Область техники, к которой относится изобретение
Изобретение относится к области катализа и может быть использовано в качестве катализаторов для получения эфиров акриловой кислоты по реакции метатезиса малеатов с этиленом.
Уровень техники
Известен высокоактивный катализатор Ховейды-Блехерта для метатезиса электронодефицитных олефинов, содержащий фенильный заместитель в орто-положении к изопропоксигруппе бензилиденового фрагмента (US 2004/0176608 А1, 2004).
Известен катализатор Ховейды-Грелы, содержащий элетроноакцепторные группы в мета- и пара-положениях к изопропоксигруппе бензилиденового фрагмента, такие, как нитро-группа (WO 2004/035596 А1, 2004). Высокая активность этого катализатора связана с большой скоростью инициирования, обусловленной частичной потерей электронной плотности на изопропокси-группе и, вследствие этого, слабым удерживанием хелатирущего заместителя рутениевым центром.
Недостатком указанных катализаторов является недостаточный срок эффективной работы, вызванный малым числом оборотов в реакциях метатезиса эфиров малеиновой кислоты (малеатов) с этиленом.
Раскрытие изобретения
Задача, решаемая данным изобретением, состоит в разработке катализатора для реакции метатезиса диалкилмалеатов с этиленом, сохраняющего свою активность в течение длительного периода работы.
Технический результат состоит в увеличении числа оборотов катализатора для реакции метатезиса диалкилмалеатов с этиленом.
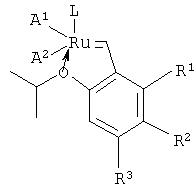
Техническии результат достигается тем, что катализатор для получения эфиров акриловой кислоты по реакции метатезиса диалкилмалеатов с этиленом имеет формулу:

где А1, А2 - хлор,
L - дигидроимидазольный лиганд,
R1, R2, R3 - одинаковые или разные, заместители выбранные из группы, содержащей водород, алкил-, триалкилсилил- или алкокси-группы.
Введение в карбеноидный фрагмент катализатора электронодонорных заместителей, таких как алкил-, алкокси- или триметилсилил- позволяет, увеличить число оборотов за счет снижения скорости инициирования катализатора: снижается скорость разложения активных частиц по бимолекулярному механизму, благодаря уменьшению их концентрации в реакционной смеси.
В качестве дигидроимидазольного лиганда L может использоваться лиганд (IMesH2) формулы:

В частном случае изобретения R1 и R3 представляют собой водород, R2 - триметилсилил, а А1 и А2 - хлор. Тогда катализатор имеет формулу:
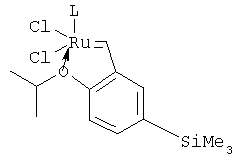
Использование хлорид-ионов в качестве анионов и введение триметилсилильной группы в пара-положение бензилиденового заместителя способствуют увеличению числа оборотов катализатора.
В другом частном случае изобретения R1 и R2 представляют собой водород, R3 - триметилсилил, а А1, А2 - хлор. Тогда катализатор имеет формулу:
Как и в предыдущем случае, введение триметилсилильной группы в мета-положение бензилиденового заместителя способствует увеличению числа оборотов катализатора.
Еще одним вариантом изобретения, при котором также достигается указанный выше технический результат, предлагается катализатор формулы:

При использовании предложенных катализаторов в каталитической композиции, содержащей в качестве второго компонента 2-изопропоксистирол или его производное, достигается увеличение не только числа оборотов катализатора, но и времени его жизни. Соотношение катализатора и 2-изопропоксистирола или его производного составляет: на 1 моль предложенного катализатора приходится от 5 до 100 мольных эквивалентов 2-изопропоксистирола или его производного. Такие каталитические системы наиболее эффективны при 100-120°С в реакциях с отгонкой образующегося акрилата.
В качестве производного 2-изопропоксистирола может быть выбран 2-изопропокси-5-триметилсилилстирол. Благодаря наличию в его структуре электронодонорного заместителя (триметилсильная группа), время жизни каталитической системы и ее число оборотов значительно возрастают.
Осуществление изобретения
Разработка катализатора, обладающего повышенным числом оборотов, для метатезиса диалкилмалеатов с этиленом проводилась на основе катализатора Ховейды второго поколения (1), который является наиболее эффективным в реакции этенолиза малеатов. Проверяемые модификации включали в себя варьирование заместителей в бензилиденовом фрагменте, варьирование имидазольного лиганда и анионов.

