Область техники
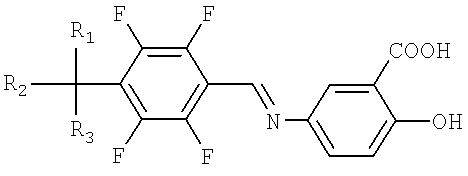
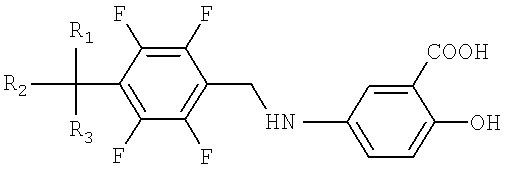
Настоящее изобретение относится к способу получения производных тетрафторбензил-5-аминосалициловой кислоты, представленных Формулой I, и их фармацевтически приемлемых солей, которые являются пригодными для профилактики и лечения острых и хронических нейродегенеративных заболеваний.
Формула I

где М+ представляет собой литий, натрий или калий; и R1, R2 и R3, независимо друг от друга, могут быть водородом или галогеном.
Уровень техники
Заявки на патент Кореи №2003-0097706 и 2004-0066639 раскрывают терапевтическую эффективность тетрафторбензильных производных в профилактике и лечении острых и хронических нейродегенеративных заболеваний и возможность их эффективного использования для профилактики и лечения хронических нейродегенеративных заболеваний, таких как болезнь Альцгеймера, болезнь Паркинсона и болезнь Хантингтона; вызванная судорогами болезнь мозга, такая как эпилепсия; и ишемическая болезнь головного мозга, такая как инсульт.
Способ получения производных тетрафторбензил-5-аминосалициловой кислоты описан в заявке на патент Кореи №2003-0097706 и проиллюстрирован в следующей схеме реакции II.
Схема реакции II
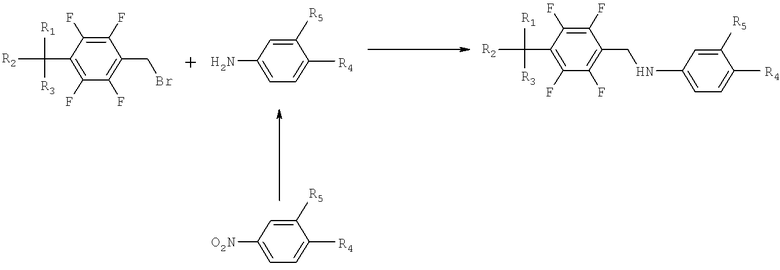
где R1, R2 и R3 являются водородом или галогеном соответственно; R4 представляет собой гидрокси, алкил, алкокси, галоген, алкокси, замещенный галогеном, алканоилокси или нитро; и R5 представляет собой карбоновую кислоту или сложный эфир карбоновой кислоты, замещенный C1-C4 алкилом, карбоксиамид, сульфокислоту, галоген или нитрогруппу.
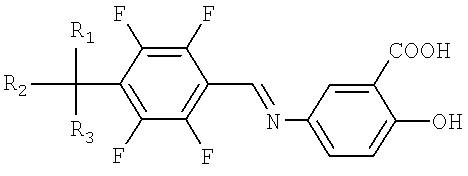
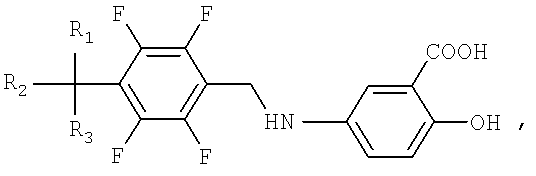
Схема реакции II иллюстрирует способ получения производных тетрафторбензил-5-аминосалициловой кислоты. Вначале нитробензол подвергают гидрированию, полученный анилин взаимодействует с соединением тетрафторбензилбромида в присутствии триэтиламина и диметилформамида, с образованием желаемого производного тетрафторбензил-5-аминосалициловой кислоты.
Способ, однако, приводит также к образованию димера (формула III) в количествах более чем 1% от общей массы продукта. Димер образуется в результате побочной реакции между исходным веществом, соединением тетрафторбензилбромида и вторичной аминовой группой производного тетрафторбензил-5-аминосалициловой кислоты. Димер удаляется с трудом обычными методами перекристаллизации, и требуется более сложный процесс очистки, чтобы поддерживать стандарт по примеси 0.1%. Таким образом, данная схема реакции является неприемлемой для применения в промышленности. Она приводит к пониженному выходу желаемого продукта и таким образом увеличивает производственные затраты.
