Область техники
Настоящее изобретение в целом относится к способу получения трийодированных ароматических производных, включающему конденсацию аминосульфинильного производного с α-гидроксикислотой с получением соответствующих амидо-производных, которые представляют собой подходящие промежуточные соединения для синтеза рентгеновских контрастирующих агентов.
Уровень техники
Контрастирующие агенты, также известные как контрастирующая среда, часто используют во время медицинских визуализирующих исследований, чтобы подкрасить конкретные части тела и сделать их более доступными для наблюдения. В их числе находятся рентгеновские контрастирующие среды и йодированные неионные контрастирующие среды, например, такие как диатризоат, иоталамат, иокситаламат, метризоат, иогексол, йомепрол (The Merck Index, XIII Ed., 2001, № 5071), иопамидол (The Merck Index, XIII Ed., 2001, № 5073), йопентол, йопромид, йоверсол, йоксилан, йодиксанол, йозаркол, иогуламид, иоглунид, иоглуамид, ацетризоат, йодамид, иоцетамид, иоксаглат, йотролан, йотазул, йодипамид, йокармат, йодоксамат, йотроксат, йотролан и т.д. Другие примеры похожих йодированных контрастирующих агентов также описаны, например, в публикации WO 94/14478 (Bracco).
Такие соединения могут быть получены множеством синтетических способов, некоторые из которых характеризуются превращением ароматических амино-производных в соответствующие карбоксамиды по реакции с подходящим производным α-гидрокси-кислоты. Один из типичных примеров такой реакции можно найти в способах получения иопамидола, которые описаны, например, в публикациях WO 02/44132, WO 96/37459, WO 96/37460, US 5362905, WO 97/47590, WO 98/24757, WO 98/28259 и WO 99/58494. В качестве общей ссылки на способ получения иопамидола, смотрите, например, метод синтеза на представленной ниже схеме I:
Схема I

В соответствии со схемой I амино-производное формулы (3) соответствующим образом вводят в реакцию с (2S)-2-(ацетилокси)пропаноилхлоридом (4) с получением соединения (5).
Соединение формулы (3), в свою очередь, затем может быть получено при взаимодействии исходного материала формулы (1) с подходящим хлорирующим агентом, например с тионилхлоридом. Удивительно и в соответствии с тем, что указано в публикации WO 96/37459, промежуточное сульфинильное производное формулы (2) может быть получено во время стадии хлорирования.
Соединение формулы (5), полученное таким образом, далее вводят в реакцию с выбранным аминоспиртом, в данном случае с 2-амино-1,3-пропандиолом, хорошо известным как серинол, и подвергают снятию защитной группы у гидроксигруппы так, что получают иопамидол формулы (6).
Обе такие последние реакции проводят в соответствии с обычными способами, известными в данной области техники, или для получения карбоксамидов реакцией ацилхлоридных производных с аминосоединениями или для отщепления ацетильной защитной группы от гидроксильной функциональности.
Согласно приведенной выше схеме I соединение (4) представляет собой активированную реакционно-способную форму (2S)-2-гидроксипропановой кислоты, общеизвестной как L-молочная кислота, где гидроксигруппа соответствующим образом защищена, как показано. В этой связи необходимо отметить, что получение соединения (4) является затратным по времени, так как требует дополнительной синтетической стадии, начиная от коммерчески доступной натриевой соли (2S)-2-гидроксипропановой кислоты (7), как это показано на приведенной ниже схеме II:
Схема II
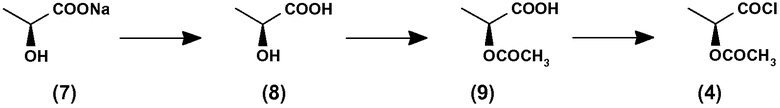
Согласно схеме II первая стадия представлена превращением соединения (7) в L-молочную кислоту (8) при работе в кислых условиях. Соединение, полученное таким образом, затем защищают по гидроксигруппе, что приводит к соответствующему ацетилированному промежуточному соединению (9), которое после выделения и очистки затем превращают в целевой ацетилхлорид (4) путем соответствующей функционализации карбоксильной группы с помощью подходящего хлорирующего агента, как правило, с помощью тионилхлорида.
Кроме временных затрат методика в соответствии со схемой II приводит к образованию желаемого соединения (4) в способе, который предполагает некоторое количество стадий перегонки или для выделения, или для очистки промежуточных соединений. Кроме того, несмотря на тот факт, что каждая из приведенных выше стадий может быть проведена в соответствии с обычными способами, использование чувствительных к воздуху и/или влаге реагентов, например тионилхлорида, может, таким образом, требовать выбора реакционных условий, достаточно обременительных, по меньшей мере, при работе с большими количествами веществ и реагентов в промышленном масштабе.
