Настоящее изобретение относится к способу получения акриловой кислоты гетерогенно катализируемым парциальным окислением пропана в газовой фазе, при котором содержащую пропан, молекулярный кислород и, по меньшей мере, один газ-разбавитель реакционную смесь при повышенной температуре пропускают над массой оксидов мультиметаллов общей стехиометрии
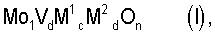
где М1=Те и/или Sb,
М2=по меньшей мере один элемент из группы, включающей Nb, Та, W, Ti, Al, Zr, Cr, Mn, Ga, Fe, Ru, Co, Rh, Ni, Pd, Pt, La, Bi, B, Ce, Sn, Zn, Si и In,
b=0,01 до 1,
с=>0 до 1,
d=>0 до 1 и
n=число, которое определяется валентностью и количеством отличных от кислорода элементов в (I),
и при этом пропан парциально окисляют в акриловую кислоту.
Способ получения акриловой кислоты, важного мономера для получения полимеров, посредством гетерогенно катализируемого парциального окисления пропана на оксидах металлов общей стехиометрии I в общем известны (см., например, DE-A 10119933, DE-A 10051419, DE-A 10046672, DE-A 10033121, ЕР-А 1090684, ЕР-А 962253, ЕР-А 895809, DE-A 19835247, ЕР-А 608838, WO 00/29105 и WO 00/29106).
Общий признак известных из уровня техники решений (см., например, ЕР-В 608838, стр.4, строки 39/40) и полученный результат заключаются в том, что применение значительного количества водяного пара в исходной реакционной смеси дает преимущество как относительно активности катализатора, так и селективности образования акриловой кислоты.
Недостатком этого известного из уровня техники приема является, однако, то, что при окислительном, катализируемом получении акриловой кислоты образуется не чистая акриловая кислота, а газовая смесь, от которой акриловая кислота должна отделяться.
Обычно газовая смесь, полученная гетерогенно катализируемым парофазным парциальным окислением пропана с получением акриловой кислоты, содержит наряду с непрореагировавшим пропаном такие побочные компоненты, как пропен, акролеин, СО2, СО, уксусная кислота и пропионовая кислота, а также применяемые в качестве газов-разбавителей компоненты, такие, как например, водяной пар, от которых акриловая кислота должна отделяться, чтобы получать чистый продукт.
Вследствие выраженного сродства акриловой кислоты с водой (при применяемых для детских пеленок суперабсорберах речь идет о полимере на базе акриловой кислоты) вышеупомянутая задача отделения тем сложнее, чем больше водяного пара применяют при парофазном окислении в качестве газа-разбавителя.
Присутствие повышенных количеств водяного пара способствует, кроме того, образованию пропионовой кислоты в качестве побочного продукта, который вследствие своего сходства с акриловой кислотой с трудом может от нее отделяться.
Поэтому предпочтительным являлся бы способ гетерогенно катализируемого парциального окисления пропана с получением акриловой кислоты, который или совсем не требует, или требует только малого количества водяного пара и при котором активность катализатора и селективность образования акриловой кислоты все-таки удовлетворительны.
В соответствие с этим был разработан способ получения акриловой кислоты гетерогенно катализируемым парциальным окислением пропана в газовой фазе, при котором реакционную газовую смесь, содержащую пропан, молекулярный кислород и, по меньшей мере, один газ-разбавитель, при повышенной температуре пропускают над массой оксидов мультиметаллов общей стехиометрии I
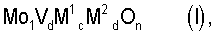
где М1=Те и/или Sb,
М2=по меньшей мере один элемент из группы, включающей Nb, Та, W, Ti, AI, Zr, Cr, Mn, Ga, Fe, Ru, Co, Rh, Ni, Pd, Pt, La, Bi, B, Ce, Sn, Zn, Si и In,
b=0,01 до 1,
c=>0 до 1,
d=>0 до 1 и
n=число, которое определяется валентностью и количеством отличного от кислорода элемента в (I),
и при этом парциально окисляют пропан с получением акриловой кислоты, причем этот способ отличается тем, что состав исходной реакционной газовой смеси во время проведения процесса, по меньшей мере, один раз изменяют таким образом, что содержащаяся до изменения в исходной реакционной газовой смеси молярная доля газа-разбавителя, водяного пара, в пересчете на содержащееся в исходной реакционной газовой смеси молярное количество пропана, после изменения меньше.
Основой способа по изобретению является тот неожиданно установленный факт, что оксиды металлов стехиометрии I в значительном объеме сохраняет свою способность образовывать акриловую кислоту при каталитическом окислении пропана в газовой фазе в присутствии повышенного количества водяного пара в качестве газа-разбавителя с высокой активностью и селективностью, если в исходной реакционной газовой смеси в течение определенного времени применяют определенную долю водяного пара, в пересчете на содержащийся в исходной реакционной газовой смеси пропан, и эту долю потом снижают. Т.е. также и после произведенного снижения доли водяного пара в исходной реакционной газовой смеси гетерогенно катализируемое окисление в газовой фазе пропана в остальном проходит при неизменных условиях с более высокой селективностью образования акриловой кислоты, чем если бы гетерогенно катализируемое окисление в газовой фазе осуществлялось постоянно с меньшим количеством водяного пара в исходной реакционной газовой смеси. Это имеет место также и тогда, когда снижение доли водяного пара производят до такой степени, что реакционная смесь после осуществленного снижения больше не содержит водяного пара.
Обычно при способе по изобретению снижение доли водяного пара, по меньшей мере, частично компенсируют повышением доли отличных от водяного пара, пригодных для применения при способе по изобретению газов-разбавителей. Компенсацию, однако, можно и не производить. Такими другими газами-разбавителями являются, в частности, молекулярный кислород, окиси углерода, такие, как СО и СО2, а также благородные газы, такие, как He или Ar. Сам пропан может быть газом-разбавителем. В этом случае при способе по изобретению пропан применяют в сверхстехиометрическом количестве, в пересчете на применяемое количество молекулярного кислорода.
Обычно применяются смеси вышеприведенных газов-разбавителей. Они обычно действуют как инертные газы. Это означает то, что при проведении способа по изобретению они химически в основном не изменяются.
При способе по изобретению можно полностью компенсировать или даже сверхкомпенсировать молярное снижение доли водяного пара посредством эквимолярного повышения молярной доли отличных от водяного пара инертных газов-разбавителей. Целесообразным образом при способе по изобретению в качестве замены для водяного пара применяют молекулярный азот.
Как правило, способ по изобретению проводят при температуре, например, от 200 до 550°С или от 230 до 480°С, соответственно от 300 до 440°С. При способе по изобретению температура перед и после снижения доли водяного пара может быть одинаковой. Она может быть ниже или выше после снижения доли водяного пара, чем до снижения доли водяного пара.
Рабочее давление (абсолютное) при способе по изобретению может составлять как 1 атм, так и менее чем 1 атм или более чем 1 атм. Типичным рабочим давлением при способе по изобретению является от 1,5 до 10 бар, часто от 1,5 до 4 бар. Рабочее давление во время проведения процесса может быть как константным, так и изменяющимся. Это означает то, что рабочее давление перед снижением доли водяного пара может быть выше или ниже, чем после понижения.
В рамках способа по изобретению требуемое изменение состава исходной реакционной газовой смеси может многократно осуществляться периодически последовательно. Это означает то, что после определенной продолжительности работы при сниженной доле водяного пара доля водяного пара в реакционной смеси может определенное время снова повышаться и после этого снова снижаться и т.д.
В качестве источника кислорода для способа по изобретению может применяться как чистый кислород, так и воздух или обогащенный кислородом, соответственно бедный кислородом воздух.
К применяемому для способа по изобретению пропану не предъявляются никакие особенно высокие требования относительно его чистоты. Может применяться также и пропан, содержащий пропен в качестве сопровождающего газа. В типичном случае состав исходной реакционной газовой смеси для способа по изобретению является следующий (молярные соотношения):
пропан : кислород : H2O : прочие газы-разбавители=1:(0,1-10):(0-50):(0-50).