Формула III
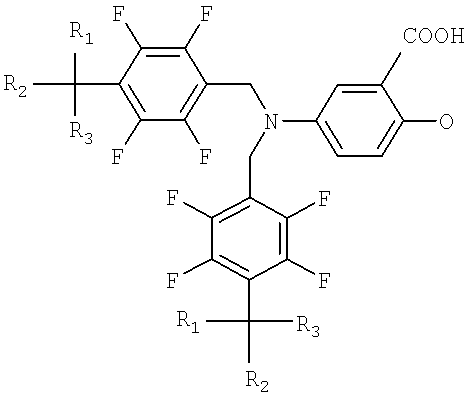
где R1, R2 и R3, независимо друг от друга, могут быть водородом или галогеном.
Кроме того, заявка на патент Кореи №2003-0097706 не раскрывает соли производного тетрафторбензил-5-аминосалициловой кислоты более детально. Таким образом, требуется другое исследование состава вышеупомянутого соединения с точки зрения стабильности.
Интенсивное и полное исследование показало, что образования димера при получении производного тетрафторбензил-5-аминосалициловой кислоты можно избежать при использовании производного тетрафторбензилидин-5-аминосалициловой кислоты в качестве промежуточного соединения. Также стабильность желаемого соединения была улучшена посредством получения соединения в виде соли с использованием щелочных металлов, таким образом, приводя к настоящему изобретению.
Раскрытие изобретения
Настоящее изобретение должно обеспечить новый способ получения производных тетрафторбензил-5-аминосалициловой кислоты, предотвращающий образование примесей, таких как димер, представленный Формулой III.
В частности, целью настоящего изобретения является новый способ получения производных тетрафторбензил-5-аминосалициловой кислоты при использовании производного тетрафторбензилидин-5-аминосалициловой кислоты, представленного Формулой II, в качестве промежуточного соединения.
Далее, другой целью настоящего изобретения является получение солей производных тетрафторбензил-5-аминосалициловой кислоты, которые имеют повышенную стабильность и меньшую токсичность.
Сущность изобретения
Способ получения тетрафторбензил-5-аминосалициловой кислоты показан на схеме реакции I. Способ получения включает следующие стадии:
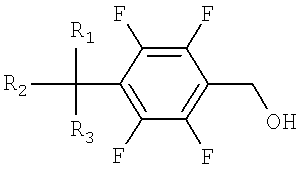
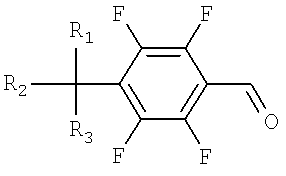
a) окисление тетрафторбензилового спирта, представленного Формулой 1, до тетрафторбензальдегида, представленного Формулой 2;
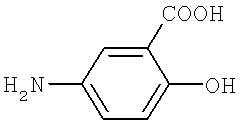
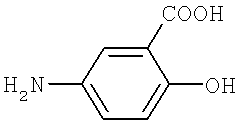
b) превращение тетрафторбензальдегида в производное тетрафторбензилидин-5-аминосалициловой кислоты, представленное Формулой II посредством реакции дегидратации-конденсации между тетрафторбензальдегидом и 5-аминосалициловой кислотой, представленной Формулой 3, и
c) гидрирование производного тетрафторбензилидина-5-аминосалициловой кислоты до тетрафторбензил-5-аминосалициловой кислоты, представленной Формулой I.
Схема реакции I
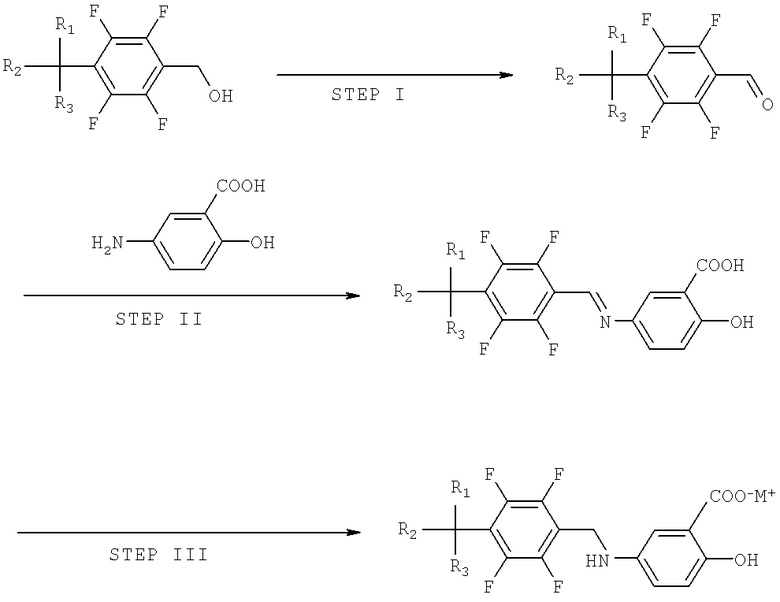
где М+ представляет литий, натрий или калий; и R1, R2 и R3 могут, независимо друг от друга, быть водородом или галогеном.