Тем самым особенно полезно будет найти альтернативный способ получения иопамидола с высоким выходом и с высокой степенью чистоты с помощью способа, включающего взаимодействие любого подходящего промежуточного предшественника с реагентами, альтернативными соединению (4).
В этой связи в литературе представлены примеры получения рассматриваемых ароматических карбоксамидо-соединений при взаимодействии сульфинильных производных с α-гидроксикислотами. Смотрите, например, публикации Bioorg. Med. Chem. Lett., 2006, 16, 4784-4787; и Bioorg. Med. Chem. Lett., 2007, 17, 6261-6255, где остаток α-гидроксикислоты вводят в реакцию с ароматическими стерически незатрудненными сульфинильными субстратами. Также реакция рассматриваемых α-гидроксикислот с подходящими сульфинильными производными, называемыми в данном случае N-сульфиниламином или даже производными N-сульфиниланилина, также представлена в публикациях Tetrahedron Lett. 2000, 41, 6017-6020; и Tetrahedron Lett. 1986, 27, 1921-1924.
Интересно, что все из N-сульфиниланилинов, представленных в упомянутых выше документах предшествующего уровня техники и настоящее время называемых как Ar-N=S=O соединения, по существу основаны на довольно простых молекулах, получаемых из:
- самого анилина, где Ar соответствует фенилу;
- моно-замещенных анилинов, где Ar соответствует п-хлор-С6Н4-, п-метил-С6Н4-, п-нитро-С6Н4-; или
- дизамещенных анилинов, где Ar представляет собой 2-фтор-4-метоксикарбонил-С6Н3- или 2,4-дихлор-С6Н3.
По сведениям заявителей, однако, такой метод синтеза никогда не был раскрыт для получения рассматриваемых карбоксамидов на одной стадии, исходя из стерически затрудненных соединений, в особенности из полностью замещенных N-сульфиниланилинов, а именно пентазамещенных по ароматическому кольцу. В этой связи неплохо отметить, что в рамках способа получения иопамидола ароматическое кольцо описанных выше сульфинильных производных формулы (2) полностью замещено в положениях 3 и 5 двумя карбонильными остатками (например, группами -СОСl), и, что замечательно, в положениях 2, 4 и 6 тремя атомами йода, которые, как известно, являются объемными заместителями.
Таким образом, установлено, что стерически затрудненные полностью замещенные N-сульфиниланилины способны вступать в реакцию на одной стадии и при мягких рабочих условиях, с подходящими α-гидроксикислотами или их солями, давая желаемые карбоксамидо-производные, в соответствии с альтернативным способом синтеза.
Суть изобретения
Таким образом, настоящее изобретение предлагает весьма выгодный способ получения иопамидола, исходя из коммерчески доступных α-гидроксикислот или их солей, исключая за счет этого описанные выше недостатки, относящиеся к синтезу соединения (4).
Следовательно, объектом настоящего изобретения является способ получения соединения формулы
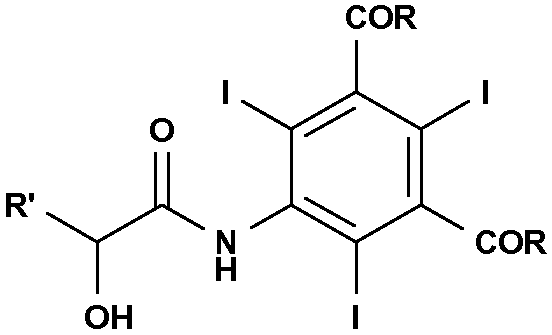
и этот способ включает взаимодействие соединения формулы (II) с α-гидроксикислотой формулы (III) или ее солью в присутствии подходящего основания:
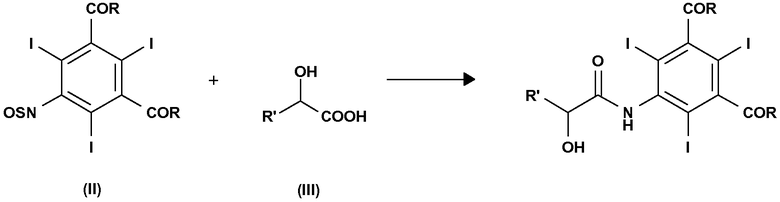
где:
R' представляет собой атом водорода или линейную или разветвленную С1-С6-алкильную группу, необязательно замещенную одной или несколькими защитными гидроксигруппами;
R, одинаковые в каждом случае, выбирают из группы, включающей алкоксигруппу (-OR1), аминогруппу (-NH2 или -NHR1) или атом хлора; и где R1 представляет собой линейную или разветвленную С1-С6-алкильную группу, необязательно замещенную одной или несколькими защищенными гидроксигруппами.
