Область изобретения
Настоящее изобретение относится к новому способу получения 7-хлор-1-циклопропил-6-фтор-4-оксо-1,4-дигидро-1,8-нафтиридин-3-карбоновой кислоты. Настоящее изобретение предлагает способ получения 7-хлор-1-циклопропил-6-фтор-4-оксо-1,4-дигидро-1,8-нафтиридин-3-карбоновой кислоты (5) высокой чистоты из этилового эфира 3-(2,6-дихлор-5-фторпиридин-3-ил)-3-оксопропановой кислоты (1) с применением единственного растворителя в одном реакционном сосуде, как показано на схеме 1.
Схема реакции 1
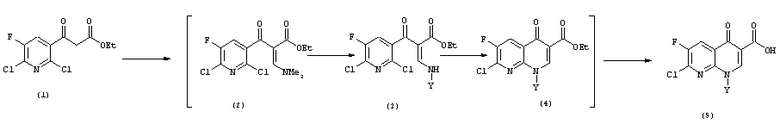
в которой
Y представляет собой неразветвленный, разветвленный или циклический алкил, имеющий от 1 до 5 атомов углерода, и незамещенный или замещенный галогеном, или представляет собой незамещенный или замещенный галогеном фенил.
Предшествующие уровни техники.
Эффективное получение соединения (5) по вышеприведенной схеме 1 имеет решающее значение для экономического производства антибиотиков на основе фторхинолонов, применяемых при лечении микробной инфекции. В предшествующем способе, известном из опубликованной заявки JP 3-74231, соединение (5) получается в следующих трех стадиях:
Стадия 1: Этиловый эфир 3-(2,6-дихлор-5-фторпиридин-3-ил)-3-оксо-пропановой кислоты взаимодействует с триэтилортоформиатом и уксусным ангидридом с получением этилового эфира 2-[(2,6-дихлор-5-фторпиридин-3-ил)]-3-этоксиакриловой кислоты;
Стадия 2: Полученное соединение вступает в реакцию с циклопропиламином и
Стадия 3: Образовавшийся циклопропиленамин циклизуется гидридом натрия с получением нафтиридинового эфира.
Однако в этом способе промежуточные соединения выделяют на каждой стадии и поэтому в целом синтез усложняется и приводит к низким выходам.
Другой способ получения соединения (5) описан в опубликованной заявке JP 2002-155081. Способ является усовершенствованным по сравнению с вышеописанным способом, раскрытым в опубликованной заявке JP 3-74231, и вышеописанные стадии 1-3 проводятся в одном и том же растворителе без промежуточных процессов выделения. Это изобретение может быть представлено более детально в виде следующих трех стадий.
Стадия 1: соединение (1) взаимодействует с триэтилортоформиатом при нагревании в присутствии уксусного ангидрида, что сопровождается удалением побочных продуктов, этанола и уксусной кислоты. После завершения реакции остаточный триэтилортоформиат следует полностью испарить при пониженном давлении для предотвращения образования побочных продуктов на следующей стадии.
Стадия 2: полученный таким образом остаток охлаждают и растворяют в толуоле. К этому раствору по каплям добавляют циклопропиламин для получения енаминового соединения.
Стадия 3: Затем к раствору енаминового соединения добавляют каталитическое количество бромида тетрабутиламмония и затем для циклизации добавляют водный раствор гидроксида натрия. Кристаллический сложный эфир нафтиридин-3-карбоновой кислоты отфильтровывают, промывают и сушат и затем гидролизуют с образованием соединения (5) с выходом около 85%.
Этот способ имеет преимущество в том, что нафтиридиновый эфир получается без промежуточного выделения, но также имеет следующий недостаток.
Реакция стадии (1) известна по ряду ссылок и широко используется в качестве основного способа (cf.: WO89/06649, EP 0 160578 A1). Однако промышленное применение этой стадии осложняется двумя обстоятельствами. Первое состоит в том, что после завершения реакции следует удалить триэтилортоформиат с помощью дистилляции при пониженном давлении. Этот способ требует длительного времени. Второе обстоятельство состоит в том, что если триэтилортоформиат не полностью удален, остаток триэтилортоформиата взаимодействует с вводимым в следующую стадию циклопропиламином с образованием побочного продукта, который трудно отделить.
Раскрытие изобретения.
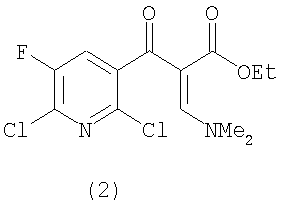
Авторы настоящего изобретения проводили широкие исследования, для того чтобы решить проблемы, описанные в опубликованной заявке JP 2002-155081. В результате мы нашли, что соединение из настоящего изобретения, которое является производным 1,8-нафтиридин-3-карбоновой кислоты, может быть получено с высоким выходом и очищено за короткое время без необходимости проводить сложные операции, просто применяя диалкилацеталь диметилформамида вместо триэтилортоформиата на первой стадии. Соответственно получение промежуточных соединений (2), (3) и (4) осуществляется в одну стадию с применением одного и того же растворителя без каких-либо промежуточных процессов выделения.
