Изобретение относится к органической химии, конкретно к новым биологически активным пептидам общей формулы:
A-L-Asp-L-Glu-B
где A-H - остаток аминокислоты или ацил;
B-OH - остаток аминокислоты или любого органического соединения, способного образовывать амидную или сложноэфирную химическую связь, обладающим цитопротекторной активностью, которые могут найти применение в фармакологии и медицине при создании новых лекарственных средств.
Известен ряд коротких пептидов, содержащих в своей структуре производные дипептида Y-L-Glu-L-Asp-X и имеющие свойства межклеточных медиаторов [1-5]. Недостатком этих соединений является их химическая неустойчивость и склонность к самопроизвольным перегруппировкам.
В случае, если X - остаток глицина, происходит самопроизвольная миграция этого остатка от α-карбоксила аспарагиновой кислоты к β-карбоксилу. И хотя это превращение может происходить в случае любой аминокислоты, скорость этой реакции сильно зависит от стерических препятствий [6-7]. В случае, если X - остаток пролина или любое другое органическое соединение с вторичной аминогруппой, то происходит самопроизвольный разрыв связи между этими остатками [8].
Следует отметить, что эти превращения приводят к частичной рацемизации остатка аспарагиновой кислоты.
Целью заявленного технического решения является получение химически стабильных пептидов, обладающих цитопротекторной активностью. Поставленная цель достигается описываемыми пептидами общей формулы:
A-L-Asp-L-Glu-B
где A-=H - остаток аминокислоты или ацил;
B-=ОН - остаток аминокислоты или любого органического соединения, способного образовывать амидную или сложноэфирную химическую связь.
Синтез заявленных соединений осуществляют с использованием общепринятых в химии биологически активных пептидов методических приемов [9-10] и может быть легко масштабирован до промышленных объемов.
Заявленные соединения могут найти применение в пищевой, косметологической или фармацевтической промышленности. Для веществ, используемых в этих областях, одним из основных критериев применимости является химическая стабильность при хранении, а также стабильность в составе конечных продуктов. Принятые условные сокращения:
Boc - трет-бутилоксикарбонил
Bzl - бензил
Z - бензилоксикарбонил
Bu - трет-бутил
DCHA - дициклогексил амин
TFA - трифторуксусная кислота
ONP - пара-нитрофенил
ONSu - N-оксисукценил
NMM - N-метилморфолин
DMF - диметилформамид
Tos - пара-толуолсульфонат
DCHA - дициклогексиламин
Ala (A) - остаток аланина
Asp (D) - остаток аспарагиновой кислоты
Glu (E) - остаток глутаминовой кислоты
Lys (K) - остаток лизина
Gly (G) - остаток глицина
Arg (R) - остаток аргинина
Trp (W) - остаток триптофана
Leu (L) - остаток лейцина
DMEM - Dulbecco's Modified Eagle's Medium, среда Игла, модифицированная Дульбекко
FBS - fetal bovine serum, сыворотка плода коровы
PBS - phosphate buffer saline, фосфатный солевой буфер, pH 7,4
HeLa (ATCC Number - CCL-2,2, subclone S3) - клетки аденокарциномы матки человека эпителиальной морфологии
NIH 3T3 (ATCC Number - CRL-1658) - эмбриональные фибробласты мыши
DMSO - диметилсульфоксид
EDTA -этилендиаминтетраацетат
Изобретения иллюстрируется следующими примерами:
Пример 1. H-Asp-Glu-OH (DE)
1. Boc-Asp(OBzl)-Glu (OBzl)-OBzl_
К охлажденному до -20°C раствору 48,5 г (150 ммоль) Boc-Asp (Bzl)-OH в 300 мл ДМФ добавляли 16,5 мл (150 ммоль) NMM и 19,5 мл (150 ммоль) изобутилхлороформиата и перемешивали в течение 15 мин. Смесь перемешивали при той же температуре 20 мин. Далее прибавляли охлажденный до -20°C раствор 74,5 г (155 ммоль) аминокомпонента H-Glu(Bzl)-OBzlTos в 200 мл ДМФ, содержащий 17 мл NMM (155 ммоль). Реакционную смесь перемешивали в течение 1 часа при -10°C, в течение 2 часов при комнатной температуре. Упаривали, остаток растворяли в этилацетате (500 мл) и последовательно промывали 5% NaHCO3, H2O, 2% H2SO4, Н2О (дважды по 200 мл каждого), упаривали, вновь упаривали с изопропиловым спиртом. К образовавшемуся маслу приливали 500 мл гексана и 50 мл эфира, и образовавшееся масло затирали до образования твердых частиц. Продукт кристаллизуется в холодильнике. Осадок отфильтровывали, промывали на фильтре гексаном, сушили. Получено 89 г (89%) хроматографически однородного продукта.
2. H-Asp-Glu-OH
12,64 г (20 ммоль) защищенного дипептида Boc-Asp(Bzl)-Glu(Bzl)-OBzl растворяли в 100 мл хлороформа и к полученному раствору при 0°C прибавляли 100 мл трифторуксусной кислоты, смесь выдерживали 1 час при комнатной температуре и растворители упаривали в вакууме. Остаток растворяли в 300 мл этилацетата и промывали 5% раствором бикарбоната 3 раза по 200 мл. Этилацетат упаривали, остаток растворяли в 500 мл 80% уксусной кислоты и гидрировали над 5% Pd/C при комнатной температуре. После завершения гидрирования (контроль по ТСХ Rf система: хлороформ: метанол: 32% уксусная кислота - 60:45:20) катализатор отфильтровывали, фильтрат упаривали, остаток растворяли в 300 мл воды, повторно упаривали до половины объема и лиофилизовали. Получили 4,8 г (91%) целевого дипептида. По данным ВЭЖХ чистота продукта больше 95%.
Пример 2. H-Ala-Asp-Glu-OH (ADE)
1. Z-Ala-Asp(OBzl)-Glu(OBzl)2
19 г (30 ммоль) Boc-Asp(OBzl)-Glu(OBzl)2 растворяли в 200 мл TFA, через 1 час упаривали, остаток растворяли в 500 мл сухого DMF и прибавляли порциями Na2CO3 до прекращения выделения CO2. Осадок отфильтровывали, промывали 100 мл DMF, прибавляли 3,33 мл (30 ммоль) N-метилморфолина и 9,72 г (30 ммоль) Z-Ala-ONSu и оставляли на 15 часов при комнатной температуре. Реакционную смесь упаривали на 80%, к образовавшемуся сиропообразному остатку прибавляли 800 мл изопропилового спирта, выдерживали 2 часа при +4°C, мелкий осадок отфильтровывали, промывали изопропиловым спиртом, эфиром, сушили. Выход 20,3 г (92%) продукта.
2. H-Ala-Asp-Glu-OH
20,3 г (28 ммоль) защищенного трипептида Z-Ala-Asp(OBzl)-Glu(OBzl)2 при нагревании до +50°C растворяли в 1000 мл АсОН + 100 мл воды и гидрировали над 5% Pd/C, периодически подогревая реакционную смесь. Ход реакции контролировали с помощью ТСХ. После окончания катализатор отфильтровывали, фильтрат упаривали до консистенции жидкого масла и прибавляли 800 мл изопропилового спирта. Образовавшийся осадок отфильтровывали, промывали изопропиловым спиртом, эфиром, сушили. Вещество растворяли в деионизированной воде, упаривали остатки изопропилового спирта и лиофилизировали. Выход 7,67 г (85%) продукта. ВЭЖХ более 95%.
В спектре ПМР:
Ala - 3,81 (α-СН); 1,35 (β-СН3); 8,08 (NH2)
Asp - 4,62 (α-СН); 2,70; 2,53 (β-СН2); 8,63 (NH)
Glu - 4,20 (α-СН); 1,97; 1,78 (β-СН2); 2,26 (γ-СН2); 8,19 (NH).
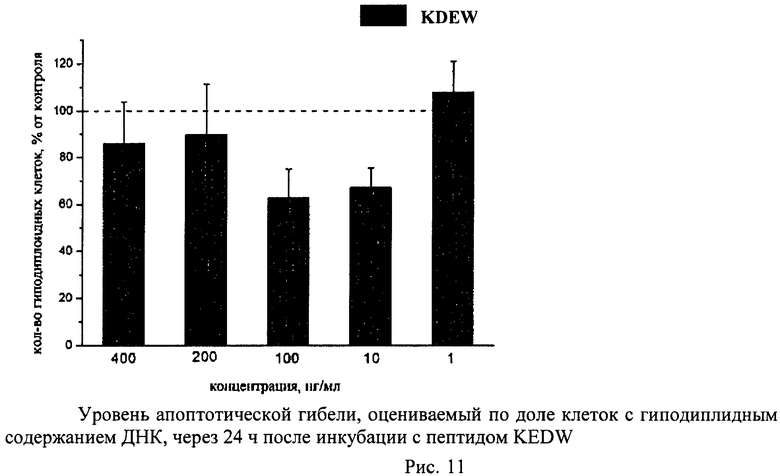
Пример 3. H-Lys-Asp-Glu-OH (KDE)
1. Z-Lys (Z)-Asp(OBzl)-Glu(OBzl)2
19 г (30 ммоль) Boc-Asp(OBzl)-Glu(OBzl)2 (1) растворяли в 200 мл TFA, через час упаривали, остаток растворяли в 500 мл сухого DMF и прибавляли порциями Na2CO3 до прекращения выделения CO2. Осадок отфильтровывали, промывали 100 мл DMF, прибавляли 3,33 мл (30 ммоль) N-метилморфолина и 15,36 г (30 ммоль) Z-Lys(Z)-ONSu и оставляли при комнатной температуре на 15 часов. Реакционную смесь упаривали, образовавшийся сиропообразный остаток растворяли в 300 мл этилацетата и промывали последовательно 2% серной кислотой, водой до нейтральной реакции, 5% раствором соды и снова водой. Этилацетат упаривали и к маслообразному остатку прибавляли 500 мл изопропилового спирта, выдерживали 2 часа при +4°C, осадок отфильтровывали, промывали изопропиловым спиртом, эфиром, сушили. Выход 21,6 г (90%) продукта.
2. H-Lys-Asp-Glu-OH
21,6 г (27 ммоль) соединения Z-Lys (Z)-Asp(OBzl)-Glu(OBzl)2, полученного как описано выше, растворяли при нагревании в 0,5 л ледяной уксусной кислоты, добавляли 50 мл воды и гидрировали в присутствии 5 г палладиевого катализатора. Катализатор отфильтровывали и упаривали растворители. Кристаллизовали из смеси уксусная кислота-этанол. При необходимости продукт очищают ионообменной хроматографией на колонке с SP-сефадексом, наносят в воде, смывают 0,04 М пиридин-ацетатным буфером (рН 5,4). Фракции, содержащие искомое соединение, упаривали. Сиропообразный остаток растворяли в 300 мл воды, упаривали до половины объема, обрабатывали активированным углем и лиофилизовали. Выход 8,95 г (85%) продукта. Чистота продукта по данным ВЭЖХ более 95%.
