Изобретение относится к новым производным бензо(b)нафтиридина-1,8 общей формулы
R N
N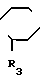 N
N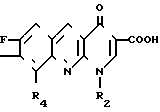
(I) в которой
R1 является атомом водорода или радикалом гидрокси- или алкильным;
R2 является атомом водорода или радикалом алкильным, фторалкильным, циклоалкильным, содержащим от 3 до 6 атомов углерода, алкилокси-;
R3 является радикалом фенильным или алкилфенильным, возможно замещенным одним или несколькими атомами галогена или радикалами алкильным, циклоалкильным, содержащим от 3 до 6 атомов углерода, алкилокси-, циано-, амино-, алкиламино-, диалкиламино-, алкилоксиалкильным, гидроксиалкильным, гидроксиалкилокси-, метилендиокси-, аминоалкильным, алкиламино- алкильным или диалкиламиноалкильным или диалкильноалкильным, у которого алкильные части могут образовывать с атомом азота, с которым они связаны, 5- или 6-членный гетероцикл, или является 5-членным гетероциклическим радикалом, содержащим 1 или 2 гетероатома, выбранных среди азота, кислорода или серы, и
R4 является атомом водорода или атомом фтора, и в которой алкильные радикалы являются неразветвленными или разветвленными и содержат от 1 до 4 атомов углерода, в виде его изомеров или их смесей, а также его солей с металлами, его солей присоединения с азотированными основаниями, его солей присоединения с кислотами и его гидратированных форм, проявляющие бактериостатическую активность.
В патентах США [1,2] были описаны производные нафтиридина следующей структуры:
в которой Х может быть кислородом, а два прилегающих радикала от R1 до R5 могут образовывать бензольный цикл.
Эти продукты используются в качестве ингибиторов выделения желудочного сока.
Заявка на патент ДЕ 3 302 126 описывает вещества, понижающие кровяное давление с общей формулой:
Y
в которой радикалы Х, Y и Z могут означать атом кислорода или радикал NR4 или CR5=CR5, где радикалы R5 могут образовывать бензольный цикл.
Целью изобретения является разработка на основе известных приемов новых соединений, обладающих высокой бактерицидной активностью.
Продукты с общей формулой (I) могут существовать в гидратированной форме и эти гидраты также входят в рамки предлагаемого изобретения.
В общей формуле (I), когда R3 является гетероциклическим радикалом, этот последний может быть выбран среди фурильного, тиекильного, пирролильного, N-алкилпирролильного, имидазолильного, пиразолильного или тиазолильного радикалов.
Продукты общей формулы (I) могут быть получены путем реакции замещения пиперазина общей формулы:
R N
N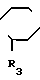 NH (II) в которой R1 и R3 определены, как и ранее, с 1,8-бензо (b) нафтиридином общей формулы:
NH (II) в которой R1 и R3 определены, как и ранее, с 1,8-бензо (b) нафтиридином общей формулы: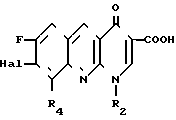 (III) в которой R2 определен выше, Hal является атомом фтора, хлора или брома, если R4 это водород, или же Hal и R4 являются одновременно атомами фтора, причем в случае необходимости, если R1 является атомом водорода и если хотят получить производное бензо (b) нафтиридина-1,8, в котором R1 является метильным радикалом, после замещения следует превращение полученного продукта в 8-/4-метил-1-пиперазинил-(бензо)b/нафтиридин.
(III) в которой R2 определен выше, Hal является атомом фтора, хлора или брома, если R4 это водород, или же Hal и R4 являются одновременно атомами фтора, причем в случае необходимости, если R1 является атомом водорода и если хотят получить производное бензо (b) нафтиридина-1,8, в котором R1 является метильным радикалом, после замещения следует превращение полученного продукта в 8-/4-метил-1-пиперазинил-(бензо)b/нафтиридин.
Обработка производного пиперазина общей формулы (II) происходит обычно в присутствии избытка этого производного как кислотного акцептора в органических растворителях. Возможно оперировать как с растворителем, так и без него при температурах, заключенных между 30 и 120оС. Когда работают в присутствии растворителя, то реакция с успехом протекает в таких растворителях, как пиридин, диметилформамид, диметилсульфоксид или ацетонитрил.
Предпочитают также работать в присутствии кислотного акцептора, например азотированного органического основания (триэтиламина), карбоната щелочного металла, например карбоната натрия, или гидроксида щелочного или щелочно-земельного металла.
При этом, когда радикал R2 продукта общей формулы (III) является атомом водорода или когда R3 содержит амино-, алкиламино-, аминоалкил- или алкиламиноалкильный заместитель, предпочитают предварительно защищать исходный продукт. Защита и отщепление защищающего радикала осуществляются в соответствии с обычными методами.
Защита может быть осуществлена с помощью любой совместимой группы, введение которой и отщепление не затрагивают остальной части молекулы. В частности, можно использовать методику, описанную в книгах: Т.У.Грин. Защитные группы в органическом синтезе. Вилей-Интерсайенс Публикэйшн, 1981 или Мак-Оми, Защитные группы в органической химии, Пленум Пресс, 1973.
В качестве примера защитные группы могут быть выбраны из радикалов: триметилсилильного, бензгидрильного, тетрагидропиранильного, формильного, ацетильного, хлорацетильного, трихлорацетильного, трифторацетильного, этоксикарбонильного, трет-бутоксикарбонильного, трихлориэтоксикарбонильного.
В случае необходимости последующая операция метилирования пиперазинильного радикала протекает с успехом при действии формалина в присутствии муравьиной кислоты. Работают обычно в водной среде при температуре, заключенной между 90 и 100оС.
Производные бензо-[b]-нафтиридина-1,8 общей формулы (I) могут быть также получены из соответствующего сложного эфира общей формулы:
R N
N N
N
 (IV) в которой R1, R3 и R4 определены, как описано выше, R2 определен, как описано выше, или является защищенным алкиламинорадикалом, а Алк является алкильным радикалом, содержащим от 1 до 4 атомов углерода в неразветвленной или разветвленной цепи, любым известным методом получения кислоты из сложного эфира, не затрагивая остальной части молекулы, с последующим, в случае необходимости, отщеплением защитной группы алкиламинорадикала, и/или, если был получен продукт с общей формулой (I), в которой R1 является атомом водорода и если хотят получить соответствующий продукт, в котором R1 является метильным радикалом, то осуществляют превращение последнего.
(IV) в которой R1, R3 и R4 определены, как описано выше, R2 определен, как описано выше, или является защищенным алкиламинорадикалом, а Алк является алкильным радикалом, содержащим от 1 до 4 атомов углерода в неразветвленной или разветвленной цепи, любым известным методом получения кислоты из сложного эфира, не затрагивая остальной части молекулы, с последующим, в случае необходимости, отщеплением защитной группы алкиламинорадикала, и/или, если был получен продукт с общей формулой (I), в которой R1 является атомом водорода и если хотят получить соответствующий продукт, в котором R1 является метильным радикалом, то осуществляют превращение последнего.
Для получения изомеров производных бензонафтиридина общей формулы (I) проводят разделение изомерных форм пиперазинов общей формулы (II) по любому известному и совместимому с молекулой методу. В качестве примера разделение осуществляется при ацилировании посредством кислоты или реакционно-способного производного хиральной кислоты, разделением изомеров методом высокоэффективной жидкостной хроматографии, затем деацилированием в результате кислотного гидролиза.
В последующих примерах изомером (-) называют изомер (s) производного бензонафтиридина общей формулы (I), у которого вращательная способность в растворе уксусной кислоты отрицательна, а изомер, полученный, исходя из производного пиперазина, у которого вращательная способность в этаноле положительна, называют изомер (+).
Можно также осуществить непосредственно синтез хирального пиперазина, как описано ниже в примерах 20 и 21, изомер (R) производного бензонафтиридина общей формулы (I), у которого вращательная способность в растворе уксусной кислоты положительна, получен из производного пиперазина, у которого вращательная способность в растворе этанола отрицательна.
Новые продукты согласно настоящему изобретению, так же, как и полупродукты их синтеза, могут быть в случае необходимости очищены такими физическими методами, как кристаллизация или хроматография.
Продукты могут быть превращены в соли металлов или в соли присоединения с азотированными основаниями в соответствии с известными методами. Эти соли могут быть получены при действии основания металла (например, щелочного или щелочно-земельного), аммиака или амина на продукт по изобретению в подходящем растворителе таком,как спирт, простой эфир или вода, или по реакции обмена с солью органической кислоты. Полученная соль осаждается после концентрирования (в случае необходимости) из своего раствора, она выделяется путем фильтрования, декантации или лиофилизации (вакуумной сушки).
Новые продукты согласно изобретению могут быть также превращены в соли присоединения с кислотами. Продукты с общей формулой (I), полученные в виде этих солей, могут быть выделены и превращены в соли других кислот в соответствии с обычным методами.
В качестве примеров фармацевтически пригодных солей можно указать соли щелочных металлов (натрия, калия, лития) или щелочно-земельных (магния, кальция), соль аммония, соли азотированных оснований (этаноламина, диэтаноламина, триметиламина, триэтиламина, метиламина, пропиламина, диизопропиламина, N, N-диметилэтаноламина, бензиламина, дициклогексиламина, N-бензил- β -фенетиламина, N, N'-дибензилэтилендиамина, дифенилендиамина, бензгидриламина, хинина, холина, аргинина, лизина, лейцина, дибензиламина.
Следующие примеры, не являющиеся ограничивающими, иллюстрируют предлагаемое изобретение.
П р и м е р 1. Суспензия, состоящая из 1,84 г 8-хлор-7-фтор-1-метил-4-оксо-1,4-дигидро-1,8-бензо[b]нафтиридин- 3-карбоновой кислоты в 20 см3 пиридина, 5,82 г 2-фенилпиперазина (RS) и 1,24 г триэтиламина, нагревают при температуре, близкой к 115оС, в течение 5 ч. После охлаждения при примерно 20оС образованный осадок центрифугируют, промывают 2 раза в 5 см3 пиридина, 2 раза в 5 см3 изопропилового спирта, 2 раза в 5 см3 этанола и 1 раз в 20 см3 этилового эфира. После одной перекристаллизации из смеси 40 см3 диметилформамида и 40 см3 этанола получают 0,920 г 7-фтор-1-метил-4-оксо-8-(3-фенил-1- пиперазинил)-1,4-дигидро-1,8-бензо[b]нафтири- дин-3-карбоновой кислоты (RS) в виде твердого вещества желтого цвета, плавящегося при 265оС.
8-Хлор-7-фтор-1-метил-4-оксо-1,4-дигид- ро-1,8-бензо[b] нафтиридин-3- карбоновая кислота может быть получена следующим способом.
суспензию, состоящую из 15 г 8-хлор-3-этоксикарбонил-7-фтор-1-метил-4-оксо-1,4-дигидро-1,8-бензо [b]нафтиридина в 150 см3 уксусной кислоты и 150 см3 17,5%-ного раствора хлороводородной кислоты, нагревают при температуре, близкой к 100оС, в течение 4 ч при перемешивании. После охлаждения при температуре, близкой к 20оС, продукт центрифугируют, промывают 2 раза в 100 см3 воды, промывают 2 раза в 150 см3 этанола, затем 2 раза в 100 см3 этилового эфира. Получают 12,7 г 8-хлор-7-фтор-1-метил-4-оксо-1,4-дигидро-1,8-бензо[b] нафти-ридин-3- карбоновой кислоты в виде твердого вещества бежевого цвета, возгоняющегося при 400-450оС, которое применяется без дополнительной очистки в последующих стадиях. 8-Хлор-3-этоксикарбонил-7-фтор-1-ме-тил-4-оксо-1,4-дигидро-1,8-бензо [b]-нафтиридин приготавливается в последующих стадиях.
При перемешивании в суспензию, состоящую из 19,3 г 2-(2,7-дихлор-6-фтор-3-хинолинкарбонил)-3-диметиламиноэтилакри- лата в 250 см3 этанола, выдерживаемую при температуре между 10 и 15оС, барботируют метиламин до поглощения 16 г газа. Позволяют температуре подняться до примерно 20оС, добавляют 0,8 г диаза-1,8-бицикло-[5,4.0. индецена-7 (ДБУ) и нагревают при температуре, близкой к 75оС, в течение 2 ч. После охлаждения до примерно 20оС продукт центрифугируют, промывают 2 раза 150 см3 этанола и 2 раза 100 см3 этилового эфира. Получают 15 г 8-хлор-3-этоксикарбонил-7-фтор-1-метил-4-оксо-1,4-дигидро-1,8-бензо [b]нафтиридина в виде твердого вещества желтого цвета, плавящегося при 360-362оС, которое применяется без дополнительной очистки в последующих стадиях.
2-(2,7-Дихлор-6-фтор-3-хинолинкарбо-нил)-3-диметиламиноэтилакрилат приготавливается следующим способом:
суспензию, состоящую из 16,5 г 3-(2,7-дихлор-6-фтор-3-хинолил)-3- оксоэтилпропионата в 160 см3 этилацетата и 19 см3 диметилацеталя N,N-диметилформамида, нагревают при температуре, близкой к 75оС, в течение 2 ч при перемешивании. Реакционную смесь концентрируют досуха при пониженном давлении (20 кПа) и при 50оС. Сухой экстракт растворяют в 50 см3 изопропилового эфира, центрифугируют, промывают 2 раза в 10 см3 изопропилового эфира. Получают 16,57 г 2-(2,7-дихлор-6-фтор-3-хинолинкарбонил)-3- диметиламиноэтилакрилата в виде твердого вещества оранжевого цвета, плавящегося при 122оС. Этот продукт применяется без дополнительной очистки в последующих стадиях.
3-(2,7-Дихлор-6-фтор-3-хинолил)-3-оксо- этилпропионат приготавливается следующим способом:
суспензию, состоящую из 38,75 г 2,7-дихлор-6-фтор-хинолин-3-карбоновой кислоты в 410 см3 трихлорметана и 24 см3 хлорида тионила, нагревают при температуре, близкой к 60оС, в течение 6 ч при перемешивании. Полученный раствор концентрируют досуха при пониженном давлении (20 кПа) и при 50оС. Сухой экстракт извлекают за 2 раза 200 см3 (в сумме) толуола и снова концентрируют при пониженном давлении в тех же условиях, что и ранее. Полученное твердое вещество желтого цвета, плавящееся при 124оС, растворяют в 230 см3 безводного тетрагидрофурана. Полученный раствор вводят по капле при перемешивании между 5 и 10оС за 30 мин в 200 см3 раствора магниевого хелата в тетрагидрофуране. Позволяют температуре подняться до 20оС и перемешивают при этой температуре в течение 15 ч. Полученный раствор вводится по капле при сильном перемешивании и температуре, близкой к 5оС, в 1 л 0,5 н.серной кислоты. Позволяют температуре полученной суспензии подняться до 20оС и перемешивают еще 2 ч при этой температуре.
Проводят экстракцию посредством 1 л этилацетата, фильтруют водную и органическую фазы через диатомированный кремнезем для фильтрования, что позволяет удалить легкую нерастворимую часть, экстрагируют водную фазу еще два раза 500 см3 этилацетата. Объединенные органические экстракты промывают 2 раза 500 см3 воды, сушат на сульфате магния, фильтруют и концентрируют досуха при пониженном давлении (20 кПа) и при 40оС. Остаток извлекают посредством 100 см3 изопропилового эфира при 20оС, центрифугируют и промывают 2 раза 30 см3 изопропилового эфира. Получают 40,55г 3-(2,7-дихлор-6-фтор-3-хинолил)-3-оксоэтилпропионата в виде твердого вещества бежевого цвета, плавящегося при 112-114оС. Этот продукт применяется без дополнительной очистки в последующих стадиях.
Приготовление магниевого хелата моноэтилмалоната:
к 6,9 г магниевых опилок добавляют последовательно 5 см3 абсолютного этанола, 0,2 см3 тетрахлорметана и 2 г моноэтилмалоната. После нагрева добавляют за 15 мин раствор 23,8 г моноэтилмалоната в 450 см3 этанола. Смесь нагревают 20 ч при температуре, близкой к 78оС, концентрируют при пониженном давлении (20 кПа) и при 50оС. Остаток извлекают за 2 раза 100 см3 толуола и концентрируют при пониженном давлении в тех же самых условиях, что и ранее. Полученный серый порошок переводят в раствор добавлением безводного тетрагидрофурана так, чтобы получить суммарный объем 200 см3.
Моноэтилмалонат был приготовлен в соответствии с методом, описанным D.S. Breslow, Baumgarten, C. R.Hauser, I.Am.Chem.Soc. 66, 1287 (1944) и перегонялся при пониженном давлении (температура кипения 132оС (2,7 кПа).
2,7-Дихлор-6-фторхинолин-3-карбоно-вая кислота приготавливается следующим способом:
при перемешивании к охлажденной до 10оС суспензии, состоящей из 69,5 г 2,7-дихлор-6-фтор-3-формил-1,4-дигидрохинолина, в 282 см3 2 н. водной щелочи калия и 282 см3 воды, добавляют за 1 ч, поддерживая температуру между 10 и 14оС, раствор 89,3 г перманганата калия в 1,4 л воды. Позволяют температуре подняться до примерно 20оС и перемешивают еще 30 мин при этой температуре. Прибавляют 27 г дитионата натрия, перемешивают 10 мин при температуре, близкой к 20оС, фильтруют через диатомированный кремнезем для фильтрования, промывают 2 раза 250 см3 воды. Фильтрат и водные фазы после промывания объединяют и прибавляют 90 см3 35%-ного водного раствора хлороводородной кислоты. Полученный осадок экстрагируют 4 раза 500 см3 этилдиацетата. Органические вытяжки объединяют, промывают 3 раза 500 см3 воды, сушат на сульфате магния, фильтруют и концентрируют при пониженном давлении (20 кПа) и при 50оС. Остаток извлекают посредством 350 см3 этилового эфира, центрифугируют, промывают 2 раза 200 см3 этилового эфира. Получают 45 г 2,7-дихлор-6-фтор-хинолин-3-карбоновой кислоты в виде твердого вещества бежевого цвета, плавящегося при 230оС, которое применяется без дополнительной очистки в последующих стадиях. При сильном перемешивании прибавляют смесь 250 см3 воды и 250 г толченого льда. Полученное твердое вещество центрифугируют при примерно -5оС и промывают 4 раза 125 см3 воды при 5оС. Полученный влажный продукт и 58 г ацетата натрия прибавляют одновременно за 1 ч к 500 см3 воды при 90оС так, чтобы поддерживать рН около 6. Перемешивают еще 15 мин при 90оС, дают температуре опуститься до примерно 50оС, центрифугируют при этой температуре и промывают 3 раза 250 см3 воды примерно 20оС. Получают 54,3 г 2,7-дихлор-6-фтор-3-формил-1,4-дигидрозинолина в виде твердого вещества желтого цвета, плавящегося при 260оС, которое применяется в таком виде в последующих стадиях.
7-Хлор-6-фтор-3,4-дигидрокарбостирил приготавливается следующим способом:
к 174,4 г 3'-хлор-4'-фтор-3-N-хлорпропионанилида прибавляют при сильном перемешивании за 5 мин 350 г хлорида алюминия. Твердую смесь нагревают за 30 мин примерно до 60оС. Температура самостоятельно поднимается примерно до 80оС и реакционная смесь становится жидкой. Затем нагревают до 110оС за 15 мин и выдерживают между 110 и 120оС в течение 3 ч. Реакционную смесь (примерно при 110оС) выливают за 10 мин при сильном перемешивании в смесь 550 см3 35%-ной хлороводородной кислоты и 500 г толченого льда. Дают температуре подняться до 20оС, центрифугируют, промывают 6 раз 500 см3 воды.
Влажный продукт перекристаллизовывают из 1,2 л этанола. Получают 108 г 7-хлор-6-фтор-3,4-дигидрокарбостирила в виде твердого вещества бежевого цвета, плавящегося при 215оС.