Ниже настоящее изобретение изложено более детально.
Наилучший способ выполнения изобретения
Настоящее изобретение относится к новому способу получения производного 1,8-нафтиридин-3-карбоновой кислоты следующей формулы (5), проводимому в одном реакционном сосуде с применением единственного растворителя без какого-либо промежуточного выделения:
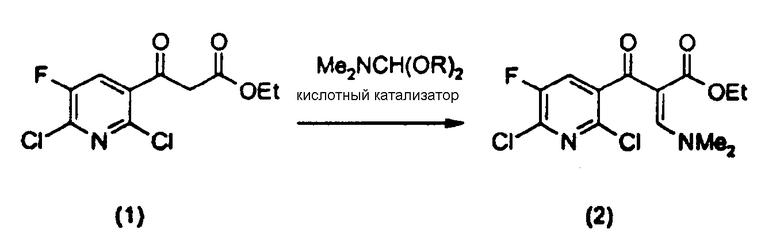
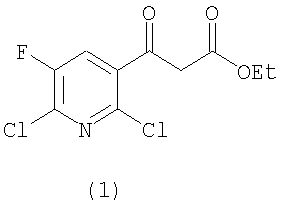
a) на первой стадии соединение формулы (1)
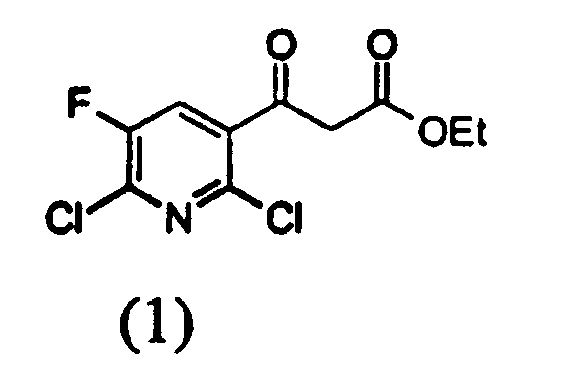
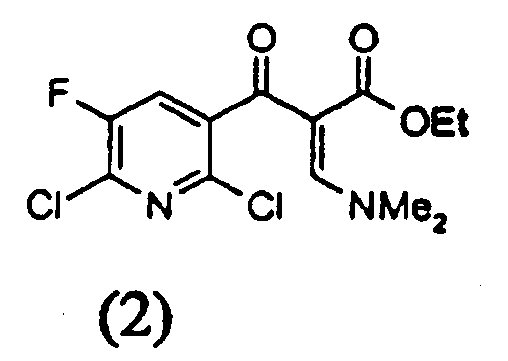
вводят в реакцию диалкилацеталем диметилформамида, имеющим формулу Me2NCH(OR)2 (где R представляет собой неразветвленный, разветвленный или циклический алкил, имеющий от 1 до 9 атомов углерода, или бензил) в растворителе в присутствии кислотного катализатора для получения соединения следующей формулы (2)
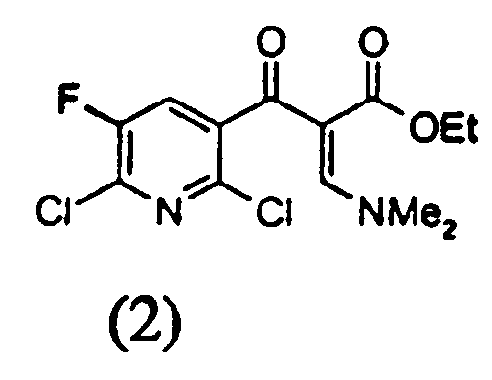
b) на второй стадии полученное соединение формулы (2)
вводят в реакцию с амином формулы YNH2 с получением соединения следующей формулы (3),
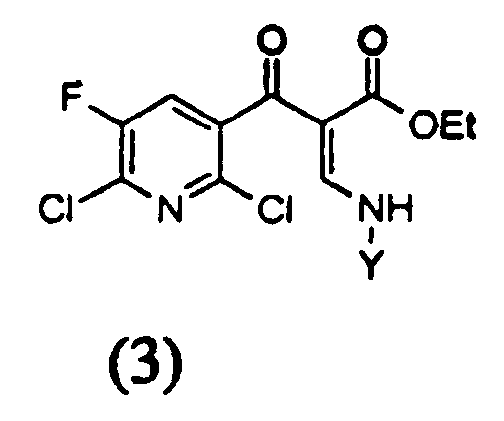
в которой Y представляет собой неразветвленный, разветвленный или циклический алкил, имеющий от 1 до 5 атомов углерода и незамещенный или замещенный галогеном, или представляет собой незамещенный или замещенный галогеном фенил,
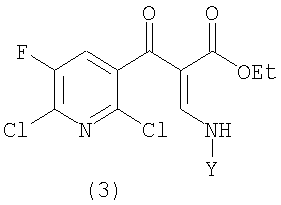
