ОБЛАСТЬ ТЕХНИКИ
Настоящее изобретение относится к новым соединениям - потенциальным ингибиторам клеточной адгезии, опосредованной αLβ2 интегринами, которые могут быть использованы для лечения воспалительных заболеваний, опосредованных αLβ2 интегринами.
ПРЕДШЕСТВУЮЩИЙ УРОВЕНЬ ТЕХНИКИ
Интегрины лейкоцитов и внутриклеточная адгезия молекул (ICAMs) играют основную роль в адгезии лейкоцитов к целевым клеткам (клеткам-мишеням) и внеклеточным матриксам. Интегрин β2 (CD18) в своей структуре имеет четыре связанные составляющие, но различные α-цепи образуют нековалентно связанную пару с CD18: αLβ2 интегрин (LFA-1, CD11a/CD18), αMβ2 интегрин (Мас-1, CD11b/CD18), αXβ2 интегрин (р150/95, CD11c/CD18) и αDβ2 интегрин (CD11a/CD18) (Bochner ed., Adhesion Molecules in Allergic Disease, Marcel Dekker, Inc. pp.1-24 (1997)). Было показано, что среди них центральным в клеточной адгезии и межэндотелиальной миграции Т-клеток, эозинофилов и других лейкоцитов воспаленных тканей является LFA-1 (Garmberg, Curr. Opin. Cell Biology, 9, 643-650 (1997); Panes et al., Br. J. Pharmacology, 126, 537-550 (1999)). LFA-1 связывается с группой молекул ICAM (ICAM-1, -2, -3, -4, -5), экспрессированных на разнообразных типах клеток, таких как эндотелиальные клетки сосудов, дендритные клетки, эпителиальные клетки, макрофаги и Т-лимфобласты (Dustin et al., J. Immunology, 137, 245-254 (1986)). В добавление, следует отметить, что при взаимодействии LFA-1/ICAM-1 и LFA-1/ICAM-3 могут выступать в качестве ко-стимулирующих сигналов, необходимых для активации Т-клеток (Wingren et al., Crit. Rev. In Immunology, 15, 235-253(1995)).
Клеточная миграция и ко-активация Т-клеток являются важнейшими процессами в ряду воспалительных болезненных состояний. Доминирующая роль LFA-1 в опосредовании воспалительных явлений, показана на нескольких различных животных моделях воспалительных заболеваний, в которых антитела к LFA-1 или ICAM-1 в значительной мере ингибируют развитие терапевтических конечных точек (Rothlein et al., Kidney International, 47, 617 (1992); J. Pharmacol. and Immunology, 147, 4167 (1991); Bennet et al., J. Pharmacol. and Exp.Therapeutics, 280, 988 (1997)). Кроме того, следует отметить, что гуманизированные моноклональные антитела к CD11a (α-цепь LFA-1) показали эффективность у пациентов с псориазом (Gottlieb et al., J. Am. Acad. Dermatology, 42, 428-35 (2000)).
Более того, было показано, что антитела против LFA-1 подавляют отторжение после трансплантации органа (Poston et al., Transplantation 69, 2005-2013 (2000); Nakakura et al., Transplantation 62, 547-552 (1996)). В публикации международной заявки WO 94/04188 описано применение моноклональных антител, направленных против αLβ2-интегринов для всех видов трансплантации.
СУЩНОСТЬ ИЗОБРЕТЕНИЯ
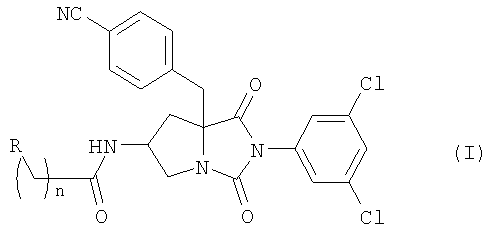
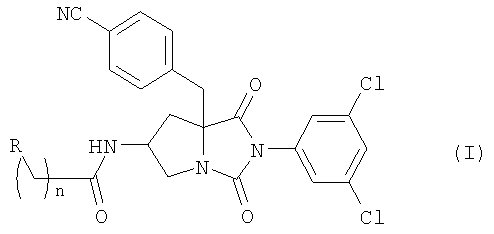
Настоящее изобретение относится к новым соединениям общей формулы (I):
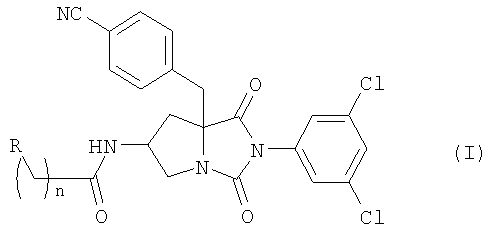
где R является атомом водорода, гидроксильной группой или карбамоильной группой и n равно 1 или 2; или их фармацевтически приемлемым солям.
КРАТКОЕ ОПИСАНИЕ ГРАФИЧЕСКИХ МАТЕРИАЛОВ
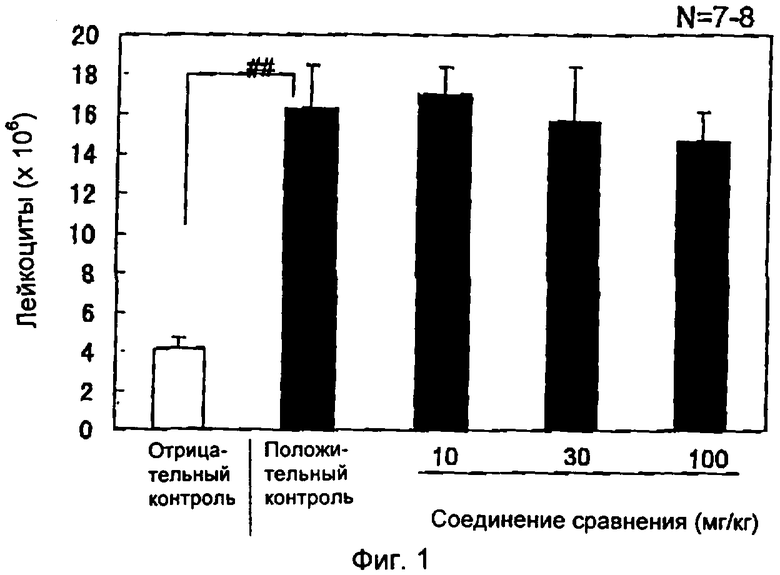
На фиг.1 показано влияние соединения сравнения на аккумуляцию лейкоцитов при перитоните, индуцированном тиогликолятом, у кроликов. Группа отрицательного контроля (белый столбик) получала фосфатный буфер интраперитонеально, группа положительного контроля и лечебная группа (черные столбики) получали тиогликолят интраперитонеально. Через 4 часа после инъекции тиогликолята оценивали аккумуляцию лейкоцитов в брюшной полости.
Результаты выражали как среднее значение ± стандартное отклонение (S.E.M.) (n=7-8). ##Р<0,001 относительно отрицательного контроля (студенческий т-тест/StatView).
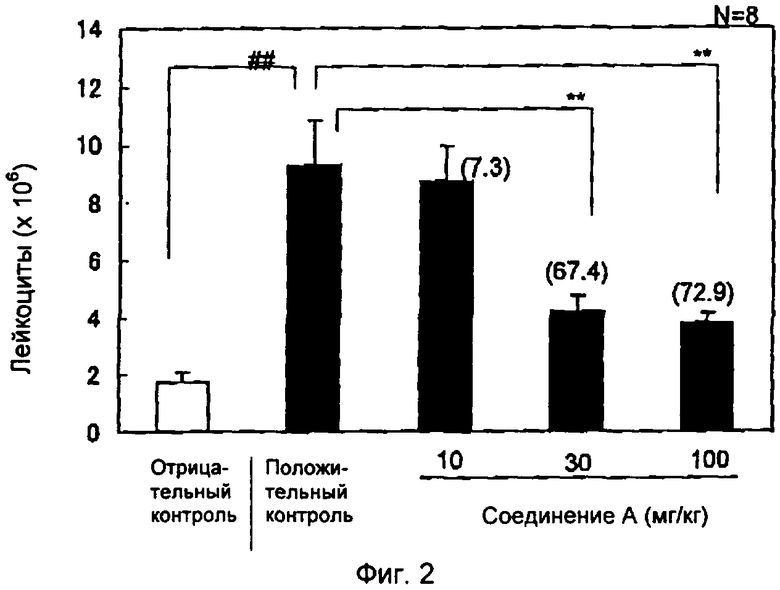
На фиг.2 показано влияние соединения А на аккумуляцию лейкоцитов при перитоните, индуцированном тиогликолятом, у кроликов. Группа отрицательного контроля (белый столбик) получала фосфатный буфер интраперитонеально, группа положительного контроля и лечебная группа (черные столбики) получали тиогликолят интраперитонеально. Через 3 часа после инъекции тиогликолята оценивали аккумуляцию лейкоцитов в брюшной полости. Результаты выражали как среднее значение±стандартное отклонение (S.E.M.) (n=7-8). % ингибирования показан в скобках. ##Р<0,001 относительно отрицательного контроля (студенческий т-тест/StatView). **P<0,01 относительно положительного контроля (двусторонний тест Дуннетта/StatView).
