагидрофуране в присутствии NaOH в течение 2-х суток при комнатной температуре (3).
Основным недостатком этого метода является предварительное получение соответствующего триметилового эфира, синтез которого представляет собой достаточно сложную синтетическую задачу. В процессе щелочного омыления трех сложноэфирных остатков имеется возможность присутствия в реакционной смеси многочисленных продуктов неполного омыления, т.е. моно- и диметиловых эфиров, а для исчерпывающего гидролиза сложноэфирных остатков необходимо продолжительное время, которое отрицательно влияет на устойчивость хлорина е6 в щелочных средах.
Известен также способ получения смеси щелочных солей (натриевых или калиевых) хлоринов переменного состава, состоящей из солей хлорина е6, пурпурина 5 и хлорина р6, согласно которому биомассу спирулины обрабатывают ацетоном для извлечения хлорофилла а с последующей обработкой ацетонового экстракта соляной кислотой для превращения хлорофилла а в феофитин а, его выделением центрифугированием после нейтрализации раствора, гидролизом сложноэфирного остатка в системе соляная кислота - ацетон-гексан, выделением фильтрацией «сырого» феофорбида а, его частичной очисткой путем переосаждения из смеси ацетон-вода, высушиванием на воздухе, проведением первоначального щелочного гидролиза в системе ацетон - 1%-ная водная щелочь, повторного гидролиза при добавлении большого количества щелочи при нагревании до 40-60°С в течение неопределенного интервала времени (20-90 минут), нейтрализацией продукта соляной кислотой, его выделением центрифугированием, переосаждением из ацетона для отделения от продуктов деструкции и высушиванием в широком интервале температур (40-100°С) от 1 часа до 30 суток (4). В результате такой сложной и неоднозначной переработки микроводорослей спирулины получается продукт неопределенного состава, физико-химические и фотофизические характеристики которого, а также, соответственно, и биологические свойства будут зависеть от множества факторов. Использование такого нестандартизованного продукта в качестве фармакологического средства не представляется возможным.
Известен способ получения хлорина е6 (18-карбокси-20-(карбоксиметил)-8-этенил-13-этил2б3-дигидро-3,7,12,17-тетраметил-21Н, 23Н-порфин-2-пропионовой кислоты) путем экстракции хлорофилла а из биомассы спирулины водно-спиртовым раствором щелочи с последующей нейтрализацией кислотой и получением осадка феофитина а, его очисткой путем повторного растворения в ацетоне, проведением щелочного гидролиза очищенного феофитина а с последующей нейтрализацией кислотой до образования осадка хлорина е6 (5). В данном способе состав и взаимное содержание хлоринов не определено, так как феофитин а осаждается вместе с гидрофобными липидами и каротиноидами. Кроме того, на последней стадии гидролиза феофитина а до хлорина е6 происходит отщепление фитола. В результате чего конечный продукт может быть загрязнен примесями фитола и родственными хлорину е6 соединениями. Помимо этого, в используемой биомассе сырой спирулины не стандартизованно содержание основных компонентов - тетрапирролов, в частности хлорофилла а; фикобилипротеинов и фикобилицианинов; липидов и белков, что также приводит к различному содержанию примесей в конечном продукте.
Наиболее близким к описываемому способу по технической сущности является метод получения хлорина е6 с последующим переводом в его бис-N-метилглюкаминовую соль путем обработки лиофильно высушенной спирулины в метиловом спирте в присутствии серной кислоты с последующей фильтрацией, упариванием метанола, добавлением к маслянистому остатку гексана, отделением гексанового слоя, добавлением к нижнему слою хлористого метилена, разбавлением смеси холодной водой, отделением органического слоя, промыванием водой, упариванием, хроматографированием на колонке с силикагелем в системе хлороформ-эфир, упариванием фракции, содержащей метилфеофорбид а, кристаллизацией метилфеофорбида а из смеси хлороформ-метанол. К ацетоновому раствору выделенного метилфеофорбида а при интенсивном перемешивании в токе инертного газа добавляют раствор NaOH, нагревают до 50°С, вновь добавляют водный раствор NaOH, выдерживают при 50-60°С, охлаждают, нейтрализуют до рН 4,5-5,0. Полученный после центрифугирования осадок, содержащий хлорин е6, промывают водой, суспендируют в апирогенной воде, 2-3 раза центрифугируют и переводят в N-метил-D-глюкаминовую соль (6). Способ имеет ряд существенных недостатков, которые не позволяют осуществлять его в промышленном масштабе. Так, например, получение метилфеофорбида а в количествах, превышающих несколько граммов, невозможно, поскольку проведение 2-х или 3-кратной экстракции гексаном из большого объема (несколько литров) вязкого фильтрата, содержащего метанол, серную кислоту и многочисленные экстрактивные продукты из спирулины, крайне сложно по следующим причинам:
а) из-за трудности нахождения границы раздела двух фаз, поскольку они имеют практически черный цвет и малопрозрачны;
б) для полноты экстракции гексаном требуется интенсивное встряхивание, что возможно лишь при использовании делительных воронок небольшого объема (не более 0,5-1 л);
в) интенсивное встряхивание приводит к значительному увеличению времени расслаивания двух фаз (1-2 часа);
г) после отделения гексанового слоя нижний слой после добавления хлористого метилена и воды и последующего встряхивания очень трудно расслаивается из-за того, что в водный слой переходят белковые продукты и другие поверхностно активные вещества;
д) для полноты отделения органического слоя в хлористом метилене от метанола и серной кислоты требуется неоднократная промывка водой, что заметно увеличивает время операции промывки;
е) перед упариванием до минимального объема отделенного органического слоя необходимо высушивание его от воды, поскольку присутствие воды недопустимо при проведении хроматографической очистки на селикагеле.