В спектре ПМР:
Lys - 3,76 (α-СН); 1,72 (β-СН2); 1,36; 1,52 (γ-СН2); 2,74 (ε-СН2); 8,11 (NH2)
Asp - 4,63 (α-СН); 2,71; 2,54 (β-СН2); 8,68 (NH)
Glu - 4,19 (α-СН); 1,98; 1,78 (β-СН2); 2,28 (γ-СН2); 8,31 (NH).
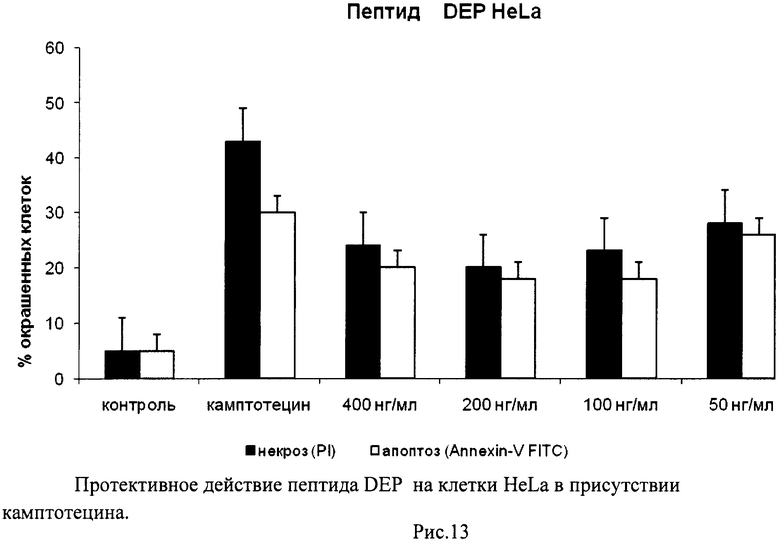
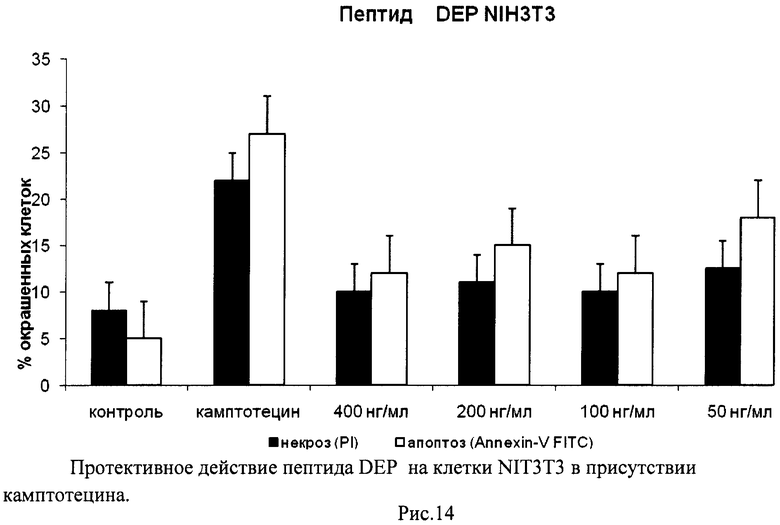
Пример 4. H-Asp-Glu-Pro-OH (DEP)
1. Z-Asp(OBut)-Glu(OBut)OH·DCHA
17,5 г (100 ммоль) H-Glu(OBut)-OH растворяли (суспендировали) в метаноле, добавляли 50 мл 40% Triton В в метаноле и после полного растворения упаривали в вакууме. Остаток растворяли в 300 мл диметилфорамида, упаривали на четверть и прибавляли 42,0 г (100 ммоль) Z-Asp(OBut)-ONSu. Через 12 часов (контроль - ТСХ в системе хлороформ: метанол: уксусная кислота - 9:1:0.5 «Б») реакционную смесь упаривали, остаток растворяли в этилацетате, промывали последовательно 2% серной кислотой, водой, сушили над Na2SO4, упаривали. Остаток растворяли в 150 мл эфира, прибавляли 20 мл (100 ммоль) дициклогексиламина и оставляли при комнатной температуре до полного выпадения осадка. Осадок отфильтровывали, промывали эфиром, высушивали на воздухе. Выход 60 г (87%) хроматографически однородного вещества.
2. H-Asp-Glu-Pro-OH
28 г (40 ммоль) Z-Asp(OBut)-Glu(OBut)-ОН·DCHA суспендировали в 500 мл этилацетата, промывали 2% серной кислотой, водой, сушили над Na2SO4, осушитель отфильтровывали, фильтрат упаривали, остаток растворяли в 150 мл диметилформамида, охлаждали до -25°C, к охлажденному раствору добавляли 4,4 мл (40 ммоль) N-метилморфолина и 5,2 мл (40 ммоль) изобутилхлорформиата, выдерживали при -20°C в течение 15 мин и добавляли охлажденный до -25°C раствор 5,6 г (44 ммоль) N-гидроксисукцинимида 25°C в 50 мл диметилформамида. Через 30 мин добавляли раствор 10,6 г (44 ммоль) H-Pro-OBzl HCl и 5 мл N-метилморфолина в 50 мл диметилформамида и оставляли при комнатной температуре на ночь. Реакционную смесь упаривали, остаток растворяли в этилацетате и промывали обычным способом. Этилацетат упаривали, остаток упаривали с толуолом. Образовавшееся масло растворяли в TFA, выдерживали в течение часа, упаривали, остаток обрабатывали смесью эфир - гексан 1:1, выпавший осадок отфильтровывали, промывали на фильтре смесью эфир - гексан 1:1, гексаном, сушили в эксикаторе над щелочью.
Полученный продукт растворяли в смеси этанол - вода - уксусная кислота (600:100:100) и гидрировали над 5% Pd\С. Катализатор отфильтровывали, фильтрат упаривали. Остаток переосаждали из уксусной кислоты изопропиловым спиртом, фильтровали, промывали на фильтре изопропиловым спиртом, эфиром, сушили. Выход 12,2 г (85%). Чистота по данным ВЭЖХ более 95%.
В спектре ПМР:
Asp - 4,13 (α-СН); 2,79; 2,66 (β-СН2); 8,14 (NH2)
Glu - 4,59 (α-СН); 1,93; 1,72 (β-СН2); 2,36 (γ-СН2); 8,67 (NH)
Pro - 4,23 (α-СН); 2,14; 1,84 (β-СН2); 1,90 (γ-СН2); 3,63 (δ-СН2).
Пример 5. H-Asp-Glu- Arg-OH (DER)
1. Z-Glu(OtBu)-ArgOH
К раствору 104,4 г (228 ммоль) Z-Glu(OtBu)ONP в 0,5 л диметилформамида прибавляли 38,3 г (228 ммоль) Н-Arg-OH. Реакционную смесь перемешивали при комнатной температуре 24 часа. Затем выдерживали при температуре -4°C четыре часа, образовавшийся осадок отфильтровывали и промывали на фильтре 0,4 л смеси диметилформамид-эфир (1:2), 0,45 л эфира, 0,4 л гексана. Сушили. Выход: 91,7 г (82,3%) хроматографически однородного Z-Glu(OtBu)ArgOH (контроль в системе: хлороформ-метанол-уксусная кислота (5:3:1)).
2. H-Asp-Glu- Arg-OH
Раствор 90,6 г (185 ммоль) ZGlu(OtBut)ArgOH в 1,2 л этилового спирта гидрировали над 10 г 5% Pd/C. После окончания гидрирования (контроль по ТСХ в системе: хлороформ-метанол-уксусная кислота (5:3:1)) катализатор отфильтровывали, растворитель упаривали. Полученный сиропообразный продукт растворяли в 500 мл диметилформамида и к полученному раствору прибавляли 75,9 г (185 ммоль) Вос-Asp(OtBu)-ONP. Реакционную смесь оставляли при комнатной температуре на 15 часов до завершения реакции (контроль в системе: хлороформ-метанол-уксусная кислота (5:3:1)). Растворитель упаривали, образовавшееся масло растворяли в 150 мл хлороформа и наносили на колонку с силикагелем (400 г), промывали хлороформом до схода нитрофенола, 10% этиловым спиртом в хлороформе, смывали 30% этиловым спиртом в хлороформе, упаривали (контроль в системе: хлороформ-метанол-уксусная кислота (5:3:1)). К остатку приливали 400 мл трифторуксусной кислоты и смесь выдерживали 2 часа при комнатной температуре. Кислоту упаривали, к остатку приливали 1000 мл диэтилового эфира, образовавшийся аморфный осадок отфильтровывали, промывали на фильтре эфиром два раза по 150 мл и сушили. Вещество растворяли в 1500 мл воды, упаривали до масла, повторно растворяли в 1000 мл воды и наносили на колонку с SP-сефадексом. Колонку промывали двумя объемами 0,05 М пиридин-ацетатного буфера и смывали вещество тем же 0,2 М буфером. Фракции, содержащие продукт, объединяли, растворители упаривали, повторно упаривали с водой для удаления остатков пиридин-ацетата и лиофилизовали. Получили 65 г (75%) продукта с чистотой по данным ВЭЖХ более 95%.
В спектре ПМР:
Asp - 4,16 (α-СН); 2,82; 2,65 (β-СН2); 8,14 (NH2)
Glu - 4,35 (α-СН) 1,93; 1,87 (β-СН2); 2,30 (γ-СН2); 8,60 (NH)
Arg - 4,16 (α-СН); 1,75; 1,62 (β-СН2); 1,54; 1,50 (γ-СН2); 3,10 (δ-СН2); 7,58 (ε-NH2); 8,28 (NH).
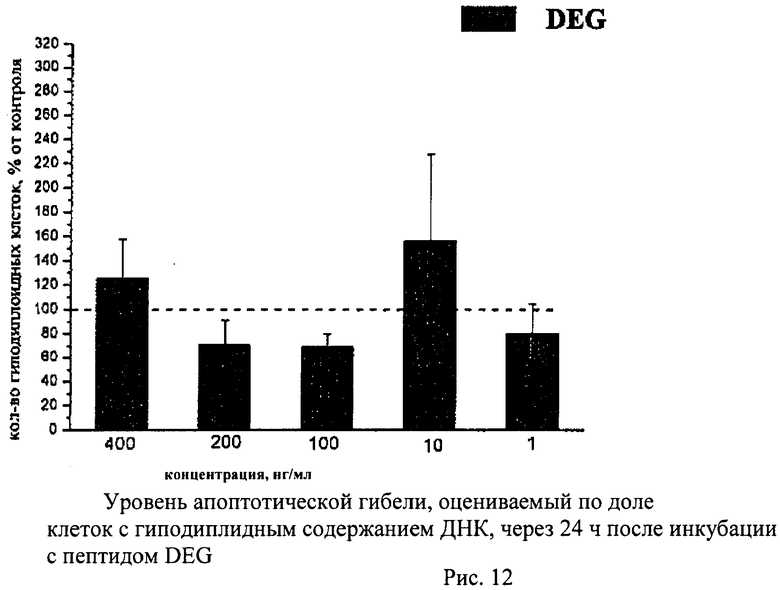
Пример 6. H-Asp-Glu-Gly-OH (DEG)
1. Boc-Glu (BzI)-Gly-Bzl
К охлажденному до -20°C раствору 8,43 г (25 ммоль) Boc-Glu (Bzl)-OH в 100 мл ДМФ добавляли 2,77 мл (25 ммоль) NMM и 3,25 мл (25 ммоль) изобутилхлороформиата, перемешивали в течение 15 мин и прибавляли охлажденный до -20°C раствор 7,97 г (25 ммоль) аминокомпонента H-Gly-OBzl×Tos в 50 мл ДМФ, содержащий 3 мл NMM (27 ммоль). Реакционную смесь перемешивали в течение 1 часа при -10°C, в течение 2 часов при комнатной температуре. Упаривали, остаток растворяли в этилацетате (200 мл) и последовательно промывали 5% NaHCO3, Н2О, 2% H2SO4, H2O (дважды по 200 мл каждого), упаривали, вновь упаривали с изопропиловым спиртом. Продукт кристаллизуется из смеси изопропиловый спирт-гексан 1:1 при +4°C в холодильнике. Осадок отфильтровывали, промывали на фильтре гексаном, сушили. Получено 11 г (91%) хроматографически однородного продукта.
