Настоящее изобретение относится к группе особенных производных диазабициклоалканов, имеющих интересную антагонистическую активность к рецептору нейрокинина NK1.
Изобретение относится также к способу получения новых соединений и к фармацевтическим композициям, содержащим фармакологически активное количество по меньшей мере одного из таких соединений в качестве активного ингредиента.
Из европейской патентной заявки ЕР 0655442 известны производные пиперазина с антагонистической активностью к нейрокинину. Новые производные пиперазина, разделяющие такую биологическую активность, раскрыты в ЕР 0899270, где описан ряд производных 2-(3-индолилметил)-1-бензоил-4-[(2-(бензиламино)этил)аминокарбонил]пиперазина, имеющих NK1-антагонистическую активность.
Неожиданно было обнаружено, что производные пиперазина также являются NK1-антагонистами, когда пиперазиновое кольцо и его N-4 заместитель являются конденсированными, образуя производные 1,4-диазабицикло[4.3.0]нонана, 1,4-диазабицикло[4.4.0]декана или 1,4-диазабицикло[4.5.0]ундекана.
Изобретение относится к соединениям общей формулы (1):
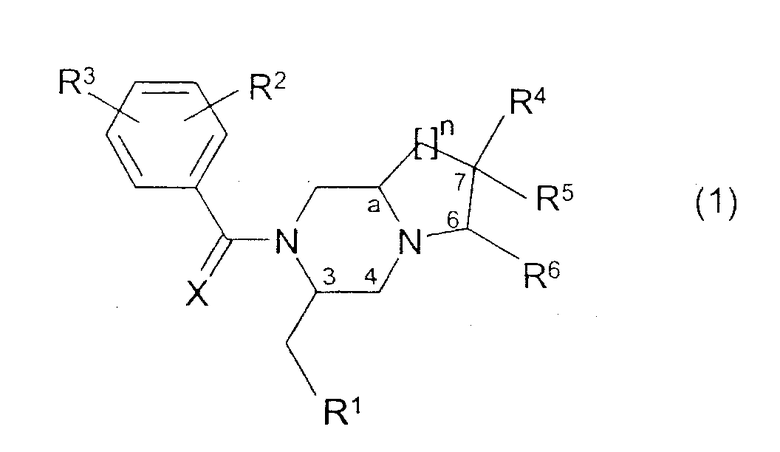
где:
- R1 представляет фенил, 2-индолил, 3-индолил, 3-индазолил или бензо[b]тиофен-3-ил, указанные группы могут быть замещены галогеном или алкилом (1-3С);
- R2 и R3 независимо представляют галоген, Н, ОСН3, СН3 или CF3;
- R4, R5 и R6 независимо представляют Н, ОН, О-алкил(1-4С), СН2ОН, NH2, диалкил(1-3C)N, пирролидин-1-ил, пиперидин-1-ил, морфолин-4-ил или морфолин-4-ил, замещенный одной или двумя метильными или метоксиметильными группами, морфолин-4-иламино, морфолин-4-илметил, имидазол-1-ил, тиоморфолин-4-ил, 1,1-диоксотиоморфолин-4-ил или 3-окса-8-азабицикло[3.2.1]окт-8-ил, R4 и R5 вместе могут представлять кетогруппу 1,3-диоксан-2-ил или 1,3-диоксолан-2-ил;
- X представляет O или S;
n имеет значения 1, 2 или 3;
a является асимметричным атомом углерода 8а, 9а или 10а, когда n равно 1, 2 или 3 соответственно,
и их фармацевтически приемлемым солям.
Все соединения, имеющие формулу (1), в которой заместители у асимметричных атомов углерода 3 и "а", а также у потенциально асимметричных атомов углерода 6 и 7, находятся в R-конфигурации или в S-конфигурации, охватываются изобретением.
К изобретению принадлежат также пролекарства, т.е. соединения, которые, будучи введены человеку любым известным способом, превращаются в результате метаболизма в соединения, имеющие формулу (1). В частности, это относится к соединениям, в которых R4, R5 R6 представляет гидрокси или гидроксиметильную группу, типичным примером чего является 3,5-бис(трифторметил)фенил[6-гидроксиметил-3-(1Н-индол-3-илметил)гексагидропирроло[1,2-a]пиразин-2-ил]метанон (соединение 1 и его энантиомеры, см. ниже). Такие соединения могут быть этерифицированы, давая соединения, которые могут быть превращены в результате метаболизма в соединения, имеющие формулу (1).
Изобретение, в частности, относится к соединениям, имеющим формулу (1), где R1 представляет 3-индолил, R2 и R3 являются группами CF3 в положениях 3 и 5, X представляет кетогруппу, n имеет значения 1 или 2 и "a", R4, R5 и R6 имеют значения, указанные выше, включая все возможные стереоизомеры, как указано выше.
Еще более предпочтительными являются соединения по изобретению, как оно описано выше, в которых R4 или R6 представляют или содержат или морфолино, или гидроксиметильную группу, R5 представляет водород, и стереохимией которых является 3R.
Соединения, имеющие формулу (1), и их соли могут быть получены согласно по меньшей мере одному из следующих способов, известных для соединений такого типа.
Соединения по настоящему изобретению, в которых n = 1 и R4 и R5 представляют водород, могут быть получены по общей методике, показанной на схеме 1:
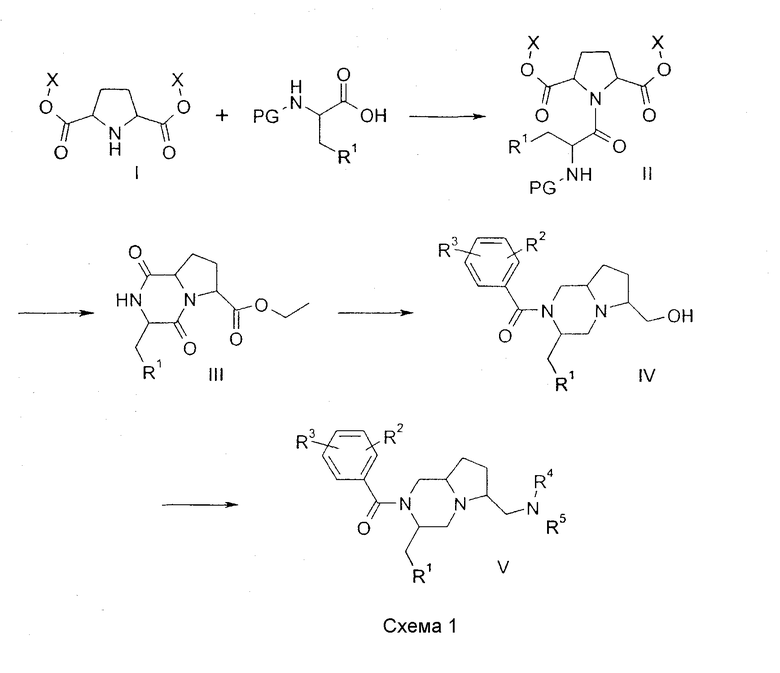
Так, диэфир дикарбоновой кислоты I (полученный аналогично методу G.Cignnarella, G.Nathansohn J. Org. Chem, 1961, 26, 1500), может быть введен в реакцию сочетания с должным образом защищенной аминокислотой по стандартным методикам сочетания, какие описаны в M.Bodanzsky, A.Bodanzsky, The Practice of Peptide Synthesis, Springer-Verlag, 1994, ISBN: 0-387-57505-7, для получения амида II. Защитная группа в II (обозначенная в формуле II как PG) может быть удалена с использованием известных методик (T.W.Greene, P.G.M.Wuts Protective groups in organic synthesis, 3d ed., John Wiley & Sons, 1999). Последующая циклизация дает замещенный дикетопиперазин III, использование хиральной аминокислоты будет приводить к образованию диастереомеров, которые могут быть разделены на этой стадии с использованием стандартных хроматографических методов. Восстановление III активным гидридным реагентом, таким как гидрид лития-алюминия, приводит к аминоспирту, который может быть ацилирован подходящим хлорангидридом в условиях, которые общеизвестны в практике, для получения IV. Превращение спирта в подходящую подвижную группу, такую как метансульфонат, и последующая реакция с амином дают соединения V. Подробные сведения о соединениях, синтезированных данным путем, приведены ниже в примере 1.
