Область изобретения
Настоящее изобретение относится к способу мечения фосфорилированных пептидов, способу селективной адсорбции фосфорилированных пептидов, к комплексным соединениям, используемым в таких способах, к способу получения комплексных соединений и к соединениям, используемым в качестве исходных веществ в способе получения.
Предпосылки изобретения
Известны in vivo ферменты, содержащие сериновый, треониновый или тирозиновый остаток на специфическом участке, соответствующем активному центру или аллостерическому участку. Ферментативная активность таких ферментов регулируется фосфорилированием или дефосфорилированием гидроксильных групп в таких остатках ферментом, называемым киназой, и т.п. Также известны ферменты, ферментативная активность которых регулируется фосфорилированием или дефосфорилированием аминогруппы или иминогруппы лизина, аргинина или гистидина или карбоксильной группы аспарагиновых кислот или глутаминовых кислот.
Одним из примеров метаболических систем, регулируемых указанным выше фосфорилированием-дефосфорилированием, является система подавления синтеза гликогена и его разложения. Прежде всего, происходит регулирование пути активации такой метаболической системы процессами фосфорилирования-дефосфорилирования.
Недавно проведенные исследования ясно показали, что фосфорилирование-дефосфорилирование играет существенную роль в связанных с заболеваниями метаболических системах.
Например, считается, что одной из причин клеточного карциногенеза является аномальное фосфорилирование-дефосфорилирование. Конкретно, развитие и остановка клеточного цикла регулируется фосфорилированием или дефосфорилированием различных ферментов, т.е. белков. Циклин и циклинзависимая киназа (CDK) являются релевантными факторами в фосфорилировании или дефосфорилировании. Если механизм, связанный с циклином и CDK, нарушен, фосфорилирование или дефосфорилирование может быть неконтролируемым, тем самым запускается механизм аномальной пролиферации клеток.
Кроме того, известны факты, что протеинкиназа C связана с дегрануляцией гистамина, что является причиной аллергических заболеваний, таких как атопический дерматит и сенная лихорадка, и что фосфорилированный tau-белок является причиной нейрофибриллярного сплетения в мозге у пациентов с болезнью Альцгеймера.
В свете вышеизложенного, понимание условий фосфорилирования-дефосфорилирования белков обеспечивает полезный инструмент для различных измерений не только при исследовании экспрессии генов в клетках живых тканей и определении ферментативной активности клеток, но также и в диагностике заболеваний или медицинском лечении.
Традиционные способы идентификации фосфорилированных белков или дефосфорилированных белков имеют разные недостатки.
Например, хотя ферментный иммуноанализ является предпочтительным для анализа образца мишеневого белка в очень малом количестве, очень трудно получить антитела мишеневого белка в достаточном количестве. Кроме того, в случае, когда уровень мишеневого белка составляет несколько кДа или ниже, невозможно получить антитела, надежно связывающиеся с сайтом в белке, где происходит фосфорилирование.
Предложен способ детекции белка, специфически связанного фосфорной кислотой, с использованием фосфорной кислоты, меченной радиоактивным изотопом 32P. Однако следует быть особо внимательным при обращении с радиоактивными изотопами, и необходимо соблюдать соответствующие правила введения и ликвидации жидких отходов радиоактивных изотопов.
Предложена идея применения двумерного электрофореза с учетом того, что происходит дифференциация электрических зарядов между фосфорилированными белками и дефосфорилированными белками. Однако очень трудно определить полосу или пятно фосфорилированного или дефосфорилированного белка при анализе образца, полученного из живого организма, т.к. образец содержит множество белков. Кроме того, использование радиоактивного изотопа для определения полосы или пятна связано с вышеуказанными проблемами.
Документ Morio YASHIRO, et al. [Preparation and Study of Dinuclear Zinc(II) Complex for the Efficient Hydrolysis of the Phosphodiester Linkage in a Diribonucleotide], Journal of the Chemical Society, Chemical Communications, pp.1793-1794 (1995), раскрывает комплекс цинка. Комплекс цинка обладает такой функцией, что два цинковых иона в комплексе разлагают группу фосфорной кислоты, а именно сложного диэфира фосфорной кислоты, из динуклеотида. Однако комплекс цинка, как раскрыто в указанном документе, имеет просто функцию катализатора. Указанный документ не раскрывает способность комплекса цинка координировано связываться с группой фосфорной кислоты. Эксперименты, проведенные авторами изобретения, показали, что константа диссоциации комплекса цинка относительно группы фосфорной кислоты, расположенной между двумя нуклеозидами в виде сэндвича, а именно сложного диэфира фосфорной кислоты, чрезвычайно высока. Другими словами, комплекс цинка обладает низкой координационной способностью в отношении группы сложного диэфира фосфорной кислоты.
Кроме того, документ Hidekazu ARII, et al. [A novel diiron complex as a functional model for hemerythrin], Journal of Inorganic Biochemistry, 82, pp.153-162 (2000), раскрывает комплекс железа, имеющий структуру, аналогичную комплексу цинка. Комплекс железа, однако, является продуктом, синтезированным как модель гемеритрина, а именно белка-носителя, несущего молекулы кислорода. Как и в случае с указанным выше документом, этот документ также не раскрывает и даже не содержит никаких отдаленных предположений относительно координационной связи комплекса железа с группой сложного моноэфира фосфорной кислоты.
Описание изобретения
В свете вышеизложенного целью настоящего изобретения является преодоление проблем предшествующего уровня техники. Еще одной целью настоящего изобретения является обеспечение способа мечения фосфорилированных пептидов, а именно белков, для более легкой детекции и способ селективной адсорбции фосфорилированных пептидов для очистки и т.п.
Еще одной целью настоящего изобретения является обеспечение соединений, обладающих высокой координационной способностью в отношении фосфорилированных пептидов, которые можно использовать в способе мечения и в способе селективной адсорбции, способа получения соединений и соединений, являющихся исходными веществами, используемыми в способе получения.
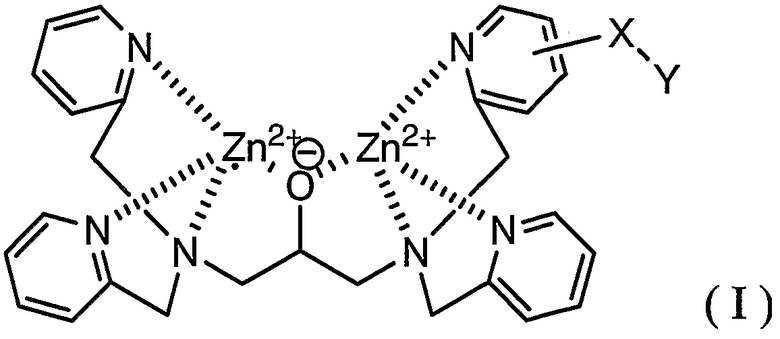
В соответствии с одним аспектом настоящего изобретения обеспечивается способ мечения фосфорилированного пептида комплексным соединением, представленным формулой (I):
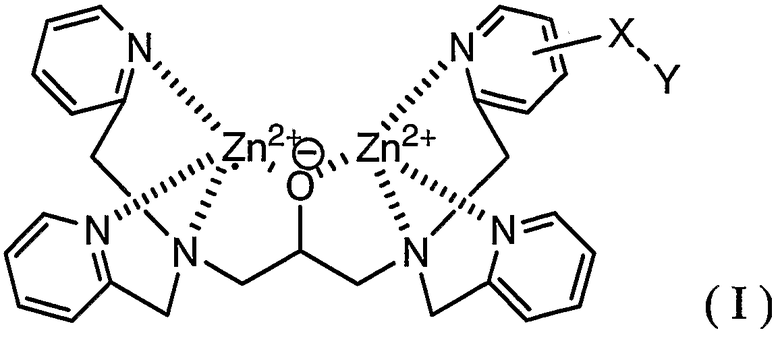
где X представляет собой линкерную группу и Y представляет собой группу мечения.
В соответствии с другим аспектом настоящего изобретения обеспечивается способ селективной адсорбции фосфорилированного пептида с использованием указанного выше комплексного соединения.
В соответствии со следующим аспектом настоящего изобретения обеспечивается указанное выше комплексное соединение.
В соответствии со следующим аспектом настоящего изобретения обеспечивается способ получения соединения (I), включающий схему 1.
Схема 1
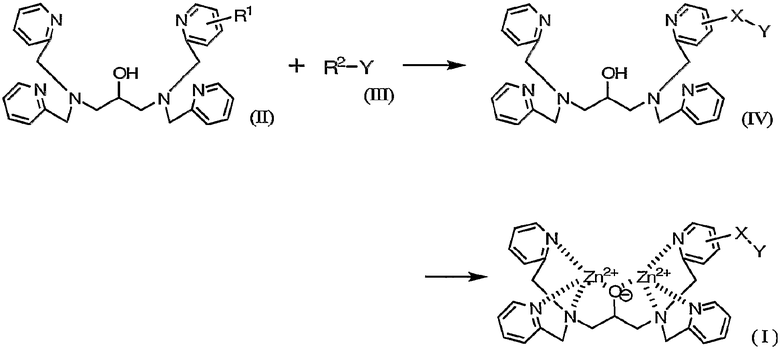
где R1 и R2 каждый представляет собой реакционноспособную группу для образования линкерной группы X и Y представляет собой группу мечения.
В соответствии со следующим аспектом настоящего изобретения обеспечивается соединение, представленное формулой (II):
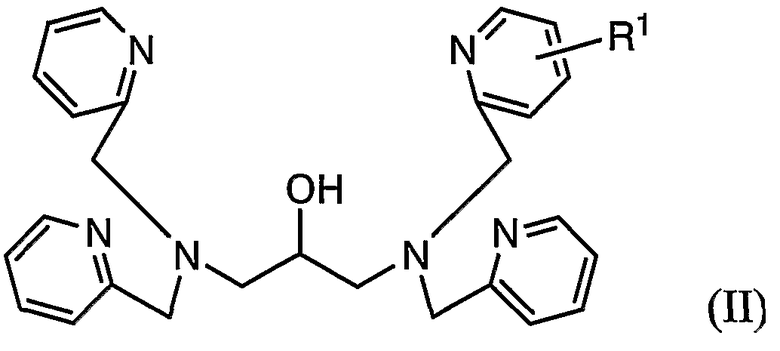
где R1 представляет собой реакционноспособную группу, за исключением аминометильной группы, гидроксиметильной группы, аминогруппы и карбоксильной группы.
В соответствии со способом мечения по настоящему изобретению фосфорилированный пептид, а именно белок, может быть легко обнаружен. Таким образом, настоящее изобретение является полезным для диагностики заболеваний или т.п. с использованием образцов, полученных из живого организма или т.п.
Кроме того, поскольку соединение (I) по настоящему изобретению демонстрирует уникальную координационную связь с двумя гидроксигруппами в группе сложного моноэфира фосфорной кислоты или фосфорного иона, соединение (I) по настоящему изобретению является полезным в качестве соединения, которое можно использовать в способе по настоящему изобретению. Также соединение (I) по настоящему изобретению является полезным для очистки или концентрирования фосфорилированного пептида и получения химической информации о фосфорилированном пептиде.
Эти и другие цели, признаки и преимущества настоящего изобретения будут более очевидны при прочтении представленного ниже подробного описания изобретения и прилагаемых чертежей.
Краткое описание чертежей
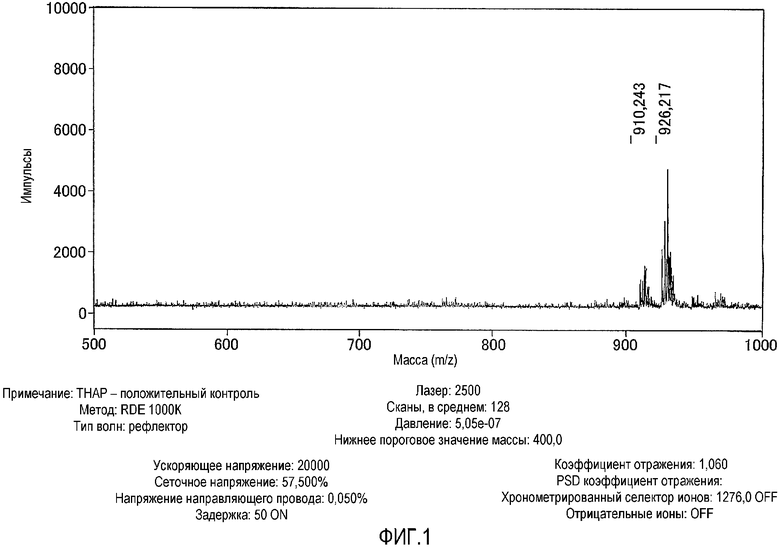
На фиг.1 представлен результат исследования комплекса цинка по настоящему изобретению, полученный с использованием масс-спектрометра MALDI-TOF.
На фиг.2 представлены гели для электрофореза, окрашенные комплексом цинка (A) по настоящему изобретению и другим традиционным красителем (B). Эта иллюстрация наглядно демонстрирует, что способом по настоящему изобретению можно идентифицировать только фосфорилированный пептид, тогда как при использовании традиционного способа окрашиваются все пептиды.
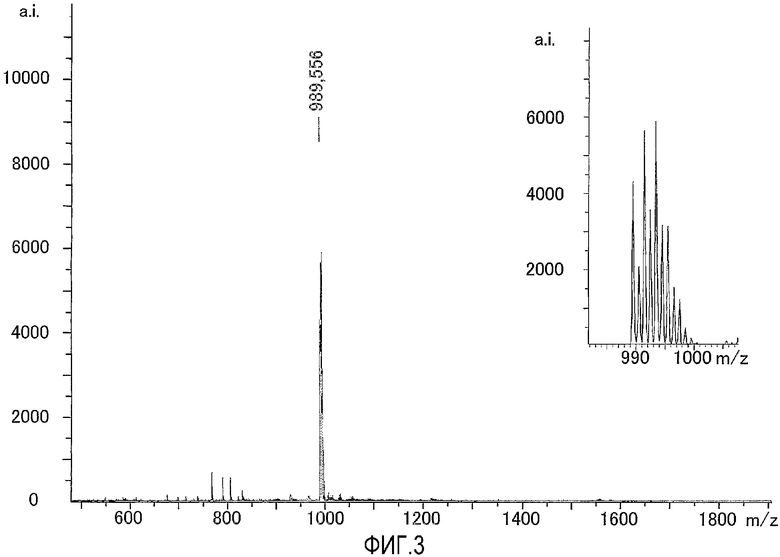
На фиг.3 представлен результат исследования комплекса цинка по настоящему изобретению, полученный с использованием масс-спектрометра MALDI-TOF, а также представлена увеличенная картина некоторой области, которая получена в результате исследования.
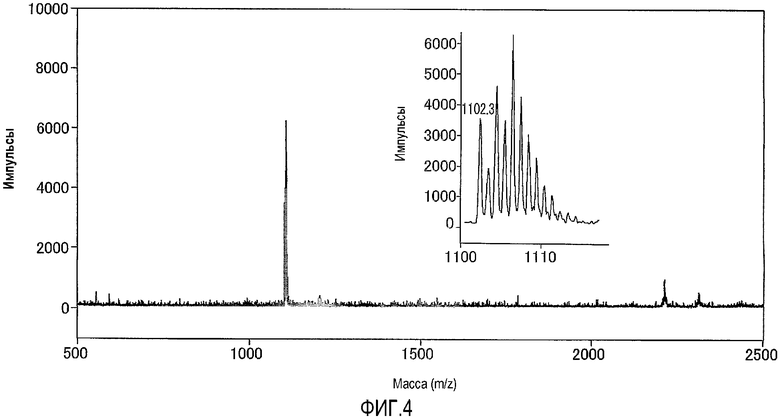
На фиг.4 представлен результат исследования комплекса цинка по настоящему изобретению, полученный с использованием масс-спектрометра MALDI-TOF, а также представлена увеличенная картина некоторой области, полученная в результате исследования.