3'-Хлор-4'-фтор-3-N-хлорпропионани- лид был приготовлен следующим способом:
при температуре, близкой к 55оС, к раствору, состоящему из 291 г 3-хлор-4-фторанилина в 500 см3 ацетона, прибавляют при перемешивании за 35 мин раствор 127 г хлорида 3-хлор-пропионовой кислоты в 200 см3 ацетона и выдерживают при этой температуре в течение 2 ч. После охлаждения до примерно 20оС нерастворимую часть удаляют фильтрованием и промывают 2 раза 200 см3 ацетона. Фильтрат и промывочные смеси объединяют, выливают при перемешивании в смесь 2 л воды и 1 кг льда. Дают температуре подняться примерно до 20оС, экстрагируют 2 раза 500 см3 дихлорметана. Объединенные органические вытяжки промывают 3 раза 500 см3, сушат на сульфате магния, перемешивают 15 мин с 6 г древесного угля, фильтруют через диатомированный кремнезем для фильтрования и концентрируют при пониженном давлении (2,7 кПа) и при 50оС. Полученное твердое вещество перекристаллизовывают из смеси 133 см3 циклогексана и 67 см3 изопропилового эфира. Получают 176 г 3'-хлор-4'-фтор-3-N-хлорпропионанилида в виде твердого вещества бежевого цвета, плавящегося при 94оС, которое применяется в таком виде в последующих стадиях.
П р и м е р 2. 1-Циклопропил-7-фтор-4-оксо-8-(3-фенил-1-пиперазинил)-1,4-дигид-ро-1,8- бензо[b]нафтиридин-3-карбоновую кислоту (RS) готовят в условиях приведенного ниже примера 11, но исходя из 2 г 8-хлор-1-циклопропил-7-фтор-4-оксо-1,4-дигидро-1,8-бензо[b] нафтиридин- 3-карбоновой кислоты, 4,9 г 2-фенилпиперазина (RS) и 1,7 см3 триэтиламина. После перекристаллизации из смеси 40 см3 диметилформамида и 50 см3 этанола получают 1,16 г 1-циклопропил-7-фтор-4-оксо-8-(3-фенил-1-пиперазинил)-1,4- дигидро-1,8- бензо[b] нафтиридин-3-карбоновой кислоты (RS) в виде твердого вещества желтого цвета, плавящегося при 254оС.
8-Хлор-1-циклопропил-7-фтор-4-оксо-1,4- дигидро-1,8-бензонафтиридин-3- карбоновую кислоту готовят в условиях примера 1, но исходя из 6,1 г 8-хлор-1-циклопропил-3-этоксикарбонил-7-фтор-4-оксо-1,4-дигидро- 1,8- бензо[b] нафтиридина. Получают 4,85 г 8-хлор-1-циклопропил-7-фтор-4-оксо-1,4-дигидро-1,8-бензо[b] нафтиридин-3-карбоновой кислоты в виде твердого вещества желтого цвета, плавящегося при 330оС, которое применяется без дополнительной очистки в последующих стадиях.
8-Хлор-1-циклопропил-3-этоксикарбо- нил-7-фтор-4-оксо-1,4-дигидро-1,8- бензо[b]нафтиридин приготавливается в следующих условиях:
раствор, состоящий из 20,6 г 2-(2,7-дихлор-6-фтор-3-хинолинкарбонил)-3-диметил- аминоэтилакрилата и 6 г циклопропиламина в 100 см3 трихлорметана, перемешивают при температуре, близкой к 20оС, в течение 24 ч. Реакционную смесь концентрируют при пониженном давлении (2,7 кПа) и при 50оС. Остаток извлекают посредством 180 см3 этанола и 10 г ДБУ, а полученный раствор нагревают при температуре, близкой к 78оС, в течение 4 ч. После охлаждения при температуре, близкой к 20оС,полученный осадок центрифугируют и промывают 2 раза 60 см3 этанола. Получают 13,65 г 8-хлор-1-циклопропил-3-этоксикарбонил-7-фтор-4-оксо-1,4-дигидро-1,8- бензо-[b] нафтиридина в виде твердого вещества бледно-желтого цвета, плавящегося при 256оС, которое применяется без дополнительной очистки в последующих стадиях.
П р и м е р 3. 7-Фтор-1-(2-фторэтил)-4-оксо-8-(3-фенил-1-пиперазинил)-4-оксо-1,4- дигидро-1,8-бензо[b] нафтиридин-3-карбо-новую кислоту (RS) готовят в следующих условиях:
суспензию, состоящую из 0,095 г 7,8-дифтор-1-(2-фтор-этил-оксо-1,4-дигидро-1,8- бензо[b] нафтиридин-3-карбоновой кислоты и 1,05 г 2-фенилпиперазина (RS) в 10 см3 диметилсульфоксида, нагревают при температуре, близкой к 100оС, при перемешивании в течение 20 мин. После охлаждения примерно при 20оС нерастворимую часть центрифугируют и промывают 3 раза 10 см3 этанола примерно при 70оС. Получают 1,3 г ожидаемого продукта в виде твердого вещества желтого цвета, разлагающегося при 305оС.
7,8-Дифтор-1-(2-фторэтил)-4-оксо-1,4- дигидро-1,8-бензонафтиридин-3- карбоновую кислоту готовят следующим образом:
суспензию, состоящую из 2,3 г 3-этоксикарбонил-7,8-дифтор-1-(2-фторэтил)-4-оксо- 1,4-дигидро-1,8-бензо [b]нафтиридина в 20 см3 уксусной кислоты и 20 см3 5 н. хлороводородной кислоты, нагревают при перемешивании и при температуре, близкой к 100оС, в течение 1 ч. Нерастворимую часть центрифугируют примерно при 70оС и промывают 3 раза 10 см3 воды и 3 раза 10 см3 этанола. Получают 1,47 г ожидаемого продукта в виде твердого вещества бежевого цвета, плавящегося при 291оС.
3-Этоксикарбонил-7,8-дифтор-1-(2-фтор- этил)-4-оксо-1,4-дигидро-1,8-бензо [b]-нафтиридин готовят следующим способом:
при перемешивании к смеси 1,46 г хлоргидрата 2-фторэтиламина и 2,06 см3 триэтиламина в 30 см3 трихлорметана прибавляют при примерно 10оС 2,58 г 2-(2-хлор-6,7-дифтор-3-хиноликарбонил)-3- диметиламиноэтилакрилата. После 16-часового перемешивания примерно при 20оС смесь концентрируют при пониженном давлении (20 кПа) при температуре, близкой к 50оС. Остаток переводят в раствор посредством 30 см3 этанола с добавкой 2,3 см3 триэтиламина. Смесь нагревают при перемешивании примерно до 75оС. Нерастворимую часть центрифугируют и промывают 3 раза 5 см3 этанола. Получают 2,3 г ожидаемого продукта в виде твердого вещества бежевого цвета, плавящегося при 266оС, которое применялось без дополнительной очистки в последующих стадиях.
2-(2-Хлор-6,7-дифтор-3-хинолинкарбо-нил)-3-диметиламиноэтилакрилат может быть получен, как описано ниже в примере 39.
П р и м е р 4. Суспензию, состоящую из 1,6 г 8-хлор-7-фтор-1-метил-4-оксо-1,4-дигидро-1,8-бензо[b] нафтиридин-3- карбоновой кислоты и 3,7 г 2-(4-фторфенил)-пиперазина (RS) в 16 см3 пиридина, нагревают при температуре, близкой к 115оС, в течение 24 ч. Реакционную смесь концентрируют при пониженном давлении (20 кПа) и при температуре примерно 60оС. Остаток извлекают посредством 40 см3 этанола и вновь концентрируют при пониженном давлении в указанных выше условиях. Полученное твердое вещество извлекают посредством 10 см3 воды, смешивают с 1,75 см3 10%-ной уксусной кислоты, центрифугируют, промывают 2 раза 10 см3 воды и 2 раза 10 см3 этанола. После двух перекристаллизаций каждый раз из 10 см3 диметилформамида получают 1,1 г 7-фтор-[(4-фторфенил)-3-пиперазинил-1]-8- метил-1,4-оксо-1,4-дигидро- 1,8-бензо[b] нафтиридин-3-карбоновой кислоты (RS) в виде твердого вещества желтого цвета, плавящегося при 270-275оС.
2-(4-Фторфенил)-пиперазинин (RS) был приготовлен согласно методу, описанному для 2-фенилпиперазина (RS) в работе R.Roderick et coll.I.Med.Chem.9, 181 (1966).
Исходя из 20 г 2-(4-фторфенил)-3-оксо-пиперазина (RS) и 7,8 галюмогидрида лития в 1,5 л этилового эфира получают 11 г 2-(4-фторфенил)-пиперазина (RS) в виде твердого вещества бежевого цвета, плавящегося при 110-112оС.
Исходя из 60 г 1-бром-1-(4-фторфенил)-этилацетата и 30 г этилендиамина получают 30 г 2-(4-фторфенил)-3-оксо-пиперазина (RS) в виде бесцветного твердого вещества, плавящегося при 115оС.
1-Бром-1-(4-фторфенил)-этилацетат был приготовлен согласно I.W.Epstein et.coll. I.Med.Chem. 24, 181 (1981), исходя из (4-фторфенил)-этилацетата.
(4-Фторфенил)-этилацетат был приготовлен согласно методу, описанному I. W.Corse et coll. I.Am.Chem.Soc. 70, 2837 (1948).
П р и м е р 5. Раствор, состоящий из 2,1 г 7-фтор-8-[3-(4-фторфенил)-1-пиперазинил)-1-метил-4-оксо-1,4-дигидро- 1,8-бензо[b] нафтиридин-3-карбоновой кислоты (RS) в 1,8 см3 98%-ной муравьиной кислоты и 4,4 см3 30%-ного водного раствора формальдегида, нагревают при температуре, близкой к 100оС, в течение 2 ч. Реакционную смесь концентрируют при пониженном давлении (20 кПа) и при примерно 50оС, смешивают с 10 см3 воды и 1,2 см3 2 н. водного раствора щелочи калия, и нагревают при примерно 100оС в течение 2 мин. После охлаждения при температуре, близкой к 20оС, нерастворимую часть центрифугируют и промывают 2 раза 20 см3 воды. После двух перекристаллизаций каждый раз в 15 см3 диметилформамида получают 1,3 г 7-фтор-8-[4-фторфенил)- 4-метил-1-пиперазинил(1-метил-4-оксо-1,4-дигидро- 1,8-бензо[b]нафтиридин-3-карбоновой кислоты (RS) в виде твердого вещества желтого цвета, разлагающегося при 213-314оС.
П р и м е р 6. Проводят реакцию в условиях примера 16, но исходя из 1,3 г 7,8-дифтор-1-метил-4-оксо-1,4-дигидро-1,8-бензо[b] нафтиридин-3-карбоновой кислоты и 1,8 г 2-[3-фторфенил]-пиперазина (RS). После одной перекристаллизации в 50 см3 50%-ного раствора диметилформамида в этаноле получают 1,74 г 7-фтор-8-[3-(3-фторфенил)-1-пиперазинил)-1-метил-4-оксо-1,4- дигидро- 1,8-бензо[b]нафтиридин-3-карбоновой кислоты (RS) в виде твердого вещества желтого цвета, плавящегося при 254оС.
П р и м е р 7. Проводят реакцию в условиях примера 4, но исходя из 1,5 г 8-хлор-1-этил-7-фтор-4-оксо-1,4-дигидро-1,8-бензо [b]нафтипиридин-3- карбоновой кислоты и 3,4 г 2-(4-фторфенил)-пиперазина (RS) в 15 см3 пиридина. Получают 0,92 г 1-этил-7-фтор-4-оксо-8-[3-(4-фторфенил-1-пиперази- нил)-1,4-дигидро- 1,8-бензо[b]нафтиридин-3-карбоновой кислоты (RS) в виде твердого вещества желтого цвета, плавящегося при 298оС.
8-Хлор-1-этил-7-фтор-4-оксо-1,4-дигид- ро-1,8-бензо[b] нафтиридин-3- карбоновую кислоту готовят в условиях примера 1, но исходя из 10,5 г 8-хлор-7-фтор-3-этоксикарбонил-1-этил-4-оксо-1,4-дигидро- 1,8-бензо[b]нафтиридина. Получают 9,3 г 8-хлор-1-этил-7-фтор-4-оксо-1,4-дигидро-, 8-бензо[b] нафтиридин-3- карбоновой кислоты в виде твердого вещества бежевого цвета, плавящегося при 380оС, которое применяется без дополнительной очистки в последующих стадиях.
8-Хлор-7-фтор-3-этоксикарбонил-1-этил-4-оксо-1,4-дигидро-1,8-бензо [b] нафтиридин готовят следующим способом:
при перемешивании в суспензию, состоящую из 13,5 г 2-(2,7-дихлор-6-фтор-3-хинолинкарбонил)-3-диметиламиноэтилакрилата в 135 см3 этанола, прибавляют за 5 мин между 10 и 15оС 16 г этиламина, дают температуре подняться примерно до 20оС, добавляют 0,5 г ДБУ и нагревают при перемешивании в течение 2 ч при температуре, близкой к 75оС. После охлаждения до температуры, близкой к 20оС, осадок центрифугируют, промывают 2 раза 100 см3 этанола и 2 раза 100 см3 этилового эфира.
Получают 10,4 г 8-хлор-7-фтор-3-этоксикарбонил-1-этил-4-оксо-1,4-дигидро-1,8-бензо [b]нафтиридина в виде твердого вещества желтого цвета, плавящегося при 300-301оС, которое применяется без дополнительной очистки в последующих стадиях.
П р и м е р 8. Проводят реакцию в условиях примера 16, но исходя из 1,5 г 1-этил-7,8-дифтор-4-оксо-1,4-дигидро-1,8-бензо[b] нафтиридин-3-карбоновой кислоты и 1,98 г 2-[3-фторфенил]-пиперазина (RS), получают 2 г 1-этил-7-фтор-8-[3-(3-фторфенил)-1-пиперазинил] -4-оксо-1,4-дигидро- 1,8-бензо(b)нафтиридин-3-карбоновой кислоты (RS) в виде вещества желтого цвета, плавящегося при 284оС.
1-Этил-7,8-дифтор-4-оксо-1,4-дигидро-1,8-бензо(b)-нафтипиридин-3- карбоновую кислоту получают в тех же самых условиях, что и в примере 17, но исходя из 8 г 3-этоксикарбонил-1-этил-7,8-дифтор-4-оксо-1,4-дигидро-1,8-бензо[b] нафтипиридина. Получают 6,70 г ожидаемого продукта в виде твердого вещества желтого цвета, разлагающегося при 330оС.
3-Этоксикарбонил-1-этил-7,8-дифтор-4-оксо-1,4-дигидро-1,8-бензо [b]нафтиридин-1,8 может быть получен, как описано ниже в примере 32.
2-(3-Фторфенил)-пиперазин (RS) был приготовлен согласно методу, описанному в заявке на французский патент 2 351 108. Исходя из 24 г 3-фторфенилглиоксаля получают 6,3 г ожидаемого продукта в виде жидкого масла желтого цвета.
3-Фторфенилглиоксаль был приготовлен согласно методу, описанному в работе Nathan Kornblum et.coll. I.Am.Chem.Soc. 79, 6562, (1957). Исходя из 40 г 3-фтор-2-бромацетофенона получают 24 г ожидаемого продукта в виде жидкого масла желтого цвета.
3'-Фтор-2-бромацетофенон был приготовлен согласно методу, описанному в работе D.V.C. Awang et. coll. Canad.I.Chem. 47, 706 (1969). Исходя из 25,8 г 3-фторацетофенона получают 40 г ожидаемого продукта в виде жидкого масла зеленоватого цвета.
П р и м е р 9. Проводя реакцию в условиях примера 16, но исходя из 1,5 г 1-этил-7,8-дифтор-4-оксо-1,4-дигидро-1,8-бензо[b] нафтиридин-3- карбоновой кислоты и 1,5 г 2-[2-фторфенил]-пиперазина (RS), получают 2 г 1-этил-7-фтор-8-[3-(2-фторфенил)-1-пиперазинил)-4-оксо-1,4-дигидро- 1,8-бензо[b] нафтиридин-3-карбоновой кислоты (RS) в виде твердого вещества желтого цвета, плавящегося при 306оС.
2(2-Фторфенил)-пиперазин (RS) был приготовлен согласно тем же самым методом, который был использован в примере 8.
Исходя из 26,8 г 2-фторфенилглиоксаля получают 6 г 2-[2-фторфенил]-пиперазина (RS), плавящегося при 70оС.
Исходя из 40,3 г 2'-фтор-2-бромацетофенона получают 26,8 г 2 фторфенилглиоксаля, используемого без дополнительной очистки в последующих стадиях.
Исходя из 20 г 2'-фторацетофенона получают 32,6 г 2'-фтор-2-бромацетофенона в виде желтого масла зеленоватого цвета, используемого без дополнительной очистки в последующих стадиях.
П р и м е р 10. 1-Циклопропил-7-фтор-8-[3-(4-фторфенил)-1-пиперазинил)] -4-оксо-1,4 -дигидро-1,8-бензо-[b]нафтиридин-3-кар-боновую кислоту (RS) готовят в условиях приведенного ниже примера 11, но исходя из 2 г 8-хлор-1-циклопропил-7-фтор-4-оксо-1,4-дигидро-1,8-бензо[b] нафтиридин-3-карбоновой кислоты и 6,534 г 2-[4-фтор- фенил]-пиперазина (RS) и 9 см3 триэтиламина. Реакционную смесь концентрируют при пониженном давлении (20 кПа) и при температуре, близкой к 60оС. Сухой экстракт извлекают посредством 20 см3 воды и 0,5 см3 уксусной кислоты. Нерастворимую часть центрифугируют и промывают 2 раза 5 см3 воды. После одной перекристаллизации из смеси 37 см3 диметилформамида и 37 см3 этанола получают 1,07 г 1-циклопропил-7-фтор-8-[3-(4-фторфенил)- 1-пиперазинил] -4-оксо-1,4-дигидро-1,8-бензо[b] нафтиридин-3-карбоновой кислоты (RS) в виде твердого вещества желтого цвета, плавящегося при 306оС.
П р и м е р 11. Суспензию, состоящую из 1,84 г 8-хлор-7-фтор-1-метил-4-оксо-1,4-дигидро-1,8-бензо[b] нафтиридин- 3-карбоновой кислоты в 20 см3 пиридина, 5,83 г 2-(4-метилфенил)-пиперазина (RS) и 1,24 г триэтиламина, нагревают при температуре, близкой к 115оС, в течение 37 ч. После охлаждения примерно до 20оС нерастворимую часть центрифугируют, промывают 2 раза 5 см3 этанола и 2 раза 5 см3 этилого эфира. После перекристаллизации из 25 см3 диметилформамида получают 0,8 г 7-фтор-1-метил-8-[3-(4-метилфенил)-1-пиперазинил] -4-оксо-1,4-дигидро- 1,8-бензо[b] нафтиридин-3-карбоновой кислоты (RS) в виде твердого вещества желтого цвета, разлагающегося при 282оС.
2-(4-Метилфенил)-пиперазин был приготовлен согласно методу, описанному в заявке на патент FP 2 351 108; исходя из 38,6 г 4-метилфенилглиоксаля (полученного, исходя из 4-метилацетофенона), получают 11,55 г 4-метил-2-фенилпиперазина (RS) в виде твердого вещества желтого цвета, плавящегося при 96-97оС.
П р и м е р 12. Суспензию, состоящую из 1,45 г 7,8-дифтор-1-метил-4-оксо-1,4-дигидро-1,8-бензо(b)нафтиридин- 3-карбоновой кислоты и 1,92 г 2-(4-метоксифенил)-пиперазина (RS) в 14 см3 диметилсульфоксида, нагревают при перемешивании и при температуре, близкой к 90оС в течение 2 ч. После охлаждения до примерно 20оС в реакционную смесь добавляют 20 см3 воды. Нерастворимую часть центрифугируют и промывают 2 раза 5 см3 воды. После одной перекристаллизации в 150 см3 диметилформамида получают 1,35 г 7-фтор-8-[3-(4-метоксифенил)-1-пиперазинил] -1- метил-4-оксо-1,4-дигидро-1,8-бензо(b) нафтиридин-3-карбоновой кислоты (RS) в виде твердого вещества желтого цвета, разлагающегося при 312оС.