2. Z-Asp(Bzl)-Glu(Bzl)-Gly-Bzl
10 г (20,7 ммоль) Boc-Glu(OBzl)-Gly-OBzl растворяли в 50 мл TFA, через час упаривали, остаток растворяли в 100 мл сухого DMF и прибавляли порциями Na2CO3 до прекращения выделения CO2. Осадок отфильтровывали, промывали 50 мл DMF, к полученному раствору прибавляли 9,53 г (21 ммоль) Z-Asp(Bzl)-ONSu и оставляли при комнатной температуре на 15 часов. Реакционную смесь упаривали, образовавшийся сиропообразный остаток растворяли в 300 мл этилацетата и промывали последовательно 2% серной кислотой, водой до нейтральной реакции, 5% раствором соды и снова водой. Этилацетат упаривали и к маслообразному остатку прибавляли 100 мл изопропилового спирта, выдерживали 2 часа при +4°C, осадок отфильтровывали, промывали изопропиловым спиртом, эфиром, сушили. Выход 12 г (80%) хроматографически однородного продукта.
3. H-Asp-Glu-Gly-OH
7,23 г (27 ммоль) Z-Asp(Bzl)-Glu(Bzl)-Gly-Bzl, полученного, как описано выше, растворяли при нагревании в 0,5 л ледяной уксусной кислоты, добавляли 50 мл воды и гидрировали в присутствии 5 г палладиевого катализатора. Катализатор отфильтровывали и растворители упаривали. Продукт переосаждали из смеси уксусная кислота-изопропиловый спирт. Получили 3,0 г (94%). Чистота продукта по данным ВЭЖХ более 95%.
В спектре ПМР:
Asp - 4,14 (α-СН); 2,87; 2,68 (β-СН2); 8,16 (NH2)
Glu - 4,35 (α-СН) 1,94; 1,80 (β-СН2); 2,31 (γ-СН2); 8,63 (NH)
Gly - 3,75 (α-СН2); 8,28 (NH).
Пример 7. H-Ala-Asp-Glu-Leu-OH (ADEL)
1. Boc-Glu(Bzl)-Leu-OBzl
К раствору 26,6 г (78 ммоль) Boc-Glu(Bzl)-OH в 200 мл диметилформамида и 8,65 мл N-метилморфолина, охлажденному до -15°C, прибавляли 10,4 мл (80 ммоль) изобутилхлорформиата, смесь перемешивали при этой температуре 20 мин и прибавляли охлажденный до -15°C раствор 30,5 г (78 ммоль) H-Leu-OBzl*Tos, рН которого предварительно был доведен до 8,5-9,0. Через час растворители упаривали, остаток растворяли в 500 мл этилацетата и промывали последовательно 400 мл 2% раствором серной кислоты, водой, 5% раствором соды и водой. Органический слой отделяли и этилацетат упаривали. Остаток кристаллизовали из смеси эфир-гексан (1:1). Получили 37,8 г (90%) хроматографически однородного вещества.
2. Boc-Asp(Bzl)-Glu(Bzl)-Leu-OBzl
27 г (50 ммоль) Boc-Glu(Bzl)-Leu-OBzl растворяли в 100 мл хлороформа и к полученному раствору прибавляли 100 мл трифторуксусной кислоты. Смесь выдерживали при комнатной температуре 2 часа и растворители упаривали. Остаток растворяли в 200 мл диметилформамида и порциями прибавляли соду до окончания выделения CO2. Осадок отфильтровывали, промывали 50 мл диметилформамида, в фильтрате выводили рН до 8,5 и охлаждали до -20°C (Раствор 1).
Одновременно к раствору 16,5 г (50 ммоль) Boc-Asp(Bzl)-OH в 100 мл диметилформамида прибавляли 5,6 мл N-метилморфолина, смесь охлаждали до - 20°C и приливали 6,5 мл (50 ммоль) изобутилхлорформиата. Смесь выдерживали при этой температуре 30 мин (Раствор 2).
Сохраняя охлаждение, оба раствора смешивали и оставляли перемешиваться до достижения комнатной температуры (примерно 2 часа.). Растворители упаривали, остаток растворяли в 300 мл этилацетата и последовательно промывали 2% раствором серной кислоты, водой, 5% раствором соды, водой и растворитель упаривали. После кристаллизации из эфира получили 32,4 г (87%) хроматографически однородного Вос-Asp(Bzl)-Glu(Bzl)-Leu-OBzl.
3. Z-Ala-Asp(Bzl)-Glu(Bzl)-Leu-OBzl
22,2 г (30 ммоль) Boc-Asp(Bzl)-Glu(Bzl)-Leu-OBzl растворяли в 100 мл хлороформа и к полученному раствору прибавляли 100 мл трифторуксусной кислоты. Смесь выдерживали при комнатной температуре 2 часа и растворители упаривали. Остаток растворяли в 200 мл диметилформамида и порциями прибавляли соду до окончания выделения CO2. Осадок отфильтровывали, промывали 50 мл диметилформамида, в фильтрате выводили рН до 8,5 и прибавляли 9,6 г (30 ммоль) Z-Ala-ONSu. Реакционную смесь оставляли на 15 часов при комнатной температуре. Растворители упаривали, остаток растворяли в 500 мл этилацетата и последовательно промывали 2% раствором серной кислоты, водой, 5% раствором соды, водой и растворитель упаривали. После кристаллизации из изопропилового спирта получили 22,4 г (87,8%) хроматографически однородного Z-Ala-Asp(Bzl)-Glu(Bzl)-Leu-OBzl.
4. H-Ala-Asp-Glu-Leu-OH
17 г (20 ммоль) защищенного тетрапептида Z-Ala-Asp(Bzl)-Glu(Bzl)-Leu-OBzl растворяли в 300 мл 80% уксусной кислоты и гидрировали над 5% Pd\C при комнатной температуре. После окончания реакции катализатор отфильтровывали, промывали на фильтре уксусной кислотой и растворители упаривали. Остаток растворяли в горячей воде, охлаждали до комнатной температуры и оставляли при +4°C на 15 часов. Выпавший осадок отфильтровывали, сушили в вакууме над КОН. Получили 8 г (89%) продукта с чистотой по данным ВЭЖХ более 95%.
В спектре ПМР:
Ala - 3,83 (α-СН); 1,34 (β-СН3); 8,08 (NH2)
Asp - 4,61 (α-СН); 2,72; 2,54 (β-СН2); 8,68 (NH)
Glu - 4,28 (α-СН); 1,91; 1,76 (β-СН2); 2,23 (γ-СН2); 7,93 (NH)
Leu - 4,17 (α-СН); 1,52 (β-СН2); 1,74; 1,62 (γ-СН2); 0,83 (δ-СН3); 0,89 (δ-СН3); 8,12 (NH).
Пример 8. Н-Asp-Glu-Leu-OH (DEL)
14,9 г (20 ммоль) защищенного трипептида Boc-Asp(Bzl)-Glu(Bzl)-Leu(Bzl) растворяли в 50 мл хлороформа и к полученному раствору прибавляли 50 мл трифторуксусной кислоты. Смесь выдерживали 1 час при комнатной температуре и растворители упаривали. Остаток растворяли в 300 мл 80% уксусной кислоты и гидрировали над 5% Pd\C при комнатной температуре. После окончания реакции катализатор отфильтровывали, промывали на фильтре уксусной кислотой и растворители упаривали. К остатку приливали 200 мл изопропилового спирта. Выпавший осадок отфильтровывали, сушили в вакууме над КОН. Получили 7 г (93%) продукта с чистотой по данным ВЭЖХ более 95%.
В спектре ПМР:
Asp - 4,13 (α-СН); 2,81; 2,65 (β-СН2); 8,15 (NH2)
Glu - 4,34 (α-СН); 1,94; 1,80 (β-СН2); 2,30 (γ-СН2); 8,58 (NH)
Leu - 4,19 (α-СН); 1,52 (β-СН2); 1,53; 1,62 (γ-СН2); 0,83 (δ-СН3); 0,89 (δ-СН3); 8,22 (NH).
Пример 9. H-Lys-Asp-Glu-Trp-NH2(KDEW-NH2)
1. Z-Glu(OtBu)-Trp-NH2
К раствору 35,4 г (100 ммоль) Z-Glu(OtBu)-OH в 200 мл диметилформамида и 11,1 мл N-метилморфолина, охлажденному до -15°C, прибавляли 13 мл (100 ммоль) изобутилхлорформиата, смесь перемешивали при этой температуре 20 мин и прибавляли охлажденный до -15°C раствор 20,4 г (100 ммоль) H-Trp-NH2, рН которого предварительно был доведен до 8,5-9,0. Через час растворители упаривали, остаток растворяли в 500 мл этилацетата и промывали последовательно 400 мл 2% раствором серной кислоты, водой, 5% раствором соды и водой. Органический слой отделяли и этилацетат упаривали. Остаток кристаллизовали из смеси эфир-гексан (1:1). Получили 45,2 г (86%) хроматографически однородного Z-Glu(OtBu)-Trp-NH2.
2. Z-Asp(OtBu)-Glu(OtBu)-Trp-NH2
К охлажденному до -20°C раствору 24,7 г (76,5 ммоль) Z-Asp (OtBu)-OH в 300 мл ДМФ добавляли 8,5 мл (76,5 ммоль) NMM и 9,95 мл (765 ммоль) iBuOCOCl и перемешивали в течение 15 мин. Смесь перемешивали при той же температуре 20 мин. Далее прибавляли охлажденный до -20°C раствор, полученный при гидрировании 40 г (76,5 ммоль) Z-Glu(OtBu)-Trp-NH2 в 200 мл ДМФ. Реакционную смесь перемешивали в течение 1 часа при -10°C, в течение 2 часов при комнатной температуре. Полноту протекания реакции контролировали в системе хлороформ: метанол: уксусная кислота - 9:1:0.5. Растворитель упаривали, остаток растворяли в этилацетате (500 мл) и последовательно промывали 5% NaHCO3, H2O, 2% H2SO4, H2O (дважды по 200 мл каждого), упаривали, вновь упаривали с изопропиловым спиртом. Продукт кристаллизовался из смеси этилацетат-гексан (2:1) при +4°C в холодильнике. Осадок отфильтровывали, промывали на фильтре гексаном, сушили. Получили 46,1 г (87%) хроматографически однородного продукта.
