Изобретение относится к области физиологически активных пептидов, конкретно к новому способу получения аналогов АКТГ (4-10) общей формулы I
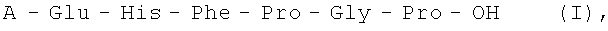
где А=Н, Met, Met(O), Lys, Ser, Trp, Ala, Gly, Thr,
и может найти применение в медицине.
Известен гептапептид Семакс формулы Met-Glu-His-Phe-Pro-Gly-Pro, являющийся стимулятором памяти [1]. В настоящее время Семакс используется в качестве субстанции при получении ноотропного лекарственного средства [2].
Известен способ получения аналогов АКТГ (4-10) общей формулы I, который основан на блочной конденсации защищенного С-концевого трипептида H-Pro-Gly-Pro-OBzl с трипептидом Z-Glu(OBut)-His-Phe-N2H3 азидным методом с последующим присоединением к полученному гексапептиду С-концевой N-защищенной аминокислоты и удалением защитных групп [3].
Существенным недостатком известного способа является низкий выход конечных продуктов и использование в процессе синтеза азидного метода конденсации пептидных фрагментов, который требует тщательного соблюдения температурного режима и использование в процессе синтеза высокотоксичных реагентов, таких как гидразин гидрат и трет-бутилнитрит, что особенно затруднительно в условиях промышленного синтеза. Кроме того, синтез осложняется нежелательными побочными реакциями [4].
Задачей изобретения является повышение выхода, упрощение процесса получения пептидов формулы I.
Поставленная задача решается описываемым способом, согласно которому пептиды общей формулы I получают путем ступенчатого наращивания пептидной цепи, начиная с С-концевого тетрапептида общей формулы
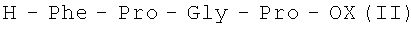
Исходный С-концевой тетрапептид формулы II получают обычными методами пептидного синтеза, например путем блочно-ступенчатого наращивания пептидной цепи в растворе с использованием активированных эфиров соответствующих защищенных аминокислот и метода смешанных ангидридов [3].
Список сокращений
АсОН - уксусная кислота;
Boc - трет-бутилоксикарбонил;
But - трет-бутил;
Bzl - бензил;
DCC - N, N'-дициклогексилкарбодиимид;
DMF - N,N-диметилформамид;
HONSu - N-гидроксисукцинимид;
HONp - п-нитрофенол;
TFA - трифторуксусная кислота;
Z - бензилоксикарбонил;
ТСХ - тонкослойная хроматография.
Экспериментальная часть
В синтезе использованы производные L-аминокислот фирмы Bachem (Швейцария). DCC, TFA, HONSu, HONp фирмы Fluka (Швейцария). DMF очищали перегонкой над нингидрином и окисью бария. Аналитическую ВЭЖХ проводили на хроматографе фирмы Gilson (Франция).
ТСХ проводили на стеклянных хроматографических пластинках Merck-Kiselgel (Германия) в следующих системах растворителей:
1Н-ЯМР - спектры снимают на спектрометре WH-500 Bruker 500 МГц (ФРГ) в DMSO-d6 при 300 К. Химические сдвиги (δ, м.д.) измеряют относительно тетраметилсилана.
Масс-спектры регистрировали на масс-спектрометре Analytical Compact MALDI 4 фирмы Kratos (Великобритания).
Описываемый способ иллюстрируется следующими примерами:
Пример 1. 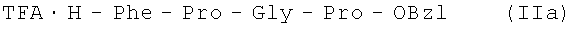
Вариант А.
I стадия: 
19,5 г (0,260 М) глицина растворяют в 260 мл 1 н. NaOH, полученный раствор добавляют к охлажденному до 0°С раствору 96,0 г (0,260 М) Z-Pro-ONp в 600 мл DMF. Реакционную смесь перемешивают при комнатной температуре в течение 16 часов, упаривают, остаток растворяют в 300 мл воды и трижды экстрагируют эфиром. Эфирный слой отделяют, к водному слою добавляют 150 мл 2 н. раствора HCl, выпавшее масло экстрагируют 600 мл этилацетата. Органический слой отделяют и промывают водой до нейтральной реакции, сушат над Na2SO4, упаривают, остаток перекристаллизовывают из эфира
В итоге получают: 67,5 г (85%) IIIa: Тпл. 122-123°С; Rf 0,67 (A); Rf 0,81 (В), Rf 0,65 (С).