Формула I
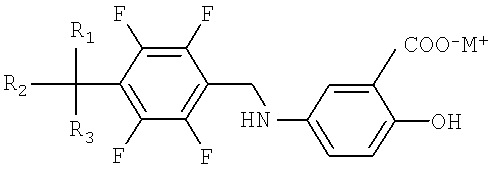
Формула II
Формула 1
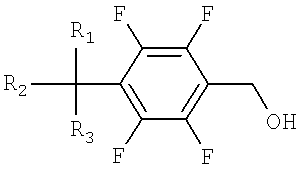
Формула 2
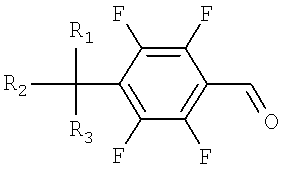
Формула 3
где M+ представляет литий, натрий или калий; и R1, R2 и R3, независимо друг от друга, могут быть водородом или галогеном.
В дополнительном аспекте настоящего изобретения заявляется 2-гидрокси-5-[(2,3,5,6-тетрафтор-4-трифторметилбензилидин)амино]бензойная кислота, представленная Формулой II, как новое промежуточное соединение для получения соединения тетрафторбензил-5-аминосалициловой кислоты, представленного Формулой I.
Соединение тетрафторбензил-5-аминосалициловой кислоты по настоящему изобретению синтезируют в соответствии со схемой реакции I следующим образом:
1) стадия I: тетрафторбензальдегид образуется посредством окисления тетрафторбензилового спирта;
2) стадия II: тетрафторбензальдегид, как получено на стадии I, и 5-аминосалициловую кислоту подвергают реакции дегидратации-конденсации в присутствии растворителя метиленхлорида при комнатной температуре с использованием молекулярных сит, для образования производного тетрафторбензилидин-5-аминосалициловой кислоты, представленной Формулой II, как соединения имина; и
3) стадия III: производное тетрафторбензилидин-5-аминосалициловой кислоты, полученное на стадии II и представленное Формулой II, гидрируют, с использованием платинового катализатора и растворителя спирта, для получения соединений тетрафторбензил-5-аминосалициловой кислоты, представленных Формулой I, посредством контактного восстановления.
Вышеупомянутые способы раскрываются более подробно, как сформулировано ниже.
Тетрафторбензальдегид на стадии I может быть легко получен посредством обычного окисления тетрафторбензилового спирта с использованием хлорхромата пиридиния и метиленхлорида.
На стадии II тетрафторбензальдегид, полученный на стадии I, и 5-аминосалициловую кислоту подвергают реакции дегидратации-конденсации при комнатной температуре, для образования производного тетрафторбензилидин-5-аминосалициловой кислоты, представленной Формулой II, согласно методикам, о которых сообщается в J. Org. Chem., 66 (6), 2001, 1992-1998.
Слабокислотные реагенты (такие, как пара-толуолсульфоновая кислота) могут быть использованы в реакции дегидратации-конденсации; как указано в Tetrahedron Lett., 45(2), 2003, 243-248. Полученное соединение перекристаллизовывают из спирта или диэтилового эфира, для получения соединения более высокого качества, представленного Формулой II. Способы согласно настоящему изобретению являются экономически выгодными, поскольку желаемое соединение может быть получено при использовании непрерывных процессов без стадии разделительной очистки.
Поэтому стадия II настоящего изобретения характеризуется предотвращением образования димера, полученного в процессе синтеза соединения тетрафторбензил-5-аминосалициловой кислоты формулы I, о котором сообщается в заявке на патент Кореи №2003-0097706.
На стадии III производное тетрафторбензилидин-5-аминосалициловой кислоты, представленное Формулой II, в качестве продукта стадии II, гидрируют с использованием катализатора палладия и растворителя спирта, для получения соединения тетрафторбензил-5-аминосалициловой кислоты, представленного Формулой I, посредством контактного восстановления.
Реакция восстановления соединения на стадии III может быть выполнена, с образованием желаемого соединения, представленного Формулой I, посредством других обычных и общих процедур восстановления, с использованием боргидрида натрия и спирта; или катализатора переходного металла и слабой кислоты, такой как уксусная кислота.