с) на третьей стадии полученное соединение формулы (3)
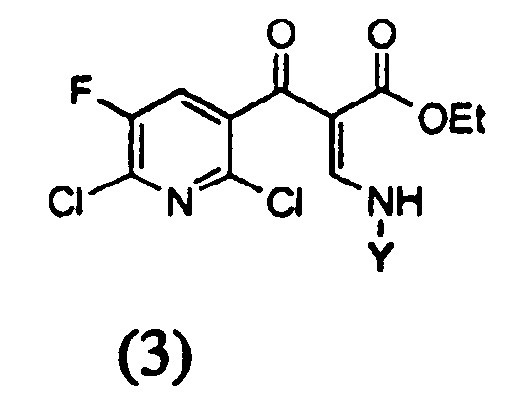
в которой Y определен выше, циклизуют в присутствии соли четвертичного аммония и основания с получением эфира 1,8-нафтиридин-3-карбоновой кислоты следующей формулы (4)
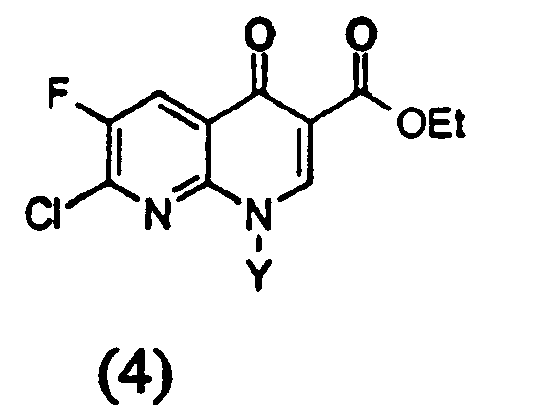
в которой Y определен выше,
d) на четвертой стадии полученное соединение формулы (4)
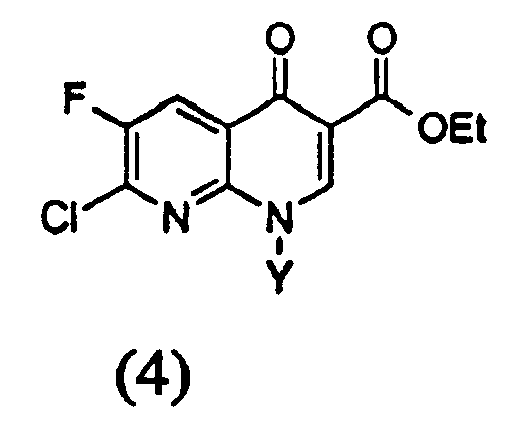
в которой Y определен выше, гидролизуют в присутствии кислоты с получением производного 1,8-нафтиридин-3-карбоновой кислоты следующей формулы (5)
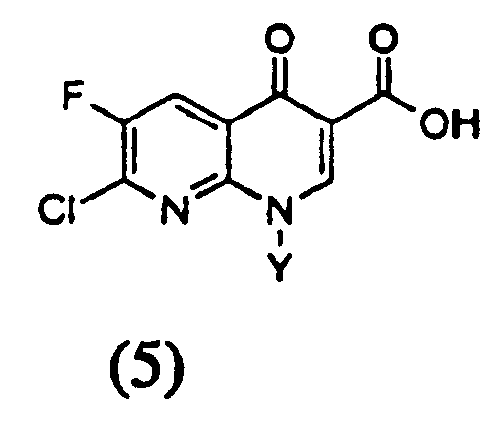
в которой Y определен выше.
Каждая вышеописанная стадия проиллюстрирована более детально следующим образом.
Стадия a)
Во-первых, растворитель, применяемый на стадиях с (a) по (d), представляет собой алкилгалогенид или углеводородный ароматический растворитель. Примерами алкилгалогенидных растворителей являются метиленхлорид, 1,2-дихлорэтан и т.д. Примерами арилгалогенидных растворителей являются бензол, хлорбензол, 1,2-дихлорбензол, толуол, ксилол и т.д., предпочтительно 1,2-дихлорбензол или толуол. Растворитель применяется в количестве от трехкратного до пятнадцатикратного (об./вес.), предпочтительно от четырехкратного до десятикратного (об./вес.), более предпочтительно в шестикратном количестве (об./вес.) по отношению к соединению (1).