3. Z-Lys(Z)-Asp(OtBu)-Glu(OtBu)-Trp-NH2
40 г (57,6 ммоль) полученного, как описано выше, защищенного трипептида
Z-Asp(OtBu)-Glu(OtBu)-Trp-NH2 растворяли в 400 мл спирта и гидрировали над 5% Pd\C. После завершения реакции катализатор отфильтровывали, спирт упаривали, остаток растворяли в 200 мл диметилформамида и к полученному раствору прибавляли 29,5 г (57,6 ммоль) Z-Lys(Z)-ONSu. Смесь оставляли на 15 часов при комнатной температуре. Растворитель упаривали, остаток растворяли в горячем этилацетате и оставляли кристаллизоваться. Образовавшийся осадок отфильтровывали, промывали смесью этилацетат-гексан (1:1), сушили. Получили 45 г (82%) хроматографически однородного вещества.
4. H-Lys-Asp-Glu-Trp-NH2
40 г (41,8 ммоль) Z-Lys(Z)-Asp (OtBu)-Glu(OtBu)-Trp-NH2 растворяли в 500 мл спирта, прибавляли 5 г 5% Pd\C в 50 мл воды и гидрировали до окончания реакции. Катализатор отфильтровывали, растворители упаривали. Остаток обрабатывали 200 мл трифтруксусной кислоты, содержащей 1% меркаптоэтанола. Через 1 час кислоту упаривали и к остатку прибавляли 500 мл эфира. Выпавший осадок отфильтровывали, промывали на фильтре эфиром и сушили. Растворяли в 100 мл воды и наносили на колонку с SP-сефадексом (или аналогичным катионообменником), уравновешенным 0,05 М пиридин-ацетатным буфером. Промывали двумя объемами стартового буфера, одним объемом 0,1 М буфера и вещество смывали 0,2 М буфером. Фракции, содержащие целевой продукт, объединяли, растворители упаривали, повторно упаривали с 300 мл воды для удаления следов пиридин-ацетатного буфера. Остаток растворяли в 400 мл воды и лиофилизовали. Получали 26 г (79%) хроматорафически однородного продукта с чистотой по данным ВЭЖХ более 95%.
В спектре ПМР:
Lys - 3,75 (α-СН), 1,70 (β-СН2), 1,51; 1,34 (γ-СН2), 2,73 (ε-СН2), 8,12 (NH2)
Asp - 4,61 (α-СН), 2,55; 2,72 (β-СН2), 8,73 (NH)
Glu - 4,19 (α-СН),1,86; 1,73 (β-СН2), 2,18 (γ-СН2), 8,16 (NH)
Trp - 4,43 (α-СН), 3,14, 2,98 (β-СН2), 7,84 (NH); 7,58; 6,96; 7,05; 7,81 (СН ароматические); 10,8 (NH индола).
Пример 10. Исследование стабильности пептидов при хранении.
Пептиды растворяли в 0,05 М ацетатаммонийном буфере с рН 4,5 и рН 7,5 с таким расчетом, чтобы их концентрация была примерно 1 мг/мл. Растворы стерильно фильтровали и хранили в стерильных, хорошо закупоренных флаконах. По истечении срока хранения вещества анализировали методом ВЭЖХ. Результаты представлены в таблице 1.
Результаты представлены в виде чистоты исследованных продуктов, выраженной в процентах.
Представленные результаты анализов свидетельствуют о том, что продукты хорошо хранятся даже в растворах при различных значениях рН.
Пример 11. Исследование биологической активности пептидных препаратов в условиях стандартного культивирования клеток
Было проведено изучение биологической активности синтезированных пептидов в пяти различных концентрациях (400, 200, 100, 10 и 1 нг/мл) с использованием в качестве модельной системы культивируемые клетки мыши и человека: HeLa S3 и NIH 3Т3.
Биологическая активность каждого пептида была определена по следующим параметрам: влиянию пептидов на пролиферативную активность клеток и способности пептидов вызывать апоптоз и некроз. Для этого были исследованы следующие показатели: доля погибших клеток, общее количество клеток во флаконе, уровень апоптотической гибели клеток.
Культивирование клеток и обработка пептидами
Культивирование фибробластов NIH ЗТЗ проводили в полной питательной среде DMEM (Панэко, РФ), содержащей 10% эмбриональной сыворотки (Панэко), гентамицин 2 ед/мл (Панэко), глютамин 29,2 мкг/мл (Панэко), в CO2-инкубаторе. Для культивирования использовали флаконы фирмы SPL (Южная Корея) с площадью 25 см2 и 175 см2. Пересев клеток осуществляли в соотношении 1:5-1:7. По достижении примерно 50% конфлюэнтности во флаконы добавляли пептиды в указанных выше конечных концентрациях. В каждом эксперименте использовали по два флакона с одинаковой концентрацией каждого пептида. В контрольные флаконы добавляли соответствующий объем полной среды без пептидов. С каждым пептидом проведено по два независимых эксперимента (по два повтора в каждом). Через 24 часа инкубации с пептидами клетки извлекали из культурального флакона с помощью обработки в смеси трипсина и версена (1:3, Панэко). Клетки из каждого флакона помещали в одинаковый объем забуференного физиологического раствора (0,01 М PBS, рН 7,2). Суспензию клеток делили на 3 равные части для исследования перечисленных выше показателей.
Культивирование клеток HeLa выполняли сходным образом за исключением того, что пересев клеток осуществляли в соотношении 1:3-1:4.
Исследование показателей осуществляли с помощью проточной цитометрии. Использовали проточный цитофлуориметр-сортировщик FACS Vantage (Becton Dickinson, США). Перед проведением каждой серии анализов прибор юстировали и калибровали по стандартным микросферам (Polyscience) в соответствии с методикой, рекомендованной фирмой-изготовителем.
Определение общего количества клеток во флаконе и доли погибших клеток
К суспензии клеток, полученной как описано выше, добавляли йодистый пропидий (prodium iodide-PI, Sigma, США) в конечной концентрации 10 мкг/мл и калибровочные частицы Calibrite (Becton Dickinson, США), меченные флуоресцеинизотиоционатом (ФИТЦем). Конечную концентрацию калибровочных частиц определяли с помощью камеры Горяева. Инкубировали 10-12 мин и немедленно анализировали на проточном цитометре по 4-м параметрам - прямое светорассеяние, боковое светорассеяние, флуоресценция PI, флуоресценция ФИТЦа. В каждом образце анализировали по 20 тыс.клеток. Данные записывали в файл. Затем проводили компьютерную обработку данных с помощью программы CellQuestPro (Becton Dickinson, США).
Определяли долю PI+клеток с поврежденной плазматической мембраной (погибшие клетки). Уровень гибели в контрольных флаконах (15-20 штук) усредняли и принимали за 100%. Возможное влияние пептидов оценивали по соотношению с контролем.
В каждом файле выявляли ФИТЦ+калибровочные частицы и определяли их количество. Затем по показателям светорассеяния идентифицировали клетки и находили их количество в тех же файлах. Рассчитывали соотношение клеток/калибровочных частиц в каждом файле (образце), по которому судили о количестве клеток во флаконе. Соотношение клеток/калибровочных частиц в контроле принимали за 100%. Количество клеток в опытных флаконах оценивали в процентах по сравнению с контролем.
Определение гибели клеток путем апоптоза
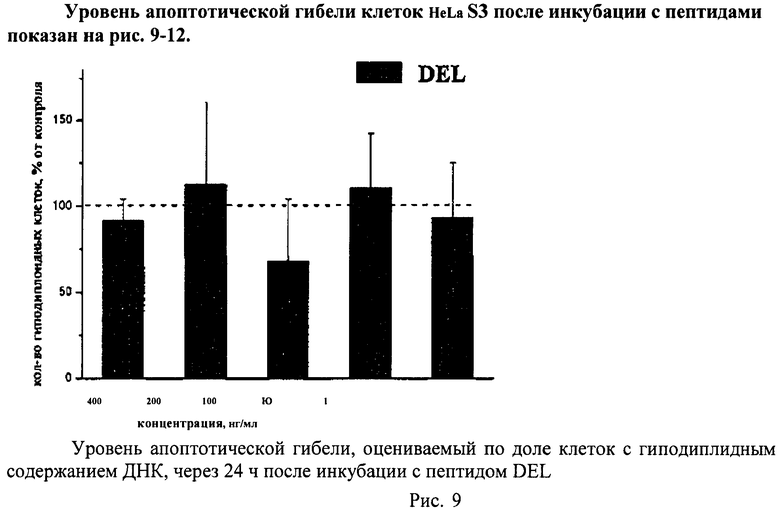
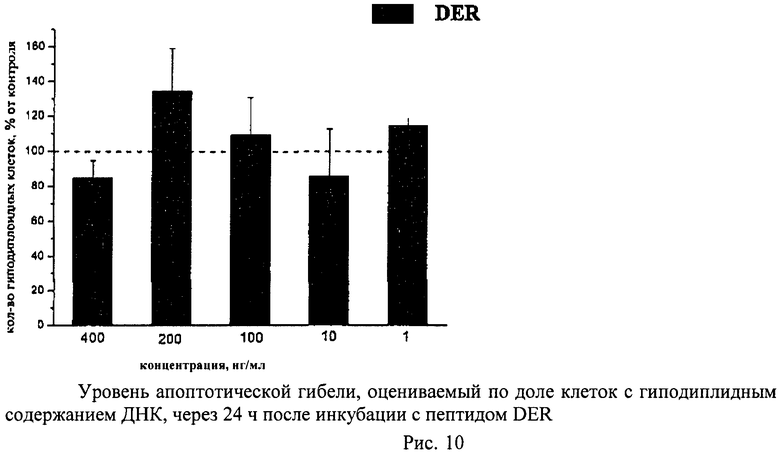
Метод основан на том, что в клетках, подвергающихся апоптотической гибели, появляется фрагментированная ДНК, которая удаляется из клеток во время обработки раствором HCl. Затем оставшуюся в клетках ДНК окрашивают специфическими красителями (например, PI) и определяют поклеточное содержание ДНК. Апоптотическую гибель клеток оценивают по доле клеток с гиподиплоидным содержанием ДНК.