II стадия: 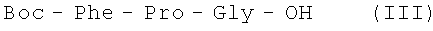
72,9 г (0.238 М) IIIa гидрируют в присутствии 7,0 г 5% Pd/C в смеси 600 мл этанола и 238 мл 1 н. NaOH. После окончания гидрирования (контроль с помощью ТСХ в системе А) катализатор отфильтровывают, промывают на фильтре этанолом, фильтрат упаривают, остаток растворяют в 700 мл DMF. К полученному раствору при 0°С прибавляют 86,0 г (0,238 М) Boc-Phe-ONp. Реакционную смесь перемешивают при комнатной температуре в течение 16 часов, упаривают, к остатку добавляют 280 мл 1 н. раствора H2SO4, выпавшее масло экстрагируют 600 мл этилацетата, органический слой промывают водой до нейтральной реакции, сушат над Na2SO4 и упаривают. Остаток перекристаллизовывают из смеси этилацетат-эфир.
В итоге получают 47,3 г (55%) III: Тпл. 139-141°С; Rf 0,69 (А); Rf 0,85 (Б); Rf 0,23 (С).
Вариант Б.

34,6 г (0,300 М) пролина растворяют в 150 мл 2 н. NaOH, полученный раствор добавляют к охлажденному до 0°С раствору 115,9 г (0,30 М) Boc-Phe ONp в 900 мл DMF. Реакционную смесь перемешивают при комнатной температуре в течение 16 часов, упаривают, остаток растворяют в 900 мл воды и трижды экстрагируют эфиром. Эфирный слой отделяют, к водному слою добавляют 180 мл 2 н. раствора H2SO4, выпавшее масло экстрагируют 700 мл этилацетата, органический слой промывают водой до нейтральной реакции, сушат над Na2SO4 и упаривают. Остаток растворяют в 600 мл DMF, к полученному раствору добавляют 42,0 г (0,330 М) п-нитрофенола. Реакционную смесь охлаждают до -15°С и при перемешивании добавляют к ней предварительно охлажденный до -15°С раствор 63,0 г (0,310 М) DCC в 300 мл DMF. Реакционную смесь выдерживают при +4°С в течение 18 часов, упаривают до постоянного веса, остаток трижды промывают гексаном и растворяют в 500 мл DMF. Полученный раствор охлаждают до -10°С и постепенно, так, чтобы температура реакционной смеси не поднималась выше +5°С, добавляют раствор 22,5 г (0,300 М) глицина в 150 мл 2 н. NaOH. Реакционную смесь перемешивают при комнатной температуре в течение 16 часов, упаривают, остаток растворяют в 500 мл воды и трижды экстрагируют эфиром. К водному слою добавляют 200 мл 2 н. раствора Н2SO4, вапавшее масло экстрагируют 900 мл этилацетата, органический слой промывают водой до нейтральной реакции, сушат над Na2SO4, упаривают. Остаток перекристаллизовывают из эфира.
В итоге получают 109 г (80%) III с t пл. 138-141°С; Rf 0,69 (A); Rf 0,85 (Б); Rf 0,23 (С).
