ССЫЛКА НА РОДСТВЕННЫЕ ЗАЯВКИ
Настоящая заявка, согласно § 119 Кодекса законов США, испрашивает приоритет предварительной заявке на патент США № 61/432990, поданной 14 января 2011 года, содержание которой включено в настоящее описание посредством ссылки.
УРОВЕНЬ ТЕХНИКИ
Резистентность бактерий к антибиотикам известна давно, и она считается сегодня во всем мире серьезной проблемой здоровья. В результате этой устойчивости, некоторые бактериальные инфекции трудно лечить антибиотиками или они даже являются неизлечимыми. Эта проблема стала особенно серьезной в связи с недавним развитием у некоторых штаммов бактерий, таких как Streptococcus pneumoniae (SP), Mycobacterium tuberculosis и Enterococcus, множественной резистентности к лекарственным препаратам. Появление ванкомицин-резистентных энтерококков вызвало особую тревогу, поскольку ранее ванкомицин был единственным эффективным антибиотиком для лечения этой инфекции, и он расценивался как "последнее средство" среди лекарственных препаратов для многих инфекций. Хотя многие другие лекарственно-резистентные бактерии не вызывают опасные для жизни заболевания, как энтерококки, существует опасение, что гены, которые вызывают резистентность, могут распространиться на более смертоносные микроорганизмы, такие как Staphylococcus aureus, для которых устойчивость к метициллину является широко распространенной (De Clerq, et al., Current Opinion in Anti-infective Investigational Drugs, 1999, 1, 1; Levy, "The Challenge of Antibiotic Resistance", Scientific American, March, 1998).
Еще одной проблемой является вопрос, как быстро может распространяться устойчивость к антибиотикам. Например, до 1960-х годов бактерии SP были полностью чувствительны к пенициллину, и в 1987 году в США только 0,02% штаммов SP были устойчивы к нему. Тем не менее, к 1995 году стало известно, что устойчивость SP к пенициллину была на уровне около семи процентов и достигала 30% в некоторых частях США (Lewis, FDA Consumer magazine (September, 1995); Gershman in The Medical Reporter, 1997).
Больницы, в частности, служат центрами для формирования и передачи лекарственно-резистентных микроорганизмов. Случаи инфицирования в больницах, известные как внутрибольничные инфекции, становятся все более серьезной проблемой. Каждый год в больницах инфицируется два миллиона американцев, и более половины из этих инфекций являются устойчивыми по крайней мере к одному антибиотику. Центр по контролю и профилактике заболеваний США сообщил, что в 1992 году более 13000 больничных пациентов умерло от бактериальных инфекций, которые были резистентны к лечению антибиотиками (Lewis, "The Rise of Antibiotic-Resistant Infections", FDA Consumer magazine, September 1995).
В результате необходимости борьбы с лекарственно-резистентными бактериями и увеличения неэффективности доступных лекарственных средств, наблюдается возрождение интереса к разработке новых антибиотиков. Одной из привлекательных стратегий разработки новых антибиотиков является ингибирование ДНК гиразы и/или топоизомеразы IV, бактериальных ферментов, необходимых для репликации ДНК и, следовательно, необходимых для роста бактерий и деления клеток. Активности гиразы и/или топоизомеразы IV также связаны с событиями при транскрипции, репарации и рекомбинации ДНК.
Гиразы являются представителями топоизомеразы, группы ферментов, которые катализируют взаимопревращение топологических изомеров ДНК (см., в общем, Kornberg and Baker, DNA Replication, 2d Ed., Chapter 12, 1992, W. H. Freeman and Co.; Drlica, Molecular Microbiology, 1992, 6, 425; Drlica and Zhao, Microbiology and Molecular Biology Reviews, 1997, 61, pp. 377-392). Гираза сама по себе контролирует суперспирализацию ДНК и снимает топологический стресс, который возникает, когда в процессе репликации раскручиваются нити ДНК родительского дуплекса. Гираза также катализирует конверсию раскрученных замкнутых кольцевых дуплексных ДНК в отрицательную суперспиральную форму, которая более благоприятна для рекомбинации. Механизм сверхспиральной реакции включает охватывание гиразой области ДНК, двойное разрушение нити в этой области, пропускание второй области ДНК через этот разрыв и повторное соединение разорванных нитей. Такой механизм расщепления характерен для топоизомеразы типа II. Реакция суперспирализации вызывается связыванием АТФ с гиразой. Затем, в ходе реакции, АТФ подвергается гидролизу. Это связывание и последующий гидролиз АТФ вызывают конформационные изменения в ДНК-связанной гиразе, необходимые для ее активности. Кроме того, было обнаружено, что уровень суперспирализации ДНК (или степень раскрученности) зависит от соотношения АТФ/АДФ. В отсутствие АТФ, гираза способна только к раскручиванию суперспиральной ДНК.
Бактериальные ДНК-гиразы представляют собой белковый тетрамер с массой 400 кДа, состоящий из двух субъединиц А (GyrA) и двух субъединиц В (GyrB). Связывание и расщепление ДНК связано с GyrA, в то время как АТФ связывается и гидролизуется белком GyrB. GyrB состоит из амино-концевого домена, который обладает АТФазной активностью, и карбокси-концевого домена, который взаимодействует с GyrA и ДНК. В отличие от этого, эукариотические топоизомеразы типа II являются гомодимерами, которые могут раскручивать отрицательные и положительные суперспирали, но не могут формировать отрицательные суперспирали. В идеале, антибиотик, основанный на ингибировании бактериальной ДНК-гиразы и/или топоизомеразы IV, будет селективным для этих ферментов и будет относительно неактивным против эукариотической топоизомеразы типа II.
В первую очередь, топоизомеразы IV разрывают связанные димеры хромосом на заключительной стадии репликации ДНК.
Широко распространенные антибиотики хинолонового ряда ингибируют бактериальную ДНК-гиразу (GyrA) и/или топоизомеразу IV (ParC). Примеры хинолонов включают давно известные соединения, такие как налидиксовая кислота и оксолиновая кислота, а также позднее, более мощные фторхинолоны, такие как норфлоксацин, ципрофлоксацин и тровафлоксацин. Эти соединения связываются с GyrA и/или Parc, стабилизируя расщепленный комплекс, ингибируя, таким образом, общую функцию гиразы, что приводит к гибели клеток. Фторхинолоны ингибируют каталитические субъединицы гиразы (GyrA) и/или Топоизомеразы IV (ParC) (см. Drlica and Zhao, Microbiology and Molecular Biology Reviews, 1997, 61, 377-392). Однако лекарственная резистентность также признается как проблема для этого класса соединений (WHO Report, "Use of Quinolones in Food Animals and Potential Impact on Human Health", 1998). В случае хинолонов, как и с другими классами антибиотиков, бактерии, на которые воздействовали давно известные соединения, часто формируют перекрестную резистентность к более эффективным соединениям того же класса.
Связанные субъединицы, отвечающие за поставку энергии, необходимы для каталитического оборота/восстановления ферментов при гидролизе АТФ, представляют собой GyrB (гираза) и ParE (топоизомераза IV), соответственно (см.: Champoux, J.J., Annu. Rev. Biochem., 2001, 70, pp. 369-413). Соединения, для которых мишенью являются эти же сайты связывания АТФ в субъединицах GyrB и ParE, были бы полезны для лечения различных бактериальных инфекций (см.: Charifson et al., J. Med. Chem., 2008, 51, pp. 5243-5263).
Известно меньшее число ингибиторов, которые связываются с GyrB. Их примеры включают кумарины, новобиоцин и коумермицин A1, циклотиалидин, цинодин и клероцидин. Как было показано, кумарины очень сильно связываются с GyrB. Например, новобиоцин образует сеть водородных связей с белком и формирует несколько гидрофобных контактов. В то же время новобиоцин и АТФ формируют связи внутри сайта связывания АТФ с минимальным перекрытием в ориентации связей этих двух соединений. Перекрывающиеся части представляют собой сахаридные единицы в новобиоцине и аденин в АТФ (Maxwell, Trends in Microbiology, 1997, 5, 102).
Для кумарин-резистентных бактерий наиболее распространенной точкой мутации является остаток аргинина на поверхности, который связывается с карбонилом кумаринового кольца (Arg136 в GyrB кишечной палочки). В то же время ферменты с этой мутацией показывают более низкую суперспирализацию и АТФазную активность, и они также менее чувствительны к ингибированию лекарственными препаратами кумаринового ряда (Maxwell, Mol. Microbiol., 1993, 9, 681).
Несмотря на то, что кумарины являются мощными ингибиторами суперспирализации гиразы, они не были широко использованы в качестве антибиотиков. Как правило, они непригодны из-за их низкой проницаемости в отношении бактерий, токсичности в отношении эукариотов и из-за плохой растворимости в воде (Maxwell, Trends in Microbiology, 1997, 5, 102). Было бы желательно, чтобы новый эффективный ингибитор GyrB и ParE преодолел бы эти недостатки и, предпочтительно, в своей активности не был бы основан на исключительном связывании с Arg136. Такой ингибитор будет привлекательным кандидатом-антибиотиком, без проблем в части резистентности, которые мешают другим классам антибиотиков.
Поскольку резистентность бактерий к антибиотикам стала важной проблемой для общественного здравоохранения, существует постоянная потребность в разработке новых и более мощных антибиотиков. В частности, существует потребность в антибиотиках, которые представляют собой новый класс соединений, который ранее не использовался для лечения бактериальных инфекций. Соединения, которые нацелены на сайты связывания АТФ в субъединицах как GyrB (гираза), так и ParE (топоизомераза IV), были бы полезны для лечения различных бактериальных инфекций. Такие соединения были бы особенно полезны при лечении внутрибольничных инфекций в лечебных заведениях, где формирование и трансмиссия резистентных бактерий становится все более распространенной.
СУЩНОСТЬ ИЗОБРЕТЕНИЯ
В одном варианте осуществления настоящее изобретение относится к способу получения соединения формулы (I)
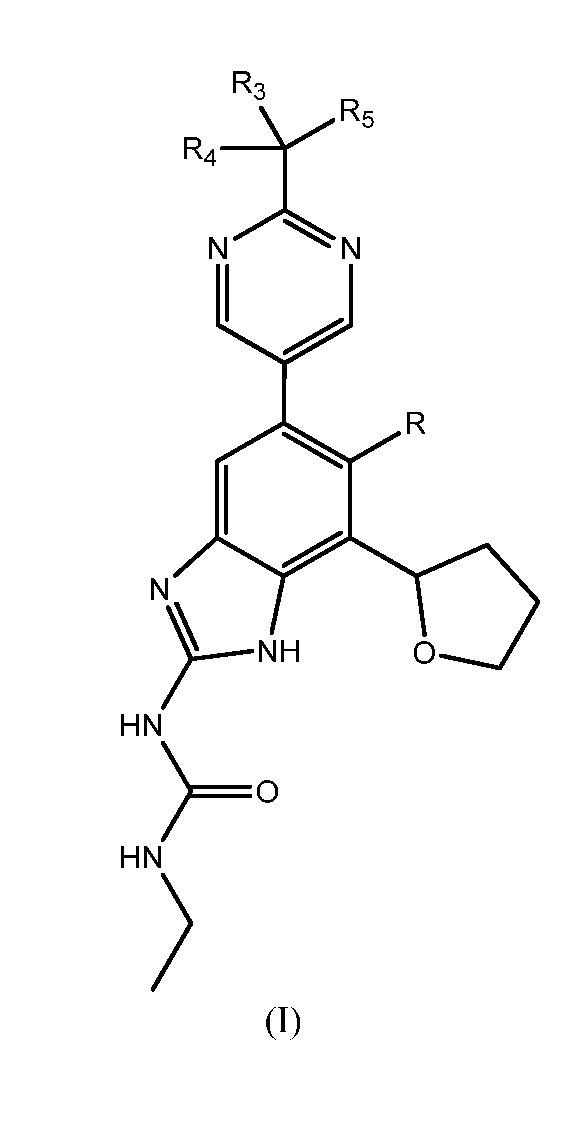
или к его фармацевтически приемлемой соли, где R представляет собой Н или F, и каждый из R3, R4 и R5 независимо представляет собой необязательно замещенный алкил или необязательно защищенную гидроксильную группу.
Способ включает получение фенилпиримидинового соединения формулы (II)

где R представляет собой Н или F, и каждый из R3, R4 и R5 независимо представляет собой необязательно замещенный алкил или необязательно защищенную гидроксильную группу;
и взаимодействие фенилпиримидинового соединения формулы (II) с производным мочевины формулы А или В:
 ,
,
где R6 представляет собой необязательно замещенный алкил, необязательно замещенный арил, необязательно замещенный насыщенный или ненасыщенный карбоцикл или необязательно замещенный насыщенный или ненасыщенный гетероцикл, с получением соединения формулы (I), и, необязательно, взаимодействие соединения формулы (I) с подходящей кислотой, с получением фармацевтически приемлемой соли соединения формулы (I).
В некоторых случаях данного варианта выполнения изобретения, R6 может представлять собой метил, этил, бензил или п-нитробензил. В дополнительном варианте выполнения изобретения, реакция может быть проведена в смеси диоксана и буфера при температуре от 75°С до 125°С. В одном дополнительном варианте выполнения изобретения, буфер может иметь рН 3,5, и реакция может быть проведена при температуре дефлегмации.
Во втором варианте осуществления настоящее изобретение относится к способу получения фенилпиримидинового соединения формулы (II)

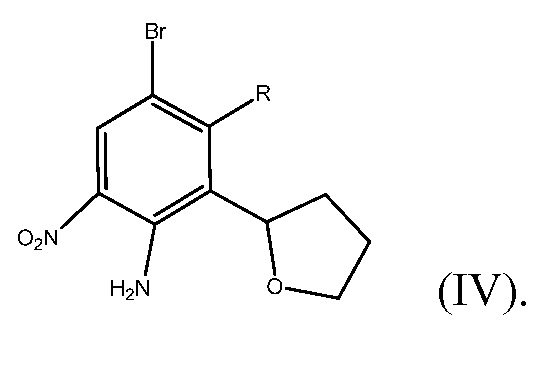
Способ включает получение фенилтетрагидрофуранового производного формулы (IV)

где R представляет собой Н или F, и
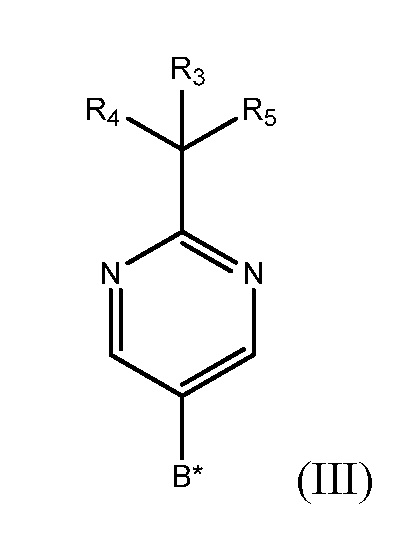
взаимодействие фенилтетрагидрофуранового производного формулы (IV) с производным бороновой кислоты формулы (III)

где каждый из R3, R4 и R5 независимо представляет собой необязательно замещенный алкил или необязательно защищенную гидроксильную группу, и
В* представляет собой 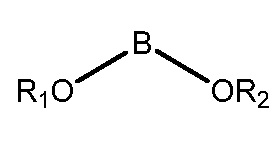 ,
,
где каждый из R1 и R2 независимо представляет собой алкил или H, или OR1 и OR2 вместе с атомом B, к которому они присоединены, образуют необязательно замещенное 5-, 6- или 7-членное кольцо или BF3X, где X может представлять собой любой одновалентный катион, в присутствии палладиевого катализатора в полярном растворителе с получением фенилпиримидинового соединения формулы (V)

где R представляет собой Н или F, и каждый из R3, R4 и R5 независимо представляет собой необязательно замещенный алкил или необязательно защищенную гидроксильную группу; и обработку фенилпиримидинового соединения формулы (V) подходящим восстанавливающим агентом с получением фенилпиримидинового соединения формулы (II).
В некоторых случаях этого варианта осуществления изобретения В* может представлять собой
 ,
,
где каждый из атомов углерода в кольце может быть незамещенным или замещенным одной или двумя метильными или этильными группами. В дополнительном варианте выполнения изобретения В* может представлять собой

В третьем варианте осуществления настоящее изобретение относится к способу получения фенилтетрагидрофуранового соединения формулы (IV)
Способ включает получение соединения формулы (VI)

где R представляет собой Н или F,
и нитрование соединения формулы (VI) соответствующим нитрующим агентом с получением фенилтетрагидрофуранового соединения формулы (IV).
В четвертом варианте осуществления настоящее изобретение относится к альтернативному способу получения фенилтетрагидрофуранового соединения формулы (IV). Способ включает получение соединения формулы (VI)

где R представляет собой Н или F;
защиту аминогруппы в соединении формулы (VI) аминозащитной группой с получением амино-защищенного соединения;
нитрование амино-защищенного соединения подходящим нитрующим агентом с получением амино-защищенного нитросоединения, и снятие защиты с защищенного амино-нитросоединения с получением фенилтетрагидрофуранового соединения формулы (IV).
В некоторых аспектах этого варианта осуществления изобретения, указанное получение фенилпиримидинового соединения формулы (II) может дополнительно включать восстановление фенилпиримидинового производного формулы (V)

где R представляет собой Н или F, и каждый из R3, R4 и R5 независимо представляет собой необязательно замещенный алкил или необязательно защищенную гидроксильную группу, подходящим восстанавливающим агентом с получением соединения формулы (II). В дополнительном варианте выполнения изобретения нитрование соединения формулы (VI) может включать взаимодействие соединения формулы (VI) с NH4NO3 в присутствии сильной кислоты, при температуре от приблизительно 20°С до приблизительно 50°С, с получением соединения (IV).
В пятом варианте осуществления настоящее изобретение относится к способу получения соединения формулы (VI)
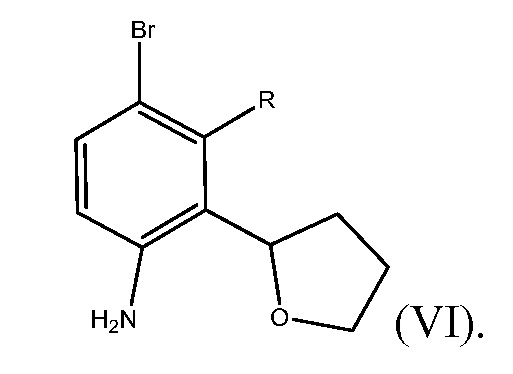
Способ включает получение соединения формулы (VII)
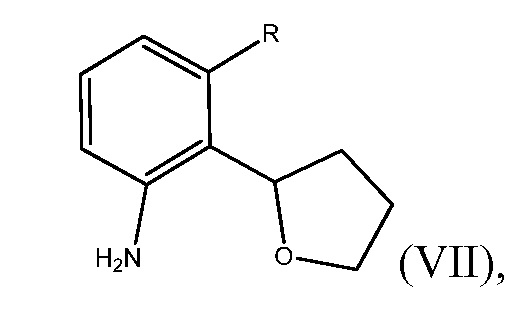
где R представляет собой Н или F, и взаимодействие соединения формулы (VII) с бромирующим агентом в полярном апротонном растворителе с получением соединения формулы (VI).
В некоторых вариантах этого выполнения изобретения, соединение формулы (VII) может быть энантиомерно обогащенным.
В шестом варианте осуществления настоящее изобретение относится к способу получения соединения формулы (VII)