7,8-Дифтор-1-метил-4-оксо-1,4-дигид-ро-1,8-бензо(b)нафтиридин-3- карбоновая кислота приготавливается следующим способом:
суспензию, состоящую из 8 г 3-этоксикарбонил-7-8-дифтор-1-метил- 4-оксо-1,4-дигидро-1,8-бензо(b) нафтиридина в 80 см3 17,5%-ного водного раствора хлороводородной кислоты и 80 см3 уксусной кислоты, нагревают при перемешивании и при температуре,близкой к 100оС, в течение 1,5 ч. После охлаждения до примерно 20оС твердое вещество центрифугируют и промывают 6 раз 100 см3 воды. После одной перекристаллизации в 160 см3 диметилформамида получают 6,44 г 7,8-дифтор-1-метил-4-оксо-дигидро-1,8-бензо(b) нафтиридин- 3-карбоновой кислоты в виде твердого вещества желтого цвета, разлагающегося при 360оС.
3-Этоксикарбонил-7,8-дифтор-1-метил- 4-оксо-1,4-дигидро-1,8-бензо-(b) нафтиридин может быть получен, как описано ниже в примере 19.
2-(4-Метоксифенил)-пиперазин (RS) был приготовлен согласно методу, описанному в заявке на патент FP 2 351 108, исходя из 23,4 г 2-(4-метоксифенил)-глиоксаля и 10,26 г этилендиамина, получают 6,21 г 2-(4-метоксифенил)-пиперазин (RS) в виде маслянистого продукта, который используется таким, как он есть.
4-Метоксифенилглиоксаль может быть приготовлен согласно методу, описанному в работе Nathan Kornblum et coll. I.Am.Chem.Soc. 79, 6562 (1957). Исходя из 45,4 г 2-бром-4-метоксиацетофенона в 200 см3 диметилсульфоксида получают 23,4 г 4-метоксифенилглиоксаля в виде желтого масла оранжевого цвета, которое применяется без дополнительной очистки в последующих стадиях.
2-Бром-4'-метоксиацетофенон был приготовлен согласно работе N.G.P.H.Bun. Hoi et coll. I.Chem.Soc. 255 (1951).
П р и м е р 13. Суспензию, состоящую из 2 г 7,8-дифтор-1-метокси-4-оксо-1,4-дигидро-1,8-бензо(b)нафтиридин-3- карбоновой кислоты и 2,1 г 2-фенилпиперазина (RS) в 30 см3 диметилсульфоксида, нагревают при перемешивании в течение 15 мин примерно при 50оС. После охлаждения примерно до 20оС реакционную смесь приливают к 100 см3 воды, куда добавляют 1,2 см3 уксусной кислоты. Нерастворимую часть центрифугируют, промывают 3 раза 10 см3 воды и перекристаллизовывают из 80 см3 диметил- формамида. Получают 2 г 7-фтор-1-метокси-4-оксо-8-[(3-фенил)-1-пиперазинил] -1,4-ди-гидро- 1,8-бензо[b] нафтиридин-3-карбоновой кислоты (RS) в виде твердого вещества желтого цвета, разлагающегося при 284оС.
7,8-Дифтор-1-метокси-4-оксо-1,4-дигид- ро-1,8-бензо[b] нафтиридин- 3-карбоновую кислоту готовят в следующих условиях:
суспензию, состоящую из 2,78 г 3-этоксикарбонил-7,8-дифтор-1-метокси-4-оксо-1,4- дигидро-1,8- бензо[b] нафтиридина в 30 см3 17,5%-ной хлороводородной кислоты и 30 см3 уксусной кислоты, нагревают при температуре, близкой к 100оС, в течение 1 ч. После охлаждения при примерно 20оС реакционную смесь приливают к 100 см3 воды. Образованный осадок центрифугируют, промывают 3 раза 30 см3 воды и 2 раза 5 см3 этанола. После одной перекристаллизации из 100 см3 диметилформамида с 20% этанола получают 2,03 г 7,8-дифтор-1-метокси-4-оксо-1,4-дигидро-1,8-бензо[b]нафтиридин-3- карбоновой кислоты в виде твердого вещества желтого цвета, плавящегося при 325-327оС.
3-Этоксикарбонил-7,8-дифтор-1-меток- си-4-оксо-1,4-дигидро-1,8- бензо[b] нафтиридин получают в следующих условиях:
к суспензии, состоящей из 1,7 г хлоргидрата метоксиамина в 40 см3 трихлорметана, добавляют 2,13 г триэтиламина. После 15-минутного перемешивания при температуре, близкой к 20оС, к полученному раствору добавляют 3,69 г 2-(2-хлор-6,7-дифтор-3-хинолинкарбонил)-3-диметиламиноэтилакри- лата и перемешивают в течение 4,5 ч примерно при 20оС. Реакционную смесь концентрируют досуха при пониженном давлении (20 кПа) и при температуре, близкой к 50оС. Остаток извлекают посредством 70 см3 этанола и 3,6 г триэтиламина и нагревают в течение 30 мин при температуре, близкой к 75оС. После охлаждения примерно до 20оС полученный осадок центрифугируют и промывают 3 раза 30 см3 этанола. Получают 2,67 г 3-этоксикарбонил-7,8-дифтор-1-метокси-4- оксо-1,4-дигидро-1,8-бензо(b) нафтиридина в виде твердого вещества бледно-желтого цвета, плавящегося при 266-268оС.
2-(2-Хлор-6,7-дифтор-2-хинолинкарбо-нил)-3-диметиламиноэтилакрилат может быть получен, как описано ниже в примере 20.
П р и м е р 14. 8-[3-(4-Цианофенил)-1-пиперазинил]-7-фтор-1-метил-оксо-1,4-дигид- ро- 1,8-бензо[b]нафтиридин-3-карбоновую кислоту (RS) готовят в условиях примера 11, но исходя из 1,84 г 8-хлор-7-фтор-1-метил-4- оксо-1,4-дигидро-1,8-бензо[b] нафтиридин-3-карбоновой кислоты, 5,61 г 2-(4-цианофенил)-пиперазина (RS) и 1,7 см3 триэтиламина получают 1,15 г 8-[3-(4-цианофенил)-1-пиперазинил] -7-фтор-1-ме-тил-4-оксо-1,4- дигидро-1,8-бензо[b]нафтиридин-3-карбоновой кислоты в виде твердого вещества желтого цвета, разлагающегося при 332оС.
2-(4-Цианофенил)-пиперазин (RS) может быть приготовлен согласно методу, описанному в заявке на патент FP 2 351 108, исходя из 45 г 4-цианофенилглиоксаля. Получают 9,4 г 2-(4-цианофенил)-пиперазина в виде желтого масла оранжевого цвета, которое применяется в таком виде в последующих стадиях. 4-Циано-фенилглиоксаль приготавливается, исходя из 4-цианоацетофенона.
П р и м е р 15. Проводя реакцию, как в примере 16, но исходя из 1,16 г 7,8-дифтор-1-метил-4-оксо-1,4-дигидро-1,8-бензо[b]на- фтиридин-3- карбоновой кислоты и 1,82 г 2-(4-метоксиметилфенил)-пиперазина (RS) получают 1,70 г 7-фтор-8-[3-(4-метоксиметилфенил)-1- пиперазинил] -1-метил-4-оксо-1,4-дигидро-1,8-бензо[b]нафтиридин- 3-карбоновой кислоты (RS) в виде твердого вещества желтого цвета, разлагающегося при 284оС.
2-(4-Метоксиметилфенил)-пиперазин (RS) был приготовлен согласно методу, описанному в заявке на патент FP 2 351 108, исходя из 33,5 г 4-метоксиметилфениглиоксаля и 13,48 г этилендиамина. Сырой продукт очищался методом хроматографии на 750 г двуокиси кремния (230-400 меш) в суспензии, содержащей смесь 70% дихлорметана, 26% этанола, 4% триэтиламина, при элюировании посредством 1,8 л той же смеси. После концентрировании элюата при пониженном давлении (20 кПа) и при примерно при 50оС получают 12,15 г ожидаемого продукта в виде твердого вещества оранжевого цвета, плавящегося при 79оС.
4-Метоксиметилфенилглиоксаль был приготовлен согласно методу, описанному в заявке на патент FP 2 351 108, исходя из 30,8 г 4-метоксиметилацетофенона и 24 г диоксида селена. Получают 30 г ожидаемого продукта в виде жидкого масла коричневого цвета, которое применяли без дополнительной очистки в последующих стадиях.
4-Метоксиметилацетофенон был приготовлен согласно методу, описанному в работе H.B.Rass et coll. I.Am.Chem.Soc. 71, 1767, (1949), исходя из 4-метоксиметилцианобензола, полученного исходя из 4-цианобензилбромида.
П р и м е р 16. Суспензию, состоящую из 1,015 г 7,8-дифтор-1-метил-4-оксо-1,4-дигидро-1,8-бензо[b] нафтиридин-3- карбоновой кислоты и 1,7 г 2-[4-(2-гидрокси- этокси)-фенил] -пиперазина (RS) в 15 см3 диметилсульфоксида, нагревают при перемешивании в течение 2,5 ч при температуре, близкой к 100оС. После охлаждения примерно до 20оС суспензию смешивают с 30 см3 воды, центрифугируют, промывают 3 раза 10 см3 воды. После перекристаллизации в 15 см3 диметилформамида получают 1,2 г 7-фтор-8-[3-/4-(2-гидроксиэтокси)-фенил] -1-пиперазинил/-1-метил-4- оксо-1,4-дигидро-1,8-бензо[b] нафтиридин-3-карбоновой кислоты (RS) в виде твердого вещества желтого цвета, плавящегося при 276оС.
2-[4-(2-Гидроксиэтокси)-фенил] -пипера- зин (RS) может быть приготовлен согласно методу, описанному в заявке на патент FP 2 351 108, исходя из 18,5 г 2-[4-(2-гидроксиэтокси)-фенил]-глиоксаля и 6,86 г этилендиамина. Получают 4 г 2-[4-(2-гидроксиэтокси)- фенил]-пиперазина (RS) в виде твердого вещества бежевого цвета, плавящегося при 141оС.
2-[4-(2-Гидроксиэтокси)-фенил]-глиок- саль может быть приготовлен согласно методу, описанному в работе Nathan Kornblum et coll. I.Am.Chem.Soc. 79, 6562 (1957). Исходя из 30,5 г 4-(2-гидроксиэтокси)-2-бромацетофенона получают 18,5 г 2-[2-гидроксиэтокси)-фенил]-глиоксаля в виде желтого масла желтого цвета, которое применяется без дополнительной очистки в последующих стадиях.
4'-2'-Гидроксиэтокси-(2-бромацетофенон) может быть приготовлен согласно методу, описанному в работе N.G.P.H.Buu Hoi et coll. I.Chem.Soc. 255 (1951). Исходя из 27 г 4-(2-гидроксиэтокси)-ацетофенона получают 28,85 г 4'-(2'-гидроксиэтокси)-2-бромацетофенон в виде твердого вещества бежевого цвета, плавящегося при 78оС.
4-(2-Гидроксиэтокси)-ацетофенон был приготовлен согласно работе C.Rufer et coll. I.Med.Chem. 18 (3), 263 (1975).
П р и м е р 17. Проводят реакцию в условиях примера 16, но исходя из 1,45 г 7,8-дифтор-1-метил-4-оксо-1,4-дигидро-1,8-бензо[b] нафтиридин-3- карбоновой кислоты и 2,09 г 2-(3,4-диметилфенил)-пиперазина (RS) получают 2,15 г 7-фтор-1-метил- 8-[3-(3,4-диметилфенил)-1-пиперазинил]-4-оксо- 1,4- дигидро-1,8-бензо[b] нафтиридин-3-карбоновой кислоты (RS) в виде твердого вещества желтого цвета, плавящегося при 276оС.
2-(3,4-Диметилфенил)-пиперазин (RS) был приготовлен согласно методу, описанному в заявке на патент FP 2 351 108. Исходя из 42,4 г 3,4-диметилфенилглиоксаля получают 7,9 г 2-(3,4-диметилфенил)-пиперазина (RS) в виде твердого вещества бежевого цвета, плавящегося при 98-100оС.
3,4-Диметилфенилглиоксаль может быть получен согласно методу, описанному в работе Nathan Korn blum et coll. I.Am.Chem.Soc. 79, 6562 (1957). Исходя из 60 г 3',4'-диметил-2-бромацетофенона получают 40,4 г 3,4-диметилфенилглиоксаля в виде жидкого масла.
3', 4'-Диметил-2-бромацетофенон был приготовлен согласно методу, описанному в работе R.M.Laird et R.E.Parker I.Am.Chem.Soc. 83, 4277 (1961).
П р и м е р 18. Проводя реакцию в условиях примера 16, но исходя из 1,16 г 7,8-дифтор-1-метил-4-оксо-1,4-дигидро-1,8-бензо[b] нафтиридин-3- карбоновой кислоты и 1,82 г 2-(4-амино-3-метоксифенил)-1- пиперазинил)-1-метил-4-оксо-1,4-дигидро-1,8-бензо[b] нафтиридин- 3-карбоновой кислоты (RS) в виде твердого вещества желтого цвета, разлагающегося при 259оС.
2-(4-Амино-3-метоксифенил)-пиперазин (RS) был приготовлен следующим способом:
в раствор, содержащий 18 г 2-(3-метокси-4-нитрофенил)-пиперазин (RS) в 150 см3 этанола и нагретый при перемешивании примерно 75оС, добавляют за 10 мин раствор, содержащий 53 г гидросульфата натрия в 200 см3 воды. Смесь выдерживают при перемешивании в течение 1 ч при той же температуре. Реакционную смесь концентрируют при пониженном давлении (20 кПа) и примерно при 60оС. Остаток извлекают посредством 150 см3 3,75 н. водного раствора щелочи натрия и 50 г карбоната калия. Смесь подвергают экстракции 4 раза 100 см3 трихлорметана. Объединенные органические вытяжки сушат на сульфате магния, фильтруют и концентрируют при пониженном давлении (20 кПа) и примерно при 40оС. Получают 13 г ожидаемого продукта в виде жидкого масла коричневого цвета, которое применяется без дополнительной очистки в последующих стадиях.
2-[3-Метокси-4-нитрофенил] -пиперазин (RS) был приготовлен согласно методу, описанному в заявке на патент FP 2 351 108, но исходя из 36 г 3-метокси-4-нитрофенилглиоксаля и 12,1 г этилендиамина. Сырой продукт очищают методом хроматографии на 800 г двуокиси кремния (230-400 меш) в суспензии, содержащей смесь 70% дихлорметана, 26% этанола, 4% триэтиламина, и при элюировании 2 л той же смеси. После концентрирования элюата при пониженном давлении (20 кПа) и при примерно 50оС получают 18 г ожидаемого продукта в виде желтого масла красного цвета, которое использовали без дополнительной очистки в последующих стадиях.
3-Метокси-4-нитрофенилглиоксаль был приготовлен согласно методу, описанному в заявке на патент FP 2 351 108, но исходя из 33,6 г 3-метокси-4-нитроацетофенона и 22 г диоксида селена. Получают 35 г ожидаемого продукта в виде жидкого масла коричневого цвета, которое применяли без дополнительной очистки в последующих стадиях.
3-Метокси-4-нитроацетофенон был приготовлен согласно методу, описанному в патенте Южно-Африканской Республики 67 06 465.
П р и м е р 19. Раствор, содержащий 1,07 г 3-этоксикарбонил-7-фтор-1-метил-4-оксо-8-(3-фенил-1-пиперазинил)- 1,4-дигидро-1,8-бензо[b]нафтиридина (RS) в 10 см3 уксусной кислоты и 10 см3 17,5%-ного водного раствора хлороводородной кислоты, нагревают при температуре, близкой к 100оС, в течение 40 мин. Реакционную смесь концентрируют досуха при пониженном давлении (20 кПа) и примерно при 60оС. Сухой остаток извлекают посредством 10 см3 этанола, центрифугируют, промывают 2 раза 10 см3 этанола и 2 раза 10 см3 этилового эфира. Полученное твердое вещество переводят в суспензию с 30 см3 воды и нагревают до температуры, близкой к 95оС. После добавления 2 н. водного раствора и перемешивания в течение 30 мин твердое вещество центрифугируют примерно 95оС, промывают 3 раза 20 см3 воды примерно при 80оС, 3 раза 15 см3 этанола примерно 60оС и 3 раза 20 см3 этилового эфира. После одной перекристаллизации в смеси 12 см3 диметилформамида и 6 см3 этанола получают 0,7 г 7-фтор-1-метил-4- оксо-8-(3-фенил-1-пиперазинил)-1,4-дигидро-1,8-бензо[b]на-фтиридин-3- карбоновой кислоты (RS) в виде твердого вещества желтого цвета, плавящегося при 265оС.
3-Этоксикарбонил-7-фтор-1-метил-4-ок- со-8-(3-фенил-1-пиперазинил)- 1,4-дигидро-1,8-бензо[b]нафтиридин-1,8 (RS) может быть приготовлен следующим способом.
суспензию, состоящую из 3 этоксикарбонил-7,8-дифтор-1-метил-4-оксо-1,4-дигид- ро-1,8-бензо[b] нафтиридина 0,265 г карбоната натрия и 0,5 г 2-фенилпиперазина (RS) в 25 см3 диметилсульфоксида, нагревают при температуре, близкой к 95оС, в течение 2 ч. После охлаждения примерно до 20оС реакционную смесь приливают к 75 см3 воды и экстрагируют 4 раза 50 см3 дихлорметана. Объединенные органические вытяжки промывают 3 раза 40 см3 воды, сушат на сульфате магния, фильтруют и концентрируют досуха при пониженном давлении (20 кПа) и примерно при 50оС. Получают 1,07 г 3-этоксикарбонил-7-фтор-4-оксо-8-[3-фенил- 1-пиперазинил)-1,4-дигидро- 1,8-бензо[b]нафтиридина (RS) в виде твердого вещества желтого цвета, плавящегося при 220-222оС, которое применяется без дополнительной очистки в последующих стадиях.
3-Этоксикарбонил-7,8-дифтор-1-метил- 4-оксо-1,4-дигидро-1,8-бензо(b) нафтиридин приготавливается следующим образом.
При перемешивании в суспензию, состоящую из 22,3 г 2-(2-хлор-6,7-дифтор-3-хинолинкарбонил)-3-диметиламиноэтилакри- лата в 480 см3 этанола, выдерживаемую при температуре, близкой к 0оС, прибавляют за 10 мин меду 0 и 5оС раствор, содержащий при примерно 0оС 11,3 г метиламина в 50 см3 этанола, перемешивают 1 ч между 0 и 5оС, дают температуре подняться до примерно 25оС и перемешивают еще 16 ч при той же температуре. Нерастворимую часть центрифугируют, промывают 3 раза 100 см3 этилового эфира. После одной перекристаллизации в 250 см3 диметилформамида получают 16 г 3-этоксикарбонил-7,8-дифтор-1-метил-4-ок- со-1,4-дигидро-1,8-бензо(b) нафтиридина в виде твердого вещества желтого цвета, плавящегося при 323-324оС.
2-(2-Хлор-6,7-дифтор-3-хинолинкарбо-нил)-3-диметиламиноэтиоакрилат приготавливается следующим способом:
суспензию, состоящую из 6,17 г 3-оксо-3-(2-хлор-6,7-дифтор-3-хинолил)-этилпропи- оната в 7,15 г N,N-диметилформамиддиметилацеталя и 60 см3 этилацетата, нагревают при температуре, близкой к 75оС, в течение 1 ч 15 мин. Реакционную смесь концентрируют досуха при пониженном давлении (20 кПа) и примерно 50оС. Остаток извлекают посредством 50 см3 изопропилового эфира, центрифугируют, промывают 3 раза 25 см3 того же растворителя. Получают 6,65 г 2-(2-хлор-6,7-дифтор-3-хинолинкарбонил)-3-ди- метиламиноэтилакрилата в виде твердого вещества оранжевого цвета, плавящегося при 140оС.