Подробное описание изобретения
В соответствии с первым вариантом осуществления настоящее изобретение основано на способе получения соединения формулы (I), и этот способ включает взаимодействие соединения формулы (II) с (2S)-2-гидроксипропановой кислотой или ее солями в присутствии подходящего основания:
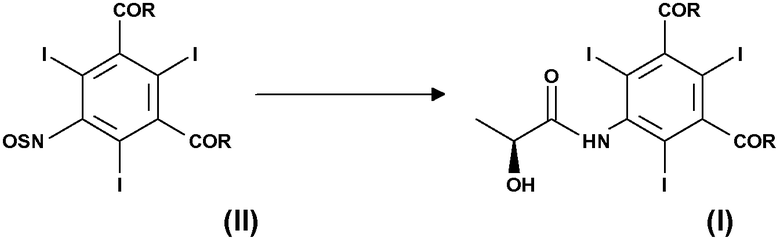
где:
R, одинаковые в каждом случае, выбирают из группы, включающей алкоксигруппу -OR1, аминогруппу -NH2 или -NHR1 или атом хлора; и где R1 представляет собой линейную или разветвленную С1-С6-алкильную группу, необязательно замещенную одной или несколькими защищенными гидроксигруппами.
Кроме того, как подробно показано в экспериментальной части, способ настоящего изобретения дает возможность получать карбоксамидо-производные со степенью конверсии, которая является неожиданно более высокой, чем конверсия, полученная с соответствующими нейодированными субстратами.
Настоящее изобретение, таким образом, основано на новом способе синтеза ароматических карбоксамидов и их производных путем введения в реакцию трийодированных пентазамещенных N-сульфиниланилинов с замещенными α-гидроксикислотами или их солями.
Описанный выше способ особенно выгоден, так как дает возможность получать соединения формулы (I) с высоким выходом и высокой чистотой без необходимости вводить в реакцию соответствующие анилино-производные с соединением (4), причем последние получают, как ранее сообщалось, через довольно обременительный многостадийный способ.
Важно, поскольку соединения формулы (I) являются оптически активными, чтобы в способе изобретения принималось во внимание их получение, исходя из соответствующего оптически активного α-гидроксикислотного предшественника или его соли, путем полного сохранения оптической конфигурации во время проведения реакции.
Очевидно, что способ охватывает получение соединений формулы (I), соответствующих любой оптической конфигурации у ассиметричного атома углерода (*), несущего гидроксигруппу,
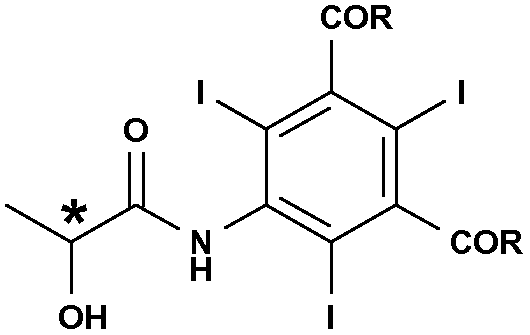
то есть любому (S) или (R) энантиомеру и даже их любой рацемической (R,S) смеси [которые также определяют как (L), (D) или (L,D)] в зависимости от конфигурации используемой молочной кислоты или ее соли.
В соответствии со способом изобретения в описанных выше соединениях R может представлять собой алкоксигруппу -OR1, где R1 представляет собой линейную или разветвленную С1-С6-алкильную группу, необязательно замещенную одной или несколькими защищенными гидроксигруппами. Кроме того, и если не предложено другое, R также может быть представлен аминогруппой -NH2 или -NHR1, где R1 принимает представленные выше значения, или атом хлора.
В данном описании, если не указано другое, определение «линейный или разветвленный С1-С6-алкил» означает линейную или разветвленную алкильную группу из 1-6 атомов углерода. Примерами таких групп являются, но не ограничиваются ими, метил, этил, н-пропил, изопропил, н-бутил, изобутил, втор-бутил, трет-бутил, н-гексил и т.д.
Аналогично, определение алкоксигруппа -OR1 означает алкилоксигруппы, где алкил имеет ранее представленные значения. Неограничивающими примерами линейных или разветвленных алкоксигрупп являются, например, метокси-, этокси-, н-пропокси-, изопропокси-, н-бутокси-, изобутокси-, втор-бутокси-, трет-бутокси-, н-гексилоксигруппа и т.д.
