ОБЛАСТЬ ИЗОБРЕТЕНИЯ
Согласно изобретению предложены новые соединения пиридилпиридонов формулы (I), фармацевтические композиции, содержащие такие соединения, и способы применения таких соединений в лечении заболеваний, включая рак и диабет II типа.
ПРЕДШЕСТВУЮЩИЙ УРОВЕНЬ ТЕХНИКИ
Ферменты, принадлежащие семейству фосфатидилинозитид-3-киназ (PI3K), представляют собой регуляторы нескольких важных клеточных событий. Данное семейство состоит из трех классов, I, II и III, и в то время как группа, принадлежащая классу I, в течение многих лет представляла собой интересную мишень для лекарственных средств, ферменты классов II и III используются в меньшей степени. PI3K класса III, вакуолярный сортирующий белок 34 (Vps34, PIK3C3), образует гетеродимер со своей регуляторной субъединицей р150 (Vps15), и этот димер участвует в работе нескольких комплексов, регулирующих события, связанные с везикулярным транспортом, такие как аутофагия, эндоцитоз, экзоцитоз и микропиноцитоз (Amaravadi et al., Clin. Cancer Res., 2011, 17: 654-666; Carpentier et al., 2013, Traffic). Данный фермент отвечает за фосфорилирование фосфатидилинозита (PI) с образованием фосфатидилинозит-(3)-фосфата (PI3P). Связывание данного лиганда с доменами РХ и FYVE приводит к рекрутированию и делокализации этих эффекторных белков, результатом чего является образование, элонгация и движение везикул (Backer et al., J. Biochem., 2008, 410: 1-17).
Аутофагия представляет собой катаболический процесс, при котором клеточные компоненты подвергаются разрушению посредством заключения их в везикулы с двухслойной мембраной, аутофагосомы, которые сливаются с содержащими протеазы лизосомами. Для клетки это является способом переработки поврежденных органелл и неправильно свернутых белков и тем самым поддержания функционирования клетки. Этот путь также является способом рециркуляции содержимого клетки с образованием новых структурных единиц (Boya et al., Nat. Cell Biol., 2013, 15: 713-720). Аутофагия представляет собой клеточный ответ на связанные со стрессом состояния, такие как нехватка питательных веществ, ацидоз и гипоксия, а также лечение лекарственными средствами. Ввиду этого ингибирование аутофагии является средством усиления действия противораковых лекарственных средств и десенсибилизации опухолей, устойчивых к лекарственным средствам (Nagelkerke et al., Semin. Cancer Biol., 2014, 31: 99-105). Опухоли на самых последних стадиях развития демонстрируют сильную активацию аутофагического потока (Leone et al. Trends in Endocrin. Metab., 2013, 24: 209-217). Общепринятым маркером для изучения аутофагического потока является обнаружение в аутофагосоме "аутофагического пятна" в форме липидированного белка LC3 (легкая цепь ассоциированного с микротрубочками белка 1 (МАР1)). Ингибирование Vps34 приводит к ингибированию аутофагии по результатам измерений перераспределения LC3 в пятне (Dowdle et al., Nat. Cell Biol., 2014, 16: 1069-79).
Как было недавно описано, удаление регуляторной субъединицы р150 приводит к повышению чувствительности к инсулину in vivo ввиду снижения интернализации инсулиновых рецепторов (Nemazanyy, Nature Commun., 2015, 6: 8283). В животной модели с гетерозиготным генотипом в отношении мутации, приводящей к инактивации киназы (kinase dead), этот результат подтвержден повышенной толерантностью к глюкозе и повышенной чувствительностью к инсулину (WO2013076501).
Ингибирование Vps34 могло бы быть выгодным при различных болезненных состояниях, включая рак, воспалительные заболевания, аутоиммунные заболевания, нейродегенеративные расстройства, сердечно-сосудистые расстройства, диабет II типа и вирусные инфекции (рассмотрены в Rubinsztein et al., Nat. Rev., 2012, 11: 709-730). Виды рака, при которых будет выгодно ингибирование Vps34, включают, но не ограничиваются этим, рак молочной железы, такой как трижды негативный рак молочной железы, рак мочевого пузыря, рак печени, рак шейки матки, рак поджелудочной железы, лейкоз, лимфому, рак почки, рак толстой кишки, глиому, рак предстательной железы, рак яичников, меланому и рак легкого, а также гипоксические опухоли. Таким образом, существует потребность в новых и сильнодействующих ингибиторах Vps34.
Предыдущие изобретения, описывающие ингибиторы Vps34 для применения при оказании воздействия на заболевания, включают WO 2015150555, WO 2015150557, WO 2015108861, WO 2015108881, WO 2012085815, WO 2012085244, WO 2013190510, Farkas, J. Biol. Chem., 2011, 286(45): 38904-12.
ОПИСАНИЕ ИЗОБРЕТЕНИЯ
Целью данного изобретения является разработка новых и сильнодействующих ингибиторов Vps34. Другой целью изобретения является разработка новых и сильнодействующих ингибиторов Vps34, которые могут быть использованы для лечения рака и других заболеваний, таких как диабет II типа.
Согласно одному из аспектов изобретения предложены соединение формулы (I),
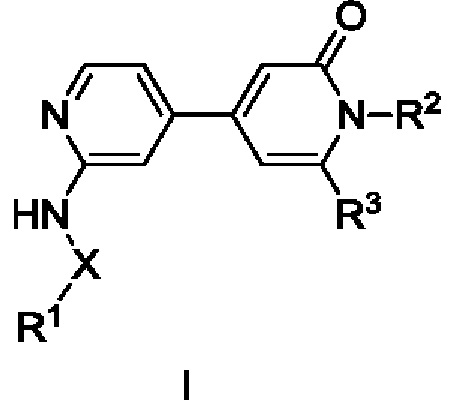
где
X представляет собой С=O или связь;
R1 выбран из Н, С1-С3алкила, С1-С3галогеналкила, С1-С3алкоксиС1-С3алкила, С3-С6циклоалкила, С3-С6циклогалогеналкила, С1-С3алкокси, С1-С3галогеналкокси, С3-С6циклоалкоксиметила, N-С1-С3алкиламино, N,N-диС1-С3алкиламино, 1-пирролидинила, 1-пиперидинила и 1-азетидинила, при условии, что когда R1 представляет собой С1-С3алкокси, С1-С3галогеналкокси, N-С1-С3алкиламино, N,N-диС1-С3алкиламино, 1-пирролидинил, 1-пиперидинил или 1-азетидинил, то X представляет собой С=O;
R2 выбран из атома водорода, С1-С3галогеналкила и С1-С3алкила;
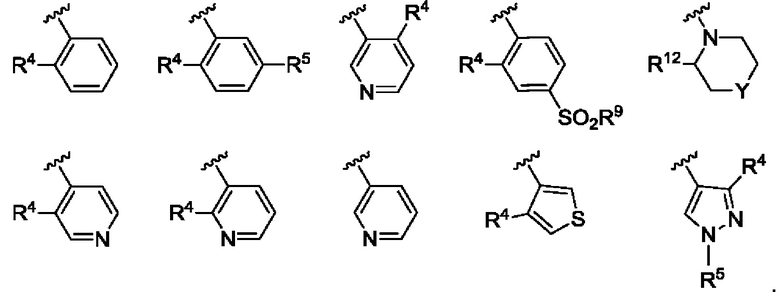
R3 выбран из А, фенила и моноциклического гетероарила, при этом указанный фенил и указанный гетероарил возможно замещены одним или более из R4, R5, R6 и R7;
R4, R5, R6 и R7 независимо выбраны из галогена, С1-С6алкила, С3-С6циклоалкила, С1-С6алкокси, С1-С3галогеналкокси, N,N-диС1-С3алкиламино, N-C1-С3алкиламино, 1-азетидинила, С1-С6галогеналкила, амино, NHSO2R8, SO2R9 и гидрокси;
R8 представляет собой С1-С3галогеналкил или С1-С3алкил;
R9 выбран из R10, С1-С6алкила, амино, N-С1-С3алкиламино, N,N-диС1-С3алкиламино и С1-С3алкоксиС1-С3алкила, при этом указанный С1-С6алкил и указанный С1-С3алкоксиС1-С3алкил возможно замещены одним R10 и/или одним или более чем одним атомом галогена;
R10 выбран из фенила, моноциклического гетероарила, С3-С6циклоалкила, гетероциклила, каждый из которых возможно замещен одним или более R11;
R11 выбран из галогена, С1-С3алкоксиС1-С3алкила, амино, N-C1-С3алкиламино, N,N-диС1-С3алкиламино, С1-С3галогеналкокси, С1-С3алкокси, С3-С6циклоалкила, С1-С3галогеналкила и С1-С3алкила;
А представляет собой
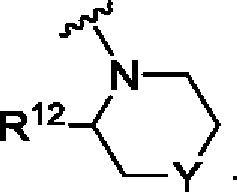
R12 выбран из атома водорода, галогена, COR13, С1-С6алкила, С1-С3алкоксиС1-С3алкила, С-С6алкокси, С3-С6циклоалкила, С1-С3цианоалкила, С1-С3галогеналкила;
R13 выбран из С1-С3алкокси, N-С1-С3алкиламино, N,N-диС1-С3алкиламино, 1-пирролидинила, 1-пиперидинила и 1-азетидинила;
Y представляет собой СН2, S, SO, SO2, NR14, NCOR9, NCOOR15, NSO2R9, NCOCH2R9, О или связь;
R14 выбран из Н, С1-С3галогеналкила, С1-С3алкоксиС1-С3алкила, С1-С3алкила, С3-С6циклоалкила;
R15 выбран из R10, С1-С6алкила и С1-С3алкоксиС1-С3алкила, при этом указанный С1-С6алкил и указанный С1-С3алкоксиС1-С3алкил возможно замещены одним R10 и/или одним или более чем одним атомом галогена;
или его фармацевтически приемлемая соль либо фармацевтически приемлемые соли.
Согласно одному из воплощений этого аспекта изобретения R2 представляет собой атом водорода или С1-С3алкил, как например, атом водорода или метил, как например, атом водорода.
Согласно одному из воплощений этого аспекта изобретения R1 выбран из Н, С1-С3алкила, С1-С3алкокси, С1-С3галогеналкокси, С1-С3алкоксиС1-С3алкила, N,N-диС1-С3алкиламино, 1-пирролидинила и С3-С6циклоалкила.
Согласно одному из воплощений этого аспекта R1 выбран из Н, метила, метокси, метоксиметила, N,N-диметиламино, 1-пирролидинила и циклопропила.
Согласно одному из воплощений этого аспекта изобретения R1 выбран из Н, метила, метоксиметила, N,N-диметиламино, 1-пирролидинила и циклопропила.
Согласно одному из воплощений этого аспекта изобретения R3 выбран из А, фенила и моноциклического гетероарила, выбранного из пиридила, тиенила, фурила, пиримидинила и пиразолила, при этом указанный фенил и указанный гетероарил возможно замещены R4 и/или R5.
Согласно одному из воплощений этого аспекта изобретения R3 выбран из А, фенила и пиридила, при этом указанный фенил и указанный пиридил возможно и независимо замещены R4 и/или R5.
Согласно одному из воплощений этого аспекта изобретения R4, R5, R6 и R7 независимо выбраны из атома фтора, атома хлора, С1-С3алкила, С3-С6циклоалкила, С1-С3фторалкила и SO2R9.
Согласно одному из воплощений этого аспекта изобретения Y представляет собой СН2, NSO2R9, О или связь.
Согласно одному из воплощений этого аспекта изобретения Y представляет собой СН2, О или связь.
Согласно одному из воплощений этого аспекта изобретения R12 выбран из атома водорода, С1-С3алкила, С1-С3алкоксиС1-С3алкила, С1-С3галогеналкила и С3-С6циклоалкила.
Согласно одному из воплощений этого аспекта изобретения R12 выбран из атома водорода, С1-С3алкила, С1-С3галогеналкила и С3-С6циклоалкила.
Согласно одному из воплощений этого аспекта изобретения R9 выбран из R10, N,N-диС1-С3алкиламино и метоксиС1-С3алкила, при этом указанный С1-С3алкил возможно замещен одним R10.
Согласно одному из воплощений этого аспекта изобретения R10 выбран из фенила, пиридила, имидазолила, изоксазолила, оксазолила, циклопропила, циклопентила, пирролидинила, тетрагидрофурила, каждый из которых возможно замещен одним или более чем одним метилом и/или атомом фтора.
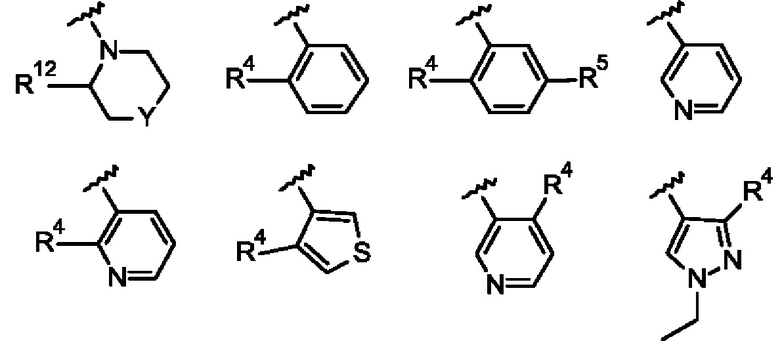
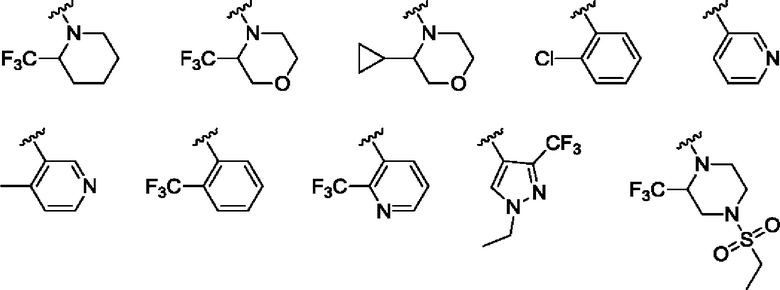
Согласно одному из воплощений этого аспекта изобретения
R3 выбран из
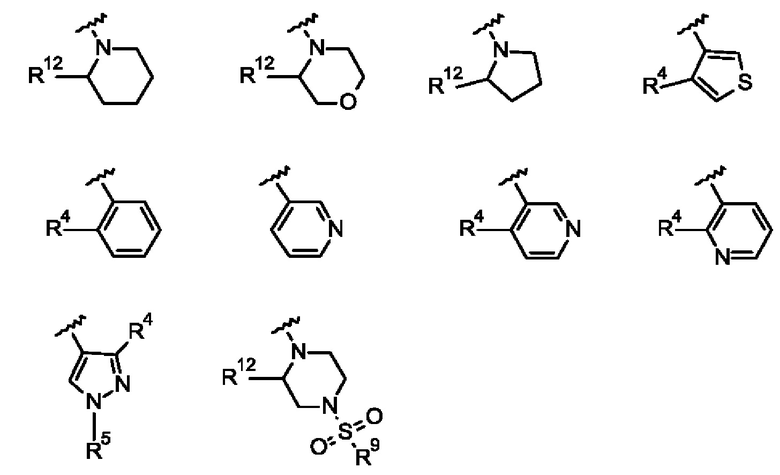
Согласно одному из воплощений этого аспекта изобретения
R3 выбран из
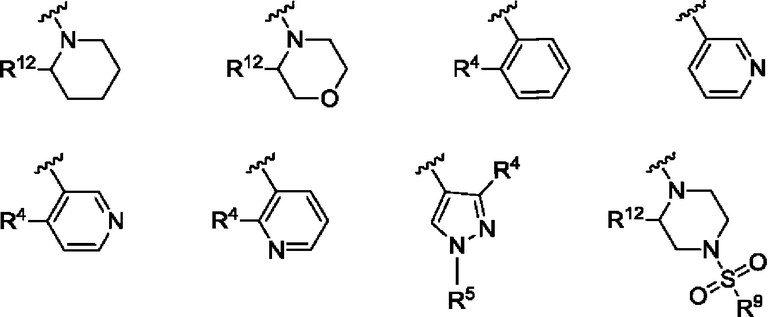
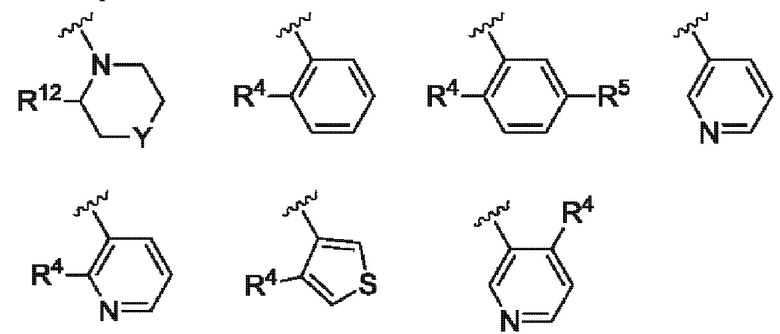
Согласно одному из воплощений этого аспекта изобретения
R3 выбран из
Согласно одному из воплощений этого аспекта изобретения
R3 выбран из
где Y выбран из СН2, О и связи;
R4 выбран из CF3, атома хлора, циклопропила и метила;
R5 представляет собой атом фтора; и
R12 выбран из атома водорода, циклопропила, метила, 1-метокси-1-метил-этила и CF3.
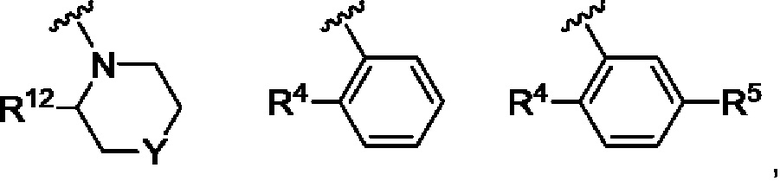
Согласно одному из воплощений этого аспекта изобретения
R3 выбран из
где Y выбран из СН2, О и связи;
R4 выбран из CF3, атома хлора, циклопропила и метила;
R5 представляет собой атом фтора; и
R12 выбран из атома водорода, циклопропила, метила и CF3.
Согласно одному из воплощений этого аспекта изобретения
R3 выбран из
где Y выбран из СН2 и О;
R4 выбран из CF3, атома хлора, циклопропила и атома хлора;
R5 представляет собой атом фтора; и
R12 представляет собой CF3 и циклопропил.
Согласно одному из воплощений этого аспекта изобретения
R1 выбран из Н, метила, метокси, метоксиметила, N,N-диметиламино, 1-пирролидинила и циклопропила;
R2 представляет собой атом водорода; и
R3 выбран из
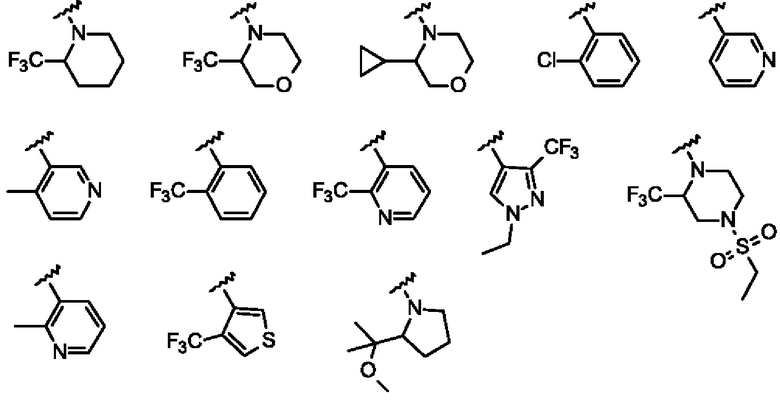
Согласно одному из воплощений этого аспекта изобретения
R1 выбран из Н, метила, метокси, метоксиметила, N,N-диметиламино, пирролидинила и циклопропила;
R2 представляет собой атом водорода; и
R3 выбран из
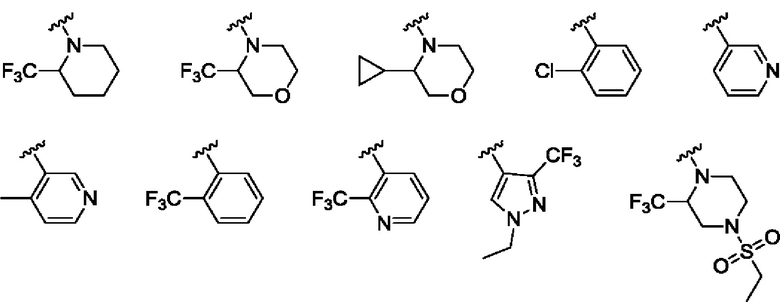
Согласно одному из воплощений этого аспекта изобретения
R1 выбран из Н, метила, метоксиметила, N,N-диметиламино, 1-пирролидинила и циклопропила;
R2 представляет собой атом водорода; и
R3 выбран из
Согласно одному из воплощений этого аспекта изобретения указанно соединение представляет собой:
N-[4-[2-(2-хлорфенил)-6-оксо-1Н-пиридин-4-ил]-2-пиридил]ацетамид;
4-(2-амино-4-пиридил)-6-(3-пиридил)-1Н-пиридин-2-он;
4-(2-амино-4-пиридил)-6-(2-хлорфенил)-1Н-пиридин-2-он;
N-[4-[2-(2-хлорфенил)-6-оксо-1Н-пиридин-4-ил]-2-пиридил]-2-метокси-ацетамид;
N-[4-[2-оксо-6-[2-(трифторметил)-1-пиперидил]-1Н-пиридин-4-ил]-2-пиридил]ацетамид;
N-[4-[2-оксо-6-[2-(трифторметил)-1-пиперидил]-1Н-пиридин-4-ил]-2-пиридил]циклопропанкарбоксамид;
N-[4-[2-оксо-6-[3-(трифторметил)морфолин-4-ил]-1Н-пиридин-4-ил]-2-пиридил]ацетамид;
метил-N-[4-[2-(2-хлорфенил)-6-оксо-1Н-пиридин-4-ил]-2-пиридил]карбамат;
метил-N-[4-[2-[1-этил-3-(трифторметил)пиразол-4-ил]-6-оксо-1Н-пиридин-4-ил]-2-пиридил]карбамат;
метил-N-[4-[2-оксо-6-[2-(трифторметил)-3-пиридил]-1Н-пиридин-4-ил]-2-пиридил]карбамат;
метил-N-[4-[2-оксо-6-[2-(трифторметил)фенил]-1Н-пиридин-4-ил]-2-пиридил]карбамат;
N-[4-[2-оксо-6-[2-(трифторметил)фенил]-1Н-пиридин-4-ил]-2-пиридил]ацетамид;
N-[4-[2-(4-метил-3-пиридил)-6-оксо-1Н-пиридин-4-ил]-2-пиридил]ацетамид;
N-[4-[2-оксо-6-[2-(трифторметил)-3-пиридил]-1Н-пиридин-4-ил]-2-пиридил]ацетамид;
N-[4-[2-[1-этил-3-(трифторметил)пиразол-4-ил]-6-оксо-1Н-пиридин-4-ил]-2-пиридил]ацетамид;
метил-N-[4-[2-оксо-6-[3-(трифторметил)морфолин-4-ил]-1Н-пиридин-4-ил]-2-пиридил]карбамат;
метил-N-[4-[2-оксо-6-[2-(трифторметил)-1-пиперидил]-1Н-пиридин-4-ил]-2-пиридил]карбамат;
N-[4-[2-(3-циклопропилморфолин-4-ил)-6-оксо-1Н-пиридин-4-ил]-2-пиридил]ацетамид;
N-[4-[2-[4-этилсульфонил-2-(трифторметил)пиперазин-1-ил]-6-оксо-1Н-пиридин-4-ил]-2-пиридил]ацетамид;
N-[4-[2-(2-метил-3-пиридил)-6-оксо-1Н-пиридин-4-ил]-2-пиридил]ацетамид;
N-[4-[2-оксо-6-[4-(трифторметил)-3-тиенил]-1Н-пиридин-4-ил]-2-пиридил]ацетамид;
1,1-диметил-3-[4-[2-оксо-6-[2-(трифторметил)фенил]-1Н-пиридин-4-ил]-2-пиридил]мочевину;
N-[4-[2-оксо-6-[2-(трифторметил)фенил]-1Н-пиридин-4-ил]-2-пиридил]пирролидин-1-карбоксамид; или
N-[4-[2-[2-(1-метокси-1-метил-этил)пирролидин-1-ил]-6-оксо-1Н-пиридин-4-ил]-2-пиридил]ацетамид.
Согласно одному из воплощений этого аспекта изобретения указанное соединение представляет собой:
4-(2-амино-4-пиридил)-6-(3-пиридил)-1Н-пиридин-2-он;
4-(2-амино-4-пиридил)-6-(2-хлорфенил)-1Н-пиридин-2-он;
N-[4-[2-(2-хлорфенил)-6-оксо-1Н-пиридин-4-ил]-2-пиридил]-2-метокси-ацетамид;
N-[4-[2-оксо-6-[2-(трифторметил)-1-пиперидил]-1Н-пиридин-4-ил]-2-пиридил]ацетамид;
N-[4-[2-оксо-6-[2-(трифторметил)-1-пиперидил]-1Н-пиридин-4-ил]-2-пиридил]циклопропанкарбоксамид;
N-[4-[2-оксо-6-[3-(трифторметил)морфолин-4-ил]-1Н-пиридин-4-ил]-2-пиридил]ацетамид;
метил-N-[4-[2-(2-хлорфенил)-6-оксо-1Н-пиридин-4-ил]-2-пиридил]карбамат;
метил-N-[4-[2-[1-этил-3-(трифторметил)пиразол-4-ил]-6-оксо-1Н-пиридин-4-ил]-2-пиридил]карбамат;
метил-N-[4-[2-оксо-6-[2-(трифторметил)-3-пиридил]-1Н-пиридин-4-ил]-2-пиридил]карбамат;
метил-N-[4-[2-(4-метил-3-пиридил)-6-оксо-1Н-пиридин-4-ил]-2-пиридил]карбамат;
метил-N-[4-[2-оксо-6-[2-(трифторметил)фенил]-1Н-пиридин-4-ил]-2-пиридил]карбамат;
N-[4-[2-оксо-6-[2-(трифторметил)фенил]-1Н-пиридин-4-ил]-2-пиридил]ацетамид;
N-[4-[2-(4-метил-3-пиридил)-6-оксо-1Н-пиридин-4-ил]-2-пиридил]ацетамид;
N-[4-[2-оксо-6-[2-(трифторметил)-3-пиридил]-1Н-пиридин-4-ил]-2-пиридил]ацетамид;
N-[4-[2-[1-этил-3-(трифторметил)пиразол-4-ил]-6-оксо-1Н-пиридин-4-ил]-2-пиридил]ацетамид;
метил-N-[4-[2-оксо-6-[3-(трифторметил)морфолин-4-ил]-1Н-пиридин-4-ил]-2-пиридил]карбамат;
метил-N-[4-[2-оксо-6-[2-(трифторметил)-1-пиперидил]-1Н-пиридин-4-ил]-2-пиридил]карбамат;
метил-N-[4-[2-[4-этилсульфонил-2-(трифторметил)пиперазин-1-ил]-6-оксо-1Н-пиридин-4-ил]-2-пиридил]карбамат;
метил-N-[4-[2-(3-циклопропилморфолин-4-ил)-6-оксо-1Н-пиридин-4-ил]-2-пиридил]карбамат;
N-[4-[2-(3-циклопропилморфолин-4-ил)-6-оксо-1Н-пиридин-4-ил]-2-пиридил]ацетамид;
N-[4-[2-[4-этилсульфонил-2-(трифторметил)пиперазин-1-ил]-6-оксо-1Н-пиридин-4-ил]-2-пиридил]ацетамид;
3-[4-[2-(2-хлорфенил)-6-оксо-1Н-пиридин-4-ил]-2-пиридил]-1,1-диметил-мочевина; или
N-[4-[2-(2-хлорфенил)-6-оксо-1Н-пиридин-4-ил]-2-пиридил]пирролидин-1-карбоксамид.
Согласно одному из воплощений этого аспекта изобретения указанное соединение представляет собой:
1N-[4-[2-(2-хлорфенил)-6-оксо-1Н-пиридин-4-ил]-2-пиридил]ацетамид;
4-(2-амино-4-пиридил)-6-(3-пиридил)-1Н-пиридин-2-он;
4-(2-амино-4-пиридил)-6-(2-хлорфенил)-1Н-пиридин-2-он;
N-[4-[2-(2-хлорфенил)-6-оксо-1Н-пиридин-4-ил]-2-пиридил]-2-метокси-ацетамид;
N-[4-[2-оксо-6-[2-(трифторметил)-1-пиперидил]-1Н-пиридин-4-ил]-2-пиридил]ацетамид;
N-[4-[2-оксо-6-[2-(трифторметил)-1-пиперидил]-1Н-пиридин-4-ил]-2-пиридил]циклопропанкарбоксамид;
N-[4-[2-оксо-6-[3-(трифторметил)морфолин-4-ил]-1Н-пиридин-4-ил]-2-пиридил]ацетамид;
метил-N-[4-[2-(2-хлорфенил)-6-оксо-1Н-пиридин-4-ил]-2-пиридил]карбамат;
метил-N-[4-[2-[1-этил-3-(трифторметил)пиразол-4-ил]-6-оксо-1Н-пиридин-4-ил]-2-пиридил]карбамат;
метил-N-[4-[2-оксо-6-[2-(трифторметил)-3-пиридил]-1Н-пиридин-4-ил]-2-пиридил]карбамат;
метил-N-[4-[2-оксо-6-[2-(трифторметил)фенил]-1Н-пиридин-4-ил]-2-пиридил]карбамат;
N-[4-[2-оксо-6-[2-(трифторметил)фенил]-1Н-пиридин-4-ил]-2-пиридил]ацетамид;
N-[4-[2-(4-метил-3-пиридил)-6-оксо-1Н-пиридин-4-ил]-2-пиридил]ацетамид;
N-[4-[2-оксо-6-[2-(трифторметил)-3-пиридил]-1Н-пиридин-4-ил]-2-пиридил]ацетамид;
N-[4-[2-[1-этил-3-(трифторметил)пиразол-4-ил]-6-оксо-1Н-пиридин-4-ил]-2-пиридил]ацетамид;
метил-N-[4-[2-оксо-6-[3-(трифторметил)морфолин-4-ил]-1Н-пиридин-4-ил]-2-пиридил]карбамат;
метил-N-[4-[2-оксо-6-[2-(трифторметил)-1-пиперидил]-1Н-пиридин-4-ил]-2-пиридил]карбамат;
N-[4-[2-(3-циклопропилморфолин-4-ил)-6-оксо-1Н-пиридин-4-ил]-2-пиридил]ацетамид; или
N-[4-[2-[4-этилсульфонил-2-(трифторметил)пиперазин-1-ил]-6-оксо-1Н-пиридин-4-ил]-2-пиридил]ацетамид.