3-Оксо-3-(2-хлор-6,7-дифтор-3-хинолил) -этилпропионат приготавливается следующим способом:
суспензию, состоящую из 14,13 г 2-хлор-6,7-дифторхинолин-3-карбоновой кислоты в 29 см3 хлорида тионила и 220 см3 трихлорметана, нагревают при температуре, близкой к 60оС, в течение 4 ч. Полученный раствор концентрируют досуха при пониженном давлении (20 кПа) и примерно при 60оС. Полученный остаток извлекают посредством 75 см3 н-гексана, центрифугируют, промывают 2 раза 60 см3 того же растворителя. 14,4 г полученного вещества желтого цвета переводят в раствор в 115 см3 тетрагидрофурана.
Этот раствор вводят по капле при перемешивании за 35 мин между 5 и 10оС в 70 см3 раствора магниевого хелата моноэтилмалоната в тетрагидрофуране, который был приготовлен в условиях, описанных ниже. Температуре дают подняться примерно до 20оС и перемешивают еще 2 ч при тех же условиях. Полученный раствор вводят по капле при перемешивании за 30 мин при температуре, близкой к 5оС, в 560 см3 0,5 н.серной кислоты. Температура полученной суспензии самопроизвольно повышается до 20оС и перемешивают еще 1,5 ч при этой температуре. Проводят экстракцию 3 раза 250 см3 этилацетата. Объединенные органические вытяжки промываются 2 раза 250 см2 воды, сушат на сульфате магния, фильтруют и концентрируют досуха при пониженном давлении (20 кПа) и при 50оС. Полученный остаток извлекают посредством 50 см3 н-гексана с 20% изопропилового эфира, центрифугируют, промывают 10 см3 той же смеси и перекристаллизовывают из 60 см3 изопропанола с 30% н-гексана. Получают 11,84 г 3-оксо-3-(2-хлор-6,7-дифтор-3-хинолил)-этилпропионата в виде твердого вещества кремового цвета, плавящегося при 107оС.
Приготовляют магниевый хелат моноэтилмалоната следующим образом.
К 2,78 г магниевых опилок добавляют последовательно 2 см3 абсолютного этанола, 0,1 см3 тетрахлорметана и 1 г моноэтилмалоната. После нагрева добавляют за 15 мин раствор, содержащий 9 г моноэтилмалоната в 180 см3 этанола. Смесь нагревают 20 ч при температуре, близкой к 75оС, концентрируют при пониженном давлении (20 кПа) и при 50оС. Остаток извлекают за 2 раза посредством 100 см3 толуола и концентрируют при пониженном давлении в тех же условиях, что и ранее. Полученный серый порошок переводят в раствор добавлением безводного тетрагидрофурана так, чтобы получить суммарный объем 70 см3.
2-Хлор-6,7-дифторхинолин-3-карбоно-вая кислота была приготовлена следующим образом:
при перемешивании к охлажденной до 10оС суспензии, состоящей из 70,18 г 2-хлор-6,7-дифтор-3-формил-1,4-дигидро-хи-нолина в 970 см3 1 н.водной щелочи калия, прибавляют за 1 ч, поддерживая температуру между 10 и 14оС, раствор, содержащий 115 г перманганата калия в 1,215 л воды. Дают температуре подняться примерно до 20оС и перемешивают еще 30 мин при этой температуре. Добавляют 38,5 г дитионата натрия, перемешивают 10 мин при температуре, близкой к 20оС, фильтруют через диатомированный кремнезем, промывают 3 раза 200 см3 воды. Фильтрат и промывочные водные фазы объединяют и добавляют 140 см3 35% -ного водного раствора хлороводородной кислоты. Образованный осадок экстрагируют 4 раза 800 см3 этилацетата. Объединенные органические вытяжки промывают 2 раза 500 см3 воды, сушат на сульфате магния, фильтруют и концентрируют при пониженном давлении (20 кПа) и при 50оС. Остаток извлекают посредством 400 см3 этилового эфира, центрифугируют, промывают 2 раза 200 см3 того же растворителя. Получают 49,2 г 2-хлор-6,7-дифтор- хинолин-3- карбоновой кислоты в виде твердого вещества бежевого цвета, плавящегося при 232оС, которое применяется без дополнительной очистки в последующих стадиях.
2-Хлор-6,7-дифтор-3-формил-1,4-дигид- рохинолин был приготовлен следующим способом
к смеси, содержащей 800 см3 трихлорметана и 74,35 см3 диметилформамида, прибавляют за 30 мин при перемешивании между 10 и 15оС 76,9 см3 хлорида фосфорида и перемешивают 1 ч при температуре близкой к 20оС. К полученному раствору прибавляют за 10 мин примерно 20оС и при сильном перемешивании 65,8 г 6,7-дифтор-3,4-дигидрокарбостирила. Полученную суспензию нагревают до температуры, близкой к 60оС, и выдерживают при этой температуре в течение 2 ч. Реакционную смесь концентрируют при пониженном давлении (20 кПа) и при 50оС до получения пастообразной смеси. При сильном перемешивании прибавляют смесь 500 г льда и 500 см3 воды. Полученное твердое вещество центрифугируют при 5оС и промывают 3 раза 300 см3 воды при 5оС. Полученный влажный продукт и 60 г ацетата натрия добавляют одновременно за 1 ч к 1,5 л воды при 90оС так, чтобы поддерживать рН около 6. Перемешивают еще 30 мин при 90оС, дают температуре опуститься примерно до 50оС, центрифугируют при этой температуре и промывают 3 раза 300 см3 воды примерно при 20оС. Получают 70,18 г 2-хлор-6,7-дифтор-3- формил-1,4-дигидрохинолина в виде твердого вещества желтого цвета, плавящегося при 260оС, которое применяется в таком виде в последующих стадиях.
6,7-Дифтор-3,4-дигидрокарбостирил получается следующим способом
к 67 г 3',4'-дифтор-3-N-хлорпропионанилиду прибавляют при сильном перемешивании 134 г хлорида алюминия, затем после примерно 2 мин добавляют снова маленькими порциями за 15 мин 135,9 г 3',4'-дифтор-3-N-хлорпропионанилида и 272 г хлорида алюминия. Температура самопроизвольно поднимается примерно до 60оС и реакционная смесь становится жидкой. Затем нагревают до 110оС за 20 мин и выдерживают между 110 и 120оС в течение 2 ч. Реакционную смесь (примерно при 110оС) приливают за 10 мин при сильном перемешивании к смеси 840 см3 35%-ной хлороводородной кислоты и 1 кг толченого льда. Дают температуре подняться примерно до 20оС, центрифугируют, промывают 2 раза 600 см3, 300 см3 этанола при 5оС и 2 раза 400 см3 этилового эфира примерно при 20оС. Получают 131,58 г 6,7-дифтор-1,4-дигидрокарбостирила в виде твердого вещества бежевого цвета, плавящегося при 216оС, которое применяется в таком виде в последующих стадиях.
3',4'-Дифтор-3-N-хлорпропионанилид приготавливается следующим образом
к раствору, содержащему 125 г 3,4-дифторанилина в 80 см3 пиридина и 1,5 л ацетона, нагретому до температуры, близкой к 55оС, прибавляют при перемешивании за 1,5 ч 139,16 г хлорида 3-хлорпропионовой кислоты и выдерживают при этой температуре в течение 1,5 ч. После охлаждения примерно до 20оС раствор приливается при перемешивании к смеси 1 л воды и 500 г толченого льда. Дают температуре подняться примерно до 20оС и проводят экстракцию 3 раза 500 см3 дихлорметана. Объединенные органические вытяжки промывают 500 см3 1 н. хлороводородной кислоты, 5 раз 500 см3 воды, сушат на сульфате магния, фильтруют и концентрируют при пониженном давлении (20 кПа) и примерно при 50оС. Полученное твердое вещество извлекают посредством 500 см3 н-гексана, центрифугируют, промывают 2 раза 100 см3 того же растворителя. Получают 202,9 г 3'.4'-дифтор-3-N-хлорпропионанилида в виде твердого вещества бежевого цвета, плавящегося при 76оС, которое применяется без дополнительной очистки в последующих стадиях.
2-Фенилпиперазин (RS) был приготовлен согласно способу, описанному в работе W.R.Roderick et coll. I.Med.Chem. 9, 181 (1966). перазина) (S) в виде бесцветного твердого вещества, плавящегося при 258оС.
/α/D20 -9,9о ± 0,9 (с 0,5; диметилформамид).
Исходя из 17,5 г 2-фенилэтилендиамина (S) получают 22,5 г 2-фенил-1,4-ди(п-сульфонилтолуол)-этилендиамина (S) в виде твердого вещества желтого цвета, плавящегося при 164оС.
/α/D20 +17,2 ± 0,7 (с 0,7; диметилформамид.
Исходя из 20 г α -аминофенилацетамида (S) получают 17,5 г 2-фенилэтилендиамина (S) в виде жидкого масла желтого цвета, которое немедленно применяется на следующей стадии.
Исходя из 45,1 г 2-фенилметилглицината (S) получают 21,5 г α -аминофенилацетамида (S) в виде бесцветного твердого вещества, плавящегося при 137-138оС.
/α/D20 +114о ± 2 (с 1; этанол).
Исходя из 45,3 г 2-фенилглицина (S) получают 45,2 г 2 -фенилметилглицината (S) в виде желтого масла желтого цвета.
П р и м е р 21. Проводя реакцию в условиях примера 20, но исходя из 66,3 г 3-этоксикарбонил-7-фтор-1-метил-4-оксо-8-(3-фенил-1-пиперазинил)-1,4- дигидро-1,8-бензо[b] нафтиридина (RS) получают без перекристаллизации 59,15 г 7-фтор-1-метил- 4-оксо-8-(3-фенил-1-пиперазинил)-1,4-ди- гидро-1,8-бензо [b] нафтиридин-3-карбоновой кислоты (R) в виде твердого вещества желтого цвета, разлагающегося при 276-278оС.
/α/D20 +39,8о ± 0,6 (с 1; уксусная кислота).
Проводят реакцию в условиях примера 20, но исходя из 51,6 г 3-этоксикарбонил-7,8-дифтор-1-метил-4-оксо-1,4-дигидро-1,8-бензо [b] нафтиридина и 31,7 г 2-фенилпиперазина (R) получают 66,86 г 3-этоксикарбонил-7-фтор-1-метил-4-оксо-8-(3-фенил-1- пиперазинил)- 1,4-дигидро-1,8-бензо[b]нафтиридина (R) в виде твердого вещества желтого цвета, плавящегося при 221оС.
/α/D20 +43о ± 1 (с 0,5; уксусная кислота).
2-Фенилпиперазин (R) может быть получен следующим способом
смесь, состоящую из 10 г 2-фенил-1,4-(п-сульфонилтолуол)-пиперазина (R) и 9,4 г фенола в 100 см3 48%-ный бромоводородной кислоты, нагревают при очень сильном перемешивании в течение 5 ч при температуре, близкой к 120оС. После охлаждения до примерно 80оС реакционную смесь смешивают с 250 см3 воды, охлаждают до 20оС и промывают 5 раз 100 см3 дихлорметана. Водную фазу концентрируют при пониженном давлении (20 кПа) и при примерно 80оС. Остаток извлекают посредством 100 см3 этилацетата, охлаждают до 5оС и смешивают между 5 и 20оС со 100 см3 30%-ного водного раствора щелочи натрия. Органическую фазу отделяют, а водную фазу снова подвергают экстракции 3 раза 100 см3 этилацетата.
Объединенные органические вытяжки промывают 4 раза 20 см3 5 н. водного раствора щелочи натрия, сушат на сульфате магния, фильтруют и концентрируют досуха при пониженном давлении (20 кПа) и при примерно 30оС. Полученный остаток извлекают посредством 15 см3 изопропилового эфира при примерно 0оС, центрифугируют и промывают при той же температуре 10 см3 того же растворителя. Получают 2,3 г 2-фенилпиперазина (R) в виде бесцветного твердого вещества, плавящегося при 117оС.
/ α/D20 38о ± 0,6 (с 2; этанол).
2-Фенил-4-ди(п-сульфонилтолуол)-пи-перазин (R) был приготовлен следующим образом:
смесь, состоящую из 11 г 2-фенил-1,4-ди(п-сульфонилтолуол)-этилендиамина (R) и 13,82 г карбоната калия в 110 см3 диметилформамида, нагревают при перемешивании и при 60оС в течение 30 мин, затем смешивают с 18,8 г 1,2-дибромэтана и нагревают при 115оС в течение 1 ч. После охлаждения до примерно 20оС реакционную смесь приливают к 250 см3 воды и подвергают экстракции 3 раза 200 см3 дихлорметана. Объединенные органические вытяжки промывают 3 раза 120 см3 воды, сушат на сульфате магния и концентрируют при пониженном давлении (20 кПа) и при примерно 30оС. Остаток извлекают посредством 50 см3 этилового эфира, центрифугируют, промывают 3 раза 15 см3 того же растворителя. Получают 10,55 г ожидаемого продукта в виде бесцветного твердого вещества, плавящегося при 258оС.
/ α/D20 +9,6о ±0,8 (с 0,5; диметилформамид),
2-Фенил-1,4-ди(п-сульфонилтолуол)-эти- лендиамин (R) был приготовлен согласно методу, описанному в работе Douglas G. Neilson et coll. I.Chem.Soc. 393 (1966), но исходя из 7,5 г 2-фенилэтилендиамина (R) получают 12,9 г ожидаемого продукта в виде твердого вещества желтого цвета, плавящегося при 164оС.
/α/D20 -18,2 ± 0,7 (с 0,7; диметилформамид).
2-Фенилэтилендиамин (R) был приготовлен согласно методу описанному в работе A.C.Brown et coll. I.Am.Chem.Soc. 86, 3566 (1964), но исходя из 11 г α -аминофенилацетамида (R) и 293 см3 1М раствора борана в тетрагидрофуране. Получают 7,5 г ожидаемого продукта в виде жидкого желтого цвета, который будучи нестабильным используется немедленно на следующей стадии.
α -Аминофенилацетамид (R) был приготовлен согласно методу, описанному в работе Douglas G. Neilson et coll. I.Chem.Soc. 393 (1966). Исходя из 27,9 г 2-фенилметил-глицината (R) получают после одной перекристаллизации в 135 см3 этилацетата с 26% метанола 12,93 г ожидаемого продукта в виде бесцветного твердого вещества, плавящегося при 137-138оС.
/ α/D20 -115о ± 3 (с 0,5; этанол).
2-Фенилметилглицинат (R) был приготовлен согласно методу, описанному в работе Douglas G. Neilson et coll, I.Chem.Soc. 393 (1966), но исходя из 43 г 2-фенилглицина (R) и 22,8 см3 хлорида тионила в 85 см3 метанола получают в виде жидкого масла желтого цвета.
Монометансульфонат 7-фтор-1-метил-4-оксо-8-(3-фенил-1-пиперазинил)-1,4-ди- гидро-1,8- бензо[b]нафтиридин-3-карбоновой кислоты (R) был приготовлен следующим способом
суспензию состоящую из 3,5 г 7-фтор-1-метил-4-оксо-8-(3-фенил-1-пиперазинил)-1,4 -дигидро-1,8-бензо[b] нафтиридин-кар- боновой кислоты (R) в 70 см3 воды, смешивают со 100 см3 0,1 н. водного раствора метансульфокислоты и нагревают до примерно 90оС. После охлаждения до примерно 20оС нерастворимую часть центрифугируют и промывают 3 раза 25 см3 воды. Получают 3,9 г ожидаемой соли в виде твердого вещества желтого цвета, разлагающегося при 335-340оС.
/α/D20 -19 ± 2 (с 0,2; вода с 50% этанола).
7-Фтор-1-метил-4-оксо-8-(3-фенил-1-пи- перазинил)-1,4-дигидро-1,8- бензо[b] нафтиридин-3-карбоксилат холина (R) был приготовлен следующим образом:
к суспензии, состоящей из 3,5 г 7-фтор-1-метил-4-оксо-8-(3-фенил-1-пиперазинил)-1,4-дигидро-1,8-бензо [b]нафтиридин-3-карбоновой кислоты (R) в 30 см3 метанола, прибавляют 2,72 см3 45%-ного раствора холина в метаноле. К полученному раствору добавляют 200 см3 изопропилового эфира. Осадок центрифугируют, промывают 3 раза 60 см3 того же растворителя, затем 3 раза 60 см3 ацетона. Получают 3,97 г ожидаемой соли в виде твердого вещества желтого цвета, плавящегося при 234оС.
/α/D20 -33,9о ± 0,9 (с1; метанол).
Моноизетионат 7-фтор-1-метил-4-оксо-8-(3-фенил-1-пиперазинил)-1,4-дигидро-1,8- бензо[b]нафтиридин-3-карбоновой кислоты был приготовлен следующим образом:
к суспензии, состоящей из 1 г 7-фтор-1-метил-4-оксо-8-(3-фенил-1- пиперазинил)-1,4-дигидро-1,8-бензо[b] нафтиридин-3-кар- боновой кислоты (R) в 40 см3 воды с 50% этанола, прибавляют 2,6 см3 1 н. водного раствора изетионовой кислоты. После растворения при 80оС и последующего охлаждения до 10оС нерастворимую часть центрифугируют, промывают 2 раза 15 см3 воды с 50% этанола, 2 раза 15 см3 воды и 3 раза 15 см3 этанола. Получают 1 г ожидаемой соли в виде твердого вещества желтого цвета, разлагающегося при 334оС.
/ α/D20 +111о ± 6 (с 0,1; диметилсульфоксид).
П р и м е р 22. 7-Фтор-1-метил-4-оксо-8-(3-фенил-1-пиперазинил)-1,4-дигидро-1,8-бензо [b] нафтиридин-3-карбоновую кислоту (S), изомер (-)- была приготовлена тем же способом, что и 7-фтор-1-метил-4-оксо-8-(3-фенил-1-пиперазинил)-1,4-дигидро-1,8-бен- зо [b]нафтиридин-3-карбоновая кислота (RS) в примере 10, но исходя из 4,4 г 3-этоксикарбонил-7-8-дифтор-1-метил-4-оксо-1,4-диги- дро-1,8-бензо[b] нафтиридина 2,7 г 2-фенилпиперазина (+) (/α/D20 +35,4о ± 0,3 (с 2; этанол) и 1,48 г карбоната натрия получают 6,3 г твердого вещества желтого цвета, плавящегося при 226оС.
После кислотного гидролиза в тех же условиях, которые были описаны для 3-этоксикарбонил-7-фтор-1-метил-4-оксо-8-(3-фе- нил-1-пиперазинил)-1,4- дигидро-1,8-бензо[b] нафтиридина (RS), получают 3,95 г 7-фтор-1-метил-4-оксо-8-(3-фенил-1-пиперази- нил)-1,4-дигидро-1,8[b] нафтиридин-3-карбоновой кислоты (S), изомер (-), в виде твердого вещества желтого цвета, плавящегося при 276-278оС.
/ α/D20 -40,4о ± 1 (с 1; уксусная кислота).