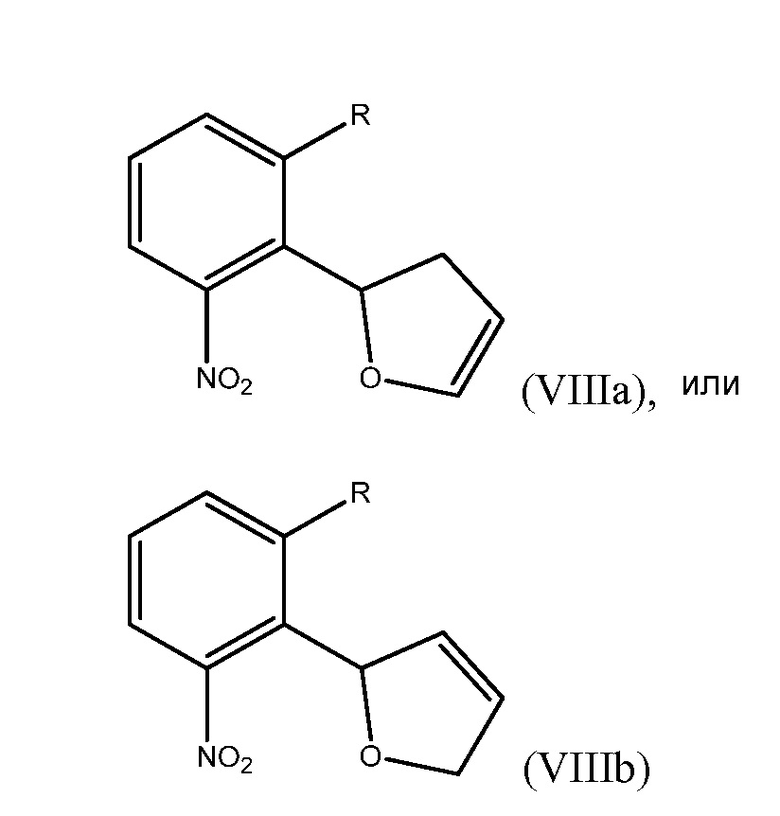
Способ включает получение дигидрофуранилнитробензольного соединения, выбранного из группы, состоящей из соединения формулы (VIIIa)

где R представляет собой Н или F,
и соединения формулы (VIIIb)

где R представляет собой Н или F, и обработку дигидрофуранилнитробензольного соединения восстанавливающим агентом с получением соединения формулы (VII).
В седьмом варианте осуществления настоящее изобретение относится к способу получения дигидрофуранилнитробензольного соединения формулы (VIIIa)

где R представляет собой Н или F,
или соединения формулы (VIIIb)

где R представляет собой Н или F,
где способ включает следующие стадии:
получение соединения формулы (IX)

где R представляет собой Н или F,
и обработка соединения формулы (IX) 2,3-дигидрофураном в присутствии палладиевого катализатора с получением дигидрофуранилнитробензольного соединения.
В восьмом варианте осуществления настоящее изобретение относится к соединению формулы
 ,
,
где R представляет собой Н или F, или его фармацевтически приемлемой соли, полученному в соответствии со способами настоящего изобретения. В некоторых вариантах осуществления изобретения соединение формулы (I) имеет формулу
 ,
,
где R представляет собой Н или F, или представляет его фармацевтически приемлемую соль. В других вариантах осуществления изобретения соединение формулы (I) может представлять собой (R)-1-этил-3-(5-(2-(2-гидроксипропан-2-ил)пиримидин-5-ил)-7-(тетрагидрофуран-2-ил)-1H-бензо[d]имидазол-2-ил)мочевину или ее фармацевтически приемлемую соль, (R)-1-этил-3-(6-фтор-5-(2-(2-гидроксипропан-2-ил)пиримидин-5-ил)-7-(тетрагидрофуран-2-ил)-1H-бензо[d]имидазол-2-ил)мочевину или ее фармацевтически приемлемую соль, кислую метансульфоновую соль (R)-1-этил-3-(5-(2-(2-гидроксипропан-2-ил)пиримидин-5-ил)-7-(тетрагидрофуран-2-ил)-1H-бензо[d]имидазол-2-ил)мочевины или кислую метансульфоновую соль (R)-1-этил-3-(6-фтор-5-(2-(2-гидроксипропан-2-ил)пиримидин-5-ил)-7-(тетрагидрофуран-2-ил)-1H-бензо[d]имидазол-2-ил)мочевины.
В девятом варианте выполнения настоящее изобретение дополнительно относится к соединению формулы (II)
 ,
,
где R представляет собой Н или F, и каждый из R3, R4 и R5 независимо представляет собой необязательно замещенный алкил или необязательно защищенную гидроксильную группу. Соединение формулы (II) может быть получено способом, включающим получение фенилтетрагидрофуранового производного формулы (IV)

и взаимодействие фенилтетрагидрофуранового производного формулы (IV) с производным бороновой кислоты формулы (III)

в присутствии палладиевого катализатора в полярном растворителе, где каждый из R3, R4 и R5 независимо представляет собой необязательно замещенный алкил или необязательно защищенную гидроксильную группу, и В* представляет собой  , где каждый из R1 и R2 независимо представляет собой алкил или H, или OR1 и OR2 вместе с атомом B, к которому они присоединены, образуют необязательно замещенное 5-, 6- или 7-членное кольцо или BF3X, где X является любым одновалентным катионом.
, где каждый из R1 и R2 независимо представляет собой алкил или H, или OR1 и OR2 вместе с атомом B, к которому они присоединены, образуют необязательно замещенное 5-, 6- или 7-членное кольцо или BF3X, где X является любым одновалентным катионом.
В некоторых случаях данного варианта осуществления способ дополнительно включает восстановление фенилпиримидинового производного формулы (V)

где R представляет собой Н или F, и каждый из R3, R4 и R5 независимо представляет собой необязательно замещенный алкил или необязательно защищенную гидроксильную группу, с получением соединения формулы (II).
В десятом варианте осуществления настоящее изобретение относится к соединению формулы (V)

где R представляет собой Н или F, и каждый из R3, R4 и R5 независимо представляет собой необязательно замещенный алкил или необязательно защищенную гидроксильную группу. Соединение формулы (V) может быть получено способом, включающим получение фенилтетрагидрофуранового производного формулы (IV)

где R представляет собой Н или F, и взаимодействие фенилтетрагидрофуранового производного формулы (IV) с производным бороновой кислоты формулы (III)
в присутствии палладиевого катализатора в полярном растворителе, где каждый из R3, R4 и R5 независимо представляет собой необязательно замещенный алкил или необязательно защищенную гидроксильную группу, и В* представляет собой  , где каждый из R1 и R2 независимо представляет собой алкил или H, или OR1 и OR2 вместе с атомом B, к которому они присоединены, образуют необязательно замещенное 5-, 6- или 7-членное кольцо или BF3X, где X является любым одновалентным катионом, с получением соединения формулы (V).
, где каждый из R1 и R2 независимо представляет собой алкил или H, или OR1 и OR2 вместе с атомом B, к которому они присоединены, образуют необязательно замещенное 5-, 6- или 7-членное кольцо или BF3X, где X является любым одновалентным катионом, с получением соединения формулы (V).
В одиннадцатом варианте осуществления настоящее изобретение относится к способу получения соединения формулы (I)

или его фармацевтически приемлемой соли, где R представляет собой Н или F, и каждый из R3, R4 и R5 независимо представляет собой необязательно замещенный алкил или необязательно защищенную гидроксильную группу. Способ включает получение дигидрофуранилнитробензольного соединения формулы
 ,
,
где R представляет собой Н или F, и превращение соединения формулы (VIIIa) или (VIIIb) или их комбинации в соединение формулы (I) или его фармацевтически приемлемую соль.
В двенадцатом варианте осуществления настоящее изобретение относится к способу получения дигидрофуранилнитробензольного соединения формулы (VIIIa) или (VIIIb). Способ включает взаимодействие соединения формулы (IX)

где R представляет собой Н или F, с 2,3-дигидрофураном в присутствии палладиевого катализатора с получением дигидрофуранилнитробензольного соединения формулы (VIIIa) или (VIIIb).
В одном варианте этого осуществления способ дополнительно включает взаимодействие соединения формулы (VIIIa) или (VIIIb) с восстанавливающим агентом с получением соединения формулы (VII)
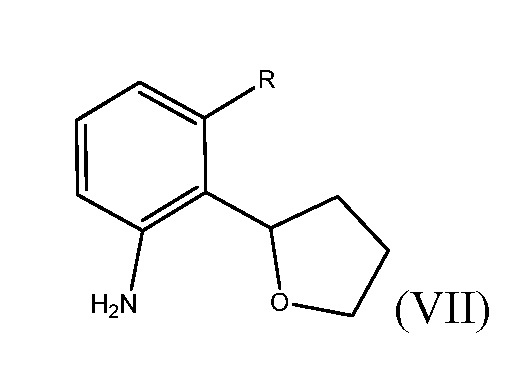 ,
,
где R представляет собой Н или F. В другом варианте выполнения изобретения способ дополнительно включает взаимодействие соединения формулы (VII) с бромирующим агентом в полярном апротонном растворителе с получением соединения формулы (VI). В еще одном варианте выполнения изобретения способ дополнительно включает нитрование соединения формулы (VI) соответствующим нитрующим агентом с получением фенилтетрагидрофуранового соединения формулы (IV). В еще одном дополнительном варианте выполнения изобретения, способ дополнительно включает взаимодействие фенилтетрагидрофуранового соединения формулы (IV) с производным бороновой кислоты формулы (III)

где каждый из R3, R4 и R5 независимо представляет собой необязательно замещенный алкил или необязательно защищенную гидроксильную группу, и В* представляет собой  , где каждый из R1 и R2 независимо представляет собой алкил или H, или OR1 и OR2 вместе с атомом B, к которому они присоединены, образуют необязательно замещенное 5-, 6- или 7-членное кольцо или BF3X, где X является любым одновалентным катионом, в присутствии палладиевого катализатора в полярном растворителе с получением фенилпиримидинового соединения формулы (V)
, где каждый из R1 и R2 независимо представляет собой алкил или H, или OR1 и OR2 вместе с атомом B, к которому они присоединены, образуют необязательно замещенное 5-, 6- или 7-членное кольцо или BF3X, где X является любым одновалентным катионом, в присутствии палладиевого катализатора в полярном растворителе с получением фенилпиримидинового соединения формулы (V)

где R представляет собой Н или F, и каждый из R3, R4 и R5 независимо представляет собой необязательно замещенный алкил или необязательно защищенную гидроксильную группу; и взаимодействие фенилпиримидинового соединения формулы (V) с подходящим восстанавливающим агентом с получением фенилпиримидинового соединения формулы (II). Способ может дополнительно включать взаимодействие фенилпиримидинового соединения формулы (II) с производным мочевины формулы А или В:
 ,
,
где R6 представляет собой необязательно замещенный алкил, необязательно замещенный арил, необязательно замещенный насыщенный или ненасыщенный карбоцикл или необязательно замещенный насыщенный или ненасыщенный гетероцикл, с получением соединения формулы (I) и, необязательно, взаимодействие соединения формулы (I) с подходящей кислотой с получением фармацевтически приемлемой соли соединения формулы (I).
КРАТКОЕ ОПИСАНИЕ ФИГУР
Фиг. 1 представляет собой графическое изображение двух симметрично независимых молекул соединения 12, полученных способом эллипсоида тепловых колебаний.
Фиг. 2 представляет собой графическое изображение двух симметрично независимых молекул соединения 23, полученных способом эллипсоида тепловых колебаний.
ПОДРОБНОЕ ОПИСАНИЕ
Настоящее изобретение относится к способу получения соединений и их фармацевтически приемлемых солей, полезных в качестве ингибиторов гиразы и антибактериальных агентов. Ингибиторы гиразы по настоящему изобретению, в целом охватываемых патентом США № RE40245 E, и могут быть представлены соединениями формулы (I) или их солями:
 ,
,
где R представляет собой Н или F, и каждый из R3, R4 и R5 независимо представляет собой необязательно замещенный алкил или необязательно защищенную гидроксильную группу.
В конкретном варианте осуществления соединения по настоящему изобретению могут быть представлены соединениями формулы (Ia) или их солями:
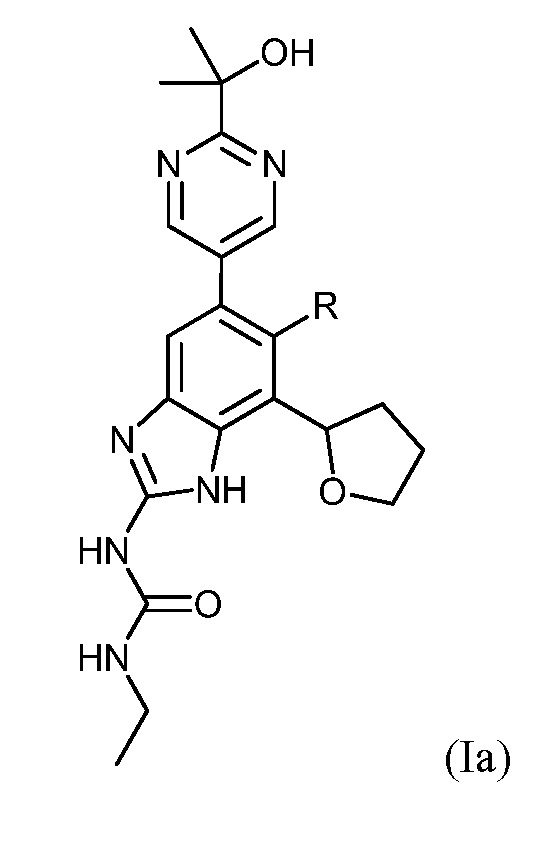 ,
,
где R представляет собой Н или F.
В некоторых вариантах осуществления соединение формулы (I) представляет собой соединение следующей структуры:
 .
.
В дополнительных вариантах осуществления изобретения, соединение формулы (I) представляет собой соединение следующей структуры:
 .
.
В дополнительных вариантах осуществления изобретения, соединение формулы (I) представляет собой соединение следующей структуры:

Способ получения соединения формулы (I) отражен на Схеме 1.
Схема 1

На стадии А бромнитробензол (IX), где R представляет собой Н или F, обрабатывают 2,3-дигидрофураном в присутствии подходящего палладиевого катализатора и подходящего основания. Арилирование реакцией Хека при помощи 2,3-дигидрофурана приводит к получению смеси дигидрофуранилнитробензолов формулы (VIIIa) и формулы (VIIIb), где R представляет собой Н или F.
Палладиевый катализатор, используемый в реакции арилирования Хека, может быть любым подходящим катализатором на основе палладия, из числа известных специалистам в данной области. Примеры палладиевых катализаторов, пригодных для реакции арилирования Хека между бромнитробензолом формулы (IX) и 2,3-дигидрофураном, включают комплексы Pd(II), Pd(I) и Pd(0). В одном варианте осуществления комплекс Pd(II), подходящий для целей настоящего изобретения, имеет общую формулу PdX2(фосфин)2, где Х представляет собой одновалентную отрицательно заряженную группу, такую как галоидная группа, а фосфин, как используется здесь, относится к классу соединений, в котором один, два или три атома водорода в соединении PH3 заменены на соответствующее число таких групп, как фенил (Ph), циклогексил (Су), трет-бутил (tBU) или изопропил (iPr). Примеры фосфиновых лигандов для Pd-катализаторов по настоящему изобретению включают ди-трет-бутилметилфосфин, ди-трет-бутилнеопентилфосфин, дициклогексил-(2-метилфенил)фосфин, дициклогексил-(2,4,6-триметилфенил)фосфин, трициклопентилфосфин, трет-бутилдифенилфосфин, циклогексилдифенилфосфин, трис(4-хлорфенил)фосфин, бензилдифенилфосфин, три(м-толил)фосфин, трис(4-метоксифенил)фосфин, 1,3-бис(дифенилфосфино)пропан (dppp), 1,2-бис(дифенилфосфино)этан (dppe), 1,4-бис(дифенилфосфино)бутан (dppb), мезо-2,4-бис(дифенилфосфино)пентан (mbdpp) и 1,3-бис(диизопропилфосфино)пропан (dippp). В другом варианте осуществления Pd-катализаторы, пригодные для реакции арилирования Хека, включают PdCl2(PPh3)2, PdCl2(PCy3)2, PdCl2(PiPr3)2, PdCl2(PhCN)2, Pd(N,N-диметил-β-аланинат)2, PdCl2{PR2(Ph-R’)}2, где R представляет собой трет-бутил и R’ представляет собой 4-диметиламино группу, бис(дибензилиденацетон)палладий(II), ацетат палладия(II), PdCl2(бис-гидразон), дихлорид [1,3-бис(2,6-диизопропилфенил)имидазол-2-илиден](3-хлорпиридил)палладия(II), дихлорид ди(2-пиридил)метанолпалладия, [1,1’-бис(ди-трет-бутилфосфино)ферроцен]дихлорпалладий(II), [1,1’-бис(дифенилфосфино)ферроцен]дихлорпалладий(II) (Pd(dppf)Cl2), (NHC)Pd(аллил)Cl, где NHC представляет собой N-гетероциклический карбен, такой как N,N’-бис(2,6-диизопропилфенил)имидазол)-2-илиден, N,N’-бис(2,6-диизопропилфенил)-4,5-дигидроимидазол)-2-илиден, N,N’-бис(2,4,6-триметилфенил)имидазол)-2-илиден и N,N’-бис-трет-бутил-имидазол)-2-илиден. В некоторых вариантах выполнения изобретения, одни или несколько лигандов Pd могут быть связаны с субстратом, таким как частица. Примеры таких катализаторов включают (Ar’Ph2P)2PdCl2, где Ar’ группа является частью полимера таким образом, что катализатор представляет собой полимерный палладиевый катализатор. В другом варианте осуществления комплексные соединения Pd(0), полезные для целей настоящего изобретения, включают Pd(фосфин)4 (например, Pd(PPh3)4, Pd(PCy3)4, Pd(PiPr3)4, Pd(tBu3P)2 и трис(дибензилиденацетон)дипалладий(0). В еще одном варианте осуществления Pd(I)-катализаторы, полезные для целей настоящего изобретения, включают Pd2X2(фосфин)2, где X представляет собой одновалентный анион, такой как галоидное соединение. Пример такого Pd(I)-катализатора включает Pd2Br2(tBu3P)2.
В некоторых вариантах осуществления изобретения палладиевый катализатор, используемый в реакции арилирования Хека, может быть любым подходящим хиральным палладиевым катализатором, из числа известных специалистам в данной области. Примеры хиральных палладиевых катализаторов, подходящих для реакции арилирования Хека между бромнитробензолом формулы (IX) и 2,3-дигидрофураном, включают комплексы Pd(II) и Pd(0) с 2,2’-бис(дифенилфосфино)бинафтилом (ΒIΝΑΡ), с другими лигандами типа ΒIΝΑΡ, c JosiPhos, с другими лигандами типа JosiPhos, PhanePhos, SynPhos, DifluoroPhos, SegPhos, P-Phos, TunePhos, 2,4-бис(дифенилфосфино)пентаном и Phox. Реакции арилирования Хека, которые проводились с использованием хиральных палладиевых катализаторов, могут привести к получению энантиомерно обогащенного дигидрофуранилнитробензольного соединения(ий) формулы (VIIIa) и/или формулы (VIIIb), где R представляет собой Н или F. В некоторых вариантах осуществления изобретения, энантиомерный избыток дигидрофуранилнитробензольного соединения(ий) формулы (VIIIa) и/или формулы (VIIIb) может составлять приблизительно 5-100%, приблизительно 10-100%, приблизительно 20-100%, приблизительно 30-100%, приблизительно 40-100%, приблизительно 50-100%, приблизительно 60-100%, приблизительно 70-100%, приблизительно 80-100%, приблизительно 85-100%, приблизительно 90-100%, приблизительно 91-100%, приблизительно 92-100%, приблизительно 93-100%, приблизительно 94-100%, приблизительно 95-100%, приблизительно 96-100%, приблизительно 97-100%, приблизительно 98-100%, приблизительно 99-100% или составлять приблизительно 100%. Таким образом, любое хиральное соединение, полученное из энантиомерно обогащенного соединения(ий) формулы (VIIIa) и/или (VIIIb), также может содержать избыток одного из двух энантиомеров.
Основание, используемое в реакции арилирования Хека, может быть любым подходящим основанием, из числа известных специалистам в данной области. Примеры оснований, подходящих для реакции арилирования Хека между бромнитробензолом формулы (IX) и 2,3-дигидрофураном, включают карбонат калия, карбонат натрия, карбонат цезия, бикарбонат натрия, фосфат калия, трет-бутоксид натрия, трет-бутоксид калия, триэтиламин, диизопропилэтиламин, 1,8-бис(диметиламино)нафталин, дициклогексиламин, дициклогексилметиламин, 2,6-лутидин, ацетат натрия и ацетат калия.
В качестве растворителя для реакции арилирования Хека может использоваться любой подходящий растворитель, из числа известных специалистам в данной области техники. Примеры растворителей, подходящих для реакции арилирования Хека между бромнитробензолом формулы (IX) и 2,3-дигидрофураном, включают 1,4-диоксан, тетрагидрофуран, 1,2-диметоксиэтан, толуол, Ν,Ν-диметилформамид, диметилсульфоксид, ацетонитрил и N,N-диметилацетамид.
Реакцию арилирования Хека можно проводить при любой подходящей температуре в диапазоне от 0°С до 200°С. В некоторых вариантах осуществления изобретения, реакция может быть проведена при температуре между 50 и 150°С. В других вариантах осуществления реакция может быть проведена при температуре между 75 и 125°С. В других вариантах осуществления реакция может быть проведена при температуре между 90 и 110°С.
На стадии B одно из дигидрофуранилнитробензольных соединений (VIIIa) и (VIIIb), или смеси дигидрофуранилнитробензольных соединений (VIIIa) и (VIIIb), где R представляет собой Н или F, обрабатывают газообразным водородом в присутствии катализатора на основе переходного металла и основания. Каталитическое гидрирование ароматических нитро заместителей и двойной связи в дигидрофуранильном заместителе приводит к получению тетрагидрофураниланилинового соединения формулы (VII), где R представляет собой Н или F.
Катализатор на основе переходного металла, используемый в реакции каталитического гидрирования, может быть любым подходящим катализатором, из числа известных специалистам в данной области техники. Примеры катализаторов на основе переходных металлов, пригодных для каталитического гидрирования дигидрофуранилнитробензольных соединений (VIIIa) и (VIIIb), включают палладий на угле, платину на угле, оксид платины и т.п.
Основание, используемое в реакции каталитического гидрирования, может быть любым подходящим основанием, из числа известных специалистам в данной области. Примеры оснований, пригодных для каталитического гидрирования дигидрофуранилнитробензольных соединений (VIIIa) и (VIIIb), включают карбонат калия, карбонат натрия, карбонат цезия, бикарбонат натрия, фосфат калия, триэтиламин, диизопропилэтиламин и другие аминооснования, такие как пиридин, 2,6-лутидин, дициклогексилметиламин, пирролидин и метилпирролидин.
Каталитическое гидрирование может быть выполнено в любом подходящем растворителе, из числа известных специалистам в данной области. Примеры растворителей, подходящих для каталитического гидрирования дигидрофуранилнитробензольных соединений (VIIIa) и (VIIIb), включают метанол, этанол, изопропанол, тетрагидрофуран, 1,4-диоксан, 1,2-диметоксиэтан, этилацетат, гексан и толуол, а также любые их смеси.
Каталитическую реакцию гидрирования можно проводить при любой подходящей температуре в диапазоне от -50 до 100°С. В некоторых вариантах осуществления реакция может быть проведена при температуре в диапазоне от 0 до 50°С. В других вариантах осуществления реакция может быть проведена при температуре между 10 и 40°С. В других вариантах осуществления реакция может быть проведена при температуре между 20 и 30°С.
Каталитическая реакция гидрирования может быть проведена при любом подходящем давлении водорода, составляющего от 15 фунтов на квадратный дюйм до 100 фунтов на квадратный дюйм (1-7 атм.). В некоторых вариантах осуществления реакция может быть проведена при давлении между 20 и 55 фунтов на квадратный дюйм (1,4-3,9 атм.). В других вариантах осуществления реакцию можно проводить при давлении между 25 и 50 фунтов на квадратный дюйм (1,8-3,5 атм.). В других вариантах осуществления реакцию можно проводить при давлении между 30 и 45 фунтов на квадратный дюйм (2,1-3,2 атм.).
На стадии С тетрагидрофураниланилиновое соединение формулы (VII), где R представляет собой Н или F, обрабатывают подходящим бромирующим агентом в подходящем полярном апротонном растворителе. Бромированием тетрагидрофураниланилинового соединения (VII) в пара-положении получают броманилиновое соединение формулы (VI), где R представляет собой Н или F.
Бромирующий агент, используемый в реакции бромирования, может быть любым подходящим бромирующим агентом, из числа известных специалистам в данной области. Примеры бромирующих агентов, подходящих для бромирования тетрагидрофураниланилинового соединения (VII), включают N-бромсукцинимид, бром и бромамин-T.
Растворитель, используемый в реакции бромирования, может быть любым подходящим полярным апротонным растворителем, из числа известных специалистам в данной области. Примеры растворителей, подходящих для бромирования тетрагидрофураниланилинового соединения (VII), включают ацетонитрил, N,N-диметилформамид, диметилсульфоксид, гексаметилфосфорилтриамид (НМРА), а также смеси вышеуказанных растворителей с эфирными растворителями, такими как тетрагидрофуран, диэтиловый эфир, 1,4-диоксан, 1,2-диметоксиэтан и метил-трет-бутиловый эфир.
Реакцию бромирования можно проводить при любой подходящей температуре в диапазоне от -78 до 75°С. В одном варианте выполнения изобретения реакцию проводят при температуре между -50 и 50°С. В другом варианте реакцию проводят при температуре между -35 и +35°С. В еще одном варианте реакцию проводят при температуре между -20 и 10°С.
На стадии D соединение формулы (VI) нитруют с получением соединения формулы (IV). Аминогруппа соединения формулы (VI), где R представляет собой Н или F, может быть защищена с помощью любой из доступных различных защитных групп. Примеры обычных защитных групп для аминогруппы включают алкоксикарбонил (например, трет-бутоксикарбонил, известный как BOC), 1,1-диоксобензо[6]тиофен-метоксикарбонил (Bsmoc), замещенный алкоксикарбонил (например, галогенированные алкоксикарбонильные группы, такие как 2,2,2-трихлорэтоксикарбонил), трет-бутилсульфонил (BUS), циклоалкоксикарбонил, бицилиалкоксикарбонил, алкенилоксикарбонил и арилалкоксикарбонил. Примеры таких защитных групп представляют собой этоксикарбонил, циклопентилоксикарбонил, циклогексилоксикарбонил, 2,2,2-трихлорэтоксикарбонил, аллилоксикарбонил, 1-адамантилоксикарбонил, трет-бутилоксикарбонил, трет-амилоксикарбонил, бензилоксикарбонил (Cbz), п-нитробензилоксикарбонил, п-метоксибензилоксикарбонил, дифенилметоксикарбонил. Другие азотзащитные группы включают ацильные группы, такие как формил, ацетил, пропионил, пивалоил, трет-бутилацетил, 2-хлорацетил, 2-бромацетил, трифторацетил, трихлорацетил, фталил, о-нитрофеноксиацетил, α-хлорбутирил, бензоил, 4-хлорбензоил, 4-бромбензоил и 4-нитробензоил. Дополнительные схемы защиты аминов описаны в Green, T. W. and Wuts, P. G. M., Protective Groups in Organic Synthesis, John Wiley & Sons, Inc., pp. 494-653 (1999).
N-защищенное соединение формулы (VI) нитруют с использованием различных нитрующих агентов, из числа известных специалистам в данной области. Может быть использован любой реагент, который, при соответствующих условиях, может ввести нитрогруппы в фенильное кольцо соединения формулы (VI). Примеры методов нитрования могут быть найдены в источнике March, Advanced Organic Chemistry, John Wiles & Sons, 2001, pp. 696-699, содержание которого включено сюда посредством ссылки.
В одном варианте осуществления изобретения нитрующий агент может представлять собой смесь ангидрида трифторуксусной кислоты и нитрата. В этом варианте избыточное количество трифторуксусного ангидрида может быть использовано для защиты аминогруппы соединения формулы (VI). После стадии защиты выполняется добавление соли нитрата для получения нитрующей смеси in situ. Например, для получения активного нитрующего агента может быть добавлен нитрат аммония. В некоторых вариантах осуществления изобретения нитрат аммония может быть добавлен в небольших порциях при температуре приблизительно 30°С, принимая меры к тому, чтобы температура оставалась в пределах от 30 до 40°С.
Альтернативно, N-защищенное соединение формулы (VI) может быть подвергнуто взаимодействию со смесью сильной кислоты и нитрата. Например, для нитрования N-защищенного соединения формулы (VI) может быть использована смесь трифторуксусной кислоты и NH4NO3.
Другие нитрующие агенты включают другие неорганические нитраты, органические нитраты, нитрат серебра/трифенилфосфин/оксид бромид, нозилаты лантаноидов, соли N-нитро-пиридина и хинолина, и азотную кислоту. Возможно также проведение нитрования соединения формулы (VI) без предварительной защиты анилина. В конкретном варианте осуществления соединение формулы (VI) может быть подвергнуто взаимодействию с нитрующим агентом при температуре между 0°С и 50°С, между 5°С и 50°С, между 10°С и 50°С, между 15°С и 50°С, между 20°С и 50°С, между 22°С и 50°С, между 24°С и 45°С, между 25°С и 45°С, между 27°С и 45°С, между 28°С и 45°С, между 28°С и 44°С, между 28°С и 43°С, между 28°С и 42°С, между 30°С и 45°С, между 30°С и 44°С, между 30°С и 43°С или между 30°С и 42°С.
Защита нитрованного N-защищенного соединения формулы (VI) может быть удалена с помощью любого способа, из числа известных специалистам в данной области техники. Например, защита N-трифторметилкарбонил-защищенного соединения формулы (VI) может быть удалена путем кипячения с обратным холодильником N-защищенного соединения в растворе сильной кислоты, такой как H2SO4 или HCl. Кроме того, в случае, когда R=Н, удаление защитной группы может быть осуществлено путем кипячения N-защищенного соединения с обратным холодильником в присутствии основания, такого как гидроксид натрия, карбонат калия или ацетат натрия. По завершении стадии D, из соединения формулы (VI) по настоящему изобретению получают соединение формулы (IV)

На стадии E соединение формулы (IV) подвергают реакции сочетания типа Suzuki с производным бороновой кислоты формулы (III)

в присутствии палладиевого катализатора, где каждый из R3, R4 и R5 независимо представляет собой необязательно замещенный алкил или необязательно защищенную гидроксильную группу, и В* представляет собой  , где каждый из R1 и R2 независимо представляет собой алкил или H, или OR1 и OR2 вместе с атомом B, к которому они присоединены, образуют необязательно замещенное 5-, 6- или 7-членное кольцо или BF3X, где X является любым одновалентным катионом.
, где каждый из R1 и R2 независимо представляет собой алкил или H, или OR1 и OR2 вместе с атомом B, к которому они присоединены, образуют необязательно замещенное 5-, 6- или 7-членное кольцо или BF3X, где X является любым одновалентным катионом.
Продукт реакции сочетания типа Suzuki подвергают каталитическому гидрированию с получением соединения формулы (II), где R представляет собой Н или F, и каждый из R3, R4 и R5 независимо представляет собой необязательно замещенный алкил или необязательно защищенную гидроксильную группу:

Как используется здесь, термин "производное бороновой кислоты" относится к бороновом кислотам, сложным боронатным эфирам и к трифторборатам. Бороновые кислоты и боронатные эфиры включают любое соединение бора, содержащее группу:
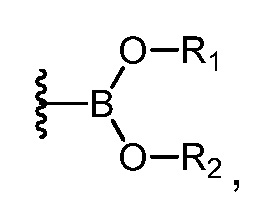
где R1 и R2 независимо представляют собой H или алкил, или OR1 и OR2 вместе с атомом B, к которому они присоединены, образуют необязательно замещенное 5-, 6- или 7-членное кольцо.
Примеры циклических боронатных эфиров включают следующее соединение формулы (III), с кольцами, образованными OR1, OR2 и атомом B:
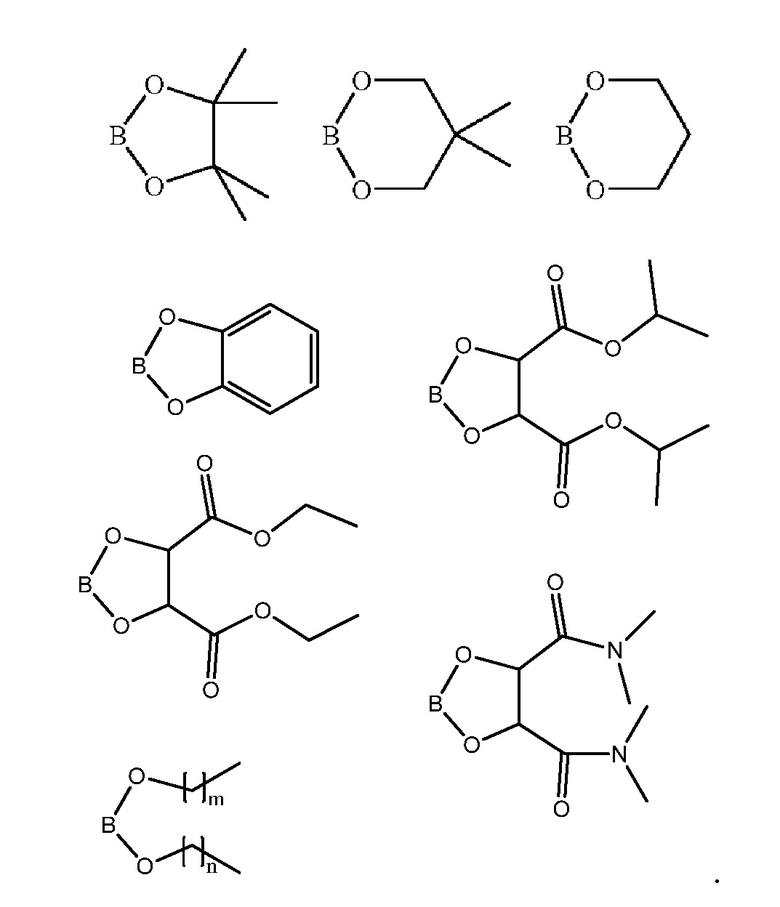
Соединение 7 является специфическим боронатным эфиром. Эти примеры производных бороновой кислоты являются только примерами возможных вариаций замещений для R1 и R2. Специалист в данной области техники может представить много дополнительных возможных модификаций, и все эти модификации охватываются настоящим изобретением, если такие модификации не мешают реакции сочетания между соединениями формулы (III) и формулы (IV), катализируемой палладием.
Трифторбораты включают любое соединение бора, содержащее группу -BF3X, где Х обозначает любой одновалентный катион. В некоторых вариантах Х может представлять собой K+, Na+, Li+, Rb+ или Cs+.
Каждый из R3, R4 и R5 в соединениях формулы (II) и формулы (III) может независимо представлять собой необязательно замещенную алкильную или гидрокси группу, где гидроксильная группа может быть защищена любой защитной группой, из числа известных специалистам в данной области техники. В одном варианте осуществления изобретения каждый из R3, R4 и R5 представляет собой С1-C6-алкильную группу или гидроксильную группу. В конкретном варианте осуществления изобретения, каждый из R3, R4 и R5 независимо друг от друга может представлять собой метил, этил, пропил, изопропил или гидрокси.
В одном варианте осуществления изобретения соединение формулы (III) имеет следующую структуру:

Настоящее изобретение также охватывает использование псевдо-галогенида (например, трифлата) вместо группы брома в соединении формулы (IV)
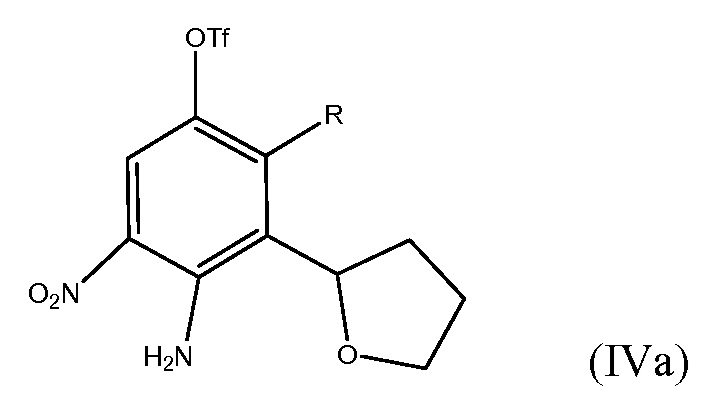 .
.
Также может быть использована любая другая уходящая группа, которая не мешает реакции сочетания. Другие уходящие группы включают хлорид, йодид, тозилат и мезилат.
Палладиевый катализатор, используемый в реакции сочетания типа Suzuki между соединениями формулы (III) и формулы (IV), может быть любым подходящим палладиевым катализатором, из числа известных специалистам в данной области. Примеры Pd-катализаторов, подходящих для реакции сочетания типа Suzuki между соединениями формулы (III) и формулы (IV), включают комплексы Pd(II), Pd(I) и Pd(0). В одном варианте осуществления изобретения комплекс Pd(II), подходящий для целей настоящего изобретения, имеет общую формулу PdX2(фосфин)2, где Х представляет собой одновалентную отрицательно заряженную группу, такую как галоидная группа, а фосфин, как используется здесь, относится к классу соединений, в котором один, два или три атома водорода в соединении PH3 заменены на соответствующее число таких групп, как фенил (Ph), циклогексил (Су), трет-бутил (tBU) или изопропил (iPr). Примеры фосфиновых лигандов для Pd-катализаторов по настоящему изобретению включают ди-трет-бутилметилфосфин, ди-трет-бутилнеопентилфосфин, дициклогексил-(2-метилфенил)фосфин, дициклогексил-(2,4,6-триметилфенил)фосфин, трициклопентилфосфин, трет-бутилдифенилфосфин, циклогексилдифенилфосфин, трис(4-хлорфенил)фосфин, бензилдифенилфосфин, три(м-толил)фосфин, трис(4-метоксифенил)фосфин, 1,3-бис(дифенилфосфино)пропан (dppp), 1,2-бис(дифенилфосфино)этан (dppe), 1,4-бис(дифенилфосфино)бутан (dppb), мезо-2,4-бис(дифенилфосфино)пентан (mbdpp) и 1,3-бис(диизопропилфосфино)пропан (dippp). В другом варианте осуществления Pd-катализаторы, подходящие для реакции арилирования Хека, включают PdCl2(PPh3)2, PdCl2(PCy3)2, PdCl2(PiPr3)2, PdCl2(PhCN)2, Pd(N,N-диметил-β-аланинат)2, PdCl2{PR2(Ph-R’)}2, где R представляет собой трет-бутил и R’ представляет собой 4-диметиламино группу, бис(дибензилиденацетон)палладий(II), ацетат палладия(II), PdCl2(бис-гидразон), дихлорид [1,3-бис(2,6-диизопропилфенил)имидазол-2-илиден](3-хлорпиридил)палладия(II), дихлорид ди(2-пиридил)метанолпалладия, [1,1’-бис(ди-трет-бутилфосфино)ферроцен]дихлорпалладий(II), [1,1’-бис(дифенилфосфино)ферроцен]дихлорпалладий(II) (Pd(dppf)Cl2), (NHC)Pd(аллил)Cl, где NHC представляет собой N-гетероциклический карбен, такой как N,N’-бис(2,6-диизопропилфенил)имидазол)-2-илиден, N,N’-бис(2,6-диизопропилфенил)-4,5-дигидроимидазол)-2-илиден, N,N’-бис(2,4,6-триметилфенил)имидазол)-2-илиден и N,N’-бис-трет-бутил-имидазол)-2-илиден. В некоторых вариантах осуществления изобретения одни или несколько лигандов Pd могут быть связаны с субстратом, таким как частица. Примеры таких катализаторов включают (Ar’Ph2P)2PdCl2, где Ar’ группа является частью полимера таким образом, что катализатор представляет собой полимерный палладиевый катализатор. В другом варианте осуществления комплексные соединения Pd(0), полезные для целей настоящего изобретения, включают Pd(фосфин)4 (например, Pd(PPh3)4, Pd(PCy3)4, Pd(PiPr3)4, Pd(tBu3P)2 и трис(дибензилиденацетон)дипалладий(0). В еще одном варианте осуществления Pd(I)-катализаторы, полезные для целей настоящего изобретения, включают Pd2X2(фосфин)2, где X представляет собой одновалентный анион, такой как галоидное соединение. Пример такого Pd(I)-катализатора включает Pd2Br2(tBu3P)2.
Продукт реакции сочетания Сузуки подвергают каталитическому гидрированию в условиях, указанных для стадии B.
На стадии F соединение формулы (II) обрабатывают соединением формулы А или формулы В, получая при этом бензимидазольное соединение формулы (I):
 ,
,
где R6 может представлять собой необязательно замещенный алкил, бензил или п-нитробензил. В конкретных вариантах R6 представляет собой метил или этил.
В одном варианте осуществления изобретения соединение формулы (I) имеет следующую структуру:

Соединение формулы (Ia) содержит один хиральный центр в положении С-2 тетрагидрофурильного кольца. Таким образом, соединение формулы (Ia) может быть представлено в виде рацемической смеси или может содержать избыток одного из двух энантиомеров этого соединения.
Рацемическая смесь соединения формулы (Ia) может быть энантиомерно обогащена с помощью любого способа, из числа известных специалистам в данной области. Примеры способов энантиомерного обогащения, из числа известных в данной области, включают преобразование из энантиомеров в диастереомеры и использование различных физических свойств диастереомеров для разделения энантиомеров или обогащения одним энантиомером. В некоторых вариантах осуществления изобретения, рацемическая смесь (или смесь, обогащенная одним энантиомером, когда желательно повысить содержание конкретного энантиомера) соединения формулы (Ia) может быть энантиомерно обогащена с помощью препаративной колоночной хроматографии, подходящей для выделения чистого энантиомера из рацемической смеси или из смеси, обогащенной энантиомером, когда желательно повысить содержание конкретного энантиомера. В вариантах осуществления изобретения, когда энантиомерная смесь является энантиомерно обогащенной, избыток одного из двух энантиомеров может составлять между приблизительно 5-100%, приблизительно 10-100%, приблизительно 20-100%, приблизительно 30-100%, приблизительно 40-100%, приблизительно 50-100%, приблизительно 60-100%, приблизительно 70-100%, приблизительно 80-100%, приблизительно 85-100%, приблизительно 90-100%, приблизительно 91-100%, приблизительно 92-100%, приблизительно 93-100%, приблизительно 94-100%, приблизительно 95-100%, приблизительно 96-100%, приблизительно 97-100%, приблизительно 98-100%, приблизительно 99-100% или составлять приблизительно 100%.
Соединения, содержащие одну или несколько защищенных гидроксильных групп, могут быть получены с использованием методов, из числа известных специалистам в данной области техники. Примеры схем защиты гидрокси групп описаны в работе Green, T. W. and Wuts, P. G. M., Protective Groups in Organic Synthesis, John Wiley & Sons, Inc., pp. 17-245 (1999).
Термин "алкил", используемый здесь, относится к прямой и разветвленной цепи, содержащей до десяти атомов углерода. Примеры алкильных групп, подходящих для целей настоящего изобретения, включают линейные и разветвленные С1-12-алкильные группы. Как используется здесь, термин "короткоцепочечный алкил" относится к алкильной цепи, содержащей до 4 атомов углерода. Используемый здесь термин "алкильная цепь средней длины" относится к алкильной цепи, имеющей 5-7 атомов углерода. Примеры алкильных групп включают группы метила, этила, пропила, изопропила, бутила, втор-бутила, трет-бутила, 3-пентила, гексила и октила, которые могут быть необязательно замещенными.
Арильные группы, пригодные для способов по настоящему изобретению, включают С6-14-арил, предпочтительно - С6-10-арил. Типичные С6-10-арильные группы включают группы фенила, нафтила, фенантренила, антраценила, инденила, азуленила, бифенила, бифениленила и флуоренила.
Термин "карбоцикл", как используется здесь, включает циклоалкильные и частично насыщенные карбоциклические группы. Примером циклоалкильных групп является С3-7-циклоалкил. Типичные циклоалкильные группы включают циклопропил, циклобутил, циклопентил, циклогексил и циклогептил.
Термин "гетероцикл", используемый здесь, относится к насыщенной или частично насыщенной 3-7-членной моноциклической или 7-10-членной бициклической кольцевой системе. Кольцевая гетероциклическая система может состоять из атомов углерода и от одного до четырех гетероатомов, выбранных из группы, состоящей из О, N и S, где гетероатомы азота и серы могут быть окислены, атом азота может быть кватернизирован, и где гетероциклическое кольцо может быть замещено по атому углерода или азота, если такое соединение является стабильным.
Необязательные заместители для алкила, арила, насыщенного или ненасыщенного карбоцикла и насыщенного или ненасыщенного гетероцикла включают один или несколько из числа галогена, С1-6-галогеналкила, С6-10-арила, С4-7-циклоалкила, С1-6-алкила, С2-6-алкенила, С2-6-алкинила, С6-10-арил-С1-6-алкила, С6-10-арил-С2-6-алкенила, С6-10-арил-С2-6-алкинила, С1-6-гидроксиалкила, нитро, амино, С1-6-алкиламино, циано, С1-6-ациламино, гидрокси, сульфанила, сульфонила, сульфоксида, С1-6-ацилокси, С1-6-алкокси и карбокси.
Если не указано иное, то структуры, представленные здесь, также включают все стереохимические формы представленной структуры, т.е. R и S конфигурации для каждого асимметричного центра. Следовательно, отдельные стереохимические изомеры, а также энантиомерные и диастереомерные смеси соединений по настоящему изобретению, входят в объем притязаний настоящего изобретения. Меченные изотопами формы соединений, приведенные здесь, где один или несколько атомов заменены атомами, имеющими атомную массу или массовое число, отличные от атомной массы или массового числа, обычно обнаруживаемых в природе, также входят в объем притязаний настоящего изобретения. Примеры изотопов, которые могут быть включены в соединения по изобретению, включают изотопы водорода, углерода, кислорода и фтора, такие как 2H, 3H, 13С, 14С, 15N, 18О и 17О. Такие радиоактивные соединения, или иначе называемые соединения, меченные изотопами, являются полезными, например, в качестве исследовательских инструментов для ингибиторов гиразы и/или топоизомеразы IV с улучшенным терапевтическим профилем.
В одном варианте осуществления изобретения соединения формулы (I) включают соединения формулы (Ib)
 ,
,
где R представляет собой Н или F.
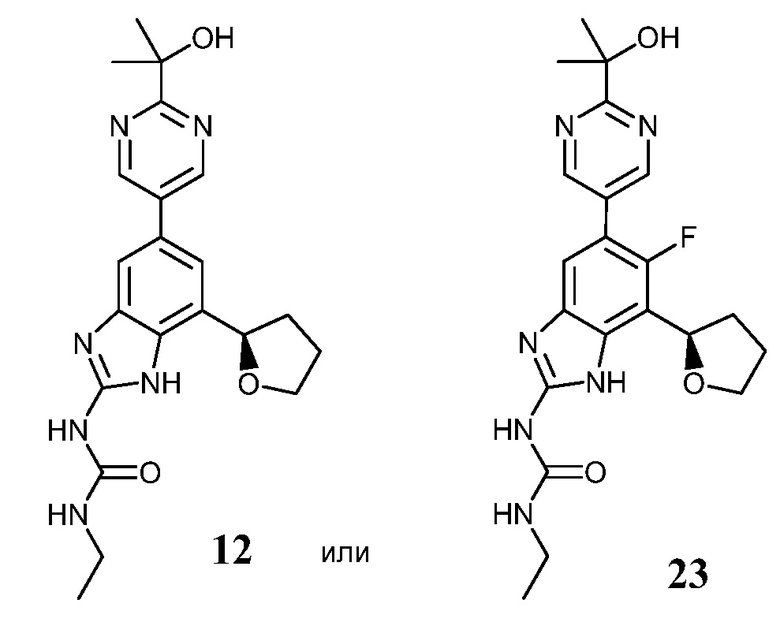
В другом варианте осуществления изобретения соединения формулы (I) включают соединения 12 и 23, представленные ниже:

(R)-1-этил-3-(5-(2-(2-гидроксипропан-2-ил)пиримидин-5-ил)-7-(тетрагидрофуран-2-ил)-1H-бензо[d]имидазол-2-ил)мочевина или его фармацевтически приемлемая соль; и

(R)-1-этил-3-(6-фтор-5-(2-(2-гидроксипропан-2-ил)пиримидин-5-ил)-7-(тетрагидрофуран-2-ил)-1H-бензо[d]имидазол-2-ил)мочевина или его фармацевтически приемлемая соль.
Несмотря на то, что соединения по изобретению эффективны в виде свободного основания, соединения формулы (I) могут быть введены в виде фармацевтически приемлемых кислотно-аддитивных солей. Соединения формулы (I) могут быть превращены в соответствующие кислотно-аддитивные соли с использованием способов, хорошо известных специалистам в данной области. Примеры нетоксичных кислотно-аддитивных солей соединения формулы (I), содержащих фармацевтически приемлемые анионы, включают соли соединения формулы (I), такие как ацетат, бензолсульфонат (также известный как бесилат), бензоат, бикарбонат, битартрат, бромид, эдентат кальция, камсилат, карбонат, хлорид, цитрат, дигидрохлорид, эдетат, эдисилат, эстолат, фумарат, глюцептат, глюконат, глутамат, гликоллиларсанинат, гексилрезорцинат, гидрабамин, гидробромид, гидроксинафтоат, йодид, изетионат, лактат, лактобионат, малат, малеат, манделат, метансульфонат (также известный как мезилат), метилбромид, метилнитрат, метилсульфат, мукат, напсилат, никотинат, нитрат, памоат (эмбонат), пантотенат, фосфат, дифосфат, полигалактуронат, салицилат, стеарат, субацетат, сукцинат, сульфат, бисульфат, таннат, тартрат, теоклат, п-толуолсульфонат (также известный как тозилат), а также триэтиодид соединения формулы (I).
Один вариант осуществления этого изобретения относится к способу лечения бактериальной инфекции у млекопитающего, нуждающегося в этом, где способ включает введение указанному млекопитающему терапевтически эффективного количества соединения, имеющего формулу (I), или его фармацевтически приемлемой соли.
Для того, чтобы это изобретение было более понятным, приведены нижеследующие примеры. Эти примеры представлены с целью иллюстрации изобретения и не должны быть истолкованы как ограничивающие каким-либо образом объем притязаний.
Примеры
Пример 1: Схема синтеза соединений 11, 12 и 13
Схема 2 представляет схему способа получения соединений 11, 12 и 13.
Схема 2

Пример 1.a
Получение 2-(2-нитрофенил)-2,5-дигидрофурана (соединение 3а) и 2-(2-нитрофенил)-2,3-дигидрофурана (соединение 3b)
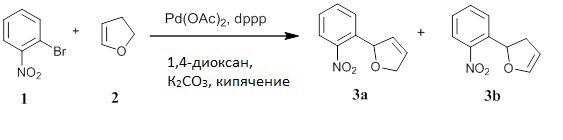
В реакционном сосуде смешивали 1-бром-2-нитробензол (1) (600 г, 99%, 2,941 моль, Alfa Aesar A11686), 1,3-бис(дифенилфосфино)пропан (62,50 г, 97%, 147,0 ммоль; Alfa Aesar A12931), 1,4-диоксан (2,970 л, Sigma-Aldrich 360481), карбонат калия (812,9 г, 5,882 моль, JT-Baker 301201) и 2,3-дигидрофуран (2) (1,041 кг, 99%, 1,124 л, 14,70 моль, Aldrich 200018). Через перемешиваемую смесь в течение 4 часов барботировали азот, затем добавляли ацетат палладия(II) (16,51 г, 73,52 ммоль, Strem 461780) и продолжали деоксигенацию в течение 10 минут. Реакционную смесь перемешивали при кипячении с обратным холодильником под током азота в течение ночи (ЯМР и анализ аликвоты показали полное израсходование арилбромида). Реакционной смеси давали охладиться, затем разбавляли гексаном (1 л), фильтровали через короткий объемный фильтр Florisil® (500 г, 200 меш) и элюировали этилацетатом. Фильтрат концентрировали под пониженным давлении (т.к. 2-(2-нитрофенил)-2,3-дигидрофуран (3b) является летучим соединением в условиях высокого вакуума, и он может быть при комнатной температуре в определенной степени нестабильным) с получением смеси соединений (3а) и (3b) в виде темно-коричневого масла (654,0 г). Сырой продукт хранился в холодильнике и передавался для дальнейших манипуляций без дополнительной очистки.
Пример 1.а.1
Асимметричное получение 2-(2-нитрофенил)-2,5-дигидрофурана (соединение 3а) и 2-(2-нитрофенил)-2,3-дигидрофурана (соединение 3b)
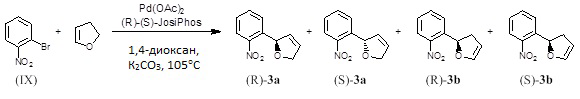
В реакционной пробирке смешивали 1-бром-2-нитробензол (50,0 мг, 98%, 0,2426 ммоль, Aldrich 365424), карбонат калия (67,1 мг, 0,4852 ммоль, JT-Baker 301201), этанольный аддукт (R)-(-)-1-[(S)-2-(дифенилфосфино)ферроценил]этилдициклогексилфосфина ((R)-(S)-JosiPhos, 7,8 мг, 0,01213 ммоль, Strem 261210), 2,3-дигидрофуран (1,0 мл, 99%, 13,08 ммоль; Aldrich 200018) и 1,4-диоксан (0,98 мл). Через перемешиваемую смесь в течение 20 минут барботировали азот, затем добавляли ацетат палладия(II) (1,36 мг, 0,006065 ммоль, Strem 461780). Пробирку закупоривали, и реакционную смесь перемешивали при 105°С в течение ночи. ВЭЖХ сырой реакционной смеси показал почти полное израсходование бромистого арила и образование смеси (1:1) из 2-(2-нитрофенил)-2,5-дигидрофурана (соединение 3а) и 2-(2-нитрофенил)-2,3-дигидрофурана (соединение 3b). Реакционную смесь охлаждали, разбавляли гексаном (2 мл), отфильтровывали и промывали этилацетатом. Отфильтрованный раствор концентрировали на роторном испарителе, получая коричневое масло (51 мг). Этот материал не помещали под высокий вакуум из-за проблем с летучестью и стабильностью. Сырая реакционная смесь была идентифицирована анализом 1Н-ЯМР как смесь 1:1 соединений 3а и 3b. Масло очищали хроматографией на силикагеле, элюируя смесью EtOAc (от 0 до 38%) в гексане (или от 0 до 100% CH2Cl2 в гексане) с получением чистых образцов (3а) и (3b). Аналитические данные для этих образцов были следующими.
2-(2-Нитрофенил)-2,5-дигидрофуран (соединение 3а) был получен в виде желтого твердого вещества (чистота по ВЭЖХ 97%, энантиомерный избыток 97,0%): ЖХ-МС: (колонка C18, элюент 10-90% MeOH/вода, градиент от 3 до 5 мин, модификатор муравьиная кислота) M+1: 192,05 (3,40 мин); ВЭЖХ: время удерживания 4,2 мин (колонка YMC ODS-AQ 150×3,0 мм, элюент смесь 10-90% CH3CN/вода, градиент в течение 8 минут, модификатор 0,1% TFA, скорость потока 1 мл/мин); аналитическая хиральная ВЭЖХ: время удерживания 7,4 мин (основной энантиомер) и 8,1 мин (минорный энантиомер), элюент 10% изопропиловый спирт/гексан, колонка CHIRALCEL® OJ® 4,6×250 мм, скорость потока 1 мл/мин, температура 30°С; 1H ЯМР (300 МГц, CDCl3) δ 8,02 (д, J=8,2 Гц, 1H), 7,73 (д, J=7,9 Гц, 1H), 7,64 (т, J=7,6 Гц, 1H), 7,45-7,38 (м, 1H), 6,37-6,30 (м, 1H), 6,11-6,06 (м, 1H), 6,04-5,98 (м, 1H), 5,02-4,83 (м, 2H) м.д.; 13C ЯМР (75 МГц, CDCl3) δ 146,97, 139,11, 133,95, 129,58, 128,10, 128,09, 126,78, 124,38, 84,28, 76,42 м.д.; 13C DEPT ЯМР (75 МГц, CDCl3) δ 133,95 (CH), 129,58 (CH), 128,10 (CH), 128,09 (CH), 126,78 (CH), 124,38 (CH), 84,28 (CH), 76,42 (CH2) м.д.
2-(2-Нитрофенил)-2,3-дигидрофуран (соединение 3b) был получен в виде желтого масла (чистота по ВЭЖХ 79-90%, энантиомерный избыток 44,0%): ЖХ-МС: (колонка C18, элюент 10-90% MeOH/вода, градиент с 3 по 5 мин, модификатор муравьиная кислота) M+1: 192,05 (3,72 мин); ВЭЖХ: время удерживания 4,8 мин (колонка YMC ODS-AQ 150×3,0 мм, элюент 10-90% CH3CN/вода, градиент 8 мин, модификатор 0,1% TFA, скорость потока 1 мл/мин,); аналитическая хиральная ВЭЖХ: время удерживания 5,96 мин (основной энантиомер) и 6,35 мин (минорный энантиомер), элюент 10% изопропиловый спирт/гексан, колонка CHIRALCEL® OJ ® 4,6×250 мм, скорость потока 1 мл/мин, температура 30°С; 1H ЯМР (300 МГц, CDCl3) δ 8,08 (д, J=8,2 Гц, 1H), 7,73 (д, J=7,8 Гц, 1H), 7,65 (т, J=7,6 Гц, 1H), 7,48-7,39 (м, 1H), 6,50 (кв, J=2,4 Гц, 1H), 6,10 (дд, J=10,9, 7,4 Гц, 1H), 4,95 (кв, J=2,5 Гц, 1H), 3,46-3,35 (м, 1H), 2,50-2,39 (м, 1H) м.д.; 13C ЯМР (75 МГц, CDCl3) δ 146,60, 144,98, 139,73, 133,93, 128,07, 127,11, 124,85, 99,29, 78,45, 38,29 м.д.; 13C DEPT ЯМР (75 МГц, CDCl3) δ 144,98 (CH), 133,93 (CH), 128,07 (CH), 127,11 (CH), 124,85 (CH), 99,29 (CH), 78,45 (CH), 38,29 (CH2) м.д.
Соединения 3a и 3b были подвергнуты стадии восстановления с получением 2-тетрагидрофуран-2-ил-анилина (соединение 4), как описано в примере 1.b (см. ниже). Анализ этого материала показал, что оба соединения 3a и 3b были получены в виде основных энантиомеров, при энантиомерном избытке, равном 70%. Абсолютная стереохимия основных энантиомеров ((R) или (S)) не была определена.
Пример 1.b
Получение 2-тетрагидрофуран-2-ил-анилина (соединение 4)

В сосуд Парра, в атмосфере азота, помещали 5%-ый палладий на угле (16,3 г, влажность 50%, 3,83 ммоль, Aldrich 330116) и затем MeOH (100 мл, JT-Baker 909333). Сырую смесь 2-(2-нитрофенил)-2,5-дигидрофурана и 2-(2-нитрофенил)-2,3-дигидрофурана (соединения 3а и 3b) (163 г) растворяли в МеОН (389 мл) и добавляли в сосуд Парра, после чего добавляли NEt3 (237,6 мл, 1,705 моль, Sigma-Aldrich 471283). Сосуд помещали в шейкер Парра и насыщали водородом. Водород добавляли до давления 30 фунтов на квадратный дюйм (2,1 атм.), затем сосуд подвергали встряхиванию до тех пор, пока исходный материал не был полностью израсходован (ЖХ-МС и ЯМР показали завершение реакции). Реакционную смесь продували азотом, отфильтровали через Celite™ и промывали этилацетатом. Фильтрат концентрировали на роторном испарителе с получением коричневого масла. Реакцию повторяли три раза в том же масштабе, и партии были объединены для дальнейшей очистки. Сырой продукт подвергали перегонке под вакуумом (при давлении приблизительно 15 торр), собирали дистиллят при 108-129°С с получением соединения 4 в виде прозрачного бледно-желтого масла (427,9 г, средний выход 84%, чистота по ГХ-МС 98%). ЖХ-МС: (колонка C18, элюент 10-90% CH3CN/вода, градиент 5 минут, модификатор муравьиная кислота) M+1: 163,95 (1,46 мин). 1H ЯМР (300 МГц, CDCl3) δ 7,15-7,04 (м, 2H), 6,77-6,62 (м, 2H), 4,85-4,77 (м, 1H), 4,18 (с, 2H), 4,12-4,02 (м, 1H), 3,94-3,85 (м, 1H), 2,25-1,95 (м, 4H) м.д.
Пример 1.с
Получение 4-бром-2-тетрагидрофуран-2-ил-анилин (соединение 5).

