Область техники
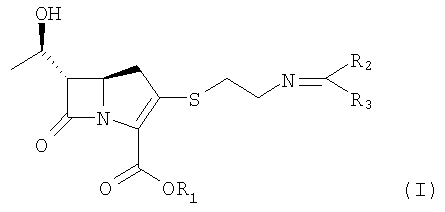
Данное изобретение относится к соединению формулы II, приведенной ниже:
Формула II
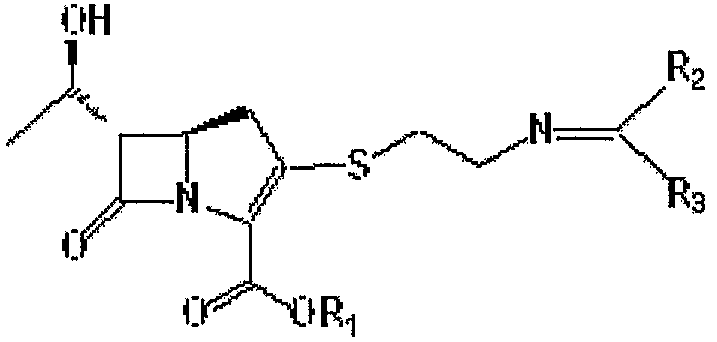
где
R1 является п-нитробензильной или п-метоксибензильной группой; и R2 и R3 могут быть одинаковыми или разными, и каждый независимо представляет С1-6 алкильную или арильную группу,
или его производному, и способу получения соединения формулы II.
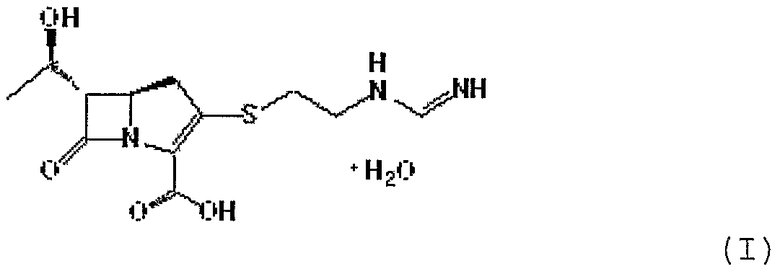
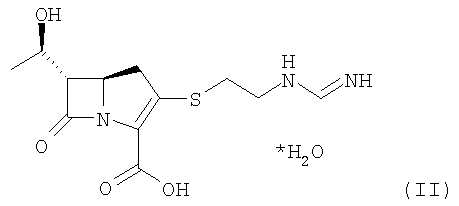
Данное изобретение относится также к способу получения имипенема формулы I, приведенной ниже:
Формула I
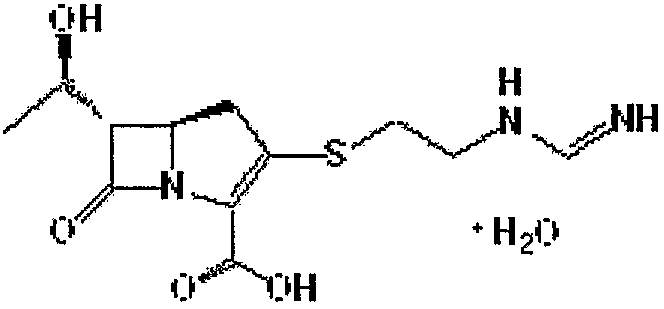
путем использования соединения формулы II.
Имипенем формулы I является карбапенемным антибиотиком, который является представителем бета-лактамных антибиотиков.
Уровень техники
Первым карбапенемным антибиотиком, который был открыт, был тиенамицин, выделенный из существующих в природе Streptomyces cattleya в Merck Co., U.S.A. в 1976 г.
Так как тиенамицин является химически очень нестабильным, несмотря на превосходные фармакологические свойства, лекарственные препараты на его основе не были разработаны. Были предприняты многочисленные попытки по преодолению химической нестабильности тиенамицина с сохранением в то же время его фармакологического действия. Например, имипенем, который является новым производным тиенамицина, синтезированным в Merck Co., получен путем модификации аминогруппы тиенамицина в N-формимидоильную группу. Имипенем является новой концепцией антибиотика с улучшенной стабильностью. Имипенем широко использовался в качестве терапевтического средства до настоящего времени. Имипенем как карбапенемный антибиотик является первым доступным соединением среди бета-лактамных антибиотиков нового типа, имеющих систему карбапенемового кольца, и проявляет высокую стабильность даже в присутствии бета-лактамазы. Кроме того, имипенем демонстрирует чрезвычайно широкий спектр антибиотической активности против грамположительных и грамотрицательных аэробных и анаэробных видов. Имипенем получают только путем полного химического синтеза в отличие от обычных цефалоспориновых антибиотиков.
О первом промышленном синтезе имипенема было сообщено в 1981 г. Начиная с 1989 г., предлагались улучшенные способы синтеза имипенема.
В патенте США № 4292436 описан способ получения моногидрата имипенема in situ путем активации бициклического сложного кетоэфира, взаимодействия активированного сложного эфира с защищенным по амину N-формимидоил-2-аминоэтантиольным соединением с последующим каталитическим гидрированием с использованием оксида платины в качестве катализатора для удаления 2-карбоксила и защищающих амин групп без выделения какого-либо промежуточного соединения, как изображено на схеме реакций 1, приведенной ниже:
Схема реакций 1

где R является водородом или защитной группой; и Х является уходящей группой.
Однако этот способ имеет недостатки, состоящие в том, что имипенем получают из бициклического сложного кетоэфира с таким низким выходом, как 35%, и способ дополнительно включает четыре стадии получения защищенного N-формимидоил-2-аминоэтантиольного соединения. Кроме того, другим недостатком данного способа является необходимость большого избытка воды и растворителей (660-кратное количество от исходного материала) для экстрагирования после промывания, после того как вводят N-формимидоил-2-аминоэтантиольное соединение, что экономически невыгодно.
С другой стороны, в патентах США № 4845261 и 4894450 описан новый способ непрерывного получения имипенема из бициклического сложного кетоэфира в четыре стадии без выделения и очистки каких-либо промежуточных соединений. Процедура данного способа представлена на схеме реакций 2, приведенной ниже:
Схема реакций 2

где R1 является п-нитробензильной группой.