2-Фенилпиперазин (R), изомер (-), и 2-фенилпиперазин (S), изомер (+), были приготовлены при кислотном гидролизе двух диастереоизомерных соединений (R и S), соответствующих структуре 1-(2'-трифторметил-2-метоксифенилацетид)-3-фенилпипера- зина, называемых А и В. Эти два последних соединения получены при ацилировании 2'-фенилпиперазина (RS) хлоридом хиральной кислоты; хлоридом 2-трифторметил-2-метоксифенилуксусной кислоты (R), изомер (-). Получение происходит следующим образом:
к охлажденному до примерно -25оС раствору, содержащему 12,77 г 2-фенилпиперазина (RS) в 220 см3 трихлорметана, прибавляют по капле за 25 мин при перемешивании раствор, содержащий 19,89 г хлорида 2-трифторметил-2-метоксифенил- уксусной кислоты (R), изомер (-), в 80 см3 того же растворителя, поддерживая температуру между -30 и -25оС. После 15-минутного перемешивания в этих условиях дают температуре подняться до примерно 0оС, добавляют между 0 и 5оС 50 см3 2 н. водного раствора щелочи натрия, дают температуре подняться до примерно 20оС и добавляют 150 см3 воды. Органическую фазу отделяют, а водную фазу снова экстрагируют 2 раза 200 см3 трихлорметана. Объединенные органические вытяжки промывают один раз 200 см3 0,5 н. раствора щелочи натрия, 4 раза 200 см3 воды, сушат на сульфате магния, фильтруют и концентрируют досуха при пониженном давлении (20 кПа) и при примерно 40оС. Остаток (29,8 г), содержащий 2 диастереоизомера А и В, переводят в раствор в 500 см3 дихлорметана. Эту смесь разделяют методом высокоэффективной жидкостной хроматографии на 2 колонках с диаметром 57 мм и длиной 300 мм, каждая из которых содержит 300 г диоксида кремния (55-105 меш), путем введения 5 проб по 100 см3. Элюирование осуществляется смесью дихлорметана с 2% этанола. Диастереоизомера А элюируют 1 л этой смеси во фракции, заключенной между 2,5 и 3,5 л. Диастереизомер В элюируют 2 л той же смеси во фракции, заключенной между 4 и 6 л. После концентрирования при пониженном давлении (20 кПа) и при примерно 50оС каждой из двух фракций получают соответственно 13,62 г соединения А в виде бесцветного твердого вещества, плавящегося при 102оС, и 14 г соединения В того же вида, плавящегося при 130оС. 13,62 г соединения А извлекают посредством смеси 140 см3 48%-ного водного раствора бромоводородной кислоты и 26 см3 уксусной кислоты и нагревают в течение 30 ч при температуре, близкой к 110оС. Реакционную смесь концентрируют досуха при пониженном давлении (20 кПа) и при примерно 80оС. Полученный остаток извлекают посредством 200 см3 этилацетата. Полученную суспензию охлаждают примерно до 10оС и смешивают между 10 и 20оС со 160 см3 30%-ного водного раствора щелочи натрия. Органическую фазу отделяют, а водная фаза снова экстрагируют 3 раза 100 см3 этилацетата. Объединенные органические вытяжки промывают один раз 80 см3 4 н. водного раствора щелочи натрия, 3 раза 80 см3 30%-ного водного раствора хлорида натрия и концентрируют досуха при пониженном давлении. После извлечения полученного остатка посредством 80 см3 10%-ного водного раствора метансульфокислоты и экстрагирования 3 раза 100 см3 этилацетата водную фазу смешивают со 120 см3 30%-ного водного раствора щелочи натрия и экстрагируют 4 раза 150 см3 этилацетата. Объединенные органические вытяжки промывают 3 раза 80 см3 30%-ного водного раствора хлорида натрия, сушат на сульфате магния, фильтруют и концентрируют при пониженном давлении (20 кПа) и при температуре, близкой к 50оС. Полученный остаток извлекают посредством 30 см3 изопропилового эфира, центрифугируют и промывают 10 см3 того же растворителя. Получают 2,45 г 2-фенилпиперазина, изомер (+) в виде бесцветного твердого вещества, плавящегося при 114оС.
/α/D20 +35,4о ± 0,5 (с 2; этанол).
14 г соединения В, обработанных при тех же условиях, что и соединение А, приводят к получению 3,08 г 2-фенилпиперазина, изомер (-), в виде бесцветного твердого вещества, плавящегося при 114оС.
/α/D20 -37,5о ± 0,5 (с 2; этанол).
П р и м е р 23. 7-Фтор-1-метил-4-оксо-8-(3-фенил-1-пиперазинил)-1,4-дигидро-1,8- бензо(b)нафтиридин-3-карбоновая кислота (+) была получена тем же способом, что и 7-фтор-1-метил-4-оксо-8-(3-фенил-1-пипера- зинил)-1,4-дигидро-1,8- бензо(b)нафтиридин-3-карбоновая кислота (RS) в примере 19, исходя из 4,4 г 3-этоксикарбонил-7,8-дифтор-1-метил-4-оксо-1,4-дигидро-1,8- бензо(b)нафтиридина, 2,7 г 2-фенилпиперазина (-) /α/D20 -37о ±0,5 (с этанол) и 1,48 г карбоната натрия получают 6,3 г твердого вещества желтого цвета, плавящегося при 226оС. После кислотного гидролиза в тех же условиях, которые описаны в примере 12 для 3-этоксикарбонил-7-фтор-1-метил-4-оксо-8-(3-фенил-1-пиперазинил)- 1,4-дигидро-1,8-бензо(b)нафтиридина (RS), получают 4,26 г 7-фтор-1-метил-4-оксо-8-(3-фенил-1-пиперазинил)-1,4-дигидро-1,8- бензо(b)нафтиридин(3-карбоновой кислоты (RS), изомер (+), в виде твердого вещества желтого цвета, плавящегося при 276-278оС.
/α/D20 +40,6о ± 1 (с 1; уксусная кислота).
П р и м е р 24. Суспензию, состоящую из 2 г 3-этоксикарбонил-7,9- дифтор-1-метил-4-оксо-8-(3-фенил-1(пиперазинил)-1,4-дигид-ро-1,8-бензо (b)нафтиридина (RS) в 25 см3 этанола и 15 см3 1 н. водного раствора щелочи калия, нагревают при перемешивании в течение 2 ч при температуре, близкой к 75оС. Полученный раствор при примерно 75оС смешивают с 9 г 10%-ного водного раствора уксусной кислоты. Полученную часть центрифугируют при температуре, близкой к 75оС, и промывают 3 раза 30 см3 воды при примерно 20оС. После одной перекристаллизации в 40 см4 диметилформамида получают 1,5 г 7,9- дифтор-1-метил-4-оксо-8-(3-фенил-1-пиперазинил)-1,4-дигидро-1,8- бензо(b)нафтиридин-3-карбоновой кислоты (RS) в виде твердого вещества оранжевого цвета, плавящегося при 298оС. 3-Этоксикарбонил-7,9-дифтор-1-метил-4-оксо-8-(3-фенил-1-пиперазинил) 1,4-дигидро-1,8-бензо(b)нафтиридин (RS) приготавливается следующим образом:
суспензию, состоящую из 1,8 г 3-этоксикарбонил-7,8,9-трифтор-1- метил-4-оксо-1,4-дигидро-1,8-бензо-(b)нафтиридина и 3,2 г 2-фенилпиперазина (RS) в 30 см3 диметилсульфоксида, нагревают при температуре, близкой к 100оС, и при перемешивании в течение 1,5 ч. Полученный раствор при 100оС приливают при перемешивании к смеси, содержащий 150 см3 воды и 50 г льда. Полученную суспензию экстрагируют 3 раза 40 см3 трихлорметана при 20оС. Объединенные органические вытяжки промывают 2 раза 50 см3 воды,сушат на сульфате магния, фильтруют и концентрируют досуха при пониженном давлении (20 кПа) и при 50оС. Полученное вещество перекристаллизовывают из смеси 40 см3 диметилформамида и 40 см4 этанола. Получают 2 г 3-этоксикарбонил-7,9-дифтор-1-метил-4-оксо-8-(3-фенил-1-пиперазинил)- 1,4-дигидро-1,8-бензо(b)нафтиридина-(RS) в виде твердого вещества желтого цвета, плавящегося при 216оС.
3-Этоксикарбонил-7,8,9-трифтор-1-метил-4-оксо-1,4-дигидро-1,8- бензо(b)нафтиридин приготавливается следующим способом
при перемешивании в суспензию, состоящую из 19,3 г 2-(2-хлор-6,7,8-трифтор-3-хинолинкарбонил)3-диметиламиноэтилакрилата в 150 см3 этанола, поддерживая температуру, близкую к 50оС, прибавляют за 10 мин между 5 и 10оС раствор, содержащий при примерно 5оС 10 г метиламина в 50 см3 этанола, перемешивают 1 ч между 5 и 10оС и дают температуре подняться до примерно 20оС. Полученный раствор смешивают с 7,6 г ДБУ и нагревают в течение 1 ч при примерно 30оС. После охлаждения до температуры, близкой к 20оС, продукт центрифугируют, промывают 2 раза 100 см3 этанола и 2 раза 100 см3 изопропилового эфира. Получают 13,4 г 3-этоксикарбонил-7,8,9-трифтор-1-метил-4-оксо-1,4-дигидро-1,8-бензо (b)нафтиридина в виде твердого вещества желтого цвета, плавящегося при 320оС, которое применяется без дополнительной очистки в последующих стадиях. этанола и 15 см3 1 н. водного раствора щелочи калия, нагревают при перемешивании в течение 2 ч при температуре, близкой к 75оС. Полученный раствор при примерно 75оС смешивают с 9 г 10%-ного водного раствора уксусной кислоты. Полученную часть центрифугируют при температуре, близкой к 75оС, и промывают 3 раза 30 см3 воды при примерно 20оС. После одной перекристаллизации в 40 см4 диметилформамида получают 1,5 г 7,9- дифтор-1-метил-4-оксо-8-(3-фенил-1-пиперазинил)- 1,4-дигидро-1,8- бензо(b)нафтиридин-3-карбоновой кислоты (RS) в виде твердого вещества оранжевого цвета, плавящегося при 298оС.
3-Этоксикарбонил-7,9-дифтор-1-метил- -4-оксо-8-(3-фенил-1-пиперазинил) 1,4-дигидро-1,8-бензо(b)нафтиридин (RS) приготавливается следующим образом:
суспензию, состоящую из 1,8 г 3-этоксикарбонил-7,8,9-трифтор-1- метил-4-оксо-1,4-дигидро-1,8-бензо-(b)нафтиридина и 3,2 г 2-фенилпиперазина (RS) в 30 см3 диметилсульфоксида, нагревают при температуре, близкой к 100оС, и при перемешивании в течение 1,5 ч. Полученный раствор при 100оС приливают при перемешивании к смеси, содержащей 150 см3 воды и 50 г льда. Полученную суспензию экстрагируют 3 раза 40 см3 трихлорметана при 20оС. Объединенные органические вытяжки промывают 2 раза 50 см3 воды,сушат на сульфате магния, фильтруют и концентрируют досуха при пониженном давлении (20 кПа) и при 50оС. Полученное вещество перекристаллизовывают из смеси 40 см3 диметилформамида и 40 см4 этанола. Получают 2 г 3-этоксикарбонил-7,9-дифтор-1-метил-4-оксо-8-(3-фенил-1-пиперазинил)- 1,4-дигидро-1,8-бензо(b)нафтиридина-(RS) в виде твердого вещества желтого цвета, плавящегося при 216оС.
3-Этоксикарбонил-7,8,9-трифтор-1-ме-тил-4-оксо-1,4-дигидро-1,8- бензо(b)нафтиридин приготавливается следующим способом:
при перемешивании в суспензию, состоящую из 19,3 г 2-(2-хлор-6,7,8-трифтор-3-хинолинкарбонил)3-диметиламиноэтилак-рилата в 150 см3 этанола, поддерживая температуру, близкую к 50оС, прибавляют за 10 мин между 5 и 10оС раствор, содержащий при примерно 5оС 10 г метиламина в 50 см3 этанола, перемешивают 1 ч между 5 и 10оС и дают температуре подняться до примерно 20оС. Полученный раствор смешивают с 7,6 г ДБУ и нагревают в течение 1 ч при примерно 30оС. После охлаждения до температуры, близкой к 20оС, продукт центрифугируют, промывают 2 раза 100 см3 этанола и 2 раза 100 см3 изопропилового эфира. Получают 13,4 г 3-этоксикарбонил-7,8,9-трифтор-1-метил-4-оксо-1,4-дигидро-1,8-бензо (b)нафтиридина в виде твердого вещества желтого цвета, плавящегося при 320оС, которое применяется без дополнительной очистки в последующих стадиях. 2-(2-Хлор-6,7,8-трифтор-3-хинолинкар-бонил)-3-диметиламиноэтилакрилат может быть приготовлен следующим способом:
суспензию, состоящую из 26,7 г 3-(2-хлор-6,7,8-трифтор-3-хинолин)-3-оксо-этил- пропионата в 270 см3 этилацетата и 32 см3 N,N диметилформамиддиметилацеталя, нагревают при температуре, близкой к 75оС, и при перемешивании в течение 2 ч.
Реакционную смесь концентрируют досуха при пониженном давлении (20 кПа) и при примерно 50оС. Сухой остаток извлекают посредством 175 см3 изопропилового эфира, центрифугируют, промывают 2 раза 35 см3 того же растворителя. Получают 19,32 г 2-(2-хлор-6,7,8-трифтор-3-хинолинкарбонил)-3-диметиламиноэтилакрилата в виде твердого вещества оранжевого цвета, плавящегося при 118оС, которое применяется без дополнительной очистки в последующих стадиях.
3-(2-Хлор-6,7,8-трифтор-3-хинолин)-3- оксоэтилпропионат приготавливается следующим способом:
суспензию, состоящую из 46,3 г 2-хлор-6,7,8-трифторхинолин-3-карбоновой кислоты в 640 см3 трихлорметана и 84 см3 хлорида тионила, нагревают при перемешивании в течение 6 ч при температуре, близкой к 60оС. Полученный раствор концентрируют досуха при пониженном давлении (20 кПа) и при примерно 50оС. Полученный сухой остаток извлекают посредством 140 см3 петролейного эфира (40-60), центрифугируют, промывают 2 раза 60 см3 того же растворителя. 47,61 г полученного твердого вещества желтого цвета переводят в раствор в 400 см3 тетрагидрофурана. Этот раствор по капле вводят при перемешивании за 1,5 ч между 5 и 10оС в 250 см3 раствора магниевого хелата моноэтилмалоната в тетрагидрофуране, который был приготовлен в условиях примера 19. Дают температуре подняться до примерно 20оС и перемешивают еще 2 ч в этих условиях. Полученный раствор вводится по капле при сильном перемешивании за 1 ч при температуре, близкой к 5оС, в 1750 см3 0,5 н. серной кислоты. Перемешивают еще 2 ч при этой температуре, экстрагируют при примерно 5оС 3 раза 600 см3 этилового эфира. Объединенные органические фазы промывают 3 раза 500 см3 воды, сушатся на сульфате магния и концентрируют при пониженном давлении (20 кПа) и при температуре, близкой к 30оС. Сухой остаток извлекают посредством смеси 135 см3 изопропилового эфира и 15 см3 н-гексана, центрифугируют при 5оС, промывают 2 раза 115 см3 той же смеси при той же температуре. Получают 47,4 г 3-(хлор-6,7,8-трифтор-3-хинолин)-3-оксоэтилпропионата в виде твердого вещества бежевого цвета, плавящегося при 78-80оС, которое используется без дополнительной очистки в последующих стадиях.
2-Хлор-6,7,8-трифтор-хинолин-3-карбо- новая кислота приготавливается следующим способом:
при перемешивании к охлажденной до примерно 10оС суспензии, состоящей из 45,7 г 2-хлор-6,7,8-трифтор-3-фтормил-1,4-дигидро-хинолина в 585 см3 1 н. щелочи калия прибавляют за 1 ч, поддерживая температуру между 10 и 14оС, раствор, содержащий 69,65 г перманганата калия в 730 см3 воды. Перемешивают еще 30 мин при примерно 10оС. Добавляют 12 г дитионата натрия, перемешивают 10 мин при температуре, близкой к 10оС, фильтруют через диатомированный кремнезем и промывают 3 раза 400 см3 воды. Фильтрат и промывочные воды объединяют и добавляют к 70 см3 35%-ного водного раствора хлороводородной кислоты. Образованный осадок экстрагируют 3 раза 500 см3 этилацетата. Объединенные органические вытяжки сушат на сульфате магния, фильтруют и концентрируют при пониженном давлении (20 кПа) и при 50оС. Остаток извлекают посредством смеси 100 см3 этилового эфира и 100 см3 изопропилового эфира, центрифугируют, промывают 100 см3 той же смеси. Получают 46,43 г 2-хлор-6,7,8- трифтор-хино- лин-3-карбоновой кислоты в виде бесцветного твердого вещества, разлагающегося при 225-230оС, которое используется без дополнительной очистки в последующих стадиях.
2-Хлор-6,7,8-трифтор-3-формил-1,4-ди-гидрохинолин приготавливается следующим способом:
к смеси 525 см3 трихлорметана и 49 см3 диметилформамида прибавляют за 40 мин при перемешивании между 5 и 10оС 50 см3 хлорида фосфорила, перемешивают 15 мин при этой температуре и дают температуре подняться до примерно 20оС. К полученному раствору прибавляют за 20 мин при примерно 20оС при сильном перемешивании постепенно 46,8 г 6,7,8-трифтор-3,4-дигидрокарбостирила. Перемешивают 30 мин при температуре, близкой к 20оС, нагревают до примерно 60оС и выдерживают при этой же температуре в течение 2,5 часов. Реакционную смесь концентрируют при пониженном давлении (20 кПа) и при примерно 50оС. Маслянистый остаток приливают при сильном перемешивании к 500 г льда. Добавляют небольшими порциями за 30 мин 100 г ацетата натрия. Полученную суспензию приливают за 15 мин при сильном перемешивании к 1 л воды, предварительно нагретой до примерно 90оС, и перемешивают еще 15 мин при той же температуре. Нерастворимую часть центрифугируют при примерно 90оС и промывают 3 раза 250 см3 воды. Получают 47,7 г 2-хлор-6,7,8-трифтор-3-формил-1,4-дигидрохинолина в виде бесцветного твердого вещества, разлагающегося при 220оС.
6,7,8-Трифтор-3,4-дигидрокарбостирил готовится следующим способом:
24,35 г 6,7,8-трифторкарбостирила в виде суспензии в смеси 450 см3 этанола и 150 см3 диметилформамида гидрируют при перемешивании и при примерно 50оС в присутствии 5 г никеля Ренея при давлении в 1 атм до прекращения абсорбции водорода. Никель Ренея, используемый с качеством W-2, предварительно промывают 50 см3 2%-ного водного раствора уксусной кислоты, 2 раза 50 см3 и 3 раза 50 см3 этанола. Реакционную смесь смешивают с 250 см3 диметилформамида, фильтруют при примерно 50оС через диатомированный кремнезем. Фильтрат концентрируют при пониженном давлении (20 кПа) и при примерно 70оС. Сухой остаток извлекают посредством 150 см3 воды, центрифугируют и промывают 2 раза 50 см3 воды. Получают 23,6 г 6,7,8-трифтор-3,4-дигидрокарбростирила в виде твердого вещества светло-бежевого цвета, плавящегося при 217оС, которое используется без дополнительной очистки в последующих стадиях.
6,7,8-Трифторкарбостирил готовится следующим способом:
суспензию, состоящую из 60,83 г 4-хлор-6,7,8-трифторкарбостирила в 520 см3 уксусной кислоты и 38,15 см3 триэтиламина, гидрируют при давлении в 1 атм, в присутствии 5,25 г палладия на угле (10%-ного) до прекращения поглощения водорода при температуре, близкой к 25оС. Реакционную смесь затем нагревают до примерно 40оС, фильтруют при той же температуре через диатомированный кремнезем для фильтрования. Фильтрат концентрируют при пониженном давлении (20 кПа) и при температуре, близкой к 50оС. Сухой остаток извлекают посредством 400 см3 воды. Нерастворимую часть центрифугируют, промывают 4 раза 170 см3 воды, 2 раза 110 см3 этанола и 2 раза 110 см3 изопопилового эфира. Получают 48,35 г 6,7,8-трифторкарбостирила в виде бесцветного твердого вещества, возгоняющегося при 288оС, которое используется без дополнительной очистки в последующих стадиях.
4-Хлор-6,7,8-трифторкарбостирил готовится следующим способом:
суспензию, состоящую из 70,4 г 4-хлор-2-этокси-6,7,8-трифторхинолина в 170 см3 35%-ного водного раствора хлороводородной кислоты, 420 см3 уксусной кислоты и 250 см3 воды, нагревают при перемешивании и при температуре, близкой к 100оС, в течение 2,5 ч. После охлаждения до примерно 20оС реакционную смесь приливают к 1100 см3 воды при примерно 5оС, перемешивают 15 мин при той же температуре, затем нерастворимую часть центрифугируют и промывают 3 раза 220 см3 воды. Получают 61 г 4-хлор-6,7,8-трифторкарбостирила в виде твердого вещества бежевого цвета, плавящегося при 213оС, которое используется без дополнительной очистки в последующих стадиях.