Согласно одному из аспектов изобретения предложено соединение по настоящему изобретению для применения в лечении или профилактике заболевания.
Согласно одному из аспектов изобретения предложено соединение по настоящему изобретению для применения в лечении рака. Обычно указанный рак выбран из рака молочной железы, такого как трижды негативный рак молочной железы, рака мочевого пузыря, рака печени, рака шейки матки, рака поджелудочной железы, лейкоза, лимфомы, рака почки, рака толстой кишки, глиомы, рака предстательной железы, рака яичников, меланомы и рака легкого, а также гипоксических опухолей.
Согласно одному из аспектов изобретения предложено соединение по настоящему изобретению для применения в лечении диабета II типа.
Согласно одному из аспектов изобретения предложено соединение по настоящему изобретению для применения в лечении заболевания, выбранного из воспалительных заболеваний, аутоиммунных заболеваний, нейродегенеративных расстройств, сердечно-сосудистых расстройств и вирусных инфекций.
Согласно одному из аспектов изобретения предложено применение соединения по настоящему изобретению для приготовления лекарственного средства для лечения рака. Обычно указанный рак выбран из рака молочной железы, такого как трижды негативный рак молочной железы, рака мочевого пузыря, рака печени, рака шейки матки, рака поджелудочной железы, лейкоза, лимфомы, рака почки, рака толстой кишки, глиомы, рака предстательной железы, рака яичников, меланомы и рака легкого, а также гипоксических опухолей.
Согласно одному из аспектов изобретения предложено применение соединения по настоящему изобретению для приготовления лекарственного средства для лечения диабета II типа.
Согласно одному из аспектов изобретения предложено применение соединения по настоящему изобретению для приготовления лекарственного средства для лечения заболевания, выбранного из воспалительных заболеваний, аутоиммунных заболеваний, нейродегенеративных расстройств, сердечно-сосудистых расстройств и вирусных инфекций.
Согласно одному из аспектов изобретения предложен способ лечения рака, включающий введение терапевтически эффективного количества соединения по настоящему изобретению пациенту, нуждающемуся в этом. Обычно указанный рак выбран из рака молочной железы, такого как трижды негативный рак молочной железы, рака мочевого пузыря, рака печени, рака шейки матки, рака поджелудочной железы, лейкоза, лимфомы, рака почки, рака толстой кишки, глиомы, рака предстательной железы, рака яичников, меланомы и рака легкого, а также гипоксических опухолей.
Согласно одному из аспектов изобретения предложен способ лечения гипоксических опухолей, включающий введение терапевтически эффективного количества соединения по настоящему изобретению пациенту, нуждающемуся в этом.
Согласно одному из аспектов изобретения предложено соединение по настоящему изобретению для применения в лечении рака, при этом указанное лечение рака дополнительно включает лучевую терапию.
Согласно одному из аспектов изобретения предложен способ лечения рака, включающий введение терапевтически эффективного количества соединения по настоящему изобретение пациенту, нуждающемуся в этом, вместе с лучевой терапией.
Соединения по настоящему изобретению также могут быть использованы для лечения рака вместе с лучевой терапией и/или хирургическим вмешательством. Обычно, применение цитотоксических и/или цитостатических агентов в комбинации с соединением или композицией по настоящему изобретению будет способствовать:
(1) достижению лучшей эффективности в снижении роста опухоли или даже устранению опухоли по сравнению со случаем введения каждого из агентов по отдельности;
(2) обеспечению введения меньших количеств применяемых химиотерапевтических агентов;
(3) предоставлению химиотерапевтического лечения, которое хорошо переносится пациентом, при этом количество вредных фармакологических осложнений будет меньше наблюдаемого в случае химиотерапии одним агентом и некоторыми другими видами комбинированной терапии;
(4) обеспечению лечения более широкого спектра разных типов рака у млекопитающих, в особенности у людей;
(5) достижению более высокого значения коэффициента ответа среди подвергаемых лечению пациентов;
(6) обеспечению более продолжительного периода времени выживания среди подвергаемых лечению пациентов по сравнению со стандартными методами химиотерапии;
(7) обеспечению задержки прогрессирования опухолей; и/или
(8) получению по меньшей мере таких же удовлетворительных результатов по эффективности и переносимости, как и в случаях применения агентов, используемых по-отдельности, по сравнению с известными случаями, когда другие комбинации противораковых агентов оказывают антагонистические эффекты.
Согласно одному из аспектов изобретения предложен способ лечения диабета II типа, включающий введение терапевтически эффективного количества соединения по настоящему изобретению, пациенту, нуждающемуся в этом.
Согласно одному из аспектов изобретения предложен способ лечения заболевания, выбранного из воспалительных заболеваний, аутоиммунных заболеваний, нейродегенеративных расстройств и вирусных инфекций, включающий введение терапевтически эффективного количества соединения по настоящему изобретению, пациенту, нуждающемуся в этом.
Согласно одному из аспектов изобретения предложена фармацевтическая композиция, содержащая соединение по настоящему изобретению и фармацевтически приемлемый разбавитель, носитель и/или эксципиент.
Согласно одному из аспектов изобретения предложена фармацевтическая композиция, содержащая терапевтически эффективное количество соединения по настоящему изобретению и другого противоракового средства, выбранного из алкилирующих агентов, антиметаболитов, противораковых производных камптотецина, противораковых средств растительного происхождения, антибиотиков, ферментов, координационных комплексов платины, ингибиторов тирозинкиназ, гормонов, антагонистов гормонов, моноклональных антител, интерферонов и модификаторов биологического ответа.
Использованный в данном описании термин "С1-С6алкил" означает насыщенные углеводородные группы как с линейной, так и разветвленной цепью, состоящие из 1-6 атомов углерода. Примеры С1-С6алкильных групп включают метильную, этильную, н-пропильную, изопропильную, н-бутильную, изобутильную, втор-бутильную, трет-бутильную, н-пентильную, 4-метил-бутильную, н-гексильную, 2-этил-бутильную группы. Среди неразветвленных С1-С6алкильных групп типичными являются метильная, этильная, н-пропильная, н-бутильная, н-пентильная и н-гексильная группы. Среди разветвленных алкильных групп можно упомянуть изопропильную, изобутильную, втор-бутильную, трет-бутильную, 4-метил-бутильную и 2-этил-бутильную группы.
Использованный в данном описании термин "С1-С3алкил" означает насыщенные углеводородные группы как с линейной, так и разветвленной цепью, состоящие из 1-3 атомов углерода. Примеры С1-С3алкильных групп включают метильную, этильную, н-пропильную и изопропильную группы.
Использованный в данном описании термин "С1-С6алкокси" означает группу О-алкил, при этом используют "С1-С6алкил", как он описан выше. Примеры групп С1-С6алкокси включают, но не ограничиваются этим, группы метокси, этокси, изопропокси, н-пропокси, н-бутокси, н-гексокси, 3-метил-бутокси.
Использованный в данном описании термин "С1-С3алкокси" означает группу О-алкил, при этом используют "С1-С3алкил", как он описан выше. Примеры групп С1-С3алкокси включают, но не ограничиваются этим, группы метокси, этокси, изопропокси и н-пропокси.
Использованный в данном описании термин "С1-С6галогеналкил" означает насыщенные углеводородные группы как с линейной, так и разветвленной цепью, состоящие из 1-6 атомов углерода, и в которых от 1 до всех атомов водорода замещены атомом(ами) галогена разного или одного и того же типа. Примеры (V С6 галогеналкильных групп включают метильные группы, замещенные 1-3 атомами галогена, этильные группы, замещенные 1-5 атомами галогена, н-пропильные или изопропильные группы, замещенные 1-7 атомами галогена, н-бутильные или изобутильные группы, замещенные 1-9 атомами галогена, и втор-бутильные или трет-бутильные группы, замещенные 1-9 атомами галогена.
Использованный в данном описании термин "С1-С3 галогеналкил" означает насыщенные углеводородные группы как с линейной, так и разветвленной цепью, состоящие из 1-3 атомов углерода, и в которых от 1 до всех атомов водорода замещены атомом(ами) галогена разного или одного и того же типа. Примеры С1-С3 галогеналкильных групп включают метил, замещенный 1-3 атомами галогена, этил, замещенный 1-5 атомами галогена, и н-пропил или изопропил, замещенный 1-7 атомами галогена.
Использованный в данном описании термин "С1-С3галогеналкокси" означает насыщенные группы алкокси как с линейной, так и разветвленной цепью, состоящие из 1-3 атомов углерода, и в которых от 1 до всех атомов водорода замещены атомом(ами) галогена разного или одного и того же типа. Примеры групп С1-С3 галогеналкокси включают группу метокси, замещенную 1-3 атомами галогена, группу этокси, замещенную 1-5 атомами галогена, и группу н-пропокси или изопропокси, замещенную 1-7 атомами галогена.
Использованный в данном описании термин "С1-С3фторалкил" означает насыщенные углеводородные группы как с линейной, так и разветвленной цепью, состоящие из 1-3 атомов углерода, и в которых от 1 до всех атомов водорода замещены атомом(ами) фтора. Примеры С1-С3фторалкильных групп включают метил, замещенный 1-3 атомами фтора, этил, замещенный 1-5 атомами фтора, и н-пропил или изопропил, замещенный 1-7 атомами фтора.
Использованный в данном описании термин "С1-С3фторалкокси" означает насыщенные группы алкокси как с линейной, так и разветвленной цепью, состоящие из 1-3 атомов углерода, и в которых от 1 до всех атомов водорода замещены атомом(ами) фтора. Примеры групп С1-С3фторалкокси включают группу метокси, замещенную 1-3 атомами фтора, группу этокси, замещенную 1-5 атомами фтора, и группу н-пропокси или изопропокси, замещенную 1-7 атомами фтора.
Использованный в данном описании термин "С3-С6циклоалкил" означает циклическую насыщенную углеводородную группу, состоящую из 3-6 атомов углерода. Примеры С3-С6циклоалкильных групп включают циклопропил, циклобутил, циклопентил и циклогексил.
Использованный в данном описании термин "С1-С3алкоксиС1-С3алкил" означает насыщенную углеводородную группу как с линейной, так и разветвленной цепью, состоящую из 1-3 атомов углерода, замещенную группой алкокси из 1-3 атомов углерода. Примеры С1-С3алкоксиС1-С3алкильных групп приведены ниже:
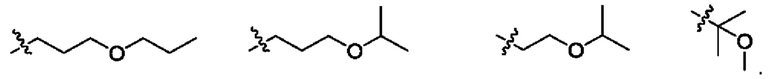
Использованный в данном описании термин "С1-С3цианоалкил" означает циано(CN)-производное как с линейной, так и разветвленной цепью, содержащее от одного до трех атомов углерода, включая атом углерода, который составляет часть цианогруппы. Примеры С1-С3цианоалкильных групп приведены ниже:
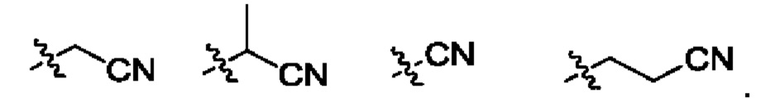
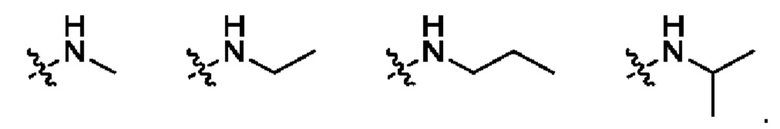
Использованный в данном описании термин "N,N-С1-С3алкиламино" означает амино-содержащий заместитель, несущий одну С1-С3алкильную группу, как она определена выше. Примеры N,N-С1-С3алкиламино приведены ниже:
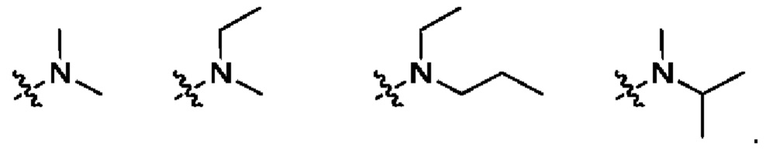
Использованный в данном описании термин "N,N-диС1-С3алкиламино" означает амино-содержащий заместитель, несущий две С1-С3алкильные группы, как они определены выше. Примеры N,N-диС1-С3алкиламино приведены ниже:
Использованный в данном описании термин "галоген" означает фтор, хлор, бром или йод. Использованный в данном описании термин "атом галогена" означает атом фтора, хлора, брома или йода.
Использованный в данном описании термин "гетероарил" означает моноциклическую ароматическую группу, состоящую из атомов углерода, в которой от одного до трех атомов углерода заменены на один или несколько гетероатомов, независимо выбранных из атомов азота, кислорода или серы. В бициклическом ариле одно из колец может быть частично насыщенным.
Использованный в данном описании термин "моноциклический гетероарил" означает моноциклическую ароматическую группу, состоящую из атомов углерода, в которой от одного до трех атомов углерода заменены на один или несколько гетероатомов, независимо выбранных из атомов азота, кислорода или серы.
Примеры моноциклических гетероарильных групп включают, но не ограничиваются этим, фурил, тиенил, пирролил, оксазолил, тиазолил, имидазолил, оксадиазолил, тиадиазолил, пиридил, триазолил, триазинил, пиридазил, изотиазолил, изоксазолил, пиразинил, пиразолил и пиримидинил.
Использованный в данном описании термин "гетероциклил" означает циклическую группу, состоящую из атомов углерода, в которой от одного до трех атомов углерода заменены на один или несколько гетероатомов, независимо выбранных из атомов азота, кислорода или серы. Примеры гетеро цикл ильных групп включают, но не ограничиваются этим, тетрагидрофурил, тетрагидропиранил, пирролидинил, пиперидинил, пиперазинил, морфолинил и диоксанил.
В зависимости от заместителей, имеющихся в соединениях формулы (I), соединения могут образовывать соли, которые находятся в пределах объема настоящего изобретения. Соли соединений формулы (I), подходящие для применения в медицине, представляют собой соли, где противоион является фармацевтически приемлемым.
Подходящие соли по изобретению включают соли, образованные при взаимодействии с органическими или неорганическими кислотами или основаниями. В частности, подходящие соли, образованные при взаимодействии с кислотами согласно изобретению, включают соли, образованные при взаимодействии с минеральными кислотами, с сильными органическими карбоновыми кислотами, такими как алканкарбоновые кислоты, состоящие из 1-4 атомов углерода, которые являются незамещенными или замещенными, например, атомом галогена, такими как насыщенные или ненасыщенные дикарбоновые кислоты, такими как гидроксикарбоновые кислоты, такими как аминокислоты, или с органическими сульфоновыми кислотами, такими как (С1-С4)алкил- или -арилсульфоновые кислоты, которые являются незамещенными или замещенными, например, атомом галогена. Фармацевтически приемлемые соли присоединения кислоты включают соли, образованные из соляной, бромистоводородной, серной, азотной, лимонной, винной, уксусной, фосфорной, молочной, пировиноградной, уксусной, трифторуксусной, янтарной, перхлорной, фумаровой, малеиновой, гликолевой, молочной, салициловой, щавелевоуксусной, метансульфоновой, этансульфоновой, п-толуолсульфоновой, муравьиной, бензойной, малоновой, нафталин-2-сульфоновой, бензолсульфоновой, изэтионовой, аскорбиновой, яблочной, фталевой, аспарагиновой и глутаминовой кислот, лизина и аргинина.
Фармацевтически приемлемые соли с основаниями включают соли аммония, соли щелочных металлов, например, соли калия и натрия, соли щелочноземельных металлов, например, соли кальция и магния, и соли с органическими основаниями, например, дициклогексиламином, N-метил-О-глюкамином, морфолином, тиоморфолином, пиперидином, пирролидином, с низшими моно-, ди- или триалкиламинами, например, этил-, трет-бутил-, диэтил-, диизопропил-, триэтил-, трибутил- или диметил-пропиламином, либо с низшим моно-, ди- или три-гидроксиалкиламином, например, моно-, ди- или три-этаноламином. Кроме того, могут быть образованы соответствующие внутренние соли.
Соединения по изобретению можно использовать для профилактики и/или лечения в том виде, как они есть, или в форме фармацевтической композиции. Несмотря на существующую возможность введения активного ингредиента как такового, также возможно его нахождение в составе фармацевтической композиции. Соответственно, согласно изобретению предложена фармацевтическая композиция, содержащая соединение формулы (I) и фармацевтически приемлемый разбавитель, эксципиент и/или носитель. Фармацевтические композиции по изобретению могут быть в форме фармацевтической композиции, которая описана ниже.
Типичные композиции для перорального введения включают суспензии, которые могут содержать, например, микрокристаллическую целлюлозу для придания объема, альгиновую кислоту или альгинат натрия в качестве суспендирующего агента, метилцеллюлозу в качестве усилителя вязкости и подсластители или ароматизаторы, как например известные в данной области техники; и таблетки с немедленным высвобождением, которые могут содержать, например, микрокристаллическую целлюлозу, двузамещенный фосфат кальция, крахмал, стеарат магния, сульфат кальция, сорбит, глюкозу и/или лактозу и/или другие эксципиенты, связующие вещества, сухие наполнители, разрыхлители, разбавители и смазывающие вещества, как например известные в данной области техники. Подходящие связующие вещества включают крахмал, желатин, природные сахара, такие как глюкоза или бета-лактоза, сахаристые вещества из кукурузы, природные и синтетические камеди, такие как аравийская камедь, трагакантовая камедь или альгинат натрия, карбоксиметилцеллюлозу, полиэтилен гликоль, воски и тому подобное. Разрыхлители включают, без ограничения, крахмал, метилцеллюлозу, агар, бентонит, ксантановую камедь и тому подобное. Доставку соединений формулы (I) также можно осуществлять через ротовую полость посредством сублингвального и/или трансбуккального введения. Формованные таблетки, прессованные таблетки или сублимационно высушенные таблетки представляют собой типичные формы, которые могут быть использованы. Типичные композиции включают композиции, в состав которых входит(ят) соединение(я) по настоящему изобретению с быстро растворяющимися разбавителями, такими как маннит, лактоза, сахароза и/или циклодекстрины. В такие композиции также могут быть включены высокомолекулярные эксципиенты, такие как целлюлозы (авицел) или полиэтиленгликоли (ПЭГ). Такие композиции также могут включать в себя эксципиент, способствующий адгезии к слизистой оболочке, такой как гидроксипропилцеллюлоза (НРС), гидроксипропилметилцеллюлоза (НРМС), натриевая соль карбоксиметилцеллюлозы (SCMC), сополимер на основе малеинового ангидрида (например, Gantrez) и агенты, регулирующие высвобождение, такие как сополимер на основе полимера акриловой кислоты (например, карбопол 934). Для простоты изготовления и применения также могут быть добавлены смазывающие вещества, скользящие вещества, ароматизаторы, красители и стабилизаторы. Смазывающие вещества, используемые в этих лекарственных формах, включают олеат натрия, стеарат натрия, стеарат магния, бензоат натрия, ацетат натрия, хлорид натрия и тому подобное. Для перорального введения в жидкой форме компоненты перорального лекарственного средства могут быть объединены с любым пригодным для перорального введения нетоксичным, фармацевтически приемлемым инертным носителем, таким как этанол, глицерин, вода и тому подобное.
Композиции по настоящему изобретению, подходящие для перорального введения, могут быть представлены в виде дискретных единиц, таких как капсулы, облатки, пилюли или таблетки, каждая из которых содержит предварительно заданное количество активного ингредиента; в виде порошка или гранул; в виде раствора или суспензии в водной жидкости или неводной жидкости, например, в виде эликсиров, настоек, суспензий или сиропов; или в виде жидкой эмульсии типа масло-в-воде либо жидкой эмульсии типа вода-в-масле. Активный ингредиент также может быть представлен в виде болюса, электуария или пасты.
Таблетка может быть изготовлена путем прессования или формования, возможно с одним или несколькими дополнительными ингредиентами. Прессованные таблетки могут быть изготовлены в подходящей машине путем прессования активного ингредиента в свободно текучей форме, такой как порошок или гранулы, возможно в смеси со связующим веществом, смазывающим веществом, инертным разбавителем, поверхностно-активным веществом или диспергирующим агентом. Формованные таблетки могут быть изготовлены в подходящей машине путем формования смеси измельченного соединения, увлажненного инертным жидким разбавителем. Возможно, что таблетки могут иметь покрытие или риску и могут быть изготовлены с тем, чтобы обеспечить медленное или регулируемое высвобождение из нее активного ингредиента. Например, соединения по настоящему изобретению можно вводить в форме, подходящей для немедленного высвобождения или продолжительного высвобождения. Немедленное высвобождение или продолжительное высвобождение может быть осуществлено посредством применения подходящих фармацевтических композиций, содержащих соединения по настоящему изобретению, или, особенно в случае продолжительного высвобождения, посредством применения таких устройств, как подкожные имплантаты или осмотические насосы. Соединения по настоящему изобретению также можно вводить с использованием липосом.
Типичными композициями в стандартной лекарственной форме являются такие, которые содержат активный ингредиент в эффективной дозе, как приведено ранее, или в соответствующей ее доле.
Следует понимать, что помимо ингредиентов, в частности, упомянутых выше, композиции по данному изобретению могут включать в себя другие агенты, традиционно применяемые в данной области техники с учетом типа рассматриваемой композиции, например, те, которые подходят для перорального введения, могут включать в себя ароматизаторы.
Данные композиции могут быть представлены в стандартной лекарственной форме и могут быть приготовлены любым из способов, хорошо известных в области фармацевтики. Такие способы могут включать стадию объединения активного ингредиента с носителем, который состоит из одного или нескольких дополнительных ингредиентов. Композиции могут быть приготовлены путем непрерывного и равномерного объединения активного ингредиента с жидкими носителями или тонкоизмельченными твердыми носителями либо с ними обоими и затем, при необходимости, придания продукту формы желаемой композиции.
Соединения по настоящему изобретению также можно вводить в составе систем доставки с использованием липосом, таких как малые однослойные везикулы, большие однослойные везикулы и многослойные везикулы. Липосомы могут быть образованы из разнообразных фосфолипидов, 1,2-дипальмитоилфосфатидилхолина, фосфатидилэтаноламина (цефалина), фосфатидилсерина, фосфатидилинозита, дифосфатидилглицерина (кардиолипина) или фосфатидилхолина (лецитина).
Композиции для парентерального введения включают водные и неводные стерильные растворы для инъекций, которые могут содержать антиоксиданты, буферы, бактериостатические средства и растворенные вещества, делающие композицию изотоничной крови предполагаемого реципиента; и водные и неводные стерильные суспензии, которые могут включать в себя суспендирующие агенты и загустители. Данные композиции могут быть представлены в однодозовых или многодозовых контейнерах, например, герметично запаянных ампулах и герметично закрытых флаконах, и могут храниться в сублимационно высушенном (лиофилизированном) состоянии, требующем только добавления стерильного жидкого носителя, например, физиологического раствора или воды для инъекций, непосредственно перед применением. Из стерильных порошков, гранул и таблеток описанного ранее вида могут быть приготовлены экстемпоральные растворы и суспензии для инъекций. Типичные композиции для парентерального введения включают инъекционные растворы или суспензии, которые могут содержать, например, подходящие нетоксичные парентерально приемлемые разбавители или растворители, такие как полиэтиленгликоль, этанол, 1,3-бутандиол, вода, раствор Рингера, изотонический раствор хлорида натрия, или другие подходящие диспергирующие или увлажняющие и суспендирующие агенты, включая синтетические моно-или диглицериды, и жирные кислоты, в том числе олеиновую кислоту или кремофор.
Типичные композиции для назального, аэрозольного или ингаляционного введения включают растворы в физиологическом растворе, которые могут содержать, например, бензиловый спирт или другие подходящие консерванты, стимуляторы всасывания для усиления биодоступности и/или другие солюбилизирующие или диспергирующие агенты, например, агенты, известные в данной области техники.
Композиции для ректального введения могут быть представлены в виде суппозитория с типичными носителями, такими как масло какао, синтетические эфиры на основе глицерина (глицериды) или полиэтиленгликоль. Как правило, такие носители являются твердыми при обычных температурах, но расплавляются и/или растворяются в прямой кишке с высвобождением лекарственного средства.
Композиции для местного введения в полость рта, например, трансбуккально или сублингвально, включают пастилки, содержащие активный ингредиент в ароматизированной основе, такой как сахароза и аравийская или трагакантовая камедь, и пастилки, содержащие активный ингредиент в такой основе, как желатин и глицерин или сахароза и аравийская камедь. Типичные композиции для местного введения включают в себя носитель для местного применения, такой как Plastibase® (минеральное масло с загустителем полиэтиленом).
Соединения формулы (I) можно вводить в виде единственного фармацевтического агента или в комбинации с одним или более чем одним дополнительным терапевтическим агентом, при этом данная комбинация не оказывает никаких неприемлемых, неблагоприятных эффектов. Такая фармацевтическая композиция подразумевает введение единой композиции фармацевтических средств, которая содержит соединение формулы (I) и один или более чем один дополнительный терапевтический агент, а также введение соединения формулы (I) и каждого дополнительного терапевтического агента в составе своей собственной отдельно вводимой фармацевтической композиции. Например, соединение формулы (I) и терапевтический агент можно вводить пациенту вместе в единой перорально вводимой композиции, такой как капсула или таблетка, или каждый агент можно вводить в композициях с отдельной дозировкой.
В случае использования композиций для раздельного введения введение соединения формулы (I) и одного или более чем одного дополнительного терапевтического агента может быть осуществлено по существу параллельно (например, одновременно) или с разнесением по времени (например, последовательно).
Количество активного ингредиента, которое необходимо для достижения терапевтического эффекта, несомненно, будет варьировать в зависимости от конкретного соединения, пути введения, подвергаемого лечению субъекта, в том числе типа, вида, возраста, массы, пола и состояния здоровья данного субъекта и функции почек и печени у данного субъекта, и конкретного подвергаемого лечению расстройства или заболевания, а также его тяжести. Врач, ветеринар или врач-клиницист, имеющий обычную квалификацию, может легко определить и прописать эффективное количество лекарственного средства, необходимое для предотвращения, противодействия или задержки прогрессирования данного состояния.
Пероральные дозировки по настоящему изобретению, в случае использования для достижения указанных эффектов, будут находиться в диапазоне от примерно 0,01 мг на кг массы тела в сутки (мг/кг/сутки) до примерно 100 мг/кг/сутки, предпочтительно от 0,01 мг на кг массы тела в сутки (мг/кг/сутки) до 10 мг/кг/сутки и наиболее предпочтительно от 0,1 до 5,0 мг/кг/сутки для взрослого человека. В случае перорального введения композиции могут быть приготовлены в форме таблеток или в других формах представления, приготовленных в дискретных единицах, содержащих 0,01, 0,05, 0,1, 0,5, 1,0, 2,5, 5,0, 10,0, 15,0, 25,0, 50,0, 100 и 500 миллиграммов активного ингредиента, для корректировки дозировки симптоматической терапии пациента, подлежащего лечению. Обычно лекарственное средство содержит от примерно 0,01 мг до примерно 500 мг активного ингредиента, предпочтительно от примерно 1 мг до примерно 100 мг активного ингредиента. Если внутривенно, то наиболее предпочтительные дозы будут находиться в диапазоне от примерно 0,1 до примерно 10 мг/кг/минута при инфузировании с постоянной скоростью. Соединения по настоящему изобретению можно вводить в однократной суточной дозе, или общую суточную дозировку можно вводить в разделенных дозах в режиме двух, трех или четырех раз в сутки. Кроме того, соединения по настоящему изобретению можно вводить в интраназальной форме посредством местного применения вместе с подходящими интраназальными наполнителями или можно вводить трансдермальными путями, используя такие формы трансдермальных накожных пластырей, которые хорошо известны специалистам средней квалификации в данной области техники. В случае введения в форме трансдермальной системы доставки введение дозы, несомненно, будет непрерывным, а не прерывистым в ходе всего режима дозирования.
Получение соединений
Соединения по настоящему изобретению могут быть получены в виде свободного основания или его фармацевтически приемлемой соли описанным ниже способом. При последующем описании таких способов подразумевается, что там, где это целесообразно, к различным реагентам и промежуточным соединениям будут добавлены и впоследствии удалены из них подходящие защитные группы методом, совершенно очевидным специалисту в области органического синтеза. Традиционные методики использования таких защитных групп, а также примеры подходящих защитных групп описаны, например, в Protective Groups in Organic Synthesis под редакцией T.W. Greene, P.G.M Wutz, в 4м издании, Wiley-lnterscience, New York, 2006. Очевидно, что для альтернативного нагревания реакционных смесей можно использовать микроволновое излучение.