Предпочтительно такое соотношение составляет 1:(0,5-5):(1-30):(0-30).
Это соотношение согласно изобретению лежит перед снижением доли водяного пара в диапазоне А=1:(0,1-10):(0,1-50):(0-50), и после снижения доли водяного пара в диапазоне В=1:(0,1-10):(0-30):(0-30).
Согласно изобретению диапазон А включает молярные соотношения 1:(0,55):(2-30):(0-30) и диапазон В молярные соотношения 1:(0,5-5):(0-20):(0-30).
Приведенные диапазоны действительны, в частности, тогда, когда в качестве прочих газов-разбавителей применяют в основном молекулярный кислород. Во время применения диапазона А для исходной реакционной газовой смеси температура реакции составляет целесообразным образом от 250 до 550°С и во время применения диапазона В для исходной реакционной газовой смеси температура реакции составляет также от 250 до 550°С.
Способ по изобретению может быть осуществлен описанным в уровне техники образом, т.е. катализаторный слой может быть как неподвижным, так и движущимся или псевдоожиженным.
При этом применяемые в способе по изобретению оксиды металлов I могут применяться как таковые (например, размельченные в порошок или щебень) или же в виде формованных тел.
Получение пригодных для способа по изобретению оксидов металлов I может осуществляться известным из вышеприведенного уровня техники методом. В зависимости от выбранного способа получения структура получаемых оксидов металлов может быть скорее аморфной (например, как описано в WO 00/29105 или в WO 00/29106) или кристаллической (как, например, описано в ЕР-А 608838, ЕР-А 962253, ЕР-А 895809 и DE-A 19835247).
Получение, как правило, осуществляется таким образом, что при атмосферном давлении (1 атм) из источников исходных элементарных химических элементов для оксидов металлов получают по возможности тщательно перемешанную, предпочтительно тонкую сухую массу и ее затем термической обработкой в окисляющей (например, воздух), восстанавливающей, инертной (например, N2) или имеющей пониженное давление атмосфере переводят в активный оксид. Само собой разумеется для способа по изобретению могут применяться полученные гидротермическим путем оксиды металлов I, описанным в DE-A 10033121.
Формование оксидов металлов I в пригодные для способа по изобретению тела может осуществляться, например, как описано в DE-A 10118814 и в DE-A 10119933.
В качестве оксидов металлов I предпочтительно применяют такие, при которых М1=Те. Далее для способа по изобретению пригодны такие массы оксидов металлов I, при которых М2=Nb, Та, W и/или титан. Предпочтительно М2=Nb. Стехиометрический коэффициент b применяемых согласно изобретению активных оксидов металлов I составляет предпочтительно 0,1 до 0,6. Соответствующим образом предпочтительный диапазон для стехиометрического коэффициента с составляет от 0,01 до 1, соответственно от 0,05 до 0,4, и пригодные значения для коэффициента d составляют от 0,01 до 1, соответственно 0,1 до 0,6. Особенно пригодными, применяемыми согласно изобретению активными оксидами металлов являются такие, при которых стехиометрические коэффициенты b, с и d лежат в вышеприведенных пределах. Другими пригодными стехиометрическими значениями являются описанные в вышеприведенных в качестве уровня техники публикациях.
Далее для способа по изобретению предпочтительно применяются такие оксиды металлов I, рентгеновские дифрактограммы которых имеют дифракционные отражения h и i, пики которых лежат при углах дифракции (2Θ) 22,2±0,50 (h) и 27,3±0,50 (i). Полуширина пика этих дифракционных отражений может при этом быть очень малой или же очень четко выраженной.
Особенно предпочтительны для способа по изобретению оксиды металлов I, рентгеновская дифрактограмма которых имеет дополнительно к дифракционным отражениям h и i дифракционное отражение k, пик которого составляет 28,2±0,50 (k). Среди таких оксидов металлов предпочтительны согласно изобретению такие, при которых дифракционное отражение h на рентгеновской дифрактограмме является самым интенсивным, а также имеет полуширину пика максимально 0,5, полуширина дифракционного отражения i и дифракционного отражения k составляет одновременно ≤1°, интенсивность Pk дифракционного отражения k и интенсивность Рi дифракционного отражения i удовлетворяют условию 0,65≤R≤0,85, в котором R представляет собой определяемое формулой
R=Pi/(Pi+Pk)
соотношение интенсивностей дифракционных отражений. Предпочтительно рентгеновская дифрактограмма этих оксидов металлов I не имеет дифракционных отражений, максимум которых лежит при 2Θ=50±0,3.
Среди этих оксидов металлов I для способа по изобретению предпочтительны такие, при которых 0,67≤R≤0,75 и особенно предпочтительны такие, при которых R=0,70 до 0,75, соответственно R=0,72.
Получение оксидов металлов I, которые имеют дифракционное отражение i и дифракционное отражение k с полушириной пика ≤1 и которые одновременно удовлетворяют соотношению 0,65≤R≤0,85 и в случае необходимости не имеют дифракционного отражения, максимум которого составляет 2Θ=50±0,3, может осуществляться, как описано в DE-A 10118814 и DE-A 10119933.
При этом речь идет об оксидах металлов I, которые относительно своего дифракционного отражения i содержат так называемую i-фазу. Другой кристаллической фазой, в которой могут иметься оксиды металлов I, является, например, так называемая k-фаза. Она характерна наличием дифракционных отражений h и k и наличием дифракционного отражения с максимумом при 50±0,3°.
Способ получения применяемых согласно изобретению оксидов металлов I, в которых доминирует i-фаза, описывается, например, в публикациях JP-A 11-43314, DE-A 10118814, DE-A 10119933 и в более ранней заявке DE-A 10046672, в которых оксиды металлов I рекомендуются в качестве катализаторов для гетерогенно катализируемого гидрирования этана с получением этилена, а также в качестве катализаторов для гетерогенно катализируемого газофазного окисления пропана, соответственно пропена с получением акриловой кислоты.
Согласно этим известным способам, раскрытым также и во многих публикациях, (например, в более ранней заявке DE-A 10033121) сначала получают массу оксидов металлов стехиометрии (I), которая представляет собой смесь из i-фазы и других фаз (например, k-фазы). В этой смеси доля i-фазы может быть повышена за счет того, что другие фазы, например k-фазу, отбирают под микроскопом или активную оксиды металлов промывают пригодными жидкостями. Такой жидкостью могут быть, например, водные растворы органических кислот (например, щавелевой кислоты, муравьиной кислоты, уксусной кислоты, лимонной и винной кислот), неорганических кислот (например, азотной кислоты), спирты и водные растворы перекиси водорода. Далее также и в JP-A 7-232071 описывается способ получения применяемых согласно изобретению оксидов металлов I с четко выраженной долей i-фазы.
Менее систематичным образом применяемые согласно изобретению оксиды металлов I с четко выраженной долей i-фаз могут получаться описанным в DE-A 19835247 образом. Согласно этому решению от подходящего источника их элементарных структур получают по возможности тщательно перемешанную, предпочтительно тонкую сухую смесь и ее термически обрабатывают при температуре от 350 до 700°С, соответственно от 400 до 650°С или от 400 до 600°С. Термическую обработку можно в принципе осуществлять как в окисляющей, восстанавливающей, так и в инертной атмосфере. В качестве окисляющей атмосферы пригодны, например, воздух, обогащенный молекулярным кислородом воздух или обедненный кислородом воздух. Предпочтительно термическую обработку осуществляют в инертной атмосфере, т.е. в атмосфере молекулярного азота и/или благородного газа. Обычно термическая обработка происходит при атмосферном давлении (1 атм). Само собой разумеется термическая обработка может проходить под вакуумом или при повышенном давлении.
Если термическая обработка осуществляется в газовой атмосфере, то последняя может быть неподвижной или текучей. В общем термическая обработка может проходить до 24 часов или более.