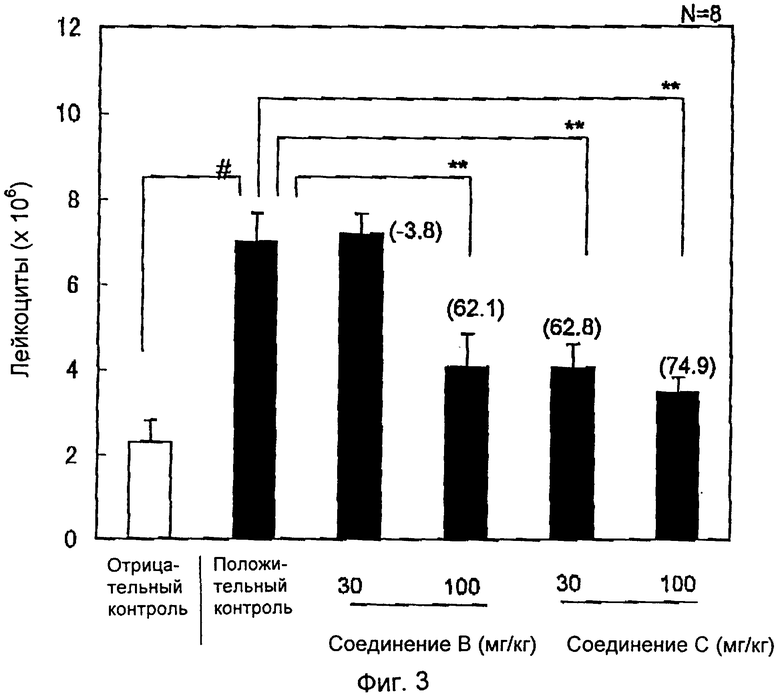
На фиг.3 показано влияние соединений В и С на аккумуляцию лейкоцитов при перитоните, индуцированном тиогликолятом, у кроликов. Группа отрицательного контроля (белый столбик) получала фосфатный буфер интраперитонеально, группа положительного контроля и лечебная группа (черные столбики) получали тиогликолят интраперитонеально. Через 3 часа после инъекции тиогликолята оценивали аккумуляцию лейкоцитов в брюшной полости. Результаты выражали как среднее значение±стандартное отклонение (S.E.M.) (n=7-8). % ингибирования показан в скобках. ##Р<0,001 относительно отрицательного контроля (студенческий т-тест/StatView). **P<0,01 относительно положительного контроля (двусторонний 30 тест Дуннетта/StatView).
СВЕДЕНИЯ, ПОДТВЕРЖДАЮЩИЕ ВОЗМОЖНОСТЬ ОСУЩЕСТВЛЕНИЯ ИЗОБРЕТЕНИЯ
Желаемые соединения согласно настоящему изобретению могут существовать в виде оптических изомеров, в основе которых лежат ассиметричные атомы этих соединений, и настоящее изобретение включает также эти оптические изомеры и их смеси.
В примере осуществления настоящего изобретения - стерическая конфигурация с не нуждающейся в фиксации связью. Соединения по настоящему изобретению могут быть соединением с единственной конфигурацией или смесью соединений нескольких различных конфигураций.
В предпочтительном примере осуществления изобретения, в соединении формулы (I) R является атомом водорода.
В другом предпочтительном примере осуществления изобретения в соединении формулы (I) R является гидроксильной группой.
В еще одном предпочтительном примере осуществления изобретения в соединении формулы (I) R является карбамоильной группой.
В ином предпочтительном примере осуществления изобретения в соединении общей формулы (I) n равно 1.
В другом предпочтительном примере осуществления изобретения в соединении формулы (I) n равно 2.
В более предпочтительном примере осуществления соединений общей формулы (I), R является атомом водорода, а n равно 1.
В другом более предпочтительном примере осуществления соединений общей формулы (I), R является гидроксильной группой, а n равно 1.
В еще одном предпочтительном примере осуществления соединений общей формулы (I), R является карбамоильной группой, а n равно 2.
Наиболее предпочтительные по настоящему изобретению соединения могут быть выбраны из нижеследующих:
(5S,7S)-5-(4-цианобензил)-3-(3,5-дихлорфенил)-7-ацетиламино-1,3-диазабицикло[3.3.0]октан-2,4-дион;
(5S,7S)-5-(4-цианобензил)-3-(3,5-дихлорфенил)-7-[(2-гидроксиацетил)амино]-1,3-диазабицикло[3.3.0]октан-2,4-дион;
(5S,7S)-5-(4-цианобензил)-3-(3,5-дихлорфенил)-7-[(3-карбамоилпропионил)амино]-1,3-диазабицикло[3.3.0]октан-2,4-дион.
Характеристикой данных соединений является комбинация ациламиногруппы в 7-м положении и 4-цианобензильной группы в 5-м положении 1,3-диазобицикло[3.3.0] октанового ядра, причем эта характеристика не была конкретно описана в предшествующих публикациях.
Соединения по настоящему изобретению обладают сильной ингибиторной активностью по отношению к обоим LFA-1, вызывающим клеточную адгезию и ко-активацию Т-клеток, а также проявляют превосходную биодоступность после орального введения, что отражает общее повышение а) связывания белков плазмы, б) растворимости в воде и в) липофильности.
Поэтому соединения по настоящему изобретению проявляют превосходную способность действовать in vivo против неблагоприятных условий, возникающих в результате клеточной адгезии, опосредованной LFA-1.
В добавление, следует отметить, что соединения по настоящему изобретению также обладают сильной антагонистической активностью к рецептору вещества Р, т.е. рецептору нейрокинина-1 (NK1). Антагонисты рецептора вещества Р считаются полезными для лечения таких воспалительных заболеваний, как астма, ревматоидные артриты, воспалительные заболевания кишечника, циститы и другие гастрорасстройства (Kraneveld et al., Int. Immunopharmacology, 1, 1629-1650 (2001); Swain et al., Ann. Rep. Med. Chem., 34, 51-60 (1999); Ohnmacht Jr. et al., Ann. Rep. Med. Chem., 33, 71-80 (1998)). Так, соединения по настоящему изобретению обладают превосходными терапевтическими возможностями против нежелательных состояний, вызываемых или опосредуемых рецептором вещества Р. Кроме того, соединения согласно настоящему изобретению могут проявлять превосходный эффект при лечении или профилактике воспалительных заболеваний, благодаря двойственной активности ингибирования клеточной адгезии, опосредованной LFA-1, и антагонизма к рецептору вещества Р.
Кроме того, соединения общей формулы (I) имеют пониженную цитотоксичность и низкую ингибиторную активность цитохрома Р450 по сравнению с описанными ранее соединениями и, следовательно, соединения по настоящему изобретению могут иметь пониженное потенциальное побочное действие.
Соединения по настоящему изобретению клинически могут применяться как в свободной форме, так и в форме фармацевтически приемлемых солей. Фармацевтически приемлемые соли включают соль с кислотой - неорганической или органической, и соль с неорганическим основанием, или органическим основанием, или с аминокислотой. Фармацевтически приемлемые соли также включают внутримолекулярные соли, или их сольваты или гидраты.
Соединения по настоящему изобретению могут входить в состав фармацевтических композиций, включающие терапевтически эффективное количество соединения, как оно определено выше, и фармацевтически приемлемый носитель или разбавитель. Фармацевтически приемлемым носителем или разбавителем могут быть, например, связующие вещества (например - сироп, аравийская камедь, желатин, сорбит, трагакант, поливинилпирролидон), наполнители (например - лактоза, сахароза, кукурузный крахмал, фосфат калия, сорбит, глицин), вещества, улучшающие скольжение (например - стеарат магния, тальк, полиэтиленгликоль, диоксид кремния), дезинтеграторы (например - картофельный крахмал), увлажняющие вещества (например - лаурилсульфат натрия) и тому подобные.
Желаемые соединения по настоящему изобретению или их фармацевтически приемлемые соли могут быть введены или орально, или парентерально, а также могут быть применяться в качестве специального фармацевтического препарата. Эти фармацевтические препараты, при введении перорально, могут быть в твердой форме, такой как таблетки, гранулы, капсулы и порошок или в жидкой форме, такой как раствор, суспензия и эмульсия. При парентеральном введении фармацевтический препарат может быть в форме суппозитория, инъекционного препарата или препарата для внутривенной капельницы, при использовании для инъекции дистиллированную воду, физиологический раствор, водный раствор глюкозы и так далее, и ингаляции традиционным способом.
Доза желаемых соединений или их фармацевтически приемлемых солей по настоящему изобретению меняется в зависимости от способа введения, возраста, пола, массы тела и состояния пациента, но в основном дневная доза предпочтительно составляет от 0,1 до 100 мг/кг/день, особенно предпочтительна доза от 1 до 100 мг/кг/день.
Соединения по настоящему изобретению могут применяться для лечения или профилактики состояний, опосредованных LFA-1, у пациентов, например, у людей. Соединения по настоящему изобретению также могут применяться для лечения пациентов страдающих от или имеющих повышенную восприимчивость к состояниям, опосредованным LFA-1. Состояния, опосредованные LFA-1, включают воспалительные, аутоиммунные и аллергические заболевания.
Соединения по настоящему изобретению могут применяться также для лечения или профилактики состояний у пациентов, вызываемых или опосредованных веществом Р, а также для лечения пациентов страдающих от или имеющих повышенную восприимчивость к таким состояниям. Примерами вышеуказанных состояний могут быть воспалительные заболевания.
Соединения по настоящему изобретению могут применяться для лечения или профилактики таких заболеваний, как ревматоидный артрит, астма, хроническая легочная обструкция, аллергические состояния, синдром респираторного дистресс-синдрома взрослых, СПИД, заболевания сердечно-сосудистой системы, тромбоз, патологическая агрегация тромбоцитов, реокклюзия после тромболизиса, реперфузионные поражения, кожные воспалительные заболевания (например - псориаз, экзема, контактный и атопический дерматиты), остеопороз, остеоартрит, атеросклероз, артериосклероз, включая артериосклероз, обусловленный трансплантацией, неопластические заболевания, включая метастазирование неопластического (опухолевого) или злокачественного (ракового) роста, раны, отслоение сетчатки, сахарный диабет 1 типа, рассеянный склероз, системная красная волчанка (СКВ), воспалительные состояния глаз, воспалительные заболевания кишечника (например - болезнь Крона и язвенный колит), циститы, желудочные расстройства (гастрорасстройства), местный энтерит, синдром Шегрена и другие аутоиммунные заболевания.