Согласно другому аспекту настоящего изобретения предложен способ получения соединения формулы (I) или его фармацевтически приемлемой соли, где R1, R2, R3 и X являются такими, если не указано иное, как определено в данном описании. Указанный способ включает:
(1) образование соответствующего соединения формулы (I)
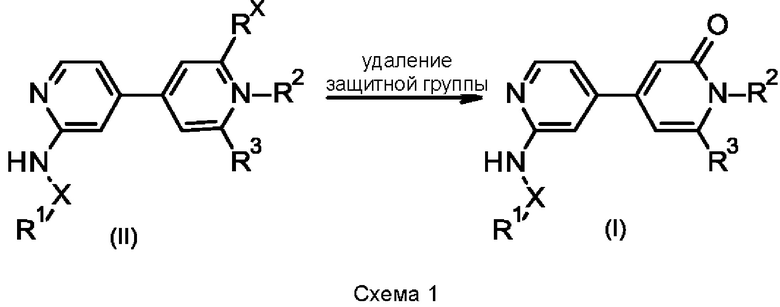
Соединение формулы (I) может быть получено (схема 1) исходя из, например, соединения формулы (II), где Rx может представлять собой F, ОСН3, ОС(СН3)3 или OSiR'R''R''' (где R', R'' и R''' независимо представляют собой арил (такой как фенил) или алкил (такой как метил или трет-бутил)). Если Rx представляет собой F, то превращение в соединение (I) может быть осуществлено посредством, например, кислотного гидролиза с использованием водного раствора HCl. Если Rx представляет собой ОСН3, то превращение в соединение (I) может быть осуществлено в результате взаимодействия, например, с триметилсилилиодидом в подходящем растворителе, таком как хлороформ, или в результате взаимодействия с HBr в подходящем растворителе, таком как уксусная кислота, или в результате взаимодействия с BBr3 в подходящем растворителе, таком как дихлорметан. Если Rx представляет собой ОС(СН3)3, то превращение в соединение (I) может быть осуществлено в результате взаимодействия, например, с трифторуксусной кислотой в подходящем растворителе, таком как дихлорметан. Если Rx представляет собой OSiR'R''R''', то превращение в соединение (I) может быть осуществлено с использованием, например, HCl в подходящем растворителе, таком как метанол, или с использованием фторида тетрабутиламмония в тетрагидрофуране. Если в этой реакции используется энантиомерно чистое или обогащенное соединение (II), то получают энантиомерно чистое или энантиомерно обогащенное соединение (I).
Соединения формулы (II) имеются в продаже или известны из литературных источников, либо их получают стандартными способами, известными в данной области техники. Соединение формулы (I) или (II) может быть разделено на свои энантиомеры стандартными способами, известными в данной области техники, например, посредством хроматографии на хиральной неподвижной фазе.
Общие методы
Все использованные растворители были сорта "чистый для анализа", и имеющиеся в продаже безводные растворители в рабочем порядке использовали для проведения реакций. Исходные вещества были получены из коммерческих источников или получены в соответствии с опубликованными в литературе методиками. Комнатная температура означает +20-25°С. Составы смесей растворителей приведены в объемных процентах или в объемных соотношениях.
Нагревание микроволновым излучением выполняли в микроволновом резонаторе Initiator от Biotage, осуществляющем непрерывное облучение при 2,45 ГГц. Очевидно, что для нагревания реакционных смесей можно использовать микроволновое излучение.
Нормально-фазовую хроматографию выполняли в ручном варианте на силикагеле 60 (0,040-0,063 мм) от Merck или в автоматизированном варианте с использованием системы ISCO Combiflash® Companion™, применяя колонки SiliaSep™ для нормально-фазовой флэш-хроматографии и указанную систему растворителей.
Спектры ядерного магнитного резонанса (ЯМР) регистрировали на ЯМР-спектрометре при 400 МГц (или в более сильном поле), оснащенном датчиком подходящей конфигурации. Спектры регистрировали при температуре окружающей среды, если не указано иное. Химические сдвиги приведены в млн-1 в сторону слабого и сильного поля по отношению к сигналу TMS (тетраметилсилан) (0.00 млн-1). Использовали следующие референсные сигналы: остаточный сигнал растворителя DMSO-d6 δ 2.5, CDCl3 δ 7.26 или метанола-d4 δ 3.31. Мультиплетности резонансных сигналов обозначаются как s, d, t, q, m и br для синглетного, дублетного, триплетного, квартетного, мультиплетного и уширенного сигналов, соответственно.
Жидкостную хроматографию высокого давления (HPLC) проводили на колонке с обращенной фазой. Применяли линейный градиент, используя, например, подвижную фазу А (водный 0,1%-ный раствор NH3, или водный 0,1%-ный раствор уксусной кислоты, или водный 0,1%-ный раствор муравьиной кислоты) и В (ацетонитрил или метанол). Масс-спектрометрические (MS) анализы проводили в режиме положительных ионов, используя ионизацию электрораспылением (ES+).
Препаративную хроматографию проводили на Gilson-PREP GX271 или GX281 с использованием Trilution Ic в качестве программного обеспечения на колонке с обращенной фазой. Применяли линейный градиент, используя, например, подвижную фазу А (водный 0,1%-ный раствор NH3, или водный 0,1%-ный раствор уксусной кислоты, или водный 0,1%-ный раствор муравьиной кислоты) и В (ацетонитрил или метанол).
Препаративную хиральную хроматографию для разделения энантиомеров проводили на системе Thar для SFC, используя сверхкритическую жидкостную хроматографию (SFC) на хиральной неподвижной фазе. Применяли линейный градиент, используя подвижную фазу А (двуокись углерода) и В (ацетонитрил, или метанол, или этанол, или 2-пропанол, или любые их смеси). Могут быть использованы вспомогательные вещества (такие как диэтиламин, или изопропиламин, или аммиак, или муравьиная кислота, или TFA).
Названия соединений приведены с использованием BIOVIA Draw 16.1.
Сокращения
Amphos - (4-(N,N-диметиламино)фенил)ди-трет-бутилфосфин;
безв. - безводный;
водн. - водный;
BuLi - бутиллитий;
DCM - дихлорметан;
DMAc - N,N-диметилацетамид;
DME - 1,2-диметоксиэтан;
DMF - N,N-диметилформамид;
DMSO - диметилсульфоксид;
EtOAc - этилацетат;
EtOH - этанол;
ч - час(ы);
HPLC - высокоэффективная жидкостная хроматография (или высокого давления);
KOtBu - трет-бутилат калия;
LCMS - жидкостная хроматография в сочетании с масс-спектрометрией;
MeCN - ацетонитрил;
2-MeTHF - 2-метил-тетрагидрофуран;
MeOH - метанол;
мин - минута(ы);
ЯМР - ядерный магнитный резонанс;
PEPPSI-iPr - [1,3-бис(2,6-диизопропилфенил)имидазол-2-илиден](3-хлорпиридил)палладия(II) дихлорид;
Pd(OAc)2 - ацетат палладия(II);
PdCl2(dppf) - [1,1'-бис(дифенилфосфино)ферроцен]-дихлорпалладий(II);
колич. - количественный;
КТ - комнатная температура;
насыщ. - насыщенный;
S-Phos - 2-дициклогексилфосфино-2',6'-диметоксибифенил;
TFA - трифторуксусная кислота;
THF - тетрагидрофуран.
Пример 1
N-[4-[2-(2-Хлорфенил)-6-оксо-1Н-пиридин-4-ил]-2-пиридил]ацетамид
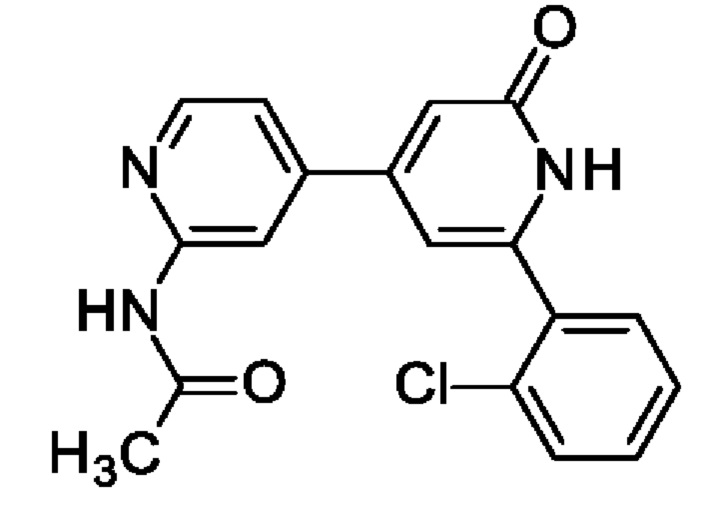
2,6-Дихлор-4-иод-пиридин (0,5 г; 1,83 ммоль), N-[4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-2-пиридил]ацетамид (0,53 г; 2,01 ммоль), K2CO3 (0,5 г; 3,65 ммоль) и PdCl2(dppf) (0,07 г; 0,09 ммоль) растворяли в DME (3 мл) и воде (1 мл) и смесь перемешивали при 80°С в течение 1 ч. Добавляли (2-хлорфенил)бороновую кислоту (0,29 г; 1,83 ммоль), K2CO3 (0,5 г; 3,65 ммоль) и PdCl2(dppf) (0,07 г; 0,09 ммоль) и смесь перемешивали в течение 4 ч при 100°С. Органический слой отделяли, фильтровали и концентрировали. Неочищенное вещество переносили в толуол (4 мл), добавляли KOtBu (0,141 г; 1,26 ммоль) и смесь перемешивали при 100°С в течение 30 мин. После охлаждения до КТ смесь концентрировали, остаток растворяли в смеси MeOH/DMF, фильтровали и очищали препаративной HPLC, получая продукт в виде твердого вещества (6 мг; 4%). 1Н ЯМР (500 МГц, метанол-d4) δ млн-1 1.92 (s, 1Н) 2.22 (s, 3Н) 6.75 (s, 1Н) 6.87 (s, 1H) 7.43 (dd, 1H) 7.46-7.52 (m, 1 H) 7.55 (td, 1H) 7.57-7.67 (m, 2H) 8.39-8.53 (m, 2H). MS ES+ m/z 341 [M+H]+.
Пример 2
N-[4-(2,6-Дихлор-4-пиридил)-2-пиридил]ацетамид
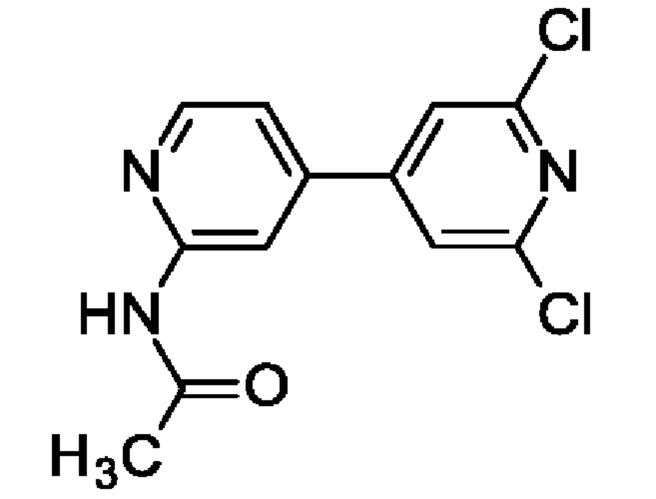
2,6-Дихлор-4-иод-пиридин (1 г; 3,65 ммоль), N-[4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-2-пиридил]ацетамид (1,2 г; 4,58 ммоль), PdCl2(PPh3)2 (128 мг; 0,18 ммоль) и K2CO3 (1,51 г; 10,95 ммоль) переносили в смесь 1,4-диоксан : H2O : EtOH (6:3:1; 15 мл) и через смесь барботировали азот в течение 5 мин, после чего нагревали до 80°С в течение 2 ч. После охлаждения до КТ добавляли воду (10 мл), рассол (10 мл) и EtOAc (25 мл), смесь энергично перемешивали в течение 5 мин и органический слой отделяли. Водный слой экстрагировали EtOAc (3 × 20 мл) и объединенные органические экстракты промывали рассолом, обрабатывали Na2SO4, фильтровали и концентрировали. После перекристаллизации из MeCN получали продукт в виде твердого вещества (760 мг; 74%). MS ES+ m/z 282 [М+Н]+.
Пример 3
N-[4-(2-Бензилокси-6-хлор-4-пиридил)-2-пиридил]ацетамид
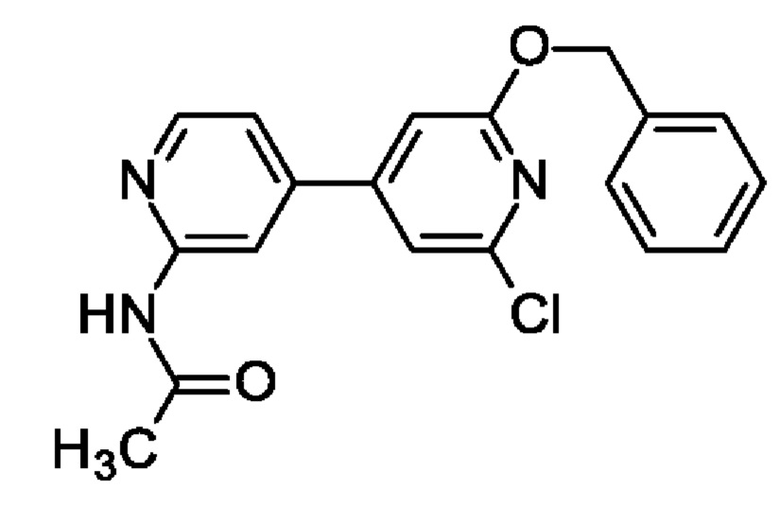
Фенилметанол (0,7 мл; 6,73 ммоль) добавляли к 60%-ной суспензии NaH (300 мг; 7,83 ммоль) в 2-MeTHF (5 мл) и DMF (5 мл) при 0°С в атмосфере азота. Смесь перемешивали при КТ в течение 20 мин, после чего добавляли раствор N-[4-(2,6-дихлор-4-пиридил)-2-пиридил]ацетамида (760 мг; 2,69 ммоль) в 2-MeTHF (10 мл) и DMF (10 мл) и полученную смесь перемешивали при 60°С в течение 1,5 ч. После охлаждения до КТ добавляли воду (40 мл) и EtOAc (20 мл) и органический слой отделяли. Водный слой экстрагировали EtOAc (2 × 15 мл) и объединенные органические экстракты промывали водой (2 × 15 мл), рассолом, обрабатывали Na2SO4, фильтровали, концентрировали и очищали на колонке с силикагелем с элюированием 20-75% EtOAc в гептане, получая продукт в виде масла, которое отвердевало при стоянии (430 мг; 45%). MS ES+ m/z 354 [М+Н]+.
Пример 4
N-[4-[2-Бензилокси-6-(3-пиридил)-4-пиридил]-2-пиридил]ацетамид
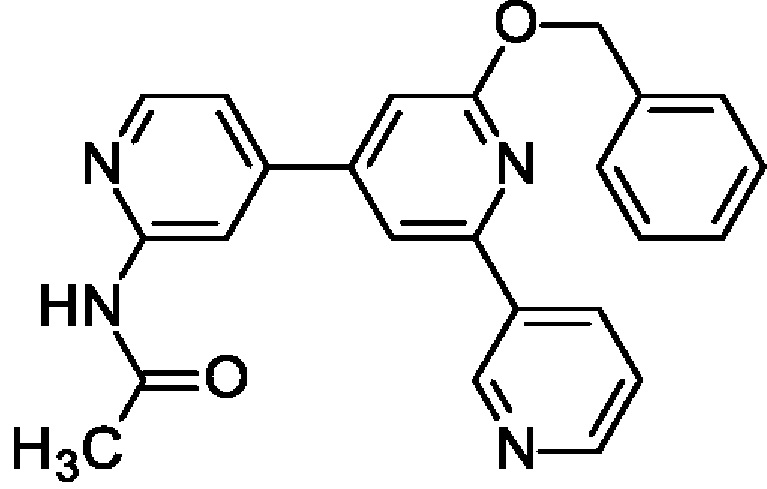
N-[4-(2-Бензилокси-6-хлор-4-пиридил)-2-пиридил]ацетамид (500 мг; 1,41 ммоль), 3-пиридилбороновую кислоту (208 мг; 1,7 ммоль), PdCl2(PPh3)2 (50 мг; 0,07 ммоль) и K2CO3 (585 мг; 4,24 ммоль) переносили в MeCN (15 мл) и воду (5 мл). Полученную смесь перемешивали при 80°С в течение ночи. После охлаждения до КТ смесь фильтровали и органический слой отделяли. Водный слой экстрагировали EtOAc (2 × 10 мл) и объединенные органические экстракты обрабатывали Na2SO4, фильтровали и концентрировали, получая продукт в виде твердого вещества (440 мг; 79%). MS ES+ m/z 397 [М+Н]+.
Пример 5
4-(2-Амино-4-пиридил)-6-(3-пиридил)-1Н-пиридин-2-он
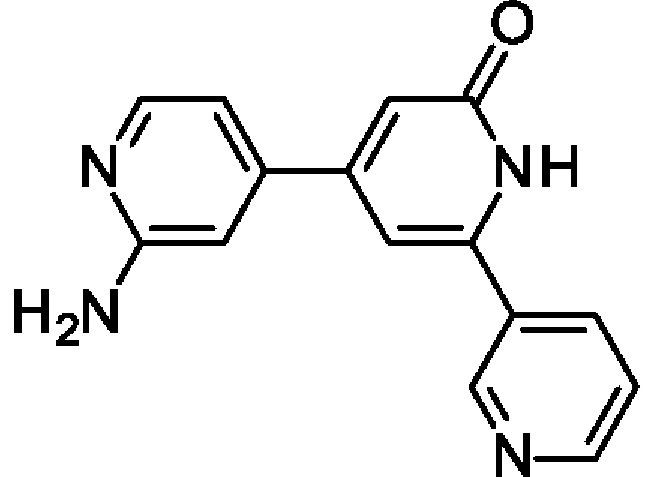
N-[4-[2-Бензилокси-6-(3-пиридил)-4-пиридил]-2-пиридил]ацетамид (440 мг; 1,11 ммоль) переносили в 1,4-диоксан (5 мл) и 2 М водн. раствор HCl (4 мл) и полученную смесь перемешивали при 90°С в течение ночи. После охлаждения до КТ добавляли 2 М водн. раствор NaOH до тех пор, пока значение рН не становилось немного выше 7. Смесь перемешивали в течение 1 ч и полученный осадок отфильтровывали, промывали водой, затем 1,4-диоксаном и сушили. Неочищенный продукт суспендировали в MeCN (10 мл), перемешивали при КТ в течение 1 ч, отфильтровывали, промывали MeCN и сушили, получая продукт в виде твердого вещества (160 мг; 55%). 1Н ЯМР (500 МГц, DMSO-d6) δ млн-1 11.84 (br.s., 1Н), 9.11 (br.s., 1Н), 8.66 (d, 1Н), 8.30 (d, 1Н), 8.07-8.02 (m, 1Н), 7.54 (dd, 1Н), 7.20 (s, 1Н), 7.03 (d, 1Н), 6.96 (s, 1Н), 6.79-6.63 (m, 3Н). MS ES+ m/z 265 [М+Н]+.
Пример 6
6-(2-Хлорфенил)-4-гидрокси-1Н-пиридин-2-он
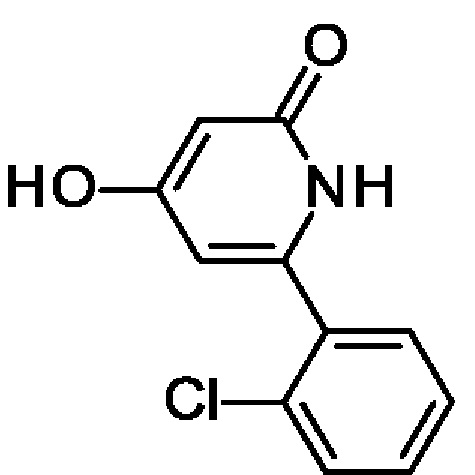
Этил-3-оксобутаноат (6,33 мл; 50 ммоль) по каплям добавляли к 60%-ной суспензии NaH (1,92 г; 50 ммоль) в 2-MeTHF (60 мл) при-78°С в атмосфере азота. Через 5 мин охлаждающую баню удаляли и смесь перемешивали при КТ в течение 20 мин. Смесь еще раз охлаждали до-78°С и медленно в течение 20 мин добавляли 1,6 М раствор н-BuLi (31,25 мл; 50 ммоль). Полученный раствор перемешивали при-78°С в течение 30 мин. Добавляли одной порцией 2-хлорбензонитрил (6,88 г; 50 ммоль) в виде твердого вещества и реакционную смесь перемешивали в охлаждающей бане стающим льдом в течение ночи. Смесь охлаждали до 0°С и медленно добавляли МеОН (15 мл). Охлаждающую баню удаляли, смесь перемешивали при КТ в течение 30 мин и затем еще раз охлаждали до 0°С. Смесь нейтрализовали, медленно добавляя конц. HCl, и полученный осадок отфильтровывали, промывали EtOH, пентаном и сушили, получая продукт в виде твердого вещества (11,08 г; 87%). MS ES+ m/z 222 [М+Н]+.
Пример 7
2,4-Дихлор-6-(2-хлорфенил)пиридин
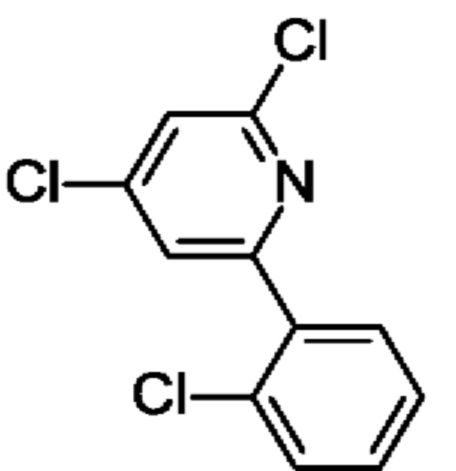
6-(2-Хлорфенил)-4-гидрокси-1Н-пиридин-2-он (5 г; 22,56 ммоль) переносили в POCl3 (40 мл) и медленно добавляли N,N-диметиланилин (5,5 мл; 43,4 ммоль). Полученную смесь кипятили с обратным холодильником в течение ночи. После охлаждения до КТ смесь выливали на лед (600 мл) и перемешивали при КТ в течение 30 мин. Осадок отфильтровывали и промывали водой. Твердое вещество растворяли в EtOAc (100 мл), обрабатывали Na2SO4, фильтровали и концентрировали, получая продукт в виде твердого вещества (7 г; 83%). MS ES+ m/z 258 [М+Н]+.
Пример 8
4-Хлор-6-(2-хлорфенил)-1Н-пиридин-2-он
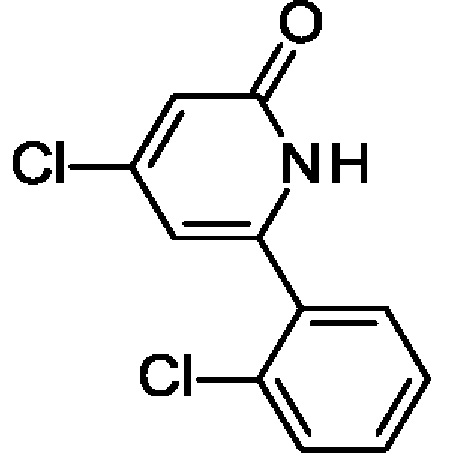
2,4-Дихлор-6-(2-хлорфенил)пиридин (5,7 г; 22,05 ммоль) и KOtBu (6,19 г; 55,12 ммоль) переносили в толуол (75 мл) и полученную смесь перемешивали при 100°С в течение 2 ч. После охлаждения до КТ добавляли воду (40 мл) и органический слой отделяли. Водный слой слегка подкисляли, используя конц. HCl, и экстрагировали EtOAc (2 × 40 мл). Объединенные органические экстракты промывали рассолом (50 мл), обрабатывали Na2SO4, фильтровали и концентрировали. Полученный остаток переносили в DCM (30 мл) и добавляли TFA (5 мл; 67,3 ммоль). Реакционную смесь перемешивали при КТ в течение 1 ч, концентрировали и полученный остаток переносили в МеОН (25 мл). Добавляли 30%-ный раствор NH4OH (20 мл) и воду (20 мл) и смесь перемешивали при КТ в течение ночи. Образовавшийся осадок отфильтровывали, промывали водой, EtOH, пентаном и сушили, получая продукт в виде твердого вещества (4,13 г; 78%). MS ES+ m/z 240 [М+Н]+.
Пример 9
N-(4-Хлор-2-пиридил)-2-метокси-ацетамид
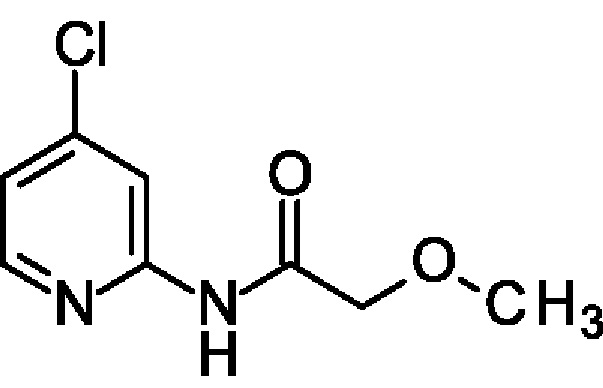
4-Хлорпиридин-2-амин (500 мг; 3,89 ммоль) и Et3N (1,1 мл; 7,78 ммоль) переносили в DCM (10 мл) при КТ и медленно добавляли 2-метоксиацетилхлорид (0,53 мл; 5,83 ммоль). Полученную смесь перемешивали при КТ в течение 15 мин. Добавляли 0,5 М водн. раствор HCl (10 мл) и органический слой отделяли. Водный слой экстрагировали DCM (5 мл) и объединенные органические экстракты обрабатывали Na2SO4, фильтровали и концентрировали, получая продукт в виде масла (750 мг; 96%). MS ES+ m/z 201 [М+Н]+.
Пример 10
2-Метокси-N-[4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-2-пиридил]ацетамид
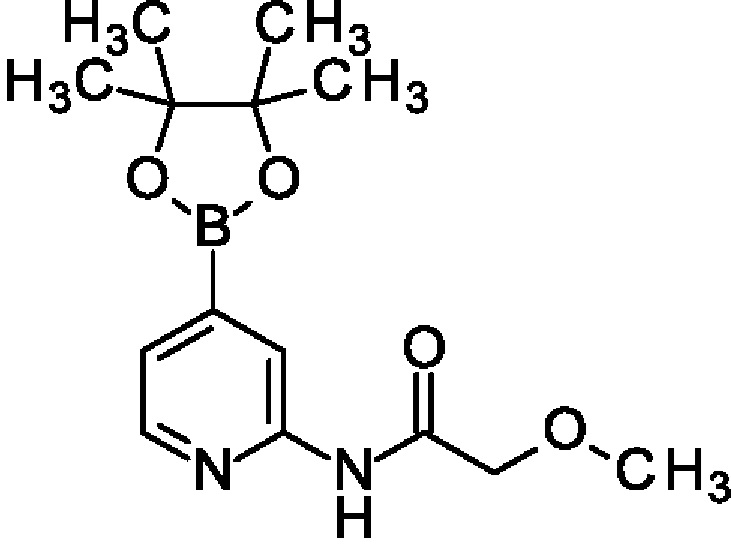
N-(4-Хлор-2-пиридил)-2-метокси-ацетамид (750 мг; 3,74 ммоль), 4,4,5,5-тетраметил-2-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-1,3,2-диоксаборолан (1,23 г; 4,86 ммоль) и KOAc (1,1 г; 11,22 ммоль) переносили в 1,4-диоксан (10 мл) и через смесь барботировали азот в течение 5 мин. Добавляли S-Phos (92 мг; 0,22 ммоль) и Pd(OAc)2 (25 мг; 0,11 ммоль) и полученную смесь перемешивали при 90°С в течение 3 ч. S-Phos (92 мг; 0,22 ммоль) и Pd(OAc)2 (25 мг; 0,11 ммоль) добавляли еще раз и перемешивание продолжали в течение 2 ч. После охлаждения до КТ смесь фильтровали через целит и осадок на фильтре промывали EtOAc. Фильтрат разбавляли водой и органический слой отделяли. Водный слой экстрагировали EtOAc (2 × 10 мл) и объединенные органические экстракты промывали рассолом, обрабатывали Na2SO4, фильтровали и концентрировали, получая продукт в виде масла, которое использовали без дополнительной очистки (1,5 г; колич.). MS ES+ m/z 293 [М+Н]+.