Предпочтительно термическая обработка осуществляется сначала в окисляющей (содержащей кислород) атмосфере (например, в атмосфере воздуха) при температуре от 150 до 400°С, соответственно от 250 до 350°С. В заключение термическая обработка продолжается в атмосфере инертного газа при температуре от 350 до 700°С, соответственно от 400 до 650°С или от 400 до 600°С. Само собой разумеется термическая обработка может происходить таким образом, что масса предшественника катализатора перед ее термической обработкой сначала (в случае необходимости после порошкования) таблетируется (в случае необходимости при добавке от 0,5 до 2 мас.% тонкого графита), потом происходит термическая обработка и затем масса снова раскалывается.
Тщательное перемешивание исходных соединений в рамках получения применяемых согласно изобретению оксидов металлов I может происходит в сухой или влажной форме.
Если это происходит в сухой форме, то исходные соединения применяются в качестве тонкого порошка и после смешения и в случае необходимости уплотнения подвергаются кальцинированию (термической обработке).
Предпочтительно тщательное смешение во влажной форме. Обычно исходные соединения при этом смешиваются друг с другом в форме водного раствора и/или суспензии. После этого водную массу сушат и после сушки кальцинируют. Целесообразным образом при водной массе речь идет о водном растворе или водной суспензии. Предпочтительно процесс сушки проходит непосредственно после процесса получения водной смеси и посредством распылительной сушки (выходная температура составляет, как правило, от 100 до 150°С; распылительная сушка может проходить как прямотоком, так и пртивотоком), которая обуславливает особенно тщательное сухое перемешивание, прежде всего тогда, когда при подлежащей распылительной сушке водной массе речь идет о водном растворе.
В качестве источников исходных элементов в рамках получения применяемых согласно изобретению оксидов металлов пригодны все те, которые при нагревании (в случае необходимости на воздухе) способны образовывать оксиды и/или гидроксиды. Само собой разумеется в качестве таких исходных соединений могут также использоваться уже оксиды и/или гидроксиды исходных элементов или же исключительно такие.
Согласно изобретению подходящим источником для элемента Mo являются, например, оксиды молибдена, такие как триокись молибдена, молибдаты, такие, как гептамолибдаттетрагидрат аммония и галогениды молибдена, такие, как молибденхлорид.
Пригодными, применяемыми согласно изобретению исходными соединениями для элемента V являются, например, ванадилацетилацетонат, ванадаты, такие, как метаванадат аммония, ванадиноксиды, такие, как ванадинпероксид (V2O5), ванадингалогениды, такие, как ванадинтетрахлорид (VCl4) и ванадиноксигалогениды, такие, как VOCl3. При этом в качестве исходных соединений ванадия могут также применяться такие, которые содержат ванадий в стадии окисления +4.
В качестве источника элемента теллур пригодны согласно изобретению оксиды теллура, такие, как диоксид теллура, металлический теллур, галогениды теллура, такие, как TeCl2, а также теллуровые кислоты, такие, как ортотеллуровая кислота H6TeO6.
Предпочтительными исходными соединениями сурьмы являются галогениды сурьмы, такие, как SbCl3, оксида сурьмы, такие, как триоксид сурьмы (Sb2О3), сурьмяная кислота, такая, как HSb(OH)6, а также оксидные соли сурьмы, такие, как оксидный сульфат сурьмы (SbO)2SO4.
Пригодными согласно изобретению источниками ниобия являются, например, оксиды ниобия, такие, как пентоксид ниобия (Nb2O5), оксидгалогениды ниобия, такие, как NbOCl3, галогениды ниобия, такие, как NbCl5, а также комплексные соединения из ниобия и органических карбоновых кислот и/или дикарбоновых кислот, как, например, оксалаты и алкоголяты. Само собой разумеется в качестве источников ниобия могут быть применяемые в ЕР-А 895809, содержащие Nb растворы.
Относительно всех других элементов М2 пригодны в качестве исходных соединений согласно изобретению прежде всего их галогениды, нитраты, формиаты, оксалаты, ацетаты, карбонаты и/или гидроксиды. Пригодными исходными соединениями являются также их оксисоединения, например вольфраматы, соответственно, отводимые от них кислоты. Часто в качестве исходных соединений применяются также и аммониевые соли.
Далее в качестве исходных соединений для получения масс оксидов металлов I пригодны также полианионы типа Андерсона, которые описаны, например, в публикации Polyhedron, Vol.6, No. 2, стр.213-218, 1987. Другим литературным источником для полианионов типа Андерсона может служить Kinetics and Catalysis, Vol.40, No. 3, 1999, стр.401-404.
Другие, пригодные в качестве исходных соединений полианионы являются полианионы типа Dawson или Keggin. Предпочтительно согласно изобретению применяются такие исходные соединения, которые при повышенной температуре или в присутствии кислорода или при исключении кислорода, в случае необходимости при высвобождении газообразных соединений, превращаются в свои оксиды.
Получаемые, как описано выше, применяемые согласно изобретению оксиды металлов I могут применяться для способа по изобретению как таковые [например, в качестве порошка или после таблетировния порошка (часто при добавке тонкого графита в количестве от 0,5 до 2 мас.%) и последующего раскалывания в щебень] или же в виде формованных тел. При этом формование может осуществляться, например, нанесением на носитель, как это описано в более ранней заявке DE-A 10051419.
Применяемые согласно изобретению для оксидов металлов I носители являются предпочтительно химически инертными. Это означает то, что они в основном не принимают участие в процессе каталитического газофазного окисления пропана в акриловую кислоту, катализируемого оксидами металлов по изобретению.
В качестве материала для носителей пригодны согласно изобретению, в частности, оксид алюминия, диоксид кремния, такие силикаты, как глина, каолин, стеатит, пемза, силикат алюминия и силикат магния, карбид кремния, диоксид циркония и диоксид тория.
Поверхность носителя может быть как гладкой, так и шероховатой. С преимуществом применяют шероховатую поверхность, так как повышенная поверхностная шероховатость, как правило, обуславливает повышенную адгезионную прочность нанесенного покрытия активной массы.
Часто шероховатость поверхности Rz носителя лежит в пределах от 5 до 200 мм, особенно часто в диапазоне от 20 до 100 мм (определено согласно стандарту Германии DIN 4768 лист 1 тестером "Hommel" для измерительных величин поверхностей по стандартам DIN-ISO фирмы Hommelwerke, DE).
Далее материал носителя может быть пористым или непористым. Целесообразным образом материал носителя непористый (общий объем пор в пересчете на объем носителя ≤1 об.%).
Толщина находящегося на носителе покрытия активной оксидной массы составляет обычно от 10 до 1000 мм. Она может также составлять от 50 до 700 мм, от 100 до 600 мм или от 150 до 400 мм. Возможная толщина покрытия составляет также от 10 до 500 мм, от 100 до 500 мм или от 150 до 300 мм.
В принципе для способа по изобретению пригодна любая геометрия носителя. Их продольная длина составляет, как правило, от 1 до 10 мм. Предпочтительно в качестве носителей применяются шарики или цилиндры, в частности полые цилиндры. Подходящими диаметрами для шариковых носителей являются диаметры от 1,5 до 4 мм. Если применяют в качестве носителей цилиндры, то их длина составляет предпочтительно от 2 до 10 мм и их внешний диаметр составляет предпочтительно от 4 до 10 мм. В том случае, если применяют кольцеобразные носители, толщина их стенок обычно составляет от 1 до 4 мм. Пригодные согласно изобретению кольцеобразные носители могут также иметь длину от 3 до 6 мм, внешний диаметр от 4 до 8 мм и толщину стенок от 1 до 2 мм. Возможна также геометрия носителя с размерами 7 мм × 3 мм × 4 мм или 5 мм × 3 мм × 2 мм (внешний диаметр × длина × внутренний диаметр).