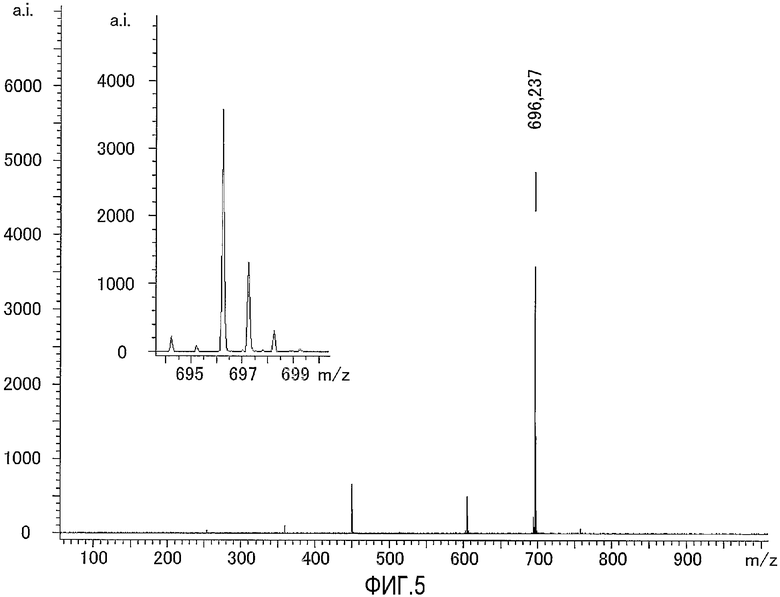
На фиг.5 представлен результат исследования, полученный с использованием масс-спектрометра MALDI-TOF, исходного соединения, используемого для получения соединения по настоящему изобретению, содержащего биотин в качестве группы мечения, а также представлена увеличенная картина некоторой области, полученная в результате исследования.
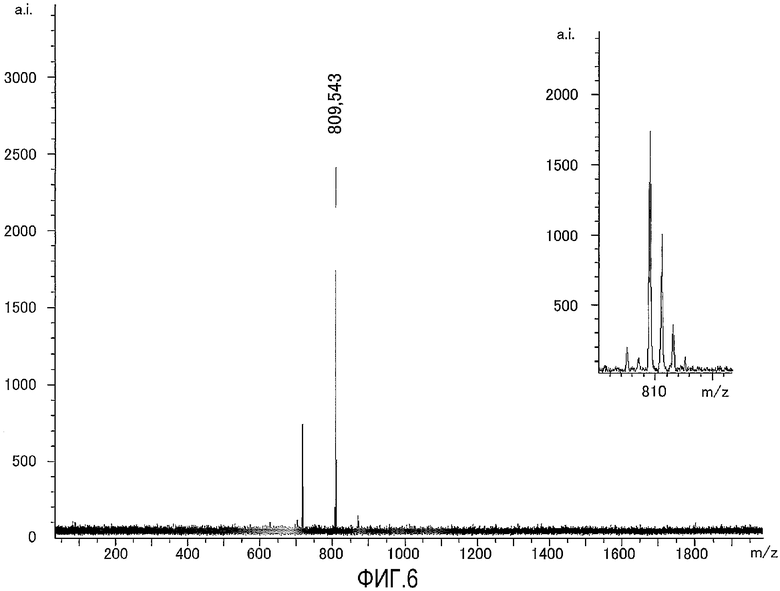
На фиг.6 представлен результат исследования, полученный с использованием масс-спектрометра MALDI-TOF, исходного соединения, используемого для получения соединения по настоящему изобретению, содержащего биотин в качестве группы мечения, а также представлена увеличенная картина некоторой области, полученная в результате исследования.
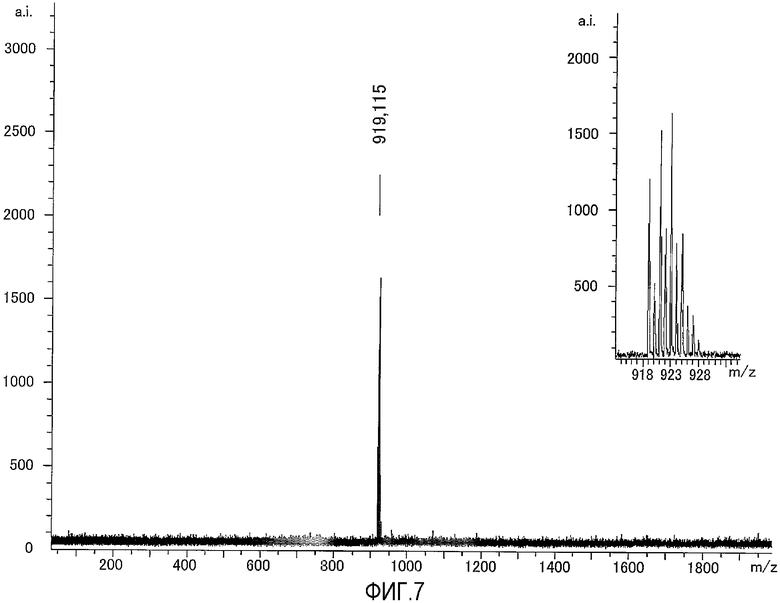
На фиг.7 представлен результат исследования комплекса цинка по настоящему изобретению, полученный с использованием масс-спектрометра MALDI-TOF, а также представлена увеличенная картина некоторой области, полученная в результате исследования.
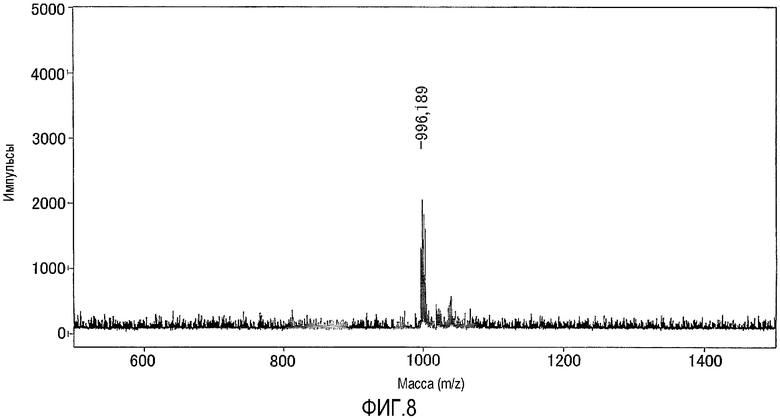
На фиг.8 представлен результат исследования комплекса цинка по настоящему изобретению, полученный с использованием масс-спектрометра MALDI-TOF.
На фиг.9 представлены гели для электрофореза, окрашенные комплексом цинка (A) по настоящему изобретению и другим традиционным красителем (B). Эта иллюстрация наглядно демонстрирует, что способом по настоящему изобретению можно идентифицировать только фосфорилированный пептид, тогда как при использовании традиционного способа окрашиваются все пептиды.
Лучший способ осуществления изобретения
Основным отличительным признаком способа по настоящему изобретению является то, что фосфорилированный пептид можно легко идентифицировать путем образования сложного соединения, в котором комплексное соединение, содержащее группу мечения, представленное формулой (I), специфически связано с фосфорилированным пептидом.
До настоящего времени были известны различные комплексы металлов, способные связываться с группой фосфорной кислоты. Однако соединение, которое является аналогичным соединению, представленному формулой (I), и которое содержит группу мечения, не было известно. Авторы настоящего изобретения обнаружили, что использование комплексного соединения, представленного формулой (I), является выгодным для обеспечения легкой детекции и идентификации фосфорилированного пептида даже в образце, полученном из живого организма, содержащего множество различных пептидов, и таким образом было создано настоящее изобретение.
Ниже описан способ мечения фосфорилированных пептидов в соответствии с одним вариантом осуществления настоящего изобретения.
Сначала получают образец, содержащий, по существу, все возможные виды пептидов, составляющих клетку ткани, которую нужно исследовать. Такое получение осуществляют в соответствии с традиционным способом, широко используемым в биохимии.
Затем пептиды, содержащиеся в образце, разделяют. Способ разделения конкретно не ограничен, и можно использовать такой традиционный способ разделения, как электрофорез.
При осуществлении электрофореза гель после электрофореза погружают в раствор, содержащий комплексное соединение, представленное формулой (I), для мечения фосфорилированного пептида, и затем фосфорилированный пептид определяют способом детекции в зависимости от типа группы мечения.
Растворитель, который используют в растворе, содержащем комплексное соединение, представленное формулой (I), конкретно не ограничен при условии, что он не должен мешать детекции фосфорилированного пептида. Примерами такого растворителя являются вода, включающая буфер, и раствор, содержащий соль, отличную от буфера; спирты, такие как метанол и этанол; и смешанный растворитель, содержащий указанные компоненты. Предпочтительно, используют водный растворитель, в основном, в целях предотвращения денатурирования пептида.
Ниже описано соединение (I), используемое в указанном выше способе.
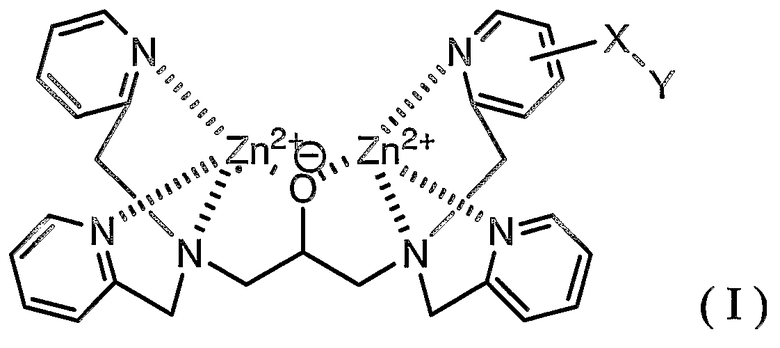
где X представляет собой линкерную группу и Y представляет собой группу мечения.
В формуле (I) Zn выбран в качестве координационного металла, поскольку Zn обладает высокой координационной способностью в отношении группы фосфорной кислоты в фосфорилированном белке, а именно сложного моноэфира фосфорной кислоты.
Линкерная группа в настоящем описании и формуле изобретения означает группу, способную связываться с основным скелетом и с группой мечения. Линкерная группа способствует получению соединения (I) и ингибирует действие группы мечения, препятствующее координации соединения (I) с группой фосфорной кислоты, связанной с пептидом. Учитывая это обстоятельство, линкерная группа может представлять собой координационную связь, непосредственно связывающую основной скелет с группой мечения, если можно легко получить исходное соединение, в котором группа мечения непосредственно связана с основным скелетом, в синтезе соединения (I) или если группа мечения имеет такой небольшой размер, что можно легко предотвратить действие, препятствующее координации соединения (I) с группой фосфорной кислоты.
Тип линкерной группы в настоящем описании и формуле изобретения конкретно не ограничен, при условии, что линкерная группа обладает указанными выше функциями. Примерами линкерных групп являются следующие: C1-C6 алкиленовая группа, аминогруппа (-NH-), группа простого эфира (-O-), тиоэфирная группа (-S-), карбонильная группа (-C(=O)-), тионильная группа (-C(=S)-), группа сложного эфира, амидная группа, группа мочевины (-NHC(=O)NH-), группа тиомочевины (-NHC(=S)NH-); C1-C6 алкиленовая группа, содержащая на одном конце группу, выбранную из группы, включающей аминогруппу, группу простого эфира, тиоэфирную группу, карбонильную группу, тионильную группу, группу сложного эфира, амидную группу, группу мочевины и группу тиомочевины; C1-C6 алкиленовая группа, содержащая на противоположных концах две группы, выбранные из группы, включающей аминогруппу, группу простого эфира, тиоэфирную группу, карбонильную группу, тионильную группу, группу сложного эфира, амидную группу, группу мочевины и группу тиомочевины, где группы, расположенные на противоположных концах, являются одинаковыми или отличными друг от друга; и группа, в которой две или более двух групп выбраны из группы, включающей аминогруппу, группу простого эфира, тиоэфирную группу, карбонильную группу, тионильную группу, группу сложного эфира, амидную группу, группу мочевины и группу тиомочевины, и C1-C6 алкиленовая группа являются линейно связанными.
C1-C6 алкиленовая группа означает двухвалентную алифатическую углеводородную группу, содержащую 1-6 атомов углерода, которая является линейной или разветвленной, например метиленовая, этиленовая, пропиленовая, тетраметиленовая, гексаметиленовая, метилэтиленовая, метилпропиленовая и диметилпропиленовая группа, предпочтительно представляет собой C1-C4 алкиленовую группу и более предпочтительно представляет собой C1-C2 алкиленовую группу.
Группа мечения в настоящем описании и формуле изобретения конкретно не ограничена при условии, что она является группой, традиционно используемой в биохимии. Однако соединение, содержащее радиоизотоп, не является предпочтительным с точки зрения обращения с ним. Примерами группы мечения являются флуоресцентная группа, группа, содержащая радикал окиси азота, и биотин.
Флуоресцентная группа представляет собой заместитель, способный стабильно генерировать флуоресценцию с относительно длинной длиной волны, и группу, обычно используемую в биохимии, можно неограниченно использовать в качестве флуоресцентной группы, независимо от того, является ли соединение растворимым в воде или в масле. Примерами флуоресцентных групп являются аминометилкумарин и его производные, флуоресцеин и его производные, тетраметилродамин и его производные, антранилоил и его производные, нитробензоксадиазол и его производные и диметиламинонафталин и его производные.
Группа, содержащая радикал окиси азота, представляет собой группу, содержащую стабильный радикал, и является способной к детекции фосфорилированного пептида посредством электронного спинового резонанса (ESR). Как правило, биологическая молекула не демонстрирует электронный спиновый резонанс, поскольку она не содержит непарный электрон. С другой стороны, поскольку пептид, связанный с соединением (I), содержащим радикал окиси азота, координированный с ним, демонстрирует электронный спиновый резонанс, фосфорилированный пептид может быть идентифицирован.
Биотин обладает специфической и высокой аффинностью к авидину, полученному из альбумина, и к стрептоавидину, полученному из actinomycetes. В свете этого комплексное соединение может специфически связываться с ферментом посредством биотина и авидина или стрептоавидина путем связывания авидина или стрептоавидина с соединением (I), содержащим биотин в качестве группы мечения, и затем связывания с биотинилированным ферментом. Фосфорилированный пептид можно идентифицировать с использованием фермента, такого как щелочная фосфатаза, пероксидаза и люцифераза, и с использованием агента окрашивания, в зависимости от типа фермента. Например, если сначала фосфорилированный пептид связывают при помощи комплексного соединения по настоящему изобретению, после чего комплексное соединение связывают при помощи щелочной фосфатазы в качестве фермента через стрептоавидин, затем добавляют нитросиний тетразолий и 5-бром-4-хлор-3-индолилфосфат в качестве агентов окрашивания и смеси дают прореагировать в течение нескольких часов, фосфорилированный пептид становится пурпурного цвета, что дает возможность идентифицировать фосфорилированный пептид. Кроме того, стрептоавидин, меченный флуоресцентным пигментом, таким как родамин, является коммерчески доступным. Использование стрептоавидина, меченного флуоресцентным пигментом, дает возможность идентифицировать фосфорилированный пептид известным способом анализа флуоресцентного изображения.