4-Хлор-2-этокси-6,7,8-трифторхинолин готовится следующим способом:
суспензию, состоящую из 69,5 г 2-этокси-6,7,8-трифтор-4-гидроксихинолина в 430 см3 хлорида фосфорила, нагревают при перемешивании и при температуре, близкой к 100оС, в течение 30 мин. Полученный раствор концентрируют при пониженном давлении (20 кПа) и при 60оС до объема 100 см3. Остаток извлекают посредством 750 см3 этилацетата. Полученный раствор приливают при перемешивании за 10 мин к смеси 400 см3 воды и 200 г льда и перемешивают при этих же условиях в течение 30 мин. После отделения органической вытяжки водную фазу снова экстрагируют 2 раза 250 см3 этилацетата. Объединенные органические вытяжки промывают 2 раза 250 см3 воды, сушат на сульфате магния и концентрируют при пониженном давлении (20 кПа) и при 40оС. Полученный маслянистый остаток извлекают посредством 370 см3 петролейного эфира (40-60). После фильтрования через диатомированный кремнезем фильтрат концентрируют досуха при пониженном давлении (20 кПа) и при примерно 30оС. Получают 70,7 г 4-хлор-2-этокси-6,7,8-трифторхинолина в виде твердого вещества бежевого цвета, плавящегося при 45оС, которое используется без дополнительной очистки в последующих стадиях.
2-Этокси-6,7,8-трифтор-4-гидроксихи-нолин может быть приготовлен следующим образом:
раствор, содержащий 122 г 2,3,4-трифтор-N-[(1'-этокси-2'-этоксикарбонил)-этили- ден]-анилина в 120 см2 оксида фенила, вводят по каплям за 25 мин при перемешивании в 600 см3 оксида фенила при температуре, близкой к 250оС, постоянно удаляя образующийся этанол методом дистилляции. После перемешивания в течение 15 мин при той же температуре раствор охлаждают до 20оС и смешивают с 750 см3 н-гексана. Образованный осадок центрифугируют и промывают 3 раза 200 см3 н-гексана. Получают 69,5 г 2-этокси-6,7,8-трифтор-4-гидроксихинолина в виде твердого вещества бежевого цвета, плавящегося при 171оС, которое используется без дополнительной очистки в последующих стадиях.
2,3,4-Трифтор-N-[(1'-этокси-2'-этокси- карбонил)этилиден] -анилин может быть приготовлен следующим способом
к раствору, содержащему 90 г хлоргидрата 2-этоксикарбонил-1-этоксиэтилиденамина в 820 см3 этанола, прибавляют за один раз при перемешивании 58,8 г 2,3,4-трифторанилина. После 48-часового перемешивания при температуре, близкой к 20оС, полученную суспензию фильтруют и фильтрат концентрируют при пониженном давлении (20 кПа) и при температуре, близкой к 50оС.
Маслянистый остаток извлекают посредством 250 см3 воды. Полученную смесь экстрагируют 3 раза 200 см3 этилового эфира. Объединенные органические вытяжки промывают 4 раза 150 см3 воды, сушат на сульфате магния и концентрируют при пониженном давлении (20 кПа) и при 30оС. Получают 122 г 2,3,4-трифтор-N-[(1'-этокси- 2'-этоксикарбонил)-этилиден] -аналина в виде жидкого масла желтого цвета, которое используется без дополнительной очистки в последующих стадиях.
Хлоргидрат 2-этоксикарбонил-1-этоксиэтилиденамина был приготовлен согласно методу, описанному в работе A.Pinner et coll. Ber Dtsch.Chem.Ges. 28, 478 (1895).
П р и м е р 25. Проводя реакцию в условиях примера 39, но исходя из 0,978 г 3-этоксикарбонил-7,9-дифтор-1-метил-оксо-8-(3-фенил-1-пиперазинил)- 1,4-дигидро-1,8-бензо(b)нафтиридина-(R) получают после одной перекристаллизации в 25 см3 диметилформамида 0,540 г 7,9-дифтор-1-метил-4-оксо-8-(3-фенил-1-пиперазинил)-1,4-дигидро- 1,8-бензо(b)нафтиридин-3-карбоновой кислоты (R) в виде твердого вещества желтого цвета, разлагающегося при 270-272оС.
/α/D20 +113о ± 5 (с 2; трихлорметан).
Проводя реакцию в условиях примера 30, но исходя из 1,21 г 3-этоксикарбонил-7,8,9-трифтор-1-метил-4-оксо-1,4-дигидро-1,8- бензо(b)нафтиридина и 0,7 г 2-фенилпиперазина (R) после перекристаллизации в 22 см3 этанола с 30% диметилформамида получают 1,02 г 3-этоксикарбонил-7,9-дифтор-1-метил-4-оксо-8-(3-фенил-1-пиперазинил)- 1,4-дигидро-1,8-бензо(b)нафтиридина в виде твердого вещества желтого цвета, плавящегося при 216оС.
/α/D20 +98о ± 2 (с 0,5; уксусная кислота).
П р и м е р 26. Проводя реакцию в условиях примера 25, но исходя из 1,33 г 3-этоксикарбонил-7,9-дифтор-1-метил-4-ок- со-8-(4-фенил-1-пиперазинил)- 1,4-дигидро-1,8-бензо(b)нафтиридина-(s) получают 0,779 г 7,9-дифтор-1-метил-4-оксо-8-(3-фенил-1-пиперазинил)-1,4-дигидро-1,8- бензо(b)нафтиридин-3-карбоновой кислоты (s) в виде твердого вещества желтого цвета, разлагающегося при 270-272оС.
/α/D20 +118о ± 5 (с 0,2; трихлорметан).
Проводя реакцию в условиях примера 25, но исходя из 1,38 г 3-этоксикарбонил-7,8,9-трифтор-1-метил-4-оксо-1,4-дигидро-бензо(b) нафтиридина-1,8 и 0,8 г 2-фенилпиперазина (s), получают 1,02 г 3-этоксикарбонил-7,9-дифтор-1-метил-4-оксо-8-(3-фенил)-1-пиперазинил)- 1,4-дигидро-1,8-бензо(b)нафтиридина-(s) в виде твердого вещества желтого цвета, плавящегося при 216оС.
/α/D20 -99о ± 2 (с 0,5; уксусная кислота).
П р и м е р 27. 1-Циклопропил-7,9-дифтор-4-оксо-8-(3-фенил-1-пиперазинил)-1,4-дигидро- 1,8-бензо(b)нафтиридин-3-карбоновую кислоту (RS) получают в условиях примера 24, но исходя из 1,8 г 1-циклопропил-3-этоксикарбонил-7,9-ди- фтор-4-оксо-8-(3-фенил-1- пиперазинил)-1,4-дигидро-1,8-бензо(b)нафтиридина-(RS). После перекристаллизации из смеси 30 см3 диметилформамида и 20 см3 этанола получают 1,2 г 1-циклопропил-7,9-дифтор-4-оксо-8-(3-фенил-1-пиперазинил)-1,4-дигидро- 1,8-бензо(b)нафтиридин-3-карбоновой кислоты (RS) в виде твердого вещества желтого цвета, плавящегося при 274оС.
1-Циклопропил-3-этоксикарбонил-7,9-дифтор-4-оксо-8-(3-фенил-1- пиперазинил)-1,4-дигидро-1,8-бензо(b)нафтиридин-(RS) получают в условиях примера 19, но исходя из 1,8 г 1-циклопропил-3-этоксикарбонил-7,8,9-трифтор-4-оксо-1,4-дигидро-1,8- бензо(b)нафтиридина и 3,2 г 2-фенилпипе- разина (RS). После перекристаллизации из смеси 5 см3 диметилформамида и 40 см3 этанола получают 1,8 г 1-циклопропил-3-этоксикарбонил-7,9-дифтор-4-оксо-8-(3-фе- нил-1- пиперазинил)-1,4-дигидро-1,8-бензо(b)нафтиридина-(RS) в виде твердого вещества желтого цвета, плавящегося при 242оС.
1-Циклопропил-3-этоксикарбонил-7,8,9-трифтор-4-оксо-1,4-дигидро- 1,8-бензо(b)нафтиридин может быть получен в следующих условиях:
в раствор, содержащий 7 г 2-(2-хлор-6,7,8-трифтор-3-хинолинкарбонил)-3-диме-тиламиноэтилакрилата в 100 см3 трихлорметана, выдерживаемый при температуре, близкой к 20оС, прибавляют за 5 мин 4,12 г циклопропиламина и перемешивают еще 4 ч при той же температуре. Реакционную смесь концентрируют при пониженном давлении (20 кПа) и при 50оС. Полученный маслянистый остаток извлекают посредством 100 см3 этанола и 3 г ДБУ. Смесь нагревают до 80оС и при перемешивании выдерживают при этой температуре в течение 1,5 ч. После охлаждения до примерно 20оС нерастворимую часть центрифугируют, промывают 2 раза 30 см3 этанола и 2 раза 30 см3 изопропилового эфира. Получают 4,5 г 1-циклопропил-3-этокси-(карбонил-7,8,9- три- фтор-4-оксо-1,4-дигидро-1,8-бензо(b)нафти- ридина в виде бесцветного твердого вещества, плавящегося при 260оС.
П р и м е р а 28. Проводя реакцию в условиях примера 39, но исходя из 2,4 г 3-этоксикарбонил-7,9-дифтор-1-(2-фторэтил) -4-оксо-8-(3-фенил-1- пиперазинил)-1,4-дигидро-1,8-бензо(b)нафтиридина (RS) получают 1,6 г 7,9-дифтор-1-(2-фторэтил)- 4-оксо-8-(3-фенил-1-пиперазинил)- 1,4-дигидро-1,8-бензо(b)нафтиридин-3 карбоновой кислоты (RS) в виде твердого вещества желтого цвета, разлагающегося при 307-310оС.
3-Этоксикарбонил-7,9-дифтор-1-(2-фтор- этил-4-оксо-8-)3-фенил-1- (пиперазинил)-1,4-дигидро-1,8-бензо(b)нафтиридин (RS) был приготовлен в следующих условиях:
суспензию, состоящую из 2,8 г 3-этоксикарбонил-1-(2-фторэтил)-7,8,9-трифтор-4- оксо-1,4-дигидро-1,8- бензо(b)нафтиридина, 1,7 г 2-фенилпиперазина (RS) и 1,1 г карбоната натрия в 40 см3 диметилсульфоксида, нагревают при перемешивании в течение 2 ч при температуре, близкой к 100оС. После охлаждения до примерно 20оС реакционную смесь приливают к 200 см3 воды и экстрагируют 3 раза 100 см3 трихлорметана. Объединенные органические вытяжки промывают 1 раз 50 см3 воды и концентрируют при пониженном давлении (20 кПа) и при температуре, близкой к 40оС. Маслянистый остаток извлекают посредством 100 см3 этанола и концентрируют снова при пониженном давлении в описанных выше условиях. Полученное твердое вещество перекристаллизовывают из 100 см3 этанола с 20% диметилформамида, центрифугируют и промывают 2 раза 20 см3 этанола. Получают 2,4 г целевого продукта в виде твердого вещества желтого цвета, плавящегося при 208оС.
3-Этоксикарбонил-1-(2-фторэтил-7,8,9-трифтор-4-оксо-1,4-дигидро- 1,8-бензо(b)нафтиридин был приготовлен в условиях, близких к примеру 3, но исходя из 3,9 г 2-(2-хлор-6,7,8-трифтор-3 хинолинкарбонил)-диметиламиноэтилакрилата, 3 г хлоргидрата 2-фторэтиламина, 4,2 см2 триэтиламина в 100 см3 этанола. После добавления 1,8 см3 ДБУ раствор нагревают 2 ч при 80оС. Получают в результате обработки реакционной смеси, как в примере 24, 2,8 г 3-этоксикарбонил-7,8,9-трифтор-1-(2-фторэтил)-4-оксо-1,4-дигидро- 1,8-бензо(b)нафтиридина в виде твердого вещества желтого цвета, плавящегося при 302-304оС.
2-(2-Хлор-6,7,8-трифтор-3-хинолинкар-бонил)-диметиламиноэтилакрилат может быть приготовлен, как описано в примере 24.
П р и м е р 29. Суспензию, состоящую из 2,65 г 8-(3-бензил-1- пиперазинил)-3-этоксикарбонил-7-фтор-1-метил-4-оксо-1,4-дигид- ро- 1,8-бензо(b)нафтиридина-(RS) в 14 см3 1 н. водной щелочи калия и 20 см3 этанола, нагревают при температуре, близкой к 80оС, в течение 20 мин, смешивают при этой температуре с 32 см3 5%-ной уксусной кислотой и перемешивают в течение 20 мин. Нерастворимую часть центрифугируют, промывают 2 раза 20 см3 воды, 2 раза 20 см3 этанола и 2 раза 20 см3 этилового эфира. После двух перекристаллизаций в 50 см3 (каждый раз) диметилформамида получают 2,05 г 8-(3-бензил-1-пиперазинил)-7-фтор-1-метил-4-оксо- 1,4-дигидро-1,8-бензо(b)нафтиридин-3-карбоновой кислоты (RS) в виде твердого вещества желтого цвета, разлагающегося при 270-275оС.
8-(3-Бензил-1-пиперазинил)-3-этокси- карбонил-7-фтор-1-метил-4-оксо- 1,4-дигидро-1,8-бензо(b)нафтиридин-(RS) готовится следующим способом:
суспензию, состоящую из 1,3 г 3-этоксикарбонил-7,8-дифтор-1-метил-4-оксо-1,4- дигидро-1,8-бензо(b) нафтиридина и 1,3 г 2-бензилпиперазина (RS) в 25 см3 диметилсульфоксида, нагревают при примерно 90оС и при перемешивании в течение 1 ч 45 мин. После охлаждения до температуры,близкой к 20оС, реакционную смесь добавляют к 100 см3 воды, экстрагируют 3 раза 30 см3 трихлорметана. Объединенные органические вытяжки промывают 3 раза 30 см3 воды, сушат на сульфате магния, фильтруют и концентрируют досуха при примерно 50оС при пониженном давлении (20 кПа). После одной перекристаллизации из смеси 4 см3 диметилформамида и 1 см3 этанола получают 1,6 г 8-(3-бензил-1-пиперазинил)-3-этоксикарбонил-7-фтор-1-метил-4-оксо-1,4- дигидро-1,8-бензо(b)нафтиридина-(RS) в виде твердого вещества желтого цвета, плавящегося при 190оС.
2-Бензилпиперазин (RS) может быть приготовлен гидрированием 2-бензилпиразина, полученного согласно методу, описанному в работе 26, 3379 (1961).
Раствор, содержащий 8 г 2-бензилпиперазина в 60 см3 уксусной кислоты, смешивают с 0,8 г диоксида платины и гидрируют при давлении в 1 атм. при примерно 20оС. После окончания поглощения водорода катализатор удаляют фильтрованием через диатомированный кремнезем. Раствор концентрируют досуха при пониженном давлении (20 кПа) и при примерно 60оС. Сухой остаток переводят в суспензию в 40 см3 этанола, смешивают при примерно 20оС с раствором этилата натрия, приготовленным исходя из 1,49 г натрия в 40 см3 этанола. После 2 ч перемешивания при примерно 20оС суспензию концентрируют досуха при пониженном давлении (20 кПа) и при 30оС. Сухой остаток извлекают посредством 60 см3 этилового эфира. Нерастворимую часть удаляют фильтрованием через диатомированный кремнезем. Фильтрат концентрируют досуха при пониженном давлении в условиях, аналогичных предыдущим. Сухой остаток извлекают посредством 20 см3 изопропилового эфира, центрифугируют и промывают 2 раза 5 см3 того же растворителя. Получают 5,32 г 2-бензилпиперазина (RS) в виде твердого вещества светло-бежевого цвета, плавящегося при 90оС. нафтипиридина и 1,08 г 2-(2-фторфенил)-пиперазина (RS). После перекристаллизации в 60 см3 диметилформамида с 50% этанола получают 1,3 г ожидаемого продукта в виде твердого вещества желтого цвета, плавящегося при 228оС.
П р и м е р 31. 7,9-Дифтор-8-[3-(4-фторфенил)-1-пиперазинил]-1-метил-4-оксо-1,4- дигидро-1,8-бензо(b)нафтипиридин-3-кар-боновая кислота (RS) была приготовлена в условиях примера 24, но исходя из 1,5 г 3-этоксикарбонил-7,9-дифтор-8-[3-(4-фтор- фенил)-1-пиперазинил] -1- (метил-4-оксо-1,4-дигидро-1,8-бензо(b)нафтипиридина-(RS). После одной перекристаллизации из 30 см3 диметилформамида получают 1 г 7,9-дифтор-8-[3-(4-фторфенил)-1-пиперазинил] -1- метил-4-оксо-1,4- дигидро-1,8-бензо(b)нафтипиридин-3-карбоновой кислоты (RS) в виде твердого вещества желтого цвета, плавящегося при 266оС. 3-Этоксикарбонил-7,9-дифтор-3-[3-(4-фторфенил)-1-пиперазинил)-1-метил- 4-оксо-1,4-дигидро-1,8-бензо(b)нафтипиридин-(RS) был приготовлен в условиях примера 39, но исходя из 2 г 3-этокси-карбонил-7,8,9-трифтор-1-метил-4-оксо-1,4-дигидро-1,8- бензо(b)нафтипиридина и 4 г 2-(4-фторфенил)-пиперазина (RS). После одной перекристаллизации из смеси 3,5 см3 диметилформамида и 31,5 см3 этанола получают 2,1 г 3-этокси(карбонил)-7,9-дифтор-8-[3-(4-фторфенил)-1-пиперазинил]-1-метил- 4-оксо-1,4-дигидро-1,8-бензо(b)нафтипири- дина (RS) в виде твердого вещества бежевого цвета, плавящегося при 242оС.
П р и м е р 32. 1-Этил-7-фтор-8-[8-(4-метоксифенил)-1-пиперазинил]-4-оксо-1,4- дигидро-1,8-бензо(b)нафтипиридин-3-карбоно- вая кислота (RS) была приготовлена в условиях, описанных ниже в примере 33, но исходя из 1,5 г 3-этоксикарбонил-1-этил-7- фтор-8-[3-(4-метоксифенил)-1- пиперазинил)-4-оксо-1,4-дигидро-1,8-бензо(b)нафти- пиридина (RS). После перекристаллизации в 30 см3 диметилформамида получают 0,85 г 1-этил-7-фтор-[3-(4-метоксифенил)-1-пипе- разинил] -4-оксо-1,4-дигидро- 1,8-бензо(b)нафтипиридин-3-карбоновой кислоты (RS) в виде твердого вещества желтого цвета, плавящегося при 270оС.
3-Этоксикарбонил-1-этил-7-фтор-8-[3- (4-метоксифенил)-1-пиперазинил] 4-оксо-1,4-дигидро-1,8-бензо(b)нафтипиридин-(RS) был получен в условиях примера 33, но исходя из 1,99 г 3-этоксикарбонил-1-этил-7,8-дифтор-4-оксо-1,4-дигидро-1,8-бензо(b) нафтипиридина и 2,3 г 2-(4-метоксифенил)-пиперазина (RS). Получают 2,2 г 3-этоксикарбонил-1-этил-7-фтор-8-(3-4-метоксифе-нил-1-пиперазинил)- 4-оксо-1,4-дигидро-1,8-бензо(b)нафтипиридина (RS) в виде твердого вещества бежевого цвета, плавящегося при 210-212оС.
3-Этоксикарбонил-1-этил-7,8-дифтор-4-оксо-1,4-дигидро-1,8-бензо(b) нафтипиридин был приготовлен в следующих условиях
в суспензию, состоящую из 20 г 2-(2-хлор-6,7-дифтор-3-хинолинкарбонил)-3-ди- метиламиноэтилакрилата в 200 см3 этанола при 2оС, прибавляют за 10 мин между 2 и 5оС при перемешивании раствор с температурой примерно 2оС, содержащий 14,6 г этиламина в 200 см3 этанола, перемешивают еще 40 мин между 2 и 5оС, затем дают температуре подняться до примерно 20оС за 2 ч. После 24-часового выдерживания при примерно 20оС нерастворимую часть центрифугируют, промывают 2 раза 30 см3 этанола и 2 раза 50 см3 изопропилового эфира. Получают 16,35 г 3-этоксикарбонил-1-этил-7,8-дифтор-4-оксо-1,4-дигидро-1,8-бензо(b) нафтипиридина в виде твердого вещества бежевого цвета, плавящегося при температуре 290оС.