Введение в бензилиденовый фрагмент электроноакцепторных заместителей, таких как нитро-группа, привело к получению катализатора (3), обладающего максимальной среди протестированных комплексов продуктивностью (около 2220 ч-1, таблица 2). Однако время жизни этого катализатора составило всего около 70 минут, а число его оборотов не превысило 1500 (таблицы 1 и 2).



Напротив, введение в орто- и(или) пара-положения бензилиденового фрагмента катализатора (1) электронодонорных заместителей, таких, как алкил-, алкокси- или триалкилсилил-, приводит к катализаторам, обладающим продуктивностью, временем жизни и числом оборотов, сравнимыми с таковыми для комплекса (1). Следует отметить, что такие катализаторы обладают более низкой скоростью инициирования по сравнению с комплексами (1) и (3), что обусловлено повышенной электронной плотностью на изопропокси-группе и, как следствие, более прочной связью Ru-О. Наблюдаемый эффект увеличения числа оборотов таких катализаторов обусловлен более низкой концентрацией активных частиц в реакционной смеси.
Наибольшим числом оборотов обладают катализаторы с триметилсилильными группами, такие, как (4) и (5) (1760 и 1900 соответственно, таблицы 1 и 2). Близкие значения продуктивности и числа оборотов были получены для катализатора (6), содержащего метальную группу в бензилиденовом фрагменте (2040 ч-1, 1750 оборотов, таблицы 1 и 2). Числа оборотов для катализаторов (7) и (8), содержащих изопропокси-группу, были несколько ниже, чем, даже, у комплекса (1) (1600 и 1670 оборотов для комплексов 7 и 8, соответственно, таблицы 1 и 2). Время жизни катализаторов во всех случаях составляет около 100-120 минут.
Варьирование в гетероциклическом лиганде влияет на время жизни и число оборотов катализатора в большей степени. Так, применение имидазольного лиганда (как в комплексе 9), вместо дигидроимидазольного (комплекс 1) приводит к уменьшению времени жизни катализатора до 50 минут, а его числа оборотов с 1740 до 600 (таблицы 1 и 2). Увеличение стерических препятствий у рутениевого центра заместителями ароматических колец гетероциклического лиганда способствует росту времени жизни катализатора. Например, катализатор (10), содержащий изопропильные группы в гетероциклическом лиганде, обладает одним из продолжительных времен жизни среди протестированных комплексов (более 5 часов, таблицы 1 и 2), хотя его продуктивность и число оборотов составляют лишь 1080 ч-1 и 1310 соответственно (таблицы 1 и 2). Напротив, катализатор (11), в котором рутениевый центр менее блокирован гетероциклическим лигандом, обладает значительно меньшим временем жизни (около 50 минут, таблицы 1 и 2). Продуктивность и число оборотов при этом также низкие. Наиболее оптимальным является использование в качестве гетероциклического лиганда 2,5-N,N'-ди(2,4,6-триметилфенил)-3,4-дигидроимидазолидена (IMesH2).
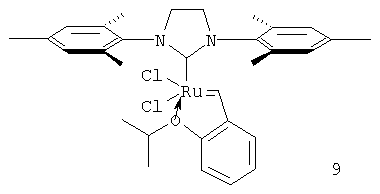

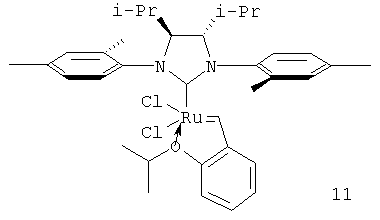
Увеличение размера аниона при рутениевом центре также приводит к росту времени жизни катализатора, хотя продуктивность и его число оборотов при этом снижаются. Так, время жизни катализатора (12) в тестируемых условиях составляет более 5 часов, а число его оборотов менее 100 (таблицы 1 и 2).

