Настоящее изобретение касается способа получения из пропана акролеина, или акриловой кислоты, или их смеси, при котором
А) - в первую реакционную зону А подают по меньшей мере два газообразных потока подачи, содержащих пропан, по меньшей мере один из которых содержит свежий пропан, с образованием реакционного газа А;
- в реакционной зоне А реакционный газ А проводят по меньшей мере через один слой катализатора, в котором путем частичного гетерогенно-катализируемого дегидрирования пропана образуются молекулярный водород и пропилен;
- в реакционную зону А подают молекулярный кислород, который окисляет в реакционной зоне А по меньшей мере часть содержащегося в реакционном газе А молекулярного водорода до водяного пара; и
- отбирают из реакционной зоны А газ-продукт А, содержащий пропилен, пропан и водяной пар;
B) содержащийся в газе-продукте А водяной пар при необходимости полностью или частично путем прямого и/или непрямого охлаждения отделяют в первой зоне разделения I методом конденсации с сохранением газа-продукта А*;
C) в реакционной зоне В, подавая молекулярный кислород, используют газ-продукт А или газ-продукт А* для подачи по меньшей мере в один реактор окисления с реакционным газом В, содержащим пропан, пропилен и молекулярный кислород, и подвергают содержащийся в нем пропилен гетерогенно-катализируемому частичному окислению в газовой фазе с образованием акролеина, или акриловой кислоты, или их смеси в качестве целевого продукта, а также газа-продукта В, содержащего непревращенный пропан;
D) из реакционной зоны В выводят газ-продукт В и отделяют содержащийся в нем целевой продукт во второй зоне разделения II, причем остается остаточный газ, содержащий пропан;
Е) при необходимости возвращают часть остаточного газа, имеющую состав остаточного газа, в качестве потока подачи, содержащего пропан, в реакционную зону А;
F) в зоне разделения III пропан, содержащийся в не возвращенном в реакционную зону А остаточном газе, из которого ранее при необходимости конденсацией удалили, возможно, содержавшийся в нем водяной пар и/или посредством разделительной мембраны полностью или частично удалили содержавшийся в нем молекулярный водород, отделяют путем абсорбции в органическим растворителем (абсорбенте) с образованием содержащего пропан абсорбата; и
G) в разделительной зоне IV отделяют пропан от абсорбата и возвращают в реакционную зону А в виде содержащего пропан потока подачи.
Акриловая кислота - важный основной химический продукт, в числе прочего находящий применение при производстве полимеров, которые используют, например, при нахождении в дисперсионном распределении в водной среде в качестве связующего агента. Еще одна область применения полимеров акриловой кислоты - это суперабсорбер в области гигиены и других областях. Акролеин - важный промежуточный продукт, например, для производства глутарового диальдегида, метионина, 1,3-пропандиола, 3-пиколина, фолиевой кислоты и акриловой кислоты (ср., например, заявки на патент США US-A 6166263 и US-A 6187963).
Способ получения акролеина, или акриловой кислоты, или их смеси из пропана, при котором в первой реакционной зоне осуществляют частичное гетерогенно-катализируемое дегидрирование пропана до пропилена, а затем образовавшийся пропилен в присутствии не превращенного пропана (т.е. не отделяя предварительно пропилен от оставшегося в дегидрировании пропана, как это рекомендовано, например, в европейской заявке на патент ЕР-А 1617931 (ср. также с международной заявкой WO 04/09041)) гетерогенно-катализируемо частично окисляют до акролеина, или до акриловой кислоты, или до их смеси, известны (ср., например, с немецкой заявкой на патент DE-A3313573, европейской заявкой на патент ЕР-А 117146, заявкой на патент США US-A 3161670, немецкой заявкой на патент DE-A 10 2004 032129, европейской заявкой на патент ЕР-А 731077, немецкими заявками на патент DE-A 10 2005 049699, DE-A 10 2005 052923, международными заявками WO 01/96271, WO 03/011804, WO 03/076370, WO 01/96270, немецкими заявками на патент DE-A 10 2005 009 891, DE-A 10 2005 013039, DE-A 10 2005 022798, DE-A 10 2005 009885, DE-A 10 2005 010111, DE-A 10245585, DE-A 10316039, немецкой заявкой №10 2005 056377.5, немецкой заявкой №10 2005 049699.7, немецкой заявкой №10 2006 015235.2, немецкой заявкой №10 2005 061626.7 и описанным в этих публикациях известным уровнем техники).
Общий признак описанных там способов состоит в том, что после отделения целевого продукта акролеина, или акриловой кислоты, или акролеина и акриловой кислоты из итоговой газовой смеси, полученной частичным окислением пропилена от остающегося при этом остаточного газа, содержащего пропан, по меньшей мере часть содержащегося в остаточном газе пропана возвращают в виде циркуляционного газа в гетерогенно-катализируемое частичное дегидрирование пропана до пропилена.
В остальном вышеописанные способы уровня техники можно в основном разделить на две группы. Способы одной из этих групп отличаются тем, что, прежде чем подвергнуть пропилен в присутствии остающегося пропана частичному окислению до желательного целевого продукта, отличающиеся от пропана и пропилена компоненты газовой смеси, полученной гетерогенно-катализируемым частичным дегидрированием, отделяют путем абсорбции (немецкая заявка №10 2005 013039.9). Недостатком такого способа является то, что он требует, как правило, по меньшей мере трех зон, в которых необходимо проводить сжатие газа. Во-первых, прежде чем подвергнуть газ-продукт, полученный гетерогенно-катализируемым частичным дегидрированием пропана, абсорбционному отделению содержащегося в нем пропилена и пропана, обычно необходимо его сжатие, поскольку в норме абсорбционное отделение можно эффективно реализовать только под давлением. Дополнительно, как правило, необходимо сжатие источника кислорода, применяемого для гетерогенно-катализируемого частичного окисления, а сверх того обычно требуется сжатие пропана, остающегося при гетерогенно-катализируемом частичном окислении пропилена и возвращаемого в гетерогенно-катализируемое частичное дегидрирование. Способы, относящиеся ко второй из этих двух групп, отличаются тем, что они рекомендуют применять для гетрогенно-катализированного частичного окисления газовую смесь, полученную гетерогенно-катализируемым частичным дегидрированием как таковую (ср., например, с немецкой заявкой на патент DE-A 10316039). В основе этого способа лежит, в том числе, то предположение, что молекулярный водород, образовавшийся при гетерогенно-катализируемом частичном дегидрировании пропана, не оказывает неблагоприятного влияния при последующем гетерогенно-катализируемом частичном окислении пропилена (в отличие от гетерогенно-катализируемого оксидегидрирования, протекающего экзотермически, к проведению которого понуждает наличие кислорода, и при котором водород на промежуточном этапе не образуется (водород, отщепляемый от подлежащего дегидрированию углеводорода, отщепляется непосредственно в виде воды (Н2О)) или же не обнаруживается, в настоящей заявке под гетерогенно-катализируемым дегидрированием (которое, например, необходимо проводить в реакционной зоне А) подразумевают (”обычное”) дегидрирование, обладающее в отличие от оксидегидрирования эндотермическим тепловым режимом (в качестве следующего этапа в реакционной зоне А происходит экзотермическое сжигание водорода), и при котором по меньшей мере на промежуточном этапе образуется свободный молекулярный водород; для этого, как правило, необходимы другие условия реакции и иные катализаторы, нежели для оксидегидрирования; т.е. гетерогенно-катализируемое частичное дегидрирование пропана в реакционной зоне А при реализации способа согласно изобретению обязательно протекает с образованием H2. Собственные исследования, однако, показали, что присутствие в реакционной газовой смеси для гетерогенно-катализируемого частичного окисления пропилена повышенных количеств молекулярного водорода хотя и не оказывает заметного влияния на спектр побочных продуктов частичного окисления, но (предположительно, по причине выраженного восстановительного потенциала молекулярного водорода) заметно влияет на срок службы катализаторов, применяемых для гетерогенно-катализируемого частичного окисления в газовой фазе.
Дополнительно, реакционный газ В, отягощенный молекулярным водородом, компонентом, способным к формированию гремучего газа и обладающим особым поведением при диффузии (определенные конструкционные материалы проницаемы для молекулярного водорода). С вышеописанными проблемами нераздельно связано благоприятное для продления срока годности катализатора в частичном окислении в реакционной зоне В использование избытка молекулярного кислорода (относительно стехиометрического соотношения целевой реакции), что естественным образом влечет за собой повышенную взрывоопасность в присутствии молекулярного водорода в реакционной зоне В (если акриловая кислота представляет собой целевой продукт, а в реакционный газ В заранее добавляют совокупное необходимое количество молекулярного кислорода, то обычно требуется молярное соотношение содержащегося в реакционном газе В молекулярного кислорода к содержащемуся там же пропилену, по меньшей мере превышающее 1,5 (при этом уже учтена небольшая доля полного сгорания пропилена)).
Принимая во внимание описанный уровень техники, задача настоящего изобретения состояла в том, чтобы представить соответствующий преамбуле настоящей публикации улучшенный способ, который если и обладает описанными недостатками, то во всяком случае в сниженной степени.
Соответственно, был обнаружен способ получения акролеина, или акриловой кислоты, или их смеси из пропана, при котором
А) - в первую реакционную зону А подают по меньшей мере два газообразных потока подачи, содержащих пропан, по меньшей мере один из которых содержит свежий пропан, с образованием реакционного газа А;
- в реакционной зоне А реакционный газ А проводят по меньшей мере через один твердый слой катализатора, в котором путем частичного гетерогенно-катализируемого дегидрирования пропана образуются молекулярный водород и пропилен;
- в реакционную зону А подают молекулярный кислород, который окисляет в реакционной зоне А по меньшей мере часть содержащегося в реакционном газе А молекулярного водорода до водяного пара; и
- отбирают из реакционной зоны А газ-продукт А, содержащий пропилен, пропан и водяной пар;
B) содержащийся в газе-продукте А водяной пар при необходимости полностью или частично путем прямого и/или непрямого охлаждения отделяют в первой зоне разделения I методом конденсации с сохранением газа-продукта А*;
C) в реакционной зоне В, подавая молекулярный кислород, используют газ-продукт А или газ-продукт А* для подачи по меньшей мере в один реактор окисления с реакционным газом В, содержащим пропан, пропилен и молекулярный кислород, и подвергают содержащийся в нем пропилен гетерогенно-катализируемому частичному окислению в газовой фазе с образованием акролеина, или акриловой кислоты, или их смеси в качестве целевого продукта, а также газа-продукта В, содержащего непревращенный пропан;
D) из реакционной зоны В выводят газ-продукт В и отделяют содержащийся в нем целевой продукт во второй зоне разделения II, причем остается остаточный газ, содержащий пропан;
Е) при необходимости возвращают часть остаточного газа, имеющую состав остаточного газа, в качестве потока подачи, содержащего пропан, в реакционную зону А;
F) в зоне разделения III пропан, содержащийся в не возвращенном в реакционную зону А остаточном газе, из которого ранее при необходимости конденсацией удалили, возможно, содержавшийся в нем водяной пар и/или посредством разделительной мембраны полностью или частично удалили содержавшийся в нем молекулярный водород, отделяют путем абсорбции в органическим растворителем (абсорбенте) с образованием содержащего пропан абсорбата; и
G) в разделительной зоне IV отделяют пропан от абсорбата и возвращают в реакционную зону А в виде содержащего пропан потока подачи,
отличающийся тем, что в реакционной зоне А окисляют до водяного пара по меньшей мере столько молекулярного водорода, чтобы это окисленное в реакционной зоне А до водяного пара количество водорода составляло по меньшей мере 20 моль-% количества молекулярного водорода, образовавшегося в реакционной зоне А.
Согласно изобретению целесообразно также использовать по меньшей мере часть теплоты, вырабатывающейся при окислении (сгорании) молекулярного водорода в реакционной зоне А, для того чтобы путем непрямого теплообмена с реакционным газом А и/или газом-продуктом А, используемым в качестве теплоносителя, нагревать компоненты реакционного газа А подачи реакционного газа А (газообразные потоки подачи, подаваемые в реакционную зону А).
Согласно изобретению целесообразно, чтобы количество водорода, окисляемого в реакционной зоне А до водяного пара, составляло по меньшей мере 25 моль-%, желательно - по меньшей мере 30 моль-%, предпочтительно - по меньшей мере 35 моль-%, особо предпочтительно - по меньшей мере 40 моль-%, еще более предпочтительно - по меньшей мере 45 моль-%, а крайне предпочтительно - по меньшей мере 50 моль-%, в каждом случае отнесенного на массу образующегося в реакционной зоне А молекулярного водорода.
Т.е. к способам согласно изобретению относятся также те, в которых в реакционной зоне А (отнесенного на массу образующегося в реакционной зоне А молекулярного водорода) окисляется до водяного пара до 55 моль-%, или до 60 моль-%, или до 65 моль-%, или до 70 моль-%, или до 75 моль-%, или до 80 моль-%, или до 85 моль-%, или до 90 моль-%, или до 95 моль-%, или до 100 моль-% молекулярного водорода.
Если в дополнение к воздействию молекулярного водорода в качестве компонента реакционного газа В на срок службы катализатора в частичном окислении принять во внимание, что сжигание (окисление) молекулярного водорода до воды обеспечивает выделение примерно вдвое большего количества теплоты, чем расходуется для его образования в процессе дегидрирования пропана, то с точки зрения теплового баланса целесообразно, чтобы количество молекулярного водорода, сжигаемого в реакционной зоне А в процессе реализации способа согласно изобретению, составляло относительно образующегося в реакционной зоне А молекулярного водорода от 30 до 70 моль-%, желательно - от 40 до 60 моль-%.
Ограниченное содержание молекулярного водорода в реакционном газе В (т.е. в реакционном газе В, еще содержащем молекулярный водород) в некотором смысле благоприятно еще и с той точки зрения, что молекулярный водород обладает преимуществом самой высокой теплопроводности среди газов. Согласно Walter J. Moore, Physikalische Chemie, издательство WDEG, Berlin (1973), стр.171, при нормальных условиях она более чем в десять раз выше, чем теплопроводность диоксида углерода, и примерно в восемь раз, чем таковая молекулярного азота или молекулярного кислорода.
Именно эта повышенная теплопроводность ответственна, согласно Таблице I немецкой заявки на патент DE-A 3313573, за то, что в реакционной зоне В избирательность формирования СОх в присутствии молекулярного водорода существенно ниже, чем в отсутствие молекулярного водорода. Вследствие ускоренного отвода тепла от места протекания реакции в реакционной зоне В она обусловливает более низкие температуры поверхности катализатора и, следовательно, меньшую долю полного сгорания пропилена. В особенности это касается тех случаев, когда, как в способе согласно изобретению, молекулярный водород, обладающий крайне высокой теплопроводностью, и пропан, имеющий чрезвычайно высокую теплоемкость, совместно, синергическим образом, участвуют в теплоотводе.
В принципе, реакционный газ В при реализации способа согласно изобретению может, однако, вовсе не содержать молекулярного водорода.
В принципе, для подачи по меньшей мере в один реактор окисления в зоне реакции В можно применять продукт-газ А как таковой, отводимый от реакционной зоны А и содержащий пропилен, пропан и водяной пар. Согласно изобретению, однако, целесообразно отделять от продукта-газа А конденсацией в первой зоне разделения I по меньшей мере часть водяного пара, содержащегося в продукте-газе А, путем прямого и/или непрямого охлаждения продукта-газа А, причем остается продукт-газ А*, содержание водяного пара в котором ниже, чем таковое в продукте-газе А.
Обоснование этого состоит, во-первых, в том, что итоговые продукты акролеин и/или акриловая кислота обладают значительно более высокой аффинностью к воде, чем компоненты продукта-газа А, ввиду чего отделение целевых продуктов от продукта-газа В, содержащего повышенное количество водяного пара, влечет за собой значительную потребность в энергии. Во-вторых, значительные количества водяного пара в реакционном газе В обычно снижают срок службы применяемых в частичном окислении катализаторов, как правило, содержащих молибден в форме оксидов, поскольку Н2О способствует сублимации оксидов молибдена. Дополнительно, отделение водяного пара между реакционной зоной А и реакционной зоной В снижает общий объем реакционного газа В и, следовательно, потребность в энергии, расходуемой на перемещение его через реакционную зону В.
При реализации способа согласно изобретению целесообразно отделять конденсацией в зоне разделения I по меньшей мере 5 моль-%, более целесообразно - по меньшей мере 10 моль-%, еще лучше - по меньшей мере 15 моль-%, особо целесообразно - по меньшей мере 20 моль-%, еще более целесообразно - по меньшей мере 25 моль-% или по меньшей мере 30 моль-%, еще более целесообразно - по меньшей мере 35 моль-% или по меньшей мере 40 моль-%, еще лучше - по меньшей мере 45 моль-% или по меньшей мере 50 моль-%, особо выгодно - по меньшей мере 55 моль-% или по меньшей мере 60 моль-%, предпочтительно - по меньшей мере 65 моль-% или по меньшей мере 70 моль-%, особо предпочтительно - по меньшей мере 75 моль-% или по меньшей мере 80 моль-%, крайне предпочтительно - по меньшей мере 85 моль-% или по меньшей мере 90 моль-%, во многих случаях - по меньшей мере 95 моль-% или в предельном случае - до 100 моль-% (нередко, однако, не более 80 моль-%, или не более чем 85 моль-%, или не более чем 90 моль-%, или не более чем 95 моль-%, или не более чем 98 моль-%) содержащегося в продукте-газе А водяного пара. Ограниченное (но не исчезающе малое) содержание водяного пара в реакционном газе В выгодно, поскольку он обычно благоприятно воздействует на активность применяемых в реакционной зоне В катализаторов (особенно в случае мультиметаллических оксидов).
Для реализации способа согласно изобретению выгодно осуществлять отделение воды между реакционной зоной А и реакционной зоной В путем простой конденсации.
Вода, отделяемая между реакционной зоной А и реакционной зоной В, может при этом представлять собой не только воду, возникшую из-за окисления водорода в реакционной зоне А. Гораздо чаще возможно, что в реакционной зоне А водяной пар применяли также в качестве инертного разбавляющего газа, который при необходимости также частично или полностью отделяют в зоне разделения I.
Ввиду сравнительно высокой температуры кипения воды, особенно при повышенном давлении, вышеупомянутой конденсации обычно можно добиться простым прямым и/или непрямым охлаждением продукта-газа А. В принципе, конденсацию можно осуществлять одноступенчатым или многоступенчатым охлаждением. Согласно изобретению целесообразно, чтобы конденсационное отделение воды осуществляли многоступенчатым охлаждением. С точки зрения техники применения при этом целесообразно применять прямое и непрямое охлаждение в комбинации. В качестве непрямых теплообменников для целей, согласно изобретению, в принципе, можно применять все известные типы непрямых теплообменников. Среди таковых предпочтительно применять теплообменники в виде пучка труб и пластинчатые теплообменники. В качестве примера на первой ступени охлаждения можно проводить отобранный из реакционной зоны А горячий (типичная температура продукта-газа А составляет, например, от 400 до 700°С, предпочтительно - от 500 до 600°С; типичные значения рабочего давления продукта-газа А составляют, как правило, более 1 или от 1,5 до 5 бар, либо же от 2 до 5, предпочтительно - от 1,5 или 2 до 3 бар) продукт-газ А через первый (в самом общем случае при реализации способа согласно изобретению отобранный из реакционной зоны А продукт-газ А можно применять для того, чтобы путем непрямого теплообмена, при необходимости, в серии подключенных последовательно друг за другом теплообменников (например, двух последовательно подключенных друг за другом теплообменников) нагреть по меньшей мере часть компонентов или все компоненты газовой смеси загрузки реакционного газа А (в каждом случае до желательной температуры) и одновременно охладить продукт-газ А) непрямой теплообменник, например, прямотоком или противотоком, что и поток газа (циркуляционный газ пропана), содержащий пропан и выделенный из абсорбата в зоне разделения IV, чтобы довести этот поток газа, применяемый в качестве одного из поступающих в реакционную зону А содержащих пропан потоков подачи, до исходного уровня температуры, желательного для реакционной зоны А. Этот уровень температуры может колебаться в типичных при реализации способа согласно изобретению пределах, например от 400 до 600°С, предпочтительно - от 450 до 550°С. Через следующий за ним второй непрямой теплообменник целесообразно проводить уже, как описано, охлажденный продукт-газ А, например, прямотоком или противотоком продукту-газу А, от которого ранее уже в желательном количестве отделили содержавшийся в нем водяной пар (т.е. проводку осуществляют в том же направлении или противотоком продукту-газу А*), чтобы снова поднять его температуру до уровня, соответствующего последующему частичному окислению содержащегося в нем пропилена и целесообразного для этого окисления. Температура продукта-газа А на выходе из второго непрямого теплообменника, как правило, остается выше 100°С. В третьем непрямом теплообменнике, который обычно выполнен как воздушный радиатор (например, проведение потока продукта-газа А в том же направлении или противотоком к воздуху), температуру продукта-газа А снижают до уровня менее 100°С. Затем, в подключенном последовательно прямом охладителе можно осуществить отделение водяного пара. При этом прямой охладитель (прямой конденсатор) может иметь конструкцию, известную как таковая (например, соответствовать ректификационной колонне), и иметь обычные (например, для ректификационных колонн) внутренние устройства для увеличения площади поверхности теплообмена. Как правило, однако, обходятся без таких внутренних устройств. Обычно корпус колонны прямого конденсатора не снабжают теплоизоляцией. Предпочтительны любые мероприятия, которые повышают теплопотери через стенку колонны, например приваренные параллельно поверхности колонны перфорированные пластины или ребра охлаждения. В качестве хладагента для целей прямого охлаждения продукта-газа А целесообразно применять водную фазу, ранее отделенную от другого продукта-газа А путем конденсации, которую целесообразно перед использованием для прямого охлаждения охладить с помощью охлаждающей воды (предпочтительно с точки зрения техники применения - воды с поверхности) в непрямом теплообменнике до температур не выше 35°С, предпочтительно - не выше 30°С. В прямом холодильнике возможно проведение прямотоком или противотоком продукта-газа А и прямого водного хладагента в тонкодисперсном распылении. Как правило, предпочтительно проведение в одном направлении.
Отделять водную фазу и остаточную газообразную фазу друг от друга можно с помощью известных сепараторов (например, механических каплеуловителей). После охлаждения отделенной водной фазы, которое следует проводить так, как уже описано, ее снова можно подавать на прямое охлаждение продукта-газа А.
Температуру остающейся газообразной фазы (в описанном случае она образует продукт-газ А*) можно с помощью последующего (уже описанного выше) непрямого теплообмена с еще подлежащим освобождению от водяного пара продуктом-газом А поднять до уровня, благоприятного для последующего частичного окисления пропилена. В принципе, при реализации способа согласно изобретению возможно также отделять путем конденсации водяной пар, содержащийся в продукте-газе А, только от части этого газа. Образующийся благодаря этому продукт-газ А* можно впоследствии применять для подачи на гетерогенно-катализируемое частичное окисление пропилена либо как таковой, либо в смеси с частью продукта-газа А, не прошедшей обработку конденсацией (в смысле изобретения эта смесь тоже образует газ продукции А*).
Еще одно благоприятное последствие прямого охлаждения продукта-газа А (например, с помощью водной фазы, ранее отделенной из продукта-газа А и предпочтительно охлажденной путем непрямого теплообмена) состоит в общем случае в том, что при прямом охлаждении одновременно обеспечивают отделение, возможно, присутствующих в продукте-газе А твердых частиц, которые могут, например, представлять собой частицы катализатора дегидрирования, имеющие происхождение в гетерогенно-катализируемом частичном дегидрировании пропана, способные, согласно теоретическому изложению немецкой заявки на патент DE-A 10316039, создавать помехи последующему гетерогенно-катализируемому частичному окислению пропилена. Последнее существенно особенно тогда, когда продукт-газ А либо продукт-газ А*, применяемый для подачи на частичное окисление, еще содержит молекулярный водород. Т.е. согласно изобретению вообще целесообразно, чтобы между реакционной зоной А и реакционной зоной В имела место операция механической сепарации согласно немецкой заявке на патент DE-A 10316039, пусть даже ее осуществляют лишь в форме прямого охлаждения реакционного газа А, проводимой, как это описано.
Поэтому отделение водяного пара от продукта-газа А в зоне разделения I способом конденсации особо целесообразно тогда, когда оно включает в себя прямое охлаждение продукта-газа А водной фазой, предпочтительно - ранее отделенной из продукта-газа А и охлажденной (предпочтительно непрямым теплообменом).
В водной фазе, отделенной, как это описано, путем конденсации, как правило, еще содержится растворенный пропан, который можно простым способом, например путем нагревания водной фазы (при необходимости - при пониженном давлении), вернуть и снова направить в реакционную зону А в качестве еще одного потока подачи, содержащего пропан. Прямое и/или непрямое охлаждение продукта-газа А в целях отделения конденсацией содержащейся в продукте-газе А воды связано, как правило, с потерей давления в размере не более 1 бар, преимущественно - не более 0,5 бар, предпочтительно - не более 0,3 бар.
Окисление молекулярного водорода до воды в реакционной зоне А можно проводить после самого гетерогенно-катализируемого частичного дегидрирования пропана и/или в процессе гетерогенно-катализируемого частичного дегидрирования пропана, например, тогда, когда достигнуты определенные значения доли относительно оборота пропана, который получается при однократном прохождении реакционного газа А через реакционную зону А. Согласно изобретению целесообразно, чтобы по меньшей мере часть водорода, окисляемого в реакционной зоне А до воды, уже окислилась до воды, прежде чем будет достигнуто значение оборота пропана, желательное (и достигнутое итоговое значение оборота) для реакционной зоны А (относительно однократного прохождения реакционного газа А через реакционную зону А).
Во-первых, этот способ работы выгоден, потому что при удалении молекулярного водорода из равновесного положения гетерогенно-катализируемого дегидрирования положение равновесия сдвигается в пользу желательного образования пропилена. Дополнительная выгода состоит еще и в том, что теплота, выделяющаяся при реакции окисления водорода, компенсирует теплоту, потребленную при происшедшем ранее дегидрировании, и может быть использована для последующего дегидрирования. Согласно изобретению целесообразно сжигать молекулярный водород (по меньшей мере частично) с помощью молекулярного кислорода в реакционной зоне А самое позднее тогда, когда уже образовалось 90 моль-%, а лучше - после того как образовалось уже 75 моль-% общего количества формирующегося в реакционной зоне А молекулярного водорода. Согласно изобретению предпочтительно сжигать молекулярный водород (по меньшей мере частично) с помощью молекулярного кислорода в реакционной зоне А самое позднее, после того как образовалось 65 моль-%, или 55 моль-%, или 45 моль-% общего количества формирующегося в реакционной зоне А молекулярного водорода.
Влиять на время сжигания молекулярного водорода в реакционной зоне А можно, во-первых, посредством того, где (когда) в реакционную зону А подают молекулярный кислород.
Если в реакционную зону А вводят часть (состав которой соответствует таковому остаточного газа) остающегося в зоне разделения II содержащего пропан остаточного газа как такового (т.е. без прохождения разделительных зон III/IV), то включать в свой состав молекулярный кислород может уже газовая смесь подачи реакционной зоны А, поскольку вышеуказанный остаточный газ, как правило, еще содержит молекулярный кислород, используемый в реакционной зоне В в избытке.
Кроме того, влияние на него (время сжигания) можно оказывать, меняя способ загрузки катализатора в реакционную зону А. Так, в реакционную зону А можно локально загружать катализатор, который чрезвычайно избирательно катализирует только окисление молекулярного водорода молекулярным кислородом до воды. Другие катализаторы обладают способностью чрезвычайно избирательно катализировать дегидрирование. Как правило, однако, те катализаторы, которые в состоянии катализировать дегидрирование, обладают также способностью катализировать и сжигание водорода, причем в норме они обладают особо выраженной способностью снижать энергию активации последней из упомянутых реакций.
Под свежим пропаном в настоящем тексте подразумевают пропан, который еще не участвовал в дегидрировании в реакционной зоне А. Как правило, его подают как компонент сырого пропана (который предпочтительно соответствует спецификации согласно немецким заявкам на патент DE-A 10246119 и DE-A 10245585), содержащего также в небольших количествах отличные от пропана компоненты.
Такой сырой пропан получают, например, по способу, описанному в немецкой заявке на патент DE-A 102005 022798. При реализации способа согласно изобретению в реакционную зону А, как правило, в качестве другого потока, содержащего пропан, подают кроме свежего пропана только пропан, отделенный в зоне разделения IV от абсорбата, а также, при необходимости, часть остаточного циркуляционного газа.
При реализации способа согласно изобретению предпочтительно осуществлять подачу свежего пропана исключительно внутрь реакционной зоны А в качестве компонента газовой смеси подачи для реакционной зоны А. В принципе, однако, из соображений взрывобезопасности часть свежего пропана можно вводить и в газовые смеси подачи первого и/или второго этапа окисления реакционной зоны В либо же на любом другом этапе в рамках реализации способа.
В настоящем тексте под ”нагрузкой слоя катализатора, катализирующего этап реакции, реакционным газом” подразумевают количество реакционного газа в нормальных литрах (= Нл; объем в литрах, который соответствующее количество реакционной смеси газов занимало бы при нормальных условиях, т.е. при 0°С и 1 атм), которое за один час проходит через один литр слоя катализатора (например, твердого слоя катализатора).
Нагрузка может также быть рассчитана только для одного компонента реакционного газа. В этом случае это тот объем этого компонента в Нл/л·ч, который проходит через литр слоя катализатора в час (чистую засыпку из инертного материала не включают при расчете в твердый слой катализатора). Если слой катализатора состоит из смеси катализатора и инертных формованных изделий разбавления, нагрузка, если это соответствующим образом упомянуто, может быть также рассчитана только на единицу объема имеющегося катализатора.
При этом в настоящем тексте под инертным газом в общем случае подразумевают компонент реакционного газа, который в условиях соответствующей реакции в основном ведет себя химически инертным образом и при рассмотрении каждого инертного компонента реакционного газа отдельно остается химически неизменным более чем на 95 моль-%, предпочтительно более чем на 97 моль-% либо же более чем на 99 моль-%.
Часть остаточного газа, соответствующую по составу остаточному газу, возвращают в качестве содержащего пропан потока подачи в реакционную зону А, и это количество остаточного газа составляет до 10 вес.-% от общего количества остаточного газа.
В качестве источника молекулярного кислорода, необходимого в реакционной зоне А, помимо остаточного циркуляционного газа происхождением из зоны разделения II, рассматривают также молекулярный кислород как таковой или смесь молекулярного кислорода и газа, обладающего в реакционной зоне А химической инертностью (например, благородные газы, как то: аргон, молекулярный азот, водяной пар, диоксид углерода и т.д., либо же смесь таких инертных газов). Т.е. молекулярный кислород можно подавать в реакционную зону А в виде газа, который содержит не более чем 50 об.-%, предпочтительно - не более чем 40 об.-% или не более чем 30 об.-%, целесообразно - не более чем 25 об.-% или не более чем 20 об-%, особо целесообразно - не более чем 15 об.-% или не более чем 10 об.-%, а крайне предпочтительно - не более чем 5 об.-% или не более чем 2 об.-% других (отличных от молекулярного кислорода) газов (компонентов). Поскольку при реализации способа согласно изобретению потребность в кислороде в реакционной зоне А меньше по сравнению с таковой в реакционной зоне В, выгодно, особенно по соображениям экономичности, также и применение воздуха в качестве источника кислорода при реализации способа согласно изобретению для удовлетворения потребности в реакционной зоне А. Однако из соображений минимизации объемов перемещаемого и подлежащего сжатию газа предпочтительна подача в реакционную зону А чистого кислорода. Возвращаемый в реакционную зону А циркуляционный газ с пропаном (поступающий из зоны разделения IV), как правило, содержит лишь следы молекулярного кислорода.
Сказанное выше в основном справедливо и в отношении подачи молекулярного кислорода, необходимого в реакционной зоне В. Т.е. подача чистого молекулярного кислорода как для удовлетворения потребности в кислороде реакционной зоны А, так и реакционной зоны В целесообразна, в частности, потому что в этом случае газовые потоки способа, согласно изобретению, не нагружены сверх необходимого инертным газом, который при дальнейшей циркуляции снова придется выпускать.
Типичные реакционные газы В, которые при реализации способа согласно изобретению можно подавать в реакционную зону В, характеризуются следующим составом:
от 4 до 25 об.-% пропилена,
от 6 до 70 об.-% пропана,
от 0 до 60 об.-% H2O,
от 5 до 60 об.-% O2 и
от 0 до 20, предпочтительно - до 10 об.-% Н2.
Предпочтительные согласно изобретению реакционные газы В характеризуются следующим составом:
от 7 до 25 об.-% пропилена,
от 10 до 40 об.-% пропана,
от 1 до 10 об.-% Н2О,
от 10 до 30 об.-% O2
и от 0 до 10 об.-% Н2.
Крайне предпочтительные согласно изобретению реакционные газы В для вышеуказанной подачи характеризуются следующим составом:
от 15 до 25 об.-% пропилена,
от 20 до 40 об.-% пропана,
от 2 до 8 об.-% H2O,
от 15 до 30 об.-% O2 и
от 0 до 5 об.-% H2.
Целесообразность среднего уровня содержания пропана в реакционном газе В следует, например, из немецкой заявки на патент DE-A 10245585.
В рамках приведенных выше пределов состава выгодно, чтобы молярное соотношение V1 содержащегося в реакционном газе В пропана к содержащемуся в реакционном газе В пропилену составляло от 1 до 9. Кроме того, в рамках приведенных выше пределов состава целесообразно, чтобы молярное соотношение V2 содержащегося в реакционном газе В молекулярного кислорода к содержащемуся в реакционном газе В пропилену составляло от 1 до 2,5. Также в смысле настоящего изобретения в рамках приведенных выше пределов состава целесообразно, чтобы молярное соотношение V3 содержащегося в реакционном газе В молекулярного водорода к содержащемуся в реакционном газе В пропилену составляло от 0 до 0,80, либо от 0 до 0,60, или от 0,1 до 0,50. Зачастую соотношение V3 также составляет от 0,4 до 0,6. Также в рамках приведенных выше пределов состава целесообразно, чтобы молярное соотношение V4 содержащегося в реакционном газе В водяного пара к общему количеству молей содержащихся в реакционном газе В пропана и пропилена составляло от 0 либо 0,001 до 10.