Примеры 11 и 12
4-(2-Амино-4-пиридил)-6-(2-хлорфенил)-1Н-пиридин-2-он и N-[4-[2-(2-хлорфенил)-6-оксо-1Н-пиридин-4-ил]-2-пиридил]-2-метокси-ацетамид
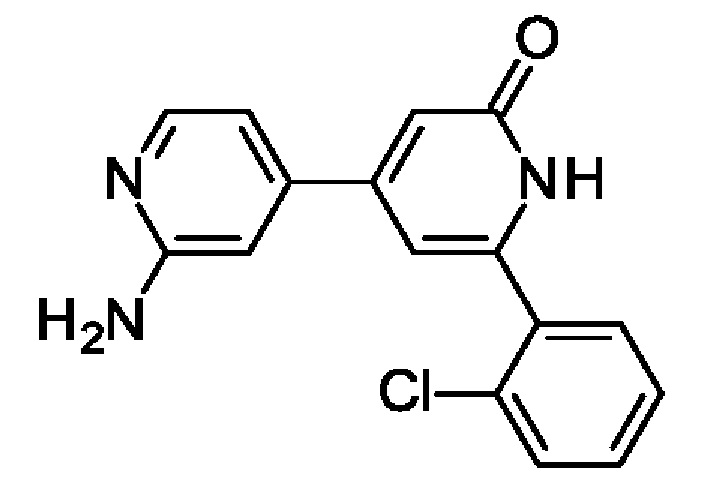
2-Метокси-1N-[4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-2-пиридил]ацетамид (183 мг; 0,62 ммоль), 4-хлор-6-(2-хлорфенил)-1Н-пиридин-2-он (100 мг; 0,42 ммоль), Pd(PPh3)4 (0,02 г; 0,02 ммоль) и K2CO3 (0,17 г; 1,25 ммоль) переносили в смесь 1,4-диоксан : H2O : EtOH (6:3:1; 1,5 мл) и смесь нагревали в микроволновом реакторе при 150°С в течение 15 мин. Органический слой отделяли и очищали препаративной HPLC, получая продукты. Твердое вещество (12 мг; 10%). 1Н ЯМР (500 МГц, DMSO-d6) δ млн-1 7.98 (d, 1Н), 7.63-7.53 (m, 2Н), 7.53-7.40 (m, 3Н), 6.80 (dd, 1Н), 6.73 (s, 1Н), 6.62 (d, 1Н), 6.54 (s, 1Н), 6.06 (s, 2Н). MS ES+ m/z 298 [М+Н]+.
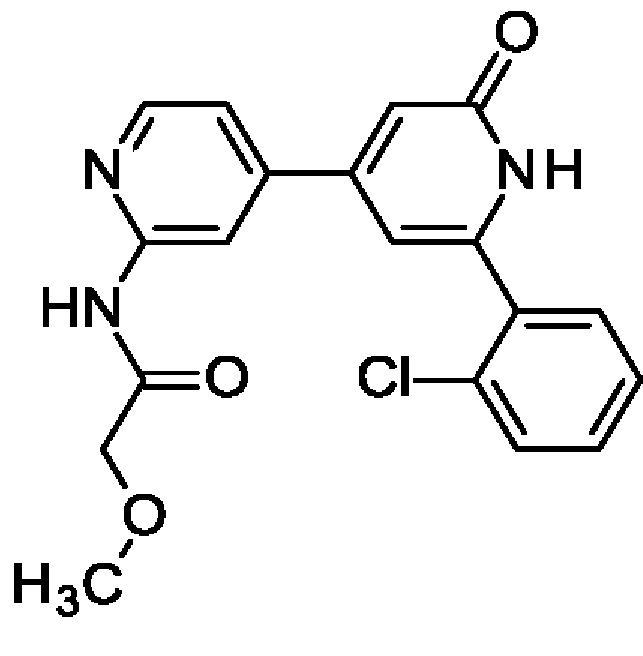
Твердое вещество (25 мг; 16%). 1Н ЯМР (500 МГц, DMSO-d6) δ млн-1 10.23 (br s, 1Н), 8.43 (d, 1Н), 8.36 (s, 1Н), 7.60 (m, 2Н), 7.50-7.54 (m, 3Н), 6.73 (br s, 1Н), 6.54 (s, 1Н), 4.07-4.12 (m, 2Н), 3.35-3.39 (m, 3Н). MS ES+ m/z 370 [М+Н]+.
Пример 13
4-Бензилокси-2,6-дихлор-пиридин
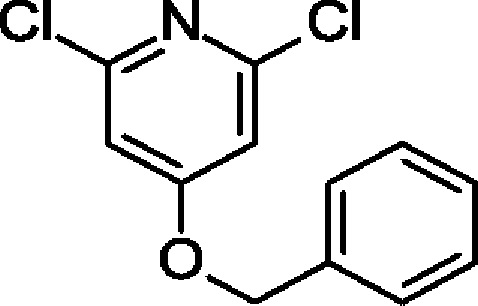
60%-ную суспензию NaH (945 мг; 24,7 ммоль) добавляли порциями к раствору 2,4,6-трихлорпиридина (4,5 г; 24,7 ммоль) в DMF (25 мл) при 0°С. Через 20 мин по каплям добавляли фенилметанол (2,7 г; 24,7 ммоль) и смесь перемешивали в течение 3 ч. Добавляли воду (30 мл) и осадок отфильтровывали. Твердое вещество растворяли в EtOAc, обрабатывали MgSO4, фильтровали и концентрировали, получая продукт в виде твердого вещества (5 г; 80%). MS ES+ m/z 254 [М+Н]+.
Пример 14
4-Бензилокси-2-трет-бутокси-6-хлор-пиридин
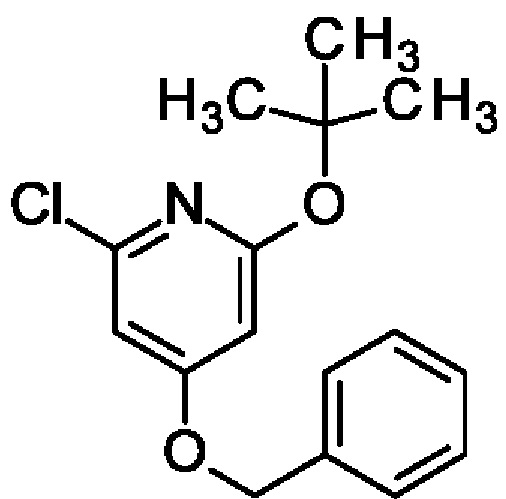
4-Бензилокси-2,6-дихлор-пиридин (5 г; 19,7 ммоль) и KOtBu (2,2 г; 19,7 ммоль) растворяли в 2-MeTHF (25 мл) и смесь перемешивали при 70°С в течение 2 ч. После охлаждения до КТ смесь фильтровали, концентрировали и очищали на колонке с силикагелем с элюированием 30%-ным EtOAc в гептане, получая продукт (4 г; 70%). MS ES+ m/z 292 [М+Н]+.
Пример 15
4-Бензилокси-2-трет-бутокси-6-[2-(трифторметил)-1-пиперидил]пиридин
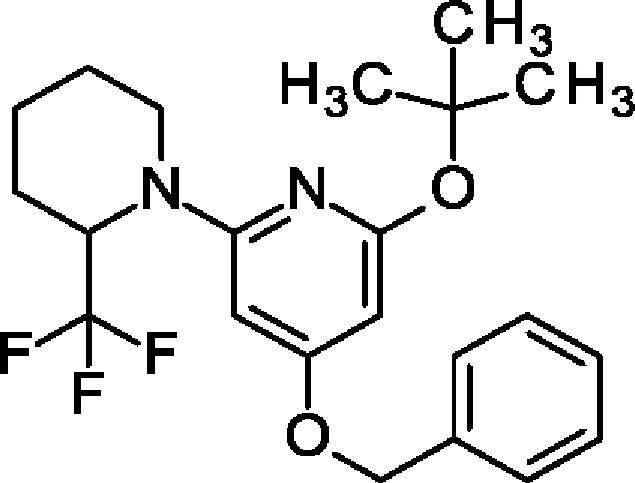
4-Бензилокси-2-трет-бутокси-6-хлор-пиридин (4 г; 13,7 ммоль), 2-(трифторметил)пиперидин (2,3 г; 15,1 ммоль), PEPPSI-iPr (146 мг; 1,37 ммоль) и KOtBu (3,85 г; 34,3 ммоль) переносили в 1,4-диоксан (30 мл) и смесь перемешивали при 90°С в течение 2 ч. После охлаждения до КТ добавляли воду и EtOAc и органический слой отделяли, фильтровали, концентрировали и очищали на колонке с силикагелем с элюированием 30%-ным EtOAc в гептане, получая продукт (4,1 г; 73%). MS ES+ m/z 409 [М+Н]+.
Пример 16
2-трет-Бутокси-6-[2-(трифторметил)-1-пиперидил]пиридин-4-ол
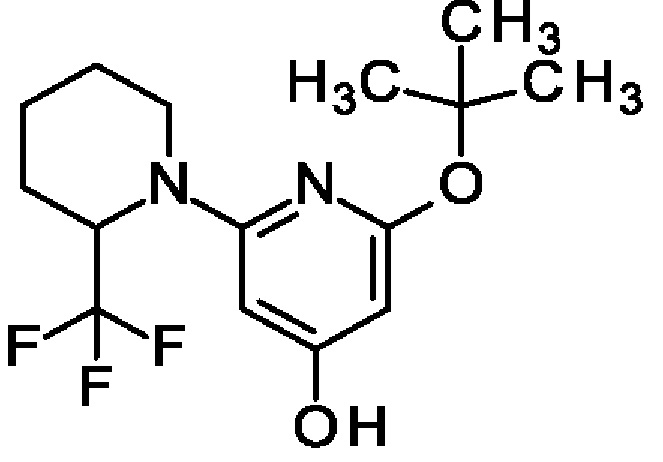
Смесь 4-бензилокси-2-трет-бутокси-6-[2-(трифторметил)-1-пиперидил]пиридина (3,5 г; 8,57 ммоль) и 10%-ного Pd/C (600 мг; 0,56 ммоль) в МеОН и EtOAc гидрировали (1,5 бар (150 кПа)) при КТ в течение 2 ч. Смесь фильтровали через целит и концентрировали, получая продукт (2,7 г; колич.). MS ES+ m/z 319 [M+H]+.
Пример 17
[2-трет-Бутокси-6-[2-(трифторметил)-1-пиперидил]-4-пиридил]-трифторметансульфонат
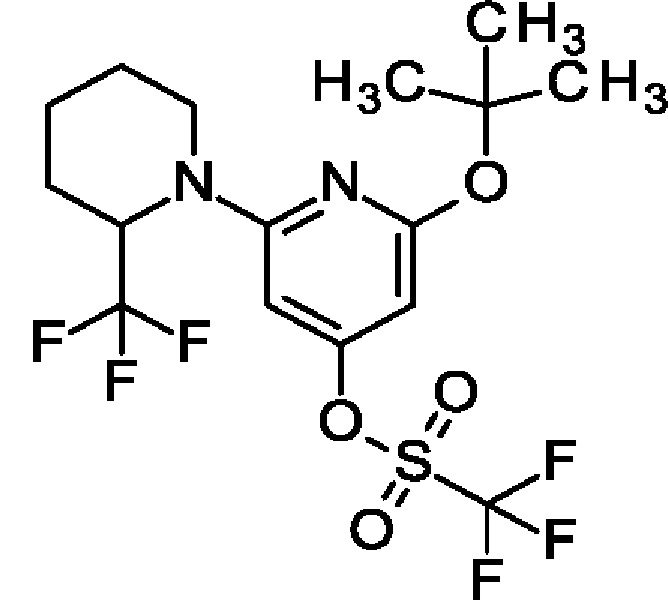
2-трет-Бутокси-6-[2-(трифторметил)-1-пиперидил]пиридин-4-ол (2,7 г; 8,48 ммоль) и Et3N (1,66 мл; 11,9 ммоль) переносили в DCM (20 мл) при 0°С. По каплям в течение 5 минут добавляли трифторметилсульфонил-трифторметансульфонат (2,54 мл; 11,9 ммоль) и перемешивали в течение 1 ч. Смесь промывали насыщ. водн. раствором NaHCO3 (2 × 20 мл), концентрировали и очищали на колонке с силикагелем с элюированием 20%-ным EtOAc в гептане, получая продукт (3,5 г; 92%). MS ES+ m/z 451 [М+Н]+.
Пример 18
2-трет-Бутокси-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-6-[2-(трифторметил)-1-пиперидил]пиридин
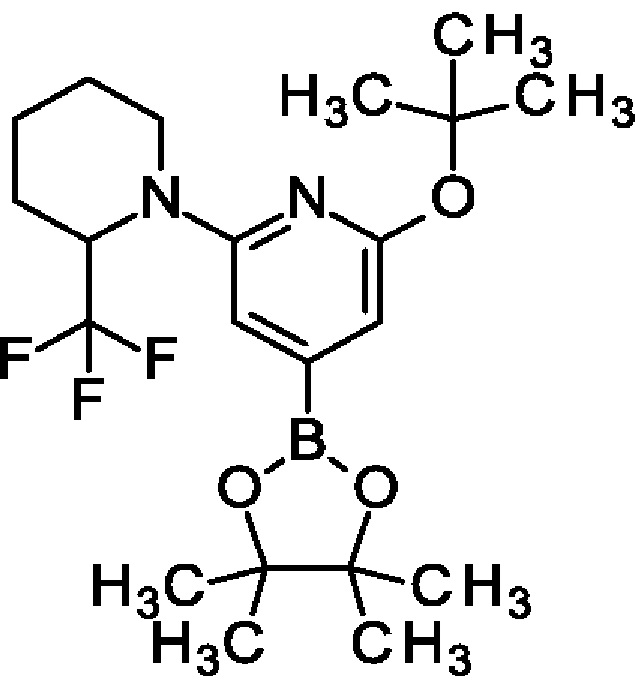
4,4,5,5-Тетраметил-2-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-1,3,2-диоксаборолан (2,96 г; 11,7 ммоль), [2-трет-бутокси-6-[2-(трифторметил)-1-пиперидил]-4-пиридил]трифторметансульфонат (3,5 г; 7,77 ммоль), KOAc (1,14 г; 11,7 ммоль) и PdCl2(dppf) (215 мг; 0,29 ммоль) переносили в толуол (10 мл) и перемешивали при 90°С в течение 5 ч. После охлаждения до КТ смесь концентрировали и остаток растворяли в EtOAc, промывали водой, концентрировали и очищали на колонке с силикагелем с элюированием 0-60% EtOAc в гептане, получая продукт (2,15 г; 65%). MS ES+ m/z 347 [М+Н]+.
Пример 19
N[4-[2-Оксо-6-[2-(трифторметил)-1-пиперидил]-1Н-пиридин-4-ил]-2-пиридил]ацетам ид
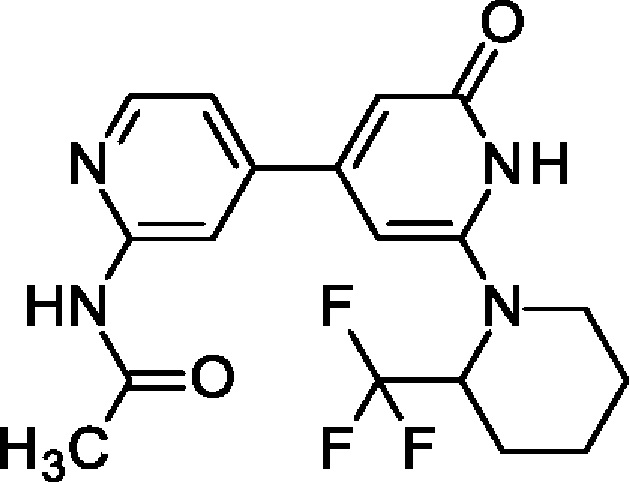
2-трет-Бутокси-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-6-[2-(трифторметил)-1-пиперидил]пиридин (100 мг; 0,23 ммоль), N-(4-хлор-2-пиридил)ацетамид (52 мг; 0,3 ммоль), K2CO3 (81 мг; 0,58 ммоль) и PdCl2(dppf) (26 мг; 0,035 ммоль) растворяли в 1,4-диоксане (3 мл) и воде (1 мл) и смесь перемешивали при 90°С в течение 3 ч. Добавляли воду и EtOAc, органический слой отделяли и концентрировали. Остаток растворяли в DCM и добавляли TFA (0,35 мл; 4,67 ммоль). Смесь перемешивали при КТ в течение 30 мин, концентрировали и очищали препаративной HPLC, получая продукт в виде твердого вещества (10 мг; 11%). 1Н ЯМР (500 МГц, метанол-d4) δ млн-1 1.52-1.66 (m, 1Н), 1.69-1.77 (m, 2Н), 1.76-1.78 (m, 1Н), 1.77-1.88 (m, 2Н), 2.08 (br d, 1Н), 2.20 (s, 3Н), 3.22 (br t, 1Н), 3.31 (dt, 2Н), 3.85 (br d, 1Н), 5.12-5.26 (m, 1Н), 6.22 (s, 2Н), 7.25 (dd, 1Н), 8.26-8.36 (m, 2Н). MS ES+ m/z 381 [М+Н]+.
Пример 20
4-[2-трет-Бутокси-6-[2-(трифторметил)-1-пиперидил]-4-пиридил]пиридин-2-амин
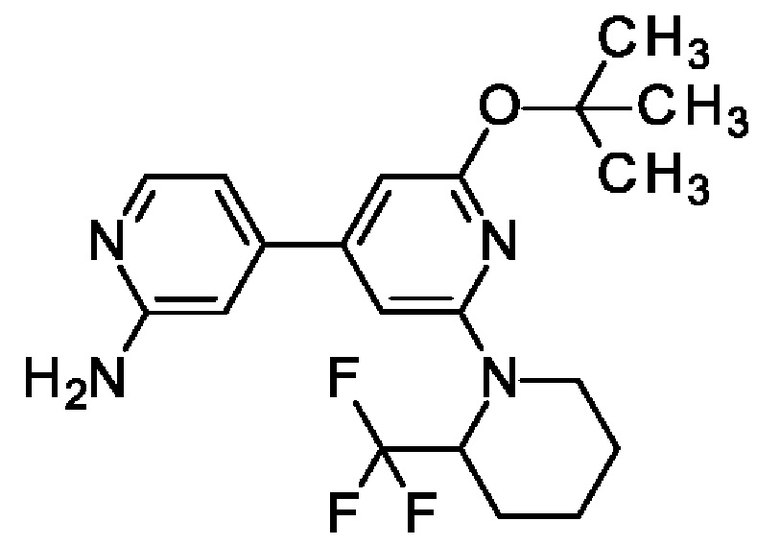
2-трет-Бутокси-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-6-[2-(трифторметил)-1-пиперидил]пиридин (700 мг; 1,63 ммоль), 4-бромпиридин-2-амин (339 мг; 1,96 ммоль), K2CO3 (565 мг; 4,09 ммоль) и PdCl2(dppf) (120 мг; 0,16 ммоль) переносили в диоксан (10 мл) и воду (3 мл) и смесь перемешивали при 90°С в течение 4 ч. После охлаждения до КТ добавляли воду и EtOAc, органический слой отделяли, концентрировали и очищали на колонке с силикагелем с элюированием 0-20% МеОН в DCM, получая продукт в виде твердого вещества (280 мг; 37%). MS ES+ m/z 395 [М+Н]+.
Пример 21
N-[4-[2-Оксо-6-[2-(трифторметил)-1-пиперидил]-1Н-пиридин-4-ил]-2-пиридил] циклопропан карбоксамид
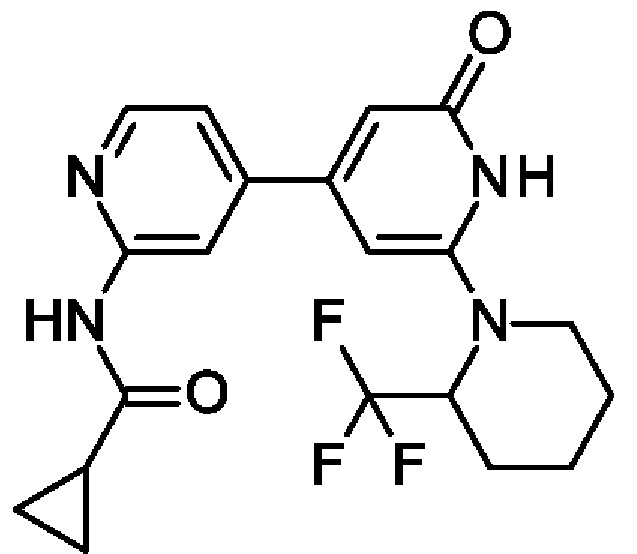
К раствору 4-[2-трет-бутокси-6-[2-(трифторметил)-1-пиперидил]-4-пиридил]пиридин-2-амина (85 мг; 0,22 ммоль) в DCM (3 мл) добавляли циклопропанкарбонилхлорид (0,022 мл; 0,24 ммоль), затем Et3N (0,03 мл; 0,22 ммоль) и смесь перемешивали при КТ в течение 1 ч. Смесь фильтровали и медленно добавляли TFA (0,08 мл; 1,08 ммоль). Через 1 ч выдерживания при КТ смесь концентрировали и очищали препаративной HPLC, получая продукт в виде твердого вещества (8 мг; 9%). 1Н ЯМР (500 МГц, DMSO-d6) δ млн-1 0.81-0.87 (m, 4Н), 1.49 (br d, 1Н), 1.68 (br s, 2Н), 1.71-1.82 (m, 2Н), 1.98-2.07 (m, 2Н), 3.04 (br t, 1Н), 4.17 (brs, 1Н), 5.56 (brs, 1Н), 6.19 (s, 1Н), 6.50-6.79 (m, 1Н), 6.55 (br s, 1Н), 7.41 (dd, 1Н), 8.34 (s, 1Н), 8.39 (dd, 1Н), 10.42 (br s, 1Н), 10.90 (s, 1Н). MS ES+ m/z 407 [М+Н]+.
Пример 22
4-(4-Бензилокси-6-трет-бутокси-2-пиридил)-3-(трифторметил)морфолин
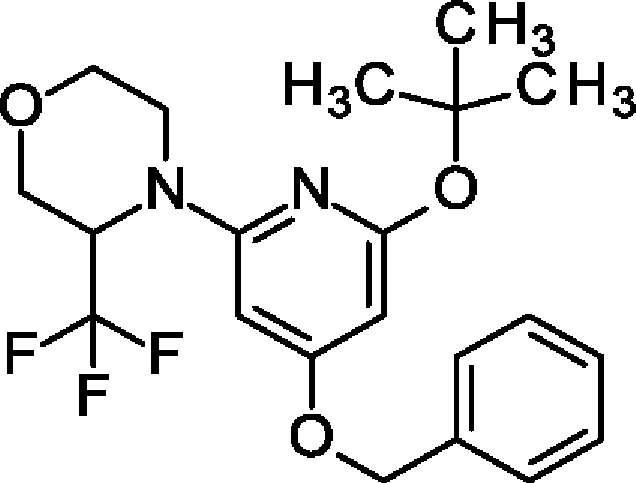
Указанное в заголовке соединение получали, как описано в примере 15, с использованием 3-(трифторметил)морфолина вместо 2-(трифторметил)пиперидина, получая продукт в виде масла (1 г; 50%). MS ES+ m/z 411 [М+Н]+.
Пример 23
2-трет-Бутокси-6-[3-(трифторметил)морфолин-4-ил]пиридин-4-ол
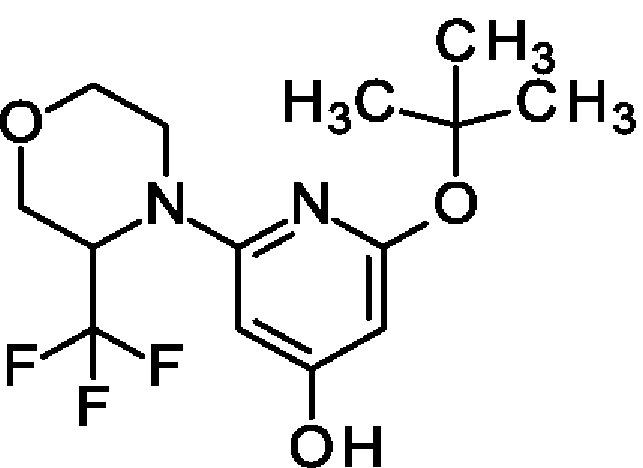
Указанное в заголовке соединение получали, как описано в примере 16, с использованием 4-(4-бензилокси-6-трет-бутокси-2-пиридил)-3-(трифторметил)морфолина вместо 4-бензилокси-2-трет-бутокси-6-[2-(трифторметил)-1-пиперидил]пиридина, получая продукт (780 мг; 99%). MS ES+ m/z 321 [М+Н]+.
Пример 24
[2-трет-Бутокси-6-[3-(трифторметил)морфолин-4-ил]-4-пиридил]-трифторметансульфонат
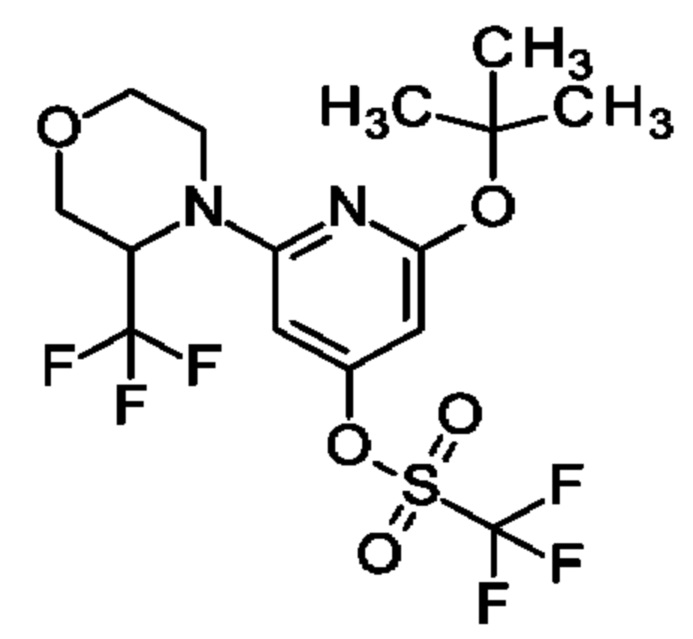
Указанное в заголовке соединение получали, как описано в примере 17, с использованием 2-трет-бутокси-6-[3-(трифторметил)морфолин-4-ил]пиридин-4-ола вместо 2-трет-бутокси-6-[2-(трифторметил)-1-пиперидил]пиридин-4-ола, получая продукт в виде масла (800 мг; 81%). MS ES+ m/z 453 [М+Н]+.
Пример 25
4-[6-трет-Бутокси-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-2-пиридил]-3-(трифторметил)морфолин
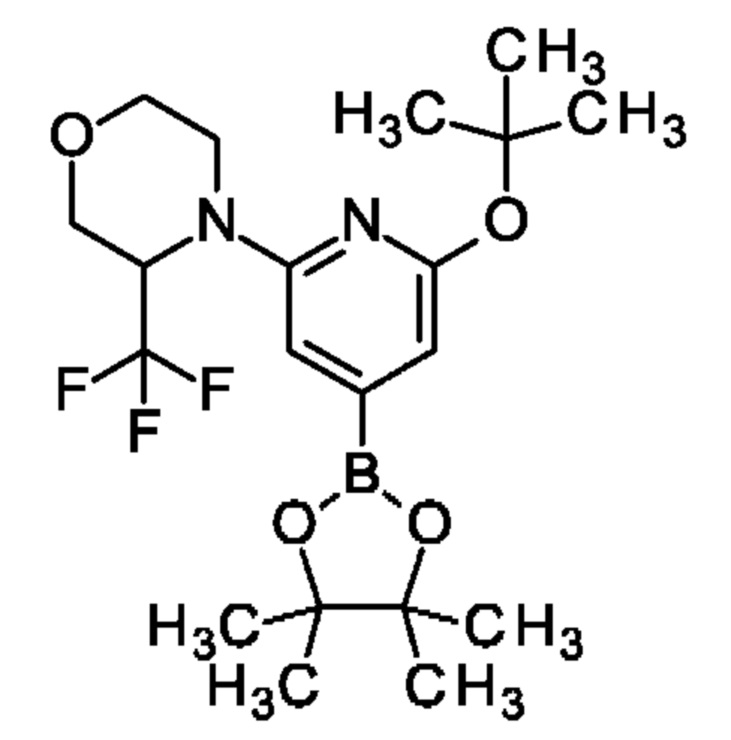
Указанное в заголовке соединение получали, как описано в примере 18, с использованием [2-трет-бутокси-6-[3-(трифторметил)морфолин-4-ил]-4-пиридил]-трифторметансульфоната вместо [2-трет-бутокси-6-[2-(трифторметил)-1-пиперидил]-4-пиридил]трифторметансульфоната, получая продукт (270 мг; 33%). MS ES+ m/z 431 [М+Н]+.
Пример 26
N-[4-[2-Оксо-6-[3-(трифторметил)морфолин-4-ил]-1Н-пиридин-4-ил]-2-пиридил]ацетамид
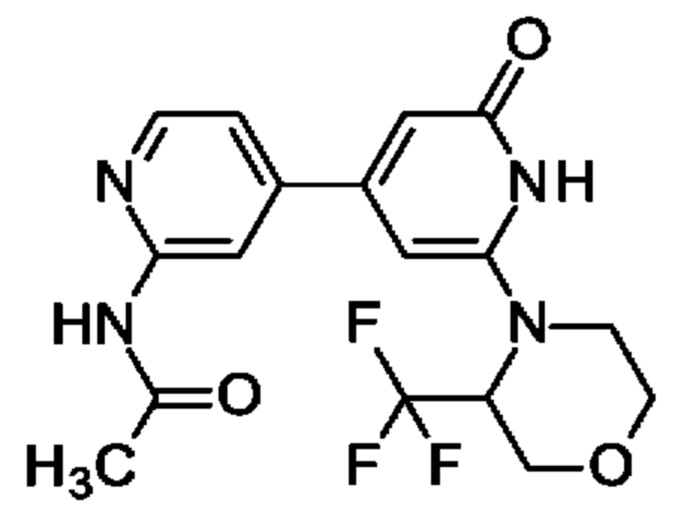
Указанное в заголовке соединение получали, как описано в примере 19, с использованием 4-[6-трет-бутокси-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-2-пиридил]-3-(трифторметил)морфолина вместо 2-трет-бутокси-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-6-[2-(трифторметил)-1-пиперидил]пиридина, получая продукт в виде твердого вещества (20 мг; 25%). 1Н ЯМР (500 МГц, DMSO-d6) δ млн-1 2.13 (s, 3Н), 3.25-3.31 (m, 1Н), 3.51-3.57 (m, 1Н), 3.72-3.78 (m, 1Н), 3.94-4.04 (m, 2Н), 4.19 (d, 1Н), 5.31 (br d, 1Н), 6.25 (s, 1Н), 6.56 (br s, 1Н), 7.41 (d, 1Н), 8.34 (s, 1Н), 8.39 (dd, 1Н), 10.55 (br s, 1Н), 10.60 (s, 1Н). MS ES+ m/z 383 [М+Н]+.