Получение применяемых согласно изобретению катализаторов может осуществляться таким образом, что образуют оксидную массу общей формулы (I), ее переводят в тонкоизмельченную форму и затем с помощью жидкого связующего наносят на поверхность носителя. Для этого поверхность носителя увлажняют с помощью жидкого связующего и приведением в контакт с тонкоизмельченной активной массой общей формулы (I) на увлажненной поверхности укрепляют слой активной массы. После этого носитель с нанесенным влажным слоем подвергают сушке. Затем для получения повышенной плотности слоя процесс периодически повторяют. В этом случае носитель с нанесенным слоем становится новым "носителем" и т.д.
Само собой разумеется, что тонкость подлежащей нанесению на поверхность носителя каталитически активной оксидной массы общей формулы (I) подгоняется к желаемой толщине покрытия. Для диапазона толщин покрытия от 100 до 500 мкм пригодны, например, такие порошки активной массы, из которых, по меньшей мере, 50% общего числа порошковых частичек просеивается через сито размером отверстий от 1 до 20 мкм. При этом числовая доля частиц с продольным размером свыше 50 мкм составляет менее чем 10%. Как правило, распределение продольных размеров порошковых частиц соответствует распределению Гаусса. Часто распределение размеров частиц имеет следующую характеристику:
При этом
D=диаметр частицы,
х=процентная доля частиц, диаметр которых ≥D, и
у=процентная доля частиц, диаметр которых <D.
Для проведения описанного способа нанесения покрытия в технических масштабах рекомендуется, например, применение описанных в DE-A 2909671, а также в DE-A 10051419 методов. Согласно этим методам подлежащие покрытию носители загружают в наклонную (угол наклона составляет, как правило, ≥0° и ≤90°, в большинстве случаев ≥30° и ≤90°, углом наклона при этом является угол средней оси вращающейся емкости относительно горизонтали) вращающуюся емкость (например, вращающуюся тарелку дражировочного барабана). Во вращающейся емкости носители, например шарообразные или цилиндрические, подводятся к двум последовательно расположенным на определенном расстоянии друг от друга дозирующим устройствам. Первое из обоих дозирующих устройств целесообразным образом соответствует соплу (например, приводимому сжатым воздухом распылительному соплу), посредством которого перемещающиеся во вращающейся емкости носители опрыскиваются жидким связующим и контролировано увлажняются. Второе дозирующее устройство находится вне конуса опрыскивания жидкого связующего и служит для подвода тонкой активной оксидной массы (например, через качающийся лоток или шнек). Контролировано увлажненные носители воспринимают подаваемый порошок активной массы, который вследствие движения уплотняется на внешней поверхности носителя в сплошное покрытие.
При необходимости снабженный таким образом основным слоем носитель в течение последующего вращения снова подпадает под распылительное сопло, для контролированного увлажнения, и при дальнейшем перемещении наносится другой слой тонкой активной оксидной массы и т.д. (промежуточная сушка, как правило, не требуется). Тонкая активная оксидная масса и связующее при этом, как правило, подаются непрерывно и синхронно.
Удаление жидкого связующего может осуществляться после окончания нанесения покрытия, например, путем воздействия горячих газов, таких, как N2 или воздух. Следует заметить, что описанный способ нанесения покрытия обеспечивает полностью удовлетворительную адгезию как следующих друг за другом слоев, так и основного слоя на поверхности носителя.
Существенным для вышеописанного способа нанесения покрытия является то, что увлажнение подлежащей нанесению покрытия поверхности носителя производится контролировано. Короче говоря, поверхность носителя увлажняют таким образом, что хотя она адсорбирует жидкое связующее, однако, на поверхности носителя не имеется определяемая визуально жидкая фаза. Если поверхность носителя слишком влажная, то тонкая каталитически активная оксидная масса агломерирует с образованием отдельных агломератов, вместо того чтобы равномерно наноситься на поверхность. Подробные сведения об этом можно найти в DE-A 2909671 и DE-A 10051419.
Вышеупомянутое заключительное удаление применяемого жидкого связующего может осуществляться контролировано, например, испарением и/или сублимацией. В самом простом случае это может происходить воздействием горячего газа соответствующей температуры (например, от 50 до 300°С, часто 150°С). Воздействием горячего газа можно обеспечить только предварительную сушку. Окончательная сушка может происходить, например, в сушильной печи любого типа (например, в конвейерной сушилке) или в реакторе. Воздействующая температура при этом не должна превышать применяемой для получения активной оксидной массы температуры кальцинирования. Само собой разумеется, что сушка может быть производиться исключительно в сушильной печи.
В качестве связующего для процесса нанесения покрытия могут применяться независимо от формы носителя следующие вещества: вода, одноатомные спирты, такие, как этанол, метанол, пропанол и бутанол, многоатомные спирты, такие, как этиленгликоль, 1,4-бутандиол, 1,6-гександиол или глицерин, одно- или многовалентные органические карбоновые кислоты, такие, как пропионовая кислота, щавелевая кислота, малоновая кислота, глутаровая кислота или малеиновая кислота, аминоспирты, такие, как этаноламин или диэтаноламин, а также одно- или многовалентные органические амиды, такие, как формамид. Пригодными связующими являются также и растворы, содержащие от 20 до 90 мас.% воды и от 10 до 80 мас.% растворенного в воде органического соединения, точка кипения которого или температура сублимации которого при атмосферном давлении (1 атм) составляет >100°С, предпочтительно >150°С. С успехом выбирается органическое соединение из вышеприведенного перечня возможных органических связующих. Органическая доля вышеприведенных водных растворов связующего составляет предпочтительно от 10 до 50 мас.% и особенно предпочтительно от 20 до 30 мас.%. При этом в качестве органических компонентов пригодны также и моносахариды и олигосахариды, такие, как глюкоза, фруктоза, сахароза или лактоза, а также полиэтиленоксиды и полиакрилаты.
Получение пригодных согласно изобретению катализаторов на носителе может осуществляться не только способом нанесения тонкоизмельченной активной оксидной массы общей формулы (I) на увлажненную поверхность носителя. Вместо активной оксидной массы на увлажненную поверхность носителя может также наноситься ее тонкоизмельченная масса-предшественник (с применением того же способа нанесения слоев связующего) и затем кальцинироваться после сушки покрытого слоями носителя.
В качестве такой массы-предшественника пригодна такая масса, которая может быть получена таким образом, что из источников составных частей желаемой активной оксидной массы общей формулы (I) сначала получают по возможности тщательно перемешанную, предпочтительно тонкую сухую смесь (например, распылительной сушкой водной суспензии или раствора источников) и эту тонкую сухую смесь (в случае необходимости после таблетирования с добавкой от 0,5 до 2 мас.% тонкого графита) при температуре от 150 до 350°С, предпочтительно от 250 до 350°С, термически обрабатывают (несколько часов) под окислительной (содержащей кислород) атмосферой (например, под воздухом) и затем при необходимости подвергают помолу.
После нанесения на носитель массы-предшественника проводят кальцинирование предпочтительно в атмосфере инертного газа (также возможны все другие атмосферы) при температуре от 360 до 700°С, соответственно от 400 до 650°С или от 400 до 600°С.
Само собой разумеется, что формование применяемых согласно изобретению оксидов металлов формулы I может осуществляться также и экструзией и/или таблетированием как тонкой массы оксидов металлов I, так и тонкой массы-предшественника оксидов металлов I.
Геометрическая форма носителей может быть при этом шаровидной, цилиндрической или может представлять собой полые цилиндры (кольца). Продольный размер названных форм составляет при этом, как правило, от 1 до 10 мм. В случае цилиндров длина составляет предпочтительно от 2 до 10 мм, а их внешний диаметр составляет предпочтительно от 4 до 10 мм. В случае колец толщина стенок составляет обычно от 1 до 4 мм. Пригодные согласно изобретению кольцеобразные сплошные катализаторы, т.е. катализаторы без носителя, могут иметь длину от 3 до 6 мм, внешний диаметр от 4 до 8 мм и толщину стенок от 1 до 2 мм. Также возможны сплошные катализаторы с размерами 7 мм × 3 мм × 4 мм или 5 мм × 3 мм × 2 мм (внешний диаметр × длина × внутренний диаметр).