Ниже описан способ селективной адсорбции фосфорилированных пептидов в соответствии с вариантом осуществления настоящего изобретения.
В соответствии со способом селективной адсорбции фосфорилированных пептидов биотин или подобное вещество, способное к специфическому связыванию со специфическим соединением, используют в качестве группы мечения. Например, коммерчески доступным является агарозный гель со связанным с ним стрептоавидином. Агарозный гель с комплексным соединением по настоящему изобретению, связанным с ним, можно получить путем соединения его с комплексным соединением, содержащим биотин в качестве группы мечения, для взаимодействия. Фосфорилированный пептид в смешанном образце можно селективно адсорбировать в комплексное соединение по настоящему изобретению путем нанесения смешанного образца на агарозный гель. После селективной адсорбции добавление фосфорнокислого буфера или подобного соединения, способного к десорбции фосфорилированного пептида из комплексного соединения по настоящему изобретению, обеспечивает возможность эксклюзивного получения фосфорилированного пептида. Таким же образом, использование агарозного геля является выгодным для очистки или концентрации фосфорилированного пептида без использования электрофореза. Также, коммерчески доступны магнитные бусинки со связанным с ними стрептоавидином. Использование магнитных бусинок также делает возможным очистку или концентрацию фосфорилированного пептида. Кроме того, коммерчески доступны пластины со связанным с ними стрептоавидином для измерения поверхностного плазмонного резонанса (SPR). Пластину с комплексным соединением по настоящему изобретению, связанным с ней, для измерения поверхностного плазмонного резонанса можно получить нанесением на эту пластину комплексного соединения по настоящему изобретению, содержащего биотин в качестве группы мечения. Присутствие или отсутствие фосфорилированного пептида в образце можно определить путем измерения поверхностного плазмонного резонанса (SPR) пластины.
Можно синтезировать соединение, эквивалентное соединению (I), в котором в пиридиновое кольцо введена метильная группа или подобная группа, для обеспечения, по существу, такого же действия и эффектов, как и в описанном варианте воплощения настоящего изобретения. Такое соединение, эквивалентное соединению (I), входит в объем настоящего изобретения.
Расположение (-X-Y) группы в соединении по настоящему изобретению конкретно не ограничено. Группа (-X-Y) может быть расположена на участке, как показано в соединении (I').
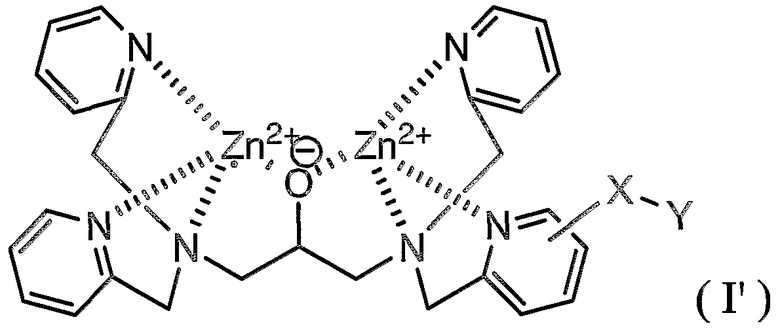
Соединение (I) и соединение (I') являются, по существу, эквивалентными. Хотя и непонятно, какое соединение синтезируется, соединение (I) или соединение (I'), что действительно понятно, это то, что синтезируется смесь соединения (I) и соединения (I'). Нет необходимости указывать, что соединение (I') входит в объем настоящего изобретения.
Комплексное соединение, представленное формулой (I), можно легко получить способом, включающим схему 1.
Схема 1
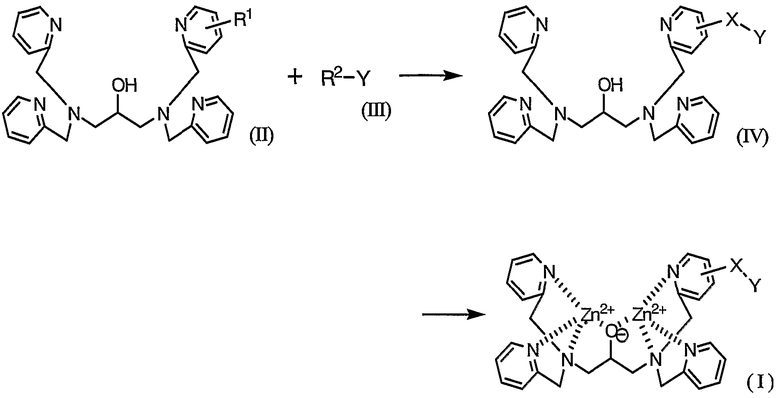
где X и Y определены выше и R1 и R2 каждый представляет собой реакционноспособную группу для образования линкерной группы X.
На указанной выше схеме группу мечения Y вводят в основной скелет через линкерную группу X путем взаимодействия реакционноспособных групп R1 и R2.
Тип групп R1, R2, растворитель, температура реакции, реагент, отличный от указанных выше, способ очистки и другие факторы в основном определяются типом группы X. Например, в случае введения группы мечения через аминогруппу (вторичную или третичную аминогруппу), в качестве комбинации R1 и R2, комбинация группы, содержащей аминогруппу (первичную или вторичную аминогруппу) на ее удаленном конце и удаляемой группы, такой как атом галогена. Конденсация R1 и R2 в присутствии основных групп в растворителе является примером общего условия реакции. В случае, если R1 является активной группой, можно легко ввести группу мечения.
Затем соединение (I) можно синтезировать путем добавления соли металла к раствору, содержащему соединение (IV). В качестве соли металла можно добавлять нитрат цинка (II) или ацетат цинка (II). В случае добавления ацетата цинка (II) получают соединение формулы (V), представленной ниже, где уксусная кислота является временно координированной.
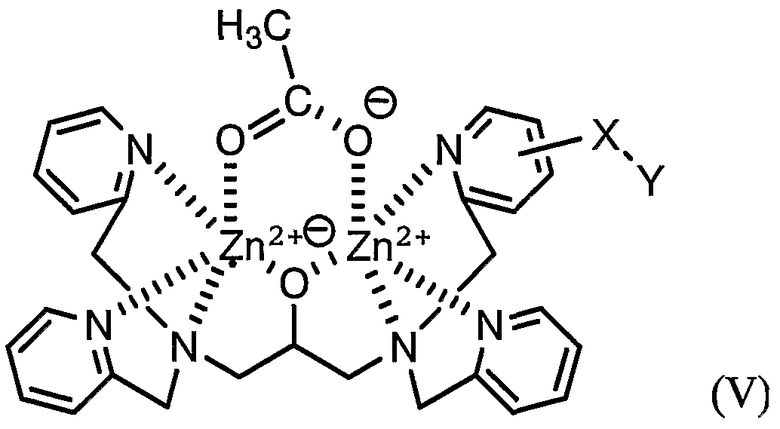
Соединение (V) является более химически стабильным, чем соединение (I) и, следовательно, полезно для хранения. Соединение (V) является эквивалентным соединению (I) и может быть использовано таким же образом, как и соединение (I). Конкретно, путем добавления соединения (V) к смеси, содержащей пептид, можно осуществить детекцию фосфорилированного пептида, поскольку группа сложного моноэфира фосфорной кислоты взаимозаменяемым образом координируется к соединению (I) вместо уксусной кислоты.
Соединение (II), т.е. исходное соединение для соединения (I), можно синтезировать в соответствии с представленной ниже схемой 2.
Схема 2
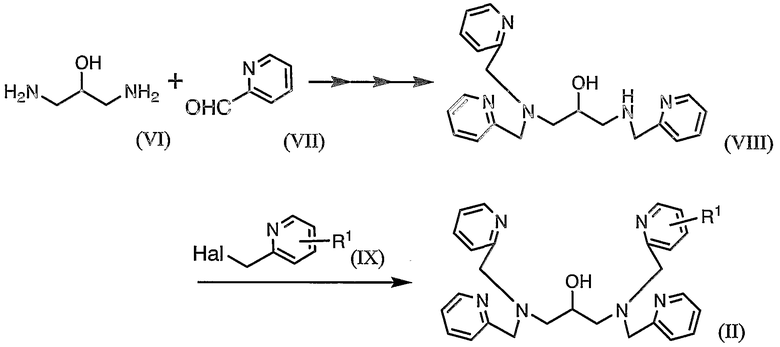
где R1 определен выше и "Hal" представляет собой атом галогена и, предпочтительно, бром.
Соединение (VI), т.е. 1,3-диамино-2-пропанол, как соединение, являющееся исходным веществом, может быть коммерчески доступным. Кроме того, поскольку соединение (VII) и соединение (IX) оба имеют относительно простую структуру, соединения (VII) и (IX) могут быть коммерчески доступными или их можно синтезировать способом, хорошо известным специалистам в данной области.
На схеме 2 сначала соединение (VI) и соединение (VII) подвергают взаимодействию друг с другом в присутствии катализатора конденсации с получением соединения (VIII). Такое взаимодействие может быть осуществлено постадийно путем введения соединения (VII). Альтернативно, соединение (VIII) можно получить в одну стадию, используя 3 или более эквивалентов соединения (VII) относительно соединения (VI).
На схеме 2 восстановительное аминирование осуществляют как реакцию конденсации. Растворитель, используемый в восстановительном аминировании, конкретно не ограничен при условии, что растворитель способен существенно растворять соединение (VI) и соединение (VII) и не ингибирует аминирование. Например, спирты, такие как метанол, этанол и изопропанол; простые эфиры, такие как диэтиловый эфир, тетрагидрофуран и диоксан; вода или смешанный растворитель, содержащий два или более из указанных компонентов, могут быть использованы в качестве растворителя.
Восстановительное аминирование можно осуществить с использованием традиционного восстановителя после конденсации соединения (VI) и соединения (VII) в присутствии концентрированной хлористоводородной кислоты в качестве катализатора.
Оптимальные условия, касающиеся температуры реакции и времени реакции, могут быть выбраны, необязательно, в зависимости от типа исходного соединения или других факторов. Например, реакцию можно осуществить при температуре реакции от 20 до 80°С в течение времени от 12 до 100 часов.
После завершения реакции растворитель и т.п. отгоняют при пониженном давлении до того, как добавляют воду. После добавления воды полученную смесь экстрагируют растворителем, нерастворимым в воде, и органический слой сушат над безводным сульфатом магния или подобным веществом. Затем растворитель отгоняют при пониженном давлении. После этого остаток очищают хорошо известным способом, таким как колоночная хроматография на силикагеле, с получением соединения (VIII).
Способ получения соединения (VIII) не ограничивается способом, показанным на схеме 2. Альтернативно, соединение (VIII) можно синтезировать с использованием, например, соединения (VI) и галогенсодержащего соединения.
Затем соединение (II) можно синтезировать путем взаимодействия соединения (VIII) с соединением (IX). Такое взаимодействие можно осуществить известным способом синтеза третичных аминов. Например, соединение (VIII) и соединение (IX) конденсируют в присутствии основания в растворителе. На стадии конденсации защитную группу можно вводить и отщеплять, в зависимости от типа R1, как это необходимо. Альтернативно, соединение (II) можно синтезировать при осуществлении стадии конденсации с использованием соединения, содержащего неактивную группу заместителя, вместо использования R1 в соединении (IX), и путем замещения неактивного заместителя группой R1 путем преобразования функциональных групп. Например, стадию конденсации осуществляют с использованием соединения, содержащего в качестве неактивного заместителя нитрогруппу, и замещением нитрогруппы аминогруппой как группой, являющейся реакционноспособной.
Представленное ниже комплексное соединение (X) можно использовать в способе по настоящему изобретению вместо комплексного соединения (I).
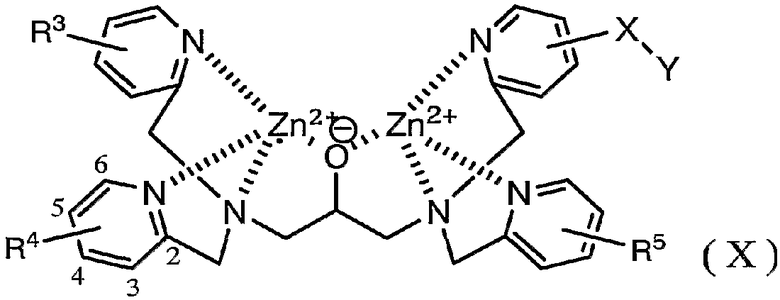
где X, Y определены выше, и R3 - R5 каждый представляет собой группу электронно-донорного заместителя в положении 4 или 6 пиридинового кольца.
Комплексное соединение (X), используемое в способе по настоящему изобретению, является электрообогащенным азотом пиридина посредством электронно-донорной группы заместителя, введенной в соответствующее положение для замещения. Следовательно, комплексное соединение (X), используемое в способе по настоящему изобретению, является высококоординированным к цинку, обеспечивая, таким образом, возможность получения комплексного соединения (X) простым способом с обеспечением при этом стабильности.
Способ использования комплексного соединения (X), способ получения комплексного соединения (X) и исходное соединение для комплексного соединения (X) являются, по существу, такими же, как указано для комплексного соединения (I).
В указанных выше способах и соединении (I) предпочтительно использовать в качестве группы мечения биотин при получении комплексного соединения. Использование биотина является предпочтительным, поскольку биотин не представляет трудностей в обращении с ним, и он нашел широкое применение, поскольку демонстрирует различные реакции окрашивания. Использование биотина является эффективным для легкой идентификации фосфорилированного пептида.
Ниже представлены примеры получения и экспериментальные примеры для более подробного описания настоящего изобретения. Однако настоящее изобретение не ограничивается приведенными примерами.
Примеры
Пример получения 1-1: Метил 6-бромметилникотинат
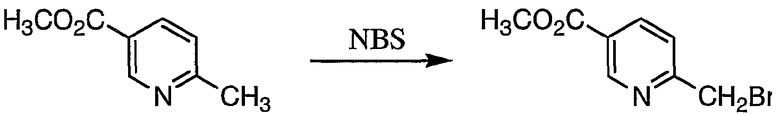
К раствору метил 6-метилникотината (50 г, 331 ммоль) в тетрахлориде углерода (625 мл) добавляли N-бромсукцинимид (59 г, 331 ммоль). Затем добавляли 1,0 г бензоилпероксида, реакцию смеси осуществляли при температуре от 40 до 50°С в течение 24 часов при облучении светом от прожектора.
После охлаждения реакционной смеси осажденные кристаллы отделяли фильтрованием. Фильтрат промывали водным раствором, содержащим гидрокарбонат натрия, и концентрировали. Остаток, полученный в результате концентрации, очищали колоночной хроматографией на силикагеле с получением 37 г целевого соединения.
1H-ЯМР (CDCl3, 300 МГц): δ 3,96 (3H, с, OCH3), 4,58 (2H, с, CH2Br), 7,54 (1H, д, Py), 8,30 (1H, дд, Py), 9,17 (1H, д, Py).
Пример получения 1-2: N,N,N'-три(2-пиридилметил)-1,3- диаминопропан-2-ол
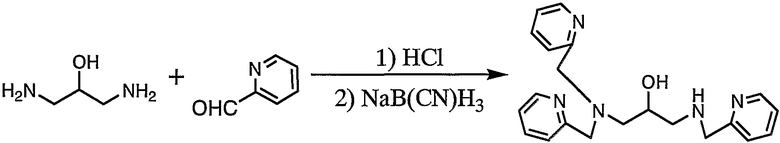
К раствору 1,3-диаминопропан-2-ола (32,6 г, 362 ммоль) в метаноле (2400 мл) добавляли 60 мл концентрированной хлористоводородной кислоты. Затем добавляли по каплям 2-пиридинальдегид (116,3 г, 1,09 моль), а затем добавляли цианоборгидрид натрия (50,16 г, 798 ммоль). По завершении добавления реакцию смеси осуществляли при комнатной температуре в течение 3 дней.