Частными примерами R в диметилформамиддиалкилацетале [Me2NCH(OR)2], применяемом в качестве реагента, являются метил, этил, пропил, изопропил, бутил, третбутил, неопентил, бензил, циклогексил и т.д., предпочтительно метил, этил, пропил, изопропил и т.д. и более предпочтительно метил. Диметилформамиддиалкилацеталь применяют в количестве от 1 до 3 моль эквивалентов, предпочтительно от 1 до 1,5 моль эквивалентов и более предпочтительно от 1,05 до 1,15 моль эквивалентов на моль соединения (1).
Примерами кислотного катализатора являются органические карбоновые кислоты, такие как уксусная, пропановая и бутановая кислота и т.д., предпочтительно уксусная кислота. Среди них уксусная кислота применяется в количествах от 0,05 до 0,6 моль эквивалентов, более предпочтительно от 0,2 до 0,3 моль эквивалентов на моль соединения (1).
На стадии (а) соединение (1), диметилацеталь диметилформамида, кислотный катализатор и растворитель можно объединять в любом порядке для удобного проведения реакции, т.к. порядок их объединения не влияет на реакцию. Температура реакции находится между 0 и 50°С, предпочтительно между 10 и 40°С и более предпочтительно между 20 и 30°С.
В частности, стадию (а) согласно настоящему изобретению наиболее предпочтительно проводить как реакцию соединения (1) с количеством диметилацеталь диметилформамида от 1,05 до 1,15 моль эквивалентов и уксусной кислотой в количестве от 0,2 до 0,3 моль эквивалентов при 6-кратном разбавлении (об./вес.) толуолом по отношению к соединению (1) при температуре от 20 до 30°С.
Енаминовая структура, показанная в соединении (2) и образованная на стадии (а), может быть представлена как смесь E и Z формы и настоящее изобретение включает обе формы.
Стадия (b)
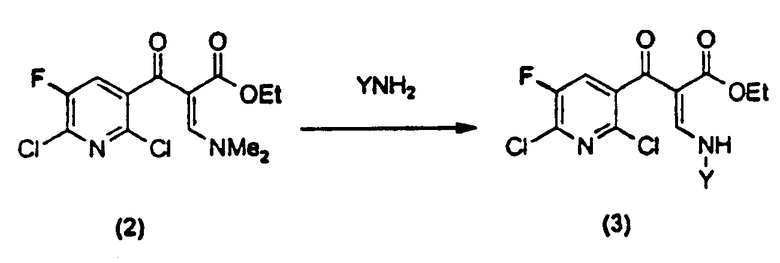
Амин формулы YNH2 добавляют к неочищенной реакционной смеси соединения (2), полученного на стадии (а). Здесь типичными примерами Y являются такие замещенные или незамещенные алкилы, как метил, этил, пропил, изопропил, бутил, втор-бутил, трет-бутил, пентил и т.д. Среди них неразветвленные или разветвленные алкилы, имеющие от 1 до 4 атомов углерода, являются предпочтительными, а метил, этил и пропил являются более предпочтительными. В случае галогензамещенной алкильной группы фтор, хлор, бром, иод и т.д. могут быть введены в качестве галогенидных заместителей, и среди них фтор и хлор являются предпочтительными. Типичными примерами галогензамещенных алкильных групп являются хлорметил, 2-хлорметил, фторметил, 1-фторэтил, 2-фторэтил и т.д., среди которых фторметил и 2-фторэтил являются предпочтительными. Примерами циклоалкилов могут служить циклопропил, циклобутил, циклопентил, циклогексил и т.д. Эти циклические алкилы могут замещаться в любых положениях галогенами, такими как фтор, хлор, бром и иод, предпочтительно хлор и фтор. В качестве предпочтительных примеров циклических алкилов могут быть упомянуты 2-фторциклопропил, 2,2-дифторциклопропил, 2-хлор-2-фторциклопропил и т.д., и в качестве более предпочтительного примера циклического алкила можно привести циклопропил или 2-хлор-2-фторциклопропил и т.д. Типичными примерами фенилов, незамещенных или замещенных галогенами, являются фенил и 2,4-дифторфенил, предпочтительно 2,4-дифторфенил. В настоящем изобретении циклопропил является наиболее предпочтительным.
При получении соединения формулы (3) с применением вышеописанного амина амин формулы YNH2 применяют в количестве от 0,9 до 2 моль эквивалентов, предпочтительно от 1 до 1,5 моль эквивалентов, более предпочтительно от 1,1 до 1,3 моль эквивалентов и наиболее предпочтительно от 1,1 до 1,2 моль эквивалентов на моль соединения (1). В этой реакции для удобства перемешивания один и тот же растворитель можно добавлять в реакционный раствор на всех стадиях, и количество растворителя составляет от 0 до пятикратного (об./вес.) по отношению к соединению (1), но желательно не добавлять растворитель, для того чтобы не снижать скорость реакции и не увеличивать реакционный объем. Здесь температура реакции находится между 0 и 50°С, предпочтительно между 10 и 40°С и более предпочтительно между 20 и 30°С.