Само собой разумеется, что для осуществления способа по изобретению пригодны все формы оксидов металлов I, приведенные в DE-A 10101695.
Существенным для изобретения является то, что предпочтительно применяемые согласно изобретению оксиды металлов I имеют рентгеновскую дифрактограмму (в настоящей заявке в отношении Cu-Kα-излучения), которая имеет дифракционные отражения h, i и k, пики которых лежат в углах дифракции (20) 22,2±0,4° (h), 27,3±0,4° (i) и 28,2±0,4° (k), причем
- отражение дифракции h внутри рентгеновской дифрактограммы является самым интенсивным и имеет полуширину пика максимально 0,5°,
- интенсивность Рi дифракционного отражения i и интенсивность Рk дифракционного отражения k удовлетворяют условию 0,65≤0,85, где R является определяемым формулой
R=Рi/(Рi+Pk)
соотношением интенсивностей, и
- полуширина пика дифракционного отражения i и дифракционного отражения k составляет <1°.
Определение интенсивности дифракционного отражения на рентгеновской дифракторгамме соответствует приведенному в DE-A 19835247, а также в DE-A 10051419 и DE-A 10046672 определению.
Это означает то, что если А1 обозначает пик дифакционного отражения 1 и В1 обозначает на линии рентгеновской дифрактограммы при рассмотрении вдоль вертикальной к оси 2Θ оси интенсивности ближайший выраженный минимум (свидетельствующие о минимумах плечи отражения не учитываются) слева от пика А1 и В2 обозначает соответствующим образом ближайший выраженный минимум справа от пика А1 и С1 обозначает точку, в которой проходящая от пика А1 вертикально к оси 2Θ прямая пересекает соединяющую точки В1 и В2 прямую, то интенсивность дифракционного отражения 1 является длиной участка прямой А1C1, который проходит от пика А1 к точке С1. Термин минимум при этом означает точку, в которой градиент наклона прилегающей к кривой в базовой зоне отражения 1 касательной переходит с отрицательного значения в положительное значение, или означает точку, в которой градиент наклона подходит к нулю, причем для установки градиента наклона берутся координаты оси 2Θ и оси интенсивности.
Полушириной пика является в упомянутых публикациях соответствующим образом длина участка прямой, который имеется между обеими точками пересечения Н1 и Н2, если в середине участка прямой А1С1 проходит параллельная к оси 2Θ линия, причем Н1, Н2 представляют собой каждая первую точку пересечения этой параллельной линии с вышеопределенной линией рентгеновской дифрактограммы слева и справа от А1.
Пример определения полуширины пика и интенсивности показывает фиг.6 заявки DE-A 10046672.
Наряду с дифракционными отражениями h, i и k рентгеновская дифрактограмма предпочтительных применяемых согласно изобретению оксидов металлов формулы I содержит, как правило, еще другие дифракционные отражения, пики которых лежат при нижеприведенных углах дифракции (2Θ):
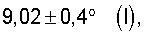
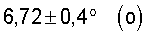 и
и
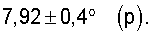
Преимущественным является то, что рентгеновская дифрактограмма каталитически активной оксидной массы общей формулы (1) содержит дополнительно дифракционное отражение, пик которого лежит при следующих углах дифракции (2Θ):
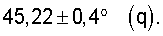
Часто рентгеновская дифрактограмма оксидов металлов формулы (1) содержит также и дифракционные отражения 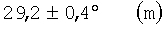 и
и 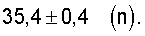
Если каталитически активная оксидная масса общей формулы (I) содержит k-фазу, то ее рентгеновская дифракторгамма, как правило, содержит еще другие дифракционные отражения, пик которых лежит при углах (2Θ):
36,2±0,4° и
50,0±0,4°.
Если дифракционному отражению h придают интенсивность 100, то согласно изобретению преимущественно, если дифракционные отражения i, I, m, n, о, р, q по той же шкале интенсивности имеют следующие значения:
Если рентгеновская дифракторграмма содержит вышеприведенные дифракционные отражения, то их полуширина пика, как правило, ≤10.
Все относящиеся к рентгеновской дифрактограмме данные относятся к полученной при применении Cu-Kα-излучения в качестве рентгеновского излучения дифракторгамме (дифрактометр фирмы Siemens Theta-Theta D-5000, напряжение 40 кВт, ток 40 мА, апертурная диафрагма V20 (варьируемая), диафрагма рассеянного излучения V20 (варьируемая), вторичная монохроматичная диафрагма (0,1 мм), детекторная диафрагма (0,6 мм), измерительный интервал (2Θ) 0,020, время измерения за одну стадию 2,4 сек, детектор: сцинтилляционная счетная труба).
Удельная поверхность применяемых согласно изобретению оксидов металлов (I) часто составляет от 1 до 30 м2/г (поверхность по БЭТ, азот).
При осуществлении способа по изобретению свежеизготовленные катализаторы сначала подвергают воздействию смеси исходного реакционного газа, которая имеет повышенную долю водяного пара в пересчете на содержащийся пропан (т.е. находится в области состава А).
Если способ по изобретению проводится в неподвижном слое, то целесообразно его осуществляют в кожухотрубном реакторе, контактные трубы которого загружены катализатором. Вокруг контактных труб в нормальном случае пропускают в качестве теплоносителя жидкость, как правило, солевую ванну.
Рассматривая по реактору, реакционная газовая смесь пропускается через контактные трубы прямотоком или противотоком к солевой ванне. Поток солевой ванны может быть чисто параллельным по отношению к контактным трубам. Само собой разумеется, что на него может быть наложен поперечный поток. В общем поток солевой ванны вокруг контактных труб может иметь формы меандра. При этом, рассматривая по реактору, солевая ванна может направляться прямотоком или противотоком к реакционной газовой смеси. Пригодные для способа по изобретению кожухотрубные реакторы описаны, например, в ЕР-А 700714 и ЕР-А 700893. Нагрузка слоя катализатора пропаном может при способе по изобретению составлять, например, от 10 до 500 нл/л·ч. Нагрузка реакционной газовой смесью часто составляет от 100 до 10000 нл/л·ч, часто от 500 до 5000 нл/л·ч.
Исходная реакционная газовая смесь при способе по изобретению может подаваться в содержащую катализатор реакционную зону предварительно нагретой до температуры реакции.
Само собой разумеется, что способом по изобретению получают газовую смесь продукта, которая не состоит исключительно из акриловой кислоты. Она содержит наряду с непрореагировавшим пропаном побочные компоненты, такие, как пропен, акролеин, CO2, CO, Н2О, уксусную кислоту, пропионовую кислоту и т.п., от которых акриловая кислота должна отделяться.
В связи с тем, что способ по изобретению позволяет ограничивать как содержащееся в газовой смеси продукта количество водяного пара, так и образование побочных продуктов, а именно уксусной кислоты и пропионовой кислоты, то отделение акриловой кислоты может осуществляться тем же образом, что и при гетерогенно катализируемом газофазном окислении пропена в акриловую кислоту.
Т.е. из газовой смеси продукта акриловая кислота может выделяться, например, абсорбцией с помощью высококипящего инертного гидрофобного органического растворителя (например, смеси из простого дифенилового эфира и дифенила, которая в случае необходимости может еще содержать добавки, такие, как диметилфталат). Результирующая при этом газовая смесь из абсорбента и акриловой кислоты может обрабатываться известным образом ректификацией, экстракцией и/или кристаллизацией до получения чистой акриловой кислоты. Альтернативно отделение акриловой кислоты от реакционной газовой смеси может происходить также и фракционированной конденсацией, как это описано в DE-A 19924532.
Получаемый при этом конденсат акриловой кислоты может очищаться далее, например, фракционированной кристаллизацией (например, кристаллизацией в суспензии и/или слоистой кристаллизацией).