После добавления к раствору концентрированной хлористоводородной кислоты и доведения pH раствора до 6 полученный раствор концентрировали до некоторой степени. Затем добавляли 0,1 н. водный раствор гидроксида натрия для доведения pH раствора до 7, а затем экстрагировали хлороформом. Экстракты собирали и сушили, полученное вещество концентрировали. Остаток, полученный в результате концентрации, очищали колоночной хроматографией на силикагеле с получением 34 г целевого соединения.
1H-ЯМР (CDCl3, 300 МГц): δ 2,59-2,83 (4H, м, CH2), 3,86-4,01 (7H, м, NCH2Py, CH), 7,15 (3H, дд, Py), 7,23-7,32 (3H, м, Py), 7,56-7,65 (3H, м, Py), 8,53 (3H, дд, Py).
Пример получения 1-3: N,N,N'-три(2-пиридилметил)-N'-(5- метоксикарбонил-2-пиридилметил)-1,3-диаминопропан-2-ол
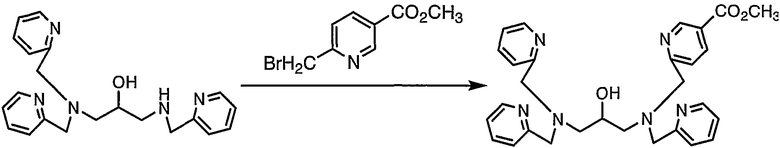
К раствору N,N,N'-три(2-пиридилметил)-1,3-диаминопропан-2-ола (18,2 г, 50 ммоль), полученного в примере получения 1-2, в безводном диметилформамиде (150 мл) добавляли карбонат калия (13,8 г, 100 ммоль), а затем добавляли по каплям раствор метил 6-бромметилникотината (11,5 г, 50 ммоль), полученного в примере получения 1-1, в безводном диметилформамиде (75 мл). После завершения добавления по каплям смесь подвергали взаимодействию при 50°С в течение 1 часа.
По завершении реакции раствор охлаждали. Затем охлажденный раствор выливали в 750 мл воды и pH раствора доводили до 8 путем добавления 1 н. раствора хлористоводородной кислоты. После экстракции этилацетатом экстракты собирали, промывали водой и насыщенным солевым раствором и концентрировали. Остаток, полученный в результате концентрации, очищали колоночной хроматографией на силикагеле с получением 21,5 г целевого соединения.
1H-ЯМР (CDCl3, 300 МГц): δ 2,58-2,73 (4H, м, CH2), 3,83-3,95 (12H, м, OCH3, NCH2Py, CH), 7,10-7,14 (3H, м, Py), 7,34 (3H, д, Py), 7,50-4,60 (4H, м, Py), 8,17 (1H, дд, Py), 8,50 (3H, д, Py), 9,09 (1H, д, Py).
Пример получения 1-4: N,N,N'-три(2-пиридилметил)-N'-[5-N"-(2-аминоэтил)карбамоил-2-пиридилметил]-1,3-диаминопропан-2-ол
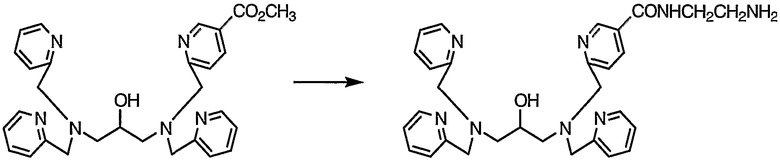
К раствору N,N,N'-три(2-пиридилметил)-N'-(5-метоксикарбонил-2-пиридилметил)-1,3-диаминопропан-2-ола (9,7 г, 18,9 ммоль), полученного в примере получения 1-3, в метаноле (100 мл) добавляли по каплям этилендиамин (22,7 г, 378 ммоль). После завершения добавления по каплям смесь подвергали взаимодействию при комнатной температуре в течение 3 дней.
По завершении реакции раствор концентрировали и остаток, полученный в результате концентрации, очищали колоночной хроматографией на силикагеле, с получением 9,72 г целевого соединения.
1H-ЯМР (CDCl3, 300 МГц): δ 2,54-2,71 (4H, м, CH2), 2,94 (2H, т, CH2N), 3,49 (2H, дт, CH2N), 3,80-3,99 (9H, м, NCH2Py, CH), 7,12 (3H, ддд, Py), 7,35 (3H, д, Py), 7,45 (1H, д, Py), 7,58 (3H, ддд, Py), 8,02 (1H, дд, Py), 8,49 (3H, ддд, Py), 8,89 (1H, д, Py).
Пример получения 1-5: N,N,N'-три(2-пиридилметил)-N'-[5-N"-(7-нитро-2,1,3-бензоксадиазол-4-иламиноэтил)карбамоил-2-пиридилметил]-1,3-диаминопропан-2-ол
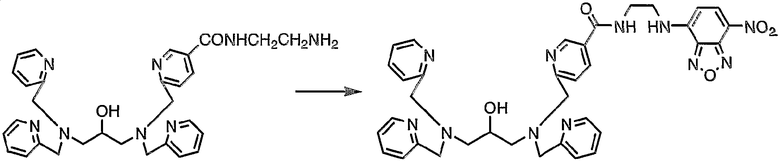
К раствору N,N,N'-три(2-пиридилметил)-N'-[5-N"-(2-аминоэтил)карбамоил-2-пиридилметил]-1,3-диаминопропан-2-ола (200 мг, 0,37 ммоль), полученного в примере получения 1-4, в ацетонитриле (20 мл) добавляли гидрокарбонат натрия (336 мг, 4,0 ммоль), затем добавляли 4-хлор-7-нитро-2,1,3-бензоксадиазол (73,8 мг, 0,37 ммоль). Смесь подвергали взаимодействию при комнатной температуре в течение 2 часов.
По завершении реакции раствор концентрировали. Затем добавляли 50 мл дихлорметана и 50 мл воды и органический слой и водный слой разделяли. Органический слой сушили над безводным сульфатом натрия, концентрировали и полученный остаток очищали колоночной хроматографией на силикагеле с получением 71,2 мг целевого соединения.
lH-ЯМР (CDCl3, 300 МГц): δ 2,46-2,69 (4H, м, CH2), 3,65-3,95 (13H, м, NCH2CH2N, NCH2Py, CH), 6,06 (1H, д, Ar), 7,08-7,13 (3H, м, Py), 7,32 (3H, д, Py), 7,42 (1H, д, Py), 7,56 (3H, ддд, Py), 7,96 (1H, дд, Py), 8,44-8,48 (3H, м, Py), 8,19 (1H, д, Ar), 8,83 (1H, д, Py).
Пример получения 1-6: Раствор, содержащий комплекс цинка по настоящему изобретению
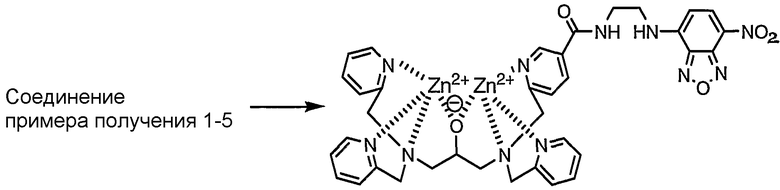
Получали 50 мкМ водного раствора, содержащего соединение, полученное в примере получения 1-5, затем к раствору добавляли 2 эквивалента нитрата цинка. Таким образом, получали раствор, содержащий комплекс цинка по настоящему изобретению.
Комплекс цинка идентифицировали в соответствии со следующим способом. А именно, соединение, полученное в примере получения 1-5, растворяли в фосфорнокислом буфере (pH=6,86) с получением 50 мкМ раствора, затем к раствору добавляли 2 эквивалента нитрата цинка. Комплекс цинка по настоящему изобретению в растворе показывал следующую структуру, определенную при помощи масс-спектрометра MALDI-TOF (матрица: 2',4',6'-тригидроксиацетофенон, тип волн: рефлектор, ускоряющее напряжение: 20000 В, сеточное напряжение: 57,500%, лазер: 2500, сканы, в среднем: 128, давление: 5.05e-07).
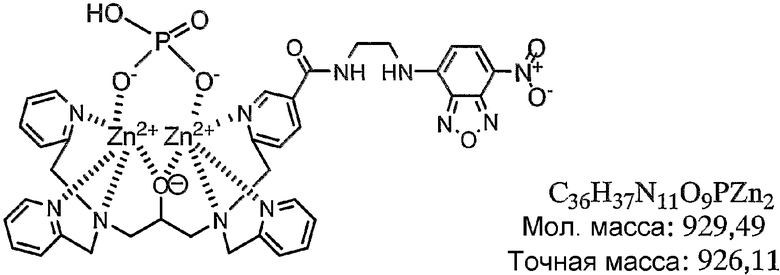
Результаты измерений, полученные при помощи масс-спектрометра MALDI-TOF, представлены на фиг.1. Как показано на фиг.1, наблюдали молекулярный ионный пик при 926,2 (точная масса: 926,11). Считается, что появление молекулярного ионного пика при 910,2 вызвано отщеплением кислорода в группе оксадиазола.
Пример получения 2-1: N,N,N'-три(2-пиридилметил)-N'-[5-N"-(2-D-биотинамидоэтил)карбамоил-2-пиридилметил]-1,3-диаминопропан-2-ол
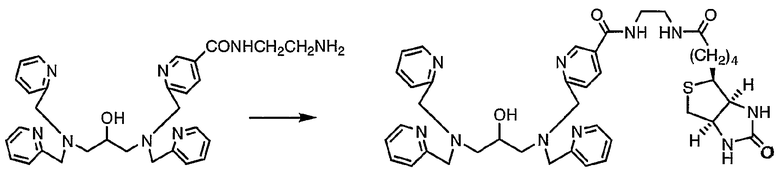
К раствору D-биотина (137 мг, 0,56 ммоль) в диметилформамиде (10 мл) добавляли 1,1'-карбонилдиимидазол (116 мг, 0,72 ммоль). Смесь подвергали взаимодействию при комнатной температуре в течение 12 часов. Затем раствор охлаждали льдом, после чего добавляли по каплям раствор N,N,N'-три(2-пиридилметил)-N'-[5-N"-(2-аминоэтил)карбамоил-2-пиридилметил]-1,3-диаминопропан-2-ола (260 мг, 0,48 ммоль), полученного в примере получения 1-4, в диметилформамиде (3 мл). Охлаждающую баню удаляли и реакцию осуществляли при комнатной температуре в течение 2 часов.
По завершении реакции раствор выливали в 50 мл воды, а затем дважды экстрагировали 50 мл хлороформа. Затем экстракты концентрировали, неочищенный продукт очищали колоночной хроматографией на силикагеле с получением 265 мг целевого соединения.
1H-ЯМР (CDC13, 300 МГц): δ 1,27-1,47 (2H, м, CH2), 1,50-1,75 (4H, м, CH2), 2,13-2,26 (2H, м, COCH2), 2,52-2,74 (5H, м, NCH2, SCH2), 2,79-2,88 (1H, м, SCH2), 3,01-3,12 (1H, м, SCH), 3,43-3,65 (4H, м, NCH2CH2N), 3,80-4,02 (9H, м, NCH2Py, CHO), 4,22-4,28 (1H, м, NCH), 4,42-4,49 (1H, м, NCH), 5,83 (1H, шир.с, NHCO), 6,60 (1H, шир.с, NHCO), 7,07-7,18 (3H, м, Py), 7,31-7,38 (3H, м, Py), 7,44 (1H, д, Py), 7,59 (3H, ддд, Py), 8,03 (1H, дд, Py), 8,15 (1H, шир.с, NHCO), 8,42-8,58 (3H, м, Py), 8,94 (1H, д, Py).
Пример получения 2-2: Раствор, содержащий комплекс цинка по настоящему изобретению
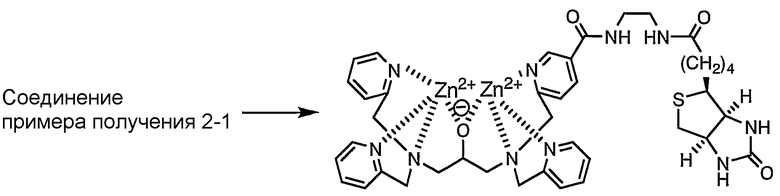
Соединение, полученное в примере получения 2-1, растворяли в фосфорнокислом буфере (pH=6,86) с получением 3 мМ раствора, затем к раствору добавляли 2 эквивалента нитрата цинка.
Комплекс цинка по настоящему изобретению в растворе показывал следующую структуру, определенную при помощи масс-спектрометра MALDI-TOF (матрица: 2',4',6'-тригидроксиацетофенон).
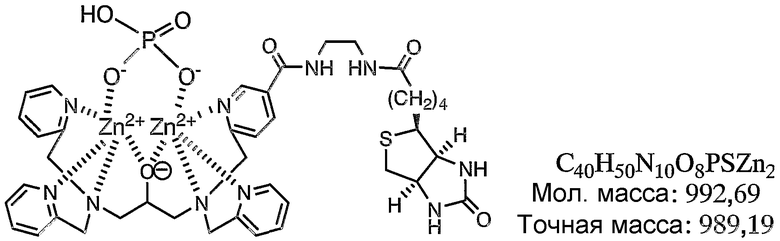
Результаты измерений, полученные при помощи масс-спектрометра MALDI-TOF, представлены на фиг.3. Как показано на фиг.3, наблюдали молекулярный ионный пик при 989,6 (точная масса: 989,19).
Экспериментальный пример 1: Электрофорез в 10% нативном полиакриламидном геле
Сначала получали гели для электрофореза, pH буфер для электрофореза и окрашивающий раствор для растворения образцов в указанных ниже условиях.
Концентрирующий гель:
125 мМ буфера Трис-хлористоводородная кислота (pH=6,8)
4,5% (масс./об.) полиакриламида (акриламид:бисакриламид = 30:1)
Разделяющий гель:
375 мМ буфера Трис-хлористоводородная кислота (pH=8,8)
10% (масс./об.) полиакриламидного (акриламид:бисакриламид = 30:1)
pH буфера для электрофореза (pH=8,3):
25 мМ Трис
190 мМ глицина
Окрашивающий раствор для растворения образцов (3×концентрированный раствор):
195 мМ буфера Трис-хлористоводородная кислота (pH=6,8)
10% (масс./об.) глицерина
0,1% (масс./об.) бромфенолового синего (BPB), используемого в электрофорезе в качестве окрашивающего маркера
Затем 2 мкг каждого из 1: бычьего сывороточного альбумина, 2: β-казеина (фосфорилированный), и 3: β-казеина (дефосфорилированный) растворяли в окрашивающем растворе для получения образцов. Соответствующие образцы наносили на гель. Затем сообщали постоянный электрический ток 4 мА до тех пор, пока окрашивающий маркер не вытек.