Особо целесообразно, чтобы V1 в реакционном газе В, применяемом для подачи в реакционную зону В (в исходной смеси реакционного газа В), составлял от 1 до 7 или до 4, либо от 2 до 6, а крайне выгодно - от 2 до 5, либо от 3,5 до 4,5. Кроме того, для исходной смеси реакционного газа В предпочтительно, чтобы V2 составляло от 1,2 до 2,0, либо 1,4, либо 1,8. Соответственно, в исходной смеси реакционного газа В V4 предпочтительно находится в пределах от 0,015 до 5, более желательно - от 0,01 до 3, целесообразно - от 0,01 до 1, а особо целесообразно - от 0,01 до 0,3 либо до 0,1. Согласно изобретению предпочтительны не взрывчатые исходные смеси реакционного газа В.
Для ответа на вопрос, является ли исходная смесь реакционного газа В взрывчатой или нет, определяют, распространяется ли в смеси, находящейся в определенных условиях (давление, температура), сгорание (воспламенение, взрыв), инициируемое местным источником возгорания (например, раскаленной платиновой проволокой (ср. DIN 51649 и с описанием исследования в международной заявке WO 04/007405). Если распространение происходит, смесь здесь называют взрывчатой. Если распространение не происходит, то в данной публикации смесь считают взрывобезопасной. Если исходная реакционная газовая смесь частичного окисления согласно изобретению не взрывчата, то это обычно касается и реакционных газовых смесей, образующихся из нее в процессе частичного окисления (ср. с международной заявкой WO 04/007405).
В самом общем случае для способа согласно изобретению характерно, что в исходной газовой смеси реакционного газа В общее содержание компонентов, отличных от пропилена, молекулярного водорода, водяного пара, молекулярного азота и молекулярного кислорода большей частью составляет не более 20 об.-%, либо не более 15 об.-%, либо не более 10 об.-%, либо не более 5 об.-%. До 80 об.-% этих прочих компонентов исходной газовой смеси реакционного газа В могут составлять этан и/или метан. В остальном к таким компонентам могут относиться прежде всего оксиды углерода (CO2, СО) и инертный газ, а также следы побочных продуктов окисления, как то: формальдегида, бензальдегида, метакролеина, уксусной кислоты, пропионовой и метакриловой кислоты и т.д. Само собой разумеется, что к этим возможным прочим компонентам исходной газовой смеси реакционного газа В относятся также этилен, изобутен, н-бутан и н-бутены. Содержание молекулярного азота в исходной газовой смеси реакционного газа В может варьировать в широких пределах. В принципе содержание азота может составлять до 70 об.-%. Как правило, содержание молекулярного азота в исходной газовой смеси реакционного газа В составляет не более 65 об.-%, либо не более 60 об.-%, либо не более 50 об.-%, либо не более 40 об.-%, либо не более 30 об.-%, либо не более 20 об.-%, либо не более 10 об.-%, либо не более 5 об.-%. В принципе, содержание N2 в исходной газовой смеси реакционного газа В может быть исчезающе малым. Минимизация содержания N2 в исходной газовой смеси реакционного газа В минимизирует также объемы перемещаемого и подлежащего сжатию газа при реализации способа согласно изобретению.
Наоборот, разбавление реакционного газа В молекулярным азотом снижает масштабы образования побочного продукта - пропионовой кислоты в реакционной зоне В.
Для гетерогенно-катализируемого частичного дегидрирования пропана в реакционной зоне А можно, в принципе использовать все известные в уровне техники способы гетерогенно-катализируемого частичного дегидрирования пропана до пропилена, как, например, описанные в международных заявках WO 03/076370, WO 01/96271, европейской заявке на патент ЕР-А 117146, международной заявке WO 03/011804, европейской заявке на патент ЕР-А 731077, заявке на патент США US-A 316260, международной заявке WO 01/96270, немецких заявках на патент DE-A 3313573, DE-A 10245585, DE-A 10316039, DE-A 10 2005 009891, DE-A 10 2005 013039, DE-A 10 2005 022798, DE-A 10 2005 009885, DE-A 10 2005 010111, DE-A 10 2005 049699, DE-A 10 2004 032129, немецкой заявке №10 2005 056377.5, немецкой заявке №10 2006 017623.5, немецкой заявке №10 2006 015235.2, немецкой заявке №10 2005 061626.7 и в немецкой заявке №10 2005 057197.2, а также известные из упомянутого в этих публикациях уровня техники. Это обусловлено тем фактом, что окисление (сгорание) молекулярного водорода с молекулярным кислородом в реакционной зоне А в принципе может следовать за любым способом гетерогенно-катализируемого дегидрирования пропана. В принципе, гетерогенно-катализируемое частичное дегидрирование пропана и сжигание молекулярного водорода в пределах реакционной зоны также можно осуществлять в отличных друг от друга, например последовательно подключенных в пространственном отношении, реакторах. При этом целесообразно, что исходная газовая смесь реакционного газа В при реализации способа согласно изобретению не обязательно содержит молекулярный кислород. В самом общем случае реализация способа согласно изобретению в реакционной зоне А возможна как с применением одного единственного реактора, так и, например, с последовательным подключением более чем одного реактора.
В расчете относительно однократного прохождения пропана, поданного в реакционную зону А, через реакционную зону А в принципе возможно обеспечить изотермическое состояние реакционной зоны А путем целенаправленного теплообмена с протекающими за пределами реакционной зоны А текучими (т.е. жидкими или газообразными) теплоносителями. Также возможно, с тем же основанием для расчета, адиабатическое состояние, т.е. в основном без такого целенаправленного теплообмена с протекающими за пределами реакционной зоны А теплоносителями. В последнем случае, применяя меры, рекомендованные в вышеприведенных публикациях, а также те, которые еще предстоит описать, тепловой эффект брутто, рассчитанный относительно однократного прохождения пропана, поданного в реакционную зону А, через реакционную зону А, можно сделать как эндотермическим (отрицательным) или аутотермическим (в основном нулевым), так и экзотермическим (положительным). Равным образом при реализации способа согласно изобретению можно применять все катализаторы, рекомендованные в вышеописанных публикациях, включая описанный в них уровень техники. В принципе, гетерогенно-катализируемое дегидрирование пропана в реакционной зоне А, вне зависимости от того, осуществляют ли его в адиабатическом и/или изотермическом режиме (вдоль реакционной зоны А возможны также изменения от адиабатического режима к изотермическому и наоборот), можно проводить как в реакторе с твердым слоем (или в нескольких, например, подключенных последовательно), так и в реакторе с подвижным слоем, текучим слоем или кипящим слоем (ввиду обратного смешивания последний особенно пригоден для нагрева исходной газовой смеси реакционного газа А до температуры реакции в реакционной зоне а посредством сжигания водорода в реакционном газе А, если уже в исходную смесь реакционного газа А подан молекулярный кислород).
Как правило, для частичного дегидрирования по меньшей мере пропана до пропилена необходимы сравнительно высокие температуры реакции. Получаемый при этом оборот пропана обычно достигает пределов термодинамического равновесия. Обычно температура протекания реакции находится между 300 и 800°С, либо же между 400 и 700°С, либо же между 450 и 650°С. На молекулу пропана, дегидрируемого до пропилена, при этом вырабатывается одна молекула водорода. Согласно изобретению целесообразно, чтобы рабочее давление во всей реакционной зоне А составляло от 1 до 5 бар, предпочтительно - от 1,5 до 4 бар, и выгодно - от 2 до 3 бар. В принципе, рабочее давление в реакционной зоне А может, однако, иметь и более высокие или более низкие значения, чем указано выше. Удаление продукта реакции Н2 и высокие температуры благоприятствуют сдвигу равновесия дегидрирования в пользу пропилена, формирующегося в реакционной зоне А.
Поскольку реакция гетерогенно-катализируемого дегидрирования проходит с увеличением объема, оборот можно повысить, понижая парциальное давление продуктов дегидрирования. Простой способ добиться этого состоит, например, в дегидрировании при пониженном давлении (проведение его при повышенном давлении, однако, как правило, благоприятно сказывается на сроке службы катализатора) и/или в добавлении в основном инертных разбавляющих газов, например водяного пара, который в обычном случае представляет собой газ, инертный относительно реакции дегидрирования. Еще одно преимущество, которое, как правило, дает разбавление водяным паром, состоит в снижении коксования применяемого катализатора, поскольку водяной пар реагирует с образовавшимся коксом по принципу газификации угля. Теплоемкость водяного пара также позволяет компенсировать часть эндотермического характера дегидрирования.
В то время как в ограниченных количествах водяной пар полезен для активности катализаторов частичного дегидрирования в следующей реакционной зоне В, наличие более значительных [его] количеств в реакционной зоне В по уже упомянутым причинам неблагоприятно. Сверх того, согласно изобретению в реакционной зоне А должно происходить окисление по меньшей мере части водорода до водяного пара. Поэтому согласно изобретению предпочтительно, чтобы количество водяного пара, подаваемое в реакционную зону А, составляло относительно реакционного газа А, подаваемого на засыпку катализатора реакционного зоны А, не более 20 об.-%, предпочтительно - не более 15 об.-%, а особо предпочтительно - не более 10 об.-%. Как правило, однако, количество водяного пара, подаваемого в реакционную зону А, рассчитанное таким же образом, составляет не менее 1 об.-%, нередко - не менее 2 об.-%, или не менее 3 об.-%, а чаще - не менее 5 об.-%.
Прочие средства разбавления, пригодные для гетерогенно-катализируемого дегидрирования пропана, - это, например, азот, благородные газы, как то: гелий, неон и аргон, но также и такие соединения, как СО, CO2, метан и этан. Применять все указанные средства разбавления возможно либо сами по себе, либо в форме различных смесей. Постольку поскольку вышеупомянутые газы разбавления образуются при реализации способа с циркуляционным газом согласно изобретению в качестве побочного продукта, либо же их подают в виде свежего газа или компонента свежего газа, при реализации способа согласно изобретению необходим выпуск в соответствующем количестве, ввиду чего соответствующая подача свежего газа согласно изобретению менее предпочтительна. Возможность такого выпуска имеется в различных зонах разделения, в особенности в зонах разделения III и IV.
В принципе, для гетерогенно-катализируемого дегидрирования пропана можно использовать все катализаторы дегидрирования, известные из уровня техники. Их можно грубо подразделить на две группы. Во-первых, это таковые, имеющие оксидную природу (например, оксид хрома и/или оксид алюминия), а во-вторых, это таковые, состоящие из осажденного на носителе, как правило, оксидном (например, оксиде циркония, оксиде алюминия, диоксиде кремния, диоксиде титана, оксиде магния, оксиде лантана и/или оксиде церия), металла, как правило, сравнительно благородного (например, платины). Помимо прочего можно применять все катализаторы дегидрирования, которые рекомендованы в международной заявке WO 01/96270, европейской заявке на патент ЕР-А 731077, немецких заявках на патент DE-A 10211275, DE-A 10131297, международной заявке WO 99/46039, заявке на патент США US-A 4788371, европейской заявке на патент ЕР-А 705136, международной заявке WO 99/29420, заявках на патент США US-A 4220091, US-A 5430220, US-A 5877369, европейской заявке на патент ЕР-А 117146, немецких заявках на патент DE-А 19937196, DE-A 19937105, заявках на патент США US-A 3670044, US-A 6566573 и в международной заявке WO 94/29021, а также в немецкой заявке на патент DE-A 19937107. В частности можно применять катализаторы из примеров 1, 2, 3 и 4 немецкой заявки на патент DE-А 19937107. Наконец, в качестве катализаторов, пригодных к применению для дегидрирования в реакционной зоне А, следует рекомендовать катализаторы, указанные в немецкой заявке №10 2005 044916. Эти катализаторы могут быть единственными, применяемыми в пределах реакционной зоны А при реализации способа согласно изобретению, поскольку они, как правило, катализируют также и горение молекулярного водорода с молекулярным кислородом с образованием воды.
В принципе, засыпка с каталитической активностью в реакционной зоне А может состоять исключительно из формованных изделий, обладающих каталитической активностью. Разумеется, засыпка с каталитической активностью в реакционной зоне А может также состоять и из формованных изделий, обладающих каталитической активностью, разбавленных инертными формованными изделиями. Эти инертные формованные изделия можно изготавливать, например, из обожженных глин (алюмосиликатов) или стеатита (например, С 220 производства фирмы CeramTec), или прочих (предпочтительно - в основном не имеющих пор) материалов класса высокотемпературной керамики, как то: оксидов алюминия, диоксида кремния, диоксида титана, оксида магния, оксида лантана, оксида церия, смешанного оксида цинка и алюминия, диоксида тория, диоксида циркония, карбида кремния или прочих силикатов, как то: силиката магния, или смесей вышеупомянутых материалов.
Вышеупомянутые материалы можно при реализации способа согласно изобретению использовать также для инертной покровной, а при необходимости и для замыкающей засыпки твердого слоя катализатора.
Речь при этом идет о катализаторах дегидрирования, которые содержат от 10 до 99,9 вес.-% оксида циркония, от 0 до 60 вес.-% оксида алюминия, диоксида кремния и/или диоксида титана и от 0,1 до 10 вес.-% по меньшей мере одного элемента первой или второй главной группы, одного из элементов третьей побочной группы, одного элемента восьмой побочной группы периодической системы элементов, лантан и/или олово, с тем условием, чтобы сумма весовых процентных долей составляла 100 вес.-%.
Особо удобен в применении также катализатор дегидрирования, использованный в примерах исполнения настоящей публикации.
В предпочтительной форме исполнения вышеупомянутые катализаторы дегидрирования содержат по меньшей мере один элемент VIII побочной группы, по меньшей мере один элемент из I и II основной группы, по меньшей мере один элемент из III и/или IV основной группы и по меньшей мере один элемент III побочной группы, включая лантаноиды и актиноиды. Предпочтительно, чтобы в качестве элемента VIII побочной группы активная масса катализаторов дегидрирования содержала платину и/или палладий, особо предпочтительно - платину. Предпочтительно, чтобы в качестве элементов I и II основной группы активная масса вышеупомянутых катализаторов дегидрирования содержала калий и/или цезий. В качестве элементов III побочной группы, включая лантаноиды и актиноиды, активная масса вышеупомянутых катализаторов дегидрирования предпочтительно содержит лантан и/или церий. В качестве элементов III и/или IV основной группы активная масса вышеупомянутых катализаторов дегидрирования предпочтительно содержит один или несколько элементов, принадлежащих к группе, включающей бор, галлий, кремний, германий, олово и свинец, особо предпочтительно - олово.
Крайне предпочтительно, чтобы активная масса вышеупомянутых катализаторов дегидрирования содержала по меньшей мере по одному представителю вышеупомянутых групп элементов.
В общем случае катализаторы дегидрирования могут представлять собой нити (обычный диаметр составляет от 0,1 либо же 1 до 10 мм, предпочтительно - от 1,5 до 5 мм; обычная длина от 1 до 20 мм, предпочтительно - от 3 до 10 мм), таблетки (предпочтительно тех же размеров, что и нити) и/или катализаторные кольца (внешний диаметр и высота в каждом случае обычно от 2 до 30 мм или до 10 мм, целесообразная толщина стенок - от 1 до 10 мм, или до 5 мм, или до 3 мм). Для проведения гетерогенно-катализируемого дегидрирования в кипящем слое (либо же в текучем или подвижном слое) следует, соответственно, применять более тонкодисперсный катализатор. Согласно изобретению для реакционной зоны А предпочтительно применение твердых слоев катализатора.
При реализации способа согласно изобретению нет принципиальных ограничений ни относительно геометрических параметров катализаторов (особенно в случае катализаторов на носителях), ни относительно геометрических параметров инертных формованных изделий. Особо часто применяют сплошные цилиндры, полые цилиндры (кольца), шарики, конусы, пирамиды и кубы, а также нити, колеса, звездочки и монолиты.
Максимальный размер формованных изделий из катализатора, а также инертных формованных изделий (самый длинный отрезок прямой, соединяющий две точки, находящиеся на поверхности формованного изделия) при этом может составлять от 0,5 мм до 100 мм, часто - от 1,5 мм до 80 мм, и во многих случаях от 3 мм до 50 мм либо же до 20 мм.
Как правило, когда катализаторы дегидрирования (в частности, таковые, рекомендованные, а особенно - приведенные в качестве примеров в немецкой заявке на патент DE-A 19937107), обычно способны катализировать как дегидрирование пропана, но также и реакцию горения молекулярного водорода и пропана (пропилена) с молекулярным кислородом. При этом сжигание водорода проходит и значительно быстрее, чем дегидрирование пропана, и значительно быстрее, чем сгорание такового в случае конкуренции за эти катализаторы (т.е. при таких условиях они обеспечивают самую низкую - со значительным запасом - энергию активации для сгорания молекулярного водорода). В свою очередь, сгорание пропана (пропилена) на этих катализаторах опять же, как правило, проходит быстрее, чем дегидрирование.
Вышеописанная связь позволяет при реализации способа согласно изобретению обойтись в реакционной зоне А всего одним типом катализатора.
При этом целесообразно следить в реакционной зоне А за тем, чтобы в тех случаях, когда реакционный газ А содержит молекулярный кислород и соприкасается с вышеописанными катализаторами, реакционный газ А содержал по меньшей мере вдвое большее количество молекулярного водорода, чем молекулярного кислорода, поскольку в ином случае возрастает риск сгорания пропана и/или пропилена, а это снижает выход конечного продукта относительно оборота сырья (пропана).
Само собой разумеется, что в качестве катализаторов, применяемых в реакционной зоне А для сжигания молекулярного водорода с образованием воды, можно также использовать и те, которые специфически катализируют эту реакцию. Такие катализаторы описаны, например, в заявках на патент США US-A 4788371, US-A 4886928, US-A 5430209, US-A 5530171, US-A 5527979 и US-A 5563314.
Для реализации гетерогенно-катализируемого дегидрирования пропана, в принципе, можно применять все известные из уровня техники типы реакторов и варианты способа.
Описания таких вариантов способа содержатся, например, во всех цитированных в связи с катализаторами дегидрирования публикациях уровня техники, а также принадлежат к самому уровню техники, описанному в начале настоящего текста.
Сравнительно подробное описание способов дегидрирования, годных к применению согласно изобретению, содержит также Catalytica® Studies Division, Oxidative Deyhdrogenation and Alternative Dehydrogenation Process, Study Number 4192 OD, 1993, 430 Ferguson Drive, Montain View, California, 94043-5272 U.S.A.
Поскольку для гетерогенно-катализируемого дегидрирования пропана необходимы сравнительно высокие температуры реакции, при реализации способа согласно изобретению в реакционной зоне А, как правило, в небольших количествах образуются высококипящие высокомолекулярные органические соединения, вплоть до углерода, которые осаждаются на поверхности катализатора и тем самым деактивируют ее. Чтобы свести к минимуму это побочное явление, целесообразно разбавлять содержащий пропан реакционный газ А, направляемый при повышенной температуре на поверхность катализатора для гетерогенно-катализируемого дегидрирования, водяным паром. При реализации способа согласно изобретению этого можно особо просто и изящно добиться, проводя отделение пропана от абсорбата в зоне разделения IV методом отгонки (стриппинга) с помощью водяного пара или содержащего водяной пар газа (например, инертного газа), при этом возвращая стрип-газ, нагруженный пропаном (нагруженный пропаном водяной пар), в реакционную зону А в виде циркуляционного газа пропана. Использование водяного пара позволяет при неизменных в остальном условиях полностью или частично удалить осаждающийся углерод по принципу газификации угля и продлить таким образом срок службы катализатора.
Еще одна возможность удалить осаждающиеся соединения углерода состоит в том, чтобы время от времени промывать катализатор дегидрирования (а также, при необходимости, катализатор, отдельно применяемый для сжигания водорода) при повышенной температуре газом, содержащим кислород (целесообразно - в отсутствие углеводородов), и фактически сжигать таким образом осажденный углерод. Образование углеродных отложений можно, однако, также в определенной степени подавить (и таким образом продлить срок службы катализатора), добавляя молекулярный водород к пропану, подлежащему гетерогенно-катализируемому дегидрированию, до прохождения пропана через катализатор дегидрирования. При реализации способа согласно изобретению этого, например, просто добиться, отделяя через разделительную мембрану в зоне разделения III (или после зоны разделения III) от остаточного газа, получаемого в зоне разделения II, еще, возможно, содержащийся в этом газе молекулярный водород и возвращая водород в реакционную зону А. Разумеется, здесь можно также применять молекулярный водород и из других источников. Возможно, например, наличие молекулярного водорода и в возвращаемом (возможно) в реакционную зону А остаточном циркуляционном газе.
Разумеется, также существует возможность добавлять к подлежащему дегидрированию пропану смесь из водяного пара и молекулярного водорода.
Добавление молекулярного водорода к гетерогенно-катализируемому дегидрированию пропана также снижает образование таких побочных продуктов как аллен (пропандиен), пропин и ацетилен.
Таким образом, в реакционную зону А для формирования реакционного газа А, подлежащего проведению по меньшей мере через один слой катализатора (в настоящей публикации этот газ также называют газовой смесью загрузки реакционной зоны А или исходной газовой смесью реакционной зоны А), в простейшем случае можно добавлять только свежий пропан и газ циркуляции пропана. Последний при этом уже может содержать то количество молекулярного кислорода (например, в тех случаях, когда пропан в зоне разделения IV отделяют от абсорбата методом стриппинга, а стрипп-газ содержит добавленный молекулярный кислород, например, в виде воздуха), которое необходимо, чтобы обеспечить сгорание водорода, потребное согласно изобретению в реакционной зоне А. Исходная газовая смесь реакционного газа А может, однако, состоять также только из свежего пропана, газа циркуляции пропана и остаточного циркуляционного газа, причем последний также может содержать молекулярный кислород.
В указанных выше случаях целесообразно, чтобы газ циркуляции пропана содержал как раз такое количество водорода, чтобы он, совместно с водяным паром, образовавшимся в реакционной зоне А при сгорании водорода, мог проявить свои полезные для всего процесса свойства. В вышеописанных случаях в подаче в реакционную зону А прочих газообразных потоков острой необходимости нет. Желательное химическое преобразование в реакционной зоне А происходит при обычном прохождении через нее реакционного газа А.
Разумеется, чтобы обеспечить благоприятный эффект, описанный в настоящей публикации, для образования реакционного газа А, подлежащего проведению через по меньшей мере один твердый слой катализатора, в дополнение к свежему пропану, газу циркуляции пропана и, при необходимости, остаточному циркуляционному газу можно также дополнительно подавать водяной пар и/или дополнительный молекулярный водород. Молярное соотношение молекулярного водорода и пропана в газовой смеси загрузки реакционной зоны А, как правило, не превышает 5, большей частью не больше 3, во многих случаях не более 1 или 0,1.
Молярное соотношение водяного пара и пропана в газовой смеси загрузки реакционной зоны А может составлять, например, от 0 до 30. Целесообразно, чтобы оно составляло от 0,05 до 2, а выгодно - от 0,1 до 0,5.
Кроме того, в зависимости от потребности в газовую смесь загрузки реакционной зоны А можно дополнительно подавать молекулярный кислород (в чистом виде и/или в виде смеси с инертным газом) и/или дополнительный инертный газ. Желательное химическое преобразование в реакционной зоне А опять же происходит при обычном прохождении через нее реакционного газа А (газа загрузки реакционной зоны А) без подачи других газообразных потоков на протяжении пути реакции. В настоящей публикации под ”путем реакции” в реакционной зоне А подразумевают в зависимости от оборота гетерогенно-катализированного дегидрирования путь протекания через реакционную зону А того пропана, который добавляют в реакционный газ А перед первым проходом реакционного газа А через по меньшей мере один слой катализатора реакционной зоны А.
Надлежащая форма реактора для такого гетерогенно-катализированного дегидрирования пропана и с простым прохождением газовой смеси загрузки через реакционную зону А, и без промежуточной подачи газа - это, например, трубчатый реактор с твердым слоем или реактор в виде пучка труб. При этом катализатор дегидрирования находится в реакторной трубе или в пучке реакторных труб в виде твердого слоя. Если сгорание водорода в реакционной зоне А, необходимое согласно изобретению, настроено так, что происходящая в реакционной зоне А реакция брутто имеет эндотермический тепловой баланс, то согласно изобретению целесообразно обогревать реакционные трубы снаружи (разумеется, при необходимости возможно также охлаждение). Это можно обеспечить, например, сжигая в окружающем реакторные трубы пространстве газ, наприме, углеводород, как то: метан. Выгодно использовать эту прямую форму нагрева контактных труб только на первых 20-30% засыпки твердого слоя, а остальную длину засыпки нагревать до необходимой температуры реакции посредством выделяемого при сгорании теплового излучения. Таким образом можно добиться примерно изотермического протекания реакции. Надлежащие значения внутреннего диаметра реакторной трубы составляют примерно от 10 до 15 см. Типичный реактор дегидрирования в виде пучка труб включает в себя от 300 до 1000 труб или более. Температура внутри реакторных труб находится в диапазоне от 300 до 800°С, предпочтительно - в диапазоне от 400 до 700°С. Целесообразно подавать исходную смесь реакционного газа А в трубчатый реактор, предварительно разогрев ее до температуры реакции. При выходе продукта-газа (газовой смеси) А из реакторной трубы ее температура может быть на 50-100°С ниже. Эта температура выхода может, однако, быть и выше или находиться на том же уровне. В рамках вышеупомянутого способа работы целесообразно применение оксидных катализаторов дегидрирования на основе оксида хрома и/или оксида алюминия. Катализатор дегидрирования применяют большей частью в неразбавленном виде. В промышленном масштабе можно параллельно эксплуатировать несколько таких реакторов в виде пучка труб и использовать их газы продукции А в виде смеси для подачи в реакционную зону В. При необходимости в режиме дегидрирования могут находиться только два из этих реакторов, в то время как в третьем реакторе проводят регенерацию катализатора.
Простое прохождение газа загрузки через реакционную зону А можно также осуществлять и в реакторе подвижным слоем, текучим слоем или кипящим слоем, как это, например, описано в немецкой заявке на патент DE-A 10245585, а также в процитированной в этой публикации по этому поводу литературе.
В принципе, реакционная зона А способа согласно изобретению может также состоять, например, из двух участков. Такое строение реакционной зоны А рекомендуется, в частности, тогда, когда в состав газа загрузки реакционной зоны А не входит молекулярный кислород.
В этом случае на первом участке может происходить гетерогенно-катализируемое дегидрирование как таковое, а на втором участке, после промежуточной подачи молекулярного кислорода и/или смеси молекулярного кислорода с инертным газом, необходимое согласно изобретению гетерогенно-катализируемое сгорание водорода.
В самом общем случае эксплуатацию реакционной зоны А при реализации способа согласно изобретению осуществляют так, чтобы при расчете на однократное прохождение реакционной зоны А подвергать реакции дегидрирования в реакционной зоне А от не менее чем 5 моль-% до не более чем 60 моль-%, предпочтительно - от не менее чем 10 моль-% до не более чем 50 моль-%, особо предпочтительно - от не менее чем 15 моль-% до не более чем 40 моль-%, а крайне предпочтительно - от не менее чем 20 моль-% до не более чем 35 моль-% общего количества пропана, поданного в реакционную зону А. Оборот в реакционной зоне А, ограниченный до такой степени, обычно достаточен согласно изобретению, поскольку остающееся количество не прошедшего преобразование пропана исполняет в следующей реакционной зоне В, в основном, роль разбавляющего газа, а при дальнейшей работе по способу согласно изобретению ее можно практически без потерь вернуть в реакционную зону А. Преимущество способа работы с низким оборотом пропана состоит в том, что при однократном прохождении реакционного газа А через реакционную зону А количество теплоты, необходимое для эндотермического дегидрирования, сравнительно низко, а для достижения этого оборота достаточны сравнительно невысокие температуры реакции.
Согласно изобретению может быть, как уже сказано, целесообразно проводить дегидрирование пропана в реакционной зоне А (например, со сравнительно низким оборотом пропана) в адиабатическом или практически адиабатическом режиме. Это означает, что газовую смесь загрузки реакционной зоны А, как правило, сначала нагревают до температуры от 500 до 700°С (либо же от 550 до 650°С), например, путем прямой обработки окружающих ее стенок пламенем. В норме при этом одного единственного адиабатического прохождения через слой катализатора будет достаточно, чтобы добиться как желательного оборота дегидрирования, так и потребного согласно изобретению сгорания водорода, причем реакционный газ, в зависимости от количественного соотношения эндотермического дегидрирования и экзотермического сгорания водорода, с точки зрения баланса брутто будет нагреваться, охлаждаться или вести себя термически нейтрально. Согласно изобретению предпочтителен адиабатический способ работы, при котором реакционный газ охлаждается при однократном прохождении на 30-200°С. При необходимости возможно дожигание молекулярного водорода, образовавшегося в рамках гетерогенно-катализируемого дегидрирования на втором участке реакционной зоны А с использованием подданного на промежуточном этапе молекулярного кислорода. Это сжигание также можно проводить в адиабатическом режиме.
Следует отметить, что при этом, в особенности при адиабатическом режиме эксплуатации, достаточен один реактор в виде шахтной печи, эксплуатируемый как реактор с твердым слоем, через который в осевом и/или радиальном направлении протекает реакционный газ А.
В простейшем случае речь идет о сплошном закрытом объеме реактора, например о баке, внутренний диаметр которого составляет от 0,1 до 10 м, возможно также - от 0,5 до 5 м, и в котором на несущей конструкции (например, на решетке) расположен твердый слой катализатора. При этом через загруженный катализатором объем реактора, который при адиабатическом режиме эксплуатации в основном снабжен теплоизоляцией, в осевом направлении протекает горячий реакционный газ А, содержащий пропан. Частицы катализатора при этом могут иметь как шарообразную, так и кольцевидную или нитевидную форму. Поскольку в этом случае объем реакции обеспечивают с помощью очень дешевого аппарата, предпочтительны все геометрические параметры катализатора, обеспечивающие минимальное падение давления. В первую очередь, это геометрические параметры, обеспечивающие создание больших полостей или структурированность, как, например, монолиты или тела, имеющие строение сот. Для обеспечения радиального потока содержащего пропан реакционного газа А реактор может состоять, например из двух заключенных в оболочку цилиндрических решеток, концентрически вставленных друг в друга, а засыпка катализатора может быть размещена в кольцевой щели. В случае эксплуатации в адиабатическом режиме металлическую оболочку при необходимости также снабжают термоизоляцией.
В качестве засыпки катализатора для гетерогенно-катализируемого дегидрирования пропана согласно изобретению особо удобно применять катализаторы, описание которых изложено в немецкой заявке на патент DE-A 19937107, прежде всего, приведенные в ней в качестве примеров, а также смеси их с формованными изделиями, инертными относительно гетерогенно-катализируемого дегидрирования.
Вышеупомянутые катализаторы можно, например, после длительной эксплуатации простым способом регенерировать, сначала (на первом этапе регенерации) проводя через слой катализатора разбавленный азотом и/или (предпочтительно) водяным паром воздух, имеющий на входе температуру от 300 до 600°С, нередко - от 400 до 450°С. При этом нагрузка слоя катализатора регенерационным газом (например, воздухом) может составлять, например, от 50 до 10000 ч-1, а содержание кислорода в регенерационном газе - от 0,1 или 0,5 до 20 об.-%.
Затем, на следующих этапах регенерации, при в остальном неизменных условиях регенерации в качестве регенерационного газа можно использовать воздух. С точки зрения техники применения перед регенерацией рекомендуется продуть катализатор инертным газом, например N2.
Затем, как правило, полезно продолжить регенерацию с использованием чистого молекулярного водорода или молекулярного водорода, разбавленного инертным газом (предпочтительно водяным паром и/или азотом, содержание водорода должно удовлетворять условию ≥1 об.-%) при в остальном неизменных условиях регенерации.
Во всех случаях гетерогенно-катализируемое дегидрирование пропана в реакционной зоне А способа согласно изобретению можно проводить со сравнительно низким оборотом пропана (не более 30 моль-%) при той же нагрузке (слоя) катализатора (как реакционным газом вообще, так и содержащимся в таковом пропаном), что и в вариантах с высоким оборотом пропана (более 30 моль-%). Эта нагрузка реакционным газом А может составлять, например, от 100 до 40000 или до 10000 ч-1, часто - от 300 до 7000 ч-1, то есть во многих случаях - от 500 до 4000 ч-1. Указанные значения нагрузки можно также использовать и для слоев катализатора, при необходимости специфически используемых для сжигания водорода.
Особо изящно можно реализовать в реакционной зоне А гетерогенно-катализируемое дегидрирование пропана (особенно при значениях оборота пропана от 15 до 35 моль-% на однократное прохождение) в многоступенчатом реакторе, ввиду чего предпочтительно, чтобы в состав реакционной зоны А входил по меньшей мере один такой реактор.
Такой реактор включает в себя более одного твердого слоя катализатора, катализирующего дегидрирование, слои которого расположены последовательно друг за другом. Число слоев катализатора может составлять, например, от 1 до 20, целесообразно - от 2 до 8 либо же от 3 до 6. При возрастании числа ступеней все легче повышать значения оборота пропана. Предпочтительно располагать слои катализатора друг за другом радиально или по оси. С точки зрения техники применения целесообразно применять в таком многоступенчатом реакторе тип твердого слоя катализатора.
В простейшем случае твердые слои катализатора в реакторе, имеющем форму шахтной печи, располагают по оси или в кольцевых щелях между цилиндрическими решетками, концентрически вставленным друг в друга. Однако можно также размещать кольцевые щели в сегментах друг над другом и проводить реакционный газ после радиального прохода одного сегмента в следующий, расположенный выше или ниже.
На пути от одного слоя катализатора к другому в многоступенчатом реакторе целесообразно подвергать реакционный газ А промежуточному нагреву, например, проводя его над теплообменными поверхностями (например, ребрами), нагретыми горячими газами, или проводя его через нагретые горящими газами трубы. При необходимости также возможно подобное же промежуточное охлаждение.
Если шахтный реактор в остальном работает в адиабатическом режиме, то для оборотов пропана, не превышающих 30 моль-%, прежде всего при использовании катализаторов, описанных в немецкой заявке на патент DE-А 19937107, в частности приведенных в качестве примеров, достаточно вводить в реактор дегидрирования реакционную газовую смесь, предварительно нагрев ее до температуры от 450 до 550°С (предпочтительно - от 450 до 500°С), и поддерживать ее температуру внутри многоступенчатого реактора в этом диапазоне. Это значит, что все дегидрирование пропана можно таким образом проводить при чрезвычайно низких температурах, что особо благоприятно сказывается на сроке службы твердых слоев катализатора между двумя процедурами регенерации. Для более высоких оборотов пропана реакционную газовую смесь целесообразно вводить в реактор дегидрирования, предварительно нагрев ее до более высокой температуры (она может достигать 700°С), и поддерживать ее температуру внутри многоступенчатого реактора в этом диапазоне повышенной температуры.