П р и м е р 33. 1-Циклопропил-7-фтор-3-[3-(4-метоксифенил)-1-пиперазинил]-4-оксо- 1,4-дигидро-1,8-бензо(b)нафтипиридин-3- карбоновая кислота-(RS) была приготовлена в условиях примера 29, но исходя из 1,5 г 1-циклопропил-3-этоксикарбонил-7-фтор-8-[3-(4-метоксифенил)-1- пиперазинил]-4-оксо-1,4-дигидро-1,8-бензо(b)нафтипиридина-(RS) в 18,4 см3 1 н. водного раствора щелочи калия и 18,4 см3 этанола. После одной перекристаллизации из 50 см3 диметилформамида получают 1,5 г 1-циклопропил-7-фтор- 8-[3-(4-метоксифенил)-1- пиперазинил)-4-оксо-1,4-дигидро-1,8-бензо(b)нафтипиридин-3- карбоновой кислоты-(RS) в виде твердого вещества желтого цвета, разлагающегося при 287оС.
1-Циклопропил-3-этоксикарбонил-7- фтор-8-[3-(4-метоксифенил)-1- пиперазинил] -4-оксо-1,4-дигидро-1,8-бензо(b)нафти- пиридин-(RS) был приготовлен следующим способом:
суспензию, состоящую из 2 г 1-циклопропил-3-этоксикарбонил-7,8-дифтор-4-ок- со-1,4-дигидро-1,8- бензо(b)нафтипиридина и 2,3 г 3-(4-метоксифенил)-пиперазина (RS) в 20 см3 диметил-сульфоксида, нагревают при температуре, близкой к 90оС, в течение 1,5 ч. После охлаждения до примерно 20оС нерастворимую часть центрифугируют, промывают 3 раза 15 см3 воды и перекристаллизовывают из смеси 100 см3 этанола и 25 см3 диметилформамида. Получают 2,1 г 1-циклопропил-3-этоксикарбонил-7-фтор-8-[3-(4-метоксифенил)-1- пиперазинил] -4-оксо-1,4-дигидро-1,8-бензо(b)нафтипиридина- (RS) в виде твердого вещества желтого цвета, плавящегося при 199оС.
1-Циклопропил-3-этоксикарбонил-7,8-дифтор-4-оксо-1,4-дигидро-1,8- бензо(b)нафтипиридин был приготовлен следующим способом:
раствор, содержащий 20 г 2-(2-хлор-6,7-дифтор-3-хинолинкарбонил)- 3-диметиламиноэтилакрилата и 9,25 г циклопро- пиламина в 80 см3 трихлорметана, перемешивают при температуре, близкой к 20оС, в течение 2 ч. Раствор концентрируют при пониженном давлении (20 кПа) и при 40оС. Остаток извлекают посредством 300 см3 этанола, смешивают с 8,2 г ДБУ и нагревают при перемешивании и при примерно 75оС в течение 30 мин. После охлаждения до 20оС нерастворимую часть центрифугируют, промывают 2 раза 30 см3 этанола. Получают 11,1 г 1-циклопропил-3-этоксикарбонил-7,8-дифтор-4-оксо-1,4-дигидро-1,8- бензо(b)нафтипиридина в виде твердого вещества желтого цвета, плавящегося при температуре 230оС.
П р и м е р 34. 7,9-Дифтор-8-[3-(4-метоксифенил)-1-пиперазинил]-1-метил-4-оксо- 1,4-дигидро-1,8-бензо[b]нафтиридин-3-кар- боновая кислота (RS) была получена в условиях, близких к изложенным в примере 39, исходя из 2,8 г 3-этоксикарбонил-7,9-дифтор-8-[3-(4-метоксифенил)-1- пиперазинил]-1-метил-4-оксо-1,4-дигидро-1,8-бензо[b] наф- типиридина-(RS) в 24 см3 1 н. водного раствора щелочи калия и 25 см3 этанола. Получают 2 г 7,9-дифтор-8-[3-(4-метоксифенил)- 1-пиперазинил] -1-метил-4-оксо-1,4- дигидро-1,8-бензо[b]нафтипиридин-3-карбоно- вой кислоты-RS в виде твердого вещества желтого цвета, плавящегося при температуре 288-290оС.
3-Этоксикарбонил-7,9-дифтор-8-[3-(4-метоксифенил)-1-пиперазинил]-1- метил-4-оксо-1,4-дигидро-1,8-бензо[b]нафтипири- дин-(RS) был приготовлен в следующих условиях.
Cуспензию, состоящую из 2,5 г 3-этоксикарбонил-7,8,9-трифтор-1- метил-4-оксо-1,4-дигидро-1,8-бензо[b] нафтипиридина и 3,5 г 2-(4- метоксифенил)-пиперазина (RS) в 40 см3 диметилсульфоксида, нагревают при перемешивании и при температуре, близкой к 95оС, в течение 2 ч. После обработки реакционной смеси согласно условиям, описанным для примера 39, получают 2,8 г 3-этоксикарбонил-7,9-дифтор-8-[3-(4-метокси- фенил)-1-пиперазинил]-1- метил-4-оксо-1,4-дигидро-1,8-бензо[b] нафтипиридина-(RS) в виде твердого вещества желтого цвета, плавящегося при 209оС, которое используется без дополнительной очистки в последующих стадиях.
3-Этоксикарбонил-7,8,9-трифтор-1-ме-тил-4-оксо-1,4-дигидро-бензо[b] нафтиридин-1,8 может быть получен, как описано в примере 24.
2-(4-Метоксифенил)-пиперазин (RS) был приготовлен согласно методу, описанному в примере 12.
П р и м е р 35. Проводя реакцию в условиях примера 39, но исходя из 0,9 г 8-[3-(4-аминофенил)-1-пиперазинил] -3-этоксикар-бонил-7-фтор-1-метил- 4-оксо-1,4-дигидро-1,8-бензо[b] нафтипиридина-(RS), получают 0,7 г 8-[3-(4-аминофенил)-1-пиперазинил] -7-фтор-1-метил-4-оксо-1,4-дигидро- 1,8-бензо[b] нафтиридин-3-карбоновой кислоты (RS) в виде твердого вещества желтого цвета, разлагающегося при 315оС.
8-[3-(4-Аминофенил)-1-пиперазинил] -3-этоксикарбонил-7-фтор-1-метил- 4-оксо-1,4-дигидро-1,8-бензо[b] нафтиридин-(RS) был приготовлен в следующих условиях:
суспензию, состоящую из 0,7 г 3-этоксикарбонил-7-фтор-1-метил-8-[3-(4-нитрофе-нил)-1-пиперазинил] -4- оксо-1,4-дигидро-1,8-бензо[b]нафтипиридина-(RS) и 5 г никеля Ренея в 150 см3 этанола, гидрируют при давлении в 1 атм при температуре примерно 20оС в течение 7 ч, что позволяет поглотить теоретически необходимое количество водорода (100 см3 при 20оС и 1 атм). После добавления 50 см3 диметилформамида катализатор удаляют фильтрованием и фильтрат концентрируют досуха при пониженном давлении (20 кПа) и при 50оС. Полученный остаток (0,550 г) хроматографируют на 15 г диоксида кремния (230-400 меш) в суспензии, состоящей из хлороформа с 20% этанола, и элюируют 200 см3 той же смеси растворителей. После концентрирования досуха в условиях, аналогичным предыдущим, получают 0,3 г 8-[3-(4-аминофенил)-1-пиперазинил)-3-этоксикарбонил-7-фтор-1-метил- 4-оксо-1,4-дигидро-1,8-бензо[b]нафтиридина-(RS) в виде твердого вещества, плавящегося при 180-182оС.
3-Этоксикарбонил-7-фтор-1-метил-8-[3-(4-нитрофенил)-1-пиперазинил] 4-оксо-1,4-дигидро-1,8-бензо[b] нафтиридин-(RS) был приготовлен в условиях примера 39, но исходя из 0,636 г 3-этоксикарбонил-7,8-дифтор-1-метил-4-оксо-1,4-дигидро-1,8-бензо[b] нафтиридина и 0,5 г 2-(нитрофенил)-пиперазина (RS). После кристаллизации в 10 см3 диметилформамида получают 0,7 г целевого продукта в виде твердого вещества желтого цвета, разлагающегося при примерно 300оС.
2-(4-Нитрофенил)-пиперазин (RS) был приготовлен в соответствии с теми же методами, которые использовались ранее в примере 8.
Исходя из 18 г 4-нитрофенилглиоксаля получают 6,2 г ожидаемого продукта в виде жидкого масла коричневого цвета, которое используется без дополнительной очистки в последующих стадиях.
П р и м е р 36. Проводя реакцию в условиях примера 39, но исходя из 2,2 г 3-этоксикарбонил-7,9-дифтор-8-{ 3-[4-(2-гид- роксиэтокси)- фенил]-1-пиперазинил} -1-метил-4-оксо-1,4-дигидро-1,8-бензо[b] нафтиридина (RS) получают 1,6 г 7,9-дифтор-8-{3-[4-(2-гидроксиэтокси)-фе- нил]-1-пиперазинил}-1-метил- 4-оксо-1,4-дигидро-1,8-бензо[b] нафтиридин-3-карбо-новой кислоты (RS) в виде твердого вещества желтого цвета, плавящегося при температуре 226-228оС.
3-Этоксикарбонил-7,9-дифтор-8-{3-[4- (2-гидроксиэтокси)-фенил]-1- пиперазинил} -1-метил-4-оксо-1,4-дигидро-1,8-бензо[b]нафтиридин-(RS) был приготовлен в условиях примера 39, но исходя из 1,9 г 3-этоксикарбонил-7,8,9-трифтор-1-метил-4-оксо-1,4-ди- гидро-1,8-бензо[b] нафтиридина и 1,6 г 2-[4(2-гидроксиэтокси)-фенил]-пиперазина- (RS) получают 2,2 г целевого продукта в виде твердого вещества желтого цвета, плавящегося при 183оС.
3-Этоксикарбонил-7,8,9-трифтор-1-ме-тил-4-оксо-1,4-дигидро-бензо[b] нафтиридин-1,8 может быть получен, как описано выше в примере 24.
П р и м е р 37. Проводя реакцию в условиях, близких к условиям примера 39, но исходя из 2,4 г 1-циклопропил-3-этоксикарбонил-7,9-дифтор-8-{3-[4-(2-гидрокси- этокси)- фенил]-пиперазинил}-4-оксо-1,4-дигидро-1,8-бензо[b] нафтиридина-(RS), прибавляя 100 см3 этанола с 50% воды к реакционной смеси до введения метансульфокислоты, получают 1,4 г 1-циклопропил-7,8,9-{3-[4-(2-гидроксиэтокси)-фенил] -1-пи-перазинил} -4- оксо-1,4-дигидро-1,8-бензо[b] нафтиридин-3-карбоновой кислоты-(RS) в виде твердого вещества желтого цвета, плавящегося при температуре 228-230оС. 1-Циклопропил-3-этоксикарбонил-7,9-дифтор-8-{ 3-[4-(2-гидроксиэтокси)- фенил]-1-пиперазинил}-4-оксо-1,4-дигидро-1,8-бен- зо[b] нафтиридин-(RS) был приготовлен в условиях примера 39, но исходя из 2 г 1-циклопропил-3-этоксикарбонил-7,8,9-трифтор- -4-оксо-1,4-дигидро-1,8- бензо[b]нафтиридина и 1,6 г 2-[4-(2-гидроксиэтокси)-фенил]-пиперазина (RS) получают 2,4 г целевого продукта в виде твердого вещества желтого цвета, плавящегося при 225-226оС. 1-Циклопропил-3-этоксикарбонил-7,8,9-трифтор-4-оксо-1,4-дигидро-бензо[b] нафтиридин-1,8 приготавливается, как было описано в примере 27.
2-[4-(2-гидроксиэтокси)-фенил] -пипера- зин (RS) может быть получен, как описано в примере 16.
П р и м е р 38. Проводя реакцию в условиях примера 39, но исходя из 2,1 г 3-этоксикарбонил-7-фтор-1-метил-8-{3-[(3,4-мети- лендиокси)-фенил]-1- пиперазинил} -4-оксо-1,4-дигидро-1,8-бензо[b]нафтиридина-1,8 (RS) получают после перекристаллизации в 30 см3 диметилформамида 1,6 г 7-фтор-1-метил-8-{ 3-[(3,4-метилендиокси)-фенил]-1-пи-перазинил}-4-оксо- 1,4-дигидро-1,8-бензо[b] нафтиридин-3-карбоновой кислоты-(RS) в виде твердого вещества желтого цвета, разлагающегося при 255оС.
3-Этоксикарбонил-7-фтор-1-метил-8-{ 3-[(3,4-метилендиокси)-фенил] -1- пиперазинил} -4-оксо-1,4-дигидро-1,8-бензо[b] нафти- ридин был приготовлен в условиях, близких к условиях примера 39, но исходя из 1,59 г 3-этоксикарбонил-7,8-дифтор-1-метил-4-ок- со-1,4-дигидро-1,8-бензо[b] нафтиридина 1,25 г 2-(3,4-диоксиметиленфенил)-пиперазина-(RS) и 0,64 г карбоната натрия получают 2,15 г 3-этоксикарбонил-7-фтор-1-ме- тил-8-{3-[(3,4-метилендиокси)-фенил] -1-пи- перазинил} -4-оксо-1,4-дигидро-1,8-бензо[b] нафтиридина (RS) в виде твердого вещества желтого цвета, плавящегося при 252оС.
2-(3,4-Метилендиоксифенил)-пиперазин (RS) был приготовлен согласно способу, описанному в заявке на патент FP 2 351 108.
П р и м е р 39. Суспензию, состоящую из 1,3 г 3-этоксикарбонил-7-фтор-8-[3-(2-фурил)-1-пиперазинил] -1-метил-4- оксо-1,4-дигидро-1,8-бензо[b]нафтиридина-(RS) в 12 см3 0,5 н. водного раствора поташа и 12 см3 этанола, нагревают в течение 1 ч при температуре, близкой к 80оС, затем смешивают при той же температуре с 6 см3 1 н. водного раствора метансульфокислоты. После охлаждения до примерно 20оС нерастворимую часть центрифугируют, промывают 3 раза 10 см3 воды, 3 раза 10 см3 этанола, 3 раза 10 см3 этилового эфира и перекристаллизовывают из смеси 13 см3 этанола и 27 см3 диметилформамида. Получают 0,570 г 7-фтор-8-[3-(2-фурил)-1-пиперазинил]-1-ме- тил-4-оксо-1,4-дигидро- 1,8-бензо[b]нафтиридин-3-карбоновой кислоты (RS) в виде твердого вещества желтого цвета, плавящегося при 258-260оС.
3-Этоксикарбонил-7-фтор-8-[3-(2-фурил) -1-пиперазинил] -1-метил-4- оксо-1,4-дигидро-1,8-бензо[b] нафтиридин-(RS) был приготовлен следующим способом:
суспензию, состоящую из 0,96 г 3-этоксикарбонил-7,8-дифтор-1-метил-4-оксо-1,4-дигидро-1,8-бензо[b] нафтиридина 0,6 г 2-(2-фурил)-пиперазинил-(RS) и 0,38 г карбоната натрия в 20 см3 диметилсульфоксида, нагревают при примерно 95оС в течение 5,5 ч. После охлаждения до 20оС смесь приливают к 50 см3 воды при температуре, близкой к 5оС, и экстрагируют 3 раза 50 см3 воды, сушат на сульфате магния, фильтруют и концентрируют досуха при пониженном давлении (20 кПа) и при 40оС. Полученный остаток извлекают посредством 20 см3 этанола, центрифугируют, промывают 2 раза 20 см3 этанола и 3 раза 30 см3 этилового эфира. Получают 1,3 г 3-этоксикарбонил-7-фтор-8-[3-(2-фурил)-1-пиперазинил] -1-метил-4-ок-со- 1,4-дигидро-1,8-бензо[b]нафтиридин приготавливается, как было описано в примере 19.
2-(2-Фурил)-пиперазин (RS) был приготовлен согласно методу, описанному в заявке на патент FP 230 053.
П р и м е р 40. Проводя реакцию в условиях примера 39, но исходя из 1,25 г 1-циклопропил-3-этоксикарбонил-7-фтор-8-[3-(2-фурил)-1- пиперазинил]-4-оксо-1,4-дигидро-1,8-бензо[b] нафтиридина-(RS), получают 0,95 г 1-циклопропил-7-фтор-8-[3- (2-фурил)-1-пиперазинил] -4-оксо-1,4-дигид- ро- 1,8-бензо(b)нафтиридин-3-карбоновой кислоты (RS) в виде твердого вещества желтого цвета, плавящегося при 240-241оС.
1-Циклопропил-3-этоксикарбонил-7- фтор-8-[3-(2-фурил)-1-пиперазинил] 4-оксо-1,4-дигидро-1,8-бензо(b)нафтиридин-(RS) был приготовлен в условиях примера 39, но исходя из 1,05 г 1-циклопропил-7,8-дифтор-3-этоксикарбонил-4-оксо-1,4-дигидро-1,8- бензо(b)нафтиридина и 0,6 г 2-(2-фурил)-пиперазина (RS), получают 1,3 г 1-циклопропил-3-этоксикарбонил-7-фтор-8-[3-(2-фурил) -1- пиперазинил]-4-оксо-1,4-дигидро-1,8-бензо(b) нафтиридина в виде твердого вещества желтого цвета, плавящегося при 182оС.
1-Циклопропил-3-этокси-7,8-дифтор-4-оксо-1,4-дигидро-1,8-бензо(b) нафтиридин был приготовлен следующим способом:
раствор, содержащий 20 г 2-(2-хлор-6,7-дифтор-3-хинолинкарбонил)-3-диметилами- ноэтилакрилата и 9,25 г циклопропиламина в 80 см3 трихлорметана, перемешивают при температуре близкой к 20оС, в течение 5 ч. Раствор концентрируют при пониженном давлении (20 кПа) и при 40оС. Остаток извлекают посредством 300 см3 этанола, смешивают с 8,2 г ДБУ и нагревают при перемешивании и при примерно 75оС в течение 30 мин. После охлаждения до 20оС нерастворимую часть центрифугируют, промывают 2 раза 30 см3 этанола. Получают 11,1 г 1-циклопропил-3-этоксикарбонил-7,8-дифтор-4-оксо-1,4-дигидро-1,8- бензо(b)нафтиридина в виде твердого вещества желтого цвета, плавящегося при температуре 230оС.
П р и м е р 41. Проводя реакцию в условиях примера 39, но исходя из 1,58 г 3-этоксикарбонил-7,9-дифтор-8-[3-(2-фурил)-1-пи- перазинил)-1-метил-4- оксо-1,4-дигидро-1,8-бензо(b)нафтиридина (RS) получают 1,07 г 7,9-дифтор-8-[3-(2-фурил)-1-пиперазинил]-1-метил-4-оксо-1,4-дигидро- 1,8-бензо(b) нафтиридин-3-карбоновой кислоты (RS) в виде твердого вещества желтого цвета, плавящегося при 295-296оС.
3-Этоксикарбонил-7,9-дифтор-8-[3-(2-фурил)-1-пиперазинил]-1-метил- 4-оксо-1,4-дигидро-1,8-бензо(b)нафтиридин-(RS) был приготовлен в условиях примера 39, но исходя из 1,85 г 3-этоксикарбонил-7,8,9-трифтор-1-метил-4-оксо-1,4-дигидро-1,8-бензо(b) нафтиридина и 1 г 2-(2-фурил)-пиперазина (RS). После концентрирования при пониженном давлении объединенных органических вытяжек (20 кПа) примерно 40оС твердый остаток перекристаллизовывают из 100 см3 этанола. Получают 1,72 г 3-этоксикарбонил-7,9-дифтор-8-[3-(2-фурил)-1-пи- перазинил] -1-метил-4- оксо-1,4-дигидро-бензо[b]нафтиридина-(RS) в виде твердого вещества желтого цвета, плавящегося при температуре 208-210оС.