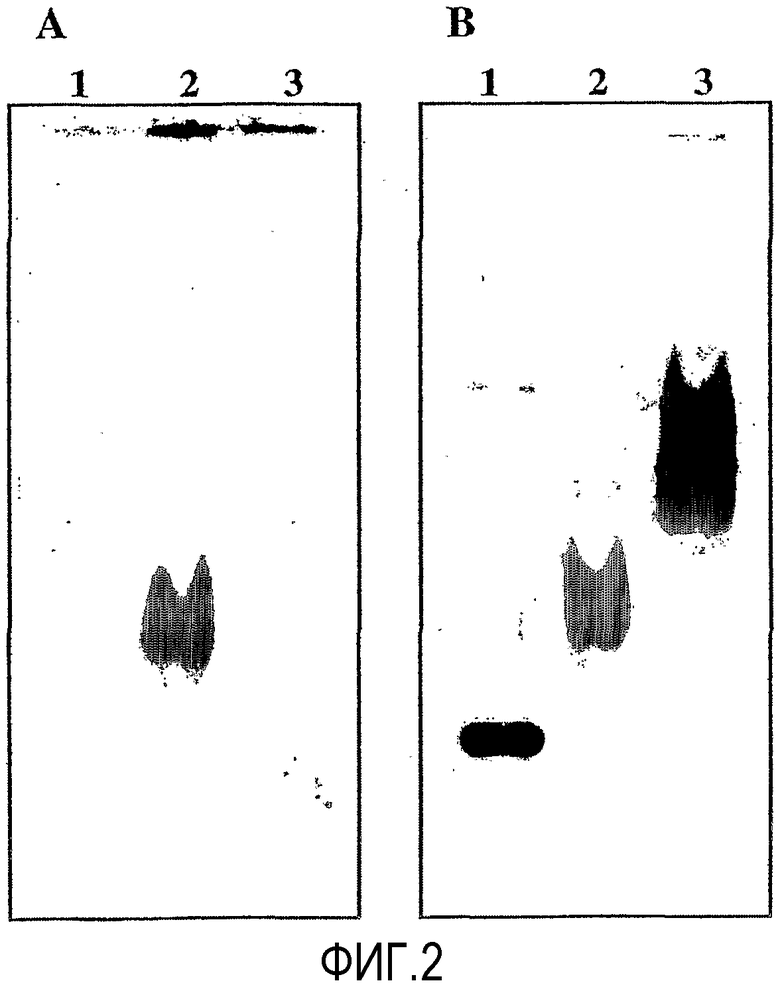
Гель погружали в раствор, содержащий комплекс цинка (50 мкМ), полученный в примере получения 1-6, примерно на 30 минут. Затем гель извлекали из раствора и фотографировали при облучении УФ-лампой. Затем гель окрашивали кумасси бриллиантовым голубым в соответствии с традиционным способом окрашивания и окрашенный гель фотографировали. Гель, окрашенный раствором, содержащим комплекс цинка, указан как "гель A", а гель, окрашенный кумасси бриллиантовым голубым, указан как "гель B", оба эти геля показаны на фиг.2.
Как видно из фиг.2, в соответствии со способом по настоящему изобретению β-казеин, связанный с фосфорной кислотой, может быть отдельно идентифицирован. Таким образом, очевидно, что способ по настоящему изобретению является выгодным для идентификации исключительно только фосфорилированных пептидов в образцах, полученных из живых организмов.
Пример получения 3-1: N,N,N'-три(2-пиридилметил)-N'-[5-N"- 2-(6-D-биотинамидогексакарбоксиамидоэтил)карбамоил-2-пиридилметил]-1,3-диаминопропан-2-ол
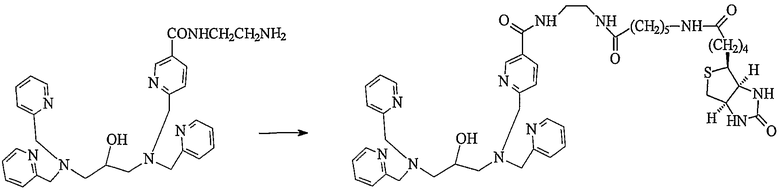
К раствору N,N,N'-три(2-пиридилметил)-N'-[5-N"-(2-аминоэтил)карбамоил-2-пиридилметил]-1,3-диаминопропан-2-ола (113 мг, 0,21 ммоль), полученного в примере получения 1-4, в ацетонитриле (10 мл) добавляли по каплям раствор 5-(N-сукцинимидилоксикарбонил)пентил-D-биотинамида (95 мг, 0,21 ммоль) в диметилсульфоксиде (2 мл).
После взаимодействия смеси при комнатной температуре в течение 6 часов реакционную смесь концентрировали и неочищенный продукт, полученный в результате концентрации, очищали колоночной хроматографией на силикагеле, с получением 136 мг целевого соединения (выход: 76%).
1H-ЯМР (CDCl3, 500 МГц): δ 1,25-1,32 (2H, м, CH2), 1,38-1,47 (4H, м, CH2), 1,56-1,67 (5H, м, CH2), 1,68-1,77 (1H, м, CH2), 2,11-2,22 (4H, м, COCH2), 2,58 (2H, дд, J=8,0 и 13,3 Гц, NCH), 2,66 (1H, дд, J=3,9 и 13,3 Гц,NCH), 2,68 (1H, дд, J=3,9 и 13,3 Гц, NCH),2,71 (1H, дд, J=3,0 и 13,3 Гц, SCH),2,88 (1H, дд, J=5,2 и 13,0 Гц, SCH), 3,07-3,21 (3H, м, NCH, SCH), 3,45-3,59 (4H, м, NCH2), 3,82-3,91 (8H, м, NCH2Py), 3,94 (1H, тт, J=3,9 и 8,0 Гц, OCH), 4,28-4,32 (1H, м, NCH), 4,46-4,50 (1H, м, NCH), 5,62 (1H, шир.с, NHCO), 6,40 (1H, шир.с, NHCO), 6,62 (1H, т, J=5,7 Гц, NHCO), 7,10-7,14 (3H, м, Py), 7,23 (1H, т, J=6,0 Гц, NHCO), 7,32-7,36 (3H, м, Py), 7,43 (1H, д, J=8,2 Гц, Py), 7,56-7,61 (3H, м, Py), 8,06 (1H, дд, J=2,3 и 8,2 Гц, Py), 8,17 (1H, т, J=5,0 Гц, NHCO), 8,46-8,50 (3H, м, Py), 8,96 (1H, д, J=2,3 Гц, Py).
13C-ЯМР (CDCl3, 125 МГц): δ 25,0 (CH2), 25,5 (CH2), 26,2 (CH2), 27,7 (CH2), 27,8 (CH2), 29,0 (CH2), 35,6 (CH2CO), 36,1 (CH2CO), 39,0 (CH2NH), 39,3 (CH2NH), 40,7 (CH2S), 41,0 (CH2NH), 55,6 (CH2S), 59,2 (CH2N), 60,2 (CHNH), 60,7 (CH2Py), 60,8 (CH2Py), 61,0 (CH2Py), 61,8 (CHNH), 67,4 (CHOH), 122,1 (Py), 122,8 (Py), 123,2 (Py), 128,5 (Py), 135,6 (Py), 136,5 (Py), 148,0 (Py), 149,0 (Py), 159,3 (Py), 159,4 (Py), 162,6 (Py), 163,9 (NCON), 166,3 (CONH), 173,4 (COM), 174,9 (CONH).
Пример получения 3-2: Раствор, содержащий комплекс цинка по настоящему изобретению
В фосфорнокислотном буфере (pH=6,86) растворяли 200 нмоль соединения, полученного в примере получения 3-1, для получения 3 мМ раствора, содержащего соединение, затем к раствору добавляли 2 эквивалента нитрата цинка. Таким образом, получали раствор, содержащий комплекс цинка по настоящему изобретению.
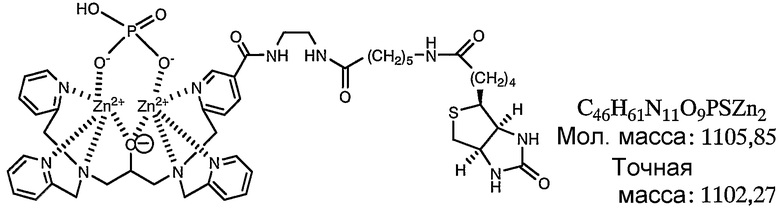
Раствор анализировали при помощи масс-спектрометра MALDI-TOF (матрица: 2',4',6'-тригидроксиацетофенон) и идентифицировали комплекс цинка. Результаты измерений, полученные при помощи масс-спектрометра MALDI-TOF, представлены на фиг.4. Как показано на фиг.4, наблюдали молекулярный ионный пик при 1102,3 (точная масса: 1102,27).
Экспериментальный пример 2
Суспензию (0,3 мл), содержащую стрептавидинагарозу (количество связанной группы биотина: 60-120 нмоль/мл) и буфер (Sigma-Aldrich Co.), загружали в центрифужную фильтровальную установку (объем: 0,5 мл, диаметр пор фильтра: 0,22 мкм). Фильтровальную установку подвергали центрифугированию для осуществления разделения при центрифужном ускорении 2000g в течение 15 секунд, после чего суспензию фильтровали. В фильтровальную установку загружали 5,0 мМ Трис-ацетатного буфера (pH=7,4, 0,30 мл), затем осуществляли разделение, подвергая фильтровальную установку центрифугированию при центрифужном ускорении 2000g в течение 15 секунд, и фильтрацию для промывки. Такую стадию центрифужного разделения и фильтрации для промывки повторяли 5 раз.
В фильтровальную установку загружали 5,0 мМ Трис-ацетатного буфера (pH=7,4, 0,30 мл) с растворенными в нем 120 нмоль/мл соединения, полученного в примере получения 3-1, и 500 нмоль/мл ацетата цинка. Фильтровальную установку приводили в состояние равновесия в течение 5 минут и затем осуществляли разделение центрифугированием при 2000g в течение 15 секунд и фильтрацию. Затем в фильтровальную установку загружали 5,0 мМ Трис-ацетатного буфера (pH=7,4, 0,30 мл) и осуществляли центрифужное разделение при 2000g в течение 15 секунд и фильтрацию для промывки. Стадию центрифужного разделения и фильтрации для промывки циклически повторяли 5 раз. Затем в фильтровальную установку загружали 5,0 мМ Трис-ацетатного буфера (pH=7,4, 0,30 мл), в котором было растворено 10 нмоль/мл ацетата цинка, и осуществляли центрифужное разделение при 2000g в течение 15 секунд и фильтрацию для промывки. Стадию центрифужного разделения и фильтрации для промывки циклически повторяли 5 раз.
В фильтровальную установку загружали 5,0 мМ Трис-ацетатного буфера (pH=7,4, 0,30 мл) с растворенным в нем нефосфорилированным пептидом p60c (p60c-src пептид 521-533, 14 нмоль/мл) и фосфорилированным пептидом P-p60c (O-фосфорил p60c-src пептид 521-533, 12 нмоль/мл), в качестве образцов. Фильтровальную установку приводили в состояние равновесия в течение 5 минут и затем осуществляли разделение центрифугированием при 2000g в течение 15 секунд и фильтрацию. Полученный фильтрат извлекали как фракцию 1.
Затем в фильтровальную установку загружали 5,0 мМ Трис-ацетатного буфера (pH=7,4, 0,50 M NaNO3, 0,30 мл) и осуществляли центрифужное разделение при 2000g в течение 15 секунд и фильтрацию (3 раза). Фильтрат, полученный после первой фильтрации, извлекали как фракцию 2, фильтрат после второй фильтрации извлекали как фракцию 3, и фильтрат после третьей фильтрации извлекали как фракцию 4.
Кроме того, в фильтровальную установку загружали 1,0 мМ буфера, содержащего фосфорную кислоту-гидроксид натрия (pH=7,4, 0,50 M NaNO3, 0,30 мл), и осуществляли центрифужное разделение при 2000g в течение 15 секунд и фильтрацию (2 раза). Фильтрат, полученный после первой фильтрации, извлекали как фракцию 5, и фильтрат после второй фильтрацию извлекали как фракцию 6.
Количество пептида в соответствующих фракциях 1-6 определяли методом высокоэффективной жидкостной хроматографии (ВЭЖХ). Показатели выделения/извлечения соответствующих пептидов относительно общего количества представлены в таблице 1.
Условия анализа ВЭЖХ:
колонка: Capcell Pak C18 type UG80, 4,6 мм (диаметр)
подвижная фаза: ацетонитрил:вода = 14:86 (об./об.), 0,1% (об./об.) трифторуксусной кислоты
скорость потока: 1 мл/мин
температура колонки: 40°С
способ детекции: УФ 266 нм
время удерживания: P-p60c (5,3 минуты), p60c (13,4 минуты)
Представленные выше результаты показывают, что комплексное соединение по настоящему изобретению может селективно адсорбировать фосфорилированный пептид, и что использование комплексного соединения по настоящему изобретению является выгодным для селективного выделения фосфорилированного пептида в смешанном образце фосфорилированного пептида и нефосфорилированного пептида.
Сравнительный пример 1
Суспензию (0,3 мл), содержащую стрептавидинагарозу (количество связанной группы биотина: 60-120 нмоль/мл) и буфер (Sigma-Aldrich Co.), загружали в центрифужную фильтровальную установку (объем: 0,5 мл, диаметр пор фильтра: 0,22 мкм). Фильтровальную установку подвергали центрифужному разделению при 2000g в течение 15 секунд, затем суспензию фильтровали. Затем в фильтровальную установку загружали 5,0 мМ Трис-ацетатного буфера (pH=7,4, 0,30 мл) и осуществляли центрифужное разделение при 2000g в течение 15 секунд и фильтрацию для промывки. Стадию центрифужного разделения и фильтрации для промывки циклически повторяли 5 раз.
Осуществляли способ, аналогичный способу экспериментального примера 2, с использованием образцов, эквивалентных тем, которые использовали в экспериментальном примере 2, используя фильтровальную установку (в этом сравнительном примере не использовали комплексное соединение по настоящему изобретению). Таким образом, получали фракции 7-12.
ВЭЖХ для анализа фракций 7-12 осуществляли таким же способом, как и в экспериментальном примере 2. Результаты анализа представлены в таблице 2.
Результаты, представленные в таблице 2, показывают, что отделение фосфорилированного пептида от нефосфорилированного пептида не было успешным, даже при осуществлении способа, аналогичного способу экспериментального примера 2, поскольку не было использовано комплексное соединение по настоящему изобретению.
Пример получения 4-1: 2-ацетоксиметил-4-нитропиридин
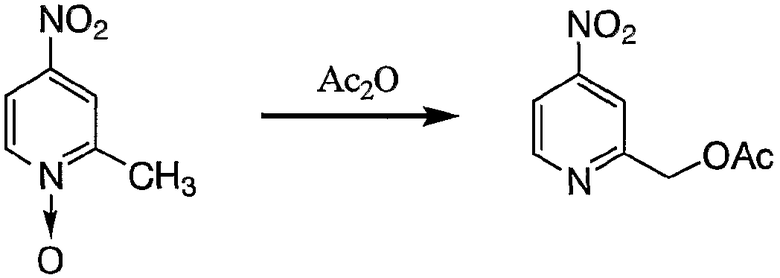
200 мл уксусного ангидрида нагревали до 100°С, после чего постепенно добавляли 2-метил-4-нитропиридин N-оксид (25,0 г, 162 ммоль). После добавления смесь постепенно нагревали и поддерживали при 130°С в течение 20 минут. Затем реакционную смесь охлаждали до 80°С, после чего добавляли по каплям 200 мл этанола для остановки реакции.
Затем реакционную смесь концентрировали, остаток переносили в воду (500 мл) и pH смеси доводили до 9 путем добавления гидрокарбоната натрия. После экстракции этилацетатом 2 раза (500 мл для первой экстракции и 200 мл для второй экстракции) полученные органические слои промывали 200 мл воды и 200 мл насыщенного солевого раствора. После концентрации органического слоя полученный неочищенный продукт очищали колоночной хроматографией на силикагеле с получением 7,68 г целевого соединения.