Определение «защищенная гидроксигруппа» относится, если не оговорено другое, к гидроксигруппе, соответствующим образом защищенной остатком, выбранным из числа хорошо известных в данной области, особенно предназначенных для предупреждения случаев любой нежелательной реакции, вовлекающей саму свободную (то есть незащищенную) гидроксигруппу. Указанные выбранные защищающие группа или остаток не должны создавать помехи во время всего процесса и затем должны быть легко снимаемыми, чтобы получать конечный продукт с соответствующей гидроксигруппой (или группами) в незащищенной форме. Неограничивающими примерами указанных защитных групп являются, наряду с другими, ацильные группы, в том числе алкилкарбонильная, арилкарбонильная или арилалкил-карбонильная группа, где указанный алкил определен выше. Предпочтительно гидрокси-защищающая группа представляет собой алкилкарбонильную группу, такую как ацетил (-СОСН3).
Более того, при подходящих случаях одна единственная защитная группа одновременно может быть эффективно использована для защиты более чем одной гидроксигруппы. Кроме того, например, в случае вицинальных диолов последние могут быть подходящим образом защищены за счет образования внутримолекулярных циклических ацеталей или кеталей, которые при известных условиях могут быть легко расщеплены соответствующим образом.
Для общих сведений по защитным группам в органической химии смотрите, например, публикацию T.W. Green, Protective Groups in Organic Synthesis (Willey N.Y. 1981).
Соответственно, также в случае карбоксамидов (I) и (II), где R представляет собой (-NHR1), указанная группа R1 необязательно может быть замещена одной или несколькими, например одной или двумя, защищенными гидроксигруппами, описанными ранее.
Таким образом, с учетом описанного выше, в рамках формул (I) и (II) определяют несколько соединений, например соединения, которые можно отнести к эфирам изофталевой кислоты (например, когда R=-OR1) или даже к дихлориду изофталевой кислоты (например, когда R=-Сl).
Аналогично, когда R представляет собой аминогруппу, в соответствии с настоящим изобретением подразумеваются карбоксамиды изофталевой кислоты (например, когда R=-NH2), в том числе N-алкилкарбоксамиды (например, когда R=-NHR1).
В соответствии с предпочтительным вариантом осуществления настоящее изобретение относится к способу получения соединения формулы (I), начиная от соединения формулы (II), где R представляет собой атом хлора.
В соответствии с другим вариантом осуществления настоящее изобретение относится к способу получения соединения формулы (I), исходя из соединения формулы (II), где R представляет собой -OR1 и R1 имеет определенные выше значения. Даже более предпочтительно R1 представляет собой линейную или разветвленную С1-С4-алкильную группу, в особенности метил.
В соответствии с еще одним предпочтительным вариантом осуществления настоящее изобретение относится к способу получения соединения формулы (I), исходя из соединения формулы (II), где R представляет собой -NH2 или -NHR1, а R1 имеет определенные выше значения.
Более предпочтительно R1 представляет собой линейную или разветвленную С1-С4-алкильную группу, необязательно замещенную одной или несколькими защищенными гидроксигруппами.
Даже более предпочтительно в рамках данного класса R1 представляет собой группу -СН(СН2ОН)2, где гидроксигруппы соответствующим образом защищены.
Соединения формулы (I) являются подходящими контрастирующими агентами, которые используют при диагностике, или их предшественниками, при соответствующем снятии защиты с любой необязательной гидроксигруппы в соответствии с известными способами.
Как правило, например, при синтезе иопамидола последний может быть получен путем снятия защиты с соответствующего соединения формулы (I), где R представляет собой -NHR1 и R1 представляет собой -СН(СН2ОН)2, где обе гидроксигруппы защищены, например, в виде ацетилированных промежуточных соединений:

Как указывалось ранее и в соответствии с другим аспектом изобретения, настоящий способ проводят путем соответствующего введения в реакцию соединения формулы (II) с (2S)-2-гидрокси-пропановой кислотой или ее солью.
Подходящими примерами упоминаемых выше солей являются соли щелочных металлов или щелочноземельных металлов, предпочтительно соли натрия, лития или калия.
Даже более предпочтительно способ изобретения проводят в присутствии натриевой соли, коммерчески доступной с высокой степенью оптической чистоты.
В соответствии со стереохимией реакции мольное отношение между сульфинильным субстратом формулы (II) и α-гидрокси-кислотой или ее солью должно составлять по меньшей мере 1:1.
Предпочтительно, однако, проводить реакцию в присутствии избытка выбранной α-гидроксикислоты или ее соли.
Как ароматический субстрат формулы (II), так и выбранную α-гидроксикислоту или ее соль вводят в реакцию в присутствии основания, такого как, например, любое подходящее органическое основание.
Предпочтительными примерами такого основания являются, например, имидазол, 1Н-бензотриазол или 1,2,4-триазол, причем последний является особенно предпочтительным.
Как правило, мольное количество основания относительно исходного материала (II) будет находиться в интервале от приблизительно 10 до приблизительно 150%, например составляет приблизительно 110%.