В случае проведения реакции с отгонкой образующегося продукта катализатор (1) имеет число оборотов 930. Результаты испытаний других катализаторов представлены в таблице 3. Максимальное число оборотов показал комплекс (13), содержащий нафтилиденовый заместитель.

Число оборотов различных катализаторов в реакции метатезиса диметилмалеата с этиленом в условиях отгонки продукта из реакционной смеси.
Для получения эфиров акриловой кислоты по реакции этенолиза малеатов в условиях отгонки образующегося продукта при атмосферном давлении в ходе реакции более дешевым и эффективным оказалось проведение метатезиса в присутствии каталитической системы, состоящей из заявленного катализатора и производных 2-изопропоксистирола.
В качестве второго компонента каталитической системы тестировались 2-изопропоксистирол (14) и его производные (15) и (16), на основе которых ранее были получены катализаторы, обладающие максимальным числом оборотов. Результаты этих экспериментов представлены в таблице 4.



Влияние количества производного 2-изопропоксистирола на выход метилакрилата и число оборотов катализатора в реакции этенолиза диметилмалеата в условиях отгонки продукта из реакционной смеси.
Наиболее эффективными добавками оказались стиролы (14) и (15) в количестве от 5 до 100 мольных эквивалентов. При использовании этих соединений в количестве от 8 до 25 мольных эквивалентов (по отношению к катализатору) число оборотов катализатора возрастает более чем в 1.6 раза и в тестируемых условиях достигает 1525. В присутствие стирола (16) во всех случаях наблюдалось уменьшение числа оборотов катализатора (таблица 4), несмотря на то, что катализатор (13), полученный из этого олефина, наиболее активен в реакции с отгонкой акрилата.
Изобретение иллюстрируется следующими примерами.
Пример 1.
Синтез катализаторов, реакции с применением бутиллития и реактивов Гриньяра, реакции малеатов с этиленом проводят по стандартным методикам в условиях, исключающих попадание влаги и воздуха в реакционную систему, используя реакторы Шленка, подсоединенные к вакуумной и этиленовой линиям. Катализаторы (1), (2), (3), (6), (7), (8), (9), (10), (12), (IMesH2)(Cl)2(C5H5N)2RuC=CHPh (катализатор Граббса третьего поколения), (Cy3Р)2(Cl)2RuC=CHPh (катализатор Граббса первого поколения) и 2-изопропоксистирол (14) получают по опубликованным методикам. Этилен полимеризационной чистоты (ГОСТ 25070-87) перед использованием высушивают, последовательно пропуская через колонны, наполненные окисью алюминия и цеолитом 13Х, прокаленными при 400°С. Коммерческие диэтилмалеат (97%, фирмы "Aldrich"), диметилмалеат (96%, фирмы "Acros") и дибутилмалеат (97%, фирмы "Acros") очищают перегонкой в вакууме и высушивают над молекулярными ситами. Анализ реакционной смеси ведут методом ГЖХ, отбирая 0.1 мл реакционной смеси и обрабатывая ее 1 мл винилэтилового эфира с целью дезактивирования активных каталитических частиц. При вычислении выхода продукта используются калибровочные коэффициенты, полученные из анализа искусственных смесей соответствующих акрилатов, малеатов и фумаратов с последующим пересчетом отношений площадей на хроматограмме к массовым соотношениям составных компонентов.