Сложноэфирные пролекарства соединений IV могут быть получены ацилированием хлорангидридами в растворителе, таком как ацетонитрил, в присутствии основания, такого как диизопропилэтиламин, при температурах между 20°С и 80°С (см. пример 6 ниже).
Соединения по изобретению, в которых n = 1 и R6 = H, могут быть получены по общей методике, показанной на схеме 2. Так, реакция эфира аминокислоты с должным образом защищенным производным 4-гидроксипролина по стандартным методикам сочетания, какие описаны в M.Bodanzsky, A.Bodanzsky, The Practice of Peptide Synthesis, Springer-Verlag, 1994, ISBN: 0-387-57505-7, дает дипептид VI. Защитная группа в VI может быть удалена с использованием известных методик (T.W.Greene, P.G.M.Wuts Protective groups in organic synthesis, 3d ed., John Wiley & Sons, 1999). Последующая циклизация дает замещенный дикетопиперазин VII, данная реакция может быть осуществлена путем перемешивания в смеси ацетонитрила и пиперидина. Защита гидроксигруппы соединения VII образованием силилового простого эфира по стандартным методикам, какие описаны в T.W.Greene, P.G.M.Wuts Protective groups in organic synthesis, 3d ed., John Wiley & Sons, 1999, и последующее восстановление активным гидридным реагентом, таким как гидрид лития-алюминия, приводит к аминоспиртам, подобным VIII. Соединения VIII могут быть ацилированы подходящим хлорангидридом в условиях, которые общеизвестны в практике, для получения IX. Превращение спирта в подходящую подвижную группу, такую как метансульфонат, и последующая реакция с амином дают соединения X. Подробные сведения о соединениях, синтезированных данным способом, приведены ниже в примере 2.
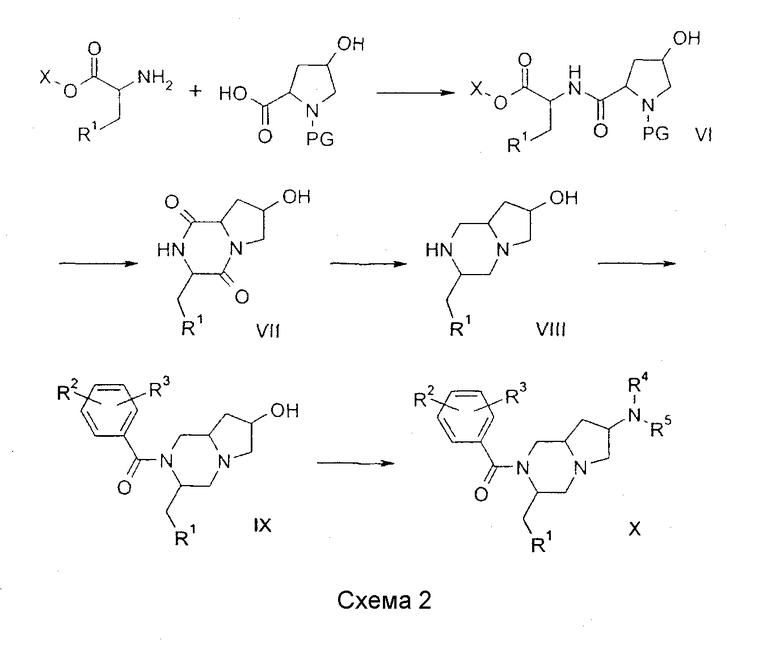
Соединения по изобретению, в которых n = 2 и R6 = H, могут быть получены по общей методике, показанной на схеме 3. Так, реакция эфира аминокислоты с 1-бензиловым эфиром 5-оксопиперидин-1,2-дикарбоновой кислоты (H.C.Beyerman, P.Boekee Recl. Trav. Chim. Pays-Bas, 1959, 79, 648) по стандартным методикам сочетания, какие описаны в M.Bodanzsky, A.Bodanzsky, The Practice of Peptide Synthesis, Springer-Verlag, 1994, ISBN: 0-387-57505-7, дает дипептиды XI; использование хиральных аминокислот будет приводить к образованию диастереомеров, и они могут быть разделены на этой (или на последующей) стадии с использованием стандартных хроматографических методов. Защита кетона в XI в виде циклического или ациклического кеталя, таких как описаны в T.W.Greene, P.G.M.Wuts Protective groups in organic synthesis, 3d ed., John Wiley & Sons, 1999, дает соединения формулы XII, в которых А и В представляют циклический или ациклический кеталь. Данная реакция может быть проведена, следуя обычным методикам, которые общеизвестны специалистам. Удаление бензилоксикарбонильной группы в восстановительных условиях (H2, Pd/C) в растворителе, таком как метанол, с последующей катализируемой кислотой циклизацией дает дикетопиперазины формулы XIII. Восстановление XIII активным гидридным реагентом, таким как гидрид лития-алюминия, приводит к амину, который может быть ацилирован подходящим хлорангидридом в условиях, которые общеизвестны в практике, для получения XIV. Гидролиз кеталя для получения XV может быть проведен методами, которые описаны в T.W.Greene, P.G.M.Wuts Protective groups in organic synthesis, 3d ed., John Wiley & Sons, 1999. Восстановительное аминирование XV подходящим амином в растворителе, таком как 1,2-дихлорэтан, с восстанавливающим агентом, таким как триацетоксиборгидрид натрия, дает соединения формулы XVI.
Альтернативно, соединение XVI может быть получено из XV восстановлением кетона до спирта XVII восстанавливающим агентом, таким как триацетоксиборгидрид натрия, в растворителе, таком как уксусная кислота. Спирт может быть превращен в подвижную группу (L), такую как хлор, бром или метансульфонат, в условиях, общеизвестных специалистам, давая XVIII. Замещение подвижной группы в XVIII подходящим амином в растворителе, таком как ацетонитрил, дает соединения формулы XVI. Подробные сведения о соединениях, синтезированных данным способом, приведены ниже в примерах 3, 4 и 5.
Промежуточные соединения VIII (схема 2), XIII (схема 3) и XVIII (схема 3) являются новыми соединениями. Изобретение относится также к этим новым соединениям.
Подходящие соли присоединения кислот могут быть образованы с неорганическими кислотами, такими как соляная кислота, серная кислота, фосфорная кислота и азотная кислота, или с органическими кислотами, такими как лимонная кислота, фумаровая кислота, малеиновая кислота, винная кислота, уксусная кислота, трифторуксусная кислота, бензойная кислота, п-толуолсульфокислота, метансульфоновая кислота и нафталинсульфокислота.