П р и м е р 42. Проводя реакцию в условиях примера 39, но исходя из 2 г 3-этоксикарбонил-7-фтор-1-метил-4-оксо-8-[3-(2-тие- нил)-1- пиперазинил)-1,4-дигидро-бензо(b)нафтиридина-(RS) получают без перекристаллизации 1,8 г чистой 7-фтор- 1-метил-4-оксо-8-[3-(2-тиенил)-1-пиперази-нил)-1,4-дигидро-1,8- бензо(b) нафтиридин-3-карбоновой кислоты-(RS) в виде твердого вещества желтого цвета, плавящегося при 268-270оС.
3-Этоксикарбонил-7-фтор-1-метил-4-ок- со-8-[3-(2-тиенил)-1-(пиперазинил)- 1,4-дигидро-1,8-бензо(b) нафтиридин-(RS) был приготовлен в условиях примера 39, но исходя из 1,59 г 3-этоксикарбонил-7,8-дифтор-1-метил-4-оксо-1,4-дигидро-1,8-бензо(b) нафтиридина и 1,02 г 2-(2-тиенил)-пиперазина-(RS). Получают 2,25 г 3-этоксикарбонил-7-фтор-1-метил-4-оксо-8-[3-(2-тиенил)-1-пи- перазинид)- 1,4-дигидро-1,8-бензо(b)нафтиридина (RS) в виде твердого вещества желтого цвета, плавящегося при 228оС.
2-(2-Тиенил)-пиперазин (RS) был приготовлен в соответствии с условиями, описанными в работе Ieffrey W. H. Watthey et coll. I.Med.Chem. 26, 1116 (1983).
2-(2-Тиенил)-пиперазин (RS) был приготовлен в соответствии с условиями, описанными в работе Ieffrey W. H. Watthey et coll. I.Med.Chem. 26, 1116 (1983).
П р и м е р 43. Проводя реакцию в условиях примера 39, но исходя из 2 г 1-циклопропил-3-этоксикарбонил-7-фтор-4-оксо-8-[3-(2-тиенил)- 1-пиперазинил] -1,4-дигидро-1,8-бензо(b)нафтиридина-(RS) получают 1,75 г 1-циклопропил-7-фтор-4-оксо-8-[3-(2-тиенил)-1-пиперазинил]-1,4- дигидро-1,8-бензо(b)нафтиридин-3-карбоновой кислоты-(RS) в виде твердого вещества желтого цвета, плавящегося при 245оС.
1-Циклопропил-3-этоксикарбонил-7- фтор-4-оксо-8-[3-(2-тиенил)-1- пиперазинил] -1,4-дигидро-1,8-бензо(b)нафтиридин-(RS) был приготовлен в условиях примера 39, но исходя из 1,7 г 1-циклопропил-7,8-дифтор-3-этоксикарбонил-4-оксо-1,4-дигид- ро-1,8- бензо(b) нафтиридина и 1 г 2-(2-тиенил)-пиперазина (RS) получают 2,2 г 1-циклопропил-3-этоксикарбонил-7-фтор-4-оксо- 8-[3-(2-тиенил)- 1-пиперазинил] -1,4-дигидро-бензо(b)нафтиридина-1,8 (RS) в виде твердого желтого цвета, плавящегося при 210-212оС.
П р и м е р 44. Проводя реакцию в условиях примера 39, но исходя из 1,6 г 3-этоксикарбонил-7,9-дифтор-1-метил-4-ок- со-8-[3-(2-тиенил)-1 -пиперазинил] -1,4-дигидро-1,8-бензо(b)нафтиридина-(RS), получают 0,75 г 7,9-дифтор-1-метил-4-оксо- 8-[3-(2-тиенил)-1-пиперазинил] 1,4-дигидро-бензо(b) нафтиридин-3-карбоновой кислоты (RS) в виде твердого вещества желтого цвета, плавящегося при 292-294оС.
3-Этоксикарбонил-7,9-дифтор-1-метил-4-оксо-8-[3-(2-тиенил)-1- пиперазинил] -1,4-дигидро-1,8-бензо(b)нафтиридин-(RS) был приготовлен в условиях примера 39, но исходя из 1,68 г 3-этоксикарбонил-7,8,9-трифтор-1-метил-4-оксо-1,4-дигидро-1,8- бензо(b) нафтиридина и 1,01 г 2-(2-тиенил)-пиперазина-(RS) получают таким образом 1,68 г 3-этоксикарбонил-7,9-дифтор-1-метил-4-оксо-8-[3-(2-тиенил)-1- пиперазинил]-1,4-дигидро-бензо(b)нафтиридина (RS) в виде твердого вещества желтого цвета, плавящегося при 220оС.
П р и м е р 45. Проводя реакцию в условиях примера 39, но исходя из 3,2 г 3-этоксикарбонил-7-фтор-8-[4-гидрокси-1-пиперазинил] -1-метил-4- оксо-1,4-дигидро-1,8-бензо(b) нафтиридина получают 2,86 г 7-фтор-8-[4-гидрокси-1-пиперазинил] -1-ме- тил-4-оксо-1,4-дигидро-1,8- бензо(b)нафтиридин-3-карбоновой кислоты в виде твердого вещества желтого цвета, плавящегося при температуре 290оС.
3-Этоксикарбонил-7-фтор-8-[4-гидро- кси-1-пиперазинил] -1-метил-4-оксо- 1,4-дигидро-1,8-бензо(b) нафтиридин был приготовлен в условиях примера 39, но исходя из 3 г 3-этоксикарбонил-7,8-дифтор-1-метил- 4-оксо-1,4-дигидро-1,8-бензо(b)нафтиридина, 1,97 г дихлоргидрата 1-гидроксипиперазина и 1,99 г карбоната натрия получают 3,25 г 3-этоксикарбонил-7-фтор-8-[4-гидрокси-1-пиперазинил]-1-метил-4-оксо- 1,4-дигидро-1,8-бензо(b)нафтиридина в виде твердого вещества желтого цвета, плавящегося при 258-260оС.
Дихлоргидрат-1-гидроксипиперазин был приготовлен согласно способу, описанному в работе Toschio Uno et coll. I.Het.chem. 26, 393 (1988).
П р и м е р 46. Проводя реакцию в условиях примера 39, но исходя из 1,4 г 3-этоксикарбонил-7,9-дифтор-8-[4-гидрокси-1-пи- перазинил]-1-метил-4- оксо-1,4-дигидро-1,8-бензо(b)нафтиридина получают 0,5 г 7,9-дифтор-8-[4-гидрокси-1-пиперазинил] -1-ме- тил-4-оксо-1,4-дигидро- 1,8-бензо(b)нафтиридин-3-карбоновой кислоты в виде твердого вещества желтого цвета, плавящегося при температуре 285-288оС.
3-Этоксикарбонил-7,9-дифтор-8-[4-гид- рокси-1-пиперазинил] -1-метил-4- оксо-1,4-дигидро-1,8-бензо(b)нафтиридин был приготовлен в условиях примера 39, но исходя из 1,9 г 3-этоксикарбонил-7,8,9-трифтор-1-метил-4-оксо-1,4-дигидро-1,8-бензо(b) нафтиридина, 1 г дихлоргидрата 1-гидроксипи- перазина и 1,6 г карбоната калия получают 1,4 г 3-этоксикарбонил-7,9-дифтор-8-[4-гидрокси-1-пиперазинил] -1-метил-4- оксо-1,4-дигидро-1,8-бензо(b)нафтиридина в виде твердого вещества желтого цвета, плавящегося при 255-258оС.
П р и м е р 47. Работают, как в примере 16, но используют 1,16 г 7,8-дифтор-1-метил-4-оксо-1,4-дигидро-1,8-бензо(b)нафти-ридин-43- карбоновой кислоты и 1,8 г (b) 2-(4-диметиламинофенил)-пиперазина получают после нагревания в течение около 55 мин или температуре около 100оС с последующей перекристаллизацией в 70 см3 ДФА, 1,6 г 7-фтор-8-3(4-диметиламинофенил)-1-пиперазинил-1-метил-4-оксо-1,4- дигидро-1,8-бензо(b)нафтиридин-3-карбоно-вой кислоты (RS) в форме твердого желтого продукта, плавящегося при 322оС.
(RS) 2-(4-диметиламинофенил)-пиперазин может быть получен по методу, описанному в заявке на патент Франции 2 351 108, но используя в качестве исходного 4,7 г 4-диметиламино-фенилглиоксаля и 2,1 см2 этилендиамина получается 3,1 г (RS) 2-(4-диметиламинофенил)-пиперазино в форме твердого продукта желтого цвета, плавящегося при 122оС.
Новые производные бензо(b) нафтиридина-1,8 общей формулы (I) и их фармацевтически пригодные соли проявляют особенно интересные бактерицидные свойства. Они проявляют значительную активность in vitro и in vivo на бактерии грам-положительные и обычную активность по отношению к бактериям, ответственным за большинство инфекций верхних и нижних дыхательных путей.
В тестах in vitro продукты общей формулы (I) оказываются активными при концентрации, заключенной между 0,12 и 4 мкг/см3, по отношению к золотистому стафилококку IP 8203.
В тестах in vivo продукты с общей формулой (I) оказываются активными по отношению к экспериментальным заражениям золотистым стафилококком IP 8203 мыши при дозах, заключенных между 2 и 200 мг/кг при подкожном введении, или при дозах, заключенных между 4 и 150 мг/кг при оральном введении.
Другой интересной особенностью продуктов является их слабая токсичность. Их ДЛ50 обычно выше 500 мг/кг при подкожном введении у мыши.
С другой стороны, продукты оказываются особенно интересными для профилактики и лечения СПИДа (синдрома приобретенного иммунного дефицита) и связанных с ним синдромов (ARC (AIDS related complex).
Изученные продукты замедляют цитопатогенный эффект вируса ВИЧ (HIV) в клеточной культуре при концентрациях, устраняющих цитотоксический и цитостатический эффект.
Активность по отношению к цитопатогенному эффекту вируса ВИЧ. Продукты в порошкообразном виде были введены в раствор в количестве 2 мг продукта на 1 мл (примерно 4.10-3 М) в смесь диметилсульфоксида (ДМСО и L-лизина (основание) в объемном соотношении 1:19. Сначала добавляют 1 объем ДМСР и растворяют продукт как можно лучше, затем добавляют 19 объемов раствора основания L-лизина (4.10-3 М) в дистиллированной воде. Смесь выдерживается 15 мин при 60оС. Таким способом получают запас раствора продукта с концентрацией ДМСО 5% и с содержанием молярного соотношения продукт/лизин, близким к 1. Тест осуществляется на потомстве лимфобластоида СЕМ клон 13. На микропластинку с 96 лунками помещают 25 мкл/лунку тестируемого раствора продукта в изотоническом фосфатном буфере (ИФБ) или один ИФБ в случае контроля. Продукты изучаются при различных концентрациях (обычно 8) при норме 6 лунок на концентрацию. Затем добавляют 125 мкл клеточной суспензии СЕМ (между 5 и 8.104 клеток на 1 мл) в среде RPMI, содержащей 10% сыворотки телячьего зародыша (эмбриона), 100 ЕД/мл пенициллина, 100 мкг/мл стрептомицина и 2 мкмоль/мл глутамина, и микропластинки выдерживаются 1 ч при 37оС в атмосфере, содержащей 5% углекислого газа. Для каждой концентрации опыт разделен на две части: одна часть (3 лунки) для зараженных клеток с целью определения противовирусной активности и другая часть (3 лунки) для незараженных клеток с целью определения цитотоксичности продуктов. Затем заражают первую серию посредством ВИЧ-1 (100 мкл на лунку суспензии вируса LAV-I-ВRU, содержащей 200-300 ТС1Д50), тогда как другая серия получает 100 мкл среды RPMI, которая была определена ранее.
В конце 7-го дня инкубации 150 мкл клеток берутся на анализ для измерения клеточной жизнеспособности (определяемой в соответствии с модифицированным методом, описанным R.Pouwels et coll. I. Virol.Meth. 20, 309-321 (1988). Добавляют во взятую пробу 10 мкл раствора, содержащего 7 мг МГТ (бромид 4,5-диметил-тиазол-2-ил-2, 5-дифенилтетразолия) на 1 мл изотонического фосфатного буфера. После 3 ч инкубации при 37оС всплывшую часть удаляют.
МТТ превращается в соль формазана (голубого) внутри живых клеток и пропорционально их числу, тогда как ничего не происходит в лунках, содержащих только мертвые клетки. Затем добавляют 100 мкл изопропанола (содержащего 0,04 моль/л хлороводородной кислоты), и микропластинки встряхивают до растворения голубого формазана. Поглощение при 540 нм регистрируется при помощи автоматического прибора ELISA на микропластинках. Это поглощение пропорционально количеству живых клеток.
Результаты, полученные на 14-й день, приведены ниже в табл.1.
Биологические испытания in vitro. В серию пробирок, содержащих определенный объем (20 см3) пригодной питательной среды (агар Муллера-Хинтона) добавляли 1/10 от этого объема серий, разбавленный в геометрической прогрессии (отношение 2), испытываемый продукт. Пробирки инокулировали при помощи многоточечного инокулятора, который давал пятно 104 колонки образованных единиц микроорганизма в соевом бульоне, инкубированном в течение 18 ч при 37оС и разбавленном в отношении 1/100 той же средой.
После инокулирования пробирки инкубировали в течение 24 ч при 37оС.
Минимум тормозящей концентрации представляет самую низкую концентрацию, при помощи которой тормозят развитие микроорганизмов.
Активность против внутрибрюшинных инфекций у мышей. Мышам была сделана внутрибрюшинная инъекция 0,5 см3 подходящей культуры возрастом 18 ч испытываемого микроорганизма в "Brain Heart Infusion" среде (Difco), разбавленной 5% муцина свиньи. Эта инокуляция вызывала смерть контрольных животных в течение 24-48 ч. Это испытываемое соединение вводили подкожно дважды в интервале 5 ч в день инокуляции, первую дозу давали через 1 ч после инокуляции микроорганизмом. Использовали единичные дозы, содержащие по объему 50 см3/кг.
Лечебная доза 50% (СД50) представляет дозу испытываемого соединения, каждое введение которой вызывает у половины обрабатываемых животных выживание в течение испытываемого периода (8 дней).
Активность в пробирке и на живом организме показана в табл.2, активность in vitro и in vivo в табл.3.
Предлагаемое изобретение относится также к фармацевтическим составам, пригодным к использованию в медицине, в частности в ветеринарии, которые содержат в качестве активного продукта, по меньшей мере, один продукт с общей формулой (I) в чистом состоянии (в свободном виде или в виде соли) или в виде смеси с одним или несколькими разбавителями или добавками, являющимися совместными и фармацевтически допустимыми. Эти составы могут применяться оральным, парентеральным или ректальным путями.
В качестве твердых составов орального назначения могут использоваться таблетки, пилюли, порошки или гранулы. В этих составах активный продукт смешивается с одним или несколькими разбавителями или инертными добавками такими, как сахароза, лактоза или крахмал. Эти составы могут также содержать кроме разбавителей другие вещества, например смазывающие такие, как стеарат магния.
В качестве жидких составов орального назначения можно использовать фармацевтически допустимые эмульсии, растворы, суспензии, сиропы и эликсиры, содержащие инертные разбавители такие, как вода или парафиновое масло. Эти составы могут также содержать кроме разбавителей другие вещества, например смачивающие, смягчающие или ароматизирующие продукты.
Составы парентерального назначения могут быть стерильными водными или неводными растворами, суспензиями или эмульсиями. В качестве растворителя или связующего можно использовать пропиленгликоль, полиэтиленгликоль, жидкие растительные масла, особенно оливковое масло, или органические сложные эфиры, которые могут впрыскиваться, например этилеат. Эти составы могут также содержать добавки, особенно смачивающие, эмульгирующие или диспергирующие агенты. Стерилизация может осуществляться несколькими способами, например, при помощи бактериологического фильтра, внедряя в состав стерилизующие агенты, при облучении или при нагреве. Они могут быть также приготовлены в виде стерильных твердых составов, которые в момент использования будут растворяться в стерильной воде или любой другой стерильной среде, которая может впрыскиваться.
Составами ректального назначения являются ректальные свечи или капсулы, которые могут содержать помимо активного продукта индифферентные вещества такие, как масло какао или воск.
В терапевтическом лечении человека или ветеринарии составы особенно полезны при лечении инфекций бактериального происхождения.
В общем случае врач будет определять дозировку, которую он сочтет самой подходящей в зависимости от возраста, веса, степени заражения и других факторов, свойственных пациенту, подлежащему лечению. Обычно дозы заключаются между 0,2 и 1 г активного продукта два раза в день оральным или парентеpальными путями для взрослого.
Следующие примеры иллюстрируют составы согласно изобретению.
П р и м е р А. Готовят в соответствии с обычными методами таблетки с дозой 250 мг активного продукта, имеющие следующие состав: (в мг) 7-фтор-1-метил-4-оксо-8-(3-фенил-1-пиперазинил)-1,4-дигидро- бензо(b)нафтиридин-1,8-3-карбоновая кислота. (R) 250 Крахмал 50 Лактоза 35 Тальк 15
П р и м е р В. Готовят в соответствии с обычными методами таблетки с дозой 250 мг активного продукта, имеющие следующий состав: (в мг) 7-фтор-8-[3-(2-фурил)-1-пиперазинил] -1-метил-4- оксо-1,4-дигидро-бензо(b) нафтиридин-1,8-3-карбоновая кислота (R) 250 Крахмал 50 Лактоза 35 Тальк 15
Продукты с общей формулой (I) также представляют интерес в области агрохимии для антибактериальной обработки растений.
С другой стороны, продукты с общей формулой (I) могут также применяться в качестве агентов консервации и дезинфекции органических или неорганических веществ. В том числе при производстве красителей, жиров, бумаги, древесины, полимеров или также в текстильной промышленности, в пищевой промышленности или при обработке воды.




Использование: в медицине в качестве бактерицидного агента. Сущность изобретения: продукты: производные 1,8-бензо-(в)нафтиридина ф-лы 1, где R1 водород, гидроксигруппа или алкил, R2 водород, алкил, фторалкил, циклоалкил, содержащий от 3 до 6 атомов углерода, алкилокси- или алкиламино-группа, R3 фенил или алкилфенил, возможно замещенный одним или несколькими атомами галогена или радикалами: алкил, циклоалкил, циано, амино, R4 водород или фтор. Реагент 1: пиперазин ф-лы 2. Реагент 2: нафтиридин ф-лы 3. Условия реакции: в среде органического растворителя в присутствии хлористого водорода. 2 с. п. ф-лы. Структура соединений ф-л 1, 2, 3: 
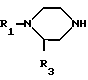
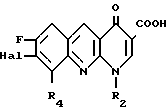

где R-водород, гидроксигруппа или алкил;
R2 водород, алкил, фторалкил, циклоалкил, содержащий 3 6 атомов углерода, алкилокси- или алкиламиногруппа;
R3 фенил или алкилфенил, возможно замещенный одним или несколькими атомами галогена или радикалами, алкил, циклоалкил, содержащий 3 - 6 атомов углерода, алкилокси-, циано-, амино-, алкиламино-, диалкиламино-, алкилоксиалкил, гидроксилалкил, гидрооксилкилокси-, метилендиокси-, аминоалкил, алкиламиноалкил или диалкиламиноалкил или является фурилом или тиенилом;
R4 водород или фтор,
причем алкильные радикалы являются неразветвленными или разветвленными и содержат 1 4 атомов углерода,
в виде их изомеров или смеси изомеров, или их соли с металлами, или соли, полученные присоединением азотсодержащих оснований или кислот, или их гидротированные формы в качестве бактерицидного агента.
Приоритет по признакам:
30.10.89 при R1 водород, алкил; R2 водород, алкил, фторалкил, циклоалкил, содержащий 3-6 атомов углерода, алкилокси- или алкиламиногруппа; R3 фенил или алкилфенил, возможно замещенный одним или несколькими атомами галогена или радикалами: алкил, циклоалкил, содержащий 3 6 атомов углерода, алкилокси-, циано-, амино-, алкиламино или диалкиламино; R4 водород или фтор.
| Патент США N 4292317, кл | |||
| Устройство для сортировки каменного угля | 1921 |
|
SU61A1 |
| Приспособление для изготовления в грунте бетонных свай с употреблением обсадных труб | 1915 |
|
SU1981A1 |