Что касается рабочих условий и если не представлено другое, то настоящий способ проводят в присутствии подходящего растворителя, предпочтительно апротонного растворителя из числа растворителей, обычно принятых в органическом синтезе. Например, можно назвать N,N-диметилацетамид (ДМА), N,N-диметилформамид (ДМФА), диметилсульфоксид (ДМСО), N-метил-2-пирролидон (N-МП), ацетонитрил (СН3CN), дихлорметан (ДХМ) и т.д., также допустимы их смеси.
Предпочтительно реакцию проводят в присутствии ДМА или ДМСО.
В соответствии с настоящим способом получения рентгеновских контрастирующих агентов или их промежуточных соединений реакцию соединений (II) с получением продуктов (I) проводят при температуре в интервале от приблизительно 0 до приблизительно 40ºС, более предпочтительно при комнатной температуре, то есть при температуре от приблизительно 20 до приблизительно 25°С.
Время реакции может меняться от приблизительно 1 до приблизительно 10 часов, хотя обычно реакция завершается за приблизительно 4 часа.
В соответствии с необязательным аспектом изобретения способ получения соединений формулы (I) также может быть проведен в присутствии подходящего катализатора, который, когда бы он не потребовался, может быть использован для повышения степени конверсии соединения (II) до соединения (I). Подходящими примерами катализаторов являются катализаторы фазового перехода, такие как, например, четвертичные аммонийные соли, однако предпочтительным является тетрабутил-аммонийбромид ([N(Bu)]4 +Br-).
Исходные материалы настоящего изобретения известны в данной области техники и могут быть получены в соответствии с обычными методами, например, так, как представлено в следующих разделах. Аналогично, любой другой реагент или растворитель, включая также необязательный катализатор, являются известными и обычно принятыми в органических синтезах.
В практической предпочтительной экспериментальной методике способ настоящего изобретения может быть проведен следующим образом.
В реакторе, оборудованном магнитной мешалкой и термопарой, находящемся в атмосфере инертного газа, в системе подходящего апротонного растворителя при выбранной температуре растворяют соответствующее количество соединения (II).
Подходящее количество основания, необязательно вместе с подходящим катализатором, добавляют к реакционной смеси и затем перемешивают до получения раствора. Необходимое количество (L)-молочной кислоты или ее соли добавляют к смеси и полученную в результате суспензию перемешивают в течение некоторого периода времени, например в течение нескольких часов, до окончания реакции.
Протекание реакции можно контролировать в соответствии с обычными методами (например, с помощью ВЭЖХ).
Требуемое соединение формулы (I) получают с высоким выходом и чистотой, и оно может быть выделено и очищено обычными способами. С другой стороны, оно может быть напрямую переработано в конечное соединение, предназначенное для применения в диагностике в зависимости от обстоятельств.
В этой связи неплохо отметить, что соединения формулы (I), где R представляют собой атомы хлора, в качестве приемлемых промежуточных соединений для синтеза контрастирующих агентов, в частности иопамидола, может быть получено в соответствии с данным способом или исходя из соответствующего соединения формулы (II), как подробно обсуждено выше, или, с другой стороны, исходя из его предшественника 5-амино-2,4,6-трийодизофталеновой кислоты без необходимости какой-либо очистки промежуточного соединения формулы (II), полученного таким образом.
Следовательно, еще одним объектом настоящего изобретения является способ получения дихлорида 5-[(2S)-2-гидроксипропиониламино]-2,4,6-трийодизофталевой кислоты, причем указанный способ включает:
(а) взаимодействие 5-амино-2,4,6-трийодизофталевой кислоты с подходящим хлорирующим агентом с получением сырого продукта, содержащего дихлорид 5-сульфиниламино-2,4-6-трийодизофталевой кислоты; и
(b) обработку сырого продукта стадии (а) (2S)-2-гидроксипропановой кислотой или ее солью в присутствии подходящего основания, в соответствии с представленной ниже схемой:
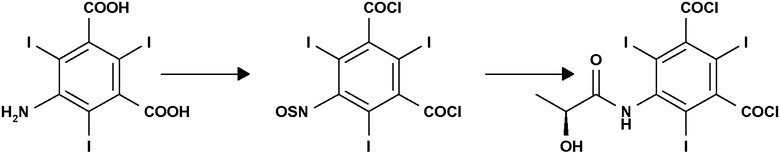
Приведенный выше способ особенно выгоден, так как он дает возможность получать соединение формулы (I), где R представляет собой атомы хлора, из предшественника 5-амино-2,4,6-трийодизофталевой кислоты, широко известного промежуточного соединения для синтеза контрастирующих агентов.
Хлорирующий агент обеспечивает как конверсию карбоксильных групп в соответствующие ацилхлоридные группы, так и образование N-сульфинильной реакционной группы.