Соединения по настоящему изобретению также могут применяться при отторжении трансплантата (т.е. хронического и острого отторжения трансплантата), после трансплантации органа, включая отторжение аллотрансплантата (болезни «хозяин против трансплантата») и болезни «трансплантат против хозяина».
Соединения по настоящему изобретению предпочтительно могут применяться для лечения или профилактики псориаза, ревматоидного артрита, воспалительных заболеваний кишечника (например - болезнь Крона и язвенный колит), системной красной волчанки, атопического дерматита, синдрома Шегрена и отторжении после трансплантации органа (отторжение аллотрансплантата и болезни «трансплантат против хозяина»).
Кроме того, соединения по настоящему изобретению предпочтительно могут быть использованы для лечения и профилактики ревматоидного артрита, астмы, хронических обструктивных легочных заболеваний, псориаза, рассеянного склероза и отторжении после трансплантации органа.
Предпочтительно соединения по настоящему изобретению могут применяться для лечения или профилактики воспалительных заболеваний, таких как астма, воспалительные заболевания кишечника, циститы и другие желудочные расстройства.
Согласно настоящему изобретению желаемые соединения (I) могут быть получены в соответствии с одним из следующих способов.
Способ А:
Среди соединений по настоящему изобретению, соединение формулы (I-а):
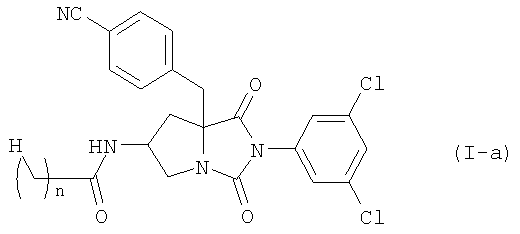
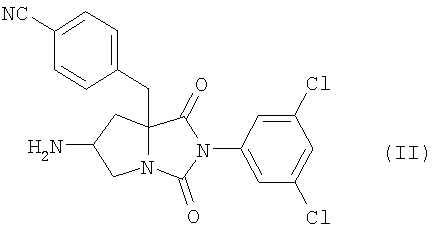
где n является таким же, как определено выше, или его фармацевтически приемлемая соль могут быть получены конденсацией соединения (II)
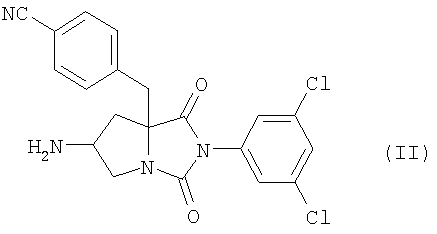
или его соли с соединением формулы (III-а):
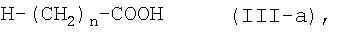
где n является таким же, как определено выше, или с его солью, или с его реакционноспособным производным, с последующим превращением полученного соединения в его фармацевтически приемлемую соль, если это желательно.
Соль соединений (II) и (III-а) может быть, например, солью с неорганической или органической кислотами (например - трифторацетат, гидрохлорид, сульфат) или с неорганическими основаниями (например - соли щелочных металлов, такие как натриевая или калиевая соли, соли щелочноземельных металлов, такие как соли бария и кальция).
Реакция конденсации соединения (II) или его соответствующей соли с соединением (III-а) или с его соответствующей солью может быть проведена в присутствии конденсирующего реагента, с основанием или в его отсутствие, в подходящем растворителе.
Конденсирующий реагент может быть выбран среди традиционных конденсирующих реагентов, которые используются для пептидных синтезов, например BOP-CI, ВОР реагент, DCC, EDC или CDI. Предпочтительно использовать конденсирующий реагент с активатором (например - НОВТ).
Основание может быть выбрано среди традиционных органических оснований, таких как алкиламины (например - DIEA, Et3N), циклические амины (например - DBU, DBN, 4-метилморфолин), пиридины (например - пиридин, DMAP) и традиционных неорганических оснований, таких как карбонаты щелочных металлов (например - Na2CO3, К2СО3), гидрокарбонаты щелочных металлов (например - NaHCO3, КНСО3), гидроксиды щелочных металлов (например - NaOH, КОН) и тому подобные.
Растворитель должен быть выбран таким образом, чтобы он не препятствовал протеканию реакции конденсации. В качестве растворителя могут быть выбраны, например, сложные эфиры (например - метилацетат, этилацетат), галогеналканы (например - CHCl3, СН2Cl2), простые эфиры (например - диэтиловый эфир, THF, DME, диоксан), амиды (например - DMF, N-метилпирролидон), кетоны (например - ацетон, метилэтилкетон), СН3CN, DMSO, H2O и смеси этих растворителей. Реакция может быть проведена при температуре от -50 до 50°С, предпочтительно проведение при температуре от 0°С до комнатной температуры.
Конденсация соединения (II) или его соответствующей соли с реакционноспособным производным соединения (III-а) проводится в присутствии или отсутствии основания в подходящем растворителе или без растворителя.
Примерами реакционноспособных производных соединения (III-а) являются галогенангидрид (например - хлорангидрид), реакционноспособный эфир (например - эфир с п-нитрофенолом), его ангидрид, и смешанный ангидрид с другой карбоновой кислотой (например - смешанный ангидрид с изобутановой кислотой) и тому подобное.
Основание может быть выбрано среди традиционных органических оснований, таких как алкиламины (например - DIEA, Et3N), циклические амины (например - DBU, DBN, 4-метилморфолин), пиридины (например - пиридин, DMAP), и традиционных неорганических оснований, таких как карбонаты щелочных металлов (например - Na2СО3, К2СО3), гидрокарбонаты щелочных металлов (например - NaHCO3, КНСО3), гидроксиды щелочных металлов (например - NaOH, КОН) и тому подобные.
Растворитель должен быть выбран таким образом, чтобы он не препятствовал протеканию реакции конденсации. В качестве растворителя могут быть выбраны, например, сложные эфиры (например - метилацетат, этилацетат), галогеналканы (например - CHCl3, CH2Cl2), простые эфиры (например - диэтиловый эфир, THF, DME, диоксан), амиды (например - DMF, N-метилпирролидон), кетоны (например - ацетон, метилэтилкетон), СН3CN, DMSO, H2O и смеси этих растворителей.
Конденсация может быть проведена при температуре от -30°С до комнатной температуры.
Способ Б:
Среди соединений по настоящему изобретению, соединение формулы (I-б):
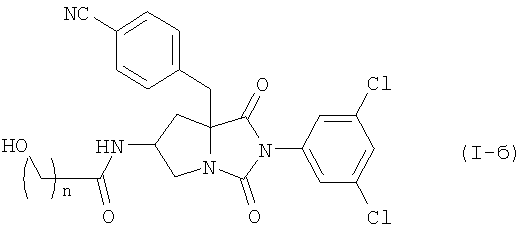
где n является таким же, как определено выше, или его фармацевтически приемлемая соль, могут быть получены конденсацией соединения (II) или его соли с соединением формулы (III-б):
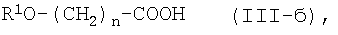
где R1O является защищенной или незащищенной гидроксильной группой и n является таким же, как определено выше, или с его солью, или с его реакционноспособным производным, с последующим удалением защитной группы и, при желании - с последующим превращением конечного соединения в его фармацевтически приемлемую соль.
Соль соединения (III-б) может быть, например, солью с неорганическим основанием (например - солью щелочных металлов, таких как натриевая соль или калиевая соль, и солью щелочноземельных металлов, таких как бариевая соль и кальциевая соль).
Защитная группа для гидроксильной группы может быть выбрана среди традиционных защитных групп, использующихся для защиты гидроксильных групп. Защитная группа должна быть легко удаляемой традиционными методами. Примеры таких защитных групп включают триалкилсилильные группы (например - триметилсилильная, триэтилсилильная и третбутилдиметилсилильная группы), бензильную группу, метильную группу и тетрагидропиранильную группу.
Реакция конденсации соединения (II) или его соли с соединением (III-б) (где R1O является защищенной гидроксильной группой) или с его солью или с реакционноспособным производным может быть проведена с использованием процедуры, подобной той, которая описана в Способе А.
Удаление защитной группы может быть проведено обычным методом, выбранным в соответствии с принадлежностью удаляемой защитной группы, например, путем гидролиза, обработки кислотой или BBr3, каталитической редукции.
Гидролиз может быть проведен с использованием неорганических оснований, таких как гидроксиды щелочных металлов (например - LiOH, NaOH и КОН) в подходящем растворителе, таком как эфиры (например - диэтиловый эфир, диоксан и THF), спирты (например - МеОН, EtOH), СН3CN, DMSO, H2O и тому подобное, при комнатной температуре или при нагревании.
Обработка кислотой может быть проведена с использованием неорганической или органической кислот, таких как хлористоводородной, бромистоводородной, уксусной, п-толуолсульфоновой, трифторуксусной кислот в подходящем растворителе, таком как эфиры (например - диэтиловый эфир, диоксан и THF), галогеналканы (например - CHCl3, CH2Cl2), спирты (например - МеОН, EtOH), СН3CN, DMSO, H2O и тому подобное, при комнатной температуре или при нагревании.
Каталитическое восстановление может быть проведено с использованием катализатора, такого как палладий на подложке из активированного углерода, никель Ренея (скелетный никелевый катализатор гидрирования) в атмосфере водорода, при комнатной температуре или при нагревании в подходящем растворителе, таком как простые эфиры (например - диэтиловый эфир, диоксан и THF), сложные эфиры (например - метилацетат, этилацетат), спирты (например - МеОН, EtOH), СН3CN, AcOH, Н2O и тому подобное.
Обработка BBr3, используемая для деметилирования, может быть проведена в подходящем растворителе (например - THF, CH2Cl2, AcOH) при температуре от -78 до 50°С.