Хлорин е6 с сопутствующими соединениями переводят в соответствующую N-метилглюкаминовую соль в виде раствора, который и используют в качестве фотосенсибилизатора. Основным недостатком данного метода является получение исключительно водных растворов N-метилглюкаминовой соли хлорина е6, что не позволяет в максимальной степени использовать широкие возможности применения хлорина е6 для медицинских целей, что связано с недостаточной стабильностью водных растворов при хранении. Во всех вышеперечисленных способах в результате последовательных химических операций получают хлорин е6 только в виде водного раствора. Хлорин е6 не был выделен в сухом виде и охарактеризован физико-химическими методами, не описаны его спектральные свойства. В то же время результаты специальных исследований свидетельствуют о том, что индивидуальный хлорин е6 обладает максимальным фотосенсибилизирующим действием (7).
Целью данного изобретения является разработка способа получения хлорина е6 высокой степени чистоты, на основе которого возможно получение высокоактивных лекарственных форм. Для получения высокоочищенного, постоянного по составу и стабильного при хранении хлорина е6 предлагается способ получения хлорина е6 исходя из стандартизованного исходного продукта - метилфеофорбида а, который получают из биомассы спирулины по улучшенной технологии, позволяющей получать метилфеофорбид а в больших количествах и с использованием меньшего количества органических растворителей.
В соответствии с изобретением описывается способ получения хлорина е6, характеризующийся тем, что суспензию лиофильно высушенной спирулины в метиловом или абсолютном этиловом спирте обрабатывают концентрированной серной кислотой в количестве 5-10% от объема раствора в течение 10-12 часов при перемешивании с последующей фильтрацией, полученный раствор разбавляют водой, нейтрализуют водной щелочью до рН 4,5-5,0, выпавший осадок алкилфеофорбида отделяют фильтрацией через слой целита с последующим промыванием горячей водой и легким петролейным эфиром, продукт элюируют с целита ацетоном, раствор упаривают, хроматографируют на силикагеле системой хлористый метилен-эфир, перекристаллизовывают из смеси хлористый метилен-метанол, алкилфеофорбид а отделяют фильтрованием, растворяют в ацетоне при интенсивном перемешивании в токе инертного газа, добавляют водный раствор щелочи и нагревают при 40-60°С в течение 1,5 часов, раствор охлаждают, подкисляют соляной кислотой до рН 4,0-5,0, выпавший осадок отделяют фильтрацией через слой целита, промывают дистиллированной водой, продукт смывают с целита водным раствором аммиака, раствор лиофилизируют, остаток растворяют в водном растворе щелочи с последующей фильтрацией через слой сефадекса G-25, фракцию, содержащую хлорин е6, подкисляют соляной кислотой до рН 4,0-5,0, осадок хлорина е6 отделяют центрифугированием, суспендируют в воде и лиофилизируют.
Процесс легко масштабируется без изменения времени протекания обработки спирулины. Нейтрализация фильтрата после отделения спирулины и разбавления холодной водой протекает без заметного экзотермического эффекта; выпадение осадка происходит быстро, в течение нескольких минут, а для его «созревания» для улучшения фильтрации через целит требуется не более 1-2 часов. Использование горячей воды для промывки отфильтрованного на целите осадка позволяет получить в конце этой операции сыпучий продукт, удобный для промывки его петролейным эфиром и практически полного отделения от липидов, фитола и других продуктов, затрудняющих хроматографическую очистку. Из отмытого таким образом целита легко извлекается метилфеофорбид а путем промывки ацетоном.