Настоящее изобретение обеспечивает способ получения производного тетрафторбензил-5-аминосалициловой кислоты, который предотвращает образование примесей, таких как димер, представленный Формулой III.
Конкретное воплощение состоит в том, что настоящее изобретение характеризуется использованием производного тетрафторбензилидин-5-аминосалициловой кислоты (формула II) в качестве промежуточного соединения, для предотвращения образования димера.
Дополнительный аспект настоящего изобретения обеспечивает фармацевтически приемлемые соли тетрафторбензил-5-аминосалициловой кислоты, представленной Формулой I. Фармацевтически приемлемые соли желаемого соединения согласно настоящему изобретению включают щелочные металлы, такие как натрий, калий и литий.
Соли соединения, представленного Формулой I, могут быть получены непосредственно кристаллизацией или лиофилизацией с использованием неорганического реагента, такого как гидроксид лития, гидрооксид натрия или гидроксид калия в присутствии спирта, ацетона, ацетонитрила и других органических растворителей.
Фармацевтически приемлемые соли в настоящем изобретении были выбраны согласно следующим критериям.
Был проведен скрининг различных видов фармацевтически приемлемых солей желаемого соединения, представленного Формулой I, таких как щелочные металлы; щелочноземельные металлы, такие как кальций; фармацевтически приемлемые нетоксичные соли, такие как образованные с соляной кислотой, фосфорной кислотой, серной кислотой, азотной кислотой, малеиновой кислотой, уксусной кислотой или лимонной кислотой; и органические соли, такие как образованные с N,N-дибензилэтилендиамином или этилендиамином.
Кислотно-аддитивные соединения и их соли, такие как натриевая, калийная и литиевая, как получено в Примерах 3-6, перемешивают в смесителе на водяной бане при 25°С, при 100 об/мин в течение 48 часов, для измерения их растворимости в воде. Наблюдают, что кислотные (свободное основание) соединения являются плохо растворимыми в воде, тогда как соли натрия, калия и лития имеют растворимость 25~80 мг/мл, 590 мг/мл и 500 мг/мл соответственно, в зависимости от метода кристаллизации.
Если используют нетоксичные кислоты, связанные с амином, то реакции не наблюдают; и желаемый продукт не образовывался. В экспериментах, при использовании органических солей, наблюдалась реакция, и был получен желаемый продукт, но они были плохо растворимыми в воде.
Поэтому способы получения желаемого соединения с использованием различных кислот или органических солей были исключены из настоящего изобретения. Фармацевтически приемлемые соли желаемого соединения, которые включают щелочноземельные металлы, такие как кальций, были также исключены из-за их недостаточной растворимости в воде.
Таким образом, настоящее изобретение относится к солям натрия, калия и лития, имеющим лучшую растворимость и стабильность среди фармацевтически приемлемых солей желаемого соединения, представленного формулой I.
Эти соли настоящего изобретения имеют лучший профиль стабильности при пероральной форме дозировки с меньшей токсичностью.
Преимущества изобретения
Как упомянуто выше, настоящее изобретение обеспечивает способ получения производного тетрафторбензил-5-аминосалициловой кислоты, который предотвращает образование примесей, таких как димер, представленный Формулой III.
Настоящее изобретение обеспечивает способ получения производного тетрафторбензил-5-аминосалициловой кислоты при использовании производного тетрафторбензилидин-5-аминосалициловой кислоты, представленного Формулой II, в качестве промежуточного соединения.
Другой объект настоящего изобретения - это соли производного тетрафторбензил-5-аминосалициловой кислоты с лучшей стабильностью желаемого соединения и с меньшей токсичностью.
Краткое описание чертежей
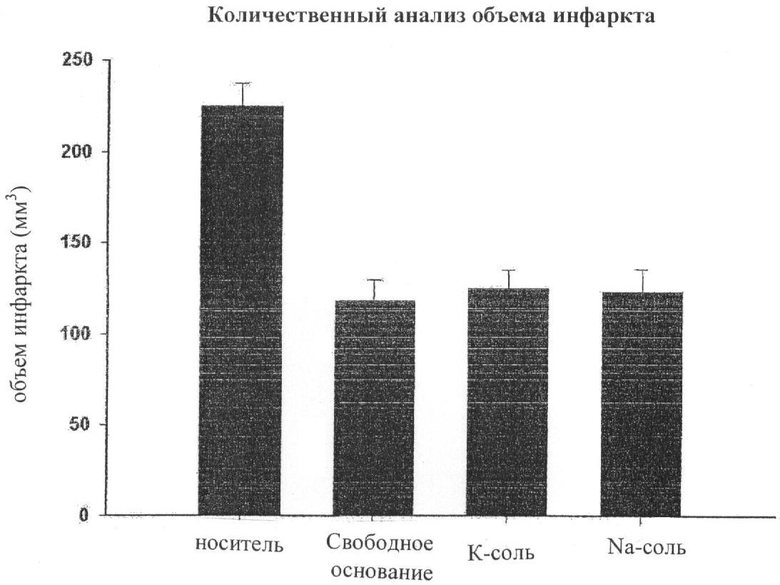
На чертеже показаны защитные эффекты соединения тетрафторбензил-5-аминосалициловой кислоты, так же как и защитные эффекты форм калиевой и натриевой соли тетрафторбензил-5-аминосалициловой кислоты на крысах с очаговой ишемией головного мозга, вызванной окклюзией средней мозговой артерии.