Еще выгоднее проводить представленный выше промежуточный нагрев прямым образом (это позволяет работать в аутотермическом режиме). Для этого в реакционный газ А либо уже до протекания через первый слой катализатора (например, в качестве компонента остаточного циркуляционного газа и/или газа циркуляции пропана, в этом случае целесообразно, чтобы исходная смесь реакционного газа А содержала добавку молекулярного водорода), и/или между следующими друг за другом слоями катализатора в ограниченном объеме добавляют молекулярный кислород. Таким образом можно обеспечить сгорание (как правило, катализируемое самими катализаторами дегидрирования) содержащегося в реакционном газе А молекулярного водорода, образовавшегося в процессе гетерогенно-катализируемого дегидрирования пропана или добавленного в реакционный газ А, в особо целенаправленном и контролируемом режиме (с точки зрения техники применения может оказаться целесообразным ввести в многоступенчатый реактор слои катализатора, загрузка которых особо специфичным (избирательным) образом катализирует сгорание водорода. В качестве таких катализаторов можно использовать, например, катализаторы, описанные в публикациях заявок на патент США US-A 4788371, US-A 4886928, US-A 5430209, US-A 5530171, US-A 5527979 и US-A 5563314; такие катализаторы могут, например, располагаться в многоступенчатом реакторе в слоях, перемежающихся со слоями, содержащими катализатор дегидрирования; эти слои также пригодны для описанного выше сжигания водорода на втором участке реакционной зоны А). Выделяющаяся при этом теплота реакции позволяет, в зависимости от количества сожженного молекулярного водорода, вести гетерогенно-катализируемое дегидрирование пропана в общем в экзотермическом, или в общем в аутотермическом (тепловой баланс брутто в основном равен нулю), или в общем в эндотермическом режиме. При возрастании выбранного времени пребывания реакционного газа в слое катализатора такое дегидрирование пропана возможно при снижении температуры или поддержании ее в основном на постоянном уровне, что особенно продлевает срок службы между двумя этапами регенерации и согласно изобретению предпочтительно.
В частности, в тех случаях, когда молекулярный водород сжигают как в процессе гетерогенно-катализируемого дегидрирования пропана (фаза 1), так и после гетерогенно-катализируемого дегидрирования пропана (фаза 2) согласно изобретению целесообразно, чтобы реакционный газ реакционной зоны А после гетерогенно-катализируемого дегидрирования в том количестве, в котором его подают в реакционную зону А на окончательное сжигание водорода (часть его можно возвращать в качестве циркуляционного газа дегидрирования в реакционную зону А как один из потоков подачи, содержащих пропан), сначала охладили путем непрямого теплообмена с компонентами смеси подачи реакционного газа А, отличными от циркуляционного газа пропана и циркуляционного газа дегидрирования, а после завершающего сжигания молекулярного водорода в реакционной зоне А также путем непрямого теплообмена, но уже с газом циркуляции пропана охлаждали газ продукта А, прежде чем подвергнуть его дальнейшей обработке. Это справедливо особенно тогда, когда общий режим реакционной зоны А экзотермический.
В общем случае подачу кислорода, описанную выше, следует осуществлять так, чтобы содержание кислорода в реакционном газе А относительно содержащегося в нем же молекулярного водорода составляло от 0,5 до 50, либо же до 40, либо же до 30, а предпочтительно - от 10 до 25 об.-%. В качестве источника кислорода, как уже было указано ранее, можно использовать чистый молекулярный кислород (предпочтительно согласно изобретению) или молекулярный кислород, разбавленный инертным газом, например СО, CO2, N2, и/или инертными газами, в особенности - воздух. Согласно изобретению предпочтительно подавать молекулярный кислород в виде газа, который содержит не более чем 30 об.-%, предпочтительно - не более чем 25 об.-%, целесообразно - не более чем 20 об.-%, особо целесообразно - не более чем 15 об.-%, еще более благоприятно - не более чем 10 об.-%, а особо предпочтительно - не более чем 5 об.-% других (отличных от молекулярного кислорода) газов. Особо целесообразно осуществлять указанную подачу кислорода в чистом виде.
Поскольку сгорание 1 моль молекулярного водорода с образованием H2O дает примерно вдвое больше (ок. 240 кДж/моль) энергии, чем требуется на дегидрирование 1 моль пропана с образованием пропилена и Н2 (ок. 120 кДж/моль), аутотермический режим реакционной зоны А, описанный выше, в адиабатическом многоступенчатом реакторе хорошо согласуется с желательными преимуществами способа работы согласно изобретению, для этого требуется сжигание количества водорода, соответствующего примерно 50 моль-% молекулярного водорода, образовавшегося в рамках дегидрирования в реакционной зоне А.
Выгода способа согласно изобретению заметна не только тогда, когда в реакционной зоне А сжигают водород в количестве примерно 50 моль-% от образовавшегося в реакционной зоне А. Эта выгода имеется уже тогда, когда в реакционной зоне А сжигают водород в количестве от 25 до 95 моль-%, либо же до 100 моль-%, или от 30 до 90 моль-%, или от 35 до 85 моль-%, или от 40 до 80 моль-%, или от 45 до 75 моль-%, или от 50 до 70 моль-%, или от 35 до 65 моль-%, или от 40 до 60 моль-%, или от 45 до 55 моль-% от образовавшегося в реакционной зоне А количества водорода образованием воды (предпочтительно - работая в описанном выше адиабатическом режиме многоступенчатого реактора).
Как правило, подачу кислорода, описанную выше, следует осуществлять так, чтобы содержание кислорода в реакционном газе А относительно содержащегося в нем же молекулярного пропана и пропилена составляло от 0,01 или 0,5 до 3 об.-%.
Изотермические качества гетерогенно-катализируемого частичного дегидрирования пропана можно также улучшить посредством размещения в многоступенчатом реакторе между слоями катализатора замкнутых встроенных структур (например, в виде труб), которые перед заполнением целесообразно, но не обязательно, подвергнуть эвакуации. Такие встроенные структуры можно также размещать в конкретном слое катализатора. Эти структуры содержат надлежащие твердые вещества или жидкости, которые испаряются или плавятся при превышении определенной температуры и таким образом потребляют тепло, а когда температура опускается ниже этого значения, снова конденсируются и при этом выделяют тепло.
Возможность нагрева газовой смеси загрузки для гетерогенно-катализируемого дегидрирования пропана до необходимой температуры реакции в реакционной зоне А состоит также в том, чтобы при вхождении в реакционную зону А сжечь часть содержащегося в ней пропана и/или Н2 с помощью содержащейся в газовой смеси загрузки молекулярного кислорода А (например, на надлежащих катализаторах горения со специфическим действием, например, путем простого проведения над ними и/или через них) и посредством выделившейся при этом теплоты сгорания обеспечить нагрев до желательной для дегидрирования температуры реакции (такой способ работы (как уже упомянуто) целесообразен, в частности, в реакторе с кипящим слоем).
Согласно изобретению предпочтительно адиабатическое (с теплоизоляцией от наружной среды) исполнение реакционной зоны А. Соответственно вышеупомянутому, реакционную зону А способа согласно изобретению можно организовать, как это описано в немецких заявках на патент DE-A 10 2004 032129 и DE-А 102005 01339, с той, однако, разницей, что в качестве газовой смеси загрузки реакционной зоны А применяют смесь из свежего пропана, газа циркуляции пропана, содержащего водяной пар, и остаточного циркуляционного газа, при необходимости содержащего молекулярный кислород. При этом реакционную зону А реализуют в виде многоступенчатого (предпочтительно адиабатического) реактора, в котором слои катализатора располагаются друг за другом радиально или по оси. Целесообразно, чтобы число ступеней катализаторных слоев в таком многоступенчатом реакторе равнялось трем. При этом предпочтительно, чтобы гетерогенно-катализируемое дегидрирование пропана осуществляли в аутотермическом режиме. Для этого к смеси загрузки реакционной зоны А после прохождения первого (твердого) слоя катализатора, а также между последующими в направлении потока твердыми слоями катализатора в направлении потока реакционного газа в ограниченном количестве добавляют молекулярный кислород или содержащую таковой смесь с инертным газом (например, как это описано в немецкой заявке №10 2006 017623.5). Таким образом обеспечивают катализируемое самими катализаторами дегидрирования ограниченное сгорание водорода, образованного в процессе гетерогенно-катализируемого дегидрирования пропанам (а также, при необходимости, в чрезвычайно малых количествах пропана и/или пропилена), экзотермический тепловой баланс какового сгорания в основном поддерживает температуру дегидрирования.
При этом целесообразно проводить гетерогенно-катализированное дегидрирование пропана катализом в основном в распределенном на три ступени катализатора так, чтобы оборот вводимого в реактор пропана исходя из однократного прохождения реактора составлял ок. 20 моль.-% (разумеется, он может составлять при реализации способа согласно изобретению также и 30 моль.-%, или 40 моль.-%, или 50 моль.-%, или более). При этом в норме достигают значения избирательности образования пропилена, составляющего ок. 90 моль.-%. Максимальный вклад отдельной ступени в оборот с ростом длительности эксплуатации возрастает спереди кзади в направлении потока. Как правило, регенерацию засыпки катализатора проводят, прежде чем третья ступень в направлении потока станет давать максимальный вклад. Целесообразно проводить регенерацию тогда, когда достигнута одинаковая степень коксообразования всех ступеней.
В завершение в одном из (твердых) слоев катализатора, подключенных вслед за дегидрированием, после происшедшего ранее промежуточного ввода кислорода можно осуществлять в основном чистое сгорание молекулярного водорода. В принципе, его можно доводить до той степени, когда газ продукта А, выходящий из реакционной зоны А, более в основном вообще не содержит молекулярного водорода.
В самом общем случае для описанного выше гетерогенно-катализированного частичного дегидрирования пропана выгодно, если нагрузка общего количества катализатора (сумма по всем слоям) общим количеством пропана и пропилена составляет не менее 500 Нл/л·ч и не более 20000 Нл/л·ч (типичные значения - от 1500 Нл/л·ч до 2500 Нл/л·ч). Максимальную температуру реакции в пределах конкретного твердого слоя катализатора при этом целесообразно поддерживать в пределах 500°С-600°С (либо же 650°С).
Недостаток описанного выше способа с использованием смеси загрузки реакционного газа А, уже содержащей молекулярный кислород, состоит в том, что практически все катализаторы, которые катализируют дегидрирование пропана, катализируют также и сгорание пропана и пропилена с молекулярным кислородом (полное окисление пропана и пропилена до оксидов углерода и водяного пара).
На это, помимо уже описанной меры с добавлением в газовую смесь загрузки реакционного газа А газа, содержащего молекулярный водород в соответствии с немецкой заявкой на патент DE-A 10211275, можно также ответить, разделяя отбираемый из зоны дегидрирования (за которой в реакционной зоне А может следовать еще одна зона для сжигания молекулярного водорода) газ продукции на две части идентичного состава, с тем чтобы подавать на частичное окисление в качестве продукта-газа А или продукта-газа А* лишь одну из этих частей (при необходимости - после осуществленного ранее частичного или полного сжигания содержащегося в ней молекулярного водорода), а вторую часть возвращать на дегидрирование в качестве компонента реакционного газа А (как правило - газовой смеси загрузки реакционного газа А).
Молекулярный водород, содержащийся в этом циркуляционном газе дегидрирования, происходящем из дегидрирования, должен в этом случае защитить пропан и, возможно, пропилен, содержащийся в газовой смеси загрузки для реакционной зоны А, от содержащегося в этой же смеси молекулярного кислорода. Эта защита основана на том, что, как уже сказано, сжигание молекулярного водорода с образованием воды, катализируемое теми же самыми катализаторами, в смысле кинетики более предпочтительно, чем полное сгорание пропана и/или пропилена.
Согласно теоретическому изложению немецкой заявки на патент DE-A 10211275 целесообразно вводить циркуляционный газ дегидрирования по принципу струйного насоса (этот принцип называют также петлеобразным режимом; роль ведущего потока может исполнять газ циркуляции пропана). Сверх того в указанной публикации упомянута возможность добавлять в газовую смесь загрузки реакционной зоны А в качестве еще одной степени защиты от окисления дополнительные количества молекулярного водорода (это также может быть, например, молекулярный водород, отделенный с помощью разделительной мембраны от остаточного газа, поступающего с зоны разделения II и еще содержащего молекулярный водород).
При реализации способа согласно изобретению типичные смеси загрузки реакционного газа А (образующиеся в реакционной зоне А реакционные газы А) характеризуются следующим составом:
от 55 до 80 об.-% пропана,
от 0,1 до 20 об.-% пропилена,
от 0 до 10 об.-% Н2,
от 0 до 5 об.-% O2,
от 0 до 20 об.-% N2 и
от 5 до 15 об.-% H2O.
Предпочтительные для способа согласно изобретению смеси загрузки реакционного газа А (газовая смесь, входящая в первый находящийся в реакционной зоне А твердый слой катализатора в направлении потока реакционного газа А) характеризуются следующим составом:
от 55 до 80 об.-% пропана,
от 0,1 до 20 об.-% пропилена,
от 2 до 10 об.-% Н2,
от 1 до 5 об.-% CO2,
от 0 до 20 об.-% N2 и
от 5 до 15 об.-% H2O.
Доля компонентов смесей загрузки реакционного газа А, отличных от компонентов, приведенных в списке, в совокупности, как правило, составляет не более 10 об.-%, а предпочтительно - не более 6 об.-%. Типичные продукты-газы А характеризуются следующим составом:
от 30 до 50 об.-% пропана,
от 15 до 30 об.-% пропилена,
от 0 до 10 об.-% H2, предпочтительно - от 0 до 6 об.-% Н2,
от 10 до 25 об.-% H2O,
от 0 до 1 об.-% CO2 и
от 0 до 35 об.-% N2.
Доля компонентов реакционного газа А, отличных от компонентов, приведенных в списке, в совокупности, как правило, составляет не более 10 об.-%.
Температура продукта-газа А, выходящего из реакционной зоны А, обычно составляет от 300 до 800°С, предпочтительно - от 400 до 700°С, а особо предпочтительно - от 450 до 650°С.
Давление продукта-газа А, выходящего из реакционной зоны А, составляет от 1 до 5 бар, предпочтительно - от 1,5 до 4 бар, и выгодно - от 2 до 3 бар.
Согласно изобретению продукт-газ А либо без дальнейшего отделения побочных продуктов (предпочтительно, однако, после механического отделения содержащихся в нем частиц твердого вещества согласно теоретическому изложению немецкой заявки на патент DE-A 10316039), либо после отделения путем конденсации (обусловленного прямым и/или непрямым охлаждением) отделения части или всего количества содержащегося в продукте-газе А водяного пара (т.е. в виде продукта-газа А*) используют для загрузки по меньшей мере одного реактора окисления в реакционной зоне В с реакционным газом В.
Вне зависимости от того, предпринимают ли отделение воды из продукта-газа А путем конденсации, согласно изобретению целесообразно по меньшей мере охладить горячий продукт-газ А перед дальнейшим применением его в реакционной зоне В путем непрямого теплообмена с газом циркуляции пропана, а также, при необходимости, с другими компонентами смеси загрузки реакционного газа А (обычно за исключение циркуляционного газа дегидрирования), и таким образом нагреть газ циркуляции пропана (а также, при необходимости, другие компоненты смеси загрузки реакционного газа А).
В остальном для применения продукта-газа А либо продукта-газа А* для загрузки по меньшей мере одного реактора окисления в реакционной зоне В достаточно добавить к продукту-газу А либо продукту-газу А* то количество молекулярного кислорода, которое необходимо для достижения поставленной цели в реакционной зоне В.
В принципе, эту добавку можно, как уже было сказано, осуществлять с помощью чистого кислорода или с помощью смеси (например, воздуха) молекулярного кислорода с одним или несколькими газами, демонстрирующими в реакционной зоне В химически инертное поведение (например N2, H2O, благородными газами, CO2). В настоящей публикации их также называют первичными источниками кислорода. Согласно изобретению предпочтительно осуществлять подачу в виде газа, который содержит не более чем 30 об.-%, предпочтительно - не более чем 25 об.-%, целесообразно - не более чем 20 об.-%, особо целесообразно - не более чем 15 об.-%, еще более благоприятно - не более чем 10 об.-%, а особо предпочтительно - не более чем 5 об.-% либо же не более чем 2 об.-% других (отличных от молекулярного кислорода) газов. Крайне предпочтительно здесь применение чистого кислорода.
Обычно количество добавляемого молекулярного кислорода соразмеряют так, чтобы в газе загрузки для реакционной зоны В (исходной смеси реакционного газа В) молярное соотношение содержания молекулярного кислорода к содержанию пропилена было более или равно 1 и менее или равно 3. Перед добавлением к продукту-газу А либо продукту-газу А* газа, содержащего молекулярный кислород, их температуру целесообразно довести до значения, составляющего от 250 до 370°С, а особо целесообразно - от 270 до 320°С. Газ, содержащий молекулярный кислород, при добавлении к продукту-газу А либо же продукту-газу А* сжат и находится под давлением, которое обычно превышает давление реакционного газа А либо же А* (при необходимости на величину до 1 бар), так что газ, содержащий молекулярный кислород, можно просто впрыснуть в реакционный газ А либо же А* (если в качестве источника кислорода используют воздух, для его сжатия обычно применяют радиальный компрессор, как это описано, например, в немецкой заявке на патент DE-A 10353014). Температуру газа, содержащего молекулярный кислород, при этом обычно задают так, чтобы в смеси загрузки реакционного газа В установилась желательная температура (нередко от 240 до 300°С). Как правило, эта температура газа, содержащего молекулярный кислород, составляет от 100 до 200°С.
Типичные смеси загрузки реакционного газа В содержат:
от 10 до 40 об.-% пропана,
от 5 до 25 об.-% пропилена,
от 0 до 10 об.-% H2,
от 10 до 30 об.-% О2 и
от 1 до 10 об.-% H2O.
Предпочтительные смеси загрузки реакционного газа В содержат:
от 15 до 40 об.-% пропана,
от 7 до 20 об.-% пропилена,
от 0 до 6 об.-% Н2,
от 15 до 30 об.-% CO2 и
от 1 до 7 об.-% H2O.
Целесообразно, чтобы давление на входе реакционного газа В по меньшей мере в один реактор окисления реакционной зоны В при реализации способа согласно изобретению составляло от 1 бар до 4 бар, преимущественно - от 1,3 бар до 3 бар, а во многих случаях - от 1,5 до 2,5 бар.
В принципе, гетерогенно-катализируемое частичное окисление пропилена до акриловой кислоты молекулярным кислородом в газовой происходит по способу, известному как таковой, в два следующих друг за другом вдоль координаты реакции этапа, первый из которых ведет к образованию акролеина, а второй - акриловой кислоты из акролеина.
Это проведение реакции в два последовательных этапа открывает способом, известным как таковой, возможность прерывать реализацию способа согласно изобретению в реакционной зоне В на стадии акролеина (на этапе преимущественного образования акролеина) и проводить на этом этапе отделение конечного продукта, либо же продолжать реализацию способа согласно изобретению до преимущественного формирования акриловой кислоты и лишь тогда проводить отделение конечного продукта.
Если реализацию способа согласно изобретению проводят до преимущественного формирования акриловой кислоты, то согласно изобретению целесообразно реализовывать способ двухступенчатым образом, т.е. в два следующих друг за другом этапа окисления, причем целесообразно использовать на каждом из обоих этапов окисления наиболее подходящий твердый слой катализатора и предпочтительно - также оптимальные прочие условия реакции, например, температуру твердого слоя катализатора.
Хотя катализаторы первого этапа окисления (пропилен → акролеин), представляя собой активные массы наиболее подходящих мультиметаллических оксидов, содержащих элементы Мо, Fe, Bi, в состоянии в определенной мере также катализировать и второй этап окисления (пропилен → акриловая кислота), но на втором этапе окисления обычно предпочитают катализаторы, активная масса которых представляет собой мультиметаллический оксид, содержащий по меньшей мере один из элементов Мо и V.
Таким образом, подлежащий проведению в реакционной зоне В согласно изобретению способ гетерогенно-катализируемого частичного окисления акролеина на твердых слоях катализатора, активная масса которых содержит мультиметаллический оксид, содержащий по меньшей мере один из элементов Мо, Fe и Bi, особо пригоден в виде одноступенчатого способа для получения акролеина (а также, при необходимости, акриловой кислоты), или же в качестве первого этапа реакции двухступенчатого получения акриловой кислоты.
При этом реализацию одноступенчатого гетерогенно-катализируемого частичного окисления пропилена до акролеина, а также, при необходимости, акриловой кислоты либо двухступенчатого гетерогенно-катализируемого частичного окисления пропилена до акриловой кислоты с использованием производимого согласно изобретению реакционного газа В можно осуществлять так, как это в подробностях описано в публикациях европейских заявок на патент ЕР-А 700714 (первый этап реакции; как описано в тексте, но также и в режиме противотока соляной ванны и исходной реакционной газовой смеси по реактору в виде пучка труб), ЕР-А 700893 (второй этап реакции; как описано в тексте, но также и в соответствующем режиме противотока), международных заявок WO 04/085369 (в частности, указанный текст рассматривают как составную часть настоящей публикации) (в виде двухэтапного способа), WO 04/85363, немецкой заявки на патент DE-A 10313212 (первый этап реакции), европейских заявок на патент ЕР-А 1159248 (в виде двухэтапного способа), ЕР-А 1159246 (второй этап реакции), ЕР-А 1159247 (в виде двухэтапного способа), немецких заявок на патент DE-A 19948248 (в виде двухэтапного способа), DE-A 10101695 (в один или два этапа), международной заявки WO 04/085368 (в виде двухэтапного способа), немецкой заявки на патент DE 10 2004 021764 (в два этапа), международных заявок WO 04/085362 (первый этап реакции), WO 04/085370 (второй этап реакции), WO 04/085365 (второй этап реакции), WO 04/085367 (в два этапа), европейских заявок на патент ЕР-А 990636, ЕР-А 1007007 и ЕР-А 1106598.
В особенности это касается всех приведенных в указанных публикациях примеров исполнения. Их можно проводить так, как описано в этих публикациях, с той, однако, разницей, что в качестве исходной реакционной газовой смеси для первого этапа реакции (преобразование пропилена в акролеин) применяют образованный согласно изобретению реакционный газ В. В том, что касается остальных параметров, действуют так, как описано в примерах исполнения указанных публикаций (в частности, касаемо твердых слоев катализаторов и нагрузки твердых слоев катализаторов реагентами). Если в указанных примерах исполнения уровня техники работу проводят в два этапа, а между двумя этапами реакции осуществляют ввод вторичного кислорода (согласно изобретению вторичный воздух менее предпочтителен), то это осуществляют соответствующим же образом, регулируя его количество, однако, так, чтобы молярное соотношение молекулярного кислорода и акролеина в газовой смеси загрузки второго этапа реакции соответствовало таковому в примерах исполнения указанных публикаций.
Согласно изобретению целесообразно подбирать количества кислорода в реакционной зоне В так, чтобы продукт-газ В содержал не прошедший преобразование молекулярный кислород (целесообразно от 0,5 до 6 об.-%, выгодно - от 1 до 5 об.-%, предпочтительно - от 2 до 4 об.-%). В случае двухэтапного режима работы сказанное выше действительно для каждого из двух этапов окисления.
Катализаторы на основе мультиметаллических оксидов, особо пригодные для конкретного этапа окисления, многократно описаны ранее и хорошо известны специалисту. Например, в европейской заявке на патент ЕР-А 253409 на странице 5 содержится ссылка на соответствующие патенты США.
Удобные катализаторы для конкретного этапа окисления приведены также в немецких заявках на патент DE-A 4431957, DE-A 10 2004 025445 и DE-А 4431949. В особенности это касается таковых с общей формулой I в обеих из приведенных старых публикаций. Особо целесообразные катализаторы для конкретного этапа окисления описаны в немецких заявках на патент DE-A 10325488, DE-A 10325487, DE-A 10353954, DE-A 10344149, DE-A 10351269, DE-A 10350812 и DE-A 10350822.
Для этапа реакции частичного окисления в газовой фазе пропилена до акролеина, или акриловой кислоты, или их смеси согласно изобретению можно, в принципе, использовать в качестве активной массы все содержащие Мо, Bi и Fe мультиметаллические оксидные массы.
В частности, это мультиметаллические активные оксидные массы общей формулы I немецкой заявки на патент DE-A 19955176, мультиметаллические активные оксидные массы общей формулы I немецкой заявки на патент DE-A 19948523, мультиметаллические активные оксидные массы общих формул I, II и III немецкой заявки на патент DE-A 10101695, мультиметаллические активные оксидные массы общих формул I, II и III немецкой заявки на патент DE-A 19948248 и мультиметаллические активные оксидные массы общих формул I, II и III немецкой заявки на патент DE-A 19955168, а также мультиметаллические активные оксидные массы, указанные в европейской заявке на патент ЕР-А 700714.
Кроме того, для этого этапа окисления пригодны катализаторы на основе мультиметаллических оксидных масс, содержащие Мо, Bi и Fe, описанные в публикациях Research Disclosure Nr.497012 от 29.08.2005 (в приведенных там примерах исполнения удельную поверхность по оплошности указали в см2/г вместо правильной единицы м2/г при том же численном значении), немецких, европейских, международных и японских заявках DE-А 10046957, DE-A 10063162, DE-C 3338380, DE-A 19902562, EP-A 15565, DE-C 2380765, EP-A 807465, EP-A 27 93 74, DE-A 3300044, EP-A 575897, US-A 4438217, DE-A 19855913, WO 98/24746, DE-A 19746210 (имеющие общую формулу II), JP-A 91/294239 и в ЕР-А 293224 и EP-A 700714. В особенности это касается примеров форм исполнения в этих публикациях, среди которых особо предпочтительны такие европейские заявки на патент EP-A 15565, EP-A 575897, немецкие заявки на патент DE-A 19746210 и DE-A 19855913. В связи с этим следует особо подчеркнуть катализатор согласно примеру 1с из европейской заявки на патент EP-A 15565, а также катализатор, производимый таким же образом, однако имеющий состав активной массы Mo12Ni6,5Zn2Fe2Bi1P0,0065K0,06Ox·10 SiO2. Кроме того, следует подчеркнуть пример №3 из немецкой заявки на патент DE-A 19855913 (стехиометрия: Mo12Co7Fe3Bi0,6K0,08Si1,6Ox), представляющий собой полный катализатор в виде полых цилиндров с геометрией 5 мм × 3 мм × 2 мм, (наружный диаметр × высота × внутренний диаметр), а также полный катализатор на основе мультиметаллической оксидной массы II согласно примеру 1 немецкой заявки на патент DE-A 19746210. Далее необходимо назвать катализаторы на основе мультиметаллической оксидной массы из заявки на патент США US-A 4,438,217. Последнее, в частности, действительно тогда, когда таковые имеют геометрию полого цилиндра с размерами 5,5 мм × 3 мм × 3,5 мм, или 5 мм × 2 мм × 2 мм, или 5 мм × 3 мм × 2 мм, или 6 мм × 3 мм × 3 мм, или 7 мм × 3 мм × 4 мм (в каждом случае наружный диаметр × высота × внутренний диаметр). Прочие возможные в этом контексте геометрические параметры катализатора - это тяжи (например, длиной 7,7 мм и диаметром 7 мм; или длиной 6,4 мм и диаметром 5,7 мм).
Множество мультиметаллических оксидных активных масс, пригодных для катализаторов, пригодных для этапа пропилен → акролеин, и при необходимости акриловую кислоту, можно объединить под общей формулой IV,
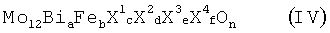 ,
,
в которой переменные имеют следующие значения:
X1 = никель и/или кобальт,
X2 = таллий, щелочной металл и/или щелочноземельный металл,
X3 = цинк, фосфор, мышьяк, бор, сурьма, олово, церий, свинец и/или вольфрам,
X4 = кремний, алюминий, титан и/или цирконий,
а = от 0,5 до 5,
b = от 0,01 до 5, предпочтительно - от 2 до 4,
с = от 0 до 10, предпочтительно - от 3 до 10,
d = от 0 до 2, предпочтительно - от 0,02 до 2,
е = от 0 до 8, предпочтительно - от 0 до 5,
f = 0-10 и
n = число, определяемое валентностью и частотой встречаемости отличных от кислорода элементов в формуле IV.
Способ их получения сам по себе известен (см., например, немецкую заявку на патент DE-A 4023239), и обычно этим массам придают форму шаров, колец или цилиндров либо же применяют в виде оболочечных катализаторов, т.е. заранее формованных инертных носителей, покрытых активной массой. Разумеется, применять их в качестве катализаторов можно также и в порошковой форме (например, в реакторах с кипящим слоем).
В принципе, существует простой способ изготовления активных масс с общей формулой IV, состоящий в создании по возможности гомогенной, предпочтительно тонкодисперсной сухой смеси элементарных компонентов, составленной соответственно их стехиометрии, из надлежащих источников и ее кальцинировании при температурах 350-650°С. Кальцинацию можно проводить как в атмосфере инертного газа, так и окислительной атмосфере, как, например, в воздухе (смеси инертного газа и кислорода), а также в восстановительной атмосфере (например, смеси из инертного газа, NH3, СО и/или H2). Продолжительность кальцинирования может составлять от нескольких минут от нескольких часов, и с ростом температуры она обычно сокращается. В качестве источников элементарных составляющих мультиметаллических оксидных масс IV можно рассматривать такие соединения, которые уже представляют собой оксиды, и/или такие соединения, которые можно превратить в оксиды посредством нагрева, по крайней мере, в присутствии кислорода.
Помимо оксидов такими исходными соединениями (источниками) являются прежде всего галогениды, нитраты, формиаты, оксалаты, цитраты, ацетаты, карбонаты, аминные комплексы, соли аммония и/или гидроксиды (такие соединения, как NH4OH, (NH4)2CO3, NH4NO3, NH4CHO2, СН3СООН, NH4CH3CO2 и/или оксалат аммония, которые самое позднее при последующем кальцинировании распадаются на газообразные летучие соединения и/или могут быть подвергнуты разложению - можно дополнительно вводить в гомогенную сухую смесь).
Гомогенное смешивание исходных соединений для получения мультиметаллических оксидных масс IV можно осуществлять в сухом или в мокром виде. Если его осуществляют в сухом виде, то целесообразно использовать исходные соединения в виде тонкодисперсных порошков, а после смешивания и при необходимости уплотнения подвергать их кальцинированию. Предпочтительно, однако, проводить гомогенное смешивание в мокром виде. Обычно при этом исходные соединения смешивают друг с другом в форме водного раствора и/или суспензии. Особо гомогенные сухие смеси получают при описанном способе смешивания, когда источники элементарных составляющих присутствуют исключительно в растворенном виде. В качестве растворителя предпочтительно использовать воду. Затем полученную водную массу сушат, причем процесс сушки предпочтительно проводить путем распылительной сушки водной смеси при температуре выхода из сопла, составляющей 100-150°С.
Мультиметаллические оксидные активные массы общей формулы IV на этапе ”пропилен → акролеин (и при необходимости акриловая кислота)” можно применять как в виде порошка, так и придав катализатору определенную геометрическую форму, причем формообразование можно проводить до заключительного кальцинирования или после него. Например, из активной массы в форме порошка или ее не кальцинированной и/или частично кальцинированной массы-предшественника можно изготовить полный катализатор путем прессования с получением желаемой геометрической формы (например, путем таблетирования, экструзии или ленточного прессования), причем при необходимости можно добавлять вспомогательные компоненты, как, например, графит или стеариновую кислоту в качестве средства, способствующего скольжению, и/или вспомогательные средства формообразования и армирующие агенты, как то: микроволокна из стекла, асбест, карбид кремния или титанат калия. Вместо графита в качестве вспомогательного средства при формообразовании можно применять также гексагональный нитрид бора, как это рекомендовано в немецкой заявке на патент DE-A 10 2005 037678. К надлежащим геометрическим формам катализаторов относятся, например, сплошные или полые цилиндры с внешним диаметром и длиной от 2 до 10 мм. В случае полых цилиндров целесообразна толщина стенки, составляющая от 1 до 3 мм. Разумеется, полные катализаторы могут также иметь форму шариков, причем диаметр шариков может составлять от 2 до 10 мм. Особо удобная геометрическая форма - это полые цилиндры с размерами 5 мм × 3 мм × 2 мм (внешний диаметр × длина × внутренний диаметр), особенно в случае полных катализаторов.
Разумеется, формообразование порошкообразной активной массы или ее не кальцинированной и/или частично кальцинированной порошкообразной массы-предшественника можно также осуществлять, нанося ее на предварительно формованные инертные носители катализаторов. Покрытие носителей для получения оболочечных катализаторов, как правило, осуществляют в надлежащей вращающейся емкости, как это известно, например, из немецкой заявки на патент DE-A 2909671, из европейских заявок на патент ЕР-А 293859 или ЕР-А 714700. Для покрытия носителей целесообразно увлажнить подлежащую нанесению порошковую массу, а после нанесения снова высушить ее, например, посредством горячего воздуха. Толщину нанесенной на носители порошковой массы целесообразно выбирать в пределах 10-1000 мкм, предпочтительно - в диапазоне 50-500 мкм, а особо предпочтительно - в диапазоне 150-250 мкм.
При этом в качестве материалов-носителей можно применять обычные пористые или не имеющие пор оксиды алюминия, оксид кремния, оксид тория, оксид циркония, карбид кремния или силикаты, как то: силикат магния или алюмосиликат. В отношении желаемой реакции, относящейся к способу согласно изобретению, они демонстрируют в основном инертное поведение. Изделия-носители могут иметь единообразную или неправильную форму, причем носители правильной формы с явно выраженной шероховатостью поверхности, например шары или полые цилиндры, предпочтительны. Можно применять в основном не имеющие пор обладающие шероховатой поверхностью шарообразные носители из стеатита, диаметр которых составляет от 1 до 10 или до 8 мм, предпочтительно - от 4 до 5 мм. Также, однако, можно применять в качестве изделий-носителей цилиндры, длина которых составляет от 2 до 10 мм, а наружный диаметр - от 4 до 10 мм. Кроме того, в случае применения в качестве изделий-носителей пригодных согласно изобретению колец толщина их стенок составляет обычно от 1 до 4 мм. Кольцевидные изделия-носители, использование которых согласно изобретению предпочтительно, имеют длину от 2 до 6 мм, наружный диаметр от 4 до 8 мм и толщину стенок от 1 до 2 мм. Прежде всего пригодны к применению согласно изобретению в качестве изделий-носителей согласно изобретению также кольца с геометрическими размерами 7 мм × 3 мм × 4 мм (диаметр × длина × внутренний диаметр). Тонкость (дисперсность) подлежащих нанесению на поверхность изделия-носителя каталитически активных оксидных масс, разумеется, подбирают в соответствии с желательной толщиной оболочки (ср. европейскую заявку на патент ЕР-А 714700).