Изобретение иллюстрируется следующими примерами.
Пример 1. К суспензии 200 г лиофильно высушенной спирулины в 600 мл метанола при интенсивном перемешивании приливают 30 мл концентрированной серной кислоты, перемешивают 10-12 часов, полученный сине-зеленый продукт, состоящий из спирулины, отфильтровывают через стеклянный фильтр №2, разбавляют 600 мл воды, нейтрализуют водным раствором щелочи до рН 4,5-5,0, выпавший рыхлый осадок отфильтровывают через слой целита 545, целит промывают горячей водой, затем - легким петролейным эфиром, продукт элюируют с целлита ацетоном, раствор упаривают до минимального объема, остаток хроматографируют на колонке с силикагелем G-25 в системе хлористый метилен - эфир (95:5), основную фракцию упаривают, вещество перекристаллизовывают из смеси хлористый метилен-метанол. Получают 0,155 г метилфеофорбида а.
УФ спектр в хлороформе, λmax нм (ε. 10-3): UV-VIS λmax (CHCl3) 668 nm (e 45.3), 610 (13.1), 536 (14.0), 506 (15,8), 412 (104.8). (выход по хл. около 1-1,5%).
Пример 2. Аналогично, из 200 г лиофильно высушенной спирулины в 600 мл абсолютного этанола получают 0,120 г этилфеофорбида а.
ПМР (400 Мгц, CDCl3): 9.47 (1Н, с, 10-Н); 9.32 (1Н, с, 5-Н); 8.56 (1Н, с, 20-Н); 7.95 (1Н, м, 31-Н); 6.25 (1Н, дд, 32-H); 6,17 (1Н, дд, 32-Н); 3.67 (1Н, к, 81-Н); 3.68 (3Н, с, 121); 3.39 (3Н, с, 21); 3.21 (3Н, с, 71); 1.80 (д, Н-181); 1.68 (т, Н-82); 4.47 (к, Н-18); 4.20 (дд, Н-17); 2.62 (дт, На-171); 2.31 (дт, На-171); 2.50 (дт, Нb-172); 2.24 (т, Нb′-172); 0.53 (1H ушир с, NH); -1.65 (1Н, с, NH).
Пример 3. К раствору 1 г метилфеофорбида а в 100 мл ацетона при интенсивном перемешивании в токе инертного газа добавляют 120 мл 10% водного раствора едкого калия при температуре 50°С в течение 1,5 часа, контролируя процесс образования хлорина е6 методом ТСХ (на селикагеле Merck (0,04-0,063 мм), в системе хлористый метилен-эфир 9:1). Раствор охлаждают до 10-15°С, подкисляют разбавленной соляной кислотой (1:3) до рН 4,0-5,0, выпавший объемный осадок отфильтровывают через слой целита 545, целит промывают дистиллированной водой до отсутствия в маточнике ионов хлора и ацетона; хлорин е6 смывают 100 мл воды, содержащей 1 мл концентрированного водного аммиака, раствор лиофилизируют и получают 0,85 г (87%) «сырого» хлорина е6 в виде темно-зеленого воздушного порошка. Полученный хлорин е6 растворяют в 20 мл 1% раствора NaOH, очищают на колонке с сефадексом G-25, элюируя 1% раствором щелочи, основную фракцию отделяют, подкисляют разбавленной соляной кислотой до рН 4,0-5,5, осадок центрифугируют, промывают дистиллированной водой до отсутствия ионов хлора в фильтрате, осадок суспендируют в 50 мл дистиллированной воды и лиофилизируют. Получают 0,76 г (77%) хлорина е6 (C34H36N4O6 м.м. 596.68) в виде воздушного порошка темно-зеленого цвета с содержанием основного вещества не менее 98% (по данным ВЭЖХ).
Спектр поглощения в воде при рН 9,0 λmax нм (ε 10-3): 654 (36.750).