К перемешиваемому раствору 2-тетрагидрофуран-2-ил-анилина (4) (53,45 г, 327,5 ммоль) в метил-трет-бутиловом эфире (MTBE, 641,4 мл) и ацетонитриле (213,8 мл), охлажденному до 2°С, добавляли N-бромсукцинимид (NBS, 58,88 г, 99%, 327,5 ммоль, Aldrich B81255) четырьмя порциями, поддерживая внутреннюю температуру ниже приблизительно 8°С. Реакционную смесь перемешивали при охлаждении на ледяной бане в течение 30 минут (ЯМР отобранной аликвоты показал полное израсходование исходного материала). Водный 1н раствор Na2S2O3 (330 мл) добавляли к реакционной смеси, удаляли охлаждающую баню и перемешивали в течение 20 минут. Смесь разбавляли EtOAc, и разделяли слои. Органическую фазу промывали насыщенным водным раствором NaHCO3 (2x), водой, насыщенным солевым раствором, высушивали над MgSO4, фильтровали через тонкий слой диоксида кремния, элюировали с помощью EtOAc и концентрировали при пониженном давлении с получением соединения 5 в виде масла темно-янтарного цвета (82,25 г, чистота по ВЭЖХ 77-94%). Дальнейшие манипуляции выполняли без дополнительной очистки. ЖХ-МС (колонка C18, элюент 10-90% CH3CN/вода, градиент 5 минут, модификатор муравьиная кислота) M+1: 242,10 (2,89 мин). 1H ЯМР (300 МГц, CDCl3) δ 7,22 (д, J=2,3 Гц, 1H), 7,16 (дд, J=8,4, 2,3 Гц, 1H), 6,54 (д, J=8,4 Гц, 1H), 4,79-4,73 (м, 1H), 4,15 (с, 2H), 4,10-4,01 (м, 1H), 3,93-3,85 (м, 1H), 2,26-2,13 (м, 1H), 2,12-1,97 (м, 3H) м.д.
Пример 1.d
Получение N-(4-бром-2-нитро-6-тетрагидрофуран-2-ил-фенил)-2,2,2-трифтор-ацетамида (соединение 6)

К трифторуксусному ангидриду (455,3 мл, 3,275 моль, Sigma-Aldrich 106232), перемешиваемому при 2°С, медленно добавляли в течение 15 минут через капельную воронку 4-бром-2-тетрагидрофуран-2-ил-анилин (соединение 5) (79,29 г, 327,5 ммоль) в виде густого масла (температура реакции повышалась до 14°С). Оставшееся масло ополаскивали безводным 2-метилтетрагидрофуран (39,6 мл, Sigma-Aldrich 414247) и вводили в реакционную смесь. Холодную баню удаляли и добавляли нитрат аммония (34,08 г, 425,8 ммоль, Aldrich 467758). Реакционная температура в течение приблизительно 30 минут поднималась до 40°С, после чего использовали баню с холодной водой для контроля экзотермической реакции и доведения температуры реакции до комнатной температуры. Затем убирали охлаждающую баню, и перемешивание продолжали в течение еще 40 минут (ВЭЖХ показала небольшое количество материала, не подвергшегося нитрованию). Реакционную смесь медленно выливали в перемешиваемую смесь измельченного льда (800 г). Твердый осадок собирали фильтрованием, промывали водой, насыщенным водным раствором NaHCO3 (рН 8), и затем еще раз промывали водой и гексаном. Влажное твердое вещество сначала высушивали в конвекционной печи при температуре 50°С в течение нескольких часов, а затем при пониженном давлении в печи при 40°С в течение ночи с получением соединения 6 в виде светло-коричневого твердого вещества (77,86 г, выход 62%, чистота по ВЭЖХ 98%). ЖХ-МС: (колонка C18, элюент 10-90% CH3CN/вода, градиент 5 минут, модификатор муравьиная кислота) M+1: 383,19 (3,27 мин). 1H ЯМР (300 МГц, CDCl3) δ 9,81 (с, 1H), 8,08 (д, J=2,2 Гц, 1H), 7,73 (д, J=2,2 Гц, 1H), 4,88 (дд, J=9,0, 6,5 Гц, 1H), 4,17-4,08 (м, 1H), 4,03-3,95 (м, 1H), 2,45-2,34 (м, 1H), 2,17-2,06 (м, 2H), 1,96-1,83 (м, 1H) м.д.
Пример 1.е
Получение 4-бром-2-нитро-6-тетрагидрофуран-2-ил-анилина (соединение 6а).
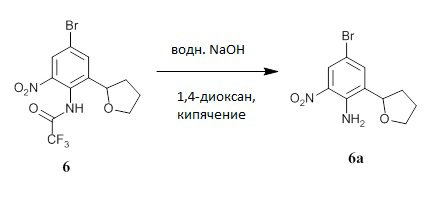
N-(4-бром-2-нитро-6-тетрагидрофуран-2-ил-фенил)-2,2,2-трифтор-ацетамид (соединение 6) (54,00 г, 140,9 ммоль) растворяли в 1,4-диоксане (162 мл) и добавляли водный 6М раствор NaOH (70,45 мл, 422,7 ммоль, JT-Baker 567202). Реакционную смесь перемешивали при кипячении с обратным холодильником в течение 2 дней (ВЭЖХ показала полную конверсию). Смесь охлаждали, разбавляли МТВЕ (800 мл) и промывали водой (2×200 мл), насыщенным водным раствором NH4C1, водой и насыщенным солевым раствором. Смесь высушивали над MgSO4, фильтровали и концентрировали при пониженном давлении с получением соединения 6а в виде темно-янтарного масла (40,96 г, выход 93%, чистота по ВЭЖХ с ЯМР 92%). ЖХ-МС: (колонка C18, элюент 10-90% МеОН/вода, градиент с 3 по 5 мин, модификатор муравьиная кислота) M+1: 287,28 (3,44 мин). 1H ЯМР (300 МГц, CDCl3) δ 8,24 (д, J=2,4 Гц, 1H), 7,41 (д, J=2,3 Гц, 1H), 6,91 (с, 2H), 4,80 (т, J=7,2 Гц, 1H), 4,14-4,05 (м, 1H), 3,98-3,90 (м, 1H), 2,36-2,19 (м, 1H), 2,15-2,01 (м, 3H) м.д.
Пример 1.f
Получение 2-[5-(4-амино-3-нитро-5-тетрагидрофуран-2-ил-фенил)пиримидин-2-ил]пропан-2-ола (соединение 8).

Смешивали 4-бром-2-нитро-6-тетрагидрофуран-2-ил-анилин (соединение 6а) (40,40 г, 92%, 129,5 ммоль), 1,4-диоксан (260 мл, Sigma-Aldrich 360481), 2-[5-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)пиримидин-2-ил]пропан-2-ол (соединение 7) (41,05 г, 155,4 ммоль) и водный 2,7М раствор Na2CO3 (143,9 мл, 388,5 ммоль). Через перемешиваемую смесь пропускали ток азота в течение 1 ч, и затем добавляли тетракис(трифенилфосфин)палладий(0) (7,48 г, 6,47 ммоль, Strem 462150). Реакционную смесь перемешивали при кипячении с обратным холодильником в течение 2 часов (ВЭЖХ показала завершение реакции), охлаждали и разбавляли EtOAc. Смесь промывали водой, насыщенным водным раствором NH4C1 и насыщенным солевым раствором, фильтровали через короткий объемный фильтр Florisil® и элюировали с использованием EtOAc. Фильтрат концентрировали при пониженном давлении с получением темно-коричневого масла. Полученное масло растворяли в CH2Cl2 и элюировали через короткий объемный фильтр из силикагеля с помощью CH2Cl2, а затем с помощью EtOAc. Целевую фракцию концентрировали на роторном испарителе до образования осадка с получением густой коричневой суспензии, которую растирали с МТВЕ. Твердое вещество собирали путем фильтрации, промывали МТВЕ и высушивали под высоким вакуумом с получением соединения 8 в виде твердого вещества желтого цвета (35,14 г, чистота по ВЭЖХ более 99%). ЖХ-МС: (колонка C18, элюент 10-90% CH3CN/вода, градиент 5 мин, модификатор муравьиная кислота) M+1: 345,00 (2,69 мин). 1H ЯМР (300 МГц, CDCl3) δ 8,88 (с, 2H), 8,36 (д, J=2,2 Гц, 1H), 7,56 (д, J=2,1 Гц, 1H), 7,09 (с, 2H), 4,92 (т, J=7,2 Гц, 1H), 4,62 (с, 1H), 4,20-4,11 (м, 1H), 4,03-3,94 (м, 1H), 2,39-2,26 (м, 1H), 2,23-2,08 (м, 3H), 1,64 (с, 6H) м.д. Фильтрат концентрировали и очищали с помощью хроматографии на силикагеле ISCO, при этом элюция с помощью 0-80% EtOAc/гексан дала вторую порцию продукта 8 в виде твердого вещества янтарного цвета (4,46 г, общий выход 88%, чистота по ВЭЖХ 88%).
Пример 1.g
Получение 2-[5-(3,4-диамино-5-тетрагидрофуран-2-ил-фенил)пиримидин-2-ил]пропан-2-ола (соединение 9)
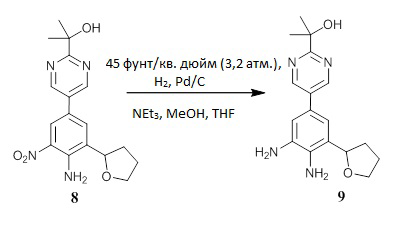
В сосуд Парра, содержащий суспензию 2-[5-(4-амино-3-нитро-5-тетрагидрофуран-2-ил-фенил)пиримидин-2-ил]пропан-2-ол (соединение 8) (30,10 г, 87,41 ммоль) и THF (90 мл), в атмосфере азота, добавляли 5%-ую суспензию палладия на угле (3,01 г, влажность 50%, 0,707 ммоль, Aldrich 330116) в МеОН (90 мл, JT-Baker 909333), с последующим добавлением NEt3 (24,37 мл, 174,8 ммоль, Sigma-Aldrich 471283). Сосуд помещали в шейкер Парра и насыщали водородом. Водород добавляли до давления 45 фунтов на квадратный дюйм (3,2 атм.), затем сосуд подвергали встряхиванию до тех пор, пока исходный материал не был полностью израсходован (ВЭЖХ показала полную конверсию). Реакционную смесь продували азотом, отфильтровали через Celite™ и промывали этилацетатом. Фильтрат повторно фильтровали через 0,5-микронный стекловолоконный фильтр, зажатый между двумя бумажными пластинами Р5, и концентрировали при пониженном давлении, получая соединение 9 в виде светло-коричневого вспененного продукта (28,96 г, выход 98%, чистота по ЯМР 93%). ЖХ-МС: (колонка C18, элюент 10-90% CH3CN/вода, градиент 5 мин, модификатор муравьиная кислота) M+1: 315,32 (1,54 мин). 1H ЯМР (300 МГц, CDCl3) δ 8,83 (с, 2H), 6,92 (д, J=1,8 Гц, 1H), 6,88 (д, J=1,8 Гц, 1H), 4,90 (дд, J=7,9, 6,2 Гц, 1H), 4,72 (с, 1H), 4,18 (с, 2H), 4,17-4,08 (м, 1H), 3,99-3,89 (м, 1H), 3,46 (с, 2H), 2,34-2,19 (м, 1H), 2,17-2,05 (м, 3H), 1,63 (с, 6H) м.д.
Пример 1.h
Получение 1-этил-3-[5-[2-(1-гидрокси-1-метил-этил)пиримидин-5-ил]-7-тетрагидрофуран-2-ил-1Н-бензимидазол-2-ил]мочевины (соединение 11).

К перемешиваемому раствору 2-[5-(3,4-диамино-5-тетрагидрофуран-2-ил-фенил)пиримидин-2-ил]пропан-2-ола (соединение 9) (32,10 г, 102,1 ммоль ) в 1,4-диоксане (160,5 мл, Sigma-Aldrich 360481) добавляли буфер (рН 3,5, 240,8 мл), полученный растворением тригидрата NaOAc (34,5 г) в водном 1н растворе H2SO4 (240 мл). К реакционной смеси добавляли 1-этил-3-(N-(этилкарбамоил)-C-метилсульфанил-карбоимидоил)мочевину (соединение 10) (28,46 г, 122,5 ммоль, CB Research and Development) и перемешивали при кипячении с обратным холодильником в течение ночи (ВЭЖХ показала израсходование 99% исходного диамина). Реакционную смесь охлаждали до комнатной температуры и выливали порциями (из-за вспенивания) в перемешиваемый водный раствор насыщенного NaHCO3 (480 мл) и воды (120 мл), имеющий рН 8-9. Эту смесь перемешивали в течение 30 минут, твердое вещество собирали путем фильтрации, обильно промывали водой до нейтрального значения рН, а затем более аккуратно промывали этанолом. Твердое вещество высушивали при пониженном давлении, получая соединение 11 в виде твердого вещества не совсем белого цвета (34,48 г, выход 82%, чистота по ВЭЖХ 99,4%). ЖХ-МС: (Колонка С18, элюент 10-90% CH3CN/вода, градиент 5 мин, модификатор муравьиная кислота) M+1: 411,41 (1,73 мин). 1H ЯМР (300 МГц, MeOD) δ 9,02 (с, 2H), 7,62 (с, 1H), 7,37 (с, 1H), 5,31 (с, 1H), 4,23 (дд, J=14,5, 7,3 Гц, 1H), 4,01 (дд, J=15,0, 7,1 Гц, 1H), 3,38-3,28 (м, 2H), 2,58-2,46 (м, 1H), 2,16-2,05 (м, 2H), 2,02-1,88 (м, 1H), 1,63 (с, 6H), 1,22 (т, J=7,2 Гц, 3H) м.д.
Пример 1.i
Хиральное хроматографическое выделение 1-этил-3-[5-[2-(1-гидрокси-1-метил-этил)пиримидин-5-ил]-7-[(2R)-тетрагидрофуран-2-ил]-1H-бензимидазол-2-ил]мочевины (соединение 12)