Остающаяся при основном отделении акриловой кислоты остаточная газовая смесь содержит, в частности, прореагировавший пропан и, в случае необходимости, непрореагировавший пропен. Он может отделяться от остаточной газовой смеси, например, посредством фракционированной ректификации под давлением и затем возвращаться на газофазное окисление согласно изобретению. Однако более выгодно контактирование в экстракционном устройстве остаточного газа с гидрофобным органическим растворителем (например, путем пропускания через него), который способен предпочтительно абсорбировать пропан и, в случае необходимости, пропен.
Последующей десорбцией и/или стриппингом воздухом абсорбированный пропан и, в случае необходимости, абсорбированный пропен может снова высвобождаться и возвращаться в процесс по изобретению. Таким образом обеспечивается экономичная конверсия всего пропана. Акриловая кислота может, однако, также отделяться от газовой смеси продукта описанным в DE-A 10059122 способом.
Достойным замечания эффектом способа по изобретению является то, что при сниженной потребности в водяном паре обеспечивается высокая селективность образования акриловой кислоты.
Само собой разумеется, что применяемые согласно изобретению оксиды металлов формулы I могут также использоваться в форме разбавленной тонкими, например, коллоидальными, материалами, такими, как диоксид кремния, диоксид титана, оксид алюминия, оксид циркония и оксид ниобия.
Массовое соотношение может при этом составлять до 9 (разбавитель) : 1 (активная масса). Возможные соотношения разбавления составляют, например, 6 (разбавитель) : 1 (активная масса) и 3 (разбавитель) : 1 (активная масса). Врабатывание разбавителя может происходить перед и/или после кальцинирования. Если врабатывание происходит до кальцинирования, разбавитель должен выбираться таким образом, что он в основном сохраняется при кальцинировании. Это, как правило, имеет место в случае полученных при соответственно высоких температурах оксидов.
Применяемые при способе по изобретению катализаторы могут многократно регенерироваться при температуре ≤ температуры реакции посредством обработки кислородсодержащими газами, например обогащенными или обедненными кислородом газами, к которым может также добавляться водяной пар.
Примеры
А) Получение применяемых согласно изобретению в качестве катализатора оксидов металлов формулы I.
В емкости из нержавеющей стали в 44,6 л воды при 80°С растворяют при перемешивании 1287,25 г метаванадата аммония (77,5 мас.% V2O5, фирма G.f.E, Нюрнберг, DE). Образуется прозрачный желтоватый раствор, который охлаждают до 60°С и потом последовательно при сохранении температуры 60°С сначала примешивают 1683,75 г теллуровой кислоты (99 мас.% Н6ТеО6, фирма Fluka) и потом 5868,0 г гептамолибдата аммония (81,5 мас.% MoO3, фирма Starck). Получают темно-красный раствор А. Во второй емкости из нержавеющей стали при 60°С в 8,3 л воды растворяют 1599,0 г ниобоксалат аммония (21,1 мас.% Nb, фирма Starck, Гослар, DE) с получением раствора В. Оба раствора А и В охлаждают до 30°С и при этой температуре объединяют друг с другом, причем раствор В вмешивают в раствор А. Вмешивание производят непрерывно в течение 10 минут. Образуется оранжевая водная суспензия, которую сушат распылением (Темкости≥30°С, ТВХ=240°С, ТВЫХ=110°С, продолжительность сушки 1,5 ч, сушильная башня фирмы Nipolosa). Полученный продукт имеет также оранжевый цвет. В продукт распылительной сушки вмешивают 1 мас.% тонкого графита (ситовый анализ: мин, 50 мас.%<24 мм, макс. 10 мас.% >24 мм и <48 мм, макс. 5 мас.%>48 мм, поверхность по БЭТ 6 - 13 м2/г).
Получаемую смесь спрессовывают в полые цилиндры (кольца) с размерами 16 мм × 2,5 мм × 8 мм (внешний диаметр х высота х внутренний диаметр) таким образом, что предел прочности при боковом сжатии колец составляет около 10 Н.
200 г этих колец по две порции в 100 г последовательно кальцинируют во вращающейся шаровой печи из кварцевого стекла 1 л внутренней емкости согласно чертежу (1 - корпус печи, 2 - вращающийся поршень, 3 - обогреваемая камера, 4 - поток азота/воздуха). Для этого содержимое вращающейся печи нагревают в потоке воздуха 50 нл/ч с линейной характеристикой нагрева в течение 27,5 мин от 25 до 275°С и эту температуру при сохранении воздушного потока поддерживают в течение 1 часа. Затем в течение 32,5 минут нагревают с линейной характеристикой от 275 до 600°С, причем воздушный поток заменяют потоком азота в 50 нл/ч. Температуру 600°С и поток азота сохраняют в течение 2 часов и всю печь оставляют охлаждаться до 25°С с сохранением потока азота. Получают черные таблетки следующего состава: MO1,0V0,33 Te0,15, Nb0,11, Ox.
В мельнице фирмы Retsch таблетки в сухом состоянии размалывают до размера частиц <100 мм. 150 г размолотого материала размешивают в 1500 мл 10 мас.%-ного водного раствора HNO3 при 70°С в течение 7 часов с рециркуляцией выделяющейся жидкой фазы, твердое вещество отфильтровывают от получаемой суспензии и промывают водой до отсутствия нитрата. Фильтрат сушат в течение ночи воздухом при 110°С в муфельной печи. Полученная активная масса имеет следующий состав: MO0,1V0,27Te0,12, 12Nb0,13Ох.
75 г полученной порошковой активной массы наносят на 300 г шарообразного носителя с диаметром 2,2-3,2 мм (материал носителя=стеатит фирмы Ceramtec, DE, общий объем пор носителя v 1 об.% в пересчете на общий объем носителя; Rz=45 мм). Для этого носитель помещают в дражировочный барабан с 2 л внутреннего объема (угол наклона средней оси барабана к горизонтали 30°). Барабан вращается со скоростью 25 об/мин. Распылительным соплом с мощностью 300 нл/ч сжатого воздуха в течение 60 минут наносят на носитель приблизительно 30 мл смеси глицерина и воды (весовое соотношение глицерина и воды=1:3). Сопло при этом установлено таким образом, что распылительный конус увлажняет транспортируемые поводковыми листами в самую верхнюю точку наклоненного барабана носители только в верхней половине участка, по которому перемещаются носители. Тонкоизмельченный порошок активной массы вводят в барабан порошковым шнеком, причем точка подачи порошка лежит в пределах участка, по которому перемещаются носители, однако под распылительным конусом. Периодическим повторением увлажнения и дозировки порошка покрытый основным слоем носитель при последующих повторных увлажнениях становится сам носителем.
После окончания нанесения покрытия носитель сушат в течение 16 часов при 120°С в вентиляционном сушильном шкафу (фирмы Binder, DE, внутренний объем 53 л).
Глицерин удаляют двухчасовой термообработкой при 150°С в атмосфере воздуха. Получают применяемый согласно изобретению катализатор на носителе, содержащий 20 мас.% активной массы.
Б) Проведение способа по изобретению и сравнительного способа
По 35 г свежеизготовленного катализатора помещают в трубчатый реактор с одной трубой (длина трубы 140 см, внутренний диаметр 8,5 мм, внешний диаметр 60 мм, V2A сталь, длина слоя катализатора 54,5 см, перед которым размещают слой длиной 30 см из стеатитных шариков фирмы Ceramtec (2,2-3,2 мм диаметр), предназначенный для подогрева исходной реакционной газовой смеси, а трубу реактора заполняют теми же шариками после катализаторной зоны), который обогревается электрическими нагревательными матами. При температуре матов 350°С катализатор вводят в трубчатый реактор.