1H-ЯМР (CDCl3, 300 МГц): δ 2,23 (3H, с, COCH3), 3,36 (2H, с, CH2Py), 7,97 (1H, дд, Py), 8,07 (1H, д, Py), 8,90 (1H, д, Py).
Пример получения 4-2: 2-гидроксиметил-4-нитропиридин
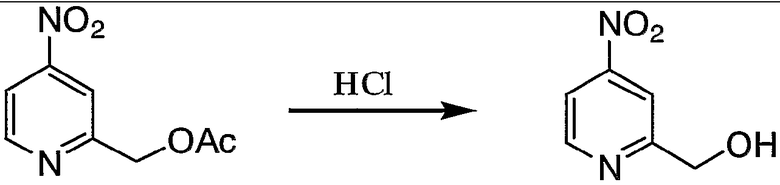
К 2-ацетоксиметил-4-нитропиридину (7,68 г, 39,2 ммоль), полученному в примере получения 4-1, добавляли 30 мл 10% раствора хлористоводородной кислоты. Реакцию смеси осуществляли при 50°С в течение 30 минут. После охлаждения реакционной смеси раствор выливали в 200 мл воды и pH смеси доводили до 9 путем добавления гидрокарбоната натрия. Смесь экстрагировали 100 мл этилацетата (три раза) и полученный органические слои концентрировали. Остаток, полученный в результате концентрации, очищали колоночной хроматографией на силикагеле с получением 4,70 г целевого соединения.
1H-ЯМР (CDCl3, 300 МГц): δ 3,36 (1H, т, OH), 4,94 (2H, д, CH2Py), 7,95 (1H, ддт, Py), 8,09 (1H, дт, Py), 8,86 (1H, д, Py).
Пример получения 4-3: 2-бромметил-4-нитропиридин
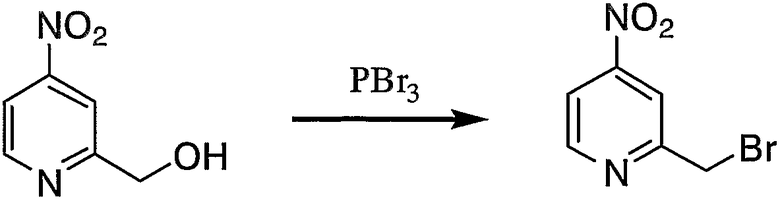
К суспензии 2-гидроксиметил-4-нитропиридина (1,00 г, 6,49 ммоль), полученного в примере получения 4-2, в безводном простом эфире (40 мл) добавляли по каплям трибромид фосфора (0,88 г, 3,24 ммоль) при 0°С. После завершения добавления по каплям реакцию смеси осуществляли при 0°С в течение 2 часов, после чего реакцию осуществляли при 20°С в течение 64 часов.
Реакционную смесь выливали в 250 мл охлажденной льдом воды и pH водного слоя доводили до 8 путем добавления гидрокарбоната натрия. Затем эфирный слой и водный слой разделяли, водный слой дважды экстрагировали 100 мл этилацетата. Органические слои собирали и сушили над безводным сульфатом натрия. Затем растворитель отгоняли, неочищенный продукт очищали колоночной хроматографией на силикагеле с получением 0,80 г целевого соединения.
1H-ЯМР (CDCl3, 300 МГц): δ 4,66 (2H, с, CH2Py), 7,96 (1H, дд, Py), 8,19 (1H, дд, Py), 8,88 (1H, дд, Py).
Пример получения 4-4: N,N,N'-три(2-пиридилметил)-N'-(4-нитро-2-пиридилметил)-1,3-диаминопропан-2-ол
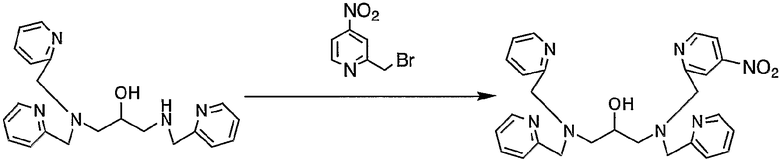
К раствору N,N,N'-три(2-пиридилметил)-1,3-диаминопропан-2-ола (2,65 г, 7,28 ммоль), полученного в примере получения 1-2, в безводном диметилформамиде (40 мл) добавляли безводный карбонат калия (2,02 г, 14,6 ммоль) и смесь нагревали до 55°С. Затем добавляли по каплям раствор 2-бромметил-4-нитропиридина (1,58 г, 7,28 ммоль), полученного в примере получения 4-3, в безводном диметилформамиде (20 мл) при той же температуре, т.е. 55°С. После чего реакцию смеси осуществляли при 55°С в течение 1,5 часа и смесь охлаждали.
Реакционную смесь выливали в 200 мл воды и pH смеси доводили до 8 путем добавления 1 н. раствора хлористоводородной кислоты. После экстракции 400 мл этилацетата (3 раза) органический слой промывали 250 мл насыщенного солевого раствора. После сушки над безводным сульфатом магния органический слой концентрировали. Затем отгоняли растворитель, неочищенный продукт очищали колоночной хроматографией на силикагеле с получением 3,30 г целевого соединения.
1H-ЯМР (CDCl3, 300 МГц): δ 2,60-2,78 (4H, м, NCH2), 3,84-3,96 (6H, м, CH2Py), 3,96-4,03 (1H, м, OCH), 4,07 (2H, с, CH2Py), 7,09-7,14 (3H, м, Py), 7,34 (3H, т, Py), 7,55-7,62 (3H, м, Py), 7,80 (1H, дд, Py), 8,22 (1H, д, Py), 8,49-8,52 (3H, м, Py), 8,76 (1H, дд, Py).
Пример получения 4-5: N,N,N'-три(2-пиридилметил)-N'-(4-азид- 2-пиридилметил)-1,3-диаминопропан-2-ол
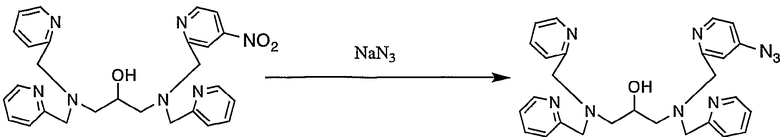
К раствору N,N,N'-три(2-пиридилметил)-N'-(4-нитро-2-пиридилметил)-1,3-диаминопропан-2-ола (1,01 г, 2,00 ммоль), полученного в примере получения 4-4, в безводном тетрагидрофуране (17 мл) добавляли по каплям при комнатной температуре азид натрия (156 мг, 2,40 ммоль) в воде (3,3 мл). Реакционную смесь нагревали до 50°С и подвергали взаимодействию при той же температуре, т.е. 50°С, в течение 17 часов.
Затем смесь охлаждали, водный слой дважды экстрагировали 10 мл хлороформа. Экстракты собирали и концентрировали и полученный неочищенный продукт очищали колоночной хроматографией на силикагеле с получением 980 мг целевого соединения.
1H-ЯМР (CDCl3, 300 МГц): δ 2,58-2,74 (4H, м, NCH2), 3,83-3,92 (8H, м, CH2Py), 3,92-4,01 (1H, м, OCH), 6,74 (1H, дд, Py), 7,09-7,14 (3H, м, Py), 7,20 (1H, д, Py), 7,32-7,36 (3H, м, Py), 7,55-7,60 (3H, м, Py), 8,39 (1H, д, Py), 8,49-8,52 (3H, м, Py).
Пример получения 4-6: N,N,N'-три(2-пиридилметил)-N'-(4-амино-2-пиридилметил)-1,3-диаминопропан-2-ол
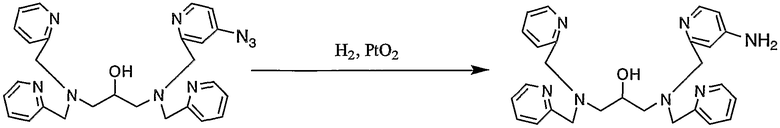
К раствору N,N,N'-три(2-пиридилметил)-N'-(4-азид-2-пиридилметил)-1,3-диаминопропан-2-ола (980 мг, 1,98 ммоль), полученного в примере получения 4-5, в этаноле (20 мл) добавляли 100 мг оксида платины. Реакцию смеси осуществляли при давлении водорода 30 кПа в течение 2 часов. Затем катализатор (оксид платины) удаляли фильтрованием, фильтрат концентрировали и полученный неочищенный продукт очищали колоночной хроматографией на силикагеле с получением 690 мг целевого соединения.
1H-ЯМР (CDCl3, 300 МГц): δ 2,43-2,71 (4H, м, NCH2), 3,66-3,92 (8H, м, CH2Py), 3,92-4,01 (1H, м, OCH), 6,35 (1H, дд, Py), 6,63 (1H, д, Py), 7,08-7,14 (3H, м, Py), 7,27-7,39 (3H, м, Py), 7,54-7,62 (3H, м, Py), 8,10 (1H, д, Py), 8,49-8,50 (3H, м, Py).
Пример получения 4-7: N,N,N'-три(2-пиридилметил)-N'-(4-D- биотинамидо-2-пиридилметил)-1,3-диаминопропан-2-ол
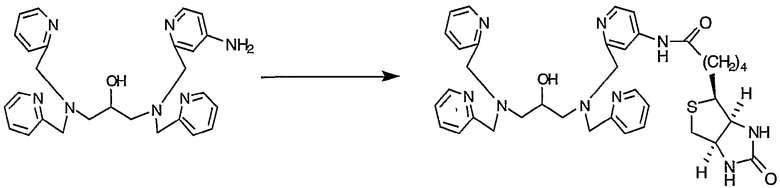
62,4 мг D-биотина (0,26 ммоль) суспендировали в 5 мл дихлорметана, затем добавляли при комнатной температуре 5,2 мг 4-диметиламинопиридина (0,04 ммоль) и 35,8 мкл триэтиламина (0,26 ммоль). К полученной смеси добавляли по каплям при комнатной температуре раствор N,N,N'-три(2-пиридилметил)-N'-(4-амино-2-пиридилметил)-1,3-диаминопропан-2-ола (100 мг, 0,21 ммоль), полученного в примере получения 4-6, в дихлорметане (2 мл). После завершения добавления по каплям добавляли при комнатной температуре гидрохлорид 1-этил-3-(3-диметиламинопропил)карбодиимида (49,0 мг, 0,26 ммоль). Реакционную смесь нагревали и реакцию осуществляли при кипячении с обратным холодильником в течение 5 часов. К реакционной смеси добавляли D-биотин (6,0 мг, 0,02 ммоль), гидрохлорид 1-этил-3-(3-диметиламинопропил)карбодиимида (10,0 мг, 0,05 ммоль) и 2 мл дихлорметана, после чего реакцию осуществляли при кипячении с обратным холодильником в течение 6 часов.
Затем реакционную смесь охлаждали и выливали в 40 мл воды. После экстракции 30 мл хлороформа (3 раза) органические слои промывали 30 мл воды (два раза) и 30 мл насыщенного солевого раствора. Затем органический слой сушили над безводным сульфатом магния и концентрировали, полученный неочищенный продукт очищали ВЭЖХ с получением 62,5 мг целевого соединения.
1H-ЯМР (CDCl3, 300 МГц): δ 1,37-1,45 (2H, м, CH2), 1,53-1,71 (4H, м, CH2), 2,19-2,25 (2H, м, COCH2), 2,62-2,75 (5H, м, NCH2, SCH2), 2,87-2,93 (1H, дд, SCH2), 3,09-3,15 (1H, м, SCH), 3,67-3,80 (9H, м, OCH, NCH2Py), 4,28-4,32 (1H, м, NCH), 4,48-4,52 (1H, м, NCH), 5,15-5,40 (2H, м, NHCO), 5,89 (1H, шир.с, NHCO) 6,48-6,50 (1H, м, Py), 6,72-6,76 (1H, м, Py), 7,13-7,17 (3H, м, Py), 7,27-7,29 (1H, м, Py), 7,37 (2H, д, Py), 7,56-7,66 (3H, м, Py), 8,00 (lH, дд, Py), 8,49-8,50 (3H, м, Py).
Это соединение анализировали при помощи масс-спектрометра MALDI-TOF с использованием α-циано-4-гидроксикоричной кислоты (CHCA) в качестве матрицы. Результат измерений представлен на фиг.5. Как видно из фиг.5, наблюдали молекулярный ионный пик 696,2 иона протонного аддукта (M++1).
Пример получения 5-1: Гидрохлорид метил 6-аминогексаноата
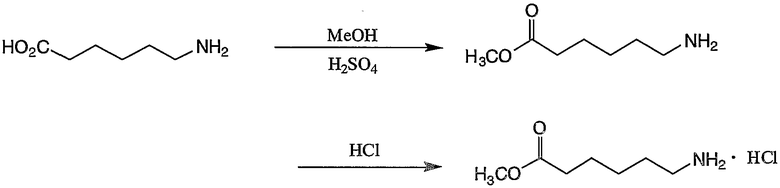
К суспензии 6-аминогексановой кислоты (10,0 г, 76,2 ммоль) в метаноле (300 мл) добавляли 5,51 г концентрированной серной кислоты. Смесь нагревали и осуществляли ее взаимодействие в течение 8 часов, отгоняя при этом воду, образуемую в ходе реакции этерификации, при помощи метанола, и 60 мл метанола добавляли поочередно четыре раза. Реакционную смесь охлаждали и pH смеси доводили до 6,2 путем добавления 28% раствора метилата натрия в метаноле. Растворитель отгоняли, остаток переносили в 500 мл воды и pH смеси доводили до 10,3 путем добавления карбоната натрия. После экстракции 350 мл дихлорметана (3 раза) экстракт сушили над безводным сульфатом магния. Затем отгоняли растворитель с получением метил 6-аминогексаноата.
Полученный метил 6-аминогексаноат растворяли в 40 мл диоксана, затем добавляли 7,1 мл 4-н-гидрохлоридного раствора диоксана. После этого смесь перемешивали при комнатной температуре в течение 30 минут, растворитель отгоняли, затем к остатку добавляли простой эфир. Осажденные кристаллы выделяли фильтрованием и сушили при пониженном давлении с получением 3,3 г целевого соединения.
1H-ЯМР (ДМСО-d6, 300 МГц): δ 1,26-1,36 (2H, м, CH2), 1,48-1,61 (4H, м, CH2), 2,31 (2H, т, CH2CO), 2,73 (2H, т, CH2N), 3,59 (3H, с, CH3), 8,1 (3H, шир.с, NH3).
Пример получения 5-2: 5-карбометоксипентил-D-биотинамид
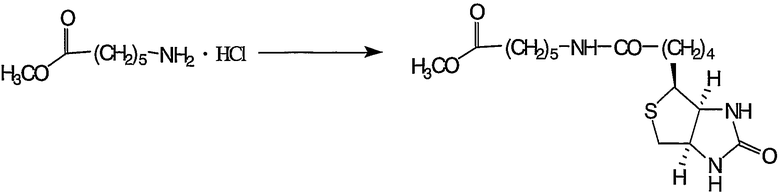
К раствору гидрохлорида метил 6-аминогексаноата (1,00 г, 5,50 ммоль), полученного в примере получения 5-1, в диметилформамиде (40 мл) добавляли триэтиламин (1,20 г, 11,8 ммоль) и 4-диметиламинопиридин (137 мг, 1,1 ммоль). Кроме того, добавляли D-биотин (1,34 г, 5,50 ммоль) и гидрохлорид 1-этил-3-(3-диметиламинопропил)карбодиимида (1,42 г, 7,42 ммоль). Реакцию смеси осуществляли при температуре от 35 до 45°С в течение 23 часов.