Соединения по изобретению общей формулы (1), а также их соли, имеют NK1-антагонистическую активность и показывают хорошую биодоступность. Они являются полезными при лечении расстройств, в которые вовлечены нейрокинины, которые взаимодействуют с NK1-рецепторами, например, нейрокинин-1 (вещество Р), или расстройств, которые могут подвергаться лечению путем манипуляции с данными системами. Примерами являются острые и хронические боли, рвота, воспалительные заболевания, такие как менингит, артрит, астма, псориаз и (солнечные) ожоги; желудочно-кишечные расстройства, в частности синдром раздраженной толстой кишки, воспалительные заболевания желудка (болезнь Крона), язвенный колит; расстройства, связанные с чрезмерной подвижностью мочевого пузыря или желудочно-кишечного тракта, воспаление мочевых путей; аллергические реакции, такие как экзема и ринит; сердечно-сосудистые расстройства, такие как гипертония, атеросклероз, отек, ангина, гистаминовая головная боль и мигрень; кожные заболевания, такие как крапивница, красная волчанка и зуд; респираторные расстройства, включая хроническую обструктивную болезнь легких, спазмы бронхов, бронхопневмонию, бронхит, респираторный дистресс-синдром и синдром фиброзно-кистозной дегенерации; различные опухолевые заболевания; психиатрические и/или неврологические расстройства, такие как шизофрения и другие психотические расстройства; расстройства настроения, такие как биполярные I, биполярные II и униполярные депрессивные расстройства, подобные легкой депрессии, сезонному аффективному расстройству, посленочной депрессивной дистимии и сильной депрессии; расстройства, связанные с беспокойством, включая паническое нарушение (с агорафобией или без нее), социальные фобии, расстройство навязчивой компульсивности (со связанным с заболеванием тиком или без него или с шизотипальным расстройством), посттравматический стресс и общее состояние беспокойства; нарушения, расстройства, связанные с веществами, включая расстройства, связанные с приемом лекарств (подобные зависимости и злоупотреблению) и вызванные лекарствами расстройства (подобные синдрому отмены лекарства); расстройства общего развития, включая аутизм и болезнь Ретта; расстройства, связанные с дефицитом внимания и разрушительным поведением, такие как дефицит внимания и гиперактивность; нарушения контроля импульсов, подобные агрессии, патологической привязанности к игре; расстройства питания, подобные нервной анорексии и нервной булимии, тучности; нарушения сна, подобные бессоннице; тиковые нарушения, подобные синдрому Туретта; синдрому беспокойства ног; расстройства, характеризуемые ослаблением познавательной способности и памяти, такие как болезнь Альцгеймера, болезнь Крейтцфельда-Якоба, болезнь Хантингтона, болезнь Паркинсона и нейрореабилитация (посттравматические повреждения мозга).
NK1-антагонистические свойства соединений по изобретению проверяли с использованием описанных ниже методик.
ФАРМАКОЛОГИЧЕСКИЕ МЕТОДЫ
Связывание с рецептором для NK1-рецептора человека
Афинность соединений к NK1-рецепторам человека оценивали, используя методы связывания с меченым рецептором. Мембранные препараты готовили из клеток фибробластов яичника китайского хомячка (СНО), в которые был стабильно экспрессирован рецептор NK1 человека. Мембраны инкубировали с [3H]-веществом Р в отсутствие или в присутствии заданных концентраций соединений, разбавленных в подходящем буфере в присутствии ингибитора пептидазы, в течение 10 минут при 25оС. Отделение связанной радиоактивности от свободной проводили фильтрацией через стекловолоконные фильтры Whatman GF/B с двумя 5 сек промывками. Связанную радиоактивность подсчитывали жидкостным сцинтилляционным счетчиком, используя счетчик Betaplate. Измеренную радиоактивность наносили на график как функцию концентрации замещающего испытуемого соединения и рассчитывали кривые замещения, используя четырехпараметрическую логистическую регрессию, дающую величины IC50, т.е. концентрации замещающего соединения, при которых замещено 50% радиолиганда. Значения афинности pK1 рассчитывали, корректируя величины IC50 в соответствии с концентрацией радиолиганда и его афинностью к NK1-рецептору человека по уравнению Чена-Прусова:
pK1 = -log[IC50/(1+S/Kd)],
в котором IC50 является таковым, как описано выше, S представляет концентрацию [3H]-вещества Р, использованную при анализе, выраженную в моль/л, и Kd - есть константа равновесной диссоциации [3H]-вещества Р для NK1-рецептора человека (в моль/л).
Измерения сАМР
Эффект испытуемых соединений при образовании циклической АМФ (сАМР) оценивали, используя клетки фибробластов СНО, стабильно экспрессированные клонированными рецепторами NK1 человека. В дополнение к сочетанию с фосфолипазой С NK1-рецепторы человека способны также стимулировать аденилатциклазу, которая превращает ATP в cAMP. Для испытаний клетки культивировали в 24-луночных планшетах. Перед экспериментами среду заменяли не содержащей сыворотки культуральной средой α-DMEM, содержащей [3H]-аденин, который захватывается клетками и последовательно превращается в меченый радиоактивностью аденозин, AMP, ADP и, наконец, в меченый радиоактивностью ATP. Спустя 2 часа клетки дважды промывают забуференным фосфатом солевым раствором (pH 7,4) в присутствии 1 мМ изобутилметилксантина (IBMX, ингибитора фосфодиэстераз, которые гидролизуют cAMP в AMP). Вслед за этим клетки стимулировали 10 нМ веществом Р в отсутствие или присутствии испытуемых соединений в соответствующих разведениях в PBS/IBMX в течение 20 мин. После стимуляции среду отсасывали и клетки экстрагировали 5% трихлоруксусной кислотой. Радиомеченные ATP и cAMP извлекали из экстракта, используя последовательную колоночную хроматографию. Экстракты разделяли ионообменной хроматографией на колонках DOWEX 50WX4, позволяющих извлекать ATP. Колонки последовательно помещали на верх колонок с оксидом алюминия и элюировали водой. Извлечение cAMP проводили элюированием колонок с оксидом алюминия 100 мМ имидазолом (pH 7,4). Во фракциях и ATP, и cAMP определяли радиоактивность, используя жидкостной сцинтиляционный счетчик, и рассчитывали коэффициенты превращения как:
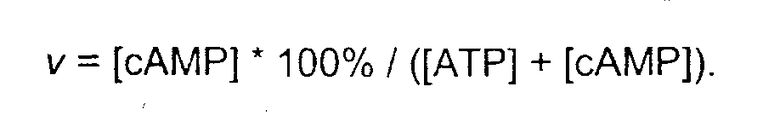
Соотношения «концентрация-отклик» строили нанесением на график превращения cAMP как функции концентрации соединения, и рассчитывали значения IC50 четырехпараметрической логистической регрессией. Величины антагонистической активности (pA2) рассчитывали, используя уравнение:
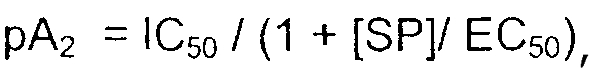
в котором IC50 испытуемого соединения получали из соотношения концентрация-эффект, [SP] представляет концентрацию вещества Р (в моль/л, обычно 10 нМ), и EC50 представляет активность вещества Р по отношению к клонированным NK1-рецепторам человека.
Индуцированное NK1-агонистом подергивание лап у песчанок
Способность NK1-антагонистов антагонизировать подергивание ног, вызванное центральным введением NK1-агонистов, была показана ранее (Rupniak and Williams, 1994 (Eur. J. Pharmacol. 265: 179), Bristow and Young, 1994 (Eur. J. Pharmacol. 254: 245)). Поэтому авторы использовали данную модель для оценки in vivo активности соединений по изобретению.