Как представлено на схеме реакций 1, так как процесс, состоящий из четырех стадий, происходит in situ без проведения какого-либо выделения и очистки, конечный продукт неизбежно содержит большие количества примесей, что делает трудными выделение и очистку конечного продукта. Кроме того, процесс сопровождается использованием дорогостоящего бис(дихлорфенил)фосфорхлоридата с целью активации предшественника, бициклического сложного кетоэфира.
Так как используемый в качестве растворителя для реакции по схеме реакций 2 дорогой N-этилпирролидинон является высокополярным органическим растворителем, трудно удалить данный растворитель из получающегося водного раствора после завершения данной реакции. Кроме того, использование растворителя для реакции в большом количестве (200-кратное количество от исходного материала), создает экономические препятствия для осуществления способа в промышленном масштабе.
В патенте США № 4373772 предложен полусинтетический способ получения моногидрата имипенема с использованием тиенамицина, выделенного из Streptomyces cattleya, в качестве исходного вещества. Вся процедура этого способа представлена на схеме реакций 3, приведенной ниже:
Схема реакций 3
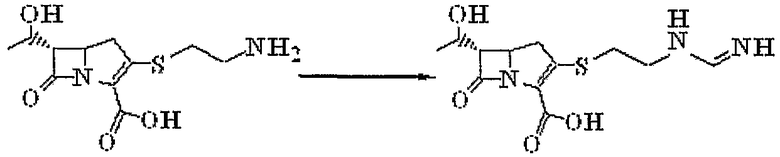
Как представлено на схеме реакций 3, однако, так как химически нестабильный тиенамицин получают из микроорганизма в малом количестве, данный способ является невыгодным из-за плохой экономической эффективности. Кроме того, использование большого количества воды (214-кратное количество от исходного вещества) в качестве растворителя при реакции создает трудности при реакции, выделении и очистке. Кроме того, данный способ имеет тот недостаток, что вместе с целевым продуктом реакции, имипенемом, образуется 5% или более димера бистиенамицинформамидина в качестве нежелательного побочного продукта реакции.
Dr. Ranbaxy подал международную РСТ заявку (WO 02/36594), относящуюся к способу получения имипенема. Этот способ похож на способ Merck Co., за исключением того, что в качестве растворителя для реакции используют смешанный растворитель из тетрагидрофурана и дорогостоящего 1,3-диметил-3,4,5,6-тетрагидро-(2Н)-пиримидинона. Однако данная публикация не касается катализатора гидрирования. Кроме того, конечный продукт, имипенем, получают в кристаллической форме с очень низким выходом, равным 23% от бициклического сложного кетоэфира в качестве исходного вещества путем адсорбционной хроматографии.
Сущность изобретения
Как указано выше, в соответствии с общепринятыми способами получения имипенема, так как промежуточные соединения невозможно выделить и очистить, или они нестабильны, продукт, имипенем, получают путем непрерывных реакционных стадий (реакции in situ). В результате неизбежно образование больших количеств примесей.
По этим причинам большие количества примесей создают затруднения при обработке, выделении и очистке, приводя к низким выходу и чистоте конечного продукта.
Другими словами, в соответствии с общепринятыми способами синтеза имипенема, так как в целом стадии реакций протекают непрерывно без какого-либо выделения промежуточных соединений, из-за химической нестабильности промежуточных соединений выделение и очистку имипенема выполняют в присутствии примесей. Кроме того, выделение путем кристаллизации затруднено из-за присутствия примесей, что приводит к низким выходу и чистоте конечного продукта.
К тому же, так как общепринятые способы требуют использования дорогостоящих растворителей для реакции и большого количества растворителей (200-кратные количества по исходным веществам), с ними связаны проблемы в плане экономической эффективности и промышленного применения.
Поэтому данное изобретение было создано с учетом представленных выше проблем, и объектом данного изобретения является получение нового защищенного по амину соединения тиенамицина формулы II, пригодного для получения моногидрата имипенема формулы I, и способ получения соединения тиенамицина формулы II.
Другим объектом данного изобретения является создание способа получения имипенема с использованием защищенного по амину соединения тиенамицина формулы II.
Как объяснено выше, исходя из соединения формулы II или его производного данного изобретения, разные защитные группы эффективно вводят по карбоксильной группе и аминогруппе тиенамицина с получением защищенного по амину соединения тиенамицина формулы II в качестве промежуточного соединения имипенема, которое используют для получения моногидрата имипенема формулы I. По способу данного изобретения может быть решена проблема низкого выхода моногидрата имипенема из-за наличия больших количеств примесей в результате отсутствия выделения и очистки промежуточных соединений. К тому же моногидрат имипенема с высокой чистотой может быть получен из нового промежуточного соединения формулы II простым способом. Кроме того, выход и качество моногидрата имипенема формулы I могут быть значительно улучшены.
Так как при способе данного изобретения используются обычные органические растворители и вода, данные растворители легко удалить после завершения реакции. Кроме того, так как используют палладиевый катализатор, содержащий большое количество воды, при гидрировании для удаления защитных групп, опасность манипуляций значительно снижена, что позволяет проводить данный процесс в мягких условиях реакции. Соответственно, способ данного изобретения является экономически выгодным и делает возможным получение имипенема без затруднений.