В случае использования для реакции конденсации соединения (III-б), где R1O является гидроксильной группой, следует защитить гидроксильную группу соединения (III-б) in situ перед осуществлением реакции конденсации.
Защита гидроксильной группы может быть проведена взаимодействием соединения (III-б) с триалкилсилилгалидами в подходящем растворителе в присутствии основания. Примеры триалкилсилилгалидов включают триметилсилилхлорид, триэтилсилилхлорид и третбутилдиметилсилилхлорид. Основание может быть выбрано среди оснований, традиционно использующихся для защиты гидроксильной группы, например, триэтиламин, имидазол, пиридин.
Растворитель должен быть выбран таким образом, чтобы он не препятствовал протеканию реакции. В качестве растворителя могут быть выбраны, например, сложные эфиры (например - метилацетат, этилацетат), ароматические углеводороды (бензол, толуол), галогеналканы (например - CHCl3, CH2Cl2), простые эфиры (например - диэтиловый эфир, THF, DME, диоксан), амиды (например - DMF, N-метилпирролидон), кетоны (например - ацетон, метилэтилкетон), СН3CN, DMSO и смеси этих растворителей. Реакция может быть проведена при температуре от -50 до 50°С, предпочтительно проведение при температуре от 0°С до комнатной температуры. При необходимости, соединение с защищенной гидроксильной группой может быть выделено посредством обычной процедуры.
Способ В:
Среди соединений по настоящему изобретению, соединение формулы (I-в):
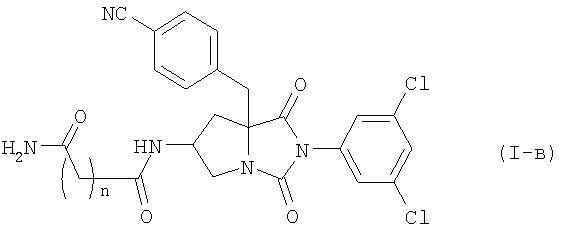
где n является таким же, как определено выше, или его фармацевтически приемлемая соль могут быть получены конденсацией соединения (II) с соединением (III-в):
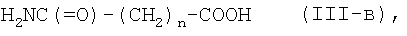
где n является таким же, как определено выше, или с его солью, или с его реакционноспособным производным, с последующим превращением полученного соединения в его фармацевтически приемлемую соль, если это желательно.
Соль соединения (III-в) может быть, например, солью с неорганическим основанием (например - солью щелочных металлов, таких как натриевая соль или калиевая соль и солью щелочноземельных металлов, таких как бариевая соль и кальциевая соль).
Взаимодействие соединения (II) или его соли с соединением (III-в) или с его солью может быть проведено с использованием процедуры, подобной описанной в Способе А.
Исходное соединение формулы (II) может быть получено в соответствии с описанием WO 01/30781 по следующей схеме.
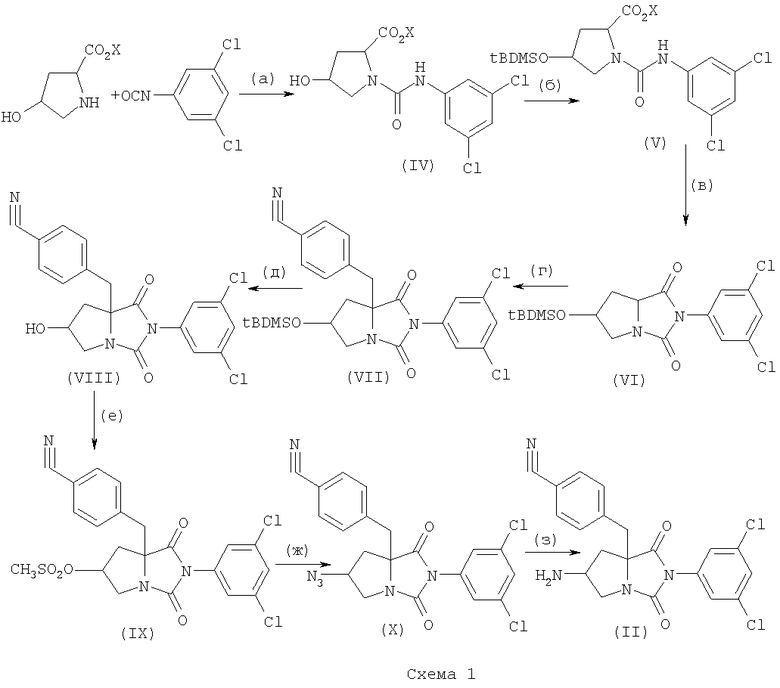
(На вышеуказанной схеме Х является C1-6 алкильной группой или бензильной группой; tBDMSO является третбутилдиметилсилилокси группой.)
Стадия (а): Соединение (IV) может быть получено взаимодействием C1-6 алкилового или бензилового эфира 4-гидроксипролина с 3,5-дихлорфенилизоцианата в присутствии основания в подходящем растворителе.
Основание может быть выбрано среди традиционных органических оснований, таких как алкиламины (например - Et3N, DIEA), пиридин и неорганических оснований, таких как гидрокарбонаты щелочных металлов (например - NaHCO3, КНСО3) и карбонаты щелочных металлов (например - Na2CO3, К2СО3).
Растворитель должен быть выбран таким образом, чтобы он не препятствовал протеканию реакции конденсации. В качестве растворителя могут быть выбраны, например, СН2Cl2, DME, THF, НМРА или их смесь. Реакция может быть проведена при температуре от -78°С до комнатной температуры.
Стадия (б): Соединение (V) может быть получено посредством защиты гидроксильной группы соединения (IV). Защита может быть проведена обычным способом, например, взаимодействием соединения (IV) с третбутилдиметилсилилхлоридом в присутствии имидазола в подходящем растворителе, таком как СН3CN. Реакцию проводят при температуре от 0°С до точки кипения растворителя, наиболее предпочтительной является комнатная температура.
Стадия (в): Соединение (VI) может быть получено циклизацией соединения (V). Циклизация может быть проведена в присутствии или в отсутствие основания, в подходящем растворителе.
Основание может быть выбрано среди традиционных неорганических оснований, таких как алкоксиды щелочных металлов (например - NaOEt, NaOMe), карбонаты щелочных металлов (например - К2СО3, Na2CO3) и гидрокарбонаты щелочных металлов (например - NaHCO3) и органических оснований, таких как пиридин, DMAP, Et3N, и DIEA.
Растворитель должен быть выбран таким образом, чтобы он не препятствовал протеканию реакции циклизации. В качестве растворителя могут быть выбраны, например, толуол, DME, CH2Cl2, THF, CH3CN, DMF, спирты (например - MeOH, EtOH) или их смесь. Реакцию проводят при температуре от 0°С до точки кипения растворителя, наиболее предпочтительной является температура от 50 до 100°С.
Стадия (г): Соединение (VII) может быть получено конденсацией соединения (VI) с соединением формулы (XI):
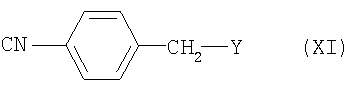
где Y является удаляемой группой.
Удаляемая группа может быть выбрана среди атомов галогенов (например - атом хлора, атом брома, атом йода), п-толуолсульфонилоксигруппы и метилсульфонилоксигруппы.
Реакция конденсации может быть проведена в присутствии основания в подходящем растворителе.
Основание может быть выбрано среди традиционных оснований, таких как амиды щелочных металлов (например - LDA, KHMDS с или без LiCl).
Растворитель должен быть выбран таким образом, чтобы он не препятствовал протеканию реакции конденсации. В качестве растворителя могут быть выбраны, например, диэтиловый эфир, DME, THF, DMF, НМРА или их смесь. Реакцию проводят при температуре от -78°С до комнатной температуры.
Стадия (д): Соединение (VIII) может быть получено посредством удаления защитной группы соединения (VII).
Удаление защитной группы может быть проведено обычным методом, например обработкой соединения смесью HF/пиридин, H-Bu4NF или кислотой (например - HCl, AcOH, TFA, p-TsOH) в подходящем растворителе или без растворителя.
Растворитель должен быть выбран таким образом, чтобы он не препятствовал протеканию реакции конденсации. В качестве растворителя могут быть выбраны, например, СН3CN, THF, DMF, спирты (например - МеОН, EtOH) или их смесь. Реакцию проводят при температуре от -78°С до комнатной температуры.
Стадия (е): Соединение (IX) может быть получено взаимодействием соединения (VIII) с метилсульфонилхлоридом в присутствии основания в подходящем растворителе.
Основание может быть выбрано среди традиционных оснований, таких как Et3N, DIEA, пиридин, NaHCO3, КНСО3, Na2СО3 и K2СО3.
Растворитель должен быть выбран таким образом, чтобы он не препятствовал протеканию реакции. В качестве растворителя могут быть выбраны, например, CH2Cl2, THF, DMF, CH3CN, толуол. Реакцию проводят при температуре от -20 до 50°С.
Стадия (ж): Соединение (X) может быть получено взаимодействием соединения (IX) с азидом щелочного металла (например - NaN3).
Реакция замещения может быть проведена при температуре от 0 до 100°С в органическом растворителе.
Растворитель должен быть выбран таким образом, чтобы он не препятствовал протеканию реакции. В качестве растворителя могут быть выбраны, например, CH2Cl2, THF, DMF, СН3CN и толуол.
Стадия (з): Соединение (II) может быть получено восстановлением соединения (X). Восстановление проводят посредством каталитического восстановления, например, в присутствии палладиевого или платинового катализатора (например - Pd-C, PtO2), в подходящем растворителе, в атмосфере водорода при комнатной температуре.
Растворитель должен быть выбран таким образом, чтобы он не препятствовал протеканию реакции. В качестве растворителя могут быть выбраны, например, EtOAc, MeOH и EtOH.