Кроме того, мультиметаллические окислительные массы для этапа ”пропилен-акролеин (а также при необходимости акриловая кислота)” это также массы с общей формулой V,
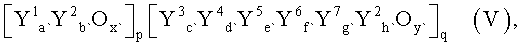
в которой переменные имеют следующие значения:
Y1 = только висмут или висмут и по меньшей мере один из элементов теллур, сурьма, олово и медь,
Y2 = молибден или вольфрам, или молибден и вольфрам,
Y3 = щелочной металл, таллий и/или самарий,
Y4 = щелочноземельный металл, никель, кобальт, медь, марганец, цинк, олово, кадмий и/или ртуть,
Y5 = железо или железо и по меньшей мере один из элементов хром и церий,
Y6 = фосфор, мышьяк, бор и/или сурьма,
Y7 = редкоземельный металл, титан, цирконий, ниобий, тантал, рений, рутений, родий, серебро, золото, алюминий, галлий, индий, кремний, германий, свинец, торий и/или уран,
а' = от 0,01 до 8,
b' = от 0,1 до 30,
с' = от 0 до 4,
d = от 0 до 20,
е' > от 0 до 20,
f = от 0 до 6,
g' = от 0 до 15,
h'' = от 8 до 16,
х', y' = числа, определяемые валентностью и частотой встречаемости отличных от кислорода элементов в формуле V, а
p, q = числа, соотношение которых p/q составляет от 0,1 до 10,
содержащие протяженные в трех измерениях, отграниченные от своего локального окружения в силу своего отличного от локального окружения состава, области с химическим составом,  , чей наибольший диаметр (длиннейший отрезок, проходящий через центр тяжести области и прямо соединяющий две точки, находящиеся на поверхности (границе раздела) области) составляет от 1 нм до 100 мкм, часто - от 10 нм до 500 нм или от 1 мкм до 50 либо же 25 мкм.
, чей наибольший диаметр (длиннейший отрезок, проходящий через центр тяжести области и прямо соединяющий две точки, находящиеся на поверхности (границе раздела) области) составляет от 1 нм до 100 мкм, часто - от 10 нм до 500 нм или от 1 мкм до 50 либо же 25 мкм.
Особо целесообразно использовать такие мультиметаллические оксидные массы согласно изобретению II, в которых Y1 - это только висмут.
Среди них, в свою очередь, предпочтительны те, что соответствуют общей формуле III,
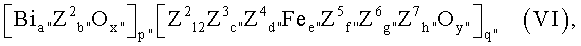
в которой переменные имеют следующие значения:
Z2 = молибден или вольфрам, или молибден и вольфрам,
Z3 = никель и/или кобальт,
Z4 = таллий, щелочной металл и/или щелочноземельный металл,
Z5 = фосфор, мышьяк, бор, сурьма, олово, церий и/или свинец,
Z6 = кремний, алюминий, титан и/или цирконий,
Z7 = медь, серебро и/или золото,
а = от 0,1 до 1,
b = от 0,2 до 2,
с = от 3 до 10,
d = от 0,02 до 2,
е = от 0,01 до 5, предпочтительно - от 0,1 до 3,
f = от 0 до 5,
g = от 0 до 10,
h'' = от 0 до 1,
х'', y'' = числа, определяемые валентностью и частотой встречаемости отличных от кислорода элементов в формуле VI, а
p'', q'' = числа, соотношение которых p''/q'' составляет от 0,1 до 5, предпочтительно - от 0,5 до 2,
причем крайне предпочтительны те массы VI, в которых Z2 b''=(вольфрам)b'' и Z2 12=(молибден)12.
Кроме того, целесообразно, чтобы по меньшей мере 25 моль-% (предпочтительно - по меньшей мере 50 моль-%, а особо предпочтительно - по меньшей мере 100 моль-%) общей доли 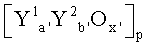
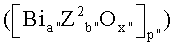 годных к применению согласно изобретению мульти металлических оксидных масс V (мультиметаллических оксидных масс VI) было представлено в годных к применению согласно изобретению мультиметаллических оксидных массах V (мультиметаллических оксидных массах VI) в форме протяженных в трех измерениях, отграниченных от своего локального окружения в силу своего отличного от локального окружения состава, областей с химическим составом
годных к применению согласно изобретению мульти металлических оксидных масс V (мультиметаллических оксидных масс VI) было представлено в годных к применению согласно изобретению мультиметаллических оксидных массах V (мультиметаллических оксидных массах VI) в форме протяженных в трех измерениях, отграниченных от своего локального окружения в силу своего отличного от локального окружения состава, областей с химическим составом 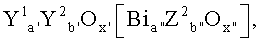 наибольший диаметр которых лежит в пределах от 1 нм до 100 мкм.
наибольший диаметр которых лежит в пределах от 1 нм до 100 мкм.
В отношении формообразования к катализаторам на основе мультиметаллических оксидных масс V относится то же самое, что уже было сказано относительно катализаторов на основе мультиметаллических оксидных масс IV.
Изготовление активных масс мультиметаллических оксидных масс II описано, например, в европейской заявке на патент ЕР-А 575897, а также в немецкой заявке на патент DE-A 19855913.
Рекомендованные выше инертные материалы-носители можно в т.ч. также использовать в качестве инертных материалов для разбавления и/или отграничения соответствующего твердого слоя катализатора или для его защитной предварительной засыпки, разогревающей газовую смесь.
Для катализаторов второго этапа реакции, частичного окисления акролеина до акриловой кислоты, можно, как уже было сказано, в принципе, использовать в качестве активных масс все необходимые содержащие Мо и V мультиметаллические оксидные массы, например, как в немецкой заявке на патент DE-A 10046928.
Ряд этих масс, например, таких как в немецкой заявке на патент DE-А 19815281, можно объединить под общей формулой VII,
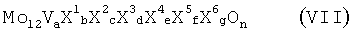
в которой переменные имеют следующие значения:
X1 = W, Nb, Та, Cr и/или Се,
X2 = Cu, Ni, Co, Fe, Mn и/или Zn,
X3 = Sb и/или Bi,
X4 = один или несколько щелочных металлов,
X5 = один или несколько щелочных металлов,
X6 = Si, Al, Ti и/или Zr,
а = от 1 до 6,
b = от 0,2 до 4,
с = от 0,5 до 18,
d = от 0 до 40,
е = от 0 до 2,
f = от 0 до 4,
g = 0-40 и
n = число, определяемое валентностью и частотой встречаемости отличных от кислорода элементов в формуле VII.
Предпочтительные согласно изобретению формы исполнения в рамках определения мультиметаллических оксидов VII - это такие, охваченные следующими значениями переменных общей формулы VII:
X1 = W, Nb, и/или Cr,
X2 = Cu, Mi, Co, и/или Fe,
X3 = Sb,
X4 = Na и/или К,
X5 = Са, Sr и/или Ва,
X6 = Si, Al, и/или Ti,
а = от 1,5 до 5,
b = от 0,5 до 2,
с = от 0,5 до 3,
d = от 0 до 2,
е = от 0 до 0,2,
f = 0-1 и
n = число, определяемое валентностью и частотой встречаемости отличных от кислорода элементов в формуле VII.
Крайне предпочтительные согласно изобретению мультиметаллические оксиды VII - это, однако, таковые общей формулы VIII,
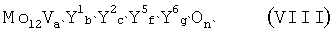
где
Y1 = W и/или Nb,
Y2 = Cu и/или Mi,
Y5 = Са и/или Sr,
Y6 = Si и/или Al,
а' = от 2 до 4,
b' = от 1 до 1,5,
с' = от 1 до 3,
f' = от 0 до 0,5,
g' = 0-8 и
n' = число, определяемое валентностью и частотой встречаемости отличных от кислорода элементов в формуле VIII.
Пригодные к применению согласно изобретению мультиметаллические оксидные массы (VII) можно получать методами, известными как таковые, например, описаны в немецкой заявке на патент DE-A 4335973 или в европейской заявке на патент ЕР-А 714700.
В принципе, существует простой способ изготовления пригодных для этапа ”акролеин → акриловая” мультиметаллических оксидных активных масс, в особенности таковых с общей формулой VII, состоящий в создании по возможности гомогенной, предпочтительно тонкодисперсной сухой смеси элементарных компонентов, составленной соответственно их стехиометрии, из надлежащих источников и ее кальцинировании при температурах 350-600°С. Кальцинацию можно проводить как в атмосфере инертного газа, так и окислительной атмосфере, как, например, в воздухе (смеси инертного газа и кислорода), а также в восстановительной атмосфере (например, смеси из инертного газа и восстанавливающих газов, как то: H2, NH3, СО, метана и/или акролеина или указанных газов с восстанавливающим действие как таковых). Продолжительность кальцинирования может составлять от нескольких минут от нескольких часов, и с ростом температуры она обычно сокращается. В качестве источников элементарных составляющих мультиметаллических оксидных масс VII можно рассматривать такие соединения, которые уже представляют собой оксиды и/или такие соединения, которые можно превратить в оксиды посредством нагрева, по крайней мере, в присутствии кислорода.
Гомогенное смешивание исходных соединений для получения мультмметаллических оксидных масс VII можно осуществлять в сухом или в мокром виде. Если его осуществляют в сухом виде, то целесообразно использовать исходные соединения в виде тонкодисперсных порошков, а после смешивания и, при необходимости, уплотнения подвергать их кальцинированию. Предпочтительно, однако, проводить гомогенное смешивание в мокром виде.
Обычно при этом исходные соединения смешивают друг с другом в форме водного раствора и/или суспензии. Особо гомогенные сухие смеси получают при описанном способе смешивания, когда источники элементарных составляющих присутствуют исключительно в растворенном виде. В качестве растворителя предпочтительно использовать воду. Затем полученную водную массу сушат, причем процесс сушки предпочтительно проводить путем распылительной сушки водной смеси при температуре выхода из сопла, составляющей 100-150°С.
Полученные мультиметаллические оксидные массы, в частности таковые общей формулы VII, можно использовать для окисления акролеина как в виде порошка, так и придав катализатору определенную геометрическую форму, причем формообразование можно проводить до заключительного кальцинирования или после него. Например, из активной массы в форме порошка или ее не кальцинированной и/или частично кальцинированной массы-предшественника можно изготовить полный катализатор путем прессования с получением желаемой геометрической формы (например, путем таблетирования, экструзии или ленточного прессования), причем при необходимости можно добавлять вспомогательные компоненты, как, например, графит или стеариновую кислоту в качестве средства, способствующего скольжению, и/или вспомогательные средства формообразования и армирующие агенты, как то: микроволокна из стекла, асбест, карбид кремния или титанат калия. К надлежащим геометрическим формам катализаторов относятся, например, сплошные или полые цилиндры с внешним диаметром и длиной от 2 до 10 мм. В случае полых цилиндров целесообразна толщина стенки, составляющая от 1 до 3 мм. Разумеется, полные катализаторы могут также иметь форму шариков, причем диаметр шариков может составлять от 2 до 10 мм, например 8,2 мм или 5,1 мм.
Разумеется, формообразование порошкообразной активной массы или ее не кальцинированной и/или частично кальцинированной порошкообразной массы-предшественника можно также осуществлять, нанося ее на предварительно формованные инертные носители катализаторов. Покрытие носителей для получения оболочечных катализаторов, как правило, осуществляют в надлежащей вращающейся емкости, как это известно, например, из немецкой заявки на патент DE-A 2909671, из европейских заявок на патент ЕР-А 293859 или ЕР-А 714700.
Для покрытия носителей целесообразно увлажнить подлежащую нанесению порошковую массу, а после нанесения снова высушить ее, например, посредством горячего воздуха. Толщину нанесенной на носители порошковой массы целесообразно выбирать в пределах 10-1000 мкм, предпочтительно - в диапазоне 50-500 мкм, а особо предпочтительно - в диапазоне 150-250 мкм.
При этом в качестве материалов-носителей можно применять обычные пористые или не имеющие пор оксиды алюминия, оксид кремния, оксид тория, оксид циркония, карбид кремния или силикаты, как то: силикат магния или алюмосиликат. Изделия-носители могут иметь единообразную или неправильную форму, причем носители правильной формы с явно выраженной шероховатостью поверхности, например шары или полые цилиндры с накладкой мелкокускового материала, предпочтительны. Можно применять в основном не имеющие пор, обладающие шероховатой поверхностью шарообразные носители из стеатита, диаметр которых составляет от 1 до 10 или до 8 мм, предпочтительно - от 4 до 5 мм. Т.е. шарики с надлежащими геометрическими параметрами могут иметь диаметр 8,2 мм или 5,1 мм. Также, однако, можно применять в качестве изделий-носителей цилиндры, длина которых составляет от 2 до 10 мм, а наружный диаметр - от 4 до 10 мм. Кроме того, в случае применения в качестве изделий-носителей надлежащих колец толщина их стенок составляет обычно от 1 до 4 мм. Кольцевидные изделия-носители, использование которых предпочтительно, имеют длину от 2 до 6 мм, наружный диаметр от 4 до 8 мм и толщину стенок от 1 до 2 мм. Прежде всего пригодны к применению в качестве изделий-носителей согласно изобретению также кольца с геометрическими размерами 7 мм × 3 мм × 4 мм (наружный диаметр × длина × внутренний диаметр). Тонкость (дисперсность) подлежащих нанесению на поверхность изделия-носителя каталитически активных оксидных масс, разумеется, подбирают в соответствии с желательной толщиной оболочки (ср. европейскую заявку на патент ЕР-А 714700).
Мультиметаллические окислительные массы, целесообразные к применению на этапе ”акролеин → акриловая кислота” - это также массы с общей формулой IX,
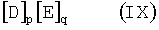 ,
,
в которой переменные имеют следующие значения:
D = Mo12Va”N1 b”N2 c”N3 d”N4 e”N5 f”N6 g”Ox”,
Е = Z7 12Cuh”Hi”Oy”,
Z1 = W, Nb, Та, Cr и/или Се,
Z2 = Cu, Ni, Co, Fe, Mn и/или Zn,
Z3 = Sb и/или Bi,
Z4 = Li, Na, К, Rb, Cs и/или Н,
Z5 = Mg, Са, Sr и/или Ва,
Z6 = Si, Al, Ti и/или Zr,
Z7 = Mo, W, V, Nb и/или Та, предпочтительно - Mo и/или W,
а'' = от 1 до 8,
b'' = от 0,2 до 5,
с'' = от 0 до 23,
d'' = от 0 до 50,
е'' = от 0 до 2,
f'' = от 0 до 5,
g'' = от 0 до 50,
h'' = от 4 до 30,
i'' = 0-20 и
х'', y'' = числа, определяемые валентностью и частотой встречаемости отличных от кислорода элементов в формуле IX, а
p, q = отличные от нуля числа, соотношение которых p/q составляет от 160:1 до 1:1,
и которые получают следующим образом: мультиметаллическую оксидную массу Е
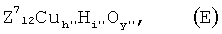 ,
,
формируют в мелкодисперсной форме отдельно (исходная масса 1), а затем предварительно сформированную твердую исходную массу 1 вводят в желаемом количественном соотношении p:q в водный раствор, водную суспензию или мелкодисперсную сухую смесь источников элементов Мо, V, Z1, Z2, Z3, Z4, Z5, Z6, содержащую вышеупомянутые элементы в стехиометрическом соотношении D
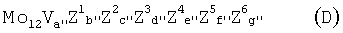 ,
,
(исходная масса 2), получаемую при этом водную смесь при необходимости сушат, а полученную сухую массу-предшественник до или после сушки кальцинируют при температурах 250-600°С, получая катализатор в необходимой геометрической форме.
Предпочтительны мультиметаллические оксидные массы IX, при получении которых введение предварительно сформированной твердой исходной массы 1 в исходную массу 2 на водной основе происходит при температуре ниже 70°С. Подробное описание изготовления катализаторов на основе мультиметаллических оксидных масс IX содержится, например, в европейской заявке на патент ЕР-А668104, в немецких заявках на патент DE-A 19736105, DE-A 10046928, DE-A19740493 и DE-A 19528646.
В отношении формообразования к катализаторам на основе мультиметаллических оксидных масс IX относится то же самое, что уже было сказано относительно катализаторов на основе мультиметаллических оксидных масс VII.
Катализаторы на основе мультиметаллических оксидов, замечательным образом применимые на этапе ”акролеин → акриловая кислота”, это согласно немецкой заявке на патент DE-A 19815281, в частности, с мультиметалическими оксидными активными массами общей формулы I этой публикации.
Для этапа преобразования пропилена в акролеин целесообразно применение сплошных колец из катализирующего материала, а для этапа преобразования акролеина в акриловую кислоту - оболочечных катализирующих колец.
Реализация частичного окисления в рамках способа согласно изобретению пропилена до акролеина (а также, при необходимости, акриловой кислоты) возможна с использованием описанных катализаторов в однозонном реакторе со множеством контактных труб и твердым слоем, описанном, например, в немецкой заявке на патент DE-A 4431957. При этом проведение реакционной газовой смеси и теплоносителя (агента теплообмена) по реактору возможно прямотоком или противотоком.
Давление реакции обычно находится в пределах от 1 до 3 бар, а общая нагрузка твердого слоя катализатора реакционным газом В предпочтительно составляет от 1500 до 4000 либо же 6000 Нл/л·ч или более. Типичная нагрузка пропиленом (твердого слоя катализатора) составляет от 90 до 200 Нл/л·ч, или до 300 Нл/л·ч, или более. Значения нагрузки пропиленом, превышающие 135 Нл/л·ч, либо же составляющие не менее 140 Нл/л·ч, или не менее 150 Нл/л·ч, или не менее 160 Нл/л·ч, особо предпочтительны согласно изобретению, поскольку исходная реакционная газовая смесь для реакционной зоны В согласно изобретению, ввиду присутствия не прошедшего преобразование пропана и, возможно, молекулярного водорода, обеспечивает выгодные параметры точки перегрева (все вышесказанное справедливо также вне зависимости от выбора конкретного реактора с твердым слоем).
Газовую смесь загрузки предпочтительно вводить в однозонный реактор со множеством контактных труб и твердым слоем сверху. В качестве теплоносителя целесообразно применять соляной расплав, предпочтительно состоящий из 60 вес.-% нитрата калия (KNO3) и 40 вес.-% нитрита натрия (NaNO2), или из 53 вес.-% нитрата калия (KNO3), 40 вес.-% нитрита натрия (NaNO2) и 7 вес.-% нитрата натрия (NaNO3).
Как уже сказано, проведение реакционной газовой смеси и соляного расплава по реактору возможно прямотоком или противотоком. Предпочтительно, чтобы поток самого соляного расплава двигался вокруг контактных труб по траектории в форме меандров.
Если поток вводят в контактные трубы сверху вниз, то целесообразно осуществлять загрузку контактных труб катализатором (снизу вверх) в следующем порядке (если поток вводят снизу вверх, то целесообразно использовать обратную последовательность загрузки):
- сначала, на протяжении 40-80 или до 60% длины контактной трубы - либо только катализатор, либо смесь катализатора с инертным материалом, причем массовая доля последнего составляет в смеси 30 либо же до 20 вес.-% (участок С);
- затем, на протяжении 20-50 или до 40% общей длины трубы - либо только катализатор, либо смесь катализатора с инертным материалом, причем массовая доля последнего составляет в смеси до 40 вес.-% (участок В); и
- на заключительном этапе, составляющем от 10 до 20% общей длины трубы, - засыпка из инертного материала (участок А), который предпочтительно выбирать так, чтобы он обеспечивал по возможности малое падение давления.
Предпочтительно, чтобы на участке С разбавления не было.
Описанный выше вариант загрузки целесообразен, в частности, тогда. когда в качестве катализаторов применяют таковые согласно Research Disclosure Nr. 497012 от 29.08.2005, или согласно примеру 1 немецкой заявки на патент DE-A 10046957, или согласно примеру 3 немецкой заявки на патент DE-A 10046957, а в качестве инертного материала - кольца из стеатита с геометрическими характеристиками 7 мм × 7 мм × 4 мм (внешний диаметр × длина × внутренний диаметр). В отношении температуры соляной бани справедливо сказанное в немецкой заявке на патент DE-A 4431957.
Частичное окисление в реакционной зоне В пропилена с образованием акролеина (и, при необходимости, акриловой кислоты) с помощью описанных катализаторов, однако, можно также проводить, например, и в двухзонном реакторе с множественными контактными трубами и твердым слоем, как это описано в немецких заявках на патент DE-A 19910506, DE-A 10 2005 009885, DE-A 10 2004 032129, DE-A 10 2005 013039 и DE-A 10 2005 009891, а также DE-A 10 2005 010111. В обоих вышеописанных случаях (равно как и при реализации способа согласно изобретению вообще) достигнутые при простом прохождении значения оборота пропена обычно составляют не менее 90 моль-%, либо же не менее 95 моль-%, а избирательность образования акролеина - не менее 90 моль-%. Согласно изобретению целесообразно проводить частичное окисление пропена с образованием акролеина, или акриловой кислоты, или их смеси так, как это описано в европейской заявке на патент ЕР-А 1159244, а крайне предпочтительно - так, как это описано в международной заявке WO 04/085363, а также в WO 04/085362.
Публикации европейской заявки на патент ЕР-А 1159244 и международных заявок WO 04/085363 и WO 04/085362 считаются составной частью настоящей публикации.
Т.е. подлежащее проведению в реакционной зоне В частичное окисление пропилена с образованием акролеина (и, при необходимости, акриловой кислоты) особо целесообразно проводить на твердом слое катализатора с повышенной нагрузкой пропиленом, имеющем по меньшей мере две температурные зоны.
В связи с этим дана ссылка, например, на европейскую заявку на патент ЕР-А 1159244 и международную заявку WO 04/085362.
В случае двухступенчатого частичного окисления пропилена с образованием акролеина реализация второго этапа, а именно частичное окисление акролеина до акриловой кислоты, возможно с использованием описанных катализаторов в однозонном реакторе со множеством контактных труб и твердым слоем, описанном, например, в немецкой заявке на патент DE-A 4431949. При этом возможно проведение реакционной газовой смеси и теплоносителя с одинаковым относительно реактора направлением потоков. Как правило, газовую смесь продукции предшествующего частичного окисления пропилена до акролеина в принципе подают на второй этап реакции, т.е. на частичное окисление акролеина, как таковую, т.е. без отделения побочных продуктов, при необходимости - после промежуточного охлаждения. Охлаждение можно проводить непрямым или прямым способом, например, добавляя вторичный кислород (согласно изобретению вторичный воздух менее предпочтителен).
При этом молекулярный кислород, необходимый для частичного окисления акролеина, может уже находиться в составе исходной смеси реакционного газа В для частичного окисления пропилена до акролеина. Возможно, однако, его частичное или полное добавление непосредственно к газовой смеси продукции первого этапа реакции, т.е. частичного окисления пропилена до акролеина (это можно осуществлять в виде вторичного воздуха, предпочтительно, однако, в форме чистого кислорода или смесей инертного газа и кислорода (в концентрации предпочтительно не более 50 об.-%, или не более 40 об.-%, или не более 30 об.-%, или не более 20 об.-%, или не более 10 об.-%, или не более 5 об.-%, или не более 2 об.-%)). В отношении давления и температуры вторичного источника кислорода справедливо сказанное относительно первичного источника кислорода. Вне зависимости от способа работы целесообразно, чтобы газовая смесь загрузки (исходная реакционная газовая смесь) такого частичного окисления акролеина до акриловой кислоты обладала следующим составом:
от 3 до 25 об.-% акролеина,
от 5 до 65 об.-% молекулярного кислорода,
от 6 до 70 об.-% пропана,
от 0 до 20 об.-% молекулярного водорода,
от 8 до 65 об.-% водяного пара, а также
от 0 до 70 об.-% молекулярного азота.
Предпочтительно, чтобы вышеуказанная исходная реакционная газовая смесь обладала следующим составом:
от 5 до 20 об.-% акролеина,
от 5 до 15 об.-% молекулярного кислорода,
от 10 до 40 об.-% пропана,
от 0 до 10 об.-% молекулярного водорода и
99 от 5 до 30 об.-% водяного пара.
Крайне предпочтительно, чтобы вышеуказанная исходная реакционная газовая смесь обладала следующим составом:
от 10 до 20 об.-% акролеина,
от 7 до 15 об.-% молекулярного кислорода,
от 20 до 40 об.-% пропана,
от 3 до 10 об.-% молекулярного водорода и
от 15 до 30 об.-% водяного пара.
Как правило, содержание азота в вышеуказанных смесях составляет не более 20 об.-%, предпочтительно - не более 15 об.-%, особо предпочтительно - не более 10 об.-%, а крайне предпочтительно - не более 5 об.-%. Согласно изобретению целесообразно, чтобы соотношение содержащихся в газовой смеси загрузки для второго этапа окисления О2 и акролеина, О2: акролеин, как правило, было не менее 0,5 и не более 2, нередко не менее 1 и не более 1,5.
Содержание СО2 в газовой смеси загрузки для второго этапа окисления, как правило, составляет не более 5 об.-%.
Как и на первом этапе реакции (пропилен → акролеин), давление на втором этапе реакции (акролеин → акриловая кислота) обычно находится в пределах от 1 до 3 бар, предпочтительно - от 1,3 до 3 бар, во многих случаях - от 1,5 до 2,5 бар, а общая нагрузка твердого слоя катализатора (исходной) реакционной газовой смесью предпочтительно составляет от 1500 до 4000 либо же 6000 Нл/л·ч или более. Типичная нагрузка акролеином (твердого слоя катализатора) составляет от 90 до 190 Нл/л·ч, или до 290 Нл/л·ч, или более. Значения нагрузки акролеином, превышающие 135 Нл/л·ч, либо не менее 140 Нл/л·ч, либо не менее 150 Нл/л·ч, либо не менее 160 Нл/л·ч особо предпочтительны, поскольку подлежащая применению согласно изобретению исходная реакционная газовая смесь ввиду присутствия пропана и, возможно, молекулярного водорода, также обеспечивает выгодные параметры точки перегрева.
Температура газовой смеси загрузки для второго этапа окисления, как правило, составляет от 220 до 270°С.
Целесообразно, чтобы оборот акролеина, рассчитанный относительно однократного прохождения реакционной газовой смеси через твердый слой катализатора второго этапа реакции, в норме составлял не менее 90 моль-%, а связанная с этим избирательность формирования акриловой кислоты - не менее 90 моль-%.
Газовую смесь загрузки также предпочтительно вводить в однозонный реактор со множеством контактных труб и твердым слоем сверху. В качестве теплоносителя на втором этапе реакции также применяют соляной расплав, предпочтительно состоящий из 60 вес.-% нитрата калия (KNO3) и 40 вес.-% нитрита натрия (NaNO2), или из 53 вес.-% нитрата калия (KNO3), 40 вес.-% нитрита натрия (NaNO2) и 7 вес.-% нитрата натрия (NaNO3). Как уже сказано, проведение реакционной газовой смеси и соляного расплава по реактору возможно прямотоком или противотоком. Предпочтительно, чтобы поток самого соляного расплава двигался вокруг контактных труб по траектории в форме меандров.
Если поток вводят в контактные трубы сверху вниз, то целесообразно осуществлять загрузку контактных труб катализатором снизу вверх в следующем порядке:
- сначала, на протяжении 50-80 или до 70% длины контактной трубы - либо только катализатор, либо смесь катализатора с инертным материалом, причем массовая доля последнего составляет в смеси до 30 (либо же до 20) вес.-% (участок С);
- затем, на протяжении 20-40% общей длины трубы - либо только катализатор, либо смесь катализатора с инертным материалом, причем массовая доля последнего составляет в смеси до 50 или до 40 вес.-% (участок В); и
- на заключительном этапе, составляющем от 5 до 20% общей длины трубы - засыпка из инертного материала (участок А), который предпочтительно выбирать так, чтобы он обеспечивал по возможности малое падение давления.
Предпочтительно, чтобы на участке С разбавления не было. Как и вообще при гетерогенно-катализируемом частичном окислении акролеина до акриловой кислоты в газовой фазе (в особенности при высоких значениях нагрузки слоя катализатора акролеином и высоких значениях содержания водяного пара в газовой смеси загрузки), участок В также может состоять из двух последовательных ступеней разбавления катализатора (в целях минимизации температуры перегрева и чувствительности к температуре перегрева). Снизу вверх сначала до 30 (либо же 20) вес.-% инертного материала, а затем количество инертного материала превышает 20 вес.-% и достигает 50 либо же 40 вес.-%. В этом случае предпочтительно, чтобы на участке С разбавления не было.
Если поток вводят в контактные трубы снизу вверх, то целесообразно использовать обратную последовательность загрузки контактных труб.
Описанный выше вариант загрузки целесообразен, в частности, тогда, когда в качестве катализаторов применяют таковые согласно примеру получения 5 немецкой заявки на патент DE-A 10046928, или согласно немецкой заявке на патент DE-A 19815281, а в качестве инертного материала - кольца из стеатита с геометрическими характеристиками 7 мм × 7 мм × 4 мм или 7 мм × 7 мм × 3 мм (внешний диаметр × длина × внутренний диаметр). В отношении температуры соляной бани справедливо сказанное в немецкой заявке на патент DE-A 4431949. Ее, как правило, выбирают так, чтобы в норме достигать при однократном прохождении оборота акролеина не менее 90 моль-%, либо не менее 95 моль-%, либо не менее 99 моль-%.
Частичное окисление акролеина до акриловой кислоты с помощью описанных катализаторов, однако, можно также проводить, например, и в двухзонном реакторе с множественными контактными трубами и твердым слоем, как это описано в немецкой заявке на патент DE-A 19910508. В отношении оборота акролеина справедливо сказанное выше. В случае проведения описанного выше частичного окисления акролеина в виде второго этапа реакции двухступенчатого частичного окисления пропилена до акриловой кислоты в двухзонном реакторе с множественными контактными трубами и твердым слоем для создания газовой смеси загрузки (исходной реакционной газовой смеси) также целесообразно непосредственно применять газовую смесь продукции частичного окисления, проведенного на первом этапе (пропилен → акролеин), возможно, после осуществления непрямого или прямого (например, посредством добавления вторичного кислорода) охлаждения таковой, как это уже было описано выше. Необходимый для частичного окисления акролеина кислород предпочтительно добавлять в виде чистого молекулярного кислорода (при необходимости, однако, также и в виде воздуха или в виде смеси молекулярного кислорода с инертным газом с долей инертного газа предпочтительно не более 50 об.-%, особо предпочтительно - не более 40 об.-%, или не более 30 об.-%, или не более 20 об.-%, еще лучше - не более 10 об.-%, или не более 5 об.-%, или не более 2 об.-%) и вводят его, например, непосредственно в газовую смесь продукции первого этапа двухступенчатого частичного окисления (пропилен → акролеин). Также, однако, как уже описано, он может уже присутствовать в исходной реакционной газовой смеси для первого этапа реакции.
При двухступенчатом частичном окислении пропилена до акриловой кислоты с немедленным дальнейшим использованием газовой смеси продукции первого этапа частичного окисления для загрузки второго этапа частичного окисления, как правило, подключают последовательно друг за другом два однозонных реактора со множеством контактных труб и твердым слоем (при высокой нагрузке слоя катализатора реагентами, как это справедливо в общем случае, предпочтителен способ работы с одинаковым направлением потоков реакционного газа и соляной бани (теплоносителя) по реактору в виде пучка труб) или два двухзонных реактора со множеством контактных труб и твердым слоем. Также возможно смешанное последовательное подключение (однозонный/двухзонный или наоборот).
Между реакторами может находиться промежуточный охладитель, который при необходимости может содержать инертные засыпки, исполняющие функцию фильтров. Для первого этапа двухступенчатого частичного окисления пропилена до акриловой кислоты температура соляной бани реакторов со множеством контактных труб, как правило, составляет от 300 до 400°С. Для второго этапа двухступенчатого частичного окисления пропилена до акриловой кислоты, для частичного окисления акролеина до акриловой кислоты, температура соляной бани реакторов со множеством контактных труб большей частью составляет от 200 до 350°С. Кроме того, обычно теплообменники (предпочтительно соляные расплавы) проводят через надлежащие реакторы со множеством контактных труб и твердым слоем в таких количествах, чтобы разность между их температурами на входе и на выходе, как правило, не превышала 5°С. Как уже упомянуто, оба этапа частичного окисления пропилена до акриловой кислоты можно, однако, также проводить в одном реакторе на одной засыпке, как это описано в немецкой заявке на патент DE-A 10121592.
Здесь также следует упомянуть, что часть исходной газовой смеси реакционного газа В для первого этапа (”пропилен → акролеин”) может представлять собой остаточный газ, поступающий из зоны разделения II.
В целях полноты изложения следует отметить, что как в газовую смесь загрузки для первого этапа окисления, так и в газовую смесь загрузки для второго этапа окисления, либо же только в одну из них можно дополнительно добавлять свежий пропан. Это не является предпочтительным согласно изобретению, но может, однако, в ряде случаев быть целесообразным, чтобы исключить способность газовых смесей загрузки к воспламенению.
В совокупности реактор в виде пучка труб, внутри которого загрузка катализатором соответствующим образом изменяется на протяжении длины отдельных контактных труб с окончанием первого этапа реакции (изложение подобных вариантов двухэтапного частичного окисления пропилена в так называемом „Single-Reactor” содержится, например, в европейских заявках на патент ЕР-А 911313, ЕР-А 979813, ЕР-А 990636 и в немецкой заявке на патент DE-A 2830765) представляет собой простейшую форму реализации двух этапов окисления для обоих этапов частичного окисления пропилена до акриловой кислоты. При необходимости засыпку контактных труб катализатором в таких случаях прерывают засыпкой из инертного материала.
Предпочтительно, однако, реализовывать оба этапа окисления в форме последовательно подключенных друг за другом систем в виде пучков труб. Они могут находиться в одном реакторе, причем переход от одного пучка труб к другому пучку труб образует размещенная вне контактной трубы засыпка из инертного материала, причем целесообразно, чтобы к ней существовал доступ. Хотя теплоноситель, как правило, омывает контактные трубы, засыпки из инертного материала, размещенной так, как это описано выше, он не достигает. Поэтому целесообразно, чтобы оба пучка контактных труб располагались в реакторах, отделенных друг от друга в пространстве. При этом, как правило, между обоими реакторами в виде пучка труб находится промежуточный охладитель, предназначенный для того, чтобы снизить степень дожигания акролеина в газовой смеси продукции, покидающей первую зону окисления. Температура реакции на первом ее этапе (пропилен → акролеин), как правило, лежит в диапазоне от 300 до 450°С, предпочтительно - от 320 до 390°С. Температура реакции на втором этапе реакции (акролеин → акриловая кислота), как правило, лежит в диапазоне от 200 до 370°С, предпочтительно - от 220 до 330°С.Целесообразно, чтобы давление реакции на обоих этапах реакции составляло от 1 до 4, а предпочтительно - от 1,3 до 3 бар. Нагрузка (Нл/л·ч) катализаторов окисления реакционным газом на обоих этапах реакции нередко составляет от 1500 до 2500 Нл/л·ч либо же до 4000 Нл/л·ч. При этом нагрузка акролеином может составлять от 100 до 200 или 300 и более Нл/л·ч.