Краткое описание чертежей
Представленные выше и другие объекты, особенности и другие преимущества данного изобретения могут быть более ясно поняты из следующего далее подробного описания в соединении с сопровождающими чертежами, на которых:
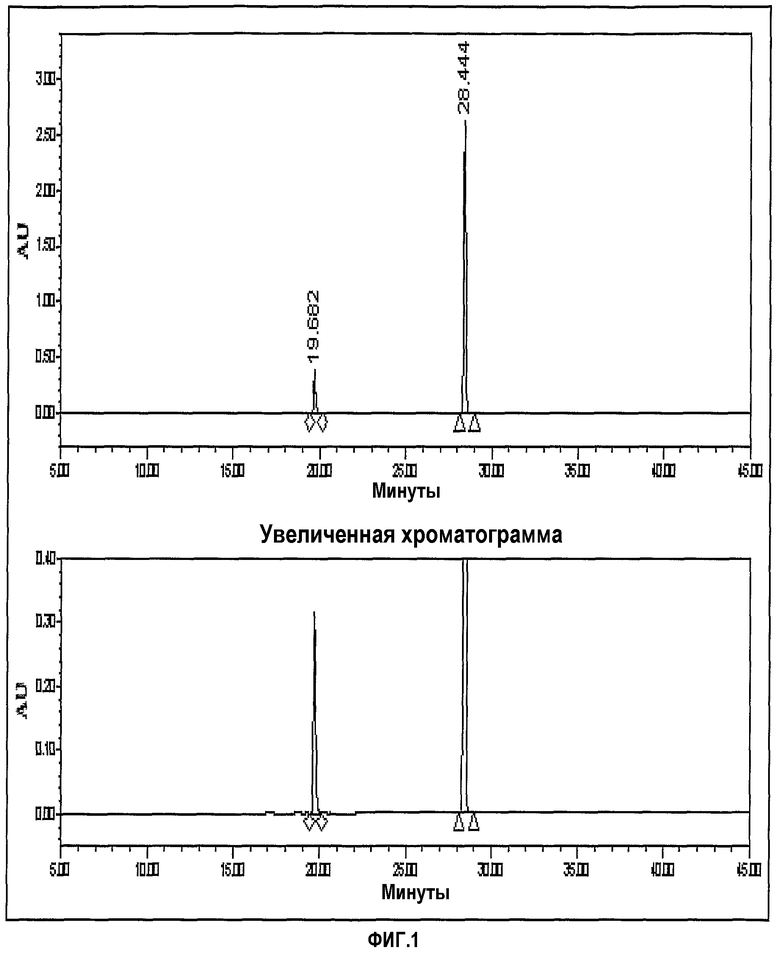
фиг.1 представляет хроматограмму ЖХ, показывающую чистоту кетонового соединения;
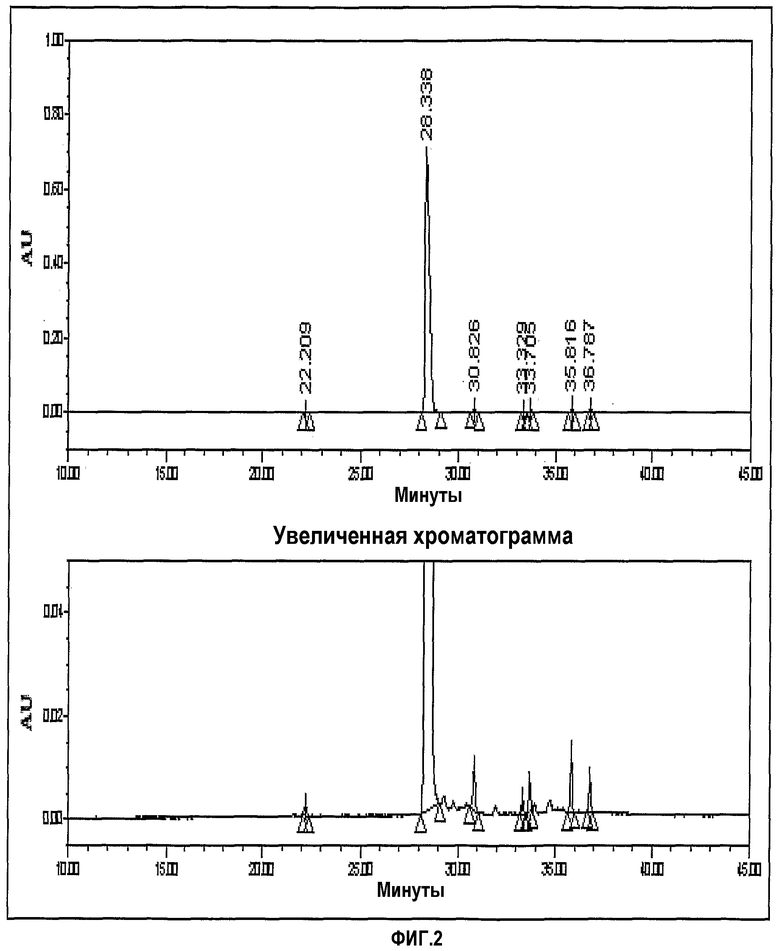
фиг.2 представляет хроматограмму ЖХ, показывающую чистоту иминосоединения;