На протяжении настоящего описания и формулы изобретения C1-6 алкильная группа означает линейную цепь или разветвленную цепь алкильной группы, имеющую от 1 до 6 атомов углерода, предпочтительно - от 1 до 4 атомов углерода, например, метальную, этильную, пропильную, изопропильную, бутильную, изобутильную группы и подобные.
Сокращения:
ОПИСАНИЕ ПРИМЕРОВ ОСУЩЕСТВЛЕНИЯ ИЗОБРЕТЕНИЯ
Соединения по настоящему изобретению поясняются следующими примерами, однако они не ограничивается только лишь указанными примерами.
Пример 1: (5S,7S)-5-(4-цианобензил)-3-(3,5-дихлорфенил)-7-ацетиламино-1,3-диазабицикло[3.3.0]октан-2,4-дион
К раствору (5S,7S)-5-(4-цианобензил)-3-(3,5-дихлорфенил)-7-ацетиламино-1,3-диазабицикло[3.3.0]октан-2,4-диона (78,5 мг) в ТГФ (5 мл) добавляли уксусный ангидрид (1,0 мл). Реакционную смесь перемешивали 2 часа при 45°С, затем концентрировали и очищали с помощью препаративной тонкослойной хроматографии (силикагель, CH2Cl2), для получения целевого соединения, указанного в заголовке (выход - 84 мг). Масс-спектрометрия (m/z) 478,8 [MNa+].
Пример 2: (5S,7S)-5-(4-цианобензил)-3-(3,5-дихлорфенил)-7-[(3-карбамоилпропионил)амино]-1,3-диазабицикло[3.3.0]октан-2,4-дион.
Смесь (5S,7S)-5-(4-цианобензил)-3-(3,5-дихлорфенил)-7-амино-1,3-диазабицикло[3.3.0]октан-2,4-диона (82,7 мг), полуамида янтарной кислоты (45,86 мг), EDC (93,12 мг), НОВТ (61,24 мг), DIEA (104,79 мкл) в ТГФ (5 мл) перемешивали при комнатной температуре в течение ночи. Реакционную смесь концентрировали и очищали с использованием высокоэффективной жидкостной хроматографии (ВЭЖХ) (колонка Beckman 5μ С18; градиент элюента H2O/MeCN (10-100%)/0,1% TFA), выход целевого соединения - 72 мг. Масс-спектрометрия (m/z) 536 [MNa+].
Пример 3: (5S,7S)-5-(4-цианобензил)-3-(3,5-дихлорфенил)-7-[(2-карбамоилацетил)амино]-1,3-диазабицикло[3.3.0]октан-2,4-дион.
Смесь (5S,7S)-5-(4-цианобензил)-3-(3,5-дихлорфенил)-7-амино-1,3-диазабицикло[3.3.0]октан-2,4-диона (200 мг), полуамида малоновой кислоты (59,5 мг), EDC (112 мг), НОВТ (97,5 мг), DIEA (168 мкл) в ТГФ (5 мл) перемешивали при комнатной температуре в течение ночи. Реакционную смесь испаряли. Остаток растворяли в этилацетате, полученный раствор промывали водой и нейтрализовали водным раствором NaHCO3, рассолом, затем сушили (Na2SO4) и концентрировали, выход целевого соединения - 212 мг. Масс-спектрометрия (m/z) 500 [MH+].
Пример 4: (5S,7S)-5-(4-цианобензил)-3-(3,5-дихлорфенил)-7-[(3-гидроксипропионил)амино]-1,3-диазабицикло[3.3.0]октан-2,4-дион.
Стадия 1: Смесь (5S,7S)-5-(4-цианобензил)-3-(3,5-дихлорфенил)-7-амино-1,3-диазабицикло[3.3.0]октан-2,4-диона (0,300 г), 3-метоксипропионовой кислоты (0,209 мкл), EDC (0,224 г), НОВТ (0,221 г), DIEA (0,38 мкл) в ТГФ (15 мл) перемешивали при комнатной температуре в течение ночи. Реакционную смесь испаряли. Остаток очищали с помощью ВЭЖХ (колонка Beckman 5μ C18; градиент элюента H2O/MeCN (10-100%)/0,1% AcOH) с получением пены, которая была растворена в этилацетате, полученный раствор промывали водой и нейтрализовали водным раствором NaHCO3, рассолом, затем сушили (Na2SO4) и концентрировали с получением 0,259 г (5S,7S)-5-(4-цианобензил)-3-(3,5-дихлорфенил)-7-[(3-метоксипропионил)амино]-1,3-диазабицикло[3.3.0]октан-2,4-диона. Масс-спектрометрия (m/z) 501 [MH+].
Стадия 2: BBr3 (3 мл 1 М раствора в СН2Cl2) добавляли к раствору (5S,7S)-5-(4-цианобензил)-3-(3,5-дихлорфенил)-7-[(3-метоксипропионил)амино]-1,3-диазабицикло[3.3.0]октан-2,4-диона (0,16 г) в CH2Cl2 (15 мл) при температуре -78°С, затем смесь перемешивали при этой температуре в течение 8 часов. Реакционную смесь испаряли. Остаток очищали с помощью ВЭЖХ (колонка Beckman 5μ С18; градиент элюента H2O/MeCN (10-100%)/0,1% АсОН) с получением пены, которая была растворена в этилацетате, полученный раствор промывали водой и нейтрализовали водным раствором NaHCO3, рассолом, затем сушили (Na2SO4) и концентрировали с получением 0,199 г целевого соединения. Масс-спектрометрия (m/z) 487 [MH+].
Пример 5: (5S,7S)-5-(4-цианобензил)-3-(3,5-дихлорфенил)-7-[(2-гидроксиацетил)амино]-1,3-диазабицикло[3.3.0]октан-2.4-дион.
Стадия 1: К раствору (5S,7S)-5-(4-цианобензил)-3-(3,5-дихлорфенил)-7-амино-1,3-диазабицикло[3.3.0]октан-2,4-диона (150 мг) и DIEA (189 мкл) в ТГФ (4 мл) добавляли раствор бензилоксиацетилхлорида (57 мкл) в ТГФ (2 мл). Реакционную смесь перемешивали при комнатной температуре в течение ночи. Реакционную массу концентрировали и остаток экстрагировали этилацетатом. Полученный раствор промывали рассолом, сушили (Na2SO4), фильтровали и концентрировали. Остаток очищали с помощью ВЭЖХ (колонка Beckman 5μ C18; градиент элюента H2O/MeCN (10-100%)/0,1% АсОН) с получением пены, которая была растворена в этилацетате, полученный раствор промывали водой и нейтрализовали водным раствором NaHCO3, рассолом, затем сушили (Na2SO4) и концентрировали с получением 0,135 г (5S,7S)-5-(4-цианобензил)-3-(3,5-дихлорфенил)-7-[(2-бензилоксиацетил)амино]-1,3-диазабицикло[3.3.0]октан-2,4-диона.
Масс-спектрометрия (m/z) 563,4 [MH+].
Стадия 2: Через раствор 0,125 г (5S,7S)-5-(4-цианобензил)-3-(3,5-дихлорфенил)-7-[(2-бензилоксиацетил)амино]-1,3-диазабицикло[3.3.0]октан-2,4-диона, полученного на Стадии 1, в 10 мл этанола, содержащий Pd/C (5%, 15 мг), пропускали водород, реакционную смесь перемешивали в атмосфере водорода при комнатной температуре в течение ночи. К реакционной смеси дополнительно добавляли катализатор 5% Pd/C (10 мг) и перемешивали в атмосфере водорода при комнатной температуре в течение ночи. Реакционную смесь фильтровали через слой целита, фильтрат концентрировали. Остаток очищали с помощью ВЭЖХ (колонка Beckman 5μ С18; градиент элюента H2O/MeCN (10-100%)/0,1% TFA) с получением 0,023 г целевого соединения. Масс-спектрометрия (m/z) 473 [MH+], 495 [MNa+].
Эталонный пример 1: (5S,7S)-5-(4-цианобензил)-3-(3,5-дихлорфенил)-7-амино-1,3-диазабицикло[3.3.0]октан-2,4-дион.
Целевое соединение получено в соответствии с нижеследующей схемой:
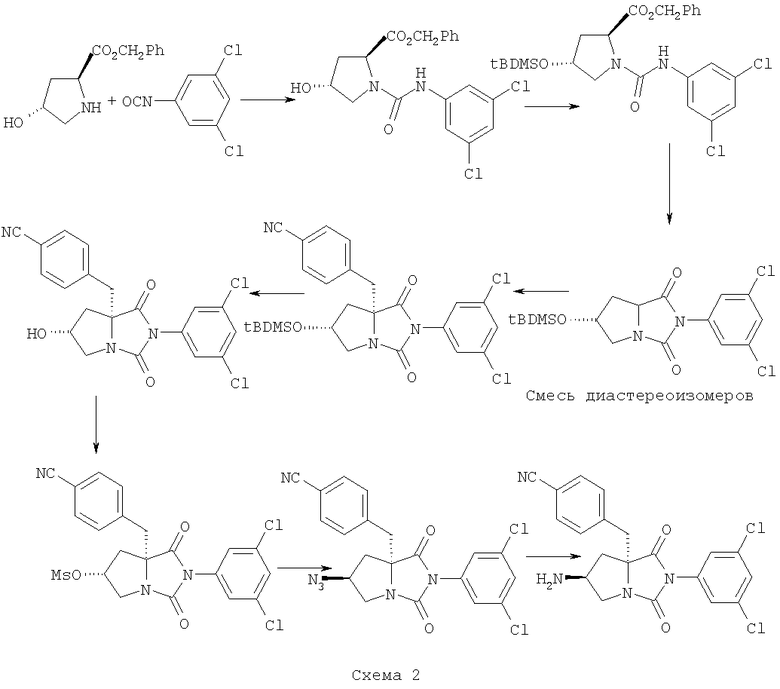
(На схеме tBDMSO является третбутилдиметилсилилокси группой, MsO является метилсульфонилокси группой.)