Осуществление изобретения
Настоящее изобретение далее раскрыто в следующих примерах и экспериментах, которые являются всего лишь иллюстративными и не должны рассматриваться как ограничивающие объем настоящего изобретения.
Пример 1: Получение 2,3,5,6-тетрафтор-4-трифторметилбензальдегида
2,3,5,6-Тетрафторбензиловый спирт (49.62 г, 0.200 моль) и метиленхлорид (500 мл) перемешивают при комнатной температуре для полного растворения и затем медленно добавляют хлорхромат пиридиния (73.29 г, 0.340 моль) к реакционной смеси. Реакционную смесь нагревают в колбе при кипении с обратным холодильником в течение 4 часов, в то время как температуре реакции позволяют увеличиваться; и затем охлаждают. После того как нерастворенный материал отфильтровывают при пониженном давлении, осадок промывают 500 мл очищенной воды и 500 мл насыщенного физиологического солевого раствора. Отделенный масляный слой высушивают над MgSO4 (5 г) и концентрируют при пониженном давлении, с получением желаемого соединения; 48.23 г 2,3,5,6-тетрафторбензальдегида (0.196 моль, выход 98.0%) в виде масла желтого цвета.
1H ЯМР (CDCI3, 300 МГц): 10.35 (м, 1Н)
Пример 2: Получение 2-гидрокси-5-[(2,3,5,6-тетрафтор-4-трифторметилбензилидин) амино]бензойной кислоты
5-Аминосалициловую кислоту (33.02 г, 0.216 моль) добавляют к раствору 2,3,5,6-тетрафторбензальдегида (48.23 г, 0.196 моль), полученного в Примере 1, и метиленхлорида (500 мл). Реакционную смесь перемешивают в течение 10 минут и после добавления 4Å молекулярных сит (5.0 г) перемешивают при комнатной температуре в течение 16 часов. После того как нерастворенный материал был отфильтрован при пониженном давлении, осадок промывают 500 мл очищенной воды и 500 мл насыщенного физиологического солевого раствора. Отделенный масляный слой высушивают над MgSO4 (5 г) и отфильтровывают при пониженном давлении. Масляный раствор концентрируют при пониженном давлении для получения 2-гидрокси-5-[(2,3,5,6-тетрафтор-4-трифторметилбензилидин)амино]бензойной кислоты в виде раствора желтого масла. Осадок перекристаллизовывают из этилового спирта (200 мл) и высушивают при пониженном давлении, для образования 65.97 г (0.173 моль, выход 88.3%) желаемого белого твердого продукта.
1Н ЯМР (CDCI3, 300 МГц): 7.12 (дд, 1Н), 7.57 (д, 1Н), 7.95 (д, 1Н), 8.39 (с, 2Н)
Пример 3: Получение 2-гидрокси-5-(2,3,5,6-тетрафтор-4-трифторметилбензиламино)бензойной кислоты
5-Аминосалициловую кислоту (33.02 г, 0.216 моль) добавляют к раствору 2,3,5,6-тетрафторбензальдегида (48.23 г, 0.196 моль), полученного в Примере 1, и метиленхлорида (500 мл). Реакционную смесь перемешивают в течение 10 минут и после добавления 4Å молекулярных сит (5.0 г) перемешивают при комнатной температуре в течение 16 часов. Нерастворенный материал отфильтровывают при пониженном давлении и высушивают над MgSO4 (10 г). Раствор концентрируют при пониженном давлении и затем добавляют к осадку этиловый спирт (600 мл). Платиновый катализатор (9.65 мг) прибавляют к раствору и размешивают при 20-25°С в течение 2 часов при 4 атм. Нерастворенный материал отфильтровывают при пониженном давлении и концентрируют для удаления растворителя. Этилацетат (500 мл) и очищенную воду (500 мл) прибавляют к концентрированному осадку. Раствор охлаждают до 5-10°С и затем рН раствора доводят до значения 1.0-1.5 с использованием соляной кислоты; в то время как поддерживают температуру, установившуюся при значении 5-10°С. Раствор перемешивают в течение 30 минут при комнатной температуре и оставляют в покое. Отделенный масляный слой промывают 500 мл очищенной воды и 500 мл насыщенного физиологического солевого раствора в указанном порядке. Активированный уголь (5 г) добавляют к отделенному масляному слою и перемешивают в течение 1 часа при комнатной температуре. Масляный слой отфильтровывают при пониженном давлении и концентрируют, для получения осадка в виде светлого твердого продукта белого цвета. Этилацетат (100 мл) прибавляют к концентрированному осадку и допускают повышение температуры до 50°С для полного растворения. Н-гексан (400 мл) прибавляют к реакционной смеси, затем раствор охлаждают до 5-10°С и перемешивают в течение 6 часов. Осажденные кристаллы отфильтровывают и высушивают при пониженном давлении, с образованием 63.99 г (0.167 моль, выход 85.2%) 2-гидрокси-5-(2,3,5,6-тетрафтор-4-трифторметилбензиламино)бензойной кислоты в виде желаемого соединения. (Чистота по ВЭЖХ: более чем 99.8%.)