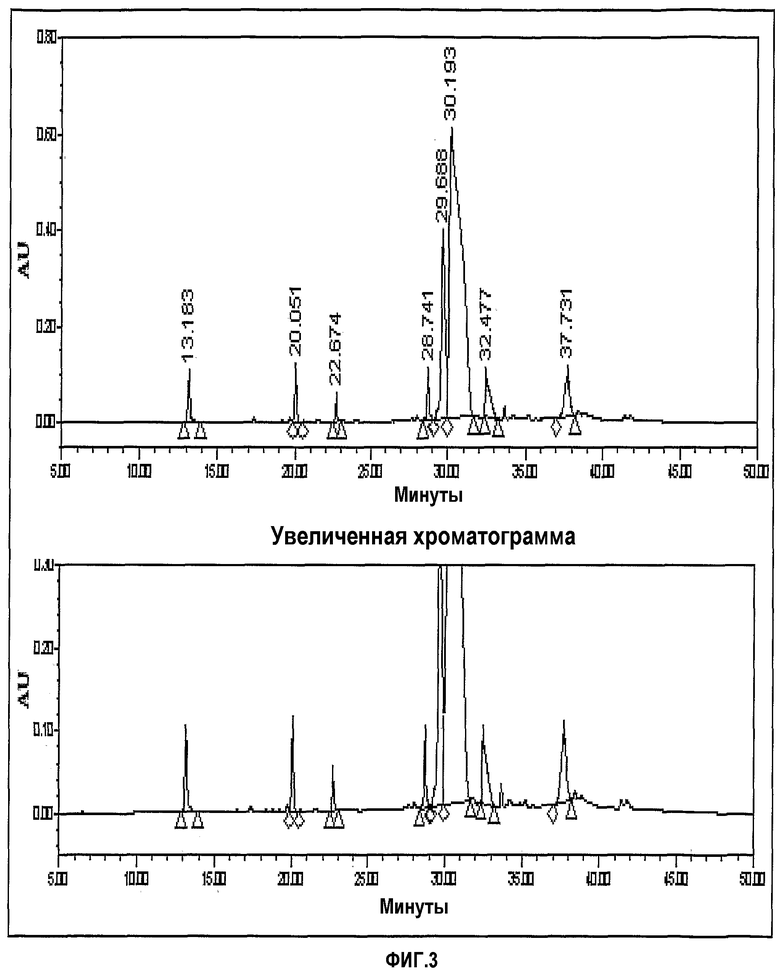
фиг.3 представляет хроматограмму ЖХ, показывающую формимидирование по Merck;
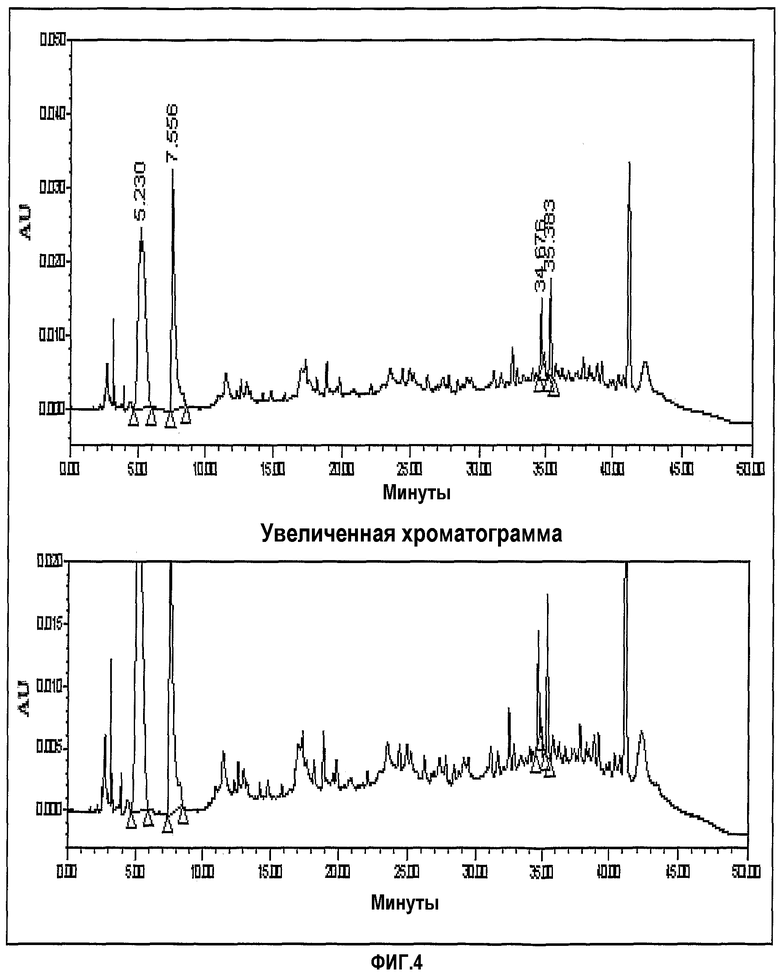
фиг.4 представляет хроматограмму ЖХ, показывающую гидрирование по Merck;
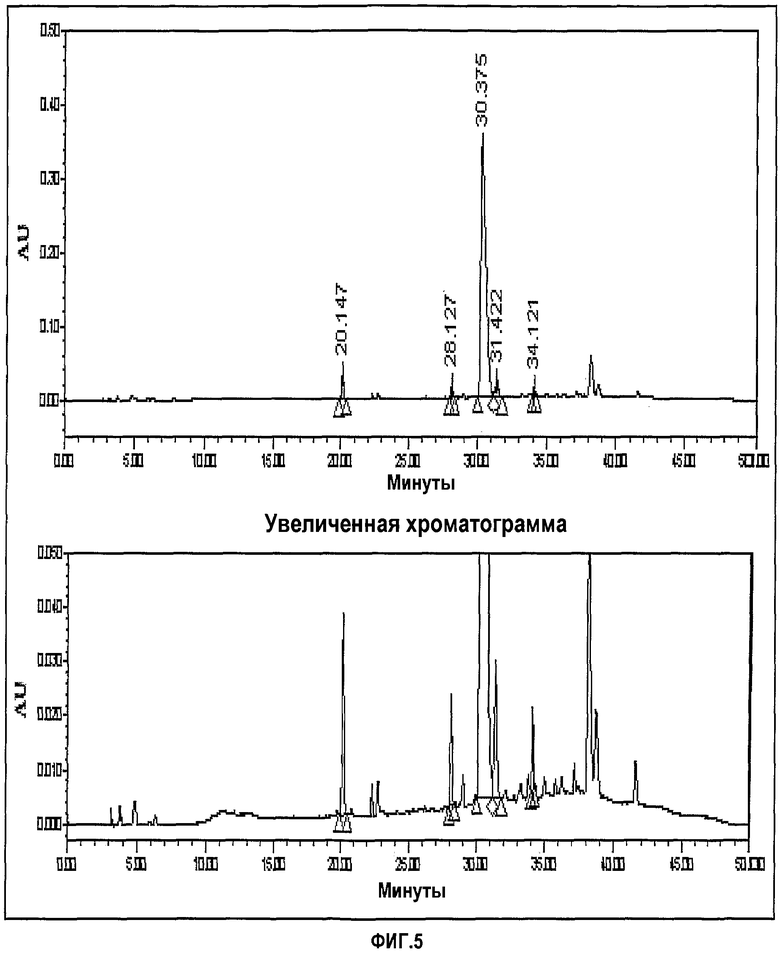
фиг.5 представляет хроматограмму ЖХ, показывающую CWP формимидирование (по Choongwae Pharma Corp.); и
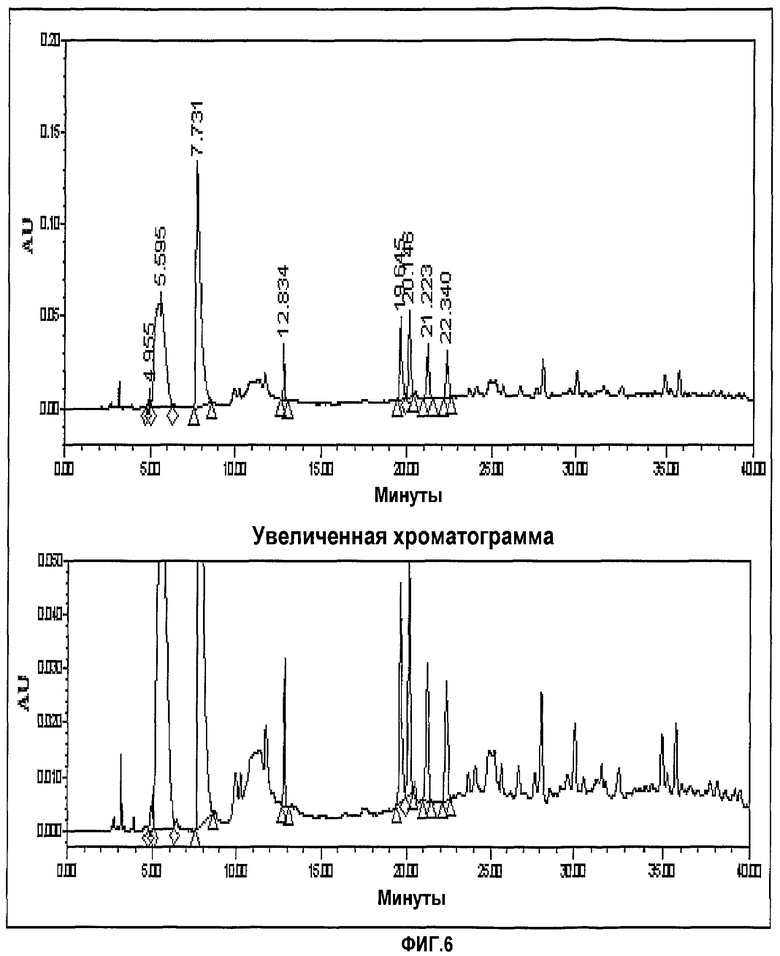
фиг.6 представляет хроматограмму ЖХ, показывающую CWP гидрирование.