В колбу Шленка на 25 мл, снабженную магнитной мешалкой и обратным холодильником, помещают 212 мг (0.25 ммоль) катализатора Граббса второго поколения (2) и 37 мг (0.375 ммоль) хлорида меди (I). Колбу заполняют аргоном и прибавляют раствор 117 мг (0.500 ммоль) 2-изопропокси-5-триметилсилилстирола (15) в сухом дихлорометане (4 мл). Реакционную смесь кипят 1 час, после чего охлаждают до комнатной температуры. Далее все операции выполняются на воздухе. Растворитель упаривают в вакууме, продукт выделяют с помощью препаративной хроматографии на силикагеле, элюируя смесью циклогексан: этилацетат=4:1. Получают 130 мг (выход 74%) комплекса (4) в виде зеленых кристаллов.
Спектр 1Н ЯМР (500 МГц, в CD2Cl2): 0.25 (9Н, с), 1.23 (6Н, д, J 6.0 Гц), 2.42 (6Н, с), 2.44 (12Н, с), 4.18 (4Н, с), 4.89 (1Н, септет, J 6.0 Гц), 6.85 (1Н, д, J 8.8 Гц), 7.08 (5Н, м), 7.70 (1Н, дд, J 8.8, 1.9 Гц), 16.40 (1Н, с).
Пример 2.

В колбу Шленка на 25 мл, снабженную магнитной мешалкой и обратным холодильником, помещают 212 мг (0.25 ммоль) катализатора Граббса второго поколения (2) и 37 мг (0.375 ммоль) хлорида меди (I). Колбу заполняют аргоном и прибавляют раствор 117 мг (0.500 ммоль) 2-изопропокси-5-триметилсилилстирола в сухом дихлорометане (4 мл). Реакционную смесь кипят 1 час, после чего охлаждают до комнатной температуры. Далее все операции выполняются на воздухе. Растворитель упаривают в вакууме, продукт выделяют с помощью препаративной хроматографии на силикагеле, элюируя смесью циклогексан: этилацетат=4:1. Получают 100 мг (выход 57%) комплекса (5) в виде зеленых кристаллов.
Спектр 1Н ЯМР (500 МГц, в CD2Cl2): 0.23 (9Н, с), 1.23 (6Н, д, J 5.6 Гц), 2.41 (6Н, с), 2.44 (12Н, с), 4.15 (4Н, с), 4.94 (1Н, септет, J 5.6 Гц), 6.93 (1Н, д, J 7.7 Гц), 6.95 (1Н, с), 7.07 (1Н, д, J 7.7 Гц), 7.07 (4Н, с), 16.51 (1Н, с).
Пример 3.