Пробу рацемата 1-этил-3-[5-[2-(1-гидрокси-1-метил-этил)пиримидин-5-ил]-7-тетрагидрофуран-2-ил-1Н-бензимидазол-2-ил]мочевины (соединение 11) (24,60 г) разделяли на колонке CHIRALPAK® IC® (от Chiral Technologies) с использованием элюента CH2C12/MeOH/TEA (60/40/0,1) при температуре 35°С с получением целевого энантиомера (соединение 12) в виде белого твердого вещества (11,35 г, выход 45%, чистота по ВЭЖХ свыше 99%, энантиомерный избыток свыше 99%). Аналитическая хиральная ВЭЖХ: время удерживания 6,2 мин (колонка CHIRALPAK® IC® 4,6×250 мм, скорость потока 1 мл/мин, температура 30°С).
Структура и абсолютная стереохимия соединения 12 была подтверждена рентгеноструктурным анализом монокристаллов. Данные дифракции для монокристалла были получены на дифрактометре Bruker Apex II, оснащенном Cu К-альфа источником в герметичной ампуле (Cu Кα излучение, γ=1,54178 Å) и детектором Apex II CCD. Был использован кристалл размерами 1/2×0,05×0,05 мм, он был очищен с помощью минерального масла, установлен в держатель MicroMount и выровнен по центру системы в дифрактометре Bruker APEX II. Были получены три серии из 40 кадров в различной пространственной ориентации, которые обеспечили получение матрицы ориентации и начальных параметров ячейки. Окончательные параметры ячейки были получены и уточнены после сбора полного набора данных. Принимая во внимание отсутствие систематической ошибки и мощность статистики, была определена и уточнена структура, как ацентрическая пространственная группа Р21.
Набор дифракционных данных для различных пространственных ориентаций был получен с разрешением 0,9 Å, используя шаг 0,5°, при экспозиции 60 секунд для каждого кадра. Данные были получены при 100°К. Интегрирование интенсивности и уточнение параметров элементарной ячейки было осуществлено с использованием программного обеспечения APEXII. Анализ кристалла после сбора данных не показал никаких признаков разложения. Как показано на Фиг. 1, существуют две молекулы с независимой симметрией структуры, и обе молекулы с независимой симметрией представляют собой R-изомеры.
Данные по дифракции были собраны, уточнены и преобразованы с помощью программного обеспечения Apex II. Структура была установлена с помощью программного обеспечения SHELXS97 (Sheldrick, 1990); режим(ы) считывания данных и уточнения структуры выполнен при помощи программного обеспечения SHELXL97 (Sheldrick, 1997). Кристалл имеет моноклинную элементарную ячейку с пространственной группой Ρ21. Параметры решетки являются следующими: а=9,8423(4) Å, B=10,8426(3) Å, с=19,4441(7) Å, β=102,966(3)°. Объем=2022,09(12) Å3.
Пример 1.j
Получение соли метансульфоновой кислоты и 1-этил-3-[5-[2-(1-гидрокси-1-метил-этил)пиримидин-5-ил]-7-[(2R)-тетрагидрофуран-2-ил]-1H-бензимидазол-2-ил]мочевины (соединение 13)
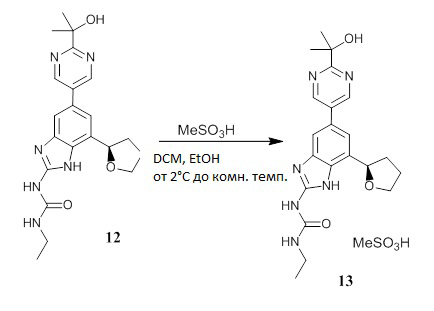
Перемешиваемую суспензию 1-этил-3-[5-[2-(1-гидрокси-1-метил-этил)пиримидин-5-ил]-7-[(2R)-тетрагидрофуран-2-ил]-1H-бензимидазол-2-ил]мочевины (соединение 12) (9,32 г, 22,71 ммоль) в абсолютном этаноле (93,2 мл) охлаждали в водяной бане со льдом. После добавления метансульфоновой кислоты (1,548 мл, 23,85 ммоль, Sigma-Aldrich 471356) охлаждающая баня была удалена, и смесь перемешивали при комнатной температуре в течение 20 минут. Смесь концентрировали в роторном испарителе при 35°С до состояния густой суспензии, разбавляли EtOAc, собирали фильтрацией твердое вещество, промывали EtOAc и высушивали при пониженном давлении с получением первой порции продукта (соединение 13) в виде белого твердого вещества (8,10 г). Фильтрат концентрировали на роторном испарителе, с получением желтоватого прозрачного вспененного вещества, которое затем растворяли в EtOH, концентрировали до состояния густой суспензии твердого вещества, растирали с EtOAc/Et2O и собирали фильтрованием. Твердое вещество промывали EtOAc/Et2O, объединяли с первой порцией и высушивали при пониженном давлении с получением соединения 13 в виде белого твердого вещества (9,89 г, выход 86%, чистота по ВЭЖХ более 99%, энантиомерный избыток 99%). Аналитическая хиральная ВЭЖХ показала присутствие одного энантиомера с временем удерживания 6,3 мин при использовании в качестве элюента CH2C12/MeOH/TEA (60/40/0,1) на колонке CHIRALPAK ® IC® 4,6×250 мм при скорости потока 1 мл/мин, при температуре 30°С. ЖХ-МС: (колонка С18, элюент 10-90% CH3CN/вода, градиент 5 мин, модификатор муравьиная кислота) M+1: 411,53 (1,74 мин). 1H ЯМР (300 МГц, MeOD) δ 9,07 (с, 2H), 7,79 (с, 1H), 7,62 (с, 1H), 5,30 (т, J=7,3 Гц, 1H), 4,24 (дд, J=14,6, 7,3 Гц, 1H), 4,04 (дд, J=15,0, 7,6 Гц, 1H), 3,40-3,30 (м, 2H), 2,72 (с, 3H), 2,65-2,54 (м, 1H), 2,20-2,07 (м, 2H), 2,04-1,90 (м, 1H), 1,64 (с, 6H), 1,23 (т, J=7,2 Гц, 3H) м.д.
Пример 1.1
Снятие защиты/реакция Suzuki без выделения промежуточных продуктов.
Получение 2-[5-(4-амино-3-нитро-5-тетрагидрофуран-2-ил-фенил)пиримидин-2-ил]пропан-2-ола (соединение 8)
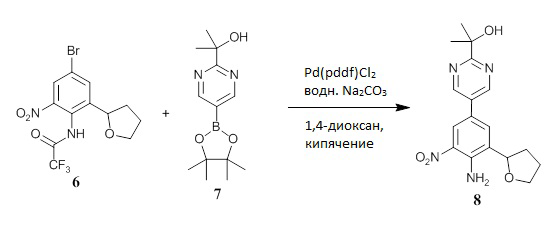
Смешивали N-(4-бром-2-нитро-6-тетрагидрофуран-2-ил-фенил)-2,2,2-трифтор-ацетамид (соединение 6) (19,00 г, 49,59 ммоль), 2-[5-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)пиримидин-2-ил]пропан-2-ол (соединение 7) (14,41 г, 54,55 ммоль), водный 2,7М раствор карбоната натрия (73,48 мл, 198,4 ммоль) и 1,4-диоксан (190 мл, Sigma-Aldrich 360481). Через перемешиваемую смесь в течение 40 минут барботировали азот, с последующим добавлением аддукта [1,1’-бис(дифенилфосфино)ферроцен]дихлорпалладия с дихлорметаном (2,025 г, 2,480 ммоль, Strem 460450). Реакционную смесь перемешивали при кипячении с обратным холодильником под азотом в течение 7 часов, затем добавили еще 50 мл насыщенного водного раствора карбоната натрия и еще кипятили с обратным холодильником в течение 16 часов. Реакционную смесь оставляли охлаждаться, затем ее разбавляли EtOAc (500 мл) и водой (200 мл). Слои разделяли, и водную фазу экстрагировали с помощью EtOAc (200 мл). Объединенную органическую фазу промывали водой (500 мл), насыщенным солевым раствором (500 мл), высушивали над Na2SO4, фильтровали через твердый фильтр Florisil® и концентрировали на роторном испарителе с получением сырого продукта (8) в виде масла оранжевого цвета. Полученный продукт очищали с помощью хроматографии на силикагеле ISCO, используя в качестве элюента 20-90% EtOAc/гексан, с получением соединения 8, в виде твердого вещества оранжевого цвета (15,00 г, чистота 81-88%). ЖХ-МС: (колонка С18, элюент 10-90% CH3CN/вода, градиент 5 мин, модификатор муравьиная кислота) M+1: 345,35 (2,68 мин). 1H ЯМР (300 МГц, CDCl3) δ 8,88 (с, 2H), 8,36 (д, J=2,2 Гц, 1H), 7,56 (д, J=2,1 Гц, 1H), 7,09 (с, 2H), 4,92 (т, J=7,2 Гц, 1H), 4,62 (с, 1H), 4,20-4,11 (м, 1H), 4,03-3,94 (м, 1H), 2,39-2,26 (м, 1H), 2,23-2,08 (м, 3H), 1,64 (с, 6H) м.д.
Пример 2
Схема синтеза соединений 22, 23 и 24
Схема 3 представляет собой схему способа получения соединений 22, 23 и 24.
Схема 3

Пример 2.a
Получение 2-(2-фтор-6-нитро-фенил)-2,3-дигидрофурана (соединение 15A) и 2-(2-фтор-6-нитро-фенил)-2,5-дигидрофурана (соединение 15B)
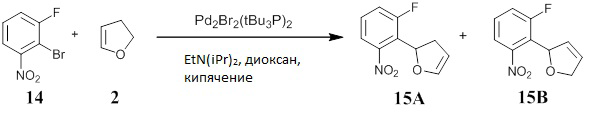
В реакционную колбу загружали 2-бром-1-фтор-3-нитробензол (соединение 14) (200,3 г, 98%, 892,3 ммоль, Bosche F6657), 1,4-диоксан (981,5 мл, Sigma-Aldrich 360481) и 2,3-дигидрофуран (соединение 2) (341,1 мл, 99%, 4,462 моль, Aldrich 200018), а затем димер N,N-диизопропилэтиламина (155,4 мл, 892,3 ммоль, Sigma-Aldrich 550043) и бром(три-трет-бутилфосфин)палладия(I) (6,936 г, 8,923 ммоль, Johnson Matthey С4099). Полученную смесь перемешивали при кипячении с обратным холодильником в течение 2 часов (ВЭЖХ показала израсходование 98% исходного арилбромида). Реакционной смеси давали охладиться, после чего осадок удаляли фильтрованием, промывали EtOAc, и фильтрат концентрировали в вакууме до получения темного красновато-коричневого полутвердого маслянистого вещества. Полутвердое маслянистое вещество растворяли в CH2Cl2, элюировали через твердый фильтр из двуокиси кремния с CH2Cl2 и концентрировали в вакууме с получением смеси соединений 15А и 15В в виде темного масла янтарного цвета (291,3 г). Сырой продукт использовали далее без дополнительной очистки. Основной продукт представлял собой 2-(2-фтор-6-нитро-фенил)-2,3-дигидрофуран (соединение 15A) (96%). ЖХ-МС: (колонка С18, элюент 10-90% CH3CN/вода, градиент 5 мин, модификатор муравьиная кислота) M+1: 210,23 (3,13 мин). 1H ЯМР (300 МГц, CDCl3) δ 7,54 (дт, J=8,0, 1,2 Гц, 1H), 7,43 (тд, J=8,2, 5,2 Гц, 1H), 7,32 (ддд, J=9,7, 8,3, 1,3 Гц, 1H), 6,33 (дд, J=4,9, 2,4 Гц, 1H), 5,80 (т, J=10,9 Гц, 1H), 5,06 (кв, J=2,4 Гц, 1H), 3,18-3,07 (м, 1H), 2,94-2,82 (м, 1H) м.д.
Минорный продукт представлял собой 2-(2-фтор-6-нитрофенил)-2,5-дигидрофуран (соединение 15B) (4%): ГХ-МС: (колонка Agilent HP-5MS 30 м×250 мкм×0,25 мкм, нагрев от 60°С до 300°С в течение 2 мин через 15 мин, скорость потока 1 мл/мин) M+1: 210 (11,95 мин). 1H ЯМР (300 МГц, CDCl3) δ 7,47 (д, J=8,0 Гц, 1H), 7,43-7,34 (м, 1H), 7,30-7,23 (м, 1H), 6,21-6,15 (м, 1H), 6,11-6,06 (м, 1H), 5,97-5,91 (м, 1H), 4,89-4,73 (м, 2H) м.д.
Пример 2.b
Получение 3-фтор-2-тетрагидрофуран-2-ил-анилина (соединение 16)

В сосуд Парра в атмосфере азота помещали 5%-ый палладий на угле (37,3 г, влажность 50%, 8,76 ммоль, Aldrich 330116), а затем MeOH (70 мл, JT-Baker 909333). Затем в сосуд Парра добавляли сырую смесь 2-(2-фтор-6-нитро-фенил)-2,3-дигидрофурана и 2-(2-фтор-6-нитро-фенил)-2,5-дигидрофурана (соединения 15А и 15В) (186,6 г, 892,1 ммоль), растворенную в МеОН (117 мл), после чего добавили NEt3 (124,3 мл, 892,1 ммоль, Sigma-Aldrich 471283). Сосуд помещали в шейкер Парра и насыщали водородом. Водород добавляли до давления 45 фунтов на квадратный дюйм (3,2 атм.), затем сосуд подвергали встряхиванию до тех пор, пока исходный материал не был полностью израсходован (ВЭЖХ и ЖХ-МС показали завершение реакции). Реакционную смесь продували азотом, отфильтровали через Celite™ и промывали этилацетатом. Фильтрат концентрировали на роторном испарителе с получением продукта в виде коричневого масла, которое растворяли в Et2O и промывали водой (2х). Эфирную фазу экстрагировали водным 1н раствором HCl (5×250 мл), которую промывали Et2O (3х) и затем подщелачивали водным 6н раствором NaOH до величины рН 12-14. Основную водную фазу экстрагировали дихлорметаном (CH2Cl2, 4x), и объединенный органический экстракт промывали насыщенным водным раствором NH4C1, высушивали над MgSO4 и фильтровали через слой двуокиси кремния, с элюированием продукта с помощью CH2Cl2 в 25% EtOAc/гексан. Целевой фильтрат концентрировали при пониженном давлении, получая продукт 16 в виде светло-коричневого масла (121,8 г, чистота по ГХ-МС с ЯМР 84%). ГХ-МС: (колонка Agilent HP-5MS 30 м×250 мкм×0,25 мкм, нагрев от 60°С до 300°С в течение 2 мин через 15 мин, скорость потока 1 мл/мин) M+1: 182,0 (11,44 мин). ЖХ-МС: (колонка С18, элюент 10-90% CH3CN/вода, градиент 5 мин, модификатор муравьиная кислота) M+1: 182,10 (2,61 мин). 1H ЯМР (300 МГц, CDCl3) δ 6,97 (тд, J=8,1, 6,3 Гц, 1H), 6,43-6,35 (м, 2H), 5,21-5,13 (м, 1H), 4,54 (с, 2H), 4,16-4,07 (м, 1H), 3,90-3,81 (м, 1H), 2,23-2,00 (м, 4H) м.д.
Дополнительные продукты были получены следующим образом: объединенную эфирную фазу промывали насыщенным водным раствором NaHCO3, насыщенным солевым раствором, высушивали над Na2SO4, декантировали и концентрировали при пониженном давлении. Полученное масло перегоняли под вакуумом (приблизительно 15 торр), собирали дистиллята при температуре 101-108°С. К перемешиваемому раствору перегнанного масла в EtOH (1 объем) при 2°С медленно добавляли 5М раствор HCl (1 экв) в iPrOH. Полученную суспензию нагревали до комнатной температуры, разбавляли EtOAc (3 объема, об./об.) и перемешивали в течение 2 часов. Белое твердое вещество собирали фильтрованием, промывали EtOAc и высушивали при пониженном давлении с получением второй порции продукта в виде HCl соли. Маточный раствор концентрировали до получения суспензии, разбавляли этилацетатом, и твердое вещество собирали фильтрованием, промывали EtOAc и высушивали в вакууме с получением третьей порции продукта в виде HCl соли. ЖХ-МС: (колонка С18, элюент 10-90% CH3CN/вода, градиент 5 мин, модификатор муравьиная кислота) M+1: 182,10 (2,58 мин). 1H ЯМР (300 МГц, CDCl3) δ 10,73 (уш. с, 3H), 7,66 (д, J=8,1 Гц, 1H), 7,33 (тд, J=8,2, 5,9 Гц, 1H), 7,13-7,05 (м, 1H), 5,26 (дд, J=9,0, 6,5 Гц, 1H), 4,38-4,28 (м, 1H), 4,00-3,91 (м, 1H), 2,59-2,46 (м, 1H), 2,30-1,95 (м, 3H) м.д. Общий выход продуктов в третьей порции составил 76%.
Пример 2.c
Получение 4-бром-3-фтор-2-тетрагидрофуран-2-ил-анилина (соединение 17).

К перемешиваемому раствору 3-фтор-2-тетрагидрофуран-2-ил-анилина (соединение 16) (131,9 г, 92%, 669,7 ммоль) в смеси метил-трет-бутилового эфира (1,456 л) и ацетонитрила (485 мл), охлажденному до -20°С, добавляли тремя порциями N-бромсукцинимид (NBS, 120,4 г, 99%, 669,7 ммоль, Aldrich B81255), поддерживая температуру приблизительно ниже -15°С. После завершения добавления ингредиентов, перемешивание продолжали при температуре от -15 до -10°С в течение 30 минут. 1H-ЯМР реакционной аликвоты показал израсходование 96% исходного анилина. К реакционной смеси добавляли еще одну порцию NBS (4,82 г) и перемешивали при температуре -10°С в течение 30 минут. К реакционной смеси добавляли водный 1н раствор Na2S2O3 (670 мл). Холодную баню удаляли, смесь перемешивали в течение 20 минут и затем разбавляли этилацетатом. Слои разделяли. Органическую фазу промывали насыщенным водным раствором NaHCO3 (2x), водой и насыщенным солевым раствором, высушивали над Na2SO4, декантировали и концентрировали при пониженном давлении с получением темно-янтарного масла. Остаток разбавляли гексаном и элюировали через тонкий слой двуокиси кремния при помощи смеси 25%-50% EtOAc/гексан. Целевой фильтрат концентрировали в вакууме, получая соединение 17 в виде темного масла янтарного цвета (182,9 г, выход 90%, чистота по ЯМР 86%). ЖХ-МС: (колонка С18, элюент 10-90% AcN/вода, градиент 5 мин, модификатор муравьиная кислота) M+1: 260,12 (3,20 мин). 1H ЯМР (300 МГц, CDCl3) δ 7,15 (дд, J=8,6, 7,6 Гц, 1H), 6,30 (дд, J=8,7, 1,3 Гц, 1H), 5,19-5,12 (м, 1H), 4,58 (с, 2H), 4,16-4,07 (м, 1H), 3,90-3,81 (м, 1H), 2,23-1,99 (м, 4H) м.д.
Пример 2.d
Получение N-(4-бром-3-фтор-6-нитро-2-тетрагидрофуран-2-ил-фенил)-2,2,2-трифтор-ацетамида (соединение 18).

К трифторуксусному ангидриду (565,3 мл, 4,067 моль, Sigma-Aldrich 106232), перемешиваемому при 2°С, медленно добавляли в течение 20 минут через капельную воронку чистый 4-бром-3-фтор-2-тетрагидрофуран-2-ил-анилин (соединение 17) (123,0 г, 86%, 406,7 ммоль) в виде густого масла (температура реакции возрастала до 13°С). Оставшееся масло ополаскивали безводным THF (35 мл) и вводили в реакционную смесь. Холодную баню удаляли, и реакционную смесь нагревали до 35°С, а затем в течение 2,5 часов порциями добавляли NH4NO3 (4,88 г, 20 порций, 1,22 моль, Sigma-Aldrich A7455), поддерживая температуру реакции между 30 и 41°С, с использованием водяной бани со льдом для контролирования экзотермической реакции только по мере необходимости. После завершения добавления реакционную смесь перемешивали в течение еще 10 минут (ВЭЖХ показала полноту реакции на 99%). Реакционную смесь медленно выливали в измельченный лед (1,23 кг) и перемешивали в течение 1 часа для того, чтобы получить фильтруемый твердый осадок, который собирали и промывали водой и, аккуратно, насыщенным водным раствором NaHCO3, а затем снова водой (до рН 7). Полученный продукт высушивали в конвекционной печи в течение ночи при 40°С, а затем при пониженном давлении в печи при 50°С в течение ночи с получением соединения 18 в виде твердого вещества бежевого цвета (152,5 г, выход 90%; чистота по ВЭЖХ 96%). ЖХ-МС: (колонка С18, элюент 10-90% CH3CN/вода, градиент 5 мин, модификатор муравьиная кислота) M+1: 401,30 (3,41 мин). 1H ЯМР (300 МГц, CDCl3) δ 10,56 (с, 1H), 8,19 (д, J=6,6 Гц, 1H), 5,22 (дд, J=10,3, 6,4 Гц, 1H), 4,22 (дд, J=15,8, 7,2 Гц, 1H), 3,99 (дд, J=16,1, 7,5 Гц, 1H), 2,50-2,38 (м, 1H), 2,22-2,11 (м, 2H), 1,86-1,71 (м, 1H) м.д.
Пример 2.e
Получение 4-бром-3-фтор-6-нитро-2-тетрагидрофуран-2-ил-анилина (соединение 19).

В реакционную колбу загружали N-(4-бром-3-фтор-6-нитро-2-тетрагидрофуран-2-ил-фенил)-2,2,2-трифтор-ацетамид (соединение 18) (242,3 г, 604,1 ммоль), 1,4-диоксан (1,212 л) и водный 2М раствор серной кислоты (362,4 мл, 724,9 ммоль), и смесь кипятили с обратным холодильником при перемешивании в течение 5 дней (ВЭЖХ показала 98% конверсии). Реакционную смесь охлаждали, разбавляли этилацетатом, нейтрализовывали насыщенным водным раствором NaHCO3, разделяли слои, и водную фазу повторно экстрагировали этилацетатом (2х). Объединенные органические фазы промывали концентрированным солевым раствором (2х), высушивали над MgSO4, фильтровали и концентрировали в вакууме, получая соединение 19 в виде твердого вещества зеленовато-коричневого цвета (181,7 г, выход 94%, чистота по ВЭЖХ 95%). Продукт использовали на следующей стадии без дополнительной очистки. ЖХ-МС: (колонка С18, элюент 10-90% CH3CN/вода, градиент 5 мин, модификатор муравьиная кислота) M+1: 305,20 (3,63 мин). 1H ЯМР (300 МГц, CDCl3) δ 8,35 (д, J=7,3 Гц, 1H), 7,45 (с, 2H), 5,23-5,16 (м, 1H), 4,23-4,14 (м, 1H), 3,93-3,84 (м, 1H), 2,31-1,96 (м, 4H) м.д.
Пример 2.f
Получение 2-[5-(4-амино-2-фтор-5-нитро-3-тетрагидрофуран-2-ил-фенил)-2-пиримидин-2-ола (соединение 20).