Лучший способ осуществления изобретения
Пример 1
Получение (5R,6S)-п-нитробензил-3-(дифенилфосфоно)-6-[(1R)-1-гидроксиэтил]-1-азабицикло[3,2,0]гепт-2-ен-7-он-2-карбоксилата
20,0 г (5R,6S)-п-нитробензил-6-[(1R)-1-гидроксиэтил]-1-азабицикло[3,2,0]гепт-3,7-дион-2-карбоксилата формулы III, приведенной ниже, растворяли в смеси раствора ацетонитрила (100 мл) и тетрагидрофурана (100 мл). Температуру реакционной смеси снижали до 0 - -10°С. К реакционной смеси последовательно добавляли 11,1 г N,N-диизопропилэтиламина и 18,5 г дифенилхлорфосфата. Полученную смесь перемешивали в течение 1,5-2 часов при сохранении температуры реакции -10°С с получением енолфосфата формулы IV, приведенной ниже. Енолфосфат формулы IV использовали на следующей стадии без дополнительного выделения.
Формула III
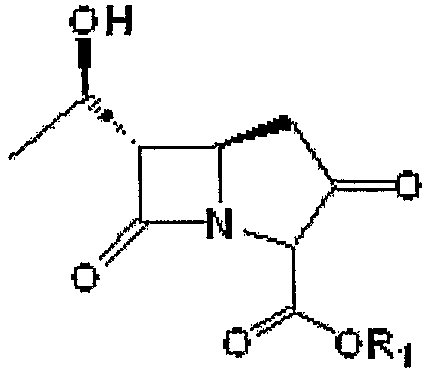
где R1 является п-нитробензилом.
Формула IV
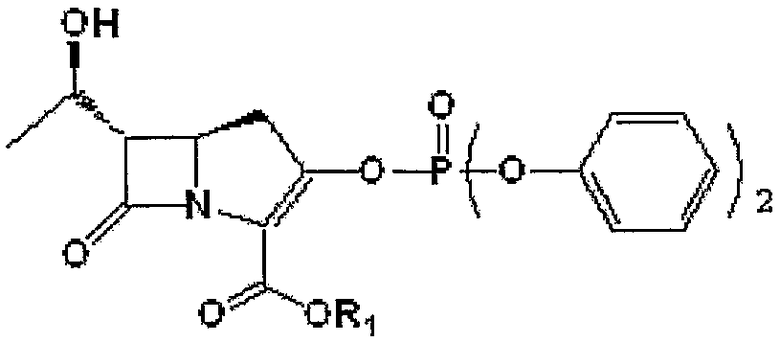
где R1 является п-нитробензилом.
Пример 2
Получение (5R,6S)-п-нитробензил-6-[(1R)-1-гидроксиэтил]-3-({2-[(1-изопропилиден)амино]этил}тио)-1-азабицикло[3,2,0]гепт-2-ен-7-он-2-карбоксилата
Температуру реакционного раствора енолфосфатного производного формулы IV, полученного в примере 1, снижали с -40 до -60°С, и затем туда последовательно добавляли 7,8 г гидрохлорида 2-аминоэтантиола и 11,1 г диизопропиламина. Реакционную смесь перемешивали в течение 0,5-1 часа при той же температуре. К реакционной смеси добавляли растворитель кетонового типа в присутствии основания, перемешивали и кристаллизовали. Полученный осадок отфильтровывали, промывали гексаном и сушили при пониженном давлении при комнатной температуре, с получением 20,5 г (выход: 80,0%) защищенного по амину тиенамицина формулы II.
1Н-ЯМР (ДМСО-d6, 300 МГц, м.д.) δ: 1,14 (д, J=6,3 Гц, 3H), 1,78 (с, 3H), 1,90 (с, 3H), 3,12 (м, 2H), 3,28-3,32 (м, 2H), 3,37-3,40 (м, 2H), 3,94 (м, 1H), 4,13 (дт, J=2,4, 8,7 Гц, 1H), 5,07 (д, J=5,1 Гц, 1H), 5,27 (д, J=14,1 Гц, 1H), 5,43 (д, J=14,1 Гц, 1Н), 7,70 (д, J=8,7 Гц, 2Н), 8,22 (д, J=8,7 Гц, 2Н).
Масса: 447,51
Т.пл.: 148-151°С
Цвет: бледно-желтый
Пример 3
Получение (5R,6S)-п-нитробензил-3-[[2-[(формимидоиламино) этил]тио]-6-[(1R)-1-гидроксиэтил]-1-азабицикло[3,2,0]гепт-2-ен-7-он-2-карбоксилата
После того как 20,0 г промежуточного соединения формулы II, полученного в примере 2, добавляли к смешанному растворителю из дистиллированной воды и тетрагидрофурана, температуру реакционной смеси снижали с 5 до -5°С или ниже. К реакционной смеси добавляли 18 мл N-метилморфолина и 30,8 г гидрохлорида бензилформимидата. Полученную смесь перемешивали при 0-10°С в течение 2-3 часов, получая соединение формулы V, которое использовали на следующей стадии без дополнительного выделения.