Ряд подходящих хлорирующих агентов известен в данной области и может быть использован в настоящем способе. Из них особенно предпочтительным является тионилхлорид; смотрите, для общих сведений публикации: GB 1472050; Pillai et al., J. Org. Chem., 1994, 59, 1344-50; и Harrie J.M., Org. Proc. Res. Dev., 1999, 3, 38-43.
Рабочие условия стадии (а) широко представлены в данной области и включают, например, взаимодействие исходного материала с тионилхлоридом, необязательно в присутствии третичного амина, как описано, например, в публикации Bull. Korean Chem. Soc., 1990, 11, 494-496. Аналогично, Chidambaram с соавторами (Organic Process Research & Development 2002, 6, 632-636) описывают синтез 4-аминосульфинильного соединения реакцией этилового эфира 4-амино-3-фторбензойной кислоты с SOCl2, тогда как в публикации DE1085648В (Lentia GMBH) раскрыто получение в качестве промежуточного соединения хлорангидрида 3-сульфиниламино-2,4,6-трийодбензойной кислоты реакцией 3-амино-2,4,6-трийодбензойной кислоты с тионилхлоридом.
Ряд растворителей может быть использован на стадии (а), и предпочтительно реакцию проводят в апротонном растворителе, выбранном из растворителей, широко известных в органическом синтезе, таких как, например, дихлорметан (ДХМ), толуол и т.д. (смотрите для общих сведений публикацию WO 96/37459).
С другой стороны, стадию (а) можно проводить в присутствии тионилхлорида, действующего и как реагент, и как растворитель. Очевидно, что в конце реакции любой избыток тионилхлорида необходимо соответствующим образом удалять обычными средствами, например вакуумной отгонкой.
Следующую стадию (b) затем проводят так, как подробно описано ранее.
Исходный материал настоящего изобретения известен и может быть легко получен обычными способами, например гидрированием или даже химическим восстановлением коммерчески доступной 5-нитроизофталевой кислоты, после чего следует йодирование ароматического кольца (смотрите для общих сведений публикацию WO 96/37458).
Кроме того, настоящее изобретение относится к способу, который может быть эффективно применен при промышленном получении контрастирующих агентов, в частности иопамидола. Такой удобный способ, как полагают, может привести к конечному продукту прямым путем за счет меньшего числа всех стадий, если сравнивать с путем синтеза, который представлен на схеме I.
Таким образом, еще одним объектом настоящего изобретения является способ получения иопамидола реакцией дихлорида 5-[(2S)-2-гидроксипропиониламино]-2,4,6-трийодизофталевой кислоты, причем последний получат так, как описано ранее, с 2-амино-1,3-пропандиолом, и указанную реакцию проводят в соответствии с обычными способами; смотрите для общих сведений, наряду с другими, публикации WO 96/037460, US 5362905, WO 97/047590, WO 98/24747, WO 98/028259 и WO 99/058494. Способ настоящего изобретения имеет общее применение, и, следовательно, он может быть эффективно применен к ряду ароматических субстратов, которые соответствующим образом вводят в реакцию с выбранными α-гидроксикислотами или их солями.
Способ настоящего изобретения, который дает возможность получать иопамидол, в соответствии с рабочими условия представлен подробно.
Как ранее указывалось, установлено, что стерически затрудненные субстраты при взаимодействии с α-гидрокси-кислотным остатком (например, с молочной кислотой) или ее солями дают продукты конденсации даже при более высоком выходе, если сравнивать с соответствующим нейодированным и частично замещенным ароматическим исходным материалом. Как показано в примерах 1 и 2 в приведенной ниже экспериментальной части, конверсия сульфинильных производных до соответствующих амидопроизводных падает от 88,8%, когда в качестве исходного материала используют трийодированный субстрат, до 33,6% в случае менее затрудненного нейодированного субстрата.
Следующие примеры в данном описании предназначены для более хорошей иллюстрации способа настоящего изобретения, не ограничивая его.
Экспериментальная часть
Продукты реакции следующих примеров анализируют с помощью методики ВЭЖХ следующим образом:
Колонка: FLUOPHASE PFP (перфторфенил) 100 Å, 5 мкм, 250×4,6 мм.
Температура: 40ºС;
Подвижная фаза: А - вода; В - ацетонитрил/метанол, 85:15; элюирование с градиентом;
Определение (УФ): 245 нм.
Пример 1
Получение соединения (I) из соединения (II), где R=-ОСН 3
В реакторе объемом 100 мл, оборудованном магнитным размешивателем и термопарой и находящемся в атмосфере азота, в ДМА (45 мл) при 20-25ºС растворяют соединение (II) (10,44 г, 0,0165 моль). После нагревания раствора до 40ºС добавляют 1,2,4-триазол (1,20 г, 0,0175 моль) и смесь перемешивают до получения раствора. К раствору добавляют натриевую соль (2S)-2-гидроксипропионовой кислоты ((L)-лактат натрия, 1,96 г, 0,0175 моль) и полученную суспензию перемешивают при 40ºС в течение 5 часов.