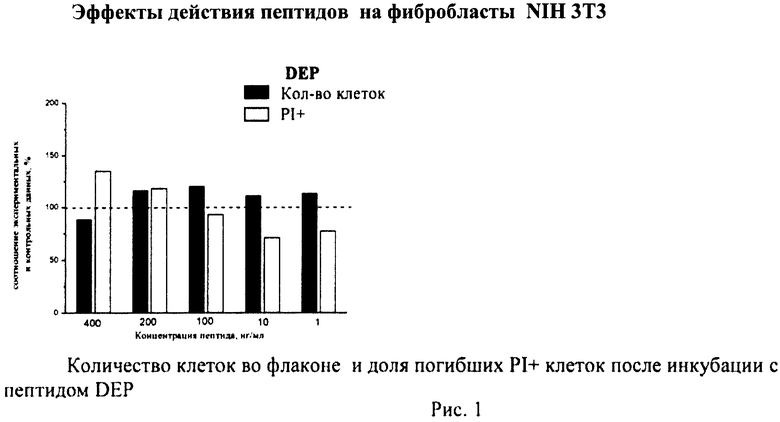
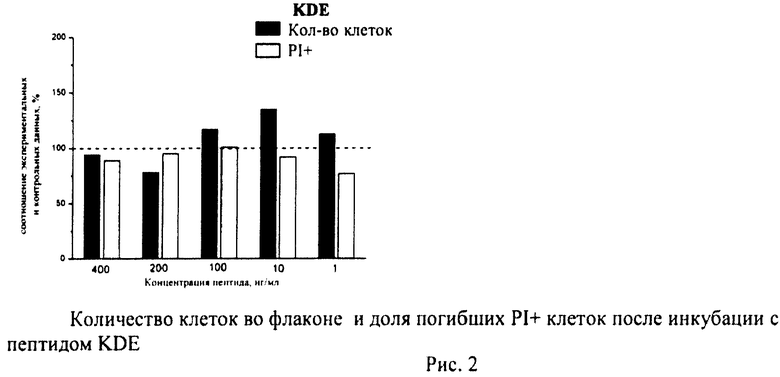
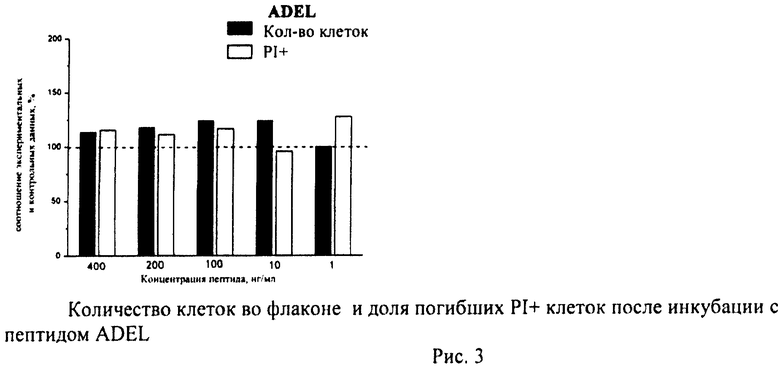
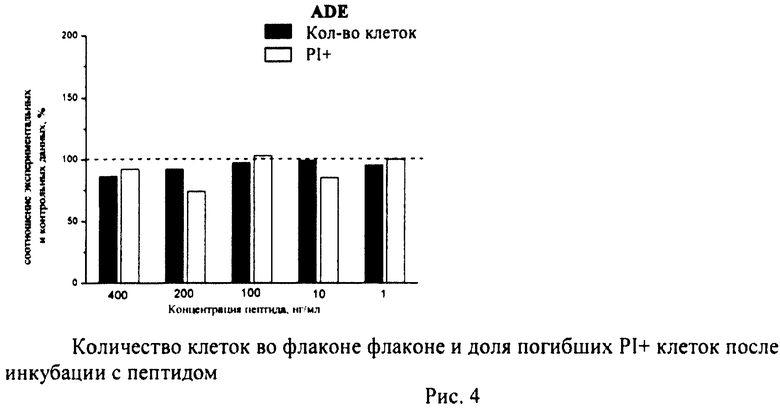
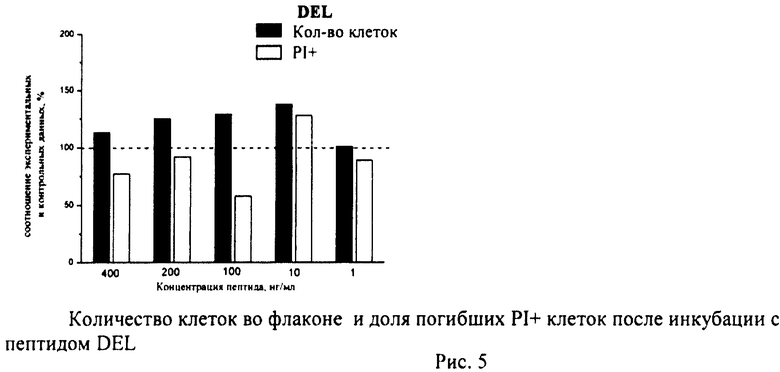
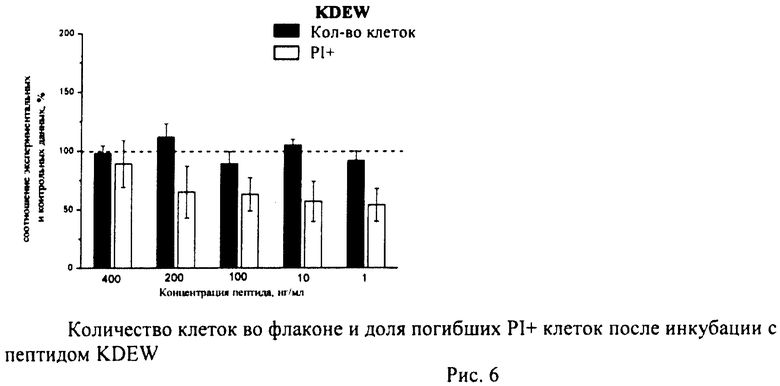
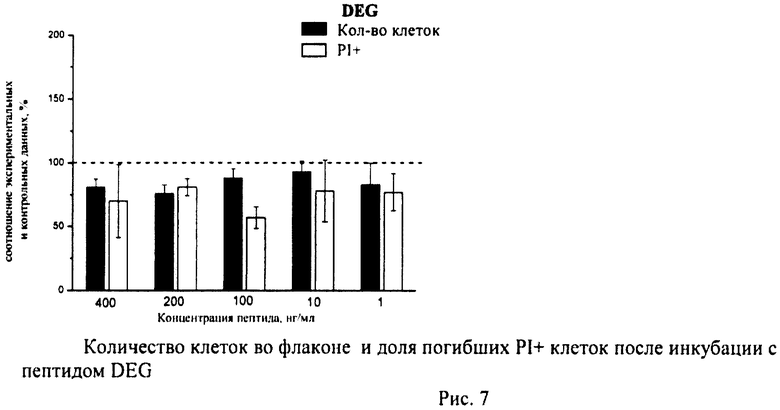
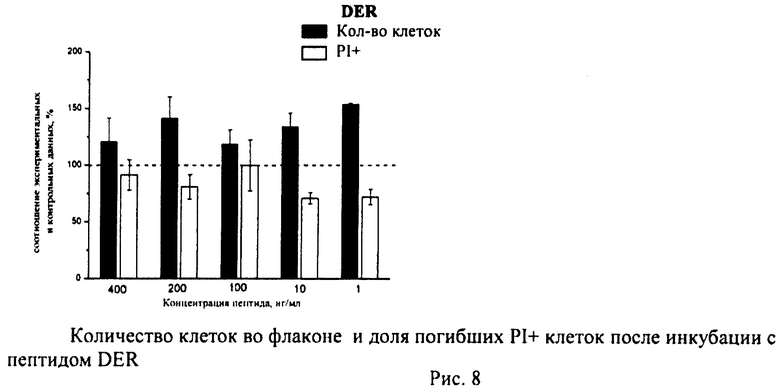
Суспензию клеток, полученную, как описано выше, охлаждали до +4°C. К суспензии клеток добавляли охлажденный этанол в объемном соотношении 1:3 при постоянном перемешивании. Клетки хранили 1-2 недели до анализа. При помощи центрифугирования удаляли этанол из образцов, затем удаляли фрагментированную ДНК из клеток с помощью 0,6М HCl (37°C, 10 мин). Далее клетки отмывали PBS и окрашивали в 0,3 мл раствора PI (50 мкг/мл) в PBS, выдерживали 15 мин при комнатной температуре и анализировали на проточном цитометре. PI в данной концентрации позволяет проводить определение поклеточного содержания ДНК и выявлять фракцию клеток с фрагментированной ДНК с высоким разрешением. Для измерения флуоресценции PI использовали узкополосные фильтры 585/42 нм, мощность лазера составляла 57 мВт. В каждом образце анализировали 5·103 клеток, затем проводили компьютерную обработку с использованием программы CellQuestPro (Becton Dickinson, США). На первом этапе анализа выделяли регион клеток по светорассеянию, затем в этом регионе оценивали флуоресценцию PI. Маркер гиподиплоидного содержания ДНК выставляли по контрольным образцам. Результаты приведены на Рис.1 - Рис.12.
Пример 12. Изучение протекторных свойств пептидов
В экспериментах по изучению влияния пептидов на апоптоз и некроз использовали набор реагентов ApoTarget Annexin-V FITC Apoptosis Kit (Invitrogen, США).
Апоптоз - программируемая клеточная гибель, являющаяся результатом реализации генетической программы или ответом на внешние факторы и требующая затрат энергии и синтеза макромолекул. Апоптоз сопровождается появлением характерных цитологических признаков (маркеров апоптоза) и молекулярных процессов. В живых клетках, в частности, наблюдается асимметричное распределение различных фосфолипидов между внутренним и наружным монослоями цитоплазматической мембраны: фосфолипиды, содержащие холин, такие как фосфатидилхолин и сфингомиелин, локализованы в основном в наружном монослое, а фосфатидилэтаноламин и фосфатидилсерин (аминофосфолипиды) - во внутреннем. В процессе апоптоза цитоплплазматическая мембрана претерпевает изменения, одним из которых является переход фосфатидилсерина из внутреннего ее монослоя в наружный, где фосфатидилсерин оказывается доступным для связывания с Annexin-V, меченным флуоресцентным красителем FITC. Annexin-V - кальций-зависимый, фосфолипид-связывающий белок с мол. массой 35-36 кДа, обладающий высоким сродством к фосфатидилхолину (Кд равна 5×10-10 M). Annexin-V-связывающий анализ основан на быстром и высоко специфическом выявлении клеток, содержащих фосфатидилхолин в наружном монослое цитоплазматической мембраны, т.е. клеток, находящихся на ранней стадии апоптоза.
Помимо Annexin-V, меченного флуоресцентным красителем FITC, набор реагентов ApoTarget Annexin-V FITC Apoptosis Kit содержит еще один краситель - пропидиум йодид (PI). PI обладает способностью проникать только в те клетки, у которых нарушена целостность цитоплазматической мембраны, что является одним из характерных признаков поздних стадий апоптоза или некроза. Проникнув в клетки с поврежденной клеточной мембраной, PI связывается с ДНК. «Красная» флуоресценция образовавшихся комплексов выявляется с помощью поточного цитофлуориметра.
Таким образом, используя процедуру двойного окрашивания клеток (Annexin-V FITC плюс PI) и метод поточной цитофлуорометрии, можно различать 3 популяции клеток и определять их процентное соотношение: 1) неапоптотические клетки (Annexin-V-негативные и PI-негативные), 2) клетки, находящиеся на ранней стадии апоптоза (Annexin-V-позитивные и PI-негативные), 3) некротические клетки или клетки, находящиеся на поздней стадии апоптоза (Annexin-V-позитивные и PI-позитивные). По увеличению доли апоптотических и некротических клеток можно судить о степени влияния добавляемых в среду культивирования соединений на упомянутые процессы. В качестве положительного контроля в экспериментах использовали рекомендуемое производителем соединение камптотецин (Camptothecin, Sigma, США), обладающее ярко выраженной способностью вызывать апоптоз и некроз клеток, в особенности опухолевых.