III стадия: 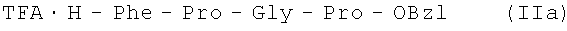
66,4 г (0,153 М) III растворяют в 500 мл DMF, к полученному раствору добавляют 17,0 мл (0,153 М) N-метилморфолина. Реакционную смесь при перемешивании охлаждают до -15°С и постепенно (с такой скоростью, чтобы температура реакционной смеси не поднималась выше -10°С), прибавляют 19,85 мл (0,153 М) изобутилхлорформиата, перемешивают при -10°С в течение 10 минут, затем добавляют охлажденный до -10°С раствор 64,4 г (0,290 М) H-Pro-OBzl в 200 мл DMF. Через 30 минут реакционную смесь упаривают, остаток растворяют в 600 мл этилацетата, полученный раствор промывают последовательно 5% раствором NaHCO3, водой, 2% раствором Н2SO4, водой, сушат над Na2SO4 и упаривают. Оставшееся масло растворяют в 250 мл TFA, выдерживают в течение часа, упаривают. Остаток растворяют в 300 мл воды, добавляют 300 мл эфира и выдерживают при +4°С в течение 2 часов. Выпавший кристаллический осадок отфильтровывают, промывают на фильтре водой, эфиром и высушивают до постоянного веса.
В итоге получают 88,58 г (92%) IIa 99,0% чистоты по данным аналитической ВЭЖХ: ТR=25,81 мин. Колонка Ultropac TSK ODS-120T 4,6×250 мм, 5 мкм. Буфер А: 0,05 М КН2PO4, рН 3,0; буфер В: 70% ацетонитрил + 30% Н2O; градиент: 10%-70% В за 30 минут. Т пл. 167-169°С, Rf 0,60 (А); Rf 0,85 (Б); Rf 0,68 (С).
Аминокислотный анализ: Phe 1,00 (1), Pro 1,93 (2), Gly 1,05 (1).
MALDI-MS m/z, найдено: 507,6. [М+Н]+ вычислено: 506,6
1Н-ЯМР - спектр (DMSO-d6, δ, м.д.):
Phe - 8,1 (NH, 1H); 4,34 (α-СН, 1Н); 3,12/2,94 (β-CH, 2H)
Pro - 4,49 (α-CH, 1H); 1,90/2,05 (β-CH, 2H)
Gly - 8,02 (NH, 1H); 4,09/3,85 (α-CH, 1H)
Pro - 4,42 (α-CH, 1H); 2,17/1,83 (β-CH, 2H)
Пример 2. 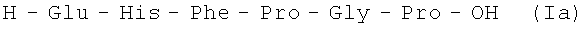
I стадия: 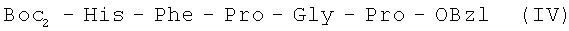
126,9 г (0,200М) IIa растворяют в 600 мл DMF, к полученному раствору прибавляют 22,2 мл (0,200М) N-метилморфолина и 95,47 г (0,200 М) Boc2-His-ONp, pH реакционной смеси поддерживают в интервале 8,0-8,5 добавлением N-метилморфолина. Полноту протекания реакции контролируют с помощью ТСХ в системе (А). Через 16 часов реакционную смесь упаривают, остаток растворяют в 700 мл этилацетата, промывают несколько раз 2% водным раствором аммиака, водой, 5% водным раствором лимонной кислоты, водой. Органический слой сушат Na2SO4, упаривают. Остаток переосаждают из смеси эфир-гексан.
В итоге получают 151,90 г (90%) IV с Rf 0,80 (A); Rf 0,88 (Б); Rf 0,81 (С).
II стадия: 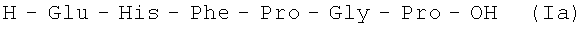
168,78 г (0,200 М) IV растворяют в 800 мл TFA, выдерживают в течение 1 часа, упаривают, остаток обрабатывают эфиром, выпавший осадок отфильтровывают, промывают на фильтре эфиром, сушат. Полученный продукт растворяют в 600 мл DMF, к раствору прибавляют 22,2 мл (0,200 М) N-метилморфолина и 86,9 г (0,200 М) Boc-Glu(OBzl)-ONSu., pH реакционной смеси поддерживают в интервале 8,0-8,5 добавлением в случае необходимости дополнительного количества N-метилморфолина. Полноту протекания реакции контролируют с помощью ТСХ в системе (А). Через 16 часов реакционную смесь упаривают, остаток растворяют в 700 мл этилацетата, промывают водой, 2% водным раствором H2SO4, водой. Органический слой сушат над Na2SO4 и упаривают. Остаток (Rf 0,74 (А); Rf 0,92 (Б); Rf 0,79 (С)) растворяют в 700 мл этилового спирта и гидрируют в присутствии 14,0 г 5% Pd/C. Процесс гидрирования контролируют с помощью ТСХ в системах А и Б. После окончания гидрирования катализатор отфильтровывают, промывают на фильтре этиловым спиртом, фильтрат упаривают до постоянного веса, остаток растворяют в 300 мл TFA, выдерживают в течение 1 часа, добавляют 200 мл 2,5 н. HCl в этилацетате, упаривают, остаток обрабатывают эфиром, выпавший осадок отфильтровывают, промывают на фильтре эфиром и сушат.