За 60 мин до анестезии N2O (0,8 л/мин), галотаном (3%) и O2 (0,8 л/мин) самцам песчанки (40-60 г, Charles River) вводили носитель или испытуемое соединение (перорально). После успешной общей анестезии устанавливали подачу анестезирующего агента: N2O (0,6 л/мин), галотан (1,5%) и О2 (0,6 л/мин), и делали надрез по средней линии скальпа. GR 73632 вводили в церебровентрикулярную область (АР - 0,5 мм, L - 1,2 мм и вертикаль - 4,5 мм от темени). После выхода из анестезии (около 3-4 мин) в течение 5 минут регистрировали реакцию подергивания лап. Выбранным критерием антагонизма данной реакции было определено замедление подергивания в течение ≥ 5 мин.
Соединения по изобретению обладают высокой афинностью к NK1-рецепторам с величиной рКi ≥ 7,0 в описанном выше опыте по связыванию. Соединения по изобретению являются также активными в сАМР тесте, причем значения их рА2 находятся в линейной связи со значениями их рКi. Некоторые из соединений, охватываемых изобретением, проникают через гематоэнцефалический барьер, о чем свидетельствует их активность в испытании на индуцированном агонистом нейрокинина подергивании лап у песчанок. Данное свойство делает их полезными для лечения расстройств ЦНС.
Изобретение будет дополнительно пояснено следующими частными примерами.
Пример 1 (см. схему 1)
Стадия 1: К смеси гидрохлорида диэтилового сложного эфира транс-пирролидин-2,5-дикарбоновой кислоты (4,2 г) и Nα-карбобензилокси-D-триптофана (10,0 г) в ацетонитриле (200 мл) добавляли 1,3-диизопропилкарбодиимид (2,6 мл) и затем 4-диметиламинопиридин (˜20 мг). Реакционную смесь перемешивали в течение ночи при комнатной температуре, затем добавляли диизопропилэтиламин (2,5 мл) и перемешивание продолжали еще одни сутки. Реакцию гасили NaOH (водн., 2 н.), и реакционную смесь экстрагировали этилацетатом. Органический слой сушили (Na2SO4), фильтровали и концентрировали в вакууме. Остаток очищали флэш-хроматографией (SiO2, MTBE), получая смесь (2R,5R,2'R) и (2S,5S,2'R) диэтилового сложного эфира 1-[2-бензилоксикарбониламино-3-(1Н-индол-3-ил)пропионил]пирролидин-2,5-дикарбоновой кислоты (8,25 г, 90%). Rf 0,62 (EtOAc) (промежуточные соединения 1 и 2).
Стадия 2: Смесь (2R,5R,2'R) и (2S,5S,2'R) диэтилового эфира 1-[2-бензилоксикарбониламино-3-(1Н-индол-3-ил)пропионил]пирролидин-2,5-дикарбоновой кислоты (8,5 г), палладия на угле (10%, ˜250 мг) и этанола (250 мл) гидрировали в течение ночи Н2 (1 атм). Катализатор удаляли фильтрацией через целит и оставшийся раствор концентрировали в вакууме. Полученные два диастереомера могут быть разделены флэш-хроматографией (SiO2, CH2Cl2/MeOH/NH4OH 95:4,5:0,5), получая этиловый сложный эфир (3R,6S,8aS)-3-(1H-индол-3-илметил)-1,4-диоксооктагидропирроло[1,2-a]пиразин-6-карбоновой кислоты (2,3 г) и этилового сложного эфира (3R,6R,8aR)-3-(1Н-индол-3-илметил)-1,4-диоксооктагидропирроло[1,2-a]пиразин-6-карбоновой кислоты (1,75 г) (промежуточные соединения 3 и 4).
Стадия 3: К суспензии гидрида литий-алюминия (1,2 г) в ТГФ (50 мл) добавляли раствор этилового эфира (3R,6R,8aR)-3-(1Н-индол-3-илметил)-1,4-диоксооктагидропирроло[1,2-a]пиразин-6-карбоновой кислоты (1,75 г) в ТГФ (50 мл). Полученную смесь нагревали при кипении с обратным холодильником в течение 3 часов, затем добавляли по каплям смесь воды (3 мл) и ТГФ (30 мл), после чего добавляли 50% гидроксид натрия (водн., 0,5 мл) и нагревание при кипении с обратным холодильником продолжали в течение 2 часов. После охлаждения до комнатной температуры добавляли диизопропилэтиламин (4 мл) и 3,5-бис(трифторметил)бензоилхлорид (2,4 мл), и перемешивание продолжали в течение ночи. Добавляли этилацетат и смесь экстрагировали бикарбонатом натрия (5% водн. раствор). Органический слой сушили (Na2SO4), фильтровали и концентрировали в вакууме. Остаток очищали флэш-хроматографией (SiO2, CH2Cl2/MeOH/NH4OH 95:4,5:0,5), получая 3,5-бис(трифторметил)фенил[(3R,6R,8aR)-6-гидроксиметил-3-(1Н-индол-3-илметил)гексагидропирроло[1,2-a]пиразин-2-ил]метанон (1,49 г). MH+ 526; Rf 0,20 (EtOAc) (соединение 1).
Стадия 4: Смесь (3,5-бис(трифторметил)фенил)-[(3R,6R,8aR)-6-гидроксиметил-3-(1Н-индол-3-илметил)гексагидропирроло[1,2-a]пиразин-2-ил]метанона (1,30 г), трифенилфосфина (1,2 г), четыреххлористого углерода (4 мл) и ацетонитрила (40 мл) нагревали при 70°С в течение 6 часов. После охдаждения до комнатной температуры растворитель удаляли в вакууме. Остаток очищали ионообменной хроматографией в колонке с сильным катионным ионообменником (SCX), получая 3,5-бис(трифторметил)фенил[(3R,6R,8aR)-6-хлорметил-3-(1Н-индол-3-илметил)гексагидропирроло[1,2-a]пиразин-2-ил]метанон (1,06 г) (промежуточное соединение 5).
Стадия 5: К раствору (3,5-бис(трифторметил)фенил)-[(3R,6R,8aR)-6-хлорметил-3-(1Н-индол-3-илметил)гексагидропирроло[1,2-a]пиразин-2-ил]метанона (1,06 г) в диметилформамиде (15 мл) добавляли морфолин (15 мл). Полученную смесь нагревали при 120°С в течение 6 часов. После охдаждения до комнатной температуры добавляли этилацетат и 5% водный бикарбонат натрия и слои разделяли. Органический слой сушили и концентрировали в вакууме. Остаток очищали флэш-хроматографией (SiO2, MTBE/MeOH/NH4OH 95:4,5:0,5), получая 3,5-бис(трифторметил)фенил[(3R,6R,8aR)-3-(1Н-индол-3-илметил)-6-(морфолин-4-илметил)гексагидропирроло[1,2-a]пиразин-2-ил]метанона (407 мг). Rf 0,46 (MTBE/MeOH/NH4OH 95:4,5:0,5) (соединение 2).
Подобным образом были получены следующие соединения:
Соединение 3: MH+ 595, Rf 0,71 (CH2Cl2/MeOH/NH4OH 90:10:1)
Соединение 4: Rf 0,38 (CH2Cl2/MeOH/NH4OH 90:10:1)
Пример 2 (см. схему 2)
Стадия 1: К смеси Nα-9-фторенилметоксикарбонил-L-транс-4-гидроксипролина (10,2 г), гидрохлорида метилового сложного эфира D-триптофана (8,1 г) и гексафторфосфата бензотриазол-1-илокситрис(пирролидино)фосфония (15 г) в ацетонитриле (200 мл) добавляли диизопропилэтиламин (15 мл) при 0оС. Полученную смесь перемешивали в течение ночи при комнатной температуре. Растворитель удаляли в вакууме, остаток растворяли в этилацетате и экстрагировали дважды водой и дважды 2 М соляной кислотой. Органический слой сушили (Na2SO4), фильтровали и концентрировали в вакууме. Остаток растворяли в ацетонитриле (200 мл) и добавляли пиперидин (25 мл), спустя 2 часа при комнатной температуре образовавшийся 1-(9Н-фторен-9-илметил)пиперидин удаляли фильтрацией, а оставшийся раствор оставляли на 72 часа. Образовавшееся кристаллическое вещество отбирали путем фильтрации, получая (3R,7R,8aS)-7-гидрокси-3-(1Н-индол-3-илметил)гексагидропирроло[1,2-a]пиразин-1,4-дион (7,4 г) (промежуточное соединение 6).