К перемешиваемому раствору 4-бром-3-фтор-6-нитро-2-тетрагидрофуран-2-ил-анилина (соединение 19) (525,0 г, 1,721 моль, Bridge Organics Co.) в 1,4-диоксане (4,20 л, Sigma-Aldrich 360481) добавляли водный 1,2М раствор NaHCO3 (4,302 л, 5,163 моль). Ток азота барботировали через перемешиваемую смесь в течение 2 часов, затем добавляли 2-[5-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)пиримидин-2-ил]пропан-2-ол (соединение 7) (545,4 г, 2,065 моль, Bridge Organics Co.) и аддукт [1,1’-бис(дифенилфосфино)ферроцен]дихлорпалладия с дихлорметаном (42,16 г, 51,63 ммоль, Strem 460450). Реакционную смесь перемешивали при кипячении с обратным холодильником в течение ночи, охлаждали, разбавляли EtOAc (8,4 л) и разделяли слои. Органическую фазу промывали насыщенным водным раствором NH4C1 и затем насыщенным солевым раствором. Водную фазу повторно экстрагировали этилацетатом (4 л), и этот органический экстракт промывали насыщенным солевым раствором. Объединенную органическую фазу высушивали над MgSO4, фильтровали через короткий плотный фильтр Florisil®, элюировали с помощью EtOAc, и фильтрат концентрировали на роторном испарителе с получением влажного темно-коричневого твердого вещества. Его растворяли в CH2Cl2, помещали на фильтр из силикагеля, элюировали гексаном, затем смесью 25% EtOAc/гексан и затем 50%EtOAc/гексан. Целевой фильтрат концентрировали на роторном испарителе до состояния густой суспензии, твердое вещество собирали путем фильтрации, растирали с МТВЕ и высушивали в вакууме, получая соединение 20 в виде твердого вещества ярко-желтого цвета (выход 55,8%, чистота по ВЭЖХ 90-97%). Фильтрат концентрировали, и повторением указанной выше очистки получали вторую порцию соединения 20 в виде твердого вещества ярко-желтого цвета (выход 19,7%). Фильтрат снова концентрировали с получением темно-коричневого масла, и оно было загружено на колонку с силикагелем с толуолом и минимальным количеством CH2Cl2. Продукт элюировали смесью EtOAc/гексан (от 0% до 50%). Целевые фракции концентрировали до состояния густой суспензии и разбавляли смесью МТВЕ/гексан. Твердое вещество собирали путем фильтрации и промывали минимальным количеством МТВЕ с получением третьей порции соединения 20 в виде твердого вещества ярко-желтого цвета (выход 4,9%), с общим выходом 80% из трех порций. ЖХ-МС: (колонка С18, элюент 10-90% CH3CN/вода, градиент 5 мин, модификатор муравьиная кислота) M+1: 363,48 (2,95 мин). 1H ЯМР (300 МГц, CDCl3) δ 8,84 (д, J=1,6 Гц, 2H), 8,27 (д, J=8,0 Гц, 1H), 7,62 (с, 2H), 5,31-5,24 (м, 1H), 4,63 (с, 1H), 4,27-4,18 (м, 1H), 3,97-3,87 (м, 1H), 2,33-2,05 (м, 4H), 1,64 (с, 6H) м.д.
Пример 2.g
Получение 2-[5-(4,5-диамино-2-фтор-3-тетрагидрофуран-2-ил-фенил)пиримидин-2-ил]-2-ола (соединение 21).

В сосуд Парра, в атмосфере азота, помещали 5%-ый палладий на угле (14,21 г, 50% влажность, 3,339 ммоль, Aldrich 330116), а затем добавляли MeOH (242 мл, JT-Baker 909333) и NEt3 (46,54 мл; 333,9 ммоль, Sigma-Aldrich 471283). В горячем THF (360 мл) растворяли 2-[5-(4-амино-2-фтор-5-нитро-3-тетрагидрофуран-2-ил-фенил)пиримидин-2-ил]пропан-2-ол (соединение 20) (121,0 г, 333,9 ммоль), давали охладиться, и полученный раствор добавляли к реакционной смеси, остаточное количество соединения 20 смывали другой порцией THF (124 мл). Сосуд помещали в шейкер Парра и насыщали водородом. После добавления водорода до давления 45 фунтов на квадратный дюйм (3,2 атм.) сосуд подвергали встряхиванию до израсходования в реакции соединения 20 (ВЭЖХ и ЖХ-МС показали полное завершение реакции). Реакционную смесь продували азотом, фильтровали через Celite™ и промывали EtOAc. Продукт вновь фильтровали через бумагу (микростекловолокно), и фильтрат концентрировали в вакууме. Реакцию повторяли три раза в том же масштабе, и порции продукта были объединены с получением соединение 21 в виде твердого вещества коричневого цвета (447 г, выход 99%, чистота по ВЭЖХ 93%). ЖХ-МС: (колонка С18, элюент 10-90% CH3CN/вода, градиент 5 мин, модификатор муравьиная кислота) M+1: 333,46 (1,79 мин). 1H ЯМР (300 МГц, CDCl3) δ 8,81 (д, J=1,4 Гц, 2H), 6,69 (д, J=7,3 Гц, 1H), 5,27-5,20 (м, 1H), 4,73 (с, 1H), 4,70 (с, 2H), 4,23-4,14 (м, 1H), 3,94-3,86 (м, 1H), 3,22 (с, 2H), 2,32-2,22 (м, 1H), 2,18-1,99 (м, 3H), 1,63 (с, 6H) м.д.
Пример 2.h
Получение 1-этил-3-[6-фтор-5-[2-(1-гидрокси-1-метил-этил)пиримидин-5-ил]-7-тетрагидрофуран-2-ил-1Н-бензимидазол-2-ил]мочевины (соединение 22)
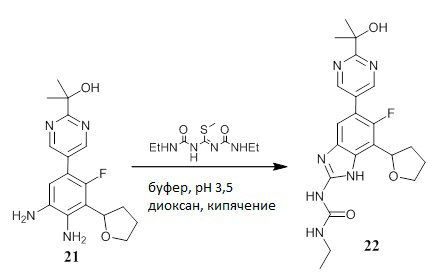
К перемешиваемой суспензии 2-[5-(4,5-диамино-2-фтор-3-тетрагидрофуран-2-ил-фенил)пиримидин-2-ил]пропан-2-ола (соединение 21) (111,3 г, 334,9 ммоль) и 1,4-диоксана (556,5 мл, Sigma-Aldrich 360481) добавляли 1-этил-3-(N-(этилкарбамоил)-C-метилсульфанил-карбонимидоил)мочевину (соединение 10) (93,36 г; 401,9 ммоль, CB Research and Development), а затем буфер рН 3,5 (1,113 л), полученный путем растворения тригидрата NaOAc (158,1 г) в водном 1н растворе H2SO4 (1,100 л). Реакционную смесь перемешивали при кипячении с обратным холодильником в течение ночи (ВЭЖХ показала полную конверсию), охлаждали до комнатной температуры и выливали порциями (для уменьшения вспенивания) к перемешиваемому насыщенному водному раствору NaHCO3 (2,23 л) с достижением значения рН 8-9. Полученную смесь перемешивали в течение 30 минут, твердое вещество собирали путем фильтрации, обильно промывали водой до нейтрального значения рН, а затем более аккуратно промывали EtOH. Твердое вещество высушивали при пониженном давлении, получая соединение 22 в виде твердого вещества не совсем бело-желтоватого цвета (135,2 г, выход 94%, чистота по ВЭЖХ 99%). ЖХ-МС: (колонка С18, элюент 10-90% CH3CN/вода, градиент 5 мин, модификатор муравьиная кислота) M+1: 429,58 (2,03 мин). 1H ЯМР (300 МГц, MeOD) δ 8,95 (д, J=1,6 Гц, 2H), 7,45 (д, J=6,5 Гц, 1H), 5,38 (уш. с, 1H), 4,27 (дд, J=14,9, 7,1 Гц, 1H), 4,01 (дд, J=15,1, 7,0 Гц, 1H), 3,37-3,29 (м, 2H), 2,55 (уш. с, 1H), 2,19-2,07 (м, 2H), 2,02-1,82 (уш. с, 1H), 1,63 (с, 6H), 1,21 (т, J=7,2 Гц, 3H) м.д.
Пример 2.i
Хиральное хроматографическое выделение 1-этил-3-[6-фтор-5-[2-(1-гидрокси-л-метил-этил)пиримидин-5-ил]-7-[(2R)-тетрагидрофуран-2-ил]-1Н-бензимидазол-2-ил]мочевины (соединение 23)
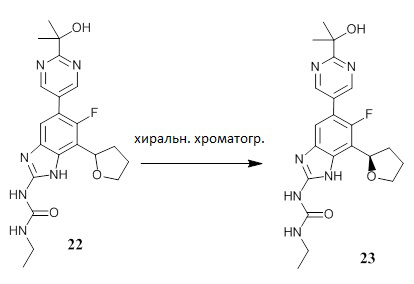
Рацемическую пробу 1-этил-3-[6-фтор-5-[2-(1-гидрокси-1-метил-этил)пиримидин-5-ил]-7-тетрагидрофуран-2-ил-1Н-бензимидазол-2-ил]мочевины (соединение 22) (133,60 г) разделяли на колонке CHIRALPAK® IC® (от Chiral Technologies), с использованием в качестве элюента смесь CH2Cl2/MeOH/TEA (60/40/0,1) при температуре 25°С, с получением целевого энантиомера (соединение 23) в виде белого твердого вещества (66,8 г, выход 45%, чистота по ВЭЖХ 99,8%, энантиомерный избыток 99%). Аналитическая хиральная ВЭЖХ: время удерживания 7,7 мин (колонка CHIRALPAK® IC®, 4,6×250 мм, скорость потока 1 мл/мин, температура 30°С). Твердое вещество суспендировали в смеси 2:1 EtOH/ Et2O (5 объемов), перемешивали в течение 10 минут, собирали фильтрацией, промывали смесью 2:1 EtOH/Et2O и высушивали при пониженном давлении, получая белое твердое вещество (60,6 г ).
Структура и абсолютная стереохимия соединения 23 была подтверждена рентгеноструктурным анализом монокристаллов. Данные дифракции для монокристалла были получены на дифрактометре Bruker Apex II, оснащенном Cu К-альфа источником в герметичной ампуле (Cu Кα излучение, γ=1,54178 Å) и детектором Apex II CCD. Был использован кристалл размерами 0,15×0,15×0,10 мм, он был очищен с помощью минерального масла, установлен в держатель MicroMount и выровнен по центру системы в дифрактометре Bruker APEX II. Были получены три серии из 40 кадров в различной пространственной ориентации, которые обеспечили получение матрицы ориентации и начальных параметров ячейки. Окончательные параметры ячейки были получены и уточнены после сбора полного набора данных. Принимая во внимание отсутствие систематической ошибки и мощность статистики, была определена и уточнена структура, как ацентрическая пространственная группа Р21.
Набор дифракционных данных для различных пространственных ориентаций был получен с разрешением 0,85 Å, используя шаг 0,5°, при экспозиции 60 секунд для каждого кадра. Данные были получены при 100°К.
Интегрирование интенсивности и уточнение параметров элементарной ячейки было осуществлено с использованием программного обеспечения APEXII. Анализ кристалла после сбора данных не показал никаких признаков разложения. Как показано на Фиг. 2, существуют две молекулы с независимой симметрией структуры, и обе молекулы с независимой симметрией представляют собой R-изомеры.
Данные по дифракции были собраны, уточнены и преобразованы с помощью программного обеспечения ApexII. Структура была установлена с помощью программного обеспечения SHELXS97 (Sheldrick, 1990); режим(ы) считывания данных и уточнения структуры выполнен при помощи программного обеспечения SHELXL97 (Sheldrick, 1997). Кристалл имеет моноклинную элементарную ячейку с пространственной группой Ρ21. Параметры решетки являются следующими: а=9,9016(2) Å, b=10,9184(2) Å, с=19,2975(4) Å, β=102,826(1)°. Объем=2034,19(7) Å3.
Пример 2.j
Получение соли метансульфоновой кислоты и 1-этил-3-[6-фтор-5-[2-(1-гидрокси-1-метил-этил)пиримидин-5-ил]-7-[(2R)-тетрагидрофуран-2-ил]-1Н-бензимидазол-2-ил]мочевины (соединение 24)

К перемешиваемой суспензии 1-этил-3-[6-фтор-5-[2-(1-гидрокси-1-метил-этил)пиримидин-5-ил]-7-[(2R)-тетрагидрофуран-2-ил]-1Н-бензимидазол-2-ил]мочевины (соединение 23) (15,05 г, 35,13 ммоль) в дихлорметане (60 мл, JT-Baker 931533) и абсолютном этаноле (15 мл, Pharmco-AAPER 111000200) добавляли метансульфоновую кислоту (2,392 мл, 36,89 ммоль, Sigma-Aldrich 471356). Перемешивали при комнатной температуре до получения визуально наблюдаемого прозрачного раствора. Медленно, в течение приблизительно 1 часа, добавляли гептан (300 мл), и затем, путем фильтрации, собирали твердый осадок (используя бумагу из стекломикроволокна Whatman GF/F, с помещенной на ней сверху бумагу Whatman #3). Высушивали при пониженном давлении в вакуумной печи (обезвоживание было выполнено над сульфатом кальция и гидроксидом калия) в течение ночи при 40°С, с получением соединения 24 в виде белого твердого вещества (13,46 г, чистота по ВЭЖХ более 99%, энантиомерный избыток 99%). Аналитическая хиральная ВЭЖХ показала присутствие одного энантиомера с временем удерживания 8,6 мин при элюции с помощью смеси CH2Cl2/MeOH/TEA (60/40/0,1) со скоростью потока 1 мл/мин на колонке CHIRALPAK® IC® 4,6×250 мм при температуре 30°С. Вторую порцию белого твердого продукта 24 (4,36 г, чистота по ВЭЖХ 98%, энантиомерный избыток более 99%) получали из фильтрата. ЖХ-МС: (колонка С18, элюент 10-90% CH3CN/вода, градиент 5 мин, модификатор муравьиная кислота) M+1: 429,58 (2,03 мин). 1H ЯМР (300 МГц, MeOD) δ 9,00 (д, J=1,6 Гц, 2H), 7,67 (д, J=6,1 Гц, 1H), 5,39 (т, J=7,7 Гц, 1H), 4,30 (дд, J=14,9, 6,9 Гц, 1H), 4,03 (дд, J=14,8, 7,7 Гц, 1H), 3,40-3,31 (м, 2H), 2,72 (с, 3H), 2,70-2,60 (м, 1H), 2,21-2,08 (м, 2H), 1,98-1,84 (м, 1H), 1,65 (с, 6H), 1,22 (т, J=7,2 Гц, 3H) м.д.
| название | год | авторы | номер документа |
|---|---|---|---|
| МОДУЛЯТОРЫ АТФ-СВЯЗЫВАЮЩИХ ТРАНСПОРТЕРОВ | 2010 |
|
RU2552353C2 |
| ПРОИЗВОДНЫЕ 2-УРЕИДО-6-ГЕТЕРОАРИЛ-3Н-БЕНЗИМИДАЗОЛ-6-КАРБОНОВОЙ КИСЛОТЫ И РОДСТВЕННЫЕ СОЕДИНЕНИЯ В КАЧЕСТВЕ ИНГИБИТОРОВ ГИРАЗЫ И/ИЛИ ТОПОИЗОМЕРАЗЫ IV ДЛЯ ЛЕЧЕНИЯ БАКТЕРИАЛЬНЫХ ИНФЕКЦИЙ | 2003 |
|
RU2333208C2 |
| СПОСОБЫ ПОЛУЧЕНИЯ ПРОИЗВОДНЫХ ДИГИДРО-2,3-БЕНЗОДИАЗЕПИНА И ПРОМЕЖУТОЧНЫЕ СОЕДИНЕНИЯ | 1995 |
|
RU2151149C1 |
| СПОСОБЫ ПОЛУЧЕНИЯ РАЦЕМИЧЕСКОГО АМИНОЗАМЕЩЕННОГО 5,6,7,8-ТЕТРАГИДРОХИНОЛИНА ИЛИ РАЦЕМИЧЕСКОГО АМИНОЗАМЕЩЕННОГО 5,6,7,8-ТЕТРАГИДРОИЗОХИНОЛИНА, СПОСОБЫ ИХ РАЗДЕЛЕНИЯ И РАЦЕМИЗАЦИИ, СПОСОБ ПОЛУЧЕНИЯ КЕТОЗАМЕЩЕННОГО 5,6,7,8-ТЕТРАГИДРОХИНОЛИНА ИЛИ КЕТОЗАМЕЩЕННОГО 5,6,7,8-ТЕТРАГИДРОИЗОХИНОЛИНА, СПОСОБ ПОЛУЧЕНИЯ ЭНАНТИОМЕРА КОНДЕНСИРОВАННОГО БИЦИКЛИЧЕСКОГО КОЛЬЦА, ЗАМЕЩЕННОГО ПЕРВИЧНЫМ АМИНОМ, ПРОИЗВОДНЫЕ 5,6,7,8-ТЕТРАГИДРОХИНОЛИНА | 2002 |
|
RU2308451C2 |
| ПЕРВИЧНЫЕ КАРБОКСАМИДЫ В КАЧЕСТВЕ ИНГИБИТОРОВ BТK | 2014 |
|
RU2708395C2 |
| ТРИЦИКЛИЧЕСКИЕ ПИРРОЛО ПРОИЗВОДНЫЕ, СПОСОБ ИХ ПОЛУЧЕНИЯ И ИХ ПРИМЕНЕНИЕ В КАЧЕСТВЕ ИНГИБИТОРОВ КИНАЗЫ | 2012 |
|
RU2591191C2 |
| ПИРИМИДИНОВЫЕ ИНГИБИТОРЫ ГИРАЗЫ И ТОПОИЗОМЕРАЗЫ IV | 2012 |
|
RU2609259C2 |
| СОЕДИНЕНИЯ ДЛЯ ЛЕЧЕНИЯ НЕКОТОРЫХ ЛЕЙКОЗОВ | 2019 |
|
RU2804709C2 |
| ПРОИЗВОДНЫЕ ПИРРОЛИДИНА | 2016 |
|
RU2740019C1 |
| МОДУЛЯТОРЫ ТРАНСПОРТЕРОВ АТФ-СВЯЗЫВАЮЩЕЙ КАССЕТЫ | 2005 |
|
RU2528046C2 |


Данная заявка относится к способу получения соединений формулы (I)

или их фармацевтически приемлемых солей, где R представляет собой H или F и каждый из R3, R4, и R5 независимо представляет собой С1-С6 алкил или гидроксильную группу, а также к промежуточным соединениям и способам их получения. Соединения формулы (I) или их соли ингибируют бактериальную гиразу и/или топоизомеразу IV и полезны при лечении бактериальных инфекций. 4 н. и 14 з.п. ф-лы, 2 ил., 2 пр.
1. Способ получения соединения формулы (I)