Пример 27
N-[4-(2-трет-Бутокси-6-хлор-4-пиридил)-2-пиридил]ацетамид
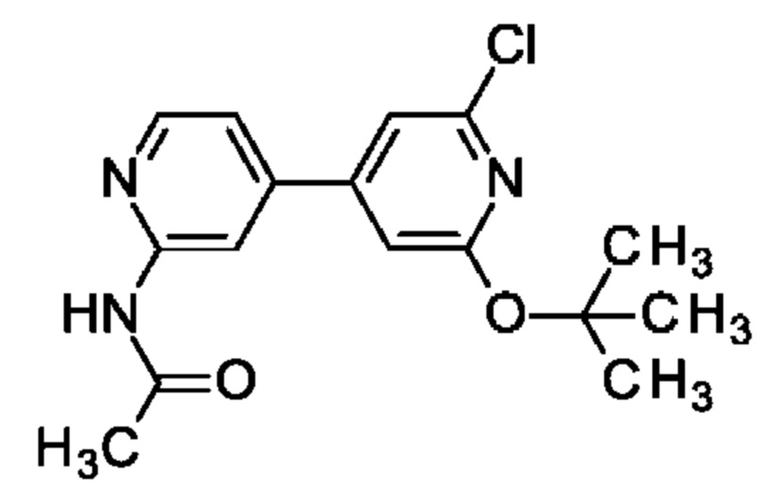
Смесь N-[4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-2-пиридил]ацетамида (3 г; 9,62 ммоль), 2-трет-бутокси-6-хлор-4-иод-пиридина (Bioorganic & Medicinal Chemistry Letters (2012), 22 (5), 1940-1943; 2,52 г; 9,62 ммоль) и Na2CO3 (3,05 г; 28,86 ммоль) в 1,4-диоксане (20 мл) и воде (4 мл) продували азотом в течение 20 мин. Добавляли PdCl2(dppf) (351 мг; 0,48 ммоль) и полученную смесь нагревали и перемешивали при 110°С в течение ночи. После охлаждения до КТ добавляли EtOAc, смесь фильтровали через целит, концентрировали и очищали препаративной HPLC, получая продукт в виде твердого вещества (1,58 г; 22%). MS ES+ m/z 320 [М+Н]+.
Пример 28
1-Этилсульфонил-3-(трифторметил)пиперазин
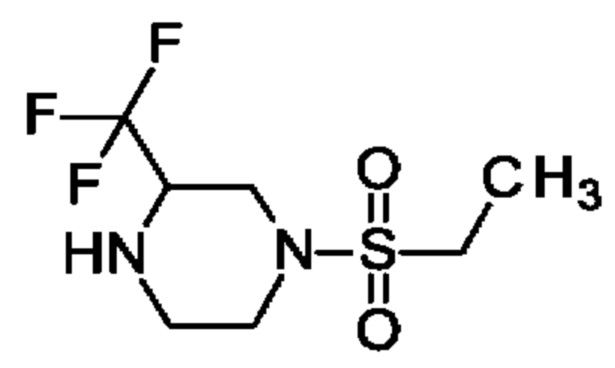
Этансульфонилхлорид (1,18 мл; 12,5 ммоль) медленно добавляли к раствору 2-(трифторметил)-пиперазина (1,93 г; 12,5 ммоль) и триэтиламин (TEA; 2,09 мл; 15 ммоль) в DCM (40 мл) при 0°С и полученную смесь перемешивали при КТ в течение ночи. Добавляли DCM (50 мл) и воду (60 мл), органический слой отделяли и водный слой экстрагировали DCM (2×50 мл). Объединенные органические экстракты дважды промывали рассолом, обрабатывали Na2SO4, фильтровали и концентрировали, получая продукт в виде твердого вещества (3 г; 98%). 1Н ЯМР (500 МГц, DMSO-d6) δ млн-1 1.21 (t, 3Н), 2.64-2.70 (m, 1Н), 2.86-3.03 (m, 4Н), 3.07-3.16 (m, 2Н), 3.36-3.38 (m, 1Н), 3.41-3.49 (m, 1Н), 3.53 (dd, 1Н).
Пример 29
N-[4-[2-трет-Бутокси-6-[4-этилсульфонил-2-(трифторметил)пиперазин-1-ил]-4-пиридил]-2-пиридил]ацетамид
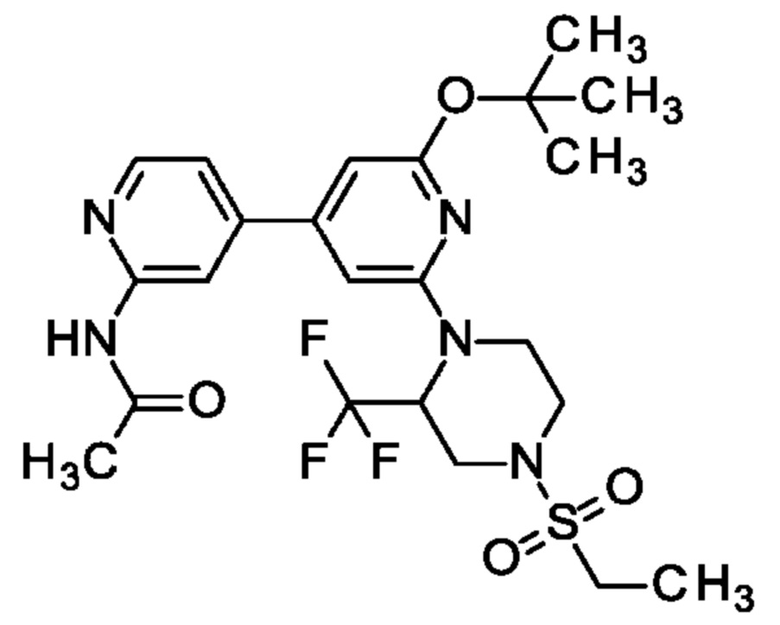
Смесь N-[4-(2-трет-бутокси-6-хлор-4-пиридил)-2-пиридил]ацетамида (96 мг; 0,3 ммоль), 1-этилсульфонил-3-(трифторметил)пиперазина (81 мг; 0,33 ммоль), Cs2CO3 (98 мг; 0,3 ммоль), Xantphos (174 мг; 0,3 ммоль) и Pd(OAc)2 (67 мг; 0,3 ммоль) в 1,4-диоксане (2 мл) перемешивали в атмосфере аргона в герметично закрытой пробирке при 100°С в течение 6 ч. После охлаждения до КТ добавляли воду и смесь экстрагировали EtOAc. Объединенные органические экстракты промывали рассолом, обрабатывали Na2SO4, фильтровали, концентрировали и очищали на колонке с силикагелем с элюированием 0-5% МеОН в DCM, получая продукт в виде твердого вещества (110 мг; 55%). MS ES+ m/z 530 [М+Н]+.
Пример 30
N-[4-[2-[4-Этилсульфонил-2-(трифторметил)пиперазин-1-ил]-6-оксо-1Н-пиридин-4-ил]-2-пиридил]ацетамид
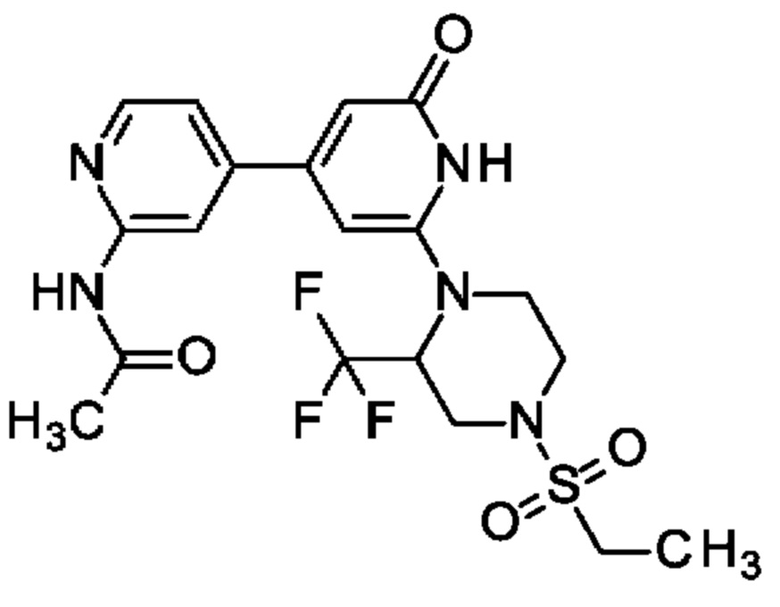
TFA (0,91 мл; 12,2 ммоль) добавляли к раствору N-[4-[2-трет-бутокси-6-[4-этилсульфонил-2-(трифторметил)пиперазин-1-ил]-4-пиридил]-2-пиридил]ацетамида (108 мг; 0,2 ммоль) в DCM (8 мл) при 0°С и полученную смесь перемешивали при 0°С в течение 1 ч. Смесь концентрировали и очищали препаративной HPLC, получая продукт в виде твердого вещества (27 мг; 28%). 1Н ЯМР (500 МГц, DMSO-d6) δ млн-1 1.23 (t, 3Н), 2.09-2.16 (m, 3Н), 2.94-3.05 (m, 1Н), 3.10-3.32 (m, 3Н), 3.66 (br d, 1Н), 3.94 (br d, 1Н), 4.11 (q, 1Н), 4.34 (br d, 1 Н), 5.65 (br s, 1H), 6.27 (s, 1H), 6.65 (s, 1H), 7.42 (dd, 1H), 8.34 (s, 1H), 8.40 (d, 1H), 10.63 (br s, 1H), 10.67 (s, 1H). MS ES+ m/z 474 [M+H]+.
Пример 31
N-[4-[2-трет-Бутокси-6-(3-циклопропилморфолин-4-ил)-4-пиридил]-2-пиридил]ацетамид
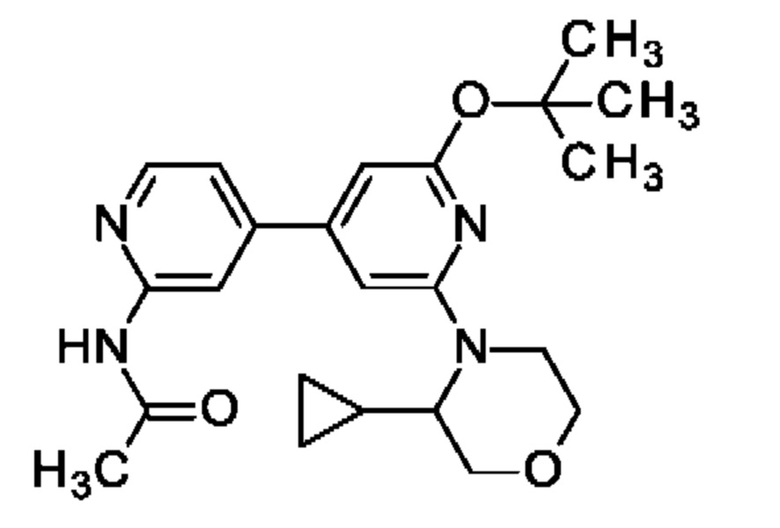
Указанное в заголовке соединение получали, как описано в примере 29, с использованием 3-циклопропилморфолина, получая продукт в виде твердого вещества (44 мг; 29%). MS ES+ m/z 411 [М+Н]+.
Пример 32
N-[4-[2-(3-Циклопропилморфолин-4-ил)-6-оксо-1Н-пиридин-4-ил]-2-пиридил]ацетамид
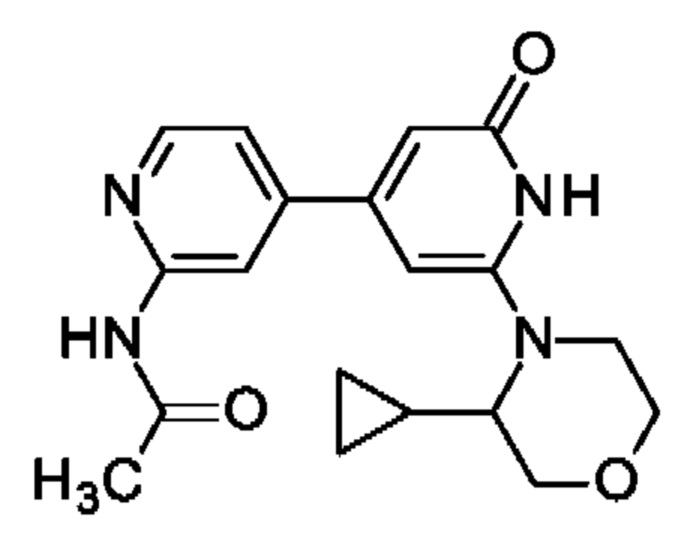
Указанное в заголовке соединение получали, как описано в примере 30, получая продукт в виде твердого вещества (8 мг; 21%). 1Н ЯМР (500 МГц, DMSO-d6) δ млн-1 0.29-0.48 (m, 4Н), 1.37-1.50 (m, 1Н), 2.11-2.17 (m, 3Н), 3.34-3.40 (m, 1Н), 3.48 (td, 1Н), 3.57 (dd, 1Н), 3.76-3.96 (m, 4Н), 6.07 (s, 1Н), 6.32 (br s, 1Н), 7.37 (d, 1Н), 8.32 (s, 1Н), 8.34-8.40 (m, 1Н), 10.02-10.44 (m, 1Н), 10.58 (br s, 1Н). MS ES+ m/z 355 [М+Н]+.
Пример 33
N-[4-[2-трет-Бутокси-6-[2-(трифторметил)фенил]-4-пиридил]-2-пиридил]ацетамид
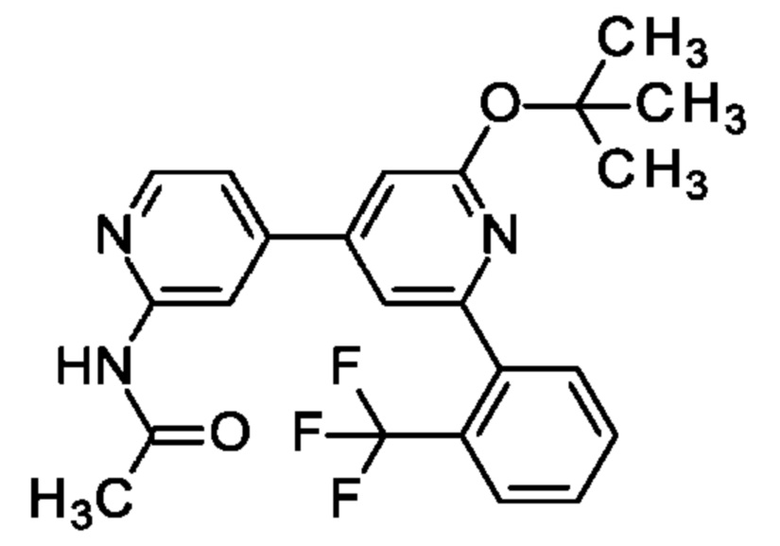
N-[4-(2-трет-Бутокси-6-хлор-4-пиридил)-2-пиридил]ацетамид (96 мг; 0,3 ммоль), [2-(трифторметил)фенил]бороновую кислоту (86 мг; 0,45 ммоль), K2CO3 (83 мг; 0,6 ммоль) и PdCl2(Amphos) (11 мг; 0,02 ммоль) переносили в 1,4-диоксан (1,5 мл) и воду (0,5 мл) и полученную смесь перемешивали при 100°С в течение 1 ч. После охлаждения до КТ добавляли рассол и смесь экстрагировали EtOAc. Объединенные органические экстракты промывали рассолом, обрабатывали Na2SO4, фильтровали, концентрировали и очищали на колонке с силикагелем с элюированием 0-100% EtOAc в изогексане, получая продукт в виде твердого вещества (105 мг; 82%). MS ES+ m/z 430 [М+Н]+.
Пример 34
N-[4-[2-Оксо-6-[2-(трифторметил)фенил]-1Н-пиридин-4-ил]-2-пиридил]ацетамид
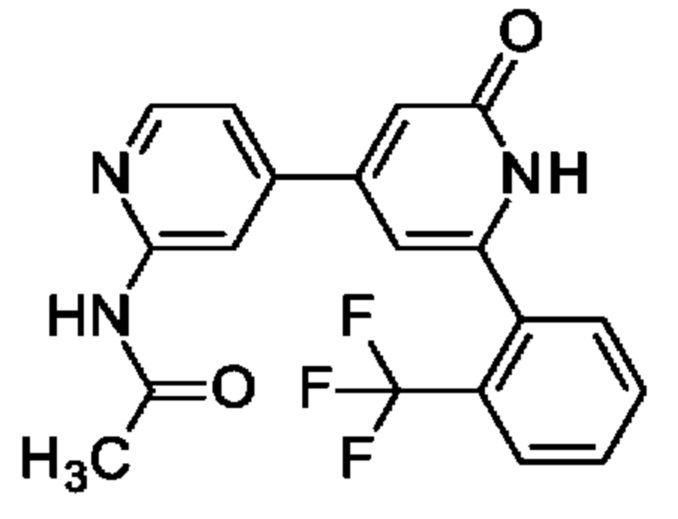
Указанное в заголовке соединение получали, как описано в примере 30, получая продукт в виде твердого вещества (42 мг; 47%). 1Н ЯМР (500 МГц, DMSO-d6) δ млн-1 2.12 (s, 3Н), 6.33-6.61 (m, 1Н), 6.74 (br s, 1Н), 7.43 (dd, 1Н), 7.66 (d, 1Н), 7.71-7.83 (m, 2Н), 7.89 (d, 1Н), 8.35-8.43 (m, 2Н), 10.64 (s, 1Н), 11.86-12.38 (m, 1Н). MS ES+ m/z 374 [М+Н]+.
Пример 35
N-[4-[2-трет-Бутокси-6-[2-(трифторметил)-3-пиридил]-4-пиридил]-2-пиридил]ацетамид
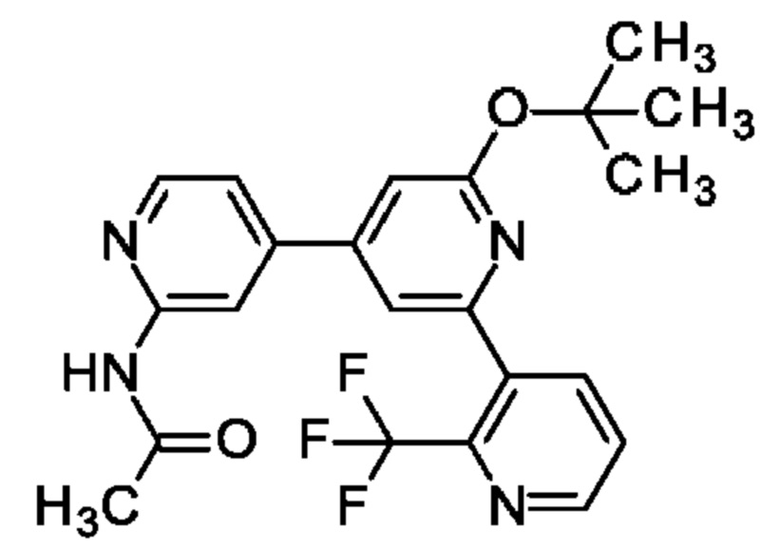
N-[4-(3-трет-Бутокси-5-хлор-фенил)-2-пиридил]ацетамид (128 мг; 0,4 ммоль), [2-(трифторметил)-3-пиридил]бороновую кислоту (84 мг; 0,44 ммоль), K2CO3 (111 мг; 0,8 ммоль) и PdCl2(Amphos) (14 мг; 0,02 ммоль) переносили в 1,4-диоксан (2,5 мл) и воду (0,5 мл) и полученную смесь перемешивали при 90°С в течение 4 ч. [2-(Трифторметил)-3-пиридил]бороновую кислоту (84 мг; 0,44 ммоль) добавляли еще раз, затем PdCl2(dppf) (29 мг; 0,04 ммоль) и смесь перемешивали при 90°С в течение ночи. После охлаждения до КТ добавляли воду и смесь экстрагировали EtOAc. Объединенные органические экстракты промывали рассолом, обрабатывали Na2SO4, фильтровали, концентрировали и очищали на колонке с силикагелем с элюированием 0-5% МеОН в DCM, получая продукт в виде твердого вещества (70 мг; 41%). MS ES+ m/z 431 [М+Н]+.
Пример 36
N-[4-[2-Оксо-6-[2-(трифторметил)-3-пиридил]-1Н-пиридин-4-ил]-2-пиридил]ацетамид
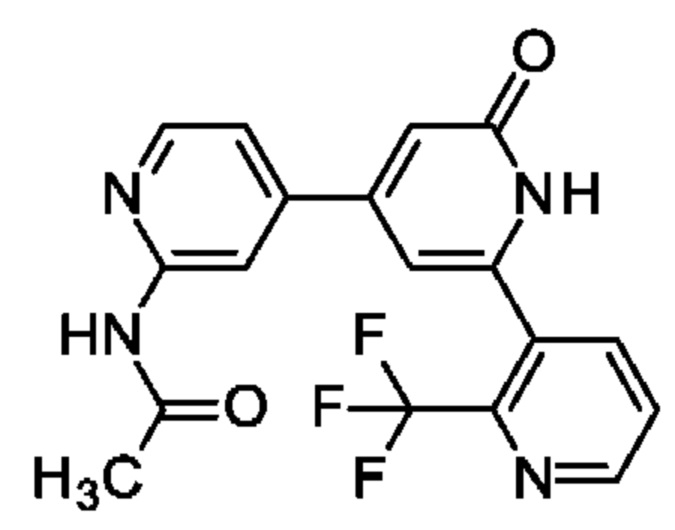
Указанное в заголовке соединение получали, как описано в примере 30, получая продукт в виде твердого вещества (30 мг; 54%). 1Н ЯМР (500 МГц, DMSO-d6) δ млн-1 2.08-2.16 (m, 3Н), 6.43-6.93 (m, 1Н), 6.81 (br s, 1Н), 7.44 (dd, 1Н), 7.86 (dd, 1Н), 8.18 (d, 1Н), 8.36-8.44 (m, 2Н), 8.87 (s, 1Н), 10.65 (s, 1Н), 11.75-12.42 (m, 1Н). MS ES+ m/z 375 [М+Н]+.
Пример 37
N-[4-[2-трет-Бутокси-6-(2-метил-3-пиридил)-4-пиридил]-2-пиридил]ацетамид
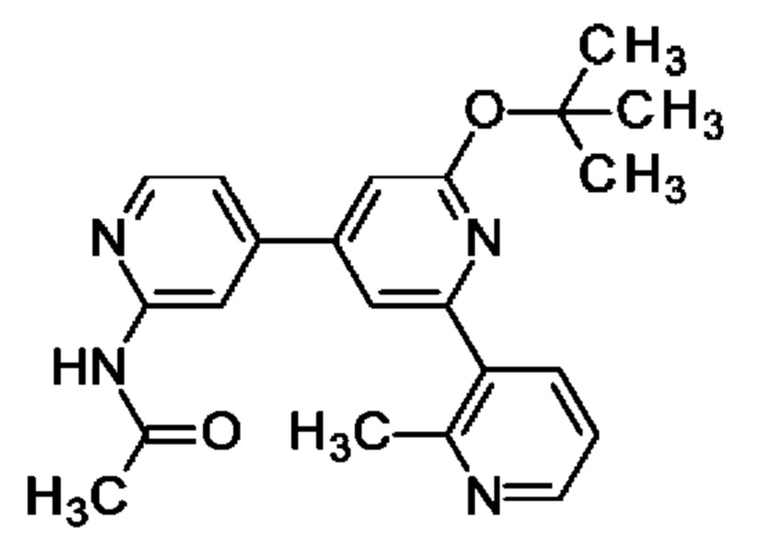
Указанное в заголовке соединение получали, как описано в примере 33, с использованием (2-метил-3-пиридил)бороновой кислоты, получая продукт в виде твердого вещества (110 мг; 93%). MS ES+ m/z 377 [М+Н]+.
Пример 38
N-[4-[2-(2-Метил-3-пиридил)-6-оксо-1Н-пиридин-4-ил]-2-пиридил]ацетамид
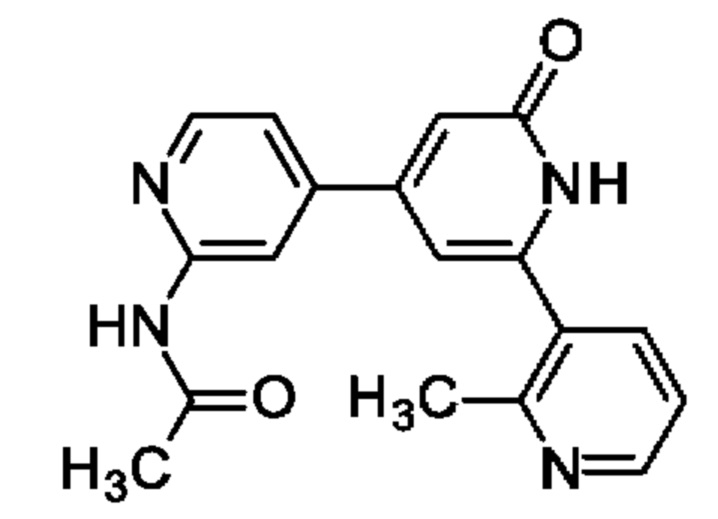
Указанное в заголовке соединение получали, как описано в примере 30, получая продукт в виде твердого вещества (32 мг; 36%). 1Н ЯМР (500 МГц, DMSO-d6) δ млн-1 2.08-2.14 (m, 3Н), 2.47 (s, 3Н), 6.58 (br s, 1Н), 6.69 (s, 1Н), 7.36 (dd, 1Н), 7.47 (dd, 1Н), 7.81 (dd, 1Н), 8.35-8.44 (m, 2Н), 8.56 (dd, 1Н), 10.67 (s, 1Н), 12.03 (br s, 1Н). MS ES+ m/z 321 [М+Н]+.
Пример 39
N-[4-[2-трет-Бутокси-6-(4-метил-3-пиридил)-4-пиридил]-2-пиридил]ацетамид
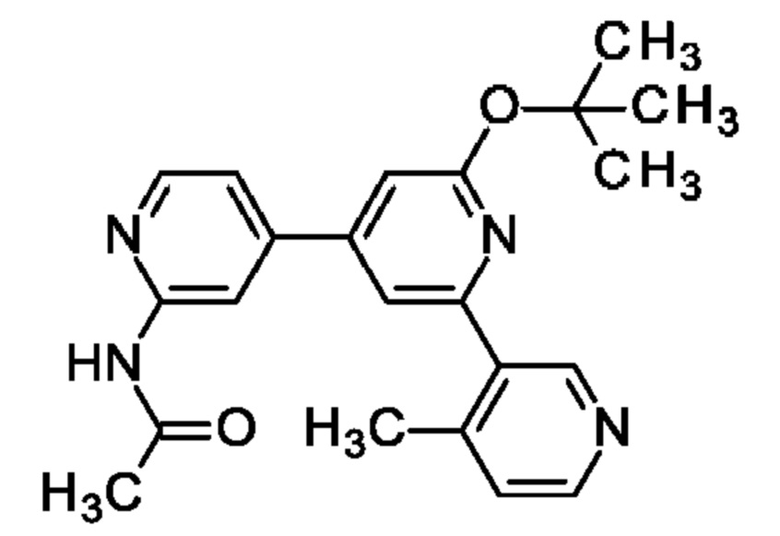
Указанное в заголовке соединение получали, как описано в примере 33, с использованием (4-метил-3-пиридил)бороновой кислоты, получая продукт в виде твердого вещества (110 мг; 78%). MS ES+ m/z 377 [М+Н]+.
Пример 40
N-[4-[2-(4-Метил-3-пиридил)-6-оксо-1Н-пиридин-4-ил]-2-пиридил]ацетамид
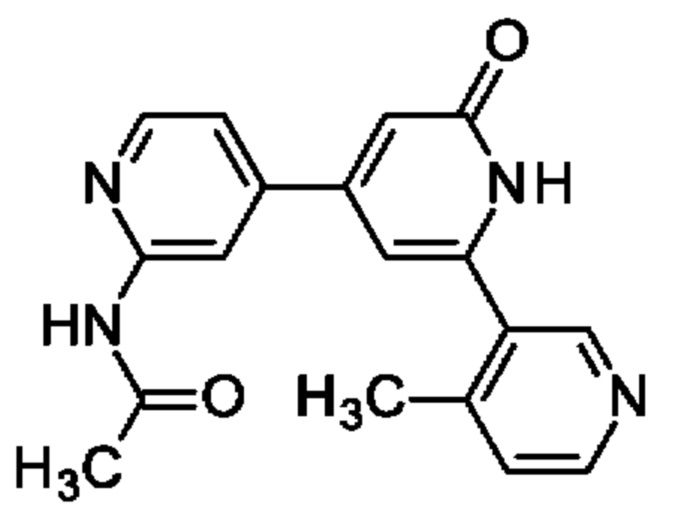
Указанное в заголовке соединение получали, как описано в примере 30, получая продукт в виде твердого вещества (40 мг; 45%). 1Н ЯМР (500 МГц, DMSO-d6) δ млн-1 2.12 (s, 3Н), 2.36 (s, 3Н), 6.60 (br s, 1Н), 6.70 (s, 1Н), 7.39 (d, 1Н), 7.48 (dd, 1Н), 8.36-8.44 (m, 2Н), 8.53 (d, 1Н), 8.55 (s, 1Н), 10.67 (s, 1Н), 12.02 (br s, 1Н). MS ES+ m/z 321 [M+H]+.