Стадия 2: К раствору (3R,7R,8aS)-7-гидрокси-3-(1Н-индол-3-илметил)гексагидропирроло[1,2-a]пиразин-1,4-диона (2,5 г) в диметилформамиде (30 мл) добавляли имидазол (1,7 г) и трет-бутилхлордифенилсилан (4,35 мл). Полученную смесь перемешивали в течение ночи при комнатной температуре и затем распределяли между водой и этилацетатом. Органический слой сушили, фильтровали и концентрировали в вакууме. Остаток очищали флэш-хроматографией (SiO2, EtOAc), получая (3R,7R,8aS)-7-(трет-бутилдифенилсиланилокси)-3-(1Н-индол-3-илметил)гексагидропирроло[1,2-a]пиразин-1,4-дион (4,6 г, 99%) (промежуточное соединение 7).
Стадия 3: К суспензии гидрида лития-алюминия (1,8 г) в ТГФ (120 мл) добавляли раствор (3R,7R,8aS)-7-(трет-бутилдифенилсиланилокси)-3-(1Н-индол-3-илметил)гексагидропирроло[1,2-a]пиразин-1,4-диона (4,6 г) в ТГФ (35 мл). Полученную смесь нагревали при кипении с обратным холодильником в течение ночи, затем по каплям добавляли смесь воды (1,8 мл) и ТГФ (30 мл), с последующим вводом гидроксида натрия (2 М, 2×1,8 мл). Образовавшиеся соли удаляли фильтрацией и оставшийся раствор концентрировали в вакууме. Остаток суспендировали в этилацетате (100 мл) и ТГФ (20 мл), добавляли диизопропилэтиламин (5 мл) и 3,5-бис(трифторметил)бензоилхлорид (2 мл), и смесь перемешивали при комнатной температуре в течение ночи. Смесь экстрагировали водой, сушили, фильтровали и концентрировали в вакууме. Остаток очищали флэш-хроматографией (SiO2, МТВЕ/гексан 2:1), получая (3R,7R,8aS)-2-(3,5-бис(трифторметил)бензоил)-3-(1Н-индол-3-илметил)октагидропирроло[1,2-a]пиразин-7-ильный сложный эфир 3,5-бис(трифторметил)бензойной кислоты (4,2 г, 65%). МН+ 752 (промежуточное соединение 8).
Стадия 4: Смесь (3R,7R,8aS)-2-(3,5-бис(трифторметил)бензоил)-3-(1Н-индол-3-илметил)октагидропирроло[1,2-a]пиразин-7-ильного эфира 3,5-бис(трифторметил)бензойной кислоты (4,2 г), 1,4-диоксана (45 мл), метанола (12 мл) и 4 М гидроксида натрия (водн., 3 мл) перемешивали в течение 30 мин при комнатной температуре. Смесь концентрировали и остаток распределяли между водой и этилацетатом. Органический слой сушили, фильтровали и концентрировали, получая 3,5-бис(трифторметил)фенил[(3R,7R,8aS)-7-гидрокси-3-(1Н-индол-3-илметил)гексагидропирроло[1,2-a]пиразин-2-ил]метанон (4 г) (соединение 5).
Стадия 5а: К раствору 3,5-бис(трифторметил)фенил[(3R,7R,8aS)-7-гидрокси-3-(1Н-индол-3-илметил)гексагидропирроло[1,2-a]пиразин-2-ил]метанона (0,54 г) в этилацетате (20 мл) добавляли диизопропилэтиламин (2 мл) и метансульфонилхлорид (0,2 мл). Полученную смесь перемешивали при комнатной температуре в течение ночи и концентрировали в вакууме. К остатку добавляли морфолин (4 мл) и смесь нагревали при 95°С в течение 4 часов. Избыток морфолина удаляли в вакууме и остаток очищали препаративной ВЭЖХ, получая 3,5-бис(трифторметил)фенил[(3R,7S,8aS)-3-(1Н-индол-3-илметил)-7-(морфолин-4-ил)гексагидропирроло[1,2-a]пиразин-2-ил]метанон (81 мг). МН+ 581 (соединение 6).
Стадия 5b: К раствору 3,5-бис(трифторметил)фенил[(3R,7R,8aS)-7-гидрокси-3-(1Н-индол-3-илметил)гексагидропирроло[1,2-a]пиразин-2-ил]метанона (0,68 г) в диметилформамиде (20 мл) добавляли диизопропилэтиламин (2 мл) и метансульфонилхлорид (0,2 мл). Полученную смесь перемешивали в течение 1 часа при комнатной температуре, затем добавляли бромид цезия (в избытке) и смесь перемешивали в течение 5 часов при 95°С. Промежуточный бромид выделяли распределением между водой и этилацетатом, концентрированием органического слоя и очисткой хроматографией. К бромиду добавляли морфолин (4 мл) и смесь нагревали при 95°С в течение 4 часов. Избыток морфолина удаляли в вакууме и остаток очищали препаративной ВЭЖХ, получая 3,5-бис(трифторметил)фенил[(3R,7R,8aS)-3-(1Н-индол-3-илметил)-7-(морфолин-4-ил)гексагидропирроло[1,2-a]пиразин-2-ил]метанон (101 мг). МН+ 581 (соединение 6).
Аналогичным образом были получены соединения 8 и 9.
Пример 3 (см. схему 3)
Стадия 1: К раствору 1-бензилового сложного эфира 5-оксопиперидин-1,2-дикарбоновой кислоты (93,5 г) в ацетонитриле (1 л) добавляли раствор диизопропилкарбодиимида (53 мл) в ацетонитриле (50 мл) и смесь охлаждали до 5°С. К полученной суспензии порциями добавляли гидрохлорид метилового сложного эфира D-триптофана (85,5 г) и по каплям раствор диизопропилэтиламина (58,6 мл) в ацетонитриле (50 мл). Полученную смесь перемешивали в течение 18 часов при комнатной температуре, затем фильтровали и концентрировали в вакууме. Остаток растворяли в дихлорметане, дважды промывали соляной кислотой (1 М) и дважды водой, сушили, фильтровали и концентрировали в вакууме, получая смесь бензиловых сложных эфиров (2R,2'R)- и (2S,2'R)-2-[2-(1H-индол-3-ил)-1-метоксикарбонилэтилкарбамоил]-5-оксопиперидин-1-карбоновой кислоты (171,2 г) (промежуточные соединения 9 и 10), которую использовали без очистки на следующей стадии.
Стадия 2: Смесь бензиловых эфиров (2R,2'R)- и (2S,2'R)-2-[2-(1H-индол-3-ил)-1-метоксикарбонилэтилкарбамоил]-5-оксопиперидин-1-карбоновой кислоты (138,9 г), щавелевой кислоты (180 г) и 1,3-пропандиола (87 мл) в ацетонитриле (1,5 л) нагревали при 40°С в течение 20 часов. После этого растворитель удаляли в вакууме и остаток очищали флэш-хроматографией (SiO2, CH2Cl2/MeOH 99:1), получая смесь бензиловых сложных эфиров (9R,2'R)- и (9S,2'R)-9-[2-(1H-индол-3-ил)-1-метоксикарбонилэтилкарбамоил]-1,5-диокса-8-азаспиро[5.5]ундекан-8-карбоновой кислоты (118 г). МН+ 536, Rf 0,07 (CH2Cl2/MeOH 99:1) (промежуточные соединения 11 и 12).