В принципе, оба этапа окисления при реализации способа согласно изобретению можно организовать так, как это описано, например, в немецких заявках DE-A 19837517, DE-A 19910506, DE-A 19910508, a также DE-A 19837519.
При этом на обоих этапах реакции избыток молекулярного кислорода сравнительно с количеством, необходимым согласно стехиометрии реакции, благоприятно сказывается на кинетике конкретного частичного окисления в газовой фазе, а также на сроке службы катализатора.
В принципе, подлежащее проведению согласно изобретению гетерогенно-катализируемое частичное окисление пропилена до акриловой кислоты в газовой фазе можно также реализовывать в одном единственном однозонном реакторе в виде пучка труб следующим образом. Оба этапа реакции проводят в одном реакторе окисления, который загружен одним или несколькими катализаторами, активная масса которых представляет собой мультиметаллический оксид, содержащий по меньшей мере один из элементов Мо, Fe и Bi, способный катализировать проведение обоих этапов реакции. Само собой, эта засыпка катализатором также может постепенно или резко меняться на протяжении координаты реакции. Разумеется, что в случае исполнения двухступенчатого частичного окисления пропилена до акриловой кислоты в форме двух последовательно подключенных друг за другом этапов окисления, от газовой смеси продукции, покидающей первый этап окисления, до направления ее на второй этап окисления при необходимости можно полностью или частично отделять содержащиеся в ней и сформировавшиеся в качестве побочного продукта на первом этапе окисления двуокись углерода и водяной пар. Согласно изобретению предпочтительно выбирать способ работы, не предусматривающий такое отделение.
В качестве источников для проводимого между двумя этапами окисления введения кислорода можно, как уже сказано, помимо воздуха (который менее предпочтителен согласно изобретению) использовать как чистый молекулярный кислород, так и молекулярный кислород, разбавленный CO2, CO, благородными газами, N2 и/или насыщенными углеводородами. Согласно изобретению предпочтительно, чтобы источник кислорода содержал не более 50 об.-%, предпочтительно - не более чем 40 об.-% особо предпочтительно - не более чем 30 об.-%, крайне предпочтительно - не более чем 20 об-%, еще лучше - не более чем 10 об.-%, или не более чем 5 об.-%, или не более чем 2 об.-% отличных от молекулярного кислорода газов.
В качестве материалов для инертных формованных изделий засыпки в слоях катализатора на обоих этапах окисления можно применять те же, которые рекомендованы и для разбавления слоев катализатора в реакционной зоне А.
В рамках способа согласно изобретению благодаря, например, введению холодного кислорода из источника в газовую смесь продукции первого этапа окисления можно обеспечить ее прямое охлаждение, прежде чем ее будут использовать как компонент исходной реакционной газовой смеси для второго этапа частичного окисления.
Согласно изобретению целесообразно проводить частичное окисление акролеина до акриловой кислоты так, как это описано в европейской заявке на патент ЕР-А 1159246, а крайне предпочтительно - так, как это описано в международной заявке WO 04/085365, а также в WO 04/085370. Согласно изобретению, однако, в качестве содержащей акролеин исходной реакционной газовой смеси предпочтительно при этом использовать реакционную исходную газовую смесь, которая представляет собой газовую смесь продукции частичного окисления пропилена до акролеина на первой ступени согласно изобретению, которую при необходимости дополнили таким количеством вторичного воздуха, что отношение молекулярного кислорода к акролеину в итоговой исходной реакционной газовой смеси, во всяком случае, составляло от 0,5 до 1,5. Публикации европейской заявки на патент ЕР-А 1159246 и международных заявок WO 04/08536 и WO 04/085370 считаются составной частью настоящей публикации.
Т.е. частичное окисление акролеина до акриловой кислоты целесообразно проводить на твердом слое катализатора с повышенной нагрузкой акролеином, имеющем по меньшей мере две температурные зоны.
В общей сложности двухступенчатое частичное окисление пропилена до акриловой кислоты предпочтительно проводить так, как это описано в европейской заявке на патент ЕР-А 1159248 либо же в международных заявках WO 04/085367 или WO 04/085369.
Продукт-газ В, выходящий с подлежащего проведению согласно изобретению частичного окисления (после первого и/или второго этапа окисления) в основном содержит акролеин, или акриловую кислоту, или их смесь в качестве конечного продукта, не прошедший преобразование пропан, в ряде случаев - молекулярный водород, водяной пар, образовавшийся как побочный продукт и/или применявшийся совместно в качестве разбавляющего газа, в ряде случаев - не прошедший преобразование молекулярный кислород (с точки зрения срока службы применяемых катализаторов обычно выгодно, если содержание кислорода в газовой смеси продукции обоих этапов частичного окисления еще составляет, например, по меньшей мере от 1,5 до 4 об.-%), а также прочие побочные продукты или компоненты, которые обладают более низкой или более высокой точкой кипения, нежели вода (при нормальном давлении, т.е. 1 бар), как, например, СО, CO2, низшие альдегиды, низшие алкановые карбоновые кислоты (например, уксусную кислоту, муравьиную кислоту и пропионовую кислоту), ангидрид малеиновой кислоты, бензальдегид, ароматические карбоновые кислоты и ангидриды ароматических карбоновых кислот (например, ангидрид фталевой кислоты и бензойную кислоту), в ряде случаев - прочие углеводороды, как, например, углеводороды с 4 атомами углерода (например, бутен-1 и, возможно, прочие бутены), а также при необходимости другие инертные разбавляющие газы, например, N2.
В качестве способа отделения содержащегося в продукте-газе В конечного продукта, с которым, как правило, одновременно связано отделение воды и менее легкокипящих, нежели вода, побочных компонентов, при реализации способа согласно изобретению в зоне разделения II можно, в принципе, использовать все известные в связи с этим на нынешнем уровне способы. В основном они отличаются тем, что конечный продукт посредством, например, мер по абсорбции и/или конденсации переводят из газовой фазы в конденсированную, а затем с целью дальнейшего отделения конечного продукта обрабатывают конденсаты или абсорбаты, применяя способы экстракции, дистилляции, кристаллизации и/или десорбции. Вместе с конечным продуктом и/или после перевода конечного продукта в конденсированную фазу при этих процессах обычно переводят в конденсированную фазу и таким образом отделяют также содержащиеся в реакционном газе В водяной пар и менее легкокипящие, нежели вода, компоненты (согласно изобретению предпочтительно, чтобы в зоне разделения II в конденсированную фазу перешло количество водяного пара, которое составляет по меньшей мере 70 моль-%, предпочтительно - по меньшей мере 80 моль-%, еще лучше - по меньшей мере 90 моль-%, а особо предпочтительно - по меньшей мере 95 моль-% образовавшегося в реакционной зоне В (либо же, особо предпочтительно, совокупно содержащегося в продукте-газе В) количества водяного пара).
В качестве абсорбентов при этом можно применять, например, воду, водные растворы и/или органические растворители (например, смеси дифенила и дифенилового эфира или дифенила, дифенилового эфира и ортодиметилфталата). В рамках этой ”конденсации” (отделения) целевого продукта, воды и побочных компонентов, имеющих точку кипения выше, нежели у воды, остается, как правило, не переходящий в конденсированную фазу остаточный газ, который включает в себя относительно тяжело поддающиеся конденсации (более легкокипящие, чем вода) компоненты продукта-газа В. Обычно это прежде всего те компоненты, точка кипения которых при нормальном давлении (1 бар) не превышает -30°С (совокупная их доля в остаточном газе, как правило, составляет не менее 60 об.-%, часто не менее 70 об.-%, во многих случаях не менее 80 об.-%, однако в большинстве случаев не более 90 об.-%). В первую очередь, к ним относятся не прошедший преобразование пропан, двуокись углерода, в ряде случаев - не прошедший преобразование пропилен, при необходимости - молекулярный водород, а также прочие более легкокипящие, чем вода, побочные компоненты, как, например, СО, этан, метан, при необходимости N2, в ряде случаев - благородные газы (например, Не, Ne, Ar и т.д.). В небольших объемах остаточный газ может также содержать акриловую кислоту, акролеин и/или Н2О. Согласно изобретению предпочтительно, чтобы остаточный газ содержал не более 10 об.-%, целесообразно - не более 5 об.-%, а особо целесообразно - не более 3 об.-% водяного пара. Вышеуказанный остаточный газ в норме образует (относительно содержащегося в нем количества пропана) основную часть (обычно по меньшей мере 80%, либо же по меньшей мере 90%, или по меньшей мере 95%, или более) остаточного газа, формируемого в зоне разделения II, и в настоящей публикации его называют (основным) остаточным газом.
В зоне разделения II, в особенности в тех случаях, когда конденсацию целевого продукта проводят путем абсорбции органическим растворителем, образуется по меньшей мере еще один остаточный газ, содержащий не прошедший преобразование пропан, а также в ряде случаев - не прошедший преобразование пропилен (по содержанию в нем пропана его количество существенно меньше, чем количество (основного) остаточного газа - как правило, не более 20%, большей частью не более 10%, или не более 5%, или не более 1%). Это обусловлено тем, что образующаяся конденсированная фаза в определенной мере также поглощает не прошедший преобразование пропан, а также в определенных случаях не прошедший преобразование пропилен.
В процессе дальнейшего выделения целевого продукта из конденсированной фазы методами экстракции, дистилляции, кристаллизации и/или десорбции (в пределах зоны разделения II) эти не прошедшие преобразование пропан и в ряде случаев пропилен обычно выделяют повторно в качестве компонента по меньшей мере еще одной газовой фазы, а в настоящей публикации их обозначают как (дополнительный) остаточный газ.
Общее количество остаточного газа, остающееся в зоне разделения II, представляет собой сумму основного остаточного газа и дополнительного остаточного газа. Если в разделительной зоне II не образуется (дополнительный) остаточный газ, то (основной) остаточный газ автоматически представляет собой все количество остаточного газа (также называемого (совокупным) остаточным газом).
Согласно изобретению предпочтительно проводить перевод конечного продукта из продукта-газа В в конденсированную фазу путем фракционированной конденсации. В особенности тогда, когда целевой продукт - акриловая кислота. В принципе, однако, для отделения целевого продукта пригодны, например, все абсорбционные и/или конденсационные процессы, описанные для ”конденсации целевого продукта” и дальнейшей обработки ”конденсата” в немецких заявках на патент DE-A 10213998, DE-A 2263496, патенте США US 3,433,840 (способы, описанные в вышеуказанных публикациях, особенно рекомендуются для отделения акролеина), европейских заявках на патент ЕР-А 1388533, ЕР-А 1388532, немецкой заявке на патент DE-A 10235847, европейской заявке на патент ЕР-А 792867, международной заявке WO 98/01415, европейских заявках на патент ЕР-А 1015411, ЕР-А 1015410, международных заявках WO 99/50219, WO 00/53560, WO 02/09839, немецкой заявке на патент DE-A 10235847, международной заявке WO 03/041833, немецких заявках на патент DE-A 10223058, DE-A 10243625, DE-A 10336386, европейской заявке на патент ЕР-А 854129, заявке на патент США US-A 4,317,926, немецких заявках на патент DE-A 19837520, DE-A 19606877, DE-A 190501325, DE-A 10247240, DE-A 19740253, европейских заявках на патент ЕР-А 695736, ЕР-А 982287, ЕР-А 1041062, ЕР-А 117146, немецких заявках на патент DE-A 4308087, DE-A 4335172, DE-A 4436243, DE-A 19924532, DE-A 10332758, а также DE-A 19924533.
Кроме того, отделение акриловой кислоты можно проводить подобно тому, как это описано в европейских заявках на патент ЕР-А 982287, ЕР-А 982289, DE-A 10336386, немецких заявках на патент DE-A 10115277, DE-A 19606877, DE-A 19740252, DE-A 19627847, европейских заявках на патент ЕР-А 920408, ЕР-А 1068174, ЕР-А 1066239, ЕР-А 1066240, международных заявках WO 00/53560, WO 00/53561, немецкой заявке на патент DE-A 10053086 и европейской заявке на патент ЕР-А 982288. Предпочтительно проводить отделение так, как это представлено на фиг.7 международной заявки WO/0196271 либо же как это описано в немецкой заявке на патент DE-A 10 2004 032129 и эквивалентных ей патентах. Целесообразные способы отделения - это также способы, описанные в публикациях международных заявок WO 35514/063138, WO 2004/035514, немецких заявок на патент DE-A 10243625 и DE-A 10235847. Дальнейшую переработку полученной сырой акриловой кислоты можно проводить, например, так, как это описано в международных заявках WO 01/77056, WO 03/041832, WO 02/055469, WO 03/078378 и WO 03/041833.
Продукт-газ В обычно подают в зону разделения II под тем же рабочим давлением, под которым он выходит из реакционной зоны В.
Общим признаком вышеупомянутых способов разделения является (как уже упомянуто) то, что, например, в верхней части разделительной колонны, содержащей конкретные устройства, обладающие разделительной способностью, в нижнюю часть такой колонны подают - обычно после предварительного прямого или непрямого охлаждения - продукт-газ В, обычно получают остаточный поток газа, который в основном содержит те компоненты продукта-газа В, точка кипения которых при нормальном давлении (1 бар) не превышает -30°С (то есть тяжело поддающиеся конденсации или же высоколетучие компоненты). Остаточный газ, однако, также может содержать, например, такие компоненты, как водяной пар и акриловая кислота.
В нижней части разделительной колонны обычно получают в конденсированной фазе компоненты продукта-газа В с меньшей летучестью, включая желаемый целевой продукт. Конденсированную водную фазу, как правило, отбирают через боковой отвод и/или из нижней части колонны.
Как правило, (основной) остаточный газ обладает следующим составом:
от 1 до 20 об.-% H2O,
от 0 до 80 об.-% N2,
от 10 до 90 об.-% пропана,
от 0 до 20 об.-% Н2
от 0 до 10 об.-% O2,
от 1 до 20 об.-% CO2
и не менее 0 и до 5 об.-% СО.
Как правило, содержание акролеина и акриловой кислоты в каждом случае менее 1 об.-%.
Предпочтительный состав (основных) остаточных газов таков:
от 1 до 5 об.-% H2O,
от 10 до 80 об.-% пропана,
от 0 до 5 об.-% N2,
от 0 до 20 об.-% Н2,
от 1 до 10 об.-% O2,
от 0 до 5 об.-% СО и
от 1 до 15 об.-% CO2.
При реализации способа согласно изобретению остаточный газ выходит из зоны разделения II под давлением не менее 1 бар и не более 3 бар, предпочтительно - не более 2,5 бар, и как правило - не более 2 бар. При этом температура остаточного газа в норме не превышает 100°С, как правило, находится не выше 50°С (обычно, однако, не менее 0°С).
С точки зрения техники применения целесообразно осуществлять абсорбцию пропана, содержащегося в остаточном газе III, при давлении от 5 до 50 бар, предпочтительно - от 10 до 25 бар. Обычно остаточный газ, выходящий из зоны разделения II, сжимают в зоне сжатия до давления, выгодного для абсорбционного отделения пропана (целесообразно, чтобы этот процесс был многоступенчатым). Согласно изобретению на первом этапе сжатия целесообразно с помощью, например, многоступенчатого турбокомпрессора (радиальный компрессор, например, типа МН4В, производства фирмы Mannesmann DEMAG, Германия, в настоящем тексте также под названием ”компрессор остаточного газа”) проводить сжатие остаточного газа до рабочего давления, которое в основном соответствует таковому в реакционной зоне А (например, с 1,20 бар до 2,40 бар; как правило, сжатие на первом этапе осуществляют до давления, которое несколько превышает рабочее давление, используемое в реакционной зоне А). При этом остаточный газ нагревается на величину, например, до 50°С. Если абсорбцию пропана проводят не из всего количества остаточного газа, целесообразно разделять остаточный газ, прошедший сжатие, как описано, на две части одинакового состава. Размер меньшей части при этом может составлять до 49%, предпочтительно - не более 40 %, или до 30%, или до 20%, или до 10% общего количества. Также он может составлять и не менее 10%, и не менее 49% общего количества.
Эта часть непосредственно готова к возврату в реакционную зону А в виде остаточного циркуляционного газа. Другую часть подвергают дальнейшему сжатию до давления, при котором предполагается проводить абсорбцию пропана. В принципе, это можно осуществлять всего на одном дополнительном этапе сжатия. Согласно изобретению, однако, целесообразно использовать для этого по меньшей мере два этапа дальнейшего сжатия. Это обусловлено тем, что сжатие газа (как уже замечено относительно первого этапа сжатия) влечет за собой повышение его температуры. Снятие давления с ”отмытого от пропана” (и при необходимости прошедшего отделение Н2 на мембране) остаточного газа (его расширение), наоборот, влечет за собой его охлаждение. Если такое расширение также проводить в несколько этапов, то в каждом случае перед следующим этапом можно провести непрямой теплообмен между нагретым, сжатым и не отмытым остаточным газом и охлажденным, расширившимся и ”отмытым от пропана” остаточным газом. Такой теплообмен (который можно дополнительно поддерживать путем непрямого воздушного охлаждения сжатого остаточного газа) можно применять уже перед первичным расширением (вследствие непрямого теплообмена может произойти конденсация водяного пара, в ряде случаев еще содержащегося в остаточном газе). С точки зрения техники применения целесообразно поводить вышеуказанное расширение в расширительных турбинах (предпочтительно также многоступенчатых).
Это предназначено для возврата энергии сжатия, т.е. механическую энергию, полученную при расширении, во всех случаях (особенно в примерах и сравнительных примерах) можно использовать как непосредственно в качестве дополнительного или основного привода компрессора, так и/или для получения электроэнергии. Разность между давлением на входе и на выходе каждого этапа сжатия или расширения может, например, составлять от 2 до 15 бар.
Прежде чем содержащийся в сжатом остаточном газе пропан будет изъят из него путем абсорбции органическим растворителем в зоне разделения III (”вымыт” из сжатого остаточного газа с помощью органического растворителя, абсорбента), еще в одной зоне разделения можно провести разделение на мембранах, чтобы удалить хотя бы часть, возможно, присутствующего еще в газе молекулярного водорода. Надлежащие разделительные мембраны для этого процесса - это, например, ароматические полиимидные мембраны, например, производства UBE Industries Ltd. В частности, из последних можно применять мембраны типов А, В-Н, С и D. Для такого разделения особо предпочтительно применение полидиимидной мембраны типа В-Н производства UBE Industries Ltd. При 60°С проницаемость молекулярного водорода Н2 составляет для этой мембраны 0,7-10-3 [(об.с.у.)·сс/см2·сек·см рт.ст]. Для этого сжатый остаточный газ можно провести через мембрану, имеющую, как правило, форму трубы (возможно также применение пластинчатого или спирального модуля), проницаемую только для молекулярного водорода. Отделенный таким образом молекулярный водород можно снова применять в рамках других процессов химического синтеза или же по меньшей мере частично возвращать в реакционную зону А (целесообразно - в качестве компонента газовой смеси загрузки реакционного газа А).
Отделение молекулярного водорода от сжатого остаточного газа на мембране целесообразно согласно изобретению по той причине, что его предпочтительно также проводить при высоком давлении (например, от 5 до 50 бар, обычно - от 10 до 25 бар). Поэтому согласно изобретению целесообразно при необходимости в мембранном отделении водорода от остаточного газа проводить отделение либо непосредственно перед абсорбцией пропана (отмыванием пропана), либо в качестве предпочтительной согласно изобретению альтернативы - непосредственно после отмывания пропана от остаточного газа (абсорбции пропана), чтобы извлечь выгоду из двойного использования повышенного уровня давления. Кроме плоских мембран, спиральных (витых) или капиллярных мембран, для отделения водорода можно в первую очередь использовать мембраны в виде шлангов (половолоконные мембраны). Их внутренний диаметр может составлять, например, от нескольких мкм до нескольких мм. При их изготовлении пучок таких половолоконных мембран концами шлангов впаивают в пластины подобно трубам реактора в виде пучка труб. Вовне шланговых мембран давление предпочтительно снижено (менее 1 бар). Находящийся под повышенным давлением остаточный газ (либо же ”отмытый от пропана” остаточный газ) подводят к одной из двух концевых пластин и под давлением перемещают по просветам шлангов к выходу, расположенному в области противоположной пластины. На протяжении пути потока, заданного внутренним пространством шланга, молекулярный водород выходит наружу через проницаемую для Н2 мембрану.
Операция абсорбции пропана (как правило, вместе с пропаном абсорбируют пропилен, не прошедший преобразование в рамках частичного окисления в реакционной зоне В) из полученного остаточного газа, в общем, ничем не ограничена. Возможность просто реализовать ее состоит, например, в том, чтобы обеспечить контакт оставшегося остаточного газа (причем согласно изобретению целесообразно, чтобы его температура составляла от 10 до 100°С, предпочтительно - от 10 до 70°С) под давлением от 5 до 50 бар, предпочтительно - от 10 до 25 бар, с органическим растворителем (предпочтительно гидрофобным), температура которого, что целесообразно, составляет от 0 до 100°С, предпочтительно - от 20 до 50°С, либо же до 40°С, и которое абсорбирует пропан и пропилен (целесообразно, чтобы абсорбция была предпочтительна по сравнению с остальными компонентами остаточного газа), например, посредством пропускания (через жидкость).
В разделительной зоне IV возможно отделение пропана и, при необходимости, пропилена в смеси из абсорбата, например, путем последующей десорбции (испарения при снятии давления), ректификации и/или стриппинга с газом, инертным в реакционной зоне А (например, N2) и/или необходимым в этой реакционной зоне в качестве реагента (например, воздухом или другой смесью молекулярного кислорода и инертного газа; предпочтительно - водяным паром или смесью водяного пара и молекулярного кислорода или смесью водяного пара и воздуха), и возврат их в качестве содержащего пропан потока подачи в реакционную зону А.
В качестве абсорбента для вышеописанного абсорбционного отделения, в принципе, можно применять все абсорбенты, которые в состоянии поглощать пропан и пропилен. Предпочтительно, чтобы абсорбент представлял собой органический растворитель, гидрофобный и/или высококипящий. Целесообразно, чтобы точка кипения этого растворителя (при нормальном давлении в 1 бар) составляла по меньшей мере 120°С, предпочтительно - по меньшей мере 180°С, целесообразно - между 200 и 350°С, в особенности - от 250 до 300°С, более предпочтительно - от 260 до 290°С.Целесообразно, чтобы температура вспышки (при нормальном давлении в 1 бар) превышала 110°С. В общем случае для использования в качестве абсорбентов можно использовать относительно неполярные органические растворители, как, например, алифатические углеводороды, которые предпочтительно не несут обращенных наружу полярных групп, но также и ароматические углеводороды. В общем случае желательно, чтобы абсорбент характеризовался по возможности более высокой точкой кипения и чтобы одновременно с этим пропан и пропилен обладали в нем высокой растворимостью. В качестве примера абсорбентов следует упомянуть алифатические растворители, например алканы или алкены с 8-20 атомами углерода, или же ароматические углеводороды, например средние нефтяные фракции дистилляции парафина или простые эфиры с объемистыми (стерически требовательными) группами на атоме кислорода, или их смеси, причем к ним можно добавить полярный растворитель, как, например, 1,2-диметилфталат, описанный в немецкой заявке на патент DE-A 4308087. Кроме того, возможно применение сложных эфиров бензойной и фталевой кислот с прямоцепочечными алканолами, содержащими 1-8 атомов углерода, как то: n-бутиловый эфир бензойной кислоты, метиловый эфир бензойной кислоты, этиловый эфир бензойной кислоты, диметиловый эфир фталевой кислоты, диэтиловый эфир фталевой кислоты, а также так называемые теплонесущие масла, например дифенил, дифениловый эфир и смеси дифенила и дифенилового эфира или их производные с хлором и триарилалкены, например 4-метил-4'-бензил-дифенилметан и его изомеры 2-метил-2'-бензил-дифенилметан, 2-метил-4'-бензил-дифенилметан и 4-метил-2'-бензил-дифенилметан и смеси таких изомеров Надлежащим абсорбентом является смесь растворителей дифенила и дифенилового эфира, предпочтительно - азеотропная смесь, в частности, включающая в себя примерно 25 вес.-% дифенила (бифенила) и примерно 75 вес.-% дифенилового эфира, например имеющийся в продаже Diphyl® (например, предлагаемый Bayer Aktiengesellschaft). Нередко эта смесь растворителей содержит добавку растворителя, например диметилфталата, в количестве от 0,1 до 25 вес.-% от всей смеси растворителей. Особо удобные к применению абсорбенты - это также октан, нонан, декан, ундекан, додекан, тридекан, тетрадекан, пентадекан, гексадекан, гептадекан и октадекан, причем наиболее удобным оказался тетрадекан. Целесообразно, чтобы применяемый абсорбент, с одной стороны, удовлетворял вышеупомянутому условию по точке кипения, а с другой - одновременно обладал не слишком высокой молекулярной массой. Выгодно, чтобы молекулярная масса абсорбента не превышала 300 г/моль. Также возможно применение парафиновых масел с 8-16 атомами углерода, описанных в немецкой заявке на патент DE-A 3313573. Примеры надлежащих коммерческих продуктов - это продукты, распространяемые фирмой Haltermann, например семейства Halpasol i, как то: Halpasol 250/340 i и Halpasol 250/275 i, а также масла для печатных красок под обозначениями PKWF и Printosol. Предпочтительны коммерческие продукты, не содержащие ароматических веществ, например, типа PKWFaf. Если имеется небольшое остаточное содержание ароматических соединений, то перед применением описанным образом его целесообразно снизить до значений существенно ниже 1000 вес. частей на миллион с помощью методов ректификации и/или адсорбции. Прочие пригодные к использованию коммерческие продукты -это n-Paraffin (с 13-17 атомами углерода), или Mihagol®5 нефтеперерабатывающего завода Erdöl-Raffinerie-Emsland GmbH, LINPAR®14-17 производства фирмы CON DEA Augusta S.p.A. (Италия), или SASOL Italy S.p.A., Normal parrafins (heavy) C14-C18 (обычные парафины, тяжелые, с 14-18 атомами углерода) производства фирмы SLONAFT в Словакии.
Значения содержания линейных углеводородов в вышеуказанных продуктах (приведенные в процентах площади по итогам анализа методом газовой хроматографии) обычно составляют:
Сумма С9-C13: менее 1%; С14: от 30 до 40%; C15: от 20 до 33%; C16: от 18 до 26%; C17: до 18%; C≥18: менее 2%.
Типичный состав продукта фирмы SASOL таков:
C13: 0,48%; C14: 39,8%; C15: 20,8%; C16: 18,9%; C17: 17,3%; C18: 0,91%; C19: 0,21%,
Типичный состав продукта фирмы Haltermann таков:
C13: 0,58%; C14: 33,4%; C15: 32,8%; C16: 25,5%; C17: 6,8%; C≥18: <0,2%.
При непрерывной эксплуатации состав абсорбента изменяется соответственно условиям процесса.
Абсорбцию можно проводить как в колоннах, так и в квенчерах. При этом возможна работа как прямотоком, так и в противотоке (предпочтительно). Надлежащие абсорбционные колонны - это, например, тарельчатые колонны (с колпачковыми и/или ситовыми тарелками), колонны со структурированными пакетами (например, пакетами металлического листа с удельной поверхностью от 100 до 1000, или до 750 м2/м3, например, Mellapak® 250 Y) и насадочные колонны (например, заполненные насадками Рашига). Однако это также могут быть градирни или скрубберы, абсорберы на основе графитовых блоков, поверхностные абсорберы, например, с толстым слоем или тонкослойные абсорберы, а также тарельчатые скрубберы, горизонтальные скрубберы с механическим перемешиванием и ротационные скрубберы. Кроме того, может быть выгодно проводить абсорбцию в барботажной колонне с внутренними устройствами или без них.
Отделение пропана и, при необходимости, пропилена от абсорбента (выделение из абсорбата) можно осуществлять посредством стриппинга, выпаривания при понижении давления и/или дистилляции (ректификации).
Предпочтительно осуществлять отделение пропана и пропилена от абсорбента посредством стриппинга и/или десорбции. Десорбцию можно обычно проводить посредством изменения давления и/или температуры, например, под давлением от 0,1 до 10 бар, или от 1 до 5 бар, или от 1 до 3 бар, и при температуре от 0 до 200°С, в особенности - от 20 до 100°С, более предпочтительно - от 30 до 70°С, особо предпочтительно - от 30 до 50°С. Предпочтительный стриппинг-газ - это водяной пар. В качестве альтернативы можно использовать молекулярный азот или смесь молекулярного азота и водяного пара. В качестве стриппинг-газов можно также согласно изобретению применять диоксид углерода или его смеси с водяным паром и/или азотом. Особо удобны в применении для стриппинга также барботажные колонны с внутренними устройствами или без них.
Отделение пропана и, при необходимости, пропилена от абсорбента (выделение из абсорбата) также можно осуществлять посредством дистилляции или ректификации, причем можно применять известные специалисту колонны с пакетами, насадками или соответствующими внутренними устройствами. Возможные условия дистилляции или ректификации - это давление от 0,01 до 5 бар, или от 0,1 до 4 бар, или от 0,1 до 3 бар, и температура (в нижней части колонны) от 50 до 300°С, в особенности - от 150 до 250°С.
Циркуляционный газ пропана, полученный из абсорбата методом стриппинга, можно перед применением для загрузки реакционной зоны А в зоне разделения IV подать еще на один этап процесса, чтобы, например, снизить потери абсорбента, также извлекаемого при стриппинге (например, отделение в сепараторах и/или фильтрах высокой проницаемости) и одновременно защитить реакционную зону от абсорбента. Такое отделение абсорбента можно проводить всеми известными специалисту методами. Форма исполнения такого отделения, предпочтительная в рамках настоящего изобретения, - это, например, квенчинг выходящего из стриппингового устройства потока водой. В этом случае абсорбент вымывают водой из загруженного им выходящего потока, а одновременно выходящий поток принимает воду. Это отмывание или квенчинг можно проводить, например, в верхней части десорбционной колонны посредством тарелки для отбора жидкости путем встречного орошения водой или в отдельном аппарате. Для поддержания эффекта разделения в пространстве квенчинга можно смонтировать внутренние устройства, увеличивающие поверхность квенчинга и известные специалисту из процессов ректификации, абсорбции и десорбции.
Снижение давления абсорбента, содержащего пропан (и в ряде случаев пропилен), до уровня, достаточного для отделения пропана (и при необходимости пропилена), при реализации способа согласно изобретению возможно посредством вентиля или обратного насоса. Механическую энергию, которая в случае использования обратного насоса при этом выделяется, целесообразно согласно изобретению применять для участия в обратном сжатии абсорбента, освобожденного (например, в десорбционной колонне или в стриппинг-колонне) от пропана (и при необходимости пропилена). В принципе, абсорбент, освобожденный от пропана, можно снова использовать (согласно изобретению целесообразно - после повторного сжатия до давления абсорбции) в зоне разделения III в качестве абсорбента для отделения пропана от остаточного газа.
Согласно изобретению особенно выгодно проводить отделение пропана от абсорбента в зоне разделения IV при давлении, составляющем от 1,5 либо же от 2 до 5 бар, предпочтительно - от 2 до 5 бар, особо предпочтительно - от 2,5 до 4,5 бар, а крайне предпочтительно - от 3 до 4 бар.
Отделение пропана от абсорбента при вышеуказанных значениях рабочего давления особо целесообразно по той причине, что образовавшийся при таком давлении газ циркуляции пропана можно непосредственно возвращать в реакционную зону А. При этом такой газ циркуляции пропана можно применять в качестве ведущей струи для проведения циркуляционного газа дегидрирования по принципу струйного насоса согласно теоретическому изложению немецкой заявки на патент DE-A 10211275. При этом согласно изобретению целесообразно предварительно нагревать газ циркуляции пропана путем непрямого теплообмена с горячим реакционным газом А и/или горячим продуктом-газом А до температур, подходящих для реакционной зоны А.
Типичный состав газов циркуляции пропана, созданных из содержавшего пропан абсорбата путем десорбции и/или стриппинга в разделительной зоне IV, может при реализации способа согласно изобретению быть следующим:
Пропан от 80 до 99,99 моль-%,
Пропилен от 0 до 5 моль-%,
H2O от 0 до 20 моль-%,
Часто состав газа циркуляции пропана следующий:
Пропан от 80 до 99 моль-%,
Пропилен от 1 до 4 моль-%,
H2O от 2 до 15 моль-%,
В том случае, когда в реакционную зону А, кроме свежего пропана (представляющего собой составную часть сырого пропана) и газа циркуляции пропана, а также циркуляционного газа дегидрирования, дополнительно подают остаточный циркуляционный газ (целесообразно - сжатый) и/или другой газ, содержащий молекулярный кислород, с точки зрения техники применения предварительно создать газовую смесь из сырого пропана и остаточного циркуляционного газа и/или другого газа, содержащего молекулярный кислород, а эту первую газовую смесь (предпочтительно после ее нагрева путем непрямого теплообмена с горячим реакционным газом А и/или продуктом-газом А) смешать с газом циркуляции пропана или его смесью с циркуляционным газом дегидрирования (предпочтительно также после соответствующего нагрева путем непрямого теплообмена с горячим реакционным газом А и/или продуктом-газом А) и подать полученную при этом итоговую газовую смесь в реакционную зону А в качестве газовой смеси загрузки реакционного газа А. При этом, прежде чем подавать итоговую газовую смесь в реакционную зону А в качестве газовой смеси загрузки реакционного газа А, выгодно добавить к этой смеси молекулярный водород.
С точки зрения техники применения при реализации способа согласно изобретению целесообразно удовлетворять потребность в газе, содержащем молекулярный кислород, из общего источника.