Пример 41
N-[4-[2-трет-Бутокси-6-[4-(трифторметил)-3-тиенил]-4-пиридил]-2-пиридил]ацетамид
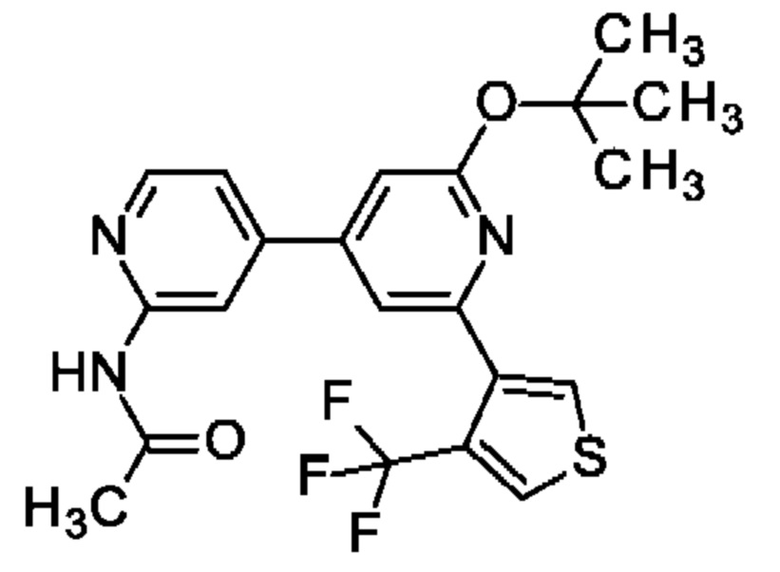
Указанное в заголовке соединение получали, как описано в примере 33, с использованием [4-(трифторметил)-3-тиенил]бороновой кислоты, получая продукт в виде твердого вещества (120 мг; 64%). MS ES+ m/z 436 [М+Н]+.
Пример 42
N-[4-[2-Оксо-6-[4-(трифторметил)-3-тиенил]-1Н-пиридин-4-ил]-2-пиридил]ацетамид
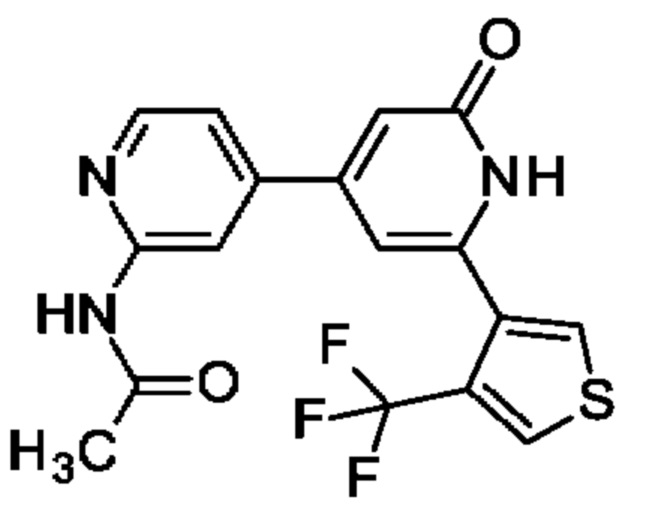
Указанное в заголовке соединение получали, как описано в примере 30, получая продукт в виде твердого вещества (27 мг; 40%). 1Н ЯМР (500 МГц, DMSO-d6) δ млн-1 2.12 (s, 3Н), 6.65 (br s, 1Н), 6.75 (br s, 1Н), 7.43 (dd, 1Н), 8.10 (d, 1Н), 8.35-8.46 (m, 3Н), 10.67 (s, 1Н), 11.90 (br s, 1H). MS ES+ m/z 380 [M+H]+.
Пример 43
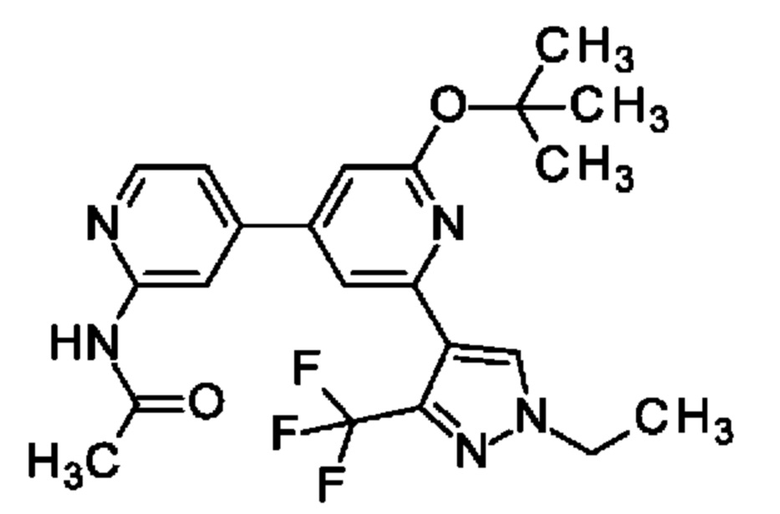
N-[4-[2-трет-Бутокси-6-[1-этил-3-(трифторметил)пиразол-4-ил]-4-пиридил]-2-пиридил]ацетамид
Указанное в заголовке соединение получали, как описано в примере 33, с использованием 1-этил-3-(трифторметил)пиразол-4-ил]бороновой кислоты, получая продукт в виде твердого вещества (62 мг; 46%). MS ES+ m/z 448 [М+Н]+.
Пример 44
N-[4-[2-[1-Этил-3-(трифторметил)пиразол-4-ил]-6-оксо-1Н-пиридин-4-ил]-2-пиридил]ацетамид
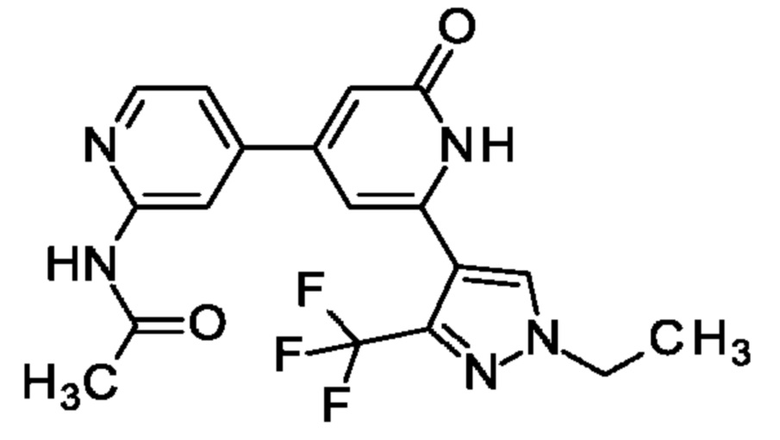
Указанное в заголовке соединение получали, как описано в примере 30, получая продукт в виде твердого вещества (18 мг; 34%). 1Н ЯМР (500 МГц, DMSO-d6) δ млн-1 1.45 (t, 3Н), 2.13 (s, 3Н), 4.28 (q, 2Н), 6.70 (br s, 2Н), 7.40 (dd, 1Н), 8.37-8.45 (m, 3Н), 10.65 (s, 1Н), 11.32-12.66 (m, 1H).MS ES+ m/z 392 [M+H]+.
Пример 45
Метил-N-[4-[2-оксо-6-[3-(трифторметил)морфолин-4-ил]-1Н-пиридин-4-ил]-2-пиридил]карбамат
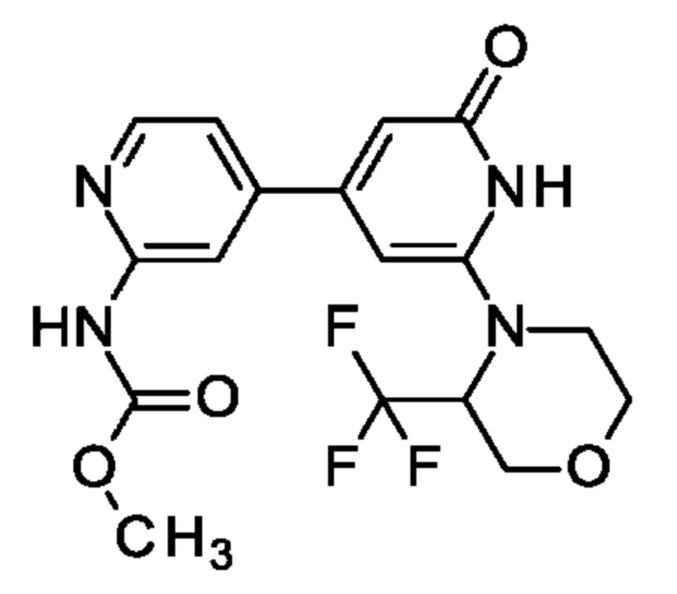
Указанное в заголовке соединение получали, как описано в примере 19, с использованием 4-[6-трет-бутокси-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-2-пиридил]-3-(трифторметил)морфолина и метил-N-(4-хлор-2-пиридил)карбамата, получая продукт в виде твердого вещества (35 мг; 36%). 1Н ЯМР (500 МГц, DMSO-d6) δ млн-1 3.24-3.37 (m, 1Н), 3.54 (td, 1Н), 3.71 (s, 3Н), 3.72-3.79 (m, 1 Н), 3.91-4.06 (m, 2Н), 4.20 (d, 1 Н), 5.31 (br d, 1 Н), 6.26 (s, 1Н), 6.57 (br s, 1Н), 7.37 (dd, 1Н), 8.08-8.10 (m, 1Н), 8.35 (dd, 1Н), 10.30 (s, 1Н), 10.56 (br s, 1Н). MS ES+ m/z 399 [М+Н]+.
Пример 46
Метил-N-[4-(2-трет-бутокси-6-хлор-4-пиридил)-2-пиридил]карбамат
Указанное в заголовке соединение получали, как описано в примере 27, с использованием метил-N-[4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-2-пиридил]карбамата, получая продукт в виде твердого вещества (940 мг; 26%). MS ES+ m/z 336 [М+Н]+.
Пример 47
Метил-N-[4-[2-трет-бутокси-6-[2-(трифторметил)фенил]-4-пиридил]-2-пиридил]карбамат
Указанное в заголовке соединение получали, как описано в примере 33, с использованием метил-N-[4-(2-трет-бутокси-6-хлор-4-пиридил)-2-пиридил]карбамата, получая продукт в виде твердого вещества (126 мг; 94%). MS ES+ m/z 446 [М+Н]+.
Пример 48
Метил-N-[4-[2-оксо-6-[2-(трифторметил)фенил]-1Н-пиридин-4-ил]-2-пиридил]карбамат
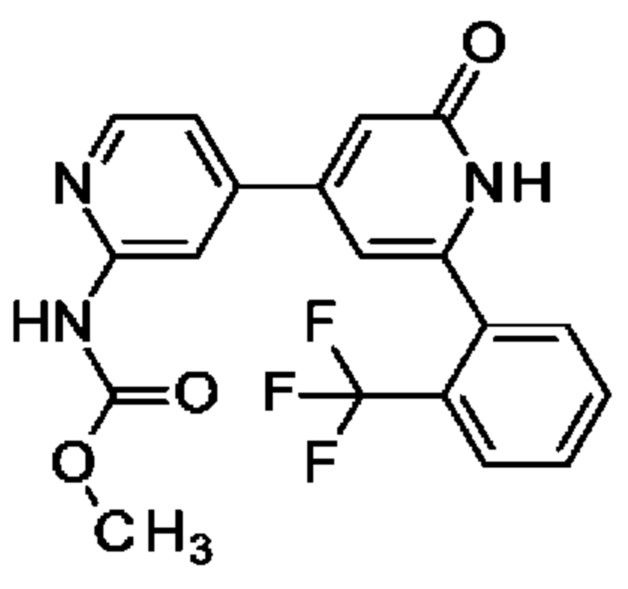
Указанное в заголовке соединение получали, как описано в примере 30, получая продукт в виде твердого вещества (40 мг; 37%). 1Н ЯМР (500 МГц, DMSO-d6) δ млн-1 3.62-3.79 (m, 3Н), 6.53 (s, 1Н), 6.75 (br s, 1Н), 7.39 (dd, 1Н), 7.66 (d, 1Н), 7.71-7.82 (m, 2Н), 7.89 (d, 1Н), 8.09-8.14 (m, 1Н), 8.35-8.39 (m, 1Н), 10.36 (s, 1Н), 12.01 (br s, 1 Н). MS ES+ m/z 390 [М+Н]+.
Пример 49
Метил-N-[4-[2-трет-бутокси-6-(2-хлорфенил)-4-пиридил]-2-пиридил]карбамат
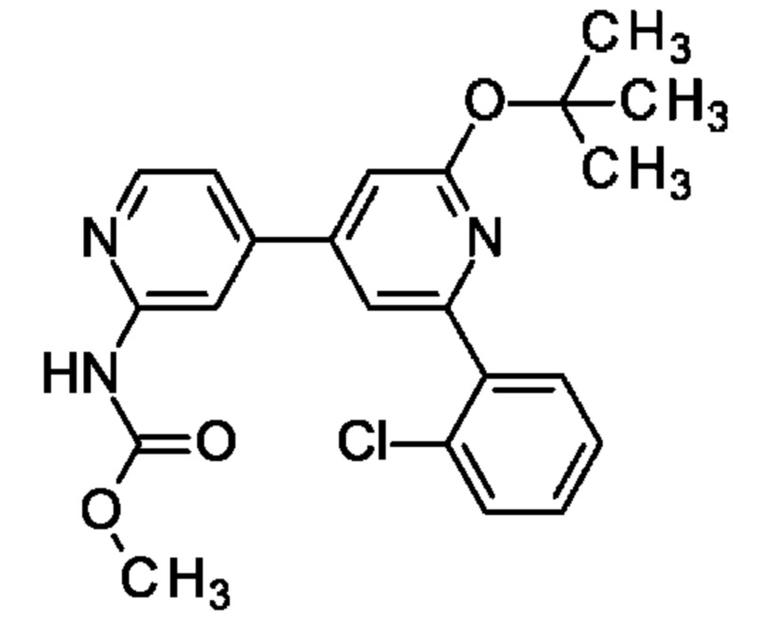
Указанное в заголовке соединение получали, как описано в примере 20, с использованием метил-N-[4-(2-трет-бутокси-6-хлор-4-пиридил)-2-пиридил]карбамата и (2-хлорфенил)бороновой кислоты, получая продукт в виде твердого вещества (82 мг; 66%). MS ES+ m/z 412 [М+Н]+.
Пример 50
Метил-N-[4-[2-(2-хлорфенил)-6-оксо-1Н-пиридин-4-ил]-2-пиридил]карбамат
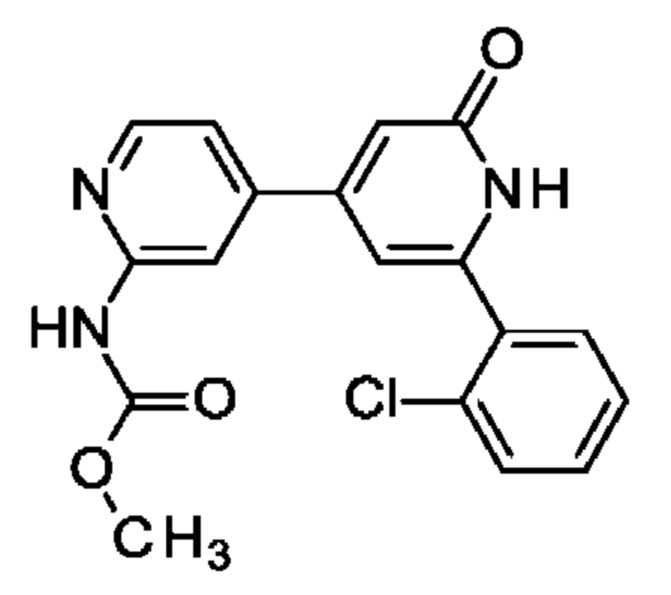
Указанное в заголовке соединение получали, как описано в примере 30, получая продукт в виде твердого вещества (8 мг; 12%). 1Н ЯМР (500 МГц, DMSO-d6) δ млн-1 3.67-3.73 (m, 3Н), 6.60 (br s, 1Н), 6.72 (br s, 1Н), 7.39-7.55 (m, 3Н), 7.58-7.63 (m, 2Н), 8.12 (s, 1Н), 8.37 (d, 1Н), 10.36 (s, 1Н), 12.00 (br s, 1Н). MS ES+ m/z 356 [М+Н]+.
Пример 51
Метил-N-[4-[2-трет-бутокси-6-[2-(трифторметил)-3-пиридил]-4-пиридил]-2-пиридил]карбамат
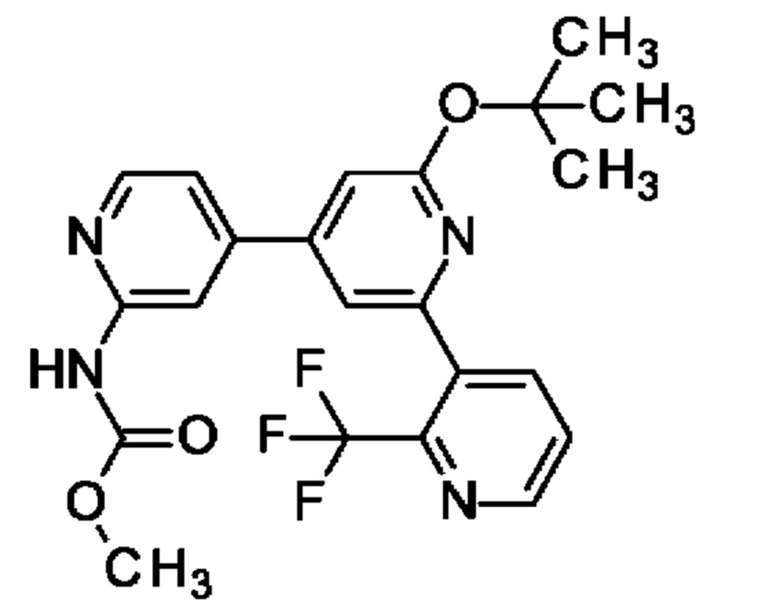
Указанное в заголовке соединение получали, как описано в примере 20, с использованием метил-N-[4-(2-трет-бутокси-6-хлор-4-пиридил)-2-пиридил]карбамата и [2-(трифторметил)-3-пиридил]бороновой кислоты, получая продукт в виде твердого вещества (57 мг; 43%). MS ES+ m/z 447 [М+Н]+.
Пример 52
Метил-N-[4-[2-оксо-6-[2-(трифторметил)-3-пиридил]-1Н-пиридин-4-ил]-2-пиридил]карбамат
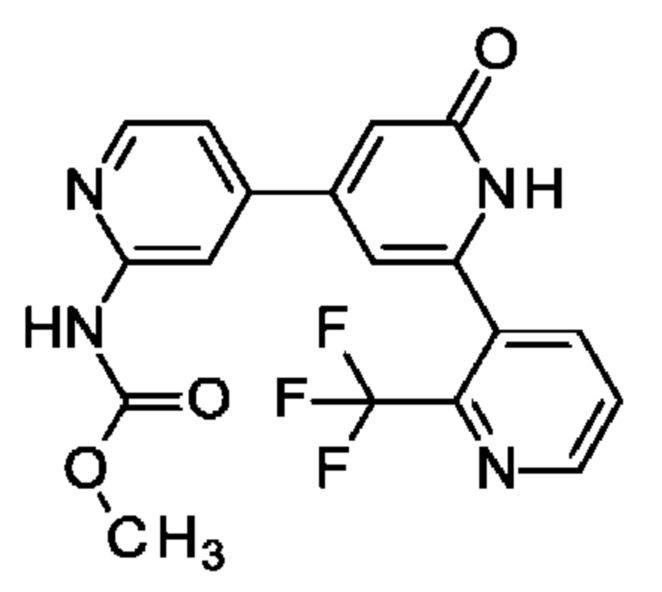
Указанное в заголовке соединение получали, как описано в примере 30, получая продукт в виде твердого вещества (18 мг; 40%). 1Н ЯМР (500 МГц, DMSO-d6) δ млн-1 3.71 (s, 3Н), 6.41-6.77 (m, 1Н), 6.82 (br s, 1Н), 7.39-7.43 (m, 1Н), 7.86 (dd, 1Н), 8.13 (s, 1Н), 8.18 (d, 1Н), 8.36-8.40 (m, 1Н), 8.88 (d, 1Н), 10.37 (s, 1Н), 11.64-12.79 (m, 1Н). MS ES+ m/z 391 [М+Н]+.
Пример 53
Метил-N-[4-[2-трет-бутокси-6-[1-этил-3-(трифторметил)пиразол-4-ил]-4-пиридил]-2-пиридил]карбамат
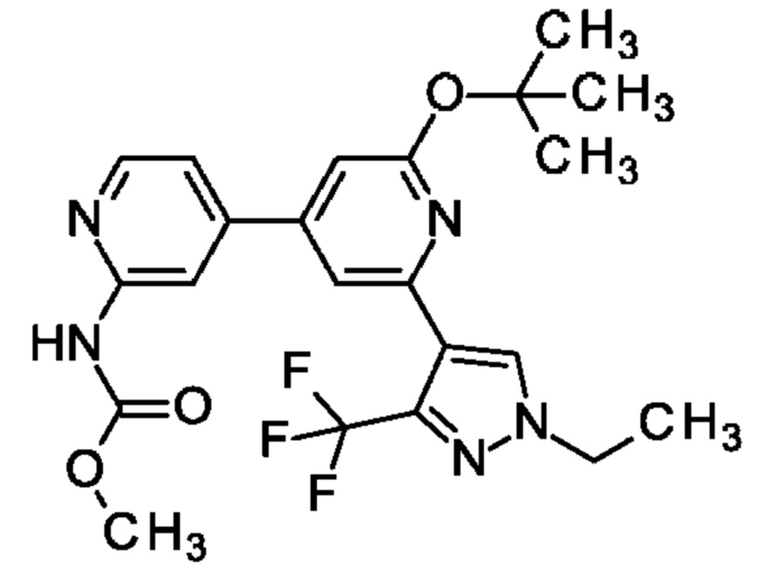
Указанное в заголовке соединение получали, как описано в примере 33, с использованием метил-N-[4-(2-трет-бутокси-6-хлор-4-пиридил)-2-пиридил]карбамата и 1-этил-3-(трифторметил)пиразол-4-ил]бороновой кислоты, получая продукт в виде твердого вещества (130 мг; 94%). MS ES+ m/z 464 [М+Н]+.
Пример 54
Метил-N-[4-[2-[1-этил-3-(трифторметил)пиразол-4-ил]-6-оксо-1Н-пиридин-4-ил]-2-пиридил]карбамат
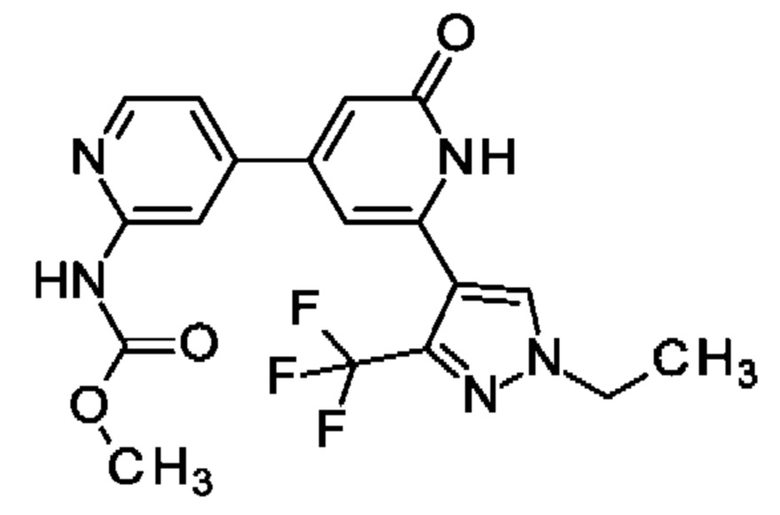
Указанное в заголовке соединение получали, как описано в примере 30, получая продукт в виде твердого вещества (6 мг; 5%). 1Н ЯМР (500 МГц, DMSO-d6) δ млн-1 1.46 (t, 3Н), 3.71 (s, 3Н), 4.29 (q, 2Н), 6.71 (br s, 2Н), 7.36 (dd, 1Н), 8.11 (s, 1Н), 8.38 (d, 1Н), 8.44 (s, 1Н), 10.37 (s, 1Н), 11.7 (br s, 1Н). MS ES+ m/z 408 [M+H]+.
Пример 55
4-Бензилокси-2-трет-бутокси-6-[2-(трифторметил)фенил]пиридин
4-Бензилокси-2-трет-бутокси-6-хлор-пиридин (1,46 г; 5 ммоль), [2-(трифторметил)фенил]бороновую кислоту (950 мг; 5 ммоль), K2CO3 (1,73 г; 12,5 ммоль) и PdCl2(dppf) (366 мг; 0,5 ммоль) растворяли в 1,4-диоксане (25 мл) и воде (5 мл) и полученную смесь перемешивали при 90°С в течение 2 ч. После охлаждения до КТ смесь разбавляли водой и EtOAc. Органический слой отделяли и водный слой экстрагировали EtOAc. Объединенные органические экстракты фильтровали через целит, обрабатывали Na2SO4, фильтровали, концентрировали и очищали на колонке с силикагелем с элюированием 0-80% EtOAc в гептане, получая продукт в виде твердого вещества (1,58 г; 79%). MS ES+ m/z 402 [М+Н]+.
Пример 56
2-трет-Бутокси-6-[2-(трифторметил)фенил]пиридин-4-ол
Указанное в заголовке соединение получали, как описано в примере 16, получая продукт (948 мг; 72%). MS ES+ m/z 312 [М+Н]+.
Пример 57
[2-трет-Бутокси-6-[2-(трифторметил)фенил]-4-пиридил]-трифторметансульфонат
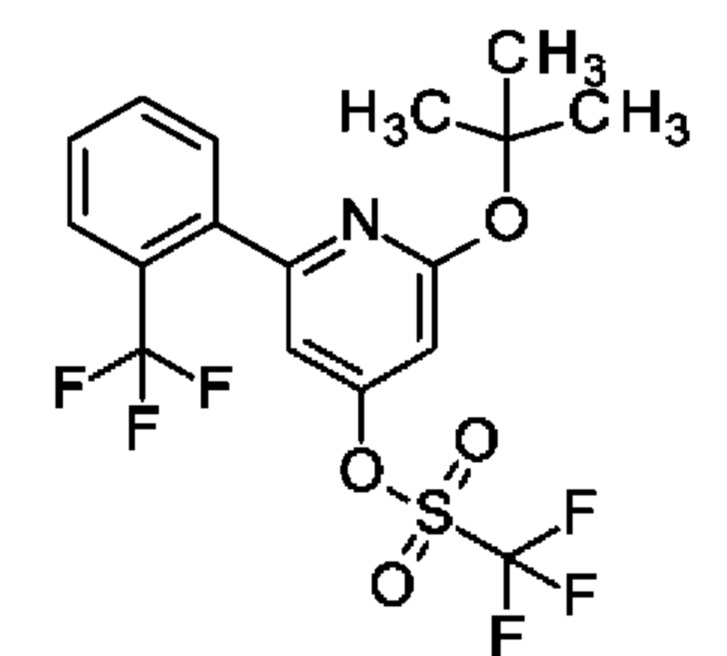
Указанное в заголовке соединение получали, как описано в примере 17, получая продукт (720 мг; 54%). MS ES+ m/z 388 [M-tBu]+.
Пример 58
2-трет-Бутокси-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-6-[2-(трифторметил)фенил]пиридин
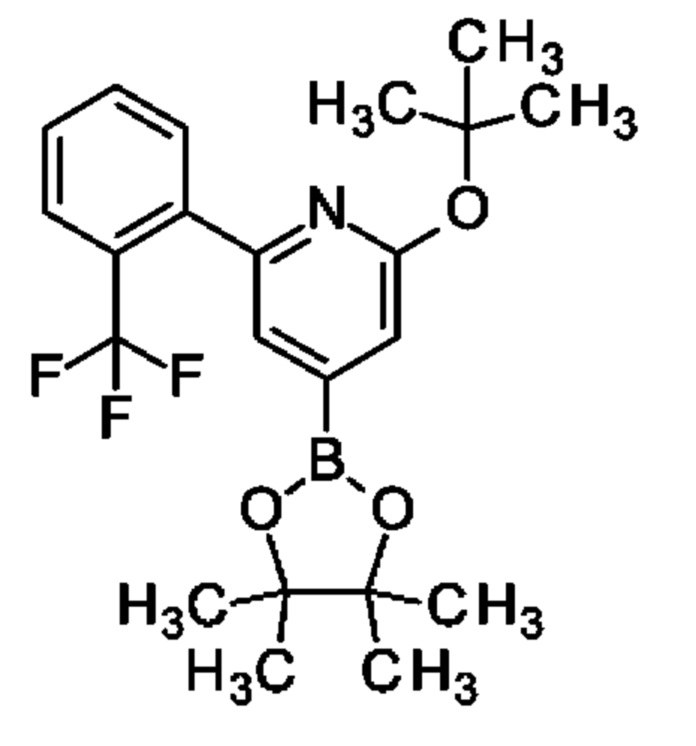
Указанное в заголовке соединение получали, как описано в примере 18, получая продукт (450 мг; 76%). MS ES+ m/z 340 [М+Н]+ (бороновая кислота).