В итоге получают 138,9 г (92%) Ia 97,2% чистоты по данным аналитической ВЭЖХ: TR=12,84 мин. Колонка Ultropac TSK ODS-120T 4,6×250 мм, 5 мкм. Буфер А: 0,05 М KH2PO4, pH 3,0; буфер В: 70% ацетонитрил + 30% H2O; градиент: 10%-70% В за 30 минут. Тпл. 118-121°С; Rf 0,21 (А); Rf 0,53 (Б).
Аминокислотный анализ: Glu 1,00 (1), His 0,98 (1), Phe 1,03 (1), Pro 1,92 (2), Gly 1,08 (1). MALDI-MS m/z, найдено: 683,8 [М+Н]+; вычислено: 682,77
1Н-ЯМР - спектр (DMSO-d6, δ, м.д.):
Glu - 8,10 (NH, 1H); 3,82 (α-СН, 1Н); 1,89 (β-CH, 2H); 2,26 (γ-СН, 2Н)
His - 8,70 (NH, 1H); 4,67 (α-CH, 1H); 3,00 (β-CH, 2H); 8,97/7,34 (С2Н)
Phe - 8,54 (NH, 1H); 4,65 (α-СН, 1Н); 3,08/2,80 (β-CH, 2H)
Pro - 4,41 (α-СН, 1Н); 1,96 (β-СН, 2Н)
Gly - 7,79 (NH, 1H); 4,01/3,82 (α-СН, 1Н)
Pro - 4,23 (α-CH, 1H); 2,13 (β-CH, 2H)
Пример 3. 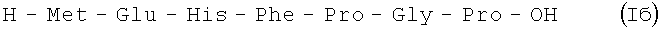
I стадия: 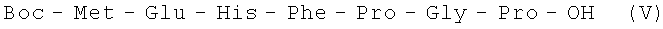
138,9 г (0,200 М) la растворяют в 400 мл воды, с помощью 20% водного раствора NaOH доводят рН реакционной смеси до 7,06, воду упаривают, остаток дважды упаривают с изопропиловым спиртом, растворяют в 600 мл DMF, к раствору прибавляют 81,1 г (0,220 М) Boc-Met-ONp.Через 16 часов реакционную смесь упаривают, остаток обрабатывают 400 мл смеси этилацетат-гексан 1:1, декантируют, оставшееся масло растворяют в 700 мл н-бутанола, трижды промывают 2% водным раствором H2SO4, водой до нейтральной реакции, упаривают, осаждают 500 мл смеси этилацетат-гексан 1:1, выпавший осадок отфильтровывают и высушивают до постоянного веса.
В итоге получают 128 г (70%) V в виде аморфного порошка с Rf 0,42 (A); Rf 0,52 (Б).
II стадия: 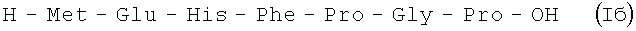
60,0 г (0.07 М) V при перемешивании обрабатывают 250 мл 3 н. раствора HCl в уксусной кислоте. После полного растворения осадка реакционную смесь выдерживают в течение часа, упаривают, к остатку добавляют 400 мл этилацетата, выпавший аморфный осадок отфильтровывают, промывают на фильтре этилацетатом, ацетонитрилом, гексаном, сушат, растворяют в воде и очищают на колонке Диасорб-130-С16Т,12 мкм; 50×250 мм производства ЗАО «БиохимМак СТ», Буфер А: 0,01 М NH4OAc (рН 4,5); буфер Б: 40% изопропиловый спирт в воде, градиент: 0% Б - 10% Б - 10 мин, далее - 0,3% в минуту. Поток V=50 мл/мин, детектор: λ=254 нм.