Стадия 3: К раствору смеси бензиловых эфиров (9R,2'R)- и (9S,2'R)-9-[2-(1H-индол-3-ил)-1-метоксикарбонилэтилкарбамоил]-1,5-диокса-8-азаспиро[5.5]ундекан-8-карбоновой кислоты (92,8 г) в метаноле (1 л) добавляли 10% палладий на угле (˜5 г). Полученную смесь гидрировали с Н2 (1 атм) в течение ночи при комнатной температуре. Катализатор удаляли фильтрацией через целит и оставшийся раствор концентрировали в вакууме, получая смесь метиловых сложных эфиров (2R,9'R)- и (2R,9'S)-2-[(1,5-диокса-8-азаспиро[5.5]ундекан-9-карбонил)амино]-3-(1H-индол-3-ил)пропионовой кислоты (69,0 г). Rf 0,24 (CH2Cl2/MeOH/NH4OH 92:7,5:0,5) (промежуточные соединения 13 и 14).
Стадия 4: Смесь метиловых эфиров (2R,9'R)- и (2R,9'S)-2-[(1,5-диокса-8-азаспиро[5.5]ундекан-9-карбонил)амино]-3-(1H-индол-3-ил)пропионовой кислоты (69,0 г) и уксусной кислоты (9 мл) в ацетонитриле (900 мл) нагревали при кипении с обратным холодильником в течение ночи. После охлаждения до комнатной температуры смесь концентрировали до примерно одной трети от ее первоначального объема. Образовавшийся осадок отделяли фильтрацией, получая (3R,9aS)-3-(1H-индол-3-илметил)гексагидроспиро[2H-пиридо[1,2-a]пиразин-1,4-дион-7,2'-[1,3]диоксан] (23,7 г, Rf 0,34 (Et2O/MeOH 9:1)). Концентрирование фильтрата и очистка остатка флэш-хроматографией (SiO2, Et2O/MeOH 9:1) дают (3R,9aR)-3-(1H-индол-3-илметил)гексагидроспиро[2H-пиридо[1,2-a]пиразин-1,4-дион-7,2'-[1,3]диоксан] (20,6 г, Rf 0,18 (Et2O/MeOH 9:1) (промежуточное соединение 15).
Стадия 5: К суспензии гидрида лития-алюминия (10,6 г) в ТГФ (500 мил) добавляли по каплям раствор (3R,9aR)-3-(1H-индол-3-илметил)гексагидроспиро[2H-пиридо[1,2-a]пиразин-1,4-дион-7,2'-[1,3]диоксана] (20,6 г) в ТГФ (100 мл), и полученную смесь нагревали при кипении с обратным холодильником в течение 2 суток. После охлаждения до 5°С по каплям добавляли воду (9,2 мл), 2 М гидроксид натрия (водн., 18,4 мл) и опять воду (9,2 мл). Полученную смесь нагревали при кипении с обратным холодильником в течение еще одного часа, охлаждали до комнатной температуры, фильтровали через целит и концентрировали в вакууме, получая (3R,9aR)-3-(1H-индол-3-илметил)октагидроспиро[2H-пиридо[1,2-a]пиразин-7,2'-[1,3]диоксан] (19,7 г, Rf 0,16 (CH2Cl2/MeOH/NH4OH 92:7,5:0,5)) (промежуточное соединение 16), которое без очистки использовали на следующей стадии.
Стадия 6: К раствору (3R,9aR)-3-(1H-индол-3-илметил)октагидроспиро[2H-пиридо[1,2-a]пиразин-7,2'-[1,3]диоксана] (19,7 г) в дихлорметане добавляли диизопропилэтиламин (9,6 мл) при комнатной температуре и при 5°С, 3,5 бис(трифторметил)бензоилхлорид (10 мл). Полученную смесь перемешивали при комнатной температуре в течение ночи, концентрировали в вакууме и очищали колоночной хроматографией (SiO2, CH2Cl2/MeOH/NH4OH 96:3,75:0,25), получая (3R,9aR)-2-[3,5-бис(трифторметил)бензоил]-3-(1H-индол-3-илметил)октагидроспиро[2H-пиридо[1,2-a]пиразин-7,2'-[1,3]диоксан] (30,0 г). Rf 0,35 (CH2Cl2/MeOH/NH4OH 96:3,75:0,25) (соединение 10).
Стадия 7: Смесь (3R,9aR)-2-[3,5-бис(трифторметил)бензоил]-3-(1H-индол-3-илметил)октагидроспиро[2H-пиридо[1,2-a]пиразин-7,2'-[1,3]диоксана] (30,0 г) в уксусной кислоте (150 мл) и 6 М соляной кислоты (150 мл) нагревали при 40°С в течение трех суток. После охлаждения до комнатной температуры добавляли дихлорметан (750 мл) и 2 М гидроксид натрия (водн., 1700 мл). Слои разделяли и водный слой дважды экстрагировали дихлорметаном. Объединенные органические слои промывали водой, концентрировали в вакууме и очищали флэш-хроматографией (SiO2, CH2Cl2/MeOH 98:2), получая (3R,9aR)-2-[3,5-бис(трифторметил)бензоил]-3-(1H-индол-3-илметил)октагидро-2H-пиридо[1,2-a]пиразин-7-он (21,7 г). Rf 0,12 (CH2Cl2/MeOH 98:2) (соединение 11).
Стадия 8: К суспензии (3R,9aR)-2-[3,5-бис(трифторметил)бензоил]-3-(1H-индол-3-илметил)октагидро-2H-пиридо[1,2-a]пиразин-7-она (5,6 г) в уксусной кислоте (75 мл) добавляли триацетоксиборгидрид натрия (6,78 г). Полученную смесь перемешивали в течение одного часа при комнатной температуре и затем выливали в воду и подщелачивали 2 М гидроксидом натрия (водн.). Образовавшийся осадок отфильтровывали, промывали водой, суспендировали в толуоле и концентрировали в вакууме, получая 3,5-бис(трифторметил)фенил[(3R,7S,9aR)-7-гидрокси-3-(1H-индол-3-илметил)октагидропиридо[1,2-a]пиразин-2-ил]метанон (5,7 г). Rf 0,35 (CH2Cl2/MeOH 9:1) (соединение 12).
Стадия 9: К суспензии 3,5-бис(трифторметил)фенил[(3R,7S,9aR)-7-гидрокси-3-(1H-индол-3-илметил)октагидропиридо[1,2-a]пиразин-2-ил]метанона (3,85 г) в ацетонитриле добавляли четырехбромистый углерод (11,6 г) и трифенилфосфин (9,18 г), полученную смесь перемешивали при комнатной температуре в течение ночи. Образовавшийся осадок удаляли фильтрацией и фильтрат концентрировали в вакууме. Остаток очищали флэш-хроматографией (SiO2, CH2Cl2/MeOH 98:2), получая 3,5-бис(трифторметил)фенил[(3R,9aR)-7-бром-3-(1H-индол-3-илметил)октагидропиридо[1,2-a]пиразин-2-ил]метанон (3,5 г). МН+ 588, Rf 0,38 (CH2Cl2/MeOH 97:3) (промежуточное соединение 17).