Пример 59
Фенил-N-(4-хлор-2-пиридил)-N-феноксикарбонил-карбамат
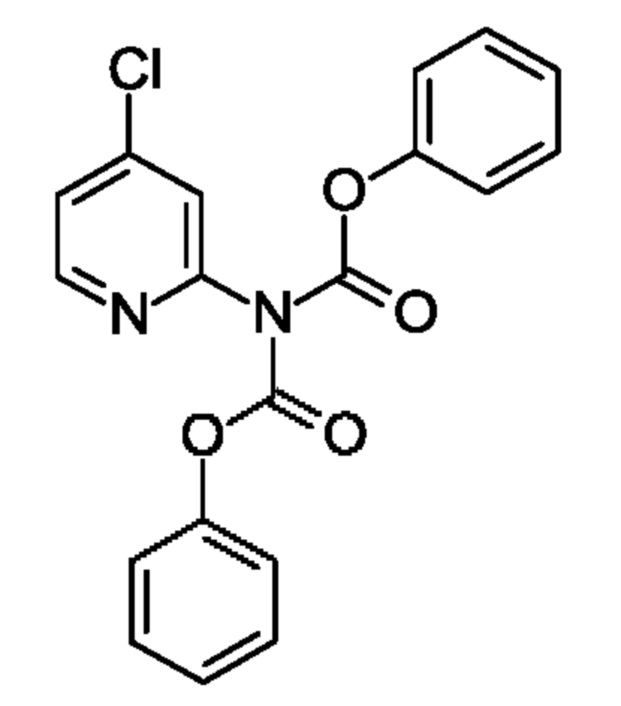
Фенилхлорформиат (0,94 мл; 7,5 ммоль) медленно добавляли к раствору 4-хлорпиридин-2-амина (386 мг; 3 ммоль) и TEA (1,25 мл; 9 ммоль) в DCM (15 мл) при 0°С и полученную смесь перемешивали при КТ в течение 2 ч. Добавляли DCM и МеОН и смесь выливали в насыщ. водн. раствор NaHCO3. После перемешивания в течение 15 мин, органический слой отделяли. Водный слой экстрагировали DCM и объединенные органические экстракты обрабатывали Na2SO4, фильтровали, концентрировали и очищали на колонке с силикагелем с элюированием 0-80% EtOAc в гептане, получая продукт в виде твердого вещества (1,1 г; 99%). MS ES+ m/z 369 [М+Н]+.
Пример 60
3-(4-Хлор-2-пиридил)-1,1-диметил-мочевина
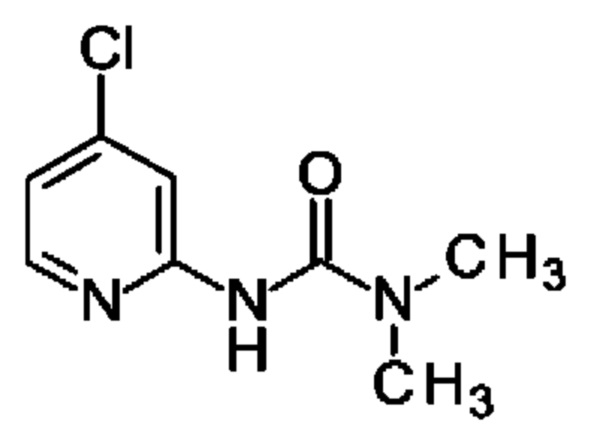
Водн. 40%-ный раствор диметиламина (1,9 мл; 15 ммоль) добавляли к раствору фенил-N-(4-хлор-2-пиридил)-N-феноксикарбонил-карбамата (1,1 г; 3 ммоль) в THF (15 мл) и полученную смесь перемешивали при КТ в течение ночи. Добавляли воду и смесь экстрагировали EtOAc. Объединенные органические экстракты обрабатывали Na2SO4, фильтровали, концентрировали и очищали на колонке с силикагелем с элюированием 0-60% EtOAc в DCM, получая продукт в виде твердого вещества (420 мг; 70%). MS ES+ m/z 200 [М+Н]+.
Пример 61
3-[4-[2-трет-Бутокси-6-[2-(трифторметил)фенил]-4-пиридил]-2-пиридил]-1,1-диметил-мочевина
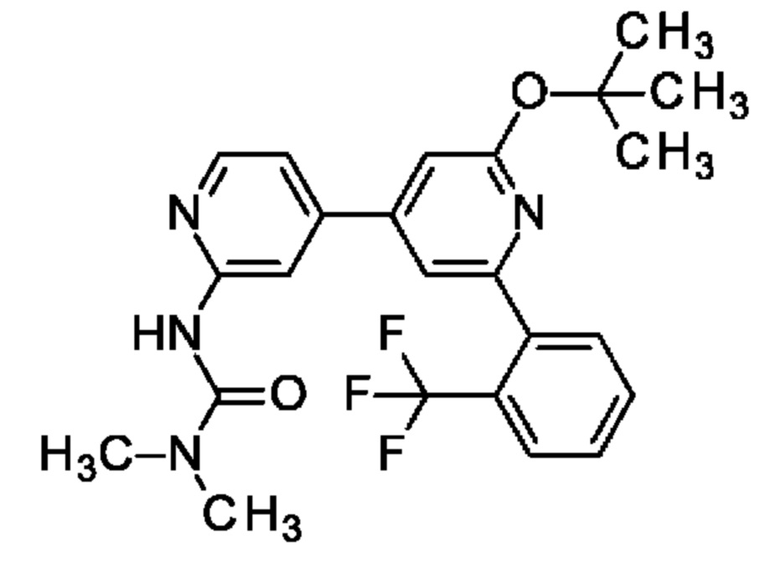
Указанное в заголовке соединение получали, как описано в примере 20, с использованием 2-трет-бутокси-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-6-[2-(трифторметил)фенил]пиридина и 3-(4-хлор-2-пиридил)-1,1-диметил-мочевины, получая продукт в виде твердого вещества (140 мг; 51%). MS ES+ m/z 459 [М+Н]+.
Пример 62
1,1-Диметил-3-[4-[2-оксо-6-[2-(трифторметил)фенил]-1Н-пиридин-4-ил]-2-пиридил]мочевина
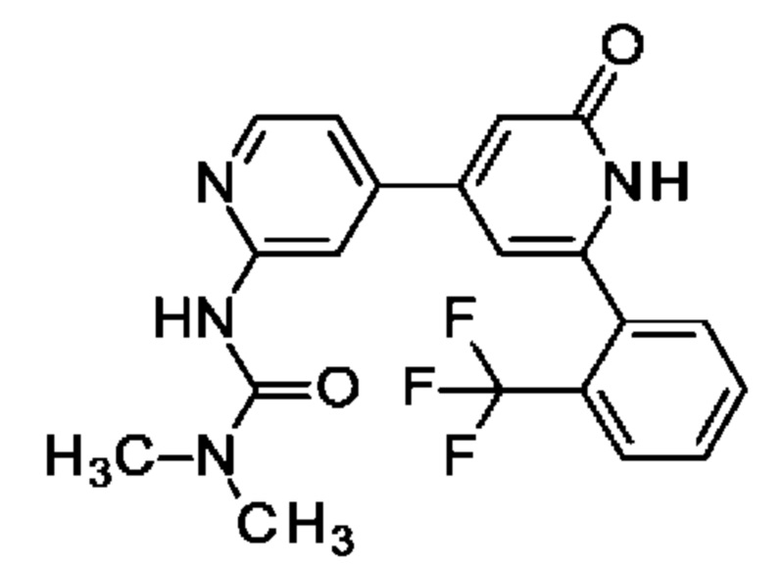
Указанное в заголовке соединение получали, как описано в примере 30, получая продукт в виде твердого вещества (28 мг; 47%). 1Н ЯМР (500 МГц, DMSO-d6) δ млн-1 2.92-3.03 (m, 6Н), 6.51 (br s, 1Н), 6.74 (s, 1Н), 7.31 (dd, 1Н), 7.64-7.85 (m, 2Н), 7.89 (d, 1Н), 8.11 (s, 1Н), 8.33 (d, 1Н), 9.02 (s, 1Н), 10.82-12.57 (m, 2Н). MS ES+ m/z 403 [М+Н]+.
Пример 63
4-Гидрокси-6-[2-(трифторметил)фенил]-1Н-пиридин-2-он
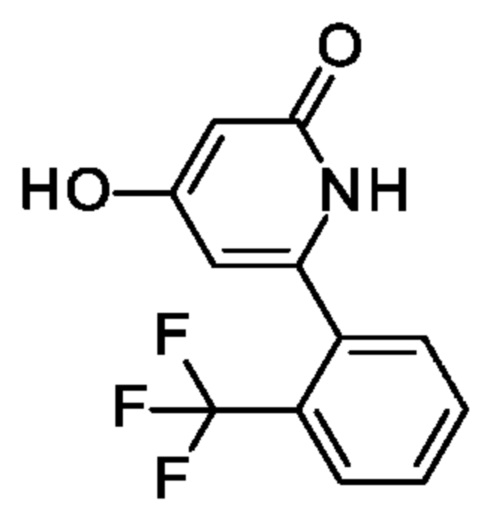
Указанное в заголовке соединение получали, как описано в примере 6, получая продукт в виде твердого вещества (2,3 г; 18%). MS ES+ m/z 256 [М+Н]+.
Пример 64
2,4-Дихлор-6-[2-(трифторметил)фенил]пиридин
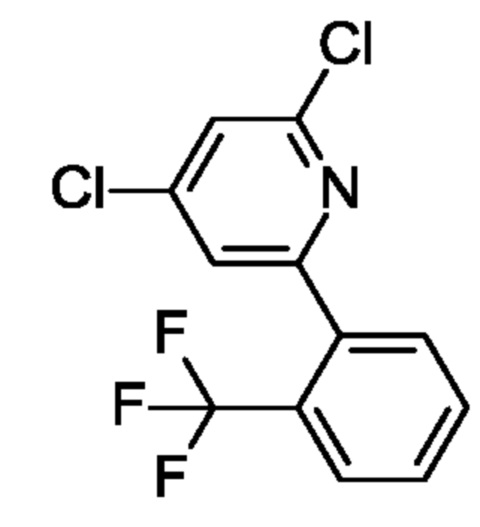
Указанное в заголовке соединение получали, как описано в примере 7, получая продукт в виде твердого вещества (1,9 г; 72%). MS ES+ m/z 292 [М+Н]+.
Пример 65
4-Хлор-6-[2-(трифторметил)фенил]-1Н-пиридин-2-он
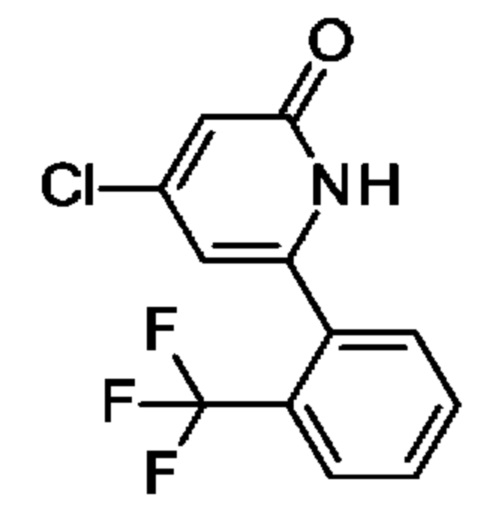
Указанное в заголовке соединение получали, как описано в примере 8, за исключением того, что раствор в TFA нейтрализовали, используя твердый NaHCO3, и экстрагировали DCM. Объединенные органические экстракты обрабатывали Na2SO4, фильтровали и концентрировали. После перекристаллизации из Et2O получали продукт в виде твердого вещества (2,05 г; 80%). MS ES+ m/z 274 [М+Н]+.
Пример 66
N-[4-[2-Оксо-6-[2-(трифторметил)фенил]-1Н-пиридин-4-ил]-2-пиридил]пирролидин-1-карбоксамид
К охлажденной во льду суспензии 4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)пиридин-2-амина (550 мг; 2,50 ммоль) в DCM (10 мл) и THF (5 мл) добавляли TEA (1,05 мл; 7,50 ммоль) и затем по каплям фенилхлорформиат (783 мг; 5 ммоль). Охлаждающую баню удаляли и полученную смесь перемешивали при КТ в течение ночи. Добавляли пирролидин (889 мг; 12,5 ммоль) и реакционную смесь перемешивали при КТ в течение 20 ч. К смеси добавляли воду и EtOAc. Водный слой отделяли и концентрировали, получая неочищенную бороновую кислоту, которую переносили в 1,4-диоксан (2,5 мл) и воду (0,5 мл). Добавляли 4-хлор-6-[2-(трифторметил)фенил]-1Н-пиридин-2-он (68 мг; 0,25 ммоль), K2CO3 (69 мг; 0,5 ммоль) и PdCl2(dppf) (27 мг; 0,04 ммоль) и полученную смесь нагревали и перемешивали при 90°С в течение 8 ч. После охлаждения до КТ смесь разбавляли 1,4-диоксаном, фильтровали через целит, концентрировали и очищали препаративной HPLC, получая продукт в виде твердого вещества (30 мг; 28%). 1Н ЯМР (500 МГц, DMSO-d6) δ млн-1 1.85 (br s, 4Н), 3.41 (br s, 4Н), 6.51 (br s, 1H), 6.74 (s, 1H), 7.32 (dd, 1H), 7.66 (d, 1H), 7.71-7.82 (m, 2H), 7.89 (d, 1H), 8.21 (s, 1H), 8.33 (d, 1H), 8.83 (s, 1H), 12.08 (br s, 1H). MS ES+ m/z 429 [M+H]+.
Пример 67
N-[4-[2-трет-Бутокси-6-[2-(1-метокси-1-метил-этил)пирролидин-1-ил]-4-пиридил]-2-пиридил]ацетамид
N-[4-(2-трет-Бутокси-6-хлор-4-пиридил)-2-пиридил]ацетамид (128 мг; 0,4 ммоль), 2-(1-метокси-1-метил-этил)пирролидин (86 мг; 0,6 ммоль), PEPPSI-iPr (14 мг; 0,02 ммоль) и KOtBu (99 мг; 0,88 ммоль) переносили в 1,4-диоксан (3 мл) и смесь перемешивали при 80°С в течение ночи. Добавляли дополнительное количество KOtBu (49 мг; 0,44 ммоль) и PEPPSI-iPr (7 мг; 0,01 ммоль) и смесь перемешивали при 90°С в течение 6 ч. После охлаждения до КТ добавляли 10%-ный водн. раствор NaCl и смесь экстрагировали EtOAc. Объединенные органические экстракты обрабатывали Na2SO4, фильтровали, концентрировали и очищали на колонке с силикагелем с элюированием 0-80% EtOAc в гептане, получая продукт в виде твердого вещества (46 мг; 27%). MS ES+ m/z 427 [М+Н]+.
Пример 68
N-[4-[2-[2-(1-Метокси-1-метил-этил)пирролидин-1-ил]-6-оксо-1Н-пиридин-4-ил]-2-пиридил]ацетамид
Указанное в заголовке соединение получали, как описано в примере 30, получая продукт в виде твердого вещества (16 мг; 41%). 1Н ЯМР (500 МГц, DMSO-d6) δ млн-1 1.12 (s, 3Н), 1.17 (br s, 3Н), 1.81-1.92 (m, 2H), 1.98 (br d, 3H), 2.12 (s, 3H), 3.19 (s, 3H), 3.39-3.52 (m, 2H), 5.77 (s, 1H), 5.86 (br s, 1H), 7.33 (dd, 1H), 8.32 (s, 1H), 8.36 (d, 1H), 10.60 (s, 1H), 10.67-11.27 (m, 1H). MS ES+ m/z 371 [M+H]+.
Пример 69
Метил-N-[4-[2-трет-бутокси-6-[2-(трифторметил)-1-пиперидил]-4-пиридил]-2-пиридил]карбамат
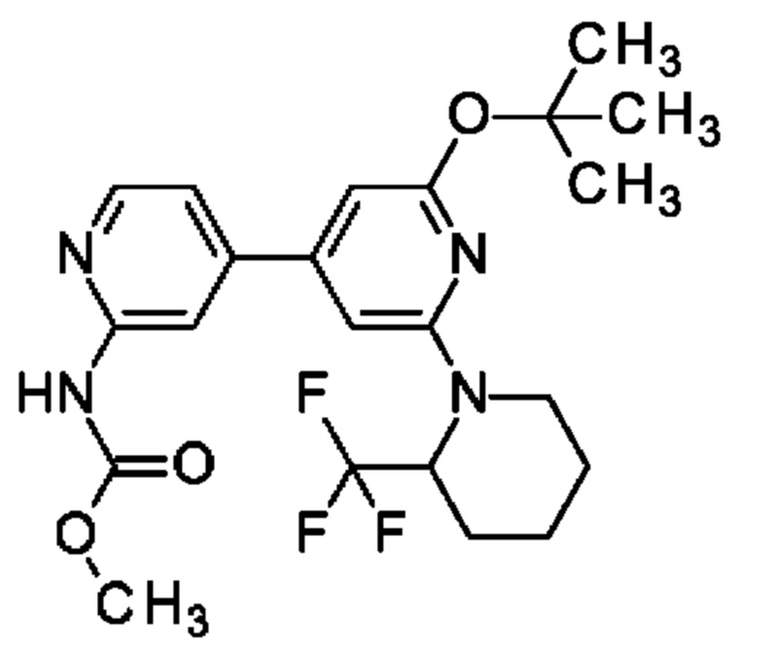
Метилхлорформиат (66 мкл, 0,86 ммоль) медленно добавляли к раствору 4-[2-трет-бутокси-6-[2-(трифторметил)-1-пиперидил]-4-пиридил]пиридин-2-амина (135 мг; 0,34 ммоль) и N,N-диизопропилэтиламина (0,3 мл; 1,71 ммоль) в DCM (15 мл) при 0°С. Полученную смесь перемешивали при 0°С в течение 30 минут, добавляли МеОН и смесь концентрировали. Полученный остаток растворяли в МеОН (10 мл), добавляли 1 М водн. раствор NaOH (2 мл) и смесь перемешивали при КТ в течение 2,5 ч. Добавляли воду и смесь экстрагировали EtOAc. Объединенные органические экстракты промывали рассолом, обрабатывали Na2SO4, фильтровали, концентрировали и очищали на колонке с силикагелем с элюированием 0-80% EtOAc в гептане, получая продукт в виде твердого вещества (150 мг; 88%). MS ES+ m/z 453 [М+Н]+.
Пример 70
Метил-N-[4-[2-оксо-6-[2-(трифторметил)-1-пиперидил]-1Н-пиридин-4-ил]-2-пиридил]карбамат
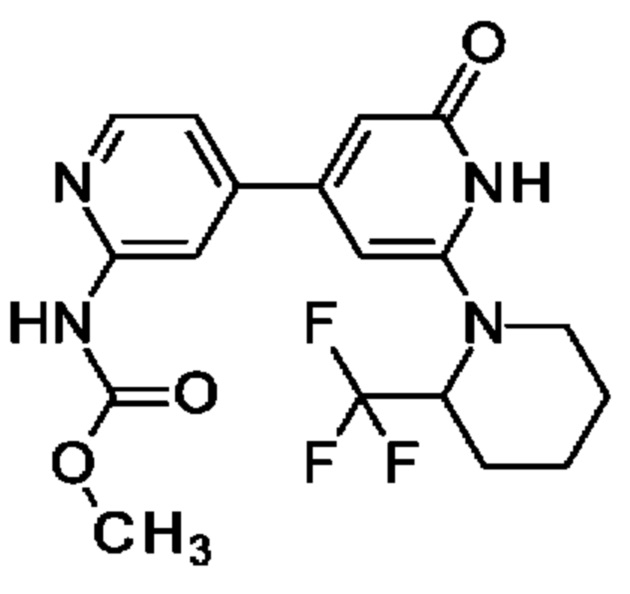
Указанное в заголовке соединение получали, как описано в примере 30, получая продукт в виде твердого вещества (36 мг; 41%). 1Н ЯМР (500 МГц, DMSO-d6) δ млн-1 1.35-1.54 (m, 1Н), 1.61-1.70 (m, 2Н), 1.70-1.83 (m, 2Н), 1.99 (br d, 1Н), 3.04 (br t, 1Н), 3.70 (s, 3Н), 4.17 (br d, 1H), 5.51-5.62 (m, 1H), 6.18 (s, 1H), 6.50 (br s, 1H), 7.37 (dd, 1H), 8.06-8.09 (m, 1H), 8.33 (dd, 1H), 10.29 (br s, 2H). MS ES+ m/z 397 [M+H]+.
Пример 71
Биохимический анализ Vps34
Готовили серии разведений соединений по изобретению в DMSO в концентрации, в 100 раз превышающей конечную концентрацию в анализе (n1=n0/3; 10 точек). Затем соединения разбавляли до концентрации, в 4 раза превышающей концентрацию в анализе, в буфере для анализа (буфере Q от Life Technologies, PV5125, разбавленном в 5 раз, дополненном 2 мМ дитиотреитом (DTT) и 2 мМ MnCl2). По 2,5 мкл разбавленных растворов соединений добавляли в 384-луночный используемый в анализе планшет, затем добавляли по 2,5 мкл 16,5 нМ раствора фермента Vps34 (Life Technologies, PV5126). Проводили предварительную инкубацию фермента и соединений при КТ в течение 15 мин. Затем в лунки, содержащие соединение и фермент, добавляли по 5 мкл субстратной смеси, содержащей 20 мкМ АТФ (Life Technologies, PV3227) и 200 мкМ субстрат PI:PS (фосфатидилсерин) (Life Technologies, PV5122) в буфере для анализа. Смешивание осуществляли посредством многократного пипетирования. Реакционную смесь инкубировали при комнатной температуре в течение 1 ч. Затем для гашения реакции добавляли по 5 мкл смеси для остановки и детектирования, приготовленной так, как описано в инструкциях к набору Adapta™ для анализа киназ (Life Technologies, PV5099), содержащей Eu-меченное антитело к АДФ Adapta™ (2,3 нМ), индикатор на АДФ Alexa Fluor 647 (9 нМ) и EDTA (этилендиаминтетрауксусная кислота; 30 мМ) в буфере для TR-FRET (резонансный перенос энергии флуоресценции с разрешением по времени). Смешивание осуществляли посредством многократного пипетирования. Затем используемый в анализе планшет инкубировали при комнатной температуре в течение 30 мин и выполняли измерения, используя микропланшетный ридер Artemis. Процент ингибирования под действием соединений рассчитывали относительно обработанных DMSO контрольных образцов. Для определения значений IC50 (концентрации, вызывающей 50%-ное ингибирование) с применением программного обеспечения Dotmatics проводили построение по точкам кривой зависимости процента ингибирования от концентрации соединений.
Соединения из примеров эффективно ингибировали Vps34, и результаты анализа показаны в Таблице 1 (средние значения IC50 в нМ; Adapta™).
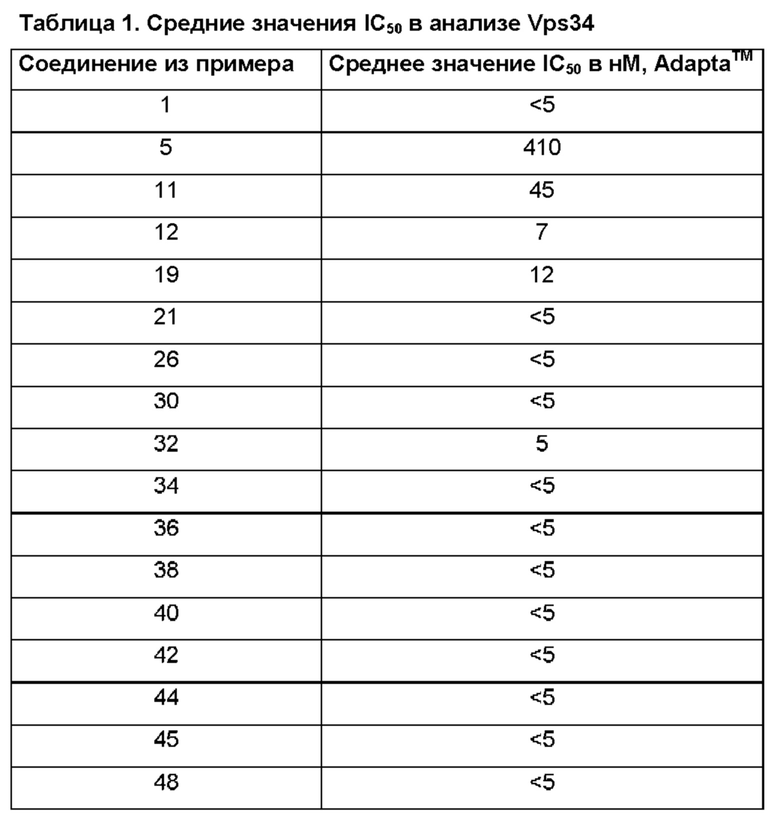
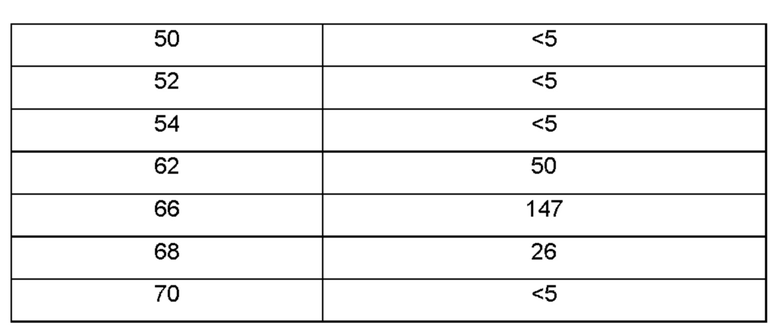
Пример 72
Одновременный многопараметрический анализ аутофагии
Для определения ингибирующего действия патентуемых соединений на аутофагию использовали клетки остеосаркомы человека (HOS), стабильно экспрессирующие меченную зеленым флуоресцентным белком (GFP) LC3 (GFP-LC3). С этой целью активировали аутофагию, используя ингибитор mTOR (мишень рапамицина у млекопитающих) KU-0063794 в концентрации 500 нМ в присутствии бафиломицина А1 (Sigma-Aldrich) в концентрации 5 нМ. Кратко, клетки высевали на ночь в 96-луночные планшеты с прозрачным дном в среде DMEM (модифицированная Дульбекко среда Игла) с высоким содержанием глюкозы (Hi-Clone, № по каталогу SH30285.01). В начале эксперимента среду удаляли и заменяли на свежую среду, содержащую ингибитор mTOR, бафиломицин А1 и разбавитель или тестируемое соединение, как указано. Через 6 часов среду удаляли, клетки дважды промывали охлажденным во льду забуференным фосфатом физиологическим раствором (PBS) и фиксировали, используя 4%-ный раствор параформальдегида в течение 20 минут при комнатной температуре. Затем клетки дважды промывали охлажденным во льду PBS, после чего добавляли краситель для окрашивания ядер Хехст 33342 в концентрации 1 мкг/мл в PBS. После инкубирования в течение ночи при 4°С клетки один раз промывали PBS для удаления избытка красителя и в каждую лунку добавляли по 100 мкл PBS. Изображения получали при 20-кратном увеличении, по 6 изображений на одну лунку, используя автоматизированный микроскоп ImageXpress (Molecular Devices Inc.), и анализировали с применением программного обеспечения MetaXpress для идентификации очагов LC3-GFP. Для получения кривой зависимости ответа от дозы использовали значения площади очагов из расчета на одну клетку, а значения IC50 рассчитывали, применяя аппроксимацию нелинейной зависимости в программном обеспечении GraphPad Prism.
Протестированные соединения из примеров эффективно ингибировали аутофагию в клетках HOS. Результаты данного анализа показаны в Таблице 2 (средние значения IC50 в мкМ; HOS-LC3).