После этого в четырех независимых друг от друга тестах W, X, У и Z четыре катализаторных слоя трубчатого реактора в течение 48 часов при температуре нагревательных матов в 350°С нагружаются исходными реакционными газовыми смесями W, X, У и Z следующего состава:
W: 3,3 об.% пропана, 10 об.% O2, 40 об.% N2, 46,7 об.% H2O;
X: 3,3 об.% пропана, 10 об.% O2, 40 об.% N2, 46,7 об.% H2O;
У: 3,3 об.% пропана, 10 об.% O2, 70 об.% N2, 16,7 об.% H2O;
Z: 3,3 об.% пропана, 10 об.% O2, 86,7 об.% N2, 0 об.% H2O.
Время пребывания (в пересчете на объем катализаторного слоя) составляет во всех случаях 2,4 сек. Рабочее давление во всех случаях составляет 2 бара (абс.).
Через 48 часов в трубы реактора далее загружают при сохранении указанного времени пребывания нижеследующие составы исходной реакционной газовой смеси и при этом через дополнительные 4 часа работы получают следующие результаты (при одноразовом проходе реактора; Тм=температура нагревательных матов):
W (согласно изобретению): 3,3 об.% пропана, 10 об.% O2, 86,7 об.% N2, 0 об.% H2O,
Тм=390°С.
Конверсия пропана при одноразовом проходе трубы реактора (Up) 25 мол.%, селективность SAS образования акриловой кислоты при одноразовом проходе трубы реактора 50 мол.%.
Z (сравнение c W): 3,3 об.% пропана, 10 об.% О2, 86,7 об.% N2, 0 об.% H2O,
Тм=390°С,
Up=25 мол.%,
Sas=40 мол.%.
х (согласно изобретению): 3,3 об.% пропана, 10 об.% О2, 70 об.% N2, 16,7 об.% Н2O
Тм=370°С,
Up=25 мол.%,
SAS=70 мол.%.
у (сравнение с Х): 3,3 об.% пропана, 10 об.% О2, 70 об.% N2, 16,7 об.% H2O,
Тм=385°С,
Up=25 мол.%,
SAS=50 мол.%.
Полученные результаты свидетельствуют о преимуществах способа по изобретению.
Аналогично примеру А по получению катализатора оксидов металлов общей формулы (I) получили следующие катализаторы на носителе, содержащие 20 мас.% активной массы.
Состав A: Mo1,0V0,28Te0,13Nb0,10Ga0,019Ox
При этом используют
раствор А:
79,65 г метаванадата аммония (78,55 мас.% V2O5, фирма G.f.E., DE, Нюрнберг) в 3000 мл воды,
122,34 г теллуровой кислоты (99 мас.% Н6ТеО6, фирма Aldrich),
400,00 г гептамолибдата аммония (82,52 мас.% МоО3, фирма Starck) и
33,40 г гидрата нитрата галия (III) в 40,00 г воды,
раствор В:
81,94 г ниобоксалата аммония (20,8 мас.% Nb, фирма Starck, DE, Гослар) в 500 мл воды.
Состав В: Mo1,0V0,29Te0,14Nb0,13Ni0,007Ox
При этом используют
раствор А:
87,61 г метаванадата аммония (78,55 мас.% V2O5, фирма G.f.E., DE, Нюрнберг) в 3040 мл воды,
117,03 г теллуровой кислоты (99 мас.% H6TeO6, фирма Aldrich),
400,00 г гептамолибдата аммония (82,52 мас.% МоО3, фирма Starck) и
25,60 г гексагидрата нитрата никелья (II) в 20,00 г воды,
раствор В:
112,67 г ниобоксалата аммония (20,8 мас.% Nb, фирма Starck, DE, Гослар) в 500 мл воды.
Состав С: Mo1,0V0,28Sb0,13Nb0,10Ga0,019O11
Получают аналогично составу А с той лишь разницей, что вместо 122,34 г теллуровой кислоты (99 мас.% H6TeO6, фирма Aldrich) используют эквимолярное количество предварительно обработанного Н2O2 дисперсного Sb2O3.
Аналогично примеру В вышеприведенные катализаторы используют в способе по изобретению при применении исходных реакционных смесей W и Z.
W: 3,3 об.% пропана, 10 об.% O2, 40 об.% N2, 46,7 об.% Н2O,
причем через 48 часов состав смеси W заменяют на
3,3 об.% пропана, 10 об.% O2, 86,7 об.% N2, 0 об.% Н2O,
и
Z: 3,3 об.% пропана, 10 об.% O2, 86,7 об.% N2, 0 об.% Н2О, причем данную смесь используют без изменений состава, т.е. вне пределов изобретения;
при этом через 4 дополнительных часа работы получают следующие результаты при одноразовом проходе реактора по селективности SAS образования акриловой кислоты (конверсия пропана при одноразовом проходе трубы реактора (Up) составляет 25 мол.%):
WA: SAS=53,2 мол.%,
WB: SAS=50,4 мол.%,
WC: SAS=49,2 мол.%,
ZA: SAS=42,8 мол.%,
ZB: SAS=40,1 мол.%,
ZC: SAS=40,3 мол.%.
Вышеприведенные данные однозначно свидетельствуют о достижении повышенной селективности SAS при изменении доли водяного пара в исходной реакционной смеси согласно данному изобретению.
| название | год | авторы | номер документа |
|---|---|---|---|
| МАССЫ ОКСИДОВ МЕТАЛЛОВ | 2003 |
|
RU2352390C9 |
| СПОСОБ ПОЛУЧЕНИЯ, ПО МЕНЬШЕЙ МЕРЕ, ОДНОГО ПРОДУКТА ЧАСТИЧНОГО ОКИСЛЕНИЯ И/ИЛИ АММОКИСЛЕНИЯ ПРОПИЛЕНА | 2003 |
|
RU2346928C9 |
| СПОСОБ ПОЛУЧЕНИЯ, ПО МЕНЬШЕЙ МЕРЕ, ОДНОГО ПРОДУКТА ЧАСТИЧНОГО ОКИСЛЕНИЯ И/ИЛИ АММОКИСЛЕНИЯ УГЛЕВОДОРОДА, ВЫБРАННОГО ИЗ ГРУППЫ, ВКЛЮЧАЮЩЕЙ АКРОЛЕИН, МЕТАКРОЛЕИН, АКРИЛОВУЮ КИСЛОТУ, МЕТАКРИЛОВУЮ КИСЛОТУ, АКРИЛОНИТРИЛ И МЕТАКРИЛОНИТРИЛ | 2004 |
|
RU2356881C2 |
| СПОСОБ ПОЛУЧЕНИЯ АКРОЛЕИНА И/ИЛИ АКРИЛОВОЙ КИСЛОТЫ | 2001 |
|
RU2285690C2 |
| СПОСОБ ПОЛУЧЕНИЯ АКРИЛОВОЙ КИСЛОТЫ ИЗ ПРОПАНА | 2006 |
|
RU2430083C9 |
| СПОСОБ ДЛИТЕЛЬНОГО ПРОВЕДЕНИЯ ГЕТЕРОГЕННО КАТАЛИЗИРУЕМОГО ЧАСТИЧНОГО ОКИСЛЕНИЯ В ГАЗОВОЙ ФАЗЕ ПРОПЕНА В АКРОЛЕИН | 2004 |
|
RU2349576C9 |
| СПОСОБ ПРИГОТОВЛЕНИЯ КАТАЛИЗАТОРА, СОСТОЯЩЕГО ИЗ НОСИТЕЛЯ И НАНЕСЕННОЙ НА ПОВЕРХНОСТЬ НОСИТЕЛЯ КАТАЛИТИЧЕСКИ АКТИВНОЙ МАССЫ | 2008 |
|
RU2464085C2 |
| СПОСОБ ПОЛУЧЕНИЯ АКРОЛЕИНА, ИЛИ АКРИЛОВОЙ КИСЛОТЫ, ИЛИ ИХ СМЕСЕЙ ИЗ ПРОПАНА | 2001 |
|
RU2312851C2 |
| СПОСОБ ПОЛУЧЕНИЯ АКРОЛЕИНА, ИЛИ АКРИЛОВОЙ КИСЛОТЫ ИЛИ ИХ СМЕСИ ИЗ ПРОПАНА | 2007 |
|
RU2452724C2 |
| СПОСОБ ПОЛУЧЕНИЯ АКРОЛЕИНА, АКРИЛОВОЙ КИСЛОТЫ ИЛИ ИХ СМЕСИ ИЗ ПРОПАНА | 2006 |
|
RU2429218C2 |
Изобретение относится к усовершенствованному способу получения акриловой кислоты гетерогенно катализируемым парциальным окислением пропана в газовой фазе, при котором исходную реакционную газовую смесь, содержащую пропан, молекулярный кислород и, по меньшей мере, один газ-разбавитель, при повышенной температуре пропускают над массой оксидов мультиметаллов общей стехиометрии 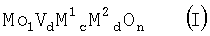 , где М1=Те и/или Sb, M2=по меньшей мере, один элемент из группы, включающей Nb, Та, W, Ti, Al, Zr, Cr, Mn, Ga, Fe, Ru, Co, Rh, Ni, Pd, Pt, La, Bi, B, Ce, Sn, Zn, Si и In, b=0,01 до 1, с=>0 до 1, d=>0 до 1 и n=числу, которое определяется валентностью и количеством отличных от кислорода элементов в (I), и при этом парциально окисляют пропан с получением акриловой кислоты, в котором состав исходной реакционной газовой смеси во время проведения процесса, по меньшей мере, один раз изменяют таким образом, что содержащаяся в исходной реакционной газовой смеси молярная доля газа-разбавителя, водяного пара, в пересчете на содержащееся в исходной реакционной газовой смеси молярное количество пропана, после изменения является меньше, чем до изменения. Применение катализаторов формулы (I) в данном способе позволяет снизить количество водяного пара в реакционной смеси с сохранением селективности и активности катализатора по целевому продукту. 4 з.п. ф-лы, 1 ил.