Спектр ПМР для триметилового эфира хлорина е6, полученного обработкой хлорина е6 эфирным раствором диазометана
ПМР 400 МГц, (CDCl3): 9.68, 9.55. 8.72 (1H, 1H, 1H, мезо-Н, все с); 8.05, 6.34, 6.13 (м,д,д, 1H, 1H, 1Н, CH=CH2); 5.34 и 5.21 (1H, 1Н, два д, CH2COOCH3); 4.43-4.38 (м, 2Н, 7,8-Н); 4.24, 3.75, 3.62, 3.56, 3.46, 3.28 (все с, 3×CH3 макроцикла и 3×СООСН3); 3.78 к и 1.70 т, (5Н, СН2СН3); 2.53 и 2.18 (два м, 4Н, CH2CH2 СООСН3); 1.73 д (8-СН3); -1.31 и -1.48 (два ушир с, 2NH).
Пример 4. К раствору 4 г метилфеофорбида а в 750 мл ацетона при интенсивном перемешивании в токе инертного газа при температуре 40-50°С прибавляют 100 мл 10%-ного NaOH, перемешивают 15 минут, прибавляют еще 350 мл 10%-ного NaOH, перемешивают 45 минут при 60°С, контролируя процесс превращения метилфеофорбида а в хлорин е6 методом ТСХ. После завершения процесса раствор охлаждают до 15-20°С, подкисляют при охлаждении разбавленной соляной кислотой (1:3) до рН 4,0-5,0. Выпавший осадок отделяют центрифугированием, промывают дважды дистиллированной водой, затем суспендируют в дистиллированной воде и лиофилизируют. Получают 3,4 г, (86%) «сырого» хлорина е6 в виде воздушного порошка темно-зеленого цвета. Выход не менее 95% основного вещества (по данным ВЭЖХ).
Пример 5. Из 4 г метилфеофорбида а после щелочной обработки и подкисления (пример 4) выпавший осадок хлорина е6 отфильтровывают через слой целита 545, целит промывают сначала дистиллированной водой для удаления неорганических солей и ацетона, затем адсорбированный на целите хлорин е6 смывают 200 мл дистиллированной воды, к которой добавляют 3 мл концентрированного водного аммиака, хроматографируют на сефадексе G-25, элюируя 1% раствором щелочи, основную фракцию собирают, подкисляют разбавленной муравьиной кислотой до рН 4,5-5,5, лиофилизируют и получают 3,04 г (77%) хлорина е6 с содержанием основного вещества не менее 98%.
Полученный хлорин е6 в виде легкого воздушного порошка темно-зеленого цвета хранят в плотно закрытых склянках, предохраняя от попадания влаги, при температуре -15°С.
Пример 6. аналогично получают хлорин е6 из этилфеофорбида а.
СПИСОК ЛИТЕРАТУРЫ
1. RU 2054476, C1, 20.02.1996.
2. RU 2144538, C1, 20.01.2000.
3. US 5002962, A, 26.03.1991.
4. RU 2183956, C1, 27.06.2002.
5. RU 2228750, C1, 20.05.2004.
6. RU 2276976, C2, 10.02.2006.
7. Ю.А.Белый, А.В.Терещенко, П.Л.Володин, М.А.Каплан, Г.В.Пономарев. Сравнительное изучение фотодинамических эффектов фотосенсибилизаторов хлоринового ряда на интактной сетчатке экспериментальных животных. Рефракционная хирургия и офтальмология, 2006, 6, (№2), 55-59.