Согласно изобретению целесообразно, чтобы испарение сырого пропана происходило с помощью получаемого в зоне разделения II конденсата (например, кислой воды) путем непрямого теплообмена. Охлажденный при этом конденсат можно применять в зоне разделения II для прямого охлаждения в целях получения нового конденсата. В качестве теплоносителей для непрямого теплообмена в целях испарения сырого пропана можно также применять другую жидкую фазу, причем целесообразно, чтобы ее температура составляла от 20 до 40°С, предпочтительно - от 25 до 35°С. Для дальнейшего нагрева сырого пропана, как правило, применяют непрямой теплообмен с горячим водяным паром (который может, например, иметь при подаче давление 5 бар и температуру 152°С), который обычно в больших количествах получают в качестве побочного продукта способа согласно изобретению (например в рамках отведения теплоты, выделяющейся в процессе частичного окисления).
При необходимости из зоны разделения IV постоянно выводят небольшое количество абсорбента и заменяют его свежим абсорбентом. Выведенный абсорбент можно переработать ректификацией с получением свежего абсорбента. Остаточный газ, прошедший вышеописанное снятие давления, предварительно ”отмытый” от пропана, а затем, при необходимости, отделение водорода на мембранах, как правило, направляют на сжигание остатков (сжигание выхлопных газов). На это сжигание, которое при необходимости поддерживают добавлением природного газа и осуществляют, применяя в качестве источника кислорода воздух, можно направлять также тяжелокипящие соединения, отделенные в зоне разделения II, а также водные конденсаты, образовавшиеся в зоне разделения II и в ряде случаев между зонами разделения II и III (например, кислую воду). Горячий газ, получаемый при этом сжигании, обычно применяют для получения водяного пара путем непрямого теплообмена с водой, а затем, как правило, выпускают в атмосферу. Обычно он состоит исключительно из молекулярного кислорода, молекулярного азота, молекулярного водорода и диоксида углерода.
Следует, наконец, отметить, что при реализации способа согласно изобретению всегда, когда образуются конденсированные фазы, содержащие акриловую кислоту и/или акролеин, к ним добавляют ингибитор полимеризации. В качестве таковых, в принципе, можно использовать все известные ингибиторы процесса. Согласно изобретению особо удобно применять, например, фенотиазин и метиловый эфир гидрохинона. Ввиду присутствия молекулярного кислорода эффективность ингибирования полимеризации возрастает.
Акролеин, синтезированный по способу согласно изобретению, можно перерабатывать в продукты акролеина, приведенные в публикациях заявок на патенты США US-A 6,166,263 и US-A 6,187,963. К ним относятся 1,3-пропандиол, метионин, глутаровый альдегид, а также 3-пиколин.
Предпочтительны способы согласно изобретению, в зоне разделения III которых рабочее давление максимально.
Целесообразно, чтобы для значений рабочего давления Р (в каждом случае определенного на входе в соответствующую зону) в различных зонах способа согласно изобретению действовали следующие соотношения:
Рреакционная зона А>Рзона разделения I>Рреакционная зона В>Рзона разделения II<Рзона разделения III>Рзона разделения IV>Рреакционная зона A,
причем зона разделения I также может отсутствовать.
Кроме того, для способа согласно изобретению целесообразно вне пределов инертных засыпок и/или засыпок катализатора поддерживать температуру реакционных газов А, в особенности тогда, когда они содержат молекулярный кислород, не выше 460°С, предпочтительно не более 440°С, чтобы минимизировать таким образом нежелательные реакции горения, а также термического разложения, в особенности пропана.
Примеры и сравнительные примеры
I. Долговременное проведение двухступенчатого частичного окисления пропилена до акриловой кислоты в отсутствие и в присутствии молекулярного водорода
А) Общая конструкция экспериментального реакторного устройства
Реактор для первого этапа окисления
Реактор состоял из цилиндра с двойными стенками из высококачественной стали (цилиндрическая направляющая труба, окруженная цилиндрической внешней емкостью). Толщина стенок во всех случаях составляла от 2 до 5 мм.
Внутренний диаметр наружного цилиндра составлял 91 мм. Внутренний диаметр направляющей трубы составлял ок. 60 мм.
Сверху и снизу цилиндр с двойными стенками был закрыт крышкой (дном).
Контактная труба (400 см общей длины, 26 мм внутреннего диаметра, 30 мм наружного диаметра, 2 мм толщины стенок, высококачественная сталь) размещена в направляющей трубе цилиндрического бака таким образом, чтобы лишь незначительно выступать за пределы крышки или дна (с уплотнением) в верхнем или нижнем конце, соответственно. Теплоноситель (соляной расплав, состоящий из 53 вес.-% нитрата калия, 40 вес.-% нитрита натрия и 7 вес.-% нитрата натрия) был заключен в цилиндрический бак. В целях обеспечения по возможности равномерных температурных краевых условий у наружной стенки контактной трубы по всей длине контактной трубы, находящейся в цилиндрическом баке (400 см), теплоноситель перекачивали осевым насосом.
Температуру теплоносителя доводили до желательного уровня с помощью электронагревателя, размещенного на внешней оболочке. В остальном применяли воздушное охлаждение.
Промежуточное охлаждение и промежуточное введение кислорода (чистый О2 в качестве вторичного газа).
Газовую смесь продукции, выходящую из первого реактора с твердым слоем в целях промежуточного охлаждения (непрямого, воздушного) пропускали через соединительную трубу (40 см длины, 26 мм внутреннего диаметра, 30 мм наружного диаметра, 2 мм толщины стенок, высококачественная сталь, обернутую 1 см теплоизолирующего материала) которая содержала центрально расположенную инертную засыпку длиной 20 см, из колец из стеатита с геометрическими параметрами 7 мм × 3 мм × 4 мм (внешний диаметр × длина × внутренний диаметр), а с контактной трубой первого этапа была непосредственно соединена фланцевым соединением.
Газовая смесь продукции всегда входила в соединительную трубу с температурой, превышавшей Твх (первый этап), и выходила из нее, имея температуру, превышавшую 200°С и лежавшую ниже 270°С.
В конце соединительной трубы к охлажденной газовой смеси продуктов добавляли молекулярный кислород, давление которого находилось на уровне такового газовой смеси продуктов. Полученную итоговую газовую смесь (газовую смесь загрузки второго этапа окисления) непосредственно направляли в контактную трубу второго этапа, с которой вышеупомянутая соединительная труба также была соединена фланцем. Количество добавляемого молекулярного кислорода подбирали так, чтобы молярное соотношение между находящимся в итоговой газовой смеси О2 и находящимся в итоговой газовой смеси акролеином составляло 1,3.
Реактор для второго этапа окисления
Применяли реактор с контактной трубой и твердым слоем, имевший конструкцию, идентичную таковой для первого этапа окисления. Направления потоков соляного расплава и газовой смеси загрузки были одинаковы относительно реактора. Соляной расплав входил снизу, газовая смесь загрузки также. Температуру входа соляного расплава Твх подбирали так, чтобы на выходе со второго этапа окисления всегда получать оборот акролеина 99,3±0,1 моль-% при простом прохождении. Твых соляного расплава превышала Твх на величину до 2°С.
Загрузка контактной трубы (снизу вверх) была следующей:
В) Результаты, полученные в зависимости от состава газовой смеси загрузки первого этапа окисления (нагрузку пропеном задали равной 150 Нл/л·ч, избирательность формирования акриловой кислоты все время была не менее 94 моль-%).
а) Состав газовой смеси загрузки первого этапа окисления в основном был следующим:
Вначале входные температуры свежезагруженного катализатором реакторного устройства составляли:
Твх (первый этап окисления): 320°С
Твх (второй этап окисления): 268°С
После эксплуатации в течение 3 месяцев температуры на входе составляли:
Твх (первый этап окисления): 330°С
Твх (второй этап окисления): 285°С
b) Состав газовой смеси загрузки первого этапа окисления в основном был следующим:
Вначале входные температуры свежезагруженного катализатором реакторного устройства составляли:
Твх (первый этап окисления): 320°С
Твх (второй этап окисления): 268°С
После эксплуатации в течение 3 месяцев температуры на входе составляли:
Твх (первый этап окисления): 324°С
Твх (второй этап окисления): 276°С
II. Первый пример способа получения акриловой кислоты из пропана (описано стационарное рабочее состояние)
Реакционная зона А представляет собой шахтный реактор, выполненный как многоступенчатый реактор и работающий в адиабатическом режиме, в котором в направлении потока друг за другом расположены три твердых слоя катализатора. Каждый твердый слой катализатора представляет собой помещенную на проволочную сетку из высококачественной стали засыпку, включающую в себя (расположение в указанной последовательности в направлении потока) инертный материал (высота засыпки 26 мм; шары из стеатита диаметром от 1,5 до 2,5 мм) и смесь из свежего катализатора дегидрирования и шаров из стеатита (диаметром от 1,5 до 2,5 мм) соотношение объемов засыпки катализатора дегидрирования и шаров из стеатита = 1:3 (в качестве альтернативы вместо этого можно использовать то же количество катализатора без разбавления).
Перед каждым твердым слоем находится статический смеситель газов. Катализатор дегидрирования - это сплав Pt/Sn, обогащенный элементами Cs, К и La в форме оксидов и нанесенный на наружную и внутреннюю поверхность тяжей-носителей их смеси оксидов ZrO2·SiO2 (средняя длина (гауссово распределение от 3 мм до 12 мм с максимумом ок. 6 мм): 6 мм, диаметр 2 мм) в стехиометрическом соотношении (соотношение масс, включая носитель) Pt0,3Sn0,6La3,0Cs0,5K0,2(ZrO2)88,3(SiO2)7,1. Изготовление предшественников катализатора и активация их до действующего катализатора - как в примере 4 немецкой заявки на патент DE-A 10219879.
В описанном многоступенчатом реакторе гетерогенно-катализируемое частичное дегидрирование пропана проводят при прямом прохождении, применяя способ работы с циркуляционным газом дегидрирования. Нагрузка пропаном общего количества катализатора (рассчитанного без инертного материала) составляет 1500 Нл/л·ч.
На первый слой катализатора в направлении потока подают 7,30 Нм3/ч газовой смеси загрузки реакционного газа А (исходная смесь; Р=2,13 бар), которая характеризуется следующим составом:
Газовая смесь загрузки реакционного газа А представляет собой смесь первой (2,34 Нм3/ч) и второй (4,96 Нм3/ч) газовой смеси, которую создают, объединяя обе смеси в статическом смесителе. Первая газовая смесь обладает температурой 400°С и находится под давлением 2,13 бар, ее состав следующий:
(Т=70°С, Р=2,40 бар),
Путем непрямого теплообмена с продуктом-газом А, направляемым из реакционной зоны А в направлении реакционной зоны В, в первом теплообменнике температуру доводят до 400°С. Вторая газовая смесь включает в себя:
3,68 Нм3/ч циркуляционного газа дегидрирования (Т=551°С, Р=2,05 бар) и
1,28 Нм3/ч газа циркуляции пропана (Т=400°С, Р=3,26 бар).
Давление в ней составляет 2,13 бар, а температура 506°С.
Температуру газа циркуляции пропана регулируют путем непрямого теплообмена с продуктом-газом А, охлажденном в первом теплообменнике до температуры 435°С. Циркуляционный газ дегидрирования проводят по принципу струйного насоса (ср. с немецкой заявкой на патент DE-A 10211275), причем газ циркуляции пропана, нагретый до 400°С, играет роль ведущей струи.
Температуру газовой смеси загрузки реакционного газа А на входе и высоту засыпки слоя катализатора, через который проходит газовая смесь загрузки реакционного газа А, подбирают так, что реакционный газ А выходит из этого слоя катализатора, имея температуру 515°С, находясь под давлением 2,11 бар и имея следующий состав:
Поток на выходе составляет 7,46 Нм3/ч.
После первого слоя катализатора к реакционному газу А добавляют 0,12 Нм3/ч молекулярного кислорода (чистота более 99 об.-%, Т=139°С, Р=2,33 бар). Добавление осуществляют введением в таком режиме давления, чтобы итоговое давление полученного реакционного газа А по-прежнему составляло 2,11 бар.
Высота засыпки второго слоя катализатора подобрана так, чтобы реакционный газ А выходил из второго слоя катализатора с температурой 533°С, под давлением 2,08 и со следующим составом:
Поток на выходе составляет 7,79 Нм3/ч.
Перед статическим смесителем, находящимся перед третьим слоем катализатора, в этот реакционный газ А добавляют 0,13 Нм3/ч молекулярного кислорода (чистота более 99 об.-%, Т=139°С), тем же способом под давлением 2,33 бар. Итоговое давление полученного реакционного газа А по-прежнему составляет 2,08 бар.
Высота засыпки третьего слоя катализатора подобрана так, чтобы газ продукции А выходил из третьего слоя катализатора с температурой 551°С, под давлением 2,05 бар и со следующим составом:
Поток на выходе составляет 8,17 Нм3/ч.
Продукт-газ А разделяют с помощью разделителя потоков на две части идентичного состава. Первый поток равен 3,68 Нм3/ч, а второй поток 4,49 Нм3/ч. Первый поток возвращают в реакционную зону А в виде составной части газовой смеси загрузки реакционного газа А. В целях проводки циркуляционного газа дегидрирования первого потока на нагретом, как это описано выше, газе циркуляции пропана работает струйный насос, причем подачу ведущей струи, выходящей с падением давления из направляющего сопла через участок смешения и диффузор, осуществляют в направлении входа реакционной зоны А, а всасывающий штуцер направлен в сторону потока первой части продукта-газа А, и связь ”всасывающий штуцер - участок смешения - диффузор” представляет собой единственную линию, соединяющую возвращаемую первую часть продукта-газа А и вход в реакционную зону А. Вторую часть продукта-газа А выводят из реакционной зоны А и сначала охлаждают с 551°С до 435°С путем непрямого теплообмена с первой газовой смесью, состоящей из сжатого остаточного циркуляционного газа, сырого пропана и молекулярного кислорода и участвующей в формировании исходной газовой смеси (газовой смеси загрузки) реакционного газа А. При этом рабочее давление в выведенном из реакционной зоны А продукте-газе А падает с 2,05 бар до 1,95 бар. При этом первая газовая смесь нагревается с 164°С до 506°С, а давление в ней падает с 2,40 бар до 2,13 бар.
За этим следует второй непрямой теплообмен с газом циркуляции пропана из зоны разделения IV, при котором продукт-газ А охлаждают с 435°С до 335°С. Рабочее давление в продукте-газе А при этом падает до 1,94 бар. Температура газа циркуляции пропана, наоборот, возрастает с 69°С до 400°С.
Затем конденсацией отделяют от охлажденного до 335°С продукта-газа А 0,76 Нм3/ч водяного пара. Для этого продукт-газ А, охлажденный до 335°С, сначала проводят через непрямой теплообмен с 3,73 Нм3/ч продукта-газа А* (Т=40°С, Р=1,68 бар), от которого соответствующее количество водяного пара уже отделили ранее, охлаждают при этом до 108°С. Температура продукта-газа А* одновременно возрастает до 305°С. В расположенном далее воздушном радиаторе газ продукции (Т=108°С, Р=1,94 бар) охлаждается до 54°С ввиду непрямого теплообмена с наружным воздухом (при этом рабочее давление продукта-газа А падает до 1,73 бар), и после этого в нем уже наблюдается разделение фаз.
Путем прямого охлаждения с помощью охлажденного (до 29°С) распыленного водного конденсата, ранее отделенного от продукта-газа А методом соответствующего прямого охлаждения, от продукта-газа А конденсацией отделяют вышеуказанное количество водяного пара и создают продукт-газ А* (Р=1,68 бар, Т=40°С, 3,73 Нм3/ч). Его, как описано выше, нагревают до 305°С. Затем в продукт-газ А* (опять же дросселированием) вводят 1,39 Нм3/ч молекулярного кислорода (чистота более 99 об.-%, Т=139°С, Р=2,33 бар). Таким образом формируют исходную газовую смесь реакционного газа В (газовую смесь загрузки), имеющую температуру 288°С и находящуюся под рабочим давлением 1,63 бар.
Исходная газовая смесь реакционного газа В (газовая смесь загрузки) характеризуется следующим составом:
Ее загружают в реакционную зону В.
Первый этап окисления в направлении потока реакционного газа В - это реактор со множеством труб, имеющий две температурные зоны. Реакционные трубы имеют следующую конструкцию: сталь V2A-Stahl; 30 мм наружного диаметра, 2 мм толщины стенок, 26 мм внутреннего диаметра, длина 350 см. Загрузка реакционных труб сверху вниз следующая:
На первых 175 см, считая сверху вниз, обеспечивали термостатичность посредством соляной бани А, перекачиваемой противотоком относительно реакционного газа В. На вторых 175 см обеспечивали термостатичность посредством соляной бани В, перекачиваемой противотоком относительно реакционного газа В.
Второй этап окисления в направлении потока реакционного газа В - это также реактор со множеством труб, имеющий две температурные зоны. Сверху вниз засыпка реакционных труб следующая:
На первых 175 см, считая сверху вниз, обеспечивали термостатичность посредством соляной бани С, перекачиваемой противотоком относительно реакционного газа. На вторых 175 см обеспечивали термостатичность посредством соляной бани D, перекачиваемой противотоком относительно реакционного газа
На обоих этапах окисления соляную баню в каждом случае направляют с помощью отклоняющих перегородок вокруг реакционных труб по траектории в виде меандров.
Между двумя этапами окисления расположен теплообменник в виде пучка труб, позволяющий охлаждать газ продукции первого этапа окисления. Перед входом на второй этап окисления находится клапан для подачи молекулярного кислорода (чистота более 99 об.-%).
Нагрузку засыпки катализатора первого этапа окисления пропилена выбирают на уровне 130 Нл/л·ч. Входные температуры соляных расплавов (53 вес.-% KNO3, 40 вес.-% NaNO2, 7 вес.-% NaNO3) следующие:
В газовую смесь продукции первого этапа окисления добавляют (дросселированием) такое количество молекулярного кислорода (139°С, 2,33 бар), чтобы молярное соотношение содержащихся в газовой смеси загрузки для второго этапа окисления O2 и акролеина, O2: акролеин составляло 0,73. Нагрузка засыпки катализатора первого этапа окисления пропилена составляет 110 Нл/л·ч. Давление на входе на второй этап окисления равно 1,57 бар. Реакционный газ В выходит из промежуточного охладителя с температурой 260°С, а температура газовой смеси загрузки на входе на второй этап окисления составляет 256°С. Газовая смесь продукции первого этапа окисления характеризуется следующим составом:
Перед входом в дополнительный охладитель температура газ продукции первого этапа окисления составляет 335°С.
Газовая смесь продукции второго этапа окисления (продукт-газ В) обладает температурой 270°С и находится под давлением 1,55 бар, ее состав следующий:
Продукт-газ В (5,15 Нм3/ч), как это описано в международной заявке WO 2004/035514, подвергают фракционированной конденсации в тарельчатой колонне (зоне разделения II).
Для сжигания остатков в качестве первого горючего подают 13,7 г/ч тяжелокипящих компонентов (полиакриловые кислоты (аддукты Михаэля) и т.д.).
Со второй тарелки отбора над местом подачи в тарельчатую колонну продукта-газа В отбирают 2,80 кг/ч конденсированной сырой акриловой кислоты, имеющей температуру 15°С и содержание акриловой кислоты 96,99 вес.-%. Ее, после добавления небольшого количества воды, подвергают кристаллизации в суспензии, как это описано в международной заявке WO 2004/035514, продукт суспензионной кристаллизации отделяют от маточного раствора в гидравлических промывных колоннах, а маточный раствор возвращают в конденсационную колонну, как это описано в международной заявке WO 2004/035514. Степень очистки (содержание акриловой кислоты) отмытого продукта кристаллизации в суспензии превышает 99,87 вес.-%, он сразу пригоден для изготовления полимеризатов - ”суперабсорберов” воды для применения в области гигиены.
Количество конденсата кислой воды, отобранного с третьей тарелки отбора над местом подачи в тарельчатую колонну продукта-газа В и не возвращаемого в конденсационную колонну, составляет 1,10 кг/ч, имеет температуру 33°С и характеризуется следующим составом:
Его также подают на сжигание остатков (в качестве источника кислорода на сжигание остатков направляют 5,49 Нм3/ч воздуха).
В верхней части конденсационной колонны зоны разделения II выходят 2,98 Нм3/ч остаточного газа при температуре 33°С, под давлением 1,20 бар и в следующем составе:
На первом этапе сжатия многоступенчатого радиального компрессора остаточный газ сжимают с давления 1,20 бар до 2,40 бар, причем температура остаточного газа возрастает до 70°С. Затем остаточный газ, прошедший сжатие, разделяют на две части одинакового состава. Первую часть (1,19 Нм3/ч) направляют в качестве остаточного циркуляционного газа (прошедшего сжатие) на создание газовой смеси загрузки для реакционной зоны А. Давление во второй части (1,79 Нм3/ч) поднимают на втором этапе сжатия с 2,40 бар до 6,93 бар. При этом газ нагревается до 130°С.
В непрямом теплообменнике он остывает до 113°С, при этом образования конденсата не происходит (хладагентом является ”отмытый от пропана” остаточный газ, который впоследствии также называют ”выхлопным газом” и который после нагрева в непрямом теплообменнике с 30°С до 86°С на первом этапе расширения многоступенчатой расширительной турбины теряет давление начиная с уровня 20 бар, и при этом остывает). При этом выхлопной газ (0,58 Нм3/ч) нагревается. На втором этапе расширения давление выхлопного газа падает до 1,10 бар (температура его при этом становится 25°С), а затем его направляют на сжигание остатков.
В непрямом воздушном радиаторе сжатый остаточный газ (Р=6,93 бар и Т=113°С) охлаждают до 59°С. На третьем этапе сжатия его сжимают с давления 6,93 бар до 20 бар, причем одновременно он нагревается до 116°С.
В следующем непрямом теплообменнике он остывает до 107°С, при этом образования конденсата не происходит (хладагентом является ”отмытый от пропана” остаточный газ, который при этом нагревается с 30°С до 86°С). Еще в одном непрямом теплообменнике этот остаточный газ с температурой 107°С остывает до 35°С (хладагент - это вода с поверхности с температурой 25°С; вместо этой воды в настоящей публикации всегда можно применять другую охлаждающую воду). При этом из остаточного газа конденсацией выделяются 29 г/ч воды. Этот водный конденсат (который предпочтительно отделять каплеуловителем) также направляют на сжигание остатков, он сгорает вместе с остальными упомянутыми остатками (при подаче 5,49 Нм3/ч воздуха, как уже указано). Горячие газы из процесса сгорания охлаждают с образованием водяного пара (52 бар, 267°С, 4,6 кг/ч) и выпускают в атмосферу.
Еще 1,75 Нм3/ч содержащего пропан остаточного газа (20 бар, 35°С) подают в нижнюю часть насадочной колонны (зона разделения III). В верхнюю часть этой отмывочной колонны в качестве абсорбента подают 12,96 кг/ч технического тетрадекана типа PKWF 4/7 производства фирмы Haltermann, Германия, при температуре 30°С (Р=20 бар). Анализ методом газовой хроматографии с плазменно-ионизационным детектором дает вначале (в свежем абсорбенте) следующий состав в % площади ГХ:
Из верхней части промывочной колонны отводят ”отмытый от пропана” остаточный газ (выхлопной газ), имеющий под давлением 20 бар, при температуре 30°С и в количестве 0,58 Нм3/ч следующий состав:
С этого газа, как описано, снимают давление, а также подвергают его непрямому теплообмену, а затем подают на сжигание остатков или сжигают его в факеле.
Из нижней части ”промывочной колонны пропана”, не имеющей ни наружного нагрева, ни внешнего охлаждения, отбирают 15,27 кг/ч абсорбата (абсорбента с продуктом), находящегося под давлением 20 бар и имеющего температуру 59°С. Он содержит 14,79 вес.-% пропана и 0,38 вес.-% пропилена,
Прежде чем подать абсорбат в верхнюю часть следующей колонны десорбции, его нагревают в непрямом теплообменнике до 69°С. В качестве теплоносителя используют жидкий сток колонны десорбции (12,96 кг/ч), который, находясь под давлением 3,26 бар, имеет температуру 80°С и содержит еще 0,09 вес.-% растворенного пропана. С помощью насоса его снова сжимают до давления 20 бар и после непрямого охлаждения водой с поверхности (25°С) до 30°С возвращают в верхнюю часть ”промывочной колонны пропана” для абсорбции пропана.
После нагрева до 69°С давление абсорбата снижают (например, с помощью обратного насоса или вентиля) до 3,26 бар (механическую энергию, которая в случае использования обратного насоса при этом выделяется, целесообразно применять для участия в обратном сжатии абсорбента (жидкого стока десорбционной колонны), освобожденного в десорбционной колонне от пропана), а образованную при этом двухфазную смесь направляют в верхнюю часть десорбционной колонны.
В десорбционную колонну (также колонна с насадками) в качестве стриппинг-газа снизу, противотоком к абсорбату, стекающему из верхней части десорбционной колонны, направляют под давлением 5 бар водяной пар при температуре 152°С в количестве 0,1 кг/ч.
В десорбционной колонне находится нагревательный змеевик, по которому противотоком к поднимающемуся внутри десорбционной колонны водяному пару также проводят водяной пар при температуре 152°С, Р 5,00 бар в количестве 0,5 кг/ч.
В верхней части десорбционной колонны отходит газ циркуляции пропана в количестве 1,28 Нм3/ч, имеющий температуру 69°С и находящийся под рабочим давлением 3,26 бар. После нагрева до 400°С посредством непрямого теплообмена с продуктом-газом А, имеющим температуру Т=435°С, газ циркуляции пропана применяют в качестве направляющей струи для работы струйного насоса, с помощью которого циркуляционный газ дегидрирования и газ циркуляции пропана подают на загрузку реакционной зоны А исходной газовой смесью реакционного газа А.
Циркуляционный газ пропана характеризуется следующим составом:
III. Второй пример способа получения акриловой кислоты из пропана (описано стационарное рабочее состояние)
Реакционная зона А представляет собой многоступенчатый реактор, описанный в разделе II. В этом многоступенчатом реакторе гетерогенно-катализируемое частичное дегидрирование пропана проводят при прямом прохождении, применяя способ работы с циркуляционным газом дегидрирования. Нагрузка пропаном общего количества катализатора (рассчитанного без инертного материала) составляет 1500 Нл/л·ч.
На первый слой катализатора в направлении потока подают 8,23 Нм3/ч газовой смеси загрузки реакционного газа А (исходная смесь; Р=1,84 бар), которая характеризуется следующим составом:
Газовая смесь загрузки реакционного газа А представляет собой смесь первой (1,15 Нм3/ч) и второй (7,08 Нм3/ч) газовой смеси, которую создают, объединяя обе смеси в статическом смесителе. Первая газовая смесь обладает температурой 450°С и находится под давлением 1,84 бар, ее состав следующий:
Путем непрямого теплообмена с продуктом-газом А, направляемым из реакционной зоны А в направлении реакционной зоны В, в первом теплообменнике температуру доводят до 450°С.
Вторая газовая смесь включает в себя:
Давление в ней составляет 1,84 бар, а температура 486°С. Температуру газа циркуляции пропана регулируют путем непрямого теплообмена с продуктом-газом А, охлажденным в первом теплообменнике до температуры 476°С. Циркуляционный газ дегидрирования проводят по принципу струйного насоса (ср. с немецкой заявкой на патент DE-A 10211275), причем газ цируляции пропана, нагретый до 400°С, играет роль ведущей струи.
Температуру газовой смеси загрузки реакционного газа А на входе и высоту засыпки слоя катализатора, через который проходит газовая смесь загрузки реакционного газа А, подбирают так, что реакционный газ А выходит из этого слоя катализатора, имея температуру 503°С, находясь под давлением 1,81 бар и имея следующий состав:
Поток на выходе составляет 8,35 Нм3/ч.
После первого слоя катализатора к реакционному газу А добавляют 0,13 Нм3/ч молекулярного кислорода (чистота более 99 об.-%, Т=143°С, Р=2,40 бар). Добавление осуществляют введением в таком режиме давления, чтобы итоговое давление полученного реакционного газа А по-прежнему составляло 1,81 бар.
Высота засыпки второго слоя катализатора подобрана так, чтобы реакционный газ А выходил из второго слоя катализатора с температурой 520°С, под давлением 1,78 и со следующим составом:
Поток на выходе составляет 8,70 Нм3/ч.
Перед статическим смесителем, находящимся перед третьим слоем катализатора, в этот реакционный газ А добавляют 0,17 Нм3/ч молекулярного кислорода (чистота более 99 об.-%, Т=143°С), находящегося под давлением 1,78 бар, тем же способом (дросселированием). Итоговое давление полученного реакционного газа А по-прежнему составляет 1,78 бар.
Высота засыпки третьего слоя катализатора подобрана так, чтобы продукт-газ А выходил из третьего слоя катализатора с температурой 540°С, под давлением 1,73 бар и со следующим составом:
Поток на выходе составляет 9,17 Нм3/ч.
Продукт-газ А разделяют с помощью разделителя потоков на две части идентичного состава. Первый поток равен 5,04 Нм3/ч, а второй поток 4,13 Нм3/ч. Первый поток возвращают в реакционную зону А в виде составной части газовой смеси загрузки реакционного газа А. В целях проводки циркуляционного газа дегидрирования первого потока на нагретом, как это описано выше, газе циркуляции пропана работает струйный насос, причем подачу ведущей струи, выходящей с падением давления из направляющего сопла через участок смешения и диффузор, осуществляют в направлении входа реакционной зоны А, а всасывающий штуцер направлен в сторону потока первой части продукта-газа А, и связь ”всасывающий штуцер - участок смешения - диффузор” представляет собой единственную линию, соединяющую возвращаемую первую часть продукта-газа А и вход в реакционную зону А.
Вторую часть продукта-газа А выводят из реакционной зоны А и сначала охлаждают с 540°С до 476°С путем непрямого теплообмена с первой газовой смесью, состоящей из сырого пропана и молекулярного кислорода и участвующей в формировании исходной газовой смеси (газовой смеси загрузки) реакционного газа А. Рабочее давление продукта-газа А при этом остается в основном неизменным.
Первую газовую смесь при этом нагревают с 235°С до 450°С, в то время как рабочее давление этой газовой смеси в основном сохраняется на уровне 1,84 бар.
За этим следует второй непрямой теплообмен с газом циркуляции пропана из зоны разделения IV, при котором продукт-газ А охлаждают с 476°С до 307°С.Температура газа циркуляции пропана, наоборот, возрастает с 69°С до 400°С.
Затем конденсацией отделяют от охлажденного до 307°С продукта-газа А 0,52 Нм3/ч водяного пара. Для этого продукт-газ А, охлажденный до 307°С, сначала проводят через непрямой теплообмен с 3,60 Нм3/ч продукта-газа А* (Т=54°С, Р=1,73 бар), от которого соответствующее количество водяного пара уже отделили ранее, охлаждают при этом до 111°С.
Температура продукта-газа А* одновременно возрастает до 277°С. Путем прямого охлаждения с помощью охлажденного (до 29°С) распыленного водного конденсата, ранее отделенного от продукта-газа А методом соответствующего прямого охлаждения, от продукта-газа А конденсацией отделяют вышеуказанное количество водяного пара и создают продукт-газ А* (Р=1,73 бар, Т=54°С, 3,60 Нм3/ч). Его, как описано выше, нагревают до 277°С. Затем в продукт-газ А* (опять же дросселированием) вводят 139 Нм3/ч молекулярного кислорода (чистота более 99 об.-%, Т=143°С, Р=2,40 бар). Таким образом формируют исходную газовую смесь реакционного газа В (газовую смесь загрузки), имеющую температуру 262°С и находящуюся под рабочим давлением 1,63 бар.
Исходная газовая смесь реакционного газа В (газовая смесь загрузки) характеризуется следующим составом:
Ее загружают в реакционную зону В, имеющую ту же конструкцию, как и в разделе II.
Нагрузку засыпки катализатора первого этапа окисления пропиленом выбирают на уровне 164 Нл/л·ч. Входные температуры соляных расплавов (53 вес.-% KNO3, 40 вес.-% NaNO2, 7 вес.-% NaNO3) следующие:
В газовую смесь продукции первого этапа окисления добавляют (дросселированием) такое количество молекулярного кислорода (143°C, 2,40 бар), чтобы молярное соотношение содержащихся в газовой смеси загрузки для второго этапа окисления O2 и акролеина, O2: акролеин, составляло 0,72. Нагрузка засыпки катализатора первого этапа окисления пропилена составляет 139 Нл/л·ч. Давление на входе на второй этап окисления равно 1,57 бар.
Реакционный газ В выходит из промежуточного охладителя с температурой 260°С, а температура газовой смеси загрузки на входе на второй этап окисления составляет 256°С.
Газовая смесь продукции первого этапа окисления характеризуется следующим составом:
Перед входом в дополнительный охладитель температура газ продукции первого этапа окисления составляет 335°С.
Газовая смесь продукции второго этапа окисления (продукт-газ В) обладает температурой 270°С и находится под давлением 1,55 бар, ее состав следующий:
Продукт-газ В (5,03 Нм3/ч), как это описано в международной заявке WO 2004/035514, подвергают фракционированной конденсации в тарельчатой колонне (зоне разделения II).
Для сжигания остатков в качестве первого горючего подают 13,9 г/ч тяжелокипящих компонентов (полиакриловые кислоты (аддукты Михаэля) и т.д.).
Со второй тарелки отбора над местом подачи в тарельчатую колонну продукта-газа В отбирают 2,82 кг/ч конденсированной сырой акриловой кислоты, имеющей температуру 15°С и содержание акриловой кислоты 96,99 вес.-%. Ее, после добавления небольшого количества воды, подвергают кристаллизации в суспензии, как это описано в международной заявке WO 2004/035514, продукт суспензионной кристаллизации отделяют от маточного раствора в гидравлических промывных колоннах, а маточный раствор возвращают в конденсационную колонну, как это описано в международной заявке WO 2004/035514. Степень очистки (содержание акриловой кислоты) отмытого продукта кристаллизации в суспензии превышает 99,87 вес.-%, он сразу пригоден для изготовления полимеризатов - ”суперабсорберов” воды для применения в области гигиены.
Количество конденсата кислой воды, отобранного с третьей тарелки отбора над местом подачи в тарельчатую колонну продукта-газа В и не возвращаемого в конденсационную колонну, составляет 1,24 кг/ч, он имеет температуру 33°С и характеризуется следующим составом:
0,73 вес.%. акролеина,
4,95 вес.%. акриловой кислоты,
5,16 вес.%. уксусной кислоты и
88,1 вес.%. воды.
В верхней части конденсационной колонны из зоны разделения II выходят 2,72 Нм3/ч остаточного газа при температуре 33°С, под давлением 1,20 бар и в следующем составе:
На первом этапе сжатия многоступенчатого радиального компрессора остаточный газ сжимают с давления 1,20 бар до 6,93 бар, причем температура остаточного газа возрастает до 127°С.