В заполненную аргоном колбу Шленка на 50 мл, снабженную магнитной мешалкой и обратным холодильником, помещают 300 мг (0.752 ммоль) E-N,N'-бис-(2,4-диметилфенил)-4,5-диизопропил-4,5-дигидро-1,3-имидазола хлорида, 0.44 мл (0.752 ммоль, 1.7 М в толуоле) трет-амилата калия, 44 мл сухого гексана и 309 мг (0.376 ммоль) катализатора Граббса первого поколения. Реакционную смесь нагревают при 50°С 17 часов, охлаждают до комнатной температуры и растворитель упаривают в вакууме. Сухой остаток растворяют в дихлорометане (10 мл), прибавляют 60 мг (0.60 ммоль) хлорида меди (I) и 150 мг (0.925 ммоль) 2-изопропоксистирола (14) и смесь перемешивают при 40°С 1 час, после чего охлаждают до комнатной температуры. Далее все операции выполняются на воздухе. Растворитель упаривают в вакууме, продукт выделяют с помощью препаративной хроматографии на силикагеле, элюируя смесью циклогексан: этилацетат=6:1. Получают 85 мг (выход 25%) комплекса (11) в виде зеленых кристаллов. Судя по спектрам 1Н ЯМР, комплекс представляет собой смесь двух изомеров в соотношении 1: 3.4.
Спектр 1Н ЯМР (300 МГц, в CD2Cl2): 0.87 (6Н, м), 1.17 (6Н, м), 1.25 (6Н, д J 5.6 Гц, минорный изомер), 1.35 (6Н, д J 5.6 Гц, основной изомер), 1.87 (1Н, м), 2.03 (1Н, м), 1.39 (6Н, д J 2.8 Гц), 1.49 (6Н, д J 4.8 Гц), 4.05 (2Н, м), 4.88 (1Н, септет, J 5.7 Гц), 6.83-6.94 (3Н, м), 7.17-7.30 (4Н, м), 7.38 (1Н, д, J 7.6 Гц), 7.54 (1Н, м), 8.50 (1Н, д, J 8.6 Гц, основной изомер), 8.59 (1Н, д, J 8.6 Гц, минорный изомер), 16.54 (1Н, с, основной изомер), 17.18 (1Н, с, минорный изомер).
Пример 4.
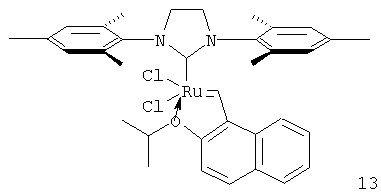
В колбу Шленка на 25 мл, снабженную магнитной мешалкой и обратным холодильником, помещают 150 мг (0.18 ммоль) катализатора Граббса второго поколения (2) и 23 мг (0.23 ммоль) хлорида меди (I). Колбу заполняют аргоном и прибавляют раствор 50 мг (0.23 ммоль) 1-винил-2-изопропоксинафталина (16) в сухом дихлорометане (6 мл). Реакционную смесь кипят 30 минут, после чего охлаждают до комнатной температуры. Далее все операции выполняются на воздухе. Растворитель упаривают в вакууме, продукт выделяют с помощью препаративной хроматографии на силикагеле, элюируя смесью циклогексан: этилацетат=6:1. Получают 55 мг (выход 49%) комплекса (13) в виде зеленых кристаллов.
Спектр 1Н ЯМР (500 МГц, в CD2Cl2): 1.33 (6Н, д, J 5.6 Гц), 2.53 (18Н, уш. с), 4.21 (4Н, с), 5.14 (1Н, септет, J 5.6 Гц), 7.09-7.29 (5Н, м), 7.36 (1Н, д, J 8.8 Гц), 7.39 (1Н, д, J 8.8 Гц), 7.62 (1Н, т, J 6.8 Гц), 7.85 (1Н, д, J 8.8 Гц), 8.27 (1Н, д, J 9.8 Гц), 18.17 (1Н, с).
Пример 5.
В колбу Шленка на 25 мл, снабженную магнитной мешалкой, помещают 11.91 г (69.2 ммоль) диэтилмалеата и замораживают жидким азотом, после чего вакуумируют до 0.1 Па. Операцию дегазации повторяют два раза. Вакуум перекрывают, реакционной смеси дают нагреться до комнатной температуры и в реакционную колбу вводят этилен, создавая давление 101325 Па, после чего смесь нагревают до 50°С. Давление этилена поддерживают постоянным на протяжении всего эксперимента. Далее в реакционную смесь вводят суспензию 4.70 мг катализатора (1) в 1.00 г (5.8 ммоль) диэтилмалеата. Контроль за ходом реакции осуществляют методом ГЖХ через 10, 20, 30, 45, 60 минут после начала эксперимента и далее через каждые 30 минут до завершения реакции (таблица 1, пример 1).
Пример 6.
Этенолиз диэтилмалеата проводят по примеру 5, но используя 6.37 мг катализатора (2) (таблицы 1 и 2, примеры 2).
Пример 7.
Этенолиз диэтилмалеата проводят по примеру 5, но вместо катализатора (1) используют 5.04 мг катализатора (3) (таблицы 1 и 2, примеры 3).
Пример 8.
Этенолиз диэтилмалеата проводят по примеру 5, но вместо катализатора (1) используют 5.24 мг катализатора (4) (таблицы 1 и 2, примеры 4).