Растворы пептидов в стерильном PBS с концентрациями 1 мг/мл готовили непосредственно перед проведением экспериментов по изучению влияния пептидов на апоптоз и некроз. Для изучения пролиферативной активности использовали те же растворы, которые после приготовления хранили в стерильных пробирках при +4°C.
В день проведения каждого эксперимента готовили рабочие разведения пептидов в среде культивирования клеток. Для этого использовали стерильный пластиковый 96-луночный планшет (Corning, США). Разведения готовили таким образом, чтобы при внесении 25 мкл каждого рабочего разведения в экспериментальную лунку с культивируемыми клетками конечные концентрации пептидов составляли 400, 200, 100 и 50 нг/мл.
Культивируемые клетки
Для культивирования клеток HeLa S3 и NIH ЗТЗ использовали реагенты компании Invitrogen (США) и пластиковые флаконы, планшеты, пипетки и фильтры компании Corning (США).
Клетки культивировали в среде DMEM, приготовленной из сухого концентрата, растворенного в апирогенной дистиллированной воде (Millipore, США). Среда содержала 10% FBS, 2 mM глутамина и пенициллин-стрептомицин (разбавленный в соответствии с рекомендациями производителя). HeLa S3 и NIH 3Т3 выращивали во флаконах Т150 при +37°C в атмосфере 5,6% CO2. Клетки постоянно поддерживали в логарифмической фазе роста, для чего рассеивали их не реже 2 раз в неделю, используя трипсин для снятия прикрепленных клеток с поверхности культуральных флаконов. Подсчет живых клеток проводили в гемацитометре после окрашивания аликвоты суспензии трипановым синим.
Изучение способности пептидов вызывать апоптоз и некроз культивируемых клеток
Клетки HeLa S3 и NIH 3Т3 снимали с поверхности культуральных флаконов, подсчитывали, доводили концентрацию до 105 кл/мл и рассеивали полученные суспензии по 1 мл в лунки 24-луночных культуральных планшетов.
На следующий день, убедившись, что клетки прикрепились к поверхности планшетов, среду культивирования во всех лунках меняли на свежую.