В непрямом теплообменнике он остывает до 114°С, при этом образования конденсата не происходит (хладагентом является ”отмытый от пропана” остаточный газ, который впоследствии также называют ”выхлопным газом” и который после нагрева в непрямом теплообменнике с 30°С до 84°С на первом этапе расширения многоступенчатой расширительной турбины теряет давление начиная с уровня 20 бар и при этом остывает). При этом выхлопной газ (0,69 Нм3/ч) нагревается. На втором этапе расширения давление выхлопного газа снижают до 1,10 бар (температура его при этом составляет 18°С), а затем направляют его на сжигание остатков.
В непрямом воздушном радиаторе сжатый остаточный газ (Р=6,93 бар и Т=114°С) охлаждают до 59°С.На следующем этапе сжатия его сжимают с давления 6,93 бар до 20 бар, причем одновременно он нагревается до 114°С. В следующем непрямом теплообменнике он остывает до 107°С, при этом образования конденсата не происходит (хладагентом является ”отмытый от пропана” остаточный газ, который при этом нагревается с 30°С до 84°С). Еще в одном непрямом теплообменнике этот остаточный газ с температурой 107°С остывает до 35°С (хладагент - это вода с поверхности с температурой 25°С).
При этом из остаточного газа конденсацией выделяются 44,8 г/ч воды. Этот водный конденсат также направляют на сжигание остатков, он сгорает в нем вместе с остальными упомянутыми остатками (при подаче 5,49 Нм3/ч воздуха). Горячие газы из процесса сгорания охлаждают с образованием водяного пара (52 бар, 267°С, 5,16 кг/ч) и выпускают в атмосферу.
Еще 2,67 Нм3/ч содержащего пропан остаточного газа (20 бар, 35°С) подают в нижнюю часть насадочной колонны (зона разделения III).
В верхнюю часть этой отмывочной колонны в качестве абсорбента подают 20,39 кг/ч технического тетрадекана производства фирмы Haltermann, Германия, при температуре 30°С (Р=20 бар), как это описано в разделе II.
Из верхней части промывочной колонны отводят ”отмытый от пропана” остаточный газ (выхлопной газ), имеющий под давлением 20 бар, при температуре 30°С и в количестве 0,69 Нм3/ч следующий состав:
С этого газа, как описано, снимают давление, а также подвергают его непрямому теплообмену, а затем подают на сжигание остатков или сжигают его в факеле.
Из нижней части ”промывочной колонны пропана”, не имеющей ни наружного нагрева, ни внешнего охлаждения, отбирают 24,27 кг/ч абсорбата (абсорбента с продуктом), находящегося под давлением 20 бар и имеющего температуру 61°С. Он содержит 15,63 вес.-% пропана и 0,41 вес.-% пропилена.
Прежде чем подать абсорбат в верхнюю часть следующей колонны десорбции, его нагревают в непрямом теплообменнике до 69°С. В качестве теплоносителя используют жидкий сток колонны десорбции (20,39 кг/ч), который, находясь под давлением 3,26 бар, имеет температуру 80°С и содержит еще 0,089 вес.-% растворенного пропана. С помощью насоса его снова сжимают до давления 20 бар и после непрямого охлаждения водой с поверхности (25°С) до 30°С возвращают в верхнюю часть ”промывочной колонны пропана” для абсорбции пропана.
После нагрева до 69°С давление абсорбата снижают (например, с помощью обратного насоса или вентиля) до 3,26 бар (механическую энергию, которая в случае использования обратного насоса при этом выделяется, целесообразно применять для участия в обратном сжатии абсорбента (жидкого стока десорбционной колонны), освобожденного в десорбционной колонне от пропана), а образованную при этом двухфазную смесь направляют в верхнюю часть десорбционной колонны.
В десорбционную колонну (также колонна с насадками) в качестве стриппинг-газа снизу, противотоком к абсорбату, стекающему из верхней части десорбционной колонны, направляют под давлением 5 бар водяной пар при температуре 152°С в количестве 0,1 кг/ч.
В десорбционной колонне находится нагревательный змеевик, по которому противотоком к поднимающемуся внутри десорбционной колонны водяному пару также проводят водяной пар при температуре 152°С, Р 5,00 бар в количестве 0,8 кг/ч.
В верхней части десорбционной колонны отходит газ циркуляции пропана в количестве 2,04 Нм3/ч, имеющий температуру 69°С и находящийся под рабочим давлением 3,26 бар. После нагрева до 400°С посредством непрямого теплообмена с продуктом-газом А, имеющим температуру Т=476°С, газ циркуляции пропана применяют в качестве направляющей струи для работы струйного насоса, с помощью которого циркуляционный газ дегидрирования и газ циркуляции пропана в смеси подают на загрузку реакционной зоны А исходной газовой смесью реакционного газа А.
Циркуляционный газ пропана характеризуется следующим составом:
IV. Третий пример способа получения акриловой кислоты из пропана (описано стационарное рабочее состояние)
Реакционная зона А представляет собой многоступенчатый реактор, описанный в разделе II.
В этом многоступенчатом реакторе гетерогенно-катализируемое частичное дегидрирование пропана проводят при прямом прохождении, применяя способ работы с циркуляционным газом дегидрирования.
Нагрузка пропаном общего количества катализатора (рассчитанного без инертного материала) составляет 1500 Нл/л·ч.
На первый слой катализатора в направлении потока подают 3,80 Нм3/ч газовой смеси загрузки реакционного газа А (исходная смесь; Т=418°С; Р=1,77 бар), которая характеризуется следующим составом:
Газовая смесь загрузки реакционного газа А представляет собой смесь первой (1,59 Нм3/ч) и второй (2,21 Нм3/ч) газовой смеси, которую создают, объединяя обе смеси в статическом смесителе. Первая газовая смесь обладает температурой 450°С и находится под давлением 1,77 бар, ее состав следующий:
Путем непрямого теплообмена с продуктом-газом А, направляемым из реакционной зоны А в направлении реакционной зоны В, в первом теплообменнике температуру доводят до 450°С.
Вторая газовая смесь - это газ циркуляции пропана (2,21 Нм3/ч; 400°С; 1,77 бар). Температуру газа циркуляции пропана регулируют путем непрямого теплообмена с продуктом-газом А, охлажденным в первом теплообменнике до температуры 500°С.
Температуру газовой смеси загрузки реакционного газа А на входе и высоту засыпки слоя катализатора, через который проходит газовая смесь загрузки реакционного газа А, подбирают так, что реакционный газ А выходит из этого слоя катализатора, имея температуру 507°С, находясь под давлением 1,76 бар и имея следующий состав:
Поток на выходе составляет 3,95 Нм3/ч.
После первого слоя катализатора к реакционному газу А добавляют 0,13 Нм3/ч молекулярного кислорода (чистота более 99 об.-%, Т=143°С, Р=2,40 бар). Добавление осуществляют введением в таком режиме давления, чтобы итоговое давление полученного реакционного газа А по-прежнему составляло 1,76 бар.
Высота засыпки второго слоя катализатора подобрана так, чтобы реакционный газ А выходил из второго слоя катализатора с температурой 544°С, под давлением 1,75 и со следующим составом:
Поток на выходе составляет 4,29 Нм3/ч.
Перед статическим смесителем, находящимся перед третьим слоем катализатора, в этот реакционный газ А добавляют 0,12 Нм3/ч молекулярного кислорода (чистота более 99 об.-%, Т=143°С), находящегося под давлением 1,75 бар, тем же способом (дросселированием). Итоговое давление полученного реакционного газа А по-прежнему составляет 1,75 бар.
Высота засыпки третьего слоя катализатора подобрана так, чтобы продукт-газ А выходил из третьего слоя катализатора с температурой 571°С, под давлением 1,73 бар и со следующим составом:
Продукт-газ А выводят из реакционной зоны А и сначала охлаждают с 571°С до 500°С путем непрямого теплообмена с первой газовой смесью, состоящей из сырого пропана, газа, содержащего молекулярный водород, и молекулярного кислорода и участвующей в формировании исходной газовой смеси (газовой смеси загрузки) реакционного газа А. Рабочее давление продукта-газа А при этом остается в основном неизменным. Первую газовую смесь при этом нагревают с 213°С до 450°С, в то время как рабочее давление этой газовой смеси в основном сохраняется на уровне 1,77 бар.
За этим следует второй непрямой теплообмен с газом циркуляции пропана из зоны разделения IV, при котором газ продукции А охлаждают с 500°С до 337°С.
Температура газа циркуляции пропана, наоборот, возрастает с 69°С до 400°С.
Затем конденсацией отделяют от охлажденного до 337°С продукта-газа А 0,79 Нм3/ч водяного пара. Для этого продукт-газ А, охлажденный до 307°С, сначала проводят через непрямой теплообмен с 3,85 Нм3/ч продукта-газа А* (Т=54°С, Р=1,73 бар), от которого соответствующее количество водяного пара уже отделили ранее, охлаждают при этом до 120°С.
Температура продукта-газа А* одновременно возрастает до 307°С. Путем прямого охлаждения с помощью охлажденного (до 29°С) распыленного водного конденсата, ранее отделенного от продукта-газа А методом соответствующего прямого охлаждения, от продукта-газа А конденсацией отделяют вышеуказанное количество водяного пара и создают продукт-газ А* (Р=1,73 бар, Т=54°С, 3,85 Нм3/ч).
Его, как описано выше, нагревают до 307°С. Затем в продукт-газ А* (опять же дросселированием) вводят 1,41 Нм3/ч молекулярного кислорода (чистота более 99 об.-%, Т=143°С, Р=2,40 бар). Таким образом формируют исходную газовую смесь реакционного газа В (газовую смесь загрузки), имеющую температуру 290°С и находящуюся под рабочим давлением 1,63 бар.
Исходная газовая смесь реакционного газа В (газовая смесь загрузки) характеризуется следующим составом:
Ее загружают в реакционную зону В, имеющую ту же конструкцию, как и в разделе II.
Нагрузку засыпки катализатора первого этапа окисления пропиленом выбирают на уровне 130 Нл/л·ч. Входные температуры соляных расплавов (53 вес.-% KNO3, 40 вес.-% NaNO2, 7 вес.-% NaNO3) следующие:
В газовую смесь продукции первого этапа окисления добавляют (дросселированием) такое количество молекулярного кислорода (143°С, 2,40 бар), чтобы молярное соотношение содержащихся в газовой смеси загрузки для второго этапа окисления O2 и акролеина, O2: акролеин составляло, 0,745. Нагрузка засыпки катализатора первого этапа окисления пропилена составляет 110 Нл/л·ч. Давление на входе на второй этап окисления равно 1,57 бар. Реакционный газ В выходит из промежуточного охладителя с температурой 260°С, а температура газовой смеси загрузки на входе на второй этап окисления составляет 256°С.
Газовая смесь продукции первого этапа окисления характеризуется следующим составом:
Перед входом в дополнительный охладитель температура газ продукции первого этапа окисления составляет 335°С.
Газовая смесь продукции второго этапа окисления (продукт-газ В) обладает температурой 270°С и находится под давлением 1,55 бар, ее состав следующий:
Продукт-газ В (5,29 Нм3/ч), как это описано в международной заявке WO 2004/035514, подвергают фракционированной конденсации в тарельчатой колонне (зоне разделения II).
Для сжигания остатков в качестве первого горючего подают 13,8 г/ч тяжелокипящих компонентов (полиакриловые кислоты (аддукты Михаэля) и т.д.).
Со второй тарелки отбора над местом подачи в тарельчатую колонну продукта-газа В отбирают 2,80 кг/ч конденсированной сырой акриловой кислоты, имеющей температуру 15°С и содержание акриловой кислоты 96,99 вес.-%. Ее, после добавления небольшого количества воды, подвергают кристаллизации в суспензии, как это описано в международной заявке WO 2004/035514, продукт суспензионной кристаллизации отделяют от маточного раствора в гидравлических промывных колоннах, а маточный раствор возвращают в конденсационную колонну, как это описано в международной заявке WO 2004/035514.
Степень очистки (содержание акриловой кислоты) отмытого продукта кристаллизации в суспензии превышает 99,87 вес.-%, он сразу пригоден для изготовления полимеризатов - ”суперабсорберов” воды для применения в области гигиены.
Количество конденсата кислой воды, отобранного с третьей тарелки отбора над местом подачи в тарельчатую колонну продукта-газа В и не возвращаемого в конденсационную колонну, составляет 1,25 кг/ч, он имеет температуру 33°С и характеризуется следующим составом:
В верхней части конденсационной колонны из зоны разделения II выходят 2,98 Нм3/ч остаточного газа при температуре 33°С, под давлением 1,20 бар и в следующем составе:
На первом этапе сжатия многоступенчатого радиального компрессора остаточный газ сжимают с давления 1,20 бар до 6,93 бар, причем температура остаточного газа возрастает до 134°С.
В непрямом теплообменнике он остывает до 123°С, при этом образования конденсата не происходит (хладагентом является ”отмытый от пропана” остаточный газ, который впоследствии также называют ”выхлопным газом” и который после отделения молекулярного водорода, а также нагрева в непрямом теплообменнике с 30°С до 102°С на первом этапе расширения многоступенчатой расширительной турбины теряет давление начиная с уровня 20 бар и при этом остывает). При этом выхлопной газ (0,59 Нм3/ч) нагревается. На втором этапе расширения давление выхлопного газа снижают до 1,10 бар (температура его при этом составляет 23°С), а затем направляют его на сжигание остатков.
В непрямом воздушном радиаторе сжатый остаточный газ (Р=6,93 бар и Т=134°С) охлаждают до 59°С. На следующем этапе сжатия его сжимают с давления 6,93 бар до 20 бар, причем одновременно он нагревается до 117°С. В следующем непрямом теплообменнике он остывает до 110°С, при этом образования конденсата не происходит (хладагентом является ”отмытый от пропана” остаточный газ, прошедший отделение Н2 на мембране, этот газ при этом нагревается с 30°С до 102°С). Еще в одном непрямом теплообменнике этой остаточный газ с температурой 110°С остывает до 35°С (хладагент - это вода с поверхности с температурой 25°С).
При этом из остаточного газа конденсацией выделяются 44,2 г/ч воды. Этот водный конденсат также направляют на сжигание остатков, он сгорает вместе с остальными упомянутыми остатками (при подаче 4,12 Нм3/ч воздуха). Горячие газы из процесса сгорания охлаждают с образованием водяного пара (52 бар, 267°С, 3,41 кг/ч) и выпускают в атмосферу.
Еще 2,93 Нм3/ч содержащего пропан остаточного газа (20 бар, 35°С) подают в нижнюю часть насадочной колонны (зона разделения III). В верхнюю часть этой отмывочной колонны в качестве абсорбента подают 21,62 кг/ч технического тетрадекана производства фирмы Haltermann, Германия, при температуре 30°С (Р=20 бар), как это описано в разделе II.
Из верхней части промывочной колонны отводят ”отмытый от пропана” остаточный газ (выхлопной газ), имеющий под давлением 20 бар, при температуре 30°С и в количестве 1,01 Нм3/ч следующий состав:
Этот ”отмытый от пропана” выхлопной газ направляют в пучок закрепленных литьем мембранных шлангов (половолоконных мембран, наружное давление 1,77 бар; полиимидные мембраны типа В-Н производства фирмы UBE Industries Ltd.), из шлангов он выходит (Р=20 бар; Т=30°С), отдавая газовый поток (поток пермеата) размером 0,41 Нм3/ч, имеющего следующий состав:
Этот газовый поток добавляют (дросселированием) в первую газовую смесь для создания газовой смеси загрузки реакционного газа А. С оставшегося выхлопного газа, как описано, снимают давление, а также подвергают его непрямому теплообмену, а затем подают на сжигание остатков или сжигают его в факеле.
Из нижней части ”промывочной колонны пропана”, не имеющей ни наружного нагрева, ни внешнего охлаждения, отбирают 25,40 кг/ч абсорбата (абсорбента с продуктом), находящегося под давлением 20 бар и имеющего температуру 59°С. Он содержит 14,40 вес.-% пропана и 0,38 вес.-% пропилена.
Прежде чем подать абсорбат в верхнюю часть следующей колонны десорбции, его нагревают в непрямом теплообменнике до 69°С. В качестве теплоносителя используют жидкий сток колонны десорбции (21,62 кг/ч), который, находясь под давлением 3,26 бар, имеет температуру 80°С и содержит еще 0,09 вес.-% растворенного пропана. С помощью насоса его снова сжимают до давления 20 бар и после непрямого охлаждения водой с поверхности (25°С) до 30°С возвращают в верхнюю часть ”промывочной колонны пропана” для абсорбции пропана.
После нагрева до 69°С давление абсорбата снижают (например, с помощью обратного насоса или вентиля) до 3,26 бар (механическую энергию, которая в случае использования обратного насоса при этом выделяется, целесообразно применять для участия в обратном сжатии абсорбента (жидкого стока десорбционной колонны), освобожденного в десорбционной колонне от пропана), а образованную при этом двухфазную смесь направляют в верхнюю часть десорбционной колонны.
В десорбционную колонну (также колонна с насадками) в качестве стриппинг-газа снизу, противотоком к абсорбату, стекающему из верхней части десорбционной колонны, направляют под давлением 5 бар водяной пар при температуре 152°С в количестве 0,2 кг/ч.
В десорбционной колонне находится нагревательный змеевик, по которому противотоком к поднимающемуся внутри десорбционной колонны водяному пару также проводят водяной пар при температуре 152°С, Р 5,00 бар в количестве 0,8 кг/ч.
В верхней части десорбционной колонны отходит газ циркуляции пропана в количестве 2,21 Нм2/ч, имеющий температуру 69°С и находящийся под рабочим давлением 3,26 бар.
После нагрева до 400°С посредством непрямого теплообмена с продуктом-газом А, имеющим температуру Т=476°С, газ циркуляции пропана подают на загрузку реакционной зоны А исходной газовой смесью реакционного газа А.
Циркуляционный газ пропана характеризуется следующим составом:
V. Четвертый пример способа получения акриловой кислоты из пропана (описано стационарное рабочее состояние)
Реакционная зона А представляет собой многоступенчатый реактор, описанный в разделе II, однако в этом примере следом за ним последовательно подключен еще один работающий в адиабатическом режиме шахтный реактор, в котором имеется только один твердый слой катализатора, конструкция которого соответствует первой ступени твердого слоя в многоступенчатом реакторе. Между двумя реакторами подключен непрямой теплообменник. В многоступенчатом реакторе гетерогенно-катализируемое частичное дегидрирование пропана проводят при прямом прохождении, применяя способ работы с циркуляционным газом дегидрирования. Нагрузка пропаном общего количества катализатора (рассчитанного без инертного материала) всех ступеней составляет 1500 Нл/л·ч. В последовательно подключенном шахтном реакторе осуществляют в основном только гетерогенно-катализируемое сжигание молекулярного водорода.
В многоступенчатом реакторе на первый слой катализатора в направлении потока подают 6,93 Нм3/ч газовой смеси загрузки реакционного газа А (исходная смесь; Р=2,46 бар), которая характеризуется следующим составом:
Газовая смесь загрузки реакционного газа А представляет собой смесь первой (1,47 Нм3/ч) и второй (5,46 Нм3/ч) газовой смеси, которую создают, объединяя обе смеси в статическом смесителе.
Первая газовая смесь обладает температурой 500°С и находится под давлением 2,46 бар, ее состав следующий:
Благодаря непрямому теплообмену (в промежуточном охладителе, теплообменнике, находящемся между двумя реакторами) с выходящим из многоступенчатого реактора и движущимся в направлении последовательно подключенного шахтного реактора реакционным газом А температуру доводят до 500°С.
Вторая газовая смесь включает в себя:
Давление в ней составляет 2,46 бар. а температура 529°С.
Температуру газа циркуляции пропана регулируют путем непрямого теплообмена с продуктом-газом А, поступающим из последовательно подключенного шахтного реактора.
Циркуляционный газ дегидрирования проводят по принципу струйного насоса (ср. с немецкой заявкой на патент DE-A 10211275), причем газ циркуляции пропана, нагретый до 500°С, играет роль ведущей струи.
Температуру газовой смеси загрузки реакционного газа А на входе и высоту засыпки слоя катализатора, через который в направлении потока в многоступенчатом реакторе проходит газовая смесь загрузки реакционного газа А, подбирают так, что реакционный газ А выходит из этого слоя катализатора, имея температуру 522°С, находясь под давлением 2,44 бар и имея следующий состав:
Поток на выходе составляет 7,18 Нм3/ч. После первого слоя катализатора многоступенчатого реактора к реакционному газу А добавляют 0,64 Нм3/ч воздуха (Т=159°С; Р=2,66 бар). Добавление осуществляют введением в таком режиме давления, чтобы итоговое давление полученного реакционного газа А по-прежнему составляло 2,44 бар. Высота засыпки второго слоя катализатора многоступенчатого реактора подобрана так, чтобы реакционный газ А выходил из второго слоя катализатора с температурой 539°С, под давлением 2,41 и со следующим составом:
Поток на выходе составляет 8,04 Нм3/ч.
Перед статическим смесителем, находящимся перед третьим слоем катализатора многоступенчатого реактора, в этот реакционный газ А добавляют 0,63 Нм3/ч воздуха (Т=159°С), тем же способом под давлением 2,66 бар. Итоговое давление полученного реакционного газа А по-прежнему составляет 2,41 бар.
Высота засыпки третьего слоя катализатора многоступенчатого реактора подобрана так, чтобы реакционный газ А выходил из третьего слоя катализатора с температурой 554°С, под давлением 2,37 и со следующим составом:
Поток на выходе составляет 8,89 Нм3/ч.
На выходе из многоступенчатого реактора реакционный газ А разделяют с помощью разделителя потоков на две части идентичного состава. Первый поток равен 3,49 Нм3/ч, а второй поток 5,40 Нм3/ч. Первый поток возвращают в реакционную зону А в виде составной части газовой смеси загрузки реакционного газа А. В целях проводки циркуляционного газа дегидрирования первого потока на нагретом, как это описано выше, газе циркуляции пропана работает струйный насос, причем подачу ведущей струи, выходящей с падением давления из направляющего сопла через участок смешения и диффузор, осуществляют в направлении входа реакционной зоны А, а всасывающий штуцер направлен в сторону потока первой части реакционного газа А, и связь ”всасывающий штуцер -участок смешения - диффузор” представляет собой единственную линию, соединяющую возвращаемую первую часть реакционного газа А и вход в реакционную зону А.
Вторую часть реакционного А выводят из реакционной зоны А и сначала охлаждают в промежуточном охладителе, находящемся между многоступенчатым реактором и следующим за ним шахтным реактором с 554°С до 473°С путем непрямого теплообмена с первой газовой смесью, состоящей из сырого пропана и молекулярного кислорода и участвующей в формировании исходной газовой смеси (газовой смеси загрузки) реакционного газа А. Первую газовую смесь при этом нагревают с 229°С до 500°С, в то время как значения рабочего давления в основном сохраняются на прежнем уровне.
Теперь к охлажденной, как это описано, второй части реакционного газа А добавляют 6406 Нм3/ч воздуха (Т=159°С, дросселированием с 2,66 бар). Затем газовую смесь проводят через последовательно подключенный шахтный реактор.
Высота засыпки находящегося в последовательно подключенном шахтном реакторе (”четвертого”) слоя катализатора подобрана так, чтобы продукт-газ А выходил из третьего слоя катализатора с температурой 580°С, под давлением 2,26 бар и со следующим составом:
За этим следует второй непрямой теплообмен с газом циркуляции пропана из зоны разделения IV, при котором продукт-газ А (5,92 Нм3/ч) охлаждают с 580°С до 388°С. Температура газа циркуляции пропана, наоборот, возрастает с 69°С до 500°С.
Затем конденсацией отделяют от охлажденного до 388°С продукта-газа А 0,82 Нм3/ч водяного пара. Для этого продукт-газ А, охлажденный до 388°С, сначала проводят через непрямой теплообмен с 5,10 Нм3/ч продукта-газа А* (Т=40°С, Р=2,01 бар), от которого соответствующее количество водяного пара уже отделили конденсационным способом ранее, охлаждают при этом до 112°С. Температура продукта-газа А* одновременно возрастает до 358°С. В расположенном далее воздушном радиаторе газ продукции (Т=112°С, Р=2,26 бар) охлаждается до 54°С ввиду непрямого теплообмена с наружным воздухом (при этом рабочее давление продукта-газа А падает до 2,06 бар), и после этого в нем уже наблюдается разделение фаз.
Путем прямого охлаждения с помощью охлажденного (до 29°С) распыленного водного конденсата, ранее отделенного от продукта-газа А методом соответствующего прямого охлаждения, от продукта-газа А конденсацией отделяют вышеуказанное количество водяного пара и создают продукт-газ А* (Р=2,01 бар, Т=40°С, 5,10 Нм3/ч). Его, как описано выше, нагревают до 358°С. Затем в продукт-газ А* дросселированием вводят 8,55 Нм3/ч воздуха (Т=159°С; Р=2,66 бар). Таким образом формируют исходную газовую смесь реакционного газа В (газовую смесь загрузки), имеющую температуру 283°С и находящуюся под рабочим давлением 1,96 бар.
Исходная газовая смесь реакционного газа В (газовая смесь загрузки) характеризуется следующим составом:
Ее загружают в реакционную зону В, имеющую ту же конструкцию, как и в разделе II.
Нагрузку засыпки катализатора первого этапа окисления пропиленом выбирают на уровне 130 Нл/л·ч. Входные температуры соляных расплавов (53 вес.-% KNO3, 40 вес.-% NaNO2, 7 вес.-% NaNO3) следующие:
В газовую смесь продукции первого этапа окисления добавляют (дросселированием) такое количество воздуха (159°С, 2,66 бар), чтобы молярное соотношение содержащихся в газовой смеси загрузки для второго этапа окисления O2 и акролеина, O2: акролеин, составляло 1,01. Нагрузка засыпки катализатора первого этапа окисления пропилена составляет 110 Нл/л·ч. Давление на входе на второй этап окисления равно 1,68 бар.
Реакционный газ В выходит из промежуточного охладителя с температурой 260°С, а температура газовой смеси загрузки на входе на второй этап окисления составляет 253°С.
Газовая смесь продукции первого этапа окисления характеризуется следующим составом:
Перед входом в дополнительный охладитель температура газ продукции первого этапа окисления составляет 335°С.
Газовая смесь продукции второго этапа окисления (продукт-газ В) обладает температурой 270°С и находится под давлением 1,55 бар, ее состав следующий:
Продукт-газ В (14,81 Нм3/ч), как это описано в международной заявке WO 2004/035514, подвергают фракционированной конденсации в тарельчатой колонне (зоне разделения II).
Для сжигания остатков в качестве первого горючего подают 13,8 г/ч тяжелокипящих компонентов (полиакриловые кислоты (аддукты Михаэля) и т.д.).
Со второй тарелки отбора над местом подачи в тарельчатую колонну продукта-газа В отбирают 2,80 кг/ч конденсированной сырой акриловой кислоты, имеющей температуру 15°С и содержание акриловой кислоты 96,99 вес.-%. Ее, после добавления небольшого количества воды, подвергают кристаллизации в суспензии, как это описано в международной заявке WO 2004/035514, продукт суспензионной кристаллизации отделяют от маточного раствора в гидравлических промывных колоннах, а маточный раствор возвращают в конденсационную колонну, как это описано в международной заявке WO 2004/035514. Степень очистки (содержание акриловой кислоты) отмытого продукта кристаллизации в суспензии превышает 99,87 вес.-%, он сразу пригоден для изготовления полимеризатов - ”суперабсорберов” воды для применения в области гигиены.
Количество конденсата кислой воды, отобранного с третьей тарелки отбора над местом подачи в тарельчатую колонну продукта-газа В и не возвращаемого в конденсационную колонну, составляет 1,19 кг/ч, он имеет температуру 34°С и характеризуется следующим составом:
В верхней части конденсационной колонны из зоны разделения II выходят 12,56 Нм3/ч остаточного газа при температуре 33°С, под давлением 1,20 бар и в следующем составе:
На первом этапе сжатия многоступенчатого радиального компрессора остаточный газ сжимают с давления 1,20 бар до 6,93 бар, причем температура остаточного газа возрастает до 217°С.
В непрямом теплообменнике он остывает до 111°С, при этом образования конденсата не происходит (хладагентом является ”отмытый от пропана” остаточный газ, который впоследствии также называют ”выхлопным газом” и который после нагрева в непрямом теплообменнике с 30°С до 138°С на первом этапе расширения многоступенчатой расширительной турбины теряет давление начиная с уровня 20 бар и при этом остывает). При этом выхлопной газ (10,39 Нм3/ч) нагревается. На втором этапе расширения давление выхлопного газа снижают до 1,10 бар (температура его при этом составляет 78°С), а затем направляют его на сжигание остатков.
В непрямом воздушном радиаторе сжатый остаточный газ (Р=6,93 бар и Т=111°С) охлаждают до 59°С. На втором этапе сжатия его сжимают с давления 6,93 бар до 20 бар, причем одновременно он нагревается до 168°С. В следующем непрямом теплообменнике он остывает до 101°С, при этом образования конденсата не происходит (хладагентом является ”отмытый от пропана” остаточный газ, который при этом нагревается с 30°С до 138°С), Еще в одном непрямом теплообменнике этот остаточный газ с температурой 101°С остывает до 35°С (хладагент - это вода с поверхности с температурой 25°С).
При этом из остаточного газа конденсацией выделяются 159,7 г/ч воды. Этот водный конденсат также направляют на сжигание остатков, он сгорает вместе с остальными упомянутыми остатками (при подаче 5,59 Нм3/ч воздуха). Горячие газы из процесса сгорания охлаждают с образованием водяного пара (52 бар, 267°С, 3,78 кг/ч) и выпускают в атмосферу.
Еще 12,37 Нм3/ч содержащего пропан остаточного газа (20 бар, 35°С) подают в нижнюю часть насадочной колонны (зона разделения III). В верхнюю часть этой отмывочной колонны в качестве абсорбента подают 66,37 кг/ч технического тетрадекана производства фирмы Haltermann, Германия, при температуре 30°С (Р=20 бар), как это описано в разделе II.
Из верхней части промывочной колонны отводят ”отмытый от пропана” остаточный газ (выхлопной газ), имеющий под давлением 20 бар, при температуре 30°С и в количестве 10,39 Нм3/ч следующий состав:
С этого выхлопного газа, как описано, снимают давление, а также подвергают его непрямому теплообмену, а затем подают на сжигание остатков или сжигают его в факеле.
Из нижней части ”промывочной колонны пропана”, не имеющей ни наружного нагрева, ни внешнего охлаждения, отбирают 70,25 кг/ч абсорбата (абсорбента с продуктом), находящегося под давлением 20 бар и имеющего температуру 41°С. Он содержит 5,41 вес.-% пропана и 0,14 вес.-% пропилена.
Прежде чем подать абсорбат в голову следующей колонны десорбции, его нагревают в непрямом теплообменнике до 69°С. В качестве теплоносителя используют жидкий сток колонны десорбции (66,37 кг/ч), который, находясь под давлением 2,97 бар, имеет температуру 80°С и содержит еще 0,03 вес.-% растворенного пропана. С помощью насоса его снова сжимают до давления 20 бар и после непрямого охлаждения водой с поверхности (25°С) до 30°С возвращают в верхнюю часть ”промывочной колонны пропана” для абсорбции пропана.
После нагрева до 69°С давление абсорбата снижают (например, с помощью обратного насоса или вентиля) до 2,97 бар (механическую энергию, которая в случае использования обратного насоса при этом выделяется, целесообразно применять для участия в обратном сжатии абсорбента (жидкого стока десорбционной колонны), освобожденного в десорбционной колонне от пропана), а образованную при этом двухфазную смесь направляют в верхнюю часть десорбционной колонны.
В десорбционной колонне находится нагревательный змеевик, по которому в том же направлении, что и у поднимающегося внутри десорбционной колонны водяного пара, также проводят водяной пар при температуре 152°С, Р 5,00 бар в количестве 1,3 кг/ч.
В верхней части десорбционной колонны отходит газ циркуляции пропана в количестве 1,98 Нм3/ч, имеющий температуру 69°С и находящийся под рабочим давлением 2,97 бар. После нагрева до 500°С посредством непрямого теплообмена с продуктом-газом А, имеющим температуру Т=580°С, газ циркуляции пропана применяют в качестве направляющей струи для работы струйного насоса, с помощью которого циркуляционный газ дегидрирования и газ циркуляции пропана подают на загрузку реакционной зоны А исходной газовой смесью реакционного газа А.
Циркуляционный газ пропана характеризуется следующим составом:
VI. Первый сравнительный пример способа получения акриловой кислоты из пропана (описано стационарное рабочее состояние)
Реакционная зона А состоит из работающего в адиабатическом режиме шахтного реактора, в котором в имеется только один твердый слой катализатора, конструкция какового слоя соответствует первой ступени твердого слоя в многоступенчатом реакторе, описанном в разделе II.
В шахтном реакторе гетерогенно-катализируемое частичное дегидрирование пропана проводят при прямом прохождении, применяя способ работы с циркуляционным газом дегидрирования. Нагрузка пропаном общего количества катализатора (рассчитанного без инертного материала) составляет 1500 Нл/л·ч.
В шахтный реактор подают 24,87 Нм3/ч газовой смеси загрузки реакционного газа А (исходная смесь; Т=450°С; Р=2,82 бар), которая характеризуется следующим составом:
Газовая смесь загрузки реакционного газа А представляет собой смесь первой (1,69 Нм3ч) и второй (23,18 Нм3ч) газовой смеси, которую создают, объединяя обе смеси в статическом смесителе.
Первая газовая смесь - это смесь из 0,013 Нм3ч водяного пара (Т=134°С; Р=3 бар), 1,06 Нм3/ч сырого пропана, включающего в себя более 98 об.-% пропана (свежего пропана, Т=122°С; Р=3,50 бар) и 0,62 Нм3/ч молекулярного водорода (чистота более 99 об.-%; Т=35°С; Р=2,20 бар), а вторая газовая смесь - это смесь 11,41 Нм3/ч циркуляционного газа дегидрирования (Т=534°С; Р=2,36 бар), 11,16 Нм3/ч сжатого остаточного циркуляционного газа (Т=444°С; Р=2,82 бар), а также 0,61 Нм3/ч сжатого воздуха (Т=175°С, Р=3,00 бар).
Путем непрямого теплообмена с продуктом-газом А, направляемым из реакционной зоны А в направлении реакционной зоны В, температуру остаточного циркуляционного газа доводят до 444°С.
Циркуляционный газ дегидрирования проводят по принципу струйного насоса (ср. с немецкой заявкой на патент DE-A 10211275), причем остаточный циркуляционный газ, нагретый до 444°С, играет роль ведущей струи. В получающуюся при этом смесь добавляют сжатый воздух (дросселированием).