Пример 9.
Этенолиз диэтилмалеата проводят по примеру 5, но вместо катализатора (1) используют 5.24 мг катализатора (5) (таблицы 1 и 2, примеры 5).
Пример 10.
Этенолиз диэтилмалеата проводят по примеру 5, но вместо катализатора (1) используют 4.80 мг катализатора (6) (таблицы 1 и 2, примеры 6).
Пример 11.
Этенолиз диэтилмалеата проводят по примеру 5, но вместо катализатора (1) используют 5.14 мг катализатора (7) (таблицы 1 и 2, примеры 7).
Пример 12.
Этенолиз диэтилмалеата проводят по примеру 5, но вместо катализатора (1) используют 5.14 мг катализатора (8) (таблицы 1 и 2, примеры 8).
Пример 13.
Этенолиз диэтилмалеата проводят по примеру 5, но вместо катализатора (1) используют 4.68 мг катализатора (9) (таблицы 1 и 2, примеры 9).
Пример 14.
Этенолиз диэтилмалеата проводят по примеру 5, но вместо катализатора (1) используют 5.33 мг катализатора (10) (таблицы 1 и 2, примеры 10).
Пример 15.
Этенолиз диэтилмалеата проводят по примеру 5, но вместо катализатора (1) используют 5.12 мг катализатора (11) (таблицы 1 и 2, примеры 11).
Пример 16.
Этенолиз диэтилмалеата проводят по примеру 5, но вместо катализатора (1) используют 5.86 мг катализатора (12) (таблицы 1 и 2, примеры 12).
Пример 17.
Получение метилакрилата по реакции метатезиса диметилмалеата с этиленом в условиях отгонки образующегося продукта при атмосферном давлении ведут в установке с объемом перегонной колбы 15 мл, не содержащей дефлегматор и включающей ловушку, подсоединенную к выходу из аллонжа вместо вакуумного насоса. В описанную установку помещают 9.81 г (68.1 ммоль) диметилмалеата. Шлиф, через который подсоединяют капилляр для ввода этилена, закрывают септой. Перегонную колбу с помещенным в нее малеатом замораживают жидким азотом и вакуумируют до 0.1 Па. Далее вакуум перекрывают и реакционной смеси дают нагреться до комнатной температуры. Операцию дегазации повторяют два раза, после чего через септу вводят металлический капилляр до дна перегонной колбы и установку заполняют этиленом, очищенным по примеру 1. Скорость потока этилена устанавливают 7.0 л/ч. Реакционную смесь нагревают с помощью масляной бани до 120°С, после чего с помощью шприца через септу в установку вводят суспензию 4.70 мг катализатора (1) в 1.00 г (6.9 ммоль) диметилмалеата. Реакцию ведут в течение 30 минут. Приемник и ловушку охлаждают до -30°С с помощью смеси спирт - жидкий азот. Выход продукта и число оборотов катализатора представлены в таблице 3, пример 1. Чистота продукта, судя по данным ГЖХ, более 99.5%.
Пример 18.
Этенолиз диметилмалеата проводят по примеру 17, но вместо катализатора (1) используют 5.24 мг катализатора (4). Выход продукта и число оборотов катализатора представлены в таблице 3, пример 2. Чистота продукта, судя по данным ГЖХ, более 99.5%.
Пример 19
Этенолиз диметилмалеата проводят по примеру 17, но вместо катализатора (1) используют 5.08 мг катализатора (13). Выход продукта и число оборотов катализатора представлены в таблице 3, пример 3. Чистота продукта, судя по данным ГЖХ, более 99.5%.
Пример 20
Этенолиз диметилмалеата проводят по примеру 17, но в установку, кроме диметилмалеата, помещают, также, 6.08 мг 2-изопропоксистирола (14). Выход продукта и число оборотов катализатора представлены в таблице 4, пример 1. Чистота продукта, судя по данным ГЖХ, более 99.5%.
Пример 21
Этенолиз диметилмалеата проводят по примеру 20, но используя 9.37 мг 2-изопропоксистирола (14). Выход продукта и число оборотов катализатора представлены в таблице 4, пример 2. Чистота продукта, судя по данным ГЖХ, более 99.5%.
Пример 22
Этенолиз диметилмалеата проводят по примеру 20, но используя 18.25 мг 2-изопропоксистирола (14). Выход продукта и число оборотов катализатора представлены в таблице 4, пример 3. Чистота продукта, судя по данным ГЖХ, более 99.5%.