В итоге получают (после очистки методом ВЭЖХ) 48,43 г (85%) Iб в виде аморфного порошка 98,9% чистоты по данным аналитической ВЭЖХ: TR=14,49 мин. Колонка Ultropac TSK ODS-120T 4,6×250 мм, 5 мкм. Буфер А: 0,05 М КН2PO4, рН 3,0; буфер В: 70% ацетонитрил + 30% H2O; градиент: 10%-70% В за 30 минут V=1 мл/мин, детектор: λ=204 нм.
MALDY-MS, m/z найдено: 814,94 [M+H]+, вычислено: 813,95.
Аминокислотный анализ: Met 1,09 (1), Glu 1,00 (1), His 1,13 (1), Phe 1,05 (1), Pro 1,93 (2), Gly 1,07(1).
1Н-ЯМР - спектр (DMSO-d6, δ, м.д.):
Met - 8,18 (NH, 1H); 3,88 (α-СН, 1Н); 1,95 (β-CH, 2H); 2,01 (S-СН3)
Glu - 8,62 (NH, 1H); 4,29 (α-CH, 1H); 1,74/1,85 (β-CH, 2H); 2,26 (γ-СН, 2Н)
His - 8,26 (NH, 1H); 4,61 (α-CH, 1H); 3,01/2,94 (β-CH, 2H); 8,97 (C2H)
Phe - 8,42 (NH, 1H); 4,64 (α-CH, 1H); 3,06/2,91 (β-CH, 2H)
Pro - 4,42 (α-CH, 1H); 1,96 (β-CH, 2H)
Gly - 7,82 (NH, 1H); 4,01 (α-CH, 1H)
Pro - 4,25 (α-CH, 1H); 2,13 (β-CH, 2H)
Пример 4. 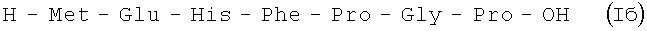
191,2 г (0.300 М) IIa растворяют в 1,5 л DMF. К полученному раствору добавляют 36 мл N-метилморфолина и 147,0 г (0,300 М) Boc2HisONp. Полученный раствор выдерживают в течение 16 часов при комнатной температуре, растворитель упаривают, остаток растворяют в 1,5 л этилацетата, промывают разбавленным водным раствором аммиака, водой, водным раствором лимонной кислоты, водой, упаривают. Полученное масло растворяют в 200 мл этилацетата, осаждают 800 мл гексана, декантируют. Последнюю процедуру повторяют еще раз. Остаток растворяют в 600 мл TFA, выдерживают в течение 1 часа, TFA упаривают, оставшееся масло затирают в эфире, образовавшийся осадок отфильтровывают, промывают на фильтре эфиром, растворяют в 1,5 л этилацетата, рН раствора доводят до 8,0 добавлением N-метилморфолина, затем к раствору добавляют 120 г (0,300 М) Boc-Glu(OBut)-ONSu и выдерживают при комнатной температуре в течение 16 часов, промывают водой, разбавленным водным раствором аммиака, 2% Н2SO4, водой до нейтральной реакции и упаривают. Остаток растирают в эфире, образовавшийся осадок отфильтровывают, промывают на фильтре эфиром, растворяют в 600 мл TFA, выдерживают в течение 1 часа, TFA упаривают, оставшееся масло затирают в эфире, образовавшийся осадок отфильтровывают, промывают на фильтре эфиром, растворяют в 1 литре воды и гидрируют в присутствии 5% Pd/C. Полноту гидрирования контролируют с помощью тонкослойной хроматографии в системе А. По окончании реакции катализатор отфильтровывают, промывают на фильтре водой, фильтрат упаривают до половины первоначального объема, доводят рН раствора до 7,0 с помощью 20% NaOH, упаривают досуха, остаток растворяют в 1,5 л DMF, к полученному раствору добавляют 11 г (0,300 М) Boc-Met-ONp и выдерживают в течение 16 часов при комнатной температуре. DMF упаривают, остаток растворяют в 1,5 л н.