Раствор, содержащий соединение формулы (3), полученное по вышеописанной реакции, включает спирт, образовавшийся в качестве побочного продукта, остаточный непрореагировавший YNH2 и диметиламин, высвобождающийся из соединения формулы (2). Таким образом, желательно, чтобы эти побочные продукты и непрореагировавшие вещества были удалены. В связи с этим применяется промывка разбавленным водным раствором кислоты, что приводит к удалению аминопроизводных в виде их солей, образующихся в водном слое. Применяемый здесь водный раствор кислоты может быть приготовлен из неорганической кислоты, такой как разбавленная серная кислота, разбавленная хлороводородная кислота, разбавленная фосфорная кислота или гидросульфат калия и т.д., или из органической кислоты, такой как винная кислота, лимонная кислота и т.д. pH во время промывки составляет от 1 до 6, предпочтительно от 2 до 5, более предпочтительно от 3 до 4. В качестве кислоты для промывки наиболее предпочтительными являются органические кислоты, такие как винная кислота, лимонная кислота и т.д. Среди них лимонная кислота является наиболее предпочтительной. Когда применяется лимонная кислота, предпочтительная концентрация водного раствора составляет от 3 до 30%, более предпочтительно от 5 до 20% и еще более предпочтительно от 10 до 15%. Температура промывки находится между 10 и 50°С, предпочтительно между 25 и 45°С и более предпочтительно между 30 и 40°С. Промывку осуществляют один или несколько раз, но если применяется предпочтительная концентрация вышеупомянутой лимонной кислоты при подходящей температуре, то достаточно одной промывки. После разделения слоев отделенный органический слой при необходимости может быть промыт один или несколько раз нейтральной водой.
Стадия c)
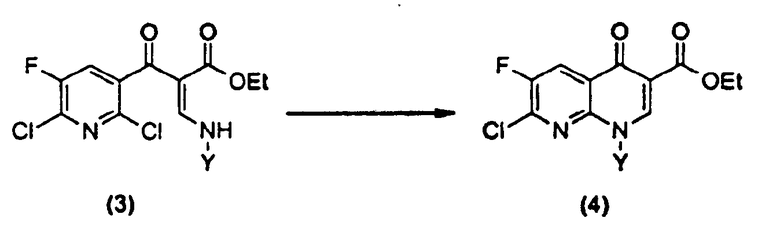
Неочищенное соединение (3) в отделенном органическом слое, полученное на стадии (b), циклизуют в присутствии соли четвертичного аммония и основания для получения соединения формулы (4).
R в соли четвертичного аммония R4NX, применяемой на данной стадии, представляет собой неразветвленный или разветвленный алкил, имеющий от 1 до 18 атомов углерода, или бензил и т.д. 4 заместителя R могут быть одинаковыми или различными; X представляет собой галогенид, HSO4 - или гидрокси, где галоген является хлором, бромом, йодом и т.д. Согласно настоящему изобретению в качестве соли четвертичного аммония на данной стадии (c) можно применять соль бензилтриалкиламмония, соль тетраметиламмония, соль тетраэтиламмония, соль тетрабутиламмония или торговые марки "Aliquat 336", "Adogen 464" и т.д. Предпочтительно применяют хлорид бензилтриэтиламмония, или бромид, или йодид; или хлорид тетраэтиламмония, или бромид, или йодид; или хлорид тетрабутиламмония, или бромид, или йодид. Более предпочтительно применение бромида тетрабутиламмония. Однако для удобства можно применять любую из вышеупомянутых солей четвертичного аммония. Соль четвертичного аммония можно применять в виде твердого вещества или водного раствора, и применяемые количества составляют от 0,001 до 1 моль эквивалента, предпочтительно от 0,01 до 0,1 моль эквивалента, более предпочтительно от 0,03 до 0,05 моль эквивалента на моль соединения (1).
Если X в применяемой здесь соли четвертичного аммония (R4NX) является галогенидом или HSO4 -, применение основания является существенным, тогда как если X является гидрокси, то соль четвертичного аммония сама по себе является основанием и поэтому применение дополнительного основания не является обязательным. Разновидностями применяемых оснований являются водные растворы гидроксида лития, гидроксида натрия, гидроксида калия, карбоната лития, карбоната натрия, гидрокарбоната натрия, гидрокарбоната калия, гидроксида четвертичного аммония и т.д.; предпочтительными являются гидроксид натрия и гидроксид калия. Среди них гидроксид натрия является наиболее предпочтительным. Применяемое количество основания в этом процессе составляет от 0,9 до 1,5 моль эквивалента, предпочтительно от 1 до 1,3 моль эквивалента, более предпочтительно от 1,1 до 1,2 моль эквивалента на моль соединения (1).