или его фармацевтически приемлемой соли,
где R представляет собой Н или F; и каждый из R3, R4 и R5 независимо представляет собой C1-С6 алкил или гидроксильную группу;
где способ включает
взаимодействие фенилпиримидинового соединения формулы (II)
 ,
,
где R представляет собой Н или F, и каждый из R3, R4 и R5 независимо представляет собой C1-С6 алкил или гидроксильную группу;
с производным мочевины формулы А или В:
 ,
,
 ,
,
где R6 представляет собой необязательно замещенный алкил, необязательно замещенный арил, необязательно замещенный насыщенный или ненасыщенный карбоцикл или необязательно замещенный насыщенный или ненасыщенный гетероцикл, с получением соединения формулы (I),
и необязательно взаимодействие соединения формулы (I) с подходящей кислотой с получением фармацевтически приемлемой соли соединения формулы (I).
2. Способ по п. 1, в котором R6 представляет собой метил, этил, бензил или п-нитробензил.
3. Способ по п. 1, в котором указанную реакцию проводят в смеси диоксана и буфера при температуре от 75°С до 125°С.
4. Способ по п. 3, в котором буфер представляет собой буфер с рН 3,5 и реакцию проводят при температуре дефлегмации.
5. Способ по п. 1, в котором фенилпиримидиновое соединение формулы (II) получают путем взаимодействия фенилтетрагидрофуранового производного формулы (IV)
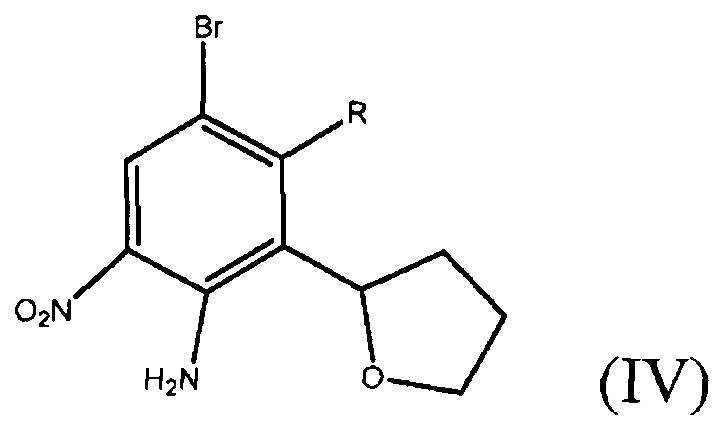 ,
,
где R представляет собой Н или F,
с производным бороновой кислоты формулы (III)
 ,
,
где каждый из R3, R4 и R5 независимо представляет собой C1-С6 алкил или гидроксильную группу и В* представляет  , где каждый из R1 и R2 независимо представляет собой алкил или Н, или OR1 и OR2 вместе с атомом В, к которому они присоединены, образуют необязательно замещенное 5-, 6- или 7-членное кольцо или BF3X, где X представляет собой любой одновалентный катион,
, где каждый из R1 и R2 независимо представляет собой алкил или Н, или OR1 и OR2 вместе с атомом В, к которому они присоединены, образуют необязательно замещенное 5-, 6- или 7-членное кольцо или BF3X, где X представляет собой любой одновалентный катион,
в присутствии палладиевого катализатора в полярном растворителе с получением фенилпиримидинового соединения формулы (V)
 ,
,
где R представляет собой Н или F, и каждый из R3, R4 и R5 независимо представляет собой C1-С6 алкил или гидроксильную группу,
и обработку фенилпиримидинового соединения формулы (V) подходящим восстанавливающим агентом с получением фенилпиримидинового соединения формулы (II).
6. Способ по п. 5, в котором В* выбирают из группы, состоящей из
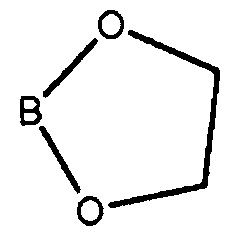 ,
,  и
и 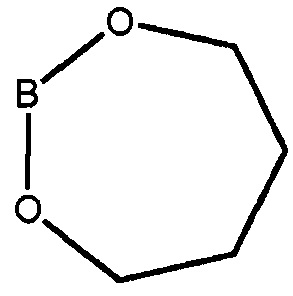 ,
,
где каждый из атомов углеродного кольца может быть незамещенным или замещенным одной или двумя метильными или этильными группами.
7. Способ по п. 6, в котором В* представляет собой
 .
.
8. Способ по п. 5, в котором указанное фенилтетрагидрофурановое соединение формулы (IV) получают путем нитрования соединения формулы (VI)
 ,
,
где R представляет собой Н или F;
соответствующим нитрующим агентом с получением фенилтетрагидрофуранового соединения формулы (IV).
9. Способ по п. 5, в котором фенилтетрагидрофурановое соединение формулы (IV) получают путем защиты аминогруппы в соединении формулы (VI) аминозащитной группой с получением аминозащищенного соединения
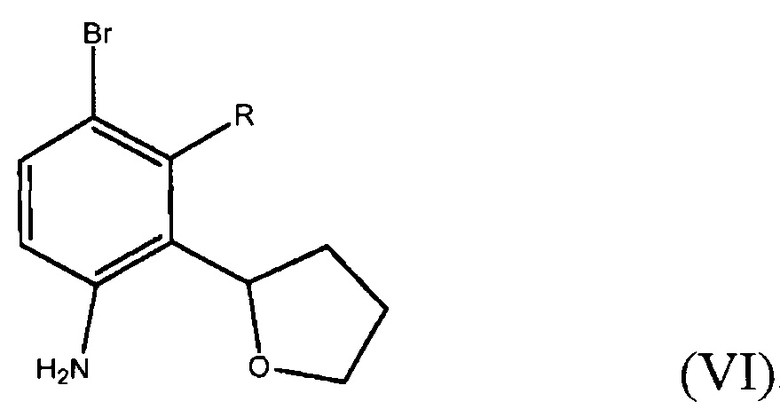 ,
,
где R представляет собой Н или F;
нитрования аминозащищенного соединения подходящим нитрующим агентом с получением аминозащищенного нитросоединения; и
снятия защиты с защищенного амино-нитросоединения с получением фенилтетрагидрофуранового соединения формулы (IV).
10. Способ по п. 1, в котором указанное фенилпиримидиновое соединение формулы (II) получают путем дополнительного восстановления фенилпиримидинового производного формулы (V)
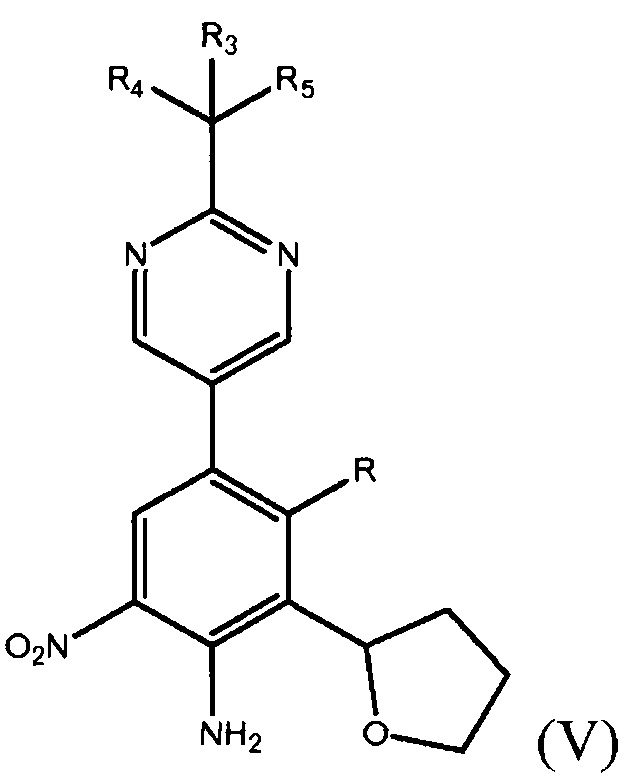 ,
,
где R представляет собой Н или F; и каждый из R3, R4 и R5 независимо представляет собой C1-С6 алкил или гидроксильную группу,
подходящим восстанавливающим агентом с получением соединения формулы (II).
11. Способ по п. 9, в котором нитрование соединения формулы (VI) включает взаимодействие соединения формулы (VI) с NH4NO3 в присутствии сильной кислоты при температуре от приблизительно 20°С до приблизительно 50°С с получением соединения (IV).
12. Способ по п. 9, в котором указанное соединение формулы (VI) получают путем взаимодействия соединения формулы (VII)
 ,
,
где R представляет собой Н или F,
с бромирующим агентом в полярном апротонном растворителе с получением соединения формулы (VI).
13. Способ по п. 12, в котором соединение формулы (VII) является энантиомерно обогащенным.
14. Способ по п. 12, в котором указанное соединение формулы (VII) получают путем обработки дигидрофуранилнитробензольного соединения, выбранного из группы, состоящей из
соединения формулы (VIIIa)
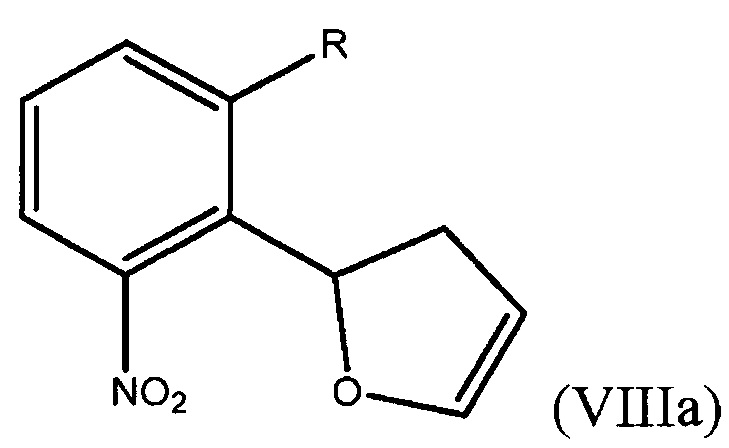 ,
,
где R представляет собой Н или F,
и соединения формулы (VIIIb)
 ,
,
где R представляет собой Н или F,
восстанавливающим агентом с получением соединения формулы (VII).
15. Способ по п. 14, в котором указанное дигидрофуранилнитробензольное соединение получают путем обработки соединения формулы (IX)
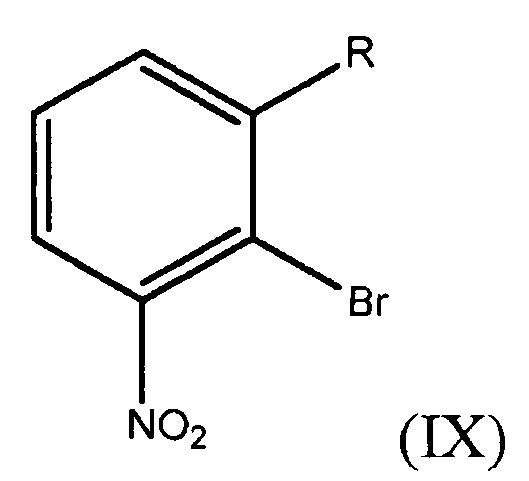 ,
,
где R представляет собой Н или F,
2,3-дигидрофураном в присутствии палладиевого катализатора с получением дигидрофуранилнитробензольного соединения.
16. Соединение формулы (II)
 ,
,
где R представляет собой Н или F, и каждый из R3, R4 и R5 независимо представляет собой C1-С6 алкил или гидроксильную группу.
17. Соединение формулы (V)
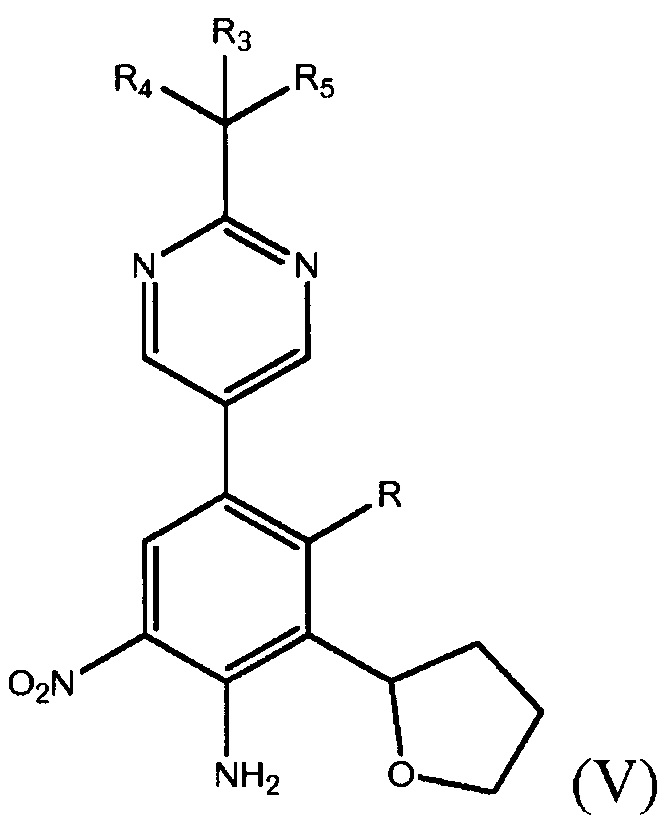 ,
,
где R представляет собой Н или F, и каждый из R3, R4 и R5 независимо представляет собой C1-С6 алкил или гидроксильную группу.
18. Способ получения соединения формулы (I)
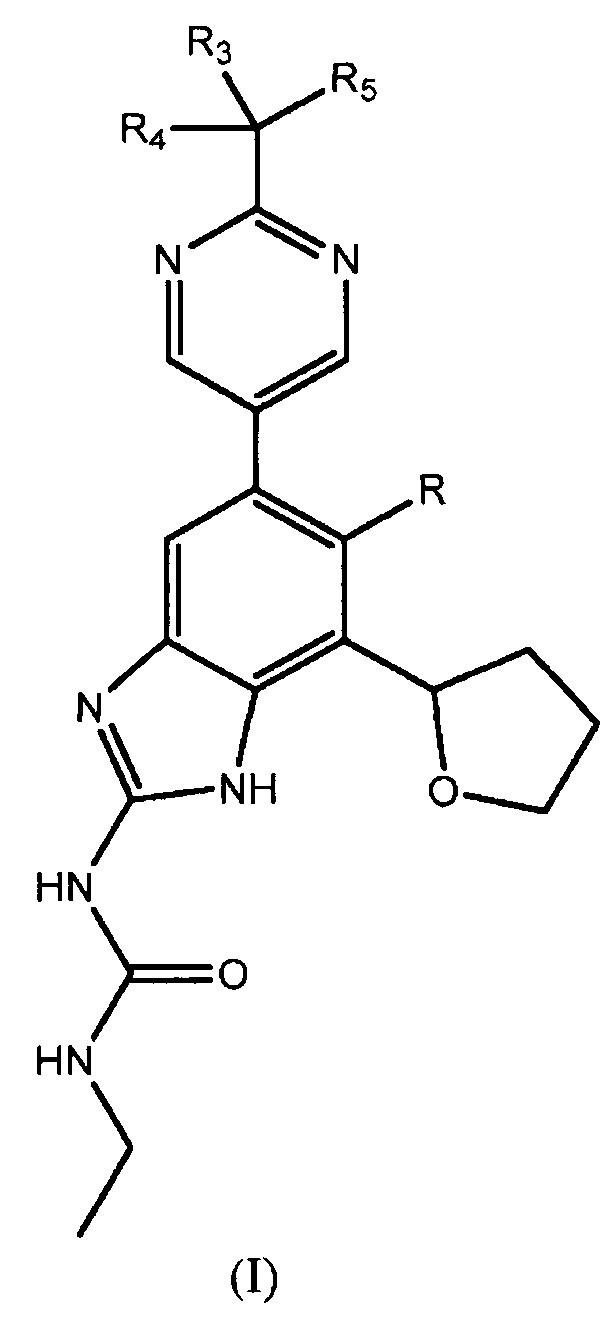
или его фармацевтически приемлемой соли,
где R представляет собой Н или F; и каждый из R3, R4 и R5 независимо представляет собой C1-С6 алкил или гидроксильную группу;
включающий взаимодействие соединения формулы (IX)
 ,
,
где R представляет собой Н или F,
с 2,3-дигидрофураном в присутствии палладиевого катализатора с получением дигидрофуранилнитробензольного соединения формулы (VIIIa)
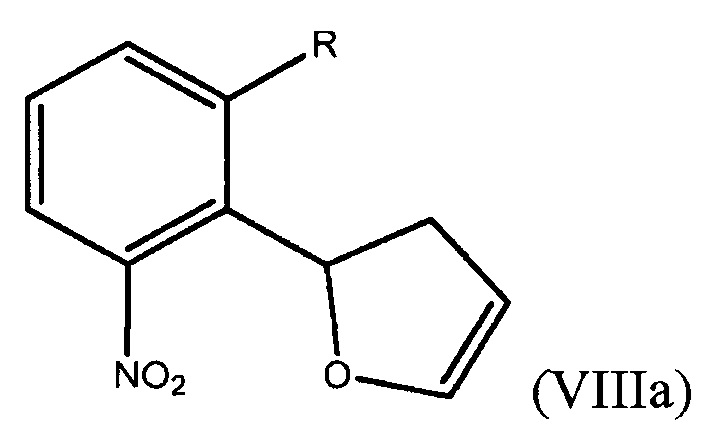
или формулы (VIIIb)
 ,
,
где R представляет собой Н или F;
взаимодействие дигидрофуранилнитробензольного соединения
формулы (VIIIa) или формулы (VIIIb) или их комбинации с восстанавливающим агентом с получением соединения формулы (VII)
 ,
,
где R представляет собой Н или F;
взаимодействие соединения формулы (VII) с бромирующим агентом в полярном апротонном растворителе с получением соединения формулы (VI)
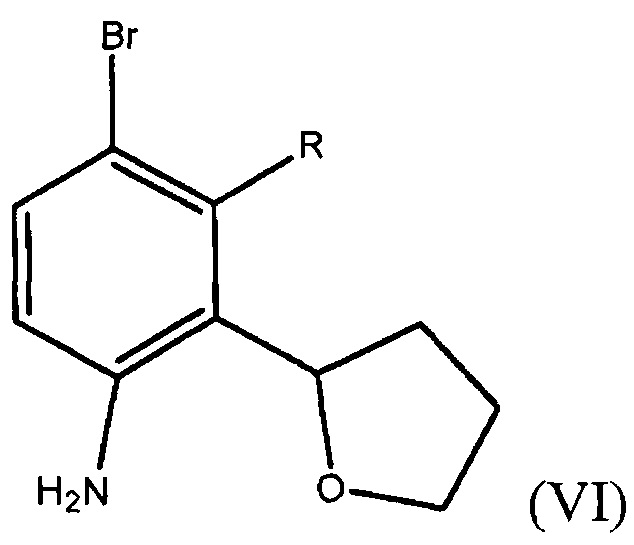 ,
,
где R представляет собой Н или F;
нитрование соединения формулы (VI) соответствующим нитрующим агентом с получением фенилтетрагидрофуранового соединения формулы (IV)
 ,
,
где R представляет собой Н или F;
взаимодействие фенилтетрагидрофуранового соединения формулы (IV) с производным бороновой кислоты формулы (III)
 ,
,
где каждый из R3, R4 и R5 независимо представляет собой C1-С6 алкил или гидроксильную группу и В* представляет собой  , где каждый из R1 и R2 независимо представляет собой алкил, или Н, или OR1 и OR2 вместе с атомом В, к которому они присоединены, образуют необязательно замещенное 5-, 6- или 7-членное кольцо или BF3X, где X представляет собой любой одновалентный катион,
, где каждый из R1 и R2 независимо представляет собой алкил, или Н, или OR1 и OR2 вместе с атомом В, к которому они присоединены, образуют необязательно замещенное 5-, 6- или 7-членное кольцо или BF3X, где X представляет собой любой одновалентный катион,
в присутствии палладиевого катализатора в полярном растворителе с получением фенилпиримидинового соединения формулы (V)
 ,
,
где R представляет собой Н или F и каждый из R3, R4 и R5 независимо представляет собой C1-С6 алкил или гидроксильную группу,
и обработку фенилпиримидинового соединения формулы (V) подходящим восстанавливающим агентом с получением фенилпиримидинового соединения формулы (II)
 ,
,
где R представляет собой Н или F и каждый из R3, R4 и R5 независимо представляет собой C1-С6 алкил или гидроксильную группу;
взаимодействие фенилпиримидинового соединения формулы (II) с производным мочевины формулы А или формулы В:
 ,
,
 ,
,
где R6 представляет собой необязательно замещенный алкил, необязательно замещенный арил, необязательно замещенный насыщенный или ненасыщенный карбоцикл или необязательно замещенный насыщенный или ненасыщенный гетероцикл, с получением соединения формулы (I);
и необязательное взаимодействие соединения формулы (I) с подходящей кислотой с получением фармацевтически приемлемой соли соединения формулы (I).
| Paul S | |||
| Charifson et al, "Dual-Targeting Benzimidazole Urea Ingibitor Gyrase and Topoisomerase", J.Medicinal Chtmistry,2008,v.51,no 17, p.5243-5263 | |||
| WO 2007056330 A1 18.05.2007 | |||
| WO 03105846 A1 24.12.2003 | |||
| ПРОИЗВОДНОЕ БЕНЗОФУРАНА | 2004 |
|
RU2328495C2 |