Высота засыпки слоя катализатора, через который проходит газовая смесь загрузки реакционного газа А, подобрана так, чтобы реакционный газ А выходил из второго слоя катализатора с температурой 534°С, под давлением 2,36 и со следующим составом:
Поток на выходе составляет 25,35 Нм3/ч.
На выходе из шахтного реактора продукт-газ А разделяют с помощью разделителя потоков на две части идентичного состава.
Первый поток равен 11,41 Нм3/ч, а второй поток 13,94 Нм3/ч. Первый поток возвращают в реакционную зону А в виде составной части газовой смеси загрузки реакционного газа А. В целях проводки циркуляционного газа дегидрирования первого потока на нагретом, как это описано выше, сжатом остаточном циркуляционном газе работает струйный насос, причем подачу ведущей струи, выходящей с падением давления из направляющего сопла через участок смешения и диффузор, осуществляют в направлении входа реакционной зоны А, а всасывающий штуцер направлен в сторону потока первой части продукта-газа А, и связь ”всасывающий штуцер - участок смешения - диффузор” представляет собой единственную линию, соединяющую возвращаемую первую часть продукта-газа А и вход в реакционную зону А.
Вторую часть продукта-газа А выводят из реакционной зоны А и сначала охлаждают в расположенном следом за этой зоной непрямом теплообменнике до 317°С путем непрямого теплообмена со сжатым остаточным циркуляционным газом (11,16 Нм3/ч; Т=131°С; Р=3,00 бар). Рабочее давление остается в основном неизменным. Остаточный циркуляционный газ при этом нагревается до 444°С, а его рабочее давление снижается до 2,82 бар.
Затем продукт-газ А путем второго непрямого теплообмена с ”отмытым от пропана” продукта-газа А (9,06 Нм3/ч; Т=30°С, Р=10,50 бар) охлаждают до 221°С, что одновременно обусловливает снижение его рабочего давления до 2,18 бар.
”Отмытый от пропана” продукт-газ А (который ниже также называют ”выхлопным газом”), при этом нагревается до 287°С, а его рабочее давление меняется на 10,10 бар. Затем давление выхлопного газа снижают на первом этапе расширения многоступенчатой расширительной турбины с 10,10 бар до 3,50 бар, при этом он охлаждается с 287°С до 176°С.Теперь выхлопной газ, покидающий первый этап расширения, находится в непрямом теплообмене с продуктом-газом А, имеющим температуру 221°С.При этом последний, сохраняя неизменным рабочее давление, охлаждается до 215°С. Выхлопной газ, сохраняя в основном свое рабочее давление без изменений, нагревается до 191°С.Затем давление выхлопного газа снижают на втором этапе расширения многоступенчатой расширительной турбины до 1,23 бар, причем он охлаждается до 89°С. Затем выхлопной газ направляют на сжигание остатков.
Продукт-газ и А, имеющий показатели 215°С и 2,18 бар, затем сначала охлаждают до 60°С в непрямом теплообменнике с воздушным охлаждением, а затем до 39°С в непрямом теплообменнике с водным охлаждением. Выделяющуюся при вызванной охлаждением конденсации воду (0,68 кг/ч) отделяют в каплеуловителе.
Оставшиеся 13,09 Нм3/ч продукта-газа А сжимают до 4,78 бар на первом этапе сжатия многоступенчатого радиального компрессора. При этом температура продукта-газа А возрастает от 39°С до 106°С. Затем продукт-газ А посредством непрямого теплообмена с воздухом охлаждают до 54°С, а выделяющуюся при этом конденсационную воду (8,0 г/ч) отделяют с помощью каплеуловителя. На втором этапе сжатия оставшиеся 13,08 Нм3/ч продукта-газа А сжимают до давления 10,50 бар. Это обусловливает нагрев продукта-газа А до 123°С. После этого продукт-газ А сначала посредством охлаждения воздухом в непрямом теплообменнике охлаждают до 54°С, а затем путем непрямого теплообмена с охлаждающей водой - до 29°С. Выделяющуюся при вызванной охлаждением конденсации воду (0,29 кг/ч) отделяют в каплеуловителе. Остающиеся 12,71 Нм3/ч продукта-газа А (рабочее давление которого по-прежнему составляет в основном 10,50 бар) направляют в нижнюю часть колонны с насадками. Состав его следующий:
В верхнюю часть отмывочной колонны пропана в качестве абсорбента подают 120,32 кг/ч технического тетрадекана производства фирмы Haltermann, Германия, при температуре 30°С (Р=10,50 бар), как это описано в разделе II.
Из верхней части промывочной колонны отводят ”отмытый от пропана” продукт-газ А (выхлопной газ), имеющий под давлением 10,50 бар, при температуре 30°С и в количестве 9,06 Нм3/ч следующий состав:
После непрямого теплообмена (подобного уже описанному) с продуктом-газом А, выведенным из реакционной зоны А, а также многоступенчатого снятия давления в многоступенчатой расширительной турбине до уровня 1,23 бар выхлопной газ подают на сжигание остатков или сжигают в факеле. С промышленной точки зрения может оказаться целесообразно, прежде чем снимать с выхлопного газа давление, отделить содержащийся в нем молекулярный водород. Это можно осуществить, например, подвергая выхлопной газ разделению на мембране, как это уже описано. Отделенный таким образом молекулярный водород можно полностью или частично вернуть в реакционную зону А (целесообразно - в качестве компонента газовой смеси загрузки реакционного газа А). В качестве альтернативы его можно, например, сжигать в топливных элементах.
Из нижней части ”промывочной колонны пропана”, не имеющей ни наружного нагрева, ни внешнего охлаждения, отбирают 127,37 кг/ч абсорбата (абсорбента с продуктом), находящегося под давлением 10,5 бар и имеющего температуру 40°С. Он содержит 3,83 вес.-% пропана и 1,54 вес.-% пропилена, 0,1 вес.-% этана и 0,1 вес.-% CO2. Прежде чем подать абсорбат в следующую колонну десорбции, давление абсорбата снижают (например, с помощью обратного насоса или вентиля) до 2,01 бар (механическую энергию, которая в случае использования обратного насоса при этом выделяется, целесообразно применять для участия в обратном сжатии абсорбента (жидкого стока десорбционной колонны), освобожденного в десорбционной колонне от пропана). Образованную при этом двухфазную смесь направляют в десорбционную колонну в ее верхней части.
В десорбционную колонну (также колонна с насадками) в качестве стриппинг-газа снизу, противотоком к абсорбату, стекающему из верхней части десорбционной колонны, направляют сжатый до давления 3,00 бар воздух (который при этом нагрелся до 175°С) в количестве 8,00 Нм3/ч.
Жидкий сток из колонны десорбции (120,32 кг/ч) насосом снова сжимают до давления 10,50 бар и после непрямого охлаждения до температуры 30°С с помощью охлаждающей воды возвращают на абсорбцию пропана в верхнюю часть колонны ”отмывки” пропана (колонны абсорбции).
В верхней части десорбционной колонны отходят 11,64 Нм3/ч газовой смеси, имеющей температуру 33°С, находящейся под давлением 2,01 бар и включающей следующие компоненты:
Путем непрямого теплообмена с водяным паром температуру газовой смеси повышают до 140°С (при этом рабочее давление снижается до 1,91 бар).
Эту смесь в качестве исходной смеси реакционного газа В (газовой смеси загрузки) загружают в реакционную зону В, имеющую ту же конструкцию, как и в разделе II.
Нагрузку засыпки катализатора первого этапа окисления пропилена выбирают на уровне 130 Нл/л·ч. Входные температуры соляных расплавов (53 вес.-% KNO3, 40 вес.-% NaNO2, 7 вес.-% NaNO3) следующие:
Из промежуточного охладителя выходит реакционный газ В в количестве 11,66 Нм3/ч при температуре 260°С, под давлением 1,68 бар и в следующем составе:
К нему добавляют (дросселированием) 2,17 Нм3/ч воздуха, сжатого в радиальном компрессоре до давления 3 бар (при этом воздух нагревается до 175°С) так, что температура входа реакционного газа на второй этап окисления составляет 252°С при давлении на входе 1,68 бар (нагрузка акролеином -110 Нл/л·ч).
Газовая смесь продукции второго этапа окисления (продукт-газ В) обладает температурой 270°С и находится под давлением 1,55 бар, ее состав следующий:
Продукт-газ В (13,42 Нм3/ч), как это описано в международной заявке WO 2004/035514, подвергают фракционированной конденсации в тарельчатой колонне.
Для сжигания остатков подают 13,8 г/ч тяжелокипящих компонентов (полиакриловые кислоты (аддукты Михаэля) и т.д.).
Со второй тарелки отбора над местом подачи в тарельчатую колонну продукта-газа В отбирают 2,82 кг/ч конденсированной сырой акриловой кислоты, имеющей температуру 15°С и содержание акриловой кислоты 96,99 вес.-%. Ее, после добавления небольшого количества воды, подвергают кристаллизации в суспензии, как это описано в международной заявке WO 2004/035514, продукт суспензионной кристаллизации отделяют от маточного раствора в гидравлических промывных колоннах, а маточный раствор возвращают в конденсационную колонну, как это описано в международной заявке WO 2004/035514. Степень очистки (содержание акриловой кислоты) отмытого продукта кристаллизации в суспензии превышает 99,87 вес.-%, он сразу пригоден для изготовления полимеризатов - ”суперабсорберов” воды для применения в области гигиены.
Количество конденсата кислой воды, отобранного с третьей тарелки отбора над местом подачи в тарельчатую колонну продукта-газа В и не возвращаемого в конденсационную колонну, составляет 1,04 кг/ч, он имеет температуру 33°С. Его также направляют на сжигание остатков.
В верхнеой части конденсационной колонны выходят 11,16 Нм3/ч остаточного газа при температуре 33°С, под давлением 1,20 бар и в следующем составе:
Все количество остаточного газа сжимают до давления 3 бар (при этом он нагревается до 131°С) и, как уже описано, после непрямого теплообмена с выходящим из реакционной зоны А продуктом-газом А возвращают в реакционную зону А в качестве остаточного циркуляционного газа (в качестве ведущей струи для струйного насоса, применяемого для подачи циркуляционного газа дегидрирования).
IV. Второй сравнительный пример способа получения акриловой кислоты из пропана (описано стационарное рабочее состояние)
Реакционная зона А состоит из работающего в адиабатическом режиме шахтного реактора, в котором в имеется только один твердый слой катализатора, конструкция какового слоя соответствует первой ступени твердого слоя в многоступенчатом реакторе, описанном в разделе II.
В шахтном реакторе гетерогенно-катализированное частичное дегидрирование пропана проводят при прямом прохождении, применяя способ работы с циркуляционным газом дегидрирования. Нагрузка пропаном общего количества катализатора (рассчитанного без инертного материала) составляет 1500 Нл/л·ч.
В шахтный реактор подают 23,79 Нм3/ч газовой смеси загрузки реакционного газа А (исходная смесь; Т=456°С; Р=2,82 бар), которая характеризуется следующим составом:
Газовая смесь загрузки реакционного газа А представляет собой смесь первой (1,69 Нм3/ч) и второй (22,10 Нм3/ч) газовой смеси, которую создают, объединяя обе смеси в статическом смесителе.
Первая газовая смесь - это смесь из
0,013 Нм3/ч водяного пара (Т=134°С, Р=3 бар),
1,08 Нм3/ч сырого пропана, содержащего более 98 об.-% пропана (свежего пропана) (Т=122°С, Р=3,50 бар), и
0,60 Нм3/ч молекулярного водорода (чистота более 99 об.-%; Т=35°С; Р=2,20 бар),
а вторая газовая смесь - это смесь из
10,93 Нм3/ч циркуляционного газа дегидрирования (Т=546°С, Р=2,82 бар).
10,52 Нм3/ч сжатого остаточного газа дегидрирования (Р=3 бар; Т=455°С)
0,65 Нм3/ч сжатого воздуха (Т=175°С, Р=3,00 бар).
Путем непрямого теплообмена с продуктом-газом А, направляемым из реакционной зоны А в направлении реакционной зоны В, температуру остаточного циркуляционного газа доводят до 455°С.
Циркуляционный газ дегидрирования проводят по принципу струйного насоса (ср. с немецкой заявкой на патент DE-A 10211275), причем остаточный циркуляционный газ, нагретый до 455°С, играет роль ведущей струи. В получающуюся при этом смесь добавляют сжатый воздух (дросселированием).
Высота засыпки слоя катализатора, через который проходит газовая смесь загрузки реакционного газа А, отрегулирована так, чтобы реакционный газ А выходил из второго слоя катализатора с температурой 546°С, под давлением 2,39 и со следующим составом:
Поток на выходе составляет 24,29 Нм3/ч.
На выходе из шахтного реактора продукт-газ А разделяют с помощью разделителя потоков на две части идентичного состава.
Первый поток равен 10,93 Нм3/ч, а второй поток 13,36 Нм3/ч. Первый поток возвращают в реакционную зону А в виде составной части газовой смеси загрузки реакционного газа А. В целях проводки циркуляционного газа дегидрирования первого потока на нагретом, как это описано выше, сжатом остаточном циркуляционном газе работает струйный насос, причем подачу ведущей струи, выходящей с падением давления из направляющего сопла через участок смешения и диффузор, осуществляют в направлении входа реакционной зоны А, а всасывающий штуцер направлен в сторону потока первой части продукта-газа А, и связь ”всасывающий штуцер - участок смешения - диффузор” представляет собой единственную линию, соединяющую возвращаемую первую часть продукта-газа А и вход в реакционную зону А.
Вторую часть продукта-газа А выводят из реакционной зоны А и сначала охлаждают в расположенном следом за этой зоной непрямом теплообменнике до 330°С путем непрямого теплообмена со сжатым остаточным циркуляционным газом (10,52 Нм3/ч; Т=135°С; Р=3,00 бар). Рабочее давление остается в основном неизменным. Остаточный циркуляционный газ при этом нагревается до 455°С, а его рабочее давление снижается до 2,82 бар.
Затем продукт-газ А путем второго непрямого теплообмена с ”отмытым от пропана” продукта-газа А (9,06 Нм3/ч; Т=30°С, Р=10,50 бар) охлаждают до 220°С, что одновременно обусловливает снижение его рабочего давления до 2,21 бар.
”Отмытый от пропана” продукт-газ А (который ниже также называют ”выхлопным газом”), при этом нагревается до 300°С, а его рабочее давление меняется на 10,10 бар. Затем давление выхлопного газа снижают на первом этапе расширения многоступенчатой расширительной турбины с 10,10 бар до 3,50 бар, при этом он охлаждается с 300°С до 187°С. Теперь выхлопной газ, покидающий первый этап расширения, находится в непрямом теплообмене с продуктом-газом А, имеющим температуру 220°С. При этом последний, сохраняя неизменным рабочее давление, охлаждается до 219°С. Выхлопной газ, сохраняя в основном свое рабочее давление без изменений, нагревается до 190°С. Затем давление выхлопного газа снижают на втором этапе расширения многоступенчатой расширительной турбины до 1,23 бар, причем он охлаждается до 88°С. Затем выхлопной газ направляют на сжигание остатков.
Продукт-газ А, имеющий показатели 219°С и 2,21 бар, затем сначала охлаждают до 60°С в непрямом теплообменнике с воздушным охлаждением, а затем до 39°С в непрямом теплообменнике с водным охлаждением. Выделяющуюся при вызванной охлаждением конденсации воду (0,68 кг/ч) отделяют в каплеуловителе.
Оставшиеся 12,52 Нм3/ч продукта-газа А сжимают до 4,81 бар на первом этапе сжатия многоступенчатого радиального компрессора. При этом температура продукта-газа А возрастает от 39°С до 108°С. Затем продукт-газ А посредством непрямого теплообмена с воздухом охлаждают до 54°С, а выделяющуюся при этом конденсационную воду (5,7 г/ч) отделяют с помощью каплеуловителя. На втором этапе сжатия оставшиеся 12,51 Нм3/ч продукта-газа А сжимают до давления 10,50 бар. Это обусловливает нагрев продукта-газа А до 126°С. После этого продукт-газ А сначала посредством охлаждения воздухом в непрямом теплообменнике охлаждают до 54°С, а затем путем непрямого теплообмена с охлаждающей водой - до 29°С. Выделяющуюся при вызванной охлаждением конденсации воду (0,28 кг/ч) отделяют в каплеуловителе. Остающиеся 12,17 Нм3/ч продукта-газа А (рабочее давление которого по-прежнему составляет в основном 10,50 бар) направляют в нижнюю часть колонны с насадками. Состав его следующий:
В верхнюю часть отмывочной колонны пропана в качестве абсорбента подают 115,96 кг/ч технического тетрадекана производства фирмы Haltermann, Германия, при температуре 30°С (Р=10,50 бар), как это описано в разделе II.
Из верхней части промывочной колонны отводят ”отмытый от пропана” продукт-газ А (выхлопной газ), имеющий под давлением 10,50 бар, при температуре 30°С и в количестве 9,06 Нм3/ч следующий состав:
После непрямого теплообмена (подобного уже описанному) с продуктом-газом А, выведенным из реакционной зоны А, а также многоступенчатого снятия давления в многоступенчатой расширительной турбине до уровня 1,23 бар выхлопной газ подают на сжигание остатков. С промышленной точки зрения может оказаться целесообразно, прежде чем снимать с выхлопного газа давление, отделить содержащийся в нем молекулярный водород. Это можно осуществить, например, подвергая выхлопной газ разделению на мембране, как это уже описано. Отделенный таким образом молекулярный водород можно полностью или частично вернуть в реакционную зону А (целесообразно - в качестве компонента газовой смеси загрузки реакционного газа А). В качестве альтернативы его можно, например, сжигать в топливных элементах.
Из нижней части ”промывочной колонны пропана”, не имеющей ни наружного нагрева, ни внешнего охлаждения, отбирают 121,93 кг/ч абсорбата (абсорбента с продуктом), находящегося под давлением 10,5 бар и имеющего температуру 40°С. Он содержит 3,14 вес.-% пропана и 1,61 вес.-% пропилена, 0,01 вес.-% этана и 0,09 вес.-% CO2. Прежде чем подать абсорбат в следующую колонну десорбции, давление абсорбата снижают (например, с помощью обратного насоса или вентиля) до 1,98 бар (механическую энергию, которая в случае использования обратного насоса при этом выделяется, целесообразно применять для участия в обратном сжатии абсорбента (жидкого стока десорбционной колонны), освобожденного в десорбционной колонне от пропана). Образованную при этом двухфазную смесь направляют в десорбционную колонну в ее верхней части.
В десорбционную колонну (также колонна с насадками) в качестве стриппинг-газа снизу, противотоком к абсорбату, стекающему из верхней части десорбционной колонны, направляют сжатый до давления 3,00 бар воздух (который при этом нагрелся до 175°С) в количестве 7,88 Нм3/ч.
Жидкий сток из колонны десорбции (115,94 кг/ч) насосом снова сжимают до давления 10,50 бар и после непрямого охлаждения до температуры 30°С с помощью охлаждающей воды возвращают на абсорбцию пропана в верхнюю часть колонны ”отмывки” пропана (колонны абсорбции).
В верхней части десорбционной колонны отходят 10,97 Нм3/ч газовой смеси, имеющей температуру 33°С, находящейся под давлением 1,98 бар и включающей следующие компоненты:
Путем непрямого теплообмена с водяным паром температуру газовой смеси повышают до 140°С (при этом рабочее давление снижается до 1,88 бар).
Эту смесь в качестве исходной смеси реакционного газа В (газовой смеси загрузки) загружают в реакционную зону В, имеющую ту же конструкцию, как и в разделе II.
Нагрузку засыпки катализатора первого этапа окисления пропилена выбирают на уровне 130 Нл/л. Входные температуры соляных расплавов (53 вес.-% KNO3, 40 вес.-% NaNO2, 7 вес.-% NaNO3) следующие:
Из промежуточного охладителя выходит реакционный газ В в количестве 10,99 Нм3/ч при температуре 260°С, под давлением 1,67 бар и в следующем составе:
К нему добавляют (дросселированием) 2,21 Нм3/ч воздуха, сжатого в радиальном компрессоре до давления 3 бар (при этом воздух нагревается до 175°С) так, что температура входа реакционного газа на второй этап окисления составляет 251°С при давлении на входе 1,67 бар (нагрузка акролеином -110 Нл/л·ч).
Газовая смесь продукции второго этапа окисления (продукт-газ В) обладает температурой 270°С и находится под давлением 1,55 бар, ее состав следующий:
Продукт-газ В (12,79 Нм3/ч), как это описано в международной заявке WO 2004/035514, подвергают фракционированной конденсации в тарельчатой колонне.
Для сжигания остатков подают 13,8 г/ч тяжелокипящих компонентов (полиакриловые кислоты (аддукты Михаэля) и т.д.).
Со второй тарелки отбора над местом подачи в тарельчатую колонну продукта-газа В отбирают 2,82 кг/ч конденсированной сырой акриловой кислоты, имеющей температуру 15°С и содержание акриловой кислоты 96,99 вес.-%. Ее, после добавления небольшого количества воды, подвергают кристаллизации в суспензии, как это описано в международной заявке WO 2004/035514, продукт суспензионной кристаллизации отделяют от маточного раствора в гидравлических промывных колоннах, а маточный раствор возвращают в конденсационную колонну, как это описано в международной заявке WO 2004/035514. Степень очистки (содержание акриловой кислоты) отмытого продукта кристаллизации в суспензии превышает 99,87 вес.-%, он сразу пригоден для изготовления полимеризатов - ”суперабсорберов” воды для применения в области гигиены.
Количество конденсата кислой воды, отобранного с третьей тарелки отбора над местом подачи в тарельчатую колонну продукта-газа В и не возвращаемого в конденсационную колонну, составляет 1,06 кг/ч, он имеет температуру 33°С. Его также направляют на сжигание остатков.
В верхней части конденсационной колонны выходят 10,52 Нм3/ч остаточного газа при температуре 33°С, под давлением 1,20 бар и в следующем составе:
Все количество остаточного газа сжимают до давления 3 бар (при этом он нагревается до 135°С) и, как уже описано, после непрямого теплообмена с выходящим из реакционной зоны А продуктом-газом А возвращают в реакционную зону А в качестве остаточного циркуляционного газа (в качестве ведущей струи для струйного насоса, применяемого для подачи циркуляционного газа дегидрирования).
Предварительная заявка на патент США №60/802786, поданная 24.05.2006, включена в настоящую заявку посредством ссылки на литературу.
В содержании вышеупомянутых публикаций возможны существенные изменения и отклонения от настоящего изобретения. Таким образом, можно считать, что изобретение в рамках прилагаемой формулы также может быть реализовано иначе, нежели конкретно указано в ней.
Изобретение относится к усовершенствованному способу получения акролеина, акриловой кислоты или их смеси из пропана, при котором А) в первую реакционную зону А подают по меньшей мере два газообразных потока подачи, содержащих пропан, по меньшей мере один из которых содержит свежий пропан, с образованием реакционного газа А; в реакционной зоне А реакционный газ А проводят по меньшей мере через один слой катализатора, в котором путем частичного гетерогенно-катализируемого дегидрирования пропана образуются молекулярный водород и пропилен; в реакционную зону А подают молекулярный кислород, который окисляет в реакционной зоне А по меньшей мере часть содержащегося в реакционном газе А молекулярного водорода до водяного пара; и отбирают из реакционной зоны А газ-продукт А, содержащий пропилен, пропан, молекулярный водород и водяной пар; В) содержащийся в газе-продукте А водяной пар при необходимости полностью или частично путем прямого и/или непрямого охлаждения отделяют в первой зоне разделения I методом конденсации с сохранением газа-продукта А*; С) в реакционной зоне В, подавая молекулярный кислород, используют газ-продукт А или газ-продукт А* для подачи по меньшей мере в один реактор окисления с реакционным газом В, содержащим пропан, пропилен и молекулярный кислород, и подвергают содержащийся в нем пропилен гетерогенно-катализируемому частичному окислению в газовой фазе с образованием акролеина, или акриловой кислоты, или их смеси в качестве целевого продукта, а также газа-продукта В, содержащего непревращенный пропан; D) из реакционной зоны В выводят газ-продукт В и отделяют содержащийся в нем целевой продукт во второй зоне разделения II, причем остается остаточный газ, содержащий пропан; Е) при необходимости возвращают часть остаточного газа, имеющую состав остаточного газа, в качестве потока подачи, содержащего пропан, в реакционную зону A; F) в зоне разделения III пропан, содержащийся в не возвращенном в реакционную зону А остаточном газе, из которого ранее при необходимости конденсацией удалили, возможно, содержавшийся в нем водяной пар и/или посредством разделительной мембраны полностью или частично удалили содержавшийся в нем молекулярный водород, отделяют путем абсорбции в органическом растворителе (абсорбенте) с образованием содержащего пропан абсорбата; и G) в разделительной зоне IV отделяют пропан от абсорбата и возвращают в реакционную зону А в виде содержащего пропан потока подачи, причем в реакционной зоне А окисляют до водяного пара по меньшей мере столько молекулярного водорода, чтобы это окисленное в реакционной зоне А до водяного пара количество водорода составляло от 30 до 70 моль-% количества молекулярного водорода, образовавшегося в реакционной зоне А, и для значений рабочего давления Р, в каждом случае определенного на входе в соответствующую зону, в различных зонах способа согласно изобретению действуют следующие соотношения: Рреакционная зона А>Рзона разделения I>Рреакционная зона В>Рзона разделения II>Рзона разделения III>Рзона разделения IV>Рреакционная зона А. 26 з.п. ф-лы, 6 пр.
1. Способ получения акролеина, акриловой кислоты или их смеси из пропана, при котором
A) - в первую реакционную зону А подают по меньшей мере два газообразных потока подачи, содержащих пропан, по меньшей мере один из которых содержит свежий пропан, с образованием реакционного газа А;
- в реакционной зоне А реакционный газ А проводят по меньшей мере через один слой катализатора, в котором путем частичного гетерогенно-катализируемого дегидрирования пропана образуются молекулярный водород и пропилен;
- в реакционную зону А подают молекулярный кислород, который окисляет в реакционной зоне А по меньшей мере часть содержащегося в реакционном газе А молекулярного водорода до водяного пара, и
- отбирают из реакционной зоны А газ-продукт А, содержащий пропилен, пропан, молекулярный водород и водяной пар;
B) содержащийся в газе-продукте А водяной пар при необходимости полностью или частично путем прямого и/или непрямого охлаждения отделяют в первой зоне разделения I методом конденсации с сохранением газа-продукта А*;
C) в реакционной зоне В, подавая молекулярный кислород, используют газ-продукт А или газ-продукт А* для подачи по меньшей мере в один реактор окисления с реакционным газом В, содержащим пропан, пропилен и молекулярный кислород, и подвергают содержащийся в нем пропилен гетерогенно-катализируемому частичному окислению в газовой фазе с образованием акролеина или акриловой кислоты или их смеси в качестве целевого продукта, а также газа-продукта В, содержащего непревращенный пропан;
D) из реакционной зоны В выводят газ-продукт В и отделяют содержащийся в нем целевой продукт во второй зоне разделения II, причем остается остаточный газ, содержащий пропан;
Е) при необходимости возвращают часть остаточного газа, имеющую состав остаточного газа, в качестве потока подачи, содержащего пропан, в реакционную зону А;
F) в зоне разделения III пропан, содержащийся в не возвращенном в реакционную зону А остаточном газе, из которого ранее при необходимости конденсацией удалили возможно содержавшийся в нем водяной пар и/или посредством разделительной мембраны полностью или частично удалили содержавшийся в нем молекулярный водород, отделяют путем абсорбции в органическом растворителе (абсорбенте) с образованием содержащего пропан абсорбата; и
G) в разделительной зоне IV отделяют пропан от абсорбата и возвращают в реакционную зону А в виде содержащего пропан потока подачи, отличающийся тем,
что в реакционной зоне А окисляют до водяного пара по меньшей мере столько молекулярного водорода, чтобы это окисленное в реакционной зоне А до водяного пара количество водорода составляло от 30 до 70 мол.% количества молекулярного водорода, образовавшегося в реакционной зоне А и для значений рабочего давления Р, в каждом случае определенного на входе в соответствующую зону, в различных зонах способа согласно изобретению действуют следующие соотношения:
Рреакционная зона А>Рзона разделения I>Рреакционная зона В>Рзона разделения II<Рзона разделения III>Рзона разделения IV>Рреакционная зона А.
2. Способ по п.1, отличающийся тем, что по меньшей мере часть теплоты, вырабатывающейся при окислении молекулярного водорода в реакционной зоне А, применяют для того, чтобы путем непрямого теплообмена с реакционным газом А, или продуктом-газом А, или реакционным газом А и продуктом-газом А, используемыми в качестве теплоносителя, нагревать газообразные потоки подачи, подаваемые в реакционную зону А.
3. Способ по п.1, отличающийся тем, что количество окисляемого до водяного пара в реакционной зоне А водорода составляет по меньшей мере 40 мол.% количества молекулярного водорода, образовавшегося в реакционной зоне А.
4. Способ по п.1, отличающийся тем, что продукт-газ А, отобранный из реакционной зоны А, применяют как таковой для загрузки по меньшей мере одного реактора окисления в реакционной зоне В.
5. Способ по п.1, отличающийся тем, что в зоне разделения I конденсацией отделяют по меньшей мере 5 мол.% содержащегося в продукте-газе А водяного пара.
6. Способ по п.1, отличающийся тем, что в зоне разделения I конденсацией отделяют по меньшей мере 25 мол.% содержащегося в продукте-газе А водяного пара.
7. Способ по п.1, отличающийся тем, что в зоне разделения I конденсацией отделяют по меньшей мере 50 мол.% содержащегося в продукте-газе А водяного пара.
8. Способ по п.1, отличающийся тем, что в зоне разделения I конденсацией отделяют от 5 до 98 мол.% содержащегося в продукте-газе А водяного пара.
9. Способ по п.1, отличающийся тем, что рабочее давление в реакционной зоне А составляет >1 до 5 бар.
10. Способ по п.1, отличающийся тем, что молекулярный водород сжигают с помощью молекулярного кислорода в реакционной зоне А самое позднее тогда, когда уже образовалось 90 мол.% общего количества формирующегося в реакционной зоне А молекулярного водорода.
11. Способ по п.1, отличающийся тем, что молекулярный водород сжигают с помощью молекулярного кислорода в реакционной зоне А самое позднее тогда, когда уже образовалось 75 мол.% общего количества формирующегося в реакционной зоне А молекулярного водорода.
12. Способ по п.1, отличающийся тем, что молекулярный водород сжигают с помощью молекулярного кислорода в реакционной зоне А самое позднее тогда, когда уже образовалось 65 мол.% общего количества формирующегося в реакционной зоне А молекулярного водорода.
13. Способ по п.1, отличающийся тем, что в зоне разделения I конденсацией отделяют по меньшей мере часть содержащегося в продукте-газе А водяного пара, и это отделение обеспечивают путем прямого или непрямого охлаждения продукта-газа А.
14. Способ по п.1, отличающийся тем, что часть остаточного газа, имеющую состав остаточного газа, возвращают в качестве потока подачи, содержащего пропан, в реакционную зону А, а остаточный газ содержит молекулярный кислород.
15. Способ по п.1, отличающийся тем, что подаваемый в реакционную зону А молекулярный кислород, также подают как составную часть воздуха.
16. Способ по п.1, отличающийся тем, что подаваемый в реакционную зону А молекулярный кислород также подают как составную часть газа, который содержит не более 50 об.% отличных от молекулярного кислорода компонентов.
17. Способ по п.1, отличающийся тем, что отделение пропана из абсорбата в зоне разделения IV проводят методом отгонки (стриппинга) с помощью содержащего водяной пар газа, а стрип-газ, нагруженный пропаном, возвращают в реакционную зону А в виде содержащего пропан потока подачи.
18. Способ по п.1, отличающийся тем, что реакционная зона А оформлена в адиабатическом режиме.
19. Способ по п.1, отличающийся тем, что при расчете на однократное прохождение реакционной зоны А реакции дегидрирования в реакционной зоне А подвергают от 10 до 40 мол.% общего количества пропана, поданного в реакционную зону А.
20. Способ по п.1, отличающийся тем, что в состав реакционной зоны А входит по меньшей мере один многоступенчатый реактор.
21. Способ по п.1, отличающийся тем, что образующийся в реакционной зоне А реакционный газ А отличается следующим составом:
от 55 до 80 об.% пропана,
от 0,1 до 20 об.% пропилена,
от 2 до 10 об.% H2,
от 1 до 5 об.% O2,
от 0 до 20 об.% N2, и
от 5 до 15 об.% H2O.
22. Способ по п.1, отличающийся тем, что продукт-газ А охлаждают путем непрямого теплообмена с отделенным в зоне разделения IV и возвращенным в реакционную зону А потоком подачи, содержащим пропан.
23. Способ по п.1, отличающийся тем, что гетерогенно-катализируемое частичное окисление в газовой фазе представляет собой двухступенчатое частичное окисление пропилена до акриловой кислоты.
24. Способ по п.1, отличающийся тем, что отделенный в зоне разделения IV поток подачи, содержащий пропан и возвращаемый в реакционную зону А, содержит следующие компоненты:
от 80 до 99,99 мол.%, пропана,
от 0 до 5 мол.%, пропилена и
от 0 до 20 мол.%, Н2О.
25. Способ по п.1, отличающийся тем, что часть остаточного газа, соответствующую по составу остаточному газу, возвращают в качестве содержащего пропан потока подачи в реакционную зону А, и это количество остаточного газа составляет до 10 вес.% от общего количества остаточного газа.
26. Способ по любому из пп.1-25, отличающийся тем, что подаваемый в реакционную зону В молекулярный кислород также подают как составную часть газа, который содержит не более 50 об.% отличных от молекулярного кислорода компонентов.
27. Способ по любому из пп.1-25, отличающийся тем, что подаваемый в реакционную зону В молекулярный кислород также подают как составную часть воздуха.
| WO 2004031107 A1, 15.04.2004 | |||
| WO 2004031106 A1, 15.04.2004 | |||
| DE 10028582 A1, 20.12.2001 | |||
| Печь для непрерывного получения сернистого натрия | 1921 |
|
SU1A1 |
| WO 00/53556 A, 14.09.2000 | |||
| WO 00/53558 A, 14.09.2000 | |||
| DE 3313573 A1, 20.10.1983 | |||
| DE 10246119 A1, 15.04.2004 | |||
| DE 10245585 A1, 08.04.2004 | |||
| RU 96104337 А, 10.08.1996. | |||