1Н ЯМР (CDCI3, 300 МГц): 4.41 (с, 2Н), 6.77 (д, 1H), 6.94 (дд, 1Н), 7.08 (д, 1Н)
Пример 4: Получение 2-гидрокси-5-(2,3,5,6-тетрафтор-4-трифторметилбензиламино) бензоата калия
<Способ 4-1>
2-Гидрокси-5-(2,3,5,6-тетрафтор-4-трифторметилбензиламино)бензойную кислоту (100 г, 0.261 моль), полученную в Примере 3, прибавляют к безводному этиловому спирту (500 мл) и затем допускают повышение температуры до 50°С для полного растворения. Полученный раствор охлаждают до 10°С. рН раствора доводят до значения 6.8-7.0 с использованием отдельно полученного 85%-раствора гидроксида калия (17.22 г, 0.261 моль) и безводного этанола (30 мл). Реакционную смесь перемешивают в течение 2 часов при комнатной температуре и затем осажденные кристаллы отфильтровывают и высушивают, для получения 100.8 г (0.239 моль, выход 91.7%) 2-гидрокси-5-(2,3,5,6-тетрафтор-4-трифторметилбензиламино)бензоата калия, в виде желаемого соединения.
<Способ 4-2>
2-Гидрокси-5-(2,3,5,6-тетрафтор-4-трифторметилбензиламино)бензойную кислоту (100 г, 0.261 моль), полученную в Примере 3, добавляют к очищенной воде (3000 мл) и затем полученный раствор охлаждают до 10°С. При той же самой температуре рН раствора устанавливают равным 6.8-7.0 с использованием 1н. раствора гидроксида калия и перемешивают в течение еще 2 часов. Раствор лиофилизуют для получения 109.9 г (0.261 моль, выход 100.0%) 2-гидрокси-5-(2,3,5,6-тетрафтор-4-трифторметилбензиламино)бензоата калия в виде желаемого соединения.
Пример 5: Получение 2-гидрокси-5-(2,3,5,6-тетрафтор-4-трифторметилбензиламино) бензоата натрия
<Способ 5-1>
Повторяют методику по Способу 4-1 Примера 4; кроме замены гидроксида калия тем же самым количеством молей гидроксида натрия; 96.75 г (0.239 моль, выход 91.5%) 2-гидрокси-5-(2,3,5,6-тетрафтор-4-трифторметилбензиламино)бензоата натрия получили в виде желаемого соединения.
<Способ 5-2>
Повторяют методику по Способу 4-2 Примера 4; кроме замены гидроксида калия тем же самым количеством молей 2-этилгексаноата натрия; 97.70 г (0.241 моль, выход 92.4%) 2-гидрокси-5-(2,3,5,6-тетрафтор-4-трифторметилбензиламино)бензоата натрия получают в виде желаемого соединения.
<Способ 5-3>
Повторяют методику по Способу 4-2 Примера 4; кроме замены гидроксида калия тем же самым количеством молей 1н. раствора гидроксида натрия; 105.7 г (0.261 моль, выход 100.0%) 2-гидрокси-5-(2,3,5,6-тетрафтор-4-трифторметилбензиламино)бензоата натрия получают в виде желаемого соединения.