С другой стороны, вместо смеси соли четвертичного аммония и основания можно применять водный раствор гидроксида четвертичного аммония, и в этом случае водный раствор гидроксида четвертичного аммония можно применять в количестве 0,9 до 1,5 моль эквивалентов, предпочтительно от 1,0 до 1,3 моль эквивалентов, более предпочтительно от 1,05 до 1,15 моль эквивалентов на моль соединения (1). Температура реакции находится между 10 и 60°С, предпочтительно между 20 и 50°С и более предпочтительно между 25 и 35°С. В заключение циклизация промежуточного соединения (3) в (4) завершается за короткое время при комнатной температуре, обеспечивая более высокую производительность, а также чистоту в сравнении с известным уровнем техники.
Стадия (d).
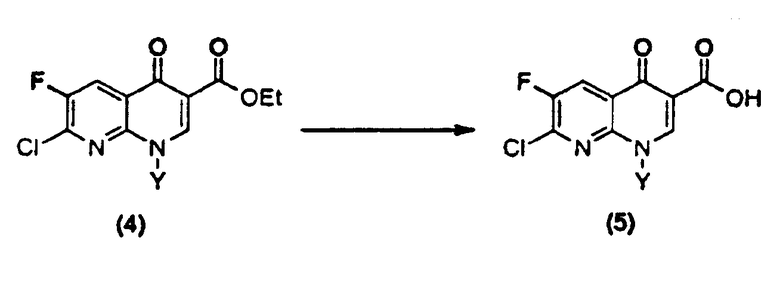
К реакционной смеси, содержащей соединение (4), полученное в процессе (c), добавляют водный раствор кислоты. Смесь нагревают для проведения гидролиза сложноэфирной группы, и образующиеся кристаллы соединения (5) отфильтровывают. Разновидностью кислоты, применяемой для гидролиза, является хлороводородная кислота или серная кислота, предпочтительно концентрированная хлороводородная кислота. Применяемое количество составляет от 1,5 до 9 моль эквивалентов, предпочтительно от 3 до 6 моль эквивалентов, более предпочтительно от 5 до 5 моль эквивалентов на моль соединения (1). В частности, в случае применения хлороводородной кислоты могут применяться водные растворы хлороводородной кислоты с концентрацией от 10 до 35%. Предпочтительно применять 35% водный раствор хлороводородной кислоты.
Реакция может быть проведена при температурах между комнатной температурой и 120°С, предпочтительно между 60 и 120°С и более предпочтительно между 110 и 120°С. После того как реакция завершается, реакционный раствор охлаждают. Полученное твердое вещество отфильтровывают, промывают водой и органическим растворителем, и полученный отфильтрованный осадок сушат, получая соединение (5) высокой чистоты. После четырех стадий выход обычно составляет 90% или выше.
Как показано выше, настоящее изобретение имеет следующие преимущества: стадии полностью осуществляют в одном сосуде без выделения промежуточных веществ, получающихся на каждой стадии, и без замены и добавления растворителя. Поскольку не требуются такие операции, как выделение, замена растворителя, смена реактора, промывание реактора и т.д., настоящее изобретение обеспечивает получение соединения (5) высокоэффективным и простым способом по таким параметрам, как длительность цикла процесса, выход и качество продукта.
Следующие примеры приведены для дальнейшей иллюстрации настоящего изобретения. Однако следует понимать, что эти примеры направлены на иллюстрацию настоящего изобретения и не могут ограничивать каким бы то ни было путем объем претензий настоящего изобретения.
Пример 1
Получение 7-хлор-1-циклопропил-6-фтор-4-оксо-1,4-дигидро-1,8-нафтиридин-3-карбоновой кислоты (с помощью гидроксида тетрабутиламмония).