В контрольные лунки (положительный контроль - стимуляция апоптоза и некроза) добавляли камптотецин до конечной концентрации 50 мкМ, которая была определена в предварительных экспериментах с клетками NIH 3Т3 и HeLa S3. В контрольные лунки (отрицательный контроль) добавляли 25 мкл свежей среды культивирования, в экспериментальные лунки добавляли по 25 мкл рабочих разведений пептидов (каждое рабочее разведение - в 2 повторах) и камптотецин в той же конечной концентрации. Инкубацию проводили при +37°C.
Через 24 часа после начала инкубации клетки снимали с поверхности 24-луночных планшетов, переносили из каждой лунки в отдельную центрифужную пробирку объемом 1,5 мл, промывали 1 мл рабочего раствора буфера для связывания красителя Annexin-V FITC и осаждали клетки центрифугированием (Sysmex Platelet Centrifuge РС-810). Супернатант сливали, к клеточному осадку добавляли 5 мкл красителя Annexin-V FITC и 10 мкл красителя пропидиум йодид (PI) и инкубировали 15 мин в темноте при комнатной температуре (+25°C). Затем в каждую пробирку добавляли 300 мкл рабочего раствора буфера для связывания Annexin-V FITC и измеряли интенсивности зеленой и красной флуоресценции.
В полученных распределениях выделяли две популяции - Annexin-V-позитивные и PI-негативные (клетки, находящиеся в стадии апоптоза) и Annexin-V-позитивные и PI-позитивные клетки (клетки, находящиеся в стадии некроза).
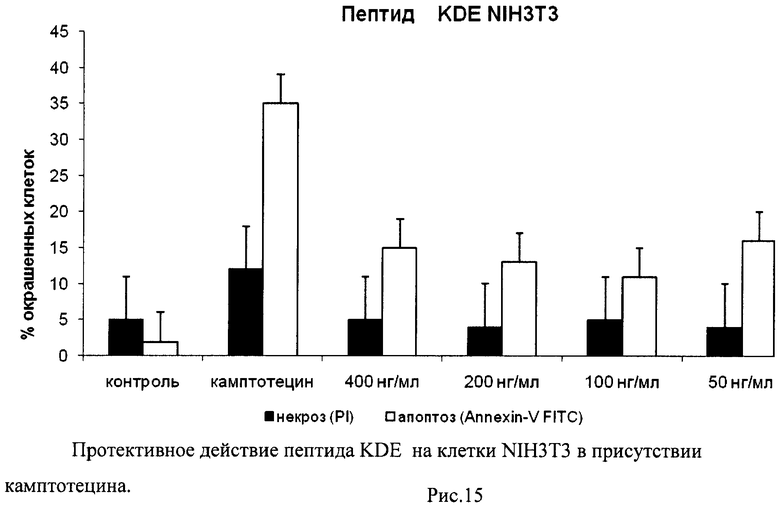
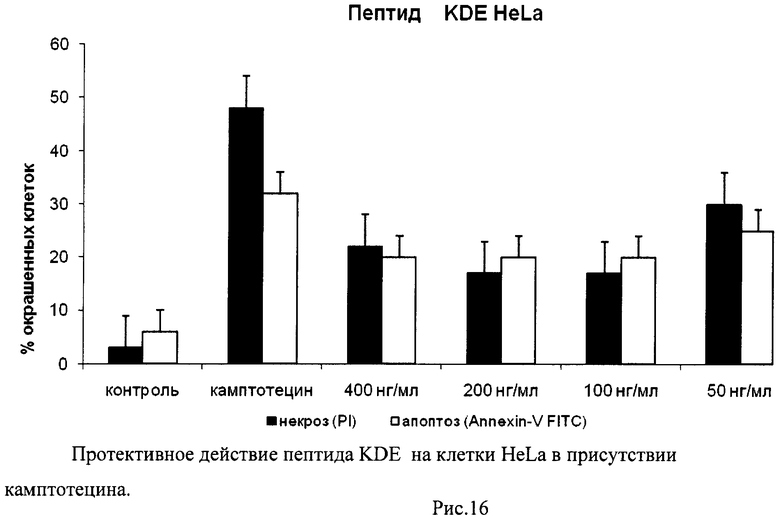
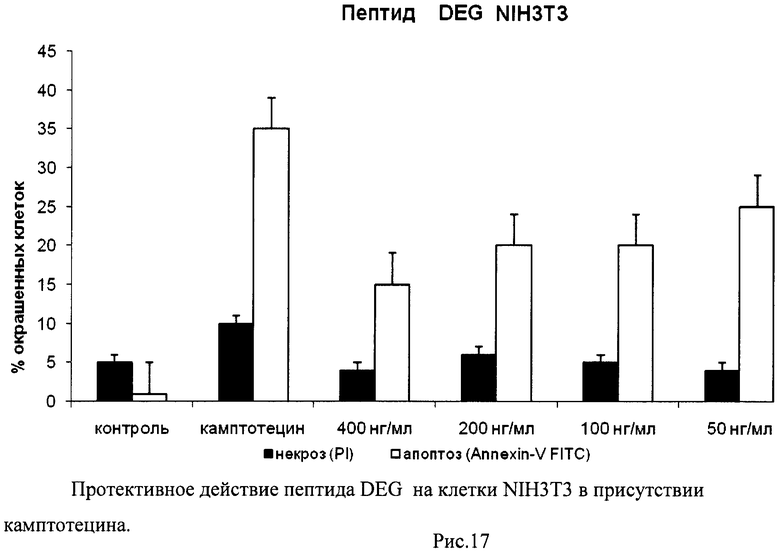
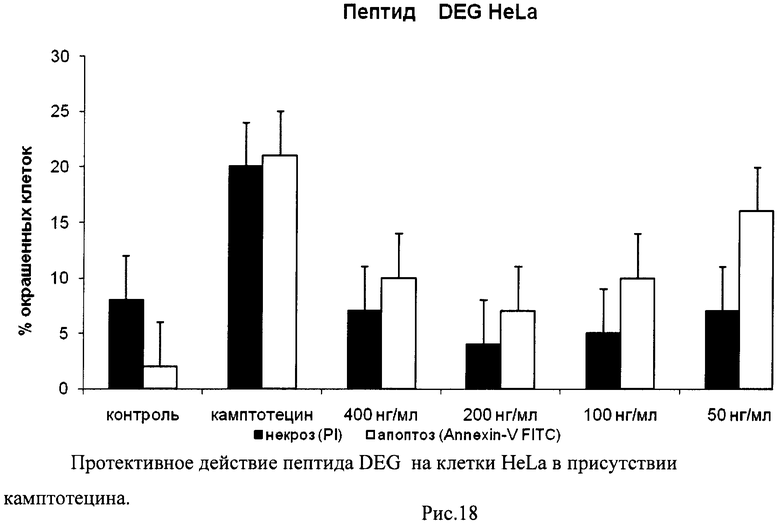
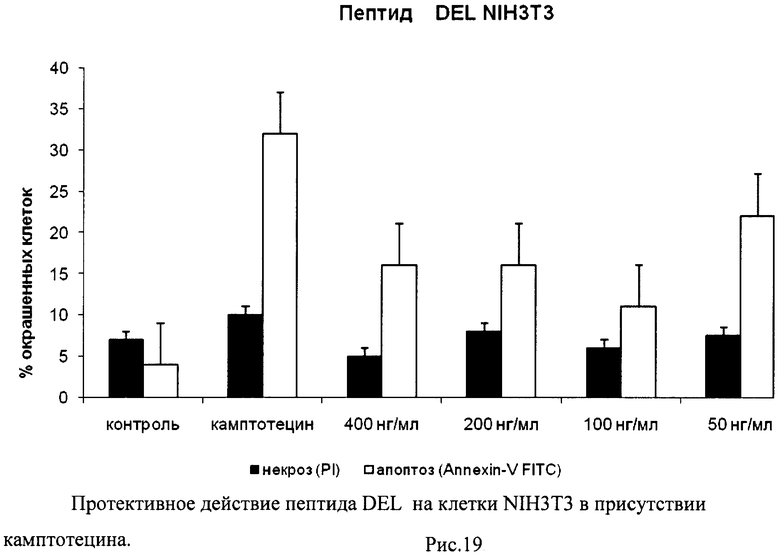
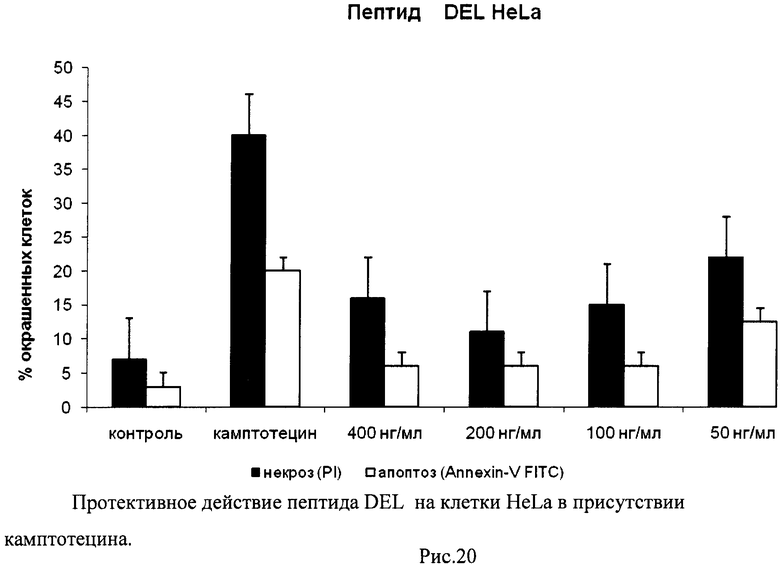
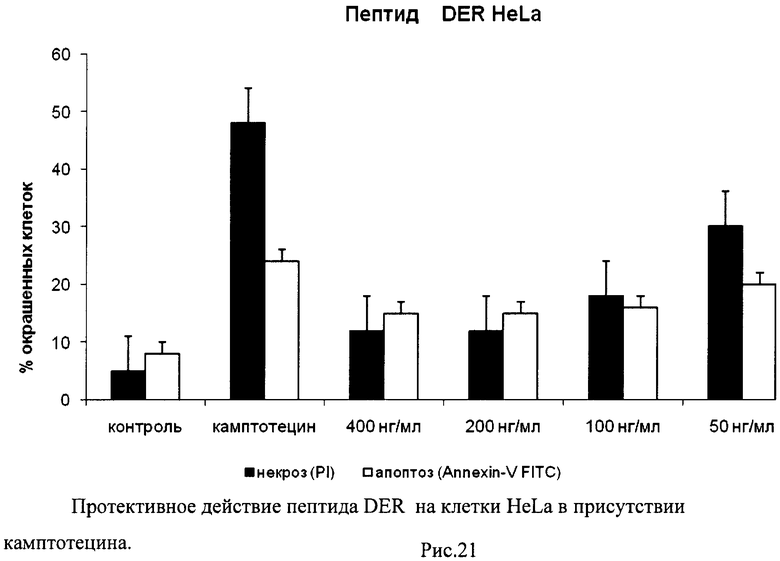
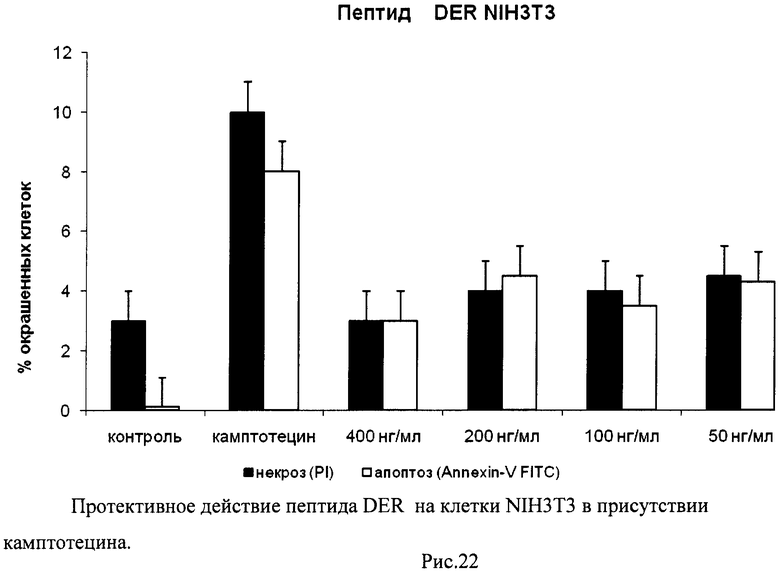
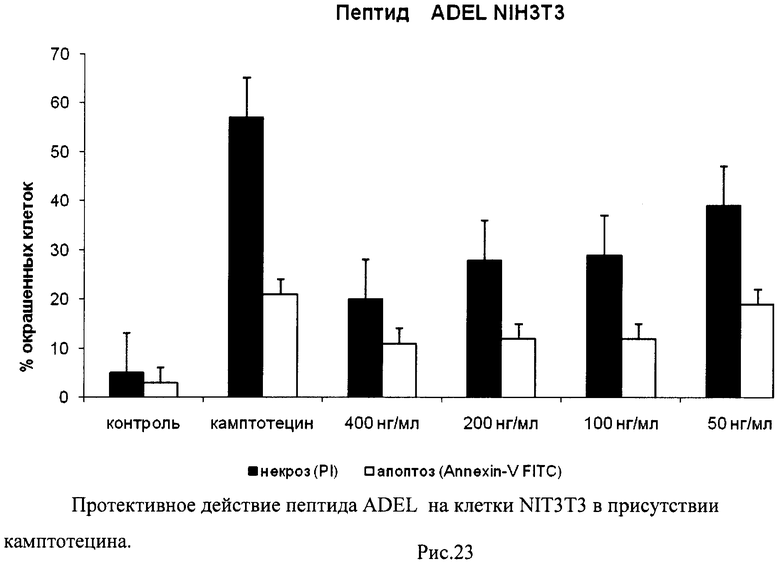
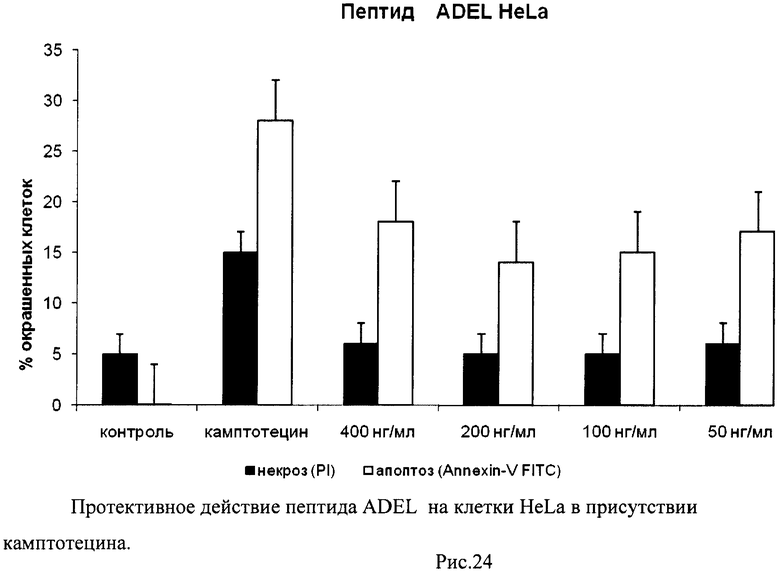
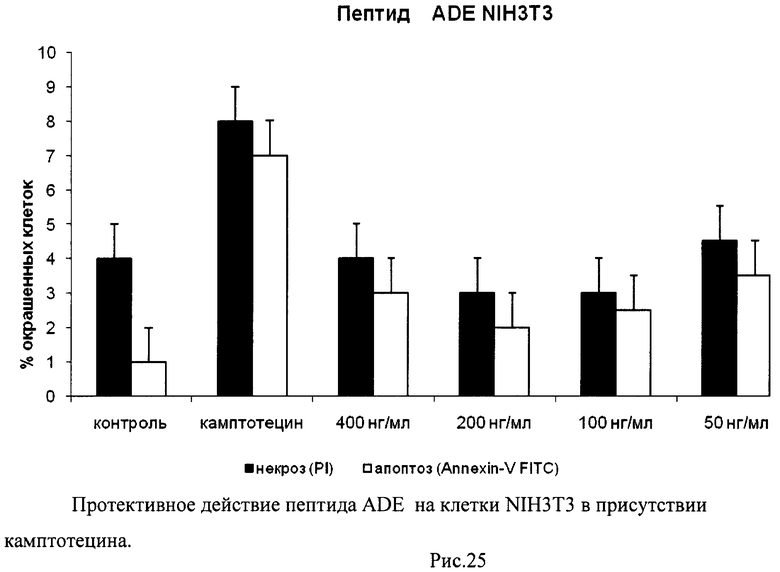
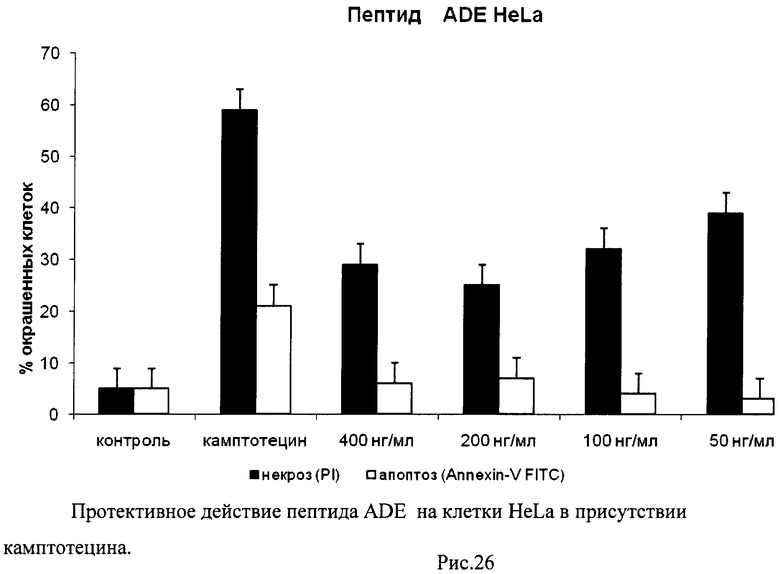
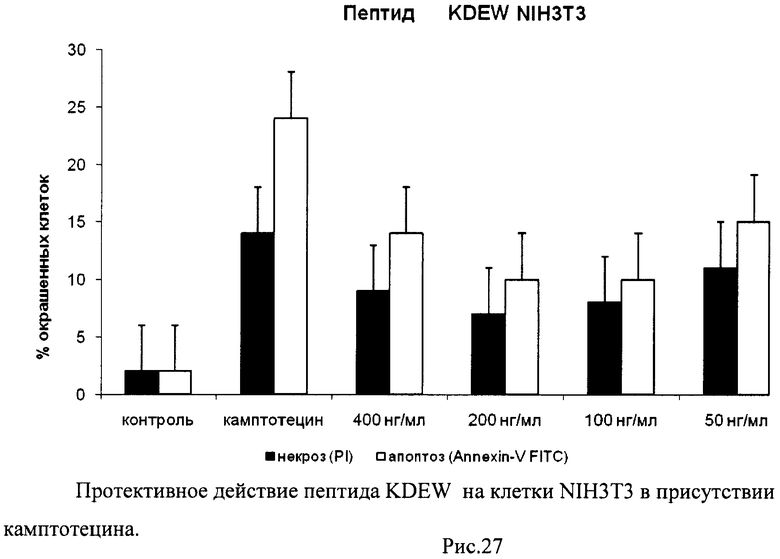
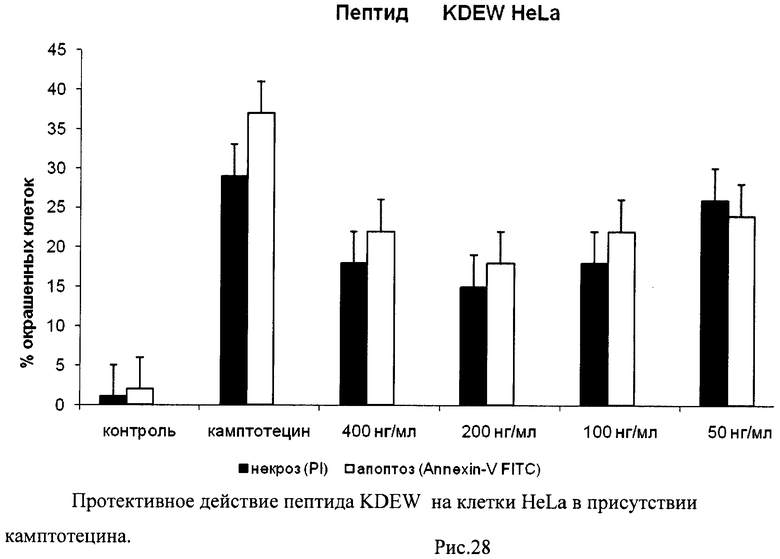
На гистограммах Рис.13 - Рис.28 представлено процентное содержание апоптотических и некротических клеток в популяциях HeLa S3 и NIH3T3 в присутствии 8 разных пептидов в 4 различных концентрациях.