Стадия 10: Смесь 3,5-бис(трифторметил)фенил[(3R,9aR)-7-бром-3-(1H-индол-3-илметил)октагидропиридо[1,2-a]пиразин-2-ил]метанона (2,94 г) и морфолина (0,92 мл) в ацетонитриле (100 мл) нагревали при 80°С в течение 40 часов. После охлаждения до комнатной температуры смесь концентрировали в вакууме и остаток очищали флэш-хроматографией (SiO2, CH2Cl2/MeOH/NH4OH 980:18,75:1,25), получая 3,5-бис(трифторметил)фенил[(3R,7R,9aR)-3-(1H-индол-3-илметил)7-морфолин-4-илоктагидропиридо[1,2-a]пиразин-2-ил]метанон (1,6 г, Rf 0,33 (CH2Cl2/MeOH/NH4OH 92:7,5:0,5)) (соединение 13) и 3,5-бис(трифторметил)фенил[(3R,7S,9aR)-3-(1Н-индол-3-илметил)-7-морфолин-4-илоктагидропиридо[1,2-a]пиразин-2-ил]метанон (1,28 г, Rf 0,27 (CH2Cl2/MeOH/NH4OH 92:7,5:0,5)) (соединение 14).
Аналогичным образом были получены соединения 15-23 и 33-40.
Пример 4 (см. схему 3)
Смесь (3R,9aR)-2-[3,5-бис(трифторметил)бензоил]-3-(1H-индол-3-илметил)октагидро-2H-пиридо[1,2-a]пиразин-7-она (0,785 г, см. пример 3, стадии 1-7), пирролидона (0,107 г), уксусной кислоты (0,09 г) и триацетоборгидрида натрия (0,47 г) в 1,2-дихлорэтане (60 мл) перемешивали в течение трех суток при комнатной температуре. Полученную смесь выливали на воду, подщелачивали бикарбонатом натрия (5% водн. р-р) и экстрагировали дихлорметаном. Органический слой концентрировали и остаток очищали колоночной хроматографией (SiO2, CH2Cl2/MeOH/NH4OH 92:7,5:0,5), получая [3,5-бис(трифторметил)фенил[(3R,7R,9aR)-3-(1H-индол-3-илметил)-7-пирролидин-1-илоктагидропиридо[1,2-a]пиразин-2-ил)метанон (0,20 г, Rf 0,23 (CH2Cl2/MeOH/NH4OH 92:7,5:0,5)) (соединение 24) и 3,5-бис(трифторметил)фенил[(3R,7S,9aR)-3-(1H-индол-3-илметил)-7-пирролидин-1-илоктагидропиридо[1,2-a]пиразин-2-ил)метанон (0,48 г, Rf 0,15 (CH2Cl2/MeOH/NH4OH 92:7,5:0,5)) (соединение 25).
Аналогичным образом были получены соединения 26-28.
Пример 5 (см. схему 3)
Стадия 1: К суспензии (3R,7S,9aR)-(3-бензил-7-гидроксиоктагидропиридо[1,2-a]пиразин-2-ил)-(3,5-бис(трифторметил)фенил)метанона (3,3 г, получен аналогично примеру 3, стадии 1-8) в дихлорметане (100 мл) добавляли диизопропилэтиламин (2,4 мл) и метансульфонилхлорид (0,8 мл) при 5°С. Полученную смесь перемешивали при комнатной температуре в течение 15 минут и концентрировали в вакууме. Остаток очищали колоночной хроматографией (SiO2, CH2Cl2/MeOH 95:5), получая (3R,7S,9aR)-3-бензил-2-(3,5-бис(трифторметил)бензоил)октагидропиридо[1,2-a]пиразин-7-ильный сложный эфир метансульфоновой кислоты (3,8 г). Rf 0,67 (CH2Cl2/MeOH 95:5) (промежуточное соединение 18).
Стадия 2: Смесь (3R,7S,9aR)-3-бензил-2-(3,5-бис(трифторметил)бензоил)октагидропиридо[1,2-a]пиразин-7-ильного эфира метансульфоновой кислоты (1,97 г) и морфолина (0,44 мл) в ацетонитриле (50 мл) нагревали при 80°С в течение 40 часов. После охлаждения до комнатной температуры смесь концентрировали в вакууме и остаток очищали флэш-хроматографией (SiO2, CH2Cl2/MeOH/NH4OH 980:18,75:1,25), получая 3,5-бис(трифторметил)фенил[(3R,7R,9aR)-3-бензил-7-морфолин-4-илоктагидропиридо[1,2-a]пиразин-2-ил]метанон (0,97 г, Rf 0,24 (CH2Cl2/MeOH/NH4OH 96:3,75:0,25)) (соединение 29) и 3,5-бис(трифторметил)фенил[(3R,7S,9aR)-3-бензил-7-морфолин-4-илоктагидропиридо[1,2-a]пиразин-2-ил]метанон (0,75 г, Rf 0,15 (CH2Cl2/MeOH/NH4OH 96:3,75:0,25)) (соединение 30).
Аналогичным образом были получены следующие соединения:
соединения 31 и 32,
соединение 41: Rf 0,11 (CH2Cl2/MeOH/NH4OH 96:3,75:0,25),
соединение 42: Rf 0,06 (CH2Cl2/MeOH/NH4OH 96:3,75:0,25),
соединение 43: Rf 0,08 (CH2Cl2/MeOH/NH4OH 96:3,75:0,25),
соединение 44: Rf 0,05 (CH2Cl2/MeOH/NH4OH 96:3,75:0,25),
соединения 45, 46, 55, 56, 58 и 59,
соединение 57: Rf 0,10 (CH2Cl2/MeOH/NH4OH 98:1,85:0,15).
Пример 6 (эфиры-пролекарства)
К раствору (3,5-бис(трифторметил)фенил)-[(3R,6R,8aR)-6-гидроксиметил-3-(1Н-индол-3-илметил)гексагидропирроло[1,2-a]пиразин-2-ил]метанона (0,52 г) в ацетонитриле (40 мл) при комнатной температуре добавляли диизопропилэтиламин (0,17 мл) и раствор ацетилхлорида (0,8 мл) в ацетонитриле (10 мл). Полученную смесь перемешивали при комнатной температуре в течение ночи и концентрировали в вакууме. Остаток очищали колоночной хроматографией (SiO2, CH2Cl2/MeOH 97:3), получая 2-(3,5-бис(трифторметил)бензоил)-3-(1Н-индол-3-илметил)гексагидропирроло[1,2-a]пиразин-6-илметиловый сложный эфир метансульфоновой-уксусной кислоты (0,32 г, Rf 0,33 (CH2Cl2/MeOH 97:3)) (соединение 47).
Аналогичным образом были получены следующие соединения:
соединение 48: Rf 0,50 (CH2Cl2/MeOH 97:3),
соединение 49: Rf 0,51 (CH2Cl2/MeOH 97:3),
соединение 50: Rf 0,50 (CH2Cl2/MeOH 97:3),
соединение 51: Rf 0,12 (CH2Cl2/MeOH 98:2),
соединение 52: Rf 0,47 (CH2Cl2/MeOH 95:5),
соединение 53: Rf 0,21 (CH2Cl2/MeOH 97:3),
соединение 54.