К перемешиваемому раствору этилового эфира 3-(2,6-дихлор-5-фторпиридин-3-ил)-3-оксопропановой кислоты (соединение (1): 10 г, 35,7 ммоль) в толуоле (60 мл) добавляли диметилацеталь диметилформамида (4,68 г, 39,3 ммоль) и уксусную кислоту (0,53 г, 8,9 ммоль) при комнатной температуре. Смесь перемешивали при комнатной температуре в течение 30 минут. После того как этиловый эфир 3-(2,6-дихлор-5-фторпиридин-3-ил)-3-оксопропановой кислоты [соединение (1)] полностью прореагировал (контроль ВЭЖХ), в раствор добавляли циклопропиламин (2,24 г, 39,3 ммоль) и смесь перемешивали в течение 30 мин. После того как соединение (2) полностью прореагировало (контроль ВЭЖХ), реакционную смесь промывали 10% водным раствором лимонной кислоты. После разделения фаз отделенный органический слой промывали дистиллированной водой и в него добавляли 25% водный раствор гидроксида тетрабутиламмония (40 г, 39,3 ммоль). Полученный раствор перемешивали в течение 1 часа. После того как соединение (3) полностью прореагировало (контроль ВЭЖХ), в реакционный раствор добавляли концентрированную хлороводородную кислоту (914,7 мл, 146 ммоль) и смесь нагревали в реакторе с обратным холодильником при кипении в течение 10 часов. Реакционный раствор охлаждали, фильтровали, промывали изопропанолом, дистиллированной водой и вновь изопропанолом и сушили до получения указанного в заголовке соединения (5) (9,4 г) в виде белых кристаллов.
Общий выход реакции:93,1%
Чистота (ВЭЖХ): 98,6%
Пример 2
Получение 7-хлор-1-циклопропил-6-фтор-4-оксо-1,4-дигидро-1,8-нафтиридин-3-карбоновой кислоты (с помощью гидроксида тетрабутиламмония и гидроксида натрия).
Согласно методике Примера 1 из этилового эфира 3-(2,6-дихлор-5-фторпиридин-3-ил)-3-оксопропановой кислоты (соединение (1): 10 г, 35,7 ммоль) в толуоле (70 мл) было получено соединение (3). К отделенному раствору соединения (3) в толуоле добавляли дистиллированную воду (10 мл) и затем добавляли 40% раствор гидроксида тетрабутиламмония (2,32 г, 3,57 ммоль) и 10 н раствор гидроксида натрия (3,93 мл, 39,3 ммоль) для циклизации в реакционном растворе. После 1,3 часа в реакционный раствор добавляли концентрированную хлороводородную кислоту (16,7 мл) и смесь нагревали в реакторе с обратным холодильником при кипении в течение 8 часов для гидролиза смеси. Реакционный раствор охлаждали и полученное твердое вещество отфильтровывали, промывали согласно методике Примера 1 и сушили до получения указанного в заголовке соединения (5) (9,3 г) в виде белого кристалла.
Общий выход реакции:92,1%
Чистота (ВЭЖХ): 99,9%
Пример 3
Получение 7-хлор-1-циклопропил-6-фтор-4-оксо-1,4-дигидро-1,8-нафтиридин-3-карбоновой кислоты
К перемешиваемому раствору этилового эфира 3-(2,6-дихлоро-5-фторпиридин-3-ил)-3-оксопропановой кислоты (соединение (1): 85 кг, 303 моль) в толуоле (808 кг) добавляли Me2NCH(OMe)2 (40,8 кг). Смесь перемешивали при комнатной температуре в течение 50 минут. К реакционной смеси добавляли циклопропиламин (22,53 кг) и смесь перемешивали при температуре от 25 до 30°С в течение 50 минут. Реакционную смесь промывали 10% водным раствором лимонной кислоты, а затем водой. После разделения фаз водный слой отбрасывали и к отделенному органическому слою добавляли бромид тетрабутиламмония (4,48 кг) и 25% водный раствор гидроксида натрия (53 кг). Полученный раствор перемешивали в течение 2 часов. 35% раствор хлороводородной кислоты (142 кг) добавляли и смесь нагревали в реакторе с обратным холодильником при кипении. Через 8 часов реакционную смесь охлаждали, в нее добавляли воду (около 50 кг) и затем водный слой отделяли. Рекционный раствор промывали водой, твердое соединение, присутствующее в отделенном органическом слое, отфильтровывали, промывали изопропанолом, водой и вновь изопропанолом. Полученное твердое вещество высушивали в вакууме до получения указанного в заголовке соединения (5) (77 кг) в виде белого кристалла.
Общий выход реакции:90%
Чистота (ВЭЖХ): 99,9%.