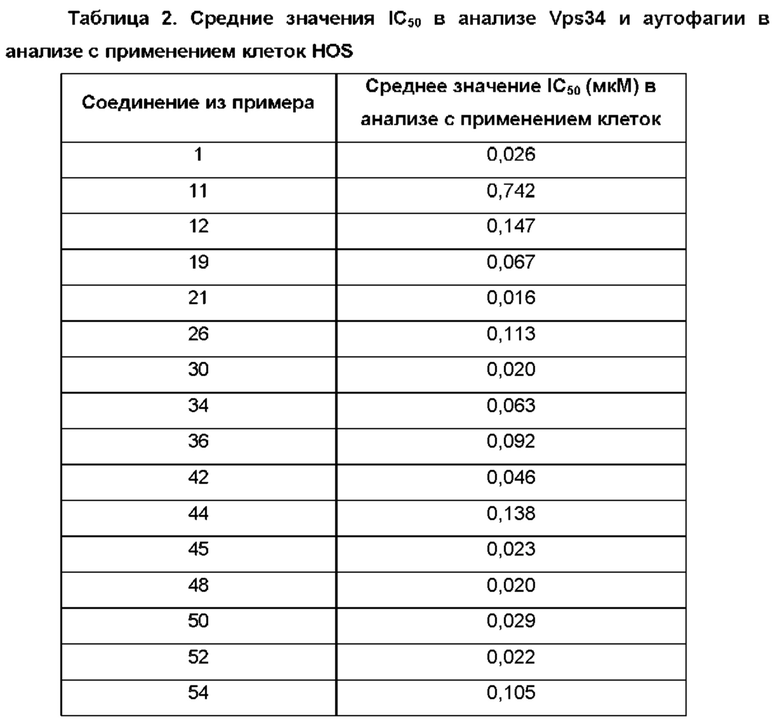
Используя подходящие замены исходного вещества и, при необходимости, адаптации реакционных условий и тому подобное, синтезируют приведенные далее соединения:
метил-N-[4-[2-(4-метил-3-пиридил)-6-оксо-1Н-пиридин-4-ил]-2-пиридил]карбамат;
метил-N-[4-[2-[4-этилсульфонил-2-(трифторметил)пиперазин-1-ил]-6-оксо-1Н-пиридин-4-ил]-2-пиридил]карбамат;
метил-N-[4-[2-(3-циклопропилморфолин-4-ил)-6-оксо-1Н-пиридин-4-ил]-2-пиридил]карбамат;
3-[4-[2-(2-хлорфенил)-6-оксо-1Н-пиридин-4-ил]-2-пиридил]-1,1-диметил-мочевина; и
N-[4-[2-(2-хлорфенил)-6-оксо-1Н-пиридин-4-ил]-2-пиридил]пирролидин-1-карбоксамид.
| название | год | авторы | номер документа |
|---|---|---|---|
| АЗАИНДОЛИЛПИРИДОНЫ И ДИАЗАИНДОЛИЛПИРИДОНЫ | 2018 |
|
RU2788659C2 |
| МОРФОЛИНИЛПИРИДОНЫ | 2018 |
|
RU2803158C2 |
| 6-ГЕТЕРОЦИКЛИЛ-4-МОРФОЛИН-4-ИЛПИРИДИН-2-ОНЫ, ПРИГОДНЫЕ ДЛЯ ЛЕЧЕНИЯ РАКА И ДИАБЕТА | 2017 |
|
RU2762968C2 |
| ПИРИДИНАМИНПИРИДОНЫ И ПИРИМИДИНАМИНПИРИДОНЫ | 2018 |
|
RU2804638C2 |
| 6-АРИЛ-4-МОРФОЛИН-1-ИЛПИРИДОНЫ, ПРИГОДНЫЕ ДЛЯ ЛЕЧЕНИЯ РАКА ИЛИ ДИАБЕТА | 2017 |
|
RU2754664C2 |
| ПОЛИЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ И СПОСОБЫ ЦЕЛЕНАПРАВЛЕННОЙ ДЕГРАДАЦИИ ПОЛИПЕПТИДОВ БЫСТРО УСКОРЕННОЙ ФИБРОСАРКОМЫ | 2019 |
|
RU2830173C2 |
| ЦИКЛИЧЕСКИЕ ДИОНЫ В КАЧЕСТВЕ ГЕРБИЦИДНЫХ СОЕДИНЕНИЙ | 2020 |
|
RU2822391C2 |
| Спироконденсированные пирролидиновые производные в качестве ингибиторов деубиквитилирующих ферментов (DUB) | 2017 |
|
RU2730552C2 |
| СОЕДИНЕНИЯ БЕНЗАЗЕПИНА ДИКАРБОКСАМИДА | 2016 |
|
RU2712248C2 |
| ПРОИЗВОДНЫЕ ПИРИДОПИРИМИДИНОНА И ИХ ИСПОЛЬЗОВАНИЕ В КАЧЕСТВЕ МОДУЛЯТОРОВ РЕЦЕПТОРА АРОМАТИЧЕСКИХ УГЛЕВОДОРОДОВ | 2021 |
|
RU2818954C1 |
Изобретение относится к соединению формулы (I) или к его фармацевтически приемлемой соли, где X представляет собой С=О или связь; R1 выбран из группы, состоящей из Н, С1-С3алкила, С1-С3алкоксиС1-С3алкила, С3-С6цикпоалкила, С1-С3алкокси, N,N-диС1-С3алкиламино и 1-пирролидинила, при условии, что, когда R1 представляет собой С1-С3алкокси, N,N-диС1-С3алкиламино или 1-пирролидинил, X представляет собой С=О; R2 представляет собой атом водорода; R3 выбран из группы, состоящей из А, фенила и моноциклического гетероарила, причем указанный моноциклический гетероарил выбран из группы, состоящей из пиридина, тиофена и пиразола, и указанный фенил и указанный гетероарил возможно замещены одним или двумя из R4, R5, R6 и R7; R4, R5, R6 и R7 независимо выбраны из группы, состоящей из галогена, С1-С6алкила, С1-С3галогеналкокси и С1-С6галогеналкила; А представляет собой 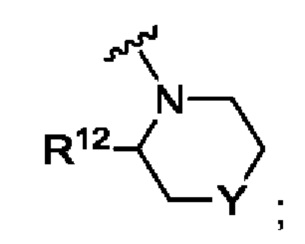 R12 выбран из группы, состоящей из С1-С3алкоксиС1-С3алкила, С3-С6циклоалкила и С1-С3 галогеналкила; Y выбран из группы, состоящей из СН2, NSO2R9, О и связи. Изобретение также относится к фармацевтической композиции для ингибирования Vps34 на основе указанных соединений. Технический результат – получены новые соединения и фармацевтическая композиция на их основе, которые могут найти применение в медицине при лечении рака. 5 н. и 20 з.п. ф-лы, 2 табл., 72 пр.
R12 выбран из группы, состоящей из С1-С3алкоксиС1-С3алкила, С3-С6циклоалкила и С1-С3 галогеналкила; Y выбран из группы, состоящей из СН2, NSO2R9, О и связи. Изобретение также относится к фармацевтической композиции для ингибирования Vps34 на основе указанных соединений. Технический результат – получены новые соединения и фармацевтическая композиция на их основе, которые могут найти применение в медицине при лечении рака. 5 н. и 20 з.п. ф-лы, 2 табл., 72 пр.
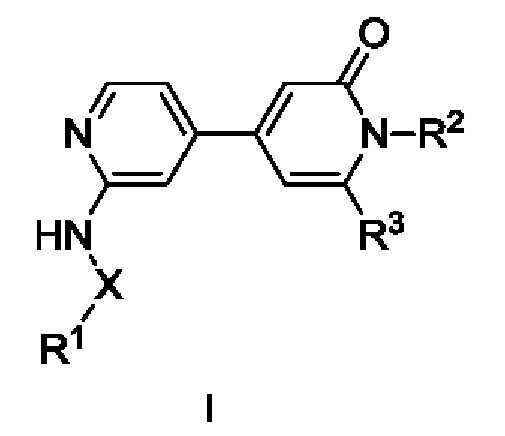
1. Соединение формулы (I),
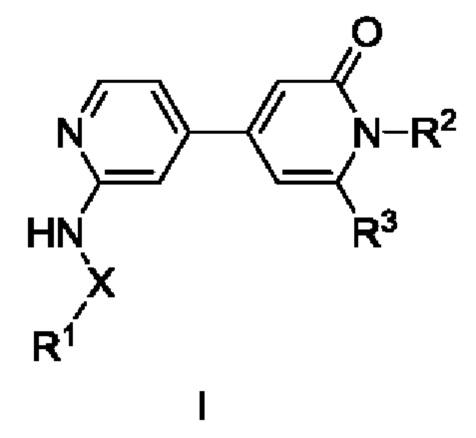
где
X представляет собой С=О или связь;
R1 выбран из группы, состоящей из Н, С1-С3алкила, С1-С3алкоксиС1-С3алкила, С3-С6циклоалкила, С1-С3алкокси, N,N-диС1-С3алкиламино и 1-пирролидинила, при условии, что, когда R1 представляет собой С1-С3алкокси, N,N-диС1-С3алкиламино или 1-пирролидинил, X представляет собой С=О;
R2 представляет собой атом водорода;
R3 выбран из группы, состоящей из А, фенила и моноциклического гетероарила, причем указанный моноциклический гетероарил выбран из группы, состоящей из пиридина, тиофена и пиразола, и указанный фенил и указанный гетероарил возможно замещены одним или двумя из R4, R5, R6 и R7;
R4, R5, R6 и R7 независимо выбраны из группы, состоящей из галогена, С1-С6алкила, С1-С3галогеналкокси и С1-С6галогеналкила;
R9 представляет собой С1-С6алкил;
А представляет собой
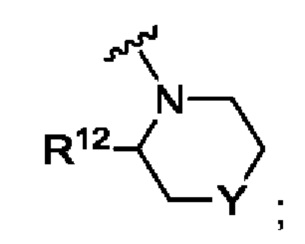
R12 выбран из группы, состоящей из С1-С3алкоксиС1-С3алкила, С3-С6циклоалкила и С1-С3 галогеналкила;
Y выбран из группы, состоящей из СН2, NSO2R9, О и связи;
или его фармацевтически приемлемая соль.
2. Соединение по п. 1, где R1 выбран из группы, состоящей из Н, метила, метокси, метоксиметила, N,N-диметиламино, 1-пирролидинила и циклопропила;
или его фармацевтически приемлемая соль.
3. Соединение по п. 1 или 2, где R1 выбран из группы, состоящей из Н, метила, метоксиметила, N,N-диметиламино, 1-пирролидинила и циклопропила;
или его фармацевтически приемлемая соль.
4. Соединение по любому из пп. 1-3, где R3 выбран из группы, состоящей из А, фенила и пиридила, при этом указанный фенил и указанный пиридил возможно и независимо замещены R4 и/или R5;
или его фармацевтически приемлемая соль.
5. Соединение по любому из пп. 1-4, где
R4, R5, R6 и R7 независимо выбраны из группы, состоящей из атома фтора, атома хлора, С1-С3алкила и С1-С3фторалкила;
или его фармацевтически приемлемая соль.
6. Соединение по любому из пп. 1-5, где Y выбран из группы, состоящей из СН2, О и связи;
или его фармацевтически приемлемая соль.
7. Соединение по любому из пп. 1-6, где R12 представляет собой С1-С3галогеналкил или С3-С6циклоалкил;
или его фармацевтически приемлемая соль.
8. Соединение по любому из пп. 1-7, где R3 выбран из группы, состоящей из
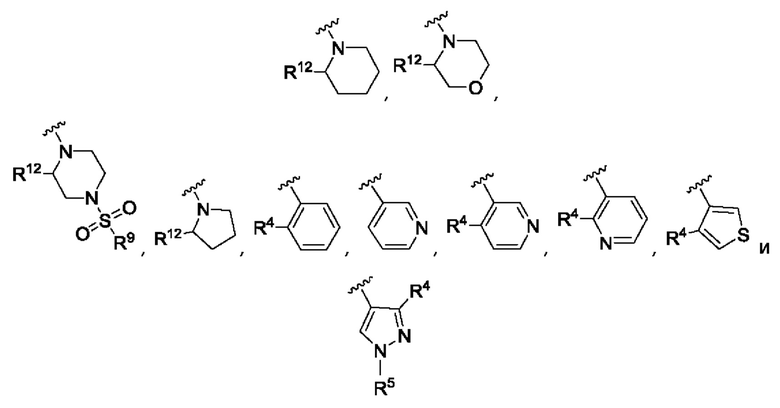
или его фармацевтически приемлемая соль.
9. Соединение по любому из пп. 1-8, где R3 выбран из группы, состоящей из
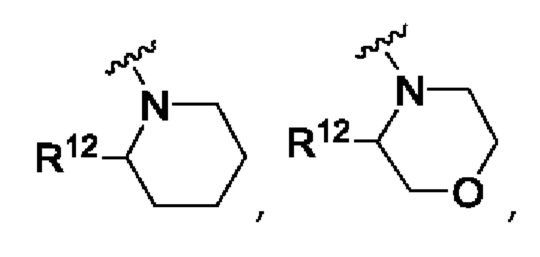
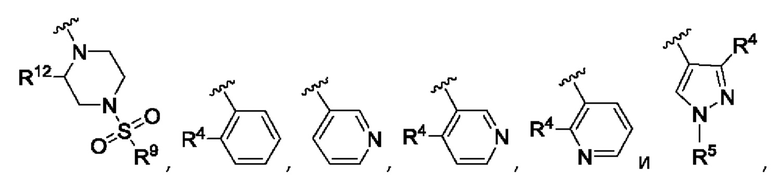
или его фармацевтически приемлемая соль.
10. Соединение по любому из пп. 1-9, где R3 выбран из группы, состоящей из
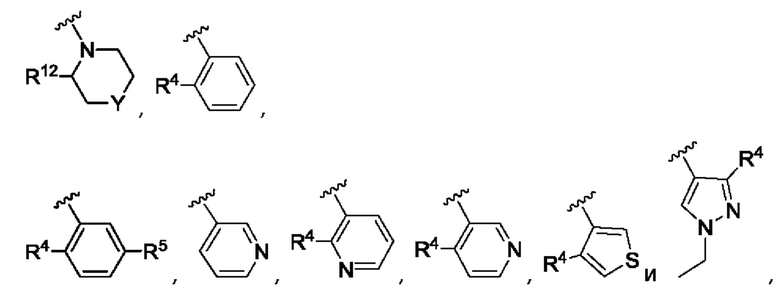
где Y выбран из СН2, О и связи;
R4 выбран из группы, состоящей из CF3, атома хлора и метила;
R5 представляет собой атом фтора; и
R12 выбран из состоящей из циклопропила, 1-метокси-1-метил-этила и CF3;
или его фармацевтически приемлемая соль.
11. Соединение по любому из пп. 1-10, где R3 выбран из группы, состоящей из
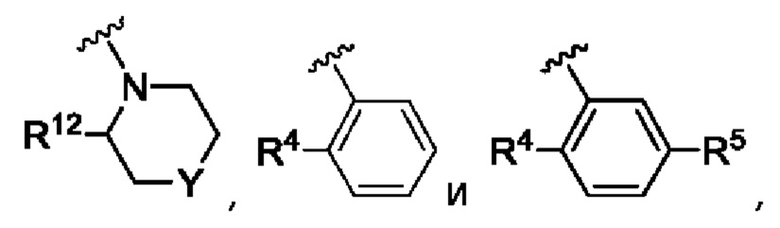
где Y представляет собой СН2 или О;
R4 представляет собой CF3 или атом хлора;
R5 представляет собой атом фтора; и
R12 представляет собой CF3 или циклопропил;
или его фармацевтически приемлемая соль.
12. Соединение по п. 1, где
R1 выбран из Н, метила, метокси, метоксиметила, N,N-диметиламино, 1-пирролидинила и циклопропила; и
R3 выбран из группы, состоящей из
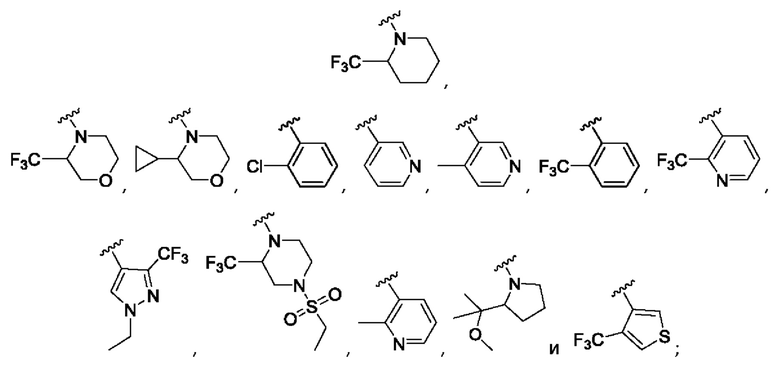
или его фармацевтически приемлемая соль.
13. Соединение по п. 1, где
R1 выбран из Н, метила, метокси, метоксиметила, N,N-диметиламино, 1-пирролидинила и циклопропила; и R3 выбран из
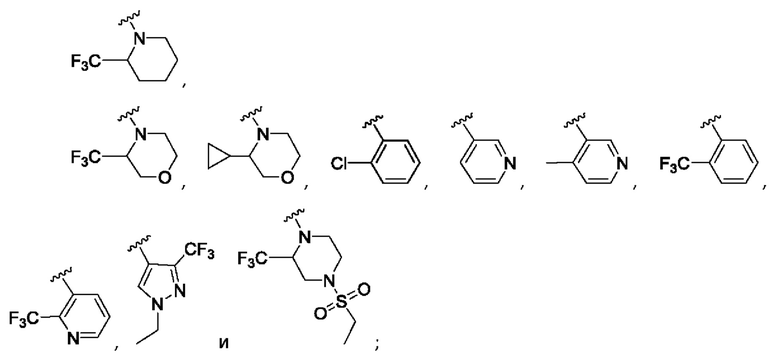
или его фармацевтически приемлемая соль.
14. Соединение по п. 1, где
R1 выбран из Н, метила, метоксиметила, N,N-диметиламино, 1-пирролидинила и циклопропила; и
R3 выбран из
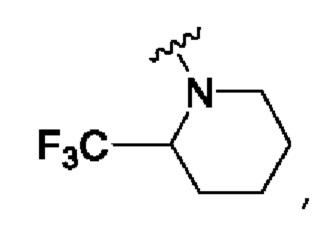
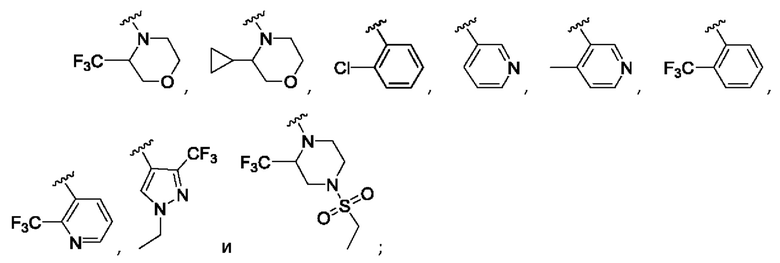
или его фармацевтически приемлемая соль.
15. Соединение по п. 1, где указанное соединение представляет собой
N-[4-[2-(2-хлорфенил)-6-оксо-1Н-пиридин-4-ил]-2-пиридил]ацетамид;
4-(2-амино-4-пиридил)-6-(3-пиридил)-1Н-пиридин-2-он;
4-(2-амино-4-пиридил)-6-(2-хлорфенил)-1Н-пиридин-2-он;
N-[4-[2-(2-хлорфенил)-6-оксо-1Н-пиридин-4-ил]-2-пиридил]-2-метокси-ацетамид;
N-[4-[2-оксо-6-[2-(трифторметил)-1-пиперидил]-1Н-пиридин-4-ил]-2-пиридил]ацетамид;
N-[4-[2-оксо-6-[2-(трифторметил)-1-пиперидил]-1Н-пиридин-4-ил]-2-пиридил]циклопропанкарбоксамид;
N-[4-[2-оксо-6-[3-(трифторметил)морфолин-4-ил]-1Н-пиридин-4-ил]-2-пиридил]ацетамид;
метил-N-[4-[2-(2-хлорфенил)-6-оксо-1Н-пиридин-4-ил]-2-пиридил]карбамат;
метил-N-[4-[2-[1-этил-3-(трифторметил)пиразол-4-ил]-6-оксо-1Н-пиридин-4-ил]-2-пиридил]карбамат;
метил-N-[4-[2-оксо-6-[2-(трифторметил)-3-пиридил]-1Н-пиридин-4-ил]-2-пиридил]карбамат;
метил-N-[4-[2-оксо-6-[2-(трифторметил)фенил]-1Н-пиридин-4-ил]-2-пиридил]карбамат;
N-[4-[2-оксо-6-[2-(трифторметил)фенил]-1Н-пиридин-4-ил]-2-пиридил]ацетамид;
N-[4-[2-(4-метил-3-пиридил)-6-оксо-1Н-пиридин-4-ил]-2-пиридил]ацетамид;
N-[4-[2-оксо-6-[2-(трифторметил)-3-пиридил]-1Н-пиридин-4-ил]-2-пиридил]ацетамид;
N-[4-[2-[1-этил-3-(трифторметил)пиразол-4-ил]-6-оксо-1Н-пиридин-4-ил]-2-пиридил]ацетамид;
метил-N-[4-[2-оксо-6-[3-(трифторметил)морфолин-4-ил]-1Н-пиридин-4-ил]-2-пиридил]карбамат;
метил-N-[4-[2-оксо-6-[2-(трифторметил)-1-пиперидил]-1Н-пиридин-4-ил]-2-пиридил]карбамат;
N-[4-[2-(3-циклопропилморфолин-4-ил)-6-оксо-1Н-пиридин-4-ил]-2-пиридил]ацетамид;
N-[4-[2-[4-этилсульфонил-2-(трифторметил)пиперазин-1-ил]-6-оксо-1Н-пиридин-4-ил]-2-пиридил]ацетамид;
N-[4-[2-(2-метил-3-пиридил)-6-оксо-1Н-пиридин-4-ил]-2-пиридил]ацетамид;
N-[4-[2-оксо-6-[4-(трифторметил)-3-тиенил]-1Н-пиридин-4-ил]-2-пиридил]ацетамид;
1,1-диметил-3-[4-[2-оксо-6-[2-(трифторметил)фенил]-1Н-пиридин-4-ил]-2-пиридил]мочевину;
N-[4-[2-оксо-6-[2-(трифторметил)фенил]-1Н-пиридин-4-ил]-2-пиридил]пирролидин-1-карбоксамид;
N-[4-[2-[2-(1-метокси-1-метил-этил)пирролидин-1-ил]-6-оксо-1Н-пиридин-4-ил]-2-пиридил]ацетамид;
или его фармацевтически приемлемая соль.
16. Соединение по п. 1, где указанное соединение представляет собой
4-(2-амино-4-пиридил)-6-(3-пиридил)-1Н-пиридин-2-он;
4-(2-амино-4-пиридил)-6-(2-хлорфенил)-1Н-пиридин-2-он;
N-[4-[2-(2-хлорфенил)-6-оксо-1Н-пиридин-4-ил]-2-пиридил]-2-метокси-ацетамид;
N-[4-[2-оксо-6-[2-(трифторметил)-1-пиперидил]-1Н-пиридин-4-ил]-2-пиридил]ацетамид;
N-[4-[2-оксо-6-[2-(трифторметил)-1-пиперидил]-1Н-пиридин-4-ил]-2-пиридил]циклопропанкарбоксамид;
N-[4-[2-оксо-6-[3-(трифторметил)морфолин-4-ил]-1Н-пиридин-4-ил]-2-пиридил]ацетамид;
метил-N-[4-[2-(2-хлорфенил)-6-оксо-1Н-пиридин-4-ил]-2-пиридил]карбамат;
метил-N-[4-[2-[1-этил-3-(трифторметил)пиразол-4-ил]-6-оксо-1Н-пиридин-4-ил]-2-пиридил]карбамат;
метил-N-[4-[2-оксо-6-[2-(трифторметил)-3-пиридил]-1Н-пиридин-4-ил]-2-пиридил]карбамат;
метил-N-[4-[2-(4-метил-3-пиридил)-6-оксо-1Н-пиридин-4-ил]-2-пиридил]карбамат;
метил-N-[4-[2-оксо-6-[2-(трифторметил)фенил]-1Н-пиридин-4-ил]-2-пиридил]карбамат;
N-[4-[2-оксо-6-[2-(трифторметил)фенил]-1Н-пиридин-4-ил]-2-пиридил]ацетамид;
N-[4-[2-(4-метил-3-пиридил)-6-оксо-1Н-пиридин-4-ил]-2-пиридил]ацетамид;
N-[4-[2-оксо-6-[2-(трифторметил)-3-пиридил]-1Н-пиридин-4-ил]-2-пиридил]ацетамид;
N-[4-[2-[1-этил-3-(трифторметил)пиразол-4-ил]-6-оксо-1Н-пиридин-4-ил]-2-пиридил]ацетамид;
метил-N-[4-[2-оксо-6-[3-(трифторметил)морфолин-4-ил]-1Н-пиридин-4-ил]-2-пиридил]карбамат;
метил-N-[4-[2-оксо-6-[2-(трифторметил)-1-пиперидил]-1Н-пиридин-4-ил]-2-пиридил]карбамат;
метил-N-[4-[2-[4-этилсульфонил-2-(трифторметил)пиперазин-1-ил]-6-оксо-1Н-пиридин-4-ил]-2-пиридил]карбамат;
метил-N-[4-[2-(3-циклопропилморфолин-4-ил)-6-оксо-1Н-пиридин-4-ил]-2-пиридил]карбамат;
N-[4-[2-(3-циклопропилморфолин-4-ил)-6-оксо-1Н-пиридин-4-ил]-2-пиридил]ацетамид;
N-[4-[2-[4-этилсульфонил-2-(трифторметил)пиперазин-1-ил]-6-оксо-1Н-пиридин-4-ил]-2-пиридил]ацетамид;
3-[4-[2-(2-хлорфенил)-6-оксо-1Н-пиридин-4-ил]-2-пиридил]-1,1-диметил-мочевину;
N-[4-[2-(2-хлорфенил)-6-оксо-1Н-пиридин-4-ил]-2-пиридил]пирролидин-1-карбоксамид;
или его фармацевтически приемлемая соль.
17. Соединение по п. 1, где указанное соединение представляет собой
N-[4-[2-(2-хлорфенил)-6-оксо-1Н-пиридин-4-ил]-2-пиридил]ацетамид;
4-(2-амино-4-пиридил)-6-(3-пиридил)-1Н-пиридин-2-он;
4-(2-амино-4-пиридил)-6-(2-хлорфенил)-1Н-пиридин-2-он;
N-[4-[2-(2-хлорфенил)-6-оксо-1Н-пиридин-4-ил]-2-пиридил]-2-метокси-ацетамид;
N-[4-[2-оксо-6-[2-(трифторметил)-1-пиперидил]-1Н-пиридин-4-ил]-2-пиридил]ацетамид;
N-[4-[2-оксо-6-[2-(трифторметил)-1-пиперидил]-1Н-пиридин-4-ил]-2-пиридил]циклопропанкарбоксамид;
N-[4-[2-оксо-6-[3-(трифторметил)морфолин-4-ил]-1Н-пиридин-4-ил]-2-пиридил]ацетамид;
метил-N-[4-[2-(2-хлорфенил)-6-оксо-1Н-пиридин-4-ил]-2-пиридил]карбамат;
метил-N-[4-[2-[1-этил-3-(трифторметил)пиразол-4-ил]-6-оксо-1Н-пиридин-4-ил]-2-пиридил]карбамат;
метил-N-[4-[2-оксо-6-[2-(трифторметил)-3-пиридил]-1Н-пиридин-4-ил]-2-пиридил]карбамат;
метил-N-[4-[2-оксо-6-[2-(трифторметил)фенил]-1Н-пиридин-4-ил]-2-пиридил]карбамат;
N-[4-[2-оксо-6-[2-(трифторметил)фенил]-1Н-пиридин-4-ил]-2-пиридил]ацетамид;
N-[4-[2-(4-метил-3-пиридил)-6-оксо-1Н-пиридин-4-ил]-2-пиридил]ацетамид;
N-[4-[2-оксо-6-[2-(трифторметил)-3-пиридил]-1Н-пиридин-4-ил]-2-пиридил]ацетамид;
N-[4-[2-[1-этил-3-(трифторметил)пиразол-4-ил]-6-оксо-1Н-пиридин-4-ил]-2-пиридил]ацетамид;
метил-N-[4-[2-оксо-6-[3-(трифторметил)морфолин-4-ил]-1Н-пиридин-4-ил]-2-пиридил]карбамат;
метил-N-[4-[2-оксо-6-[2-(трифторметил)-1-пиперидил]-1Н-пиридин-4-ил]-2-пиридил]карбамат;
N-[4-[2-(3-циклопропилморфолин-4-ил)-6-оксо-1Н-пиридин-4-ил]-2-пиридил]ацетамид;
N-[4-[2-[4-этилсульфонил-2-(трифторметил)пиперазин-1-ил]-6-оксо-1Н-пиридин-4-ил]-2-пиридил]ацетамид;
или его фармацевтически приемлемая соль.
18. Применение соединения по любому из пп. 1-17 или его фармацевтически приемлемой соли для ингибирования вакуолярного сортирующего белка 34 (Vps34) при лечении или профилактике заболевания.
19. Применение соединения по любому из пп. 1-17 или его фармацевтически приемлемой соли для ингибирования Vps34 при лечения рака.
20. Применение по п. 19, где указанный рак выбран из рака молочной железы, такого как трижды негативный рак молочной железы, рака мочевого пузыря, рака печени, рака шейки матки, рака поджелудочной железы, лейкоза, лимфомы, рака почки, рака толстой кишки, глиомы, рака предстательной железы, рака яичников, меланомы и рака легкого, а также гипоксических опухолей.
21. Применение соединения по любому из пп. 1-17 или его фармацевтически приемлемой соли для приготовления лекарственного средства для ингибирования Vps34 при лечении рака.
22. Применение по п. 21, где указанный рак выбран из рака молочной железы, такого как трижды негативный рак молочной железы, рака мочевого пузыря, рака печени, рака шейки матки, рака поджелудочной железы, лейкоза, лимфомы, рака почки, рака толстой кишки, глиомы, рака предстательной железы, рака яичников, меланомы и рака легкого, а также гипоксических опухолей.
23. Способ ингибирования Vps34 при лечении рака, включающий введение терапевтически эффективного количества соединения по любому из пп. 1-17 или его фармацевтически приемлемой соли пациенту, нуждающемуся в этом.
24. Способ по п. 23, где указанный рак выбран из рака молочной железы, такого как трижды негативный рак молочной железы, рака мочевого пузыря, рака печени, рака шейки матки, рака поджелудочной железы, лейкоза, лимфомы, рака почки, рака толстой кишки, глиомы, рака предстательной железы, рака яичников, меланомы и рака легкого, а также гипоксических опухолей.
25. Фармацевтическая композиция для ингибирования Vps34, содержащая терапевтически эффективное количество соединения по любому из пп. 1-17 или его фармацевтически приемлемую соль и фармацевтически приемлемый разбавитель, носитель и/или эксципиент.
| WO 2015108861 A1, 23.07.2015 | |||
| WO 2012085815 A1, 28.06.2012 | |||
| АЗОТОСОДЕРЖАЩИЕ ПРОИЗВОДНЫЕ ГЕТЕРОАРИЛОВ | 2011 |
|
RU2559895C2 |