| название | год | авторы | номер документа |
|---|---|---|---|
| ФОТОСЕНСИБИЛИЗАТОР И СПОСОБ ЕГО ПОЛУЧЕНИЯ | 2004 |
|
RU2276976C2 |
| ФОТОСЕНСИБИЛИЗАТОР И СПОСОБ ЕГО ПОЛУЧЕНИЯ | 2013 |
|
RU2523380C1 |
| ФОТОСЕНСИБИЛИЗАТОР И СПОСОБ ЕГО ПОЛУЧЕНИЯ | 2007 |
|
RU2416614C2 |
| СПОСОБ ПОЛУЧЕНИЯ МЕТИЛФЕОФОРБИДА (А) | 2012 |
|
RU2490273C1 |
| СПОСОБ ПОЛУЧЕНИЯ ТРИСНАТРИЕВОЙ СОЛИ ХЛОРИНА Е6 | 2019 |
|
RU2705199C1 |
| ФОТОСЕНСИБИЛИЗАТОР И СПОСОБ ЕГО ПОЛУЧЕНИЯ | 2008 |
|
RU2367434C1 |
| ФОТОСЕНСИБИЛИЗАТОР И СПОСОБ ЕГО ПОЛУЧЕНИЯ | 2014 |
|
RU2568597C1 |
| СПОСОБ ПОЛУЧЕНИЯ ХЛОРИНА Е | 2002 |
|
RU2228750C1 |
| ФОТОСЕНСИБИЛИЗАТОР И СПОСОБ ЕГО ПОЛУЧЕНИЯ | 2001 |
|
RU2183956C1 |
| СПОСОБ ФОТОДИНАМИЧЕСКОЙ ТЕРАПИИ ВНУТРИГЛАЗНЫХ НОВООБРАЗОВАНИЙ | 2011 |
|
RU2467777C1 |
Изобретение относится к фармакологии, а именно к получению биологически активных соединений, конкретно к улучшенному способу получения хлорина е6, который находит применение для получения фотосенсибилизаторов для фотодинамической терапии рака. Способ осуществляют путем обработки метанольной суспензии спирулины концентрированной серной кислотой с последующей фильтрацией, разбавлением водой, нейтрализацией щелочью, фильтрацией выпавшего осадка через слой целлита с последующим промыванием горячей водой и петролейным эфиром, элюированием с целита ацетоном, хроматографированием на силикагеле, перекристаллизацией из смеси хлористый метилен-метанол, отделением образовавшегося алкилфеофорбида, растворением его в ацетоне, обработкой щелочью, нейтрализацией соляной кислотой, фильтрацией через слой целита, промыванием водой, водным аммиаком, лиофилизацией, растворением в водной щелочи, фильтрацией через слой сефадекса, нейтрализацией, центрифугированием и лиофилизацией. Технический результат - получение чистого продукта при использовании меньшего количества реагентов.
Способ получения хлорина е6 формулы 1 из спирулины, характеризующийся
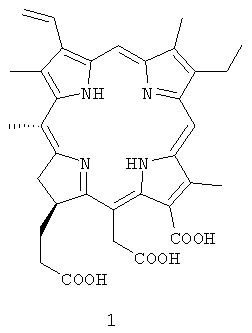
тем, что суспензию лиофильно высушенной спирулины в метиловом или абсолютном этиловом спирте обрабатывают концентрированной серной кислотой в количестве 5-10% от объема раствора в течение 10-12 ч при перемешивании с последующей фильтрацией, полученный раствор разбавляют водой, нейтрализуют водной щелочью до рН 4,5-5,0, выпавший осадок алкилфеофорбида отделяют фильтрацией через слой целита с последующим промыванием горячей водой и легким петролейным эфиром, продукт элюируют с целита ацетоном, раствор упаривают, остаток хроматографируют на силикагеле системой хлористый метилен-эфир, основную фракцию упаривают, остаток перекристаллизовывают из смеси хлористый метилен-метанол, алкилфеофорбида отделяют фильтрацией, растворяют в ацетоне, добавляют водный раствор щелочи и нагревают при 40-60°С, раствор охлаждают, подкисляют соляной кислотой до рН 4,0-5,0, выпавший осадок отделяют фильтрацией через слой целита, промывают дистиллированной водой, продукт смывают с целита водным раствором аммиака, раствор лиофилизируют, остаток растворяют в водном растворе щелочи с последующей фильтрацией через слой сефадекса, фракцию, содержащую хлорин е6, подкисляют до рН 4,0-5,5, осадок хлорина е6 отделяют центрифугированием, суспендируют в воде и лиофилизируют.
| СПОСОБ ПОЛУЧЕНИЯ 18-КАРБОКСИ-20-(КАРБОКСИМЕТИЛ)-8-ЭТЕНИЛ-13-ЭТИЛ-2,3-ДИГИДРО-3,7-12,17-ТЕТРАМЕТИЛ-21Н, 23НПОРФИН-2-ПРОПИОНОВОЙ КИСЛОТЫ ИЛИ ЕЕ СОЛЕЙ | 1993 |
|
RU2054476C1 |
| СПОСОБ ПОЛУЧЕНИЯ ВОДОРАСТВОРИМЫХ ХЛОРИНОВ | 1998 |
|
RU2144538C1 |
| US 5002962, А 26.03.1991 | |||
| ФОТОСЕНСИБИЛИЗАТОР И СПОСОБ ЕГО ПОЛУЧЕНИЯ | 2001 |
|
RU2183956C1 |
| СПОСОБ ПОЛУЧЕНИЯ ХЛОРИНА Е | 2002 |
|
RU2228750C1 |
| ФОТОСЕНСИБИЛИЗАТОР И СПОСОБ ЕГО ПОЛУЧЕНИЯ | 2004 |
|
RU2276976C2 |