Формула V
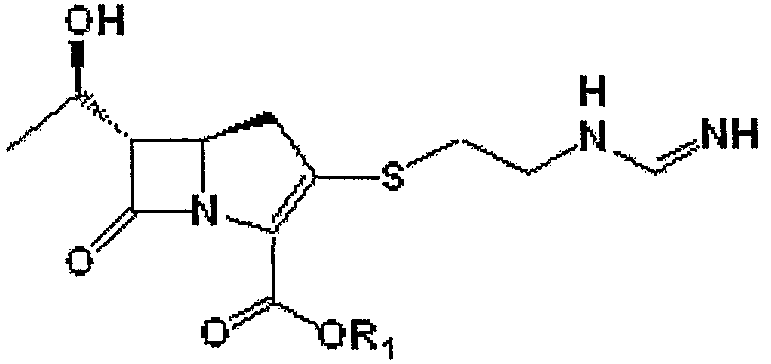
где R1 является п-нитробензилом.
Пример 4
Получение (+)-(5R,6S)-3-{[2-(формимидоиламино)этил]тио}-6-[(R)-1-гидроксиэтил]-7-оксо-1-азабицикло[3,2,0]гепт-2-ен-2-карбоновой кислоты (имипенем)
К реакционному раствору защищенного по карбоксилу N-формимидоилтиенамицина формулы V, полученного в примере 3, добавляли 16 г N-метилморфолина, чтобы довести рН до 7,0-8,0. К реакционной смеси добавляли содержащий воду палладиевый катализатор, чтобы провести реакцию по удалению защиты при 10-25°С. На этот раз реакция продолжалась в течение 3 часов при сохранении давления водорода 4-6 кг/см2, и затем катализатор удаляли фильтрованием. Анализ ВЭЖХ реакционного раствора показал, что имипенем получали с выходом 82%. Реакционный раствор промывали несколько раз этилацетатом и выпаривали под пониженным давлением для удаления остаточных органических растворителей. Полученный водный раствор очищали колоночной хроматографией с обращенной фазой с последующим концентрированием, используя методику обратного осмоса. К концентрату добавляли ацетон, перемешивали в течение 2-3 часов и кристаллизовали. Кристаллизованный моногидрат имипенема отфильтровывали, промывали и сушили при пониженном давлении, получая 8,6 г желаемого моногидрата имипенема формулы I (выход: 60%, чистота (по ВЭЖХ): 99%).
Далее способ получения имипенема по данному изобретению (далее называемый "представляемый способ") сравнили со способом Merck Co. (смотрите патенты США № 4845261 и 4894450, далее называемым "способ Merck").
Эксперименты
В соответствии со способом Merck имипенем получают без выделения каких-либо промежуточных соединений. В отличие от этого, по представленному способу иминосоединение получали из защищенного по карбоксилу тиенамицина, выделяли и использовали для получения имипенема. Затем выход и содержание имипенема сравнивали.
Экспериментальные методы
В случае способа Merck имипенем получали по методу, описанному в патентах США № 4845261 и 4894450, за исключением того, что использовали бис(дихлорфенил)фосфорхлоридат, продаваемый TCI.
Результаты
Чтобы сравнить экспериментальные результаты представляемого способа с результатами способа Merck, использовали следующие образцы:
- Кетоновое соединение (формула III)
- Иминосоединение (формула II)
- Способ Merck: формимидирование и имипенем
- Способ данного изобретения: формимидирование и имипенем
Приборы и условия для анализа ЖХ, применяемые для сравнения экспериментальных результатов представляемого способа с результатами способа Merck, были следующими:
1) Аналитический прибор
- Alliance 2695 & 2996 PDA system
- Терминал: Empower
- Колонка: С18, ODS, 4,6 мм, 5 мкм
2) Условия анализа
i) ВЭЖХ
- Скорость потока: 1,0 мл/мин
- Объем вводимой пробы: 5 мкл
- Изготовление образцов: разведения 1/10 (подвижной фазой В)
- Время цикла: 50 мин
- Температура колонки: комнатная температура
- Температура автоматического пробоотборника: 4°С
- Детектор: 254 нм
ii) Подвижные фазы
- Подвижная фаза А: буфер (NH4)2НРО4
- Подвижная фаза В: МеОН/ACN = 1:1
- Условия градиента подвижных фаз показаны в таблице 1, приведенной ниже:
Результаты эксперимента
1) Чистота кетонового соединения
Кетоновое соединение является реакционным соединением, используемым как в способе Merck, так и в представляемом способе. Кетоновое соединение, получаемое при представляемом способе, использовали при способе Merck. Чистота кетонового соединения показана в таблице 2, приведенной ниже.
Хроматограмма ЖХ, показывающая чистоту кетонового соединения, представлена на фиг.1.
2) Чистота иминосоединения:
Иминосоединение, выделяемое в соответствии с представляемым способом, является соединением, отличающим данный способ от способа Merck, и является ключевым соединением для данной экспериментальной цели. Таблица 3 представляет чистоту иминосоединения.
Хроматограмма ЖХ, показывающая чистоту иминосоединения, представлена на фиг.1.
3) Сравнение чистоты, основанной на соответствующих стадиях способа Merck и представляемого способа
Таблица 4 представляет чистоту на соответствующих стадиях в обоих способах.
4) Сравнение выхода, на основе соответствующих стадий способа Merck и представляемого способа (по отношению к теоретическим значениям)
Таблица 5 представляет выход на соответствующих стадиях при обоих способах.
Так как способ Merck не включает стадию получения иминосоединения, количество иминосоединения не могло быть вычислено. Теоретические количества на основе соответствующих стадий показывают величины, получаемые, когда соответствующие выходы составляют 100%.
Количество имипенема было вычислено на основе количества ангидрида имипенема. Содержание воды и других факторов было исключено из калибрования (так как состояние было жидким, значения факторов нельзя было точно рассчитать).
Содержание имипенема получали путем сравнения вычисленной площади пика с площадью пика стандартного реагента по USP.
Фиг.3-6 представляют хроматограммы, показывающие формимидирование по Merck, гидрирование по Merck и формимидирование по CWP и гидрирование по CWP, соответственно.