-бутанола, подкисляют 2% H2SO4 до рН 3, промывают водой до нейтральной реакции, упаривают. Остаток растворяют в 600 мл этилацетата, высаживают 600 мл гексана. Образовавшийся осадок отфильтровывают, промывают на фильтре 900 мл смеси этилацетат - гексан 2:1, гексаном и высушивают. Полученный продукт растворяют в 700 мл 3 н. HCl в уксусной кислоте, выдерживают в течение часа при комнатной температуре, упаривают, остаток растирают в этилацетате, промывают гексаном, высушивают, растворяют в воде и очищают на колонке Диасорб-130-С16Т,12 мкм; 50×250 мм производства ЗАО «БиохимМак СТ», Буфер А: 0,01 М NH4ОАс (рН 4,5); буфер Б: 40% изопропиловый спирт в воде, градиент: 0% Б - 10% Б - 10 мин, далее - 0,3% в минуту. Поток V=50 мл/мин, детектор: λ=254 нм.
В итоге получают 140 г (52%) Iб в виде аморфного порошка 98,5% чистоты по данным аналитической ВЭЖХ: TR=14,49 мин. Колонка Ultropac TSK ODS-120T 4,6×250 мм, 5 мкм. Буфер А: 0,05 М КН2PO4, рН 3,0; буфер В: 70% ацетонитрил + 30% Н2О; градиент: 10% - 70% В за 30 минут V=1 мл/мин, детектор: λ=204 нм.
MALDY-MS, m/z найдено: 814,94 [M+H]+, вычислено: 813,95.
Аминокислотный анализ: Met 0,99 (1), Glu 1,00 (1), His 0,92 (1), Phe 1,10 (1),Pro 1,93 (2), Gly 1,03(1).
1Н-ЯМР - спектр (DMSO-d6, δ, м.д.):
Met - 8,18 (NH, 1H); 3,88 (α-СН, 1Н); 1,95 (β-СН, 2Н); 2,01 (S-СН3)
Glu - 8,62 (NH, 1H); 4,29 (α-CH, 1H); 1,74/1,85 (β-CH, 2H); 2,26 (γ-СН, 2Н)
His - 8,26 (NH, 1H); 4,61 (α-CH, 1H); 3,01/2,94 (β-CH, 2H); 8,97 (C2H)
Phe - 8,42 (NH, 1H); 4,64 (α-CH, 1H); 3,06/2,91 (β-CH, 2H)
Pro - 4,42 (α-CH, 1H); 1,96 (β-CH, 2H)
Gly - 7,82 (NH, 1H); 4,01 (α-CH, 1H)
Pro - 4,25 (α-CH, 1H); 2,13 (β-CH, 2H)
Пример 5. 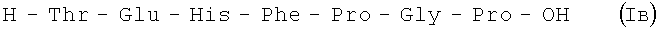
I стадия: 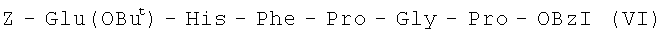
4,3 г (5 мМ) IV растворяют в 30 мл TFA, выдерживают в течение 1 часа, упаривают, остаток обрабатывают эфиром, выпавший осадок отфильтровывают, промывают на фильтре эфиром, сушат. Полученный продукт растворяют в 20 мл DMF, к раствору прибавляют 0,6 мл (5,4 мМ) N-метилморфолина и 2,17 г (5 мМ) Z-Glu(OBut)-ONSu. Полноту протекания реакции контролируют с помощью ТСХ в системе (А), рН реакционной смеси поддерживают в интервале 8,0-8,5 добавлением в случае необходимости N-метилморфолина. Через 16 часов реакционную смесь упаривают, остаток растворяют в 70 мл этилацетата, промывают водой, 2% водным раствором H2SO4, водой. Органический слой сушат над Na2SO4, упаривают и растирают с эфиром. Выпавший осадок отфильтровывают, промывают эфиром и высушивают.