Соединения по изобретению, как показано на примерах соединений 1-59, обладают высокой афинностью к NK1-рецепторам с величиной рКi ≥ 7,0 в описанном выше опыте по связыванию. Как подробно показано в приведенных примерах, некоторые из этих соединений были использованы в качестве промежуточных при синтезе других соединений. Соединения по изобретению являются активными в сАМР тесте, значения их рА2 находятся в линейной связи со значениями их рКi. Некоторые из соединений, охватываемых изобретением, проникают через гематоэнцефалический барьер, о чем свидетельствует их активность при испытании с индуцированным агонистом нейрокинина подергиванием лап у песчанок. Данное свойство делает их полезными для лечения расстройств ЦНС.
| название | год | авторы | номер документа |
|---|---|---|---|
| ПРОИЗВОДНЫЕ ГЕКСА- И ОКТАГИДРОПИРИДО[1,2-а]ПИРАЗИНА С NK-АНТАГОНИСТИЧЕСКОЙ АКТИВНОСТЬЮ | 2004 |
|
RU2349591C2 |
| ПРОИЗВОДНЫЕ ОКСИМА ПИПЕРАЗИНА, ОБЛАДАЮЩИЕ АНТАГОНИСТИЧЕСКОЙ АКТИВНОСТЬЮ ПО ОТНОШЕНИЮ К РЕЦЕПТОРУ НЕЙРОКИНИНА-1 (НК-1). | 2002 |
|
RU2273639C2 |
| ПРОИЗВОДНЫЕ 4-ФЕНИЛПИРИМИДИНА И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 2000 |
|
RU2243221C2 |
| ИМИДАЗОПИРРОЛОПИРАЗИНОВЫЕ ПРОИЗВОДНЫЕ, ПОЛЕЗНЫЕ ДЛЯ ЛЕЧЕНИЯ ЗАБОЛЕВАНИЙ, ВЫЗВАННЫХ АНОМАЛЬНОЙ АКТИВНОСТЬЮ ПРОТЕИНКИНАЗ Jak1, Jak3 ИЛИ Syk | 2010 |
|
RU2570416C2 |
| СЕЛЕКТИВНЫЕ АНТАГОНИСТЫ Н4-ГИСТАМИНОВЫХ РЕЦЕПТОРОВ ДЛЯ ЛЕЧЕНИЯ ВЕСТИБУЛЯРНЫХ НАРУШЕНИЙ | 2009 |
|
RU2589846C2 |
| ПРОИЗВОДНЫЕ ПИРРОЛИДИНА ДЛЯ ПРИМЕНЕНИЯ В КАЧЕСТВЕ ИНГИБИТОРОВ КАТЕПСИНА | 2011 |
|
RU2548684C2 |
| ПРОИЗВОДНЫЕ АРОИЛПИПЕРАЗИНА, СПОСОБ ИХ ПОЛУЧЕНИЯ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ИХ ОСНОВЕ И СПОСОБ ЛЕЧЕНИЯ | 1998 |
|
RU2258702C2 |
| Имидазопирролопиразиновые производные, полезные для лечения заболеваний, вызванных аномальной активностью протеинкиназ Jak1, Jak3 или Syk | 2010 |
|
RU2711869C2 |
| СПОСОБ ЛЕЧЕНИЯ | 2012 |
|
RU2621148C2 |
| 2-ОКСА-5-АЗАБИЦИКЛО[2.2.1]ГЕПТАН-3-ИЛЬНЫЕ ПРОИЗВОДНЫЕ | 2015 |
|
RU2697651C2 |
Настоящее изобретение относится к производным диазабициклических алканов и их фармацевтически приемлемым солям и стереоизомерам общей формулы
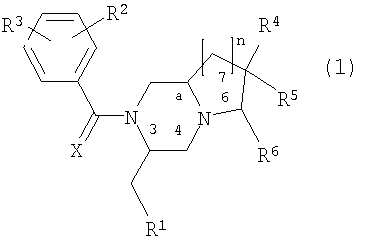
где R1 фенил, 3-индолил или бензо[b]тиофен-3-ил, указанная фенильная группа может быть замещена галогеном; R2 и R3 независимо представляют галоген, СН3 или CF3; R4 представляет Н, ОН, СН2ОН, NH2, диалкил(1-3С)N, пирролидин-1-ил, пиперидин-1-ил, морфолин-4-ил или морфолин-4-ил, замещенный одной или двумя метальными или метоксиметильными группами, морфолин-4-иламино, морфолин-4-илметил, имидазол-1-ил, тиоморфолин-4-ил, 1,1-диоксотиоморфолин-4-ил или 3-окса-8-азабицикло[3.2.1]окт-8-ил; R5 - водород; R4 и R5 вместе могут представлять кетогруппу 1,3-диоксан-2-ил или 1,3-диоксолан-2-ил; R6 представляет Н, морфолин-4-ил, замещенный метальной группой, или СН2ОН, указанная группа СН2ОН может образовывать сложный эфир с органической кислотой; Х представляет О; n имеет значения 1 или 2.
Соединения обладают антагонистической активностью по отношению к NK1-рецептору. Описаны также промежуточные соединения, используемые в синтезе соединений формулы I, фармацевтическая композиция и применение соединений для изготовления лекарственных средств для лечения, например, таких заболеваний, как хронические боли, воспалительные заболевания и расстройства центральной нервной системы. 4 н. и 3 з.п. ф-лы, 1 табл.
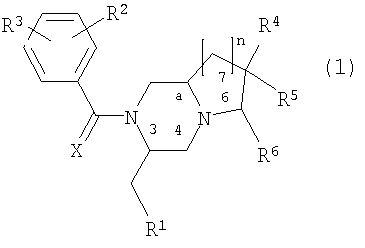
где R1 представляет фенил, 3-индолил или бензо[b]тиофен-3-ил, указанная фенильная группа может быть замещена галогеном;
R2 и R3 независимо представляют галоген, СН3 или CF3;
R4 представляет Н, ОН, СН2ОН, NH2, диалкил(1-3С)N, пирролидин-1-ил, пиперидин-1-ил, морфолин-4-ил или морфолин-4-ил, замещенный одной или двумя метальными или метоксиметильными группами, морфолин-4-иламино, морфолин-4-илметил, имидазол-1-ил, тиоморфолин-4-ил, 1,1-диоксотиоморфолин-4-ил или 3-окса-8-азабицикло[3.2.1]окт-8-ил;
R5 представляет Н, или
R4 и R5 вместе могут представлять кетогруппу 1,3-диоксан-2-ил или 1,3-диоксолан-2-ил;
R6 представляет Н, морфолин-4-ил, замещенный метильной группой, или СН2ОН, указанная группа СН2ОН может образовывать сложный эфир с органической кислотой;
n имеет значения 1 или 2; Х - представляет О;
а является асимметричным атомом углерода 8а или 9а, когда n равно 1 или 2 соответственно,
и их фармацевтически приемлемые соли, включая все возможные стереоизомеры, в которых заместители у асимметричных атомов углерода 3 и "а", а также у потенциально асимметричных атомов углерода 6 и 7, находятся в R-конфигурации или в S-конфигурации.
R6 представляет морфолин-4-илметил или СН2ОН, указанная группа СН2ОН может образовывать сложный эфир с органической кислотой;
R5 представляет водород, и стереохимией которых является 3R.
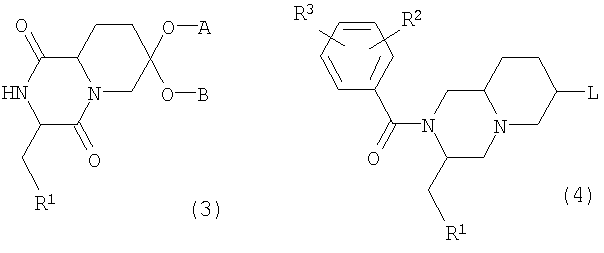
где R1, R2 и R3 имеют значения, приведенные в п.1, А и В представляют циклический или ациклический кеталь и L представляет уходящую группу, такую как хлор, бром или метансульфонат, используемые в синтезе соединений, имеющих формулу (1).
| US, 6001833, 14.12.1999 | |||
| ЕР, 1176144, A1, 30.12.2002. |