Как видно из представленных выше результатов эксперимента, представленный способ, где иминосоединение выделяют для получения имипенема, превзошел способ Merck по количеству соединения, получаемого после формимидирования, и конечного продукта, имипенема.
В соответствии с одним из аспектов данного изобретения представленные выше объекты могут быть реализованы с помощью соединения формулы II, приведенной ниже:
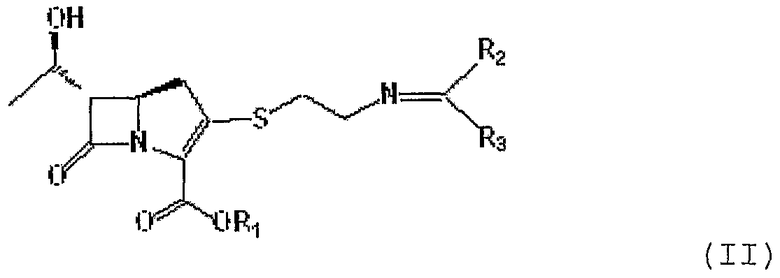
где
R1 является п-нитробензильной или п-метоксибензильной группой; и R2 и R3 могут быть одинаковыми или разными, и каждый независимо представляет С1-6 алкильную или арильную группу,
или его производного.
В соответствии с другим аспектом данного изобретения представлен способ получения соединения формулы II путем сочетания соединения формулы IV, приведенной ниже:
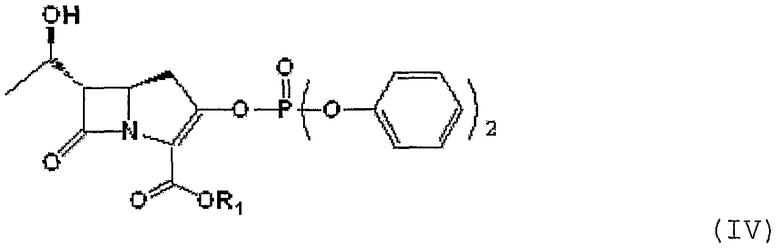
где R1 является п-нитробензильной или п-метоксибензильной группой,
или его производного с гидрохлоридом 2-аминоэтантиола в присутствии основания, с последующей реакцией с кетоном.
Кетон выбирают из группы, состоящей из ацетона, метилэтилкетона, дифенилкетона и их смесей.
Соединение формулы IV или его производное получают конденсацией соединения формулы III, приведенной ниже:
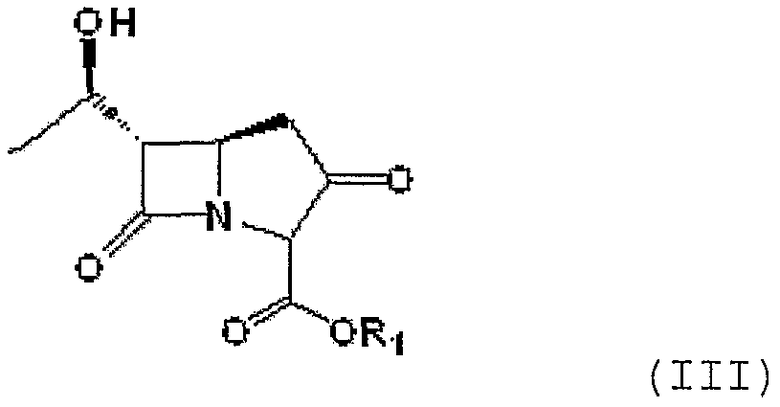
где R1 является п-нитробензильной или п-метоксибензильной группой, с дифенилхлорфосфатом в присутствии основания.
В качестве растворителя для реакции используют смешанный растворитель из ацетонитрила и тетрагидрофурана.
Температура реакции находится в интервале от 0 до -10°С.
В соответствии с еще одним аспектом данного изобретения представлен способ получения соединения формулы I, приведенной ниже:
путем взаимодействия соединения формулы II с изопропилформимидатом или бензилформимидатом в присутствии основания с получением соединения формулы V, приведенной ниже:
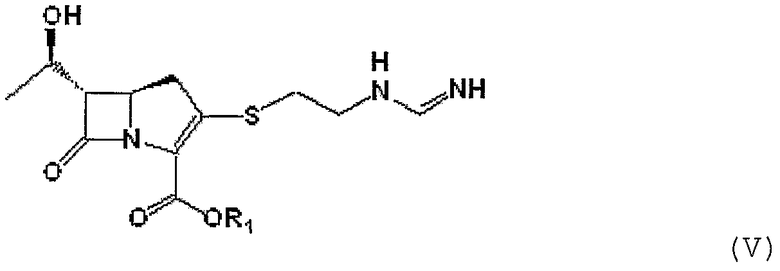
где R1 является п-нитробензильной или п-метоксибензильной группой, гидрирования соединения формулы V в присутствии катализатора-металла, выделения гидрированного соединения и кристаллизации выделенного соединения в присутствии спирта или кетона.
Гидрирование осуществляют в присутствии палладиевого катализатора, содержащего избыток воды при давлении водорода 4-6 кг/см2.
В качестве растворителя для реакции используют смешанный растворитель из воды и тетрагидрофурана.
Данное изобретение будет теперь описано более подробно.
В соответствии с данным изобретением защищенное по амину соединение тиенамицина формулы II получают путем сочетания енолфосфатного производного формулы IV с гидрохлоридом 2-аминоэтантиола с последующей реакцией с соответствующим кетоном. Защищенное по амину соединение тиенамицина, полученное таким образом, пригодно для получения моногидрата имипенема формулы I.
В соответствии с данным изобретением различные защитные группы эффективно вводят по карбоксильным и аминогруппам тиенамицинового производного, используемого в общепринятых способах получения имипенема для получения защищенного по амину соединения тиенамицина формулы II в качестве промежуточного соединения для карбапенема. Защищенное по амину соединение тиенамицина, полученное таким образом, является стабильным при комнатной температуре и может храниться при низкой температуре в течение длительного срока.
То есть по данному изобретению бициклический сложный кетоэфир формулы III конденсируют с дифенилхлорфосфатом в присутствии основания с получением енолфосфатного соединения формулы IV.