Реакцию контролируют с помощью ВЭЖХ. В конце реакции анализ смеси с помощью ВЭЖХ показывает, что количество продукта (I) соответствует выходу 88,8% (% площади ВЭЖХ).
Пример 2 (сравнительный)
Получение производного, соответствующего нейодированному соединению (I), где R=-ОCН 3
Повторяют способ примера 1 на соответствующем нейодированном субстрате, диметиловом эфире 5-(N-сульфиниламино)изофталевой кислоты (II).
В реакторе объемом 100 мл, оборудованном магнитным размешивателем и термопарой и находящемся в атмосфере азота, суспендируют нейодированное соединение (II) (4,08 г, 0,016 моль) в ДМА (90 мл) при 20-25ºС. После нагревания суспензии до 40ºС добавляют 1,2,4-триазол (1,20 г, 0,0175 моль) и (L)-лактат натрия (1,96 г, 0,0175 моль) и смесь перемешивают при 40ºС в течение 3 часов.
Реакцию контролируют с помощью ВЭЖХ. В конце реакции анализ смеси с помощью ВЭЖХ показывает, что выход нейодированного соединения (I) достигает 33,6% (% площади ВЭЖХ).
Сравнение примеров 1 и 2 однозначно показывает, что способ изобретения дает возможность получать, неожиданно, целевые соединения формулы (I) со степенью конверсии, значительно более высокой, чем степень конверсии с соответствующим менее стерически затрудненным субстратом.
Пример 3
Получение соединений формулы (I) из соответствующих производных формулы (II), где R=-ОСН 3 , -Сl или -NHCH(CH 2 ОСOCH 3 ) 2
Ряд соединений формулы (I) получен по экспериментальной методике примера 1 при реакционных условиях, указанных в таблице 1.
Экспериментальные данные
| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБ ЙОДИРОВАНИЯ ПРОИЗВОДНЫХ ФЕНОЛА | 2011 |
|
RU2563645C2 |
| Рентгеноконтрастное средство | 1980 |
|
SU1087052A3 |
| СПОСОБ ИОДИРОВАНИЯ АРОМАТИЧЕСКИХ СОЕДИНЕНИЙ | 2010 |
|
RU2506254C2 |
| Способ получения трийодированных производных 5-аминоизофталевой кислоты | 1980 |
|
SU969156A3 |
| СПОСОБ ПОЛУЧЕНИЯ ЙОДИРОВАННОГО КОНТРАСТНОГО АГЕНТА | 2009 |
|
RU2493146C2 |
| Способ получения амидов 5-оксипропиониламино-2,4,6-трийодизофталевой кислоты | 1975 |
|
SU628813A3 |
| СПОСОБ ПОЛУЧЕНИЯ ПРОИЗВОДНЫХ 2,4,6-ТРИЙОД- БЕНЗОЙНОЙ КИСЛОТЫ | 1970 |
|
SU286636A1 |
| ПРОИЗВОДНЫЕ ПИРАЗОЛО-ИМИДАЗОПИРИМИДИНА, ОБЛАДАЮЩИЕ АНТАГОНИСТИЧЕСКОЙ АКТИВНОСТЬЮ В ОТНОШЕНИИ КОРТИКОТРОПИН-РИЛИЗИНГ ФАКТОРА (CRF) | 2004 |
|
RU2377241C2 |
| СПОСОБ ПОЛУЧЕНИЯ КОНТРАСТНЫХ АГЕНТОВ | 2011 |
|
RU2566823C2 |
| ПРОИЗВОДНЫЕ ХИНАЗОЛИНА, СПОСОБ ИХ ПОЛУЧЕНИЯ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ИХ ОСНОВЕ | 1993 |
|
RU2111209C1 |
Изобретение относится к способу получения производных 2,4,6-трийодизофталевой кислоты, которые являются промежуточными соединениями для получения контрастирующих агентов, в частности йопамидола. Способ включает взаимодействие соединения формулы (II) с соединением формулы (III) или его солью в присутствии органического основания. Также изобретение относится к способу получения йопамидола, который заключается в получении соединения формулы (II) реакцией 5-амино-2,4,6-трийодизофталевой кислоты с серосодержащим хлорирующим агентом. Затем соединение формулы (II) сразу же вводят в реакцию с (2S)-2-гидроксипропионовой кислотой или ее солью с получением дихлорида 5-[(2S)-2-гидроксипропиониламино]-2,4,6-трийодизофталевой кислоты формулы (I). Полученное соединение формулы (I) подвергают взаимодействию с 2-амино-1,3-пропандиолом с получением йопамидола. Технический результат - усовершенствованный способ получения производных 2,4,6-трийодизофталевой кислоты. 2 н. и 13 з.п. ф-лы, 1 табл., 3 пр.