Пример 6: Получение 2-гидрокси-5-(2,3,5,6-тетрафтор-4-трифторметилбензиламино) бензоата лития
<Способ 1>
Повторяют методику по Способу 4-1 Примера 4; кроме замены гидроксида калия тем же самым количеством молей гидроксида лития; 90.38 г (0.232 моль, выход 89.0%) 2-гидрокси-5-(2,3,5,6-тетрафтор-4-трифторметилбензиламино)бензоата лития получают в виде желаемого соединения.
<Способ 2>
Повторяют методику по Способу 4-2 Примера 4; кроме замены гидроксида калия тем же самым количеством молей 1н. раствора гидроксида лития; 101.5 г (0.261 моль, выход 100.0%) 2-гидрокси-5-(2,3,5,6-тетрафтор-4-трифторметилбензиламино)бензоата лития получают в виде желаемого соединения.
Пример 7: Подготовка инъекции
Инъекцию готовят таким образом, что каждую из солей (Na-соль, K-соль, Li-соль), полученных в Примерах 4-6, растворяют в воде для инъекции (WFI) при концентрации 2 мг/мл. Раствором заполняют ампулу и обрабатывают в вакууме.
Экспериментальный пример 1: Испытание на стабильность препарата для инъекции
После того как получают каждый из препаратов для инъекции, как описано в Примере 7, их хранят при различных температурах (25°С, 40°С, 60°С) в течение 6 месяцев, количество активных ингредиентов измеряют с использованием ВЭЖХ.
Следующие Таблицы 1, 2 и 3 показывают остаточное количество активных ингредиентов в каждом препарате для инъекции в зависимости от времени.
Как следует из вышеуказанных Таблиц, количество каждой соли имеет тенденцию уменьшаться постепенно с течением времени, и по мере повышения температуры большие количества разлагаются.
В частности, профиль стабильности калийной соли был лучше, чем профиль стабильности натриевой и литиевой соли.
Экспериментальный пример 2: Испытание на стабильность лекарственного препарата для перорального приема
Порошок каждой из солей (натриевой соли, калиевой соли, литиевой соли), как получено в Примере 4-6, насыпают в ампулу и оставляют храниться в вакууме. Его хранят затем при различных температурах (25°С, 40°С, 60°С) в течение 6 месяцев и количество активных ингредиентов в порошке измеряют с помощью ВЭЖХ. Следующие Таблицы 4, 5 и 6 показывают оставшееся количество активных ингредиентов в порошке с течением времени.
Как следует из вышеуказанных Таблиц 4, 5 и 6, профиль стабильности образцов в порошковой форме был лучше, чем профиль стабильности в растворе. Изменения в количестве каждого образца порошка при 25°С не наблюдались.
Таким образом, результаты испытаний показывают, что калиевые и натриевые соли в порошковой форме были относительно стабильны при высоких температурах; профиль повышенной стабильности обеспечивает лекарственный препарат для перорального приема.
Экспериментальный пример 3: Испытание кратковременного токсического эффекта лекарственной формы в виде свободного основания на крысах
Используют безпатогенных SD (Sprague Dawely) самцов крыс 6-8-недельного возраста для данного эксперимента. После того как соединения растворяют в физиологическом растворе, образец вводят внутривенно в хвостовую вену животных, с использованием шприца одноразового применения. Каждый образец вводят в виде одноразовой дозы. Спустя 14 дней после введения летальности были зарегистрированы для различных уровней дозы, как показано в Таблицах 7-9.
Как отмечено в Таблицах 7-9, формы солей калия и натрия имеют меньшую токсичность, чем свободное основание. Таким образом, демонстрируется, что эти формы солей по настоящему изобретению играют роль для значительного улучшения профиля токсичности самого желаемого соединения.
Экспериментальный пример 4: Защитные эффекты соединения тетрафторбензил-5-аминосалициловой кислоты и его солей на крысах с очаговой ишемией головного мозга, вызванной окклюзией средней мозговой артерии
С использованием каждого из соединений, полученных в Примере 3-5, защитные эффекты соединений тетрафторбензил-5-аминосалициловой кислоты, представленных формулой I, и их калиевой и натриевой солей были изучены на крысах с очаговой ишемией мозга, обусловленной искусственной окклюзией средней мозговой артерии.
Беспатогенных самцов крыс SD (Sprague Dawely) подвергают очаговой ишемизации мозга путем окклюзии средней мозговой артерии в течение 60 минут и пролечивают препаратами после реперфузии. Животных умерщвляют спустя 24 часа, головной мозг извлекают для исследования. Объем зоны инфаркта был проанализирован после окрашивания срезов мозга трифенилтетразолий-хлоридом (ТТС), как показано на чертеже. Как изображено на чертеже, три соединения показали схожее защитное действие в отношении очаговой ишемии мозга.