| название | год | авторы | номер документа |
|---|---|---|---|
| ТРИЦИКЛИЧЕСКОЕ ПРОИЗВОДНОЕ СОЕДИНЕНИЕ, СПОСОБ ЕГО ПОЛУЧЕНИЯ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ, СОДЕРЖАЩАЯ ТАКОЕ СОЕДИНЕНИЕ | 2016 |
|
RU2715413C2 |
| ОКСАЗОЛИДИНОН-ХИНОЛОНГИБРИДНЫЕ АНТИБИОТИКИ | 2004 |
|
RU2371443C2 |
| ПРОИЗВОДНЫЕ 1,8-БЕНЗО(B)НАФТИРИДИНА И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ИХ ОСНОВЕ | 1992 |
|
RU2047613C1 |
| АНТАГОНИСТЫ РЕЦЕПТОРА СОМАТОСТАТИНА ПОДТИПА 5 (SSTR5) | 2014 |
|
RU2671958C2 |
| СПОСОБ ПОЛУЧЕНИЯ АЗЕТИДИНОНОВЫХ СОЕДИНЕНИЙ И ПРОИЗВОДНЫХ АЗЕТИДИНОНОВЫХ СОЕДИНЕНИЙ | 2015 |
|
RU2650687C1 |
| ПРИМЕНЕНИЕ ОКСАЗОЛИДИНОН-ХИНОЛИНОВЫХ ГИБРИДНЫХ АНТИБИОТИКОВ ДЛЯ ЛЕЧЕНИЯ СИБИРСКОЙ ЯЗВЫ И ДРУГИХ ИНФЕКЦИЙ | 2004 |
|
RU2351335C2 |
| СПОСОБ ПОЛУЧЕНИЯ ВЫСОКОЧИСТОГО ПРАЗУГРЕЛЬ ГИДРОХЛОРИДА | 2008 |
|
RU2435776C2 |
| СПОСОБЫ ПОЛУЧЕНИЯ НИТРОЗОСОЕДИНЕНИЯ И ХИНОКСАЛИНОВОГО СОЕДИНЕНИЯ | 2020 |
|
RU2791465C2 |
| СПОСОБ ПОЛУЧЕНИЯ 4-АМИНО-5-ФТОР-3-ХЛОР-6-(ЗАМЕЩЕННЫХ)ПИКОЛИНАТОВ | 2013 |
|
RU2653855C2 |
| ПРОИЗВОДНЫЕ ХИНОЛОН- И НАФТИРИДОН-КАРБОНОВОЙ КИСЛОТЫ В ВИДЕ СМЕСИ ИЗОМЕРОВ ИЛИ ОТДЕЛЬНЫХ ИЗОМЕРОВ, ИХ СОЛИ | 1994 |
|
RU2114832C1 |
Настоящее изобретение относится к способу получения 7-хлоро-1-циклопропил-6-фтор-4-оксо-1,4-дигидро-1,8-нафтиридин-3-карбоновой кислоты (5) высокой чистоты из этил 3-(2,6-дихлор-5-фторпиридин-3-ил)-3-оксопропаната (1) с применением единственного растворителя в одном реакционном сосуде. Технический результат - упрощение процесса. Эффективное получение 7-хлоро-1-циклопропил-6-фтор-4-оксо-1,4-дигидро-1,8-нафтиридин-3-карбоновой кислоты по этому способу имеет решающее значение для экономического производства антибиотиков на основе фторхинолонов, применяемых для лечения микробной инфекции. 9 з.п. ф-лы.
вводят в реакцию с диалкилацеталем диметилформамида, имеющим формулу Me2NCH(OR)2 (где R представляет собой неразветвленный, разветвленный или циклический алкил, имеющий от 1 до 9 атомов углерода, или представляет собой бензил) в растворителе в присутствии кислотного катализатора для получения соединения следующей формулы (2)
вторую стадию (b), на которой полученная реакционная смесь формулы (2)
вводят в реакцию с амином формулы YNH2 с получением соединения следующей формулы (3)
в которой Y представляет собой неразветвленный, разветвленный или циклический алкил, имеющий от 1 до 5 атомов углерода и незамещенный или замещенный галогеном, или представляет собой незамещенный или замещенный галогеном фенил,
третью стадию (3), на которой полученное соединение формулы (3)
в которой Y, определен выше, циклизуют в присутствии соли четвертичного аммония и основания с получением эфира 1,8-нафтиридин-3-карбоновой кислоты следующей формулы (4)

в которой Y определен выше,
четвертую стадию (d), на которой полученное соединение формулы (4)
в которой Y, определен выше, гидролизуют в присутствии кислоты с получением производного 1,8-нафтиридин-3-карбоновой кислоты следующей формулы (5)
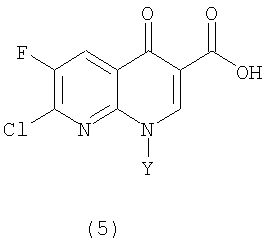
в которой Y определен выше,
причем вышеописанные стадии проводят в одном сосуде с применением единственного растворителя без выделения промежуточных веществ.
| ВЫСОКОМЕЧЕННЫЕ ТРИТИЕМ МОНОФТОРХИНОЛОНЫ | 2001 |
|
RU2191187C1 |
| ПРОИЗВОДНЫЕ ПИРИДОНКАРБОНОВОЙ КИСЛОТЫ ИЛИ ИХ СОЛИ И АНТИБАКТЕРИАЛЬНОЕ СРЕДСТВО | 1996 |
|
RU2167873C2 |
| Топчак-трактор для канатной вспашки | 1923 |
|
SU2002A1 |