, где М1=Те и/или Sb, M2=по меньшей мере, один элемент из группы, включающей Nb, Та, W, Ti, Al, Zr, Cr, Mn, Ga, Fe, Ru, Co, Rh, Ni, Pd, Pt, La, Bi, B, Ce, Sn, Zn, Si и In, b=0,01 до 1, с=>0 до 1, d=>0 до 1 и n=числу, которое определяется валентностью и количеством отличных от кислорода элементов в (I), и при этом парциально окисляют пропан с получением акриловой кислоты, в котором состав исходной реакционной газовой смеси во время проведения процесса, по меньшей мере, один раз изменяют таким образом, что содержащаяся в исходной реакционной газовой смеси молярная доля газа-разбавителя, водяного пара, в пересчете на содержащееся в исходной реакционной газовой смеси молярное количество пропана, после изменения является меньше, чем до изменения. Применение катализаторов формулы (I) в данном способе позволяет снизить количество водяного пара в реакционной смеси с сохранением селективности и активности катализатора по целевому продукту. 4 з.п. ф-лы, 1 ил.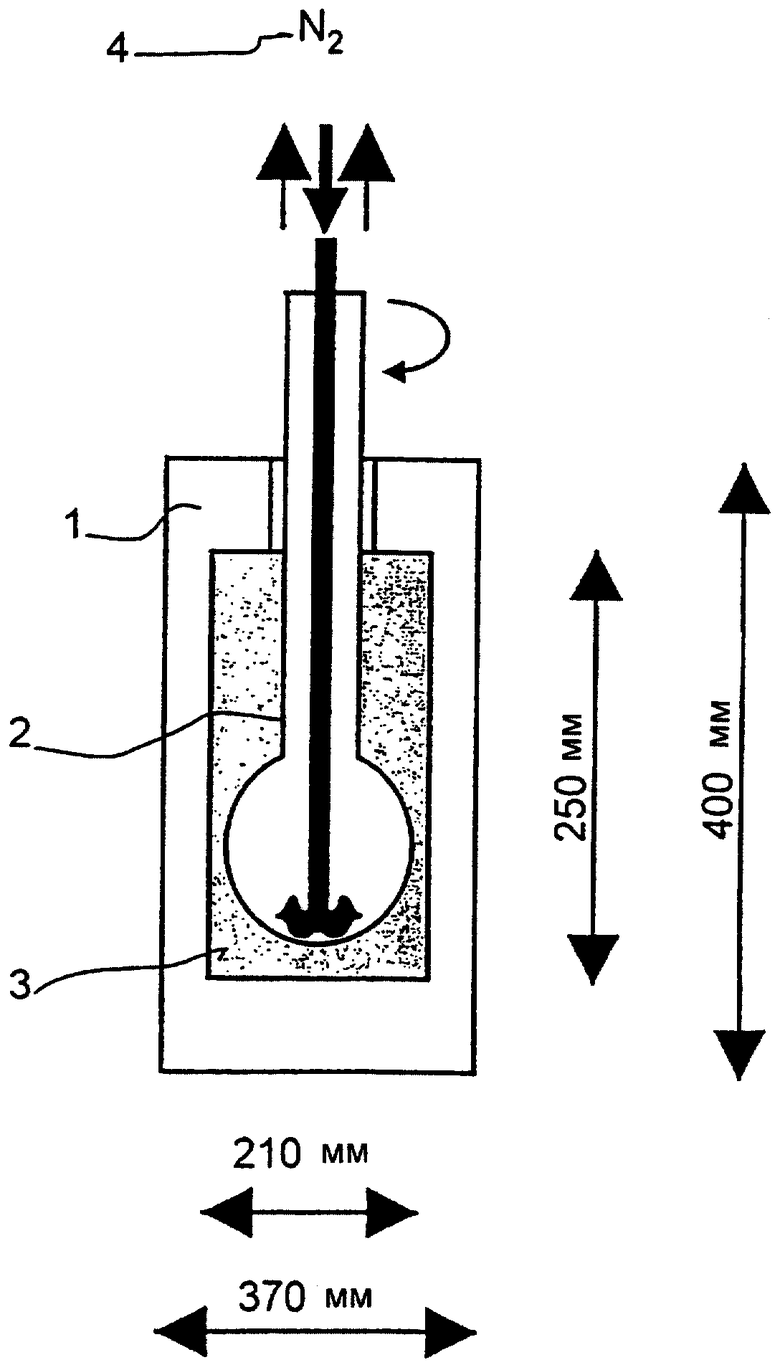
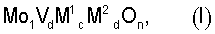
где М1=Те и/или Sb,
М2=по меньшей мере, один элемент из группы, включающей
Nb, Та, W, Ti, Al, Zr, Cr, Mn, Ga, Fe, Ru, Co, Rh, Ni, Pd, Pt, La, Bi, B, Ce, Sn, Zn, Si и In,
b=0,01 до 1,
с=>0 до 1,
d=>0 до 1,
n=число, которое определяется валентностью и количеством отличных от кислорода элементов в (I),
при этом парциально окисляют пропан с получением акриловой кислоты, отличающийся тем, что состав исходной реакционной газовой смеси во время проведения процесса, по меньшей мере, один раз изменяют таким образом, что содержащаяся в исходной реакционной газовой смеси молярная доля газа-разбавителя, водяного пара, в пересчете на содержащееся в исходной реакционной газовой смеси молярное количество пропана, после изменения является меньше чем до изменения.
| Устройство для подачи газа и сыпучих материалов | 1976 |
|
SU608838A1 |
| US 3801634 A, 02.04.1974 | |||
| US 4147885 А, 03.04.1979 | |||
| US 5705684 А, 06,01.1998 | |||
| УСТРОЙСТВО ДЛЯ КОНТРОЛИРОВАНИЯ РАБОЧИХ ПАРАМЕТРОВ И/ИЛИ ОПРЕДЕЛЕНИЯ СОСТОЯНИЯ ИЗНОСА ТРОСА ПРИ ИСПОЛЬЗОВАНИИ В ПОДЪЕМНЫХ МЕХАНИЗМАХ | 2017 |
|
RU2729841C2 |
| RU 96104337 А, 10.06.1998. | |||