Реакционную смесь выливали в 300 мл воды и экстрагировали 150 мл хлороформа (5 раз). Затем экстракты сушили над безводным сульфатом магния и растворитель отгоняли. Полученный неочищенный продукт очищали колоночной хроматографией на силикагеле с получением 1,04 г целевого соединения.
1H-ЯМР (ДМСО-d6, 300 МГц): δ 1,18-1,65 (12H, м, CH2), 2,04 (2H, т, CH2CO), 2,28 (2H, т, CH2CO), 2,57 (1H, д, CH2S), 2,82 (1H, дд, CH2S), 3,00 (1H, дт, NHCO), 3,06-3,12 (1H, м, CH2S), 3,58 (3H, с, OCH3), 4,09-4,15 (1H, м, CHN), 4,28-4,32 (1H, м, CHN), 6,35 (1H, шир.с, NHCO), 6,41 (1H, шир.с, NHCO), 7,72 (1H, т, NHCO).
Пример получения 5-3: 5-карбоксипентил-D-биотинамид
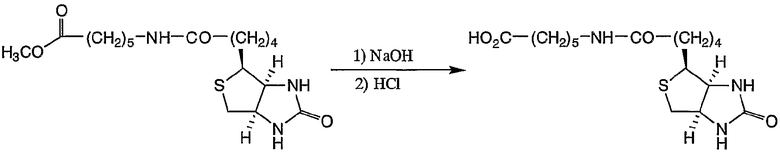
В смеси 25 мл тетрагидрофурана и 25 мл метанола растворяли 5-карбометоксипентил-D-биотинамид (800 мг, 2,15 ммоль), полученный в примере получения 5-2, затем добавляли по каплям при комнатной температуре раствор гидроксида натрия (2,33 г, 58,3 ммоль) в воде (12,5 мл). По завершении добавления смесь нагревали до 40°С и реакцию осуществляли в течение 2 часов.
После охлаждения реакционную смесь концентрировали, затем добавляли 60 мл воды. Затем добавляли 1 н. раствор хлористоводородной кислоты и pH смеси доводили до 2,0. Осажденные кристаллы выделяли фильтрованием и промывали водой. Полученные кристаллы сушили в вакууме с получением 724 мг целевого соединения.
1H-ЯМР (ДМСО-d6, 300 МГц): δ 1,17-1,67 (12H, м, CH2), 2,04 (2H, т, CH2CO), 2,19 (2H, т, CH2CO), 2,57 (1H, д, CH2S), 2,82 (1H, дд, CH2S), 3,00 (1H, дт, NHCO), 3,06-3,12 (1H, м, CH2S), 4,13 (1H, т, CHN), 4,30 (1H, т, CHN), 6,36 (1H, шир.с, NHCO), 6,43 (1H, шир.с, NHCO), 7,74 (1H, т, NHCO), 11,95 (1H, шир.с, CO2H).
Пример получения 5-4: N,N,N'-три(2-пиридилметил)-N'-[4-(6-D- биотинамидогексакарбоксиамидо)-2-пиридилметил]-1,3-диаминопропан-2-ол
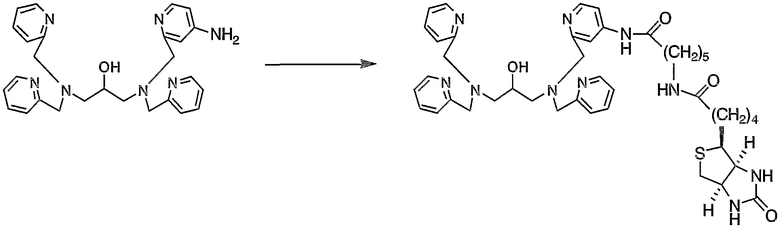
К раствору 5-карбоксипентил-D-биотинамида (99,0 мг, 0,27 ммоль), полученного в примере получения 5-3, в безводном диметилформамиде добавляли триэтиламин (38,1 мкл, 0,27 ммоль) и 4-диметиламинопиридин (5,7 мг, 0,04 ммоль), затем добавляли по каплям раствор N,N,N'-три(2-пиридилметил)-N'-(4-амино-2-пиридилметил)-1,3-диаминопропан-2-ола (100 мг, 0,21 ммоль), полученного в примере получения 4-6, в безводном диметилформамиде (1 мл). После этого добавляли гидрохлорид 1-этил-3-(3-диметиламинопропил)карбодиимида (51,7 мг, 0,27 ммоль). Реакцию смеси осуществляли при 40°С в течение 24 часов. К реакционной смеси добавляли 8,9 мкл триэтиламина (0,06 ммоль), 22,8 мг 5-карбоксипентил-D-биотинамида (0,06 ммоль) и 12,2 мг гидрохлорида 1-этил-3-(3-диметиламинопропил)карбодиимида (0,06 ммоль). Реакцию смеси осуществляли при 40°С в течение 6 часов.
После охлаждения реакционной смеси смесь выливали в 150 мл воды, затем добавляли хлорид натрия для насыщения раствора. После экстракции при помощи 40 мл хлороформа (5 раз) органические слои промывали 150 мл воды (дважды) и 150 мл насыщенного солевого раствора. Затем экстракт сушили над безводным сульфатом магния и отгоняли растворитель. Полученный неочищенный продукт очищали методом ВЭЖХ с получением 37,5 мг целевого соединения.
1H-ЯМР (CDCl3, 300 МГц): δ 1,30-1,80 (12H, м, CH2), 2,14-2,25 (4H, м, COCH2), 2,60-2,76 (5H, м, NCH2, CH2S), 2,87 (1H, дд, CH2S), 3,10-3,25 (3H, м, CH2NHCO, SCH), 3,68-3,82 (9H, м, NCH2Py, OCH), 4,28-4,33 (1H, м, NCH), 4,45-4,52 (1H, м, NCH), 5,27 (1H, шир.с, NHCO), 5,50 (1H, шир.с, NHCO), 5,60 (1H, шир.с, NHCO), 6,22 (1H, шир.с, NHCO), 6,52-6,57 (1H, м, Py), 6,72-6,75 (1H, м, Py), 7,16 (3H, т, Py), 7,27-7,30 (1H, м, Py), 7,37 (2H, д, Py), 7,57-7,66 (3H, м, Py), 7,99 (1H, д, Py), 8,46-8,52 (3H, м, Py).
Это соединение анализировали при помощи масс-спектрометра MALDI-TOF с использованием CHCA в качестве матрицы. Результат измерений представлен на фиг.6. Как видно из фиг.6, наблюдали молекулярный ионный пик 809,5 иона протонного аддукта (M++1).
Пример получения 6-1: 2-бромметил-3-гидроксипиридин
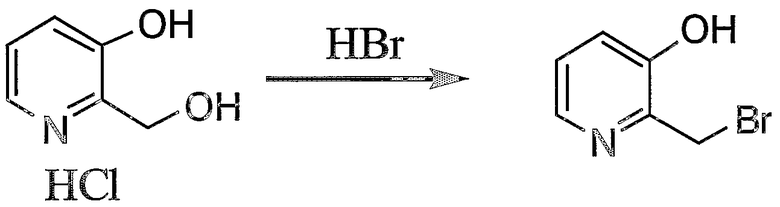
К 25% раствору бромистого водорода в уксусной кислоте (12,8 г, 39,6 ммоль) добавляли гидрохлорид 3-гидрокси-2-гидроксиметилпиридина (5,0 г, 54,7 ммоль). Реакцию смеси осуществляли при 110°С в течение 1 часа. Добавляли по каплям дополнительное количество 25% раствора бромистого водорода в уксусной кислоте (4,90 г, 15,1 ммоль) и реакцию смеси осуществляли в течение 2 часов.
После охлаждения реакционную смесь выливали в 50 мл воды и pH смеси доводили до 8 путем добавления насыщенного водного раствора гидрокарбоната натрия. После экстракции при помощи 50 мл дихлорметана (четыре раза) дихлорметановые слои концентрировали. Неочищенный продукт после концентрации очищали колоночной хроматографией на силикагеле с получением 1,36 г целевого соединения.
1H-ЯМР (CDCl3, 300 МГц): δ 5,31 (2H, с, CH2Br), 7,20-7,32 (2H, м, Py), 8,18-8,21 (1H, м, Py), 8,55 (1H, шир.с, OH).
Пример получения 6-2: N,N,N'-три(2-пиридилметил)-N'-(3- гидрокси-2-пиридилметил)-1,3-диаминопропан-2-ол
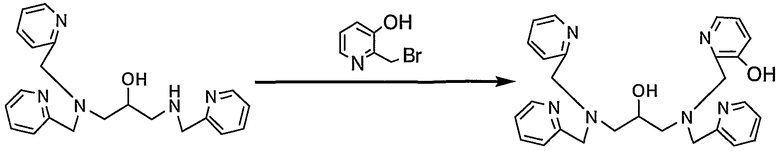
В диметилформамиде (15 мл) растворяли N,N,N'-три(2-пиридилметил)-1,3-диаминопропан-2-ол (1,00 г, 2,75 ммоль), полученный в примере получения 1-2, и 2-бромметил-3-гидроксипиридин (633 мг, 3,37 ммоль), полученный в примере получения 6-1, затем добавляли карбонат калия (760 мг, 5,50 ммоль). Реакцию смеси осуществляли при 50°С в течение 2 часов и смесь охлаждали.
Реакционную смесь выливали в 100 мл воды и pH смеси доводили до 8 путем добавления 1 н. раствора хлористоводородной кислоты. После экстракции 100 мл этилацетата (3 раза) органические слои промывали 100 мл воды и 100 мл насыщенного солевого раствора и сушили над безводным сульфатом натрия. Полученный неочищенный продукт очищали и собирали при помощи ВЭЖХ с получением 660 мг целевого соединения.
1H-ЯМР (CDCl3, 300 МГц): δ 2,51-2,69 (4H, м, NCH2), 3,71-4,08 (9H, м, CH2Py, OCH), 7,05-7,14 (4H, м, Py), 7,15-7,19 (1H, т, Py), 7,23-7,28 (3H, м, Py), 7,55 (2H, дт, Py), 7,64 (1H, дт, Py), 7,91 (1H, дд, Py), 8,48-8,55 (3H, м, Py).
Пример получения 6-3: N,N,N'-три(2-пиридилметил)-N'-(3-D- биотинилокси-2-пиридилметил)-1,3-диаминопропан-2-ол
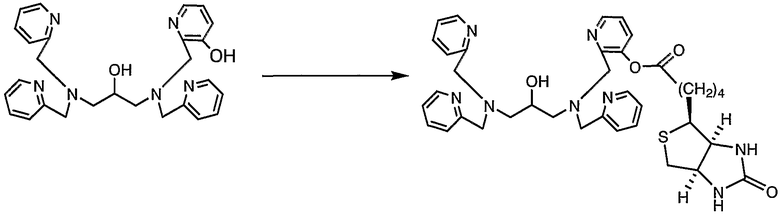
К раствору N,N,N'-три(2-пиридилметил)-N'-(3-гидрокси-2-пиридилметил)-1,3-диаминопропан-2-ола (100 мг, 0,21 ммоль), полученного в примере получения 6-2, в безводном диметилформамиде (5 мл) добавляли гидрид натрия (дисперсия в минеральном масле) (9,0 мг, 0,23 ммоль) при 3°С. После того как температура снова повышалась до комнатной, реакцию смеси осуществляли при комнатной температуре в течение 30 минут, затем добавляли по каплям раствор N-сукцинимидил D-биотината (79,8 мг, 0,23 ммоль) в безводном диметилсульфоксиде (1 мл).
После завершения добавления по каплям реакцию смеси осуществляли при комнатной температуре в течение 3,5 часов и смесь выливали в 100 мл воды. После экстракции 300 мл хлороформа (3 раза) хлороформные слои сушили над безводным сульфатом натрия. Затем растворитель отгоняли, неочищенный продукт собирали и очищали при помощи ВЭЖХ с получением 63,6 мг целевого соединения.
1H-ЯМР (CDCl3, 300 МГц): δ 1,43-1,56 (2H, м, CH2), 1,60-1,85 (4H, м, CH2), 2,53 (2H, т, COCH2), 2,58-2,68 (4H, м, NCH2), 2,71-2,77 (1H, м, SCH2), 2,91 (1H, дд, SCH2), 3,13-3,19 (1H, м, SCH), 3,76-3,99 (9H, м, OCH, NCH2Py), 4,28-4,36 (1H, м, NCH), 4,47-4,53 (1H, м, NCH), 5,16 (1H, шир.с, NHCO), 5,93 (1H, д, NHCO) 7,08-7,14 (3H, м, Py), 7,20 (1H, дд, Py), 7,33-7,42 (4H, м, Py), 7,52-7,62 (3H, м, Py), 8,42 (1H, дд, Py), 8,51 (3H, м, Py).
Пример получения 6-4: Раствор, содержащий комплекс цинка по настоящему изобретению
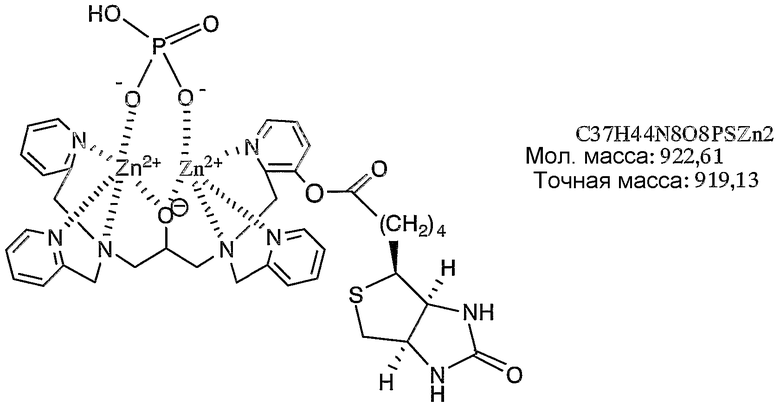
К раствору, содержащему 200 нмоль N,N,N'-три(2-пиридилметил)-N'-(3-D-биотинилокси-2-пиридилметил)-1,3-диаминопропан-2-ола, полученного в примере получения 6-3, в ацетонитриле (66,7 мкл) добавляли 40,1 мкл водного раствора нитрата цинка (100 мМ), затем добавляли 560 мкл фосфорнокислотного буфера (pH=6,86). Раствор анализировали при помощи масс-спектрометра с использованием в качестве матрицы 2',4'-6'-тригидроксиацетофенона (THAP) и идентифицировали комплекс цинка. Результаты измерений, полученные при помощи масс-спектрометра MALDI-TOF, представлены на фиг.7. Как показано на фиг.7, наблюдали молекулярный ионный пик при 919,1 (точная масса: 919,13).