Пример 23
Этенолиз диметилмалеата проводят по примеру 20, но используя 30.42 мг 2-изопропоксистирола (14). Выход продукта и число оборотов катализатора представлены в таблице 4, пример 4. Чистота продукта, судя по данным ГЖХ, более 99.5%.
Пример 24
Этенолиз диметилмалеата проводят по примеру 20, но используя 121.7 мг 2-изопропоксистирола (14). Выход продукта и число оборотов катализатора представлены в таблице 4, пример 5. Чистота продукта, судя по данным ГЖХ, более 99.5%.
Пример 25
Этенолиз диметилмалеата проводят по примеру 20, но вместо 2-изопропоксистирола (14) используют 17.58 мг стирола (15). Выход продукта и число оборотов катализатора представлены в таблице 4, пример 6. Чистота продукта, судя по данным ГЖХ, более 99.5%.
Пример 26.
Этенолиз диметилмалеата проводят по примеру 20, но вместо 2-изопропоксистирола (14) используют 23.9 мг стирола (16). Выход продукта и число оборотов катализатора представлены в таблице 4, пример 7. Чистота продукта, судя по данным ГЖХ, более 99.5%.
Пример 27
Этенолиз диметилмалеата проводят по примеру 25, но вместо катализатора (1) используют 5.24 мг катализатора (4). Выход продукта и число оборотов катализатора представлены в таблице 4, пример 8. Чистота продукта, судя по данным ГЖХ, более 99.5%.
Пример 28
Этенолиз диметилмалеата проводят по примеру 26, но вместо катализатора (1) и 2-изопропоксистирола (16) используют 8.0 мг стирола (16) и 5.08 мг катализатора (13). Выход продукта и число оборотов катализатора представлены в таблице 4, пример 8. Чистота продукта, судя по данным ГЖХ, более 99.5%.
Промышленная применимость
Производство акрилатов по реакции метатезиса олефинов из малеатов и этилена представляет интерес для промышленности в связи с низкой себестоимостью и доступностью исходных соединений, а также мягкостью условий и высокой селективностью реакции. Предложенные в изобретении катализаторы и каталитические системы для этой реакции имеют более продолжительное время жизни и обладают большим числом оборотов по сравнению с существующими катализаторами, что позволяет существенно снизить себестоимость производства акрилатов.
| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБ ПОЛУЧЕНИЯ ЭФИРОВ АКРИЛОВОЙ КИСЛОТЫ | 2006 |
|
RU2307119C1 |
| КАТАЛИТИЧЕСКАЯ КОМПОЗИЦИЯ ДЛЯ ПОЛУЧЕНИЯ ЭФИРОВ АКРИЛОВОЙ КИСЛОТЫ ПО РЕАКЦИИ МЕТАТЕЗИСА ДИАЛКИЛМАЛЕАТОВ С ЭТИЛЕНОМ | 2007 |
|
RU2326733C1 |
| СПОСОБ ПОЛУЧЕНИЯ ЭФИРОВ БИС-α, β-НЕНАСЫЩЕННЫХ ДИКАРБОНОВЫХ КИСЛОТ | 2007 |
|
RU2330015C1 |
| СПОСОБ ПОЛУЧЕНИЯ ЭФИРОВ α,β-НЕНАСЫЩЕННЫХ ЖИРНЫХ КИСЛОТ | 2006 |
|
RU2320640C1 |
| СПОСОБ ПОЛУЧЕНИЯ КОМПЛЕКСОВ РУТЕНИЯ И ИХ ПРОМЕЖУТОЧНЫХ СОЕДИНЕНИЙ И ИХ ПРИМЕНЕНИЕ В МЕТАТЕЗИСЕ ОЛЕФИНОВ | 2016 |
|
RU2735724C1 |
| РУТЕНИЕВЫЙ КАТАЛИЗАТОР, СПОСОБ ЕГО ПОЛУЧЕНИЯ И ПРИМЕНЕНИЕ В РЕАКЦИИ МЕТАТЕЗИСА | 2014 |
|
RU2578593C1 |
| РУТЕНИЕВЫЙ КАТАЛИЗАТОР МЕТАТЕЗИСНОЙ ПОЛИМЕРИЗАЦИИ ДИЦИКЛОПЕНТАДИЕНА И СПОСОБ ЕГО ПОЛУЧЕНИЯ | 2009 |
|
RU2409420C1 |
| ВЫСОКОЭФФЕКТИВНЫЕ МЕТАТЕЗИСТИЧЕСКИЕ КАТАЛИЗАТОРЫ, ВЫБИРАЕМЫЕ В РЕАКЦИЯХ ROMP И RCM | 2010 |
|
RU2546656C2 |
| РУТЕНИЕВЫЙ КАТАЛИЗАТОР МЕТАТЕЗИСНОЙ ПОЛИМЕРИЗАЦИИ ДИЦИКЛОПЕНТАДИЕНА (ВАРИАНТЫ) И СПОСОБ ПОЛУЧЕНИЯ ПОЛИДИЦИКЛОПЕНТАДИЕНА (ВАРИАНТЫ) | 2010 |
|
RU2436801C1 |
| Селенсодержащие комплексы типа Ховейды-Граббса второго поколения и способ их получения | 2023 |
|
RU2807891C1 |
Изобретение относится к органическому синтезу и касается области производства гомогенного катализатора для получения эфиров акриловой кислоты по реакции метатезиса малеатов с этиленом. Разработаны два варианта катализатора для получения эфиров акриловой кислоты по реакции метатезиса диалкилмалеатов с этиленом. Катализатор формулы:

где А1, А2 - хлор; L - дигидроимидазольный лиганд; R1, R2, R3 - одинаковые или разные, заместители выбранные из группы, содержащей водород, алкил-, триалкилсилил - или алкокси-группы. И катализатор формулы:

Использование данных катализаторов позволяет повысить число их оборотов в реакции метатезиса диалкилмалеатов с этиленом как в условиях умеренных температур (около 50°С) при проведении процесса без отгонки продукта, так и при температурах около 120°С, когда образующийся акрилат отгоняют из реакционной смеси. Разработана также каталитическая композиция, позволяющая повысить не только число оборотов, но и время жизни катализатора. Такая композиция содержит указанный катализатор и 2-изопропоксистирола или его производное при соотношении: 1 мольный эквивалент катализатора на 5-100 мольных эквивалентов 2-изопропоксистирола или его производного. 3 н. и 4 з.п. ф-лы, 4 табл.
где А1, А2 - хлор,
L - дигидроимидазольный лиганд,
R1, R2, R3 - одинаковые или разные заместители, выбранные из группы,
содержащей водород, алкил-, триалкилсилил- или алкоксигруппы.


| RU 2003131325 А, 27.01.2005 | |||
| Способ приготовления мыла | 1923 |
|
SU2004A1 |
| Grant S | |||
| Forman and Robert P | |||
| Аппарат для очищения воды при помощи химических реактивов | 1917 |
|
SU2A1 |
| Регистратор для дел | 1925 |
|
SU690A1 |
| WO 2004035596 A1, 29.04.2004 | |||
| Привод ведущего моста | 1990 |
|
SU1724489A1 |