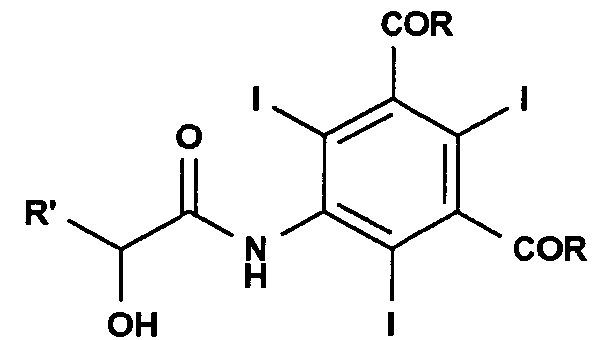



1. Способ получения соединения формулы
и этот способ включает взаимодействие соединения формулы (II)
с соединением формулы (III)
или ее солью, в присутствии органического основания, где:
R' представляет собой атом водорода или линейную или разветвленную C1-6-алкильную группу, необязательно замещенную одной или несколькими защищенными гидроксигруппами;
R, одинаковые в каждом случае, выбирают из группы, включающей алкоксигруппу (-OR1), аминогруппу (-NH2 или -NHR1) или атом хлора;
R1 представляет собой линейную или разветвленную C1-C6-алкильную группу, необязательно замещенную одной или несколькими защищенными гидроксигруппами.
2. Способ по п.1 получения соединения формулы (I)
и этот способ включает взаимодействие соединения формулы II с (2S)-2-гидроксипропионовой кислотой или ее солью в присутствии подходящего основания, где R, в каждом случае одинаковые, выбирают из группы, включающей алкоксигруппу (-OR1), аминогруппу (-NH2 или -NHR1) или атом хлора; и
R1 представляет собой линейную или разветвленную C1-C6-алкильную группу, необязательно замещенную одной или несколькими защищенными гидроксигруппами.
3. Способ по любому одному из пп.1 или 2, где R представляет собой атом хлора.
4. Способ по любому из пп.1 или 2, где R представляет собой -OR1 и R1 представляет собой линейную или разветвленную C1-C4-алкильную группу.
5. Способ по п.4, где R1 представляет собой метил.
6. Способ по любому из пп.1 и 2, где R представляет собой -NH2 или -NHR1 и R1 представляет собой линейную или разветвленную C1-C4-алкильную группу, необязательно замещенную одной или несколькими защищенными гидроксигруппами.
7. Способ по п.6, где R1 представляет собой -CH(CH2OCOCH3)2.
8. Способ по п.2, где соль (2S)-2-гидроксипропионовой кислоты выбирают из группы, включающей соль лития, натрия и калия.
9. Способ по п.8, где указанная соль (2S)-2-гидроксипропионовой кислоты представляет собой натриевую соль.
10. Способ по любому одному из пп.1, 2, 5, 7, 8 или 9, где основание представляет собой гетероциклическое основание, выбранное из имидазола, 1H-бензотриазола или 1, 2, 4-триазола.
11. Способ по п.10, где гетероциклическое основание представляет собой 1,2,4-триазол.
12. Способ по любому одному из пп.1, 2, 5, 7, 8 или 9, проводимый в присутствии катализатора фазового переноса.
13. Способ по п.12, где катализатор фазового переноса представляет собой тетрабутиламмонийбромид.
14. Способ по п.3, где соединение (II) получают реакцией 5-амино-2,4,6-трийодизофталевой кислоты с серосодержащим хлорирующим агентом, и указанное соединение (II) сразу же вводят в реакцию с (2S)-2-гидроксипропионовой кислотой или ее солью с получением дихлорида 5-[(2S)-2-гидроксипропиониламино]-2,4,6-трийодизофталевой кислоты формулы (I).
15. Способ получения йопамидола, включающий способ по п.14, который дополнительно включает взаимодействие 5-[(2S)-2-гидроксипропиониламино]-2,4,6-трийодизофталевой кислоты с 2-амино-1,3-пропандиолом.
| CHIDAMBARAM R | |||
| et al.: "REACTION OF ELECTRON-DEFICIENT N-SULFINYLANILINES WITH CHIRAL ALPHA-HYDROXY ACIDS: A NEW PROCESS FOR THE SYNTHESIS OF ENANTIOMERICALLY PURE ALPHA-HYDROXY AMIDES" Tetrahedron Letters, 2000, vol.41, no.32, p.6017-6020 | |||
| JAI MOO SHIN et al.: "NEW FACILE SYNTHESIS OF ALPHA HYDROXYAMIDES INTERMOLECULAR AND INTRAMOLECULAR CATALYSIS |