Стадия 1: п-Толуолсульфоновую кислоту (50,6 г) добавляли к суспензии L-4-трансгидроксипролина (25,25 г) в смеси бензилового спирта (100 мл) и бензола (250 мл). Смесь нагревали с ловушкой Дина Старка в течение 24 часов. Реакционную смесь концентрировали и, для осаждения осадка, добавляли диэтиловый эфир. Осадок фильтровали, промывали дополнительной порцией диэтилового эфира и сушили, выход 75 г бензилового эфира L-4-трансгидроксипролина.
Стадия 2: К суспензии соли L-4-трансгидроксипролина бензилового эфира и п-толуолсульфоновой кислоты, полученной на стадии 1, (40,43 г) в ТГФ (500 мл) и DIEA (51,3 мл) добавляли 3,5-дихлорфенилизоцианат (22,1 г). После перемешивания в течение ночи реакционную смесь концентрировали. Остаток растворяли в этилацетате, промывали 0,5 Н раствором HCl, нейтрализовали водным раствором NaHCO3, рассолом, сушили (Na2SO4), фильтровали и концентрировали. Остаток растворяли в смеси этилацетат/гексан (1:1). Белый осадок фильтровали и очищали с помощью испарительной («флэш») колоночной хроматографии (силикагель, гексан/этилацетат 2:1), выход (2S,4R)-2-[(3,5-дихлорфенил)карбамоил]-4-гидрогипролина бензилового эфира 33,07 г.
Стадия 3: К суспензии (2S,4R)-2-[(3,5-дихлорфенил)карбамоил]-4-гидрогипролина бензилового эфира (33,07 г) в CH3CN (800 мл) добавляли имидазол (11 г) и третбутилдиметилсилилхлорид (13,64 г). После перемешивания в течение 48 часов реакционную смесь концентрировали. Остаток растворяли в этилацетате, промывали 0,5 Н раствором HCl, нейтрализовали водным раствором NaHCO3, рассолом, сушили (Na2SO4), фильтровали и концентрировали. Осадок фильтровали и очищали с помощью колоночной флэш-хроматографии (силикагель, гексан/этилацетат 2:1), выход (2S,4R)-2-[(3,5-дихлорфенил)карбамоил]-4-(третбутилдиметилсилилокси)пролина бензилового эфира составил 44,45 г.
Стадия 4: К раствору (2S,4R)-2-[(3,5-дихлорфенил)карбамоил]-4-(третбутилдиметилсилилокси)пролина бензилового эфира (23,49 г) в CH3CN (500 мл) добавляли DIEA (34,44 мл) и кипятили с обратным холодильником течение 24 часов. После кипячения реакционную смесь концентрировали и очищали с помощью колоночной флэш-хроматографии (силикагель, гексан/этилацетат 1:1). В результате были выделены два диастереомера 3-(3,5-дихлорфенил)-7-(третбутилдиметилсилилокси)-1,3-диазабицикло[3.3.0]октан-2,4-диона.
Диастереомер А: 7,46 г, масс-спектрометрия (m/z) 415 [М+],
Диастереомер Б: 10,66 г, масс-спектрометрия (m/z) 415 [М+].
Стадия 5: Соединение полученное на стадии 4, диастереомеры А или Б (12,73 г) было бензилировано, как описано ниже. н-Бутил лития (30 мл 1,6 М раствора в гексане) добавляли при перемешивании к раствору диизопропиламина (6,5 мл) в ТГФ (100 мл) при температуре -78°С, в атмосфере азота. Смесь выдерживали при этой температуре еще 30 минут. Смесь добавляли через канюлю к раствору 3-(3,5-дихлорфенил)-7-(третбутилдиметилсилилокси)-1,3-диазабицикло-[3.3.0]октан-2,4-диона (12,73 г) в сухом ТГФ (100 мл) при температуре -78°С, в атмосфере азота. После перемешивания при температуре -78°С в течение 30 минут был добавлен 4-циано-а-бромтолуол (9,08 г) в ТГФ (100 мл). Реакционную смесь перемешивали при температуре -78°С в течение 2,5 часов, затем медленно нагрели до комнатной температуры и оставили стоять при комнатной температуре на полчаса. Реакционную смесь концентрировали, остаток растворили в этилацетате. Полученный раствор промыли 0,5 Н раствором HCl, нейтрализовали водным раствором NaHCO3, рассолом, сушили (Na2SO4), фильтровали и концентрировали. Остаток очищали с помощью колоночной флэш-хроматографии (силикагель, гексан/этилацетат от 24:1 до 3:1) с получением (5S,7R)- и (5R,7R)-5-(4-цианобензил)-3-(3,5-дихлорфенил)-7-(третбутилдиметилсилилокси)-1,3-диазабицикло[3.3.0]октан-2,4-диона.
(5S,7R) изомер: 7,6 г, масс-спектрометрия (m/z) 530 [М+],
(5R,7R) изомер: 1,8 г, масс-спектрометрия (m/z) 530 [М+].
Стадия 6: К раствору (5S,7R)-5-(4-цианобензил)-3-(3,5-дихлорфенил)-7-(третбутилдиметилсилилокси)-1,3-диазабицикло[3.3.0]октан-2,4-диона (1,0 г) в ТГФ (1 мл) добавляли 70% HF/пиридин (25 мл). Реакционную смесь перемешивали в течение 24 часов, затем упаривали. Остаток растворили в этилацетате, полученный раствор промыли водой, нейтрализовали водным раствором NaHCO3, рассолом, сушили (Na2SO4) и концентрировали. Остаток очищали с помощью колоночной флэш-хроматографии (силикагель, MeOH/CH2Cl2 2-7%) с получением 0,52 г (5S,7R)-5-(4-цианобензил)-3-(3,5-дихлорфенил)-7-гидрокси-1,3-диазабицикло[3.3.0]октан-2,4-диона. Масс-спектрометрия (m/z) 416 [MH+],
Стадия 7: К раствору (5S,7R)-5-(4-цианобензил)-3-(3,5-дихлорфенил)-7-гидрокси-1,3-диазабицикло[3.3.0]октан-2,4-диона, полученного на Стадии 6 (0,52 г), в CH2Cl2 (8 мл) при 0°С добавляли DIEA (0,45 мл) и метилсульфонилхлорид (0,15 мл). Реакционную смесь перемешивали в течение 1,5 часов, затем разбавили CH2Cl2. Полученную смесь промыли и нейтрализовали водным раствором NaHCO3, затем рассолом, сушили (Na2SO4), фильтровали и концентрировали. Получили 0,76 г (5S,7R)-5-(4-цианобензил)-3-(3,5-дихлорфенил)-7-метилсульфонилокси-1,3-диазабицикло[3.3.0]октан-2,4-диона. Это соединение было использовано на следующей стадии. Масс-спектрометрия (m/z) 501 [МН+].
Стадия 8: NaN3 добавили к раствору (5S,7R)-5-(4-цианобензил)-3-(3,5-дихлорфенил)-7-метилсульфонилокси-1,3-диазабицикло[3.3.0]октан-2,4-диона, полученного на предыдущей стадии (0,76 г), в DMF (5 мл). Реакционную смесь перемешивали в течение 24 часов. Реакционную смесь разделили между этилацетатом и водой. Органический слой промыли рассолом, сушили (Na2SO4), фильтровали и концентрировали. Остаток очищали с помощью колоночной флэш-хроматографии (силикагель, СН2Cl2) с получением 0,46 г (5S,7S)-5-(4-цианобензил)-3-(3,5-дихлорфенил)-7-азидо-1,3-диазабицикло[3.3.0]октан-2,4-диона. Масс-спектрометрия (m/z) 441 [MH+].
Стадия 9: Через раствор 0,42 г (5S,7S)-5-(4-цианобензил)-3-(3,5-дихлорфенил)-7-азидо-1,3-диазабицикло[3.3.0]октан-2,4-диона, полученного на предыдущей стадии, в 15 мл этанола, содержащий Pd/C (5%, 15 мг) пропускали водород, реакционную смесь перемешивали в атмосфере водорода при комнатной температуре в течение ночи. Реакционную смесь фильтровали через слой целита (Celite), фильтрат концентрировали. Остаток очищали с помощью ВЭЖХ (колонка Beckman 5μ С18; градиент элюента H2O/MeCN (10-100%)/0,1% TFA) с получением 0,21 г (5S,7S)-5-(4-цианобензил)-3-(3,5-дихлорфенил)-7-амино-1,3-диазабицикло-[3.3.0]октан-2,4-диона. Масс-спектрометрия (m/z) 415 [MH+].
Эталонный пример 2: (5S,7S)-5-(4-цианобензил)-3-(3,5-дихлорфенил)-7-амино-1,3-диазабицикло[3.3.0]октан-2,4-дион.
Стадия 1: 3-(3,5-Дихлорфенил)-7-(третбутилдиметилсилилокси)-1,3-диазабицикло[3.3.0]октан-2,4-дион был получен, следуя процедуре, аналогичной описанной в Эталонном примере 1 (стадии 1-4), заменяя соль L-4-трансгидроксипролина бензилового эфира и п-толуолсульфоновой кислоты на гидрохлорид метилового эфира L-4-трансгидроксипролина.
Стадия 2: 3-(3,5-Дихлорфенил)-7-(третбутилдиметилсилилокси)-1,3-диазабицикло[3.3.0]октан-2,4-дион, полученный в предыдущей стадии, был подвергнут процедурам, аналогичным описанным в Эталонном примере 1 (стадии 5-9) для получения целевого соединения.
Протокол клеточной адгезии.