Настоящее изобретение относится к вариантам способа получения производного тетрафторбензил-5-аминосалициловой кислоты Формулы I
 ,
,
где R1, R2 и R3 независимо друг от друга могут быть водородом или галогеном, которое может быть использовано для профилактики и лечения острых и хронических нейродегенеративных заболеваний, в частности очаговой ишемии головного мозга, и к способу получения фармацевтически приемлемых солей данного производного. Один из вариантов способа получения производного тетрафторбензил-5-аминосалициловой кислоты Формулы I включает следующие стадии:
а) окисление тетрафторбензилового спирта Формулы 1
Формула 1
до тетрафторбензальдегида Формулы 2
Формула 2
b) превращение тетрафторбензальдегида в производное тетрафторбензилидин-5-аминосалициловой кислоты Формулы II
Формула II
реакцией дегидратации-конденсации между тетрафторбензальдегидом и 5-аминосалициловой кислотой Формулы 3
Формула 3
и
с) гидрирование производного тетрафторбензилидин-5-аминосалициловой кислоты до производного тетрафторбензил-5-аминосалициловой кислоты Формулы I. 3 н. и 1 з.п. ф-лы, 9 табл., 1 ил.
1. Способ получения производного тетрафторбензил-5-аминосалициловой кислоты формулы I,
формула I
,
где R1, R2 и R3 независимо друг от друга, могут быть водородом или галогеном, включающий:
а) окисление тетрафторбензилового спирта формулы 1
формула 1
до тетрафторбензальдегида формулы 2
формула 2
b) превращение тетрафторбензальдегида в производное тетрафторбензилидин-5-аминосалициловой кислоты формулы II
формула II
реакцией дегидратации-конденсации между тетрафторбензальдегидом и 5-аминосалициловой кислотой формулы 3
формула 3
и
с) гидрирование производного тетрафторбензилидин-5-аминосалициловой кислоты до производного тетрафторбензил-5-аминосалициловой кислоты формулы I.
2. Способ по п.1, отличающийся тем, что тетрафторбензальдегид и 5-аминосалициловую кислоту подвергают реакции дегидратации-конденсации в присутствии растворителя метиленхлорида при комнатной температуре, с использованием молекулярных сит, с образованием производного тетрафторбензилидин-5-аминосалициловой кислоты формулы II.
3. Способ получения фармацевтически приемлемой соли производного тетрафторбензил-5-аминосалициловой кислоты формулы I
формула I
где R1, R2 и R3 независимо друг от друга, могут быть водородом или галогеном, включающий:
а) окисление тетрафторбензилового спирта формулы 1
формула 1
до тетрафторбензальдегида формулы 2
формула 2
b) превращение тетрафторбензальдегида в производное тетрафторбензилидин-5-аминосалициловой кислоты формулы II
формула II
реакцией дегидратации-конденсации между тетрафторбензальдегидом и 5-аминосалициловой кислотой формулы 3
формула 3
и
с) гидрирование производного тетрафторбензилидин-5-аминосалициловой кислоты до производного тетрафторбензил-5-аминосалициловой кислоты формулы I, и
d) получение солей производного тетрафторбензил-5-аминосалициловой кислоты непосредственной кристаллизацией или лиофилизацией с использованием неорганического реагента, такого как гидроксид лития, гидроксид натрия или гидроксид калия в присутствии спирта, ацетона, ацетонитрила и других органических растворителей.
4. Способ получения производного тетрафторбензил-5-аминосалициловой кислоты формулы I,
формула I
где R1, R2 и R3, независимо друг от друга, могут быть водородом или галогеном,
при котором производное тетрафторбензил-5-аминосалициловой кислоты получают гидрированием производного тетрафторбензилидин-5-аминосалициловой кислоты формулы II
формула II
.
| Способ приготовления мыла | 1923 |
|
SU2004A1 |
| Прибор, замыкающий сигнальную цепь при повышении температуры | 1918 |
|
SU99A1 |
| US 5527814 A, 18.01.1996 | |||
| ЩИТОВОЙ ДЛЯ ВОДОЕМОВ ЗАТВОР | 1922 |
|
SU2000A1 |
| ЩИТОВОЙ ДЛЯ ВОДОЕМОВ ЗАТВОР | 1922 |
|
SU2000A1 |
| Производные -/арил(алкил, алке-Нил)АМиНО/-бЕНзОйНОй КиСлОТы илифАРМАцЕВТичЕСКи пРиЕМлЕМыЕ СОли,ОблАдАющиЕ СпОСОбНОСТью СНижАТьуРОВЕНь липидОВ B СыВОРОТКЕ КРОВи | 1976 |
|
SU803856A3 |