Енолфосфатное производное формулы IV получают в полярном растворителе, выбранном из простых эфиров, например, тетрагидрофурана, диизопропилового эфира и диоксана и ацетонитрила, и затем енолфосфатное производное вводят в сочетание с гидрохлоридом 2-аминоэтантиола в присутствии основания с получением тиенамицинового производного. Затем обычный кетон, такой как ацетон, метилэтилкетон или дифенилкетон, добавляют к производному тиенамицина с получением защищенного по амину тиенамицинового соединения формулы II, которое затем выделяют и кристаллизуют.
В отличие от общепринятых способов получения имипенема различные защитные группы эффективно вводят по карбоксильным и аминогруппам тиенамицина с получением защищенного по амину соединения тиенамицина формулы II в качестве промежуточного соединения карбапенема. Соединение формулы II является новым соединением в форме тусклых белых кристаллов, которое стабильно при комнатной температуре и может храниться при низкой температуре в течение длительного срока. Кроме того, соединение формулы II пригодно в качестве промежуточного соединения для получения промежуточного соединения имипенема и для получения других карбапенемных антибиотиков.
Кроме того, в соответствии с данным изобретением защищенное по амину соединение тиенамицина в качестве нового промежуточного соединения эффективно используют для получения моногидрата имипенема формулы I.
Кроме того, по данному изобретению соединение формулы II приводят во взаимодействие с гидрохлоридом изопропилформимидата или гидрохлоридом бензилформимидата в присутствии основания с получением защищенного по карбоксильной группе имипенема формулы V, гидрированием защищенного по карбоксильной группе имипенема в присутствии катализатора-металла, для удаления защитной группы, с последующей соответствующей обработкой для получения водного раствора, который затем выделяют колоночной хроматографией с обращенной фазой и кристаллизуют из соответствующего спирта или кетона с получением моногидрата имипенема формулы I высокой чистоты с высоким выходом.
Промышленная применимость
Как объяснено выше в отношении соединения формулы II или его производного данного изобретения, различные защитные группы эффективно вводят по карбоксильным и аминогруппам тиенамицина с получением защищенного по амину соединения тиенамицина формулы II в качестве промежуточного соединения имипенема, которое используют для получения моногидрата имипинема формулы I. По способу данного изобретения проблема низкого выхода моногидрата имипенема из-за наличия большого количества примесей в результате отсутствия выделения и очистки промежуточных соединений может быть решена. Кроме того, моногидрат имипенема с высокой чистотой может быть получен из нового промежуточного соединения формулы II простым способом. Кроме того, выход и качество моногидрата имипенема формулы I могут быть значительно улучшены.
Так как при способе данного изобретения используют обычные органические растворители и воду, растворители легко удалить после завершения реакции. Кроме того, так как при гидрировании для удаления защитных групп, используют палладиевый катализатор, содержащий большое количество воды, значительно снижается опасность манипуляций, позволяя проводить процесс данного изобретения в мягких условиях реакции. Соответственно, способ данного изобретения является экономически выгодным и делает возможным получение имипенема без каких-либо затруднений.
Данное изобретение относится к соединению формулы II: где R1 представляет п-нитробензильную или п-метоксибензильную группу; и R2 и R3 могут быть одинаковыми или разными, и каждый независимо представляет C1-6 алкильную или арильную группу, или его производному, а также к способу получения соединения формулы II. Кроме того, описан способ получения имипенема формулы I с использованием соединения формулы II.
3 н. и 6 з.п. ф-лы, 5 табл., 6 ил.

где R1 представляет п-нитробензильную или п-метоксибензильную группу; R2 и R3 могут быть одинаковыми или разными и каждый независимо представляет С1-6 алкильную или арильную группу,
или его производное.
где R1 представляет п-нитробензильную или п-метоксибензильную группу; R2 и R3 могут быть одинаковыми или разными и каждый независимо представляет С1-6 алкильную или арильную группу,
путем сочетания соединения формулы IV
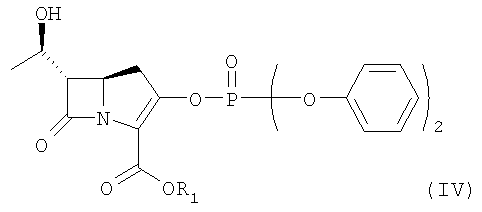
где R1 представляет п-нитробензильную или п-метоксибензильную группу
или его производного с гидрохлоридом 2-аминоэтантиола в присутствии основания с последующей реакцией с кетоном.
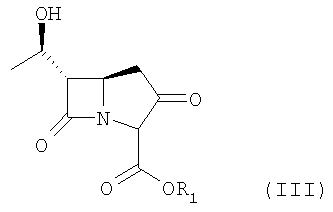
где R1 представляет п-нитробензильную или п-метоксибензильную группу,
с дифенилхлорфосфатом в присутствии основания в среде растворителя.

путем взаимодействия соединения формулы II
где R1 представляет п-нитробензильную или п-метоксибензильную группу; R2 и R3 могут быть одинаковыми или разными и каждый независимо представляет С1-6 алкильную или арильную группу,
с изопропилформимидатом или бензилформимидатом в присутствии основания в среде растворителя с получением соединения формулы V
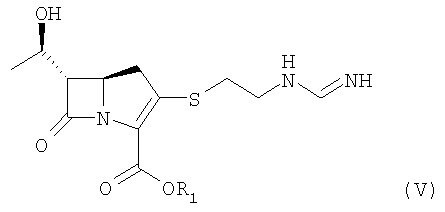
где R1 представляет п-нитробензильную или п-метоксибензильную группу,
гидрирования соединения формулы V в присутствии катализатора-металла, выделения гидрированного соединения и кристаллизации выделенного соединения в присутствии спирта или кетона.
| СПОСОБ РАЗВЕДКИ НЕФТЕГАЗОВЫХ ЗАЛЕЖЕЙ | 1996 |
|
RU2094828C1 |
| РОЗПТОЧНЫЙ КОНТАКТ | 0 |
|
SU236594A1 |
| Топчак-трактор для канатной вспашки | 1923 |
|
SU2002A1 |
| US 4894450 A, 16.01.1990 | |||
| 1-МЕТИЛКАРБАПЕНЕМ ИЛИ ЕГО ФАРМАКОЛОГИЧЕСКИ ПРИЕМЛЕМЫЕ СОЛИ, КОМПОЗИЦИЯ, СПОСОБ ПРЕДУПРЕЖДЕНИЯ ИЛИ ЛЕЧЕНИЯ БАКТЕРИАЛЬНЫХ ИНФЕКЦИЙ | 1997 |
|
RU2162088C2 |