Рекомбинантный белок ICAM-1·Fc был сконструирован из пяти внеклеточных доменов человеческого ICAM-1 при слиянии с константным регионом человеческого IgG. ICAM-1·Fc был очищен с помощью Белка А посредством аффинной хроматографии и сохранен в виде аликвотных проб при -20°С. Иммобилизованный ICAM-1·Fc был приготовлен растворением белка в PBS рН 7,5, переносом 100 мкл на лунку планшеты Falcon Probind III и последующей инкубацией при 4°С в течение ночи. Лунки планшет, покрытые BSA, служат в качестве меры неспецифической фоновой адгезии. Промытые планшеты были выдержаны в 0,25% растворе овальбумина в PBS один час при температуре 37°С. Промытые HBSS клетки Jurkat суспендировали в финальной концентрации 2,5×106/мл в TBSg адгезионном буфере (24 мМ Tris рН 7,4, 0,14 М NaCl, 2,7 мМ KCl, 2 мМ глюкозы, 0,1% HSA). 100 мкл объема клеток добавили к выдержанным, промытым и покрытым ICAM-1·Fc планшетам, содержащим 100 мкл буфера (TBSg, 10 мМ MgCl2, 2% DMSO). Адгезия наблюдалась после одного часа при 37°С. Клетки, которые не подверглись адгезии, удалили, используя отмывку для планшет EL404 (BioTek Instruments; Highland Park, VT). Число клеток, которые подверглись адгезии, было количественно определено путем измерения энзиматической активности эндогенной N-ацетилгексозаминидазы с использованием ферментного субстрата - п-нитрофенол-N-ацетил-β-D-глюкозаминида, pNAG. Количество выделившегося п-нитрофенола было измерено путем определения оптической плотности при 405 нм с использованием спектрофотометра с вертикальным входом для определения количества прикрепленных клеток (VMAX Kinetic Microplate Reader, Molecular Devices, Menio Park, CA). Для сравнительных исследований перед переносом в планшеты, покрытые ICAM-1·Fc, и серийным разведением, соединения из 100% маточных растворов DMSO были разбавлены в планшетном буфере в 2 раза от требуемой тестовой концентрации.
Тест на клеточную адгезию
Этот тест проводили, как описано выше в протоколе клеточной адгезии. Результаты представлены в Таблице 1.
Как показано в Таблице, соединения согласно изобретению демонстрируют превосходную ингибиторную активность против клеточной адгезии, опосредованной LFA-1.
Тест на связывание с человеческими рецепторами нейрокинина-1 (NK-1)
Методика проведения теста на связывание с человеческими рецепторами нейрокинина-1 подробно описана, например, Goso и др., в журнале European J. Pharmacol., 254: 221-227 (1994). Ниже приведено краткое описание метода.
Клетки человеческой лимфобластомы IM-9 выращивали в культуральной среде RPMI 1640, в которую была добавлена бычья фетальная сыворотка (10%), в атмосфере CO2 (5%). Клетки собирали центрифугированием при 900×g в течение 5 минут. Собранные клетки суспендировали в промывочном буфере (50 мМ ТРИС-HCl с рН 7,4, 150 мМ NaCl, 3 мМ MnCl2), центрифугировали при 900×g в течение 5 минут и ресуспендировали в буфере для испытаний (промывочный буфер, содержащий 0,02% БСА, 40 мкг/мл бацитрацина, 4 мкг/мл химостатина, 4 мкг/мл фосфорамидона, 4 мкг/мл лейпептина). Клетки (4×106 клеток) инкубировали с 0,3 нМ [3Н]Sar9, Met(O2)11]-вещества Р (примерное значение Kd: 0,17 нМ, как определено в двух отдельных анализах) и с увеличивающимися концентрациями (разведение 1:10) тестируемых соединений в течение 60 минут при комнатной температуре в 0,5 мл буфера для испытаний. Определяли неспецифическое связывание (NSB) в присутствии 2 мкМ L-703606. Инкубирование заканчивали вакуумной фильтрацией через стеклянный фильтр GF/C, предварительно промытый 0,3% PEI (полиэтиленимином) в течение 1-2 часов. Фильтры дважды промывали 3 мл ледяного промывочного буфера. Измеряли радиоактивность жидкостной сцинтилляционной спектрометрией. Измерения проводили в двух пробах. Ингибиторную активность рассчитывали по следующей формуле:
% ингибирования = 100% - (среднее количество распадов в минуту (dpm) для соединения - среднее количество распадов в минуту для неспецифического связывания)/(среднее количество распадов в минуту для общего связывания - среднее количество распадов в минуту для неспецифического связывания)×100%.
Значения IC50 рассчитывали исходя из данных по ингибированию при различных концентрациях с использованием метода построения кривой, а значения Ki рассчитывали по следующей формуле: Ki=IC50/(1+[Радиолиганд]/Kd).
Результаты представлены в Таблице 2:
Сравнительные эксперименты
Соединения согласно изобретению имеют улучшенные физико-химические и фармакокинетические свойства (меньшую молекулярную массу, logD - коэффициент распределения и улучшенную растворимость в водной среде) и поэтому демонстрируют повышенную эффективность in vivo, что подтверждается следующими результатами испытаний.
1. Тестируемые соединения
Тестируемые соединения имеют следующие структуры:
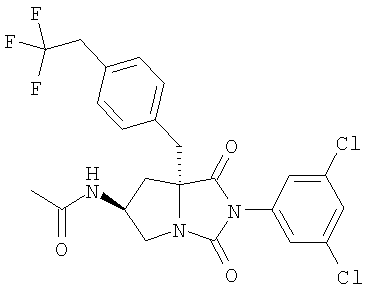
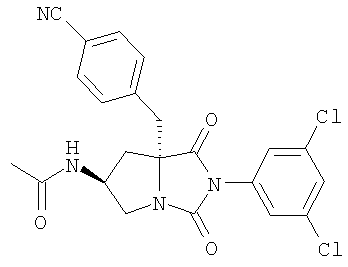
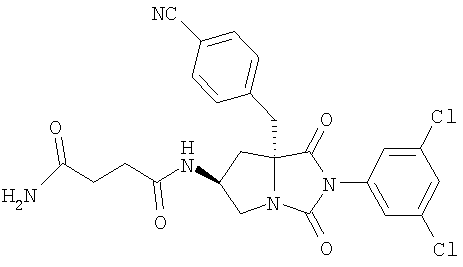
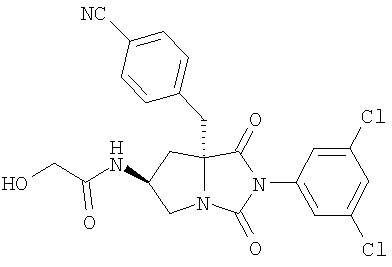
2. Методы
2.1 Растворимость
Растворимость каждого соединения определяли в водном буфере с 50 мМ КН2Р04 при рН 6,5, которое подводили с помощью 1 Н NaOH. Каждое соединение смешивали с 0,5-1,0 мл буфера, затем обрабатывали суспензию ультразвуком в течение 30 минут (UA 100, Shinmeidaikogyo Co., Япония). Затем раствор выдерживали при комнатной температуре более чем в течение 1 часа. Раствор фильтровали с использованием фильтра ULTRAFREE-MC (с порами 0,45 мкм, Millipore Со. МА), фильтрат анализировали методом ВЭЖХ.
2.2. Перитонит, индуцированный тиогликолятом
Интраперитонеальное введение тиогликолята индуцирует перитонит у новорожденных и взрослых кроликов, при котором основным веществом, ответственным за аккумуляцию лейкоцитов в брюшной полости, является LFA-1 (см. Graf et al. J Appl. Physiol. 80, 1984-1992, 1996). Мы использовали эту модель для сравнения эффективности соединений изобретения in vivo по сравнению с эффективностью соединения сравнения из WO 01/30781.
Тестируемые соединения суспендировали в 10% Gelucire® 44/14 (GATTEFOSSE, Франция), затем обрабатывали суспензии ультразвуком в течение нескольких минут на льду. Каждый раствор или эквивалентный объем растворителя вводили перорально в дозе 10 мл на кг массы тела.
Индуцировали перитонит слегка модифицированным методом Graf et al. Кратко, для индукции перитонита 4-дневным самкам нью-зеландских кроликов (Kitayama Labes, Япония) перитонеально вводили раствор тиогликолята (2 мл 3%-ного тиогликолята).
За 30 минут перед перитонеальной инъекцией тиогликолята вводили каждое из соединений или эквивалентный объем растворителя. Через 3 или 4 часа после инъекции тиогликолята оценивали ингибирующее действие соединений на аккумуляцию лейкоцитов.
Результаты выражали как среднее значение ± стандартное отклонение (S.E.M.). Различия между отрицательным и положительным контролем оценивали с использованием студенческого т-теста (StatView 5, SAS Institute Inc.). Статистическое сравнение количества перитонеального клеточного экссудата в группах с индуцированным перитонитом проводили с помощью тестов многократного сравнения Дуннетта (StatView 5, SAS Institute Inc.).
3. Результаты и обсуждение
3.1 Растворимость и clogD
В таблице 3 представлены значения рассчитанного коэффициента распределения (clogD) и растворимости тестируемых соединений. Значения clogD, рассчитанные с использованием комплекта ACD logD (Advanced Chemistry Development Inc. Канада) (рН 6,5), для соединений изобретения составляют от 1,16 до 2,31, что по меньшей мере на единицу логарифма меньше, чем коэффициент распределения соединения сравнения (3,82). Значение logD (рН 6,5) для соединения А составляет 2,43, что хорошо согласуется со значением clogD. Растворимость соединения сравнения, соединения А, соединения В и соединения С при рН 6,5 составляет 1,1, 53,2, 139,5 и 112,4 мкг/мл соответственно, что указывает на то, что соединения согласно изобретению имеют растворимость по меньшей мере в 50 раз больше, чем растворимость соединения сравнения.