Пример получения 7-1: N,N,N'-три(2-пиридилметил)-N'-[5-N"-(2-дансиламиноэтил)карбамоил-2-пиридилметил]-1,3-диаминопропан-2-ол
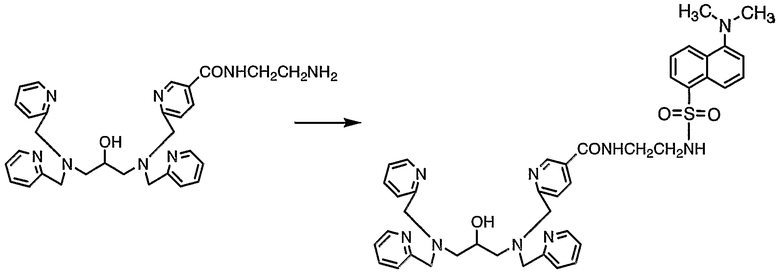
В безводном ацетонитриле (9 мл) растворяли N,N,N'-три(2-пиридилметил)-N'-[5-N"-2-(аминоэтил)карбамоил-2-пиридилметил]-1,3-диаминопропан-2-ол (1,1 г, 2,1 ммоль), полученный в примере получения 1-4, затем добавляли дансилхлорид (840 мг, 3,1 ммоль). Реакционную смесь нагревали и подвергали взаимодействию при 50°С в течение 3 часов. Затем реакционную смесь концентрировали, неочищенный продукт очищали колоночной хроматографией на силикагеле с получением 1,30 г целевого соединения.
lH-ЯМР (CDCl3, 500 МГц): δ 2,57 (1H, дд, CH2), 2,60 (1H, дд, CH2), 2,67 (1H, дд, CH2), 2,69 (1H, дд, CH2), 2,85 (6H, с, NCH3), 3,17 (2H, дд, NCH2CH2N), 3,50 (2H, дд, NCH2CH2N), 3,88 (8H, дд, NCH2Py), 3,83-3,92 (1H, м, OCH), 6,31 (1H, м, SO2NH), 7,11 (1H, д, Ar), 7,13 (3H, ддд, Py), 7,35 (3H, д, Py), 7,36 (1H, д, Py), 7,42 (1H, м, CONH), 7,46 (1H, дд, Ar), 7,49 (1H, дд, Ar), 7,60 (2H, дт, Py), 7,60 (1H, дт, Py), 7,90 (1H, дд, Py), 8,23 (1H, дд, Ar), 8,27 (1H, д, Ar), 8,49 (2H, ддд, Py), 8,50 (1H, ддд, Py), 8,51 (1H, дд, Ar), 8,80 (1H, д, Py).
13C-ЯМР (CDCl3, 125 МГц): 39,9 (NCH2CH2N), 42,7 (NCH2CH2N), 45,3 (NCH3), 59,0 (CH2), 59,1 (CH2), 60,6 (CH2Py), 60,7 (CH2Py), 61,3 (CH2Py), 67,l (CHO), 115,2 (Ar), 118,6 (Ar), 122,l (Py), 122,8 (Py), 123,2 (Ar), 123,2 (Py), 128,1 (Ar), 128,5 (Ar), 129,5 (Ar), 129,6 (Ar), 129,9 (Py), 130,6 (Ar), 134,5 (Ar), 135,4 (Py), 136,5 (Py), 136,6 (Py), 147,6 (Py), 148,9 (Py), 149,0 (Py), 152,0 (Ar), 159,1 (Py), 159,l (Py), 162,8 (Py), 166,2 (CONH).
Пример получения 7-2: Раствор, содержащий комплекс цинка по настоящему изобретению
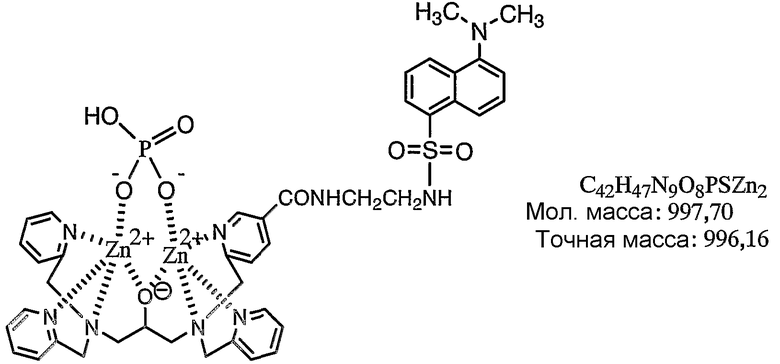
В фосфорнокислом буфере (pH=6,86) растворяли 200 нмоль N,N,N'-три(2-пиридилметил)-N'-[5-N"-(2-дансиламиноэтил)карбамоил-2-пиридилметил]-1,3-диаминопропан-2-ола, полученного в примере получения 7-1, с получением 3 мМ раствора, затем к раствору добавляли 2 эквивалента нитрата цинка. Раствор анализировали при помощи масс-спектрометра MALDI-TOF, используя THAP в качестве матрицы, и идентифицировали комплекс цинка. Результаты измерений, полученные при помощи масс-спектрометра MALDI-TOF, представлены на фиг.8. Как показано на фиг.8, наблюдали молекулярный ионный пик при 996,2 (точная масса: 996,16).
Пример получения 8-1: N,N,N'-три(2-пиридилметил)-N'-[5-N"-(2-N-5-флуоресцеинилтиоуреидоэтил)карбамоил-2-пиридилметил]-1,3-диаминопропан-2-ол
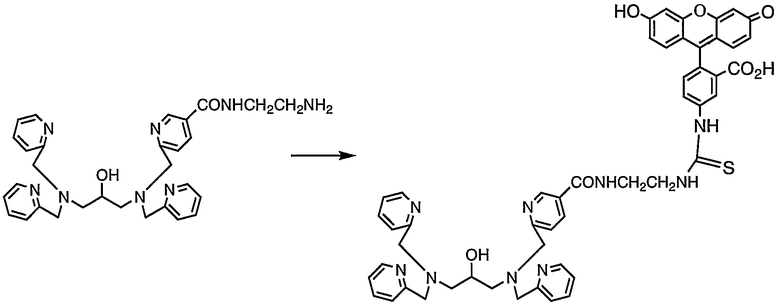
В безводном диметилформамиде (150 мл) растворяли N,N,N'-три(2-пиридилметил)-N'-[5-N"-(2-аминоэтил)карбамоил-2-пиридилметил]-1,3-диаминопропан-2-ол (950 мг, 1,76 ммоль), полученный в примере получения 1-4, затем добавляли безводный пиридин (38 мл). Затем добавляли по каплям раствор флуоресцеин-5-изотиоцианата (750 мг, 1,92 ммоль) в безводном диметилформамиде (150 мл) и реакцию смеси осуществляли при комнатной температуре в течение 3 часов. Затем реакционную смесь концентрировали, неочищенный продукт очищали колоночной хроматографией на силикагеле с получением 961 мг целевого соединения.
1H-ЯМР (ДМСО-d6, 300 МГц): δ 2,33-2,43 (2H, м, CH2), 2,50-2,61 (2H, м, CH2), 3,38-3,57 (4H, м, NCH2CH2N), 3,67-3,92 (9H, м, NCH2Py, CHO), 6,52-6,67 (6H, м, Ar), 7,16-7,23 (3H, м, Py), 7,17 (1H, д, Ar), 7,34-7,41 (3H, м, Py), 7,52 (1H, д, Py), 7,69 (3H, дт, Py), 7,68-7,77 (1H, м, Ar), 8,14 (1H, дд, Py), 8,24 (1H, д, Ar), 8,41-8,46 (3H, м, Py), 8,78 (1H, м, NHCO), 8,92 (1H, д, Py).
Экспериментальный пример 3: Электрофорез в 10% нативном полиакриламидном геле
Сначала получали гели для электрофореза, pH буфер для электрофореза и окрашивающий раствор для растворения образцов в тех же условиях, как описано в экспериментальном примере 1.
Затем по 2 мкг каждого из 1: бычьего сывороточного альбумина, 2: сывороточного альбумина человека, 3: карбонангидразы, 4: β-галактозидазы, 5: α-казеина (фосфорилированного), 6: α-казеина (дефосфорилированного), 7: β-казеина (фосфорилированного), 8: β-казеина (дефосфорилированного), 9: пепсина (фосфорилированного), и 10: пепсина (дефосфорилированного) растворяли в окрашивающем растворе для получения образцов. Соответствующие образцы наносили на гель. Затем сообщали постоянный электрический ток 40 мА до тех пор, пока окрашивающий маркер не вытек.
Полученный гель погружали на 30 минут в 10 мМ Трис-ацетатного буфера (pH=7,4), содержащего 1 мкМ N,N,N'-три(2-пиридилметил)-N'-[5-N"-(2-N-5-флуоресцеинилтиоуреидоэтил)карбамоил-2-пиридилметил]-1,3-диаминопропан-2-ола, полученного в примере получения 8-1, и 100 мкМ ацетата цинка. Затем гель извлекали из раствора. Этот гель дважды промывали 100 мл 10 мМ Трис-ацетатного буфера (pH=7,4) в течение 10 минут. Затем получали флуоресцентное изображение геля при помощи фотографии, используя анализатор флуоресцентного изображения FLA-5000 (Fuji Photo Film Co., Ltd.), при длине волны возбуждения 473 нм с использованием фильтра детекции флуоресценции 510 нм. Затем гель окрашивали кумасси бриллиантовым голубым в соответствии с традиционным способом окрашивания и окрашенный гель фотографировали. Гель, окрашенный раствором, содержащим комплекс цинка, указан как "гель A", а гель, окрашенный кумасси бриллиантовым голубым, указан как "гель B", оба эти геля показаны на фиг.9.
Как видно из фиг.9, в случае окрашивания традиционным способом все пептиды были окрашены, как показано на примере геля B. С другой стороны, только 5: α-казеин (фосфорилированный), 7: β-казеин (фосфорилированный), и 9: пепсин (фосфорилированный), все связанные с фосфорной кислотой, были идентифицированы каждый отдельно, как показано на примере геля A, который был обработан раствором, содержащим комплекс цинка по настоящему изобретению. Таким образом, очевидно, что способ по настоящему изобретению является выгодным для идентификации исключительно только фосфорилированных пептидов в образцах, полученных из живых организмов.
В основе настоящей заявки лежат Японские патентные заявки № 2003-56068, подана 3 марта 2003 г., № 2003-113707, подана 18 апреля 2003 г. и № 2003-356934, подана 16 октября 2003 г., содержание которых включено в настоящую заявку посредством ссылки.
Хотя настоящее изобретение было подробно описано при помощи примеров, следует понимать, что возможны различные изменения и модификации, очевидные для специалистов в данной области. Поэтому, если такие изменения и модификациине являются отступлением от объема настоящего изобретения, определенного далее, они должны рассматриваться как включенные в объем настоящего изобретения.
| название | год | авторы | номер документа |
|---|---|---|---|
| ГЛИКОЛИПИДНЫЕ СОЕДИНЕНИЯ И ИХ ПРИМЕНЕНИЕ В ЛЕЧЕНИИ ОПУХОЛЕЙ | 2015 |
|
RU2719486C2 |
| МАКРОГЕТЕРОЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ | 2001 |
|
RU2275373C2 |
| ЦИКЛИЧЕСКИЕ ПЕПТИДЫ, РАДИОФАРМАЦЕВТИЧЕСКИЙ ПРЕПАРАТ, ЦИКЛИЧЕСКИЕ ПЕПТИДЫ, ИМЕЮЩИЕ РАДИОАКТИВНУЮ МЕТКУ | 1994 |
|
RU2145608C1 |
| БЕНЗОПИРАНОВЫЕ И БЕНЗОКОНДЕНСИРОВАННЫЕ СОЕДИНЕНИЯ, ПРОМЕЖУТОЧНЫЕ СОЕДИНЕНИЯ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ И СПОСОБ ИНГИБИРОВАНИЯ | 1995 |
|
RU2128655C1 |
| ПЕПТИДНЫЕ СОЕДИНЕНИЯ | 2001 |
|
RU2281955C2 |
| НОВЫЕ ЦИКЛИЧЕСКИЕ ПЕПТИДНЫЕ СОЕДИНЕНИЯ | 2008 |
|
RU2436795C2 |
| НОВЫЕ СОЕДИНЕНИЯ | 2008 |
|
RU2480453C2 |
| НОВЫЕ ГЕМ-ДИФТОРИРОВАННЫЕ СОЕДИНЕНИЯ, СПОСОБЫ ИХ ПОЛУЧЕНИЯ, И ИХ ПРИМЕНЕНИЕ | 2003 |
|
RU2369612C2 |
| Бициклические гетероциклические соединения и их применения в терапии | 2012 |
|
RU2662827C2 |
| НОВОЕ СОЕДИНЕНИЕ, СПОСОБ ЕГО ПОЛУЧЕНИЯ И ЕГО ПРИМЕНЕНИЕ | 2021 |
|
RU2814103C1 |

Представлен способ мечения фосфорилированных пептидов, способ адсорбции фосфорилированных пептидов и соединения, обладающие высокой координационной способностью в отношении фосфорилированных пептидов, которые используются в таких способах, а также соединения, используемые в качестве исходных веществ. Комплексное соединение представлено формулой (I), где Х представляет собой линкерную группу и Y представляет собой флуоресцентную группу или биотин в качестве группы мечения. Соединение (I) обеспечивает легкую детекцию и идентификацию фосфорилированного пептида в образце, полученном из живого организма. 5 н. и 3 з.п. ф-лы, 2 табл., 9 ил.
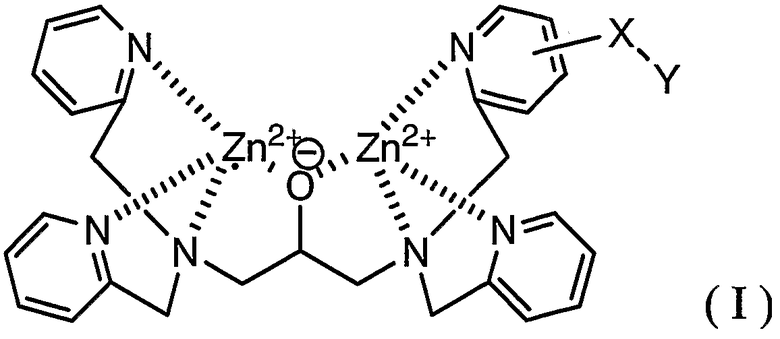
где Х представляет собой линкерную группу, и Y представляет собой флуоресцентную группу или биотин в качестве группы мечения.
где X представляет собой линкерную группу, и Y представляет собой флуоресцентную группу или биотин в качестве группы мечения.
где Х представляет собой линкерную группу, и Y представляет собой флуоресцентную группу или биотин в качестве группы мечения.
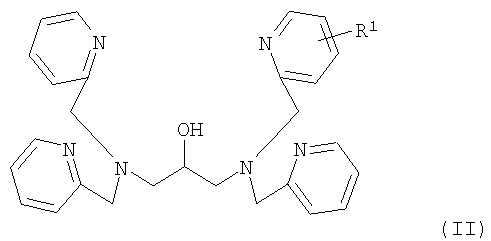
где R1 представляет собой (амино(С1-С6алкил)карбамоильную группу или гидроксигруппу.
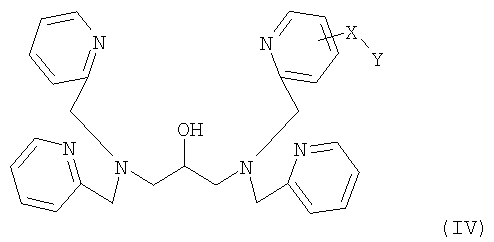
где X представляет линкерную группу, и Y представляет собой флуоресцентную группу или биотин в качестве группы мечения.
Приоритет по пунктам:
| Способ твердофазного иммуноферментного определения антител к вирусу иммунодефицита человека и меченный биотином синтетический пептид для его осуществления | 1989 |
|
SU1612264A1 |
| Adams H | |||
| et al | |||
| Печь для непрерывного получения сернистого натрия | 1921 |
|
SU1A1 |
| Chem | |||
| Soc., Dalton Trans., 2002, 925-930 | |||
| Yamaguchi K | |||
| et al | |||
| "Hydrolysis of phosphodiester with hydroxo- or carboxylate-bridged dinuclear Ni(II) and Cu(II) complexes, J | |||
| Chem | |||
| Commun., 2001, 375-376 | |||
| Yashiro | |||