В итоге получают 4,1 г (86%) VI с Rf 0,82(A); Rf 0,65 (Б); Rf 0,79 (С).
II стадия: 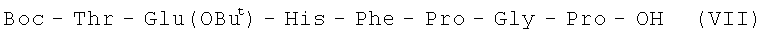
3,3 г (3,40 мМ) VI растворяют в 50 мл 95% уксусной кислоты и гидрируют над 5% Pd/C до исчезновения исходного соединения, контролируя ход реакции с помощью ТСХ в системе А. Катализатор отфильтровывают, фильтрат упаривают до объема 10 мл и добавляют 60 мл эфира. Выпавший осадок отфильтровывают, промывают на фильтре эфиром, высушивают. Полученный продукт растворяют в 30 мл DMF, добавляют 0,62 мл диизопропилэтиламина и 1,68 г (3,6 мМ) Boc-Thr-OPcp. Ход реакции контролируют с помощью ТСХ в системе А. Раствор упаривают, к остатку добавляют эфир, образовавшийся осадок отфильтровывают, промывают эфиром и сушат.
В итоге получают 3,2 г (100%) VII с Rf 0,70 (A).
III стадия: 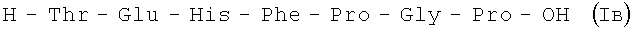
3,2 г (3,40 мМ) VII растворяют в 20 мл TFA, через 40 минут реакционную смесь упаривают до объема 5 мл, добавляют 50 мл эфира, выпавший осадок отфильтровывают и подвергают очистке методом ВЭЖХ на колонке Диасорб-130-С16Т,12 мкм; 50×250 мм производства ЗАО «БиохимМак СТ», Буфер А: 0,01 М NH4OAc (рН 4,5); буфер Б: 40% изопропиловый спирт в воде, градиент: 0%Б - 10% Б - 10 мин, далее - 0,3% в минуту. Поток V=50 мл/мин, детектор: λ=254 нм.
В итоге получают 2,5 г (94%) Iв с Rf 0,18 (A); 0,12 (D); 0,17 (В); Rf 0,5; ВЭЖХ: Rt 14,9 мин. Колонка Ultropac TSK ODS-120T 4,6×250 мм, 5 мкм. Буфер А: 0,05 М КН2РО4 рН 3,0; буфер В: 70% ацетонитрил + 30% H2O; градиент: 10%-70% В за 30 минут V=1 мл/мин, детектор: λ=204 нм.
Аналогично получают другие аналоги АКТГ (4-10), физико-химические характеристики которых представлены в таблице.
Как видно из приведенных примеров, заявленный способ позволяет масштабировать процесс, повысить выход целевых соединений.
Литература
1. Авт. свид. СССР №939440, С07С 103/52, 1981.
2. Патент РФ №2045958, А61К 38/02, 1994.
3. Патент РФ №2206573, С07К 7/06, 2003.
4. Х.-Д.Якубке, X.Ешкайт. Аминокислоты, пептиды, белки: пер. с нем. - М.: Мир, 1985.
| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБ ПОЛУЧЕНИЯ ГЕПТАПЕПТИДА И ПРОМЕЖУТОЧНЫЕ СОЕДИНЕНИЯ ДЛЯ ЕГО ПОЛУЧЕНИЯ | 2006 |
|
RU2303603C2 |
| СПОСОБ ПОЛУЧЕНИЯ ТРИПЕПТИДОВ | 2006 |
|
RU2303602C2 |
| СПОСОБ ПОЛУЧЕНИЯ ДОДЕКАПЕПТИДА И ТРИПЕПТИД ДЛЯ ЕГО ОСУЩЕСТВЛЕНИЯ | 2007 |
|
RU2340626C1 |
| СПОСОБ ПОЛУЧЕНИЯ ПЕПТИДА ЭКСЕНАТИДА | 2011 |
|
RU2458066C1 |
| ПЕПТИД И СПОСОБ ЕГО ПОЛУЧЕНИЯ | 1994 |
|
RU2067000C1 |
| ПЕПТИДЫ, ОБЛАДАЮЩИЕ АНТИКОАГУЛЯНТНО-ФИБРИНОЛИТИЧЕСКОЙ, АНТИТРОМБОЦИТАРНОЙ И АНТИТРОМБОТИЧЕСКОЙ АКТИВНОСТЬЮ | 2005 |
|
RU2286166C1 |
| ПЕПТИДЫ, ОБЛАДАЮЩИЕ ЦИТОПРОТЕКТОРНОЙ АКТИВНОСТЬЮ | 2011 |
|
RU2482128C1 |
| Способ получения пептида Ac-His-Ala-Glu-Glu-NH | 2021 |
|
RU2767030C1 |
| ГЕКСАПЕПТИД, ОБЛАДАЮЩИЙ АКТИВНОСТЬЮ СТИМУЛЯТОРА ПАМЯТИ | 1998 |
|
RU2131743C1 |
| ПРОИЗВОДНЫЕ FGF21 И ИХ ПРИМЕНЕНИЯ | 2015 |
|
RU2729011C2 |
Изобретение относится к области фармацевтической химии, конкретно к способу получения аналогов аденокортикотропного гормона (АКТГ) (4-10), обладающих нейротропной активностью. Способ получения аналогов аденокортикотропного гормона (АКТГ), последовательности (4-10), общей формулы I: A-Glu-His-Phe-Pro-Gly-Pro-OH (I), где А=Н, Met, Met(O), Lys, Ser, Trp, Ala, Gly, Thr осуществляют жидкофазным методом путем ступенчатого наращивания пептидной цепи, начиная с С-концевого защищенного тетрапептида формулы: H-Phe-Pro-Gly-Pro-OX (II), где Х - защитная группировка, с использованием соответствующих полностью защищенных аминокислот в активированной форме с последующим снятием защитных групп на каждой стадии и очисткой целевого продукта с помощью жидкостной хроматографии. Способ позволяет упростить процесс и повысить выход целевого продукта. 2 н. и 3 з.п. ф-лы, 1 табл.
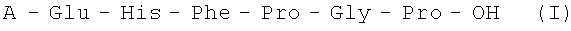
где А=Н, Met, Met(O), Lys, Ser, Trp, Ala, Gly, Thr,
отличающийся тем, что синтез осуществляют жидкофазным методом путем ступенчатого наращивания пептидной цепи, начиная с С-концевого защищенного тетрапептида формулы
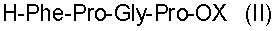
где Х - защитная группировка, с использованием соответствующих полностью защищенных аминокислот в активированной форме с последующим снятием защитных групп на каждой стадии и очисткой целевого продукта с помощью жидкостной хроматографии.
H-Phe-Pro-Gly-Pro-OBzl,
где Bzl - бензил.
H-Met-Glu-His-Phe-Pro-Gly-Pro-OH.
H-Glu-His-Phe-Pro-Gly-Pro-OH.
TFA·H-Phe-Pro-Gly-Pro-OBzl
в качестве промежуточного соединения для синтеза аналогов АКТГ (4-10) общей формулы
A-Glu-His-Phe-Pro-Gly-Pro-OH,
где А-Н, Met, Met(O), Lys, Ser, Trp, Ala, Gly, The, по п.1 формулы изобретения.
| СЕМЕЙСТВО ПЕПТИДОВ, ОБЛАДАЮЩИХ НЕЙРОТРОПНЫМИ СВОЙСТВАМИ | 2001 |
|
RU2206573C1 |
| Гептапептид в качестве стимулятора памяти пролонгированного действия | 1981 |
|
SU939440A1 |
| US 3853838 A, 12.10.1974. | |||