3.2. Перитонит, индуцированный тиогликолятом
Результаты представлены на фиг.1-3, которые выражены как среднее значение ± стандартное отклонение (S.E.M.) (n=7-8). ##Р<0,001 относительно отрицательного контроля (студенческий т-тест/StatView). **P<0,01 относительно положительного контроля (двусторонний тест Дуннетта/StatView).
Пероральное введение соединения А в дозах 30 и 100 мг/кг вызывало 67,4 и 72,9%-ное ингибирование аккумуляции лейкоцитов соответственно по сравнению с группой животных, которым вводили растворитель (Р<0,01) (фиг.2). Введение соединения В в дозе 100 мг/кг вызывало 62,1%-ное ингибирование аккумуляции лейкоцитов по сравнению с группой животных, которым вводили растворитель (Р<0,01) (фиг.3). Введение соединения С в дозах 30 и 100 мг/кг снижало аккумуляцию лейкоцитов на 62,8 и 74,9% соответственно по сравнению с группой животных, которым вводили растворитель (Р<0,01) (фиг.3).
Пероральное введение соединения сравнения в дозах вплоть до 100 мг/кг не показало существенного влияния на аккумуляцию лейкоцитов, индуцированную тиогликолятом, пероральное введение соединений А, В и С привело к более чем 60%-ному ингибированию эмиграции лейкоцитов по сравнению с группой животных, которым вводили растворитель.
| название | год | авторы | номер документа |
|---|---|---|---|
| ПРОИЗВОДНЫЕ 2-АЗА-БИЦИКЛО[3.3.0]ОКТАНА | 2008 |
|
RU2478099C2 |
| АМИДЫ ДИАЗАБИЦИКЛОАЛКАНОВ, СЕЛЕКТИВНЫЕ В ОТНОШЕНИИ АЦЕТИЛХОЛИНОВОГО ПОДТИПА НИКОТИНОВЫХ РЕЦЕПТОРОВ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ И СПОСОБЫ ЛЕЧЕНИЯ С ИХ ИСПОЛЬЗОВАНИЕМ | 2007 |
|
RU2448969C2 |
| АМИДЫ ДИАЗАБИЦИКЛОАЛКАНОВ, СЕЛЕКТИВНЫЕ В ОТНОШЕНИИ АЦЕТИЛХОЛИНОВОГО ПОДТИПА НИКОТИНОВЫХ РЕЦЕПТОРОВ | 2007 |
|
RU2517693C2 |
| 5-[(ПИПЕРАЗИН-1-ИЛ)-3-ОКСО-ПРОПИЛ]-ИМИДАЗОЛИДИН-2,4-ДИОНОВЫЕ ПРОИЗВОДНЫЕ В КАЧЕСТВЕ ИНГИБИТОРОВ ADAMTS ДЛЯ ЛЕЧЕНИЯ ОСТЕОАРТРИТА | 2015 |
|
RU2693459C2 |
| ПРОИЗВОДНЫЕ АЗАСПИРОАЛКАНОВ В КАЧЕСТВЕ ИНГИБИТОРОВ МЕТАЛЛОПРОТЕАЗ | 2004 |
|
RU2379303C2 |
| ПРОИЗВОДНЫЕ ДИАЗАБИЦИКЛИЧЕСКИХ АЛКАНОВ С NK-АНТАГОНИСТИЧЕСКОЙ АКТИВНОСТЬЮ | 2003 |
|
RU2310656C2 |
| КРИСТАЛЛИЧЕСКИЕ ФОРМЫ ПРОИЗВОДНОГО ДИАЗАБИЦИКЛООКТАНА И СПОСОБ ИХ ПОЛУЧЕНИЯ | 2014 |
|
RU2801220C2 |
| КРИСТАЛЛИЧЕСКИЕ ФОРМЫ ПРОИЗВОДНОГО ДИАЗАБИЦИКЛООКТАНА И СПОСОБ ИХ ПОЛУЧЕНИЯ | 2014 |
|
RU2695219C2 |
| КРИСТАЛЛИЧЕСКИЕ ФОРМЫ ПРОИЗВОДНОГО ДИАЗАБИЦИКЛООКТАНА И СПОСОБ ИХ ПОЛУЧЕНИЯ | 2023 |
|
RU2838718C2 |
| КРИСТАЛЛИЧЕСКИЕ ФОРМЫ ПРОИЗВОДНОГО ДИАЗАБИЦИКЛООКТАНА И СПОСОБ ИХ ПОЛУЧЕНИЯ | 2023 |
|
RU2840014C2 |
Изобретение относится к новому соединению общей формулы (I),
где R означает Н, ОН, карбамоильную группу, n равно 1 или 2, или к его фармацевтически приемлемой соли. Соединения 1 являются потенциальными ингибиторами клеточной адгезии, опосредованной интегринами, что позволяет использовать их в фармацевтической композиции для лечения или профилактики воспалительного, аутоиммунного или аллергического состояния или отторжения, опосредованного LFA-1. 8 н. и 11 з.п. ф-лы, 3 табл., 3 ил.
где R является атомом водорода, гидроксильной группой, карбамоильной группой, n равно 1 или 2, или его фармацевтически приемлемая соль.
(5S,7S)-5-(4-цианобензил)-3-(3,5-дихлорфенил)-7-ацетиламино-1,3-диазабицикло-[3.3.0]октан-2,4-дион;
(5S,7S)-5-(4-цианобензил)-3-(3,5-дихлорфенил)-7-[(2-гидроксиацетил)амино]-1,3-диазабицикло[3.3.0]октан-2,4-дион;
(5S,7S)-5-(4-цианобензил)-3-(3,5-дихлорфенил)-7-[(3-карбамоилпропионил)амино]-1,3-диазабицикло[3.3.0]октан-2,4-дион.
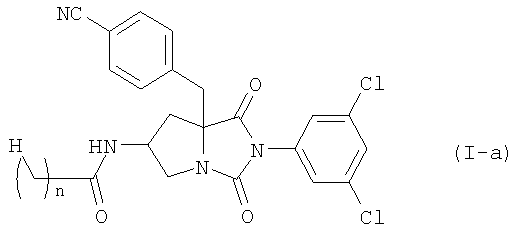
где n равно 1 или 2, или его фармацевтически приемлемой соли, включающий конденсацию соединения формулы (II)
или его соли, с соединением формулы (III-a)
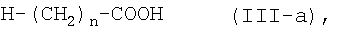
где n является таким же, как определено выше, или с его солью, или с его галогенангидридом, его реакционноспособным эфиром, его ангидридом или его смешанным ангидридом с другой карбоновой кислотой, с последующим превращением полученного соединения в его фармацевтически приемлемую соль, если это желательно.
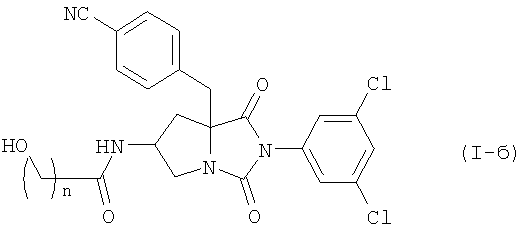
где n равно 1 или 2, или его фармацевтически приемлемой соли, включающий конденсацию соединения формулы (II)
или его соли, с соединением формулы (III-б)
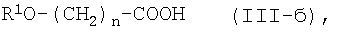
где R1O является защищенной или незащищенной гидроксильной группой, и n является таким же, как определено выше, или с его солью, или с его галогенангидридом, его реакционноспособным эфиром, его ангидридом или его смешанным ангидридом с другой карбоновой кислотой, с последующим удалением защитной группы и, при желании - с последующим превращением конечного соединения в его фармацевтически приемлемую соль.
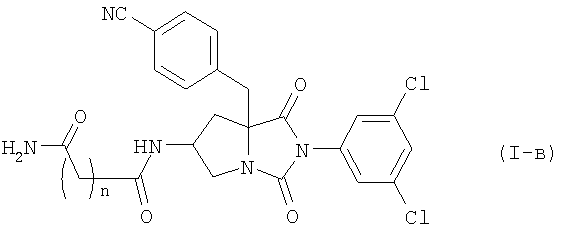
где n равно 1 или 2, или его фармацевтически приемлемой соли, включающий конденсацию соединения формулы (II)
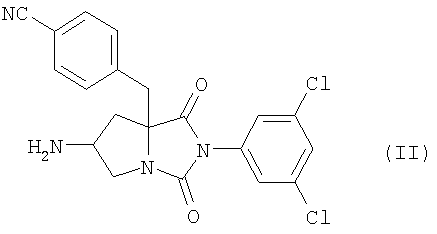
или его соли с соединением формулы (III-в)
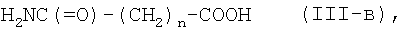
где n является таким же, как определено выше, или с его солью, или с его галогенангидридом, его реакционноспособным эфиром, его ангидридом или его смешанным ангидридом с другой карбоновой кислотой, с последующим превращением полученного соединения в его фармацевтически приемлемую соль, если это желательно.
| Печь для непрерывного получения сернистого натрия | 1921 |
|
SU1A1 |
| US 6214834, 10.04.2001 | |||
| Печь для непрерывного получения сернистого натрия | 1921 |
|
SU1A1 |
| СОЛИ ЭТИЛОВОГО ЭФИРА 3-(2-(4-(4-(АМИНОИМИНОМЕТИЛ)ФЕНИЛ)-4-МЕТИЛ-2,5-ДИОКСОИМИДАЗОЛИДИН-1-ИЛ)АЦЕТИЛАМИНО)-3-ФЕНИЛПРОПИОНОВОЙ КИСЛОТЫ, СПОСОБ ИХ ПОЛУЧЕНИЯ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ИХ ОСНОВЕ | 1997 |
|
RU2174119C2 |