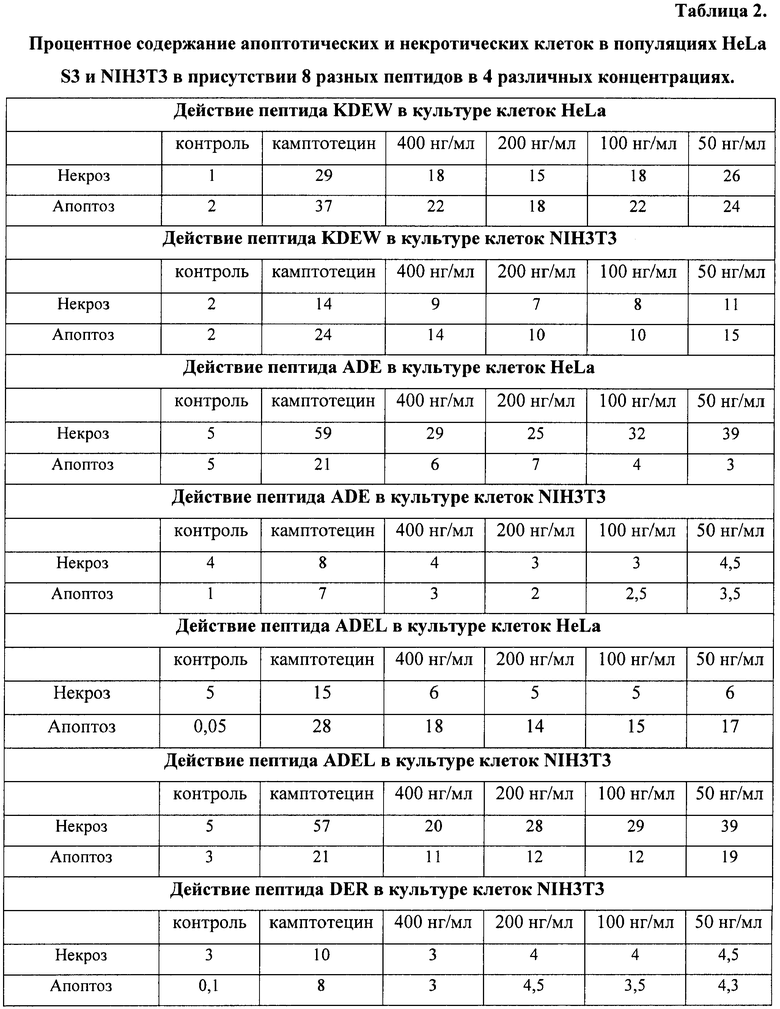
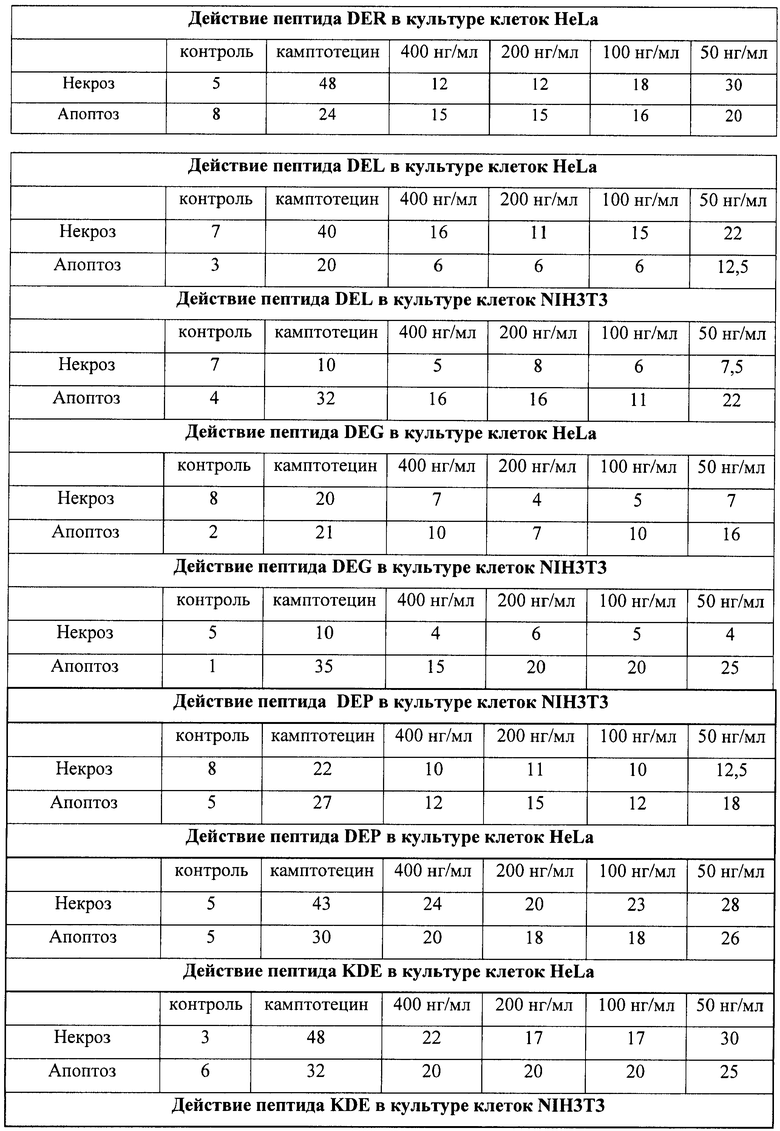

Из приведенных гистограмм (Рис. 13-28) и Таблицы 2 видно, что исследуемые пептиды в диапазоне концентраций от 50 до 400 нг/мл оказывают выраженное защитное действие на клетки HeLa S3 и NIH3T3, т.е. обладают цитопротективной активностью.
Литература
1. Патент РФ №2177802 (2002).
2. Патент РФ №2161501 (2001).
3. Патент РФ №2301074 (2007).
4 Патент РФ №2304444 (2007).
5. Патент РФ №2155063 (2000).
6. Pharmactutical Research, Vol.11, N 5, 1994, p.751-758.
7. Pharmactutical Research, Vol.7, No.8, 1990, p.787-793.
8. Synthetic peptides, sec. ed. Gregory A. Grant, Oxford Univ. Press 2002, p.283.
9. А.А.Гершкович, В.К.Кибирев, Химический синтез пептидов. Киев, Наукова думка, 1992 г.
10. Пептиды, основные методы образования пептидных связей. Москва, «Мир» 1983 г.
| название | год | авторы | номер документа |
|---|---|---|---|
| КОМПОЗИЦИЯ И ПРИМЕНЕНИЕ КОМПОЗИЦИИ ДЛЯ ПОЛУЧЕНИЯ СРЕДСТВА, ОБЛАДАЮЩЕГО ГЕПАТОПРОТЕКТИВНОЙ АКТИВНОСТЬЮ | 2012 |
|
RU2485970C1 |
| ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ, ОБЛАДАЮЩАЯ ПРОТИВОГИПОКСИЧЕСКОЙ, НЕЙРОПРОТЕКТОРНОЙ И АНТИАМНЕСТИЧЕСКОЙ АКТИВНОСТЬЮ | 2012 |
|
RU2480233C1 |
| RGD-СОДЕРЖАЩИЕ ПЕПТИДЫ | 2001 |
|
RU2214416C2 |
| СПОСОБ ПОЛУЧЕНИЯ ПЕПТИДА ЭКСЕНАТИДА | 2011 |
|
RU2458066C1 |
| ПРОИЗВОДНЫЕ ГЕМИНА И ИХ ФАРМАЦЕВТИЧЕСКИ ПРИЕМЛЕМЫЕ СОЛИ, СПОСОБ ПОЛУЧЕНИЯ, ПРИМЕНЕНИЕ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 2002 |
|
RU2238950C2 |
| Способ получения пептида Ac-His-Ala-Glu-Glu-NH | 2021 |
|
RU2767030C1 |
| Способ получения пептидов в виде кислотно-аддитивных солей | 1982 |
|
SU1277903A3 |
| ПЕПТИДЫ, ОБЛАДАЮЩИЕ РЕГЕНЕРАТИВНО-РЕПАРАТИВНЫМ ДЕЙСТВИЕМ | 1994 |
|
RU2065445C1 |
| НАЦЕЛИВАЮЩИЕ АМИНОКИСЛОТНЫЕ ЛИПИДЫ | 2013 |
|
RU2654210C2 |
| ПРОИЗВОДНЫЕ ПЕНТАПЕПТИДОВ | 1991 |
|
RU2010799C1 |
Изобретение относится к органической химии, конкретно к новым биологически активным пептидам, которые могут найти применение в фармакологии и медицине при создании новых лекарственных средств, обладающих цитопротекторной активностью. 28 ил., 2 табл., 12 пр.
Лекарственное средство, обладающее цитопротекторной активностью, представляющее собой пептид, выбранный из группы: H-Ala-Asp-Glu-OH, H-Lys-Asp-Glu-OH, H-Asp-Glu-Pro-OH, H-Asp-Glu-Arg-OH, H-Asp-Glu-Gly-OH, H-Ala-Asp-Glu-Leu-OH, H-Asp-Glu-Leu-OH, H-Lys-Asp-Glu-Trp-NH2.
| WO 2005017163 A2, 24.02.2005 | |||
| US 2009280515 A1, 12.11.2009 | |||
| US 2006024332 A1, 02.02.2006 | |||
| WO 00/04914 A1, 03.02.2000 | |||
| WO 2007124090 A2, 01.11.2007 | |||
| US 5780090 A, 14.07.1998 | |||
| ПЕПТИД, ОБЛАДАЮЩИЙ ИММУНОГЕРОПРОТЕКТОРНЫМ ДЕЙСТВИЕМ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ЕГО ОСНОВЕ И СПОСОБ ЕЕ ПРИМЕНЕНИЯ | 2006 |
|
RU2301074C1 |
| CA ACS on STN, RN 932445-09-5, 146:397532 Morisaka, Hironobu et al, "Enhanced sequence coverage in tryptic fragment analysis by two-dimensional | |||

